Предшествующий уровень техники
Область изобретения
Настоящее изобретение относится к способу получения N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида и исходных материалов для него.
Уровень техники
Соединение N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамид (альтернативно, N-гидрокси-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриламид) имеет формулу:
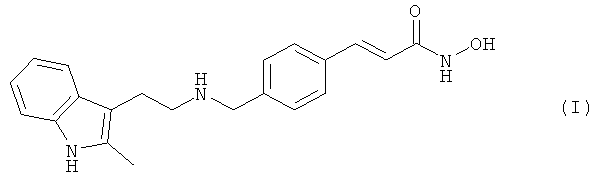
как описано в международной заявке на патент WO 02/22577. Это соединение обладает ценными фармакологическими свойствами; таким образом, могут быть использованы, например, в качестве ингибитора гистондеацетилазы, применяемого для лечения заболеваний, которые реагируют на ингибирование активности гистондеацетилазы. Предшествующие попытки изобретателей получить это соединение имели ограниченный успех из-за наличия различных примесей и побочных продуктов в продукте реакции; удаление таких примесей и побочных продуктов требует продолжительной и однообразной обработки/перекристаллизации целевого продукта. Таким образом, синтез N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида до сих пор был сложным, продолжительным и ограничен малой производительностью синтеза.
Сущность изобретения
Настоящее изобретение относится к способу получения N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида, включающему следующие стадии: (а) объединение гидроксида натрия и гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты с получением смеси при температуре ниже около -10°С; и далее (б) добавление гидроксиламина в смесь с получением N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил}фенил]-2Е-2-пропенамида. Способ также необязательно включает стадию (в) кристаллизации N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида. В предпочтительном варианте осуществления изобретения стадия (в) включает следующие подстадии: (в1) нагревание полученной на стадии (б) реакционной смеси; (в2) перемешивание реакционной смеси; (в3) добавление воды к реакционной смеси; (в4) фильтрация реакционной смеси с получением фильтрата; (в5) регулирование уровня рН фильтрата до уровня рН от 10 до 11; (в6) добавление затравочных кристаллов N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида к фильтрату; (в7) перемешивание фильтрата до образования суспензии; (в8) корректировка уровня рН суспензии до уровня рН от 8,5 до 9; и (в9) перемешивание суспензии. Способ также необязательно включает стадию (г) выделение N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида. В предпочтительном варианте получения стадия (г) включает следующие подстадии: (г1) фильтрация кристаллизованного на стадии (в) N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида; и (г2) высушивание кристаллизованного N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида.
Изобретение также относится к способу получения гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты, включающему следующие стадии: (а) объединение 2-метилтриптамина и метилового эфира (Е)-3-(4-формилфенил)акриловой кислоты с получением смеси; (б) перемешивание полученной смеси в течение и при температуре, достаточной для получения промежуточного соединения имина; (в) восстановление промежуточного соединения имина с получением метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты и добавление водного раствора хлористоводородной кислоты к реакционной смеси с получением соли гидрохлорида. В предпочтительном варианте осуществления изобретения стадия (в) включает следующие подстадии: (в1) охлаждение полученной смеси; (в2) добавление боргидрида натрия к полученной смеси; и (в3) объединение полученной смеси с хлористоводородной кислотой для кристаллизации/осаждения гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты. В следующем предпочтительном варианте осуществления подстадия (в3) включает следующие подстадии: (в3а) нагревание полученной на подстадии (в2) смеси; (в3б) добавление воды в полученную смесь и (в3в) добавление хлористоводородной кислоты в полученную смесь для кристаллизации гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты. Способ также необязательно включает стадию (г) нагревание и охлаждение суспензии гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты для расщепления полученных комплексов амин-бор в реакционной смеси. В предпочтительном варианте осуществления изобретения стадия (г) включает следующие подстадии: (г1) нагревание полученной суспензии, когда на стадии (в) образуется полученная соль гидрохлорида; (г2) перемешивание суспензии при такой же температуре, как и на стадии (г1); (г3) охлаждение суспензии и (г4) перемешивание суспензии при такой же температуре, как и на стадии (г3). Способ также необязательно включает стадию (д) выделение гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты. В предпочтительном варианте осуществления стадия (д) включает следующие подстадии: (д1) фильтрация суспензии, полученной на стадии (г); и (д2) высушивание гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты.
Настоящее изобретение также относится к способу получения 2-метилтриптамина, включающему следующие стадии: (а) получение смеси фенилгидразина и 5-хлор-2-пентанона в этаноле при начальной температуре; (б) добавление этанола в полученную смесь и кипячение смеси с обратным холодильником; (в) дистилляция этанола; (г) добавление воды в полученный раствор; (д) охлаждение полученного раствора с получением 2-метилтриптамина. В предпочтительном варианте осуществления настоящего изобретения способ также включает стадию (е) выделения и очистки 2-метилтриптамина. В предпочтительном варианте осуществления настоящего изобретения стадия (е) включает следующие подстадии: (e1) промывание полученного раствора толуолом; (е2) выделение 2-метилтриптамина; (е3) очищение 2-метилтриптамина толуолом и (е4) высушивание 2-метилтриптамина.
Подробное описание изобретения
Настоящее изобретение относится к способу получения N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида, включающему следующие стадии: (а) объединение гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты и гидроксида натрия с получением смеси при температуре ниже около -10°С; и далее (б) добавление гидроксиламина в полученную смесь с получением N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида, как показано ниже:
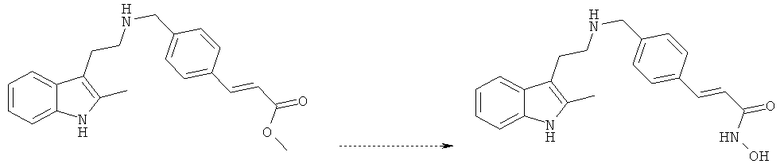
Кроме того, способ по изобретению может включать стадии кристаллизации и выделения N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида. Способ по изобретению не требует большой продолжительности, стадий сложной обработки/перекристаллизации продукта реакции. Другими словами, способ по изобретению дает возможность получить N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамид одностадийным способом с высоким выходом и достаточной чистотой.
На первой стадии способа по первому варианту осуществления гидрохлорид метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты и гидроксид натрия объединяют при температуре ниже около -10°С с получением смеси. Более предпочтительно, при температуре ниже около -15°С. В первом варианте осуществления, при температуре около 0°С. Температура может быть выбрана и поддерживаться любыми подходящими способами. Кроме того, смесь может быть получена в любом подходящем сосуде, который не должен содержать железа и тяжелых металлов, реакция проходит в инертной атмосфере. Тяжелые металлы и железо (металлический Fe и его соли) катализируют распад гидроксиламина.
Как правило, гидрохлорид метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты (также известный как метиловый эфир 3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)-(2Е)-2-пропеновой кислоты получают в форме суспензии в метаноле. Предпочтительно, гидрохлорид метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты помещают в подходящий реакционный сосуд, в который добавляли метанол, и конечную суспензию затем охлаждают до температуры ниже около -10°С. Гидрохлорид метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты может быть получен в соответствии со способом второго варианта осуществления этого изобретения. Гидрохлорид метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты предпочтительно определяют как 1,0 эквивалент. Как правило, гидроксид натрия используют в виде раствора, предпочтительно в метаноле. Гидроксид натрия является коммерчески доступным исходным материалом. Гидроксид натрия предпочтительно используется в количестве от 2,5 до 3,5 эквивалентов.
Предпочтительно метанольный раствор гидроксида натрия добавляют в суспензию гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты в подходящем реакционном сосуде в течение определенного времени. Более предпочтительно, раствор гидроксида натрия добавляют в течение 20-40 минут, предпочтительно в течение 30 минут, пока поддерживается температура ниже около -10°С.
На второй стадии способа по первому варианту осуществления, гидроксиламин добавляют в смесь, полученную на первой стадии, для получения N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида. Температура на первой стадии, а именно, ниже около -10°С, поддерживается только на этой стадии.
Как правило, гидроксиламин используют в виде раствора в воде. Гидроксиламин является коммерчески доступным материалом. Гидроксиламин предпочтительно используют в количестве от 4 до 13 эквивалентов. Предпочтительно водный раствор гидроксиламина, например, 50% водный раствор, добавляют к смеси гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты и гидроксиду натрия в подходящем реакционном сосуде в течение определенного периода времени. Более предпочтительно, раствор гидроксиламина добавляют в течение около 20-24 минут, предпочтительно около 30 минут, пока температура поддерживается на уровне ниже около -10°С. Во время протекания стадии (б) нужно осторожно добавлять к смеси гидроксиламина таким образом, чтобы устранить контакт с любым оборудованием или сосудом, которые были использованы для гидроксида натрия на стадии (а), например, не должна использоваться та же самая воронка; другими словами, реакция между гидроксиламином и гидроксидом натрия должна быть проведена до добавления гидроксиламина в смесь. Кроме того, все сосуды и переходники, которые используют для добавления гидроксиламина, не должны содержать железо и тяжелые металлы.
В предпочтительном варианте осуществления настоящего изобретения смесь перемешивают при такой же температуре, как на стадии (а) до тех пор, пока реакция не завершится или практически завершится; как правило, протекание реакции занимает около 7 часов. Процесс завершения реакции можно проконтролировать с помощью ВЭЖХ; в одном предпочтительном варианте осуществления конверсия составляет >99,5 %.
Способ первого варианта осуществления может также включать стадию (в) кристаллизации N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида. В предпочтительном варианте осуществления настоящего изобретения такая стадия может включать некоторые или все подстадии: (в1) нагревание реакционной смеси, полученной на стадии (б); (в2) перемешивание реакционной смеси; (в3) добавление воды к реакционной смеси; (в4) фильтрация реакционной смеси с получением фильтрата; (в5) корректировка уровня рН фильтрата до уровня рН от около 10 до около 11; (в6) добавление затравочных кристаллов N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида к фильтрату; (в7) перемешивание фильтрата до получения суспензии; (в8) корректировка уровня рН суспензии до уровня рН от около 8,5 до около 9; и (в9) перемешивание суспензии. Предпочтительно, все стадии (в1) - (в9) проводят при температуре нагревания стадии (в1).
На стадии (в1) реакционная смесь получается, когда гидроксиламин добавляют к исходной смеси метилового эфира и гидроксида натрия. Предпочтительно реакционную смесь нагревают до температуры от около 0°С до около 25°С. Нагревание может осуществляться любым подходящим способом. На стадии (в2), реакционную смесь перемешивают при температуре, применявшейся при выполнении стадии (в1). Предпочтительно реакционную смесь перемешивают в течение примерно 13 часов. В одном предпочтительном варианте осуществления настоящего изобретения, стадии (в1) и (в2) повторяют для обеспечения постепенного нагревания, т.е. реакционную смесь сначала нагревают до температуры от около 0°С до около 5°С и перемешивают около 4-6 часов, предпочтительно 5 часов, и затем нагревают до температуры от около 20°С до около 25°С и перемешивают дополнительно около 8-16 часов.
На стадии (в3) к реакционной смеси добавляют воду. Как правило, деминерализованную воду добавляют в течение определенного времени, более предпочтительно в течение 30 минут при температуре около 20-25°С, чтобы получить раствор.
На стадии (в4) реакционную смесь отфильтровывают для получения фильтрата. Фильтрация может проводиться любым подходящим способом или с любой подходящей средой. Стадия (в4) также необязательно включает подстадию промывки фильтровальной среды и добавления промывки к полученному фильтрату.
На стадии (в5) уровень рН фильтрата корректируется до уровня рН от 10 до 11, более предпочтительно от 10,3 до 10,7. Добавление водной хлористоводородной кислоты, как правило, применяется для достижения этой цели.
На стадии (в6) к фильтрату добавляют затравочные кристаллы N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида. Это, как правило, достигается путем введения водной суспензии кристаллов N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида.
На стадии (в7) фильтрат перемешивают до получения суспензии. Предпочтительно фильтрат перемешивают от около 30 минут до нескольких часов, пока кристаллизация не станет очевидной и видимой.
На стадии (в8) уровень рН суспензии корректируют до уровня рН от 8,5 до 9. Для этих целей, как правило, используют дополнительное количество водного раствора хлористоводородной кислоты.
На стадии (в9) суспензию перемешивают до тех пор, пока N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамид кристаллизуется. Предпочтительно суспензию перемешивают в течение от около 30 минут до нескольких часов, пока не будет завершена реакция. Способ по первому варианту осуществления может включать стадию (г) выделение N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида. В предпочтительном варианте осуществления настоящего изобретения, такая стадия может включать некоторые или все подстадии: (г1) фильтрация кристаллизованного N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида, полученного на стадии (в); и (г2) сушка кристаллизованного N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида.
На стадии (г1) кристаллизованный N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамид отфильтровывают. Фильтрация может осуществляться любыми подходящими способами. Как правило, остаток на фильтре промывают, например, смесью 1:1 деминерализованной воды и метанола.
На стадии (г2) кристаллизованный N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамид сушат. Сушка может осуществляться любым подходящим способом. Как правило, достаточно сушить при 45-50°С/1-5 мбар в течение 24 часов.
Второй вариант осуществления настоящего изобретения относится к способу получения гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты, одного из исходных материалов, которые использовались для синтеза N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил}фенил]-2Е-2-пропенамида в первом варианте осуществления. В частности, второй вариант осуществления настоящего изобретения относится к способу получения гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты, включающему следующие стадии: (а) объединение 2-метилтриптамина и метилового эфира (Е)-3-(4-формилфенил)акриловой кислоты для получения смеси; (б) перемешивание смеси в течение и при температуре, достаточной для образования промежуточного соединения имина; и (в) восстановление промежуточного соединения имина с получением гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты.
На первой стадии способ по второму варианту осуществления 2-метил/триптамин и метиловый эфир (Е)-3-(4-формилфенил)акриловой кислоты объединяют для образования смеси, предпочтительно при температуре от около 20°С до около 25°С. Смесь может быть получена в любом подходящем сосуде. Как правило, 2-метилтриптамин и метиловый эфир (Е)-3-(4-формилфенил)акриловой кислоты растворяют в растворителе, например, метаноле, для получения смеси.
2-Метилтриптамин получают в соответствии с известными способами синтеза или получают в соответствии со способом по третьему варианту осуществления изобретения, описанным ниже. 2-Метилтриптамин предпочтительно используют в количестве от 0,9 до 1,0 эквивалентов. Метиловый эфир (Е)-3-(4-формилфенил)акриловой кислоты может быть получен из коммерческих источников или получен в соответствии с известными способами синтеза. Метиловый эфир (Е)-3-(4-формилфенил)акриловой кислоты предпочтительно используют в количестве от 1,0 до 1,1 эквивалентов.
На второй стадии смесь 2-метилтриптамина и метилового эфира (Е)-3-(4-формилфенил)акриловой кислоты перемешивают в течение времени и при температуре, достаточной для получения промежуточного соединения имина:

Предпочтительно смесь перемешивают около 1 часа при температуре от около 20 до около 25°С. На стадии (в) способа по второму варианту осуществления промежуточное соединение имина восстанавливают боргидридом натрия с получением метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты, который затем кристаллизовали/осаждали как соль гидрохлорида. В предпочтительном варианте осуществления изобретения, стадия (в) включает подстадии: (в1) охлаждение смеси; (в2) добавление боргидрида натрия в смесь и (в3) объединение смеси с хлористоводородной кислотой для кристаллизации/осаждения гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты.
В первом варианте осуществления изобретения смесь разбавляют растворителем, например, метанолом, до подстадии (в1). Охлаждение на подстадии (в1) может осуществляться любым известным способом, например, в ледяной бане, охлаждающей рубашкой, и т.д. Предпочтительно смесь охлаждают при температуре от около -10 до -20°С, предпочтительно -15°С.
Добавление боргидрида натрия на подстадии (в2) предпочтительно осуществляют порциями в течение времени, поддерживая температуру подстадии (в1). Более предпочтительно, добавление осуществляют в течение 1 часа и при температуре, поддерживаемой в пределах от около -15°С до около -10°С. Предпочтительно боргидрид натрия добавляют в твердой форме и в количестве от около 0,4 до около 0,7 эквивалентов.
На подстадии (в3) смесь объединяют с кислотой, например, хлористоводородной кислотой, для осаждения гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты. Предпочтительно эту подстадию выполняют после периода перемешивания смеси на подстадии (в2). Объединение с хлористоводородной кислотой может осуществляться в соответствии с этим изобретением несколькими путями. Один предпочтительный способ включает медленное добавление смеси к предварительно охлажденному водному раствору хлористоводородной кислоты; как правило, раствор хлористоводородной кислоты охлаждают при температуре от около 0°С до около 5°С.
Другой предпочтительный способ объединения с хлористоводородной кислотой включает подстадии: (в3а) нагревание смеси, полученной на подстадии (в2); (в3б) добавление воды в смесь и (в3в) добавление хлористоводородной кислоты в смесь для осаждения гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты. На подстадии (в3а) смесь нагревают при температуре от около 20°С до около 25°С в течение 20-45 минут, предпочтительно около 25 минут. На подстадии (в3б) медленно добавляют воду, предпочтительно после этапа перемешивания смеси, полученной на подстадии (в3а). На подстадии (в3в) предпочтительно хлористоводородная кислота является водной и ее добавляют медленно порциями. Предпочтительно, первая порция хлористоводородной кислоты является такой, которая необходима для установления уровня рН реакционной смеси 8,5. Раствор хлористоводородной кислоты добавляют так, чтобы температура поддерживалась в пределах 20-25°С. Затем реакционную смесь перемешивают при этой температуре в течение по крайней мере одного часа, чтобы осуществить кристаллизацию продукта, перед тем как добавить еще хлористоводородную кислоту.
Способ по второму варианту осуществления может также включать стадию (г) нагревания и охлаждения суспензии гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты для улучшения качества и увеличения фильтрационной способности. В предпочтительном варианте осуществления такая стадия может включать некоторые или все подстадии: (г1) нагревание суспензии, полученной при восстановлении промежуточного соединения имина и добавлении хлористоводородной кислоты для получения гидрохлорида на стадии (в); (г2) перемешивание суспензии при температуре подстадии (г1); (г3) охлаждение суспензии и (г4) перемешивание суспензии при температуре подстадии (г3). Предпочтительно использовать температуру подстадии (г1) от около 60°С до около 65°С и осуществлять нагревание в течение от около 30 минут до около 45 минут. Предпочтительно перемешивание на подстадии (г2) проводят в течение от около 5 минут до около 30 минут. Предпочтительно температуру на подстадии (г3) поддерживают от около -15°С до около -10°С и проводят на протяжении от около 45 минут до около 1 часа. Предпочтительно перемешивание на подстадии (г4) проводят в течение от около 30 минут до нескольких часов. В частности, в предпочтительном варианте осуществления этого изобретения все подстадии (г1) - (г4) повторяют один или несколько раз; для достижения большей эффективности температуру осаждения поддерживают в пределах -15°С и 65°С до фильтрации продукта.
Способ по второму варианту осуществления изобретения может также включать стадию (д) выделения гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты. В предпочтительном варианте осуществления такая стадия может включать некоторые или все подстадии: (д1) фильтрование суспензии, полученной на стадии (г); и (д2) высушивание гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты.
На стадии (д1) суспензию кристаллизованного гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты, полученную на стадии (г), отфильтровывают. Фильтрация может быть проведена любым подходящим способом. Как правило, остаток на фильтре промывают, например, предварительно охлажденной (около -10°С) смесью деминерализованной воды и метанола или предварительно охлажденным (около -15°С) метанолом.
На стадии (д2) кристаллизованный гидрохлорид метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты сушат. Высушивание может осуществляться любым подходящим способом. Особенно предпочтительным является высушивание при 50°С при пониженном давлении.
Третий вариант осуществления настоящего изобретения относится к способу получения исходного материала, который используется в синтезе гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты. В частности, третий вариант осуществления настоящего изобретения относится к способу получения 2-метилтриптамина, включающему стадии: (а) получение смеси фенилгидразина и 5-хлор-2-пентанона в этаноле при первой температуре; (б) добавление этанола в смесь и кипячение смеси с обратным холодильником; (в) дистилляция этанола; (г) добавление воды к оставшемуся раствору; (д) охлаждение оставшегося раствора для получения 2-метилтриптамина.
На первой стадии способа по третьему варианту осуществления смесь фенилгидразина и 5-хлор-2-пентанона получают в этаноле при первой температуре. Фенилгидразин, 5-хлор-2-пентанон и этанол являются коммерчески доступными в качестве исходных материалов. Для целей этого изобретения предпочтительно использовать эквимолярные количества фенилгидразина и 5-хлор-2-пентанона. Таким образом, фенилгидразин предпочтительно использовать в количестве от 0,5 до 1,5, а 5-хлор-2-пентанон предпочтительно использовать в количестве от 1 до 2. В предпочтительном варианте осуществления настоящего изобретения стадия (а) включает подстадии: (al) получение раствора фенилгидразина в этаноле; (а2) нагревание раствора при температуре от около 30°С до около 40°С, более предпочтительно при температуре около 30-40°С; (а3) проведение реакции при температуре от 35°С до 45°С, пока 5-хлор-2-пентанон добавляют к реакционной смеси, как правило, в течение около 15-45 минут; и (а4) проведение реакции в течение около 30-60 минут при температуре стадии (а3). На этой стадии осторожное поддержание температуры и временных рамок являются важными условиями контроля наличия посторонних примесей.
На второй стадии способа по третьему варианту осуществления этанол добавляют в смесь и кипятят смесь с обратным холодильником. Этанол предпочтительно добавляют в количестве от 10 до 20 частей. Как правило, реакционную смесь сразу же нагревают при кипячении с обратным холодильником и выдерживают в течение минимум 50-60 минут. После кипячения с обратным холодильником реакционную смесь, как правило, охлаждают до комнатной температуры в течение примерно 20 минут.
На третьей стадии способа по третьему варианту осуществления этанол отгоняют. Дистилляция может осуществляться любым подходящим способом; для этих целей наиболее предпочтительна дистилляция в вакууме. Частичную дистилляцию этанола, как правило, осуществляют при определении объема сосуда.
На четвертой стадии способа по третьему варианту осуществления воду добавляют в оставшийся раствор. Воду предпочтительно добавляют в количестве от 10 до 20 частей. В обычном способе дистилляцию продолжают при тех же условиях путем удаления этанола и затем добавления дополнительного количества воды к остаточной смеси. Воду предпочтительно добавляют в количестве от 10 до 20 частей.
На пятой стадии способа по третьему варианту осуществления оставшийся раствор охлаждают для получения 2-метилтриптамина. Как правило, оставшийся раствор охлаждают до температуры ниже около 25°С.
Способ по третьему варианту осуществления может также включать стадию (е) выделения и очищения 2-метилтриптамина. В предпочтительных вариантах осуществления настоящего изобретения стадия (е) включает подстадии: (e1) промывка остаточного раствора толуолом; (е2) выделение 2-метилтриптамина; (е3) промывка 2-метилтриптамина толуолом и (е4) высушивание 2-метилтриптамина.
На стадии (e1) остаточный раствор промывают толуолом. На стадии (е2) выделяют 2-метилтриптамин. Выделение может достигаться любым подходящим способом. На стадии (е3) 2-метилтриптамин промывают толуолом, предпочтительно охлажденным толуолом, а именно ≤0°С. На стадии (е4) 2-метилтриптамин сушат. Высушивание может осуществляться любым подходящим способом. Высушивание в вакууме при 45°С до достижения LOD <1% наиболее предпочтительно.
Способ третьего варианта получения может использоваться для получения 2-метилтриптамина, который является исходным материалом в синтезе гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты.
Некоторые варианты осуществления изобретения будут показаны далее со ссылкой на следующие примеры. Следует понимать, что эти примеры раскрываются только лишь путем пояснения изобретения и не могут быть использованы в любом случае для ограничения объема настоящего изобретения.
Пример 1
Получение N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида
Гидрохлорид метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты (90 г, 233,8 ммоля) помещали в 4-горлую колбу для реакций и добавляли метанол (475 г). Суспензию охлаждали до -15°С. Раствор гидроксида натрия (28,2 г, 705 ммолей) в метаноле (419,2 г) добавляли к суспензии при -15°С (время добавления приблизительно 30 минут), затем добавляли раствор гидроксиламина (100,3 г 50%-го раствора в воде, соответствующего 50,15 г гидроксиламина, 1518 ммолей) при этой температуре (время добавления приблизительно 30 минут). Предупреждение: важно использовать разные капельные воронки для растворов гидроксида натрия и гидроксиламина соответственно.
Перемешивание продолжали при -15°С дополнительно в течение 7 часов, до тех пор, пока конверсия составит >99,5% в соответствии с ВЭЖХ. Реакционную смесь нагревали до 0°С, перемешивали в течение 5 часов при температуре 0-5°С, нагревали до 20°С и перемешивание продолжали в течение 8 часов при температуре 20-25°С. Деминерализованную воду (225 г) добавляли к суспензии при 20-25°С в течение 30 минут для получения раствора. Раствор отфильтровывали и фильтр, так же как и фильтрационную трубку, промывали деминерализованной водой (225 г). Уровень рН раствора корректировали до уровня 10,3-10,7 с помощью добавления водного раствора хлористоводородной кислоты (приблизительно 140 г 7,8 м/м % раствора в воде). Затравочные кристаллы добавляли в виде суспензии свободного основания N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида (80 мг) в воде (5 г), и смесь перемешивали в течение приблизительно 30 минут при температуре 20-25°С до получения суспензии. Уровень рН суспензии затем корректировали до уровня 8,5-9,0 путем добавления водного раствора хлористоводородной кислоты (приблизительно 108 г 7,8 м/м % раствора в деминерализованной воде) при температуре 20-25°С, и перемешивание продолжали по крайней мере в течение 30 минут при температуре 20-25°С. Полученное твердое вещество выделяли путем фильтрации, и остаток на фильтре промывали 1:1 (об./об.) смесью деминерализованной воды и метанола (140 мл). Сырой продукт сушили при температуре 45-50°С/5 мбар в течение 24 часов с получением N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида. Выход: 81,15 г; 99,3% от теоретического. ВЭЖХ анализ показал 97,6 % чистоту продукта, который включал вес./вес. 3,2 % воды. Содержание гидроксиламина составляло 345 ppm, что достаточно для получения соответствующего лактата с <5 ppm гидроксиламина.
Пример 2
Получение гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил]этиламино]метил}фенил)акриловой кислоты
2-Метилтриптамин (100 г, 573,8 ммоля) и метиловый эфир (Е)-3-(4-формилфенил)акриловой кислоты (115 г, 604,6 ммоля) растворяли в метаноле (1250 мл). Раствор перемешивали в течение 1 часа при температуре 20-25°С до образования промежуточного соединения имина. Раствор разбавляли метанолом (1250 мл) и охлаждали до -15°С. Боргидрид натрия (16,25 г, 429,5 ммолей) добавляли небольшими порциями в течение примерно 1 часа, поддерживая температуру от -15°С до -10°С. Реакционную смесь перемешивали дополнительно в течение 30 минут при этой температуре, и реакцию гасили медленным добавлением реакционной смеси к предварительно охлажденному раствору хлористоводородной кислоты (488 г концентрированной хлористоводородной кислоты в 337 г воды и 198 г метанола) при температуре 0-5°С. Получали суспензию. Капельную воронку промывали метанолом (40 г) и температуру повышали до 60-65°С в течение 1 часа. Суспензию перемешивали в течение 1 часа при температуре 60-65°С и затем температуру понижали до -15°С в течение 1 часа. Суспензию перемешивали в течение 1 часа при температуре от -15°С до -10°С и продукт выделяли посредством фильтрации. Сырой остаток на фильтре промывали небольшими порциями предварительно охлажденной смеси (-10°С) воды (300 мл) и метанола (600 мл). Сырой продукт сушили при температуре 50°С при пониженном давлении с получением гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты в качестве продукта. Как правило, продукт имел чистоту >99% в соответствии с ВЭЖХ. ИК, ЯМР и HR-MS подтверждали предполагаемую структуру. Температура плавления: разрушение начинается при температуре 251-252°С.
Пример 3
Получение гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты
2-Метилтриптамин (50 г, 287 ммолей) и метиловый эфир (Е)-3-(4-формилфенил)акриловой кислоты (54,6 г, 287 ммолей) суспендировали в метаноле (514 г). Раствор перемешивали в течение 1 часа при температуре 20-25°С, чтобы обеспечить образование промежуточного соединения имина. Раствор охлаждали до -15°С в течение примерно 20 минут. Боргидрид натрия (5,43 г, 143,5 ммоля) добавляли небольшими порциями в течение приблизительно 1 часа, поддерживая температуру от -15°С до -10°С. Реакционную смесь перемешивали дополнительно в течение 30 минут при этой температуре и затем температуру повышали до 20-25°С в течение примерно 25 минут. Реакционную смесь перемешивали в течение 30 минут при температуре 20-25°С и медленно добавляли воду (80 г), поддерживая температуру 20-25°С. Водный раствор хлористоводородной кислоты (70,5 г концентрированной HCl в 50 г воды) медленно добавляли в реакционную смесь, поддерживая температуру на уровне 20-25°С и контролируя выделение газа водорода. В этом случае добавление проводили в течение приблизительно 1 часа. Вторую часть водного раствора хлористоводородной кислоты (70,5 г концентрированной HCl в 50 г воды) добавляли в течение 30 минут, и температуру поднимали до 65°С в течение 30 минут. Суспензию перемешивали в течение 30 минут при температуре 65°С и затем температуру понижали до -15°С в течение 45 минут. Через 10 минут перемешивания при температуре -15°С температуру снова повышали до 65°С и суспензию перемешивали в течение 30 минут при этой температуре. В конечном итоге суспензию охлаждали до -15°С в течение 45 минут и перемешивание продолжали еще в течение 30 минут при этой температуре. Продукт выделяли путем фильтрации, и сырой остаток на фильтре промывали предварительно охлажденным (-15°С) метанолом (2×150 г). Сырой продукт сушили при температуре 50°С при пониженном давлении с получением чистого гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты в качестве продукта. Обычно продукт имел чистоту >99% в соответствии с ВЭЖХ. ИК, ЯМР и HR-MS подтверждали предполагаемую структуру. Температура плавления: разрушение начинается при температуре 251-252°С.
Пример 4
Получение гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты
2-Метилтриптамин (50 г, 287 ммолей) и метиловый эфир (Е)-3-(4-формилфенил)акриловой кислоты (54,6 г, 287 ммолей) суспендировали в метаноле (514 г). Раствор перемешивали в течение 1 часа при температуре 20-25°С, чтобы обеспечить образование промежуточного соединения имина. Раствор охлаждали до температуры -15°С в течение приблизительно 20 минут. Боргидрид натрия (5,43 г, 143,5 ммоля) добавляли несколькими порциями в течение приблизительно 1 часа, поддерживая температуру от -15°С до -10°С. Реакционную смесь перемешивали дополнительно в течение 30 минут при этой температуре и затем температуру повышали до 20-25°С в течение 25 минут. Полученную смесь перемешивали в течение 30 минут при температуре 20-25°С и воду (80 г) медленно добавляли, поддерживая температуру 20-25°С. Уровень рН реакционной смеси корректировали до уровня 8,5 путем медленного добавления водного раствора хлористоводородной кислоты (приблизительно 37,3 г 21,6 м/м % раствора в воде) при температуре 20-25°С до тех пор, пока уровень рН раствора не достигал 8,5. В этом случае добавление проводили в течение приблизительно 30 минут. После окончания добавления реакционную смесь перемешивали в течение одного часа при температуре 20-25 °С для обеспечения кристаллизации. На этой стадии в раствор могут быть добавлены затравочные кристаллы гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты для ускорения процесса кристаллизации. Получали суспензию. Вторую порцию водной хлористоводородной кислоты (36 г 21,6 м/м % HCl раствора в воде) добавляли к суспензии в течение 30 минут при температуре 20-25°С, затем добавляли третью порцию водной хлористоводородной кислоты (167,7 г 21,6 м/м % HCl раствора в воде) в течение 30 минут при температуре 20-25°С. Суспензию нагревали до 65°С. Затем суспензию охлаждали до -15°С и перемешивали в течение 30 минут при температуре от -10 до -15°С для завершения процесса кристаллизации. Продукт выделяли путем фильтрации и сырой остаток на фильтре промывали предварительно охлажденным (-15°С) метанолом (2×150 г). Сырой продукт сушили при температуре 50°С при пониженном давлении с получением чистого гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты в качестве продукта. Обычно продукт имеет чистоту >99 % в соответствии с ВЭЖХ. ИК, ЯМР и HR-MS подтверждали предполагаемую структуру. Температура плавления: разрушение начинается при температуре 251-252°С.
Пример 5
Получение 2-метилтриптамина
Фенилгидразин (64,92 г, 0,60 моль) и этанол (278 г, 350 мл, 200 proof) загружали в круглодонную колбу объемом 2 л. Раствор перемешивали в атмосфере азота и нагревали до температуры 35°С. Реакцию проводили при температуре 35-45°С, и 5-хлор-2-пентанон (74,54 г, 0,60 моль, 97%) добавляли через капельную воронку. Температуру поддерживали в пределах 35-41°С и добавление 5-хлор-2-пентанона осуществляли в течение 30 минут. Затем реакцию проводили при температуре 35-40°С в течение 30 минут. Затем в реакционную смесь добавляли этанол (556 г, 700 мл, 190 proof). Реакционную смесь незамедлительно нагревали при кипячении с обратным холодильником и выдерживали в течение 50 минут. Затем реакционную смесь охлаждали до комнатной температуры в течение 20 минут.
Затем колбу оснащали для вакуумной дистилляции. Этанол перегоняли при 35 мм Hg в водяной бане при 35-45°С до 350 мл (собирали 685 г, 820 мл дистиллята). Деионизированную воду (500 г) добавляли к остатку раствора. Дистилляцию проводили заранее при тех же условиях до 450 мл (собирали 332 г, 360 мл дистиллята). Деионизированную воду (400 г) добавляли к мутной оставшейся смеси. Смесь охлаждали до температуры ниже 25°С и полученную смесь промывали толуолом (2×347 г, 400 мл).
Продукт (2-метилтриптамин) выделяли путем фильтрации. Осадок промывали толуолом (130 г, 150 мл, охлаждали до температуры ≤0°С). Продукт сушили в вакууме при 45°С до достижения LOD <1%. Теоретический выход 104,5 г; фактически выход составил 49,2 г.
Хотя изобретение было описано со ссылкой на конкретные варианты осуществления, предполагается, что множество изменений, модификаций и вариантов может быть сделано без выхода за рамки описанного здесь изобретения. Таким образом, предполагается, что все такие изменения, модификации и варианты входят в рамки и широкий объем прилагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГИДРОКСАМАТНЫЕ ПРОИЗВОДНЫЕ, ПРИМЕНИМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ДЕЗАЦЕТИЛАЗЫ | 2001 |
|
RU2302408C2 |
| СПОСОБ ПОЛУЧЕНИЯ 5-ХЛОР-N-({(5S)-2-ОКСО-3-[4-(3-ОКСО-4-МОРФОЛИНИЛ)-ФЕНИЛ]-1, 3-ОКСАЗОЛИДИН-5-ИЛ}-МЕТИЛ)-2-ТИОФЕНКАРБОКСАМИДА | 2004 |
|
RU2383540C2 |
| КОМБИНАЦИИ ТЕРАПЕВТИЧЕСКИХ АГЕНТОВ ДЛЯ ЛЕЧЕНИЯ РАКА | 2007 |
|
RU2449788C2 |
| 2,6-ЗАМЕЩЕННЫЕ-4-МОНОЗАМЕЩЕННЫЙ АМИНО-ПИРИМИДИНЫ КАК АНТАГОНИСТЫ РЕЦЕПТОРА ПРОСТАГЛАНДИНА D2 | 2005 |
|
RU2417990C2 |
| ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ПОЛУЧЕНИЯ 4-(АЦЕТИЛАМИНО)-3-[(4-ХЛОРФЕНИЛ)ТИО]-2-МЕТИЛ-1Н-ИНДОЛ-1-УКСУСНОЙ КИСЛОТЫ | 2010 |
|
RU2551852C2 |
| ЗАМЕЩЕННЫЕ N-(ИНДОЛ-2-КАРБОНИЛ)-β-АЛАНИНАМИДЫ И ИХ ПРОИЗВОДНЫЕ, СПОСОБ ЛЕЧЕНИЯ ГЛИКОГЕНФОСФОРИЛАЗОЗАВИСИМЫХ ЗАБОЛЕВАНИЙ, СПОСОБ ПРЕДУПРЕЖДЕНИЯ ИШЕМИЧЕСКОГО ПОВРЕЖДЕНИЯ МИОКАРДА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2159613C2 |
| АЗОТСОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ ГЕТЕРОАРИЛА | 2004 |
|
RU2375350C2 |
| ПРОИЗВОДНЫЕ 2-ФЕНОКСИ И 2-ФЕНИЛСУЛЬФОНАМИДА С ССR3 АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ ДЛЯ ЛЕЧЕНИЯ АСТМЫ И ДРУГИХ ВОСПАЛИТЕЛЬНЫХ ИЛИ ИММУНОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2004 |
|
RU2380356C2 |
| АМИДЫ 3-ЗАМЕЩЕННОЙ 5- И 6-АМИНОАЛКИЛИНДОЛ-2-КАРБОНОВОЙ КИСЛОТЫ И РОДСТВЕННЫЕ АНАЛОГИ КАК ИНГИБИТОРЫ КАЗЕИНКИНАЗЫ IΕ | 2005 |
|
RU2369599C2 |
| СПОСОБ ПОЛУЧЕНИЯ 3-ГАЛОГЕНАЛКИЛ-1Н-ПИРАЗОЛОВ | 1996 |
|
RU2169143C2 |
Изобретение относится к способу получения N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида, включающего: (а) объединение при перемешивании гидроксида натрия и гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты в растворе с получением смеси при температуре ниже около -10°С; и затем (б) добавление гидроксиламина к полученной смеси с получением N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида, после чего при необходимости проведение (в) кристаллизации N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида и необязательно (г) выделение целевого продукта. Способ снижает образование побочных продуктов и позволяет получать целевой продукт с чистотой, достаточной для производства лекарственных препаратов. 15 з.п. ф-лы, 5 пр.
1. Способ получения N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида, включающий
(а) объединение при перемешивании гидроксида натрия и гидрохлорида метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты в растворе с получением смеси при температуре ниже около -10°С; и затем
(б) добавление гидроксиламина к полученной смеси с получением N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида, после чего при необходимости проводят
(в) кристаллизацию N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида и необязательно
(г) выделение целевого продукта.
2. Способ по п.1, в котором температуру на стадии (а) поддерживают ниже около -15°С.
3. Способ по п.1, в котором гидрохлорид метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты используют в виде суспензии в метаноле.
4. Способ по п.1, в котором гидроксид натрия используют в виде раствора в метаноле.
5. Способ по п.1, в котором гидроксид натрия добавляют к гидрохлориду метилового эфира (Е)-3-(4-{[2-(2-метил-1Н-индол-3-ил)этиламино]метил}фенил)акриловой кислоты в течение приблизительно 30 мин.
6. Способ по п.1, в котором гидроксид натрия используют в количестве от около 2,5 до около 3,5 эквивалентов.
7. Способ по п.1, в котором гидроксиламин используют в виде раствора в воде.
8. Способ по п.1, в котором гидроксиламин используют в количестве от около 4 до около 13 эквивалентов.
9. Способ по п.1, в котором гидроксиламин добавляют к смеси в течение приблизительно 30 мин.
10. Способ по п.1, где стадия (в) включает подстадии:
(в1) нагревание реакционной смеси, полученной на стадии (б);
(в2) перемешивание реакционной смеси;
(в3) добавление воды к реакционной смеси;
(в4) фильтрование реакционной смеси с получением фильтрата;
(в5) корректировка уровня рН фильтрата до уровня рН от около 10 до около 11;
(в6) добавление затравочных кристаллов N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида к фильтрату;
(в7) перемешивание фильтрата до получения суспензии;
(в8) корректировка уровня рН суспензии до уровня рН от около 8,5 до около 9 и
(в9) перемешивание суспензии.
11. Способ по п.10, в котором все подстадии (в1)-(в9) проводят при температуре, достигнутой при нагревании на подстадии (в1).
12. Способ по п.10, в котором реакционную смесь нагревают до температуры от около 0 до около 25°С.
13. Способ по п.10, в котором подстадии (в1) и (в2) повторяют для обеспечения постепенного нагревания.
14. Способ по п.10, в котором уровень рН фильтрата корректируют до уровня рН от около 10,3 до около 10,7 на подстадии (в5).
15. Способ по п.1, где стадия (г) включает подстадии:
(г1) фильтрование кристаллизованного N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида, полученного на стадии (в) и
(г2) высушивание кристаллизованного N-гидрокси-3-[4-[[[2-(2-метил-1Н-индол-3-ил)этил]амино]метил]фенил]-2Е-2-пропенамида.
16. Способ по п.15, в котором остаток на фильтре, полученный на подстадии (г1), промывают.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| КОВОЧНЫЕ ВАЛЬЦЫ | 0 |
|
SU233413A1 |
| Химия Гетероциклических Соединений, 1968, №8, с.875-877. | |||

