Область техники, к которой относится изобретение
Настоящее изобретение относится к соединениям, предназначенным для применения в лечении патологических состояний или расстройств, выбранных из группы, состоящей из расстройств мочевого пузыря и желудочно-кишечного тракта; воспалительных заболеваний; сердечно-сосудистых заболеваний; нефропатий; глаукомы; спастичности; рака; остеопороза; метаболических расстройств; ожирения; расстройств, связанных с пагубными пристрастиями, зависимостями, злоупотреблениями и их рецидивами; психиатрических и неврологических расстройств; нейродегенеративных расстройств; аутоиммунного гепатита и энцефалита; боли; репродуктивных расстройств, а также воспалительных и фиброзных заболеваний кожи.
Предпосылки изобретения
Рецептор CB1 является одним из семи основных мембранных рецепторов, сопряженных с G-белком (GCPR), в организме. Этот рецептор является основным GCPR мозга и, кроме того, он экспрессируется большинством тканей организма, включая, но не ограничиваясь этим, жировую ткань, печень, поджелудочную железу, мышцы, почки, мочевой пузырь и кости.
Рецептор CB1 активируется эндогенными лигандами, которые именуются эндоканнабиноидами и включают, не ограничиваясь этим, анандамид и 2-арахидонил глицерин (2-AG).
При активации эндогенными лигандами CB1 включается в регулирование большого числа физиологических функций и патологических состояний. Неполный перечень функций, в которых играет роль активация рецептора CB1, включает следующее: энергетический обмен; воспаление и иммунитет; фиброз; гомеостаз костной ткани; сохранение и накопление липидов в различных органах; формы поведения; самостоятельное введение препаратов, вызывающих зависимость, память, адаптацию, связанную со стрессом, формы поведения, опосредованные положительным подкреплением; моторику желудочно-кишечного тракта и моторику других внутренних органов, способных к сокращению; клеточную пролиферацию и дифференцировку; регулирование боли; размножение и детородную функцию. (Marsicano et al., J Endocrinol. Invest., 2006; 29(3 Suppl): 27-46 Review; Pagotto U. et al., Int J Obes. 2006, Suppl 1: S39-43 Review; Pagotto U. et al., Endocr. Rev., 2006 (1): 73-100. Review; Bifulco M., et al. Mol. Pharmacol. 2007, 71(6): 1445-56 Review).
Вследствие многообразных физиологических функций, избыточная активация рецептора CB1 вовлечена в большое число патологий, заболеваний и патофизиологических процессов. Неполный перечень примеров заболеваний и родственных им процессов, в которые вовлечена активация рецептора CB1, включает расстройства мочевого пузыря и желудочно-кишечного тракта; воспалительные заболевания; сердечно-сосудистые заболевания; нефропатии; глаукому; спастичность; рак; остеопороз; метаболические расстройства; ожирение; расстройства, связанные с пагубными пристрастиями, зависимостями, злоупотреблениями и их рецидивами; психиатрические и неврологические расстройства; нейродегенеративные расстройства; аутоиммунный гепатит и энцефалит; боль; репродуктивные расстройства, а также воспалительные и фиброзные заболевания кожи (Di Marzo et al., Nat. Rev., Drug Discov., 2004, 3: 771-784).
Рецептор CB1 является основной мишенью Δ9тетрагидроканнабинола (THC) - действующего начала препаратов, вызывающих зависимость, который получают из Cannabis sativa (конопли посевной). Именно в результате активации CB1 THC вызывает зависимость и оказывает разрушительное влияние на поведение и физиологию. Кроме того, рецептор CB1 вовлечен в опосредование действия всех других препаратов, вызывающих зависимости, включая, но не ограничиваясь этим, никотин, опиоиды, психостимуляторы и алкоголь. Помимо этого, рецептор CB1 вовлечен в опосредование привлекательных свойств нелекарственных подкрепляющих стимулов, которые способны вызывать пристрастие, включая, но не ограничиваясь этим, влечение к пище, к половым партнерам или азартным играм. Общую роль CB1 в действии препаратов, вызывающих зависимость, и другие подкрепляющих стимулов, способных вызывать пристрастия, объясняют стимулирующим действием, которое активация CB1 оказывает на активность дофаминергической передачи сигнала. Таким образом, активация дофаминергической передачи сигнала опосредует привлекательные свойства и предрасположенность к зависимости от лекарственных препаратов и нелекарственных положительных подкрепляющих стимулов. По этой причине блокирование активности CB1 было предложено в качестве способа лечения пристрастий, злоупотреблений, лекарственных зависимостей и их рецидивов (Sherma M et al., CNS Neurol Disord Drug Targets. 2008; 7(5): 468-81. Review; Wiskerke J. et al, Addict Biol. 2008; 13(2): 225-38. Review; Moreira F.A. et al., Addict Biol. 2008; 13(2): 196-212. Review; Lopez-Moreno J.A. et al, Addict. Biol. 2008; 13(2): 160-87. Review; Janero D.R. et al., Curr Psychiatry Rep., 2007; 9(5): 365-73. Review; Laviolette S.R. et al., Cell Mol. Life Sci., 2006; 63(14): 1597-613. Review; Maldonado R., et al. Trends Neurosci., 2006; 29(4): 225-32. Review; Colombo G. et al., Pharmacol. Biochem Behav., 2005; 81(2): 369-80. Review; Gardner E.L. Pharmacol. Biochem. Behav., 2005; 81(2): 263-84. Review).
Было показано, что ингибирование рецептора CB1 снижает вес и усиливает улучшение параметров кардиометаболического риска. Так, например, было показано, что антагонисты рецептора CB1 предотвращают набор лишнего веса, способствуют регулированию потребления пищи, облегчают соблюдение диеты, лечат ожирение и облегчают метаболические расстройства, часто сопровождающие ожирение, такие как диабет и дислипидимия. (Bermudez-Silva F.J. et al., 2010; Lee H.K. et al. 2009; Xie S et al., 2007).
Передача сигнала рецептора CB1 в центральной нервной системе функционально связана с моноаминергической нейротрансмиссией. Это делает антагонистов CB1 потенциальными средствами лечения психических, аффективных и когнитивных расстройств, вызываемых нарушениями в любых центральных моноаминергических системах. Кроме того, агонисты CB1 приводят к ухудшению памяти. Поэтому антагонисты CB1 являются хорошими потенциальными средствами для улучшения памяти (см. Reibaud M. et al., Eur. J. Pharmacol., 1999; 379(1): R1-2 и Terranova J.P. et al., Psychopharmacology., 1996; 126(2): 165-72). Активация CB1 может также приводить к нарушению двигательной активности и двигательным расстройствам, например, болезни Паркинсона, связанной с повышенным содержанием эндоканнабиноидов в мозге. Поэтому применение антагонистов CB1 могло бы стать хорошим потенциальным путем лечения болезни Паркинсона (см. Di Marzo V. et al., FASEB J., 2000; 14(10): 1432-8). Таким образом, антагонисты CB1 являются потенциальными средствами лечения различных психиатрических и неврологических заболеваний.
Далее, рецептор CB1 вовлечен в спастичность, как указано в работе Pryce G. et al., (Br J Pharmacol., 2007, 150(4): 519-525) и Baker D. et al. (FASEB J., 2001, 15: 300-302).
Chien F.Y. и соавторы показали, что WIN 55212-2, т.е. каннабиноидный агонист рецептора CB(1), понижает внутриглазное давление как у здоровых обезьян, так и у обезьян с глаукомой.
Рецепторы CB1, которые экспрессируются в некоторых периферических тканях, например, в нервных окончаниях желудочно-кишечного тракта, понижают моторику желудочно-кишечного тракта, главным образом, за счет ингибирования высвобождения медиатора начала сокращений. Таким образом, антагонисты рецептора CB1 могли бы найти применение при патологических состояниях, включающих пониженную моторику кишечника, например, паралитическую непроходимость кишечника, вызванную перитонитом, хирургическим вмешательством или другими неблагоприятными ситуациями (Mascolo N. et al., FASEB J., 2002 Dec; 16(14): 1973-5).
Что касается других желудочно-кишечных заболеваний, было показано, что рецепторы CB1 вовлечены в заболевания печени и, в частности, в стеатоз (жировое перерождение), стеатогепатит (NASH) и цирроз печени. Активация CB1 принимает участие в этих заболеваниях за счет двух механизмов: 1. содействие накоплению жира в печени; 2. содействие высвобождению воспалительных факторов, таких как TNFα. Ингибиторы CB1 оказывают благоприятное действие при подобных патологиях за счет того, что они уменьшают как накопление жира, так и высвобождение TNFα. Для ознакомления с примерами подобных случаев см.: 1. Mallat A. and Lotersztajn S. Diabetes and Metabolis 34(2008) 680-684; 2. Tarn J. et al., HEPATOLOGY 2011; 53: 346-355; 3. Soren V. Siegmund S.V. and Schwabe R.F. Am J Physiol. Gastrointest Liver Physiol. 294: G357-G362, 2008; 4. DeLeve D.L. et al., The American Journal of Pathology, 173, № 4, 2008; 5. Roche M. et al., Immunology, 2008, 125, 263-271; 6. Murumalla R. et al., Journal of Inflammation 2011, 8:33; 7. Croci T. et al., British Journal of Pharmacology (2003) 140, 115-122.
Рецепторы CB1 также экспрессируются в норадренергических окончаниях, иннервирующих костную ткань. Активация CB1 способна подавить высвобождение норадреналина в костной ткани, что в свою очередь повышает активность остеокластов, уменьшающих массу костной ткани и вызывающих, не ограничиваясь этим, остеопороз, связанный с менопаузой. По этой причине было предложено применять антагонисты CB1 для лечения остеопороза (Idris AI Curr Neuropharmacol. 2010 8(3): 243-53).
Рецепторы CB1 также действуют в клетках сосудистого эндотелия, где они опосредуют гипотензивное действие эндоканнабиноидов, вырабатываемых тромбоцитами и макрофагами. Антагонисты CB1 могли бы найти применение при ингибировании гипотензии, вызванной эндотоксинами или циррозом (см. Batkai S. et al., Nat Med., 2001 Jul; 7(7): 827-32), где оба указанных заболевания характеризуются повышенными уровнями эндоканнабиноидов. CB1 стимулирует также ангиогенез, вследствие чего блокирование рецептора CB1 было предложено для лечения заболеваний, при которых усиление ангиогенеза играет патофизиологическую роль, как, например, при развитии опухолей.
Кроме того, рецепторы CB1 принимают участие в патологиях сердечно-сосудистой системы, включая кардиомиопатии, например, цирротическую кардиомиопатию и кардиомиопатии, вызванные антинеопластическими препаратами, нарушение сократительной способности, инфаркт и атеросклероз. Рецепторы CB1 принимают участие в этих заболеваниях по целому ряду механизмов, которые включают регулирование кровяного давления, воспаления, накопления жиров, васкуляризацию и сократительную способность сердечной мышцы. См., например, 1. Batkai S. et al., Am J Physiol. Heart Circ Physiol. 2007, 293: H1689-H1695; 2. Seyed Ali Gaskari S.A. et al., British Journal of Pharmacology (2005) 146, 315-323; 3. Batkai S. and Pacher P. Pharmacol. Res. 2009, 60:99-106. 4. Nissen S.E. et al., JAMA. 2008; 299(13): 1547-1560. 5. Mukhopadhyay P. et al., J Am Coll Cardiol. 2007, 50: 528-536.
Далее, было показано, что рецепторы CB1 вовлечены в воспалительные заболевания, в том числе, не ограничиваясь перечисленным, воспалительные заболевания кожи, воспаление и рак кожи, вызванные УФ-излучением, фиброз кожи и заживление ран. В этом контексте было показано, что ингибирование или подавление рецептора CB1 является полезным при всех этих патологических состояниях. См., например, 1. Marquart S. et al., ARTHRITIS & RHEUMATISM, 2010, 62: 3467-3476; 2. Zheng D. et al., Cancer Res. 2008 May 15; 68(10): 3992-3998.
Помимо этого, эндоканнабиноидная передача сигнала была обнаружена при некоторых злокачественных заболеваниях у людей, а также в раковых клетках человека с высокой степенью инвазивности, в отличие от аналогичных здоровых тканей. (Sarnataro D. et al., 2006; Gazzerro P. et al., 2010; Santoro A., et al. 2009).
Далее, эндоканнабиноидная передача сигнала имеет значение для оплодотворения, предимплантации эмбриона и сперматогенеза и, следовательно, является подходящей мишенью для лечения бесплодия и репродуктивного здоровья людей.
По указанным выше причинам ингибирование рецептора CB1 было предложено в качестве способа лечения всех перечисленных патологических состояний и связанных с ними заболеваний.
Были разработаны и подвергнуты клиническому испытанию способы, направленные на блокирование активности рецептора CB1 путем ингибирования ортостерического сайта связывания, т.е. сайта, с которым связываются эндогенные лиганды при активации рецептора. Один из ортостерических ингибиторов, а именно римонабант, уже был выпущен на рынок под торговым наименованием Acomplia (Акомплиа). Акомплиа была подвергнута тестированию, и было выявлено ее полезное действие при лечении метаболических расстройств, диабета, дислипидемии и ожирения, а также, в одном из исследований, при лечении никотиновой зависимости.
К сожалению, имеющиеся ортостерические антагонисты, например, римонабант, действуют так же, как обратные агонисты рецептора CB1, т.е. они не только ингибируют активацию CB1, но также и основную активность рецептора в отсутствие эндогенного лиганда. Из-за такого действия в качестве обратного агониста и полного ингибирования активности рецептора, имеющиеся способы, основанные на введении ортостерических агонистов CB1, имеют также целый ряд нежелательных побочных эффектов. Из-за этих побочных эффектов была приостановлена коммерческая реализация Акомплии и прекращена разработка других способов ингибирования ортостерического сайта CB1.
Для многих патологий, при которых была продемонстрирована хорошая терапевтическая эффективность ортостерических антагонистов рецептора CB1, по-прежнему требуются новые эффективные способы лечения. Поэтому существует необходимость разработки способов, которые могут обеспечить ингибирование рецептора CB1, не препятствуя ортостерическому связыванию, и имеют меньше побочных эффектов, чем ортостерические антагонисты.
Таким образом, по-прежнему существует необходимость в разработке лигандов, которые позволяют ингибировать рецепторы CB1 без изменения ортостерического связывания или инициирования нежелательных эффектов.
Сущность изобретения
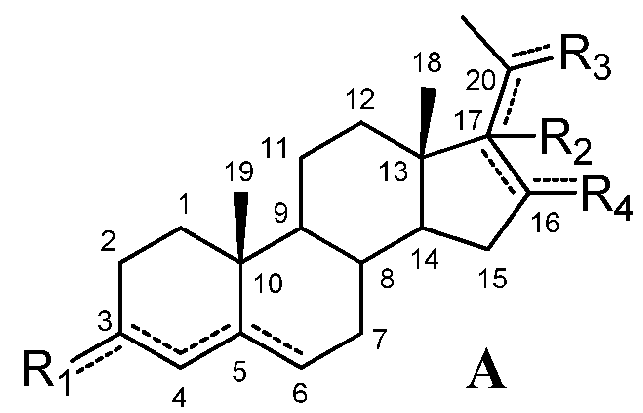
Настоящее изобретение относится к соединению формулы (A) или его фармацевтически приемлемой соли:
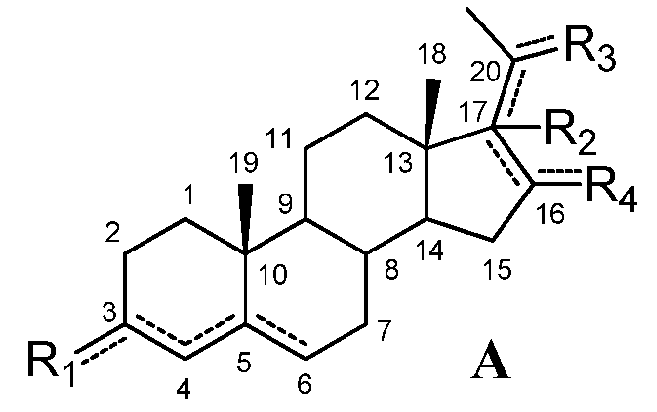 ,
,
где:
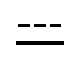 означает, что связь представляет собой простую или двойную связь,
означает, что связь представляет собой простую или двойную связь,
R1 означает, что C3 замещен
-H,
-галогеном,
-OH,
C1-8 алкокси,
Bn-O-,
Bn-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
Ph-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
=O,
-NR5R6, где каждый из R5 и R6 независимо представляет собой H, C1-8 алкил, Bn или Ph,
-O-CO-R7, где R7 означает алкил,
-O-CO-C2H4-COOH, или
-N3,
-R2 означает, что C17 замещен
-H,
-OH,
галогеном,
C1-8 алкилом,
C1-8 алкокси,
C2-6 алкенилом,
Bn-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
Ph-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, карбоксилом или галогеном, или
Bn-O-,
R3 означает, что C20 замещен
-H,
-OH,
C1-8 алкилом,
Bn,
-NR8R9, где каждый из R8 и R9 независимо представляет собой H, C1-8 алкил или Bn,
=CR10R11, где каждый из R10 и R11 независимо представляет собой H или C1-7 алкил, или
=O,
R4 означает, что C16 замещен
-H,
-OH или
=O,
при условии, что
если связь между C16 и C17 является двойной, R2 отсутствует и связь между C17 и C20 является простой, и
если связь между C17 и C20 является двойной, C20 замещен -H или -OH и R2 отсутствует,
если связь между C4 и C5 является двойной, связь между C5 и C6 является простой и наоборот,
для применения в лечении патологического состояния или расстройства, выбранного из группы, состоящей из расстройств мочевого пузыря и желудочно-кишечного тракта; воспалительных заболеваний; сердечно-сосудистых заболеваний; нефропатий; глаукомы; спастичности; рака; остеопороза; метаболических расстройств; ожирения; расстройств, связанных с пагубными пристрастиями, зависимостями, злоупотреблениями и их рецидивами; психиатрических и неврологических расстройств; нейродегенеративных расстройств; аутоиммунного гепатита и энцефалита; боли; репродуктивных расстройств, а также воспалительных и фиброзных заболеваний кожи.
Кроме того, настоящее заболевание относится к соединению формулы (II)
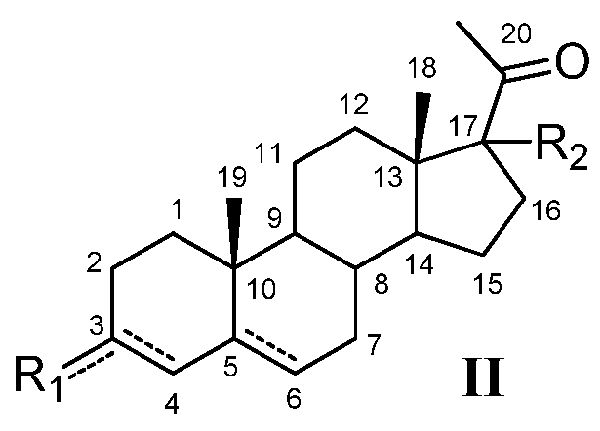
или его фармацевтически приемлемой соли, где:
означает, что связь является простой или двойной связью,
R1 означает, что C3 замещен -OH, и
-R2 означает, что C17 замещен
C3-8 алкилом,
C2-8 алкокси,
Bn-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
Ph-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, карбоксилом или галогеном, или
Bn-O-,
или где
R1 означает, что C3 замещен
-C1-8 алкокси,
Bn-O или
галогеном, и
-R2 означает, что C17 замещен
C1-8 алкилом,
C2-6 алкенилом,
C1-8 алкокси,
Bn-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
Ph-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, карбоксилом или галогеном, или
Bn-O-.
Подробное описание изобретения
Определения
Термин «агонист» относится к соединению, которое усиливает активность другого соединения или сайта рецептора.
Термины «антагонист» и «ингибитор» относятся к соединению, которое уменьшает или препятствует активности другого соединения в отношении сайта рецептора и, в более общем смысле, относятся к соединению, которое уменьшает или препятствует активации и/или активности рецептора.
Термины «лечить» или «лечение» относятся как к терапевтическому лечению, так и к профилактическим или предупреждающим мерам, цель которых заключается в предотвращении или замедлении развития целевого патологического состояния или расстройства. Субъекты, нуждающиеся в лечении, включают тех, у кого уже имеется расстройство, наряду с теми, кто склонен к расстройству, или теми, у кого необходимо предотвратить расстройство. Таким образом, у субъекта, которого предполагается лечить способом по настоящему изобретению, может быть диагностировано наличие расстройства, либо он может быть предрасположен или восприимчив к расстройству.
Термин «субъект» в настоящем описании означает млекопитающее, например, грызуна, представителя семейства кошачьих, представителя семейства псовых и примата. Предпочтительно, субъект по настоящему изобретению представляет собой человека.
Термин «терапевтически эффективное количество» используется для обозначения минимального количества действующего агента (например), которое необходимо для достижения благоприятного терапевтического или профилактического результата у субъекта. Например, «терапевтически эффективное количество» для млекопитающего означает такое количество, которое вызывает, облегчает или каким-либо другим образом приводит к улучшению патологических симптомов, прогрессирования заболевания или физиологических состояний, связанных с устойчивостью к расстройству.
Термин «рак» в настоящем описании относится или описывает физиологическое состояние субъектов, которое обычно характеризуется неуправляемым ростом или гибелью клеток. Примеры раковых заболеваний включают, не ограничиваясь этим, карциному, лимфому, бластому, саркому и лейкоз или лимфоидные злокачественные заболевания. Более конкретные примеры перечисленных раковых заболеваний включают плоскоклеточный рак (например, эпителиальный плоскоклеточный рак), рак легких, включая мелкоклеточный рак легких, немелкоклеточный рак легких, аденокарциному легких и плоскоклеточную карциному легких, рак органов брюшной полости, гепатоклеточный рак, рак желудка, включая рак желудочно-кишечного тракта, рак поджелудочной железы, глиобластому, рак шейки матки, рак яичников, рак печени, рак мочевого пузыря, гепатому, рак груди, рак ободочной кишки, рак прямой кишки, колоректальный рак, карциному эндометрия или матки, карциному слюнных желез, рак почек, рак простаты, рак вульвы, рак щитовидной железы, карциному печени, карциному анального отверстия, карциному пениса, а также рак головы и шеи.
В настоящем описании термины «пристрастие и зависимость» относятся к изменениям в поведении, заключающимся в стремлении к подкрепляющим стимулам, которые включают, не ограничиваясь этим, фармакологические вещества, пищу, половых партнеров, азартные игры, рискованное поведение. Упомянутые поведенческие изменения характеризуются одной или всеми из перечисленных характеристик: 1. неспособность индивидуума воздержаться от потребления, приема или пребывания в контакте с упомянутыми стимулами, что приводит к потреблению количеств стимула, превышающих первоначально предполагаемые, и безуспешным попыткам прекратить указанное поведение в течение продолжительного периода времени; 2. сильная мотивация получать, потреблять, принимать или находиться в контакте с упомянутыми стимулами, которая становится основой активности субъекта и которая может быть сопряжена с пренебрежением другими видами деятельности; 3. появление дискомфорта, физического или психологического, при прекращении потребления или в отсутствие подкрепляющего стимула.
В настоящем описании термин «злоупотребление» относится к физиологическому состоянию, при котором временно или постоянно нарушено нормальное состояние организма индивидуума, в результате потребления, приема или пребывания в контакте с подкрепляющим стимулом, в число которых входят, не ограничиваясь этим, фармакологические соединения, пища, половые партнеры, азартные игры. Указанные нарушения включают, не ограничиваясь этим, сердечно-сосудистые осложнения, проблемы с дыханием, заболевания печени, инфекционные заболевания, травматические повреждения. Эти нарушения нормального состояния организма могут быть связаны или не связаны с поведенческими проявлениями, которые характеризуют пристрастие и зависимость, которые описаны выше.
В настоящем описании термин «рецидив» относится к повторному проявлению влечения, зависимости или злоупотребления после периода непрерывного воздержания от употребления, приема или нахождения в контакте с подкрепляющим стимулом, в число которых входят, не ограничиваясь этим, фармакологические соединения, пища, половые партнеры, азартные игры.
В настоящем описании термин «метаболические расстройства» относится к физиологическому состоянию, при котором изменяются нормальные уровни химических веществ, используемых организмом в качестве источников энергии или, более обобщенно, в качестве компонентов, которые необходимы для обеспечения структурной или функциональной целостности организма. Упомянутые химические вещества включают, не ограничиваясь этим, углеводы (глюциды), липиды, аминокислоты и электролиты. Патологические состояния, которые обычно возникают в результате метаболических расстройств, включают, не ограничиваясь этим, диабет и дислипидемию. Метаболические расстройства могут также способствовать желудочно-кишечным и сердечно-сосудистым заболеваниям, таким как атеросклероз, NASH и цирроз. Метаболические расстройства могут быть связаны с ожирением или иметь идиопатическую природу.
«Боль» означает более или менее локализованное ощущение дискомфорта, дистресса или страданий, которое является результатом стимулирования определенных нервных окончаний. Существует много типов боли, включая, но не ограничиваясь этим, непостоянную боль, фантомную боль, стреляющую боль, острую боль, воспалительную боль, невропатическую боль, комплексную регионарную боль, невралгию, невропатию и т.п. (Dorland′s Illustrated Medical Dictionary, 28th Edition, W.B.Saunders Company, Philadelphia, Pa.). Цель лечения боли заключается в уменьшении степени тяжести боли, которую испытывает субъект, подвергаемый лечению.
Термин «воспалительные и фиброзные заболевания кожи» относится к патологиям кожи, идиопатическим или индуцированным внешним агентом, в т.ч. УФ-излучением, который вызывает изменения в коже, рак кожи и/или прекращение процесса заживления ран.
Другие наименования патологий, используемые в настоящей заявке, включая, но не ограничиваясь этим, остеопороз, нейродегенеративные заболевания, болезнь Паркинсона, болезнь Альцгеймера, шизофрению, расстройства поведения, расстройства мочевого пузыря и желудочно-кишечного тракта; воспалительные заболевания; сердечно-сосудистые заболевания; атеросклероз, стеатоз, NASH и цирроз печени, нефропатии; глаукому; спастичность; аутоиммунный гепатит и энцефалит; репродуктивные расстройства, используются в тексте описания в их исходном медицинском значении, которое указано в любом руководстве по медицине.
Выражение «атом углерода Ci замещен группой X» означает, что атом углерода в положении i химической формулы несет заместитель X, который может представлять собой атом, например, водорода или галогена, или функциональную группу.
Термин «алкил» означает одновалентный линейный или разветвленный насыщенный углеводородный фрагмент, состоящий только из атомов углерода и водорода. C1-8 алкил означает линейный или разветвленный алкил, включающий от одного до восьми атомов углерода.
Термин «алкокси» означает фрагмент формулы -OR, где R представляет собой определенный выше алкильный фрагмент.
Термин «галоген» относится к атомам фтора, хлора, брома или йода.
Термин «амино» означает фрагмент формулы -NRR′, где каждый из заместителей R и R′ независимо представляет собой водород или алкил, который определен выше по тексту.
Сокращение Bn относится к бензильной группе.
Сокращение Ph относится к фенильной группе.
Заместители, находящиеся над плоскостью молекулы, показаны жирной линией  и именуются β; заместители, находящиеся под плоскостью молекулы, показаны прерывистой линией
и именуются β; заместители, находящиеся под плоскостью молекулы, показаны прерывистой линией 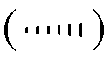 и именуются α.
и именуются α.
Термин «необязательно» означает, что следующее за ним событие или обстоятельство может, но не обязательно должно произойти, и что указанный термин включает ситуации, когда событие или обстоятельство имеет место, и ситуации, в которых событие или обстоятельство не имеют места.
Ингибирование рецептора CB1
Настоящее изобретение относится к соединению формулы (A) или его фармацевтически приемлемой соли:
 ,
,
где:
означает, что связь является простой или двойной связью,
R1 означает, что C3 замещен
-H,
-галогеном,
-OH,
C1-8 алкокси,
Bn-O-,
Bn-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
Ph-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
=O,
-NR5R6, где каждый из R5 и R6 независимо представляет собой H, C1-8 алкил, Bn или Ph,
-O-CO-R7, где R7 означает алкил,
-O-CO-C2H4-COOH или
-N3,
-R2 означает, что C17 замещен
-H,
-OH,
галогеном,
C1-8 алкилом,
C1-8 алкокси,
C2-6 алкенилом,
Bn-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
Ph-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, карбоксилом или галогеном, или
Bn-O-,
R3 означает, что C20 замещен
-H,
-OH,
C1-8 алкилом,
Bn,
-NR8R9, где каждый из R8 и R9 независимо представляет собой H, C1-8 алкил или Bn,
=CR10R11, где каждый из R10 и R11 независимо представляет собой H или C1-7 алкил, или
=O,
R4 означает, что C16 замещен
-H,
-OH или
=O,
при условии, что
если связь между C16 и C17 является двойной, R2 отсутствует и связь между C17 и C20 является простой, и
если связь между C17 и C20 является двойной, C20 замещен -H или -OH и R2 отсутствует,
если связь между C4 и C5 является двойной, связь между C5 и C6 является простой и наоборот,
для применения в лечении патологического состояния или расстройства, выбранного из группы, состоящей из расстройств мочевого пузыря и желудочно-кишечного тракта; воспалительных заболеваний; сердечно-сосудистых заболеваний; нефропатий; глаукомы; спастичности; рака; остеопороза; метаболических расстройств; ожирения; расстройств, связанных с пагубными пристрастиями, зависимостями, злоупотреблениями и их рецидивами; психиатрических и неврологических расстройств; нейродегенеративных расстройств; аутоиммунного гепатита и энцефалита; боли; репродуктивных расстройств, а также воспалительных и фиброзных заболеваний кожи.
Фактически авторы изобретения показали, что прегненолон и некоторые из его производных являются ингибиторами рецептора CB1 и способны блокировать активацию рецептора CB1, вызванную природными или синтетическими агонистами или эндогенными лигандами, без изменения ортостерического связывания.
Таким образом, соединения по настоящему изобретению действуют аналогично другим антагонистам рецептора CB1, например, римонабанту, и могут применяться в лечении патологий, при которых требуются антагонисты рецептора CB1.
Кроме того, это ингибирование является эндогенным механизмом. Поэтому оно изменяет активность рецептора более физиологичным образом, модулируя реакцию рецептора на эндогенные или экзогенные агонисты и не блокируя связывание агонистов с рецепторами. Благодаря этому более физиологичному механизму, такие ингибиторы, как прегненолон и его производные, вызывают меньшие побочные эффекты, чем ортостерические антагонисты.
Примеры расстройств мочевого пузыря и желудочно-кишечного тракта, которые можно лечить антагонистами рецептора CB1, включают, не ограничиваясь перечисленными, фиброз печени; стеатоз печени; неалкогольный стеатогепатит (NASH), цирроз печени; алкогольный стеатоз; поражение печени в результате ишемии-реперфузии, осложненное эндотоксемией; острый панкреатит; гиперактивные и болевые расстройства мочевого пузыря и изменение моторики внутренних органов, способных к сокращению.
Примеры воспалительных заболеваний, которые можно лечить антагонистами рецептора CB1, включают, не ограничиваясь перечисленным, воспаление и артрит, связанные с ожирением; хронические иммунные воспалительные заболевания и язву.
Примеры сердечно-сосудистых заболеваний, которые можно лечить антагонистами рецептора CB1, включают, не ограничиваясь перечисленным, кардиомиопатию, например, цирротическую кардиомиопатию, кардиомиопатию, вызванную антинеопластическими препаратами, дисфункцию эндотелия и смерть клеток, вовлеченных в развитие сосудистых нарушений, связанных с застойной сердечной недостаточностью; гипертензию; заболевания коронарных артерий; атеросклероз; инфаркт миокарда; заболевания, вызванные накоплением жиров, например, атеросклероз, патологии, вызванные повышенным ангиогенезом, и заболевания, включающие ангиогенез.
Примеры метаболических расстройств, которые можно лечить антагонистами рецептора CB1, включают, не ограничиваясь перечисленным, дислипидемию, диабет и осложнения диабета.
Примеры расстройств, связанных с пагубными пристрастиями, зависимостями, злоупотреблениями и их рецидивами, которые можно лечить антагонистами рецептора CB1, включают, не ограничиваясь перечисленным, лекарственную зависимость; злоупотребление лекарствами; рецидивы лекарственных зависимостей, пристрастий к лекарствам и злоупотреблений лекарствами; пристрастие к препаратам, полученным из конопли; злоупотребление препаратами, полученными из конопли; токсическое действие препаратов, полученных из конопли; психозы, вызванные препаратами, полученными из конопли.
Примеры нейродегенеративных расстройств, которые можно лечить антагонистами рецептора CB1, включают, не ограничиваясь перечисленным, болезнь Паркинсона и болезнь Альцгеймера.
Примеры психиатрических и неврологических расстройств, которые можно лечить антагонистами рецептора CB1, включают, не ограничиваясь перечисленным, шизофрению; расстройства поведения; дискинезию, вызванную L-DOPA; расстройства памяти.
Примеры репродуктивных расстройств, которые можно лечить антагонистами рецептора CB1, включают, не ограничиваясь перечисленным, бесплодие и привычный выкидыш.
Примеры воспалительных и фиброзных заболеваний, которые можно лечить антагонистами рецептора CB1, включают, не ограничиваясь перечисленными, воспаление кожи, воспаление и рак кожи, вызванные УФ-излучением, фиброз кожи и заживление ран.
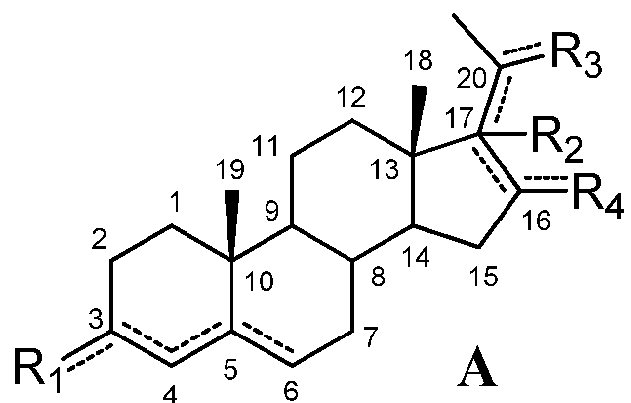
Настоящее изобретение относится к способу лечения патологического состояния или расстройства, выбранного из группы, состоящей из расстройств мочевого пузыря и желудочно-кишечного тракта; воспалительных заболеваний; сердечно-сосудистых заболеваний; нефропатий; глаукомы; спастичности; рака; остеопороза; метаболических расстройств; ожирения; расстройств, вызванных пагубными пристрастиями, зависимостями, злоупотреблениями и их рецидивами; психиатрических и неврологических расстройств; нейродегенеративных расстройств; аутоиммунного гепатита и энцефалита; боли; репродуктивных расстройств, а также воспалительных и фиброзных заболеваний кожи, у субъекта, которому это необходимо, включающему введение указанному субъекту эффективного количества соединения формулы (A) или его фармацевтически приемлемой соли:
 ,
,
где:
означает, что связь является простой или двойной связью,
R1 означает, что C3 замещен
-H,
-галогеном,
-OH,
C1-8 алкокси,
Bn-O-,
Bn-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
Ph-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
=O,
-NR5R6, где каждый из R5 и R6 независимо представляет собой H, C1-8 алкил, Bn или Ph,
-O-CO-R7, где R7 означает алкил,
-O-CO-C2H4-COOH или
-N3,
-R2 означает, что C17 замещен
-H,
-OH,
галогеном,
C1-8 алкилом,
C1-8 алкокси,
C2-6 алкенилом,
Bn-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
Ph-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, карбоксилом или галогеном, или
Bn-O-,
означает, что C20 замещен
-H,
-OH,
C1-8 алкилом,
Bn,
-NR8R9, где каждый из R8 и R9 независимо представляет собой H, C1-8 алкил или Bn,
=CR10R11, где каждый из R10 и R11 независимо представляет собой H или C1-7 алкил, или
=O,
R4 означает, что C16 замещен
-H,
-OH или
=O,
при условии, что
если связь между C16 и C17 является двойной, R2 отсутствует и связь между C17 и C20 является простой,
если связь между C17 и C20 является двойной, C20 замещен -H или -OH и R2 отсутствует, и
если связь между C4 и C5 является двойной, связь между C5 и C6 является простой и наоборот.
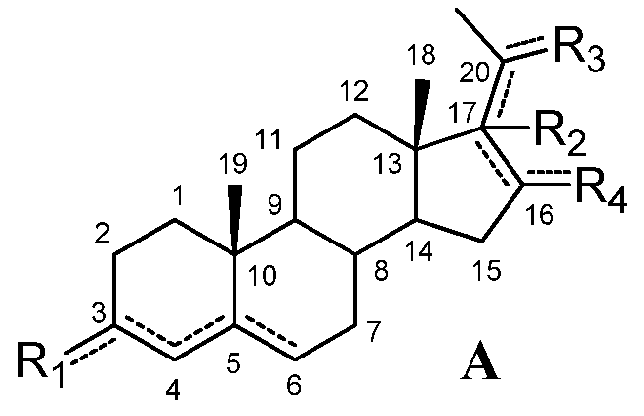
Настоящее изобретение относится к применению соединения формулы (A) или его фармацевтически приемлемой соли:
 ,
,
где:
означает, что связь является простой или двойной связью,
R1 означает, что C3 замещен
-H,
-галогеном,
-OH,
C1-8 алкокси,
Bn-O-,
Bn-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
Ph-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
=O,
-NR5R6, где каждый из R5 и R6 независимо представляет собой H, C1-8 алкил, Bn или Ph,
-O-CO-R7, где R7 означает алкил,
-O-CO-C2H4-COOH или
-N3,
-R2 означает, что C17 замещен
-H,
-OH,
галогеном,
C1-8 алкилом,
C1-8 алкокси,
C2-6 алкенилом,
Bn-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
Ph-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, карбоксилом или галогеном, или
Bn-O-,
R3 означает, что C20 замещен
-H,
-OH,
C1-8 алкилом,
Bn,
-NR8R9, где каждый из R8 и R9 независимо представляет собой H, C1-8 алкил или Bn,
=CR10R11, где каждый из R10 и R11 независимо представляет собой H или C1-7 алкил, или
=O,
R4 означает, что C16 замещен
-H,
-OH или
=O,
при условии, что
если связь между C16 и C17 является двойной, R2 отсутствует и связь между C17 и C20 является простой,
если связь между C17 и C20 является двойной, C20 замещен -H или -OH и R2 отсутствует, и
если связь между C4 и C5 является двойной, связь между C5 и C6 является простой и наоборот,
для получения лекарственного средства для лечения патологического состояния или расстройства, выбранного из группы, состоящей из расстройств мочевого пузыря и желудочно-кишечного тракта; воспалительных заболеваний; сердечно-сосудистых заболеваний; нефропатий; глаукомы; спастичности; рака; остеопороза; метаболических расстройств; ожирения; расстройств, вызванных пагубными пристрастиями, зависимостями, злоупотреблениями и их рецидивами; психиатрических и неврологических расстройств; нейродегенеративных расстройств; аутоиммунного гепатита и энцефалита; боли; репродуктивных расстройств, а также воспалительных и фиброзных заболеваний кожи.
Кроме того, настоящее изобретение относится к соединению по настоящему изобретению, где это соединение имеет формулу (I), или его фармацевтически приемлемой соли:
 ,
,
где:
означает, что связь является простой или двойной связью,
R1 означает, что C3 замещен
-H,
-галогеном,
-OH,
C1-8 алкокси,
Bn-O-,
Bn-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
Ph-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
=O,
-NR5R6, где каждый из R5 и R6 независимо представляет собой H, C1-8 алкил, Bn или Ph,
-O-CO-R7, где R7 означает алкил, или
-O-CO-C2H4-COOH,
-R2 означает, что C17 замещен
-H,
-OH,
галогеном,
C1-8 алкилом,
C1-8 алкокси,
C2-6 алкенилом,
Bn-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
Ph-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, карбоксилом или галогеном, или
Bn-O-,
R3 означает, что C20 замещен
-H,
-OH,
C1-8 алкилом,
Bn,
-NR8R9, где каждый из R8 и R9 независимо представляет собой H, C1-8 алкил или Bn,
=CR10R11, где каждый из R10 и R11 независимо представляет собой H или C1-7 алкил, или
=O,
R4 означает, что C16 замещен
-H,
-OH или
=O,
при условии, что
если связь между C16 и C17 является двойной, R2 отсутствует и связь между C17 и C20 является простой, и
если связь между C17 и C20 является двойной, C20 замещен -H или -OH и R2 отсутствует,
для применения в лечении патологического состояния или расстройства, выбранного из группы, состоящей из расстройств мочевого пузыря и желудочно-кишечного тракта; воспалительных заболеваний; сердечно-сосудистых заболеваний; нефропатий; глаукомы; спастичности; рака; остеопороза; метаболических расстройств; ожирения; расстройств, вызванных пагубными пристрастиями, зависимостями, злоупотреблениями и их рецидивами; психиатрических и неврологических расстройств; нейродегенеративных расстройств; аутоиммунного гепатита и энцефалита; боли; репродуктивных расстройств, а также воспалительных и фиброзных заболеваний кожи.
Кроме того, изобретение относится к фармацевтической композиции, включающей соединение по настоящему изобретению или его фармацевтически приемлемую соль, а также фармацевтически приемлемый носитель.
Термины «фармацевтический» или «фармацевтически приемлемый» относятся к соединениям и композициям, которые не вызывают нежелательной, аллергической или другой неблагоприятной реакции, при надлежащем введении млекопитающему, в частности человеку. Термин «фармацевтически приемлемый носитель» относится к нетоксичному твердому, полутвердому или жидкому наполнителю, разбавителю, инкапсулирующему материалу или вспомогательному компоненту любого типа.
Термин «фармацевтически приемлемый носитель» относится, не ограничиваясь этим, к нетоксичному твердому, полутвердому или жидкому наполнителю, разбавителю, связующему средству, дезинтегрирующему средству, растворителю, стабилизатору, солеобразующему агенту, смазывающему средству и инкапсулирующему материалу или вспомогательному компоненту любого типа.
Форма фармацевтической композиции, путь введения, дозировки и схемы введения, естественно, зависят от состояния пациента, подвергаемого лечению, тяжести заболевания, возраста, массы, пола и т.д.
Фармацевтические композиции по настоящему изобретению могут быть предназначены для любого пути введения, включая, но не ограничиваясь этим, пероральный, внутривенный, внутримышечный, внутриартериальный, интрамедуллярный, интратекальный, трансдермальный, местный, подкожный, интраперитонеальный, интраназальный, энтеральный, сублингвальный, вагинальный и ректальный.
Действующий ингредиент по настоящему изобретению предпочтительно вводят пероральным путем и включают его в состав дозированных лекарственных форм, например, твердых дозированных форм. Эта дозированная лекарственная форма для облегчения введения продукта взрослым или детям может представлять собой любую из следующих форм: таблетки, таблетки с покрытием, пилюли, порошки или гранулы, пакеты-саше или твердые гелевые капсулы.
В случае перорального введения в виде любой стандартной дозированной формы, композиции получают по классической методике с применением фармацевтически приемлемых эксципиентов, в число которых входят, не ограничиваясь указанными, связующие средства (например, прежелатинизированный кукурузный крахмал, поливинилпирролидон, гидроксипропилметилцеллюлоза, натрий карбоксиметилцеллюлоза, смолистые вещества, например, гуаровая камедь, каррагинан, альгиновая кислота или ее соли и т.д.); наполнители (например, лактоза, сахароза, микрокристаллическая целлюлоза, гидрофосфат кальция, дикальцийфосфат, полиолы, такие как маннит, сорбит или ксилит, фруктоза, декстрин, мальтодекстрин и т.д.); смазывающие вещества (например, стеарат магния, стеарилфумарат натрия, тальк или оксид кремния); дезинтегрирующие средства (например, картофельный крахмал или натрий крахмал гликолят, кросповидон, кроскармеллоза и т.д.), солеобразующие агенты (например, н-метилглюкамин, гидроксид натрия, гидроксид калия, хлористоводородная кислота и т.д.) или смачивающие средства (например, лаурилсульфат натрия). Таблетки или твердые гелевые капсулы можно снабжать покрытием с применением способов, известных в технике. Например, на таблетку или твердую гелевую капсулу можно нанести кишечное покрытие или покрытие, способствующее отсроченному высвобождению, которые защищают действующий ингредиент, пока он не достигнет ободочной кишки.
Другой возможностью является продолжительное или регулируемое высвобождение действующего ингредиента для доставки его в течение длительного периода времени (максимум 24 часа) и ограничения количества введений в течение дня. Таблетки или твердые гелевые капсулы можно покрывать полимером, который обеспечивает регулируемое высвобождение действующего ингредиента, или, в случае таблеток, композиция этих таблеток может включать матрицу, которая обеспечивает регулируемое высвобождение действующего ингредиента; компоненты, обеспечивающие регулируемое высвобождение, как правило, представляют собой, не ограничиваясь этим, гидрофильные полимеры (например, гидроксипропилметилцеллюлозу, натрий карбоксиметилцеллюлозу, ксантановую камедь, хитозан, полиэтиленоксид и т.д.), нерастворимые в воде и гидрофобные полимеры (например, этилцеллюлозу, поли(винилацетат)ацетат целлюлозы и т.д.), жирные кислоты (например, гидрированное растительное масло, глицерил пальмитостеарат и т.д.), спирты (например, цетиловый спирт, стеариловый спирт и т.д.), воски (например, пчелиный воск, воск карнауба и т.д.). Конкретно для прегненолона состав с продолжительным высвобождением будет также способствовать уменьшению образования активных продуктов метаболизма прегненолона.
В случае получения твердой композиции в форме таблетки, эту таблетку можно изготавливать непосредственным прессованием, способом мокрого гранулирования или способом сухого гранулирования. Какой бы из этих способов не применялся, на первом этапе действующий ингредиент смешивают со всеми описанными выше носителями, либо с частью этих носителей, затем наносят смазывающий компонент и прессуют в таблетку.
В другом варианте осуществления лекарственная форма по настоящему изобретению может применяться для буккальной доставки действующего ингредиента; соответствующие композиции могут принимать форму таблеток или лекарственных леденцов, которые изготавливают по стандартным методикам. Применяются те же ингредиенты, что и в случае обычных таблеток, но меняются соотношения между этими ингредиентами.
Предпочтительно, действующий ингредиент мог бы доставляться только для местного действия; таким таблеткам придают свойства мукоадгезивных таблеток.
С другой стороны, действующий ингредиент можно было бы доставлять непосредственно в полость рта, но с целью системной абсорбции, и, если вода недоступна, дозированная форма будет представлять собой таблетку, распадающуюся в полости рта, которая обладает тем преимуществом, что ее можно принимать без использования воды.
В другом варианте осуществления лекарственные формы по настоящему изобретению могли бы также представлять собой жидкие препараты для перорального введения. Они могут принимать форму, например, растворов, сиропов или суспензий, или же они могут представлять собой сухие продукты, предназначенные для восстановления перед применением с помощью воды или другого подходящего носителя. Эти фармацевтические формы могут представлять собой любые дозированные формы, например, ампулы, или формы, включающие несколько доз, обычно находящихся во флаконах. Упомянутые жидкие препараты можно получать стандартными способами с использованием фармацевтически приемлемых добавок, включая, но не ограничиваясь этим, суспендирующие агенты (например, сиропы сорбита или маннита, сироп сахарозы, производные целлюлозы, например, карбоксиметилцеллюлозу или гидроксипропилцеллюлозу, камеди, например, гуаровую камедь, ксантановую камедь или гуммиарабик, или гидрированные пищевые жиры); эмульгирующие агенты (например, лецитин, полисорбат, полиоксиэтилированное касторовое масло, эфиры сорбитана или полоксамеры); водные и неводные носители (например, воду, монопропиленгликоль, полиэтиленгликоль, глицерин, кунжутное масло, масло хлопчатника, соевое масло, касторовое масло, миндальное масло, сложные эфиры, имеющие форму масла, этиловый спирт, фракционированные растительные масла, триглицериды с цепями средней длины и т.д.); и консерванты (например, как правило, метил или пропил-п-гидроксибензоаты и их соли, сорбиновую кислоту и ее соли, бензойную кислоту и ее соли). Описываемые препараты при необходимости могут также содержать буферные соли, стабилизаторы, антиоксиданты, вкусоароматические добавки, красители и подсластители.
Другим подходящим путем введения препаратов по настоящему изобретению является парентеральный путь. В этом случае действующий ингредиент будет включаться в составы, подходящие для инъекций внутривенным путем, внутримышечным путем или подкожным путем; такие фармацевтические композиции могут включать носители, которые являются фармацевтически подходящими для составов, вводимых с помощью инъекций. Эти композиции могли бы иметь форму растворов или эмульсий (например, мелкодисперсных эмульсий, микроэмульсий или наноэмульсий и т.д.), и они могут быть стерильными. Описываемые композиции для придания изотоничности могли бы содержать солевые компоненты (моно- или динатрийфосфат, хлориды натрия, калия, кальция или магния и т.п. или смеси перечисленных солей). В некоторых случаях, если действующий ингредиент недостаточно стабилен, чтобы его можно было включать в раствор, этот действующий ингредиент поставляют в составе сухого препарата, в виде лиофилизованных композиций, либо в форме порошка, которые можно восстановить обратно в форму, подходящую для введения пациенту путем инъекции, добавлением, в зависимости от ситуации, стерилизованного неводного раствора для инъекций или стерилизованного водного, или физиологического солевого раствора.
Готовым продуктом обычно заполняют флаконы или ампулы.
Применяемые для введения дозировки обычно можно рассматривать, как функцию различных параметров, и, в частности, они зависят от применяемой формы введения, соответствующей патологии или, в качестве альтернативы, от желаемой продолжительности лечения.
Для поддержания необходимого уровня лекарственного средства в организме и чтобы избежать слишком частых инъекций, можно применять парентеральные составы пролонгированного действия. Эти фармацевтические формы, как правило, но не ограничиваясь этим, представляют собой микрочастицы, имплантаты или жидкости, которые после инъекции образуют in situ гель, коллоид или полутвердую матрицу. Указанные составы пролонгированного действия можно получать традиционными способами с применением фармацевтически приемлемых добавок, в число которых входят, не ограничиваясь этим, биосовместимые и биоразрушаемые полимеры (например, поли(ε-капролактон), поли(этиленоксид), поли(гликолевая кислота), поли[(молочная кислота)-(гликолевая кислота)], поли(молочная кислота) и т.д.), не биоразрушаемые полимеры (например, сополимер этилен-винилацетат, полиуретан, полиэфир(амид), поливинилхлорид и т.д.), водные и неводные носители (например, вода, кунжутное масло, масло хлопчатника, соевое масло, касторовое масло, миндальное масло, сложные эфиры, являющиеся маслами, этиловый спирт, фракционированные растительные масла, пропиленгликоль, ДМСО, ТГФ, 2-пирролидон, N-метилпирролидинон, N-винилдпирролидинон и т.д.). Конкретно для прегненолона, составы с пролонгированным высвобождением будут также способствовать уменьшению образования активных продуктов метаболизма прегненолона.
Для получения фармацевтических композиций эффективное количество соединения по настоящему изобретению можно растворить или диспергировать в фармацевтически приемлемом носителе или водной среде.
Жидкие фармацевтические формы, подходящие для инъекций, включают, не ограничиваясь этим, стерильные водные растворы или дисперсии, или неводные растворы, содержащие стерильные масляные компоненты, такие как кунжутное масло, масло арахиса, масло хлопчатника и т.д., триглицериды с цепями средней длины, триацетиновое масло, стерильный пропиленгликоль, стерильный полиэтиленгликоль, стерильный глицерин или стерильный раствор полиола.
Можно получать растворы действующих соединений в форме свободного основания или фармакологически приемлемых солей в подходящем растворителе, смешанном, если необходимо, с солюбилизирующим агентом, обычно ПАВ, например, но не ограничиваясь этим, производными полисорбатов, полиэтоксилированным касторовым маслом (например, Cremophor RH40), гидроксистеаратом ПЭГ15 (Solutol HS15), полоксамером (например, Lutrol F68), стабилизатором, например, EDTA и ее солями, с буферным агентом или с антиоксидантом (аскорбиновой кислотой и ее солью, ацетатом токоферола или метабисульфитом натрия). Указанные препараты могут содержать консерванты для предотвращения роста микроорганизмов.
Для улучшения стабильности составы можно получать в форме порошков, где указанные порошки являются стерильными, и растворять их непосредственно перед применением в водном или неводном растворителе. Указанные препараты, как правило, предназначены для немедленного введения стерильных растворов или дисперсий для инъекций.
Во всех случаях лекарственная форма должна быть стерильной и устойчивой в условиях применения, производства и хранения, и она должна быть защищена от инфицирования микроорганизмами, например, бактериями и грибками.
Соединения по настоящему изобретению
Общие формулы:
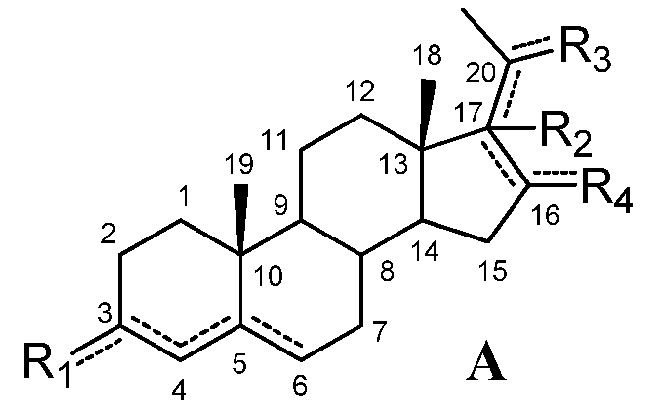
Соединения по настоящему изобретению имеют следующую формулу:
 ,
,
где:
означает, что связь является простой или двойной связью,
R1 означает, что C3 замещен
-H,
-галогеном,
-OH,
C1-8 алкокси,
Bn-O-,
Bn-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
Ph-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
=O,
-NR5R6, где каждый из R5 и R6 независимо представляет собой H, C1-8 алкил, Bn или Ph,
-O-CO-R7, где R7 означает алкил,
-O-CO-C2H4-COOH или
-N3,
-R2 означает, что C17 замещен
-H,
-OH,
галогеном,
C1-8 алкилом,
C1-8 алкокси,
C2-6 алкенилом,
Bn-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
Ph-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, карбоксилом или галогеном, или
Bn-O-,
R3 означает, что C20 замещен
-H,
-OH,
C1-8 алкилом,
Bn,
-NR8R9, где каждый из R8 и R9 независимо представляет собой H, C1-8 алкил или Bn,
=CR10R11, где каждый из R10 и R11 независимо представляет собой H или C1-7 алкил, или
=O,
R4 означает, что C16 замещен
-H,
-OH или
=O,
при условии, что
если связь между C16 и C17 является двойной, R2 отсутствует и связь между C17 и C20 является простой,
если связь между C17 и C20 является двойной, C20 замещен -H или -OH и R2 отсутствует, и
если связь между C4 и C5 является двойной, связь между C5 и C6 является простой и наоборот.
В одном из вариантов осуществления соединение по настоящему изобретению имеет формулу (I):
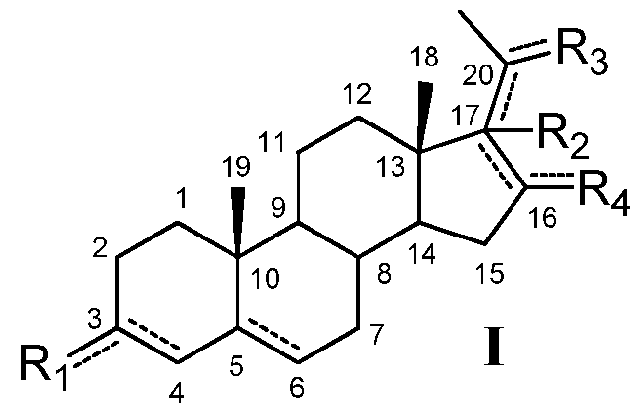 ,
,
где:
означает, что связь является простой или двойной связью,
R1 означает, что C3 замещен
-H,
-галогеном,
-OH,
C1-8 алкокси,
Bn-O-,
Bn-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
Ph-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
=O,
-NR5R6, где каждый из R5 и R6 независимо представляет собой H, C1-8 алкил, Bn или Ph,
-O-CO-R7, где R7 означает алкил, или
-O-CO-C2H4-COOH,
-R2 означает, что C17 замещен
-H,
-OH,
галогеном,
C1-8 алкилом,
C1-8 алкокси,
C2-6 алкенилом,
Bn-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
Ph-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, карбоксилом или галогеном, или
Bn-O-,
R3 означает, что C20 замещен
-H,
-OH,
C1-8 алкилом,
Bn,
-NR8R9, где каждый из R8 и R9 независимо представляет собой H, C1-8 алкил или Bn,
=CR10R11, где каждый из R10 и R11 независимо представляет собой H или C1-7 алкил, или
=O,
R4 означает, что C16 замещен
-H,
-OH или
=O,
при условии, что
если связь между C16 и C17 является двойной, R2 отсутствует и связь между C17 и C20 является простой, и
если связь между C17 и C20 является двойной, C20 замещен -H или -OH и R2 отсутствует.
Прегненолон:
В конкретном варианте осуществления соединение по настоящему изобретению представляет собой прегненолон или его фармацевтически приемлемую соль.
Прегненолон является хорошо известным стероидом (номер CAS 145-13-1). Это соединение представляет собой продукт первой стадии стероидного синтеза в мозге и других органах.
Как отмечено выше, авторы изобретения показали, что прегненолон и его фармацевтически приемлемые соли, например, ацетат или гемисукцинат прегненолона, являются ингибиторами рецептора CB1 и в силу этого могут применяться в лечении патологических расстройств и заболеваний, при которых необходимо применение антагониста рецептора CB1.
В предпочтительном варианте осуществления прегненолон вводят субъекту в такой дозировке, чтобы концентрация прегненолона в плазме субъекта не превышала 100 нг/мл. Предпочтительно, прегненолон вводят в форме составов с продолжительным высвобождением.
Фактически, при введении прегненолона в низких дозировках, которые позволяют оставаться в диапазоне эффективных концентраций (примерно 100 нг/мл или 100 нг/г ткани), уменьшается превращение прегненолона в активные продукты метаболизма. Таким образом, авторы изобретения показали, что введение прегненолона в низких концентрациях, которые не приводят к росту содержания активных продуктов метаболизма, может ингибировать эффект активации рецептора CB1. В этом заключается основное усовершенствование и отличие от документов известного уровня техники, в которых прегненолон вводят в высоких дозах для увеличения уровней активных метаболитов, действию которых приписывали наблюдаемые терапевтические эффекты. Введение прегненолона в низких дозах является предпочтительным, поскольку такой способ позволяет воздействовать на CB1-зависимые патологии без нежелательных эффектов, возникающих из-за увеличения уровней активных стероидных производных прегненолона, которые обладают прогестагенной, андрогенной, эстрогенной, глюкокортикоидной активностью или нейромодулирующими свойствами, как и другие стероиды мозга, являющиеся производными прегненолона, включая, но не ограничиваясь этим, аллопрегнанолон, тестостерон, DHEA.
Соединения с низким метаболизмом или не подвергающиеся метаболизму
В качестве альтернативы соединение по настоящему изобретению после введения субъекту практически не превращается в активные производные прегненолона.
Прегненолон обычно считают неактивным прекурсором его активных стероидных производных. Активные производные прегненолона, включая, но не ограничиваясь этим, прегненолон-сульфат, аллопрегнанолон, DHEA, DHEA-сульфат, принимают участие в регулировании различных поведенческих функций.
Однако авторы изобретения обнаружили, что ингибирование рецептора CB1 осуществляется только прегненолоном, и в нем не участвуют активные производные прегненолона.
Применение производных прегненолона, которые не превращаются или практически не превращаются в метаболиты прегненолона, позволяет избежать побочных эффектов, которые могут быть связаны с метаболитами, предшественником которых является прегненолон, и которые обладают прогестагенной, андрогенной, эстрогенной, глюкокортикоидной активностью или нейромодулирующими свойствами, как и другие стероиды мозга, являющиеся производными прегненолона, включая, но не ограничиваясь этим, аллопрегнанолон, тестостерон, DHEA.
Способность соединения по настоящему изобретению превращаться или не превращаться в активные производные прегненолона можно оценить путем введения этого соединения, например, в виде инъекции 50 мг/кг, крысе, умерщвления крысы через 30 минут, измерения концентрации аллопрегнанолона и эпиаллопрегнанолона в прилежащем ядре (центре удовольствия) мозга крысы с помощью ГХ/МС и сравнения этих концентраций с концентрациями аллопрегнанолона и эпиаллопрегнанолона у крысы, которой вводили только носитель или прегненолон.
В качестве альтернативы соединение можно вводить в любую линию клеток, которые экспрессируют фермент, вызывающий метаболизм прегненолона в культуре, затем измерять содержание аллопрегнанолона и эпиаллопрегнанолона в клетках или в культуральной среде с помощью ГХ/МС и сравнивать эти концентрации с концентрациями аллопрегнанолона и эпиаллопрегнанолона в клеточных культурах, которые обрабатывали только носителем или прегненолоном.
В одном из вариантов осуществления соединение по настоящему изобретению представляет собой соединение формулы I, где
R1 означает, что C3 замещен
-H, -галогеном, -OH, C2-8 алкокси, Bn-O-,
Bn-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
Ph-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
=O,
-NR5R6, где каждый из R5 и R6 независимо представляет собой H, C1-8 алкил, Bn или Ph,
-O-CO-R7, где R7 означает алкил, или
-O-CO-C2H4-COOH, и
-R2, R3, R4 соответствуют данным выше определениям.
Авторы изобретения протестировали широкий круг производных прегненолона для обнаружения производных, которые практически не превращаются в активные метаболиты после введения субъекту, и вследствие этого сохраняют ингибирующее действие на рецептор CB1.
Было обнаружено несколько групп таких производных:
Связи между C16 и C17 и между C17 и C20 являются простыми связями
В одном из вариантов осуществления связь между атомами C16 и C17 и связь между атомами C17 и C20 являются простыми связями.
Связь между C4 и C5 является двойной связью
В одном из вариантов осуществления связи между атомами C3 и C4 и между С5 и С6 являются простыми связями, и связь между атомами C4 и C5 является двойной.
В этом варианте осуществления соединение по настоящему изобретению отображается формулой B:
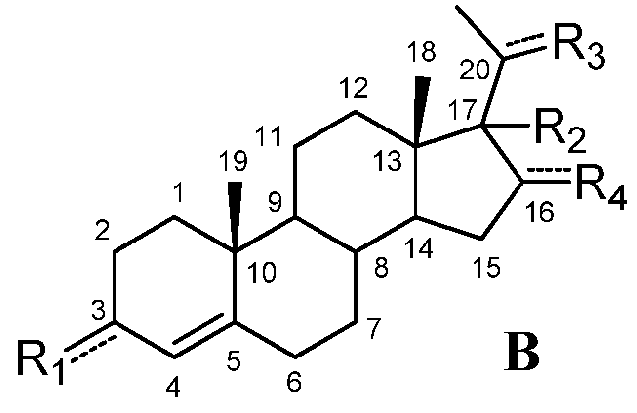 ,
,
где:
R1 означает, что C3 замещен -OH или =O,
-R2 означает, что C17 замещен -H, -OH, C1-8 алкилом, галогеном или Bn,
R3 означает, что C20 замещен -OH или =O,
R4 означает, что C16 замещен -H.
Фактически, авторы изобретения обнаружили, что производные прегненолона с двойной связью между C4 и C5 и имеющие указанные выше заместители R1, R2 и/или R3 не подвергаются метаболизму с образованием производных прегненолона.
Предпочтительно, соединение такого типа выбрано из группы, состоящей из 4-прегнен-17α,20α-диол-3-она, 4-прегнен-3β,20α-диола, 4-прегнен-20α-ол-3-она, 17α-метилпрогестерона и 17α-бензилпрогестерона.
Связи между C5 и C6 и между C4 и C5 являются простыми связями
В одном из вариантов осуществления связи между C3 и C4, C5 и C6 и между C4 и C5 являются простыми связями.
В этом варианте осуществления соединение по настоящему изобретению имеет формулу (C):
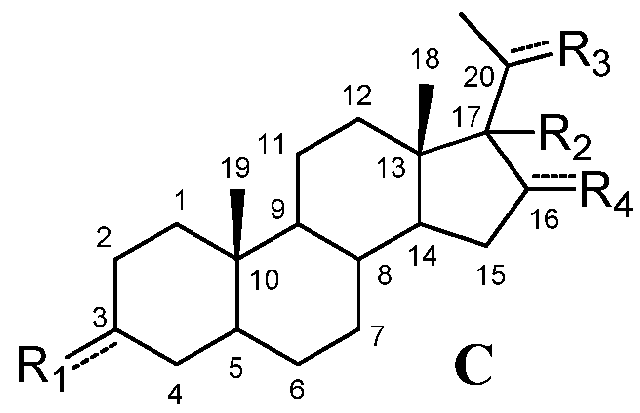 ,
,
где:
R1 означает, что C3 замещен =O или -OH,
-R2 означает, что C17 замещен -H,
R3 означает, что C20 замещен =O, и
R4 означает, что C16 замещен -H.
Предпочтительно, указанное соединение представляет собой 5β-прегнан-3,20-дион или 5β-прегнан-3β-ол-20-он.
Связь между C5 и C6 является двойной связью
В одном из вариантов осуществления связи между C3 и C4 и между C4 и C5 являются простыми, и связь между C5 и C6 является двойной связью.
В этом варианте осуществления соединение по настоящему изобретению имеет формулу (D):
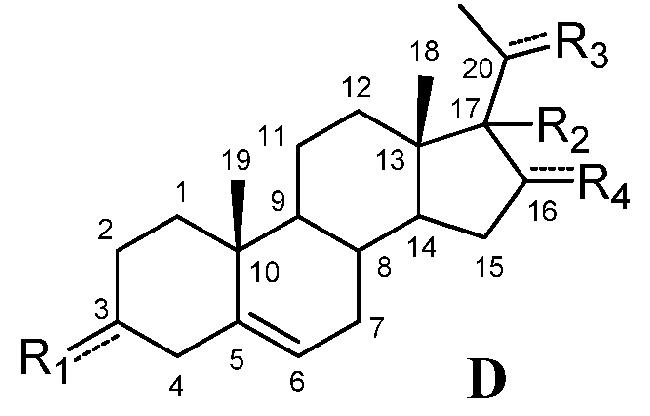 .
.
В этом варианте осуществления соединение по настоящему изобретению предпочтительно представляет собой прегненолон, модифицированный по положениям C3, C17 и/или C20.
Модификация по положению C3:
В одном из вариантов осуществления соединение представляет собой прегненолон, модифицированный по положению C3.
В этом варианте осуществления соединение имеет формулу (D),
и
R1 означает, что C3 замещен
-H, -галогеном, C1-8 алкокси, Bn-O-,
Bn-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
Ph-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
=O,
-NR5R6, где каждый из R5 и R6 независимо представляет собой H, C1-8 алкил, Bn или Ph,
-O-CO-R7, где R7 означает алкил, или
-O-CO-C2H4-COOH,
-R2 означает, что C17 замещен -H,
R3 означает, что C20 замещен =O, и
R4 означает, что C16 замещен -H.
Кроме того, в этом варианте осуществления соединение имеет формулу (D), и
R1 означает, что C3 замещен
галогеном, Bn-O- или N3,
-R2 означает, что C17 замещен -H,
R3 означает, что C20 замещен =O, и
R4 означает, что C16 замещен -H.
Предпочтительно, соединение по этому варианту осуществления выбрано из группы, состоящей из 3β-бензилоксипрегненолона, 3-азидопрегненолона и 3β-фторпрегненолона.
Модификация по положениям C3 и C17:
В одном из вариантов осуществления соединение по настоящему изобретению представляет собой прегненолон, модифицированный по положениям C3 и C17.
В этом варианте осуществления соединение имеет формулу (D), и
R1 означает, что C3 замещен C1-8 алкокси, галогеном, Bn-O- или N3,
-R2 означает, что C17 замещен Bn, -CH3 или C2-6 алкенилом,
R3 означает, что C20 замещен =O, и
R4 означает, что C16 замещен -H.
Предпочтительно, соединение по этому варианту осуществления выбрано из группы, состоящей из 3β-фтор-17α-метилпрегненолона, 17α-бензил-3β-фторпрегненолона, 17α-бензил-3β-бензилоксипрегненолона и 3β-бензилокси-17α-метилпрегненолона.
Модификация по положению C17:
В одном из вариантов осуществления соединение представляет собой прегненолон, модифицированный только по положению C17.
В этом варианте осуществления соединение имеет формулу (D), и
R1 означает, что C3 замещен -OH,
-R2 означает, что C17 замещен
-OH, галогеном, C1-8 алкилом, C1-8 алкокси, C2-6 алкенилом,
Bn-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
Ph-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, карбоксилом или галогеном, или
Bn-O-,
R3 означает, что C20 замещен =O, и
R4 означает, что C16 замещен -H.
Предпочтительно, -R2 означает, что C17 замещен C1-8 алкилом, C1-8 алкокси или Bn-.
Более предпочтительно, указанное соединение выбрано из группы, состоящей из 17α-метилпрегненолона, 17α-бензилпрегненолона, 17-метоксипрегненолона и 17α-этилпрегненолона.
Модификация по положениям C3 и/или C17
Настоящее изобретение относится также к соединению формулы (II)
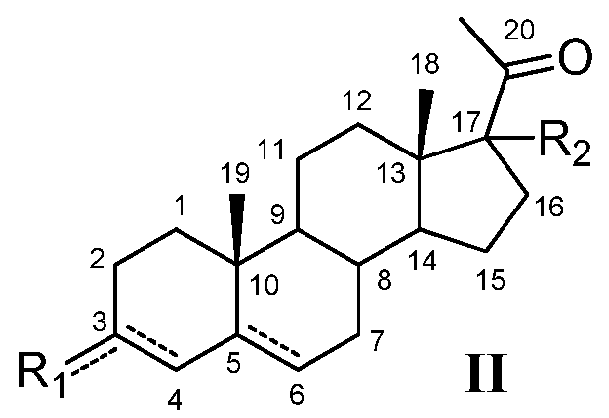
или его фармацевтически приемлемой соли, где:
означает, что связь является простой или двойной связью,
R1 означает, что C3 замещен -OH, и
-R2 означает, что C17 замещен
C3-8 алкилом,
C2-8 алкокси,
Bn-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
Ph-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, карбоксилом или галогеном, или
Bn-O-,
или где:
R1 означает, что C3 замещен
C1-8 алкокси,
Bn-O- или
галогеном, и
-R2 означает, что C17 замещен
C1-8 алкилом,
C2-6 алкенилом,
C1-8 алкокси,
Bn-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, амино, карбоксилом или галогеном,
Ph-, необязательно замещенным C1-8 алкилом, C1-8 алкокси, циано, нитро, карбоксилом или галогеном, или
Bn-O-.
Предпочтительно, соединение формулы (II) выбрано из группы, состоящей из 3β-фтор-17α-метилпрегненолона, 17α-бензил-3β-фторпрегненолона, 17α-бензил-3β-бензилоксипрегненолона, 3β-бензилокси-17α-метилпрегненолона, 17α-бензилпрегненолона, 3β-метокси-17α-метилпрегненолона, 17α-аллил-3β-метоксипрегненолона и 17α-бензил-3β-метоксипрегненолона.
Настоящее изобретение относится также к фармацевтической композиции, включающей соединение формулы (II) или его фармацевтически приемлемую соль, а также фармацевтически приемлемый носитель.
Модификации по положению C20:
В одном из вариантов осуществления соединение по настоящему изобретению представляет собой прегненолон, модифицированный по положению C20.
В этом варианте осуществления соединение имеет формулу D, где:
R1 означает, что C3 замещен -OH,
-R2 означает, что C17 замещен -H,
R3 означает, что C20 замещен
-H, -OH, C1-8 алкилом и Bn,
-NR8R9, где каждый из R8 и R9 независимо представляет собой H, C1-8 алкил или Bn,
=CR10R11, где каждый из R10 и R11 независимо представляет собой H или C1-7 алкил, или
=O, и
R4 означает, что C16 замещен -H.
Предпочтительно, R3 означает, что C20 замещен -H, -OH или -NR8R9, где каждый из R8 и R9 независимо представляет собой H или C1-8 алкил.
Более предпочтительно, указанное соединение выбрано из группы, состоящей из 5-прегнен-3β,20α-диола, 20-деоксипрегненолона и 20-метиламино-5-прегнен-3β-ола.
Модификация по положениям C20 и C16:
В одном из вариантов осуществления соединение по настоящему изобретению представляет собой прегненолон, модифицированный по положениям C20 и/или C16.
В этом варианте осуществления соединение имеет формулу D, где:
R1 означает, что C3 замещен -OH,
-R2 означает, что C17 замещен H,
R3 означает, что C20 замещен -OH или -H, и
R4 означает, что C16 замещен -OH или =O.
Связь между C16 и C17 является двойной связью, и связь между C17 и C20 является простой связью
В другом варианте осуществления связь между C16 и C17 является двойной связью, связь между C17 и C20 является простой связью, и R1 означает, что C3 замещен -H, -OH или =O, R3 означает, что C20 замещен -H, -OH или =O, и R4 означает, что C16 замещен -H.
Этот вариант осуществления отображается приведенной ниже формулой E:
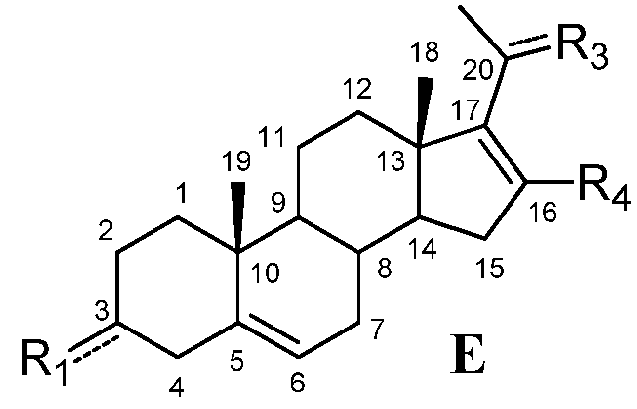 .
.
Предпочтительно, указанное соединение представляет собой 5,16-прегнадиен-20-он.
R1 находится в β-положении:
В наиболее предпочтительном варианте осуществления, если связи между C3 и R1 и между C3 и C4 являются простыми, R1 находится в β-положении.
Фактически, авторы изобретения обнаружили, что в противоположность производным, содержащим заместитель R1 в α-положении, производные, в которых R1 находится в β-положении, не оказывают влияния на рецепторы GABA и глутаматов и позволяют избежать побочных эффектов, вызываемых модификациями этих рецепторов, например, но не ограничиваясь этим, седативного действия, нарушений памяти, двигательного возбуждения. Кроме того, производные с C3 в β-положении сохраняют ингибирующее действие на рецептор CB1.
Лечение патологических состояний или расстройств:
Настоящее изобретение относится также к определенному выше соединению по настоящему изобретению или его фармацевтически приемлемой соли для применения в способе лечения по настоящему изобретению.
В одном из вариантов осуществления для применения в упомянутом способе лечения предназначено соединение формулы (II), которое определено выше, или его фармацевтически приемлемая соль.
Кроме того, настоящее изобретение относится к применению соединения по настоящему изобретению, которое определено выше, или его фармацевтически приемлемой соли для изготовления лекарственного средства.
В одном из вариантов осуществления для получения лекарственного средства предназначено соединение формулы (II), которое определено выше, или его фармацевтически приемлемая соль.
Настоящее изобретение относится также к способу лечения патологического состояния или расстройства у субъекта, которому необходимо такое лечение, включающему введение указанному субъекту эффективного количества соединения по настоящему изобретению, которое определено выше, или его фармацевтически приемлемой соли.
Помимо этого, настоящее изобретение относится к соединению по настоящему изобретению, которое определено выше, или его фармацевтически приемлемой соли для применения в лечении патологического состояния или расстройства, выбранного из группы, состоящей из расстройств мочевого пузыря и желудочно-кишечного тракта; воспалительных заболеваний; сердечно-сосудистых заболеваний; нефропатий; глаукомы; спастичности; рака; остеопороза; метаболических расстройств; ожирения; расстройств, связанных с пагубными пристрастиями, зависимостями, злоупотреблениями и их рецидивами; психиатрических и неврологических расстройств; нейродегенеративных расстройств; аутоиммунного гепатита и энцефалита; боли; репродуктивных расстройств, а также воспалительных и фиброзных заболеваний кожи.
Настоящее изобретение относится к способу лечения патологического состояния или расстройства, выбранного из группы, состоящей из расстройств мочевого пузыря и желудочно-кишечного тракта; воспалительных заболеваний; сердечно-сосудистых заболеваний; нефропатий; глаукомы; спастичности; рака; остеопороза; метаболических расстройств; ожирения; расстройств, связанных с пагубными пристрастиями, зависимостями, злоупотреблениями и их рецидивами; психиатрических и неврологических расстройств; нейродегенеративных расстройств; аутоиммунного гепатита и энцефалита; боли; репродуктивных расстройств, а также воспалительных и фиброзных заболеваний кожи, у субъекта, которому это необходимо, включающему введение указанному субъекту эффективного количества соединения по настоящему изобретению, которое определено выше, или его фармацевтически приемлемой соли.
Настоящее изобретение относится к применению соединения по настоящему изобретению, которое определено выше, для получения лекарственного средства для лечения патологического состояния или расстройства, выбранного из группы, состоящей из расстройств мочевого пузыря и желудочно-кишечного тракта; воспалительных заболеваний; сердечно-сосудистых заболеваний; нефропатий; глаукомы; спастичности; рака; остеопороза; метаболических расстройств; ожирения; расстройств, связанных с пагубными пристрастиями, зависимостями, злоупотреблениями и их рецидивами; психиатрических и неврологических расстройств; нейродегенеративных расстройств; аутоиммунного гепатита и энцефалита; боли; воспалительных и фиброзных заболеваний кожи.
Заболевания желудочно-кишечного тракта:
В предпочтительном варианте осуществления соединения по настоящему изобретению предназначены для применения в лечении желудочно-кишечных заболеваний.
В предпочтительном варианте осуществления соединение по настоящему изобретению применяется для получения лекарственного средства для лечения желудочно-кишечных заболеваний.
В предпочтительном варианте осуществления изобретение относится к способу лечения желудочно-кишечных заболеваний у субъекта, имеющего такую необходимость, включающему введение указанному субъекту эффективного количества соединения по настоящему изобретению.
Предпочтительно, соединение по настоящему изобретению предназначено для применения в лечении заболеваний печени, в частности, неалкогольного стеатогепатита (NASH) и цирроза печени.
Фактически, авторы изобретения показали, что прегненолон и его производные ингибируют накопление липидов в модели ожирения, а также выработку TNFα.
Ожирение или метаболические расстройства:
В предпочтительном варианте осуществления соединение по настоящему изобретению предназначено для применения в лечении ожирения или метаболических расстройств.
В предпочтительном варианте осуществления соединение по настоящему изобретению применяется для изготовления лекарственного средства для лечения ожирения или метаболического расстройства.
В предпочтительном варианте осуществления изобретение относится к способу лечения ожирения или метаболического расстройства у субъекта, которому это необходимо, включающему введение указанному субъекту эффективного количества соединения по настоящему изобретению.
Предпочтительно, соединение по настоящему изобретению предназначено для применения в лечении диабета и дислипидемии.
Фактически, авторы изобретения показали, что прегненолон и его производные ингибируют накопление липидов в модели ожирения, а также выработку TNFα.
Расстройства, связанные с пристрастиями, зависимостями, злоупотреблениями и их рецидивами:
В другом предпочтительном варианте осуществления соединение по настоящему изобретению предназначено для применения в лечении расстройств, связанных с пристрастиями, зависимостями, злоупотреблениями и их рецидивами.
В предпочтительном варианте осуществления соединение по настоящему изобретению применяется для изготовления лекарственного средства для лечения расстройств, связанных с пристрастиями, зависимостями, злоупотреблениями и их рецидивами.
В предпочтительном варианте осуществления изобретение относится к способу лечения расстройств, связанных с пристрастиями, зависимостями, злоупотреблениями и их рецидивами, у субъекта, которому это необходимо, включающему введение указанному субъекту эффективного количества соединения по настоящему изобретению.
Предпочтительно, соединение по настоящему изобретению предназначено для применения в лечении расстройств, связанных с пристрастиями, зависимостями, злоупотреблениями, интоксикацией, вызванных каннабиноидами, и их рецидивами.
Фактически, авторы изобретения показали, что, в частности, прегненолон и его производные ингибируют эндоканнабиноидную тетраду, стимулируемую активацией рецептора CB1 под действием THC (тетрагидроканнабинола); THC-индуцированное потребление пищи; THC-индуцированные нарушения памяти; THC-индуцированное изменение синаптической передачи; стремление к самостоятельному введению агонистов CB1.
Соединения по настоящему изобретению также применимы для лечения расстройств, связанных с пристрастием, зависимостью и злоупотреблением, вызванных алкоголем, а также их рецидивов.
Нейродегенеративные и психиатрические расстройства:
В другом предпочтительном варианте осуществления соединение по настоящему изобретению предназначено для применения в лечении нейродегенеративных и психиатрических расстройств.
В следующем предпочтительном варианте осуществления соединение по настоящему изобретению применяется для изготовления лекарственного средства для лечения нейродегенеративных и психиатрических расстройств.
В еще одном предпочтительном варианте осуществления изобретение относится к способу лечения нейродегенеративного и психиатрического расстройства у субъекта, которому это необходимо, включающему введение указанному субъекту эффективного количества соединения по настоящему изобретению.
Предпочтительно, соединение по настоящему изобретению предназначено для применения в лечении болезни Паркинсона и шизофрении.
Фактически, авторы изобретения показали, что прегненолон способен модулировать активность дофаминергической системы, повышенную в результате действия THC или кокаина, и влияние активации CB1 на возбуждающую синаптическую передачу.
Воспалительные и фиброзные заболевания кожи:
В другом предпочтительном варианте осуществления соединение по настоящему изобретению предназначено для применения в лечении воспалительных и фиброзных заболеваний кожи.
В следующем предпочтительном варианте осуществления соединение по настоящему изобретению применяется для изготовления лекарственного средства для лечения воспалительных и фиброзных заболеваний кожи.
В еще одном предпочтительном варианте осуществления изобретение относится к способу лечения воспалительных и фиброзных заболеваний кожи. Таким образом, авторы изобретения показали, что соединения по настоящему изобретению ингибируют выработку TNFα.
Предпочтительно, соединение по настоящему изобретению предназначено для применения в лечении воспаления кожи, воспаления и рака кожи, вызванных УФ-излучением, фиброза кожи и для заживления ран.
Сердечно-сосудистые заболевания:
В другом предпочтительном варианте осуществления соединение по настоящему изобретению предназначено для применения в лечении сердечно-сосудистых заболеваний.
В следующем предпочтительном варианте осуществления соединение по настоящему изобретению применяется для изготовления лекарственного средства для лечения сердечно-сосудистых заболеваний.
В еще одном предпочтительном варианте осуществления изобретение относится к способу лечения сердечно-сосудистых заболеваний.
Так, например, авторы изобретения показали, что соединения по настоящему изобретению уменьшают накопление липидов и ингибируют выработку TNFα.
Предпочтительно, соединение по настоящему изобретению предназначено для применения в лечении кардиомиопатии.
Более предпочтительно, соединение по настоящему изобретению предназначено для применения в лечении кардиомиопатии, выбранной из группы, состоящей из цирротической кардиомиопатии и кардиомиопатий, вызванных антинеопластическими препаратами, нарушения сердечных сокращений, инфаркта и атеросклероза.
Далее по тексту настоящее изобретение будет проиллюстрировано описанными ниже чертежами и примерами. Однако не стоит интерпретировать эти примеры и чертежи, как ограничивающие объем настоящего изобретения каким-либо образом.
Описание иллюстративного материала
На фиг. 1 показаны диаграммы, на которых изображены следующие параметры для крыс Wistar: (A) исходные уровни прегненолона (PREG), аллопрегнанолона (ALLO), эпиаллопрегнанолона (EPI), тестостерона (T) и дигидротестостерона (DHT) в прилежащем ядре мозга. (B) Влияние инъекции THC (3 мг/кг, ip), которая вызывает значительное и долговременное увеличение концентрации прегненолона в прилежащем ядре мозга. Влияние других препаратов, вызывающих зависимость: кокаина (20 мг/кг, ip), морфина (2 мг/кг, ip), никотина (0,4 мг/кг, ip) и этанола (1 г/кг, ip), которые вызывают существенно меньшее увеличение уровней прегненолона в прилежащем ядре. Влияние THC и других препаратов, вызывающих зависимость, на стероидные производные прегненолона: аллопрегнанолона (C), эпиаллопрегнанолона (D), тестостерона (E) и DHT (F), уровни которых были существенно меньше наблюдаемых для прегненолона. Стрелки показывают время инъекции всех препаратов. Данные выражены в виде среднее значение ± SEM (стандартное отклонение) (число животных в группе n=6-8).
На фиг. 2 показаны диаграммы, отображающие отрицательное влияние прегненолона на THC-индуцированную характерную тетраду у мышей C57B1/6. THC дозозависимым образом (по сравнению с группой, получавшей носитель) понижал (A) двигательную активность [F(3,59)=17,7, P<0,001] и (B) температуру тела (дельта T по сравнению с контролем) [F(3,59)=39,9, P<0,001], и увеличивал (C) каталепсию (время задержки до начала движения) [F(3,59)=47,5, P<0,001] и (D) аналгезию (задержку до начала ноцицептивной реакции в тесте с горячей пластиной) [F(3,59)=5,15, P<0,01]. Ингибитор P450scc, а именно, аминоглютетимид (AMG, 50 мг/кг, ip), который блокирует синтез прегненолона, усиливает все характерные эффекты THC: (A) гиполокомоцию [F(3,98)=13,8, P<0,001], (B) гипотермию [F(3,98)=4,7, P<0,01], (C) каталепсию [F(3,98)=2,1, P<0,05] и (D) аналгезию [F(3,98)=2,2, P<0,05]. Прегненолон (PREG, 6 мг/кг, sc) уменьшал эффекты THC (10 мг/кг, ip) и полностью устранял эффекты AMG (50 мг/кг, ip) на (E) локомоцию, (F) температуру тела, (G) каталепсию и (H) аналгезию. Прегненолон не оказывал влияния на животных, которые не получали THC. Данные выражены в виде среднее значение ± SEM (число животных в группе n=6-12). *=P<0,05; **=P<0,01; ***=P<0,001 по сравнению с мышами, получавшими носитель.
На фиг. 3 показаны диаграммы, отображающие подавление прегненолоном CB1-опосредованного потребления пищи. (A) Увеличение потребления пищи, вызванное THC (0,5 мг/кг, ip), у крыс Wistar, получавших пищу без ограничения, дозозависимым образом подавлялось инъекциями прегненолона [F(3,94)=3,65, P<0,02]. (B) Увеличение потребления пищи, вызванное THC (1 мг/кг, ip), у мышей C57B1/6, лишенных пищи на 24 часа, подавлялось прегненолоном (2 мг/кг, sc). (C) Прегненолон дозозависимым образом уменьшал потребление пищи у мышей C57B1/6, лишенных пищи на 24 часа. (D) Уменьшение потребления пищи, вызванное прегненолоном (PREG 6 мг/кг), у мышей C57B1/6, лишенных пищи на 24 часа, устранялось при предварительном введении антагониста рецептора CB1, а именно SR141716A (0,05 мг/кг, ip). Данные выражены в виде среднее значение ± SEM (число животных в группе n=6-12). *=P<0,05; **=P<0,01; ***=P<0,001 по сравнению с животными, получавшими носитель.
На фиг. 4 показаны диаграммы, отображающие влияние инъекций прегненолона на уровни активных стероидных производных прегненолона в мозге. (A) Введение прегненолона (s.c.) дозозависимым образом повышало уровни прегненолона в лобном отделе коры и гипоталамусе [F(3,19)=20, P<0,0001, F(3,19)=23, P<0,001, соответственно] мышей C57B1/6, лишенных пищи на 24 часа. Введение прегненолона не меняло концентраций: (B) аллопрегнанолона, (C) эпиаллопрегнанолона. Данные выражены в виде среднее значение ± SEM (число животных в группе n=7-8). *=P<0,05; ***=P<0,001 по сравнению с животными, получавшими носитель (PREG 0 мг/кг).
На фиг. 5 показаны диаграммы, отображающие ингибирование прегненолоном самостоятельного введения агониста CB1, а именно WIN 55512-2, мышами CD1. (A) Во время обучения самостоятельному введению WIN 55512-2 (инфузия 0,0125 мг/кг) число засовываний носа в активное отверстие значительно превышало соответствующее число для неактивного отверстия [F(1,18)=38,3, P<0,001]. (B) После обучения инъекция прегненолона (2 или 4 мг/кг, sc) уменьшала количество реакций на активное отверстие. (C) Прегненолон также снижал мотивацию к введению WIN 55512-2, что определяли по уменьшению точки остановки в схеме с прогрессивно нарастающей кратностью реакций. Данные выражены в виде среднее значение ± SEM (число животных в группе n=5-8). **=P<0,05; ***=P<0,001 по сравнению с животными, получавшими носитель (PREG 0 мг/кг).
На фиг. 6 показаны диаграммы, отображающие ингибирование прегненолоном неблагоприятного влияния THC на память. Как показал индекс различения в тесте на распознавание объектов, THC (10 мг/кг, ip) вызывал значительную амнезию, которая устранялась прегненолоном (PREG, 6 мг/кг, sc) [F(3,23)=24,6, P<0,001]. Данные выражены в виде среднее значение ± SEM (число животных в группе n=6-7). ***=P<0,001 по сравнению с животными, получавшими носитель.
На фиг. 7 показаны диаграммы, отображающие ингибирование прегненолоном увеличения дофаминергический активности, вызванной THC. Инъекция прегненолона крысам (PREG, 2 мг/кг, sc) понижала вызванное Δ9-тетрагидроканнабинолом (THC) увеличение (A) частоты пульсации дофаминергических нейронов вентральной области покрышки (VTA) [F(4,32)=7,14, p<0,001] и (B) увеличение исходящего потока дофамина в прилежащем ядре [F(10,80)=10,80, p<0,001]. Суммарные дозы THC (от 0,15 до 1,2 мг/кг) вводили i.v. (внутривенно) в момент времени 0 в течение 4 мин (регистрация 1 мин на дозу).
На фиг. 8 показаны диаграммы, отображающие ингибирование прегненолоном изменения дофаминергической активности, вызванной кокаином. Инъекция прегненолона (PREG, 2 мг/кг, sc) у крыс устраняла индуцированное кокаином уменьшение (A) залповой активности дофаминергических нейронов и увеличение исходящего потока дофамина в прилежащем ядре. Суммарные дозы кокаина (от 0,0125 до 0,8 мг/г) вводили i.v. в момент времени 0 в течение 4 мин (регистрация 1 мин на дозу). *=P<0,05; **=P<0,01; ***=P<0,001 по сравнению с крысами, получавшими носитель.
На фиг. 9 показаны диаграммы, отображающие влияние введения прегненолона на массу тела и потребление пищи. (A) Прегненолон 5 мг/кг (PREG5), который вводили подкожной инъекцией один раз в день до начала темновой фазы, вызывал последовательное и значимое уменьшение массы тела [F(1,29)=3,13, p<0,001] у животных, потреблявших корм с высоким содержанием жиров. (B) Тем не менее, прегненолон не изменял потребления пищи.
На фиг. 10 показаны диаграммы, отображающие влияние прегненолона на накопление жира и массу нежировой ткани у мышей с ожирением. Прегненолон, который вводили подкожно один раз в день до начала темнового цикла, дозозависимым образом блокировал увеличение жировой массы (A, C) и ослаблял понижение массы нежировой ткани (B, D), наблюдаемое при кормлении животных пищей с высоким содержанием жира. Массу жира и нежировой ткани у мышей определяли с помощью магнитного резонанса. PRE=значение, полученное до начала введения прегненолона. POST=значения, полученные через 30 дней введения прегненолона или носителя. *=P<0,05; **=P<0,01.
На фиг. 11 показаны диаграммы, отображающие ингибирование прегненолоном увеличения уровней TNFα, вызванного LPS. Бактериальный токсин LPS вводили интраперитонеально через 30 мин после инъекции прегненолона (6 мг/кг подкожно) или раствора носителя. Прегненолон вдвое уменьшал увеличение уровней TNF-альфа, вызванное инъекцией LPS. *=P<0,5.
На фиг. 12 показаны диаграммы, отображающие ингибирование прегненолоном THC-индуцированного ингибирования возбуждающих синаптических токов в прилежащем ядре крыс. Возбуждающие постсинаптические токи (EPSC), индуцируемые путем электрического стимулирования локальных аксонов, регистрировали с использованием методики фиксации потенциала пэтч-клэмп в ведущих нейронах прилежащего ядра на срезах мозга, полученных у взрослых крыс. (A) Обработка THC (20 мМ) промыванием в кювете достоверно ингибировала синаптическую передачу в контрольных срезах (ингибирование 34,3±3,7%, N=8). Действие THC значимо ослаблялось, если срезы предварительно обрабатывали 100 нМ прегненолоном (ингибирование 15,1±1,8%, N=9). (B) Остаточные синаптические токи в репрезентативных экспериментах усредняли в исходный момент времени и через 40 мин воздействия THC. ***T критерий: t=4,820, df=15, p=0,0002.
На фиг. 13 показаны диаграммы, отображающие ингибирование прегненолоном THC-индуцированного ингибирования возбуждающей синаптической передачи в прилежащем ядре мозга мышей. (A) Полевые возбуждающие постсинаптические потенциалы (fEPSP), индуцированные электрическим стимулированием локальных аксонов, регистрировали в срезах прилежащего ядра мозга, полученных у взрослых мышей. Обработка THC промыванием в кювете вызывала дозозависимое ингибирование синаптической передачи в контрольных срезах (N=5-9). Действие THC уменьшалось, если срезы предварительно обрабатывали 100 нМ прегненолоном (N=5-6). (B) Репрезентативные средние значения остаточных fEPSP регистрировали до начала эксперимента и через 40 мин после действия THC. Двухпараметрический анализ ANOVA: действие прегненолона p<0,002; действие THC p<0,0003.
На фиг. 14 представлены диаграммы, показывающие, что прегненолон является ингибитором активации рецептора CB1, который не изменяет ортостерическое связывание агонистов CB1. Прегненолон (10-12-10-4 М) не изменяет специфическое связывание агониста CB1 [3H]CP55940 с человеческим рецептором CB1, который экспрессируется клетками CHO. Данные выражены в виде среднее значение ± SEM (n=2-3 на концентрацию).
На фиг. 15 представлены диаграммы, демонстрирующие отсутствие влияния прегненолона на тревожное поведение. Тревожное поведение измеряли процентной долей входов и временем, проведенным в открытых рукавах приподнятого крестообразного лабиринта. Прегненолон не вызывал тревожного поведения даже в самых высоких дозах (10 мг/кг), существенно превышающих его эффективные дозы в диапазоне (1-6 мг/кг), соответствующие его максимальному влиянию на поведение. Ортостерический антагонист рецептора CB1 римонабант в дозировке 10 мг/кг вызывал увеличение тревожности, которое проявлялось в уменьшении входов в открытые рукава лабиринта и времени, проведенного в этих рукавах. P1, P6, P10 равно прегненолону 1, 6, 10 мг/кг. Rimo10 = римонабант 10 мг/кг. V означает носитель. *=p<0,05; ***=p<0,001.
На фиг. 16 представлены диаграммы, демонстрирующие отсутствие влияния прегненолона на токи, опосредованные рецепторами GABA-A. mIPSC регистрировали у взрослых мышей NAc PN в режиме фиксации потенциала при -80 мВ. A. Сводка амплитуд (ANOVA f=5,39, df=4,66, p<0,001) и времен затуханий (ANOVA f=24,7, df=4,66, p<0,0001) mIPSC для контрольных образцов (N=16) и срезов, предварительно обработанных прегненолоном (100 нМ: N=15; 1 мкМ: N=11) или аллопрегнанолоном (100 нМ: N=18; 1 мкМ: N=11). Апостериорные критерии: *=p<0,05; **p<0,01; ***=p<0,001. B. Результаты регистрации репрезентативных значений остаточных mIPSC. C. Средние остаточные значения mIPSC, нормализованные относительно пиков. Необходимо отметить, что только аллопрегнанолон в концентрации 1 мкМ оказывает значительное влияние на фазу затухания mIPSC. cont=контроль; preg=прегненолон; allo=аллопрегнанолон.
На фиг. 17 отображены диаграммы, демонстрирующие, что прегненолон не изменяет токи, опосредованные AMPAR и NMDAR. (A) mEPSC регистрировали у управляющих нейронов прилежащего ядра мозга в режиме фиксации потенциала при -80 мВ. a) mEPSC, зарегистрированные для контрольных (N=16) и предварительно обработанных прегненолоном (100 нМ) (N=15) срезов, продемонстрировали аналогичные амплитуды (T-критерий: t=1,16, df=29, p=0,25) и времена затухания (T-критерий: t=1,28, df=29, p=0,21). b) Средние остаточные значения mEPSC, нормализованные относительно пиковых значений, показали, что их кинетика не подвергается влиянию прегненолона. c) Результаты регистрации репрезентативных значений остаточных mIPSC. B) Токи целых клеток, зарегистрированные для NAc PN, индуцированные помещением в кювету с 25 мкМ NMDA на 1 мин. a) Токи, индуцированные NMDAR, были сравнимы для контрольных образцов (N=17) и срезов, предварительно обработанных прегненолоном 100 нМ (N=12) и 1 мкМ (N=7) (ANOVA: f=00,9, df=2,33, p=0,91). b) Репрезентативные результаты экспериментов, демонстрирующие влияние NMDA на удерживающие токи NAc PN в режиме фиксации потенциала при -30 мВ. cont=контрольные образцы; preg=прегненолон.
На фиг. 18 приведены диаграммы, демонстрирующие влияние введения высоких доз (50 мг/кг, sc) прегненолона и его C3 и C17 синтетических производных: 3-фторпрегненолона (CP1), 17-метилпрегненолона (CP2) и 3-фтор-17-метилпрегненолона (CP3) на содержание стероидов в прилежащем ядре мозга. Введение прегненолона, но не 3-фторпрегненолона, 17-метилпрегненолона или 3-фтор-17-метилпрегненолона, повышало уровни аллопрегнанолона (A) и эпиаллопрегнанолона (B) в прилежащем ядре мозга крыс Wistar. Данные выражены в виде среднее значение ± SEM (n=5-7 на концентрацию).
На фиг. 19 приведены диаграммы, демонстрирующие, что C3 и C17 синтетические производные, а именно 3-фторпрегненолон (CP1), 17-метилпрегненолон (CP2) и 3-фтор-17-метилпрегненолон (CP3), сокращают потребление пищи. (A) Увеличение потребления пищи, вызванное THC (0,5 мг/кг, sc) у крыс Wistar, имеющих неограниченный доступ к еде, значимо уменьшалось под действием 17-метилпрегненолона (8 мг/кг, sc), не статистически значимая тенденция к уменьшению наблюдалась также для 3-фторпрегненолона и 3-фтор-17-метилпрегненолона. (B) Прегненолон [F(4,28)=5,5, p<0,01], 3-фторпрегненолон [F(3,20)=3, p<0,05], 17-метилпрегненолон [F(3,20)=5,3, p<0,01] и 3-фтор-17-метилпрегненолон [F(3,20)=4, p<0,02] дозозависимым образом уменьшали потребление пищи у мышей C57B1/6, лишенных пищи на 24 часа. (C) Прегненолон, 3-фторпрегненолон, 17-метилпрегненолон и 3-фтор-17-метилпрегненолон (2 мг/кг, sc) уменьшали THC-индуцированную гипералгезию у мышей C57B1/6, лишенных пищи на 24 часа. (D) Реакция на полную дозу прегненолона и 17-метилпрегненолона при THC-индуцированной гипералгезии у мышей C57B1/6, лишенных пищи на 24 часа. Для обоих соединений первая эффективная дозировка составляла 1 мг/кг. Данные выражены в виде среднее значение ± SEM (n=6-8 на группу). *=p<0,05; **p<0,01; ***=p<0,001 по сравнению с животными, получавшими носитель. ##=p<0,01; ###=p<0,001, по сравнению с животными, получавшими THC.
На фиг. 20 приведены диаграммы, отображающие влияние введения прегненолона в стационарном режиме при помощи мининасоса Alzet на уровни прегненолона и производного метаболита аллопрегнанолона. Подкожная инъекция прегненолона вызывала кратковременное (<1 ч) увеличение уровней прегненолона (A) и, кроме того, увеличивала уровни аллопрегнанолона в течение, по крайней мере, 1 часа (D). Введение прегненолона с помощью мининасоса Alzet (B, C) дозозависимым образом повышало уровни прегненолона в плазме, но не увеличивало уровни аллопрегнанолона (E, F).
ПРИМЕРЫ
Примеры синтеза производных прегненолона
Прегненолон является хорошо известным и коммерчески доступным веществом и может применяться в качестве исходного соединения для синтеза его производных.
Пример синтеза производного прегненолона, замещенного алкилом по положению C17:
Как показано ниже, для синтеза соединения, замещенного алкилом по положению C17, на первой стадии получают соответствующий енолацетат. Затем его обрабатывают реактивом Гриньяра для получения енолята, который вводят во взаимодействие с электрофилом. Этот электрофил предпочтительно может являться йод- или бром-алкилом, -аллилом, -бензилом или -арилом.
Пример синтеза производного прегненолона, замещенного группой -OR по положению C17
На приведенной ниже схеме показано получение C17-замещенных алкокси-, бензилокси- и арилоксипрегненолонов путем взаимодействия со спиртами, катализируемого медью.
Пример синтеза производного прегненолона, замещенных галогеном по положению C3 и группой R по положению C17:
Как показано ниже, производное прегненолона, замещенное группой R по положению C17, обрабатывают DAST, осуществляя замену спиртовой группы в положении C3 на атом фтора.
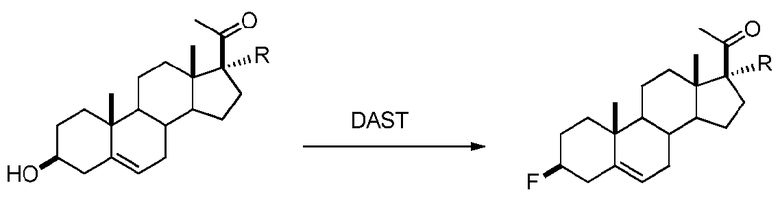
Пример синтеза производного прегненолона, замещенного группой алкокси по положению C3:
Как показано ниже, для введения фрагмента простого эфира в положение C3, на первой стадии требуется превращение спирта в соответствующий тозилат, где тозил играет роль уходящей группы. Затем тозилат обрабатывают подходящим спиртом с получением C3-алкокси прегненолона.
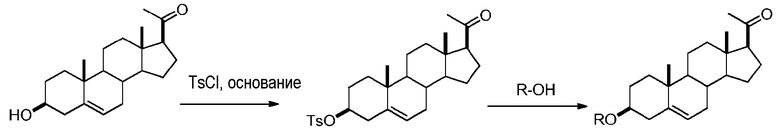
Пример синтеза производного прегненолона, замещенного =O по положению C3 и R по положению C17:
Как показано ниже, для получения производного прегненолона, замещенного =O по положению C3 и R по положению C17, C17-замещенный прегненолон обрабатывают окислителем с окислением спиртовой группы до соответствующего кетона, с последующим самопроизвольным смещением двойной связи с образованием модифицированного прогестерона.
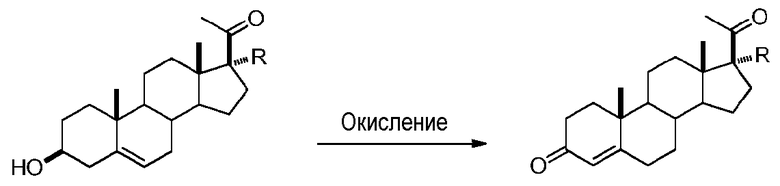
Примеры роли прегненолона и его производных в ингибировании рецептора CB1
Материалы и методики
Животные
Животных содержали индивидуально в виварии при температуре 22°C и влажности 60%, в условиях постоянного цикла свет-темнота (включение света 8:00-20:00 ч). Животные имели неограниченный доступ к пище и воде во время всех экспериментов, за исключением экспериментов по потреблению пищи и экспериментальных сессий по самостоятельному введению WIN 55212-2. После прибытия животных, их периодически брали в руки в течение двух недель перед началом проведения экспериментов. Большинство экспериментов проводили во время световой фазы, за исключением экспериментов по потреблению пищи у крыс и тестам на самостоятельное введение WIN 55212-2 у мышей CD1, которые проводили во время темновой фазы. Все эксперименты проводили в условиях, строго соответствующих рекомендациям Евросоюза (86/609/EEC).
Взрослых самцов крыс Wistar (3-4 месяца), мышей C57B1/6N (2-3 месяца), мышей C57B1/6j (2-3 месяца) и мышей CD1 (массой 25-30 г в начале эксперимента) приобретали у Charles River Laboratories (France). CB1-дефицитных (CB1-/-) и D1 CB1 мутантных (D1-CB1-/-) мышей получали в лаборатории авторов изобретения по известной методике (Marsicano et al., Nature. 2002 418:530-4; Monory et al., PLoS Biol. 2007 5(10):e269).
Препараты
Δ9-тетрагидроканнабинол (THC, Sigma-Aldrich, France) приобретали в виде раствора 30 мг/мл (масса/объем) в 100% этаноле. Перед инъекцией этот раствор растворяли в Tween 80 (1 капля/3 мл) и диметилсульфоксиде (ДМСО), разбавленном в соотношении 1:40 физиологическим раствором (2,5%). Раствор носителя содержал все ингредиенты (1 капля/3 мл Tween 80, ДМСО (2,5%) и этанол, разбавленный физиологическим раствором до конечной концентрации этанола 1,8%). Кокаин·HCl (Cooperation Pharmaceutique Française, France), морфина сульфат (Francopia, France), никотина битартрат (Sigma-Aldrich, France) и спирт USP (95%, Sigma-Aldrich, France) растворяли в физиологическом растворе. HU210, JWH133 и AM251 приобретали у Tocris, UK и WIN 55212-2, аминоглютетимид (AMG), прегненолон (5-прегнен-3β-ол-20-он), липополисахариды E. Coli 0111:B4 (LPS) у Sigma-Aldrich (France) и римонабант (SR141716A) у Cayman Chemical (Interchim, Montluçon, France).
Синтетические производные, а именно 3-фторпрегненолон, 17-метилпрегненолон и 3-фтор-17-метилпрегненолон, были синтезированы AtlanChimPharma (France). Готовили растворы препаратов в Tween 80 (1 капля/3 мл) и ДМСО (2,5%) или NMP (2,5%) и разбавляли физиологическим раствором. THC, кокаин, морфин, никотин, этанол, HU210, JWH133, AM251, WIN 55212-2, AMG и SR141716A вводили интраперитонеальной инъекцией (ip), и прегненолон или производные прегненолона вводили подкожной инъекцией (sc). Объем инъекции составлял 1 мл/кг массы тела для крыс и 10 мл/кг для мышей.
В экспериментах по SA (самостоятельному введению), WIN 55212-2 (Sigma Chemical Co., Madrid, Spain) растворяли в одной капле Tween 80 и разбавляли физиологическим раствором. Кетамина гидрохлорид (100 мг/кг) (Imalgene 1000; Rhône Merieux, Lyon, France) и ксилазина гидрохлорид (20 мг/кг) (Sigma, Madrid, Spain) смешивали и растворяли в смеси этанола (5%) и дистиллированной воды (95%). Эту анестезирующую смесь вводили интраперитонеально перед имплантацией катетера в объеме 20 мл/кг массы тела. Тиопентал натрия (5 мг/мл) (Braun Medical S.A., Barcelona, Spain) растворяли в дистиллированной воде и вводили инфузией в объеме 0,1 мл через внутривенный катетер. Для функционального анализа человеческого рецептора CB1 in vitro, прегненолон и CP55940 растворяли в ДМСО до конечной концентрации 100 мМ и хранили при -20°C.
Количественное определение нейростероидов
Отбор образцов крови и мозга
Животных умерщвляли обезглавливанием, кровь из туловища собирали в пробирки, покрытые EDTA, центрифугировали при 2000×g в течение 10 мин и хранили супернатант при -20°C. Быстро собирали мозги животных, необходимые области мозга вырезали на льду, образцы быстро замораживали на сухом льду и хранили при -80°C.
Определение содержания стероидов с помощью ГХ/МС
Уровни прегненолона (5-прегнен-3β-ол-20-она), аллопрегнанолона (3α-гидрокси-5α-прегнан-20-она), эпиаллопрегнанолона (3β-гидрокси-5α-прегнан-ол-20-она), тестостерона и 5α дигидротестостерона определяли ГХ/МС в соответствии с описанным ранее протоколом экстракции, очистки и количественного определения (George O., et al., Biol Psychiatry. 2010 68:956-63; Vallee M et al., Anal Biochem. 2000, 287:153-66).
Характерная тетрада THC
Температура тела
Температуру тела измеряли с использованием ректального зонда (зонд RET3, Physitemp instruments, USA) у мышей, находящихся в активном состоянии, и следили за температурой с помощью контрольного термометра (TH-5, Physitemp instruments, USA).
Двигательная активность
Двигательную активность измеряли с помощью автоматизированной системы «открытое поле» (размер бокса 100×100×30 см, освещение 10 люкс, система видеонаблюдения: Viewpoint, Lyon, France) или в клетках из плексигласа (длина 19 см × ширина 11 см × высота 14 см), оборудованных компьютеризированной системой лучей и фотоэлементов (Imetronic, France). Животных подвергали индивидуальному тестированию в течение 15 мин. Регистрировали суммарное горизонтальное расстояние, которое преодолевали животные в боксе.
Каталепсия
Каталепсию определяли с помощью теста на каталепсию со стержнем. Передние лапы мыши помещали на стержень диаметром 1 см, расположенный горизонтально на высоте 3,5 см от поверхности скамьи. Регистрировали задержку спуска животного со стержня.
Аналгезия
Аналгезию измеряли с помощью измерителя аналгезии с горячим поддоном (BIO-HC1.00, Bioseb, France). Поддон нагревали до температуры 52°C±0,1°C, и регистрировали время до проявления мышью первых признаков дискомфорта (облизывание или отдергивание лап или подпрыгивание на поддоне, определяемое в тексте заявки как латентность спасения). Для предотвращения поражения тканей использовали время отсечки 60 с.
Задачи на распознавание объектов
Распознавание объектов измеряли в L-лабиринте с двумя рукавами (размер каждого рукава составлял длина 30 см × ширина 4,5 см) в условиях слабого освещения (50-60 люкс). Животных тестировали индивидуально в течение трех последовательных дней в течение 9-мин сессии каждый день, причем сессии предназначались для привыкания, обучения и тестирования, соответственно. В день 1 (сессия для привыкания) мышам давали исследовать L-лабиринт, не содержащий объектов. В день 2 (сессия для обучения) в конец каждого из рукавов лабиринта помещали два идентичных объекта. Хотя при этом не проявлялось предпочтения одного из рукавов с объектом (данные не показаны), объекты распределяли по рукавам случайным образом для каждой следующей мыши/условия. В день 3 (тестовая сессия) один из знакомых объектов заменяли новым объектом и вычисляли общее время, затраченное на исследование каждого из двух объектов (нового и знакомого). Фиксировали время, затраченное в каждом рукаве, и время, затраченное на исследование объекта (знакомого или нового). Исследование объекта определяли по ориентации носа животного к объекту на расстоянии менее 2 см от него. В ходе тестовой сессии рассчитывали индекс различения, как разность между временем, затраченным на исследование нового и знакомого объекта, деленную на общее время исследования двух объектов. Считалось, что более высокий индекс различения отражает более высокую степень удерживания в памяти знакомого объекта (Puighermanal et al., 2009).
Измерение потребления пищи
Потребление пищи определяли путем измерения потребления пищи в домашних клетках животными. Каждому животному в очищенную домашнюю клетку помещали 50-100 г стандартного лабораторного корма (U.A.R., France). Оставшееся количество корма взвешивали через 1 ч и вычисляли количество съеденной пищи.
Самостоятельное введение WIN 55212-2
Эксперименты по самостоятельному внутривенному введению, в которых животных обучали осуществлять самостоятельную инфузию WIN 55212-2 (WIN), проводили в оперантных камерах для исследования мышей (модель ENV-307A-CT, Medical Associates, Georgia, VT, USA), используя ранее описанную методику (Mendizabal V, et al., Neuropsychopharmacology, 2006, 31: 1957-66).
Совместные исследования микродиализа и электрофизиологии in vivo
Общие методики: Осуществляли хирургическую операцию и обеспечивали перфузию для проведения одновременного наблюдения за электрофизиологией и микродиализом. Вкратце, проводили анестезию с использованием смеси изофлуран 2%/воздух и помещали катетер в бедренную вену для внутривенного введения препаратов. Затем животных помещали в стереотаксическую установку (David Kopf Instruments, Phymep, Paris, France), снабженную носовой маской для непрерывной подачи газовой анестезии (2% изофлуран во время хирургических процедур, 1,5% изофлуран во время экспериментов по электрофизиологии и микродиализу), и следили за температурой в прямой кишке животных, поддерживая ее на уровне 37±1°C с помощью грелки (CMA 150, Carnegie Medicin, Phymep). Микродиалитический зонд (CMA/11, длина 2 мм, внешний диаметр 240 мкм, Cuprophan, Carnegie Medicin, Phymep) и регистрирующий электрод (стеклянную микропипетку TW 150F-4, внешний диаметр 2-3 мкм, WPI-Europe, Aston Stevenage, UK) имплантировали соответственно в медиовентральную область правого прилежащего ядра, соответствующую подотделу оболочки [координаты в мм относительно брегмы: переднее-задняя (AP)=+1,7, боковая (L)=1, вентральная (V) -8] и в правую вентральную область покрышки (VTA) [координаты в мм относительно брегмы: AP=-5,4-5,8, L=0,4-0,8, V=-7,0-8,5] согласно атласу Paxinos и Watson. Через зонды с постоянной скоростью 2 мкл/мин с помощью микроперфузионного насоса (CMA 111, Carnegie Medicin, Phymep) осуществляли перфузию искусственной спинномозговой жидкости (aCSF), содержащей (в мМ): 154,1 Cl-, 147 Na+, 2,7 K+, 1 Mg2+ и 1,2 Ca2+, pH доводили до значения 7,4 2 мМ натрий-фосфатным буфером. После этого продолжали перфузию в течение 2 ч для стабилизации уровней дофамина (DA) в перфузате.
Регистрацию нейрональных импульсов DA для одиночного нейрона и мониторинг внеклеточных уровней DA начинали через 2 часа после начала перфузии через зонд (период стабилизации). Диализаты (30 мкл) собирали на льду каждые 15 мин и сразу же анализировали, для измерения базовых величин внеклеточных уровней DA, которые определяли по трем последовательным образцам, в которых содержание DA менялось менее чем на 10% (9). Поиск DA нейронов для электрофизиологических измерений производили в течение 30 минут перед введением препаратов (THC или кокаина). Частоту пульсаций DA нейронов регистрировали в течение 3-5 минут, для определения исходной частоты, определяемой отклонением менее чем на 10% от средней частоты разряда DA нейрона. Фармакологическую обработку проводили один раз, получая стабильные исходные значения частоты пульсации DA нейронов и внеклеточные уровни DA в перфузате.
Регистрация данных для DA нейронов. Активность одиночных нейронов, локализованных в VTA, регистрировали внеклеточными способами с помощью стеклянных микропипеток, заполненных 1% Fast Green, растворенным в 0,5М ацетате натрия (импеданс 2-5 Мом). Сигналы фильтровали (полоса пропускания 0,4-1 КГц), усиливали усилителем с высоким импедансом (Dagan 2400A, Dagan Corporation, USA) и выделяли отдельные импульсы с помощью строб-дискриминатора (WD-2, Dagan Corporation, USA), установленного на цифро-аналоговом осциллографе с запоминающей ЭЛТ (HM507, Hameg, Frankfurt, Germany). Затем данные экспериментов оцифровывали онлайн, пользуясь программой Spike2 (Cambridge Electronic Design, Cambridge, UK) с помощью компьютера, соединенного с интерфейсом CED 1401 (Cambridge Electronic Design, Cambridge, UK). VTA DA нейроны идентифицировали по уже опубликованным критериям (12-14). Частоту пульсации определяли как число пиков/сек.
Определение DA. Для количественного определения DA, диализаты вводили в ВЭЖХ-хроматограф, снабженный колонкой Equisil BDS с обращенной фазой (C18; 2×250 мм, размер частиц 5 мкм; Cluzeau Info Labo, Ste Foy la Grande France) и амперометрическим детектором (Antec Leyden DECADE II, Alpha-mos, Toulouse, France) с электродом из стеклоуглерода, установленным на напряжение +450 мВ, работающим в паре с электродом Ag/AgCl. Состав подвижной фазы включал (в мМ) 70 NaH2PO4, 0,1 Na2EDTA и 0,1 октилсульфоновой кислоты и 10% метанол, причем значение pH доводили до 4,8 добавлением ортофосфорной кислоты. Чувствительность методики в отношении DA составляла 0,3 пг/20 мкл, при соотношении сигнал/шум 3:1.
Гистология. В конце каждого эксперимента через электрод пропускали прямой постоянный ток (-20 мкА в течение 15 мин) для выделения красителя Fast Green, что позволяло идентифицировать участок мозга, в котором происходила регистрация данных. После этого мозг удаляли и фиксировали в растворе NaCl (0,9%)/параформальдегид (10%). Расположение электродов в VTA и микродиалитических зондов в прилежащем ядре определяли микроскопическим исследованием последовательных коронарных срезов (60 мкм), окрашенных красителем Neutral Red.
Ожирение, вызываемое рационом питания, и оценка накопления жиров
Самцам мышей C57BL/6J в возрасте 2 месяца без ограничения давали пищу с высоким 60% содержанием жиров (HFD; номер по каталогу D12492, Research Diets, New Brunswick, NJ) в течение 8 недель и затем вводили им либо прегненолон, либо носитель. Равномерное распределение животных по 3 группам обеспечивали путем подбора массы тел животных, массы жира и уровней глюкозы на голодный желудок перед началом введения препаратов.
Исследование состава организма. Оценку массы жира и нежировой ткани осуществляли in vivo, используя анализатор EchoMRI (EchoMedical Systems, Houston, TX), после чего начинали давать животным пищу с высоким содержанием жиров (HFD), и аналогичное исследование проводили непосредственно перед и сразу же после окончания курса введения прегненолона или носителя.
Измерение содержания свободных жирных кислот в плазме. Кровь мышей DIO собирали по окончании 4-недельного введения препаратов и измеряли содержание свободных жирных кислот в плазме (FFA) с использованием колориметрического реакционного набора, следуя инструкциям производителя (Abcam, номер по каталогу 65341).
Определение TNFальфа в плазме. Собирали кровь из организма мышей и центрифугировали при 3000 об/мин в течение 15 мин при 4°C. Плазму хранили при -80°C до определения TNFα. Уровни TNFα в плазме оценивали с использованием набора для анализа ELISA, следуя инструкциям производителя (Fisher Scientific, номер по каталогу E6473C).
Измерение электрофизиологических параметров срезов мозга
Получение срезов. Животных подвергали глубокой анестезии изофлураном и осуществляли перфузию через сердечную мышцу физиологического раствора, содержащего сахарозу при 4°C (компоненты раствора в мМ: 23 NaHCO3, 70 холин Cl, 75 сахароза, 25 глюкоза, 2,5 KCl, 1,25 NaH2PO4, 7 MgCl2 и 0,5 CaCl2). Извлекали мозг и нарезали срезы толщиной 250-300 мкм в коронарной плоскости, используя вибратом (Campden Instruments, Loughborough, UK). Во время нарезания срезов мозг находился в физиологическом растворе, содержащем сахарозу. Сразу же после нарезания срезов их выдерживали при 32°C в течение 40 минут в искусственной спинномозговой жидкости с низким содержанием кальция (low-Ca2+ ACSF), которая содержала (в мМ): 23 NaHCO3, 120 NaCl, 11 глюкозы, 2,5 KCl, 1,2 NaH2PO4, 2,4 MgCl2, 1,2 CaCl2. Срезы хранили в low-Ca2+ ACSF при комнатной температуре до регистрации электрофизиологических параметров.
Электрофизиология. Регистрацию осуществляли с использованием усилителя Axopatch-1D (Molecular Devices, Sunnyvale, CA). Данные фильтровали в полосе 1-2 КГц, оцифровывали при частоте выборки 10 КГц с помощью прибора Digidata 1332A (Molecular Devices), накапливали на PC, используя Clampex 9, и анализировали, используя Clampfit 10 (Molecular Devices).
Регистрация сигналов целостных клеток по методике «пэтч-клэмп» осуществляли для основных нейронов ядра NAc (PN). Клетки идентифицировали, используя инфракрасную микроскопию по способу дифференциального интерференционного контраста (микроскоп Leica DM LFSS, Leica Microsystems, Germany; Camera Till Photonics, Germany).
Для регистрации токов, опосредованных AMPAR и NMDAR, стеклянные пэтч-клэмп электроды (сопротивление 4-6 Мом) заполняли цезий-содержащим раствором следующего состава (в мМ): 125 глюконовая кислота, 125 CsOH, 10 HEPES, 10 NaCk, 0,3 EGTA, 0,05 спермин, 10 TEA-Cl, 2 MgCl2, 0,3 CaCl2, 4 Na2ATP, 0,3 NaGTP, 0,2 cAMP. Для регистрации ингибирующих токов использовали аналогичный раствор, за исключением того, что в нем содержалось (в мМ): 80 глюконовой кислоты, 80 CsOH, 30 CsCl, 20 NaCl. Более высокая концентрация хлоридов благоприятствует более высокой движущей силе при регистрации потенциалов гиперполяризации. В ходе экспериментов оценивали сопротивление доступа (Ra) с помощью 2 мВ гиперполяризующих импульсов. Ra не было скомпенсировано и клетки отбраковывались, если Ra превышало 25 Мом или изменялось >20% во время эксперимента. Перед внедрением в клетку базовый потенциал усилителя доводили до нуля.
Для регистрации fEPSP внеклеточные стеклянные регистрирующие и стимулирующие электроды заполняли ACSF. Синаптические потенциалы индуцировали с помощью двух электрических импульсов (0,1-0,25 мА, продолжительность 200 мксек), подаваемых на частоте 20 Гц каждые 10 сек. Стимулирующий электрод помещали на расстоянии >150 мкм в дорсомедиальном направлении от регистрирующего электрода.
Регистрация и анализ данных. AMPAR-опосредованные синаптические токи: рецепторы GABA-A блокировали добавлением к перфузионной среде 50 мкМ пикротоксина. Вклад NMDAR исключали путем регистрации данных при гиперполяризованных потенциалах. Для регистрации индуцированных синаптических токов и mEPSC, клетки исследовали в режиме фиксации потенциала при -70 мВ и -80 мВ, соответственно. mEPSC регистрировали в присутствии 0,5 мкМ TTX.
NMDAR-опосредованные токи: изменения удерживающих токов, индуцируемых NMDA, измеряли в клетках в режиме фиксации потенциала при -30 мВ, для освобождения NMDAR от блокирующего иона магния, которое зависит от разности потенциалов. Рецепторы AMPAR и GABA-A блокировали 20 мкМ DNQX и 50 мкМ пикротоксином, соответственно.
Токи, опосредованные рецептором GABA-A: mIPSC регистрировали в присутствии антагонистов AMPAR и NMDAR (DNQX 20 мкМ и AP-5 50 мкМ, соответственно).
Анализ mEPSC-mIPSC: Как правило, после проникновения в клетку, клетки оставляли на 20 минут для достижения равновесия, и использовали для анализа данные по mEPSC-mIPSC, зарегистрированные в течение последующих 10 минут. mEPSC-mIPSC детектировали, используя шаблон, сгенерированный путем усреднения нескольких типовых событий (Clampfit 10, Molecular Devices). Шаблон сдвигали вдоль кривой данных на одну точку за раз. В каждом положении шаблон оптимальным образом масштабировали и смещали для наилучшего соответствия данным. Применяли низший порог амплитуды 7 пА и 10 пА для mEPSC и mIPSC, соответственно, что составляло 2,5SD исходного уровня шума. Для каждой клетки измеряли кинетику mEPSC-mIPSC, исходя из усредненного события с использованием Clampfit 10. Для определения времени затухания, подгоняли две экспоненциальные кривые в диапазоне между 5 и 95% фазы затухания тока, заданных следующим уравнением y(t)=A1·e(-t/τ1)+A2·e(-t/τ2), где A означает амплитуду, t означает время и τ означает константу, соответствующую времени распада. Зачем вычисляли взвешенное значение тау.
In vitro исследование человеческого рецептора CB1
Анализ ортостерического связывания прегненолона проводили с использованием клеток яичника китайского хомячка (CHO), экспрессирующих человеческий рецептор CB1.
Связывание CB1 с прегненолоном оценивали по сродству прегненолона к агонистическому сайту человеческого каннабиноидного рецептора, экспрессируемого клетками CHO, которое определяли анализом связывания радиолиганда, а именно агониста CB1 [3H] CP 55940. Эксперименты проводились компанией CEREP Frnace, с использованием стандартных методик этого поставщика.
Приподнятый крестообразный лабиринт
Приподнятый крестообразный лабиринт состоял из центральной платформы (10×10 см), от которой отходили 4 рукава (45×10 см) под углом 90° друг к другу. Два противоположных рукава, именуемых закрытыми рукавами, имели боковые стенки высотой 50 см. Два других рукава, именуемых открытыми рукавами, не имели стенок. Лабиринт подвешивали на высоте 66 см от пола комнаты и освещали ярким светом (120 люкс). Для проведения исследования животное помещали на центральную платформу и регистрировали число входов и количество времени, проведенное в каждом из рукавов лабиринта в течение 5 минут. Число входов и время, проведенное в открытых рукавах, считалось показателем тревожного поведения, тогда как общее количество входов в рукава лабиринта считалось показателем двигательной активности.
Исследования метаболизма in vitro
При исследовании использовали линию клеток CHO-K1 (#CCL-61, ATCC-LGC, USA), полученную в виде субклона из родительской линии клеток CHO, взятых из биопсии яичника взрослой самки китайского хомячка. Клетки CHO-K1 высевали в 24-луночные планшеты (#353047, BD Biosciences, USA) в необходимой концентрации (2×104 клеток/лунку) в свежей среде, не содержащей антибиотиков и состоящей на 90% из DMEM-Glutamax (#31966-21, Life technologies, USA) и 10% из сыворотки телят (#10270-106, Life technologies, USA).
Введение прегненолона в стационарном режиме
Микроосмотические насосы (Alzet Osmotis Pumps, Charles River, France) со скоростью подачи 0,15 мкл/час (модель 2006) заполняли прегненолоном, растворенным в PEG 300 (85%) и этаноле (15%) в концентрации 125 или 250 мг/мл (соответствующей дневной дозе 12 или 24 мг/кг массы тела соответственно), и после легкой анестезии осуществляли подкожную имлантацию. Перед имплантацией все насосы заряжали намачиванием в солевом растворе при 37°C в течение 60 ч.
Статистический анализ
Статистический анализ осуществляли с использованием двухпараметрического или однопараметрического вариационного анализа (ANOVA), критерия Ньюмена-Кеулса, T-критерия Стьюдента. Все результаты выражали в виде среднее значение ± SEM (стандартная ошибка среднего). Статистический анализ проводили с помощью программы GraphPad Prism (GraphPad Software Inc., La Jolla, CA, USA) или программы Statistica 5,0© (StatSoft Inc, Tulsa, OK, USA).
Результаты:
I. Активация CB1 увеличивает синтез и концентрации прегненолона
Пример 1: THC увеличивает концентрации прегненолона в мозге в большей степени, чем другие вещества, вызывающие зависимость
В этом примере авторы изобретения проанализировали влияние введения основных веществ, вызывающих зависимость, на выработку прегненолона в организмах самцов крыс Wistar. Первая стадия стероидного синтеза во всех тканях заключается в синтезе прегненолона, который, как правило, считался неактивным предшественником активных производных стероидов. Например, в мозге два параллельных ферментных каскадных процесса, начинающихся с прегненолона, приводят к образованию, с одной стороны, аллопрегнанолона и его стереоизомера эпиаллопрегнанолона и, с другой стороны, тестостерона и его метаболита DHT. Количественное содержание этих стероидов в мозге определяли с помощью ГХ-МС, которая является единственной методикой, позволяющей дифференцировать их тонкие структурные различия. Основные классы веществ, вызывающих зависимость, вводили подкожной или интраперитонеальной инъекцией в дозировках, соответствующих ED50 для большинства из их основных безусловных поведенческих эффектов: психостимулятор кокаин (20 мг/кг), опиоид морфин (2 мг/кг), никотин (0,4 мг/кг), спирт (1 г/кг) и действующее начало конопли посевной, т.е. Δ9-тетрагидроканнабинол (THC) (3 мг/кг). Концентрации нейростероидов определяли через 15, 30 и 120 мин после инъекции в нескольких отделах мозга в восходящем порядке: вентральной части среднего мозга, гипоталамусе, комплексе полосатого тела и лобном отделе коры мозга.
Для всех исследованных отделов мозга (лобного отдела коры, полосатого тела, прилежащего ядра, вентральной части среднего мозга) были получены очень близкие результаты. Как показано в этом примере, для вентральной части полосатого тела (прилежащего ядра) исходный уровень стероидов (фиг. 1А) находился в пределах от примерно 1 нг/г ткани для прегненолона и тестостерона до 0,2 нг/г для эпиаллопрегнанолона. DHT и аллопрегнанолон имели промежуточные уровни в районе 0,4 нг/г. Все вещества, вызывающие зависимость, повышали концентрации прегненолона в мозге через 15-30 минут после инъекции (фиг. 1B). При этом следует отметить, что повышение уровней прегненолона, вызванное THC, было в несколько раз выше, чем для остальных веществ: примерно 1500% увеличение для THC по сравнению с примерно 200% увеличением для других веществ, вызывающих зависимость.
Пример 2: THC дозозависимым образом увеличивает концентрации прегненолона в мозге крысы
В этом примере авторы изобретения дополнительно охарактеризовали влияние введения THC в различных концентрациях (0,3, 0,9, 1,5, 3, 6 и 9 мг/кг) или носителя самцам крыс Wistar на концентрации прегненолона в организме, измеренные в момент максимального действия THC, которое, как показали данные предыдущего примера, наблюдалось через 30 мин после инъекции. Эти эксперименты показали, что увеличение уровней прегненолона в мозге имело дозозависимый характер, причем значение ED50 составляло приблизительно 3 мг/кг. В примере получены данные для плазмы и нескольких отделов мозга: лобного отдела коры (FCX); прилежащего ядра (ACC); полосатого тела (STR); гипоталамуса (HYP). После введения THC уровни прегненолона сопоставимым образом повышались во всех исследованных структурах мозга. Уровни прегненолона в плазме также повышались, но это увеличение было в несколько раз меньше, чем в мозге.
Пример 3: THC дозозависимым образом увеличивает концентрации прегненолона в мозге мыши
В этом примере авторы изобретения дополнительно охарактеризовали влияние введения THC на концентрации прегненолона путем исследования результатов введения THC мышам. Уровни прегненолона измеряли в момент максимального действия THC, а именно, через 30 мин после инъекции. Эти эксперименты показали, что и в случае мышей THC вызывал дозозависимое увеличение уровней прегненолона. В примере получены данные для нескольких отделов мозга: лобного отдела коры (FCX); прилежащего ядра (ACC); полосатого тела (STR). THC вызывал приблизительно одинаковое увеличение концентраций прегненолона во всех перечисленных отделах мозга.
Пример 4: Агонисты рецептора CB1 вызывают увеличение концентраций прегненолона, аналогичное их увеличению под действием THC
Действие THC в головном мозге опосредуется семейством семи мембранных рецепторов, сопряженных с G-белком (GCPR) и, главным образом, рецепторами CB1 и CB2. THC увеличивал синтез прегненолона за счет активации рецептора CB1. Так, например, инъекции независимым группам крыс (n=6-12 на группу) как синтетической смеси агонистов CB1/CB2, именуемой Win 55212, так и агониста, который обладает более высоким сродством к рецептору CB1, чем CB2, (HU210) вызывали значимое увеличение уровней прегненолона. В противоположность этому, агонист с более высоким сродством к рецептору CB2, чем CB1, (JWH 133) оказывал существенно меньшее и статистически незначимое влияние на концентрации прегненолона.
Пример 5: THC-индуцированное увеличение уровней прегненолона подавляется антагонистом рецептора CB1
Зависимость влияния THC на активацию CB1 была дополнительно продемонстрирована тем фактом, что увеличение концентраций прегненолона в прилежащем ядре головного мозга крыс Wistar, вызванное введением THC (3 мг/кг ip), блокировалось введением селективного антагониста CB1 (AM251, 8 мг/кг, ip), который вводили за 30 минут до инъекции THC (n=6 на группу).
Пример 6: THC-индуцированное увеличение уровней прегненолона подавляется у мутантных животных, у которых отсутствует рецептор CB1
В этом примере авторы изобретения дополнительно проанализировали зависимость увеличения уровней прегненолона под действием THC от рецептора CB1. Для этого авторы изобретения исследовали последствия введения THC на уровни прегненолона у мутантных мышей (n=6-8 на группу), у которых изначально отсутствует экспрессия рецептора CB1. В примере показаны данные, полученные для прилежащего ядра мозга. THC-индуцированное увеличение уровней прегненолона полностью подавлялось в организмах мутантных животных, у которых рецептор CB1 отсутствовал во всех типах клеток или селективно в популяции нейрональных клеток, экспрессирующих дофаминергический рецептор D1. У этих мутантных животных рецептор CB1 отсутствует в большинстве GABA нейронов прилежащего ядра. Последующие эксперименты показали, что THC повышает уровни прегненолона в мозге за счет действия на рецептор CB1, экспрессируемый нейронами.
Обсуждение результатов:
Данные, полученные в предыдущих примерах, согласованно подтверждают раскрытый в заявке факт, что «активация рецепторов CB1 у млекопитающих вызывает синтез прегненолона и увеличивает концентрацию этого стероида в организме». Данные предыдущих примеров позволяют разработать универсальный способ увеличения концентраций прегненолона в организме.
Приведенные в этих примерах согласующиеся факты можно суммировать следующим образом:
Во-первых, уровни прегненолона дозозависимым образом повышаются при введении трех различных агонистов рецептора CB1: THC, HU210 и Win 55212. В противоположность этому, уровни прегненолона не претерпевают значительного повышения под действием агониста, имеющего более высокое сродство к рецептору CB2, чем CB1. Увеличение концентраций прегненолона, вызванное агонистами CB1, нашло подтверждение у двух различных видов (мышей и крыс) и было обнаружено как в тканях мозга, так и в плазме.
Во-вторых, увеличение уровней прегненолона, вызванное введением THC, подавлялось введением антагониста CB1 и не проявлялось у мутантных животных, не имевших рецептора CB1.
II. Прегненолон оказывает отрицательное обратное воздействие на рецептор CB1 и ингибирует результаты активации рецептора CB1
Пример 7: THC-индуцированное увеличение уровней прегненолона оказывает отрицательное обратное воздействие на активацию рецептора CB1
В этом примере авторы изобретения исследовали потенциальную функциональную роль увеличения уровней прегненолона, вызванного активирующим действием THC на CB1. Авторы изобретения обнаружили, что прегненолон оказывает отрицательное обратное воздействие на эффекты, которые опосредованы стимулированием рецептора CB1.
Активацию рецептора CB1 обычно выявляют по четырем эффектам, обычно именуемым каннабиноидной тетрадой, в число которых входят: 1. понижение двигательной активности (гиполокомоция), 2. гипотермия, 3. каталепсия (ослабление способности начинать движение) и 4. аналгезия. Соответственно, инъекция THC (3, 10, 15 мг/кг) мышам C57Bl/6N (n=7-8 на группу) вызывала следующие дозозависимые эффекты: i) понижение двигательной активности в камере «открытое поле»; ii) понижение температуры тела; iii) увеличение задержки до начала движения (увеличенная каталепсия); и iv) увеличение болевого порога (фиг. 2A-D).
Поскольку дозировки THC, при которых наблюдается упомянутая тетрада (от 3 до 15 мг/кг THC), вызывают сильное увеличение концентраций прегненолона, авторы изобретения проанализировали действие ингибитора синтеза прегненолона-аминоглютетимида (AMG, 50 мг/кг, ip), который вводили инъекцией за 30 мин до THC, где упомянутые параметры последовательно измеряли через 30 мин после введения THC. AMG существенно усиливал все последствия введения THC (фиг. 2A-D), и это усиление полностью устранялось инъекцией экзогенного прегненолона (6 мг/кг) (фиг. 2E-H), демонстрируя зависимость наблюдаемых эффектов введения AMG от прегненолона. Эти данные демонстрируют, что секреция прегненолона, вызванная активацией рецептора CB1, способствует ингибированию эффектов, возникающих вследствие этой активации CB1, за счет петли отрицательной обратной связи.
Пример 8: Прегненолон ингибирует эндоканнабиноидную тетраду, индуцированную активацией рецептора CB1 под действием THC
В этом примере авторы изобретения исследовали может ли введение экзогенного прегненолона также ингибировать эндоканнабиноидную тетраду, индуцированную THC. Введение прегненолона (6 мг/кг) перед введением THC и в отсутствие AMG уменьшало проявления всех признаков THC-индуцированной каннабиноидной тетрады: двигательной активности, температуры тела, каталепсии и болевого порога (фиг. 2E-H). Однако введение прегненолона самого по себе в отсутствие THC не оказывало влияния на двигательную активность, температуру тела, каталепсию и болевой порог (фиг. 2E-H).
Пример 9: Прегненолон ингибирует увеличение потребления пищи, вызванное THC
Для выявления других примеров способности прегненолона ингибировать эффекты, вызванные активацией рецепторов CB1, авторы изобретения провели исследование, способен ли прегненолон (при введении с помощью инъекции за 30 мин до THC) ингибировать THC-индуцированное увеличение потребления пищи.
Было показано, что THC увеличивает потребление пищи у сытых крыс (0,5 мг/кг, n=7-8 на группу) и у мышей, которых лишали пищи на 24 часа (n=7-8 на группу, 1 мг/кг). У сытых крыс (фиг. 3А) прегненолон дозозависимым образом понижал THC-индуцированное потребление пищи, вызывая статистически значимый эффект при 2 мг/кг. Кроме того, эта доза подавляла увеличение потребления пищи, вызванное инъекцией THC мышам (фиг. 3В). В этой дозировке прегненолон не менял значимым образом исходный уровень потребления пищи (фиг. 3А, В).
Пример 10: Прегненолон ингибирует увеличение потребления пищи, вызванное лишением пищи
Активация CB1 под действием эндоканнабиноидов принимает участие в регулировании физиологического потребления пищи, т.е. потребление пищи не стимулируется экзогенными агонистами CB1, такими как THC. Поэтому авторы изобретения провели исследование, способно ли введение прегненолона модифицировать потребление пищи у лишенных ее животных, которые не получали THC. Авторы изобретения обнаружили, что прегненолон дозозависимым образом уменьшал потребление пищи у мышей, которых лишали пищи (фиг. 3C), однако первая статистически значимая доза (6 мг/кг) превышала дозу (2 мг/кг), которая могла блокировать THC-индуцированное потребление пищи.
Пример 11: Прегненолон ингибирует увеличение потребления пищи, вызванное лишением пищи, за счет CB1-зависимого механизма
Потребление пищи регулируется большим количеством физиологических систем. Поэтому в данном примере авторы изобретения проверили, зависит ли от рецептора CB1 уменьшение потребления пищи, вызванное прегненолоном, у животных, лишенных пищи, которым не вводили THC. Авторы изобретения исследовали результаты предварительного введения антагониста CB1 SR141716A (0,05 мг/кг, ip) на уменьшение потребления пищи, вызванное прегненолоном, у животных, лишенных пищи. Авторы изобретения обнаружили, что ингибирование потребления пищи, вызванное прегненолоном, у животных, лишенных пищи, зависело от рецепторов CB1. Таким образом, антагонист CB1 SR141716A при введении мышам, которым ограничивали потребление пищи, за 30 мин до введения прегненолона, подавлял уменьшение потребления пищи, вызванное введением прегненолона (фиг. 3D).
Пример 12: Прегненолон подавляет самостоятельное введение агонистов CB1
Для анализа влияния введения прегненолона на эффект положительного подкрепления активации CB1, который связан со способностью THC вызывать пристрастие, авторы изобретения использовали модель самостоятельного внутривенного введения, реализованную согласно описанным ранее протоколам (Soria et al., 2006; Mendizabal et al., 2006). Считается, что самостоятельное внутривенное введение является лучшей поведенческой моделью пристрастия. В этой модели животных обучают проявлять функциональную реакцию, в данном случае засовывать нос в отверстие для получения внутривенной инфузии препарата. Мыши легко обучались самостоятельному введению агониста CB1, а именно WIN 55212 (12,5 мкг/кг за одну инъекцию), демонстрируя явное предпочтение к отверстию, в котором установлено устройство, запускающее инфузию соединения (активному), по сравнению с отверстием, которое не вызывает таких последствий (неактивным) (фиг. 4А). Введение прегненолона в количестве 2 или 4 мг/кг по схеме латинских квадратов перед сессией самостоятельного введения WIN 55212 значительно уменьшало самостоятельное введение (фиг. 4B). Кроме того, введение прегненолона снижало мотивацию к самостоятельному введению WIN 55212, что было продемонстрировано уменьшением точки остановки в схеме с прогрессивно нарастающей кратностью PR (фиг. 4С). В этой схеме от животных требовалось осуществить все возрастающее число необходимых реакций (кратность) для получения одной инфузии препарата, причем точкой остановки считалась последняя достигнутая кратность, и ее считали надежной мерой мотивации к получению препарата. Во время сессии PR-экспериментов, необходимая кратность реакций для получения инъекции повышалась в соответствии со следующей последовательностью: 1-2-3-5-12-18-27-40-60-90-135-200-300-450-675-1000.
Пример 13: Прегненолон ингибирует ухудшение памяти, вызванное введением THC
В этом примере авторы изобретения продолжили исследование способности прегненолона ингибировать эффекты, вызванные активацией CB1. Еще одним результатом активации CB1 является инициирование нарушений памяти. Этот эффект связан с одним из нежелательных результатов использования каннабиноидов: когнитивными нарушениями, характеризующимися потерей кратковременной памяти. Активация рецептора CB1 под действием THC (10 мг/кг), который вводили инъекцией через 10 мин после обучения животных, в значительной степени нарушала запоминание в тесте на распознавание объектов у мышей (фиг. 6).
Предварительное введение прегненолона путем инъекции непосредственно после обучения (6 мг/кг) в значительной степени ослабило потерю памяти, вызванную инъекцией 10 мг/кг THC. В то же время прегненолон (6 мг/кг) не вызывал каких-либо изменений способности к запоминанию при введении в отсутствие THC (фиг. 6).
Пример 14: Прегненолон ингибирует увеличение дофаминергической активности, вызванной введением THC
В этом примере авторы изобретения продолжили исследование способности прегненолона ингибировать эффекты, вызванные активацией CB1. Считается, что каннабиноиды проявляют свою способность вызывать привыкание за счет активации рецептора CB1, что, в свою очередь, увеличивает высвобождение нейромедиатора дофамина в прилежащем ядре, т.е. области мозга, которая регулирует переход от мотивации к действию. Действие прегненолона на увеличение дофаминергической активности, вызванное THC (фиг. 7), исследовали путем параллельной регистрацией двух параметров: 1. высвобождения дофамина на уровне дофаминергических нервных окончаний в прилежащем ядре; и 2. электрической активности дофаминергических нейронов на уровне тела клетки в вентральной области покрышки (VTA). Прегненолон (при подкожной инъекции 2 мг/кг, за 30 мин до THC) в значительной степени ослаблял рост высвобождения дофамина и активность дофаминергических нейронов, вызванные введением THC. THC или кокаин вводили внутривенно в экспоненциально нарастающих кумулятивных дозах (0,15-1,2 мг/кг). После каждой дозы регистрировали пульсацию DA нейронов в течение 1 минуты и затем осуществляли следующее введение (фиг. 7).
Пример 15: Прегненолон ингибирует увеличение дофаминергической активности, вызванное введением кокаина
В этом примере авторы изобретения продолжили исследовать способность прегненолона ингибировать активацию дофаминергической системы. В предыдущем примере прегненолон показал способность противодействовать гиперактивности дофаминергической системы, вызванной THC. В настоящем примере (фиг. 8) показано, что прегненолон (при подкожной инъекции 2 мг/кг за 30 минут до кокаина) способен также противодействовать увеличению активности дофаминергической системы, вызванному психостимуляторами, такими как кокаин. Кокаин вводили внутривенно в экспоненциально возрастающих кумулятивных дозах (0,0125-0,8 мг/кг). После каждой дозы регистрировали пульсацию DA нейронов в течение 1 минуты и затем осуществляли следующее введение. Эти результаты имеют отношение к лечению шизофрении, поскольку увеличение дофаминергической активности, вызванное психостимуляторами, считается одной из экспериментальных моделей психоза. Так, например, высвобождению дофамина, вызванному психостимуляторами, приписывается развитие острого психоза, который может возникать после употребления этих препаратов людьми.
Пример 16: Прегненолон ингибирует увеличение массы тела и накопление жиров у животных, получающих питание с высоким содержанием жиров
В этом примере авторы изобретения исследовали способность прегненолона ингибировать результаты активации CB1 в контексте ожирения. Влияние прегненолона на метаболические расстройства исследовали с использованием модели ожирения, вызванного рационом питания (DIO) у мышей. В этой методике животных содержали на диете с высоким содержанием жиров (60% жира), которая вызывает прогрессирующее ожирение. Через восемь недель содержания на этой диете, когда у животных появлялась избыточная масса тела и накопление излишних жировых отложений, начинали введение прегненолона, продолжавшееся тридцать дней (один раз в день 2 мг/кг или 5 мг/кг, n=8). Прегненолон понижал массу тела с отсрочкой начала действия, которое проявлялось через 15 дней введения (фиг. 9A), но не изменял потребления пищи (фиг. 9B).
Полученные данные позволяют предположить, что влияние прегненолона на массу тела, по-видимому, не вызвано первичными метаболическими эффектами и не связано с поведенческим влиянием на потребление пищи. Это подтверждалось анализом состава тела, выполненным с помощью магнитного резонанса, который показал, что введение прегненолона оказывало различное действие на массу жира и массу нежировой ткани животных (фиг. 10). У контрольных животных, при введении пищи с высоким содержанием жиров, процентная доля жировой массы последовательно увеличивалась, тогда как процентная доля нежировой ткани сокращалась. Если животным вводили самую высокую дозу прегненолона (5 мг/кг), повышение жировой массы подавлялось, тогда как уменьшение нежировой массы ослаблялось.
Незначительное влияние прегненолона на потребление пищи в модели DIO, как кажется, противоречит результатам, наблюдаемым при использовании модели лишение пищи/возобновление питания, в которой прегненолон уменьшал потребление пищи (фиг. 3). Причина этого может заключаться в рационе питания, т.е. рационе с высоким содержанием жиров в модели DIO, по сравнению со стандартным кормом в модели лишение пищи/возобновление питания, или в возможном специфическом влиянии прегненолона на всплеск потребления пищи у голодных животных в течение первого часа возобновления доступа к пище. По этой причине в модели лишение пищи/возобновление питания потребление пищи обычно оценивают в течение одного часа, тогда как в модели DIO потребление пищи оценивают в течение 24 часов. Поэтому в следующем эксперименте действие прегненолона исследовали в течение 24 часов также и в модели лишение пищи/возобновление питания. Полученные результаты подтвердили действие прегненолона в течение первого часа возобновления питания, но в течение 24 часов значимого эффекта обнаружить не удалось. Полученные данные указывают на специфическое влияние прегненолона на всплеск потребления пищи, вызванный голоданием, которое сильно активирует эндоканнабиноидную систему и рецептор CB1. Обнаруженное незначительное влияние прегненолона на потребление пищи в течение 24 часов полностью отличается от действия известного ортостерического антагониста рецептора CB1 римонабанта, который, как было показано ранее, сильно сокращал потребление пищи в течение 24 часов. Аналогично, в случае потребления пищи с высоким содержанием жиров, римонабант, как было показано, уменьшает потребление пищи в течение первой недели введения, тогда как для прегненолона это не характерно (фиг. 9А).
Пример 17: Прегненолон ингибирует увеличение уровней TNF-альфа, вызванное LPS
В этом примере авторы изобретения продолжили исследование способности прегненолона ингибировать последствия активации рецептора CB1 в контексте воспаления и фиброза. Как было показано, активация рецептора CB1 принимает участие в воспалительном и фиброзном процессе, причем ингибирование этого рецептора ортостерическими антагонистами, например, римонабантом, понижает прирост уровней TNF-альфа, вызванный провоспалительными стимулами, например, LPS. Выработка TNF-альфа является одним из ответов клеток на воспалительные стимулы, который в большей степени вовлечен в развитие фиброза. Введение мышам прегненолона (6 мг/кг, подкожно) за 30 мин до введения LPS вдвое уменьшило прирост уровней TNF-альфа, который измеряли через 90 мин после введения LPS (фиг. 11).
Пример 18: Прегненолон ингибирует влияние активации CB1 на синаптическую передачу сигналов
В этом примере авторы изобретения продолжили исследование способности прегненолона ингибировать последствия активации CB1 в контексте синаптической передачи сигнала. Во многих литературных источниках зафиксировано, что активация рецепторов CB1 подавляет синаптическую передачу путем ингибирования высвобождения нейромедиаторов. Данное явление наблюдалось во многих отделов мозга, как для возбуждающих, так и для тормозящих синапсов. Авторы изобретения оценивали, меняет ли прегненолон способность THC ингибировать возбуждающую синаптическую передачу в прилежащем ядре (NAc). Регистрацию потенциала целой клетки по методике пэтч-клэмп проводили в NAc взрослых мышей, и AMPAR-опосредованные EPSC индуцировали путем электрического стимулирования локальных аксонов. Перфузия THC в кювете достоверно понижала амплитуду EPSC в контрольных срезах (ингибирование 34,3±3,7%). Действие THC значимо ослаблялось, если срезы предварительно обрабатывали прегненолоном 10 нМ (ингибирование 15,1±1,8%, p<0,001) (фиг. 12).
Для исследования влияния прегненолона в более широком диапазоне концентраций THC, авторы регистрировали fEPSP в срезах NAc. Поскольку стабильное измерение fEPSP возможно в течение нескольких часов, эта методика отлично подходит для построения кривых доза-реакция и ранее ее использовали для исследования функций рецептора CB1 (Robbe et al., 2001; Mato et al., 2004). Так, AMPAR-опосредованные fEPSP регистрировали с помощью электрического стимулирования локальных аксонов. Перфузия THC контрольных срезов в кювете ингибировала fEPSP дозозависимым образом (10 мкМ: 23,9±6,0%; 20 мкМ: 35,3±4,7%; 40 мкМ: 48,6±3,6%). В противоположность этому, THC вызывал менее существенное ингибирование синаптической передачи на срезах, предварительно инкубированных с 100 нМ прегненолоном (10 мкМ: 11,1±3,2%; 20 мкМ: 22,7±2,7%; 40 мкМ: 34,6±3,1%; двухпараметрический анализ ANOVA нейростероидного фактора, p=0,001) (фиг. 13).
Приведенные данные в совокупности демонстрируют, что нейростероид прегненолон уменьшает способность THC активировать CB1-зависимое модулирование возбуждающей синаптической передачи.
Обсуждение результатов:
Данные, представленные в предыдущих примерах, согласованно подтверждают раскрытые в тексте заявки данные, что: «увеличение концентраций прегненолона в организме, вызванное активацией рецептора CB1, обеспечивает эндогенное отрицательное обратное влияние на активность рецептора CB1. Эта отрицательная обратная связь приводит к тому, что прегненолон (как вырабатываемый эндогенно, так и вводимый извне) противодействует последствиям активации CB1».
Данные предыдущих примеров согласованно указывают на общий способ ингибирования последствий активации рецептора CB1 путем введения прегненолона.
Эти согласующиеся друг с другом данные можно суммировать следующим образом:
Во-первых, ингибирующее действие прегненолона на CB1-опосредованные эффекты является физиологически адекватным, поскольку эндогенное увеличение уровней прегненолона, вызванное активацией CB1, обеспечивает естественную отрицательную обратную связь, которая способствует уменьшению последствий активации CB1. Так, например, при блокировании выработки прегненолона, вызванной активацией CB1, нарастали характерные эффекты, вызванные действием THC.
Во-вторых, введение экзогенного прегненолона позволяло ингибировать многие последствия активации рецепторов CB1: 1. снижение двигательной активности (гиполокомоцию); 2. каталепсию; 3. гипотермию; 4. аналгезию; 5. повышенное потребление пищи в модели лишение пищи/возобновление питания; 6. потребление пищи, вызванное THC; 7. самостоятельное внутривенное введение агониста CB1; 8. потерю памяти, вызванную THC; 9. активацию дофаминергической системы под действием THC или кокаина; 10. накопление жиров и увеличение массы тела в модели ожирения; 11. выработку TNF-альфа; 12. ингибирование синаптической передачи, вызванное THC.
В-третьих, ингибирующее действие прегненолона на CB1-опосредованные эффекты было обнаружено у двух различных видов: крыс и мышей.
Продемонстрированное авторами изобретения согласованное влияние прегненолона на широкий круг параметров, является неожиданным и непредсказуемым с точки зрения известного уровня знаний о действии других известных стероидов, которые, напротив, должны были бы увеличивать, а не ослаблять последствия активации рецептора CB1. Например, было описано, что многие стероиды, такие как прегнанолон, аллопрегнанолон и их производные, способствуют активации рецептора GABA. Поэтому эти стероиды должны усиливать последствия активации CB1, поскольку было показано, что соединения, увеличивающие активность рецептора GABA, усиливают последствия активации CB1 при действии THC (Bellocchio L. et al., Nature Neurosci 2010, 13: 281-3; Pertwee R.G. and Wickens A.P. Neuropharmacology. 1991, 30: 237-44; Pertwee R.G., et al., Neuropharmacology. 1988 27: 1265-70). Аналогично, исходя из современного уровня знаний, можно также предсказать, что прогестерон и прогестагены, другие половые стероиды и глюкокортикоиды должны усиливать последствия активации CB1 (Anaraki D.K. et al., Europ. J. Pharmacol, 2008, 586, 186-196; Rdriguez de Fonseca F., et al., Life Sci. 1993, 54: 159-170; Becker J.B., Rudick C.N. Pharmacol. Biochem Behav. 1999, 64: 53-7; Piazza P.V. and Le Moal M. Brain Res Rev 1997, 25: 359-72).
III. Прегненолон является ингибитором рецептора CB1 с меньшими побочными эффектами и меньшим неспецифическим действием, чем ортостерические антагонисты CB1 и другие нейроактивные стероиды
Пример 19: Прегненолон не изменяет ортостерическое связывание с рецептором CB1
Предыдущие примеры показали, что прегненолон способен ингибировать все исследованные последствия активации CB1. Основываясь на этих фактах, в данном примере авторы изобретения исследовали возможные взаимодействия между прегненолоном и рецептором CB1. Авторы выясняли, может ли прегненолон действовать в качестве ортостерического антагониста CB1. Это оказалось не так, поскольку прегненолон не влиял на ортостерическое связывание агониста CB1 [3H]CP55940 с рецептором CB1, расположенным на плазматической мембране клеток CHO (фиг. 14). Хотя прегненолон не является ортостерическим антагонистом, предварительные данные, полученные авторами изобретения, показали, что он мог бы действовать, как аллостерический ингибитор.
Пример 20: Прегненолон не вызывает тревожного поведения
Одним из основных побочных действий ортостерических антагонистов CB1 является побочное влияние на поведение и, в частности, появление тревожно-депрессивного состояния. Эти эффекты регулирующие органы признали достаточно серьезными, чтобы приостановить выдачу разрешения на продажу первого ортостерического антагониста CB1 римонабанта. Для обоснования отличающегося профиля безопасности прегненолона, его действие сравнивали с действием ортостерического антагониста/обратного агониста CB1 римонабанта (оба препарата вводили подкожно), используя животную модель тревожного поведения, а именно приподнятый крестообразный лабиринт. Этот тест был выбран потому, что увеличение тревожности являлось основным нежелательным эффектом римонабанта у людей. Мышам подкожно вводили прегненолон (1, 6, 10 мг/кг), римонабант (10 мг/кг) или носитель (не менее n=7 животных на группу), через 30 минут животных помещали на центральную платформу крестообразного лабиринта и в течение 5 минут регистрировали время, проведенное в открытых и закрытых рукавах лабиринта, и количество входов в открытые и закрытые рукава. Это исследование (фиг. 15) подтвердило анксиогенное действие римонабанта, судя по уменьшению количества входов в открытые рукава приподнятого крестообразного лабиринта и сокращению времени пребывания в этих рукавах. В противоположность этому, прегненолон не вызывал увеличения тревожности (фиг. 15).
Пример 21: Прегненолон не изменяет активность рецепторов GABA-A
В этом примере авторы изобретения исследовали специфичность воздействия прегненолона на другие нейромедиаторные рецепторы и, в частности, на рецепторы GABA-A. Так, например, другие активные стероиды, такие как аллопрегнанолон и прегнанолон обладают сильным характерным действием, облегчающим активацию рецепторов GABA.
Для оценки влияния прегненолона на деятельность постсинаптических GABA авторы регистрировали mIPSC и сравнивали его амплитуду и время затухания в различных группах. Авторы установили, что амплитуда и время затухания mIPSC были близки в контрольной группе (амплитуда: 17,9±0,8 пА; время затухания: 11,1±0,3 мс) и для срезов, обработанных прегненолоном (100 нМ и 1 мкМ, соответственно; амплитуда: 17,3±0,4 пА, 15,9±0,4 пА; время затухания: 10,2±0,4 мс, 10,8±0,5 мс). В противоположность незначительному эффекту прегненолона, нейростероид аллопрегнанолон, который, как известно, является модулятором рецепторов GABA-A, значимо изменял характеристики mIPSC (100 нМ и 1 мкМ, соответственно; амплитуда: 15,9±0,5 пА, 20,2±1,2 пА; p<0,001; время затухания: 13,6±0,7 мс, 22,7±2,3 мс; p<0,0001) (фиг. 16).
В заключение, отметим, что приведенные данные позволяют предположить, что GABA-A-опосредованные синаптические токи не модулируются нейростероидом прегненолоном. Кроме того, эти данные подтверждают, что, как показано ранее (патент США 5232917), бета-заместитель в положении C3 прегненолона подавляет влияние на GABA рецептор. Авторы изобретения обнаружили, что бета-заместитель в положении C3, наоборот, придает соединению способность ингибировать активацию рецептора CB1.
Пример 22: Прегненолон не изменяет активность глутаматных рецепторов
В этом примере авторы изобретения исследовали специфичность действия прегненолона на другие нейромедиаторные рецепторы и, в частности, рецепторы NMDA и AMPA. Так, например, предполагается, что другие активные стероиды, такие как DHEA и DHEA сульфат, оказывают свое характерное действие за счет изменения активности глутаматных рецепторов.
Для оценки влияния прегненолона на токи, опосредованные рецепторами AMPA, авторы регистрировали потенциалы действия, независимые от mEPSC. В этом случае синаптические токи возникали из-за стохастического дискретного высвобождения нейромедиаторов, и предполагая, что нейростероиды не изменяли содержимого синаптических везикул, амплитуда и кинетика зарегистрированных mEPSC зависит от функции постсинаптических рецепторов.
Авторы обнаружили, что прегненолон не изменял функцию AMPAR в NAc взрослых животных (фиг. 17A). Так, например, AMPAR-опосредованный mEPSC на контрольных срезах и срезах, обработанных прегненолоном (100 нМ), продемонстрировал сходную амплитуду (контрольные срезы: 14,4±0,8 пА, прегненолон: 15,7±0,7 пА, p=0,25) и времена затухания (контрольные срезы 4,76±0,08 мс, прегненолон: 4,62±0,06 мс, p=0,21).
Для количественного определения токов, опосредованных рецепторами NMDA, авторы использовали другую стратегию. Вследствие медленной кинетики и зависимости от разности потенциалов, отдельное измерение NMDAR-опосредованных mEPSC не является таким достоверным, как AMPAR-опосредованных токов. Поэтому авторы решили регистрировать токи целых клеток, возникающие в ответ на действие экзогенного NMDA. В случае PN NAc в режиме фиксированного потенциала при -30 мВ, перфузия 25 мкМ NMDA в кювете в течение 1 минуты индуцировала входящий ток сходной величины для контрольных срезов (115±15,5 пА) и срезов, предварительно обработанных прегненолоном (100 нМ: 107±9,6 пА; 1 мкМ: 108,9±12,5 пА; p=0,91) (фиг. 17 B).
Данные этих экспериментов, взятые в совокупности, показывают, что прегненолон не влияет на функцию основных постсинаптических ионотропных глутаматергических рецепторов в NAc взрослых грызунов. Эти данные показывают также, что замена кето-группы в положении 17 стероидного цикла этаноном (метилкетоном) подавляет активность в отношении рецепторов NMDA и AMPA и придает способность ингибировать активацию рецептора CB1.
Обсуждение результатов
Данные приведенных выше примеров согласованно демонстрируют раскрытое в заявке положение о том, что: «прегненолон действует в качестве ингибитора человеческого рецептора CB1 с фармакологическим профилем, отличным от профиля ортостерических антагонистов и других нейроактивных стероидов, и это показывает, что прегненолон будет иметь меньше неспецифических и нежелательных эффектов, чем ортостерические антагонисты CB1 и другие нейроактивные стероиды.
Данные, приведенные в описанных выше примерах, согласованно позволяют разработать общий способ ингибирования активности рецептора CB1 с помощью введения прегненолона. Вследствие этого, данные предыдущих примеров позволяют разработать общий способ лечения или облегчения всех патологий, которые связаны с активацией рецептора CB1 и/или патологий, при которых может принести пользу ингибирование рецепторов CB1, путем введения прегненолона без потенциальных побочных эффектов ортостерических антагонистов CB1 и других активных стероидов, включая, но не ограничиваясь этим, DHEA, аллопрегнанолон и прегнанолон.
Эти согласующиеся друг с другом данные можно суммировать следующим образом:
Во-первых, прегненолон не изменяет связывание ортостерического агониста, но при этом ингибирует эффекты, возникающие вследствие активации рецептора CB1. Такой профиль мог бы соответствовать профилю аллостерического ингибитора.
Во-вторых, прегненолон в отличие от ортостерического антагониста не вызывает ни тревожности (пример 20), ни уменьшения потребления пищи в модели ожирения, хотя и уменьшает накопление жиров (пример 18). То, что действие прегненолона на метаболизм само по себе не включает изменения потребления пищи, позволяет также рассчитывать на меньшие побочные эффекты прегненолона. Исследование влияния ортостерических антагонистов CB1, например, римонабанта, на массу тела и метаболизм показало, что эти соединения действуют на метаболизм двумя путями, первый из которых заключается в потере массы тела вследствие уменьшения потребления пищи (примерно 50% эффекта), и второй состоит в непосредственном влиянии на метаболизм. Побочные эффекты ортостерических антагонистов рецептора CB1 и, в частности, усиление депрессии, вероятно, оказывают характерное влияние на потребление пищи. Так, хорошо известно, что все действия в отношении людей с ожирением, а именно, фармакологическое, хирургическое или поведенческое лечение, которые приводят к уменьшению потребления пищи, также вызывают у до 5% субъектов серьезные нарушения поведения и, в частности, депрессию.
Наконец, прегненолон в отличие от других нейроактивных стероидов не оказывает влияния на рецепторы GABA и глутаматов. Это является важным свидетельством в пользу того, что прегненолон не должен иметь побочных эффектов некоторых из этих стероидов, которые могут вызывать существенные изменения, включающие седативное действие, нарушения памяти, работоспособности и двигательного поведения.
IV. Производные прегненолона, для которых характерно ограниченное превращение в другие активные стероидные производные прегненолона, действуют как ингибиторы результатов активации рецептора CB1
Пример 23: Производные прегненолона, для которых характерно ограниченное превращение в другие активные стероиды, не образуют аллопрегнанолон и эпиаллопрегнанолон in vivo
В качестве примеров соединений, описываемых общей формулой A, авторы изобретения исследовали три соединения, которые получали путем:
1. замещения группы OH в положении C3 атомом фтора, что приводило к соединению, именуемому 3-фторпрегненолоном (CP1);
2. кватернизации положения C17 метильной группой, что приводило к получению соединения, именуемого 17-метилпрегненолоном (CP2);
3. замещения группы OH в положении C3 атомом фтора и кватернизации положения C17 метильной группой, что приводило к получению соединения, именуемого 3-фтор-17-метилпрегненолоном (CP3).
Ни одно из модифицированных производных прегненолона (соединений CP1, CP2, CP3) при введении в высоких дозах (50 мг/кг, n=6-7 на группу) крысам Wistar не вызывало ппродуцирования аллопрегнанолона и эпиаллопрегнанолона (фиг. 18), хотя после инъекции прегненолона уровни аллопрегнанолона и эпиаллопрегнанолона повышались (50 мг/кг) (фиг. 18). Концентрации аллопрегнанолона и эпиаллопрегнанолона измеряли в прилежащем ядре мозга индивидуальных животных с помощью ГХ/МС через 30 минут после инъекций.
Пример 24: Производные прегненолона, для которых характерно ограниченное превращение в активные стероидные производные in vitro
В этом примере авторы изобретения продолжили исследование метаболизма производных прегненолона, используя in vitro тест в клетках CHO. Эти клетки, полученные из яичников, содержат все ферменты, необходимые для метаболического превращения прегненолона в стероидные производные. В частности, введение прегненолона (1 мкМ) в эти клетки в течение 48 часов приводило к существенному увеличению содержания аллопрегнанолона и эпиаллопрегнанолона, и существенно меньшему увеличению содержания THDOC в культуральной среде (таблица 1A). В общей сложности протестировали 55 соединений плюс прегненолон, и среди этих соединений лишь для 25 наблюдалось отсутствие или значительное уменьшение метаболизма с образованием активных стероидных производных (таблица 1B-D, таблица 2A, B). Остальные соединения в большей или меньшей степени подвергались метаболизму до одного или нескольких стероидных производных прегненолона, например, аллопрегнанолона, эпиаллопрегнанолона, THDOC, тестостерона и DHEA.
Производные прегненолона с пониженной способностью к метаболизму в клетках CHO. Результаты выражены в процентах изменения по сравнению с клетками CHO, обработанными прегненолоном (таблица 2A), или в пг/мл (0 = концентрация ниже предела обнаружения). ALLO = аллопрегнанолон, EPIALLO = эпиаллопрегнанолон, PREG = прегненолон, TESTO = тестостерон
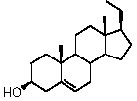
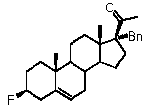
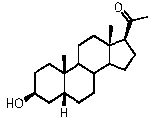
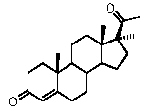
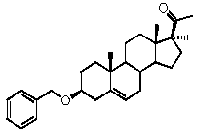
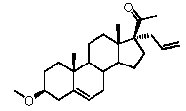
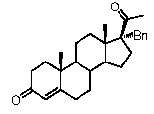
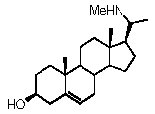

Производные прегненолона с пониженной способностью к метаболизму в клетках CHO. Результаты выражены в процентах изменения по сравнению с клетками CHO, обработанными прегненолоном (таблица 2A), или в пг/мл (0 = концентрация ниже предела обнаружения). ALLO = аллопрегнанолон, EPIALLO = эпиаллопрегнанолон, PREG = прегненолон, TESTO = тестостерон
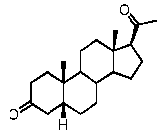
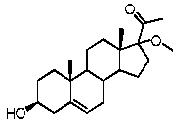
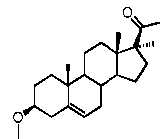
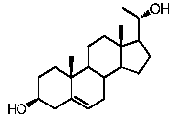
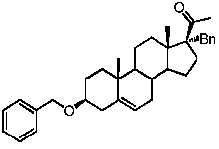
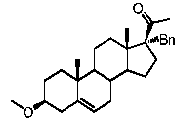
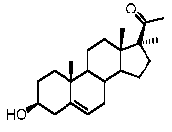
Как видно из таблицы 3, известные сведения о метаболизме стероидов не позволяют полностью предсказать, какие модификации прегненолона в заметной степени уменьшат метаболизм, а какие нет. Например, альфа-гидроксигруппа в положении C20 уменьшала метаболизм, тогда как бета-гидроксигруппа в положении C20 не уменьшала. Аналогичным образом, соединения с бета-атомом водорода в положении C5 обладали пониженной склонностью к метаболизму, тогда как соединения с альфа-атомом водорода в положении C5 легко подвергались метаболизму. Кроме того, определенные группы с положениях C3 и C17 или некоторые определенные комбинации групп блокировали метаболизм.
Сравнение модификаций производных прегненолона, которые ослабляют или сохраняют способность к метаболическому превращению в активные стероидные производные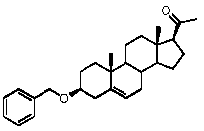
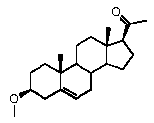
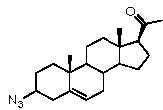
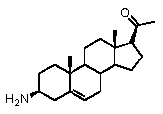
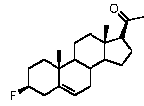
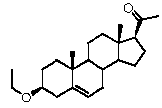
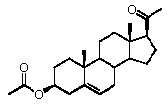
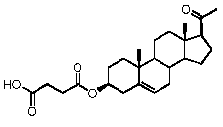
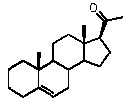
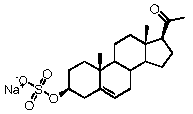
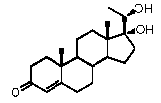

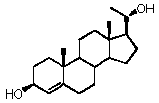
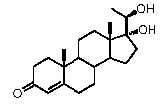
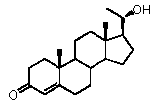
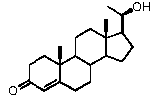
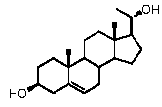
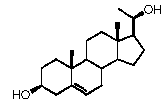
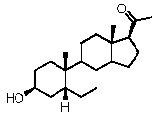

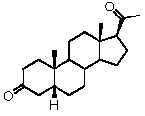
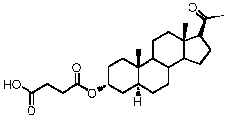
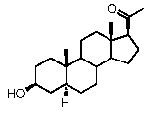
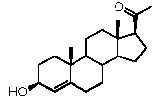
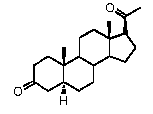
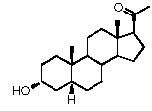
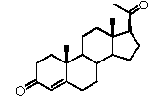
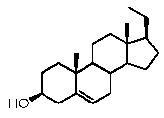
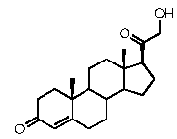

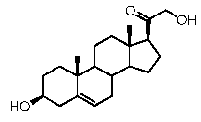
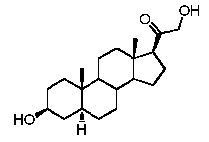
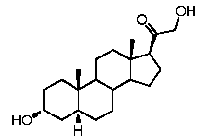
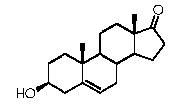
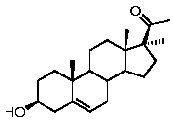
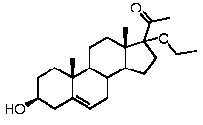
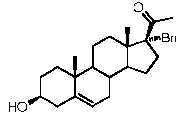
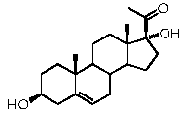
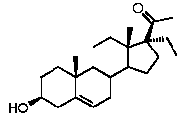
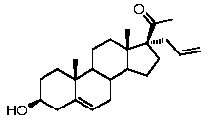
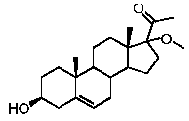
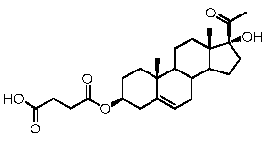
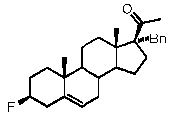
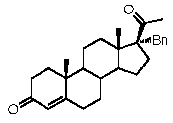
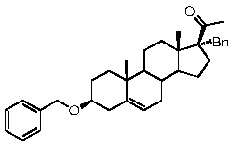
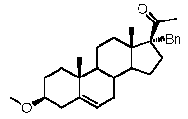
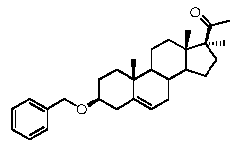
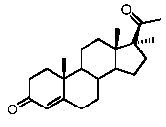
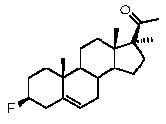
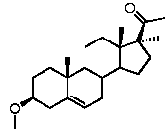
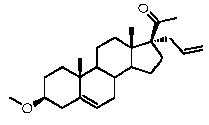
Пример 25: Производные прегненолона, для которых характерно ограниченное превращение в активные стероидные производные, ингибируют последствия активации рецептора CB1
В качестве примеров соединений общей формулы A, авторы изобретения исследовали описанные в заявке примеры влияния на потребление пищи 3-фторпрегненолона, 17-метилпрегненолона, 3-фтор-17-метилпрегненолона у крыс и мышей после стимулирования THC и/или после лишения пищи. Указанные соединения, а именно, 3-фторпрегненолон, 17-метилпрегненолон, 3-фтор-17-метилпрегненолон показали способность ингибировать влияние активации CB1 на потребление пищи. 17-Метилпрегненолон продемонстрировал большую эффективность, чем прегненолон, 3-фторпрегненолон и 3-фтор-17-метилпрегненолон, при ингибировании последствий активации рецептора CB1 (фиг. 19). 17-Метилпрегненолон проявил способность значимо понижать увеличение потребления пищи, вызванное THC, у крыс, хотя для двух остальных соединений наблюдалась только тенденция к уменьшению потребления пищи (фиг. 19А). У мышей, которым ограничивали прием пищи, все соединения понижали потребление пищи. Однако статистически значимые результаты были получены в случае самых низких доз (4 мг/кг) 17-метилпрегненолона, тогда как для получения статистически значимых результатов у прегненолона, 3-фторпрегненолона и 3-фтор-17-метилпрегненолона была необходима дозировка 6 мг/кг (фиг. 19B). Наконец, THC-индуцированное увеличение потребления пищи мышами понижалось всеми соединениями при дозировке 2 мг/кг (фиг. 19С). График зависимости реакции от дозы для THC-индуцированного увеличения потребления пищи показал, что и прегненолон, и 17-метилпрегненолон подавляли это поведение в дозировке 1 мг/кг, хотя дозировка 0,5 мг/кг оказалась неэффективной.
Другие производные прегненолона (таблица 4), которые характеризовались пониженной склонностью к метаболизму до активных стероидных производных, исследовали с точки зрения их способности ингибировать: 1. проявления, включенные в THC-индуцированную каннабиноидную тетраду (уменьшение температуры тела и двигательной активности, THC 10 мг/кг, соединение 6 мг/кг за 15-30 минут до THC), которые считаются надежным способом оценки активации рецептора CB1; 2. THC-индуцированное увеличение потребления пищи, которое является типичным результатом активации CB1 (THC 0,5-1 мг/кг, соединения 2-4 мг/кг за 30 мин до THC); 3. увеличение уровней TNF-альфа, индуцированных LPS, которое является еще одним характерным действием антагонистов CB1 (соединения 6 мг/кг, за 15 мин до LPS). Для каждого соединения и каждого теста использовали независимую группу животных (как минимум n=6 животных на соединение/на тест).
Ингибирование активации CB1 под действием производных прегненолона с пониженной способностью к метаболизму. Мышей (как минимум n=6 на группу), которым вводили производные прегненолона (2-6 мг/кг), сравнивали с соответствующим контрольными животными, которым вводили носитель. Изменения температуры тела и двигательной активности исследовали после инъекции 10 мг/кг THC, потребление пищи после инъекции 0,5-1 мг/кг THC, TNFα после системной инъекции LPS. nt = не тестировалось. Статистические исследования проводили с применением t-критерия Стьюдента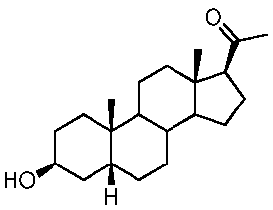
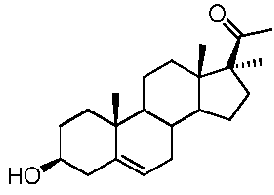
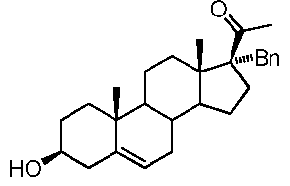
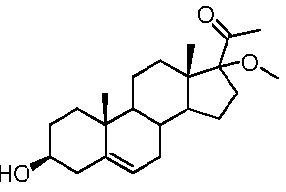

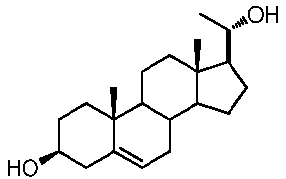
p<0,03
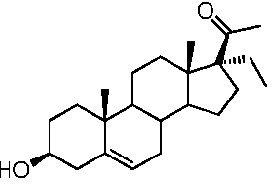
p>0,25
p>0,25
p<0,02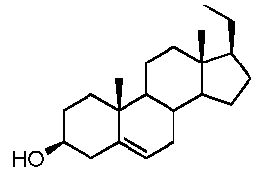
p<0,08
p>0,20
p>0,14
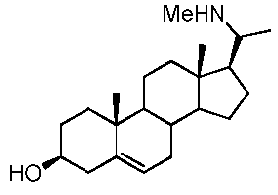
p<0,05
p>0,35
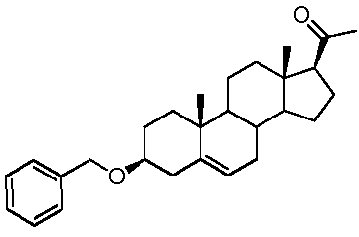
p<0,001
p<0,03
p<0,01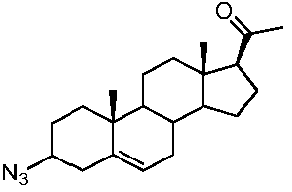
p<0,05
p<0,05
p<0,001
p<0,02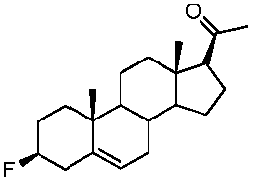
p<0,01
p<0,02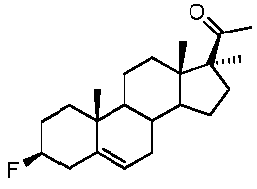
p<0,01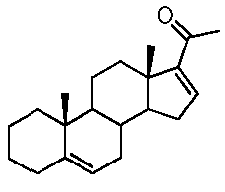
p<0,001
p<0,001
p<0,02
p<0,03
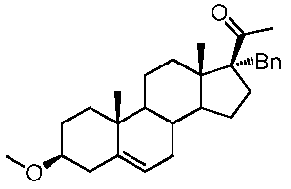
p>0,49
p>0,48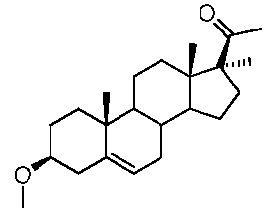
p<0,01
p>0,24
p<0,05
p>0,39
p>0,1
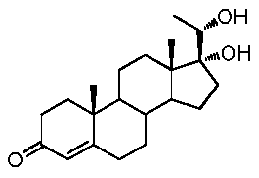
p<0,001
p<0,004
p<0,06
p<0,01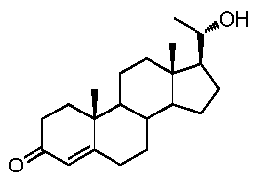
p<0,04
p<0,1
p>0,25
p<0,01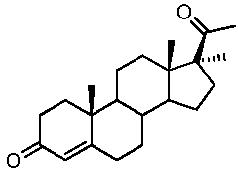
p<0,02
p<0,21
p<0,07
p<0,02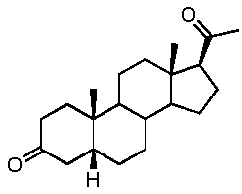
p<0,0001
p<0,05
p<0,002
p<0,003
Для соединений, которые содержат бета-гидроксильную группу в положении C3, замещение по положению C17 метильной, бензильной или метоксигруппой приводит к получению производных прегненолона, которые сохраняют хороший уровень анатагонистической активности в отношении CB1 (таблица 4A). Также хороший уровень активности наблюдается в том случае, когда положение C20 замещено альфа-гидроксильной группой и/или двойная связь C5-C6 смещена в положение C5-C4 или замещена бета-водородом в положении C5 (таблица 4A). В противоположность эффекту этильной группы в положении C17, удаление кето-группы из положения C20 или замещение этого положения метиламиногруппой значительно снижает антагонизм в отношении активности CB1 (таблица 4B). Удаление спиртовой группы в положении C3 или ее замещение фтором, азидо или бензилокси группой приводит к получению соединения с хорошим уровнем антагонизма в отношении активности рецептора CB1 (таблица 4C). В противоположность этому, замещение спиртовой группы в положении C3 метоксигруппой вызывает сильное падение активности в отношении активности CB1 (таблица 4D). Если спиртовая группа в положении C3 замещена кето-группой, наблюдается значительное снижение антагонизма в отношении активности CB1 (таблица 4E). Однако снижение активности соединений с кето-группой в положении C3 можно было ослабить модификацией положений C5, C20 и C17. Альфа-гидроксильная группа в положении C20 и C17 или замена двойной связи C5-C6 бета-водородом в положении C5 улучшает антагонистические свойства соединений с кето-группой (таблица 4E).
Обсуждение результатов:
Полученные согласующиеся данные можно суммировать следующим образом:
Во-первых, соединения общей формулы (A) представляют собой производные прегненолона, для которых характерно ограниченное превращение в активные стероидные производные прегненолона. Такие свойства соединений упомянутой формулы (A) являются неожиданными, поскольку возможное влияние химической модификации прегненолона на ослабление или сохранение склонности к метаболизму до активных стероидных производных нельзя было предсказать на основе знаний известного уровня техники или мнения специалиста в данной области.
Во-вторых, соединения общей формулы (I) и/или (II) представляют собой производные прегненолона, которые способны ингибировать последствия активации CB1 в различных системных моделях: 1. потребления пищи, вызванного введением THC, у мышей и у крыс; 2. потребления пищи, вызванного ограничением питания, у мышей; 3. явлениях, относящихся к каннабиноидной тетраде, вызванных THC; 4. увеличения выработки TNFальфа, вызванного LPS.
V. Ингибирование последствий активации рецептора CB1 является специфичным для прегненолона и не характерно для производных метаболитов
Пример 26: THC повышает концентрации прегненолона в мозге самцов крыс Wistar в большей степени, чем концентрации активных стероидных метаболитов прегненолона
В этом примере авторы изобретения показали, что введение THC (3 мг/кг sc) самцам крыс Wistar через определенный период времени вызывало значительное увеличение уровней некоторых стероидных производных прегненолона и, в частности, аллопрегнанолона и эпиаллопрегнанолона. Однако влияние введения THC на содержание прегненолона на несколько порядков величины превышало эффект, наблюдаемый для стероидных метаболитов, образующихся из прегненолона (фиг. 1C-F).
При введении THC в различных дозах самцам крыс Wistar, авторы изобретения обнаружили, что THC повышает концентрации стероидных производных прегненолона, а именно аллопрегнанолона и эпиаллопрегнанолона, хотя не наблюдалось значимого повышения уровней тестостерона и DHT. Однако даже после введения самой высокой дозы THC, увеличение концентрации других стероидов было существенно меньше, чем увеличение концентрации прегненолона.
Пример 27: THC не увеличивает уровни активных стероидных производных прегненолона у мышей
Если THC вводили в различных дозах самцам мышей, наблюдалось сильное дозозависимое увеличение концентраций прегненолона. Однако в мозге мышей не наблюдалось значимого увеличения уровней активных стероидных производных прегненолона, а именно аллопрегнанолона, эпиаллопрегнанолона, тестостерона и DHT.
Пример 28: Дозы прегненолона, которые ингибируют потребление пищи, не увеличивают концентрации активных стероидных производных прегненолона в мозге
В этом примере авторы изобретения исследовали результаты введения доз прегненолона (от 2 до 8 мг/кг), которые обладали способностью ингибировать результаты активации CB1 у мышей. Инъекции доз прегненолона в диапазоне от 2 до 8 мг/кг повышали уровни прегненолона в мозге; однако эти дозы не меняли концентраций производных метаболитов, таких как эпиаллопрегнанолон и аллопрегнанолон, в мозге мышей (фиг. 4).
Обсуждение результатов
Полученные согласующиеся данные можно суммировать следующим образом:
Во-первых, активация CB1 при введении THC у крыс вызывает значительно меньшее увеличение уровней аллопрегнанолона и эпиаллопрегнанолона по сравнению с прегненолоном. У мышей введение THC не приводит к значительному увеличению уровней аллопрегнанолона и эпиаллопрегнанолона. Таким образом, отрицательное обратное влияние на активность CB1, возникающее при увеличении концентраций эндогенного прегненолона, которое было изучено у мышей, не может возникать из-за последующего увеличения уровней активных стероидных производных, образующихся из прегненолона.
Во-вторых, введение экзогенного прегненолона в диапазоне дозировок, при которых у мышей наблюдается ингибирующее действие прегненолона на активацию CB1 (2-8 мг/кг), не вызывает повышения уровней аллопрегнанолона или эпиаллопрегнанолона в мозге (фиг. 2). Следовательно, ингибирование активности CB1, наблюдаемое после введения прегненолона, нельзя приписать последующему увеличению уровней активных стероидных производных, образующихся из прегненолона.
Наконец, производные прегненолона формул (I) и/или (II), которые не превращаются в прегнанолон, аллопрегнанолон и другие активные стероидные производные, по-прежнему способны ингибировать последствия активации CB1. Следовательно, ингибирование рецептора CB1 и/или ингибирование эффектов, вызываемых CB1, наблюдаемое после введения прегненолона, нельзя приписать активным стероидным производным, образующимся из прегненолона.
VI. Способы введения прегненолона, не вызывающие повышения уровней нейроактивных производных метаболитов
Пример 39: Введение прегненолона способами, которые имитируют составы с медленным высвобождением, позволяет уменьшить метаболизм с образованием активных стероидных производных
В данном примере авторы изобретения описали типовой способ введения прегненолона в дозировках, которые могут ингибировать активацию CB1, но не увеличивают уровни нейроактивных стероидных производных.
Сравнивали влияние способа введения на уровни прегненолона в плазме (n=5 животных на группу) при подкожном введении (6 мг/кг) или при введении с помощью микро-осмотических насосов Alzet (Alzet Osmotis Pumps, Charles River, France, model 2006), которые имитируют действие составов с продолжительным высвобождением прегненолона. С этой целью указанный мининасос имплантировали подкожно, обеспечивая высвобождение прегненолона в стационарном режиме. Использовали две концентрации прегненолона, а именно, 0,6 мг/кг/час и 1 мг/кг/час (в 10 и 6 раз меньше дозы, которую вводили подкожно). Хотя подкожное введение прегненолона мышам в количестве 6 мг/кг не увеличивало концентрацию аллопрегнанолона в мозге (фиг. 4), наблюдалось значительное увеличение содержания этого активного стероидного производного в плазме (фиг. 20).
Подкожное введение прегненолона в дозировке 6 мг/кг повышало уровни прегненолона в плазме (около 100 нг/мл), но, кроме того, вызывало увеличение уровней аллопрегнанолона. Время полужизни прегненолона при подкожном введении было достаточно коротким (полчаса), и повышенные уровни аллопрегнанолона сохранялись в течение одного часа (фиг. 20). В противоположность этому, если прегненолон вводили в стационарном режиме с помощью мининасоса Alzet в количестве 0,6 мг/кг/час, увеличение уровней прегненолона находилось в том же диапазоне, что и максимальное увеличение, наблюдавшееся после подкожной инъекции, но не происходило увеличения уровней аллопрегнанолона (фиг. 20). Кроме того, уровни прегненолона оставались в диапазоне эффективных терапевтических дозировок на протяжении двух недель. Даже если прегненолон вводили в дозировке 1 мг/кг/час, в результате чего концентрации прегненолона в плазме вдвое превышали концентрации после инъекции 6 мг/кг, содержание аллопрегнанолона не повышалось (фиг. 20).
Обсуждение результатов
Прегненолон был описан в документах известного уровня техники, как препарат для лечения психиатрических заболеваний, некоторых типов воспаления и метаболических расстройств, а также некоторых типов пристрастий, в частности, пристрастия к никотину и алкоголю. Однако во всех этих способах прегненолон применялся в высоких концентрациях с очевидной целью повышения уровней нейроактивных стероидных производных, которым приписывалось терапевтическое действие применения прегненолона. В настоящем изобретении авторы показали, что прегненолон сам по себе, без участия нейроактивных стероидных производных, в существенно меньших дозировках, чем были указаны в документах известного уровня техники, может применяться для лечения патологий, которые включают активацию рецепторов CB1. В этом контексте, поскольку прегненолон имеет очень короткое время полужизни (примерно 30 минут), составы с продолжительным высвобождением могут применяться для поддержания уровней прегненолона в терапевтическом диапазоне. Имитируя такие составы с помощью мининасоса Alzet, авторы показали, что равномерное введение прегненолона с низкой скоростью позволяет добиться двух целей: 1. создать стабильные концентрации прегненолона, которые находятся в диапазоне концентраций, способных блокировать все последствия активации CB1 (в районе 100 нг/мл); и 2. ослабить повышение уровней активных стероидных производных, например, аллопрегнанолона. Эти результаты позволяют разработать способ лечения заболеваний, в которые вовлечен CB1, включающий введение прегненолона в низких дозировках за счет применения составов прегненолона с продолжительным высвобождением, преимущество которого заключается в получении фармакологического эффекта самого прегненолона, при уменьшении нежелательных эффектов его стероидных производных.
Список литературы
В тексте настоящей заявки упоминаются различные источники, в которых описан известный уровень техники в области, к которой относится настоящее изобретение. Содержание этих ссылок включено в настоящую заявку посредством ссылок.







| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПРЕГНЕНОЛОНА | 2013 |
|
RU2667065C2 |
| 3β-(БЕНЗИЛОКСИ)-17α-МЕТИЛ-ПРЕГН-5-ЕН-20-ОН ДЛЯ ПРИМЕНЕНИЯ ПРИ ЛЕЧЕНИИ КОГНИТИВНЫХ РАССТРОЙСТВ | 2019 |
|
RU2796005C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗО-ПИРИДИНА, ОБЛАДАЮЩИЕ СРОДСТВОМ К РЕЦЕПТОРУ МЕЛАНОКОРТИНА | 2004 |
|
RU2358974C2 |
| 3-β(4-МЕТОКСИБЕНЗИЛОКСИ)ПРЕГН-5-ЕН-20-ОН ДЛЯ ПРИМЕНЕНИЯ ПРИ ЛЕЧЕНИИ НАРУШЕНИЙ, СВЯЗАННЫХ С УПОТРЕБЛЕНИЕМ КАННАБИНОИДОВ | 2019 |
|
RU2788004C2 |
| 5-СУЛЬФАНИЛ-4Н-1,2,4-ТРИАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННОГО ПРЕПАРАТА | 2002 |
|
RU2367655C2 |
| НЕЙРОАКТИВНЫЕ 19-АЛКОКСИ-17-ЗАМЕЩЕННЫЕ СТЕРОИДЫ И СПОСОБЫ ЛЕЧЕНИЯ С ИХ ПРИМЕНЕНИЕМ | 2013 |
|
RU2663665C2 |
| ПРОДУКТ, ВКЛЮЧАЮЩИЙ ПО МЕНЬШЕЙ МЕРЕ ОДНО ВЕЩЕСТВО, ИНГИБИРУЮЩЕЕ NO-СИНТАЗЫ, В КОМБИНАЦИИ ПО МЕНЬШЕЙ МЕРЕ С ОДНИМ ВЕЩЕСТВОМ, ИНГИБИРУЮЩИМ ФОСФОЛИПАЗЫ А2 | 2000 |
|
RU2256465C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И ИМИДАЗОПИРИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2004 |
|
RU2330023C2 |
| 1,3,5-ТРИЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 4,5-ДИГИДРО-1Н-ПИРАЗОЛА, ОБЛАДАЮЩИЕ СВ-АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ | 2005 |
|
RU2360904C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2004 |
|
RU2369601C2 |
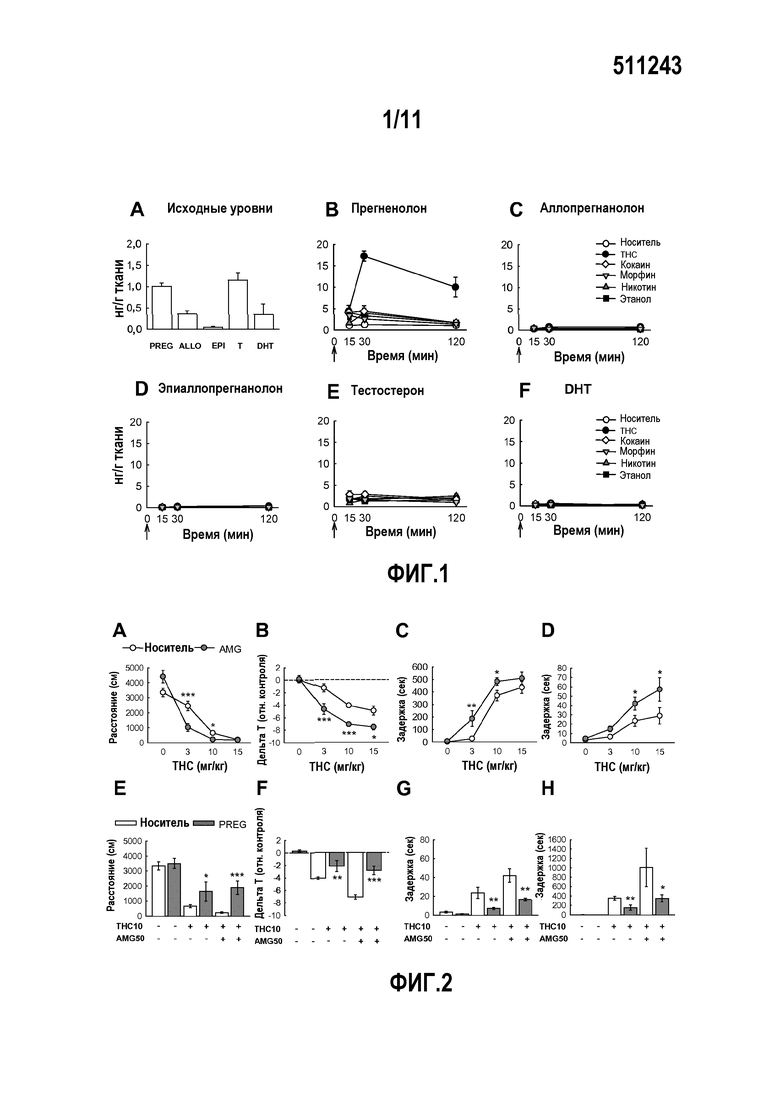
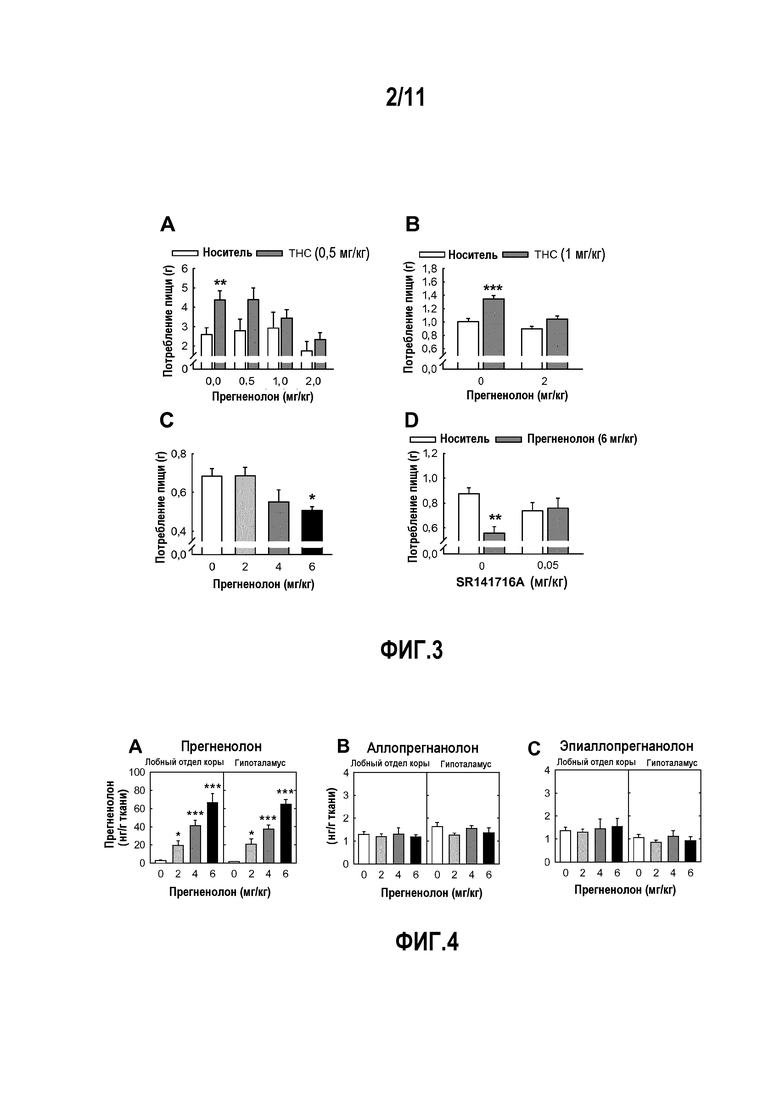
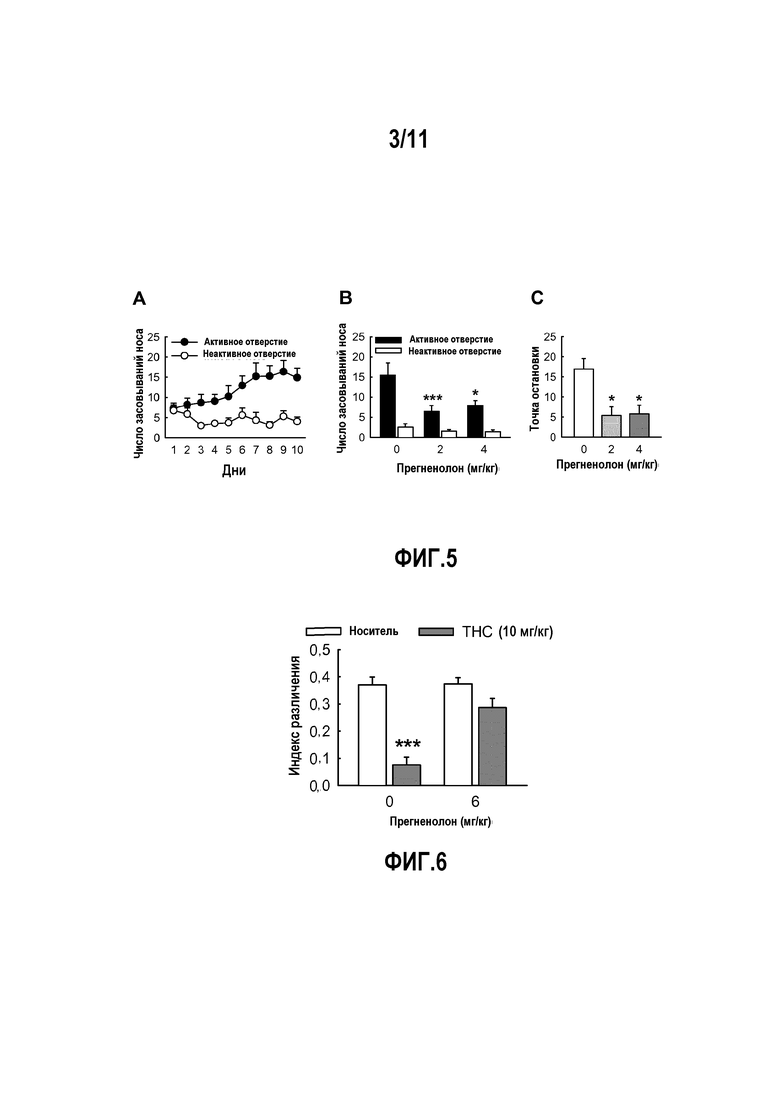
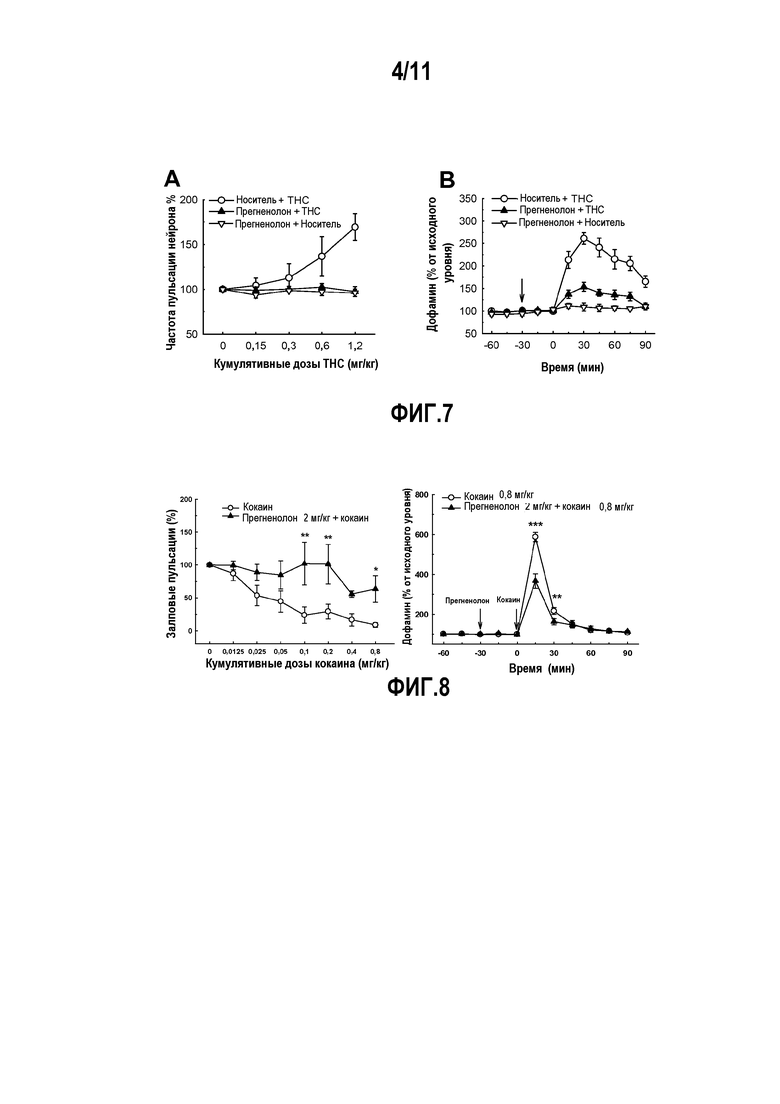
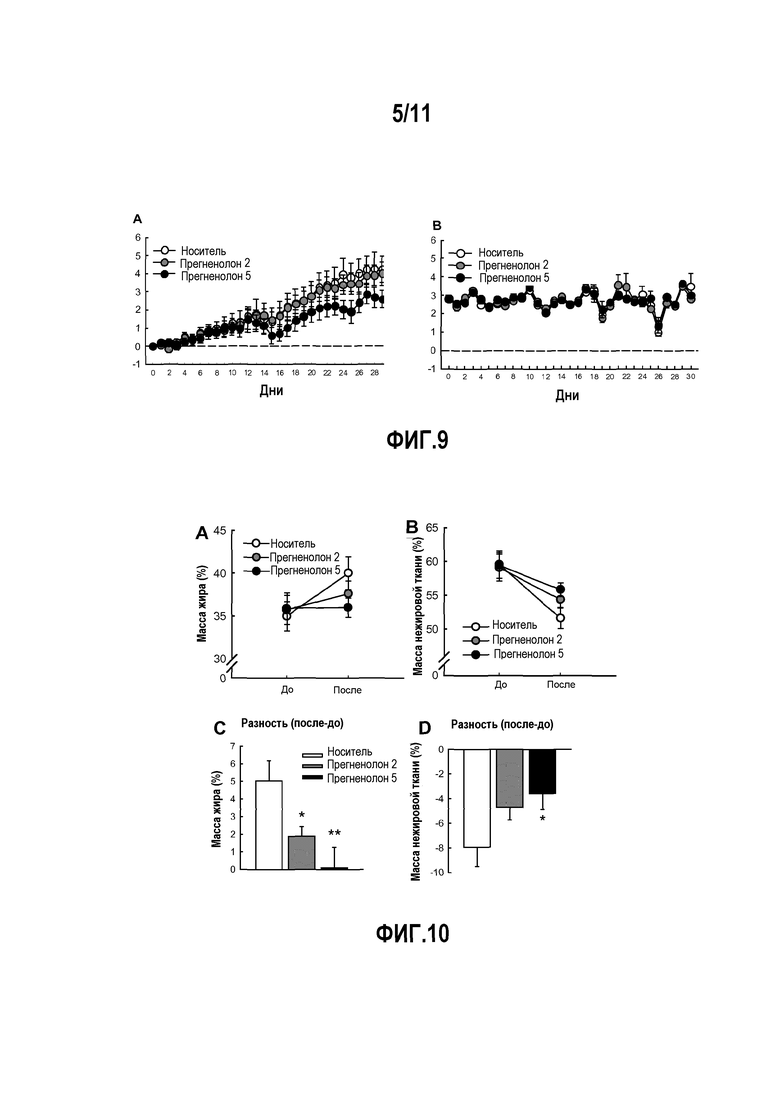
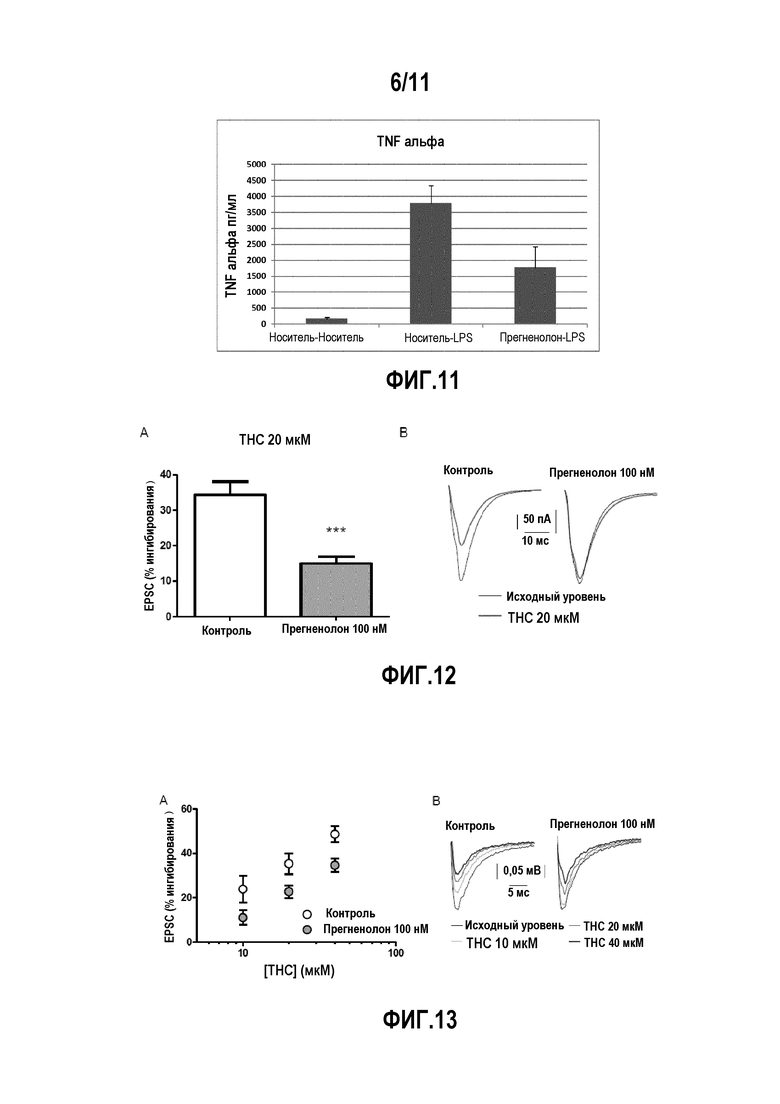
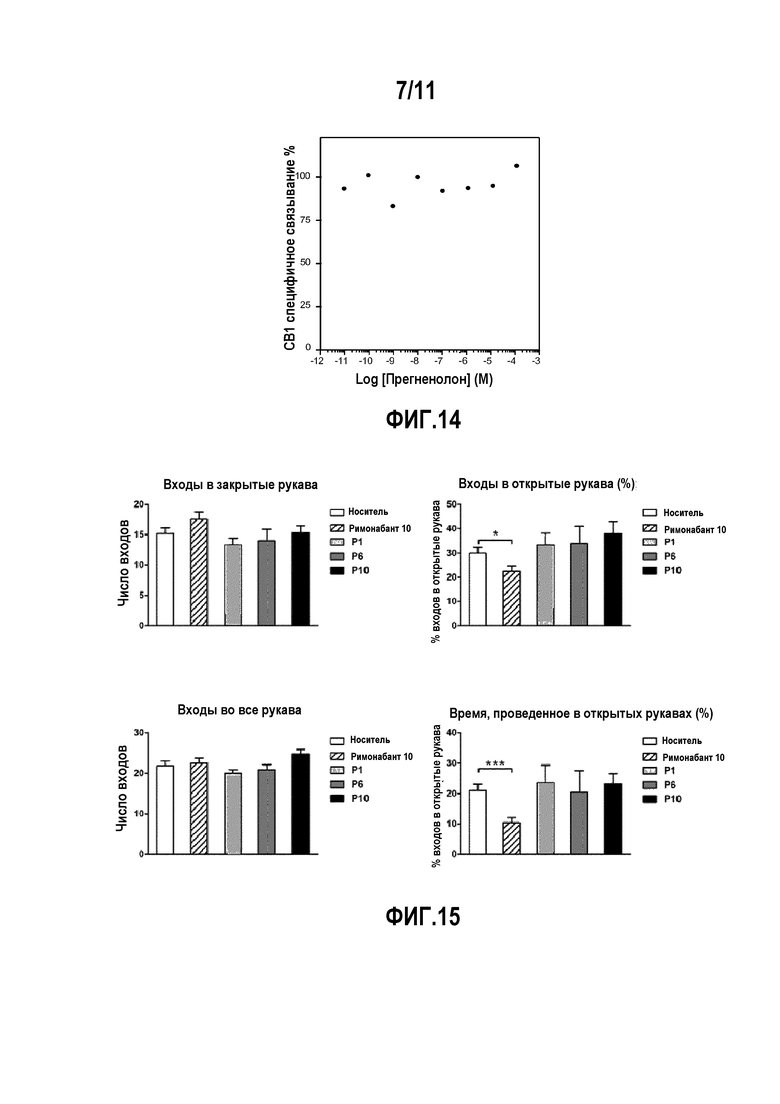


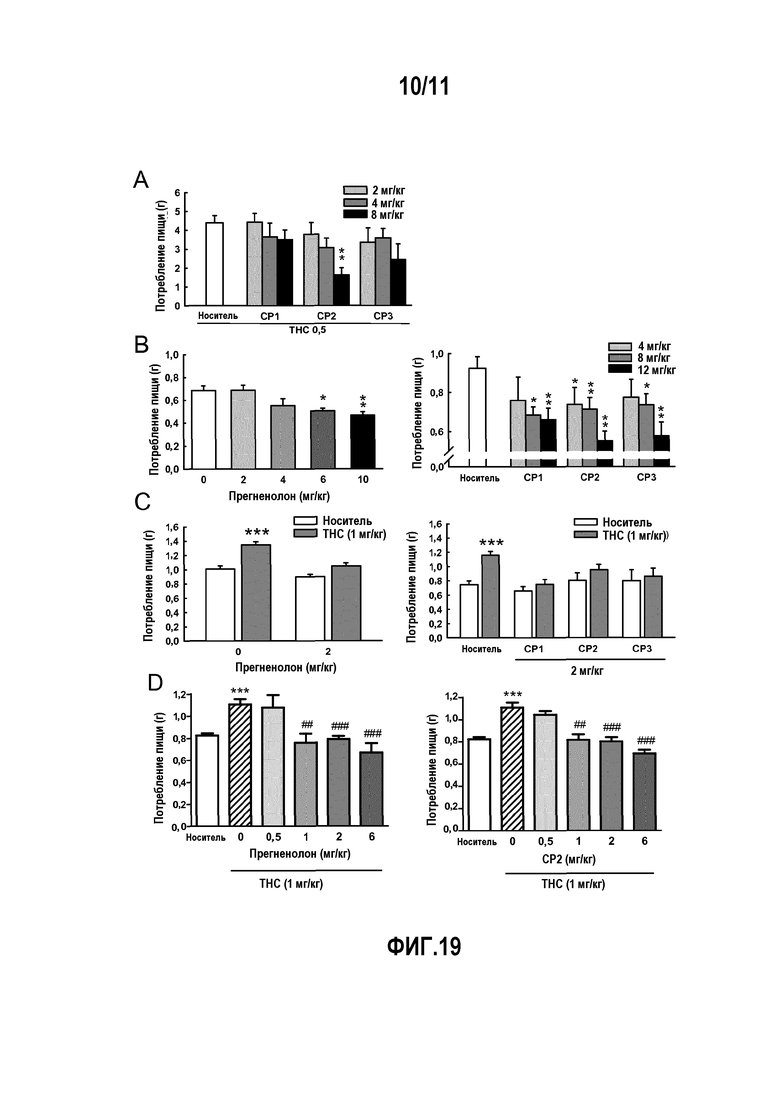
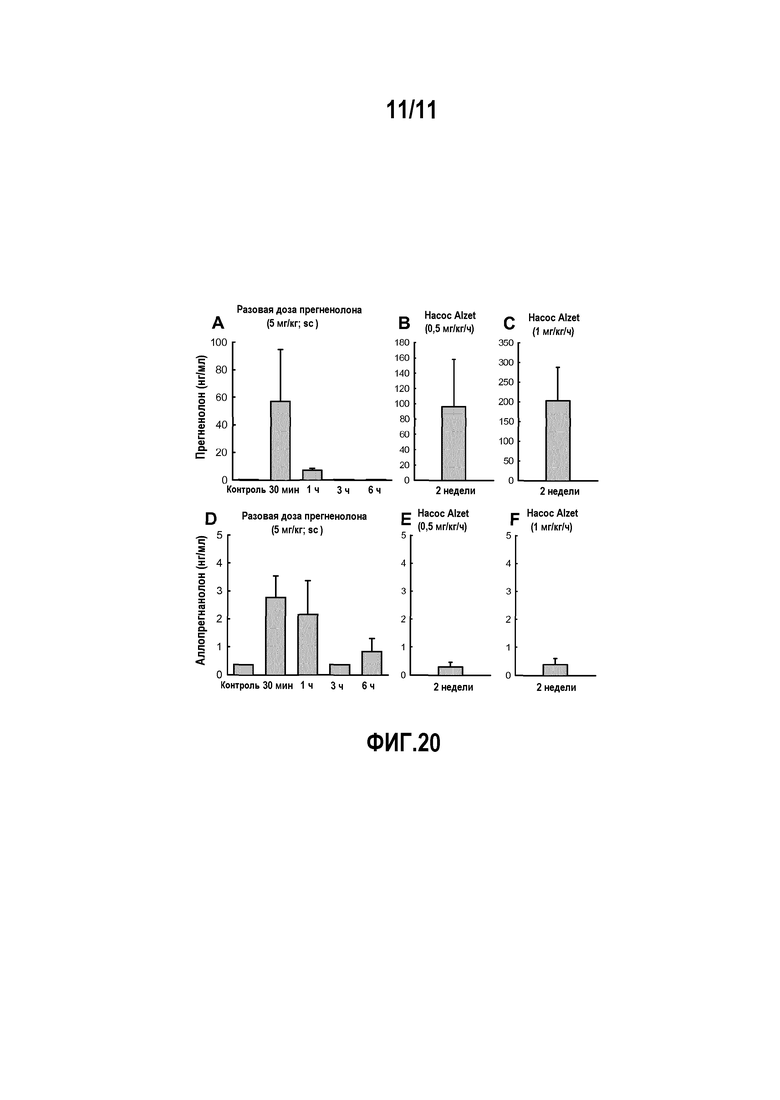
Изобретение относится к фармацевтическим композициям на основе соединений формул (B), (D), (E), обладающих свойством аллостерического ингибитора рецептора СВ1 для применения в лечении патологического состояния или расстройства, выбранного из группы, состоящей из расстройств мочевого пузыря и желудочно-кишечного тракта; воспалительных заболеваний; сердечно-сосудистых заболеваний; нефропатий; глаукомы; спастичности; рака; остеопороза; метаболических расстройств; ожирения; расстройств, вызванных пагубными пристрастиями, зависимостями, злоупотреблениями и их рецидивами; психиатрических и неврологических расстройств; нейродегенеративных расстройств; аутоиммунного гепатита и энцефалита; боли; и воспалительных и фиброзных заболеваний кожи. Указанные соединения по мере метаболизма практически не превращаются в активные производные прегненолона, что позволяет избежать многих побочных эффектов применения ортостерических антагонистов. 3 н. и 17 з.п. ф-лы, 4 табл., 20 ил., 29 пр.
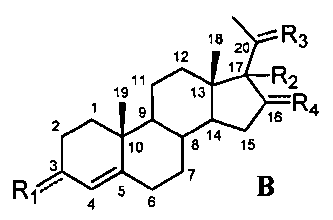
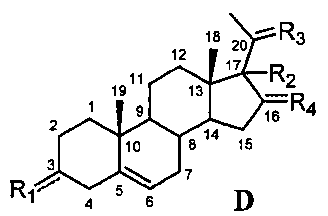
и
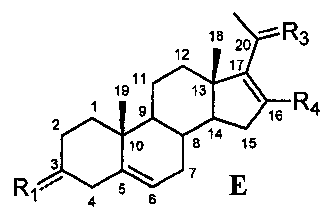
1. Фармацевтическая композиция, содержащая производное прегненолона, выбранное из следующих соединений:
соединение формулы (В)
или его фармацевтически приемлемая соль,
где
 R1 обозначает, что С3 замещен -ОН или =O,
R1 обозначает, что С3 замещен -ОН или =O,
- R2 обозначает, что С17 замещен С1-8 алкилом, галогеном или -Bn,
R3 обозначает, что С20 замещен -ОН или =O, и
R4 обозначает, что С16 замещен -Н,
соединение формулы (D)
или его фармацевтически приемлемая соль,
где
R1 обозначает, что С3 замещен -NH2, Bn-O- или -N3,
- R2 обозначает, что С17 замещен -Н,
R3 обозначает, что С20 замещен =O, и
R4 обозначает, что С16 замещен -Н,
или
R1 обозначает, что С3 замещен С1-8 алкокси, галогеном, Bn-О- или -N3,
- R2 обозначает, что С17 замещен -Bn, -СН3 или С2-6 алкенилом,
R3 обозначает, что С20 замещен =O, и
R4 обозначает, что С16 замещен -Н,
или
R1 обозначает, что С3 замещен -ОН,
- R2 обозначает, что С17 замещен С1-8 алкилом, С1-8 алкокси или Bn-,
R3 обозначает, что С20 замещен =O, и
R4 обозначает, что С16 замещен -Н,
или
R1 обозначает, что С3 замещен -ОН,
- R2 обозначает, что С17 замещен -Н,
R3 обозначает, что С20 замещен -Н, -ОН или -NR8R9, где каждый R8 и R9 независимо представляет собой Н или С1-8 алкил, и
R4 обозначает, что С16 замещен -Н,
и
соединение формулы (Е)
или его фармацевтически приемлемая соль,
где
R1 обозначает, что С3 замещен -Н,
R3 обозначает, что С20 замещен -Н, -ОН или =O, и
R4 обозначает, что С16 замещен -Н,
при условии, что в формулах В, D и Е, когда связь между С3 и R1 является простой, R1 находится в β-положении,
для применения в лечении патологического состояния или расстройства, выбранного из группы, состоящей из расстройств мочевого пузыря и желудочно-кишечного тракта; воспалительных заболеваний; сердечно-сосудистых заболеваний; нефропатий; глаукомы; спастичности; рака; остеопороза; метаболических расстройств; ожирения; расстройств, вызванных пагубными пристрастиями, зависимостями, злоупотреблениями и их рецидивами; психиатрических и неврологических расстройств; нейродегенеративных расстройств; аутоиммунного гепатита и энцефалита; боли; и воспалительных и фиброзных заболеваний кожи.
2. Фармацевтическая композиция, содержащая производное прегненолона, по п. 1, где указанное соединение, после введения субъекту, по мере метаболизма практически не превращается в активные производные прегненолона.
3. Фармацевтическая композиция, содержащая производное прегненолона, по п. 1, где указанное соединение представляет собой соединение формулы (В)
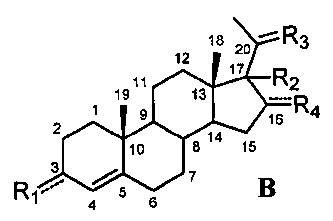
или его фармацевтически приемлемую соль,
где
R1 обозначает, что С3 замещен -ОН в β-положении или =O,
- R2 обозначает, что С17 замещен С1-8 алкилом, галогеном или -Bn,
R3 обозначает, что С20 замещен -ОН, и
R4 обозначает, что С16 замещен -Н,
или
R1 обозначает, что С3 замещен =O,
- R2 обозначает, что С17 замещен С1-8 алкилом, галогеном или - Bn,
R3 обозначает, что С20 замещен =O, и
R4 обозначает, что С16 замещен -Н.
4. Фармацевтическая композиция, содержащая производное прегненолона, по п. 3, где указанное соединение представляет собой 17α-бензилпрогестерон.
5. Фармацевтическая композиция, содержащая производное прегненолона, по п. 1, где указанное соединение представляет собой соединение формулы (D)
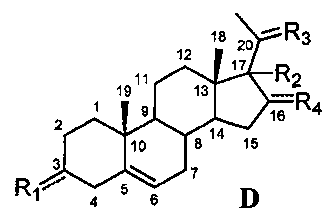
или его фармацевтически приемлемую соль,
где
R1 обозначает, что C3 замещен -NH2, Bn-О- или -N3 в β-положении,
- R2 обозначает, что С17 замещен -Н,
R3 обозначает, что С20 замещен =O, и
R4 обозначает, что С16 замещен -Н.
6. Фармацевтическая композиция, содержащая производное прегненолона, по п. 5, где указанное соединение представляет собой 5-прегнен-3β-О-бензил-20-он, 3β-аминопрегненолон или 5-прегнен-3β-азидо-20-он.
7. Фармацевтическая композиция, содержащая производное прегненолона, по п. 1, где указанное соединение представляет собой соединение формулы (D)

или его фармацевтически приемлемую соль,
где
R1 обозначает, что С3 замещен С1-8 алкокси, галогеном, Bn-О- или -N3 в β-положении,
- R2 обозначает, что С17 замещен -Bn, -СН3 или С2-6 алкенилом,
R3 обозначает, что С20 замещен =O, и
R4 обозначает, что С16 замещен -Н.
8. Фармацевтическая композиция, содержащая производное прегненолона, по п. 7, где указанное соединение представляет собой 17α-аллил-3β-метоксипрегненолон, 17α-бензил-3β-фторпрегненолон, 3β-фтор-17α-метилпрегненолон, 3β-метокси-17α-метилпрегненолон, 17α-бензил-3β-метоксипрегненолон, 3β-бензилокси-17α-метилпрегненолон или 17α-бензил-3β-бензилоксипрегненолон.
9. Фармацевтическая композиция, содержащая производное прегненолона, по п. 1, где указанное соединение представляет собой соединение формулы (D)
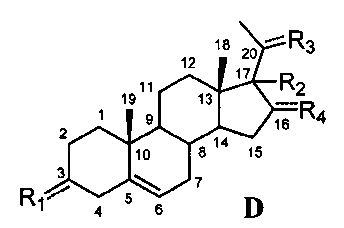
или его фармацевтически приемлемую соль,
где
R1 обозначает, что С3 замещен -ОН в β-положении,
- R2 обозначает, что С17 замещен С1-8 алкилом, С1-8 алкокси или Bn-,
R3 обозначает, что С20 замещен =O, и
R4 обозначает, что С16 замещен -Н.
10. Фармацевтическая композиция, содержащая производное прегненолона, по п. 9, где указанное соединение представляет собой 17α-бензилпрегненолон, 17α-этилпрегненолон, 17α-метилпрегненолон или 17-метоксипрегненолон.
11. Фармацевтическая композиция, содержащая производное прегненолона по п. 1, где указанное соединение представляет собой соединение формулы (D)
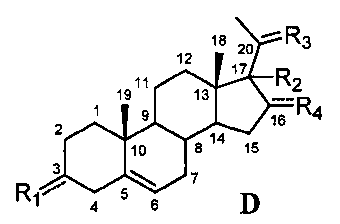
или его фармацевтически приемлемую соль,
где
R1 обозначает, что С3 замещен -ОН в β-положении,
- R2 обозначает, что С17 замещен -Н,
R3 обозначает, что С20 замещен -Н, -ОН или -NR8R9, где каждый R8 и R9 независимо представляет собой Н или С1-8 алкил, и
R4 обозначает, что С16 замещен -Н.
12. Фармацевтическая композиция, содержащая производное прегненолона, по п. 11, где указанное соединение представляет собой 20-деоксипрегненолон или 20-метиламино-5-прегнен-3β-ол.
13. Фармацевтическая композиция, содержащая производное прегненолона, по п. 1, где указанное соединение представляет собой соединение формулы (Е)
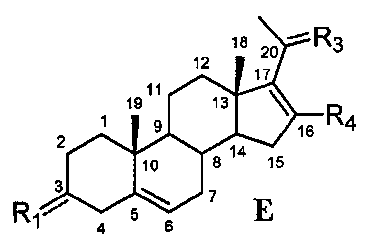
или его фармацевтически приемлемую соль,
где
R1 обозначает, что С3 замещен -Н,
R3 обозначает, что С20 замещен -Н, -ОН или =O, и
R4 обозначает, что С16 замещен -Н.
14. Фармацевтическая композиция, содержащая производное прегненолона, по п. 13, где указанное соединение представляет собой 5,16-прегнадиен-20-он.
15. Фармацевтическая композиция, содержащая производное прегненолона, по любому из пп. 1-14 для применения в лечении патологического состояния или расстройства, выбранного из группы, состоящей из расстройств мочевого пузыря и желудочно-кишечного тракта; сердечно-сосудистых заболеваний; нефропатий; спастичности; остеопороза; боли; и воспалительных и фиброзных заболеваний кожи.
16. Фармацевтическая композиция, содержащая производное прегненолона, по любому из пп. 1-14 для применения в лечении пристрастия к препаратам, полученным из конопли; зависимости от препаратов, полученных из конопли; злоупотребления препаратами, полученными из конопли; токсического действия препаратов, полученных из конопли; и рецидивами, связанными с употреблением препаратов, полученных из конопли.
17. Производное прегненолона, где указанное соединение представляет собой 17α-бензил-3β-фторпрегненолон, 17α-бензил-3β-бензилоксипрегненолон, 3β-бензилокси-17α-метилпрегненолон, 17α-бензилпрегненолон, 3β-метокси-17α-метилпрегненолон, 17α-аллил-3β-метоксипрегненолон или 17α-бензил-3β-метоксипрегненолон.
18. Фармацевтическая композиция, обладающая свойствами антагониста рецептора СВ1, содержащая производное прегненолона по п. 17 и фармацевтически приемлемый носитель.
19. Производное прегненолона по п. 17 для применения в способе лечения заболеваний, опосредованных активностью рецептора СВ1.
20. Производное прегненолона по п. 17 для лечения патологического состояния или расстройства, выбранного из группы, состоящей из расстройств мочевого пузыря и желудочно-кишечного тракта; воспалительных заболеваний; сердечно-сосудистых заболеваний; нефропатий; глаукомы; спастичности; рака; остеопороза; метаболических расстройств; ожирения; расстройств, вызванных пагубными пристрастиями, зависимостями, злоупотреблениями и их рецидивами; психиатрических и неврологических расстройств; нейродегенеративных расстройств; аутоиммунного гепатита и энцефалита; боли; репродуктивных расстройств; и воспалительных и фиброзных заболеваний кожи.
| US 5175154 A1, 29.12.1992 | |||
| US 0004628052 A1, 09.12.1986 | |||
| Взрывобезопасный электрический аппарат | 1981 |
|
SU1166189A1 |
| US 0003361744 A1, 02.01.1968 | |||
| US 0003351639 A1, 07.11.1967 | |||
| Печь для сжигания твердых и жидких нечистот | 1920 |
|
SU17A1 |
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| Прибор для исправления снимков рельефа местности | 1921 |
|
SU301A1 |
| ALLEN G R JR ET AL "New | |||

