ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Техническая область
Данное изобретение относится к препарату фармацевтически активного соединения. Более конкретно, данное изобретение относится к производству наносуспензий фармацевтически активного соединения для парентеральной или оральной доставки.
Известный уровень техники
Существует все увеличивающееся число лекарственных веществ, которым нужно создать лекарственную форму и которые плохо растворимы или нерастворимы в водных растворах. Такие лекарственные вещества представляют проблемы в отношении их доставки в инъецируемой форме, такой как посредством парентерального введения. Лекарственные вещества, которые нерастворимы в воде, могут обладать значительными преимуществами, когда изготовлены в виде стабильных суспензий субмикронных частиц. Точный контроль размера частиц является существенным для безопасности и эффективного использования этих препаратов. Частицы должны быть менее семи микрон диаметром для безопасного прохода через капилляры, не вызывая эмболии (Allen et al., 1987; Davis and Taube, 1978; Schroeder et al., 1978; Yokel et al., 1981).
Один из подходов к доставке нерастворимого лекарственного вещества описан в патенте США № 2745785. В данном патенте описан способ получения кристаллов пенициллина G, пригодных для парентерального введения. Данный способ включает стадию перекристаллизации пенициллина G из раствора формамида путем добавления воды для снижения растворимости пенициллина G. B патенте '785 дополнительно раскрывается, что частицы пенициллина G могут быть покрыты увлажняющими веществами, такими как лецитин, или эмульгаторами, поверхностно-активными и пеноуничтожающими веществами, или неполными сложными эфирами высших жирных кислот и сорбитана или полиоксиалкилена и их производными, или арилалкилполиэфирами спиртов или их солями. В патенте '785 дополнительно описано микронизирование пенициллина G с помощью струи воздуха под давлением с образованием кристаллов, находящихся в интервале от примерно 5 до 20 микрон.
Другой подход описан в патенте США № 5118528, в котором описан способ получения наночастиц. Данный способ включает стадии (1) получения жидкой фазы вещества в растворителе или смеси растворителей, к которым могут быть добавлены одно или более поверхностно-активных веществ, (2) получения второй жидкой фазы нерастворителя или смеси нерастворителей, причем нерастворитель является смешиваемым с растворителем или смесью растворителей для данного вещества, (3) добавления вместе растворов (1) и (2) при перемешивании и (4) удаления нежелательных растворителей с получением коллоидной суспензии наночастиц. В патенте '528 описано, что получаются частицы вещества менее 500 нм без подачи энергии. В частности, в патенте '528 установлено, что нежелательно использовать высокоэнергоемкое оборудование, такое как ультразвуковые излучатели и гомогенизаторы.
В патенте США № 4826689 описан метод получения частиц однородного размера из нерастворимых в воде лекарств или других органических соединений. Сначала соответствующее твердое органическое вещество растворяют в органическом растворителе, и раствор может быть разбавлен нерастворителем. Затем вливают водную осаждающую жидкость, осаждают неагрегированные частицы с одинаковым (однородным) по существу средним диаметром. Затем частицы отделяют от органического растворителя. В соответствии с данным изобретением, в зависимости от органического соединения и требуемого размера частиц, параметры температуры, отношения нерастворителя к органическому растворителю, скорость вливания, скорость перемешивания и объем могут изменяться. В патенте '689 раскрывается, что при данном процессе образуется лекарственное вещество в метастабильном состоянии, которое является термодинамически нестабильным и которое, в конечном счете, превращается в более стабильное кристаллическое состояние. В патенте '689 описано отделение лекарственного вещества в метастабильном состоянии, при котором свободная энергия занимает место между энергией раствора исходного лекарственного вещества и стабильной кристаллической формы. В патенте '689 описано использование ингибиторов кристаллизации (например, поливинилпирролидона) и поверхностно-активных веществ (например, поли(оксиэтилен)-со-оксипропилен) для получения осадка, достаточно стабильного для выделения центрифугированием, мембранной фильтрацией или обратным осмосом.
В патентах США №№ 5091188; 5091187 и 4725442 описываются (а) или покрытие мелких частиц лекарственного вещества природными или синтетическими фосфолипидами, или (b) растворение лекарственного вещества в подходящем липофильном носителе и формирование эмульсии, стабилизированной природными или полусинтетическими фосфолипидами. Один из недостатков этих подходов состоит в том, что они зависят от качества сырьевого материала лекарственного вещества, и не раскрываются стадии изменения морфологии сырьевого материала для приведения данного материала в хрупкое, более легкое для обработки состояние.
Другой подход для получения нерастворимых лекарственных веществ для парентеральной доставки описан в патенте США № 5145684. В патенте '684 описан влажный размол нерастворимого лекарственного вещества в присутствии модификатора поверхности для получения частиц лекарственного вещества, имеющих средний эффективный размер частиц менее 400 нм. В патенте '684 подчеркивается нежелательность использования каких-либо растворителей в этом способе. В патенте '684 описан модификатор поверхности, который адсорбируется на поверхности частицы лекарственного вещества в количестве, достаточном для предотвращения агломерации в более крупные частицы.
Еще одна попытка получить нерастворимые лекарственные средства для парентеральной доставки описана в патенте США №5992355. В патенте '355 описано получение частиц субмикронного размера нерастворимых лекарственных веществ с использованием комбинации модификаторов поверхности и фосфолипида с последующим снижением размера частиц при использовании таких методик, как обработка ультразвуком, гомогенизация, размол, микрофлюидизация, осаждение или перекристаллизация. В патенте '355 нет описания изменения условий процесса для получения кристаллов в более хрупкой форме.
В патенте США № 5780062 описан способ получения мелких частиц нерастворимых лекарственных веществ путем (1) растворения лекарственного вещества в смешиваемом с водой первом растворителе, (2) получения второго раствора полимера и амфифильного вещества во втором водном растворителе, в которой лекарство по существу нерастворимо, посредством чего образуется полимеро/амфифильный комплекс и (3) смешивания растворов с первой и второй стадий для осаждения агрегата лекарственного вещества и полимеро/амфифильного комплекса.
В патенте США № 5858410 описана фармацевтическая наносуспензия, пригодная для парентерального введения. В патенте '410 описана гомогенизация под высоким давлением в поршневом щелевом гомогенизаторе, по меньшей мере, одного твердого терапевтически активного соединения, диспергированного в растворителе, с образованием частиц, имеющих средний диаметр, определенный с помощью фотонной корреляционной спектроскопии (ФКС), равный от 10 до 1000 нм, причем доля частиц более 5 мкм от всего их числа составляет менее 0,1% (число распределения, определяемое с помощью счетчика частиц), без предварительного превращения в расплав, причем активное соединение является твердым при комнатной температуре и нерастворимо, только слабо растворимо или умеренно растворимо в воде, водной среде и/или органических растворителях. В примерах в патенте '410 описано струйное измельчение перед гомогенизацией.
В патенте США № 4997454 описан способ изготовления частиц однородного размера из твердых соединений. Способ по патенту '454 включает стадии растворения твердого соединения в подходящем растворителе с последующим вливанием осаждающей жидкости, причем осаждаются неагрегированные частицы с по существу однородным средним диаметром. Затем частицы отделяют от растворителя. В патенте '454 создаются помехи образованию частиц в кристаллическом состоянии, так как во время процедуры осаждения кристаллы могут растворяться и перекристаллизовываться, расширяя тем самым интервал распределения частиц по размеру. В патенте '454 поддерживаются условия во время процедуры осаждения для улавливания частиц в метастабильном состоянии.
В патенте США № 5605785 описан способ получения наноаморфных дисперсий, пригодных для фотографии соединений. Способ получения наноаморфных дисперсий включает любой известный способ эмульгирования, который дает дисперсную фазу, имеющую аморфные частицы.
КРАТКОЕ ИЗЛОЖЕНИЕ ИЗОБРЕТЕНИЯ
Данное изобретение предлагает способ получения частиц субмикронного размера органического соединения, растворимость которого больше в смешиваемом с водой первом растворителе, чем во втором растворителе, который является водным. Данный способ включает стадии (i) растворения органического соединения в смешиваемом с водой первом растворителе с образованием раствора, причем первый растворитель выбирается из группы, состоящей из N-метил-2-пирролидинона, 2-пирролидона, диметилсульфоксида, диметилацетамида, молочной кислоты, метанола, этанола, изопропанола, 3-пентанола, н-пропанола, глицерина, бутиленгликоля, этиленгликоля, пропиленгликоля, моно- и диацилированных моноглицеридов, диметилизосорбида, ацетона, диметилформамида, 1,4-диоксана, полиэтиленгликоля, сложных эфиров полиэтиленгликоля, полиэтиленгликольсорбитанов, моноалкилэфиров полиэтиленгликоля, полипропиленгликоля, полипропиленальгината, ППГ-10 бутандиола, эфира ППГ-10 и метилглюкозы, эфира ППГ-20 и метилглюкозы, стеарилового эфира ППГ-15, пропиленгликоля дикаприлага, пропиленгликоля дикапрата, пропиленгликоля лаурата; (ii) смешивания раствора со вторым растворителем для создания предсуспензии (предварительной суспензии) и (iii) подвода энергии к предсуспензии для образования частиц, имеющих средний эффективный размер частиц менее чем примерно 2 мкм.
Способ получения частиц субмикронного размера органического соединения, растворимость которого больше в смешиваемом с водой первом растворителе, чем во втором растворителе, который является водным, причем способ включает стадии (i) растворения органического соединения в смешиваемом с водой первом растворителе с получением раствора, причем первый растворитель выбирается из группы, состоящей из N-метил-2-пирролидинона, 2-пирролидона, диметилсульфоксида, диметилацетамида, молочной кислоты, метанола, этанола, изопропанола, 3-пентанола, н-пропанола, глицерина, бутиленгликоля, этиленгликоля, пропиленгликоля, моно- и диацилированных моноглицеридов, диметилизосорбида, ацетона, диметилформамида, 1,4-диоксана, этилацетата, пропилацетата, полиэтиленгликоля, сложных эфиров полиэтиленгликоля, полиэтиленгликольсорбитанов, моноалкилэфиров полиэтиленгликоля, полипропиленгликоля, полипропиленальгината, ППГ-10 бутандиола, эфира ППГ-10 и метилглюкозы, эфира ППГ-20 и метилглюкозы, стеарилового эфира ППГ-15, пропиленгликоля дикаприлата, пропиленгликоля дикапрата, пропиленгликоля лаурата; (ii) смешивания раствора со вторым растворителем для создания предсуспензии, где органическое соединение находится в аморфной форме, полукристаллической форме или в виде переохлажденной жидкости, как определено ДСК, и имеющей средний эффективный размер частиц, и (iii) отжига предсуспензии с образованием частиц, имеющих по существу тот же самый средний эффективный размер частиц предсуспензии и в более стабильной форме.
Данное изобретение дополнительно предлагает способ получения частиц субмикронного размера органического соединения, растворимость которого больше в смешиваемом с водой первом растворителе, чем во втором растворителе, который является водным. Данный способ включает стадии (i) растворения органического соединения в смешиваемом с водой первом растворителе с образованием раствора, причем первый растворитель выбирается из группы, состоящей из N-метил-2-пирролидинона, 2-пирролидона, диметилсульфоксида, диметилацетамида, молочной кислоты, метанола, этанола, изопропанола, 3-пентанола, н-пропанола, глицерина, бутиленгликоля, этиленгликоля, пропиленгликоля, моно- и диацилированных моноглицеридов, диметилизосорбида, ацетона, диметилформамида, 1,4-диоксана, полиэтиленгликоля, сложных эфиров полиэтиленгликоля, полиэтиленгликольсорбитанов, моноалкилэфиров полиэтиленгликоля, полипропиленгликоля, полипропиленальгината, ППГ-10 бутандиола, эфира ППГ-10 и метилглюкозы, эфира ППГ-20 и метилглюкозы, стеарилового эфира ППГ-15, пропиленгликоля дикаприлата, пропиленгликоля дикапрата, пропиленгликоля лаурата; (ii) смешивания раствора со вторым растворителем для создания предсуспензии частиц в хрупкой форме и (iii) подвода энергии к предсуспензии для образования частиц, имеющих средний эффективный размер частиц менее чем примерно 2 мкм.
Данное изобретение также предлагает способ получения суспензии фармацевтически активного соединения, растворимость которого больше в смешиваемом с водой первом органическом растворителе, чем во втором растворителе, который является водным. Способ включает стадии (i) растворения первого количества фармацевтически активного соединения в смешиваемом с водой первом органическом растворителе с образованием первого раствора; (ii) смешивания первого раствора со вторым растворителем для осаждения фармацевтически активного соединения; (iii) затравливание (внесение затравки) первого раствора или второго растворителя или предсуспензии. Способ дополнительно включает стадию формирования требуемого полиморфа фармацевтически активного соединения. В предпочтительной форме данного изобретения стадия затравливания включает стадию добавления затравочного соединения в первый раствор, во второй растворитель и/или в предсуспензию.
Данное изобретение, кроме того, предлагает способ получения суспензии фармацевтически активного соединения, растворимость которого больше в смешиваемом с водой первом растворителе, чем во втором растворителе, который является водным. Данный способ включает стадии (i) добавления некоторого количества фармацевтически активного соединения в первый органический растворитель для создания пересыщенного раствора, (ii) старения пересыщенного раствора для формирования обнаруживаемых кристаллов для получения затравочной смеси и (iii) смешивания затравочной смеси со вторым растворителем для осаждения фармацевтически активного соединения для создания предсуспензии.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фиг.1 и 2 представлены два варианта способов: А и В.
На фиг.3 представлены аморфные частицы перед гомогенизацией (пример 1).
На фиг.4 представлены частицы после отжига гомогенизацией.
На фиг.5 представлена рентгеновская дифрактограмма микроосажденного итраконазола с полиэтиленгликоль-660-12-гидроксистеаратом до и после гомогенизации (пример 5).
На фиг.6 представлены кристаллы карбамазепина перед гомогенизацией (пример 6).
На фиг.7 представлены микрочастицы карбамазепина после гомогенизации (Avestin С-50).
На фиг.8 представлена диаграмма способа микроосаждения для преднизолона (примеры 9-12).
На фиг.9 представлена микрофотография суспензии преднизолона перед гомогенизацией (модуляция контраста по Гофману, увеличение 1250х).
На фиг.10 представлена микрофотография суспензии преднизолона после гомогенизации (модуляция контраста по Гофману, увеличение 1250х).
На фиг.11 представлено сравнение распределения по размеру наносуспензий (данное изобретение) и коммерческой масляной эмульсии (пример 13).
На фиг.12 представлена порошковая рентгенограмма сырьевого материала итраконазола (верхний) и SMP-2-PRE (нижний) по примеру 16. Картина сырьевого материала смещена вверх для ясности.
На фиг.13а представлены данные ДСК сырьевого материала итраконазола (пример 16).
На фиг.13b представлены данные ДСК для SMP-2-PRE (пример 16).
На фиг.14 представлены данные ДСК для SMP-2-PRE, показывающие плавление менее стабильного полиморфа при нагревании до 160°С, явление перекристаллизации при охлаждении и последующее плавление более стабильного полиморфа при повторном нагревании до 180°С (пример 16).
На фиг.15 представлено сравнение образцов SMP-2-PRE после гомогенизации(пример 16). Сплошная линия = образец после затравливания сырьевым материалом итраконазола. Пунктирная линия = образец без затравливания. Сплошная линия смещена на 1 Вт/г для ясности.
На фиг.16 представлен эффект затравливания во время осаждения (пример 17). Пунктирная линия = образец без затравливания, сплошная линия = образец после затравливания сырьевым материалом итраконазола. Данные для образца без затравливания (пунктирная линия) сдвинуты вверх на 1,5 Вт/г для ясности.
На фиг.17 представлены эффект затравливания концентрата лекарственного вещества посредством старения. Верхняя рентгенограмма лучей относится к кристаллам, полученным из свежего концентрата лекарственного вещества, и соответствует стабильному полиморфу (см. фиг.12, верхний). Нижний вид относится к кристаллам, полученным для подвергнутого старению (затравленного) концентрата лекарственного вещества и соответствует метастабильному полиморфу (см. фиг.12, нижний). Верхний вид сдвинут вверх для ясности (пример 18).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Данное изобретение допускает осуществление во многих различных формах. Предпочтительные варианты осуществления данного изобретения описаны при понимании того, что данное описание должно рассматриваться в качестве иллюстраций принципов данного изобретения и не предназначено для ограничения аспектов данного изобретения в широком смысле до проиллюстрированных вариантов осуществления.
Данное изобретение предлагает методы и способы формирования частиц органического соединения, имеющих средней эффективный размер частиц, подходящий для парентерального введения, и в наиболее предпочтительной форме данного изобретения, равен менее примерно 2 мкм. Данное изобретение также пригодно для получения частиц органического соединения в форме, пригодной для орального введения. Размер частиц для оральных лекарственных форм может быть свыше 2 мкм и обычно менее примерно 7 мкм. Однако частицы могут превышать 7 мкм при условии, что частицы обладают достаточной биодоступностью и другими свойствами оральной лекарственной формы. Оральные лекарственные формы включают таблетки, капсулы, каплеты, мягкие и твердые гелевые капсулы или другие носители для доставки лекарственного вещества при оральном введении.
Способы могут быть разделены на три основных вида. Каждый из видов способов имеет стадии (1) растворения органического соединения в смешиваемом с водой органическом растворителе для получения первого раствора, (2) смешивания первого раствора со вторым водным растворителем для осаждения органического соединения для получения предсуспензии и (3) подача энергии к предсуспензии в виде перемешивания с высоким усилием сдвига или нагревания для получения стабильной формы органического соединения, имеющего желаемые пределы размера, указанные выше.
Три вида способов отличаются по физическим свойствам органического соединения, что определено исследованием с помощью дифракции рентгеновского излучения, исследованием дифференциальной сканнирующей калориметрией или другим подходящим исследованием, проводимым перед стадией подведения энергии и после стадии подведения энергии. При способе первого вида перед стадией подведения энергии органическое соединение в предсуспензии принимает аморфную форму, полукристаллическую форму или форму переохлажденной жидкости и имеет средний эффективный размер частиц. После стадии подачи энергии органическое соединение находится в кристаллической форме, имеющей средний эффективный размер частиц по существу такой же, как и размер в предсуспензии (т.е. от менее примерно 2 мкм).
При способе второго вида перед стадией подведения энергии органическое соединение находится в кристаллической форме и имеет средний эффективный размер частиц. После стадии добавления энергии органическое соединение находится в кристаллической форме, имеющей по существу тот же самый средний эффективный размер частиц, что и перед стадией добавления энергии, но кристаллы после стадии подведения энергии менее способны агрегировать. Более низкая склонность органического соединения агрегировать наблюдается с помощью рассеяния лазерного динамического света и световой микроскопии.
При третьем виде способа перед стадией подачи энергии органическое соединение находится в кристаллической форме, которая является хрупкой и имеет средний эффективный размер частиц. Под термином «хрупкая» подразумевается то, что частицы являются хрупкими и легче ломаются на более мелкие частицы.
После стадии подачи энергии органическое соединение находится в кристаллической форме, имеющей средний эффективный размер частиц, меньший, чем у кристаллов предсуспензии. Пользуясь стадиями, необходимыми для приведения органического соединения в кристаллическую форму, которая является хрупкой, последующая стадия подачи энергии может быть проведена более быстро и эффективно, по сравнению с органическим соединением с менее хрупкой кристаллической морфологией.
Стадия подачи энергии может быть осуществлена любым способом, при котором предсуспензия подвергается воздействию сил кавитации, сдвига или удара. В одной из предпочтительных форм изобретения стадия подачи энергии является стадией обжига. Отжиг в данном изобретении определяется как процесс превращения вещества, которое термодинамически нестабильно, в более стабильную форму путем единственного или повторного применения энергии (прямого нагревания или механического воздействия) с последующей тепловой релаксацией. Это снижение энергии может быть достигнуто превращением твердой формы из менее упорядоченной в более упорядоченную кристаллическую структуру. Альтернативно эта стабилизация может совершаться переупорядочением молекул поверхностно-активных веществ на поверхности раздела жидкости и твердого вещества.
Эти три вида способов (процессов) будут обсуждены отдельно ниже. Должно быть понятно, однако, что условия процесса, такие как выбор поверхностно-активных веществ или комбинация поверхностно-активных веществ, количество используемого поверхностно-активного вещества, температура реакции, скорость перемешивания растворов, скорость осаждения и тому подобное, могут выбираться так, чтобы дать возможность обработки любого лекарственного вещества при использовании любого одного из видов, обсуждаемых далее.
Способы первого вида, а также способы второго и третьего вида могут быть дополнительно разделены на два подвида, метод А и В, представленные в виде диаграммы на фиг.1 и 2.
Органическое соединение для использования в способе данного изобретения является любым органическим химическим соединением, растворимость которого снижается от одного растворителя к другому. Данное органическое соединение могло бы быть фармацевтически активным соединением из разных групп, таких как, но не ограничиваясь ими, антигиперлипидемические средства, антимикробные средства, например антибактериальные средства, такие как сульфадиазин, противогрибковые средства, такие как итраконазол; нестероидные противовоспалительные средства, например индометацин; антигиперхолестеринемические средства, например пробукол, и стероидные соединения, например дексаметазон; иммунодепрессанты, например циклоспорин А, такролимус и микофенолят мофетил. Или органическое соединение могло бы быть из группы, используемой в качестве адъювантов или эксципиентов в фармацевтических препаратах и косметических средствах, таких как, но не ограничиваясь этим, консерванты, например пропилпарабен.
Первый растворитель по данному изобретению является растворителем или смесью растворителей, в которых органическое соединение, представляющее интерес, является относительно растворимым, и которые смешиваются со вторым растворителем. Примеры таких растворителей включают, но не ограничиваются этим, поливинилпирролидон, N-метил-2-пирролидон (также называемый N-метил-2-пирролидин), 2-пирролидон, диметилсульфоксид, диметилацетамид, молочная кислота, метанол, этанол, изопропанол, 3-пентанол, н-пропанол, глицерин, бутиленгликоль (бутандиол), этиленгликоль, пропиленгликоль, моно- и диацилированные моноглицериды (такие как глицерилкаприлат), диметилизосорбид, ацетон, диметилформамид, 1,4-диоксан, полиэтиленгликоль (например, ПЭГ-4, ПЭГ-8, ПЭГ-9, ПЭГ-12, ПЭГ-14, ПЭГ-16, ПЭГ-120, ПЭГ-75, ПЭГ-150, сложные эфиры полиэтиленгликоля (например, такие как ПЭГ-4 дилаурат, ПЭГ-20 дилаурат, ПЭГ-6 изостеарат, ПЭГ-8 пальмитостеарат, ПЭГ-150 пальмитостеарат), полиэтиленгликольсорбитаны (такие как ПЭГ-20 сорбитанизостеарат), моноалкиловые простые эфиры полиэтиленгликоля (например, такие как диметиловый эфир ПЭГ-3, диметиловый эфир ПЭГ-4), полипропиленгликоль (ППГ), полипропиленальгинат, ППГ-10 бутандиол, простой эфир ППГ-10 и метилглюкозы, простой эфир ППГ-20 и метилглюкозы, простой стеариловый эфир ППГ-15, пропиленгликоля дикаприлат/дикапрат, пропиленгликоля лаурат.
Метод А
В способе А (см. фиг.1) органическое соединение («лекарство») сначала растворяют в первом растворителе с получением первого раствора. Органическое соединение может быть добавлено в количестве от примерно 0,1% (вес/об) до примерно 50% (вес/об), в зависимости от растворимости органического соединения в первом растворителе. Для обеспечения полного растворения соединения в первом растворителе может быть необходимо нагревание концентрата от примерно 30°С до примерно 100°С.
Второй водный раствор получают с добавлением к нему одного или более необязательных поверхностно-активных модификаторов, таких как анионное поверхностно-активное вещество (ПАВ), катионное ПАВ, неионное ПАВ или биологические поверхностно-активные молекулы. Подходящие анионные ПАВ включают, но не ограничиваются этим, лаурат калия, лаурилсульфат натрия, додецилсульфат натрия, сульфаты алкилполиоксиэтилена, альгинат натрия, диоктилнатрия сульфосукцинат, фосфатидилхолин, фосфатидилглицерин, фосфатидилинозин, фосфатидилсерин, фосфатидную кислоту и ее соли, сложные эфиры глицерина, натрийкарбоксиметилцеллюлозу, холевую кислоту и другие желчные кислоты (например, холевая кислота, дезоксихолевая кислота, гликохолевая кислота, таурохолевая кислота, гликодезоксихолезая кислота и ее соли (например, дезоксихолат натрия и т.д.). Подходящие катионные ПАВ включают, но не ограничиваются этим, соединения четвертичного аммония, такие как бензалкония хлорид, цетилтриметиламмония бромид, лаурилдиметилбензиламмония хлорид, ацилкарнитина гидрохлориды или алкилпиридиния галогениды. В качестве анионных ПАВ можно использовать фосфолипиды. Подходящие фосфолипиды включают, например, фосфатидилхолин, фосфатидилэтаноламин, фосфатидилсерин, фосфатидилинозин, фосфатидилглицерин, фосфатидная кислота, лизофосфолипиды, яичный или соевый фосфолипид или их сочетание. Фосфолипид может быть солевой или обессоленный, гидрированный или частично гидрированный, или природный, полусинтетический или синтетический.
Подходящие неионные ПАВ включают простые эфиры полиоксиэтилена и жирных спиртов (Macrogol и Brij), сложные эфиры полиоксиэтиленсорбитана и жирных кислот (полисорбаты), сложные эфиры полиоксиэтилена и жирных кислот (Myrj), сложные эфиры сорбитана (спен. Span), глицерина моностеарат, полиэтиленгликоли, полипропиленгликоли, цетиловый спирт, цетостеариловый спирт, стеариловый спирт, арилалкилполиэфирные спирты, полиоксиэтиленополиоксипропиленовые сополимеры (полоксамеры), полаксамины, метилцеллюлоза, гидроксицеллюлоза, гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, некристаллическая целлюлоза, полисахариды, включая крахмал и производные крахмала, такие как гидроксиэтилкрахмал (ГЭК, HES), поливиниловый спирт и поливинилпирролидон. В предпочтительном варианте данного изобретения неионное ПАВ является сополимером полиоксиэтилена и полиоксипропилена и предпочтительно блоксополимером пропиленгликоля и этиленгликоля. Такие полимеры продаются под торговым названием «Poloxamer» (полоксамер), иногда также называемым PLURONIC® (плуроник), и продаются несколькими поставщиками, включая Spectrum Chemical and Ruger. В сложные эфиры полиоксиэтилена и жирных кислот включены и те, которые имеют короткие алкильные цепи. Одним из примеров такого ПАВ является SOLUTOL® HS 15 (солутол HS 15), полиэтилен-660-гидроксистеарат, производимый фирмой BASF Aktiengesellschaft.
Поверхностно-активные биологические молекулы включают такие молекулы, как альбумин, казеин, гепарин, гирудин и другие подходящие белки.
Может быть также желательно добавлять рН регулирующее средство ко второму раствору, такое как гидроксид натрия, соляную кислоту, трис-буфер или цитрат, ацетат, лактат, меглумин и тому подобное. Второй раствор должен иметь рН в интервале от примерно 3 до примерно 11.
Для оральных лекарственных форм могут использоваться один или более из следующих эксципиентов: желатин, казеин, лецитин (фосфатиды), камедь акации, холестерин, трагакант, стеариновая кислоты, бензалкония хлорид, стеарат кальция, глицерилмоностеарат, цетостеариловый спирт, цетомакроголевый эмульгирующий воск, сложные эфиры сорбитана, полиоксиэтиленалкиловые эфиры, например простые эфиры макрогола, такие как цетомакрогол 1000, производные полиоксиэтилена и касторового масла, сложные эфиры полиоксиэтиленсорбитана и жирных кислот, например доступные для приобрегения твины ТМ, полиэтиленгликоли, полиоксиэтиленстеараты, коллоидный силикондиоксид, фосфаты, додецилсульфат натрия, кальцийкарбоксиметилцеллюлоза, натрийкарбоксиметилцеллюлоза, метилцеллюлоза, гидроксиэтилцеллюлоза, гидроксипропилцеллюлоза, фталат гидроксипропилметилцеллюлозы, некристаллическая целлюлоза, алюмосиликат магния, триэтаноламин, поливиниловый спирт (PVA, ПВС) и поливинилпирролидон (ПВП). Большинство из этих эксиципиентоз подробно описаны в Handbook of Pharmaceutical Excipients, опубликованном совместно American Pharmaceutical Association и The pharmaceutical Society of Great Britain, The Pharmaceutical Press, 1986. Модификаторы поверхности коммерчески доступны и/или могут быть получены методами, известными в данной области. Два или более модификаторов поверхности можно использовать в комбинации.
В предпочтительном варианте данного изобретения способ получения частиц органического соединения субмикронного размера включает стадии добавления первого раствора ко второму раствору. Скорость добавления зависит от размера партии и кинетики осаждения органического соединения. Обычно для процесса небольшого лабораторного масштаба (получения 1 литра) скорость добавления составляет от примерно 0,05 см3 в минуту до примерно 10 см3 в минуту. Во время добавления растворы должны постоянно перемешиваться. С помощью световой микроскопии наблюдали, что при изготовлении предсуспензии образуются аморфные частицы, полукристаллические вещества или переохлажденные жидкости. Данный способ дополнительно включает стадию проведения отжига предсуспензии для превращения аморфных частиц, переохлажденной жидкости или полукристаллического твердого вещества в кристаллическое более стабильное твердое состояние. Полученные частицы будут иметь средний эффективный размер частиц, который измеряли методами динамического рассеяния света (например, фотокорреляционной спектроскопией, лазерной диффракцией, рассеяния лазерного излучения при низком угле падения (LALLS), рассеивания лазерного света при среднем угле падения (MALLS), методы матирования света (например, метод Coulter), методом реологии или микроскопии (световой или электронной), в интервалах, представленных выше.
Стадия подведения энергии включает поставку энергии посредством воздействия ультразвуком, гомогенизации, гомогенизации в противотоке, микроожижения или другими методами получения усилия удара, сдвига или сил кавитации. Образец может охлаждаться или нагреваться во время этой стадии. В одном из предпочтительных вариантов данного изобретения стадия отжига осуществляется с помощью поршневого щелевого гомогенизатора, такого как продаваемый фирмой Avestin Inc. в виде продукта под названием EmulsiFlex-C160. В другом предпочтительном варианте данного изобретения отжиг может выполняться путем обработки ультразвуком с использованием ультразвукового процессора, такого как Vibra-Cell Ultrasonic Processor (600 Вт), производимого Sonics and Materials, Inc. В еще одном предпочтительном варианте данного изобретения отжиг может выполняться путем применения аппарата для эмульгирования, как описано в патенте США № 5720551, который включен в описание в виде ссылки и составляет часть этого.
В зависимости от скорости отжига может быть желательно регулировать температуру обрабатываемого образца до уровня в интервале от примерно -30 до 30°С. Альтернативно, чтобы выполнить желаемое фазовое изменение у обрабатываемого твердого вещества, может быть также необходимо нагреть предсуспензию до температуры в интервале от примерно 30°С до примерно 100°С во время стадии отжига.
Метод В
Метод 3 отличается от метода А в следующих отношениях.
Первое отличие состоит в том, что ПАВ или комбинацию ПАВ добавляют в первый раствор. ПАВ могут быть выбраны из групп анионных, неионных и катионных ПАВ, представленных выше.
Сравнительный пример метода А и метода В и US 5780062
В патенте США № 5780062 описан способ получения мелких частиц органического соединения путем первоначального растворения соединения в подходящем смешиваемом с водой первом растворителе. Второй раствор получают растворением полимера и амфифильного соединения в водном растворе. Первый раствор затем добавляют во второй раствор с образованием осадка, который состоит из органического соединения и комплекса полимер-амфифильное соединение. В патенте '062 не описано использования стадии подведения энергии из данного изобретения в методах А и В. Об отсутствии стабильности обычно свидетельствует быстрая агрегация и рост частиц. В некоторых случаях аморфные частицы перекристаллизовывались в виде больших кристаллов. Подведение энергии к предсуспензии способом, описанным выше, обычно дает частицы, которые проявляют сниженную скорость агрегации и роста частиц, а также отсутствие перекристаллизации при хранении продукта.
Методы А и В, кроме того, отличаются от способа патента '062 отсутствием стадии формирования комплекса полимер-амфифильное соединение перед осаждением. В методе А такой комплекс не может формироваться, так как никакого полимера не добавляется к растворителю (водной фазе). В методе В ПАВ, которое может также действовать как амфифильное соединение, или полимер, растворяют с органическим соединением в первом растворителе. Это предотвращает формирование любых комплексов амфифильного соединения-полимера перед осаждением. В патенте '062 успешное осаждение мелких частиц зависит от образования комплекса амфифильного соединения-полимера перед осаждением. В патенте '062 описано, что комплекс амфифильного соединения-полимера образует агрегаты во втором водном растворе. В патенте '062 объясняется, что гидрофобное органическое соединение взаимодействует с комплексом амфифильного соединения-полимера, снижая тем самым растворимость этих агрегатов и вызывая осаждение. В данном изобретении было продемонстрировано, что добавление ПАВ или полимера в первый раствор (метод В) приводит, при последующем добавлении во второй раствор, к формированию более однородных, более тонких частиц, чем те, которые получают по способу, описанному в патенте '062.
С этой целью были получены и проанализированы два препарата. При каждом из получений этих препаратов имеется два раствора, концентрат и водный растворитель, которые смешивают вместе и затем обрабатывают ультразвуком. Концентрат при каждом изготовлении содержит органическое соединение (итраконазол), смешиваемый с водой растворитель (N-метил-2-пирролидинон или NMP) и, возможно, полимер (полоксамер 188). Водный растворитель содержит воду, трис-буфер и, возможно, полимер (полоксамер 188) и/или ПАВ (дезоксихолат натрия). Средний диаметр частиц органического вещества измеряется перед и после обработки ультразвуком.
Первый препарат А содержит в качестве концентрата итраконазол и NMP. Водный растворитель включает воду, полоксамер 188, трис-буфер и дезоксифолат натрия. Таким образом, водный растворитель включает полимер (полоксамер 188) и амфифильное соединение (дезоксихолат натрия), которые могут образовывать комплекс амфифильного соединения-полимера, и поэтому это находится в соответствии с описанием в патенте '062. Однако, опять же в патенте '062, не описано стадии подведения энергии.
Второй препарат В содержит в качестве концентрата итраконазол, NMP и полоксамер 188. Водный растворитель включает воду, трис-буфер и дезоксихолат натрия. Этот препарат изготавливают в соответствии с данным изобретением. Так как водный растворитель не содержит комбинации полимера (полоксамера) и амфифильного соединения (дезоксихолат натрия), комплекс полимера-амфифильного соединения не может образоваться перед стадией смешивания.
В таблице 1 показаны средние диаметры частиц, измеренные с помощью лазерной дифракции на препаратах суспензии в трех повторениях. Производят первоначальное определение размера, после которого образец подвергают обработке ультразвуком в течение 1 минуты. Затем определение размера повторяют. Большое снижение размера после обработки ультразвуком по методу А было показателем агрегации частиц.
Суспензию лекарственного вещества, полученную в результате применения способов, описанных в данном изобретении, можно вводить непосредственно в виде инъецируемого раствора при условии, что в препарате используется вода для инъекций и применяются соответствующие средства для стерилизации раствора. Стерилизацию можно выполнять посредством отдельной стерилизации концентрата лекарственного средства (лекарственного вещества, растворителя и необязательно ПАВ) и разбавляющей среды (воды и необязательно буферов и ПАВ) перед смешиванием для образования предсуспензии. Методы стерилизации будут включать предфильтрацию сначала через 3,0-микронный фильтр с последующим фильтрованием через фильтр для частиц 0,45 микрон и последующей стерилизацией паром или нагреванием, или стерилизацией фильтрованием через два дублирующих 0,2-микронных мембранных фильтра.
Необязательно суспензия без растворителя может быть получена путем удаления растворителя после осаждения. Это может быть выполнено центрифугированием, диализом, диафильтрованием, фракционированием в силовом поле, фильтрованием под высоким давлением или другими методами отделения, хорошо известными в данной области. Полное удаление N-метил-2-пирролидинона обычно производили одним или тремя последовательными циклами центрифугирования; после каждого центрифугирования (18000 об/мин в течение 30 минут) супернатант декантировали и отбрасывали. К оставшимся твердым веществам добавляли объем свежего носителя для суспензии без органического растворителя и смесь диспергировали с помощью гомогенизации. Специалистам в данной области будет понятно, что и другие методы смешивания с высоким усилием сдвига могут применяться на этой стадии восстановления.
Кроме того, любые нежелательные эксципиенты, такие как ПАВ, могут быть заменены более желательным эксципиентом путем использования методов отделения, описанных в представленном выше абзаце. Растворитель и первый эксципиент могут быть удалены вместе с супернатантом после центрифугирования или фильтрования. Затем может быть добавлен объем свежего носителя для суспензии без органического растворителя и без первого эксципиента. Альтернативно может быть добавлено новое ПАВ. Например, в суспензии, состоящей из лекарственного вещества, N-метил-2-пирролидинона (растворитель), полоксамера 188 (первый эксципиент), дезоксихолат натрия, глицерина и воды может быть произведена замена на фосфолипиды (новое ПАВ), глицерин и воду после центрифугирования и удаления супернатанта.
1. Первый вид способа.
Методы первого вида способа обычно включают стадию растворения органического соединения в смешиваемом с водой первом растворителе с последующей стадией смешивания этого раствора с водным раствором для образования предсуспензии, причем органическое соединение находится в аморфной форме, полукристаллической форме или в переохлажденной жидкой форме, что определяли исследованиями рентгеновской диффракцией, ДСК, световой микроскопией или другими аналитическими методами, и имеет средний эффективный размер частиц в пределах одного из интервалов эффективного размера частиц, представленных выше. За стадией смешивания следует стадия подведения энергии и, в предпочтительном варианте данного изобретения, стадия отжига.
II. Второй вид способа.
Методы способов второго вида включают по существу те же стадии, что и стадии способов первого вида, но отличаются в следующем аспекте. Рентгеновская дифракция, ДСК или другие подходящие методы анализа предсуспензии выявляют органическое соединение в кристаллической форме и имеющее средний эффективный размер частиц. Органическое соединение после стадии подведения энергии имеет по существу тот же самый средний эффективный размер частиц, что и до стадии подачи энергии, но имеет меньшую склонность к агрегации в более крупные частицы, по сравнению с размером частиц предсуспензии. Не связывая это с какой-либо теорией, полагают, что различия в стабильности частиц могут быть связаны с переупорядочиванием молекул ПАВ на поверхности раздела жидкости и твердого вещества.
III. Третий вид способов.
При методах третьего вида изменяются первые две стадии способов первого и второго видов, чтобы обеспечить то, чтобы органическое соединение в предсуспензии находилось в хрупкой форме, имеющей средний эффективный размер частиц (например, такой как тонкие (узкие) иглы и тонкие пластинки). Хрупкие частицы могут быть сформированы с помощью выбора подходящих растворителей, ПАВ или комбинаций ПАВ, температуры конкретных растворов, скорости смешивания и скорости осаждения и тому подобного. Хрупкость может быть повышена введением дефектов пространственной решетки (например, плоскостей спайности) во время стадий смешивания первого раствора со вторым раствором. Это может возникнуть при быстрой кристаллизации, такой как кристаллизация, получаемая на стадии осаждения. На стадии подведения энергии эти хрупкие кристаллы превращают в кристаллы, которые кинетически стабилизированы и имеют средний эффективный размер частиц менее размера в предсуспензии. Кинетически стабилизированные средние частицы имеют сниженную склонность агрегировать по сравнению с частицами, которые не являются кинетически стабилизированными. В таком случае стадия подведения энергии приводит к разрушению хрупких частиц. Обеспечивая тот факт, что частицы в предсуспензии находятся в хрупком состоянии, органическое соединение может более легко и более быстро превращаться в частицы в желаемом, интервале размеров по сравнению с обработкой органического соединения, когда не было стадий для приведения их в хрупкое состояние.
Контроль полиморфов
Данное изобретение, кроме того, предлагает дополнительные стадии для контроля кристаллической структуры фармацевтически активного соединения, чтобы в итоге получить суспензию соединения с желаемым интервалом размеров частиц и с желаемой кристаллической структурой. Под термином «кристаллическая структура» понимается расположение атомов в элементарной ячейке кристалла. Фармацевтически активные соединения, которые могут кристаллизоваться в разные кристаллические структуры, называются полиморфными. Идентификация полиморфов является важной стадией при изготовлении лекарства, так как разные полиморфы одного и того же лекарственного вещества могут проявлять различия в растворимости, терапевтической активности, биодоступности и стабильности в суспензии. Соответственно, важно регулировать полиморфную форму соединения для обеспечения чистоты продукта и воспроизводимости от партии к партии.
Стадии контроля формы полиморфа соединения включают затравливание (использование затравки) первого раствора, второго раствора или предсуспензии для обеспечения образования желаемого полиморфа. Затравливание включает использование затравочного соединения или подведение энергии. В предпочтительном варианте данного изобретения затравочным соединением является фармацевтически активное соединение в желаемой полиморфной форме. Альтернативно затравочное соединение может быть также инертной примесью или органическим соединением со структурой, аналогичной структуре желаемого полиморфа, такой как желчная соль.
Затравочное соединение можно осадить из первого раствора. Этот метод включает стадии добавления фармацевтически активного соединения в количестве, достаточном, чтобы превысить растворимость фармацевтически активного соединения в первом растворителе для создания пересыщенного раствора. Пересыщенный раствор обрабатывают, чтобы осадить фармацевтически активное соединение в желаемой полиморфной форме. Обработка пересыщенного раствора включает старение раствора в течение периода времени до тех пор, пока не будет наблюдаться образование кристалла или кристаллов для создания затравочной смеси. Также возможно подавать энергию к пересыщенному раствору, чтобы вызвать осаждение из раствора фармацевтически активного соединения в желаемой полиморфной форме. Энергию можно подводить с помощью ряда способов, включая стадии подведения энергии, описанные выше. Кроме того, энергия может быть подведена за счет нагревания или воздействия на предсуспензию электромагнитной энергии, с помощью источников пучка частиц или пучка электронов. Электромагнитная энергия включает использование лазерного луча, динамической электромагнитной энергии или других источников излучения. Кроме того, предполагается использование ультразвука, статического электрического поля и статического магнитного поля в качестве источника дополнительной энергии.
В предпочтительном варианте данного изобретения метод получения затравочных кристаллов из пересыщенного раствора, подвергнутого старению, включает стадии (i) добавления некоторого количества фармацевтически активного соединения в первый органический растворитель для получения пересыщенного раствора; (ii) старение пересыщенного раствора с образованием обнаруживаемых кристаллов для создания затравочной смеси и (iii) смешивания затравочной смеси со вторым растворителем для осаждения фармацевтически активного соединения с получением предсуспензии. Предсуспензию затем можно дополнительно обработать, как подробно описано выше, с получением водной суспензии фармацевтически активного соединения в виде желаемого полиморфа и с размером частиц в желаемом интервале.
Затравливание может быть выполнено подводкой (добавлением) энергии к первому раствору, второму растворителю или предсуспензии, при условии что жидкость или жидкости, на которые производили воздействие, содержат фармацевтически активное соединение или затравочный материал. Энергия может быть поставлена таким же образом, который описан выше, в отношении пересыщенного раствора.
Соответственно, данное изобретение предлагает композицию материала фармацевтически активного соединения в желаемой полиморфной форме, по существу свободной от не установленного полиморфа или полиморфов. Один такой пример представлен в примере 16, где затравливание во время микроосаждения дает полиморф итраконазола, по существу свободный от полиморфа сырьевого материала. Предполагается, что методы данного изобретения могут применяться для селективного получения желаемого полиморфа для многочисленных фармацевтически активных соединений.
Примеры способа вида I
Пример 1. Получение суспензии итраконазола путем применения способа вида 1, метода А с гомогенизацией.
В 3-литровую колбу добавляют 1680 мл воды для инъекций. Нагревают жидкость до 60-65°С и затем медленно добавляют 44 г Pluronic F-68 (полоксамер 188) и 12 г дезоксихолата натрия, перемешивая после каждого добавления, чтобы растворить твердые вещества. После того как добавление твердых веществ завершится, перемешивают еще 15 минут при 60-65°С для обеспечения полного растворения. Получают 50 мМ трис (трометамин) буфер путем растворения 6,06 г трис в 800 мл воды для инъекций. Титруют этот раствор до рН 8,0 с использованием 0,1 М соляной кислоты. Разбавляют полученный раствор до 1 л дополнительным количеством воды для инъекций. Добавляют 200 мл трис-буфера к раствору полоксамера/дезоксихолата. Тщательно перемешивают, чтобы смешать растворы.
В 150-миллилитровый стакан добавляют 20 г интаконазола и 120 мл N-метил-2-пирролидинона. Нагревают смесь до 50-60°С и перемешивают для растворения твердых веществ. После того как полное растворение становится визуально очевидным, перемешивают еще 15 минут, чтобы обеспечить полное растворение.
Охлаждают раствор итраконазола-NMP до комнатной температуры.
Загружают в поршневой насос (два 60-миллилитровых стеклянных шприца) 120 мл раствора итраконазола, приготовленного ранее. Тем временем выливают весь раствор ПАВ в загрузочную воронку гомогенизатора, который был охлажден до 0-5°С (это может быть выполнено или применением воронки с рубашкой, через которую циркулирует хладагент, или окружая воронку льдом). Помещают механическую мешалку в раствор ПАВ так, чтобы лопасти были полностью погружены. Используя поршневой насос медленно добавляют (1-3 мл/мин) весь раствор итраконазола к перемешиваемому, охлаждаемому раствору ПАВ. Рекомендуется скорость перемешивания, по меньшей мере, 700 об/мин. Аликвоту полученной суспензии (суспензия А) анализируют с помощьз световой микроскопии (Hoffmann Modulation Contrast) и лазерной диффракции (Horiba). Суспензия А, которую изучали с помощьв световой микроскопии, состоит из приблизительно сферических аморфных частиц (до 1 микрона) или связанных друг с другом в агрегаты, или свободно движущихся в броуновском движении. См. фиг.3. Измерения динамического рассеяния света обычно дают картину бимодального распределения, означающего наличие агрегатов (10-100 микрон по размеру) и наличие одиночных аморфных частиц в интервале 200-700 нм по среднему диаметру частиц.
Суспензию сразу гомогенизируют (при 10000-30000 фунт/дюйм2) в течение 10-30 минут. В конце гомогенизации температура суспензии в загрузочной воронке не превышает 75°С. Гомогенизированную суспензию собирают в 500-миллилитровые флаконы, которые сразу же охлаждают в холодильнике (2-8°С). Эту суспензию (суспензия В) анализируют с помощью световой микроскопии и, как обнаружено, она состоит из мелких вытянутых пластин длиной от 0,5 до 2 микрон и шириной в интервале 0,2-1 микрон. См. фиг.4. Измерения динамического рассеяния света обычно показывают средний диаметр 200-700 нм.
Стабильность суспензии А («предсуспензия») (пример 1)
Во время микроскопического изучения аликвоты суспензии А непосредственно наблюдалась кристаллизация аморфного твердого вещества. Суспензию А хранили при 2-8°С в течение 12 часов и изучали с помощью световой микроскопии. Общее визуальное обследование образца выявило сильную флокуляцию с некоторым количеством осадка на дне контейнера. Микроскопическое исследование показало наличие больших, вытянутых пластинкоподобных кристаллов свыше 10 микрон длиной.
Стабильность суспензии В
В противоположность нестабильности суспензии А суспензия В была стабильна при 2-8°С в течение продолжения предварительного исследования на стабильность (1 месяц). Микроскопия образца, подвергнутого старению, ясно продемонстрировала, что никакого значительного изменения морфологии или размера частиц не происходило. Это было подтверждено измерениями по рассеянию света.
Пример 2. Получение суспензии итраконазола с применением способа вида 1, метода А и обработки ультразвуком.
В 500-миллилитровый сосуд из нержавеющей стали добавляют 252 мл воды для инъекций. Нагревают жидкость до 60-65°С и затем медленно добавляют 6,6 г Pluronic F-68 (полоксамер 188) и 0,9 г дезоксихолата натрия, перемешивая после каждого добавления, чтобы растворить твердые вещества. После того как добавление твердых веществ завершается, перемешивают в течение еще 15 минут при 60-65°С, чтобы гарантировать полное растворение. Готовят 50 мМ трис (трометаминовый) буфер путем растворения 6,06 г трис в 800 мл воды для инъекций. Титруют раствор до рН 8,0 0,1 М соляной кислотой. Разбавляют полученный раствор до 1 литра дополнительным количеством воды для инъекций. Добавляют 30 мл трис-буфера к раствору полоксамера/дезоксихолата. Тщательно перемешивают, чтобы смешать растворы.
В 30-миллилитровый контейнер добавляют 3 г итраконазола и 18 мл N-метил-2-пирролидинона. Смесь нагревают до 50-60°С и перемешивают для растворения твердых веществ. После того как полное растворение будет визуально очевидным, перемешивают еще 15 минут, чтобы гарантировать полное растворение. Охлаждают раствор итраконазола-NMP до комнатной температуры.
Загружают в поршневой насос 18 мл раствора итраконазола, приготовленного на предыдущей стадии. Помещают механическую мешалку в раствор ПАВ так, чтобы лопасти были полностью погружены. Охлаждают контейнер до 0-5°С погружением в ледяную баню. Используя поршневой насос медленно добавляют (1-3 мл/мин) весь раствор итраконазола к перемешиваемому, охлаждаемому раствору ПАЕ. Рекомендуется скорость перемешивания, по меньшей мере, 700 об/мин. Погружают ультразвуковой излучатель в полученную суспензию так, что зонд находится примерно на 1 см выше дна сосуда из нержавеющей стали. Воздействуют ультразвуком (10000-25000 Гц, при, по меньшей мере, 400 Вт) в течение 15-20 минут через 5-минутные интервалы. После первого 5-минутного воздействия ультразвуком удаляют ледяную баню и продолжают дополнительное воздействие ультразвуком. В конце обработки ультразвуком температура суспензии в сосуде не превышает 75°С.
Суспензию собирают в 500-миллилитровую стеклянную бутыль типа I, которую сразу же охлаждают в холодильнике (2-8°С). Характеристики морфологии частиц суспензии до и после обработки ультразвуком были очень похожими на те, которые наблюдались при методе А до и после гомогенизации (см. пример 1).
Пример 3. Получение суспензии итраконазола путем применения способа вида 1, метода В с гомогенизацией.
Готовят 50 мМ трис (трометаминовый) буфер растворением 6,06 г трис в 800 мл воды для инъекций. Титруют этот раствор до рН 8,0 0,1 М соляной кислотой. Разбавляют полученный раствор до 1 литра дополнительным количеством воды для инъекций. В 3-литровую колбу добавляют 1680 мл воды для инъекций. Добавляют 200 мл трис-буфера к 1680 мл воды. Тщательно перемешивают, чтобы смешать растворы.
В 150-миллилитровый стакан добавляют 44 г Pluronic F-68 (полоксамер 188) и 12 г дезоксихолата натрия к 120 мл N-метил-2-пирролидинона. Нагревают смесь до 50-60°С и перемешивают, чтобы растворить твердые вещества. После того как полное растворение станет визуально явным, перемешивают еще 15 минут, чтобы гарантировать полное растворение. К этому раствору добавляют 20 г итраконазола и перемешивают до полного растворения. Охлаждают раствор итраконазола-ПАВ-NMP до комнатной температуры.
Загружают поршневой насос (два 60-миллилитровых стеклянных шприца) 120 мл концентрированного раствора итраконазола, приготовленного ранее. Тем временем выливают разбавленный раствор трис-буфера, приготовленный, как описано выше, в загрузочную воронку гомогенизатора, который был охлажден до 0-5°С (это может быть выполнено или с помощью использования загрузочной воронки с рубашкой, через которую циркулирует хладагент, или окружая загрузочную воронку льдом). Помещают механическую мешалку в буферный раствор так, что лопасти полностью погружаются. Используя поршневой насос медленно (1-3 мл/мин) добавляют весь концентрат итраконазола-ПАВ к перемешиваемому охлаждаемому буферному раствору. Рекомендуется скорость перемешивания, по меньшей мере, 700 об/мин. Получаемую охлажденную суспензию сразу же гомогенизируют (при 10000-30000 фунт/дюйм2) в течение 10-30 минут. В конце гомогенизации температура суспензии в загрузочной воронке не превышает 75°С.
Гомогенизированную суспензию собирают в 500-миллилитровые бутыли, которые сразу же охлаждают в холодильнике (2-8°С). Характеристики морфологии частиц в суспензии до и после гомогенизации были очень похожими на характеристики, которые наблюдались в примере 1, за исключением того, что при способе вида 1 В предварительно гомогенизированный материал имел склонность образовывать меньше и меньшего размера агрегаты, что приводило в результате к значительно меньшему общему размеру частиц, который измеряли с помощью лазерной диффракции. После гомогенизации результаты динамического рассеяния света были обычно идентичными результатам, представленным в примере 1.
Пример 4. Получение суспензии итраконазола путем применения способа вида 1, метода В с обработкой ультразвуком.
В 500-миллилитровую колбу добавляют 252 мл воды для инъекций. Готовят 50 мМ трис (трометаминовый) буфер путем растворения 6,06 г трис - в 800 мл воды для инъекций. Титруют этот раствор до рН 8,0 0,1 М соляной кислотой. Разбавляют полученный раствор до 1 литра дополнительным количеством воды для инъекций. Добавляют 30 мл трис-буфера к воде. Тщательно перемешивают, чтобы смешать растворы.
В 30-миллилитровый стакан добавляют 6,6 г Pluronic F-68 (полоксамер 188) и 0,9 г дезоксихолата натрия к 18 мл N-метил-2-пирролидинона. Нагревают смесь до 50-60°С и перемешивают, чтобы растворить твердые вещества. После того как полное растворение станет визуально явным, перемешивают еще 15 минут, чтобы гарантировать полное растворение. К этому раствору добавляют 3,0 г итраконазола и перемешивают до полного растворения. Охлаждают раствор итраконазола-ПАВ-NMP до комнатной температуры.
Загружают поршневой насос (два 30-миллилитровых стеклянных шприца) 18 мл концентрированного раствора итраконазола, приготовленного ранее. Помещают механическую мешалку в буферный раствор так, что лопасти были полностью погружены. Охлаждают контейнер до 0-5°С погружением в ледяную баню. Используя поршневой насос медленно (1-3 мл/мин) добавляют весь концентрат итраконазола-ПАВ к перемешиваемому охлаждаемому буферному раствору. Рекомендуется скорость перемешивания, по меньшей мере, 700 об/мин. Получаемую охлажденную суспензию сразу же обрабатывают ультразвуком (при 10000-25000 Гц, при по меньшей мере 400 Вт) в течение 15-20 минут через 5-минутные интервалы. После 5-минутной обработки ультразвуком удаляют ледяную баню и продолжают дальнейшую обработку ультразвуком. В конце обработки ультразвуком температура суспензии в загрузочной воронке не превышает 75°С.
Полученную суспензию собирают в бутыль на 500 мл, которую сразу же охлаждают в холодильнике (2-8°С). Характеристики морфологии частиц суспензии до и после обработки ультразвуком были очень похожими на те, которые наблюдались в примере 1, за исключением того, что при способе вида 1 методе В предварительно обработанный ультразвуком материал имел склонность образовывать меньше и меньшего размера агрегаты, что давало в результате значительно меньший общий размер частиц, что измеряли по лазерной диффракции. После обработки ультразвуком результаты динамического рассеяния света были обычно идентичными результатам, представленным в примере 1.
В. Примеры способа вида 2
Пример 5. Получение суспензии итраконазола (1%) с 0,75% Solutol® HR (солутола HR) (ПЭГ-660 12-гидроксистеарат) способом вида 2, методом В.
Солутол (2,25 г) и итраконазол (3,0 г) отвешивали в стакан и добавляли 36 мл профильтрованного N-метил-2-пирролидинона (NMP). Эту смесь перемешивали при слабом нагревании (до 40°С) в течение примерно 15 минут до тех пор, пока ингредиенты раствора не растворялись. Раствор охлаждали до комнатной температуры и фильтровали через 0,2-микронный фильтр под вакуумом. Два 60-миллилитровых шприца заполняли профильтрованным концентратом лекарственного средства и помещали в поршневой насос. Насос устанавливали так, чтобы он поставлял примерно 1 мл/мин концентрата к быстро перемешиваемому (400 об/мин) водному буферному раствору. Буферный раствор состоял из 22 г/л глицерина в 5 мМ трис-буфере. Во время добавления концентрата буферный раствор выдерживали на ледяной бане при 2-3°С. В конце осаждения, после полного добавления концентрата к буферному раствору, примерно 100 мл суспензии центрифугировали в течение 1 часа, надосадочную жидкость (супернатант) отбрасывали. Осадок снова суспендировали в 20% растворе NMP в воде и снова центрифугировали в течение 1 часа. Материал сушили в течение ночи в вакуумной печи при 25°С. Высушенный материал переносили в сосуд и анализировали с помощью рентгеновской дифрактометрии, используя излучение хрома (см. фиг.5).
Другой 100 мл образец микроосажденной суспензии обрабатывали ультразвуком в течение 30 минут при 20000 Гц, 80% полной амплитуды (полная амплитуда = 600 Вт). Обработанный ультразвуков образец гомогенизировали в 3 равных аликвотах, каждую в течение 45 минут (Avestin С5, 2-5°С, 15000-20000 фунт/дюйм2). Объединенные фракции центрифугировали в течение примерно 3 часов, надосадочную жидкость удаляли и осадок снова суспендировали в 20% NMP. Повторно суспекдированную смесь снова центрифугировали (15000 об/мин при 5°С). Надосадочную жидкость декантировали и осадок сушили в вакууме в течение ночи при 25°С. Осадок анализировали рентгеновской дифрактометрией (см. фиг.5). Как видно на фиг.5, рентгенограммы обработанных образцов, перед и после гомогенизации, являются по существу идентичными, однако показывают значительно отличающиеся картины по сравнению с исходным сырьевым материалом. Негомогенизированная суспензия нестабильна и агломерирует при хранении при комнатной температуре. Стабилизация, которая происходит в результате гомогенизации, как полагают, является результатом перегруппировки ПАВ на поверхности частицы. Эта перегруппировка должна приводить в результате к более низкой склонности частиц к агрегации.
С. Примеры данного вида способа
Пример 6. Получение суспензии карбамазепина путем применения способа вида 3, метода А с гомогенизацией.
2,08 г карбамазепина растворяли в 10 мл NMP. 1,0 мл этого концентрата затем капали со скоростью 0,1 мл/мин в 20 мл перемешиваемого раствора 1,2% лецитина и 2,25% глицерина.
Температуру лецитиновой системы поддерживали на уровне 2-5°С во время всего добавления. Преддисперсию затем гомогенизировали охлажденной (5-15°С) в течение 35 минут при 15000 фунт/дюйм2. Давление повышали до 23000 фунт/дюйм2 и гомогенизацию продолжали в течение еще 20 минут. Частицы, полученные данным способом, имели средний диаметр 0,881 мкм, причем 99% частиц были менее 2,44 мкм.
Пример 7. Получение 1% суспензии карбамазепина с 0,125% Solutol® (солутола) путем применения способа вида 3, метода В с гомогенизацией.
Готовили концентрат лекарственного вещества 20% карбамазепина и 5% гликодезоксихолевой кислоты (Sigma Chemical Со.) в N-метил-2-пирролидиноне. Стадия микроосаждения включала добавление концентрата лекарства к полученному раствору (дистиллированная вода) со скоростью 0,1 мл/мин. Полученный раствор перемешивали и поддерживали при температуре приблизительно 5°С во время осаждения. После осаждения конечные концентрации ингредиентов составляли 1% карбамазепина и 0,125% солутола. Кристаллы лекарственного вещества изучали под световым микроскопом, применяя положительный фазовый контраст (400х). Осадок состоял из тонких игл диаметром примерно 2 микрона и длиной в интервале 50-150 микрон.
Гомогенизация (Avestin C-50 поршневой щелевой гомогенизатор) при примерно 20000 фунт/дюйм2 в течение примерно 15 минут дает в результате мелкие частицы размером менее 1 микрона и большей частью неагрегированные. Лазерный дифракционный анализ (Horiba) гомогенизированного материала показал, что частицы имели средний размер 0,4 микрона с 99% частиц менее 0,8 микрона. Низкоэнергетическая обработка ультразвуком, пригодная для разрушения агломерированных частиц, но с недостаточной энергией для того, чтобы вызвать дробление отдельных частиц образца перед анализом Horiba, не оказывала никакого эффекта на результаты (числа были одними и теми же с обработкой ультразвуком и без нее). Этот результат согласовывался с отсутствием агломерации частиц.
Образцы, полученные с помощью вышеприведенного способа, центрифугировали и растворы надосадочной жидкости заменяли замещающим раствором, состоящим из 0,125% солутола. После центрифугирования и замены надосадочной жидкости концентрации ингредиентов суспензии составляли 1% карбамазепина и 0,125% солутола. Образцы повторно гомогенизировали поршневым щелевым гомогенизатором и хранили при 5°С. После 4 недель хранения суспензия имела средний размер частиц 0,751 с 99% менее 1,729. Представленные числа являются данными анализа Horiba необработанных ультразвуком образцов.
Пример 8. Получение 1% суспензии карбамазепина с 0,06% гликодезоксихолата натрия и 0,06% полоксамера 188 путем применения способа вида 3, метода В с гомогенизацией.
Готовили концентрат лекарственного средства, содержащий 20% карбамазепина и 5% гликодезоксихолата в N-метил-2-пирролидиноне. Стадия микроосаждения включала добавление концентрата лекарства к полученному раствору (дистиллированная вода) со скоростью 0,1 мл/мин. Таким образом, этот и последующие примеры показывают, что добавление ПАВ или другого эксципиента к водному осаждающему раствору в методах А и В выше, является необязательным. Полученный раствор перемешивали и поддерживали при примерно 5°С во время осаждения. После осаждения конечные концентрации ингредиентов составляли 1% карбамазепина и 0,125% солутола®. Кристаллы лекарственного средства изучали под световым микроскопом, используя положительный фазовый контраст (400х). Осадок состоял из тонких игл диаметром примерно 2 микрона и длиной в интервале 50-150 микрон. Сравнение осадка с сырьевым материалом перед осаждением выявляет, что стадия осаждения в присутствии модификатора поверхности (гликодезоксихолезая кислота) дает в результате очень тонкие кристаллы, которые значительно тоньше, чем у исходного сырьевого материала (см. фиг.6).
Гомогенизация (Avestin C-50 поршневой щелевой гомогенизатор) при примерно 20000 фунт/дюйм2 в течение примерно 15 минут дает в результате мелкие частицы размером менее 1 микрона и большей частью неагрегированные (см. фиг.7). Лазерный дифракционный анализ (Horiba) гомогенизированного материала показал, что частицы имели средний размер 0,4 микрона с 99% частиц менее 0,8 микрона. Обработка образца ультразвуком перед анализом Horiba не оказывала никакого эффекта на результаты (числа были одними и теми же с обработкой ультразвуком и без нее). Этот результат согласовывался с отсутствием агломерации частиц.
Образцы, полученные с помощью вышеприведенного способа, центрифугировали и растворы надосадочной жидкости заменяли замещающим раствором, состоящим из 0,06% гликодезоксихолевой кислоты (Sigma Chemical Co.) и 0,06% полоксамера 188. Образцы повторно гомогенизировали поршневым щелевым гомогенизатором и хранили при 5°С. После 2 недель хранения суспензия имела средний размер частиц 0,531 микрон с 99% менее 1,14. Представленные числа являются данными анализа Horiba по необработанным ультразвуком образцам.
Математический анализ (пример 8) усилия, необходимого для того, чтобы разрушить осажденные частицы по сравнений с усилием, необходимым для разрушения частиц исходного сырьевого материала (карбамазепин).
Ширина самых больших кристаллов, наблюдаемых в сырьевом материале карбамазепина (фиг.6, изображение слева), приблизительно в 10 раз больше, чем ширина кристаллов в микроосажденном материале (фиг.6, изображение справа). Если предположить, что отношение толщины кристаллов (1:10 пропорционально отношению ширины кристаллов (1:10), то момент силы, необходимый для расщепления более крупных кристаллов в сырьевом материале, должен быть примерно в 1000 раз больше силы, необходимой для разрушения микроосажденного материала, так как
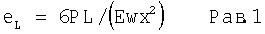
где еL - продольная нагрузка, необходимая для разрушения кристалла («деформация (yield value»);
Р - нагрузка по длине;
L - расстояние от нагрузки до точки опоры;
Е - модуль упругости;
W - ширина кристалла;
Х - толщина кристалла.
Примем, что L и Е являются одинаковыми для сырьевого материала и осажденного материала. Кроме того, примем, что w/w0=х/х0=10. Тогда
(eL)0=6P0L/(Ew0x0 2), где нижние индексы "0" относятся к сырьевому материалу,
еL=6РL/(Ewx2), относится к микроосадку
Уравнивая (еL)0 и еL,
6PL/(Ewx2)=6P0L/(Ew0x0 2).
После упрощения
P=P0(w/w0)(x/x0)2=P0(0,1)(0,1)2=0,001 P0.
Таким образом усилие деформации Р, необходимое для того, чтобы разрушить микроосажденное твердое вещество равно одной тысячной усилия, необходимого для разрушения исходного кристаллического твердого вещества. Если из-за быстрого осаждения привносятся дефекты кристаллической решетки или аморфные свойства, то модуль (Е) должен снижаться, делая микроосадок более легким для расщепления.
Пример 9. Получение 1,6% (вес/об) суспензии преднизолона с 0,05% дезоксихолата натрия и 3% N-метил-2-пирролидинона способом вида 3, методом 3.
Схема общего процесса производства представлена на фиг.8. Готовили концентрированный раствор преднизолона и дезоксихолата натрия. Преднизолон (32 г) и дезоксихолат натрия (1 г) добавляли к достаточному объему 1-метил-2-пирролидинона (NMP) с получением конечного объема 60 мл. Полученная концентрация преднизолона составляла примерно 533,3 мг/мл и концентрация дезоксихолата натрия была равна примерно 16,67 мг/мл. 60 мл концентрата NMP добавляли к 2 л воды, охлажденной до 5°С, со скоростью добавления 2,5 мл/мин при перемешивании со скоростью примерно 400 об/мин. Полученная суспензия содержала тонкие в форме игл кристаллы шириной менее 2 мкм (фиг.9). Концентрации, содержащиеся в осажденной суспензии, составляли 1,6% (вес/об) преднизолона, 0,05% дезоксихолата натрия и 3% NMP.
рН осажденной суспензии доводили до 7,5-8,5 с использованием гидроксида натрия и соляной кислоты, затем гомогенизировали (Avestin C-50 поршневой щелевой гомогенизатор) за 10 проходов при 10000 фунт/дюйм2. NMP удаляли путем осуществления двух последовательных стадий центрифугирования, заменяя каждый раз надосадочную жидкость свежим раствором ПАВ, который содержал желаемые концентрации ПАВ, необходимые для стабилизации суспензии (см. таблицу 2). Суспензию гомогенизировали в течение еще 10 проходов при 10000 фунт/дюйм2. Конечная суспензия содержала частицы со средним размером менее 1 мкм и 99% частиц менее 2 мкм. Фиг.10 является микрофотографией конечной суспензии преднизолона после гомогенизации.
На стадии центрифугирования/замены ПАВ использовали ряд различных ПАВ при различных концентрациях (см. таблицу 2). В таблице 2 приведен список сочетаний ПАВ, которые были стабильны в отношении размера частиц (средний <1 мкм, 99% <2 мкм), рН (6-8), концентрации лекарственного вещества (менее 2% потери) и способности к повторному суспендированию (повторно суспендируемые за 60 секунд или менее).
В особенности этот способ дает возможность добавления активного соединения к водному разбавителю без присутствия ПАВ или другой добавки. Этот способ является модификацией процесса метода В на фиг.2.
Перечень стабильных суспензий преднизолона, изготовленных способом микроосаждения с фигуры 8 (пример 9)
** Стабильна на протяжении по меньшей мере 6 месяцев.
Размеры частиц (по рассеянию лазерного света) в микронах:
5°С: 0,80 (средний), 1,7 (99%)
25°С: 0,90 (средний), 2,51 (99%)
40°С: 0,99 (средний), 2,03 (99%)
Различие по концентрации итраконазола между образцами, хранившимися при 5 и 25°С: <2%
Пример 10. Получение суспензии преднизолона путем применения способа вида 3, метода А с гомогенизацией.
32 г преднизолона растворяли в 40 мл NMP. Для осуществления растворения было необходимо легкое нагревание. Концентрат лекарственного средства в NMP затем капали при скорости 2,5 мл/мин в 2 л перемешиваемого раствора, который состоял из 0,1,2% лецитина и 2,2% глицерина. Никаких других модификаторов поверхности не добавляли. Систему ПАВ забуферивали при рН=8,0 5 мМ трис-буфером и температуру поддерживали при 0-5°С в течение всего процесса осаждения. Дисперсию после осаждения затем гомогенизировали охлажденной (5-15°С) за 20 проходов при 10000 фунт/дюйм2. После гомогенизации NMP удаляли центрифугированием суспензии, удаляя надосадочную жидкость и заменяя надосадочную жидкость свежим раствором ПАВ. Эту суспензию после центрифугирования затем снова гомогенизировали охлажденной (5-15°С) за еще 20 проходов при 10000 фунт/дюйм2. Частицы, полученные при этом способе, имели средний диаметр, равный 0,927 мкм, причем 99% частиц менее 2,36 мкм.
Пример 11. Получение суспензии набуметона путем применения способа вида 3, метода В с гомогенизацией.
ПАВ (2,2 г полоксамера 188) растворяли в 6 мл N-метил-2-пирролидинона. Этот раствор перемешивали при 45°С в течение 15 минут, после чего добавляли 1,0 г набуметона. Лекарственное вещество быстро растворяли. Готовили разбавитель, который состоял из 5 мМ трис-буфера с 2,2% глицерина и доводила до рН 8. Часть разбавителя (100 мл) охлаждали на ледяной бане. Концентрат лекарственного вещества медленно (примерно 0,8 мл/мин) добавляли к разбавителю при энергичном перемешивании. Эту грубую суспензию гомогенизировали при 15000 фунт/дюйм2 в течение 30 минут и затем при 20000 фунт/дюйм2 в течение 30 минут (температура 5°С). Конечная наносуспензия, как было обнаружено, являлась 930 нм по эффективному среднему диаметру (анализировали методом лазерной диффракции). 99% частиц были менее примерно 2,6 микрон.
Пример 12. Получение суспензии набуметона путем применения способа вида 3, метода В с гомогенизацией и использованием (Solutol® HS 15 (солутола HS 15) в качестве ПАВ. Замена надосадочной жидкости средой с фосфолипидом.
Набуметон (0,987 г) растворяли в 8 мл N-метил-2-пирролидона. К этому раствору добавляли 2,2 г солутола® HS 15. Эту смесь перемешивали до полного растворения ПАВ в концентрате лекарственного вещества. Готовили разбавитель, который состоял из 5 мМ трис-буфера с 2,2% глицерина и который доводили до рН 8. Разбавитель охлаждали на ледяной бане и концентрат лекарственного вещества медленно (примерно 0,5 мл/мин) добавляли к разбавителю при энергичном перемешивании. Эту грубую суспензию гомогенизировали в течение 20 минут при 15000 фунт/дюйм2 и в течение 30 минут при 20000 фунт/дюйм2.
Суспензию центрифугировали при 15000 об/мин в течение 15 минут, надосадочную жидкость удаляли и отбрасывали. Оставшийся плотный осадок снова суспендировали в разбавителе, содержащем 1,2% фосфолипидов. Эта среда была равной по объему количеству удаленной надосадочной жидкости на предыдущей стадии. Полученную суспензию затем гомогенизировали при примерно 21000 фунт/дюйм2 в течение 30 минут. Конечную суспензии анализировали с помощью лазерной диффракции, и было обнаружено, что она содержит частицы со средним диаметром 542 нм, и 99% в общем распределении частиц имели размер менее 1 микрона.
Пример 13. Получение 1% суспензии итраконазола с полоксамером с частицами со средним диаметром примерно 220 нм.
Концентрат итраконазола готовили растворением 10,02 г итраконазола в 60 мл N-метил-2-пирролидинона. Для растворения лекарственного вещества было необходимо нагревание до 70°С. Затем раствор охлаждали до комнатной температуры. Готовили часть 50 мМ трис(гидроксиметил)аминометанового буфера (трис-буфер) и рН доводили до 8,0 5 М соляной кислотой. Водный раствор ПАВ готовили объединением 22 г/л полоксамера 407, 3,0 г/л яичных фосфолипидов, 22 г/л глицерина и 3,0 г/л дигидрата холата натрия. 900 мл раствора ПАВ смешивали со 100 мл трис-буфера с получением 1000 мл водного разбавителя.
Водный разбавитель добавляли в загрузочную воронку гомогенизатора (APV Gaulin Model 15MR-8TA), который охлаждали, применяя ледяную рубашку. Раствор быстро перемешивали (4700 об/мин) и температуру контролировали. Медленно добавляли концентрат итраконазола, используя поршневой насос, со скоростью примерно 2 мл/мин. Добавление завершалось примерно через 30 минут. Полученную суспензию перемешивали в течение еще 30 минут, тогда как загрузочную воронку все еще охлаждали в ледяной рубашке, и отбирали образец для анализа с помощью световой микроскопии и динамического рассеяния света. Оставшуюся суспензию затем гомогенизировали в течение 15 минут при 10000 фунт/дюйм2. В конце гомогенизации температура повышалась до 74°С. Гомогенизированную суспензию собирали в 1-литровый стеклянный флакон типа I и герметично закрывали резиновой крышкой. Флакон, содержавший суспензию, хранили в холодильнике при 5°С.
Образец суспензии перед гомогенизацией, как показано, состоит из свободных частиц, агрегатов частиц и многослойных липидных телец. Свободные частицы не могли быть четко различимы из-за броуновского движения; однако, многие из агрегатов, по-видимому, состояли из аморфного некристаллического материала.
Гомогенизированный образец содержал свободные субмикронные частицы, имеющие превосходную гомогенность по размеру без видимых липидных пузырьков. Динамическое рассеяние света показало монодисперсное логарифмическое распределение по размеру со средним диаметром примерно 220 нм. Кумулятивный размер по 99% верхнему срезу был равен примерно 500 нм. Фиг.11 демонстрирует сравнение распределения по размеру полученной наносуспензии с распределением типичного продукта парентеральной масляной эмульсии (10% Intralipid®, Pharmacia).
Пример 14. Получение 1% наносуспензии итраконазола с гидроксиэтилкрахмалом (ГЭК).
Получение раствора А. Гидроксиэтилкрахмал (1 г, Ajinomoto) растворяли в 3 мл N-метил-2-пирролидоне (NMP). Этот раствор нагревали на водяной бане до 70-80°С в течение 1 часа. В другой контейнер добавляли 1 г итраконазола (Wyckoff). Добавляли 3 мл NMP и смесь нагревали до 70-80°С, чтобы осуществить растворение (примерно 30 минут). К этому горячему раствору добавляли фосфолипид (Lipoid S-100). Нагревание продолжали при 70-90°С в течение 30 минут, до тех пор когда фосфолипид растворялся. Раствор гидроксиэтилкрахмала объединяли с раствором итраконазола/фосфолипида. Эту смесь нагревали в течение еще 30 минут при 80-95°С, чтобы растворить смесь.
Добавление раствора А к трис-буферу. Девяносто четыре (94) мл 50 мМ трис(гидроксиметил)аминометанового буфера охлаждали на ледяной бане. В то время как трис-раствор быстро перемешивали, медленно, каплями (менее 2 см3/минуту) добавляли горячий раствор А (см. выше).
После завершения добавления полученную суспензию обрабатывали ультразвуком (ультразвуковой процессор Cole-Palmer - 20000 Гц, 80% амплитудная регулировка) при продолжающемся охлаждении на ледяной бане. Использовали однодюймовый твердый зонд. Обработку ультразвуком продолжали в течение 5 минут. Ледяную баню удаляли, зонд удаляли и перенастраивали, и зонд снова погружали в суспензию. На суспензию снова воздействовали ультразвуком в течение еще 5 минут без ледяной бани. Зонд ультразвуковой установки еще раз удаляли и перенастраивали, и после погружения зонда образец снова обрабатывали ультразвуком в течение еще 5 минут. В это время температура суспензии повышалась до 82°С. Суспензию снова быстро охлаждали на водяной бане и когда устанавливали, что ее температура ниже комнатной температуры, ее выливали в стеклянный флакон типа I и герметично закрывали. Визуальное наблюдение частиц под микроскопом показало размер отдельных частиц порядка одного микрона или менее.
После одного года хранения при комнатной температуре суспензию снова оценивали в отношении размера частиц, и обнаружено, что они имеют средний диаметр, равный примерно 300 нм.
Пример 15. Предсказывающий пример метода А с использованием ГЭК (HES).
В данном изобретении рассматривается получение 1% наносуспензии итраконазола с гидроксиэтилкрахмалом при применении метода А, следуя стадиям из примера 14, за исключением того, что ГЭК должен добавляться к трис-буферному раствору вместо раствора NMP. Водный раствор может нагреваться, чтобы растворить ГЭК.
Пример 16. Затравливания во время гомогенизации для превращения смеси полиморфов в более стабильный полиморф.
Получение образца. Наносуспензию итраконазола получали методом микроосаждения-гомогенизации следующим образом. Итраконазол (3 г) и Solutol HR (солутол HR) (2,25 г) растворяли в 36 мл N-метил-2-пирролидинона (NMP) при слабом нагревании и перемешивании с получением раствора концентрата лекарственного вещества. Раствор охлаждали до комнатной температуры и фильтровали через 0,2-микрометровый найлоновый фильтр под вакуумом для удаления нерастворенного лекарственного вещества или частиц материала. Раствор рассматривали в поляризованном свете, чтобы убедиться, что никакого кристаллического вещества не присутствует в нем после фильтрования. Раствор концентрата лекарственного вещества затем добавляют со скоростью 1,0 мл/мин к примерно 264 мл водного буферного раствора (22 г/л глицерина в 5 мМ трис-буфере). Водный раствор выдерживали при 2-3°С и непрерывно перемешивали при примерно 400 об/мин во время добавления концентрата. Примерно 100 мл полученной суспензии центрифугировали и твердые вещества снова суспендировали в предварительно профильтрованном растворе 20% NMP в воде. Эту суспензию снова центрифугировали и твердые вещества переносили в вакуумную печь для сушки в течение ночи при 25°С. Полученный твердый образец обозначали SMP 2 PRE.
Определение характеристик образца. Образец SMP 2 PRE и образец сырьевого материала итраконазола анализировали, используя порошковую рентгеновскую дифрактометрию. Определения выполняли, используя прибор Rigaku MiniFlex+ с излучением меди, размером шага 0,02° 2θ и скоростью сканирования 0,25° 2θ/мин. Полученные порошковые рентгенограммы показаны на фиг.12. Картины показывают, что SMP-2-PRE значительно отличается от сырьевого материала, что наводит на мысль о наличии другого полиморфа или псевдополиморфа.
Данные дифференциальной сканирующей калориметрии (ДСК) для образцов показаны на фиг.13а и 13b. Оба образца нагревали при скорости 2°/мин до 180°С в герметично закрытых алюминиевых сосудах.
Данные для сырьевого материала итраконазола (фиг.13а) показывают острый эндотерм при примерно 165°С.
Данные для SMP 2 PRE (фиг. 13b) показывают две эндотермы при примерно 159 и 153°С. Этот результат, в сочетании с порошковой рентгенограммой, наводит на мысль о том, что SMP 2 PRE состоит из смеси полиморфов и что преобладающей формой является полиморф, который менее стабилен, чем полиморф, присутствующий в сырьевом материале.
Дополнительное доказательство для этого заключения представлено данными ДСК на фиг.14, которые показывают, что при нагревании SMP 2 PRE в течение первого перехода, затем охлаждении и повторном нагревании менее стабильный полиморф плавится и перекристаллизовывается с образованием более стабильного полиморфа.
Затравливание. Суспензию получали объединением 0,2 г твердого SMP 2 PRE и 0,2 г сырьевого материала итраконазола с дистиллированной водой до конечного объема 20 мл (образец с затравкой (после затравливания)). Суспензию перемешивали до тех пор, пока все твердые вещества не увлажнялись. Вторую суспензию получали таким же образом, но без добавления сырьевого материала итраконазола (образец без затравки). Обе суспензии гомогенизировали при примерно 18000 фунт/дюйм2 в течение 30 минут. Конечная температура суспензий после гомогенизации была равна приблизительно 30°С. Суспензии затем центрифугировали и твердые вещества сушили в течение примерно 16 часов при 30°С.
Фиг.15 показывает данные ДСК образцов с затравкой и без затравки. Скорость нагревания для обоих образцов составляла 2°/мин до 180°С в герметически закрытых алюминиевых сосудах. Данные для образца без затравки показывают два эндотерма, указывающие на то, что смесь полиморфов все еще присутствует после гомогенизации. Данные для образца с затравкой показывают, что затравка и гомогенизация вызывают превращение твердых веществ в стабильный полиморф. Поэтому затравливание, по-видимому, влияет на кинетику перехода из менее стабильной в более стабильную полиморфную форму.
Пример 17. Затравливание во время осаждения до предпочтительной формы стабильного полиморфа.
Получение образца. Концентрат лекарственного вещества итраконазола в NMP получали растворением 1,67 г итраконазола в 10 мл NMP при перемешивании и легком нагревании. Раствор дважды фильтровали, используя 0,2-микрометровые шприцевые фильтры. Наносуспензии итраконазола затем получали добавлением 1,2 мл концентрата. лекарства к 20 мл принимающего водного раствора при примерно 3°С и перемешивании при около 500 об/мин. Наносуспензию с затравкой получали путем использования смеси примерно 0,02 г сырьевого материала итраконазола в дистиллированной воде в качестве принимающего раствора. Наносуспензию без затравки получали путем использования только дистиллированной воды в качестве принимающего раствора. Обе суспензии центрифугировали, надосадочные жидкости декантировали и твердые вещества сушили в вакуумной печи при 30°С в течение примерно 16 часов.
Определение характеристик образцов. Фиг.16 представляет сравнение данных ДСК для твердых веществ из суспензий с затравкой и без затравки. Образцы нагревали при скорости 2°/мин до 180°С в герметично закрытых алюминиевых сосудах. Пунктирная линия представляет образец без затравки, который показывает два эндотерма, указывающие на наличие полиморфной смеси.
Сплошная линия представляет образец с затравкой, который показывает только один эндотерм вблизи ожидаемой температуры плавления сырьевого материала, указывающий, что затравочный материал вызывал исключительно образование более стабильного полиморфа.
Пример 18. Контроль полиморфа посредством затравливания концентрата лекарственного вещества.
Получение образца. Экспериментально определено, что растворимость итраконазола в NMP при комнатной температуре (примерно 22°С) составляет 0,16 г/мл. Концентрированный раствор лекарственного вещества 0,20 г/мл получали растворением 2,0 г итраконазола и 0,2 г полоксамера 188 в 10 мл NMP при нагревании и перемешивании. Этому раствору затем давали охладиться до комнатной температуры с получением пересыщенного раствора. Сразу же производили эксперимент по микроосаждению, при котором 1,5 мл концентрата лекарственного вещества добавляли к 30 мл водного раствора, содержащего 0,1% дезоксихолата, 2,2% глицерина. Водный раствор выдерживали при ˜2°С и скорости перемешивания, равной 350 об/мин во время стадии добавления. Полученную предсуспензию гомогенизировали при ˜13000 фунт/дюйм2 в течение примерно 10 минут при 50°С. Суспензию затем центрифугировали, надосадочную жидкость декантировали и твердые кристаллы сушили в вакуумной печи при 30°С в течение 135 часов.
Пересыщенный концентрат лекарственного вещества затем подвергали старению путем хранения при комнатной температуре, чтобы индуцировать кристаллизацию. Через 12 дней концентрат лекарственного вещества был мутным, указывая, что произошло образование кристаллов. Суспензию итраконазола получали из концентрата лекарственного вещества таким же образом, что и в первом эксперименте путем добавления 1,5 мл - 30 мл водного раствора, содержащего 0,1% дезоксихолата, 2,2% глицерина. Водный раствор выдерживали при ˜5°С и скорости перемешивания 350 об/мин во время стадии добавления. Полученную предсуспензию гомогенизировали при ˜13000 фунт/дюйм2 в течение примерно 10 минут при 50°С. Затем суспензию центрифугировали, надосадочную жидкость декантировали и затем твердые кристаллы сушили в вакуумной печи при 30°С в течение 135 часов.
Определение характеристик образца. Порошковый рентгеноструктурный анализ использовали для определения морфологии высушенных кристаллов. Полученные изображения представлены на фиг.17. Кристаллы из первого эксперимента (с использованием свежего концентрата лекарственного вещества), как было определено, состояли из более стабильного полиморфа. В противоположность этому кристаллы из второго эксперимента (концентрат лекарственного вещества, подвергнутый старению) предпочтительно состояли из менее стабильного полиморфа с присутствием небольшого количества более стабильного полиморфа. Поэтому полагают, что старение вызывало образование кристаллов менее стабильного полиморфа в концентрате лекарственного вещества, которые затем действовали в качестве затравочного материала во время стадий микроосаждения и гомогенизации, так что предпочтительно образуется менее стабильный полиморф.
Хотя конкретные варианты осуществления были проиллюстрированы и описаны, могут быть осуществлены многочисленные модификации без отклонения от сущности данного изобретения, и объем защиты ограничивается только объемом сопровождающей формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ СУБМИКРОННЫХ ЧАСТИЦ ПАКЛИТАКСЕЛА | 2004 |
|
RU2402313C2 |
| ПОЛУЧЕНИЕ 2-(5-БРОМ-4-(4-ЦИКЛОПРОПИЛНАФТАЛИН-1-ИЛ)-4H-1,2,4-ТРИАЗОЛ-3-ИЛТИО)УКСУСНОЙ КИСЛОТЫ | 2013 |
|
RU2666549C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ ДОСТАВКИ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2009 |
|
RU2496482C2 |
| СПОСОБ ПОЛУЧЕНИЯ МАГНИТОЧУВСТВИТЕЛЬНЫХ ЛИПОСОМ | 2007 |
|
RU2357724C1 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ДОСТАВКИ С РЕГУЛИРУЕМЫМ ВЫСВОБОЖДЕНИЕМ БИОЛОГИЧЕСКИ АКТИВНЫХ СОЕДИНЕНИЙ | 2005 |
|
RU2390355C2 |
| ПРЕПАРАТЫ ГИДРОФОБНЫХ ТЕРАПЕВТИЧЕСКИХ СРЕДСТВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2013 |
|
RU2663117C2 |
| ПРЕПАРАТЫ ГИДРОФОБНЫХ ТЕРАПЕВТИЧЕСКИХ СРЕДСТВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2013 |
|
RU2789479C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВОДОРАСТВОРИМЫХ ФОРМ БИОЛОГИЧЕСКИ АКТИВНЫХ ВЕЩЕСТВ | 2007 |
|
RU2388491C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2011 |
|
RU2589842C2 |
| СПОСОБ ОБРАБОТКИ УГЛЕРОДНОГО ВОЛОКНА АППРЕТИРУЮЩИМ СОСТАВОМ, СОВМЕСТИМЫМ С ВЫСОКОТЕМПЕРАТУРНЫМИ ТЕРМОПЛАСТИЧНЫМИ СВЯЗУЮЩИМИ | 2023 |
|
RU2815005C1 |
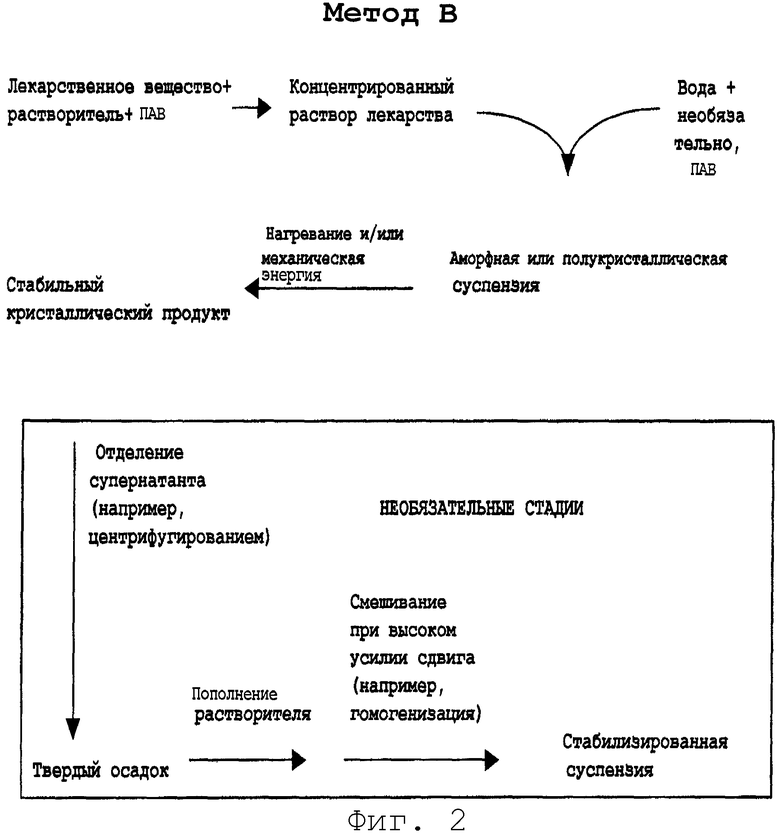


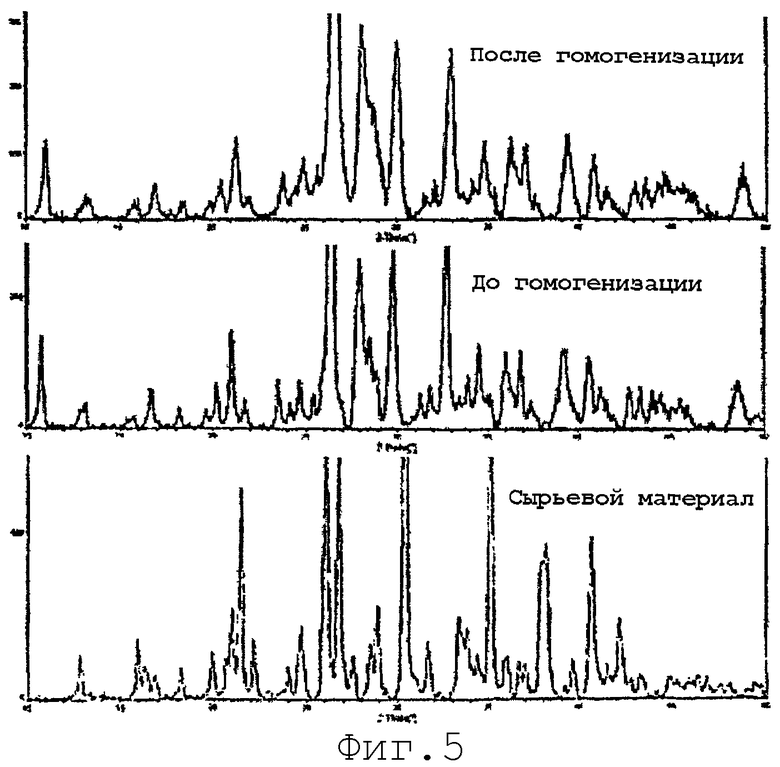
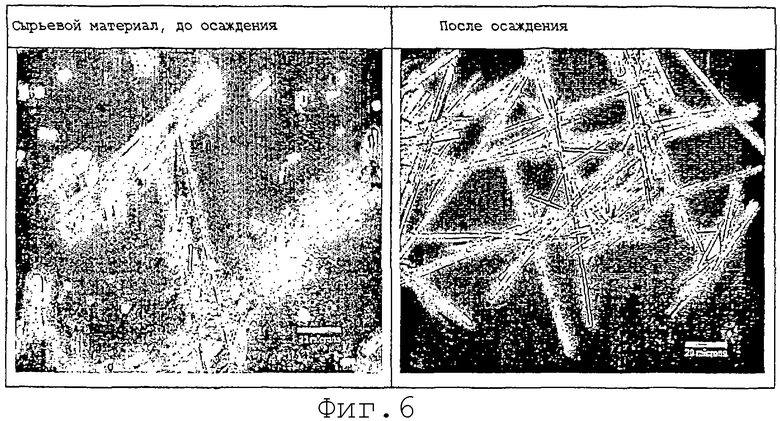
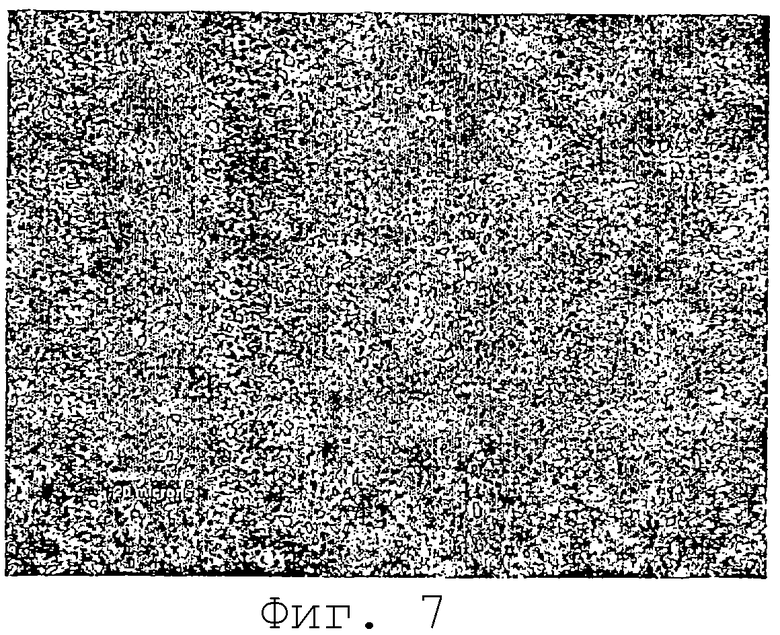
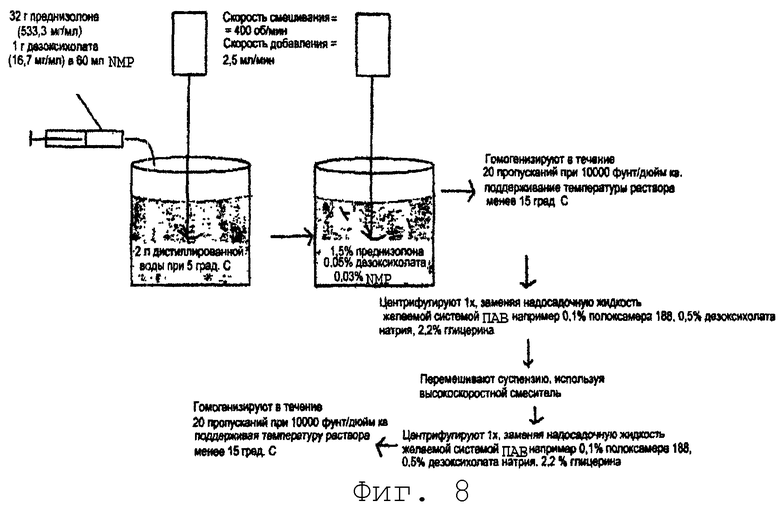


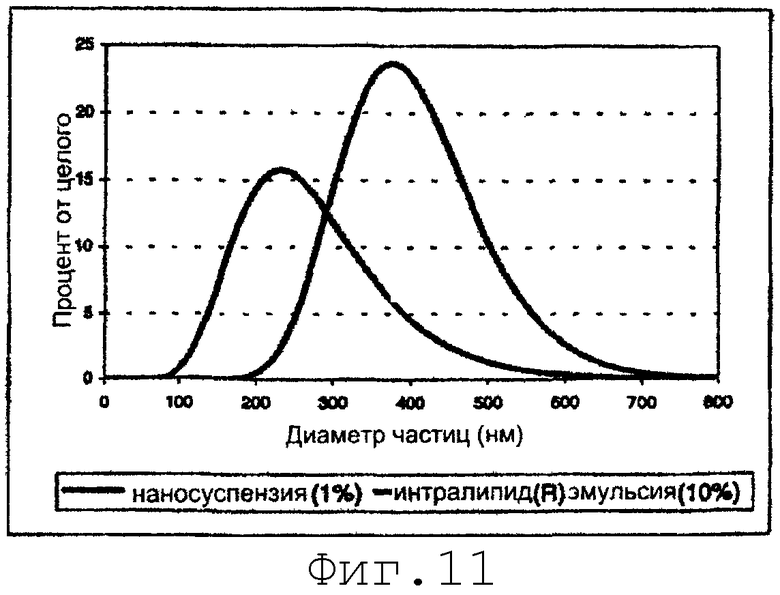
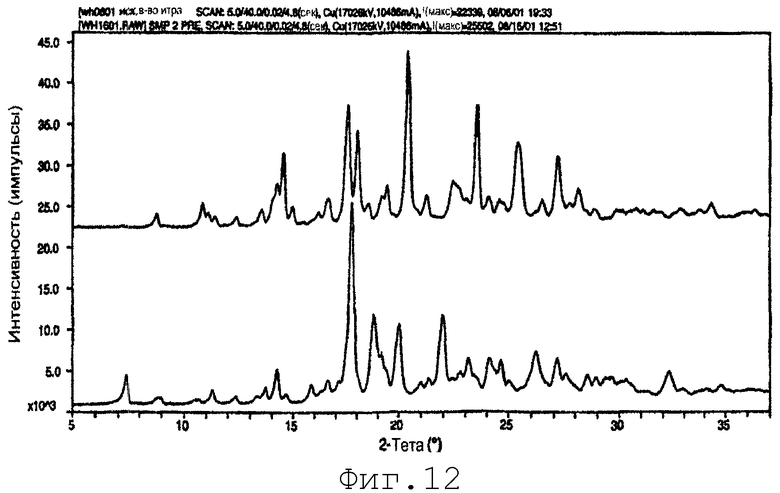
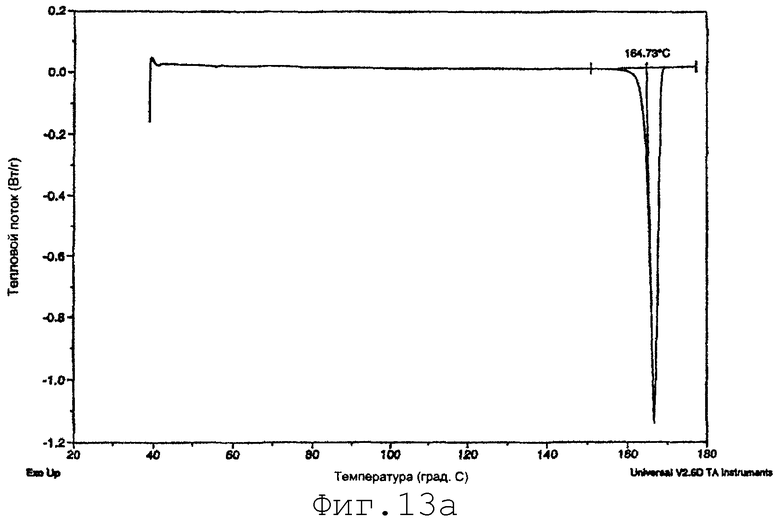
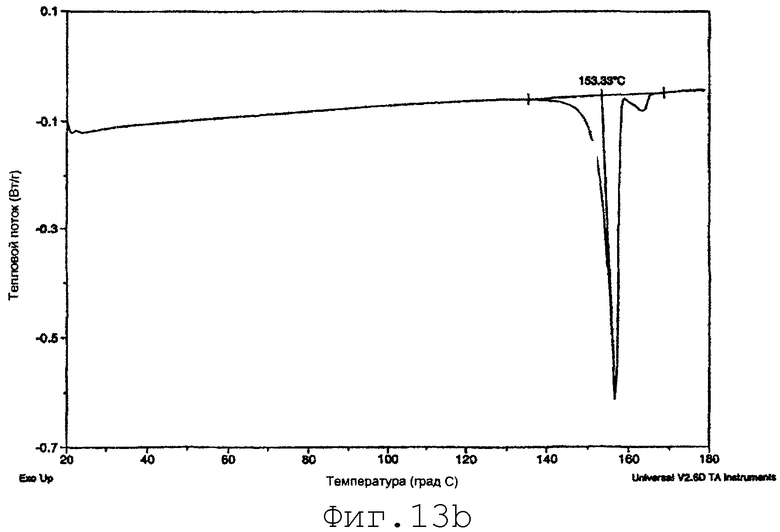
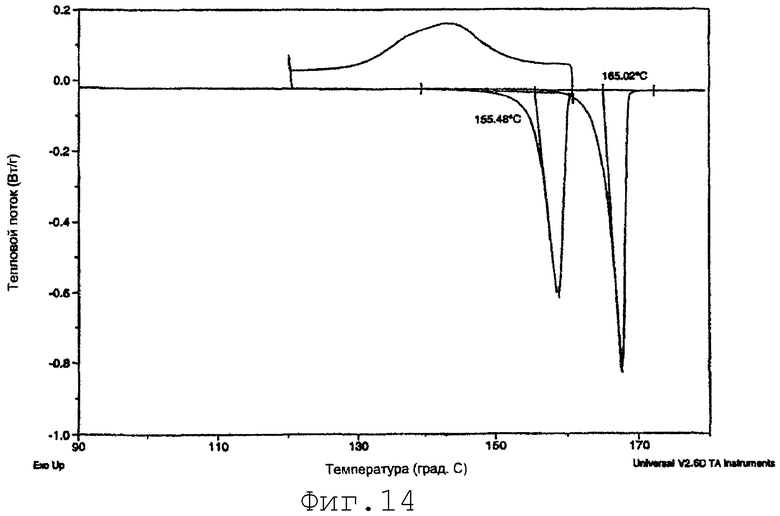
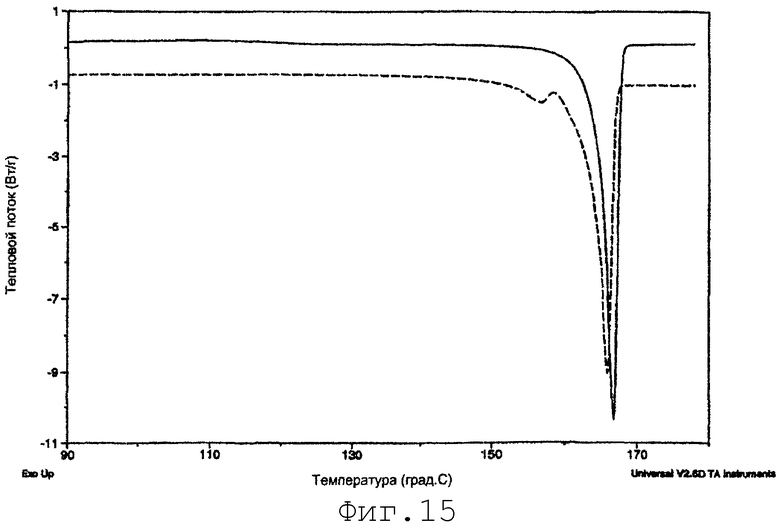
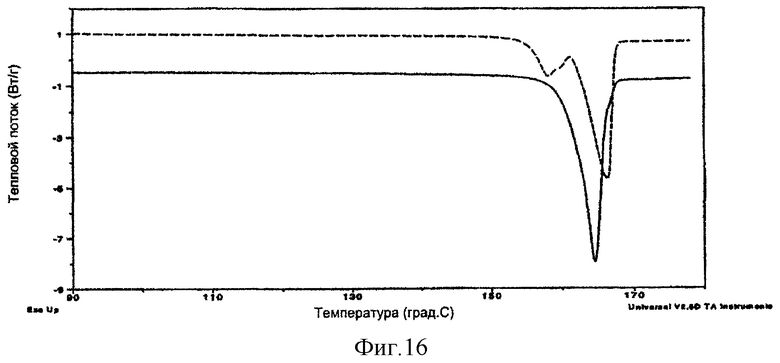
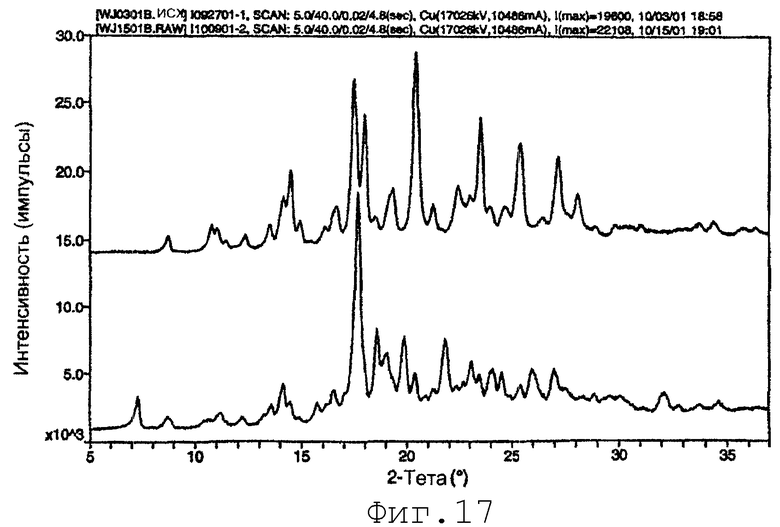
Изобретение относится к области фармацевтики и касается способа получения субмикронных частиц водонерастворимого или плохо водорастворимого органического фармацевтического соединения путем растворения этого соединения в смешиваемом с водой первом растворителе, смешивания полученного раствора со вторым растворителем для создания предсуспензии и гомогенизации полученной предсуспензии или воздействия на нее ультразвуком. Способ позволяет получить частицы размером менее 2 мкм. 2 н. и 30 з.п. ф-лы, 18 ил., 2 табл.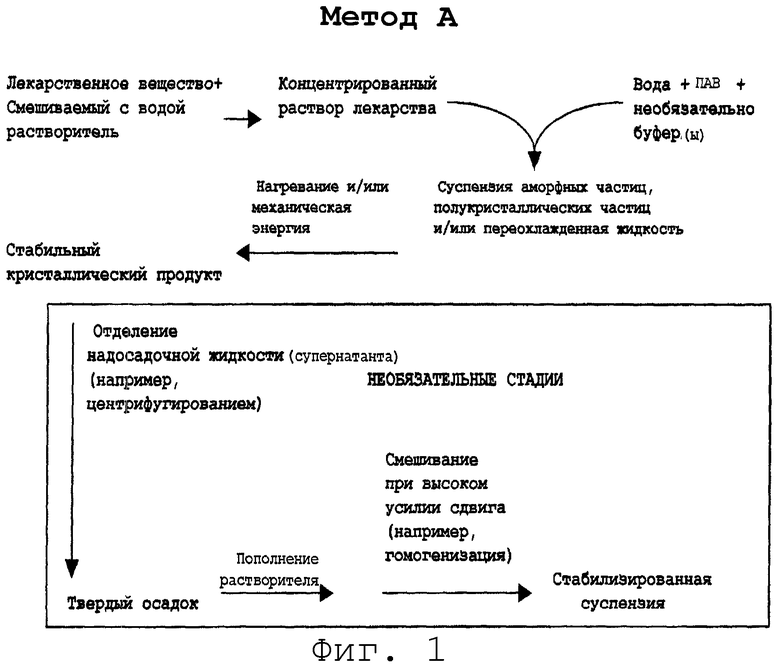
(i) растворения органического соединения в смешиваемом с водой первом растворителе с образованием раствора, причем первый растворитель выбирают из группы, состоящей из N-метил-2-пирролидинона, 2-пирролидона, диметилсульфоксида, диметилацетамида, молочной кислоты, метанола, этанола, изопропанола, 3-пентанола, н-пропанола, глицерина, бутиленгликоля, этиленгликоля, пропиленгликоля, моно- и диацилированных моноглицеридов, диметилизосорбида, ацетона, диметилформамида, 1,4-диоксана, этилацетата, пропилацетата, полиэтиленгликоля, сложных эфиров полиэтиленгликоля, полиэтиленгликольсорбитанов, моноалкилэфиров полиэтиленгликоля, полипропиленгликоля, полипропиленальгината, пропиленгликоль-10 бутандиола, эфира пропиленгликоля-10 и метилглюкозы, эфира пропиленгликоля-20 и метилглюкозы, стеарилового эфира пропиленгликоля-15, пропиленгликоля дикаприлата, пропиленгликоля дикапрата, пропиленгликоля лаурата,
(ii) смешивания раствора со вторым растворителем для создания предсуспензии, и
(iii) гомогенизации или гомогенизации в противотоке полученной предсуспензии или воздействия на нее ультразвуком для образования частиц, имеющих средний размер частиц менее примерно 2 мкм.
(i) растворения первого количества фармацевтически активного соединения в смешиваемом с водой первом органическом растворителе с образованием первого раствора, причем первый органический растворитель выбирают из группы, состоящей из N-метил-2-пирролидинона, 2-пирролидона, диметилсульфоксида, диметилацетамида, молочной кислоты, метанола, этанола, изопропанола, 3-пентанола, н-пропанола, глицерина, бутиленгликоля, этиленгликоля, пропиленгликоля, моно- и диацилированных моноглицеридов, диметилизосорбида, ацетона, диметилформамида, 1,4-диоксана, этилацетата, пропилацетата, полиэтиленгликоля, сложных эфиров полиэтиленгликоля, полиэтиленгликольсорбитанов, моноалкилэфиров полиэтиленгликоля, полипропиленгликоля, полипропиленальгината, пропиленгликоль-10 бутандиола, эфира пропиленгликоля-10 и метилглюкозы, эфира пропиленгликоля-20 и метилглюкозы, стеарилового эфира пропиленгликоля-15, пропиленгликоля дикаприлата, пропиленгликоля дикапрата, пропиленгликоля лаурата,
(ii) смешивания первого раствора со вторым растворителем для осаждения фармацевтически активного соединения для создания предсуспензии,
(iii) затравливания первого раствора или второго растворителя перед стадией смешивания или затравливания предсуспензии после стадии смешивания, причем указанное затравливание включает добавление затравочного соединения указанного органического фармацевтически активного соединения, и
(iv) получения суспензии частиц, имеющих средний размер частиц менее чем примерно 2 мкм, путем подведения энергии гомогенизации, гомогенизации в противотоке или воздействия ультразвуком.
По пп.1, 2, 5-10, 13-15, 18-21, 23-25 установлен конвенционный приоритет от 22.12.00 согласно первой заявке 60/258160, поданной в Патентное ведомство США;
по пп.3, 4, 11, 12, 16, 17, 22, 27 установлен конвенционный приоритет от 05.06.01 согласно первым заявкам 09/874799, 09/874637, 09/874499, поданным в Патентное ведомство США;
по пп.26, 28-32 установлен конвенционный приоритет от 19.10.01 согласно первой заявке 10/035821, поданной в Патентное ведомство США.
| US 4826689, А, 02.05.1989 | |||
| Г.БЕККЕР и др | |||
| Органикум | |||
| Практикум по органической химии | |||
| - М.: Мир, 1979, с.54-57 | |||
| US 5145684, А, 08.09.1998 | |||
| КАСАТКИН А.Г | |||
| Основные процессы и аппараты химической технологии | |||
| - М.: Химия, 1971, с.670-671 | |||
| Химическая энциклопедия | |||
| Под ред | |||
| И.Л.Кнунянца | |||
| - М.: Советская Энциклопедия, 1990, т.2, с.528. |

