ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новому конденсированному пиримидиновому производному, обладающему ингибирующей активностью в отношении тирозинкиназ, и к фармацевтической композиции, содержащей такое производное в качестве активного ингредиента.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
В клетках существует множество систем передачи сигналов, которые функционально соединены друг с другом для регуляции пролиферации, роста, метастазирования и апоптоза клеток (William G. Kaelin Jr., Nature Reviews Cancer 5, 689, 2005). Нарушение внутриклеточной регуляторной системы посредством генетических и внешних факторов обуславливает аномальную амплификацию или деструкцию системы передачи сигналов, что ведет к образованию опухолевой клетки (Douglas Hanahan and Robert A. Weinberg, Cell 100, 57, 2000).
Протеинтирозинкиназы играют важную роль в такой клеточной регуляции (Irena Melnikova and James Golden, Nature Reviews Drug Discovery 3, 993, 2004), и их аномальная экспрессия или мутация наблюдалась в злокачественных опухолевых клетках или при аутоиммунных заболеваниях. Протеинтирозинкиназа представляет собой фермент, который катализирует перенос фосфатных групп с АТР на остатки тирозина, расположенные в белковых субстратах. Многие белки-рецепторы факторов роста функционируют в качестве тирозинкиназ для передачи клеточных сигналов. Взаимодействие между факторами роста и их рецепторами обычно регулирует клеточный рост, а аномальная передача сигналов, обусловленная мутацией или сверхэкспрессией любого из рецепторов, обычно индуцирует различные злокачественные опухоли или аутоиммунные заболевания, такие как ревматоидный артрит.
С учетом роли указанных тирозинкиназ, был изучен целый ряд факторов роста и их рецепторов, и среди них были тщательно изучены тирозинкиназы эпидермальных факторов роста (EGF) и тирозинкиназы EGF-рецепторов (EGFR) (Nancy Е. Hynes and Heidi А. Lane, Nature Reviews Cancer 5, 341, 2005). Тирозинкиназа EGFR состоит из рецептора и тирозинкиназы и передает внеклеточные сигналы в ядро клетки через клеточную мембрану. Различные тирозинкиназы EGFR классифицируют, основываясь на их структурных различиях, на четыре подтипа, т.е. EGFR (Erb-B1), Erb-B2, Erb-B3 и Erb-B4, и известно, что активирующие EGFR мутации, такие как точечная мутация L858R в 21 экзоне и делеция внутри рамки считывания в 19 экзоне домена тирозинкиназ EGFR, являются важной причиной немелкоклеточного рака легких.
Гефитиниб (AstraZeneca) был первоначально разработан в качестве малой молекулы для ингибирования тирозинкиназ EGFR, которая селективно и обратимо ингибирует EGFR (Erb-B1). Эрлотиниб (Roche) также обладает сходными характеристиками. Указанные направленные на EGFR лекарственные средства эффективны в отношении немелкоклеточного рака легких (NSCLC) и обеспечивают терапевтическую пригодность для пациентов с активирующими EGFR мутациями.
Однако сообщалось, что развитие резистентности снижает активность конкретного лекарства, используемого в направленной на EGFR терапии. Уже сообщалось, что приблизительно у половины пациентов, которым вводили Гефитиниб или Эрлотиниб, проявлялась резистентность к действию лекарств вследствие индукции вторичной Т790М EGFR мутации (William Pao et al., Public Library of Science Medicine, 2(3), 225, 2005, Cancer Res, 67(24), 11924, 2007). Кроме того, недавно было обнаружено, что по сравнению с общепринятыми обратимыми ингибиторами, такими как Гефитиниб и Эрлотиниб, направленные на EGFR необратимые ингибиторы являются более предпочтительными для обеспечения отличной эффективности и преодоления развития резистентности (Danan Li et al., Cancer Cell 12, 81, 2007; и Anja Michalczyk et al., Bioorganic & Medicinal Chemistry 16, 3482, 2008). В результате, были разработаны необратимые ингибиторы, такие как BIBW-2992 (Афатиниб, Boeringer Ingelheim) (С Н Mom et al., British Journal of Cancer 98, 80, 2007), PF00299804 (Дакомитиниб, Pfizer) (Engelman JA, et al., Cancer Res. 67, 11924, 2007) и AV-412 (AVEO Pharmaceuticals) (Tsuyoshi Suzuki et al., Cancer Sci. 98 (12), 1977, 2007), которые в настоящее время находятся на стадии клинических испытаний. Стали известны соединения, которые образуют ковалентную связь с расположенным в АТР домене EGFR цистеином773 (Cys773), необратимо блокируя тем самым автофосфорилирование EGFR и, следовательно, эффективно ингибируя передачу сигналов в клетках рака (David W. Fry et al., Proc. Natl. Acad. Sci. U.S.A. 95, 12022, 1998), и демонстрируют более сильный ингибирующий эффект по сравнению с обратимыми ингибиторами, коммерчески доступными в виде парных ингибиторов EGFR/HER-2 или pan-HER ингибиторов, при оценке активности in vitro и в различных моделях карцином in vivo (Jeff В. Smaill et al., J. Med. Chem. 42, 1803, 1999). Тем не менее, если соединения вводят в дозе, достаточной для преодоления резистентности, индуцированной Т790М мутациями EGFR, то соединения могут вызывать серьезные побочные эффекты, такие как кожную сыпь, диарею и потерю веса вследствие высокой активности в отношении EGFR WT (дикого типа), присутствующего в нормальных клетках, и это ограничивает их клиническое применение (Martin L. Sos, et al., Cancer Res. 70, 868, 2010).
Как было подтверждено в клинических тестах необратимых ингибиторов при немелкоклеточном раке легких, соединения продемонстрировали улучшенную активность, но, тем не менее, слабый терапевтический эффект по сравнению с общепринятыми обратимыми ингибиторами при развитии резистентности у пациентов с раком. Соответственно, существует постоянная потребность в разработке нового лекарственного средства, которое является эффективным при резистентных к воздействию лекарств раках и не обладает нежелательными побочными эффектами.
Между тем, существуют различные подтверждения того, что В-клетки (В-лимфоциты) и Т-клетки (Т-лимфоциты) играют ключевую роль в патогенезе воспалительных заболеваний, аутоиммунных заболеваний и/или иммунологически опосредованных заболеваний.
Например, нарушенная передача сигналов может индуцировать нерегулируемую В-клеточную пролиферацию и дифференциацию с возникновением всех видов лимфом, включая различные острые или хронические лимфолейкозы, и может вызывать образование аутоантител, что приводит к разнообразным воспалительным заболеваниям, аутоиммунным заболеваниям и/или иммунологически опосредованным заболеваниям.
Тирозинкиназа Брутона (BTK) является представителем семейства ТЕС тирозинкиназ и играет важную роль в активации В-клеток и передаче сигналов. BTK играет существенную роль в каскаде передачи сигналов В-клетками, который связывает стимуляцию B-клеточного рецептора (BCR) на поверхности В-клеток с ответом расположенных далее в каскаде клеток. Кроме того, известно, что BTK является важнейшим регулятором развития В-клеток и активации и выживания зрелых В-клеток (Khan et al., Immunity 3, 283, 1995; Ellmeier et al., J. Exp. Med. 192, 1611, 2000; Kurosaki, Current Opinion in Immunology 12, 276, 2000; Schaeffer and Schwartzberg, Current Opinion in Immunology 12, 282, 2000). Таким образом, ингибирование ВТК может представлять собой тактику лечения с блокированием процесса заболевания, опосредованного В-клетками.
Например, известно, что дефицитные по BTK мыши являются резистентными к индуцированному коллагеном артриту, и была показана зависимая от дозы эффективность ингибиторов ВТК в модели артрита у мышей (Jansson and Holmdahl, Clin. Exp. Immunol. 94, 459, 1993; Pan et al., Chem. Med Chem. 2, 58, 2007). Таким образом, эффективные ингибиторы BTK могут быть применимы для лечения ревматоидного артрита.
В дополнение, BTK также экспрессируется отличными от В- клеток клетками, которые могут быть вовлечены в процесс заболевания, т.е. тучными клетками костного мозга. Сообщалось, что в дефицитных по ВТК тучных клетках костного мозга супрессируется индуцированная антигеном дегрануляция (Iwaki et al., J. Biol. Chem. 280, 40261, 2005). Это показывает, что ВТК могла бы быть применима для лечения патологических реакций тучных клеток, таких как аллергия и бронхиальная астма.
Кроме того, моноциты, в которых отсутствует активность BTK, продемонстрировали сниженную продукцию TNF-α в ответ на стимуляцию (Horwood et al. J Exp Med. 197, 1603, 2003). Следовательно, опосредованное TNF-α воспаление могло бы модулироваться при помощи ингибиторов BTK.
Кроме того, сообщалось, что ВТК участвует в апоптозе в качестве одного из регуляторов (Islam and Smith, Immunol. Rev. 178, 49, 2000). Таким образом, ингибиторы ВТК могли бы быть применимы для лечения некоторых B-клеточных лимфом и лейкозов (Feldhahn et al., J. Exp. Med. 201, 1837, 2005).
Между тем, Т-клетки участвуют в передаче сигналов, полученных через Т-клеточный рецептор (TCR) на поверхности клетки от антиген-презентирующих клеток, к последующим эффекторам посредством активации различных межклеточных киназ, таких как Janus киназы. При этом они секретируют различные интерлейкины (IL) или интерферон-γ для активации различных лейкоцитов, а также В-клеток. Протеинкиназы, вовлеченные в передачу сигналов в Т-клетках, представляют собой Janus киназы (JAK), такие как JAK1, JAK2, JAK3 и TYK2, индуцируемые IL-2 Т-клеточные киназы (ITK), и семейство ТЕС киназ, таких как киназы покоящихся лимфоцитов (RLK).
Janus киназы, включая JAK3, широко изучались в качестве мишени при аутоиммунных и/или воспалительных заболеваниях. Среди них, в отличие от вовлеченной в гемопоэзе и эритроцитарном гомеостазе JAK2 или экспрессирующейся в различных тканях JAK1, JAK3 экспрессируется в лимфоцитах и играет очень важную роль в передаче сигналов посредством различных цитокинов, т.е. IL-2, IL-4, IL-7, IL-9 и IL-15, что является более эффективным (Flanagan et al, Journal of medicinal Chemistry, 53, 8468, 2010). Согласно исследованиям на животных, JAK3 участвует в созревании В-клеток и Т-клеток, а также в поддержании функций Т-клеток.
Поэтому ингибиторы JAK3 могут быть применимы для лечения ревматоидного артрита, псориаза, атопического дерматита, волчанки, рассеянного склероза, сахарного диабета I типа и вызванных сахарным диабетом осложнений, рака, бронхиальной астмы, аутоиммунных нарушений щитовидной железы, язвенного колита, болезни Крона, болезни Альцгеймера, лейкоза и при других показаниях, при которых иммуносупрессия была бы желательна, таких как трансплантация органов или ксенотрансплантация (Pesu М, Laurence A, Kishore N, et al., Immunol Rev 223, 132, 2008.; Kawahara A, Minami Y, Miyazaki T, et al., Proc Natl Acad Sci USA 92, 8724, 1995; Nosaka T, van Deursen JMA, Tripp RA, et al., Science 270, 800, 1995; Papageorgiou Ac, Wikman LEK., et al., Trends Pharm Sci 25, 558, 2004).
Между тем, другие семейства ТЕС киназ также играют важную роль в активации Т-клеток (Pamela L. Schwartzberg, et al., Nature Reviews Immunology 5, 284, 2005). Например, делеция ITK, которая характеристически экспрессируется в Т-клетках, у мышей приводит к сниженной клеточной пролиферации, индуцированной стимуляцией через Т-клеточные рецепторы, и сниженной секреции различных цитокинов, таких как IL-2, IL-4, IL-5, IL-10 и IFN-γ (Schaeffer et al., Science 284, 638, 1999; Fowell et al., Immunity 11, 399, 1999; Schaffer et al., Nature Immunology 2, 1183, 2001).
Кроме того, у дефицитных по ITK мышей иммунные симптомы аллергической бронхиальной астмы были ослаблены, а легочное воспаление, эозинофильная инфильтрация и продукция слизи в ответ на провокацию аллергеном овальбумином были существенно снижены (Muller et al., Journal of Immunology 170, 5056, 2003). Это показывает, что ингибиторы ITK могли бы быть применимы для лечения астмы.
Кроме того, ПК также вовлечена в атопический дерматит. Сообщалось, что пациенты с тяжелым атопическим дерматитом имеют более высокий уровень экспрессии ее гена в Т-клетках периферической крови по сравнению с контрольными группами или с пациентами с легким атопическим дерматитом (Matsumoto et al., International archives of Allergy and Immunology 129, 327, 2002).
Между тем, RLK действует как активатор секреции IL-2, который продуцируется путем передачи сигналов через T-клеточные рецепторы спленоцитов. Таким образом, ингибирование RLK может снижать различные T-клеточные ответы (Schaeffer et al., Nature Immunology 2, 1183, 2001; Schaeffer et al., Science 284, 638, Кроме того, известно, что тирозинкиназа костного мозга (ВМХ) участвует в миграции эпителиальных и эндотелиальных клеток (Pan et al., Mol. Cell. Biol. 2002, 22, 7512). Поэтому ингибиторы BMK могут быть разработаны в качестве противораковых средств для ингибирования метастазирования раковых клеток и ангиогенеза.
Как указано выше, поскольку семейство TЕС киназ, таких как BTK, ITK, RLK, ВМХ и других, и Janus киназы, такие как JAK3, играют важнейшую роль в активации В-клеток и/или Т-клеток, которые вовлечены в патогенез воспалительных заболеваний, аутоиммунных заболеваний и иммунологически опосредованных заболеваний, соединение для эффективного ингибирования киназ может быть применимо в качестве терапевтического средства для различных воспалительных заболеваний, аутоиммунных заболеваний и иммунологически опосредованных заболеваний.
Кроме того, соединение для ингибирования BTK, вовлеченной в индуцирующую B-клеточную лимфому B-клеточную активацию, и BMX, вовлеченной в метастазирование раковых клеток, может быть применимо в качестве противоракового или противоопухолевого средства.
Таким образом, разработка соединения, которое может ингибировать вышеупомянутые киназы и селективно ингибировать вариантные EGFR, такие как при вторичных мутациях Т790М, а также при точечной мутации L858R в 21 экзоне или делеции внутри рамки считывания в 19 экзоне, представляет собой одну из наиболее важных задач.
Несмотря на то, что было высказано предположение, что необратимые ингибиторы EGFR, которые формируют ковалентную связь с расположенным на АТР домене EGFR цистеином773 (Cys773), могут проявлять ингибирующие эффекты в отношении семейства ТЕС киназ, таких как ВТК, ITK, RLK и ВМХ, в которых цистеин находится в том же положении аминокислотной последовательности, а также киназ, таких как JAK3 или BLK (Wooyoung Hur, et al., Bioorg. Med. Chem. Lett. 18, 5916, 2008), соединение, которое может необратимо, селективно и эффективно ингибировать различные EGFR, ВТК, JAK3, ITK, RLK, ВМХ и/или BLK, разработано не было.
КРАТКОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Поэтому целью настоящего изобретения является представление нового конденсированного пиримидинового производного, которое селективно и эффективно ингибирует рак или опухоли, индуцированные тирозинкиназой рецепторов эпидермального фактора роста (EGFR) или ее мутантной формой, со сниженными нежелательными побочными эффектами.
Другой целью настоящего изобретения является представление нового конденсированного пиримидинового производного, которое может лечить рак, опухоли, воспалительные заболевания, аутоиммунные заболевания или иммунологически опосредованные заболевания, опосредованные аномально активированными В-лимфоцитами, Т-лимфоцитами или обоими, посредством подавления нерецепторных тирозинкиназ, таких как семейство ТЕС киназ (например, BTK, ITK, BMX или RLK) и Janus киназ (например, JAK3).
Еще одной целью настоящего изобретения является представление фармацевтической композиции для профилактики или лечения рака, опухолей, воспалительных заболеваний, аутоиммунных заболеваний или иммунологически опосредованных заболеваний, содержащей упомянутое новое производное конденсированного пиримидина.
Согласно одному аспекту настоящего изобретения, представлено соединение формулы (I) или его фармацевтически приемлемая соль:

где
W представляет собой О или S;
X представляет собой О, NH, S, SO или SO2;
Y представляет собой атом водорода, атом галогена, С1-6алкил или C1-6алкокси;
каждый из А и В независимо представляет собой атом водорода, атом галогена или ди(С1-6алкил)аминометил;
Z представляет собой арил или гетероарил, содержащий один или несколько заместителей, выбранных из группы, состоящей из атома водорода, атома галогена, гидрокси, нитро, циано, C1-6алкила, C1-6алкокси, C1-6алкилкарбонила, C1-6алкоксикарбонила, ди(С1-6алкил)аминоС2-6алкоксикарбонила, амино, C1-6алкиламино, ди(С1-6алкил)амино, карбамоила, C1-6алкилкарбамоила, ди(С1-6алкил)карбамоила, ди(С1-6алкил)аминоС2-6алкилкарбамоила, сульфамоила, C1-6алкилсульфамоила, ди(С1-6алкил)сульфамоила, ди(С1-6алкил)аминоС2-6алкилсульфамоила, C1-6алкилсульфонила, C1-6алкилсульфинила, ди(С1-6алкил)фосфонила, гидроксиС1-6алкила, гидроксикарбонилС1-6алкила, С1-6алкоксиС1-6алкила, C1-6алкилсульфонилС1-6алкила, С1-6алкилсульфинилС1-6алкила, ди(С1-6алкил)фосфонилС1-6алкила, гидроксиС2-6алкокси, С1-6алкоксиС2-6алкокси, аминоС1-6алкила, С1-6алкиламиноС1-6алкила, ди(С1-6алкил)аминоС1-6алкила, ди(С1-6алкил)аминоацетила, аминоС2-6алкокси, С1-6алкиламиноС2-6алкокси, ди(С1-6алкил)аминоС2-6алкокси, гидроксиС2-6алкиламино, С1-6алкоксиС2-6алкиламино, аминоС2-6алкиламино, C1-6алкиламиноС2-6алкиламино, ди(С1-6алкил)аминоС2-6алкиламино, гетероарила, гетероцикла, гетероциклического окси, гетероциклического тио, гетероциклического сульфинила, гетероциклического сульфонила, гетероциклического сульфамоила, гетероциклического C1-6алкила, гетероциклического C1-6алкокси, гетероциклического амино, гетероциклического C1-6алкиламино, гетероциклического аминоС1-6алкила, гетероциклического карбонила, гетероциклического C1-6алкилкарбонила, гетероциклического карбонилС1-6алкила, гетероциклического C1-6алкилтио, гетероциклического C1-6алкилсульфинила, гетероциклического C1-6алкилсульфонила, гетероциклического аминокарбонила, гетероциклического C1-6алкиламинокарбонила, гетероциклического аминокарбонилС1-6алкила, гетероциклического карбоксамидо и гетероциклического C1-6алкилкарбоксамидо;
арил относится к C6-12циклическому или бициклическому ароматическому кольцу;
каждый из гетероарилов независимо относится к 5-12-членному циклическому или бициклическому ароматическому гетерокольцу, содержащему один или несколько N, О или S;
каждый из гетероциклов независимо относится к насыщенному или частично ненасыщенному 3-12-членному циклическому или бициклическому гетерокольцу, содержащему один или несколько N, О, S, SO или SO2, в котором атом углерода, образующий гетероцикл, необязательно содержит один или несколько заместителей, выбранных из группы, состоящей из C1-6алкила, гидрокси, гидроксиС1-6алкила, гидроксикарбонила, С1-6алкокси, амино, C1-6алкиламино, ди(С1-6алкил)амино, ди(C1-6алкил)аминоС1-6алкила, ди(С1-6алкил)аминокарбонила, гетероцикла, гетероциклического C1-6алкила и гетероарила, и в котором при условии, что гетероцикл необязательно включает атом азота, атом азота необязательно содержит заместитель, выбранный из группы, состоящей из атома водорода, C1-6алкила, моногалогенС1-6алкила, дигалогенС1-6алкила, тригалогенС1-6алкила, C3-6циклоалкила, гидроксиС2-6алкила, C1-6алкоксиС2-6алкила, C1-6алкилкарбонила, гидроксиС1-6алкилкарбонила, C1-6алкоксикарбонила, карбамоила, C1-6алкилкарбамоила, ди(C1-6алкил)карбамоила, сульфамоила, C1-6алкилсульфамоила, ди(C1-6алкил)сульфамоила, C1-6алкилсульфонила, аминоС2-6алкила, C1-6алкиламиноС2-6алкила, ди(С1-6алкил)аминоС2-6алкила, ди(С1-6алкил)аминоС1-6алкилкарбонила, гетероцикла, гетероциклического окси, гетероциклического тио, гетероциклического сульфинила, гетероциклического сульфонила, гетероциклического C1-6алкила, гетероциклического карбонила, гетероциклического C1-6алкилкарбонила, гетероциклического C1-6алкилсульфинила и гетероциклического C1-6алкилсульфонила (причем, если атом азота образует третичный амин, то он необязательно находится в форме N-оксида); и
необязательно, C1-6алкил является частично ненасыщенным или содержит С3-6циклоалкильный фрагмент, а атом углерода в гетероцикле находится в карбонильной форме.
В соответствии с другим аспектом настоящего изобретения, представлена фармацевтическая композиция для профилактики или лечения раков, опухолей, воспалительных заболеваний, аутоиммунных заболеваний или иммунологически опосредованных заболеваний, содержащая соединение формулы (I) или его фармацевтически приемлемую соль.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Представленные выше и другие цели и характерные черты настоящего изобретения станут очевидны из следующего ниже описания изобретения при рассмотрении вместе с сопроводительными чертежами, на которых соответственно представлено:
Фиг. 1: изменение размера опухолей у бестимусных мышей с ксенотрансплантированными раковыми клетками NCI-H1975 при пероральном введении соединения, полученного согласно примеру 2;
Фиг. 2: изменение массы тела у бестимусных мышей с ксенотрансплантированными раковыми клетками NCI-H1975 при пероральном введении соединения, полученного согласно примеру 2; и
Фиг. 3: изменение по шкале клинических показателей артрита в модели индуцированного коллагеном артрита (С1А) при пероральном введении соединения, полученного согласно примеру 1.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
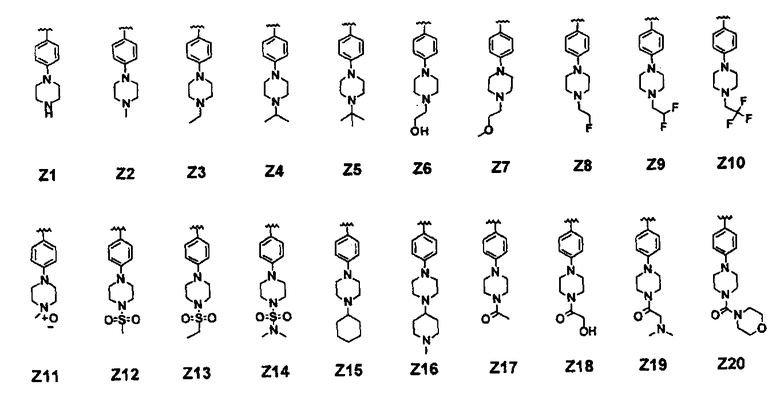
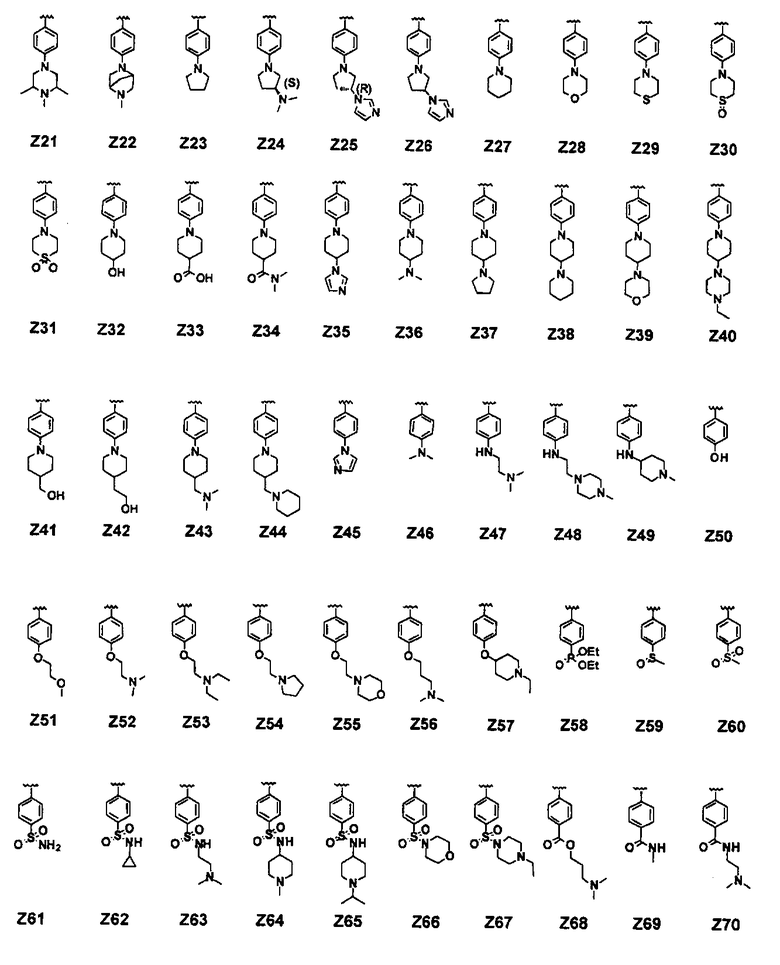
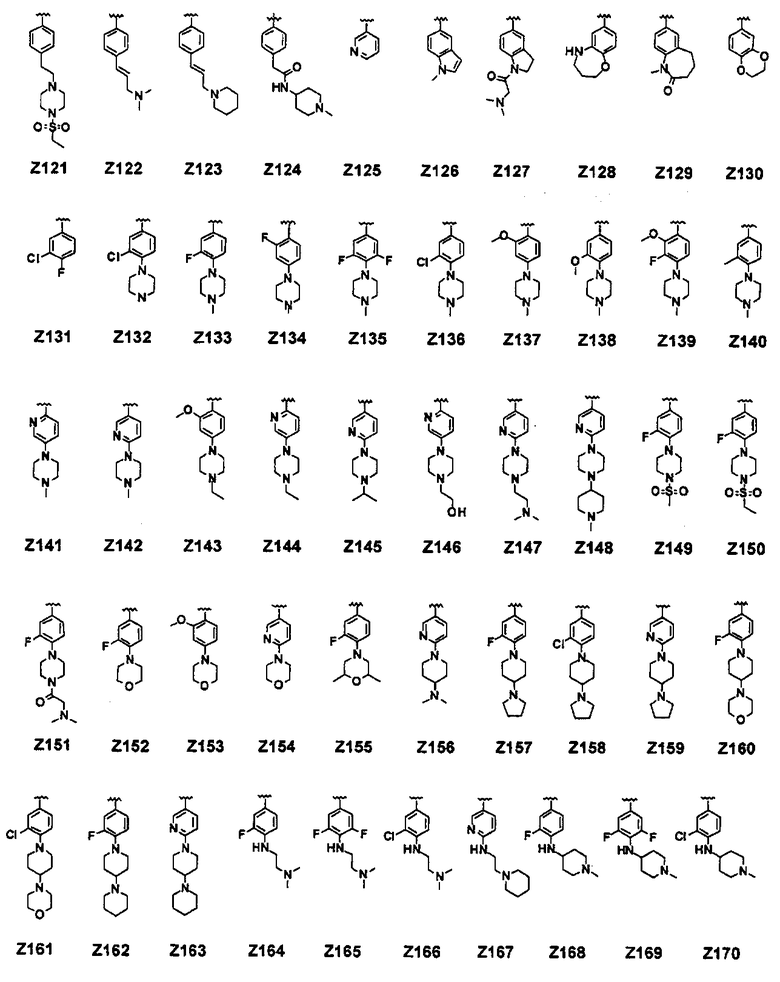
В соединении формулы (I) предпочтительные примеры Z включают заместители, выбранные из группы, состоящей из формул Z1-Z203, но они не ограничиваются ими:


Более предпочтительные примеры соединения формулы (I) согласно настоящему изобретению представляют собой следующее:
N-(3-(2-(2-метокси-4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(4-трет-бутилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(4-(2-фторэтил)пиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(4-(2,2,2-трифторэтил)пиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(4-(2-метоксиэтил)пиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(4-(2-гидроксиэтил)пиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(4-гидрокси-4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(3,4,5-триметилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(5-метил-2,5-диазабицикло[2.2.1]гепт-2-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(1-метил-2-оксо-2,3,4,5-тетрагидро-1Н-бензо[b]азепин-7-иламино)тиено[3,2-3]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(2-метокси-4-(1-метилпиперидин-4-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(2-метокси-4-(1-метилпиперидин-3-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(3-фтор-4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
диэтил(4-((4-(3-акриламидофенокси)тиено[3,2-d]пиримидин-2-ил)амино)фенил)фосфонат;
N-(3-(2-(4-[1,4′]бипиперидинил-1′-ил-3-фторфениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-((2-((3-хлор-4-(4-метилпиперазин-1-ил)фенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-(2-(4-(1-метилпиперидин-4-иламино)-3-хлорфениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(2-фтор-4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-б]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(3-метил-4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
4-((4-(3-акриламидофенокси)тиено[3,2-d]пиримидин-2-ил)амино)-2-метил-N-(1-метилпиперидин-4-ил)бензамид;
N-(4-метил-3-(2-(4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(4-фтор-3-(2-(4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(4-метокси-3-(2-(4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(5-(4-метилпиперазин-1-ил)пиридин-2-иламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
(4-(4-(3-акрилоиламинофенокси)тиено[3,2-d]пиримидин-2-иламино)фенил)амид 4-метилпиперазин-1-карбоновой кислоты;
N-(4-((4-(3-акриламидофенокси)тиено[3,2-d]пиримидин-2-ил)амино)-2-фторфенил)-4-метилпиперазин-1-карбоксамид;
N-(3-(2-(4-(4-этилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(4-изопропилпиперазин-1-ил)фениламино)тиено[3, d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(4-(2,2-дифторэтил)пиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-имидазол-1-ил-фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(пиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(4-(2-диметиламиноацетил)пиперазин-1-ил)-3-фторфениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(3-хлор-4-(пиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(4-(метилсульфонил)пиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(4-ацетилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(4-(морфолин-4-карбонил)пиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(1,4-диметил-3-оксопиперазин-2-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-морфолинофениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-((2-((4-((2-(диметиламино)этил)амино)фенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((4-((2-(4-метилпиперазин-1-ил)этил)амино)фенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-(2-(4-тиоморфолинфениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(1-оксо-1λ4-тиоморфолин-4-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
(S)-N-(3-(2-(4-(3-(диметиламино)пирролидин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(4-пирролидин-1-илпиперидин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-[1,4′]бипиперидинил-1′-илфениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
диметиламид 1-(4-(4-(3-акрилоиламинофенокси)тиено[3,2-d]пиримидин-2-иламино)фенил)пиперидин-4-карбоновой кислоты;
N-(3-(2-(4-(диметиламино)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(2-гидроксиэтил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(2-диметиламиноэтил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(3-хлор-4-фторфениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-гидроксифениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-((2-((4-ацетилфенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((4-(1,4,5,6-тетрагидропиримидин-2-ил)фенил)амино)тиено[3,2-d]пиримидин-4-ил)оксо)фенил)акриламид;
N-(3-(2-(3-фтор-2-метокси-4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(4-(4-этилпиперазин-1-ил)пиперидин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(3R-имидазол-1-илпирролидин-1-ил)фениламино]-тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(3-имидазол-1-илпирролидин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(4-имидазол-1-илпиперидин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(4-диметиламинопиперидин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси) фенил) акриламид;
N-(3-(2-(4-(4-морфолин-4-илпиперидин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(3-фтор-4-(4-пирролидин-1-илпиперидин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(3-фтор-4-(4-морфолин-4-илпиперидин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
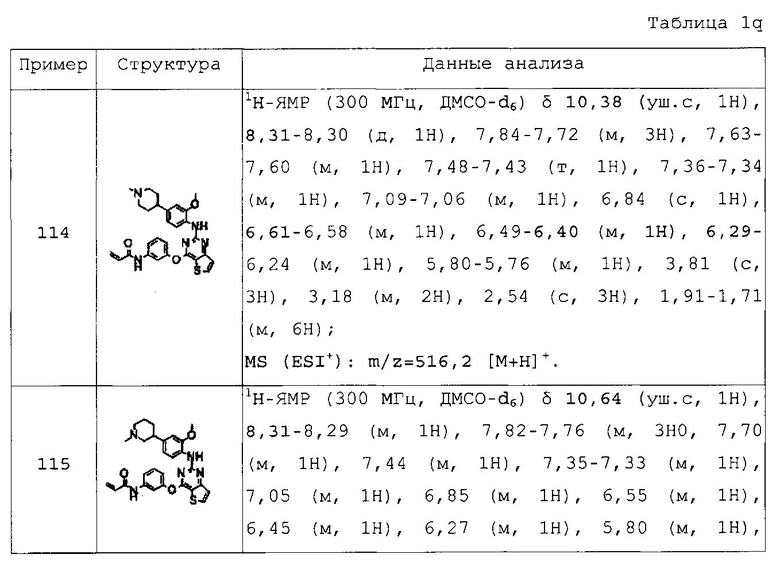
N-(3-(2-(3-хлор-4-(4-пирролидин-1-илпиперидин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(3-хлор-4-(4-морфолин-4-илпиперидин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
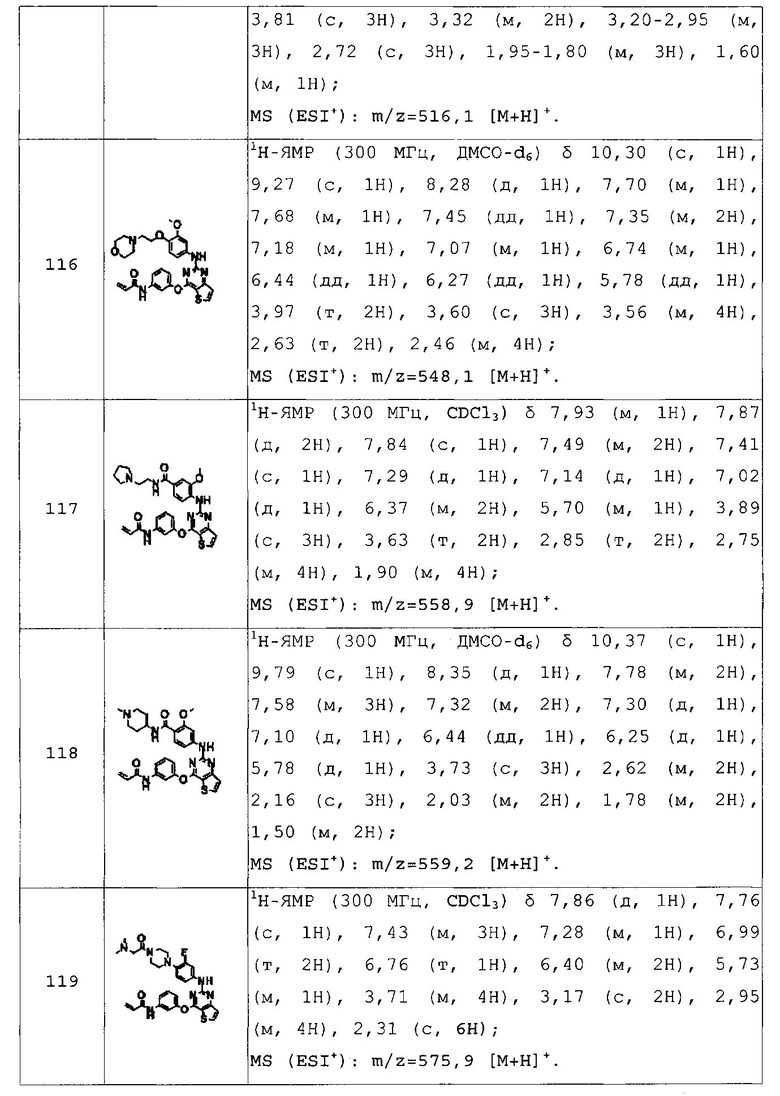
N-(3-(2-(4-(4-гидроксипиперидин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-((2-((4-(4-(гидроксиметил)пиперидин-1-ил)фенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((4-(4-(2-гидроксиэтил)пиперидин-1-ил)фенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-(2-(4-(4-(этилсульфонил)пиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-((4-этилпиперазин-1-ил)метил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-диэтиламинометилфениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(4-морфолин-4-илпиперидин-1-илметил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
(Е)-N-(3-((2-((4-(3-(диметиламино)проп-1-ен-1-ил)фенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((4-((1-метилпиперидин-4-ил)амино)фенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-(2-(4-диэтиламинометил-2-метоксифениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-((4-метилпиперазин-1-ил)метил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(3-фтор-4-(4-метилпиперазин-1-илметил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
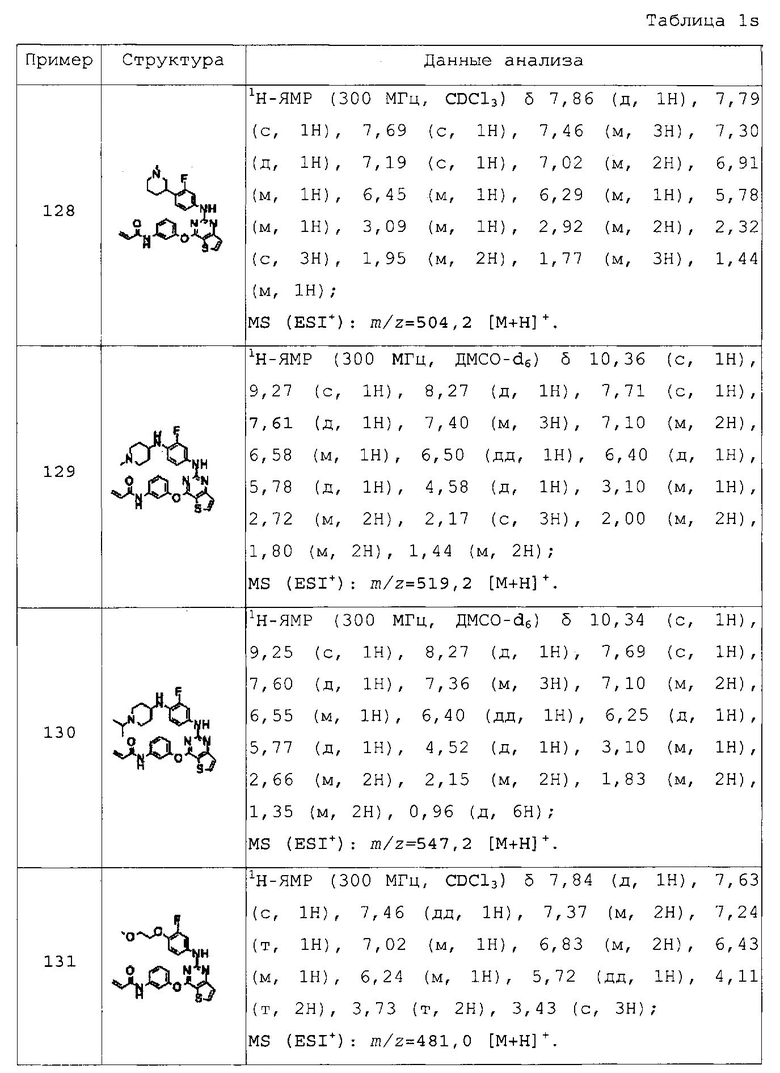
N-(3-(2-(4-(пиперидин-1-илметил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-азетидин-1-илметилфениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-пирролидин-1-илметилфениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(морфолинометил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-((2-((4-((3-(диметиламино)пирролидин-1-ил)метил)фенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((4-((4-гидроксипиперидин-1-ил)метил)фенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((4-((4-(диметиламино)пиперидин-1-ил)метил)фенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
диметил(4-((4-(3-акриламидофенокси)тиено[3,2-d]пиримидин-2-ил)амино)бензилфосфонат;
N-(3-(2-(4-((диметиламино)метил)-3-фторфениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-((3-(диметиламино)пирролидин-1-ил)метил)-3-фторфениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-((4-(диметиламино)пиперидин-1-ил)метил)-3-фторфениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-((1-метилпиперидин-4-иламино)метил)-3-фторфениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
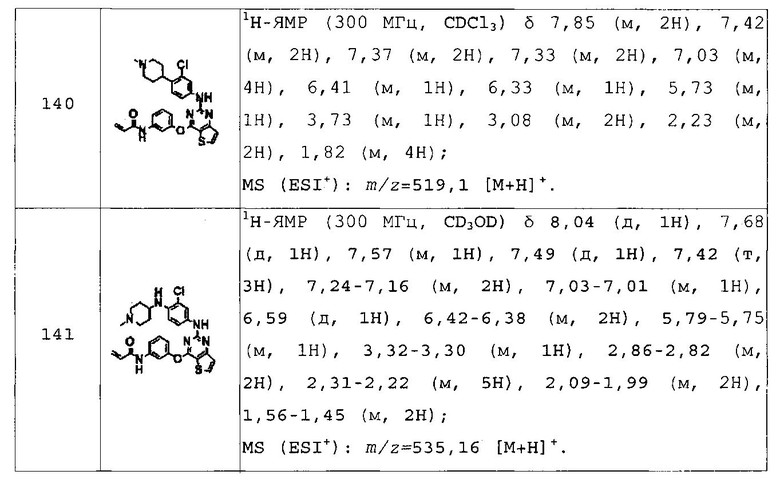
N-(3-(2-(4-диметиламинометил-2-метилфениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-((4-(циклопропилметил)пиперазин-1-ил)метил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-((4-(1-метилпиперидин-4-ил)пиперазин-1-ил)метил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-метансульфонилметилфениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(2-метансульфонилэтил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(3-хлор-4-(4-(1-метилпиперидин-4-ил)пиперазин-1-илметил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(4-(1-метилпиперидин-4-ил)пиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(4-циклогексилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(5-(4-этилпиперазин-1-ил)пиридин-2-иламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(5-(4-(2-гидроксиэтил)пиперазин-1-ил)пиридин-2-иламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(1-(4-этилпиперазин-1-ил)этил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(4-этилпиперазин-1-карбонил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(4-(2-гидроксиацетил)пиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(4-(2-диметиламиноацетил)пиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
2-(4-((4-(3-акриламидофенокси)тиено[3,2-d]пиримидин-2-ил)амино)фенил)уксусная кислота;
N-(3-((2-((4-(метилсульфинил)фенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((4-(метилсульфонил)фенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
4-((4-(3-акриламидофенокси)тиено[3,2-d]пиримидин-2-ил)амино)-N-метилбензамид;
4-((4-(3-акриламидофенокси)тиено[3,2-d]пиримидин-2-ил)амино)-N,N-диметилбензамид;
N-(3-((2-((4-(морфолин-4-карбонил)фенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((4-(4-метилпиперазин-1-карбонил)фенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-(2-(4-(4-(1-метилпиперидин-4-ил)пиперазин-1-карбонил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(4-гидроксипиперидин-1-карбонил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(3-метиламинопирролидин-1-карбонил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(3-диметиламинопирролидин-1-карбонил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
4-(4-(3-акрилоиламинофенокси)тиено[3,2-d]пиримидин-2-иламино)-N-(2-диметиламиноэтил)бензамид;
N-(3-(2-(3-хлор-4-(4-этилпиперазин-1-карбонил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-((2-((3-хлор-4-((2-(диметиламино)этил)амино)фенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
4-(4-(3-акрилоиламинофенокси)тиено[3,2-d]пиримидин-2-иламино)-2-хлор-N,N-диметилбензамид;
N-(3-(2-(3-хлор-4-(4-этансульфонилпиперазин-1-карбонил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
4-((4-(3-акриламидофенокси)тиено[3,2-d]пиримидин-2-ил)амино-2-хлор-N-(1-метилпиперидин-4-ил)бензамид;
N-(3-(2-(4-(4-этилпиперазин-1-илсульфонил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-((2-((4-((метилсульфинил)метил)фенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((4-(2-(метилсульфинил)этил)фенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((4-сульфамоилфенил)амино)тиено[3,2-бИпиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((4-(морфолиносульфонил)фенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((4-(N-циклопропилсульфамоил)фенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((4-(N-(2-(диметиламино)этил)сульфамоил)фенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((4-(N-(1-метилпиперидин-4-ил)сульфамоил)фенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((4-(N-(1-изопропилпиперидин-4-ил)сульфамоил)фенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
3-(диметиламино)пропил-4-((4-(3-акриламидофенокси)тиено[3,2-d]пиримидин-2-ил)амино)бензоат;
N-(3-(2-(4-(2-(4-этилпиперазин-1-ил)этил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(2-пиперидин-1-илэтил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(1,1-диоксо-1λ6-тиоморфолин-4-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(2-(4-этилпиперазин-1-ил)ацетил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(1-этилпиперидин-4-илокси)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(3-фтор-4-(1-метилпиперидин-4-илокси)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(2-морфолиноэтокси)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(2-метоксиэтокси)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-((2-((4-(2-(диметиламино)этокси)фенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((4-(2-(диэтиламино)этокси)фенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((4-(2-(пирролидин-1-ил)этокси)фенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((2,3,4,5-тетрагидробензо[b][1,4]оксазепин-7-ил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-(2-(2,3-дигидробензо[1,4]диоксин-6-иламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(3-фтор-4-(2-метоксиэтокси)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(2-диметиламиноэтокси)-3-фторфениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(2-диэтиламиноэтокси)-3-фторфениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(3-фтор-4-(2-(4-метилпиперазин-1-ил)этокси)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(3-метокси-4-(2-морфолин-4-илэтокси)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
(Е)-4-(диметиламино)-N-(3-(2-(4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)бут-2-енамид;
N-(3-(2-(4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-иламино)фенил)акриламид;
N-(3-(2-(4-(4-этилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-иламино)фенил)акриламид;
N-(3-(2-(4-(4-изопропилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-иламино)фенил)акриламид;
N-(3-(2-(4-(1-метилпиперидин-4-ил)фениламино)тиено[3,2-d]пиримидин-4-иламино)фенил)акриламид;
N-(3-(2-(4-(1-метилпиперидин-3-ил)фениламино)тиено[3,2-d]пиримидин-4-иламино)фенил)акриламид;
N-(3-(2-(4-диметиламинометилфениламино)тиено[3,2-d]пиримидин-4-иламино)фенил)акриламид;
N-(3-(2-(4-пиперидин-1-илметилфениламино)тиено[3,2-d]пиримидин-4-иламино)фенил)акриламид;
N-(3-(2-(4-(2-диметиламиноэтил)фениламино)тиено[3,2-d]пиримидин-4-иламино)фенил)акриламид;
N-(3-((2-((4-(2-(4-метилпиперазин-1-ил)этил)фенил)амино)тиено[3,2-d]пиримидин-4-ил)амино)фенил)акриламид;
N-(3-(2-(4-(2-диметиламиноэтокси)фениламино)тиено[3,2-d]пиримидин-4-иламино)фенил)акриламид;
N-(3-(2-(4-(3-диметиламинопропокси)фениламино)тиено[3,2-d]пиримидин-4-иламино)фенил)акриламид;
N-(3-(2-(3-фтор-4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-иламино)фенил)акриламид;
N-(3-(2-(3-фтор-4-(1-метилпиперидин-4-ил)фениламино)тиено[3,2-d]пиримидин-4-иламино)фенил)акриламид;
N-(3-(2-(3-фтор-4-(1-метилпиперидин-4-иламино)фениламино)тиено[3,2-d]пиримидин-4-иламино)фенил)акриламид;
N-(3-(2-(2-метокси-4-пиперидин-1-илметилфениламино)тиено[3,2-d]пиримидин-4-иламино)фенил)акриламид;
N-(4-фтор-3-(2-(4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-иламино)фенил)акриламид;
N-(4-фтор-3-(2-(3-фтор-4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-иламино)фенил)акриламид;
N-(3-(2-(4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илтио)фенил)акриламид;
N-(3-(2-(3-фтор-4-(1-метилпиперидин-4-ил)фениламино)тиено[3,2-d]пиримидин-4-илсульфанил)фенил)акриламид;
N-(3-(2-(3-фтор-4-морфолин-4-ил-фениламино)тиено[3,2-d]пиримидин-4-илсульфанил)фенил)акриламид;
(Е)-4-(диметиламино)-N-(3-(2-(4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илтио)фенил)бут-2-енамид;
N-(3-(2-(4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илсульфинил)фенил)акриламид;
(Z)-3-хлор-N-(3-(2-(4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
(E)-3-хлор-N-(3-(2-(4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(4-этилпиперазин-1-ил)-2-метоксифениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(2-метокси-4-морфолинофениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
4-((4-(3-акриламидофенокси)тиено[3,2-d]пиримидин-2-ил)амино)-2-метокси-N-(1-метилпиперидин-4-ил)бензамид;
N-(3-(2-(4-(пиперидин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(пирролидин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
1-(4-((4-(3-акриламидофенокси)тиено[3,2-d]пиримидин-2-ил)амино)фенил)пиперидин-4-карбоновую кислоту;
N-(3-(2-(4-(4-диметиламинометилпиперидин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(4-пиперидин-1-илметилпиперидин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(1-метил-1,2,3,6-тетрагидропиридин-4-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(1-метилпиперидин-4-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(1-этилпиперидин-4-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(1-изопропилпиперидин-4-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(1-метилпиперидин-3-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-диметиламинометилфениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(3-хлор-4-(1-метилпиперидин-4-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
4-(4-(3-акриламидофенокси)тиено[3,2-d]пиримидин-2-иламино)-N-(2-(пирролидин-1-ил)этил)бензамид;
N-(3-((2-((4-(2-((1-метилпиперидин-4-ил)амино)-2-оксоэтил)фенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-(2-(4-(3-пиперидин-1-илпропенил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(3-пирролидин-1-илпропиониламино)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
4-((4-(3-акриламидофенокси)тиено[3,2-d]пиримидин-2-ил)амино-N-(тетрагидро-2Н-пиран-4-ил)бензамид;
4-((4-(3-акриламидофенокси)тиено[3,2-d]пиримидин-2-ил)амино-N-(1-метилпиперидин-4-ил)бензамид;
4-((4-(3-акриламидофенокси)тиено[3,2-d]пиримидин-2-ил)амино)-N-(1-изопропилпиперидин-4-ил)бензамид;
4-(4-(3-акрилоиламинофенокси)тиено[3,2-d]пиримидин-2-иламино)-3-метокси-N-(2-пирролидин-1-илэтил)бензамид;
N-(3-(2-(4-(4-(N,N-диметилсульфамоил)пиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(2-(4-(этилсульфонил)пиперазин-1-ил)этил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(6-(4-метилпиперазин-1-ил)пиридин-3-иламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-((2-(пиридин-3-иламино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((6-морфолинопиридин-3-ил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((6-(4-изопропилпиперазин-1-ил)пиридин-3-ил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((6-(4-(1-метилпиперидин-4-ил)пиперазин-1-ил)пиридин-3-ил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((6-(4-(2-(диметиламино)этил)пиперазин-1-ил)пиридин-3-ил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((6-(4-(диметиламино)пиперидин-1-ил)пиридин-3-ил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((6-(4-(пирролидин-1-ил)пиперидин-1-ил)пиридин-3-ил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((6-([1,4′-бипиперидин]-1′-ил)пиридин-3-ил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((6-((4-метилпиперазин-1-ил)метил)пиридин-3-ил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((6-((2-(пиперидин-1-ил)этил)амино)пиридин-3-ил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((6-((1-изопропилпиперидин-4-ил)амино)пиридин-3-ил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((6-(метилсульфинил)пиридин-3-ил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-(2-(3-фтор-4-морфолинофениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-((2-((3-фтор-4-((1-метилпиперидин-4-ил)амино)фенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((3-фтор-4-((1-изопропилпиперидин-4-ил)амино)фенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-(2-(3-фтор-4-(4-(метилсульфонил)пиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(4-(этансульфонилапиперазин-1-ил)-3-фторфениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(4-(2,6-цис-диметилморфолино)-3-фторфениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(3-фтор-4-(1-метилпиперидин-4-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(3-фтор-4-(1-метилпиперидин-3-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(3-фтор-4-(2-морфолин-4-илэтокси)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-((2-((4-((2-(диметиламино)этил)амино)-3-фторфенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((3,5-дифтор-4-(4-метилпиперазин-1-ил)фенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((4-((2-(диметиламино)этил)амино)-3,5-дифторфенил)амино)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((3,5-дифтор-4-((1-метилпиперидин-4-ил)амино)фенил)тиено[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-(2-(4-(1-аминоциклопропил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-[1-(2-диметиламиноацетил)-2,3-дигидро-1Н-индол-5-иламино]тиено[3,2-d]пиримидин-4-илокси)фенил)акриламид;
N-(3-(2-(1-метил-1Н-индол-5-иламино)тиено[3,2-3]пиримидин-4-илокси)фенил)акриламид;
N-(3-((2-((4-(4-метилпиперазин-1-ил)фенил)амино)фуро[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((4-(4-изопропилпиперазин-1-ил)фенил)амино)фуро[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((4-морфолинофенил)амино)фуро[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((4-((диметиламино)метил)фенил)амино)фуро[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((4-((4-(диметиламино)пиперидин-1-ил)метил)фенил)амино)фуро[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((3-фтор-4-(1-метилпиперазин-4-ил)фенил)амино)фуро[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((4-(2-диметиламино)этил)амино)-3-фторфенил)амино)фуро[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-((2-((3-фтор-4-((1-метилпиперидин-4-ил)амино)фенил)амино)фуро[3,2-d]пиримидин-4-ил)окси)фенил)акриламид;
N-(3-(2-(3-метокси-4-(4-метилпиперазин-1-ил)фениламино)фуро[3,2-d]пиримидин-4-илокси)фенил)акриламид; и
N-(3-((2-((4-сульфамоилфенил)амино)фуро[3,2-d]пиримидин-4-ил)окси)фенил)акриламид.
Соединение формулы (I) согласно настоящему изобретению может быть получено согласно методике, представленной на реакционной схеме (I):
Реакционная схема (I)

где
А, В, W, X, Y и Z характеризуются теми же значениями, что и определенные выше;
R представляет собой водород, метил или этил; и
N′ представляет собой нитро или амин, защищенный трет-бутилоксикарбонилом (Boc).
Как представлено на реакционной схеме (I), соединение формулы (VIII) подвергают реакции конденсации с мочевиной в органическом растворителе (например, N,N-диметилформамид, N,N-диметилацетамид или N-метилпирролидон) при температуре в диапазоне от температуры возгонки до 200°C; или с цианатом калия в кислых условиях, таких как 6-50% водная уксусная кислота, при температуре в диапазоне от комнатной температуры до 100°C, с получением конденсированного соединения формулы (VII).
Полученное таким образом соединение формулы (VII) нагревали с обратным холодильником при перемешивании в присутствии хлорирующего агента (например, хлорангидрид фосфорной кислоты или тионилхлорид) с получением хлорированного соединения формулы (VI), с последующим проведением реакции в органическом растворителе (например, диметилсульфоксид, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, ацетонитрил, тетрагидрофуран, 1,4-диоксан, толуол или бензол) в присутствии неорганического основания (например, карбонат цезия, карбонат натрия или карбонат калия) при температуре в диапазоне от комнатной температуры до 100°C, индуцируя замещение в положении С-4 соединения формулы (VI) производным анилина, фенола или тиофенола формулы (V) с получением соединения формулы (IV).
Осуществляют взаимодействие соединения формулы (IV) с Z-NH2 в спиртовом растворе (например, 2-пропанол или 2-бутанол) в присутствии неорганической кислоты (например, соляная кислота) или органической кислоты (например, трифторуксусная кислота) при температуре в диапазоне от 70°C до температуры возгонки; или с Z-NH2 в органическом растворителе (например, 1,4-диоксан) в присутствии палладиевого катализатора (например, ацетат палладия(II) или трис(дибензилиденацетон)дипалладий(0)), и в присутствии лиганда (например, бис(дифенилфосфино)(ксантен) (Xantphos) или 2,2′-бис(дисфенилфосфино)-1,1′-бинафтил (BINAP)) и неорганического основания (например, карбонат цезия или трет-бутоксид натрия) при температуре приблизительно 100°C, с получением соединения формулы (III), содержащего группу Z-NH2.
Соединение формулы (III), в котором N′ представляет собой нитрогруппу, подвергают гидрированию с применением в качестве катализатора палладированного угля, или реакции восстановления, опосредованной Fe, с получением анилинового соединения формулы (II), нитрогруппа которого замещена аминогруппой. Соединение формулы (III), в котором N′ представляет собой аминогруппу, защищенную трет-бутилоксикарбонилом (Boc), подвергают взаимодействию с кислотой (например, трифторуксусная кислота или соляная кислота) в органическом растворителе (например, метиленхлорид), с получением анилинового соединения формулы (II) со снятыми защитными группами.
Затем анилиновое соединение формулы (II) подвергают взаимодействию с акрилоилхлоридом, замещенным А и В, в органическом растворителе (например, метиленхлорид или тетрагидрофуран) или в смешанном растворителе, таком как 50% водный тетрагидрофуран, в присутствии неорганического основания (например, бикарбонат натрия) или органического основания (например, триэтиламин или диизопропилэтиламин) при низкой температуре в диапазоне от -10°C до 10°C; или с акриловой кислотой, замещенной А и В, в пиридине с использованием агента сочетания (например, 1-этил-3-(3-диметиламинопропил)карбодиимид (EDCI) или гексафторфосфатметанамин 2-(1Н-7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония (HATU)), с получением соединения формулы (I) согласно настоящему изобретению, содержащего акриламидную группу.
Соединение формулы (I) согласно настоящему изобретению также может быть получено в форме фармацевтически приемлемой соли, образованной с неорганической или органической кислотой, такой как соляная кислота, бромистоводородная кислота, серная кислота, фосфорная кислота, азотная кислота, уксусная кислота, гликолевая кислота, молочная кислота, пировиноградная кислота, малоновая кислота, янтарная кислота, глутаровая кислота, фумаровая кислота, яблочная кислота, миндальная кислота, виннокаменная кислота, лимонная кислота, аскорбиновая кислота, пальмитиновая кислота, малеиновая кислота, гидроксималеиновая кислота, бензойная кислота, гидроксибензойная кислота, фенилуксусная кислота, коричная кислота, салициловая кислота, метансульфоновая кислота, бензолсульфоновая кислота и толуолсульфоновая кислота.
Фармацевтически приемлемая соль согласно настоящему изобретению может быть получена при помощи традиционных способов, например, посредством растворения соединения формулы (I) в смешиваемом с водой органическом растворителе, таком как ацетон, метанол, этанол и ацетонитрил, с добавлением избыточного количества органической кислоты или водного раствора неорганической кислоты, с индукцией осаждения солей из полученной смеси, с удалением растворителя и оставшейся в нем свободной кислоты и с выделением выпавших в осадок солей.
Соединение формулы (I) согласно настоящему изобретению или его фармацевтически приемлемая соль могут включать его гидрат и сольват.
Соответственно, настоящее изобретение относится к использованию соединения согласно настоящему изобретению для получения лекарственного средства для профилактики или лечения раков, опухолей, воспалительных заболеваний, аутоиммунных заболеваний или иммунологически опосредованных заболеваний.
Кроме того, настоящее изобретение относится к фармацевтической композиции для профилактики или лечения раков, опухолей, воспалительных заболеваний, аутоиммунных заболеваний или иммунологически опосредованных заболеваний, содержащей соединение согласно настоящему изобретению в качестве действующего ингредиента.
Кроме того, настоящее изобретение относится к способу профилактики или лечения раков, опухолей, воспалительных заболеваний, аутоиммунных заболеваний или иммунологически опосредованных заболеваний, включающему введение соединения согласно настоящему изобретению нуждающемуся в этом млекопитающему.
Соединение согласно настоящему изобретению формулы (I) или его фармацевтически приемлемая соль селективно и эффективно ингибирует рост раковых клеток, индуцированный тирозинкиназой рецепторов эпидермального фактора роста (EGFR) или ее мутантной формой, а также резистентность к действию лекарственных средств. Соответственно, настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль в качестве действующего ингредиента, для профилактики или лечения раков или опухолей, индуцированных тирозинкиназой EGFR или ее мутантной формой.
Типичные примеры раков или опухолей могут включать без ограничения рак печени, гепатоцеллюлярную карциному, рак щитовидной железы, колоректальный рак, рак яичка, рак кости, рак полости рта, базалиому, рак яичников, опухоль головного мозга, карциному желчного пузыря, рак желчных протоков, рак головы и шеи, карциному мочевого пузыря, рак языка, рак пищевода, глиому, глиобластому, рак почки, злокачественную меланому, рак желудка, рак молочной железы, саркому, карциному гортани, рак матки, рак шейки матки, рак предстательной железы, рак прямой кишки, рак поджелудочной железы, рак легких, рак кожи и другие солидные злокачественные опухоли.
Соединение согласно настоящему изобретению формулы (I) или его фармацевтически приемлемая соль могут обеспечить усиленные противораковые эффекты при введении их в сочетании с другим противораковым средством для лечения раков или опухолей.
Типичные примеры противораковых средств для лечения раков или опухолей могут включать без ограничения ингибиторы передачи внутриклеточных сигналов (например, иматиниб, гефитиниб, бортезомиб, эрлотиниб, сорафениб, сунитиниб, дасатиниб, вориностат, лапатиниб, темсиролимус, нилотиниб, эверолимус, пазопаниб, трастузумаб, бевацизумаб, сетуксимаб, ранибизумаб, пегаптаниб, панитумумаб и т.п.), ингибиторы митоза (например, паклитаксел, винкристин, винбластин и т.п.), алкилирующие агенты (например, цисплатин, циклофосфамид, хромабуцил, кармустин и т.п.), антиметаболиты (например, метотрексат, 5-FU и т.п.), интеркалирующие противораковые средства (актиномицин, антрациклин, блеомицин, митомицин С и т.п.), ингибиторы топоизомеразы (например, иринотекан, топотекан, тенипозид и т.п.), иммунотерапевтические средства (например, интерлейкин, интерферон и т.п.) и антигормональные средства (например, тамоксифен, ралоксифен и т.п.), и в фармацевтическую композицию согласно настоящему изобретению может быть включено, по меньшей мере, одно противораковое средство, выбранное из указанных выше.
Кроме того, соединение согласно настоящему изобретению формулы (I) или его фармацевтически приемлемая соль селективно и эффективно ингибирует тирозинкиназу Брутона (BTK), Janus киназу 3 (JAK3), индуцируемую интерлейкином-2 Т-клеточную киназу (ITK), киназу покоящихся лимфоцитов (RLK) и тирозинкиназу костного мозга (BMX), которые экспрессируются, главным образом, в аномально активированных В-лимфоцитах и/или Т-лимфоцитах. А именно, соединение согласно настоящему изобретению формулы (I) или его фармацевтически приемлемая соль может применяться для профилактики или лечения раков, опухолей, воспалительных заболеваний, аутоиммунных заболеваний или иммунологически опосредованных заболеваний, обусловленных аномально активированными В-лимфоцитами и/или Т-лимфоцитами. Поэтому настоящее изобретение также относится к фармацевтической композиции для профилактики или лечения раков, опухолей, воспалительных заболеваний, аутоиммунных заболеваний или иммунологически опосредованных заболеваний, содержащей соединение формулы (I) или его фармацевтически приемлемую соль в качестве действующего ингредиента.
Типичные примеры воспалительных заболеваний, аутоиммунных заболеваний и иммунологически опосредованных заболеваний могут включать без ограничения артрит, ревматоидный артрит, спондилоартропатию, подагрический артрит, остеоартрит, болезнь Стилла, другие артритические состояния, волчанку, системную красную волчанку (SLE), связанное с кожей заболевание, псориаз, экзему, дерматит, атопический дерматит, боль, заболевание легких, воспаление легких, респираторный дистресс-синдром у взрослых (ARDS), легочный саркоидоз, хроническое легочное воспалительное заболевание, хроническую обструктивную болезнь легких (COPD), сердечно-сосудистое заболевание, атеросклероз, инфаркт миокарда, застойную сердечную недостаточность, реперфузионное повреждение сердечной мышцы, воспалительное заболевание кишечника, болезнь Крона, неспецифический язвенный колит, синдром раздраженной толстой кишки, бронхиальную астму, синдром Шегрена, аутоиммунный тиреоидит, крапивницу, рассеянный склероз, склеродермию, отторжение пересаженных органов, гетеротрансплантацию, геморрагическую пурпуру (ITP), болезнь Паркинсона, болезнь Альцгеймера, ассоциированное с диабетом заболевание, воспаление, воспаление тазовых органов, аллергический ринит, аллергический бронхит, аллергический синусит, лейкоз, лимфому, B-клеточную лимфому, T-клеточную лимфому, миелому, острый лимфоидный лейкоз (ALL), хронический лимфоидный лейкоз (CLL), острый миелоидный лейкоз (AML), хронический миелоидный лейкоз (CML), волосатоклеточный лейкоз, болезнь Ходжкина, неходжкинскую лимфому, множественную миелому, миелодиспластический синдром (MDS), миелопролиферативные неоплазмы (MPN), диффузную крупноклеточную B-клеточную лимфому и фолликулярную лимфому.
Соединение согласно настоящему изобретению формулы (I) или его фармацевтически приемлемая соль могут обеспечить усиленные терапевтические эффекты при введении их в сочетании с другим средством для терапии воспалительных заболеваний, аутоиммунных заболеваний и иммунологически опосредованных заболеваний.
Типичные примеры средств для терапии воспалительных заболеваний, аутоиммунных заболеваний и иммунологически опосредованных заболеваний могут включать без ограничения стероидные лекарственные средства (преднизон, преднизолон, метилпреднизолон, кортизон, гидроксикортизон, бетаметазон, дексаметазон и т.п.), метотрексаты, лефлуномиды, анти-TNFα средства (например, этанерцепт, инфликсимаб, адалимунаб и т.п.), ингибиторы кальциневрина (например, такролимус, пимекролимус и т.п.) и антигистаминные средства (например, дифенгидрамин, гидроксизин, лоратадин, эбастин, кетотифен, цетиризин, левоцетириризин, фексофенадин и т.п.), и в фармацевтическую композицию согласно настоящему изобретению может быть включено, по меньшей мере, одно терапевтическое средство, выбранное из указанных выше.
Соединение формулы (I) согласно настоящему изобретению или его фармацевтически приемлемая соль могут быть введены в качестве активного ингредиента перорально или парентерально в эффективном количестве в диапазоне приблизительно от 0,1 до 2000 мг/кг, предпочтительно от 1 до 1000 мг/кг, массы тела в сутки в случае млекопитающих, включая человека (с массой тела приблизительно 70 кг), однократной дозой или в количестве до 4 раздельных доз в сутки, или on/off schedule. Доза активного ингредиента может корректироваться в зависимости от различных значимых факторов, таких как состояние подлежащего лечению субъекта, тип и серьезность заболевания, скорость введения и точка зрения врача. В определенных случаях может быть приемлемо использование меньшей, чем указано выше, дозы. Количество, превышающее указанную выше дозу, может использоваться в случае, когда оно не вызывает пагубных побочных эффектов, и такое количество может быть введено раздельными дозами в течение суток.
Фармацевтическая композиция согласно настоящему изобретению может быть приготовлена в соответствии с любым из традиционных способов в форме таблетки, гранулы, порошка, капсулы, сиропа, эмульсии или микроэмульсии для перорального введения или для парентерального введения, включая внутримышечный, внутривенный или подкожный пути введения.
Фармацевтическая композиция согласно настоящему изобретению для перорального введения может быть приготовлена путем смешивания активного ингредиента с носителем, таким как целлюлоза, силикат кальция, кукурузный крахмал, лактоза, сахароза, декстроза, фосфат кальция, стеариновая кислота, стеарат магния, стеарат кальция, желатин, тальк, поверхностно-активное вещество, суспендирующий агент, эмульгатор или разбавитель. Примерами носителя, применяемого в инъецируемой композиции согласно настоящему изобретению, являются вода, солевой раствор, раствор глюкозы, раствор глюкозоподобного вещества, спирт, гликоль, эфир (например, полиэтиленгликоль 400), масло, жирная кислота, сложный эфир жирной кислоты, глицерид, поверхностно-активное вещество, суспендирующий агент или эмульгатор.
Настоящее изобретение будет далее описано и проиллюстрировано на представленных ниже примерах, которые, тем не менее, не предназначены для ограничения объема настоящего изобретения.
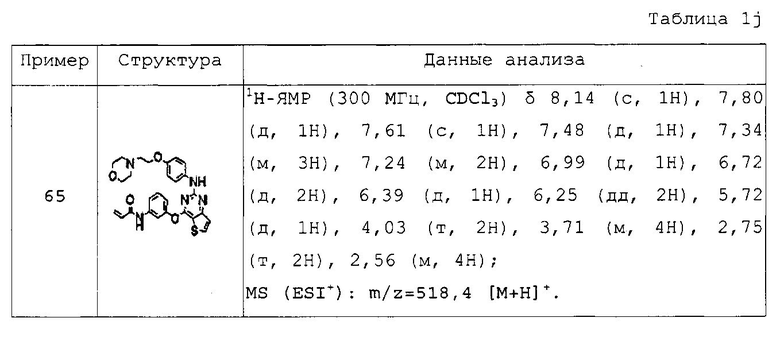
Пример 1: Получение N-(3-(2-(4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламида

Стадия 1) Получение тиено[3,2-d]пиримидин-2,4(1Н,3H)-диона

Метил-3-аминотиофен-2-карбоксилат (4,9 г, 31,3 ммоль) и мочевину (19 г, 187 ммоль) растворяли в N,N-диметилформамиде (10 мл), температуру реакционной смеси повышали до 190°C, а затем перемешивали в течение 12 часов. После завершения реакции реакционную смесь добавляли к 1н водному раствору NaOH, охлаждали до комнатной температуры и фильтровали в условиях пониженного давления для удаления нерастворимого осадка. Фильтрат подкисляли (pH 2) добавлением 2н водного раствора HCl, и фильтровали полученное твердое вещество в условиях пониженного давления с промыванием дистиллированной водой. Полученное твердое вещество сушили в условиях пониженного давления с получением указанного в заголовке соединения (выход: 3,2 г, 61,5%).
1Н-ЯМР (300 МГц, CDCl3) δ 11.59 (с, 1Н), 11.14 (с, 1Н), 8.00 (д, 1Н), 6.90 (д, 1Н).
Стадия 2) Получение 2, 4-дихлортиено[3,2-d] пиримидина
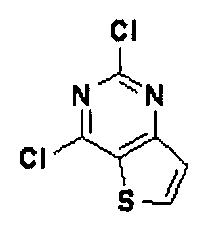
Соединение (3,2 г, 19,4 ммоль), полученное на стадии 1, растворяли в оксихлориде фосфора (12 мл),и нагревали с обратным холодильником при перемешивании в течение 3 часов при 200°C. После завершения реакции реакционную смесь охлаждали до комнатной температуры и по каплям добавляли при 4°C к дистиллированной воде при энергичном перемешивании. Полученное твердое вещество фильтровали в условиях пониженного давления с промыванием дистиллированной водой, и сушили полученное твердое вещество в условиях пониженного давления с получением указанного в заголовке соединения (выход: 2,9 г, 73,3%).
1H-ЯМР (300 МГц, ДМСО-d6] δ 8,74 (д, 1Н), 7,78 (д, 1Н).
Стадия 3) Получение 2-хлор-4-(3-нитрофенокси)тиено[3,2-d]пиримидина

Соединение (2,9 г, 14,2 ммоль), полученное на стадии 2, растворяли в N,N-диметилсульфонамиде (70 мл), добавляли к нему 3-нитрофенол (1,9 г, 14,2 ммоль) и карбонат цезия (9,2 г, 28,4 ммоль), а затем перемешивали при комнатной температуре в течение 1 часа. После завершения реакции к реакционной смеси добавляли дистиллированную воду, и фильтровали полученное твердое вещество в условиях пониженного давления при промывании дистиллированной водой. Полученное твердое вещество сушили в условиях пониженного давления с получением указанного в заголовке соединения (выход: 4,0 г, 91,8%).
1Н-ЯМР (300 МГц, CDCl3) δ 8,25-8,17 (м, 2Н), 8,08 (с, 1Н), 7,69-7,66 (м, 2Н), 7,57 (д, 1Н).
Стадия 4) Получение N-(2-метокси-4-(4-метилпиперазин-1-ил)фенил)-4-(3-нитрофенокси)тиено[3,2-d]пиримидин-2-амина

Соединение (4 г, 12,9 ммоль), полученное на стадии 3, растворяли в 2-бутаноле (70 мл), и добавляли 4-(4-метилпиперазин-1-ил)бензоламин (2,7 г, 12,9 ммоль) и трифторуксусную кислоту (1,5 мл, 12,9 ммоль). Смесь перемешивали при 100°C в течение 16 часов до завершения реакции, разбавляли дихлорметаном, а затем промывали насыщенным водным раствором NaHCO3. Органический слой сушили с безводным сульфатом натрия, а затем фильтровали и перегоняли в условиях пониженного давления. Остаток разделяли методом колоночной хроматографии (дихлорметан/метанол = 20/1 (соотношение объемов)) с получением указанного в заголовке соединения (выход: 2,67 г, 42%).
1Н-ЯМР (300 МГц, CDCl3) δ 8,20 (с, 1Н), 7,91 (м, 1Н), 7,84 (д, 1Н), 7,66 (м, 2Н), 7,36 (с, 1Н), 7,26 (м, 2Н), 6,57 (д, 1Н), 6,29 (м, 1Н), 3,82 (с, 3H), 3,19 (м, 4Н), 2,62 (м, 4Н), 2,36 (с, 3H).
Стадия 5) Получение 4-(3-аминофенокси)-N-(2-метокси-4-(4-метилпиперазин-1-ил)фенил)тиено[3,2-d]пиримидин-2-амина

Железо (1,5 г, 27,1 ммоль) и 12н водный раствор HCl (0,18 мл, 2,17 ммоль) разбавляли 50% водным раствором этанола (30 мл), а затем перемешивали при 100°C в течение 10 мин. Соединение (2,67 г, 5,42 ммоль), полученное на стадии 4, растворяли в 50% водном растворе этанола (30 мл), затем добавляли в реакционный сосуд, в котором активировали железо, а затем перемешивали при 100°C в течение 1 часа. После завершения реакции реакционную смесь фильтровали через целит для удаления железа, и перегоняли фильтрат в условиях пониженного давления. Остаток перегоняли с дихлорметаном и промывали насыщенным водным раствором NaHCO3. Органический слой сушили с безводным сульфатом натрия, а затем фильтровали и перегоняли в условиях пониженного давления. Остаток разделяли методом колоночной хроматографии (дихлорметан/метанол = 10/1 (соотношение объемов)) с получением указанного в заголовке соединения (выход: 1,7 г, 67,8%).
1Н-ЯМР (300 МГц, CDCl3) δ 8,20 (с, 1Н), 7,91 (м, 1Н), 7,84 (д, 1Н), 7,66 (м, 2Н), 7,36 (с, 1Н), 7,26 (м, 2Н), 6,57 (д, 1Н), 6,29 (м, 1Н), 3,82 (с, 3Н), 3,19 (м, 4Н), 2,62 (м, 4Н), 2,36 (с, 3Н).
Стадия 6) Получение N-(3-(2-(2-метокси-4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламида
Соединение (1,7 г, 3,69 ммоль), полученное на стадии 5, и NaHCO3 (930 мг, 11,07 ммоль) перегоняли с тетрагидрофураном (40 мл) и дистиллированной водой (6 мл), и медленно в течение 15 мин при перемешивании при 0°C добавляли акрилоилхлорид (0,36 мл, 3,69 ммоль). После завершения реакции реакционную смесь перегоняли с дихлорметаном, а затем промывали насыщенным водным раствором NaHCO3. Органический слой сушили с безводным сульфатом натрия, затем фильтровали и перегоняли в условиях пониженного давления, и разделяли остаток методом колоночной хроматографии (хлороформ/метанол = 20/1 (соотношение объемов)) с получением указанного в заголовке соединения (выход: 1,3 г, 68,2%).
1Н-ЯМР (300 МГц, CDCl3) δ 7,96 (м, 1Н), 7, 83 (д, 1Н), 7,70 (д, 1Н), 7,61 (с, 1Н), 7,45 (м, 2Н), 7,25 (м, 2Н), 7,01 (м, 1Н), 6,45 (д, 1Н), 6,35-6,32 (м, 3Н), 5,71 (дд, 1Н);
MS (ESI+): m/z=517,1 [М+Н]+.
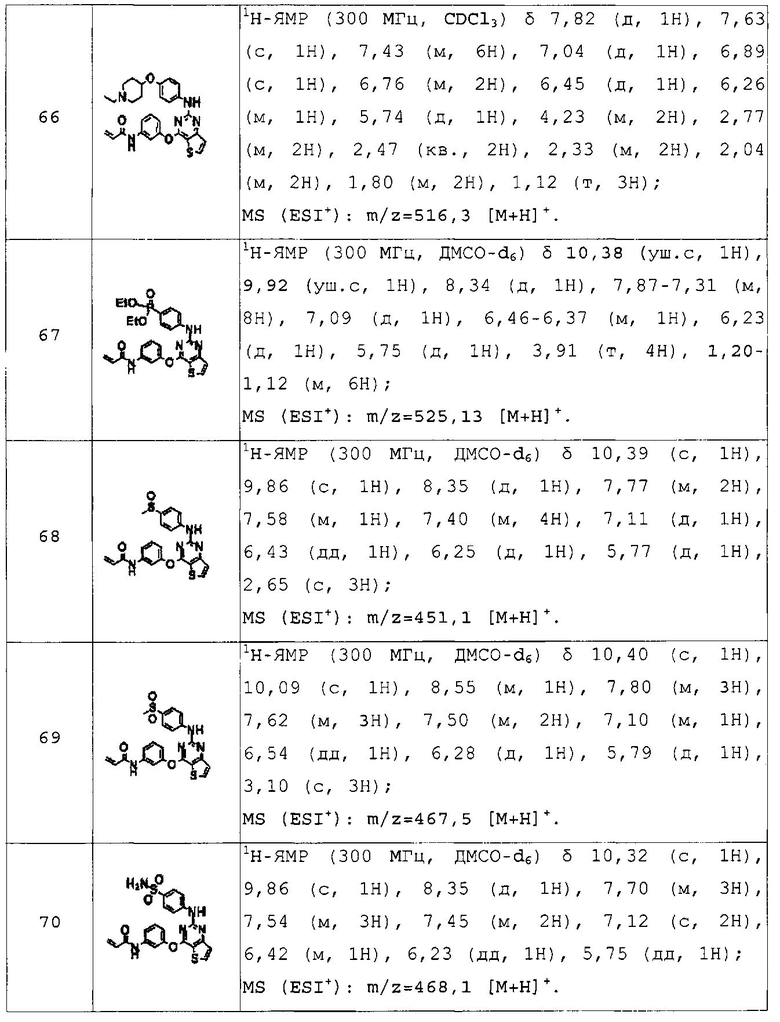
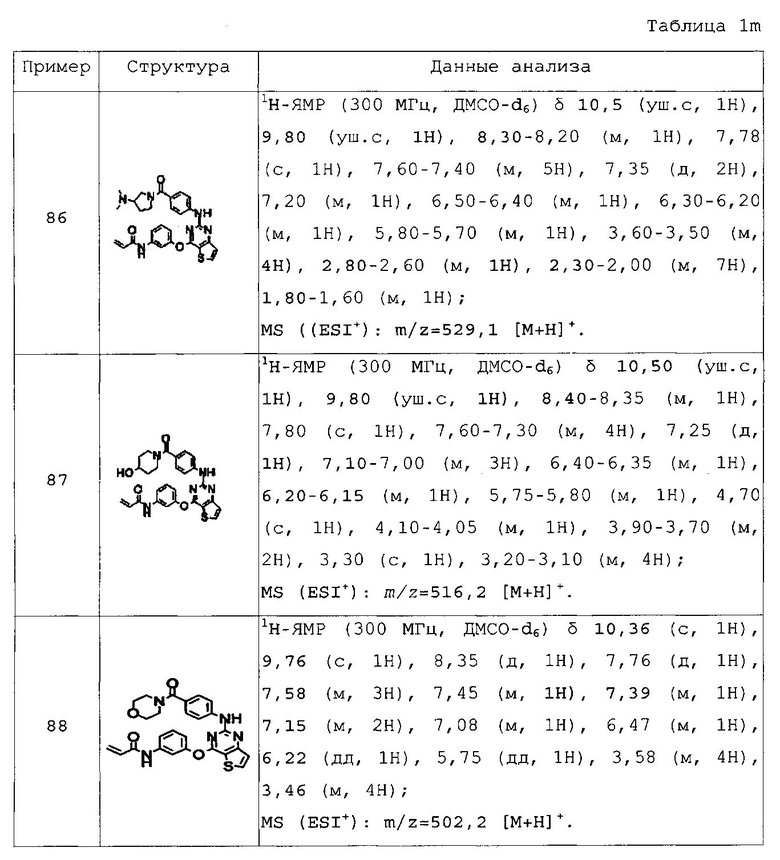
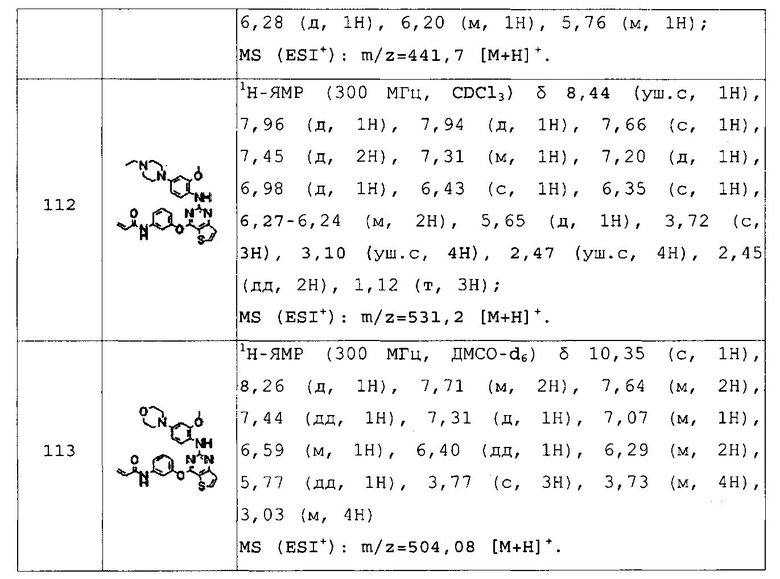
Для получения представленных ниже в таблицах 1a-1v соединений примеров 2-156 повторяли методику, описанную в примере 1, за исключением того, что вместо 4-(4-метилпиперазин-1-ил)бензоламина на стадии 4 использовали различные аминопроизводные, представленные формулой Z-NH2 (значение Z определено выше).










































Пример 157: Получение N-(3-(2-(4-(4-метил-4-оксипиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламида
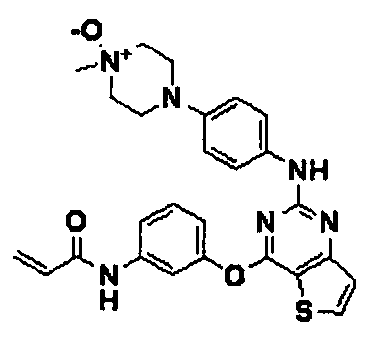
Соединение (100 мг, 0,21 ммоль), полученное согласно примеру 1, растворяли в дихлорметане (2 мл), и добавляли мета-хлорпероксибензойную кислоту (71 мг, 0,42 ммоль), а затем перемешивали при 45°C в течение 12 часов. После завершения реакции реакционную смесь разбавляли дихлорметаном и промывали насыщенным водным раствором NaHCO3. Органический слой сушили с безводным сульфатом натрия, а затем фильтровали и перегоняли в условиях пониженного давления, и разделяли остаток методом колоночной хроматографии (хлороформ с аммиаком/метанол = 4/1 (соотношение объемов)) с получением указанного в заголовке соединения (выход: 25 мг, 40%).
1Н-ЯМР (300 МГц, ДМСО-d6) δ 10,38 (с, NH), 9,27 (с, NH), 8,28 (д, 1Н), 7,74 (с, 1Н), 7,60 (д, 1Н), 7,46 (м, 3Н), 7,33 (д, 1Н), 7,05 (д, 1Н), 6,78 (д, 2Н), 6,43 (м, 1Н), 6,28 (м, 1Н), 5,76 (м, 1Н), 3,57 (м, 4Н), 2,98 (с, 3Н), 2,95 (м, 2Н), 2,50 (м, 2Н);
MS (ESI+): m/z=503,1 [M+H]+.
Пример 158: Получение N-(3-(2-(4-(пиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламида
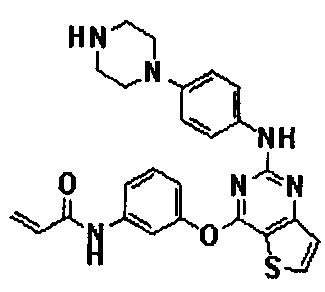
Стадия 1) Получение сложного трет-бутилового эфира 4-(4-(4-(3-акрилоиламинофенокси)тиено[3,2-d]пиримидин-2-иламино)фенил)пиперазин-1-карбоновой кислоты

Повторяли методику, описанную на стадии 4 примера 1, за исключением использования трет-бутил-4-(4-аминофенил)пиперазин-1-карбоксилата вместо 4-(4-метилпиперазин-1-ил)бензоламина с получением указанного в заголовке соединения (выход: 610 мг, 91%).
1Н-ЯМР (300 МГц, CDCl3) δ 7,82-7,80 (м, 1Н), 7,59-7,52 (м, 3Н), 7,43-7,34 (м, 3Н), 7,06-7,03 (м, 1Н), 6,92 (с, 1Н), 6,80-6,77 (м, 2Н), 6,47-6,41 (м, 1Н), 6,27-6,24 (м, 1Н), 5,79-5,75 (м, 1Н), 3,57 (м, 4Н), 3,02-2,99 (м, 4Н), 1,48 (с, 9Н).
Стадия 2) Получение N-(3-(2-(4-(пиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламида
Соединение (600 мг, 1,05 ммоль), полученное на стадии 1, растворяли в дихлорметане (10 мл), добавляли трифторуксусную кислоту (1,62 мл, 21,0 ммоль), а затем перемешивали при комнатной температуре в течение 1 часа. После завершения реакции реакционную смесь перегоняли в условиях пониженного давления для удаления растворителя, подщелачивали насыщенным водным раствором NaHCO3 (pH 8) и дважды экстрагировали хлороформом. Органический слой разделяли, промывали водой и насыщенным солевым раствором, сушили с безводным сульфатом натрия, а затем фильтровали и перегоняли в условиях пониженного давления. Остаток разделяли методом колоночной хроматографии (хлороформ/метанол = 10/1 (соотношение объемов)) с получением указанного в заголовке соединения (выход: 316 мг, 72%).
1Н-ЯМР (300 МГц, CDCl3) δ 10,28 (уш.с, 1Н), 9,15 (уш.с, 1Н), 8,26-8,24 (м, 1Н), 7,68 (с, 1Н), 7,62-7,59 (м, 1Н), 7,50-7,41 (м, 1Н), 7,31-7,29 (м, 1Н), 7,06-7,00 (м, 1Н), 6,74-6,71 (м, 2Н), 6,44-6,38 (м, 1Н), 6,27-6,21 (м, 1Н), 5,78-5,74 (м, 1Н), 3,31 (м, 4Н), 3,04-2,96 (м, 4Н);
MS (ESI+): m/z=473,4 [М+Н]+.
Повторяли методику, описанную в примере 158, за исключением использования на стадии 4 трет-бутил-4-(4-амино-2-хлорфенил)пиперазин-1-карбоксилата или сложного трет-бутилового эфира [1-(4-аминофенил)циклопропил]карбаминовой кислоты вместо трет-бутил-4-(4-аминофенил)пиперазин-1-карбоксилата, с получением соединений примеров 159 и 160, которые представлены ниже в таблице 2.

Пример 161: Получение (Z)-3-хлор-N-(3-(2-(4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламида
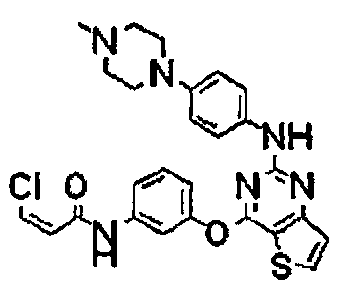
Соединение (50 мг, 0,12 ммоль), полученное на стадии 5 примера 1, растворяли в пиридине (1,5 мл), и добавляли цис-3-хлоракриловую кислоту (18 мг, 0,17 ммоль) и гидрохлорид N-(3-диметиламинопропил)-N′-этилкарбодиимида (44 мг, 0,23 ммоль), а затем перемешивали при комнатной температуре в течение 1 часа. После завершения реакции реакционную смесь разбавляли смешанным растворителем (хлороформ/2-пропанол = 3/1 (соотношение объемов)) и промывали насыщенным солевым раствором. Органический слой сушили с безводным сульфатом натрия, а затем фильтровали и перегоняли в условиях пониженного давления. Остаток разделяли методом колоночной хроматографии (дихлорметан/метанол = 6/1 (соотношение объемов)) с получением указанного в заголовке соединения (выход: 15 мг, 24%).
1Н-ЯМР (300 МГц, CDCl3) δ 8,24 (с, 1Н), 7,82 (д, 1Н), 7,62 (с, 1Н), 7,57 (д, 1Н), 7,44 (д, 1Н), 7,39 (д, 1Н), 7,35 (с, 1Н), 7,26 (д, 1Н), 7,08 (м, 1Н), 6,98 (с, 1Н), 6,81 (д, 2Н), 6,62 (д, 1Н), 6,34 (д, 1Н), 3,13 (т, 4Н), 2,59 (т, 4Н), 2,36 (с, 3Н);
MS (ESI+): m/z=521,4 [М+Н]+.
Повторяли методику, описанную в примере 161, за исключением использования транс-3-хлоракриловой кислоты и (Е)-4-(диметиламино)-2-бутеновой кислоты с получением соединений примеров 162 и 163, которые представлены ниже в таблице 3.


Пример 164: Получение N-(4-метил-3-(2-(4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламида

Осуществляли методику, аналогичную использованной в примере 1, за исключением использования на стадии 3 2-метил-5-нитрофенола (25 ммоль) вместо 3-нитрофенола, с получением указанного в заголовке соединения (30 мг, итоговый выход: 34%).
1Н-ЯМР (300 МГц, ДМСО-d6) δ 10,27 (с, 1Н), 9,21 (с, 1Н), 8,25 (д, 1Н), 7,62 (с, 1Н), 7,55 (д, 1Н), 7,33 (м, 4Н), 6,69 (м, 2Н), 6,39 (м, 1Н), 6,25 (м, 1Н), 5,75 (д, 1Н), 2,96 (м, 4Н), 2,42 (м, 4Н), 2,20 (с, 3Н), 2,07 (с, 3Н);
MS (ESI+): m/z=501,2 [M+H]+.
Осуществляли методику, аналогичную использованной в примере 164, за исключением использования 2-фтор-5-нитрофенола и 2-метокси-5-нитрофенола, с получением соединений примера 165 и примера 166, соответственно.

Пример 167: Получение N-(3-(2-(5-(4-метилпиперазин-1-ил)пиридин-2-иламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламида
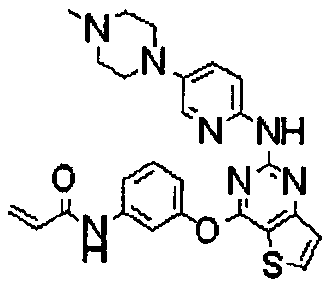
Стадия 1) Получение N-(5-(4-метилпиперазин-1-ил)пиридин-2-ил)-4-(3-нитрофенокси)тиено[3,2-d]пиримидин-2-амина
0,6 г (1,94 ммоль) соединения, полученного на стадии 3 примера 1, и 0,75 г (3,88 ммоль) 5-(4-метилпиперазин-1-ил)пиридин-2-амина растворяли в 8 мл 1,4-диоксана, добавляли 178 мг (0,2 ммоль) трис(дибензилиденацетон)дипалладия(0) и 122 мг (0,2 ммоль) 2,2′-бис(дифенилфосфино)-1,1′-бинафтил и перемешивали в течение 5 минут при комнатной температуре. Добавляли 1,27 г (3,88 ммоль) карбоната цезия и перемешивали в течение 3 часов при 100°C. После завершения реакции полученную смесь охлаждали до комнатной температуры, фильтровали через тонкий слой целита, разбавляли дихлорметаном и промывали водой. Органический слой разделяли, сушили над безводным Na2SO4, фильтровали и перегоняли в условиях пониженного давления. Полученный остаток разделяли методом колоночной хроматографии (дихлорметан/метанол (20/1, об./об.)) с получением 630 мг указанного в заголовке соединения (выход: 70%).
1Н-ЯМР (300 МГц, ДМСО-d6) δ 9,42 (с, 1Н), 8,33 (м, 2Н), 8,20 (м, 1Н), 7,91 (м, 2Н), 7,80 (м, 1Н), 7,59 (м, 1Н), 7,39 (м, 1Н), 7,05 (м, 1Н), 3,05 (м, 4Н), 2,49 (м, 4Н), 2,22 (с, 3Н).
Стадия 2) Получение N-(3-(2-(5-(4-метилпиперазин-1-ил)пиридин-2-иламино)тиено[3,2-d]пиримидин-4-илокси)фенил)акриламида
Последовательно повторяли методику, описанную на стадиях 5 и 6 примера 1, за исключением использования полученного на стадии 1 соединения (1,35 ммоль) вместо N-(4-(4-метилпиперазин-1-ил)фенил)-4-(3-нитрофенокси)тиено[3,2-d]пиримидин-2-амина, с получением 50 мг указанного в заголовке соединения (итоговый выход: 34%).
1Н-ЯМР (300 МГц, ДМСО-d6) δ 10,50 (с, 1Н), 9,37 (с, 1Н), 8,10 (д, 1Н), 7,90 (д, 1Н), 7,72 (м, 1Н), 7,64 (м, 2Н), 7,47 (дд, 1Н), 7,37 (д, 1Н), 7,09 (м, 2Н), 6,42 (дд, 1Н), 6,25 (дд, 1Н), 5,77 (дд, 1Н), 3,01 (м, 4Н), 2,42 (м, 4Н), 2,22 (с, 3H);
MS (ES+): m/z=488,3 [M+H]+.
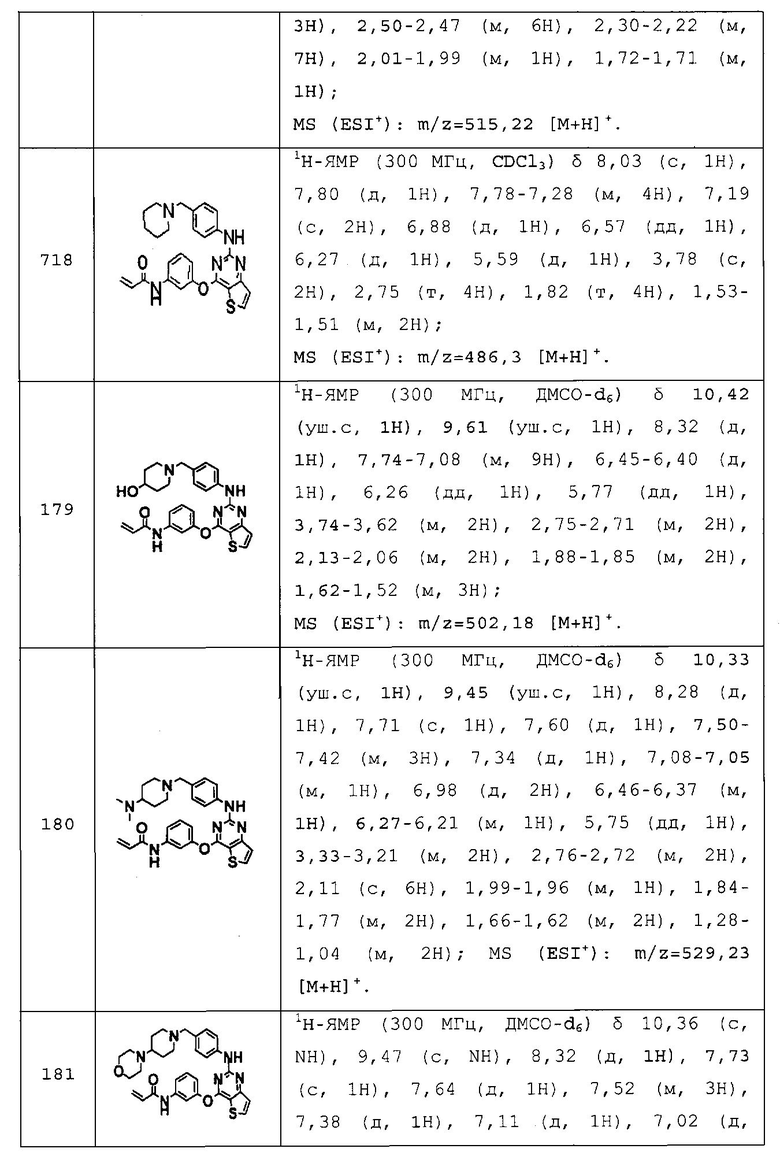
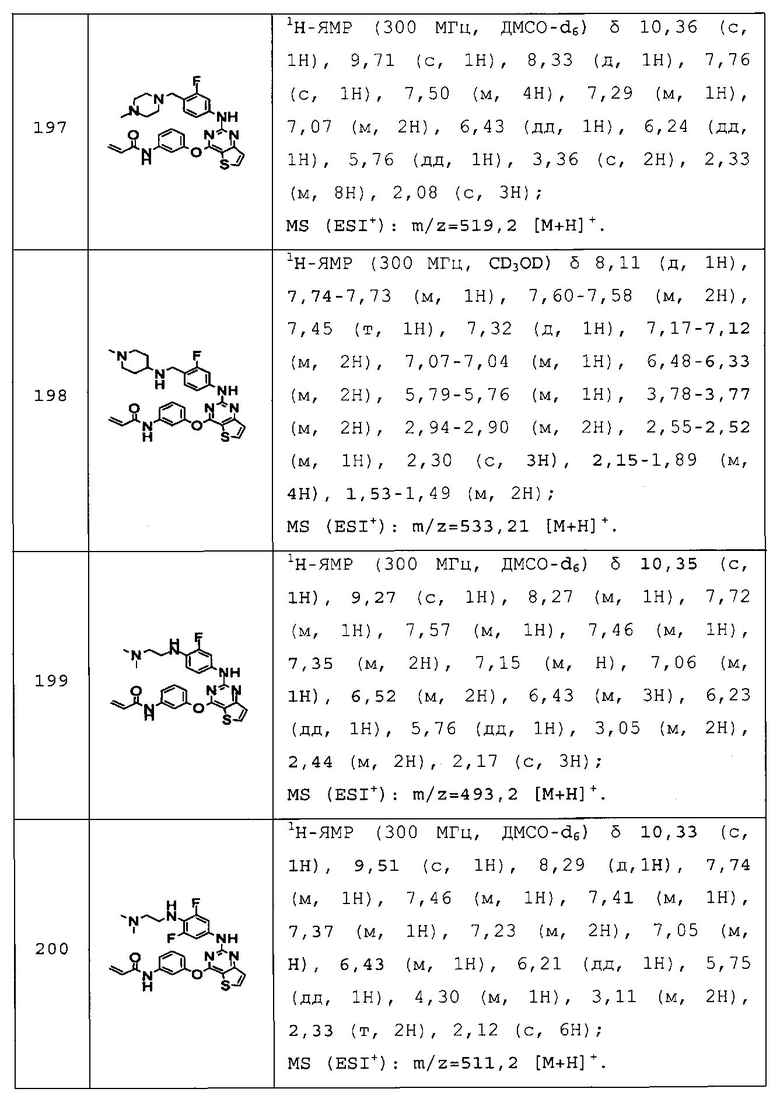
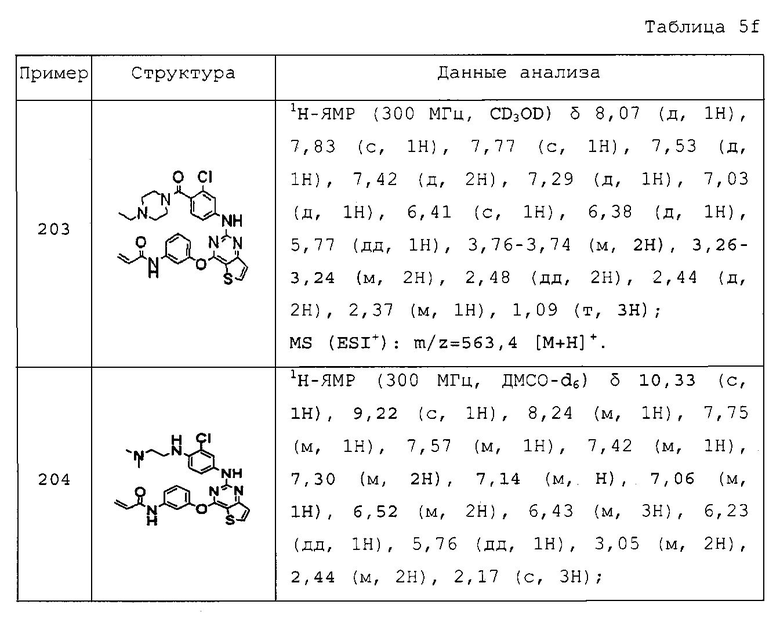
Повторяли методику, описанную в примере 167, или аналогичную методику за исключением использования на стадии 1 примера 167 различных аминопроизводных формулы Z-NH2 (Z имеет то же значение, что и определенное в настоящем изобретении) вместо 5-(4-метилпиперазин-1-ил)пиридин-2-амина, с получением указанных в заголовке соединений примеров 168-205, которые представлены в таблицах 5a-5f.











Пример 206: Получение N-(3-(2-(4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-иламино) фенил) акрил амида

Повторяли методику, описанную в примере 1, за исключением использования на стадии 3 примера 1 3-нитробензоламина (0,05 ммоль) вместо 3-нитрофенола, с получением 5 мг указанного в заголовке соединения (итоговый выход: 55%).
1Н-ЯМР (300 МГц, CDCl3) δ 8,10 (м, 1Н), 7,90 (д, 1Н), 7,51 (м, 3Н), 7,42 (м, 1Н), 7,28 (т, 1Н), 7,10 (д, 1Н), 6,89 (д, 2Н), 6,39 (м, 2Н), 5,79 (д, 1Н), 3,29 (м, 4Н), 2,68 (м, 4Н), 2,38 (с, 3Н);
MS (ESI+): m/z=486,2 [М+Н]+.
Повторяли методику, описанную в примере 206, или аналогичную методику за исключением использования в примере 1 различных аминопроизводных формулы Z-NH2 (Z имеет то же значение, что и определенное в настоящем изобретении) вместо 5-(4-метилпиперазин-1-ил)пиридин-2-амина, с получением указанных в заголовке соединений примеров 207-217, представленных в таблицах 6а и 6b.



Пример 218: Получение N-(4-фтор-3-(2-(4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-иламино)фенил)акриламида

Стадия 1) Получение N-(4-фтор-3-нитрофенил)акриламида

2 г (12,81 ммоль) 4-фтор-3-нитроанилина и 3,2 г (38,43 ммоль) бикарбоната натрия разбавляли 20 мл тетрагидрофурана и 5 мл дистиллированной воды, медленно при 0°C добавляли 1,14 мл (14,09 ммоль) акрилоилхлорида и перемешивали в течение 1 часа. После завершения реакции полученную смесь разбавляли этилацетатом и промывали насыщенным водным раствором бикарбоната натрия. Органический слой разделяли, сушили над безводным Na2SO4, фильтровали и перегоняли в условиях пониженного давления с получением 2 г указанного в заголовке соединения (выход: 74%).
1Н-ЯМР (300 МГц, ДМСО-d6) δ 10,58 (с, 1Н), 8,58 (м, 1Н), 7,91 (м, 1Н), 7,54 (т, 1Н), 6,35 (м, 2Н), 5,81 (м, 1Н).
Стадия 2) Получение N-(3-амино-4-фторфенил)акриламида

2,65 г (47,59 ммоль) железа и 0,31 мл (3,80 ммоль) 12н водной соляной кислоты разбавляли 40 мл 50% водного этанола и перемешивали в течение 1 часа при 100°C. Добавляли 2,00 г (9,51 ммоль) соединения, полученного на стадии 1, и перемешивали в течение 1 часа при 100°C. После завершения реакции полученную смесь фильтровали через тонкий слой целита для удаления железа и перегоняли в условиях пониженного давления. Полученный остаток разбавляли дихлорметаном и промывали насыщенным водным раствором бикарбоната натрия. Органический слой разделяли, сушили над безводным Na2SO4, фильтровали и перегоняли в условиях пониженного давления. Полученный остаток разделяли методом колоночной хроматографии (н-гексан/этилацетат (1/1, об./об.)) с получением 1,5 г указанного в заголовке соединения (выход: 75%).
1Н-ЯМР (300 МГц, ДМСО-d6) δ 9,87 (с, 1Н), 7,17 (м, 1Н), 6,89 (т, 1Н), 6,75 (м, 1Н), 6,39 (м, 1Н), 6,20 (м, 1Н), 5,70 (м, 1Н), 5,16 (с, 2Н).
Стадия 3) Получение N-(3-(2-хлортиено[3,2-d]пиримидйн-4-иламино)-4-фторфенил)акриламида

Соединение, полученное на стадии 2 примера 1, и 461 мг (2,22 ммоль) соединения, полученного на стадии 2, растворяли в 5 мл 1-пропанола, добавляли 0,6 мл (3,33 ммоль) диизопропилэтиламина и перемешивали в течение 24 часов при 110°C. После завершения реакции полученную смесь охлаждали до 0°C с образованием твердого вещества и фильтровали в условиях пониженного давления при промывании пропанолом. Полученное твердое вещество сушили в условиях пониженного давления с получением 270 мг указанного в заголовке соединения (выход: 36%).
1Н-ЯМР (300 МГц, ДМСО-d6) δ 10,31 (с, 1Н), 10,22 (с, 1Н), 8,25 (д, 1Н), 7,86 (м, 1Н), 7,59 (м, 1Н), 7,40 (д, 1Н), 7,32 (т, 1Н), 6,42 (м, 1Н), 6,29 (м, 1Н), 5,76 (м, 1Н).
Стадия 4) Получение N-(4-фтор-3-(2-(4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-иламино)фенил)акриламида
100 мг (0,30 ммоль) соединения, полученного на стадии 3, растворяли в 3 мл 2-бутанола, добавляли 55 мг (0,28 ммоль) 4-(4-метилпиперазин-1-ил)бензоламина и 42 мкл (0,57 ммоль) трифторуксусной кислоты и перемешивали в течение 5 часов при 100°C. После завершения реакции полученную смесь разбавляли этилацетатом и промывали насыщенным водным раствором бикарбоната натрия. Органический слой разделяли, сушили над безводным Na2SO4, фильтровали и перегоняли в условиях пониженного давления. Полученный остаток разделяли методом колоночной хроматографии (дихлорметан/метанол (10/1, об./об.)) с получением 77 мг указанного в заголовке соединения (выход: 50%).
1Н-ЯМР (300 МГц, ДМСО-d6) δ 10,26 (с, 1Н), 9,38 (с, 1Н), 8,77 (с, 1Н), 8,02 (д, 1Н), 7,82 (д, 1Н), 7,62 (м, 1Н), 7,44 (д, 2Н), 7,30 (т, 1Н), 7,15 (д, 1Н), 6,68 (м, 2Н), 6,40 (м, 1Н), 6,22 (м, 1Н), 5,73 (м, 1Н), 2,96 (м, 4Н), 2,42 (м, 4Н), 2,20 (с, 3Н); MS (ESI+): m/z=504,1 [М+Н]+.
Пример 219: Получение N-(4-фтор-3-(2-(3-фтор-4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-иламино)фенил)акриламида

Осуществляли методику, аналогичную описанной на стадии 4 примера 218, за исключением использования на стадии 4 примера 218 3-фтор-4-(4-метилпиперазин-1-ил)анилина (0,03 ммоль) вместо 4-(4-метилпиперазин-1-ил)бензоламина, с получением 8 мг указанного в заголовке соединения (итоговый выход: 50%).
1Н-ЯМР (300 МГц, ДМСО-d6) δ 10,25 (с, 1Н), 9,50 (с, 1Н), 9,08 (с, 1Н), 8,07 (д, 1Н), 7,85 (д, 1Н), 7,59 (м, 2Н), 7,26 (м, 2Н), 7,19 (д, 1Н), 6,78 (т, 1Н), 6,38 (м, 1Н), 6,27 (м, 1Н), 5,75 (м, 1Н), 2,87 (м, 4Н), 2,25 (м, 4Н), 2,21 (с, 3Н);
MS (ESI+): m/z=522,2 [М+Н]+.
Пример 220: Получение N-(3-(2-(4-диметиламинометилфениламино)тиено[3,2-d]пиримидин-4-иламино)фенил)акриламида
Осуществляли методику, аналогичную описанной на стадии 4 примера 218, за исключением использования 0,67 г (1,94 ммоль) N-(3-(2-хлортиено[3,2-d]пиримидин-4-иламино)фенил)акриламида, полученного на стадиях 1-3 примера 218, и 0,29 г (1,94 ммоль) 4-((диметиламино)метил)анилина с получением 0,69 г указанных в заголовке соединений (выход: 80%).
1Н-ЯМР (300 МГц, CDCl3) δ 8,11 (д, 2Н), 7,63 (дд, 3Н), 7,55 (м, 4Н), 7,18 (м, 2Н), 7,05 (с, 1Н), 6,45 (д, 1Н), 6,30 (кв., 1Н), 5,74 (д, 1Н), 3,38 (с, 2Н), 2,01 (с, 6Н);
MS (ESI+): m/z=467,l [М+Н]+.
Осуществляли методику, аналогичную описанной в примере 220, за исключением использования 4-(пиперидин-1-ил)метилфениламина и 2-метокси-4-(пиперидин-1-ил)метилфениламина с получением указанных в заголовке соединений примеров 221 и 222, представленных в таблице 7.

Пример 223: Получение N-(3-(2-(4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илтио)фенил)акриламида
Стадия 1) Получение трет-бутил-3-(2-хлортиено[3,2-d]пиримидин-4-илтио)фенилкарбамата

1,1 г (5,32 ммоль) соединения, полученного на стадии 2 примера 1, растворяли в 30 мл N,N-диметилсульфонамида, добавляли 1,2 г (5,32 ммоль) трет-бутил-3-меркаптофенилкарбамата и 3,4 г (10,6 ммоль) карбоната цезия и перемешивали в течение 1 часа при комнатной температуре. После завершения реакции к полученной смеси добавляли дистиллированную воду с получением твердого вещества, и фильтровали полученную смесь в условиях пониженного давления при промывании дистиллированной водой. Полученное твердое вещество сушили в условиях пониженного давления с получением 1,5 г указанного в заголовке соединения (выход: 70%).
1Н-ЯМР (300 МГц, CDCl3) δ 7, 92 (д, 1Н), 7,77 (с, 1Н), 7,56 (д, 1Н), 7,45-7,36 (м, 3H), 1,54 (с, 9Н).
Стадия 2) Получение трет-бутил-3-(2-(4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-йлтио)фенилкарбамата

1,5 г (3,72 ммоль) соединения, полученного на стадии 1, растворяли в 30 мл 2-бутанола и добавляли 0,8 г (3,72 ммоль) 4-(4-метилпиперазин-1-ил)бензоламина и 0,4 мл (3,72 ммоль) трифторуксусной кислоты. Смесь перемешивали в течение 10 часов при 100°C до завершения реакции, разбавляли дихлорметаном и промывали насыщенным водным раствором бикарбоната натрия. Органический слой сушили над безводным Na2SO4, фильтровали и перегоняли в условиях пониженного давления. Остаток разделяли методом колоночной хроматографии (дихлорметан/метанол (20/1, об./об.)) с получением 1,0 г указанного в заголовке соединения (выход: 46%).
1Н-ЯМР (300 МГц, CDCl3) δ 7,73 (д, 1Н), 7,63 (м, 1Н), 7,60 (м, 1Н), 7,39-7,30 (м, 2Н), 7,28-7,21 (м, 2Н), 7,15 (д, 1Н), 6,76 (д, 2Н), 3,25 (м, 4Н), 2,58 (м, 4Н), 2,33 (с, 3H), 1,54 (с, 9Н).
Стадия 3) Получение 4-(3-аминофенилтио)-N-(4-(4-метилпиперазин-1-ил)фенил)тиено[3,2-d]пиримидин-2-амина

1,0 г (1.82 ммоль) соединения, полученного на стадии 2, растворяли в 20 мл дихлорметана, добавляли 10 мл трифторуксусной кислоты и перемешивали в течение 2 часов при комнатной температуре. После завершения реакции полученную смесь перегоняли в условиях пониженного давления для удаления растворителя, полученный остаток подщелачивали (рН 8) насыщенным водным раствором бикарбоната натрия и экстрагировали хлороформом. Органический слой разделяли, сушили над безводным Na2SO4, фильтровали и перегоняли в условиях пониженного давления, и сушили с получением 603 мг указанного в заголовке соединения (выход: 75%).
1Н-ЯМР (300 МГц, CD3OD) δ 7,96 (д, 1Н), 7,33 (д, 2Н), 7,21 (т, 1Н), 7,17 (д, 1Н), 7,02 (м, 1Н), 6,94 (м, 2Н), 6,80 (д, 2Н), 3,14 (м, 4Н), 2,65 (м, 4Н).
Стадия 4) Получение N-(3-(2-(4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илтио)фенил)акриламида
Осуществляли методику, сходную с описанной на стадии 6 примера 1, за исключением использования соединения, полученного на стадии 3, вместо соединения, полученного на стадии 5, с получением 452 мг указанного в заголовке соединения (выход: 67%).
1Н-ЯМР (300 МГц, CDCl3) δ 7,78 (м, 1Н), 7,75 (д, 1Н), 7,46-7,41 (м, 3H), 7,20 (д, 2Н), 7,18 (д, 1Н), 6,77 (д, 2Н), 6,41 (д, 1Н), 6,21 (дд, 1Н), 5,78 (д, 1Н), 3,12 (м, 4Н), 2,60 (м, 4Н), 2,36 (с, 3H);
MS (ESI+): m/z=503,7 [М+Н]+.
Повторяли методику, описанную в примере 223, или аналогичную методику за исключением использования на стадии 2 примера 223 3-фтор-4-морфолин-4-илфениламина и 3-фтор-4-(1-метилпиперидин-4-ил)фениламина вместо 54-(4-метилпиперазин-1-ил)фениламина, с получением указанных в заголовке соединений примеров 224 и 225, представленных в таблице 8.
Пример 226: Получение (Е)-4-(диметиламино)-N-(3-(2-(4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илтио)фенил)бут-2-енамида
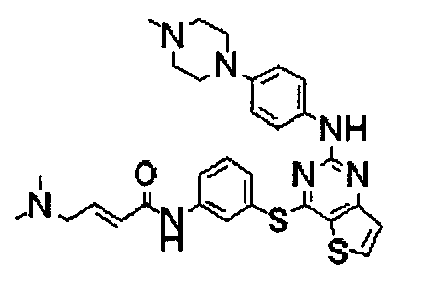
40 мг (0,09 ммоль) соединения, полученного на стадии 2 примера 223, растворяли в 1,5 мл пиридина, добавляли 22 мг (0,14 ммоль) гидрохлорида (Е)-4-(диметиламино)-2-бутеновой кислоты и 35 мг (0,18 ммоль) гидрохлорида N-(3-диметиламинопропил)-N′-этилкарбодиимида и перемешивали в течение 30 минут при 80°C. После завершения реакции полученную смесь разбавляли смесью хлороформ/2-пропанол (3/1 (об./об.)) и промывали насыщенным солевым раствором. Органический слой разделяли, сушили над безводным Na2SO4, фильтровали и перегоняли в условиях пониженного давления. Полученный остаток разделяли методом колоночной хроматографии (дихлорметан/метанол = 6/1 (об./об.)) с получением 2 мг указанного в заголовке соединения (выход: 4%).
1Н-ЯМР (300 МГц, CDCl3) δ 8,10 (м, 1Н), 8,02 (д, 1Н), 7,93 (с, 1Н), 7,50 (т, 1Н), 7,42 (м, 1Н), 7,21 (м, 3H), 6,90 (м, 1Н), 6,74 (д, 2Н), 6,28 (д, 1Н), 3,20 (д, 2Н), 3,10 (т, 4Н), 2,66 (т, 4Н), 2,39 (с, 3H), 2,17 (с, 6Н);
MS (ESI+): m/z=560,2 [М+Н]+.
Пример 227: Получение N-(3-(2-(4-(4-метилпиперазин-1-ил)фениламино)тиено[3,2-d]пиримидин-4-илсульфинил)фенил)акриламида

11 мг (0,02 ммоль) соединения, полученного согласно примеру 223, растворяли в 1,0 мл дихлорметана, добавляли 20 мг (0,04 ммоль) мета-хлорпероксибензойной кислоты и перемешивали в течение 60 минут при комнатной температуре. После завершения реакции полученную смесь разбавляли хлороформом и промывали насыщенным водным раствором бикарбоната натрия. Органический слой разделяли, сушили над безводным Na2SO4, фильтровали и перегоняли в условиях пониженного давления. Полученный остаток разделяли методом колоночной хроматографии (дихлорметан/метанол = 6/1 (об./об.)) с получением 3,0 мг указанного в заголовке соединения (выход: 25%).
1Н-ЯМР (300 МГц, CD3OD) δ 8,08 (м, 1Н), 8,01 (д, 1Н), 7,92 (м, 1Н), 7,51 (т, 1Н), 7,46 (м, 1Н), 7,22 (м, 3H), 6,73 (д, 1Н), 6,38 (м, 2Н), 5,76 (дд, 1Н), 3,63-3,56 (м, 4Н), 3,42-3,34 (м, 4Н), 3,23 (с, 3H);
MS (ESI+): m/z=519,3 [М+Н]+.
Пример 228: Получение N-(3-((2-((4-(4-метилпиперазин-1-ил)фенил)амино)фуро[3,2-d]пиримидин-4-ил)окси)фенил)акриламида
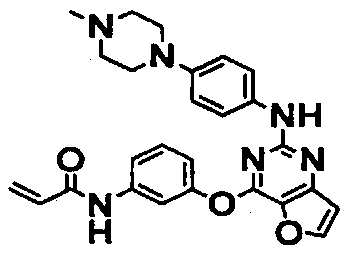
Стадия 1) Получение 2-хлор-4-(3-нитрофенокси)фуро[3,2-d]пиримидина
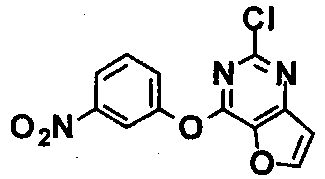
6,4 г (33,9 ммоль) 2,4-дихлорфуро[3,2-d]пиримидина (см. международные публикации WO 2008073785 и WO 2008152394) растворяли в 32 мл метанола, добавляли 5,7 г (40,6 ммоль) 3-нитрофенола и 12 мл (67,7 ммоль) диизопропилэтиламина и перемешивали в течение 24 часов при комнатной температуре. После завершения реакции полученное твердое вещество фильтровали и высушивали в условиях пониженного давления с получением 6,3 г указанного в заголовке соединения (выход: 64%).
1Н-ЯМР (300 МГц, ДМСО-d6) δ 8,61 (с, 1Н), 8,33 (с, 1Н), 8,21 (д, 1Н), 7,90 (д, 1Н), 7,79 (м, 1Н), 7,27 (с, 1Н).
Стадия 2) Получение N-[4-(4-метилпиперазин-1-ил)фенил]-4-(3-нитрофенокси)фуро[3,2-d]пиримидин-2-амина

2,5 г (8,6 ммоль) соединения, полученного на стадии 1, растворяли в 50 мл 2-бутанола и добавляли 2,0 г (10,3 ммоль) 4-(4-метилпиперазин-1-ил)анилина и 1,5 мл (8,6 ммоль) трифторуксусной кислоты. Реакционную смесь перемешивали в течение 12 часов при 100°C до завершения реакции, разбавляли дихлорметаном и промывали насыщенным водным раствором бикарбоната натрия. Органический слой разделяли, сушили над безводным Na2SO4, фильтровали и перегоняли в условиях пониженного давления и высушивали. Полученный остаток разделяли методом колоночной хроматографии (дихлорметан/метанол = 20/1 (об./об.)) с получением 2,0 г указанного в заголовке соединения (выход: 53%).
1Н-ЯМР (300 МГц, CDCl3) δ 8,20 (с, 2Н), 7,85 (с, 1Н), 7,64 (с, 2Н), 7,30 (с, 1Н), 6,79 (м, 4Н), 3,14 (м, 4Н), 2,60 (м, 4Н), 2,37 (с, 3H).
Стадия 3) Получение 4-(3-аминофенокси)-N-[4-(4-метилпиперазин-1-ил)фенил]фуро[3,2-d]пиримидин-2-амина

1,3 г (22,4 ммоль) железа и 2 мл 12н водной соляной кислоты разбавляли 10 мл 50% водного этанола и перемешивали в течение 10 минут при 100°C. 2,0 г (4,5 ммоль) соединения, полученного на стадии 2, растворяли в 10 мл 50% водного этанола, добавляли в сосуд с активированным железом и перемешивали в течение 1 часа при 100°C. После завершения реакции полученную смесь фильтровали через тонкий слой целита для удаления железа и перегоняли в условиях пониженного давления. Полученный остаток разбавляли дихлорметаном и промывали насыщенным водным раствором бикарбоната натрия. Органический слой разделяли, сушили над безводным Na2SO4, фильтровали и перегоняли в условиях пониженного давления с получением 1,8 г указанного в заголовке соединения (выход: 97%).
1Н-ЯМР (300 МГц, CDCl3) δ 7,79 (с, 1Н), 7,32 (д, 2Н), 7,24 (м, 1Н), 6,84 (м, 2Н), 6,75 (с, 1Н), 6,65 (м, 3H), 3,22 (м, 4Н), 2,60 (м, 4Н), 2,36 (с, 3H).
Стадия 4) Получение N-(3-{2-[4-(4-метилпиперазин-1-ил)фениламино]фуро[3,2-d]пиримидин-4-илокси}фенил)акриламида
1,8 г (4,3 ммоль) соединения, полученного на стадии 3, и 1,1 г (23,0 ммоль) бикарбоната натрия разбавляли 20 мл тетрагидрофурана и 5 мл дистиллированной воды, медленно при 0°C добавляли 0,4 мл (4,3 ммоль) акрилоилхлорида и перемешивали в течение 30 минут. После завершения реакции полученную смесь разбавляли дихлорметаном и промывали насыщенным водным раствором бикарбоната натрия. Органический слой разделяли, сушили над безводным Na2SO4, фильтровали и перегоняли в условиях пониженного давления и высушивали. Полученный остаток разделяли методом колоночной хроматографии (хлороформ/метанол = 20/1 (об./об.)) с получением 940 мг целевого соединения (выход: 46%).
1Н-ЯМР (300 МГц, CD3OD) δ 8,04 (с, 1Н), 7,68 (д, 2Н), 7,45 (т, 1Н), 7,32 (д, 2Н), 7,03 (д, 1Н), 6,78 (м, 3H), 6,45 (м, 2Н), 5,80 (д, 1Н), 3,08 (м, 4Н), 2,61 (м, 4Н), 2,35 (с, 3H);
MS (ESI+): m/z=470,2 [М+Н]+.
Повторяли методику, описанную в примере 228, или аналогичную методику за исключением использования на стадии 2 примера 228 различных аминопроизводных формулы Z-NH2 (Z имеет то же значение, что и определенное в настоящем изобретении) вместо 4-(4-метилпиперазин-1-ил)бензоламина, с получением указанных в заголовке соединений примеров 229-237, представленных в таблицах 9а и 9b.
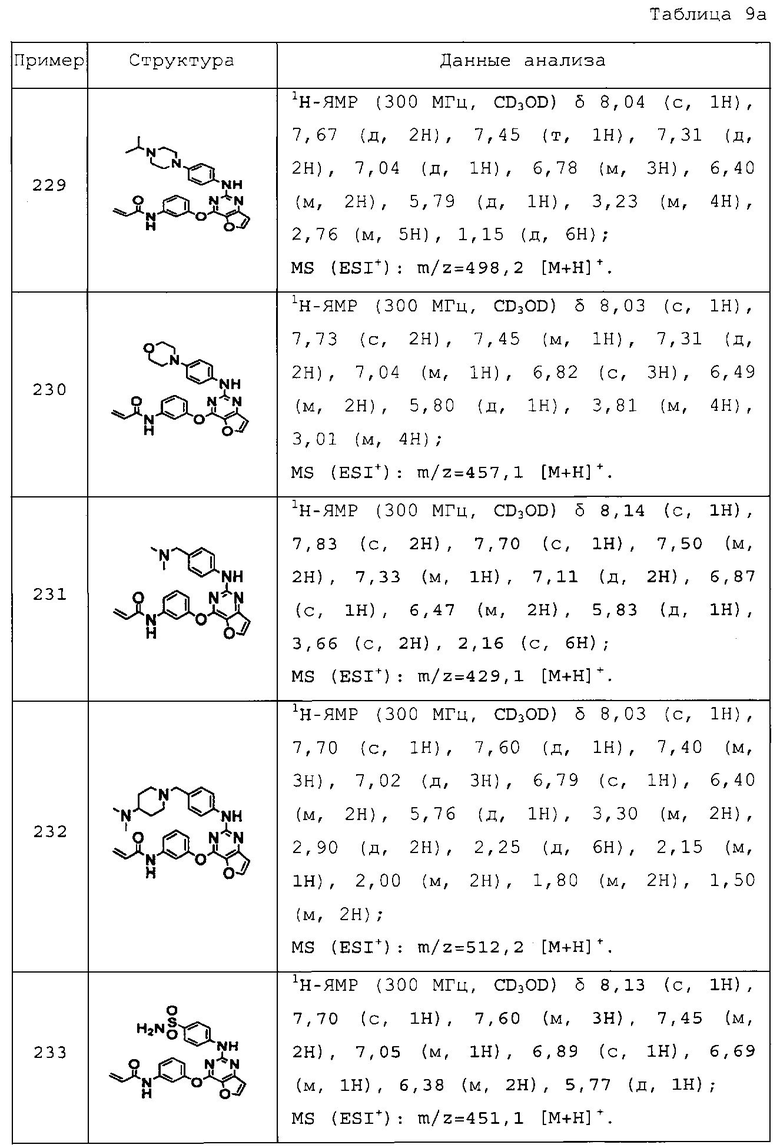
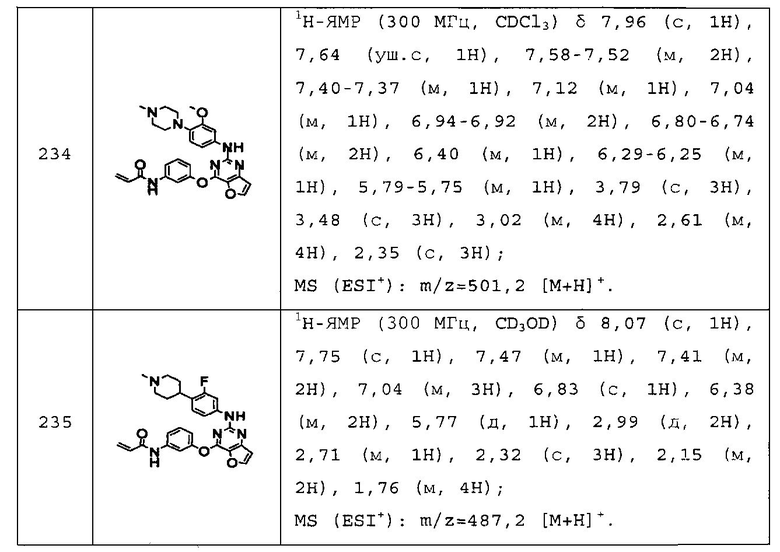
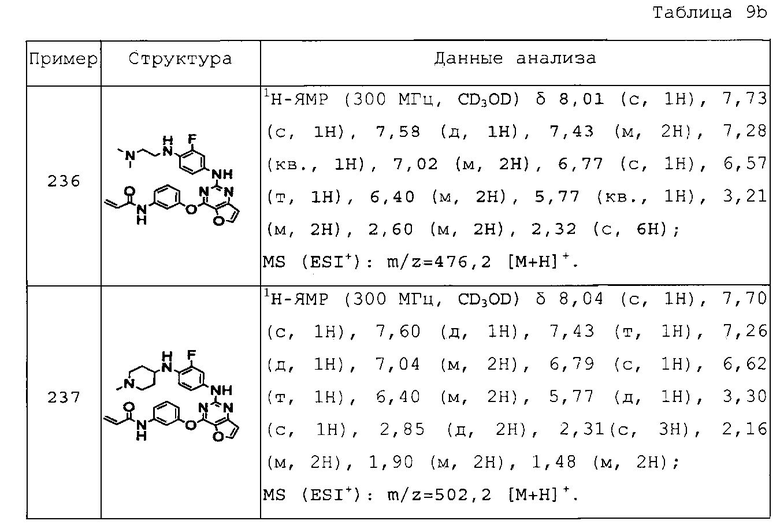
Пример приготовления 1
Таблетки для перорального введения, содержащие в качестве активного ингредиента каждое из соединений формулы (I), полученных согласно примерам 1-237, приготавливали традиционным способом, основываясь на рецептуре, представленной в таблице 10.

Пример приготовления 2
Твердые желатиновые капсулы для перорального введения, содержащие в качестве активного ингредиента каждое из соединений формулы (I), полученных согласно примерам 1-237, приготавливали традиционным способом, основываясь на рецептуре, представленной в таблице 11.
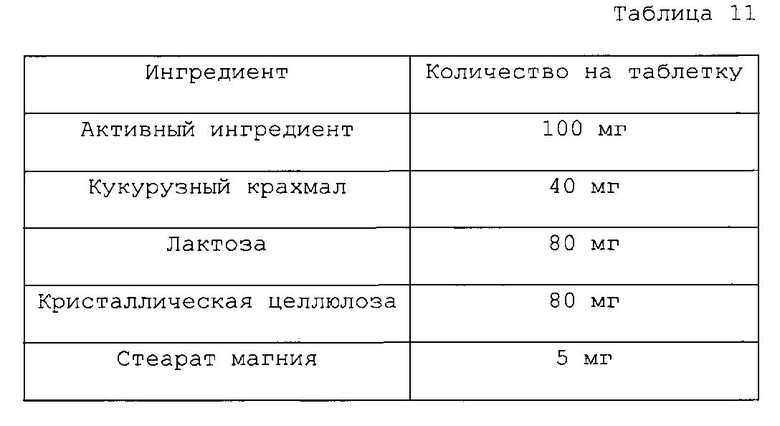
Пример приготовления 3
Инъецируемые составы, содержащие в качестве активного ингредиента каждое из соединений формулы (I), полученных согласно примерам 1-237, приготавливали традиционным способом, основываясь на рецептуре, представленной в таблице 12, причем при использовании соли соединения формулы (I) значение рН не корректировали.

Пример приготовления 4
Инъецируемые составы, содержащие в качестве активного ингредиента каждое из соединений формулы (I), полученных согласно примерам 1-237, приготавливали традиционным способом, основываясь на рецептуре, представленной в таблице 13.

Тестовый пример 1: Тест на ингибирование роста раковых клеток, экспрессирующих EGFR
Для определения того, что соединения согласно настоящему изобретению, полученные согласно примерам 1-237, селективно ингибируют рост раковых клеток, экспрессирующих мутантный EGFR, по сравнению с EGFR WT, был проведен тест на ингибирование роста раковых клеток соединениями согласно настоящему изобретению, описанный ниже. В тесте использовали линию клеток А431 злокачественной опухоли кожи со сверхэкспрессией EGFR дикого типа (WT), линию клеток НСС827 рака легких с делецией внутри рамки считывания в 19 экзоне домена тирозинкиназы EGFR и экспрессирующую мутантный EGFR L858R/T790M линию клеток NCI-Н1975, обладающих резистентностью к клинически одобренным ингибиторам EGFR, таким как гефитиниб или эрлотиниб.
Тест на ингибирование роста раковых клеток соединениями согласно настоящему изобретению проводили на линиях клеток А431 (АТСС CRL-1555), НСС827 (АТСС CRL-2868) и NCI-H1975 (АТСС CRL-5908).
Линию клеток А431 инкубировали в модифицированной по Дульбекко среде Игла с высоким содержанием глюкозы и с добавлением 10% эмбриональной бычьей сыворотки (FBS) и 1% пенициллина/стрептомицина (Gibco BRL), а линии клеток НСС827 и NCI-H1975 инкубировали в среде RPMI с добавлением 10% FBS, 1% пенициллина/стрептомицина и 1% пирувата натрия.
Каждую из линий раковых клеток, хранившихся в сосуде с жидким азотом, быстро нагревали при 37°C и центрифугировали для удаления среды. Полученную в результате клеточную массу смешивали с культуральной средой, инкубировали в культуральном флаконе при 37°C и 5% СО2 в течение 2-3 суток, и удаляли среду. Оставшиеся клетки промывали DPBS (физиологическим раствором с фосфатным буфером по Дульбекко) и отделяли от флакона с помощью трипсина-EDTA. Отделенные клетки разбавляли культуральной средой до концентрации 1×105 клеток А431/мл, а в случае клеток НСС827 и NCI-H1975 проводили разбавление до 5×104 клеток/мл. В каждую лунку 96-луночного планшета добавляли 100 мкл разбавленного раствора клеток и инкубировали при 37°C и 5% СО2 в течение 1 суток. Клетки NCI-H1975 инкубировали в обедненной среде RPMI-1640 с добавлением 0,1% FBS и 1% пенициллина/стрептомицина для максимального увеличения реакционной активности клеток в отношении тестируемых соединений на следующие сутки.
Каждое из соединений, полученных согласно примерам 1-237, растворяли в 99,5% диметилсульфоксиде (DMSO) до концентрации 25 мМ. В том случае, когда тестируемое соединение не было растворимо в DMSO, к раствору добавляли 1% HCl, и помещали его на водяную баню при 40°C на 30 мин до достижения полного растворения. Содержащий тестируемое соединение раствор в DMSO разбавляли культуральной средой до конечной концентрации 100 мкМ, а затем проводили последовательные 10-кратные разведения до 10-6 мкМ (конечная концентрация DMSO составляла менее 1%).
Из каждой лунки 96-луночного планшета удаляли среду. Затем в каждую лунку, содержащую выращенные клетки, добавляли 100 мкл тестируемого соединения, и инкубировали планшет при 37°C и 5% СО2 в течение 72 часов (за исключением клеток NCI-H1975, которые инкубировали в течение 48 часов). После удаления из планшета среды, в каждую лунку добавляли 50 мкл 10% трихлоруксусной кислоты, и выдерживали планшет при 4°C в течение 1 часа для фиксирования клеток на дне планшета. Из каждой лунки удаляли добавленный 10% раствор трихлоруксусной кислоты, планшет высушивали, добавляли в него 100 мкл раствора красителя SRB (сульфородамин-В) в концентрации 0,4%, растворенного в 1% уксусной кислоты, и оставляли полученную в результате смесь при комнатной температуре на 10 мин для прохождения реакции. После удаления раствора красителя, планшет промывали водой и тщательно высушивали. В том случае, когда раствор красителя плохо смывался водой, использовали 1% уксусную кислоту. В каждую лунку добавляли 150 мкл 10 мМ Trisma основания, и определяли оптическую плотность при длине волны 540 нм на микропланшетном ридере. В случае NCI-H1975, жизнеспособность клеток определяли как оптическую плотность при длине волны 490 нм с использованием раствора Celltiter 96 Aqueous One solution (MTS, Promega).
Концентрацию, при которой наблюдается 50% ингибирование (GI50), оценивали на основе разницы между конечной плотностью тестируемых клеток и начальной плотностью клеток, которые инкубировали в планшете без обработки тестируемым соединением, которую принимали за 100%. Расчет GI50 и анализ результатов проводили с использованием Microsoft Excel, и результаты представлены в таблицах 14a-14f, в которых А означает, что GI50≤50 нМ, В означает, что GI50 равно 50-100 нМ, С означает, что GI50 равно 100-1000 нМ, и D означает, что GI50≥1000 нМ.


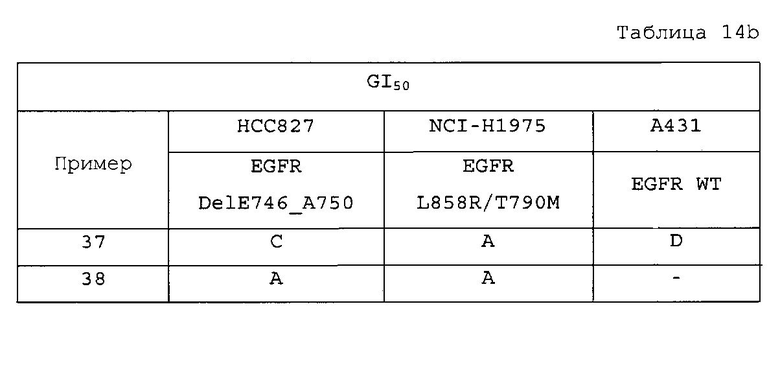
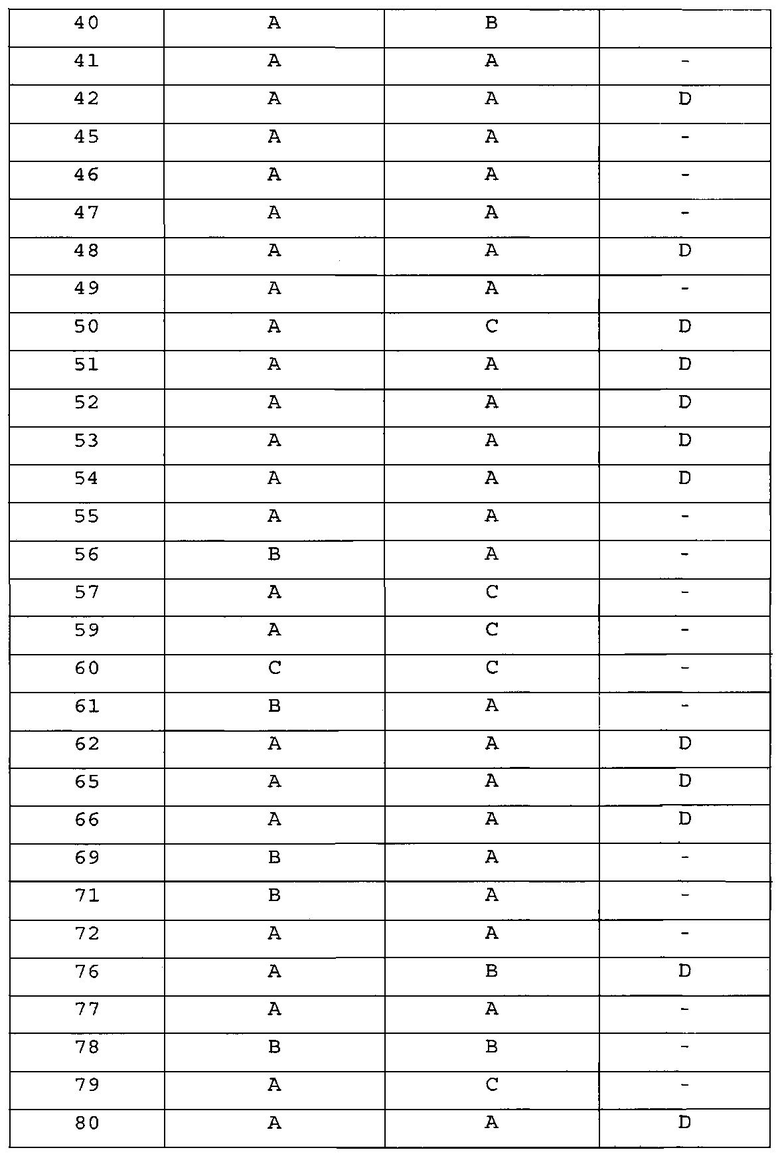



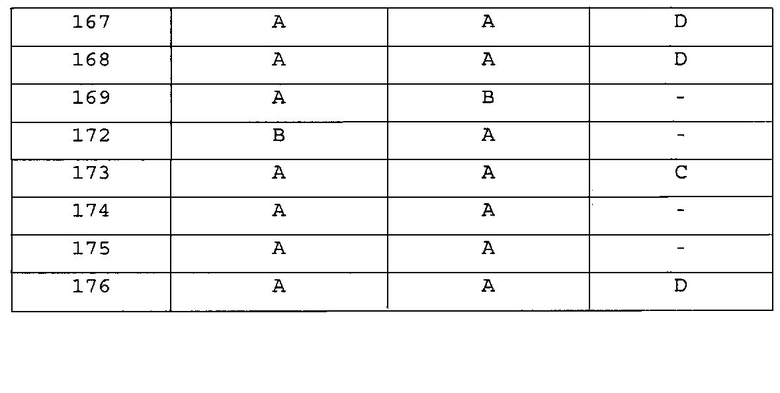
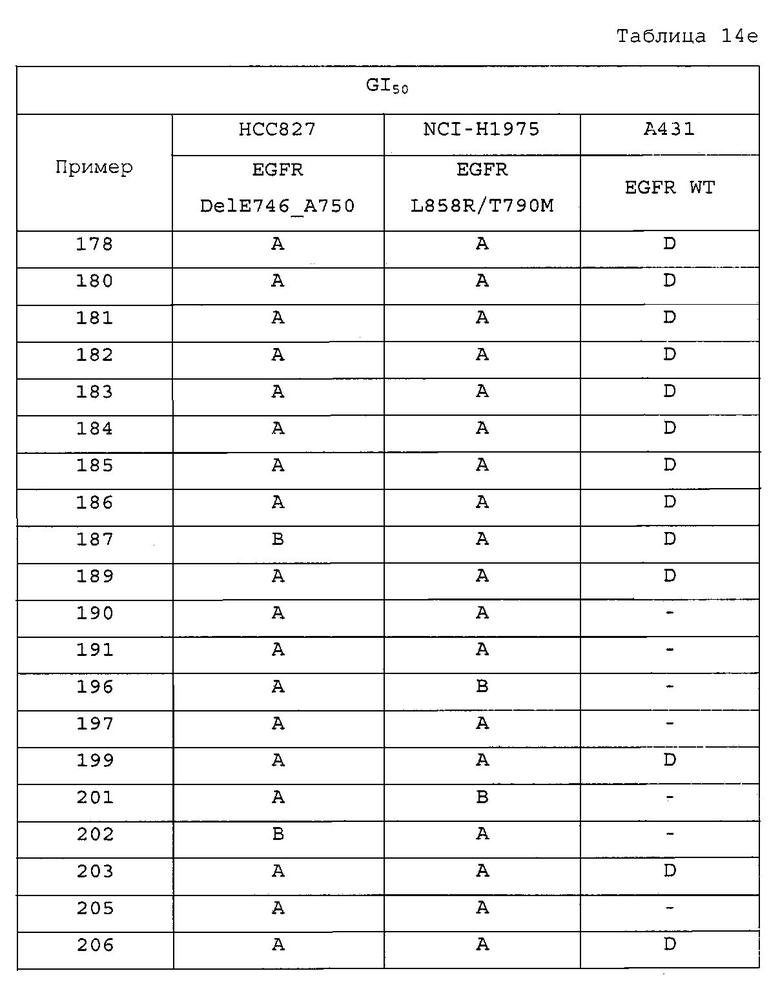
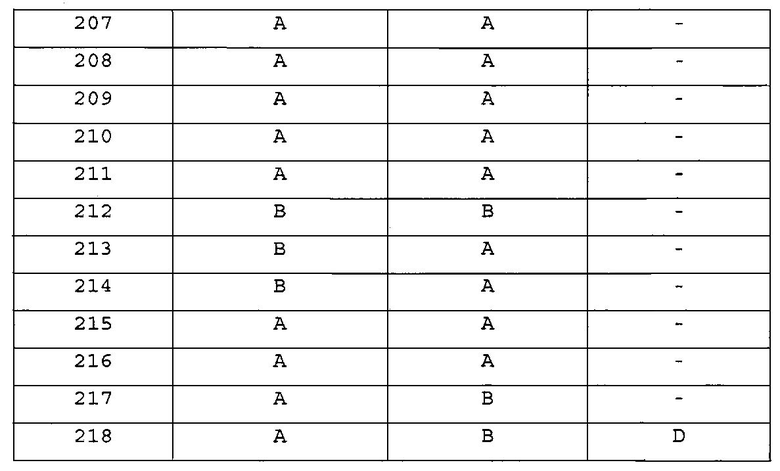
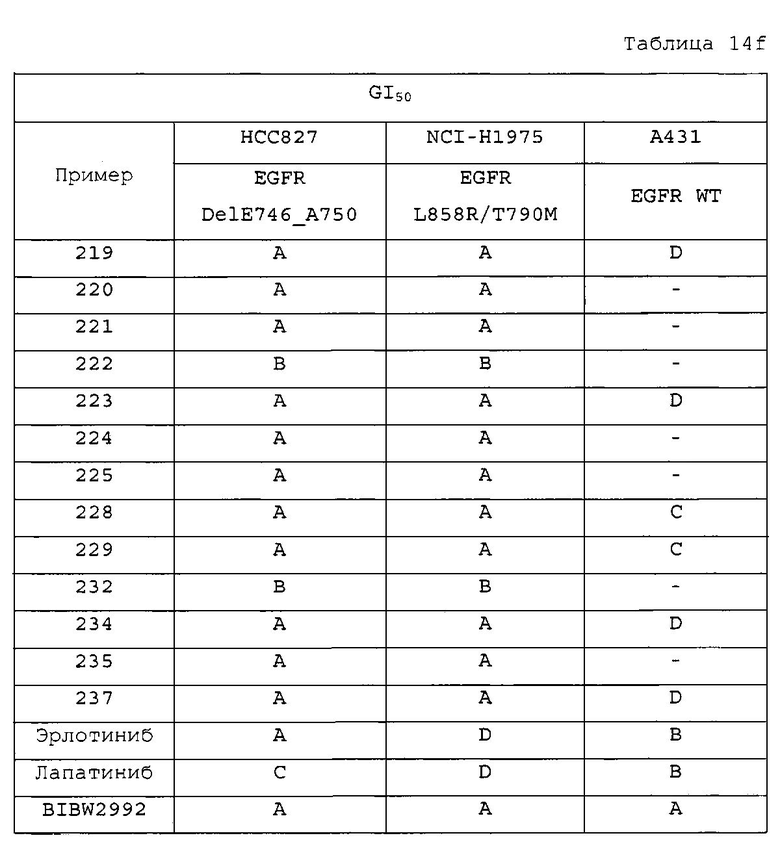
Как представлено в таблицах 14a-14f, почти все соединения согласно настоящему изобретению продемонстрировали превосходную противораковую активность посредством селективного ингибирования роста клеток НСС827 и NCI-H1975 немелкоклеточного рака легких (NSCLC), экспрессирующих мутантные EGFR (GI50=A или В), при отсутствии противораковой активности на клетках А431, экспрессирующих EGFR WT (GI50=D). Такие механизмы ингибирования соединений согласно настоящему изобретению значительно отличаются от таковых у коммерчески доступных ингибиторов тирозинкиназ EGFR (например, эрлотиниб и лапатиниб) или у разрабатываемых веществ (BIBW2992).
Как представлено в таблице 14f, эрлотиниб как ингибитор EGFR первого поколения являлся весьма эффективным ингибитором роста линии клеток NSCLC, экспрессирующих мутантные EGFR (НСС827, GI50=A), тогда как он не проявлял ингибирующую активность в отношении линии клеток NSCLC, экспрессирующих EGFR с точечной мутацией Т790М (NCI-H1975, GI50=D). Кроме того, представленный в настоящее время на рынке лапатиниб, который ингибирует как EGFR, так и HER-2, продемонстрировали слабую ингибирующую активность (НСС827, GI50=C) или ее отсутствие (NCI-H1975, GI50=D) в отношении клеточных линий NSCLC. Кроме того, BIBW2992, необратимый ингибитор с хиназолиновой структурой (Boehringer Ingelheim), находящийся в настоящее время на III фазе клинических испытаний, продемонстрировал сильную ингибирующую активность в отношении pan-HER и эффективно ингибировал рост всех линий клеток, раскрытых в таблицах 14a-14f, включая клеточную линию А431 (GI50=A). Тем не менее, такой необратимый ингибитор с хиназолиновой структурой может обладать серьезными нежелательными побочными эффектами (например, диарея, кожная сыпь и потеря массы тела) при обработке в количестве, достаточном для ингибирования EGFR Т790М, а потому все еще сохраняется потребность в разработке безопасного лекарственного средства для преодоления проблем развития резистентности EGFR Т790М. Таким образом, соединения согласно настоящему изобретению продемонстрировали существенно улучшенную ингибирующую активность в отношении мутантных EGFR, включая EGFR Т790М, при отсутствии ингибирующей активности в отношении EGFR WT, экспрессируемых нормальными клетками, что позволяет предположить, что соединения согласно настоящему изобретению могут быть применимы в качестве более эффективных и безопасных противораковых средств для пациентов с NSCLC.
Тестовый пример 2: Тест на ингибирование активности киназы EGFR WT и L858R/T790M
Ингибирующую активность соединений согласно настоящему изобретению, полученных согласно примерам 1-237, в отношении киназы EGFR WT и EGFR L858R/T790M определяли с использованием набора для оценки киназной активности z-lyte (Invitrogen, PV3191). Использованные в тесте киназы приобретали у Invitrogen.
Каждое из соединений, полученных согласно примерам 1-237, приготавливали в виде 10 мМ раствора в DMSO, из него приготавливали раствор, содержащий 4% DMSO, и разбавляли его до концентрации от 1 мкМ до 0,0001 мкМ. Затем для каждой из киназ рассчитывали приблизительное значение Kd, и разбавляли киназным буфером (50 мМ HEPES (рН 7,4), 10 мМ MgCl2, 1 мМ EGTA и 0,01% BRIJ-35) до концентрации от 1 до 100 нг/набор для анализа. Тест проводили к 384-луночных плоскодонных планшетах из полистирола. В каждую лунку добавляли 5 мкл разбавленного раствора каждого соединения, и последовательно добавляли в нее 10 мкл смеси пептидного субстрата и киназы в подходящей концентрации и 5 мкл 5-300 мкМ раствора АТР, и инкубировали планшет в мешалке при комнатной температуре в течение 60 минут. Спустя 60 мин к полученной смеси добавляли 10 мкл окрашивающего реагента для инициации флуоресцентной реакции пептидного субстрата, и для гашения реакции в планшет добавляли терминирующий раствор. В каждой лунке определяли значение интенсивности флуоресценции с помощью флуориметра (Molecular Device) при 400 нм (фильтр возбуждения) и 520 нм (фильтр испускания). Ингибирующую активность тестируемых соединений в отношении киназ определяли в виде фосфорилирования (в %) по сравнению с контрольной группой в соответствии с протоколом набора, и измеряли по оси x концентрацию, при которой наблюдалось 50% ингибирование (IC50). Расчет IC50 и анализ результатов проводили с помощью Microsoft Excel. Результаты представлены в таблице 15, в которой А означает, что IC50≤50 нМ, В означает, что IC50 равно 50-100 нМ, С означает, что IC50 равно 100-1000 нМ, и D означает, что IC50≥1000 нМ.

Как представлено в таблице 15, соединения согласно настоящему изобретению продемонстрировали относительно низкую ингибирующую активность в отношении EGFR WT, связанную с неблагоприятными эффектами (IC50=С или D), тогда как они продемонстрировали превосходную ингибирующую активность в отношении мутантных EGFR L858R/T790M, обладающих резистентностью к коммерчески доступным ингибиторам тирозинкиназ (например, эрлотиниб и лапатиниб) (IC50=А). Как и в результатах тестового примера 1, такие механизмы ингибирования соединений согласно настоящему изобретению значительно отличаются от таковых у коммерчески доступных ингибиторов тирозинкиназ EGFR (например, эрлотиниб и лапатиниб) или у разрабатываемых веществ (BIBW2992), которые сильно ингибируют EGFR WT (IC50=A или В). Таким образом, соединения согласно настоящему изобретению представляют собой эффективные и безопасные лекарственные средства, применимые для пациентов с NSCLC, демонстрируя эффективно превосходную ингибирующую активность в отношении мутантных EGFR, включая EGFR Т790М, при отсутствии ингибирующей активности в отношении EGFR WT, экспрессируемых в нормальных клетках.
Тестовый пример 3: Тест на ингибирование активности киназ BTK и JAK3
Определяли ингибирующую активность соединений согласно настоящему изобретению, полученных согласно примерам 1-237, в отношении киназ BTK и JAK3, соответственно. Повторяли методику из тестового примера 2, за исключением того, что вместо киназы EGFR использовали киназы BTK и JAK3 (Invitrogen). Результаты представлены в таблицах 16а-16с, в которых А означает, что IC50≤50 нМ, в означает, что IC50 равно 50-100 нМ, С означает, что IC50 равно 100-1000 нМ, и D означает, что IC50≥1000 нМ.





Как представлено в таблицах 16а-16с, соединения согласно настоящему изобретению продемонстрировали превосходную ингибирующую активность в отношении киназ ВТК и JAK (IC50=А или В).
Тестовый пример 4: Тест на ингибирование активности киназ BMX, ITX и RLK
Соединение, полученное согласно примеру 1, исследовали на ингибирующую активность в отношении киназ семейства ТЕС, т.е. ВМХ, ITK, ТЕХ и RLK. Измерения проводили по такой же методике, что и описанная в примере 2, за исключением того, что вместо фермента EGFR использовались ферменты BMX, TTK, TEC и RLK (Invitrogen). Результаты представлены в таблице 17. Буква «А» в таблице означает, что IC50≤50 нМ, «В» означает, что IC50 равно 50-100 нМ, «С» означает, что IC50 равно 100-1000 нМ, и «D» означает, что IC50≥1000 нМ.

Как представлено в таблице 17, соединение примера 1 согласно настоящему изобретению ингибировало киназы семейства ТЕС, такие как киназы BTK, BMX, ITK и RLK (IC50=А или В).
Тестовый пример 5: Тест на противораковую эффективность на бестимусных мышах с ксенотрансплантированными раковыми клетками NCI-H1975
Соединение согласно настоящему изобретению (пример 2) подвергали тесту на противораковую эффективность и токсичность на бестимусных мышах с ксенотрансплантированными раковыми клетками NCI-H1975, которые проявляют резистентность, обусловленную полученной точечной мутацией EGFR Т790М, к действию эрлотиниба, ранее одобренному для лечения немелкоклеточного рака легких. Для оценки противораковой эффективности и токсичности соединения согласно настоящему изобретению в тесте также использовался BIBW2992 (Boehringer Ingelheim), который в настоящее время демонстрирует превосходную активность в отношении резистентного немелкоклеточного рака легких и находится в активной разработке.
Клетки NCI-H1975 (клетки рака легких) приобретали у Американской коллекции типовых культур (АТСС). После формирования опухоли путем подкожной инъекции 1×108 клеток/0,3 мл суспензии злокачественных опухолевых клеток в спины мышей проводили пересевы клеток, и использовали в тесте опухоль, по меньшей мере, третьего поколения.
При проведении теста извлеченную из каждой отдельной мыши опухоль шестого поколения разрезали на части по 30 мг, и подкожно пересаживали в правые бока мышей с использованием троакара 12 калибра. Объем опухоли (V) рассчитывали из приведенного ниже уравнения 1 после измерения большего диаметра (L) и меньшего диаметра (S) с помощью штангенциркуля дважды в неделю в течение 18 суток проведения теста. Все тестируемые вещества вводили перорально один раз в сутки на протяжении 10 суток, и рассчитывали степень ингибирования роста опухоли (IR: степень ингибирования роста опухоли (%), рассчитанная относительно контроля с введением носителя) и максимальную потерю массы тела (mBWL: максимальная потеря массы тела, рассчитанная относительно массы тела непосредственно перед введением) с использованием приведенных ниже уравнений 2 и 3. Результаты представлены в таблице 6 и на фиг. 1 и 2.
Уравнение 1
V=L×S2/2,
где L представляет собой больший диаметр и S представляет собой меньший диаметр.
Уравнение 2
IR(%)=(1-(RTG в группе с обработкой тестируемым веществом)/(RTG в контрольной группе)) ×100,
где RTG представляет собой относительный рост опухоли, который представляет собой средний объем опухоли на конкретные сутки, основываясь на суточном среднем объеме опухоли.
Уравнение 3
mBWL(%)=(1-(средняя масса тела на x сутки/средняя масса тела непосредственно перед введением)) ×100,
где x сутки представляют собой сутки, на которые потеря массы тела является наибольшей за время проведения теста.
В следующей таблице 18 представлены результаты для IR и mBWL на модели NCI-H1975 in vivo.

1) Измерено на 16-е сутки после введения;
2) измерено на 10-е сутки после введения.
Соединение согласно настоящему изобретению не ингибировало EGFR WT и демонстрировало превосходную активность в отношении мутантных EGFR, специфических для немелкоклеточного рака легких (активный мутант: EGFR DelR746_A750, EGFR L858R; приобретенная мутация: EGFR Т790М). Как представлено в таблице 18 и фиг. 1 и 2, ингибиторы EGFR продемонстрировали сходную с BIBW2992 эффективность на NCI-H1975, животной модели, наиболее тяжелой для достижения эффективности (IR=77% в сравнении с 75%), и при этом они не продемонстрировали каких-либо неблагоприятных побочных эффектов, возникающих в результате фармакологического действия, таких как кожные заболевания и потеря массы тела (BIBW2992: 9,1% потери массы тела, соединение примера 2: 7,6% прироста массы тела при терапевтически эквивалентной дозе). Указанные экспериментальные результаты показывают, что соединения согласно настоящему изобретению селективно и эффективно ингибируют рост злокачественной опухоли и резистентность к действию лекарств, вызванную мутацией EGFR, не проявляя при этом неблагоприятных побочных эффектов.
Тестовый пример 6: Ингибирование индуцированного коллагеном артрита у мышей
Для оценки эффективности соединения согласно настоящему изобретению при ревматоидном артрите соединение подвергали тесту на ингибирование артрита в модели индуцированного коллагеном артрита (CIA) на мышах. Модель CIA является широко используемой репрезентативной моделью аутоиммунного артрита, в которой артрит индуцируется инъекцией смеси коллагена II типа и иммунологического адъюванта конкретной линии мышей, обладающей главным комплексом гистосовместимости (MHC) класса II с Н-2q или Н-2r, и тем самым аномально активируются CD4+ Т-клетки и В-клетки, специфически чувствительные к коллагену II типа.
Самцов мышей DBA/1J (в возрасте 8 недель) вначале иммунизировали внутрикожной инъекцией 0,7 мл суспензии жидкости, в которой эмульгированы равные объемы 2 мг/мл коллагена II типа и полного адъюванта Фрейнда с добавлением бактерий туберкулеза. Спустя 21 сутки мышей вторично иммунизировали, как описано выше, за исключением того, что применяли суспензию жидкости, в которой были эмульгированы равные объемы 2 мг/мл коллагена II типа и неполного адъюванта Фрейнда, не содержащего бактерии туберкулеза. Спустя 1 неделю после вторичной иммунизации, мышей оценивали по шкале клинических показателей, основываясь на таблице 10, и группировали семь животных таким образом, что средний балл в экспериментальной группе составлял от 1 до 2. Тестируемые образцы и носитель в заданных концентрациях вводили перорально в количестве 10 мл на массу тела каждый день в течение 14 суток с помощью Sonde. Артрит оценивали по шкале клинических показателей (David D Brand et al., Nature Protocol. 2(5), 1269, 2007) три раза в сутки.
Соединение примера 1 снижало отек и воспаление до последних суток (14-е сутки) теста в группах с введением 10 мг/кг и 30 мг/кг по сравнению с контрольной группой, и существенно снижало отек, воспаление и воспалительную гиперемию в группе с введением 30 мг/кг (фиг. 3).
Как представлено в таблицах 16а, 16b и 16c и на фиг. 3, соединение согласно настоящему изобретению ингибировало активность киназ BTK и JAK3, и ингибирование снижало отек, воспаление и воспалительную гиперемию, а также содержание антиколлагенового антитела в модели аутоиммунного артрита CIA по сравнению с контрольной группой, а также снижало образование паннуса в гистопатологическом тестировании. Описанные выше результаты в модели артрита на грызунах позволяют предположить, что соединение согласно настоящему изобретению может обеспечивать клинические эффекты у пациентов с ревматоидным артритом.
Кроме того, соединение согласно настоящему изобретению значительно снижало секрецию интерлейкина-6 (IL-6) и TNF-α в мононуклеарных клетках периферической крови человека (РВМС) и спленоцитах мышей, в избытке секретируемого Т-лимфоцитами, В-лимфоцитами, Cytes и макрофагами после их обработки форбол-12-миристат-13-ацетатом (РМА), фитогемагглютинином (РНА), лономицином и другими стимулирующими лимфоциты веществами по сравнению с контрольной группой. Это показывает, что соединение согласно настоящему изобретению ингибирует активацию лимфоцитов.


Хотя настоящее изобретение было описано, ссылаясь на указанные выше конкретные варианты осуществления, следует понимать, что специалисты в данной области техники могут внести в настоящее изобретение различные модификации и изменения, которые также подпадают под объем настоящего изобретения, определенный прилагаемой формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ КОНДЕНСИРОВАННЫЕ ПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ ДЛЯ ИНГИБИРОВАНИЯ ТИРОЗИНКИНАЗНОЙ АКТИВНОСТИ | 2011 |
|
RU2585177C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА ДЛЯ ТОРМОЖЕНИЯ РОСТА РАКОВЫХ КЛЕТОК И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2005 |
|
RU2362773C2 |
| ПРОИЗВОДНЫЕ ТИЕНО[3,2-d]ПИРИМИДИНА, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ ПРОТЕИНКИНАЗЫ | 2011 |
|
RU2524210C2 |
| МОДУЛЯТОРЫ ПРОТЕИН-ТИРОЗИНКИНАЗЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2014 |
|
RU2656591C2 |
| НОВЫЕ СОЕДИНЕНИЯ ПИРРОЛОПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗ | 2013 |
|
RU2645672C2 |
| ПИРАЗОЛОПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРА КИНАЗЫ | 2017 |
|
RU2714206C1 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ МОДУЛЯЦИИ КИНАЗНОЙ АКТИВНОСТИ МУТАНТОВ EGFR | 2024 |
|
RU2838180C1 |
| НОВОЕ АМИДНОЕ ПРОИЗВОДНОЕ ДЛЯ ИНГИБИРОВАНИЯ РОСТА РАКОВЫХ КЛЕТОК | 2008 |
|
RU2434010C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ МОДУЛЯЦИИ КИНАЗНОЙ АКТИВНОСТИ МУТАНТОВ EGFR | 2015 |
|
RU2822389C2 |
| ПРОИЗВОДНЫЕ ТИЕНО[3,2-d]ПИРИМИДИНА, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ ПРОТЕИНКИНАЗ | 2012 |
|
RU2625799C2 |
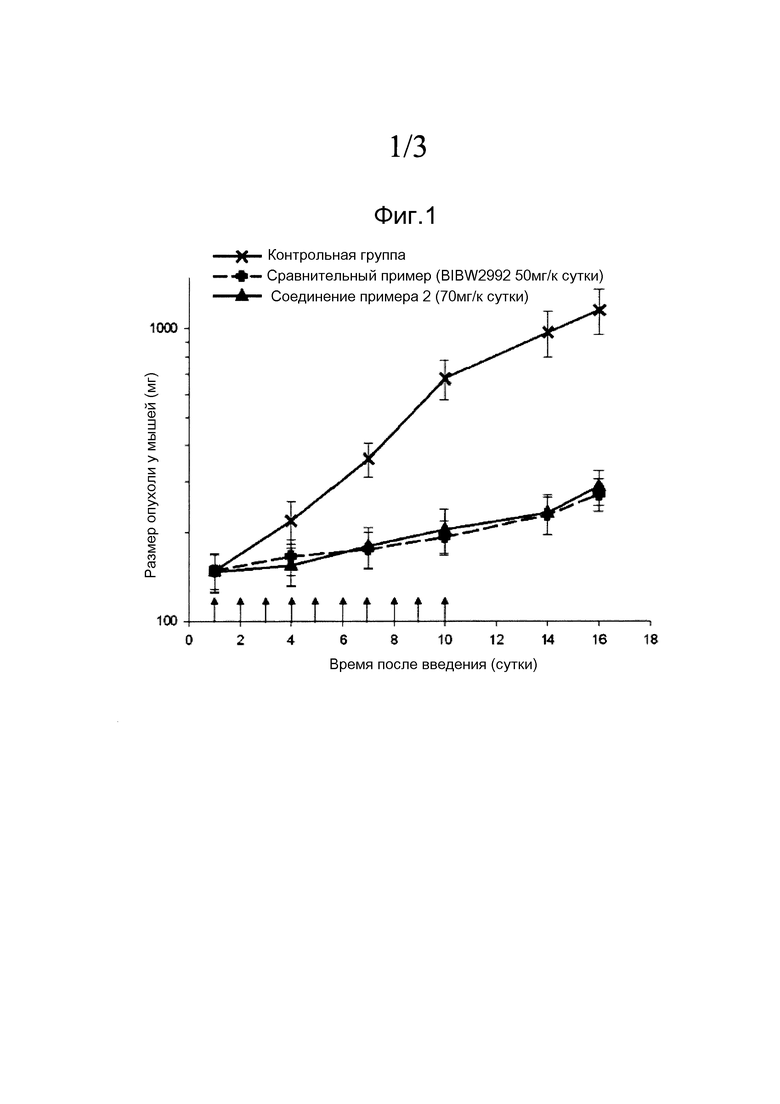


Настоящее изобретение относится к новому конденсированному пиримидиновому производному формулы (I) или его фармацевтически приемлемой соли, обладающим ингибирующей активностью в отношении тирозинкиназ Брутона (ВТК). Соединения могут найти применение для получения лекарственного средства для профилактики или лечения опосредованных тирозинкиназой Брутона (ВТК) раков и опухолей, таких как лейкоз, лимфома, В-клеточная лимфома, Т-клеточная лимфома, миелома, острый лимфоидный лейкоз (ALL), хронический лимфоидный лейкоз (CLL) и др.; воспалительных заболеваний, например, таких как артрит, ревматоидный артрит, спондилоартропатия, подагрический артрит, остеоартрит, болезнь Стилла, другие артритические состояния, волчанка и др.; аутоиммунных заболеваний или иммунологически опосредованных заболеваний, включая синдром Шенгрена, аутоиммунный тиреоидит, крапивницу, рассеянный склероз, склеродермию, отторжение пересаженных органов, гетеротрансплантацию, геморрагическую пурпуру (ITP), болезнь Паркинсона, болезнь Альцгеймера, ассоциированное с диабетом заболевание, воспаление тазовых органов, аллергический ринит, аллергический бронхит, болезнь Ходжкина, неходжкинскую лимфому, множественную миелому, миелодиспластический синдром (MDS), миелопролиферативные неоплазмы (MPN), диффузную крупноклеточную В-клеточную лимфому, фолликулярную лимфому и др. В соединении формулы (I)
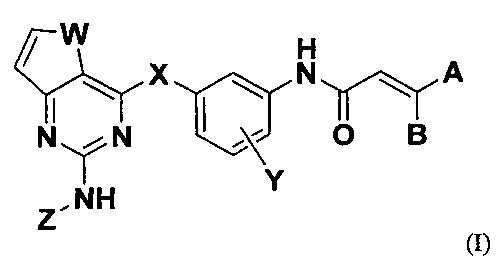
W представляет собой О; X представляет собой О, NH, S, SO или SO2; Y представляет собой атом водорода, атом галогена, С1-6алкил или С1-6алкокси, каждый из А и В независимо представляет собой атом водорода, атом галогена или ди(С1-6алкил)аминометил; Z выбирают из группы, состоящей из формул Z2, Z4, Z28, Z61, Z100, Z113, Z138, Z164, Z168 и Z189 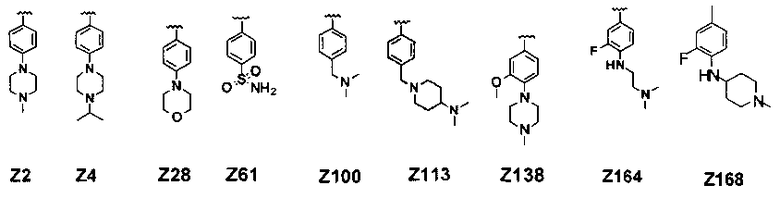
 .
.
8 н. и 2 з.п. ф-лы, 3 ил., 19 табл., 237 пр.
1. Соединение формулы (I) или его фармацевтически приемлемая соль
где
W представляет собой О;
X представляет собой О, NH, S, SO или SO2;
Y представляет собой атом водорода, атом галогена, С1-6алкил или С1-6алкокси;
каждый из А и В независимо представляет собой атом водорода, атом галогена или ди(С1-6алкил)аминометил;
Z выбирают из группы, состоящей из формул Z2, Z4, Z28, Z61, Z100, Z113, Z138, Z164, Z168 и Z189:
и .
2. Соединение по п. 1, выбранное из группы, состоящей из:


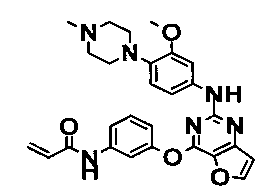
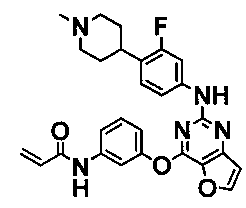
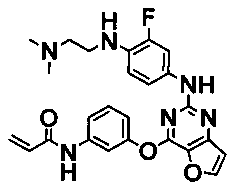 и
и
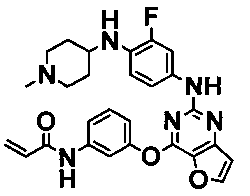
или его фармацевтически приемлемой соли.
3. Соединение формулы
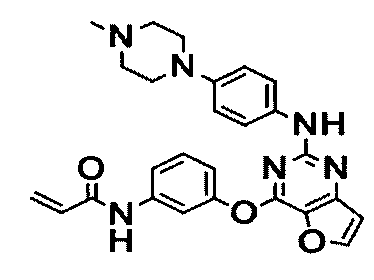
или его фармацевтически приемлемая соль.
4. Соединение формулы
 .
.
5. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении тирозинкиназы Брутона (ВТК), содержащая эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-4 и фармацевтический носитель.
6. Применение соединения по п. 1 для получения лекарственного средства для профилактики или лечения раков, опухолей, воспалительных заболеваний, опосредованных тирозинкиназой Брутона (ВТК).
7. Применение по п. 6, где воспалительные заболевания представляют собой артрит, ревматоидный артрит, спондилоартропатию, подагрический артрит, остеоартрит, болезнь Стилла, другие артритические состояния, волчанку, системную красную волчанку (SLE), связанное с кожей заболевание, псориаз, экзему, дерматит, атопический дерматит, боль, заболевание легких, воспаление легких, респираторный дистресс-синдром у взрослых (ARDS), легочный саркоидоз, хроническое легочное воспалительное заболевание, хроническую обструктивную болезнь легких (COPD), сердечно-сосудистое заболевание, атеросклероз, инфаркт миокарда, застойную сердечную недостаточность, реперфузионное повреждение сердечной мышцы, воспалительное заболевание кишечника, болезнь Крона, неспецифический язвенный колит, синдром раздраженной толстой кишки, бронхиальную астму, синдром Шегрена, аутоиммунный тиреоидит, крапивницу, рассеянный склероз, склеродермию, отторжение пересаженных органов, гетеротрансплантацию, геморрагическую пурпуру (ITP), болезнь Паркинсона, болезнь Альцгеймера, ассоциированное с диабетом заболевание, воспаление, воспаление тазовых органов, аллергический ринит, аллергический бронхит, аллергический синусит, лейкоз, лимфому, В-клеточную лимфому, Т-клеточную лимфому, миелому, острый лимфоидный лейкоз (ALL), хронический лимфоидный лейкоз (CLL), острый миелоидный лейкоз (AML), хронический миелоидный лейкоз (CML), волосатоклеточный лейкоз, болезнь Ходжкина, неходжкинскую лимфому, множественную миелому, миелодиспластический синдром (MDS), миелопролиферативные неоплазмы (MPN), диффузную крупноклеточную В-клеточную лимфому и фолликулярную лимфому.
8. Фармацевтическая композиция для профилактики или лечения раков, опухолей, воспалительных заболеваний, аутоиммунных заболеваний или иммунологически опосредованных заболеваний, содержащая эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли по п. 1 в качестве активного ингредиента.
9. Соединение формулы
или его фармацевтически приемлемая соль для применения для лечения ревматоидного артрита.
10. Соединение формулы

или его фармацевтически приемлемая соль для применения для лечения синдрома Шегрена.
| WO 2008073785 A2, ?19.08 | |||
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| SHOWALTER; HOLLIS H D; ET AL., Tyrosine Kinase Inhibitors | |||
| Устройство для электрической сигнализации | 1918 |
|
SU16A1 |
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| WO 9802438 A1,22.01.1998 | |||
| TRUMP-KALLMEYER S; et al., | |||

