УРОВЕНЬ ТЕХНИКИ
Связанные с белком G рецепторы (GPCR) образуют самое большое семейство мембранных рецепторов, содержащихся в клетке. Они преобразуют внеклеточные сигналы во внутриклеточные эффекторные системы и участвуют в целом ряде физиологических явлений, поэтому они представляют собой наиболее распространенные мишени для фармацевтических лекарственных средств, хотя имеющиеся средства лечения направлены лишь на небольшое количество GPCR.
GPCR отвечает на широкий диапазон лигандов. Благодаря развитию технологий секвенирования генома человека идентифицировано более 400 GPCR (не включая обонятельные рецепторы GPCR), примерно для 25% из которых все еще отсутствует определенный физиологически соответствующий лиганд. Эти рецепторы также известны, как "орфанные GPCR". Предполагается, что их "деорфанизация" и установление их роли in vivo приведут к пониманию новых механизмов регуляции и поэтому к обнаружению новых мишеней для лекарственных средств. Пока не ясно, является ли GPR17 таким орфанным рецептором. С точки зрения филогенетики GPR17 находится в близком родстве с нуклеотидными рецепторами P2Y и цистениллейкотриеновыми рецепторами (CysLT1, CysLT2), при идентичности последовательности аминокислот, составляющей примерно 30 и примерно 35% соответственно.
Анализ множества тканей по методике нозерн-блоттинга и с помощью ОТ-ПЦР (полимеразная цепная реакция обратной транскриптазы) указывает на то, что GPR17 экспрессируется главным образом в центральной нервной системе (ЦНС) (Ciana et al., 2006, EMBO J 25(19): 4615; Blasius et al., 1998, J Neurochem 70(4): 1357) и дополнительно в сердце и почках, т.е. в органах, обычно подвергающихся ишемическому повреждению. Идентифицированы две изоформы GPR17 человека, отличающиеся лишь длиной последовательности их аминокислот на N-конце. Короткая изоформа GPR17 кодирует белок, содержащий 339 аминокислотных остатков, при типичных для родопсина семи трансмембранных мотивах. Длинная изоформа кодирует рецептор, содержащий в последовательности аминокислот на N-конце на 28 аминокислот больше (Blasius et al., 1998). GPR17 позвоночных являются весьма сходными (идентичность последовательности аминокислот ортологов у мышей и крыс составляет ~ 90%), что может являться благоприятной характеристикой в контексте выявления лекарственных средств при разработке малых молекул -лигандов и моделей на животных.
В первоначальном сообщении о деорфанизации GPR17 идентифицировали как двойной рецептор урацилнуклеотидов и цистеиниллейкотриенов (cysLT), LTC4 и LTD4 соответственно, на основании связывания с 35SGTPγS и исследования ингибирования цАМФ (циклический аденозинмонофосфат), а также визуализации кальция в отдельной клетке (Ciana et al., 2006, тот же источник). Были представлены доказательства функционирования GPR17 в разных клеточных средах, таких как клетки 1321N1, COS7, СНО и HEK293 (Ciana et al., 2006, тот же источник). Затем в независимом исследовании была подтверждена активация GPR17 посредством урацилнуклеотидов, однако не подтверждена активация посредством CysLT (Benned-Jensen and Rosenkilde, 2010, Br J Pharmacol, 159(5): 1092). В недавних независимых сообщениях (Qi et al., 2013, J Pharmacol Ther 347,1, 38; Hennen et al.,2013, Sci Signal 6, 298) показано отсутствие ответа GPR17 и на урацилнуклеотиды, и на CysLT в разных клеточных средах, стабильно экспрессирующих GPR17 (клетки 1321N1, СНО, HEK293). Также предложена новая регуляторная роль для GPR17: при совместном экспрессировании с CysLT1 GPR17 воспроизводит рецептор CysLT1, невосприимчивый к его эндогенным липидным медиаторам LTC4 и LTD4. Для более подробного изучения фармакологических характеристик и функционирования GPR17 необходимы дополнительные исследования.
Лекарственные средства, модулирующие активность GPR17, могут обладать нейропротективным, противовоспалительным и противоишемическим воздействиями и поэтому они могут быть полезны для лечения ишемии головного мозга, сердца и почек, и удара (WO 2006/045476), и/или для улучшения восстановления после приступов этих заболеваний (Bonfanti et al, Cell Death and Disease, 2017, 8, e2871).
Полагают, что модуляторы GPR17 также участвуют в потреблении пищи, реакциях инсулина и лептина, и поэтому заявлено, что они играют роль в лечении ожирения (WO 2011/113032).
Кроме того, существует убедительное доказательство того, что GPR17 участвует в процессах миелинизации и что негативные модуляторы GPR17 (антагонисты или обратные агонисты) могут представлять собой ценные лекарственные средства, предназначенные для лечения или облегчения нарушений миелинизации, таких как рассеянный склероз или повреждение спинного мозга (Chen et al, Nature Neuroscience 2009, 12(11): 1398-406; Ceruti et al; Brain: Journal of Neurology 2009 132(Pt 8):2206-18; Hennen et al, Sci Signal, 6, 2013, 298; Simon et al J Biol Chem 291, 2016, 705; Fumagalli et al, Neuropharmacology 104, 2016, 82). Недавно две группы исследователей показали, что у взрослых мышей с удаленным GPR17 повторная миелинизация происходит быстрее, чем у однопометных мышей дикого типа, после вызванной посредством ЛФХ (лизофосфатидилхолин) демиелинизации в спинном мозге (Lu et al., Scientific Reports, 2018, 8:4502) или в мозолистом теле (Ou et al., J. Neurosci., 2016, 36(41): 10560). В отличие от этого, показано, что активация GPR17 ингибирует созревание клеток-предшественников олигодендроцитов (КПО) и, таким образом, эффективно предупреждает миелинизацию (Simon et al, приведенная выше). Это также подтверждает возможную критически важную роль GPR17 в протекании повторной миелинизации и то, что он является перспективной мишенью для лекарственных средств при лечении демиелинизирующих заболеваний. Таким образом, идентификация активных и селективных антагонистов или обратных агонистов GPR17 является существенно важной для лечения нарушений миелинизации.
Известно, что несколько тяжелых заболеваний, связанных с нарушением миелинизации, вызваны нарушениями миелинизации вследствие уменьшения количества миелина (обычно называющимся демиелинизацией) и/или вследствие неспособности организма надлежащим образом образовывать миелин (иногда называющейся дисмиелинизацией). Заболевания, связанные с нарушением миелинизации, могут быть первичными или вторичными, как ответ на определенные вызывающие их события, такие как, например, травматическое повреждение головного мозга или вирусная инфекция. Заболевания, связанные с нарушением миелинизации, главным образом могут поражать центральную нервную систему (ЦНС), однако также могут затрагивать периферическую нервную систему. Заболевания, связанные с нарушением миелинизации, включают, в частности, рассеянный склероз, нейромиелит зрительного нерва (также известный, как болезнь Девика), лейкодистрофии, синдром Гийена-Барре и многие другие заболевания, дополнительно более подробно описанные ниже (также см., например, публикации Love, J Clin Pathol, 59, 2006, 1151, Fumagalli et al, приведенную выше). Недавно установлено, что нейродегенеративные заболевания, такие как болезнь Альцгеймера, болезнь Гентингтона, болезнь Паркинсона, боковой амиотрофический склероз (БАС) и мультисистемная атрофия (МСА) сильно связаны с уменьшенной миелинизацией (см., например, публикации Ettle et al, Mol Neurobiol 53, 2016, 3046; Jellinger and Welling, Movement Disorders, 31, 2016; 1767; Kang et al, Nature Neurosci 6, 2013, 571; Bartzokis, Neurochem Res (2007) 32:1655).
Рассеянный склероз (PC) является хроническим прогрессирующим нарушением. Он является воспалительным аутоиммунным заболеванием, вызывающим поражение олигодендроцитов, демиелинизацию и, в заключение, потерю аксонов, таким образом, приводящим к возникновению широкого спектра признаков и симптомов, характерных для тяжелого неврологического заболевания, таких как, например, усталость, головокружение, затруднения движения и ходьбы, затруднения с речью и проглатыванием, боль и другие. PC проявляется в нескольких формах, при этом появление новых симптомов происходит или при отдельных приступах (рецидивирующие формы), или путем медленного накопления со временем (прогрессирующие формы). В промежутках между приступами определенные симптомы могут полностью исчезать, однако часто возникает тяжелое неврологическое нарушение, в особенности, если заболевание развивается в более прогрессирующей форме. По данным Американской ассоциации больных рассеянным склерозом в США примерно у 400000 индивидуумов диагностирован PC и во всем мире у 2,5 миллионов, при этом по оценкам ежегодно в США диагностируются 10000 новых случаев. Рассеянный склероз в 2-3 раза чаще встречается у женщин, чем у мужчин.
Не существует известного средства для этиологического лечения или излечивания рассеянного склероза, или многих других заболеваний, связанных с нарушением миелинизации. Лечение обычно является симптоматическим и при этом предпринимаются попытки улучшить функции после приступа и предотвратить новые приступы путем нацеливания лечения на воспалительный компонент заболевания. Такие иммуномодулирующие лекарственные средства обычно обладают лишь умеренной эффективностью, в особенности, если заболевание уже прогрессировало, однако могут приводить к побочным эффектам и плохо переноситься. Кроме того, большинство из имеющихся в продаже лекарственных средств, такие как β-интерфероны, глатирамерацетат или предназначенные для лечения антитела, выпускаются только в форме инъекций и/или направлены только на воспалительный компонент заболевания, а не непосредственно на демиелинизацию. Другие лекарственные средства, такие как кортикостероиды, обладают скорее неспецифическим противовоспалительным и иммунодепрессивным воздействием, таким образом, вероятно, приводящим к возникновению хронических побочных эффектов, таких как проявляющихся, например, в синдроме Кушинга.
Поэтому настоятельно необходимо безопасное и эффективное лекарственное средство, предназначенное для лечения заболеваний, связанных с нарушением миелинизации, таких как PC, предпочтительно лекарственное средство, которое является подходящим для перорального введения. В идеальном случае такое лекарственное средство должно обратить протекание демиелинизации путем уменьшения демиелинизации и/или путем обеспечения повторной миелинизации поврежденных нейронов. Химическое соединение, которое эффективно уменьшает активность рецептора GPR17, может удовлетворить этим требованиям.
Однако известны лишь немногие химические соединения, которые эффективно модулируют активность GPR17.
В WO 2005/103291 предложены эндогенные молекулы, 5-аминолевулиновая кислота (5-АЛК) и порфобилиноген (ПБГ), в качестве активирующих лигандов для GPR17, раскрыто анальгетическое действие агониста GPR17 и предложено применение агонистов GPR17 для лечения невропатической боли и в качестве средств для скрининговых исследований GPR17. Однако указанное сродство 5-АЛК и ПБГ является довольно низким и количества, необходимые для проведения исследований, являются значительными, а именно, в случае 5-АЛК они составляют сотни микромолей или в случае ПБГ составляют даже миллимоли, это делает оба соединения не особенно подходящими для применения в систематических скрининговых исследованиях и даже в терапии. Кроме того, ПБГ является химически нестабильным реакционноспособным соединением, которое быстро разлагается под воздействием воздуха и света, это делает его непригодным к использованию в регулярно проводимых исследованиях Таким образом, эти соединения не являются перспективными исходными веществами для разработки терапевтически эффективных негативных модуляторов GPR17.
Монтелукаст и пранлукаст изначально были разработаны в качестве антагонистов лейкотриенового рецептора и недавно установлено, что они также воздействуют на рецептор GPR17 (Ciana et al, EMBO J. 2006, 25, 4615-4627). Однако последующие полученные в функциональном исследовании результаты для монтелукаста являлись противоречивыми (Hennen et al., 2013, тот же источник), тогда как фармакологическое ингибирование GPR17 с помощью пранлукаста ускоряет дифференциацию первичных олигодендроцитов у мышей (Hennen et al., 2013, тот же источник) и крыс (Ou et al., J. Neurosci. 36, 2016, 10560-10573). Пранлукаст даже фенокопирует эффект подавления GPR17 в модели фокальной демиелинизации с использованием лизолецитина, поскольку и у мышей с удаленным GPR17, и у мышей дикого типа, которых лечили пранлукастом, наблюдали более раннее возникновение повторной миелинизации (Ou, тот же источник). Эти результаты сильно поддерживают предположение о том, что ингибиторы GPR17 являются перспективными для лечения демиелинизирующих заболеваний человека.
Однако сродство монтелукаста и пранлукаста к GPR17 наблюдается лишь при высоких концентрациях в микромолярном диапазоне (Köse et al, ACS Med. Chem. Lett. 2014, 5, 326-330). Принимая во внимание высокую степень связывания обоих соединений с белком и недостаточную степень их проникновения в головной мозг, маловероятно, что они могут обладать концентрациями в свободной форме, достаточно высокими для связывания с рецепторами GPR17 в количествах, подходящих для лечения человека. Кроме того, результаты, полученные с использованием этих соединений in vivo, затруднительно объяснить вследствие дополнительного высокого сродства к рецепторам CYSLTL Протекание перекрестных реакций с другими рецепторами дополнительно усложняет их использование для воздействия на GPR17.
В US 8623593 раскрыты некоторые индол-2-карбоновые кислоты, как агонисты GPR17, и их применение в скрининговых исследованиях. Однако все эти производные являются активными агонистами и не являются подходящими для подавления активности GPR17, которое необходимо для лечения нарушений миелинизации, таких как PC. Кроме того, активаторы GPR17 этого класса не обладают достаточной способностью проникать через гематоэнцефалический барьер вследствие наличия легко ионизируемых карбокси групп и поэтому они не являются подходящими основными соединениями для разработки негативных модуляторов GPR17. См. также публикации Baqi et al., Med. Chem. Commun., 2014, 5, 86 и Köse et al., 2014, тот же источник.
В WO 2013/167177 предложены некоторые фенилтриазолы и бензодиазепины, как антагонисты GPR17. Однако раскрытые соединения были выбраны лишь на основании результатов скрининга, проведенного с помощью компьютерного моделирования, и не приведены никакие результаты биологических исследований. До настоящего времени авторам настоящей заявки не удалось подтвердить, что какой-либо из заявленных лигандов, предложенных авторами предшествующей заявки на патент, обладают модулирующей активностью антагониста GPR17.
Поэтому необходимо идентифицировать активные модуляторы, предпочтительно негативные модуляторы GPR17, в особенности, обратные агонисты GPR17, которые способны эффективно уменьшать активность GPR17, предпочтительно при пероральном введении.
ЧЕРТЕЖИ:
На фиг. 1 представлено влияние соединения I-22 на экспрессирование миелина в исследовании миелинизации олигодендроцитов.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям, которые действуют, как отрицательные модуляторы рецептора GPR17. В предпочтительном варианте осуществления соединения действуют, как обратные агонисты рецептора GPR17, и, таким образом ингибируют обладающий системной активностью GPR17.
В одном варианте осуществления соединения обладают структурой формулы I

в которой
X1 обозначает N или C(R7),
R2 обозначает водород или галоген, предпочтительно водород или фтор,
R4 обозначает водород или фтор,
R5 обозначает водород или галоген,
R6 выбран из группы, включающей галоген, цианогруппу, С3-С5-циклоалкил, С3-С5-циклоалкилметоксигруппу, фенилоксигруппу, бензилоксигруппу, бензилметоксигруппу, пиридинилметоксигруппу, С1-С3-алкоксигруппу и С1-С3-алкил, где каждый из следующих: циклоалкил, бензил, пиридинил, алкил и алкоксигруппа, может содержать один или большее количество заместителей, выбранных из группы, включающей галоген, цианогруппу, С1-С2-алкоксигруппу и фтор-С1-С2-алкоксигруппу, или
R6 вместе с R7 и с атомами С, к которым они присоединены, образуют 5- или 6-членное ароматическое или неароматическое кольцо, которое может содержать 1 или 2 образующих кольцо гетероатома, где указанным кольцом предпочтительно является фенил или пиридил, и где указанное кольцо является незамещенным или замещено с помощью 1-3 остатков R13,
R7, если он содержится, выбран из группы, включающей водород, галоген, циклопропил, циклопропилоксигруппу, С1-С3-алкоксигруппу и C1-С3-алкил, где каждый из следующих: алкил и алкоксигруппа, может содержать один или большее количество заместителей, выбранных из группы, включающей галоген, С1-С2-алкоксигруппу, фтор(С1-С2)алкоксигруппу и цианогруппу, или
R7 вместе с R6 образуют кольцо, как это описано выше,
R8 выбран из группы, включающей водород, галоген, метоксигруппу, этоксигруппу, фторметоксигруппу и фторэтоксигруппу,
R10 выбран из группы, включающей водород, цианогруппу, галоген, С3-С5-циклоалкил, С3-С5-циклоалкилоксигруппу, С3-С5-циклоалкилметоксигруппу, C1-С4-алкоксигруппу и С1-С4-алкил, где каждый из следующих: циклоалкил, циклоалкилоксигруппа, алкил и алкоксигруппа, может содержать один или большее количество заместителей, выбранных из группы, включающей галоген, C1-С3-алкоксигруппу, фтор(С1-С3)алкоксигруппу и цианогруппу,
R11 выбран из группы, включающей водород, фтор и метоксигруппу,
Х2 обозначает N или C(R12),
R12 выбран из группы, включающей водород, метоксигруппу и галоген,
R13 в каждом случае независимо выбран из группы, включающей галоген, гидроксигруппу, цианогруппу, метил, метоксигруппу, фторметил и фторметоксигруппу,
и их фармацевтически приемлемые соли, сольваты, изотопы и совместные кристаллы.
В одном варианте осуществления соединений формулы I: R6 вместе с R7 и с атомами углерода, к которым R6 и R7 присоединены, образуют незамещенный или замещенный фенил, незамещенный или замещенный пиридил, или незамещенный или замещенный С5-С6-циклоалкил, где каждый заместитель, содержащийся в кольце, образованном с помощью R6 и R7, если он содержится, предпочтительно выбран из группы, включающей фтор, хлор, цианогруппу, гидроксигруппу, метил, фторметил, метоксигруппу и фторметоксигруппу,
В каждом случае, когда соединения, предлагаемые в настоящем изобретении, содержат группы R6 и R7, которые вместе с образующими кольцо атомами бициклической кольцевой системы, к которой они присоединены, образуют другое кольцо, выбранное из группы, включающей фенил и пиридил, это кольцо вместе с бициклическим фрагментом, с которым оно аннелировано, образуют трициклический фрагмент, который предпочтительно выбран из группы, включающей 1Н-бензо[g]индол-3-ил и 1Н-пирроло[3,2-h]хинолин-3-ил соответственно. В одном варианте осуществления любой заместитель, содержащийся в 1Н-пирроло[3,2-h]хинолин-3-ильном фрагменте, предпочтительно находится в положении 8, это приводит к образованию, например, 8-(фторметил)-1Н-пирроло[3,2-h]хинолина.
Один вариант осуществления относится к соединениям формулы I, в которой
X1 обозначает N или C(R7),
R2 обозначает водород или галоген, предпочтительно водород или фтор,
R4 обозначает водород или фтор,
R5 обозначает водород или галоген,
R6 выбран из группы, включающей галоген, цианогруппу, циклопропил, бензил, бензилоксигруппу, пиридинилметоксигруппу, С1-С2-алкоксигруппу и С1-С2-алкил, где каждый из следующих: циклопропил, бензил, пиридинил, алкил и алкоксигруппа, может содержать один или большее количество заместителей, выбранных из группы, включающей галоген, цианогруппу, С1-С2-алкоксигруппу и фтор-С1-С2-алкоксигруппу, или
R6 вместе с R7 и с атомами С, к которым они присоединены, образуют пиридильное кольцо таким образом, что пиридил вместе с бициклической кольцевой системой, с которой он аннелирован, образуют 1Н-пирроло[3,2-h]хинолин, где пиридильное кольцо замещено 1 или 2 остатками R13 или предпочтительно является незамещенным,
R7, если он содержится, выбран из группы, включающей водород, галоген, циклопропил, циклопропилоксигруппу, С1-С3-алкоксигруппу и С1-С3-алкил, где каждый из следующих: алкил и алкоксигруппа, может содержать один или большее количество заместителей, выбранных из группы, включающей галоген, С1-С2-алкоксигруппу, фтор(С1-С2)алкоксигруппу и цианогруппу, или
R7 вместе с R6 образуют кольцо, как это описано выше,
R8 выбран из группы, включающей водород, галоген, метоксигруппу, этоксигруппу, фторметоксигруппу и фторэтоксигруппу,
R10 выбран из группы, включающей водород, цианогруппу, галоген, С3-С5-циклоалкил, С3-С5-циклоалкилоксигруппу, С3-С5-циклоалкилметоксигруппу, С1-С4-алкоксигруппу и С1-С4-алкил, где каждый из следующих: циклоалкил, циклоалкилоксигруппа, алкил и алкоксигруппа, может содержать один или большее количество заместителей, выбранных из группы, включающей галоген, С1-С3-алкоксигруппу, фтор(С1-С3)алкоксигруппу и цианогруппу,
R11 выбран из группы, включающей водород, фтор и метоксигруппу,
Х2 обозначает N или C(R12),
R12 выбран из группы, включающей водород, метоксигруппу и галоген,
R13 в каждом случае независимо выбран из группы, включающей фтор, хлор, цианогруппу, гидроксигруппу, метил, метоксигруппу, фторметил и фторметоксигруппу,
и к их фармацевтически приемлемым солям, сольватам, изотопам и совместным кристаллам.
Один вариант осуществления относится к соединению формулы I, в которой
X1 обозначает N или C(R7),
R2 обозначает водород или галоген, предпочтительно водород или фтор,
R4 обозначает водород или фтор,
R5 обозначает водород или галоген,
R6 выбран из группы, включающей галоген, цианогруппу, С3-С5-циклоалкил, С3-С5-циклоалкилметоксигруппу, бензилоксигруппу, бензилметоксигруппу, пиридинилметоксигруппу, С1-С3-алкоксигруппу и С1-С3-алкил, где каждый из следующих: циклоалкил, бензил, пиридинил, алкил и алкоксигруппа, может содержать один или большее количество заместителей, выбранных из группы, включающей галоген, цианогруппу, С1-С2-алкоксигруппу и фтор-С1-С2-алкоксигруппу,
R7, если он содержится, выбран из группы, включающей водород, галоген, циклопропил, циклопропилоксигруппу, С1-С3-алкоксигруппу и C1-С3-алкил, где каждый из следующих: алкил и алкоксигруппа, может содержать один или большее количество заместителей, выбранных из группы, включающей галоген, С1-С2-алкоксигруппу, фтор(С1-С2)алкоксигруппу и цианогруппу, где предпочтительно, если R7 выбран из группы, включающей водород и галоген, предпочтительно из группы, включающей водород и фтор,
R8 выбран из группы, включающей водород, галоген, метоксигруппу, этоксигруппу, фторметоксигруппу и фторэтоксигруппу,
R10 выбран из группы, включающей водород, галоген, С3-С5-циклоалкил, С3-С5-циклоалкилоксигруппу, С3-С5-циклоалкилметоксигруппу, С1-С4-алкоксигруппу и С1-С4-алкил, где каждый из следующих: циклоалкил, циклоалкилоксигруппа, алкил и алкоксигруппа, может содержать один или большее количество заместителей, выбранных из группы, включающей галоген, С1-С3-алкоксигруппу, фтор(С1-С3)алкоксигруппу и цианогруппу,
R11 выбран из группы, включающей водород, фтор и метоксигруппу,
Х2 обозначает N или C(R12),
R12 выбран из группы, включающей водород, метоксигруппу и галоген,
и к его фармацевтически приемлемым солям, сольватам, изотопам и совместным кристаллам.
Один вариант осуществления относится к соединениям формулы I, в которой
X1 обозначает N или C(R7),
R2 обозначает водород или галоген, предпочтительно водород или фтор,
R4 обозначает водород или фтор,
R5 обозначает водород или галоген,
R6 выбран из группы, включающей галоген, цианогруппу, С3-С5-циклоалкил, С3-С5-циклоалкилметоксигруппу, бензилоксигруппу, бензилметоксигруппу, пиридинилметоксигруппу, C1-С3-алкоксигруппу и C1-С3-алкил, где каждый из следующих: циклоалкил, бензил, пиридинил, алкил и алкоксигруппа, может содержать один или большее количество заместителей, выбранных из группы, включающей галоген, цианогруппу, метоксигруппу и фторметоксигруппу,
R7, если он содержится, выбран из группы, включающей водород, галоген, циклопропил, циклопропилоксигруппу, C1-С3-алкоксигруппу и C1-С3-алкил, где каждый из следующих: алкил и алкоксигруппа, может содержать один или большее количество заместителей, выбранных из группы, включающей галоген, С1-С2-алкоксигруппу, фтор(С1-С2)алкоксигруппу и цианогруппу,
R8 выбран из группы, включающей водород, галоген, метоксигруппу и фторметоксигруппу,
R10 выбран из группы, включающей водород, галоген, С3-С5-циклоалкил, С3-С5-циклоалкилоксигруппу, С1-С4-алкоксигруппу и C1-C4-алкил, где каждый из следующих: циклоалкил, циклоалкилоксигруппа, алкил и алкоксигруппа, может содержать один или большее количество заместителей, выбранных из группы, включающей галоген, С1-С3-алкоксигруппу, фтор(С1-С3)алкоксигруппу и цианогруппу,
R11 выбран из группы, включающей водород, фтор и метоксигруппу,
Х2 обозначает N или C(R12),
R12 выбран из группы, включающей водород, метоксигруппу и галоген,
и к их фармацевтически приемлемым солям, сольватам, изотопам и совместным кристаллам.
Один вариант осуществления относится к соединениям формулы I, в которой
X1 обозначает N или C(R7),
R2, R4 и R5 независимо выбраны из группы, включающей водород фтор, и предпочтительно обозначают водород,
R6 выбран из группы, включающей галоген, цианогруппу, С3-С5-циклоалкил, бензилоксигруппу, пиридин-3-илметоксигруппу, пиридин-4-илметоксигруппу, C1-С3-алкоксигруппу и C1-С3-алкил, где каждый из следующих: циклоалкил, бензил, пиридинил, алкил и алкоксигруппа, может содержать один или большее количество заместителей, выбранных из группы, включающей галоген, метоксигруппу и фторметоксигруппу,
R7, если он содержится, выбран из группы, включающей водород, галоген, циклопропил, циклопропилоксигруппу, С1-С2-алкоксигруппу и С1-С2-алкил, где каждый из следующих: алкил и алкоксигруппа, может содержать один или большее количество заместителей, выбранных из группы, включающей галоген, метоксигруппу, фторметоксигруппу и цианогруппу,
R8 выбран из группы, включающей водород, галоген, и метоксигруппу,
R10 выбран из группы, включающей галоген, С3-С4-циклоалкил, С3-С4-циклоалкилоксигруппу, C1-С3-алкоксигруппу и C1-С3-алкил, где каждый из следующих: циклоалкил, циклоалкилоксигруппа, алкил и алкоксигруппа, может содержать один или большее количество заместителей, выбранных из группы, включающей галоген, С1-С2-алкоксигруппу, фтор(С1-С2)алкоксигруппу и цианогруппу,
R11 выбран из группы, включающей водород, фтор и метоксигруппу,
Х2 обозначает N или C(R12),
R12 выбран из группы, включающей водород, метоксигруппу и галоген,
и к их фармацевтически приемлемым солям, сольватам, изотопам и совместным кристаллам.
Один вариант осуществления относится к соединениям формулы I, в которой
X1 обозначает N или C(R7),
R2 обозначает водород,
R4 обозначает водород или фтор,
R5 обозначает водород или фтор,
R6 выбран из группы, включающей галоген, цианогруппу, С3-С5-циклоалкил, C1-С3-алкоксигруппу и C1-С3-алкил, где каждый из следующих: циклоалкил, алкил и алкоксигруппа, может содержать один или большее количество заместителей, выбранных из группы, включающей галоген, метоксигруппу и фторметоксигруппу,
R7, если он содержится, выбран из группы, включающей водород, галоген, циклопропил, циклопропилоксигруппу, С1-С2-алкоксигруппу и С1-С2-алкил, где каждый из следующих: алкил и алкоксигруппа, может содержать один или большее количество заместителей, выбранных из группы, включающей галоген, С1-С2-алкоксигруппу, фтор(С1-С2)алкоксигруппу и цианогруппу,
R8 выбран из группы, включающей водород, галоген, метоксигруппу и фторметоксигруппу,
R10 выбран из группы, включающей галоген, С3-С4-циклоалкил, С3-С4-циклоалкилоксигруппу, C1-С3-алкоксигруппу и C1-С3-алкил, где каждый из следующих: циклоалкил, циклоалкилоксигруппа, алкил и алкоксигруппа, может содержать один или большее количество заместителей, выбранных из группы, включающей галоген, С1-С3-алкоксигруппу, фтор(С1-С3)алкоксигруппу и цианогруппу,
R11 выбран из группы, включающей водород, фтор и метоксигруппу,
Х2 обозначает N или C(R12),
R12 выбран из группы, включающей водород, метоксигруппу и фтор,
и к их фармацевтически приемлемым солям, сольватам, изотопам и совместным кристаллам.
Один вариант осуществления относится к соединению формулы I, в которой
X1 обозначает N или C(R7),
R2 обозначает водород,
R4 обозначает водород или фтор, предпочтительно водород,
R5 обозначает водород или фтор, предпочтительно водород,
R6 выбран из группы, включающей галоген, цианогруппу, циклопропил, C1-C2-алкоксигруппу и С1-С2-алкил, где каждый из следующих: алкил и алкоксигруппа, может содержать один или большее количество заместителей, выбранных из группы, включающей галоген, метоксигруппу и фторметоксигруппу,
R7, если он содержится, выбран из группы, включающей водород, галоген, циклопропил, циклопропилоксигруппу, С1-С2-алкоксигруппу и С1-С2-алкил, где каждый из следующих: алкил и алкоксигруппа, может содержать один или большее количество заместителей, выбранных из группы, включающей галоген, С1-С2-алкоксигруппу, фтор-С1-С2-алкоксигруппу и цианогруппу,
R8 выбран из группы, включающей водород, галоген, метоксигруппу и фторметоксигруппу,
R10 выбран из группы, включающей галоген, С3-С4-циклоалкил, С3-С4-циклоалкилоксигруппу, C1-С3-алкоксигруппу и C1-С3-алкил, где каждый из следующих: циклоалкил, циклоалкилоксигруппа, алкил и алкоксигруппа, может содержать один или большее количество заместителей, выбранных из группы, включающей галоген, C1-С3-алкоксигруппу, фтор C1-С3-алкоксигруппу и цианогруппу,
R11 выбран из группы, включающей водород, фтор и метоксигруппу,
Х2 обозначает N или C(R12),
R12 выбран из группы, включающей водород, метоксигруппу и фтор,
и к его фармацевтически приемлемым солям, сольватам, изотопам и совместным кристаллам.
Один вариант осуществления относится к соединению формулы I, в которой
X1 обозначает N или C(R7),
R2 обозначает водород
R4 и R5 независимо выбраны из группы, включающей водород и фтор,
R6 выбран из группы, включающей галоген, цианогруппу, циклопропил, метоксигруппу и метил, где каждый из следующих: метоксигруппа и метил, может содержать один или большее количество заместителей, выбранных из группы, включающей галоген, метоксигруппу и фторметоксигруппу,
R7, если он содержится, выбран из группы, включающей водород, галоген, циклопропил, циклопропилоксигруппу, метоксигруппу и метил, где каждый из следующих: метоксигруппа и метил, может содержать один или большее количество заместителей, выбранных из группы, включающей фтор, метоксигруппу, фторметоксигруппу и цианогруппу,
R8 выбран из группы, включающей водород, галоген, метоксигруппу и фторметоксигруппу,
R10 выбран из группы, включающей галоген, С3-С4-циклоалкил, С3-С4-циклоалкилоксигруппу, С1-С2-алкоксигруппу и С1-С2-алкил, где каждый из следующих: циклоалкил, циклоалкилоксигруппа, алкил и алкоксигруппа, может содержать один или большее количество заместителей, выбранных из группы, включающей галоген, С1-С2-алкоксигруппу, фтор-С1-С2-алкоксигруппу и цианогруппу,
R11 выбран из группы, включающей водород, фтор и метоксигруппу,
Х2 обозначает N или C(R12),
R12 выбран из группы, включающей водород, метоксигруппу и фтор,
и к его фармацевтически приемлемым солям, сольватам, изотопам и совместным кристаллам.
Один вариант осуществления относится к соединению формулы I, в которой
X1 обозначает N или C(R7),
R2 обозначает водород
R4 и R5 независимо выбраны из группы, включающей водород и фтор,
R6 выбран из группы, включающей галоген, цианогруппу, циклопропил, С1-С3-алкоксигруппу и C1-С3-алкил, где каждый из следующих: алкоксигруппа и алкил, может являться незамещенным или замещенными одним или большим количеством атомов галогенов, предпочтительно одним или большим количеством атомов фтора,
R7, если он содержится, выбран из группы, включающей водород и галоген, предпочтительно из группы, включающей водород и фтор,
R8 выбран из группы, включающей водород, галоген, метоксигруппу, фторметоксигруппу и фторэтоксигруппу,
R10 выбран из группы, включающей галоген, С3-С5-циклоалкил, С1-С3-алкоксигруппу и C1-С3-алкил, где каждый из следующих: циклоалкил, алкил и алкоксигруппа, может содержать один или большее количество заместителей, выбранных из группы, включающей галоген, С1-С2-алкоксигруппу и галогенированную С1-С2-алкоксигруппу,
R11 выбран из группы, включающей водород, галоген и метоксигруппу,
Х2 обозначает N или C(R12),
R12 выбран из группы, включающей водород, метоксигруппу и галоген,
и к его фармацевтически приемлемым солям, сольватам, изотопам и совместным кристаллам.
Один вариант осуществления относится к соединению формулы I, в которой
X1 обозначает N или C(R7),
R2, R4 и R5 все обозначают водород,
R6 выбран из группы, включающей галоген, цианогруппу, циклопропил, метоксигруппу, метил и изопропил, где каждый из следующих: метоксигруппа и метил, могут являться незамещенным или замещенным одним или большим количеством атомов фтора,
R7, если он содержится, выбран из группы, включающей водород, фтор, и хлор,
R8 выбран из группы, включающей водород, фтор, метоксигруппу, фторметоксигруппу и фторэтоксигруппу,
R10 выбран из группы, включающей галоген, метил, циклопропил, циклопропилоксигруппу и C1-С3-алкоксигруппу, где каждая алкоксигруппа может содержать один или большее количество заместителей, выбранных из группы, включающей фтор, С1-С2-алкоксигруппу и фтор-С1-С2-алкоксигруппу,
R11 выбран из группы, включающей водород, фтор и метоксигруппу,
Х2 обозначает N или C(R12),
R12 выбран из группы, включающей водород, метоксигруппу и фтор,
и к его фармацевтически приемлемым солям, сольватам, изотопам и совместным кристаллам.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой
X1 обозначает N или C(R7),
R2, R4 и R5 все обозначают водород,
R6 выбран из группы, включающей галоген, цианогруппу, метил, фторметил, метоксигруппу и фторметоксигруппу,
R7, если он содержится, выбран из группы, включающей водород, фтор, хлор, циклопропилоксигруппу и фторметил, и предпочтительно из группы, включающей водород и фтор,
R8 выбран из группы, включающей водород, фтор и метоксигруппу,
R10 выбран из группы, включающей галоген, циклопропил и C1-C2-алкоксигруппу, где алкоксигруппа необязательно может содержать один или большее количество заместителей, выбранных из группы, включающей фтор, метоксигруппу, этоксигруппу и фтор-С1-С2-алкоксигруппу,
R11 обозначает водород или фтор,
Х2 обозначает N или C(R12), и
где R12, если он содержится, выбран из группы, включающей водород, метоксигруппу и фтор,
и к их фармацевтически приемлемым солям, сольватам, изотопам и совместным кристаллам.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой
R2, R4 и R5 все обозначают водород,
R6 обозначает хлор или фторметил,
X1 обозначает N или C(R7),
R7, если он содержится, выбран из группы, включающей водород, фтор, хлор, циклопропилоксигруппу и фторметил,
R8 выбран из группы, включающей водород, фтор и метоксигруппу,
R10 выбран из группы, включающей галоген и С1-С2-алкоксигруппу, где алкоксигруппа необязательно может содержать один или большее количество заместителей, выбранных из группы, включающей фтор, цианогруппу и фтор-С1-С2-алкоксигруппу,
R11 обозначает водород или фтор,
Х2 обозначает N или C(R12), и
где R12, если он содержится, выбран из группы, включающей водород, метоксигруппу и фтор,
и к их фармацевтически приемлемым солям, сольватам, изотопам и совместным кристаллам.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой
R2, R4 и R5 все обозначают водород,
R6 выбран из группы, включающей фтор, хлор, цианогруппу, метил, метоксигруппу, фторметоксигруппу и фторметил,
X1 обозначает N или C(R7),
R7, если он содержится, выбран из группы, включающей водород и фтор,
R8 выбран из группы, включающей фтор и метоксигруппу,
R10 выбран из группы, включающей хлор, бром, циклопропил и С1-С2-алкоксигруппу, где алкоксигруппа замещена с помощью вплоть до 3 атомов фтора или содержит один заместитель, выбранный из группы, включающей метоксигруппу, фторметоксигруппу и фторэтоксигруппу,
R11 обозначает водород или фтор,
Х2 обозначает N или C(R12),
R12, если он содержится, обозначает водород или фтор, предпочтительно водород,
и к их фармацевтически приемлемым солям, сольватам, изотопам и совместным кристаллам.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой
R2, R4 и R5 все обозначают водород,
R6 обозначает хлор или дифторметил,
X1 обозначает N или C(R7),
R7, если он содержится, выбран из группы, включающей водород, фтор, хлор и фторметил, предпочтительно обозначает водород,
R8 выбран из группы, включающей фтор и метоксигруппу,
R10 выбран из группы, включающей хлор, бром и С1-С2-алкоксигруппу, где алкоксигруппа замещена с помощью вплоть до 3 атомов фтора или одной фторметоксигруппой,
R11 обозначает водород или фтор,
Х2 обозначает N или C(R12), и
где R12, если он содержится, обозначает водород или фтор, предпочтительно водород,
и к их фармацевтически приемлемым солям, сольватам, изотопам и совместным кристаллам.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой R2 обозначает водород.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой R4 обозначает водород.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой R5 обозначает водород.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой R5 обозначает галоген, предпочтительно бром.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой R2, R4 и R5 все обозначают водород.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой R6 выбран из группы, включающей галоген, цианогруппу, фторметоксигруппу и фторметил.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой R6 обозначает изопропил.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой R6 выбран из группы, включающей хлор или фторметил, предпочтительно группы, включающей хлор и дифторметил.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой R6 обозначает фторметоксигруппу.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой R6 обозначает метоксигруппу.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой R6 обозначает цианогруппу.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой R6 и R7 вместе с атомами С, к которым они присоединены образуют фенильное или пиридильное кольцо, которое является незамещенным или замещено одним или большим количеством остатков R13, определенных в настоящем изобретении.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой R6 и R7 вместе с атомами С, к которым они присоединены образуют незамещенное пиридильное кольцо; в одном варианте осуществления указанное пиридильное кольцо вместе с бициклической системой, с которой оно аннелировано, образуют 1Н-пирроло[3,2-h]хинолиновую группу.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой R7 обозначает водород.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой R7 выбран из группы, включающей фтор, хлор, циклопропилоксигруппу, фторметил и фторметоксигруппу.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой R7 обозначает водород или фтор.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой R8 выбран из группы, включающей водород, фтор, метоксигруппу и фторметоксигруппу, предпочтительно из группы, включающей фтор и метоксигруппу.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой R8 обозначает метоксигруппу.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой R10 не обозначает водород.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой R10 выбран из группы, включающей галоген, С3-С4-циклоалкилоксигруппу, C1-С3-алкоксигруппу и C1-С3-алкил, где каждый из следующих: алкил или алкоксигруппа, может содержать один или большее количество заместителей, выбранных из группы, включающей цианогруппу, фтор, С1-С2-алкоксигруппу и фтор-С1-С2-алкоксигруппу.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой R10 выбран из группы, включающей галоген, циклопропил и С1-С2-алкоксигруппу, где алкоксигруппа необязательно может содержать один или большее количество заместителей, выбранных из группы, включающей фтор, метоксигруппу, этоксигруппу и фтор-С1-С2-алкоксигруппу.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой R10 выбран из группы, включающей галоген, циклопропил, метоксигруппу, фторметоксигруппу, метоксиэтоксигруппу, фторэтоксигруппу и фторэтоксиметоксигруппу.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой R10 выбран из группы, включающей галоген, метоксигруппу, этоксигруппу, фторметоксигруппу, фторэтоксигруппу и фторметоксиэтоксигруппу.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой R10 выбран из группы, включающей хлор, бром, метоксигруппу, дифторметоксигруппу, монофторэтоксигруппу и дифторэтоксигруппу.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой R11 обозначает водород или фтор, предпочтительно фтор.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой R11 обозначает метоксигруппу и R8 обозначает фтор.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой Х2 обозначает C(R12) и R12 обозначает водород.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой Х2 обозначает C(R12) и R12 обозначает водород, R8 обозначает метоксигруппу и R11 обозначает фтор.
Один предпочтительный вариант осуществления относится к соединениям формулы I, в которой Х2 обозначает C(R12) и R12 обозначает фтор.
Один особенно предпочтительный вариант осуществления относится к соединениям формулы I, в которой Х2 обозначает N, таким образом, обладающим структурой формулы II,

в которой все заместители являются такими, как описано выше в настоящем изобретении для формулы I.
В одном варианте осуществления в соединениях формулы II:
X1 обозначает N или C(R7)
R2, R4 и R5 все обозначают водород,
R6 выбран из группы, включающей галоген, цианогруппу, метил, фторметил, метоксигруппу, фторметоксигруппу и бензилоксигруппу,
R7, если он содержится, выбран из группы, включающей водород, фтор, хлор, циклопропилоксигруппу и фторметил, предпочтительно из группы, включающей водород и фтор,
R8 выбран из группы, включающей водород, фтор и метоксигруппу,
R10 выбран из группы, включающей галоген, циклопропил, C1-C2-алкил и С1-С2-алкоксигруппу, где алкил и алкоксигруппа необязательно могут содержать один или большее количество заместителей, выбранных из группы, включающей фтор, цианогруппу, метоксигруппу и фтор-С1-С2-алкоксигруппу,
R11 выбран из группы, включающей водород, метоксигруппу и фтор, предпочтительно из группы, включающей водород и фтор,
и их фармацевтически приемлемые соли, сольваты, изотопы и совместные кристаллы.
В одном варианте осуществления в соединениях формулы II:
X1 обозначает N или C(R7)
R2, R4 и R5 все обозначают водород,
R6 обозначает хлор или фторметил,
R7, если он содержится, выбран из группы, включающей водород, фтор, хлор, циклопропилоксигруппу и фторметил,
R8 выбран из группы, включающей водород, фтор и метоксигруппу, R10 выбран из группы, включающей галоген, C1-С2-алкил и С1-С2-алкоксигруппу, где алкил и алкоксигруппа необязательно могут содержать один или большее количество заместителей, выбранных из группы, включающей фтор, цианогруппу и фтор-С1-С2-алкоксигруппу,
R11 выбран из группы, включающей водород, метоксигруппу и фтор, предпочтительно из группы, включающей водород и фтор,
и их фармацевтически приемлемые соли, сольваты, изотопы и совместные кристаллы.
В одном варианте осуществления соединений формулы II:
X1 обозначает N или C(R7)
R2, R4 и R5 все обозначают водород,
R6 обозначает хлор или фторметил,
R7, если он содержится, выбран из группы, включающей водород, фтор, хлор, фторметил и циклопропилоксигруппу, и предпочтительно обозначает водород,
R8 выбран из группы, включающей фтор и метоксигруппу,
R10 выбран из группы, включающей хлор, бром, метоксигруппу, фторметоксигруппу, фторэтоксигруппу, фторметоксиэтоксигруппу и фторэтоксиметоксигруппу,
R11 обозначает фтор,
и их фармацевтически приемлемые соли, сольваты, изотопы и совместные кристаллы.
Один предпочтительный вариант осуществления относится к соединениям формулы II, в которой
R2, R4 и R5 все обозначают водород,
R6 обозначает хлор или дифторметил,
XI обозначает C(R7),
R7 выбран из группы, включающей водород, фтор, хлор и фторметил, и предпочтительно обозначает водород,
R8 выбран из группы, включающей фтор и метоксигруппу,
R10 выбран из группы, включающей хлор, бром и С1-С2-алкоксигруппу, где алкоксигруппа замещена с помощью вплоть до 3 атомов фтора или одной фторметоксигруппой или фторэтоксигруппой,
R11 обозначает водород или фтор,
и к их фармацевтически приемлемым солям, сольватам, изотопам и совместным кристаллам.
Один особенно предпочтительный вариант осуществления относится к соединениям формулы I, в которой Х2 обозначает C(R12), таким образом, описывающимся формулой III

в которой все заместители являются такими, как описано выше в настоящем изобретении для формулы I.
В одном варианте осуществления в соединениях формулы III:
R2, R4 и R5 все обозначают водород,
R6 выбран из группы, включающей галоген, цианогруппу, метил, метоксигруппу, фторметоксигруппу, фторметил и бензилоксигруппу,
X1 обозначает N или C(R7),
R7, если он содержится, выбран из группы, включающей водород, галоген, фторметоксигруппу и фторметил, и предпочтительно обозначает водород или фтор,
R8 выбран из группы, включающей водород, фтор, С1-С2-алкоксигруппу и фтор-C1-С2-алкоксигруппу,
R10 выбран из группы, включающей галоген, циклопропил, C1-С2-алкил и С1-С2-алкоксигруппу, где циклопропил, алкил и алкоксигруппа все необязательно могут содержать один или большее количество заместителей, выбранных из группы, включающей фтор, метоксигруппу и фтор-С1-С2-алкоксигруппу,
R11 выбран из группы, включающей водород, метоксигруппу и фтор, предпочтительно из группы, включающей водород и фтор,
R12 выбран из группы, включающей водород, метоксигруппу и фтор,
и их фармацевтически приемлемые соли, сольваты, изотопы и совместные кристаллы.
В одном варианте осуществления в соединениях формулы III:
R2, R4 и R5 все обозначают водород,
R6 выбран из группы, включающей фтор, хлор, цианогруппу, метил, метоксигруппу, фторметоксигруппу и фторметил,
X1 обозначает N или C(R7),
R7, если он содержится, обозначает водород или фтор,
R8 выбран из группы, включающей водород, фтор и метоксигруппу, предпочтительно группы, включающей фтор и метоксигруппу,
R10 выбран из группы, включающей галоген, циклопропил и С1-С2-алкоксигруппу, где алкоксигруппа необязательно может содержать один или большее количество заместителей, выбранных из группы, включающей фтор, метоксигруппу и фтор-С1-С2-алкоксигруппу,
R11 выбран из группы, включающей водород, метоксигруппу и фтор, предпочтительно группы, включающей водород и фтор,
R12 выбран из группы, включающей водород и фтор, и предпочтительно обозначает фтор,
и их фармацевтически приемлемые соли, сольваты, изотопы и совместные кристаллы.
В одном варианте осуществления в соединениях формулы III:
R2, R4 и R5 все обозначают водород,
R6 обозначает хлор или фторметил,
X1 обозначает N или C(R7),
R7, если он содержится, выбран из группы, включающей водород, фтор, хлор, циклопропилоксигруппу, фторметоксигруппу и фторметил,
R8 выбран из группы, включающей водород, фтор и метоксигруппу,
R10 выбран из группы, включающей галоген С1-С2-алкил и С1-С2-алкоксигруппу, где алкил и алкоксигруппа необязательно могут содержать один или большее количество заместителей, выбранных из группы, включающей фтор, цианогруппу и фтор-С1-С2-алкоксигруппу,
R11 выбран из группы, включающей водород, метоксигруппу и фтор, предпочтительно из группы, включающей водород и фтор,
R12 выбран из группы, включающей водород, метоксигруппу и фтор,
и их фармацевтически приемлемые соли, сольваты, изотопы и совместные кристаллы.
В предпочтительном варианте осуществления в соединениях формулы III:
R2, R4 и R5 все обозначают водород,
R6 выбран из группы, включающей хлор, метоксигруппу, фторметоксигруппу и фторметил,
X1 обозначает N или C(R7),
R7, если он содержится, выбран из группы, включающей водород и фтор,
R8 выбран из группы, включающей водород, фтор и метоксигруппу,
R10 выбран из группы, включающей хлор, бром, циклопропил, метоксигруппу, фторметоксигруппу, фторэтоксигруппу, фторметоксиэтоксигруппу и фторэтоксиметоксигруппу,
R11 обозначает водород или фтор, предпочтительно фтор, и
R12 выбран из группы, включающей водород и фтор, и предпочтительно обозначает водород,
и их фармацевтически приемлемые соли, сольваты, изотопы и совместные кристаллы.
В предпочтительном варианте осуществления в соединениях формулы III:
R2, R4 и R5 все обозначают водород,
R6 обозначает хлор или фторметил,
X1 обозначает N или C(R7),
R7, если он содержится, выбран из группы, включающей водород, фтор, хлор, фторметил и циклопропилоксигруппу,
R8 выбран из группы, включающей фтор и метоксигруппу,
R10 выбран из группы, включающей хлор, бром, метоксигруппу, фторметоксигруппу, фторэтоксигруппу, фторметоксиэтоксигруппу и фторэтоксиметоксигруппу,
R11 обозначает водород или фтор, предпочтительно фтор, и
R12 выбран из группы, включающей водород, метоксигруппу и фтор, предпочтительно обозначает водород,
и их фармацевтически приемлемые соли, сольваты, изотопы и совместные кристаллы.
В предпочтительном варианте осуществления в соединениях формулы III:
R2, R4 и R5 все обозначают водород,
R6 выбран из группы, включающей хлор, цианогруппу, метил, метоксигруппу, фторметоксигруппу и фторметил,
X1 обозначает N или C(R7),
R7, если он содержится, выбран из группы, включающей водород, фтор и хлор,
R8 выбран из группы, включающей фтор и метоксигруппу,
R10 выбран из группы, включающей хлор, бром и С1-С2-алкоксигруппу, где алкоксигруппа замещена с помощью вплоть до 3 атомов фтора или одной фторметоксигруппой или фторэтоксигруппой,
R11 обозначает водород или фтор, и
R12 обозначает водород или фтор, предпочтительно водород,
и их фармацевтически приемлемые соли, сольваты, изотопы и совместные кристаллы.
В предпочтительном варианте осуществления в соединениях формулы III:
R2, R4 и R5 все обозначают водород,
R6 обозначает хлор или дифторметил,
X1 обозначает N или C(R7),
R7, если он содержится, выбран из группы, включающей водород, фтор, хлор и фторметил, предпочтительно обозначает водород,
R10 выбран из группы, включающей хлор и С1-С2-алкоксигруппу, где алкоксигруппа замещена с помощью вплоть до 3 атомов фтора или одной фторметоксигруппой,
R11 обозначает водород или фтор, и
R12 обозначает водород или фтор, предпочтительно водород,
и их фармацевтически приемлемые соли, сольваты, изотопы и совместные кристаллы.
Один особенно предпочтительный вариант осуществления относится к соединениям формулы I, в которой XI обозначает -C(R7)-, таким образом, описывающимся формулой IV

в которой другие заместители являются такими, как описано выше в настоящем изобретении для формулы I.
В одном варианте осуществления в соединениях формулы IV:
R2, R4 и R5 все обозначают водород,
R6 выбран из группы, включающей галоген, цианогруппу, метил, метоксигруппу, фторметоксигруппу, фторметил и бензилоксигруппу,
R7 выбран из группы, включающей водород, галоген, фторметоксигруппу и фторметил, и предпочтительно обозначает водород или фтор,
Х2 обозначает N или C(R12),
R8 выбран из группы, включающей водород, фтор, С1-С2-алкоксигруппу и фтор-С1-С2-алкоксигруппу,
R10 выбран из группы, включающей галоген, циклопропил, C1-С2-алкил и С1-С2-алкоксигруппу, где циклопропил, алкил и алкоксигруппа все необязательно могут содержать один или большее количество заместителей, выбранных из группы, включающей фтор, метоксигруппу и фтор-С1-С2-алкоксигруппу,
R11 выбран из группы, включающей водород, метоксигруппу и фтор, предпочтительно из группы, включающей водород и фтор,
R12, если он содержится, выбран из группы, включающей водород, метоксигруппу и фтор,
и их фармацевтически приемлемые соли, сольваты, изотопы и совместные кристаллы.
В одном варианте осуществления в соединениях формулы IV:
R2, R4 и R5 все обозначают водород, R6 обозначает хлор или фторметил,
R7 выбран из группы, включающей водород, фтор, хлор, циклопропилоксигруппу, циклопропил, фторметоксигруппу и фторметил,
R8 выбран из группы, включающей водород, фтор и метоксигруппу,
R10 выбран из группы, включающей галоген С1-С2-алкил и С1-С2-алкоксигруппу, где алкил и алкоксигруппа необязательно могут содержать один или большее количество заместителей, выбранных из группы, включающей фтор, цианогруппу и фтор-С1-С2-алкоксигруппу,
R11 выбран из группы, включающей водород, метоксигруппу и фтор, предпочтительно из группы, включающей водород и фтор,
Х2 обозначает N или C(R12),
R12, если он содержится, выбран из группы, включающей водород, метоксигруппу и фтор,
и их фармацевтически приемлемые соли, сольваты, изотопы и совместные кристаллы.
Один предпочтительный вариант осуществления относится к соединениям формулы IV, в которой
R2, R4 и R5 все обозначают водород,
R6 выбран из группы, включающей хлор, цианогруппу, метоксигруппу, фторметоксигруппу, метил и фторметил,
R7 выбран из группы, включающей водород, галоген, фторметил и фторметоксигруппу, и предпочтительно обозначает водород или фтор,
R8 выбран из группы, включающей фтор и метоксигруппу,
R10 выбран из группы, включающей галоген, циклопропил, метоксигруппу, фторметоксигруппу, фторэтоксигруппу, фторметоксиэтоксигруппу и фторэтоксиметоксигруппу,
R11 обозначает водород или фтор, и
Х2 обозначает N или C(R12),
R12, если он содержится, обозначает водород, метоксигруппу или фтор, предпочтительно водород,
и к их фармацевтически приемлемым солям, сольватам, изотопам и совместным кристаллам.
Один предпочтительный вариант осуществления относится к соединениям формулы IV, в которой
R2, R4 и R5 все обозначают водород,
R6 обозначает хлор или фторметил,
R7 выбран из группы, включающей водород, фтор, хлор, фторметил и циклопропилоксигруппу,
R8 выбран из группы, включающей фтор и метоксигруппу,
R10 выбран из группы, включающей хлор, бром, метоксигруппу, фторметоксигруппу, фторэтоксигруппу, фторметоксиэтоксигруппу и фторэтоксиметоксигруппу,
R11 обозначает водород или фтор, и
Х2 обозначает N или C(R12),
R12, если он содержится, обозначает водород, метоксигруппу или фтор, предпочтительно водород,
и к их фармацевтически приемлемым солям, сольватам, изотопам и совместным кристаллам.
Один предпочтительный вариант осуществления относится к соединениям формулы IV, в которой
R2, R4 и R5 все обозначают водород,
R6 выбран из группы, включающей хлор, метоксигруппу, фторметоксигруппу, метил или фторметил,
R7 выбран из группы, включающей водород, фтор, хлор и фторметил, и предпочтительно обозначает водород,
R8 выбран из группы, включающей фтор и метоксигруппу,
R10 выбран из группы, включающей хлор, бром и С1-С2-алкоксигруппу, где алкоксигруппа необязательно и предпочтительно замещена с помощью 1-3 атомов фтора или одной фторметоксигруппой,
R11 обозначает водород или фтор, предпочтительно фтор,
Х2 обозначает N или C(R12), и
где R12, если он содержится, обозначает водород или фтор, предпочтительно водород,
и к их фармацевтически приемлемым солям, сольватам, изотопам и совместным кристаллам.
Один предпочтительный вариант осуществления относится к соединениям формулы IV, в которой
R2, R4 и R5 все обозначают водород,
R6 обозначает хлор или дифторметил,
R7 выбран из группы, включающей водород, фтор, хлор и фторметил, предпочтительно обозначает водород,
R8 выбран из группы, включающей фтор и метоксигруппу,
R10 выбран из группы, включающей хлор и С1-С2-алкоксигруппу, где алкоксигруппа замещена с помощью вплоть до 3 атомов фтора или одной фторметоксигруппой,
R11 обозначает водород или фтор,
Х2 обозначает N или C(R12), и
где R12, если он содержится, обозначает водород или фтор, предпочтительно водород,
и к их фармацевтически приемлемым солям, сольватам, изотопам и совместным кристаллам.
Один предпочтительный вариант осуществления относится к соединениям формулы I, II, III или IV, в которой R7 обозначает водород.
Один вариант осуществления относится к соединениям формулы I, в которой X1 обозначает N, таким образом, описывающимся формулой V

в которой все заместители являются такими, как описано выше в настоящем изобретении для формулы I.
В одном варианте осуществления в соединениях формулы V:
R2, R4 и R5 все обозначают водород,
R6 выбран из группы, включающей галоген, цианогруппу, метил, метоксигруппу, фторметоксигруппу, фторметил и бензилоксигруппу,
Х2 обозначает N или C(R12),
R8 выбран из группы, включающей водород, фтор, С1-С2-алкоксигруппу и фтор-С1-С2-алкоксигруппу,
R10 выбран из группы, включающей галоген, циклопропил, С1-С2-алкил и С1-С2-алкоксигруппу, где циклопропил, алкил и алкоксигруппа все необязательно могут содержать один или большее количество заместителей, выбранных из группы, включающей фтор, метоксигруппу и фтор-С1-С2-алкоксигруппу,
R11 выбран из группы, включающей водород, метоксигруппу и фтор, предпочтительно из группы, включающей водород и фтор,
R12, если он содержится, выбран из группы, включающей водород, метоксигруппу и фтор,
и их фармацевтически приемлемые соли, сольваты, изотопы и совместные кристаллы.
В одном варианте осуществления в соединениях формулы V:
R2, R4 и R5 все обозначают водород, R6 обозначает хлор или фторметил,
R8 выбран из группы, включающей водород, фтор и метоксигруппу,
R10 выбран из группы, включающей галоген С1-С2-алкил и С1-С2-алкоксигруппу, где алкил и алкоксигруппа необязательно могут содержать один или большее количество заместителей, выбранных из группы, включающей фтор, цианогруппу и фтор-С1-С2-алкоксигруппу,
R11 выбран из группы, включающей водород, метоксигруппу и фтор, предпочтительно из группы, включающей водород и фтор,
Х2 обозначает N или C(R12),
R12 выбран из группы, включающей водород, метоксигруппу и фтор,
и их фармацевтически приемлемые соли, сольваты, изотопы и совместные кристаллы.
Один предпочтительный вариант осуществления относится к соединениям формулы V, в которой
R2, R4 и R5 все обозначают водород,
R6 выбран из группы, включающей хлор, цианогруппу, метоксигруппу, фторметоксигруппу, метил и фторметил,
R8 выбран из группы, включающей фтор и метоксигруппу,
R10 выбран из группы, включающей галоген, циклопропил, метоксигруппу, фторметоксигруппу, фторэтоксигруппу, фторметоксиэтоксигруппу и фторэтоксиметоксигруппу,
R11 обозначает водород или фтор, и
Х2 обозначает N или C(R12),
R12, если он содержится, обозначает водород, метоксигруппу или фтор, предпочтительно водород,
и к их фармацевтически приемлемым солям, сольватам, изотопам и совместным кристаллам.
Один предпочтительный вариант осуществления относится к соединениям формулы V, в которой
R2, R4 и R5 все обозначают водород,
R6 обозначает хлор или фторметил,
R8 обозначает метоксигруппу,
R10 выбран из группы, включающей хлор, бром, метоксигруппу, фторметоксигруппу, фторэтоксигруппу, фторметоксиэтоксигруппу и фторэтоксиметоксигруппу,
R11 обозначает водород или фтор, предпочтительно фтор,
Х2 обозначает N или C(R12),
где R12, если он содержится, выбран из группы, включающей водород, метоксигруппу и фтор,
и к их фармацевтически приемлемым солям, сольватам, изотопам и совместным кристаллам.
Один предпочтительный вариант осуществления относится к соединениям формулы V, в которой
R2, R4 и R5 все обозначают водород,
R6 выбран из группы, включающей хлор, метоксигруппу, фторметоксигруппу и фторметил,
R8 выбран из группы, включающей фтор и метоксигруппу,
R10 выбран из группы, включающей хлор, бром и С1-С2-алкоксигруппу, где алкоксигруппа необязательно и предпочтительно замещена с помощью 1-3 атомов фтора или одной фторметоксигруппой или фторэтоксигруппой,
R11 обозначает водород или фтор, предпочтительно фтор,
Х2 обозначает C(R12), где R12 обозначает водород,
и к их фармацевтически приемлемым солям, сольватам, изотопам и совместным кристаллам.
Один предпочтительный вариант осуществления относится к соединениям формулы V, в которой
R2, R4 и R5 все обозначают водород,
R6 обозначает хлор или дифторметил, предпочтительно хлор,
R8 выбран из группы, включающей фтор и метоксигруппу,
R10 выбран из группы, включающей хлор и С1-С2-алкоксигруппу, где алкоксигруппа замещена с помощью вплоть до 3 атомов фтора или одной фторметоксигруппой,
R11 обозначает водород или фтор,
Х2 обозначает C(R12), где R12 выбран из группы, включающей водород, метоксигруппу и фтор,
и к их фармацевтически приемлемым солям, сольватам, изотопам и совместным кристаллам.
В одном предпочтительном варианте осуществления соединений формулы I, II, III, IV или V:
R2, R4 и R5 все обозначают водород,
R8 обозначает водород или фтор, предпочтительно фтор,
R11 обозначает метоксигруппу, Х2 обозначает C(R12) и R12 обозначает водород,
и Х1, R6 и R10 являются такими, как определено в вариантах осуществления, приведенных выше в настоящем изобретении.
Один вариант осуществления относится к соединениям формулы I, в которой R6 и R7 вместе с атомами С, к которым они присоединены, образуют 5-или 6-членное кольцо, как это представлено в формуле VI

в которой
n равно от 0 до 3, предпочтительно 0 или 1,
Х3 обозначает СН или N,
R2 обозначает водород или галоген, предпочтительно водород или фтор,
R4 обозначает водород или фтор,
R5 обозначает водород или галоген,
R8 выбран из группы, включающей водород, галоген, метоксигруппу, этоксигруппу, фторметоксигруппу и фторэтоксигруппу,
R10 выбран из группы, включающей водород, галоген, С3-С5-циклоалкил, С3-С5-циклоалкилоксигруппу, С3-С5-циклоалкилметоксигруппу, С1-С4-алкоксигруппу и С1-С4-алкил, где каждый из следующих: циклоалкил, циклоалкилоксигруппа, алкил и алкоксигруппа, может содержать один или большее количество заместителей, выбранных из группы, включающей галоген, С1-С3-алкоксигруппу, фтор(С1-С3)алкоксигруппу и цианогруппу,
R11 выбран из группы, включающей водород, фтор и метоксигруппу,
Х2 обозначает N или C(R12),
R12 выбран из группы, включающей водород, метоксигруппу и галоген,
R13 в каждом случае независимо выбран из группы, включающей галоген, цианогруппу, гидроксигруппу, метил, метоксигруппу, фторметил и фторметоксигруппу,
и к их фармацевтически приемлемым солям, сольватам, изотопам и совместным кристаллам.
Один вариант осуществления относится к соединению формулы VI, в которой
n равно 0,
Х3 обозначает N или СН,
R2 обозначает водород,
R4 обозначает водород,
R5 обозначает водород или галоген, предпочтительно водород,
R8 выбран из группы, включающей водород, фтор и метоксигруппу,
R10 выбран из группы, включающей галоген, циклопропил и С1-С2-алкоксигруппу, где алкоксигруппа необязательно может содержать один или большее количество заместителей, выбранных из группы, включающей фтор, метоксигруппу, этоксигруппу и фтор-С1-С2-алкоксигруппу,
R11 обозначает водород или фтор,
Х2 обозначает N или C(R12), и
R12, если он содержится, выбран из группы, включающей водород, метоксигруппу и фтор,
и к его фармацевтически приемлемым солям, сольватам, изотопам и совместным кристаллам.
Один вариант осуществления относится к соединению формулы VI, в которой
n равно 0, 1 или 2, предпочтительно 0 или 1,
Х3 обозначает N,
R2, R4 и R5 все обозначают водород,
R8 выбран из группы, включающей водород, фтор и метоксигруппу,
R10 выбран из группы, включающей галоген, циклопропил и С1-С2-алкоксигруппу, где алкоксигруппа необязательно может содержать один или большее количество заместителей, выбранных из группы, включающей фтор, метоксигруппу, этоксигруппу и фтор-С1-С2-алкоксигруппу,
R11 обозначает водород или фтор,
Х2 обозначает Ν,
R13 в каждом случае выбран из группы, включающей галоген, гидроксигруппу, метоксигруппу, фторметоксигруппу, метил и фторметил,
и к его фармацевтически приемлемым солям, сольватам, изотопам и совместным кристаллам.
В одном особенно предпочтительном варианте осуществления соединений формулы VI: n равно 0 и Х3 обозначает N.
В одном предпочтительном варианте осуществления соединений формулы I, II, III, IV, V или VI: R11 выбран из группы, включающей водород и фтор.
В одном предпочтительном варианте осуществления соединений формулы I, II, III, IV, V или VI: R11 обозначает фтор.
В одном предпочтительном варианте осуществления соединений формулы I, II, III, IV, V или VI: R8 обозначает метоксигруппу, R11 обозначает фтор и R12, если он содержится, обозначает водород.
В одном особенно предпочтительном варианте осуществления соединений формулы I, II, III, IV, V или VI: R2, R4 и R5 все обозначают водород, R8 обозначает метоксигруппу, R11 обозначает фтор и R12, если он содержится, обозначает водород, и другие заместители являются такими, как описано в настоящем изобретении.
В одном особенно предпочтительном варианте осуществления соединений формулы I, II, III, IV или V: R2, R4, R5 и R7, если он содержится, все обозначают водород, R8 обозначает метоксигруппу, R11 обозначает фтор и R12, если он содержится, обозначает водород, и другие заместители являются такими, как описано в настоящем изобретении.
В одном варианте осуществления соединений формулы I, II, III, IV, V или VI: R12 обозначает метоксигруппу, R8 обозначает фтор и R11 выбран из группы, включающей водород и фтор.
В одном предпочтительном варианте осуществления соединений формулы I, II, III, IV, V или VI: R12 обозначает водород.
Один вариант осуществления относится к любому конкретному модулятору GPR17, раскрытому в настоящем изобретении включая, но не ограничиваясь только ими, описанные в экспериментальном разделе и указанные в таблице 7, приведенной в настоящем изобретении.
Один предпочтительный вариант осуществления относится к соединению, выбранному из группы, включающей
6-хлор-N-[6-(2,2-дифторэтокси)-2,5-дифторпиридин-3-ил]-1H-индол-3-сульфонамид,
N-[6-(дифторметокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-пирроло[3,2-h]хинолин-3-сульфонамид,
N-[6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-пирроло[3,2-h]хинолин-3-сульфонамид,
5-бром-6-хлор-N-[6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
6-хлор-N-[6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-7-фтор-1Н-индол-3-сульфонамид,
6-циано-N-[6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-1H-индол-3-сульфонамид,
N-[6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-6-(дифторметокси)-1Н-индол-3-сульфонамид,
N-[6-[2-(дифторметокси)этокси]-5-фтор-2-метоксипиридин-3-ил]-6-(дифторметил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
6-хлор-N-[6-[2-(дифторметокси)этокси]-5-фтор-2-метоксипиридин-3-ил]-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
N-[6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-6-(дифторметил)-1H-индол-3-сульфонамид,
6-хлор-N-(6-циклопропил-5-фтор-2-метоксипиридин-3-ил)-1H-индол-3-сульфонамид,
6-хлор-N-(5-фтор-2-метоксипиридин-3-ил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
N-[6-(дифторметокси)-5-фтор-2-метоксипиридин-3-ил]-6-метокси-1H-индол-3-сульфонамид,
N-[6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-6-метокси-1H-индол-3-сульфонамид,
6-хлор-N-[6-[2-(дифторметокси)этокси]-5-фтор-2-метоксипиридин-3-ил]-1Н-индол-3-сульфонамид,
N-[6-(дифторметокси)-5-фтор-2-метоксипиридин-3-ил]-6-метил-1H-индол-3-сульфонамид,
6-циано-N-[6-(дифторметокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-индол-3-сульфонамид,
6-(дифторметил)-N-(2,5-дифтор-6-метилпиридин-3-ил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
6-(дифторметил)-N-(5-фтор-2,6-диметоксипиридин-3-ил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
6-(дифторметил)-N-[5-фтор-6-(2-фторэтокси)-2-метоксипиридин-3-ил]-1H-пирроло[2,3-b]пиридин-3-сульфонамид,
6-хлор-N-(6-циклопропил-2,5-дифторпиридин-3-ил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
6-хлор-N-[6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-индол-3-сульфонамид,
6-хлор-N-(6-циклопропил-2,5-дифторпиридин-3-ил)-1H-индол-3-сульфонамид,
N-[6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-6-(дифторметил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
6-хлор-N-[6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
N-[6-(2,2-дифторэтокси)-2-метоксипиридин-3-ил]-6-(дифторметил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
6-хлор-N-[6-(2,2-дифторэтокси)-2-метоксипиридин-3-ил]-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
N-[6-(2,2-дифторэтокси)-2,5-дифторпиридин-3-ил]-6-(дифторметил)-1Н-индол-3-сульфонамид,
6-хлор-N-[6-(дифторметокси)-4-метоксипиридин-3-ил]-1Н-индол-3-сульфонамид,
6-хлор-N-[6-(2,2-дифторэтокси)-2-метоксипиридин-3-ил]-1Н-индол-3-сульфонамид,
6-хлор-N-[2-(2,2-дифторэтокси)-6-метоксипиридин-3-ил]-1Н-индол-3-сульфонамид,
N-[6-(дифторметокси)-5-фтор-2-метоксипиридин-3-ил]-6-(дифторметил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
6-хлор-N-[6-(дифторметокси)-2-метокси-3-пиридил]-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
N-(6-хлор-5-фтор-2-метоксипиридин-3-ил)-6-(дифторметил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
6-хлор-N-[6-(2-фторэтокси)-2-метоксипиридин-3-ил]-1Н-индол-3-сульфонамид,
6-хлор-N-[5-фтор-6-(2-фторэтокси)-2-метоксипиридин-3-ил]-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
6-хлор-N-[5-фтор-6-(2-фторэтокси)-2-метоксипиридин-3-ил]-1Н-индол-3-сульфонамид,
N-[6-(дифторметокси)-5-фтор-2-метоксипиридин-3-ил]-6-(дифторметил)-1Η-индол-3-сульфонамид,
6-хлор-N-[6-(дифторметокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
6-хлор-N-(5-хлор-3-метоксипиразин-2-ил)-1Н-индол-3-сульфонамид,
6-хлор-N-[6-(дифторметокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-индол-3-сульфонамид,
N-(5-бром-3-метоксипиразин-2-ил)-6-хлор-1Н-индол-3-сульфонамид,
6-хлор-N-(2,5-дифтор-6-метилпиридин-3-ил)-1Н-индол-3-сульфонамид,
6-хлор-N-(5-фтор-2,6-диметоксипиридин-3-ил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
6-хлор-N-(6-хлор-5-фтор-2-метоксипиридин-3-ил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
6-хлор-N-(5-фтор-2,6-диметоксипиридин-3-ил)-1Н-индол-3-сульфонамид,
6-хлор-N-(2,5-дифтор-6-метоксипиридин-3-ил)-1Н-индол-3-сульфонамид,
6-хлор-N-(6-хлор-5-фтор-2-метоксипиридин-3-ил)-1Н-индол-3-сульфонамид,
6-хлор-N-(6-хлор-2,5-дифторпиридин-3-ил)-1Н-индол-3-сульфонамид,
6-хлор-N-(6-йодпиридин-3-ил)-1Н-индол-3-сульфонамид,
6-хлор-N-(6-хлор-4-фторпиридин-3-ил)-1Н-индол-3-сульфонамид,
и к его фармацевтически приемлемым солям, сольватам, изотопам и совместным кристаллам.
Другой предпочтительный вариант осуществления относится к соединениям, обладающим структурой формулы I, II, III, IV, V или VI, определенной в настоящем изобретении, или к любому соединению, отдельно раскрытому в настоящем изобретении, предпочтительно к любому из соединений I-1 - I-72, и содержащему по меньшей мере один изотоп 18F, предпочтительно в положении атома фтора, указанном в одном из соединений, раскрытых в настоящем изобретении. В качестве неограничивающего примера можно привести следующий: в соединении 6-хлор-N-(6-хлор-2,5-дифторпиридин-3-ил)-1Н-индол-3-сульфонамид, раскрытом в настоящем изобретении, по меньшей мере один из двух атомов фтора может быть представлен изотопом 18F. Это также относится к другим содержащим фтор соединениям, описанным в настоящем изобретении. Эти содержащие 18F соединения предпочтительно можно использовать в качестве предназначенных для ПЭТ (позитронная эмиссионная томография) радиоактивных индикаторов.
Другой предпочтительный вариант осуществления относится к соединениям, обладающим структурой формулы I, II, III, IV, V или VI, определенной в настоящем изобретении, или к любому соединению, отдельно раскрытому в настоящем изобретении, предпочтительно к любому из соединений I-1 - I-72, и содержащему по меньшей мере один изотоп 11С, предпочтительно в положении атома углерода, указанном в настоящем изобретении. Эти содержащие 11С соединения предпочтительно можно использовать в качестве предназначенных для ПЭТ радиоактивных индикаторов.
Другой предпочтительный вариант осуществления относится к соединениям, обладающим структурой формулы I, II, III, IV, V или VI, определенной в настоящем изобретении, или к любому соединению, отдельно раскрытому в настоящем изобретении, предпочтительно к любому из соединений I-1 - I-72, и содержащему по меньшей мере один изотоп 123I, 125I или 131I, предпочтительно в положении атома иода, указанном в настоящем изобретении. В качестве неограничивающего примера можно привести следующий: в соединении 6-хлор-N-(6-йодпиридин-3-ил)-1Н-индол-3-сульфонамид, раскрытом в настоящем изобретении, атом йода может быть представлен изотопом 123I, 125I или 131I. Содержащие 123I, 125I или 131I соединения предпочтительно можно использовать в качестве предназначенных для ОФЭКТ (однофотонная эмиссионная компьютерная томография) радиоактивных индикаторов.
Применение для лечения и диагностики
В одном варианте осуществления настоящее изобретение относится к любому из соединений, описанных в настоящем изобретении, предназначенному для применения для лечения или диагностики, предпочтительно для лечения животных, особенно предпочтительно людей.
Вследствие их способности модулировать GPR17 соединения, предлагаемые в настоящем изобретении, можно применять в качестве лекарственного средства и их можно применять для лечения и/или предупреждения различных заболеваний ЦНС.
Таким образом, одним вариантом осуществления настоящего изобретения является соединение, описанное в настоящем изобретении, предназначенное для применения в качестве лекарственного средства, предпочтительно предназначенное для применения в качестве лекарственного средства для лечения и/или предупреждения связанного с GPR17 заболевания.
Связанное с GPR17 заболевание или нарушение означает заболевание, которое связано с нарушением функции сигнальной системы GPR17, например, с сверхэкспрессированием и/или гиперактивностью рецепторов GPR17. Если не ограничиваться какими-либо теоретическими соображениями, то можно предположить, что активность GPR17 может повыситься, может расшириться ее диапазон или она другим образом может измениться в определенных тканях, например, в клетках-предшественниках олигодендроцитов (КПО) или во время созревания олигодендроцитов, вероятно, благодаря активации эндогенными стимулами, такими как, например, воспалительные факторы. Высокая активность GPR17 может препятствовать дифференциации олигодендроцитов и эффективной миелинизации и, таким образом, способствовать возникновению и дальнейшему развитию заболевания, связанного с нарушением миелинизации (см. публикацию Chen et al, приведенную выше). Таким образом, негативные модуляторы GPR17 могут способствовать миелинизации путем уменьшения или прекращения активности GPR17 и путем поддерживания созревания КПО с образованием продуцирующих миелин олигодендроцитов (см., например, публикацию Simon et al, приведенную выше).
В одном предпочтительном варианте осуществления настоящее изобретение относится к любому из соединений, описанных в настоящем изобретении, предназначенному для применения для лечения или диагностики, предназначенных для применения для предупреждения или лечения нарушения или синдрома, выбранного из числа нарушений миелинизации и/или связанных с ними, предпочтительно демиелинизирующего нарушения, например, центральной нервной системы. В одном варианте осуществления соединения, предлагаемые в настоящем изобретении, предназначены для применения для поддержки, стимуляции и/или ускорения протекания повторной миелинизации у нуждающегося в нем животного. В одном варианте осуществления повторная миелинизация, связанная с введением соединения, предлагаемого в настоящем изобретении, обеспечивает предупреждение или лечение демиелинизирующего заболевания, такого как, но не ограничиваясь только им, рассеянный склероз.
Соединения, предлагаемые в настоящем изобретении, также можно применять для лечения или предупреждения нарушения или синдрома, связанного с повреждением тканей головного мозга, церебрально-васкулярного нарушения и определенных нейродегенеративных заболеваний.
Недавно было показано, что нейродегенеративные нарушения связаны с уменьшением количества миелина. В соответствии с этим полагают, что сохранение функциональности олигодендроцитов и миелина является важным условием для предупреждения аксональной и нейронной дегенерации (см., например, публикацию Ettle et al, приведенную выше). Таким образом, негативные модуляторы GPR17 могут представлять собой превосходные возможные средства для лечения любого нейродегенеративного заболевания, связанного с демиелинизацией и/или нарушенной миелинизацией, такого как например, БАС (боковой амиотрофический склероз), МСА (мультисистемная атрофия), болезнь Альцгеймера, болезнь Гентингтона или болезнь Паркинсона.
Таким образом, в особенно предпочтительном варианте осуществления соединения, предлагаемые в настоящем изобретении, можно применять для предупреждения и/или лечения нарушения миелинизации периферической или центральной нервной системы, предпочтительно нарушения миелинизации центральной нервной системы. В одном варианте осуществления соединения, предлагаемые в настоящем изобретении, предназначены для применения для лечения и/или предупреждения и/или диагностики нарушения миелинизации путем перорального введения. В предпочтительном варианте осуществления нарушением миелинизации, которое лечат соединениями, предлагаемыми в настоящем изобретении, является демиелинизирующее нарушение.
Примерами таких нарушений миелинизации, которые лечат и/или предупреждают соединениями, раскрытыми в настоящем изобретении, являются, в частности,
- рассеянный склероз (PC), включая его различные субформы,
- нейромиелит зрительного нерва (также известный, как болезнь Девика),
- хронический рецидивирующий воспалительный неврит зрительного нерва, острый рассеянный энцефаломиелит,
- острый геморрагический лейкоэнцефалит (ОГЛ),
- перивентрикулярная лейкомаляция,
- демиелинизация вследствие вирусных инфекций, например, вызванных ВИЧ (вирус иммунодефицита человека), или прогрессивной мультифокальной лейкоэнцефалопатии,
- центральный понтинный и экстрапонтинный миелинолиз,
- демиелинизация вследствие травматического поражения тканей головного мозга, включая вызванную сжатием демиелинизацию, например, вследствие опухолей,
- демиелинизация в ответ на гипоксию, удар или ишемию, или другие сердечнососудистые заболевания,
- демиелинизация вследствие воздействия диоксида углерода, цианида или других токсинов, поражающих ЦНС,
- болезнь Шильдера,
- концентрический склероз Бало,
- перинатальная энцефалопатия,
- нейродегенеративные заболевания, включая, в частности,
 боковой амиотрофический склероз (БАС),
боковой амиотрофический склероз (БАС),
 болезнь Альцгеймера (БА),
болезнь Альцгеймера (БА),
 мультисистемную атрофию,
мультисистемную атрофию,
болезнь Паркинсона,
спинально-церебеллярную атаксию (СЦА), также известную, как спинально-церебеллярная атрофия,
болезнь Гентингтона,
- психические нарушения, такие как шизофрения и биполярное расстройство,
- заболевания, связанные с нарушением миелинизации периферической нервной системы, такие как лейкодистрофии, периферические демиелинизирующие невропатии, синдром Дежерина-Соттаса или болезнь Шарко-Мари-Тута.
Лечение или предупреждение заболевания ЦНС, такого как демиелинизирующее заболевание, также включает лечение признаков и симптомов, связанных с таким заболеванием.
Так, например, применение соединений, предлагаемых в настоящем изобретении, для лечения и/или предупреждения PC также включает лечение и/или предупреждение признаков и симптомов, связанных с PC, таких как негативные проявления со стороны зрительных нервов (потеря зрения, двойное зрение), задних столбов спинного мозга (потеря чувствительности), корково-спинномозгового пирамидного пути (спастическая слабость), церебеллярных путей (отсутствие координации, дизартрия, вертиго, нарушения познавательной способности), медиального продольного пучка (двойное зрение при боковом взгляде), спинномозгового пути тройничного нерва (онемение лица или боль), мышечная слабость (нарушение глотания, нарушение контроля над мочевым пузырем или кишечником, спазмы), или психологических нарушений, связанных с основным заболеванием, таких как депрессия, состояние тревоги или другие расстройства настроения, общая слабость или бессонница.
Таким образом, соединения, предлагаемые в настоящем изобретении, предназначены для применения для лечения признаков и симптомов заболевания, связанного с нарушением миелинизации, предпочтительно демиелинизирующего заболевания, такого как рассеянный склероз, где такие признаки и симптомы включают, но не ограничиваются только ими, выбранные из группы, включающей потерю зрения, нарушение зрения, двойное зрение, потерю или нарушение чувствительности, слабость, такую как спастическая слабость, отсутствие моторной координации, вертиго, нарушения познавательной способности, онемение лица, боль в лице, нарушение глотания, нарушение речи, нарушение контроля над мочевым пузырем и/или кишечником, спазмы, депрессию, состояние тревоги, расстройства настроения, бессонницу и усталость.
В одном предпочтительном варианте осуществления соединения, предлагаемые в настоящем изобретении, предназначены для применения для лечения рассеянного склероза. PC является заболеванием, связанным с гетерогенным нарушением миелинизации, и может проявляться в целом ряде разных форм и стадий, включая, но не ограничиваясь только ими, рецидивирующий-ремитирующий PC, вторично-прогрессирующий PC, первично-прогрессирующий PC, прогрессирующий-рецидивирующий PC, каждая из которых зависит от активности и развития заболевания. Поэтому в одном варианте осуществления соединения, предлагаемые в настоящем изобретении, предназначены для лечения рассеянного склероза, проявляющегося в разных стадиях и формах, описанных в настоящем изобретении.
В одном варианте осуществления соединения, предлагаемые в настоящем изобретении, предназначены для применения для лечения или предупреждения нейромиелита зрительного нерва (также известного, как болезнь Девика или синдром Девика). Нейромиелит зрительного нерва является сложным нарушением, характеризующимся воспалением и демиелинизацией зрительного нерва и спинного мозга. Многие из связанных с этим нарушением симптомов сходны с симптомами PC и включают мышечную слабость, в особенности, в конечностях, пониженную чувствительность и потерю контроля над мочевым пузырем.
В одном варианте осуществления соединения, предлагаемые в настоящем изобретении, предназначены для применения для предупреждения и/или лечения БАС. Недавно показано, что БАС связан с дегенерацией олигодендроцитов и повышенной демиелинизацией, это означает, что БАС является целевым заболеванием для негативных модуляторов GPR17 (см. публикации Kang et al, приведенную выше; Fumagalli et al, Neuropharmacology 104, 2016, 82).
В одном варианте осуществления соединения, предлагаемые в настоящем изобретении, предназначены для применения для предупреждения и/или лечения болезни Гентингтона. Подробно описано, что болезнь Гентингтона связана с нарушенной миелинизацией (см. публикации Bartzokis et al, приведенную выше; Huang et al, Neuron 85, 2015, 1212).
В одном варианте осуществления соединения, предлагаемые в настоящем изобретении, предназначены для применения предупреждения и/или лечения мультисистемной атрофии. Недавно показано, что МСА сильно связана с демиелинизацией (см. публикации Ettle, приведенную выше, Jellinger, приведенную выше), на основании этого предполагается, что с помощью повторной миелинизации можно лечить или предупреждать МСА.
В одном варианте осуществления соединения, предлагаемые в настоящем изобретении, предназначены для применения предупреждения и/или лечения, болезни Альцгеймера. Недавно установлено, что БА связана с усиленным некрозом клеток-олигодендроцитов и фокальной демиелинизацией и они представляют патологические процессы при БА (Mitew et al, Acta Neuropathol 119, 2010, 567).
Одним объектом настоящего изобретения является способ лечения любого из заболеваний или нарушений, описанных в настоящем изобретении, предпочтительно заболеваний, связанных с нарушением миелинизации, таких как PC, нейромиелит зрительного нерва, БАС, хорея Гентингтона, болезнь Альцгеймера или другие, путем введения нуждающемуся в нем субъекту, включая являющегося человеком пациента, соединения, предлагаемого в настоящем изобретении, в терапевтически эффективном количестве.
В другом варианте осуществления соединение, предлагаемое в настоящем изобретении, можно применять для предупреждения и лечения повреждения спинного мозга, перинатальной энцефалопатии, удара, ишемии или церебрально-васкулярного нарушения.
Одним объектом настоящего изобретения является способ предупреждения и/или лечения синдрома или нарушения, связанного с нарушением миелинизации, или синдрома или нарушения, связанного с повреждением тканей головного мозга, который включает введение нуждающемуся в нем пациенту соединения, описанного в настоящем изобретении, в терапевтически эффективном количестве. Нуждающимся в таком лечении пациентом может являться любой пациент, который страдает от повреждения тканей головного мозга, такого как механическое, химическое, вирусное или другое повреждение.
Одним объектом настоящего изобретения является способ предупреждения и/или лечения синдрома или нарушения, связанного с нарушением миелинизации, или синдрома или нарушения, связанного с ударом или другой ишемией головного мозга, который включает введение нуждающемуся в нем пациенту соединения, описанного в настоящем изобретении, в терапевтически эффективном количестве. Нуждающимся в предупреждении и/или лечении пациентом может являться любой пациент, который недавно подвергся ишемии головного мозга/удару, которые могли быть вызваны, например, непроходимостью мозговой артерии, вызванной эмболом или локальным тромбозом.
Недавно было показано, что GPR17 также связан с потреблением пищи, регулированием инсулина и ожирением. В разных публикациях описано, что негативные модуляторы GPR17 могут являться полезными для регулирования потребления пищи и для лечения ожирения (см., например, публикацию Ren et al, Diabetes 64, 2015; 3670). Поэтому в одном варианте осуществления настоящее изобретение относится к применению соединений, предлагаемых в настоящем изобретении, для предупреждения и/или лечения ожирения, и к способам лечения ожирения.
Кроме того, соединения, предлагаемые в настоящем изобретении, можно применять для лечения или предупреждения нарушения функции тканей, в которых экспрессируется GPR17, таких как, например, сердце, легкие или почки. В одном варианте осуществления соединения, предлагаемые в настоящем изобретении, можно применять для лечения или предупреждения ишемических нарушений почек и/или сердца.
Показано, что GPR17 также связан с легочным воспалением и астмой, такой как, например, вызванная клещом домашней пыли (Maekawa, J Immunol 2010, 185(3), 1846-1854). Поэтому соединения, предлагаемые в настоящем изобретении, можно применять для лечения астмы или другого легочного воспаления.
Пути лечения, предлагаемые в настоящем изобретении, могут включать введение одного из соединений, раскрытых в настоящем изобретении, в качестве "единственного средства" лечения заболевания ЦНС, предпочтительно нарушения миелинизации или связанного с ним заболевания, такого как PC или БАС. Альтернативно, соединение, раскрытое в настоящем изобретении, можно вводить вместе с другими полезными лекарственными средствами при проведении комбинированной терапии.
В качестве неограничивающего примера можно привести следующий: соединение, предлагаемое в настоящем изобретении, объединяют с другим лекарственным средством, предназначенным для лечения нарушения миелинизации, такого как PC, обладающим другим механизмом воздействия, таким как, например, противовоспалительное или иммунодепрессивное лекарственное средство. Такие соединения включают, но не ограничиваются только ими: (i) кортикостероиды, такие как преднизон, метилпреднизон или дексаметазон, (ii) бета-интерфероны, такие как интерферон бета-1а, интерферон бета-1b или пэгинтерферон бета-1a, (iii) антитела к CD20, такие как окрелизумаб, ритуксимаб и офатумумаб, (iv) соли глатирамера, такие как глатирамерацетат, (v) диметилфумарат, (vi) финголимод и другие модуляторы сфингозин-1-фосфатного рецептора, такие как понесимод, сипонимод, озанимод или лахинимод, (vii) ингибиторы дигидрооротатдегидрогеназы, такие как терифлуномид или лефлуномид, (viii) антитела к интегрину альфа-4, такие как натализумаб, (ix) антитела к CD52, такие как алемтузумаб, (х) митоксантрон, (xi) антитела к Lingo1, такие как опицинумаб, или (xii) другие иммуномодулирующие лекарственные средства, такие как мазитиниб.
Аналогичным образом, соединение, предлагаемое в настоящем изобретении, можно объединить с анальгетическим лекарственным средством, если проводят лечение болезненного патологического состояния, связанного с нарушением миелинизации. Кроме того, соединение, предлагаемое в настоящем изобретении, можно применять в комбинации с антидепрессантом для одновременного лечения психологических нарушений, связанных с основным подвергающемуся лечению заболевания, связанного с нарушением миелинизации.
При комбинированной терапии два или большее количество активных веществ могут быть включены в один и тот же препарат или представлять собой "набор компонентов", т.е. находиться в отдельных галеновых препаратах. Кроме того, два или большее количество активных веществ, включая соединения, предлагаемые в настоящем изобретении, можно вводить пациенту в одно и то же время или последовательно, например, при интервальной терапии. Дополнительное лекарственное средство можно вводить таким же путем или другим путем введения. Так, например, модулятор GPR17, предлагаемый в настоящем изобретении, можно вводить перорально, тогда как второе лекарственное средство можно вводить путем подкожной инъекции.
В одном варианте осуществления соединения, предлагаемые в настоящем изобретении, можно использовать для диагностики и/или мониторинга связанного с GPR17 заболевания, дополнительно описанного в настоящем изобретении, предпочтительно демиелинизирующего заболевания, раскрытого в настоящем изобретении, предпочтительно для диагностики и мониторинга рассеянного склероза.
В одном варианте осуществления соединения, предлагаемые в настоящем изобретении, можно применять для диагностики и/или мониторинга экспрессирования, распределения и/или активации рецептора GPR17 in vivo, например, непосредственно у субъекта, например, с использованием методик молекулярной визуализации, или in vitro, например, путем исследования любых образцов, таких как жидкости или ткани организма, взятые у субъекта. Результаты любого такого определения активности, экспрессирования и/или распределения GPR17 можно использовать для прогнозирования, диагностики и/или мониторинга (а) состояния и развития связанного с GPR17 заболевания, описанного в настоящем изобретении, предпочтительно заболевания, связанного с нарушением миелинизации, включая, но не ограничиваясь только ими, например, рассеянный склероз, и/или (b) эффективности и/или применимости лечения, проводимого в связи с любым таким связанным с GPR17 заболеванием, и/или надлежащей дозировки.
В одном варианте осуществления соединения, предлагаемые в настоящем изобретении, можно применять в качестве предназначенных для ПЭТ или ОФЭКТ радиоактивных индикаторов, дополнительно описанных в настоящем изобретении, для проведения диагностики и/или мониторинга заболевания in vivo. При этом экспрессирование, активацию и/или распределение рецептора GPR17 можно непосредственно определить у субъекта, например, путем визуализации организма человека после введения предназначенного для ПЭТ или ОФЭКТ радиоактивного индикатора GPR17, предлагаемого в настоящем изобретении. Это может ускорить надлежащую диагностику заболевания, может способствовать определению применимых возможностей лечения и/или это можно использовать для мониторинга развития заболевания и/или для мониторинга или прогнозирования успеха медицинского вмешательства, включая выбор подходящего пути введения и/или дозировки терапевтического лекарственного средства.
В одном варианте осуществления предназначенные для ПЭТ или ОФЭКТ радиоактивные индикаторы, предлагаемые в настоящем изобретении, можно использовать вместе с терапевтическим лекарственным средством, т.е. в качестве дополнительного средства для диагностики, для мониторинга и/или прогнозирования эффективности и/или безопасности указанного терапевтического лекарственного средства для конкретного субъекта или для определения подходящей дозировки лекарственного средства.
Один вариант осуществления относится к предназначенному для ПЭТ или ОФЭКТ радиоактивному индикатору, предлагаемому в настоящем изобретении, предназначенному для применения в качестве дополнительного лекарственного средства вместе с терапевтическим лекарственным средством. Терапевтическое лекарственное средство, использующееся вместе с предназначенным для ПЭТ или ОФЭКТ радиоактивным индикатором, предлагаемым в настоящем изобретении, может быть выбрано из группы, включающей (а) немеченое соединение, предлагаемое в настоящем изобретении, (b) модулирующее GPR17 соединение, которое отличается от соединений, предлагаемых в настоящем изобретении, и (с) лекарственное средство, предназначенное для лечения заболевания, связанного с нарушением миелинизации, включая, но не ограничиваясь только им, лекарственное средство, предназначенное для применения для лечения рассеянного склероза, которое не является модулятором GPR17.
Один вариант осуществления относится к набору, включающему
(a) в качестве первого компонента предназначенный для ПЭТ или ОФЭКТ радиоактивный индикатор, предлагаемый в настоящем изобретении, предпочтительно предназначенный для ПЭТ или ОФЭКТ радиоактивный индикатор, основанный на соединении, обладающем структурой, соответствующей любой из формул I, II, III, IV, V или VI, дополнительно определенных в настоящем изобретении, или обладающем структурой любого из соединений, раскрытых в настоящем изобретении, но содержащее включенный по меньшей мере один радионуклид, который является подходящим для визуализации с помощью ПЭТ или ОФЭКТ, предпочтительно радионуклид, выбранный из группы, включающей 18F, 11С, 123I, 125I и 131I.
(b) в качестве второго компонента терапевтическое лекарственное средство, выбранное из группы, включающей
i. соединение, предлагаемое в настоящем изобретении, обладающее структурой, соответствующей любой из формул I, II, III, IV, V или VI, дополнительно определенных в настоящем изобретении, или обладающее структурой любого из отдельных соединений, раскрытых в настоящем изобретении, и не содержащее включенного радионуклида,
ii. модулирующее GPR17 соединение, которое отличается от соединений, предлагаемых в настоящем изобретении, определенных в (i), и
iii. лекарственное средство, предназначенное для лечения заболевания, связанного с нарушением миелинизации, включая, но не ограничиваясь только им, лекарственное средство, предназначенное для применения для лечения рассеянного склероза, но не способные модулировать активность GPR17; такие соединения известны специалисту в данной области техники и они включают такие примеры, как дополнительно описанные выше.
Альтернативно, соединения, предлагаемые в настоящем изобретении, можно применять в проводимом in vitro диагностическом исследовании, например, для исследования любой степени экспрессирования, активности и/или распределения GPR17 в подходящих жидкостях организма субъекта, таких как, например, кровь, плазма, моча, слюна или спинномозговая жидкость.
Один вариант осуществления относится к способу лечения связанного с GPR17 заболевания, предпочтительно заболевания, связанного с нарушением миелинизации, включая, но не ограничиваясь только им, рассеянный склероз, где указанный способ включает стадии (а) определение степени экспрессирования, активности и/или распределения рецептора GPR17 у субъекта, (b) сравнение степени экспрессирования, активности и/или распределения рецептора GPR17 у указанного субъекта со степенью экспрессирования, активностью и/или распределением рецептора GPR17 у одного или большего количество здоровых субъектов или у популяции, (с) определение необходимости медицинского лечения или профилактики указанного субъекта на основании отклонения результатов, полученных для степени экспрессирования, активности и/или распределения рецептора GPR17 у указанного субъекта, от результатов, полученных для здоровых субъектов или популяции, и (d) лечение субъекта, у которого наблюдается отклонение результатов, полученных для степени экспрессирования, активности и/или распределения рецептора GPR17, путем введения терапевтического лекарственного средства указанному индивидууму, где лекарственное средство является подходящим для лечения связанных с GPR17 заболеваний или нарушений, предпочтительно путем введения модулятора GPR17, предпочтительно путем введения одного или большего количества соединений, предлагаемых в настоящем изобретении. В одном варианте осуществления (а) определение степени экспрессирования, активности и/или распределения рецептора GPR17 проводят с использованием одного из соединений, предлагаемых в настоящем изобретении, предпочтительно с использованием предназначенного для ПЭТ или ОФЭКТ радиоактивного индикатора, или путем проводимого in vitro исследования жидкостей или тканей организма указанного субъекта с использованием предназначенного для ПЭТ или ОФЭКТ радиоактивного индикатора, предлагаемого в настоящем изобретении.
В одном предпочтительном варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описанное в настоящем изобретении, и фармацевтически приемлемый носитель.
Для введения в качестве лекарственного средства соединения можно использовать в фармацевтической композиции, содержащей соединение, предлагаемое в настоящем изобретении, и фармацевтически приемлемый носитель, дополнительно определенный в настоящем изобретении. Такая фармацевтическая композиция может быть приспособлена, например, для перорального, внутривенного, внутримышечного, подкожного, назального, ректального, внутричерепного, глазного, трансбуккального или чрескожного введения и может содержать фармацевтически приемлемые носители, вспомогательные вещества, разбавители, стабилизаторы и т.п.
Так, например, соединения, предлагаемые в настоящем изобретении, можно растворить в маслах, пропиленгликоле или других растворителях, которые обычно используют для приготовления препаратов для инъекции. Примеры подходящих носителей включают, но не ограничиваются только ими, физиологический раствор, полиэтиленгликоль, этанол, растительные масла, изопропилмиристат и т.п. Соединения, предлагаемые в настоящем изобретении, можно приготовить в виде препаратов для инъекций путем растворения, суспендирования или эмульгирования в растворимом в воде растворителе, таком как физиологический раствор и 5% раствор декстрозы, или в нерастворимых в воде растворителях, таких как растительные масла, синтетические глицериды жирных кислот, эфиры высших жирных кислот и пропиленгликоль. Композиции, предлагаемые в настоящем изобретении, могут содержать любые обычные добавки, такие как растворяющие агенты, изотонические агенты, суспендирующие агенты, эмульгаторы, стабилизаторы и консерванты.
В одном варианте осуществления соединения, предлагаемые в настоящем изобретении, можно вводить перорально, например, в виде таблетки, капсулы, драже, порошка, гранулята, или в виде жидкого или полужидкого вещества, включая, но не ограничиваясь только ими, например, сиропы, суспензии, эмульсии или растворы.
Препараты для перорального введения, такие как таблетки, могут содержать, без наложения ограничений, агенты, обеспечивающие замедленное высвобождение, разрыхлители, наполнители, смазывающие вещества, стабилизаторы, антиоксиданты, вкусовые вещества, диспергирующие агенты, электролиты, буферы, красители или консерванты. Подходящие инертные наполнители и препараты известны специалистам в данной области техники и описаны в стандартных монографиях, таких как, например, Remington ("The science and practice of pharmacy", Lippincott, Williams & Wilkins, 2000), или раскрыты в других источниках, хорошо известных специалистам в данной области техники.
Таблетку можно приготовить, например, путем смешивания по меньшей мере одного соединения, предлагаемого в настоящем изобретении, по меньшей мере с одним нетоксичным фармацевтически приемлемым инертным наполнителем, таким как, например, связующее, наполнитель/разбавители, разрыхляющие агенты, пластификатор и т.п., и с необязательным растворителем (водным или неводным), и последующей обработки смеси с получением таблетки по методике, включая, но не ограничиваясь только ими, сухое прессование, сухое гранулирование, влажное гранулирование, распылительную сушку или экструзию расплава. Таблетка может не содержать покрытие или на нее можно нанести покрытие по известным методикам с целью маскировки неприятного вкуса обладающего неприятным вкусом лекарственного средства или с целью замедления распада и всасывания активного ингредиента в желудочно-кишечном тракте.
Таблетка может обеспечить немедленное высвобождение или замедленное высвобождение соединений, предлагаемых в настоящем изобретении.
Типичными агентами, обеспечивающими замедленное высвобождение, являются, например, такие, которые набухают при соприкосновении с водой, такие как поливинилпирролидон, гидроксиэтилцеллюлоза, гидроксипропилцеллюлоза, другие простые эфиры целлюлозы, крахмал, предварительно желатинизированный крахмал, полиметакрилат, поливинилацетат, микрокристаллическая целлюлоза, декстраны, и их смеси. Неограничивающие примеры разрыхлителей включают предварительно желатинизированный крахмал, натриевую соль гликолята крахмала, микрокристаллическую целлюлозу, натриевую соль карбоксиметилцеллюлозы (КМЦ-Na), сшитую КМЦ-Na и обладающую низкой степенью замещения гидроксипропилцеллюлозу, а также их смеси. Подходящие наполнители и связующие включают, но не ограничиваются только ими, микрокристаллическую целлюлозу, порошкообразную целлюлозу, лактозу (безводную или ее моногидрат), прессующийся сахар, крахмал (например, кукурузный крахмал или картофельный крахмал), предварительно желатинизированный крахмал, фруктозу, сахарозу, декстрозу, декстраны, другие сахара, такие как маннит, мальтит, сорбит, лактит и сахароза, силифицированную микрокристаллическую целлюлозу, гидрофосфат кальция, дигидрат гидрофосфата кальция, дигидрат дикальцийфосфата, трикальцийфосфат, лактат кальция или их смеси. Смазывающие вещества, антиадгезивы и/или агенты, придающие скользкость включают стеариновую кислоту, стеарат магния, стеарат кальция, лаурилсульфат натрия, гидрированное растительное масло, гидрированное касторовое масло, стеарилфумарат натрия, макроголы, глицериндибегенат, тальк, кукурузный крахмал, диоксид кремния и т.п., включая их смеси.
Соединение, предлагаемое в настоящем изобретении, можно приготовить для парентерального введения путем инъекции, например инъекции ударной дозы вещества или путем вливания. Композиции для инъекции могут поставляться в готовом к применению виде и могут находиться в таких формах, как суспензии, растворы или эмульсии в масле или водных разбавителях, и могут содержать инертные наполнители, такие как суспендирующие, стабилизирующие, консервирующие и/или диспергирующие средства. Альтернативно, активный ингредиент может находиться в порошкообразной форме для проводимого перед применением восстановления с помощью подходящего разбавителя, например, стерильной апирогенной воды.
В случае назального введения или введения путем ингаляции соединения, предлагаемые в настоящем изобретении, обычным образом можно приготовить в виде материалов для распыления с использованием в упаковках под давлением или устройствах типа небулайзер с использованием подходящего пропеллента, например, дихлордифторметана, фтортрихлорметана, дихлортетрафторэтана, диоксида углерода или другого подходящего газа или смеси газов.
В случае введения в глаза соединения, предназначенные для применения в настоящем изобретении, обычным образом можно приготовить в виде тонкоизмельченных суспензий в изотоническом, обладающем необходимым значением рН стерильном физиологическом растворе, без добавления или с добавлением консерванта, такого как бактерицидное или фунгицидное средство, например, фенилмеркурнитрат, бензилалконийхлорид или хлоргексидинацетат. Альтернативно, в случае введения в глаза соединения можно приготовить в виде мази, такой как на основе вазелинового масла.
В случае ректального введения соединения, предназначенные для применения в настоящем изобретении, обычным образом можно приготовить в виде суппозиториев. Их можно приготовить путем смешивания активного компонента с подходящим, не оказывающим раздражающего воздействия инертным наполнителем, который является твердым при комнатной температуре, но жидким при ректальной температуре и поэтому плавится в прямой кишке с высвобождением активного компонента. Такие вещества включают, например, масло какао, пчелиный воск и полиэтиленгликоли.
В одном варианте осуществления соединения можно вводить чрескожно. Этот режим введения предотвращает так называемый эффект первого прохождения, возникающий при пероральном введении, и, кроме того, обеспечивает более постоянную концентрацию в плазме, что в некоторых случаях является особенно предпочтительным. Приготовление чрескожных форм, таких как мази или кремы, или других чрескожных систем, таких как, например, пластыри или электрофорезные устройства, обычно известны из предшествующего уровня техники, см., например, публикации Venkatraman and Gale, Biomaterials 1998, Vol 19, p 1119; Prausnitz and Langer, Nat Biotechnology 2008, Vol 26.11 p 1261; WO 2001/47503; WO 2009/000262; WO 99/49852; WO 07/094876.
Предпочтительная доза соединений, предлагаемых в настоящем изобретении, зависит от ряда факторов, включая состояние здоровья и массу тела пациента, тяжесть конкретного заболевания, дозированную форму и путь и период введения, но ее могут соответствующим образом выбрать специалисты в данной области техники. В различных вариантах осуществления соединения вводят в количестве, находящемся в диапазоне от 0,001 до 10 мг/(кг массы тела) в сутки или от 0,03 до 1 мг/(кг массы тела) в сутки. Индивидуальные дозы могут находиться в диапазоне примерно от 0,1 до 1000 мг активного ингредиента в сутки, примерно от 0,2 до 750 мг/сутки, примерно от 0,3 до 500 мг/сутки, от 0,5 до 300 мг/сутки или от 1 до 100 мг/сутки. Дозы можно вводить один раз в сутки или несколько раз в сутки, при этом их разделяют на порции.
Другим объектом настоящего изобретения является набор, включающий лекарственное средство или фармацевтическую композицию, описанную в настоящем изобретении, и инструкции для его применения.
Другим объектом настоящего изобретения является упаковка, включающая по меньшей мере одну разовую дозу лекарственного средства или фармацевтической композиции, содержащей по меньшей мере одно соединение, описанное в настоящем изобретении, и инструкции для ее применения.
ОПРЕДЕЛЕНИЯ
Любое указание на соединение, предлагаемое в настоящем изобретении, также включает указание на фармацевтически приемлемые соли, сольваты, изотопы и совместные кристаллы таких соединений, если специально не указано иное.
Термин "фармацевтически приемлемые соли" означает любые соли, которые соединения, предлагаемые в настоящем изобретении, могут образовать и которые являются подходящими для введения субъектам, предпочтительно субъектам, являющимся людьми. Такие соли включают, но не ограничиваются только ими, соли присоединения с кислотами, образованные с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., или образованные с органическими кислотами, такими как уксусная кислота, пропионовая кислота, гексановая кислота, циклопентанпропионовая кислота, гликолевая кислота, пировиноградная кислота, молочная кислота, малоновая кислота, янтарная кислота, яблочная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, 3-(4-гидроксибензоил)бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, 1,2-этандисульфоновая кислота, 2-гидроксиэтансульфоновая кислота, бензолсульфоновая кислота, 4-хлорбензолсульфоновая кислота, 2-нафталинсульфоновая кислота, 4-толуолсульфоновая кислота, камфорсульфоновая кислота, 4-метилбицикло[2.2.2]окт-2-ен-1-карбоновая кислота, глюкогептоновая кислота, 3-фенилпропионовая кислота, триметилуксусная кислота, трет-бутилуксусная кислота, лаурилсерная кислота, глюконовая кислота, глутаминовая кислота, гидроксинафтойная кислота, салициловая кислота, стеариновая кислота и муконовая кислота. Другие соли включают 2,2-дихлорацетат, адипат, альгинат, аскорбат, аспартат, 2-ацетамидобензоат, капроат, капринат, камфорат, цикламат, лаурилсульфат, эдизилат, эзилат, изетионат, формиат, галактарат, гентизат, глюцептат, глюкуронат, оксоглутарат, гиппурат, лактобионат, нападизилат, ксинафоат, никотинат, олеат, оротат, оксалат, пальмитат, эмбонат, пидолат, п-аминосалицилат, себацинат, таннат, роданид, ундециленат и т.п.; или соли, образованные с кислым протоном, содержащимся в исходном соединении, замещенным, например, аммиаком, аргинином, бенетамином, бензатином, кальцием, холином, 2-диметиламиноэтанолом, диэтаноламином, диэтиламином, этаноламином, этилендиамином, меглумином, глицином, гидрабамином, имидазолом, лизином, магнием, гидроксиэтилморфолином, пиперазином, калием, эполамином, натрием, троламином, трометамином или цинком.
В объем настоящего изобретения также входят сольваты соединений, определенных в настоящем изобретении. "Сольваты" представляют собой кристаллы, образованные активным соединением и вторым компонентом (растворителем), которые в выделенном виде являются жидкими при комнатной температуре. Такие сольваты могут быть образованы с обычными органическими растворителями, например, углеводородными растворителями, такими как бензол или толуол; хлорированными растворителями, такими как хлороформ или дихлорметан; спиртовыми растворителями, такими как метанол, этанол или изопропанол; простыми эфирными растворителями, такими как диэтиловый эфир или тетрагидрофуран; или сложноэфирными растворителями, такими как этилацетат. Альтернативно, сольваты соединений, предлагаемых в настоящем изобретении, могут быть образованы с водой и в этом случае они будут являться гидратами.
В объем настоящего изобретения также входят совместные кристаллы. Термин "совместный кристалл" используют для описания случая, когда нейтральные молекулярные компоненты содержатся в кристаллическом соединении при определенном стехиометрическом соотношении. Получение фармацевтических совместных кристаллов позволяет модифицировать кристаллическую форму активного фармацевтического ингредиента, что, в свою очередь, может изменить его физико-химические характеристики без ухудшения его необходимой биологической активности. Примеры образующих совместные кристаллы соединений, которые могут содержаться в совместном кристалле вместе с активным фармацевтическим ингредиентом, включают L-аскорбиновую кислоту, лимонную кислоту, глутаровую кислоту, коричную кислоту, миндальную кислоту, мочевину и никотинамид.
В настоящее изобретение также включены все подходящие изотопозамещенные формы соединения, предлагаемого в настоящем изобретении. "Изотопозамещенная форма" или, сокращенно, "изотоп" соединения, предлагаемого в настоящем изобретении, определена, как соединение, в котором по меньшей мере один атом заменен на атом, обладающий таким же атомным номером, но атомной массой, отличающейся от атомной массы, обычно обнаруживаемой в природе, причем предпочтительным является более часто встречающийся изотоп (изотопы). Примеры изотопов, которые можно вводить в соединения, предлагаемые в настоящем изобретении, включают изотопы водорода, углерода, азота, кислорода, серы, фтора и хлора, такие как 2Η, 3Н, 11С, 13С, 14С, 15Ν, 17О, 18О, 35S, 18F и 36Cl соответственно. Некоторые изотопозамещенные формы соединений, предлагаемых в настоящем изобретении, например, такие, в которые включены такие радиоактивные изотопы, как 3Η или 14С, применимы для исследования распределения лекарственного средства и/или субстрата в тканях. Соединения, содержащие такие изотопы, как тритий, т.е. 3Н, и углерод-14, т.е. 14С, являются особенно предпочтительными вследствие легкости их получения и детектирования. Кроме того, замещение изотопами, такими как дейтерий, т.е. 2Н, может обеспечить определенные терапевтические преимущества, обусловленные их более высокой метаболической стабильностью, например, увеличенной длительностью полувыведения in vivo, возможностью использования меньших доз и поэтому в некоторых случаях это может являться предпочтительным. Изотопозамещенные формы соединений, предлагаемых в настоящем изобретении, обычно можно получить по обычным методикам с использованием соответствующих изотопозамещенных форм подходящих реагентов.
Частью настоящего изобретения также являются такие соединения, в которых по меньшей мере один атом заменен радиоактивным изотопом (радионуклидом) такого же или другого атома, которые можно использовать в методиках визуализации in vivo, таких как однофотонная эмиссионная компьютерная томография (ОФЭКТ) или позитронная эмиссионная томография (ПЭТ).
Примерами таких изотопозамещенных форм модуляторов GPR17, применимых для исследований с помощью ОФЭКТ (в настоящем изобретении такие соединения называются "предназначенными для ОФЭКТ радиоактивными индикаторами), являются соединения, в которые включен 99mTc, 111In, 82Rb, 137Cs, 123I, 125I, 131I, 67Ga, 192Ir или 201Tl и предпочтительно 123I. Так, например, для использования соединений, предлагаемых в настоящем изобретении, в качестве предназначенных для ОФЭКТ радиоактивных индикаторов в модулятор GPR17, раскрытый в настоящем изобретении можно включить изотоп 123I. В качестве неограничивающего примера можно привести следующий: для использования соединения в качестве предназначенного для ОФЭКТ радиоактивного индикатора в соединение, предлагаемое в настоящем изобретении можно включить радионуклид, выбранный из группы, включающей 123I, 125I и 131I, предпочтительным является 123I. В одном варианте осуществления предназначенный для ОФЭКТ радиоактивный индикатор, предлагаемый в настоящем изобретении, может быть основан на структуре содержащего галоген модулятора GPR17, раскрытого в настоящем изобретении, в котором в положении атома галогена, предпочтительно атома йода включен один из следующих радионуклидов: 123I, 125I и 131I.
В соответствии с этим, термин "предназначенный для ОФЭКТ радиоактивный индикатор, предлагаемый в настоящем изобретении", означает соединения, описанные в настоящей заявке на патент, и обладающие структурой, соответствующей любой из формул I, II, III, IV, V или VI, дополнительно определенных в настоящем изобретении, или другим образом отдельно раскрытые в настоящем изобретении, в которые включен по меньшей мере один радиоактивный изотоп, который является подходящим для визуализации с помощью ОФЭКТ. Он включает, но не ограничивается только ими, 99mTc, 111In, 82Rb, 137Cs, 123I, 125I, 131I, 67Ga, 192Ir или 201Tl.
Примерами производных модуляторов GPR17, применимых для исследований с помощью ПЭТ (в настоящем изобретении они называются "предназначенными для ПЭТ радиоактивными индикаторами"), являются соединения, в которые включен 11С, 13N, 15О, 18F, 76Br или 124I. Так, например, для использования соединения, предлагаемого в настоящем изобретении, в качестве предназначенного для ПЭТ радиоактивного индикатора в соединение, предлагаемое в настоящем изобретении, можно включить изотоп 18F. В одном варианте осуществления предназначенный для ПЭТ радиоактивный индикатор может быть основан на структуре содержащего фтор модулятора GPR17, раскрытого в настоящем изобретении, в котором в положении атома включен соответствующий радионуклид 18F. Это аналогичным образом относится к включению по меньшей мере одного из следующих: 11С, 13N, 15О, 76Br или 124I, вместо "немеченого" атома углерода, азота, кислорода, брома или йода соответственно (см., например, публикации Pimlott and Sutherland, Chem Soc Rev 2011, 40, 149; van der Born et al, Chem Soc Rev 2017, 46, 4709).
В соответствии с этим, термин "предназначенный для ПЭТ радиоактивный индикатор, предлагаемый в настоящем изобретении", означает соединения, описанные в настоящей заявке на патент, и обладающие структурой, соответствующей любой из формул I, II, III, IV, V или VI, дополнительно определенных в настоящем изобретении, или другим образом отдельно раскрытые в настоящем изобретении, в которые включен по меньшей мере один радиоактивный изотоп, который является подходящим для визуализации с помощью ПЭТ. Он включает, но не ограничивается только ими, 11С, 13N, 15O, 18F, 76Br или 124I.
В объем настоящего изобретения входят пролекарства соединений, предлагаемых в настоящем изобретении. Обычно такие пролекарства являются функциональными производными соединений, описанных в настоящем изобретении, которые in vivo, например, с помощью эндогенных ферментов, содержащихся в кишечнике или в крови, легко превращаются в необходимые модулирующие GPR17 соединения, описанные в настоящем изобретении. Обычные методики выбора и получения подходящих пролекарственных производных описаны, например, в публикации Design of Prodrugs, ed. Η. Bundgaard, Elsevier, 1985.
В зависимости от схемы замещения соединения, предлагаемые в настоящем изобретении, могут содержать или не содержать один или большее количество оптических стереоцентров или могут существовать или не существовать в виде разных энантиомеров или диастереоизомеров. Любые такие энантиомеры, диастереоизомеры или другие оптические изомеры входят в объем настоящего изобретения.
Соединение, предлагаемое в настоящем изобретении, также может находиться в разных кристаллических формах, т.е. в полиморфных формах, все эти формы входят в объем настоящего изобретения.
Соединения, предлагаемые в настоящем изобретении, можно включить в фармацевтическую композицию, которая также может содержать фармацевтически приемлемый носитель. "Фармацевтически приемлемый носитель" означает разбавитель, вспомогательное вещество, инертный наполнитель или носитель, или другой ингредиент, с которым вводят соединение, предлагаемое в настоящем изобретении, и которое для специалиста в данной области техники означает фармацевтически приемлемое.
Соединения, предлагаемые в настоящем изобретении, применимы для предупреждения и/или лечения определенных заболеваний, описанных в настоящем изобретении, у животных, предпочтительно у людей.
"Предупреждение" означает уменьшение вероятности возникновения заболевания или нарушения (т.е. обеспечение отсутствия проявления по меньшей мере одного из клинических симптомов заболевания у субъекта, предпочтительно человека, который может быть подвержен заболеванию или предрасположен к нему, но у которого еще не ощущаются или не проявляются симптомы заболевания).
"Лечение" любого заболевания или нарушения в одном варианте осуществления включает улучшение протекания заболевания или нарушения (т.е. остановку или ослабление развития заболевания или по меньшей мере уменьшение проявления одного из клинических симптомов заболевания). В другом варианте осуществления "лечение" означает улучшение по меньшей мере одного физического параметра, который может ощущаться или не ощущаться субъектом, предпочтительно являющимся человеком субъектом, но который является основой подвергающегося лечению заболевания или нарушения или связан с ним. В еще одном варианте осуществления "лечение" означает изменение протекания заболевания или нарушения, физическое (например, стабилизацию проявляющегося или непроявляющегося симптома), или физиологическое, (например, стабилизацию физиологического параметра), или и то, и другое. В еще одном варианте осуществления "лечение" означает задержку начала или развития, или прогрессирования заболевания или нарушения. Соответственно, "лечение" включает любое этиологическое лечение основного заболевания или нарушения (т.е. изменение протекания заболевания), а также любое лечение признаков и симптомов заболевания или нарушения (с изменением или без изменения протекания заболевания), а также любое облегчение или ослабление заболевания или нарушения, или его признаков и симптомов.
"Диагностика" или "диагностирование" заболевания или нарушения в одном варианте осуществления включают обнаружение и оценку признаков и симптомов, которые связаны с указанным заболеванием. "Диагностика" или "диагностирование" включают, но не ограничиваются только ими, обнаружение и/или оценку уменьшенного, увеличенного количества рецепторов GPR17 или другим образом неправильно (например, во времени и положении) экспрессированных, активированных или распределенных рецепторов GPR17 по сравнению с наблюдающимися у здоровых субъектов, что является показателем связанного с GPR17 заболевания или нарушения. В одном примере для такой диагностики, включая диагностику нарушения миелинизации, можно использовать лиганды для GPR17 в форме предназначенных для ПЭТ или ОФЭКТ радиоактивных индикаторов.
Термины "заболевание (заболевания)" и "нарушение (нарушения)" в настоящем изобретении в основном используют взаимозаменяемым образом.
"Мониторинг" означает наблюдение за заболеванием, патологическим состоянием или по меньшей мере одним медицинским показателем в течение определенного промежутка времени. "Мониторинг" также включает наблюдение за воздействием терапевтического лекарственного средства с помощью "дополнительного лекарственного средства".
"Дополнительное средство для диагностики" при использовании в настоящем изобретении означает соединение, которое можно использовать вместе с терапевтическим лекарственным средством для определения применимости (например, безопасности и эффективности) указанного терапевтического лекарственного средства для конкретного пациента. Применение "дополнительного средства для диагностики" может включать стадии диагностики и мониторинга.
Термины "животное (животные)" и "субъект (субъекты)" включают людей. Термины "человек", "пациент" и "являющийся человеком субъект" в настоящем изобретении используют взаимозаменяемым образом, если явно не указано иное.
Настоящее изобретение также относится к способам лечения заболевания или нарушения, более подробно описанного в настоящем изобретении, у животного, предпочтительно заболевания или нарушения у человека, которые включают введение соединений, предлагаемых в настоящем изобретении, в терапевтически эффективных количествах. "Терапевтически эффективное количество" означает такое количество соединения, которое при введении субъекту, предпочтительно являющемуся человеком субъекту, для лечения заболевания является достаточным для проведения такого лечения заболевания. "Терапевтически эффективное количество" может меняться в зависимости от соединения, заболевания и его тяжести, и состояния здоровья, возраста, массы тела, пола и т.п. подвергающегося лечению субъекта, предпочтительно являющегося человеком субъекта.
Термин "рассеянный склероз" при использовании в настоящем изобретении означает заболевание, обозначаемое кодом диагноза G35 в ICD-10-CM (Международная классификация болезней 10-го пересмотра), американская версия, 2018 г.
Термин "модуляторы GPR17" при использовании в настоящем изобретении описывает соединения, которые способны модулировать активность рецептора GPR17, предпочтительно соединения, которые способность уменьшать активность GPR17. Такие "негативные модуляторы GPR17" включают антагонисты GPR17, которые способны блокировать воздействия агонистов GPR17, а также обратные агонисты GPR17, которые также способны ингибировать обладающие системной активностью рецепторы GPR17 и варианты рецепторов. Предпочтительными модуляторами GPR17, предлагаемыми в настоящем изобретении, являются обратные агонисты GPR17.
Во всех случаях, когда после "С" в виде нижнего индекса указаны числовые значения, эти числовые значения (приведенные в скобках или без них) означают диапазон количества атомов углерода, содержащихся в соответствующей группе, приведенной непосредственно после числовых значений. Так, например, "С1-С3" и "(С1-С3)" оба обозначают группу, дополнительно точно определенную в настоящем изобретении, которая содержат от 1 до 3 атомов С.
"Алкил" включает насыщенные алифатические гидрокарбильные группы. Углеводородная цепь может являться линейной или разветвленной. Примеры "алкила" включают содержащие 1-5 атомов углерода ("С1-С5-алкил"), содержащие 1-4 атома углерода ("С1-С4алкил"), 1-3 атома углерода ("С1-С3-алкил") или 1-2 атома углерода ("С1-С2-алкил"). Примерами для этого термина являются такие группы, как метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, трет-амил и т.п. Любые количества атомов С, содержащихся в алкилах или в других группах, в настоящем изобретении может быть указано в скобках или без скобок.
"Алкилоксигруппа" и "алкоксигруппа", использующиеся в настоящем изобретении взаимозаменяемым образом (совместно алк(ил)окси), включают группу -OR, в которой R обозначает "алкил", дополнительно определенный в настоящем изобретении с приведенными примерами. Предпочтительные алк(ил)оксигруппы включают, например, мет(ил)оксигруппу, эт(ил)оксигруппу, н-проп(ил)оксигруппу, изопроп(ил)оксигруппу, н-бут(ил)оксигруппу, трет-бут(ил)оксигруппу, втор-бут(ил)оксигруппу, изобут(ил)оксигруппу и т.п.
"Галоген" включает атомы фтора, хлора, брома и йода.
"Цианогруппа" означает -C≡N.
Использующийся термин "фторалкил" означает "алкил", описанный в настоящем изобретении, который замещен одним или большим количеством атомов фтора. Типичные примеры фтор(С1-С3)алкильных групп включают, но не ограничиваются только ими CF3, -CHFCHF2 и CH2CF3. Особенно предпочтительной фторалкильной группой является дифторметил -CHF2.
Термины "фторалкилоксигруппа" или "фторалкоксигруппа", использующиеся в настоящем изобретении взаимозаменяемым образом, означают "алк(ил)оксигруппу", описанную в настоящем изобретении, которая замещена одним или большим количеством атомов фтора. Типичные примеры фтор(С1-С3)алк(ил)оксигрупп включают, но не ограничиваются только ими -OCF3, OCHFCH2F и OCH2CF3.
Термин "фторметоксигруппа" при использовании в настоящем изобретении означает метоксигруппу, которая замещена 1-3 атомами фтора. Термин "монофторметоксигруппа" означает метоксигруппу, которая замещена одним атомом фтора. Термин "дифторметоксигруппа" при использовании в настоящем изобретении означает метоксигруппу, которая замещена двумя атомами фтора. Термин "трифторметоксигруппа" означает метоксигруппу, которая замещена тремя атомами фтора.
Термин "фторэтоксигруппа" при использовании в настоящем изобретении означает этоксигруппу, которая замещена 1-3 атомами фтора. Термин "монофторэтоксигруппа" при использовании в настоящем изобретении означает этоксигруппу, которая замещена одним атомом фтора. Особенно предпочтительной монофторэтоксигруппой является группа -OCH2CH2F. Термин "дифторэтоксигруппа" при использовании в настоящем изобретении означает этоксигруппу, которая замещена двумя атомами фтора. Особенно предпочтительной дифторэтоксигруппой является группа -OCH2CHF2. Термин "трифторэтоксигруппа" означает этоксигруппу, которая замещена тремя атомами фтора. Предпочтительной трифторэтоксигруппой является группа -OCH2CF3.
Термин "фторметоксиэтоксигруппа" означает концевую фторметоксигруппу, дополнительно определенную в настоящем изобретении, которая присоединена к этоксигруппе. Предпочтительной "фторметоксиэтоксигруппой" является дифторметоксиэтоксигруппа, которая обозначена, как -OCH2CH2OCHF2.
Термин "циклоалкил" при использовании в настоящем изобретении означает одновалентную группу, образованную из насыщенного углеводорода, которая может быть незамещенной или содержать один или большее количество заместителей, дополнительно указанных в настоящем изобретении. "Циклоалкил" содержит по меньшей мере 3 и вплоть до, например, 5 образующих кольцо атомов углерода ("С3-С5-циклоалкил") или 4 образующих кольцо атома ("С3-С4-циклоалкил"). Подходящие циклоалкильные группы включают циклопропил, циклобутил и циклопентил.
Термины "бензилоксигруппа" или "фенилметоксигруппа" при использовании в настоящем изобретении означают группу, в которой фенильное кольцо связано с метоксигруппой, и означает группу -О-СН2-фенил.
Термин "бензилметоксигруппа" при использовании в настоящем изобретении означает фенилэтоксигруппу, в которой фенильное кольцо связано с этоксигруппой, и означает группу -О-СН2-СН2-фенил.
Термин "пирид(ин)илметоксигруппа" означает группу, в которой пирид(ин)ильная группа связана с метоксигруппой, и означает группу -О-СН2-пиридил, где пиридилом может являться любая пиридильная группа. В контексте настоящего изобретения предпочтительными пиридилметоксигруппами являются пиридин-3-илметоксигруппа,  и пиридин-4-илметоксигруппа,
и пиридин-4-илметоксигруппа, 
ЭКСПЕРИМЕНТАЛЬНЫЙ РАЗДЕЛ
А. ХИМИЧЕСКАЯ СЕКЦИЯ
Соединения, предлагаемые в настоящем изобретении, и пути их синтеза более подробно описаны ниже
Α-Ι. Общие методики получения соединений
Соединения формулы I, предлагаемые в настоящем изобретении, можно получить по аналогии с обычными методиками, известными специалисту в области синтетической органической химии.
Любое описание синтеза соединений общей формулы I, предлагаемых в настоящем изобретении, также является подходящим для применимых соединений субродовых формул II, III, IV и V, и соединений конкретных примеров, раскрытых в настоящем изобретении.
В одном варианте осуществления некоторые соединения общей формулы I можно получить по реакции соединения формулы XI с анилином формулы X по уравнению:

Эту реакцию можно провести с использованием хлорсульфоновой кислоты при температуре, находящейся в диапазоне от 60 до 120°С, в полярном растворителе, таком как ацетонитрил, с получением невыделенного промежуточного сульфонилхлорида XII. Затем промежуточный продукт XII непосредственно вводят в реакцию с анилином X в присутствии основания, такого как пиридин, с использованием или без использования каталитического количества 4-диметиламинопиридина (ДМАП), в полярном растворителе, таком как ацетонитрил, при температуре, предпочтительно находящейся в диапазоне от 60 до 80°С.
Альтернативно, промежуточный сульфонилхлорид XII можно получить из соединения XI в присутствии комплекса пиридин-триоксид серы в пиридине при кипячении с обратным холодильником. Промежуточную соль сульфоновой кислоты можно хлорировать в присутствии хлорирующего реагента, такого как трифенилфосфин/трихлорацетонитрил, в растворителе, таком как дихлорметан, при кипячении с обратным холодильником.
Альтернативно, некоторые соединения общей формулы I можно получить по реакции сульфонилхлорида формулы XII с анилином формулы X по уравнению:
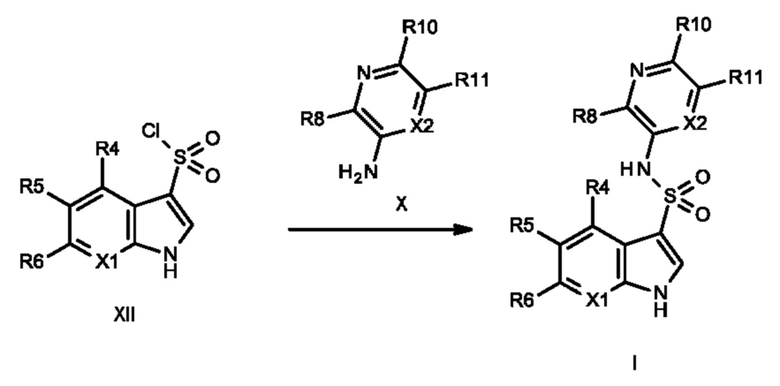
Эту реакцию можно провести в присутствии основания, такого как пиридин, использующегося в качестве растворителя, при комнатной температуре.
Альтернативно, некоторые соединения формулы I можно получить путем удаления защитной группы у соединения формулы Ι-Ρ, в которой Ρ обозначает защитную группу, такую как фенилсульфонил (PhSO2), по уравнению:

Эту реакцию можно провести в присутствии слабого основания, такого как карбонат калия или карбонат цезия, в смеси полярных растворителей, таких как метанол или диоксан и вода, при комнатной температуре или при нагревании при температуре, предпочтительно находящейся в диапазоне от 80 до 120°С. Эту реакцию можно провести в присутствии тетрабутиламмонийфторида в растворителе, таком как ТГФ (тетрагидрофуран), при нагревании при температуре, предпочтительно находящейся в диапазоне от 60 до 90°С.
Соединения формулы Ι-Ρ можно получить по реакции сульфонилхлорида формулы ХП-Р с анилином формулы X. Эту реакцию можно провести в присутствии основания, такого как пиридин, использующегося в качестве растворителя, при комнатной температуре.
Соединения формулы XII можно получить путем хлорирования соединения формулы IX по уравнению:

Эту реакцию можно провести в присутствии хлорирующего реагента, такого как оксихлорид фосфора, в полярном растворителе, таком как ацетонитрил, при температуре, находящейся в диапазоне от 50 до 100°С.
Соединения формулы IX можно получить путем сульфонилирования соединения формулы XI по уравнению:
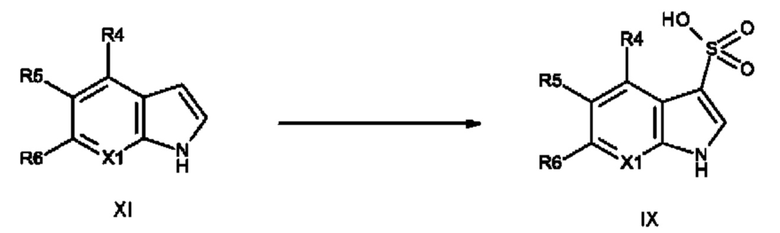
Эту реакцию можно провести в присутствии сульфонилирующего реагента, такого как комплекс пиридин-триоксид серы, в присутствии основания, такого как пиридин, использующегося в качестве растворителя, при кипячении с обратным холодильником.
Соединения формулы XII-Р, в которой Ρ обозначает защитную группу, такую как фенилсульфонил, можно получить путем хлорсульфонилирования соединения формулы ΧΙ-Ρ по уравнению:
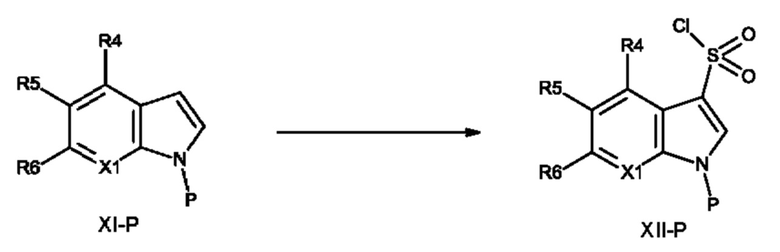
Эту реакцию можно провести в присутствии хлорсульфоновой кислоты в полярном растворителе, таком как ацетонитрил, при комнатной температуре.
Соединения формулы ΧΙ-Ρ, в которой Ρ обозначает защитную группу, такую как фенилсульфонил, можно получить путем удаления защитной группы у соединения формулы XI по уравнению:
Эту реакцию можно провести по любой методике, известной специалисту в данной области техники.
Анилины формулы X имеются в продаже или их можно получить по любой методике, известной специалисту в данной области техники, или по методикам, описанным в литературе. Альтернативно, некоторые анилины формулы X можно получить путем восстановления соединения VIII по уравнению:

Эту реакцию можно провести с использованием любого восстановительного реагента, такого как железо, в присутствии кислоты, такой как уксусная кислота, или водорода, в присутствии каталитического количества палладия на древесном угле в полярном растворителе, таком как этилацетат или метанол, или по любой методике, известной специалисту в данной области техники.
Соединения формулы VIII имеются в продаже или их можно получить по описанным в литературе методикам, или по любым другим методикам, известным специалисту в данной области техники.
Соединения формулы XI имеются в продаже или их можно получить по подходящим методикам, хорошо известным специалисту в данной области техники.
А-II. Аббревиатуры/многократно использовавшиеся реагенты
Ас: ацетил
АЦН: ацетонитрил
АсОН: уксусная кислота
Рассол: насыщенный водный раствор хлорида натрия
Boc: трет-бутоксикарбонил
nBu: н-бутил
tBu: трет-бутил
Су: циклогексил
ДАТС: диэтиламинотрифторид серы
dba: дибензилиденацетон
ДХМ: дихлорметан
ДМАП: 4-диметиламинопиридин
ДМФ: Ν,Ν-диметилформамид
ДМСО: диметилсульфоксид
Dppf: 1,1'-бис(дифенилфосфанил)ферроцен
ЭР+: ионизация электрораспылением в режиме положительных ионов
ЭР-: ионизация электрораспылением в режиме отрицательных ионов
ИЭР: ионизация электрораспылением
EtOAc: этилацетат
ч: час(ы)
ЖХ: жидкостная хроматография
ЖХМС: жидкостная хроматография-масс-спектрометрия
Me: метил
МеОН: метанол
мин: минута (минуты)
МП: микроволновая печь
NBS: Ν-бромсукцинимид
NCS: Ν-хлорсукцинимид
ЯМР: ядерный магнитный резонанс
КТ: комнатная температура
ТБАГСА: тетрабутиламмонийгидросульфат
ТБАФ: тетрабутиламмонийфторид
ТЭА: триэтиламин
АТФК: ангидрид трифторуксусной кислоты
ТГФ: тетрагидрофуран
ТСХ: тонкослойная хроматография
Xantphos: 4,5-бис(дифенилфосфино)-9,9-диметилксантен
А-III. Методики анализа
Имеющиеся в продаже растворители и реагенты обычно использовали без дополнительной очистки, включая безводные растворители, когда это являлось целесообразным (обычно продукты Sure-Seal™, выпускающиеся фирмой Aldrich Chemical Company, или AcroSeal™, выпускающиеся фирмой ACROS Organics). За протеканием реакций обычно следили с помощью тонкослойной хроматографии или жидкостной хроматографии-масс-спектрометрии.
Исследования с помощью масс-спектрометрии в режиме ЖХМС проводили с использованием разных методик и приборов следующим образом:
- ЖХМС в щелочной среде, методика 1:
Для анализа с помощью ЖХМС использовали масс-спектрометр с одной квадрупольной линзой QDA Waters. Этот спектрометр снабжен источником ИЭР и Acquity UPLC H-Class с детектором с диодной матрицей (от 200 до 400 нм). Сбор данных проводили в режиме полного сканирования в МС от 70 до 800 m/z, в режиме положительных/отрицательных ионов и при элюировании в щелочной среде. Разделение с использованием обращенной фазы проводили при 45°С с использованием колонки Waters Acquity UPLC ВЕН С18, 1,7 мкм (2,1×50 мм) для элюирования в щелочной среде. Элюирование в градиентном режиме проводили с использованием смеси вода/АЦН/формиат аммония (95/5/63 мг/л) (растворитель А) и смеси АЦН/вода/формиат аммония (95/5/63 мг/л) (растворитель В) в соответствии с таблицей 1. Инжектируемый объем: 1 мкл. Полнопоточный режим в МС.

- ЖХМС в щелочной среде, методика 2:
Масс-спектры (МС) снимали с использованием ионизации электрораспылением (ИЭР) на масс-спектрометре для ЖХМС 2010EV (Shimadzu), соединенном с ВЭЖХ (высокоэффективный жидкостный хроматограф) Modular Prominence (Shimadzu), с использованием колонки Xbridge С18, 2,1×30 мм, 2,5 мкм (Waters). Инжектировали раствор образца, обладающий концентрацией, равной примерно 1 мг/мл, объемом 3 мкл. Подвижной фазой в случае щелочной среды являлась смесь А) 5 мМ формиат аммония + 0,1% аммиака в воде и В) 5% подвижной фазы А + 0,1% аммиака в ацетонитриле. Использовавшийся градиентный режим являлся следующим: от 5:95 (В/А) до 95:5 (В/А) за 4 мин и выдерживание при 95:5 (В/А) в течение следующей 1 мин.
- ЖХМС в нейтральной среде, методика 3:
Масс-спектры (МС) снимали на приборе для ЖХМС (Applied Biosystems API 2000, ЖХ/МС/МС, ВЭЖХ Agilent 1100) по следующей методике: соединения растворяли в АЦН (растворитель А) или смеси вода (содержащая 2 мМ ацетат аммония) : МеОН состава 90:10 (растворитель В) при концентрации, равной 1,0 мг/мл, и при необходимости обрабатывали ультразвуком до обеспечения полного растворения. Затем 10 мкл раствора инжектировали в колонку для ВЭЖХ Phenomenex Luna С 18 (50×2,00 мм, размер частиц: 3 мкм) и проводили элюирование в градиентном режиме с использованием смеси вода : АЦН (градиентный режим А) или смеси вода : МеОН (градиентный режим В), от смеси состава 90:10 до смеси состава 0:100 за 10 мин, элюирование в градиентном режиме начинали через 1 мин, затем элюировали чистым органическим растворителем в течение 10 мин при скорости потока, равной 300 мкл/мин. Поглощение в УФ-области определяли при длине волны, равной от 220 до 400 нм, с использованием детектора с диодной матрицей (ДДМ).
- ЖХМС в кислой среде, методика 4:
ВЭЖХ-МС проводили с использованием системы для ЖХ-МС Agilent 1200-6120, соединенной с УФ детектором (от 230 до 400 нм и 215 нм) и масс-спектрометрическим детектором масс-спектрометра Agilent 6120 (ЭР), от 120 до 800 m/z, с использованием колонки X-Bridge C18 Waters, 2,1×20 мм, 2,5 мкм. Элюирование проводили в градиентном режиме, указанном в таблице 2, с использованием подвижной фазы А (10 мМ формиат аммония в воде + 0,1% муравьиной кислоты) и подвижной фазы В (ацетонитрил + 5% воды + 0,1% муравьиной кислоты) при скорости потока, равной 1 мл/мин.

Неочищенные материалы можно очистить с помощью хроматографии с нормальной фазой, хроматографии с обращенной фазой (в кислой или щелочной среде) или путем перекристаллизации.
Хроматографию с нормальной фазой проводили с использованием колонок с силикагелем (силикагель, 100:200 меш) или картриджей для систем для проведения флэш-хроматографии, таких как Isolera Four, выпускающиеся фирмой Biotage®, или CombiFlash®, выпускающиеся фирмой Teledyne Isco.
Препаративную хроматографию с обращенной фазой проводили с использованием двух разных приборов и в соответствии со следующими методиками:
- Препаративная ЖХМС в щелочной среде, методика 1:
Очистку с помощью ЖХМС проводили с использованием масс-спектрометра для МС-детектирования с одной квадрупольной линзой SQD или QM Waters. Этот спектрометр снабжен источником ИЭР, насосом для подачи двух компонентов Waters 2525, соединенным с пробоотборником 2767, и детектором с диодной матрицей (от 210 до 400 нм).
Параметры МС: Напряжение на капилляре ИЭР равно 3 кВ. Напряжение на конусе и экстракторе равно 10 В. Температура блока источника равна 120°С. Температура десольватации равна 300°С. Скорость потока газа на конусе равна 30 л/ч (азот). Скорость потока десольватирующего газа равна 650 л/ч. Сбор данных проводили в режиме полного сканирования в МС от 100 до 850 m/z, в режиме положительных/отрицательных ионов.
Параметры ЖХ: Разделение с использованием обращенной фазы проводили при КТ с использованием колонки XBridge prep OBD C18 (5 мкм, 30×50 мм). Элюирование в градиентном режиме проводили с использованием растворителя Al (Н2О+10 мМ NH4HCO3+50 мкл/л NH4OH) и растворителя В1 (100% АЦН) (рН ~8,5). ВЭЖХ, скорость потока: от 35 до 45 мл/мин, инжектируемый объем: 990 мкл. Отношение деления потока для МС устанавливали равным +/- 1/6000 (таблица 3).

- ОФ-ВЭЖХ (высокоэффективная жидкостная хроматография с обращенной фазой) в нейтральной среде, методика 2:
Очистку конечных продуктов с помощью ВЭЖХ проводили с помощью системы для ВЭЖХ Knauer Smartline 1050, с использованием колонки для ОФ-ВЭЖХ (Knauer, ВД (внутренний диаметр) = 20 мм, Eurospher-100 С18). Продукт растворяли в метаноле (20 мг в 8 мл) и проводили ВЭЖХ с обращенной фазой с использованием градиентного режима и смеси метанола/вода (от 70:30 до 100:0 в течение 24 мин).
Спектры ЯМР снимали с использованием разных приборов:
- тщательно экранированного спектрометра ЯМР BRUKER AVANCE III, 400 МГЦ, снабженном рабочей станцией Windows 7 Professional, использующей программное обеспечение Topspin 3.2, и широкополосным датчиком двойного резонанса, 5 мм (PABBI 1H/19F-BB Z-GRD Z82021/0075), или датчиком тройного резонанса, 1 мм (PATXI 1H/ D-13C/15N Z-GRD Z868301/004),
- спектрометра ЯМР Varian, 400 МГц, при времени накопления (at) = 2,0 с, задержке релаксации (d1) - 2,0 с и уширении линии (1b) = 0,5 Гц,
- спектрометра ЯМР Bruker Avance DRX, 500 МГц,
- спектрометра ЯМР Bruker Avance III, 600 МГц.
Химические сдвиги приведены относительно сигналов остаточных протонов дейтерированных растворителей (ДМСО-d6, бензол-d6 или CDCl3). Химические сдвиги приведены в частях на миллион (част./млн) и константы спин-спинового взаимодействия (J) приведены в герцах (Гц). Спиновые мультиплетности указаны следующим образом: широкий (br), синглет (s), дублет (d), триплет (t), квадруплет (q) и мультиплет (m).
Перед проведением окончательных анализов и биологических исследований продуктов их обычно сушили в вакууме.
A-IV: ПРИМЕРЫ СОЕДИНЕНИЙ И СИНТЕЗ
Названия приведенных ниже соединений представляют собой названия, соответствующие нормам IUPAC (Международный союз теоретической и прикладной химии), полученные с помощью программного обеспечения Biovia Draw, Version 16.1, для промежуточных продуктов формулы X, XI, XII, и с помощью программного обеспечения Pipeline Pilot 2018 с использованием, OpenEye oemetachem, version 1.4.5, для приведенных в примерах соединений формулы I.
Промежуточные продукты
Если исходные вещества имеются в продаже, то они идентифицированы с помощью их регистрационных номеров CAS.
А. Синтез промежуточных продуктов формулы Х
A.1. Синтез 2,5-дифторпиридин-3-амина Х-1:

К раствору 2,5-дифтор-3-нитропиридина (0,30 г, 1,87 ммоля) в EtOAc (40 мл) добавляли Pd/C (0,13 г, 1,27 ммоля) и реакционную смесь перемешивали при повышенном давлении водорода, при комнатной температуре в течение 8 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь фильтровали через целит, промывали с помощью EtOAc (40 мл) и фильтрат концентрировали в вакууме и получали 2,5-дифторпиридин-3-амин Х-1 (0,19 г) в виде желтого твердого вещества. Это соединение использовали в следующей реакции без дополнительной очистки.
Выход: 71%.
ЖХМС в щелочной среде, методика 2 (ЭР+): 131 (М+Н)+, чистота 90%.
1H ЯМР (400 МГц, ДМСО-d6) δ 5,81 (brs, 2Н), 6,94-6,98 (m, 1H), 7,23 (t, J = 2,69 Гц, 1Н).
А.2. Синтез 6-хлор-2,5-дифторпиридин-3-амина Х-2:

Стадия 1: Синтез 2,5-дифтор-1-оксидопиридин-1-ия Х-2а:
К раствору 2,5-дифторпиридина (3,00 г, 26,1 ммоля) в ДХМ (120 мл) добавляли гидропероксид мочевины (7,36 г, 78,2 ммоля) и реакционную смесь перемешивали при комнатной температуре в течение 10 мин. Реакционную смесь охлаждали до 0°С, затем по каплям добавляли ангидрид трифторуксусной кислоты (12 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4 ч. За протеканием реакции следили с помощью ТСХ. После завершения реакции реакционную смесь разбавляли водным раствором NaHCO3 (120 мл) и экстрагировали с помощью ДХМ (3×80 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме и получали 2,5-дифтор-1-оксидопиридин-1-ий Х-2а (1,00 г) в виде почти белого твердого вещества. Это соединение использовали в следующей реакции без дополнительной очистки.
Выход: 29%.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,39-7,47 (m, 1Н), 7,50 (m, 1H), 8,48-8,57 (m, 1H).
Стадия 2: Синтез 2-хлор-3,6-дифторпиридина X-2b:
К раствору 2,5-дифтор-1-оксидопиридин-1-ия Х-2а (0,95 г, 7,25 ммоля) в ДХМ (30 мл) при 0°С по каплям добавляли POCl3 (1,33 мл, 14,5 ммоля). Реакционную смесь перемешивали при такой же температуре в течение 5 мин, затем добавляли ДМФ (0,60 мл). Реакционную смесь перемешивали при комнатной температуре в течение 6 ч. За протеканием реакции следили с помощью ТСХ. После завершения реакции реакцию останавливали насыщенным раствором NaHCO3 (50 мл) и смесь экстрагировали с помощью EtOAc (2×50 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме и получали 2-хлор-3,6-дифторпиридин Х-2b (0,65 г) в виде бледно-коричневой жидкости. Это соединение использовали в следующей реакции без дополнительной очистки.
Выход: 60%.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,35-7,39 (m, 1Н), 8,15-8,23 (m, 1H).
Стадия 3: Синтез 2-хлор-3,6-дифтор-5-нитропиридина Х-2 с:
К раствору 2-хлор-3,6-дифторпиридина Х-2b (0,60 г, 4,01 ммоля) в дымящей HNO3 (4,19 мл, 100 ммолей) по каплям добавляли концентрированную H2SO4 (3,21 мл, 60,2 ммоля), поддерживая температуру ниже 40°С. Затем реакционную смесь нагревали при 60°С в течение 30 мин. За протеканием реакции следили с помощью ТСХ. После завершения реакции реакционную смесь охлаждали и выливали на дробленый лед и экстрагировали с помощью ДХМ (2×50 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное неочищенное вещество очищали с помощью колоночной хроматографии (диоксид кремния, 100-200 меш, 10% EtOAc в гексане) и получали 2-хлор-3,6-дифтор-5-нитропиридин Х-2 с (0,185 г) в виде бледно-желтой жидкости.
Выход: 24%.
1H ЯМР (400 МГц, ДМСО-d6) δ 9,10-9,14 (m, 1H).
Стадия 4: Синтез 6-хлор-2,5-дифторпиридин-3-амина Х-2:
К раствору 2-хлор-3,6-дифтор-5-нитропиридина Х-2с (0,18 г, 0,93 ммоля) в уксусной кислоте (9 мл) добавляли железо (0,05 г, 0,93 ммоля) и реакционную смесь нагревали при 80°С в течение 2 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь концентрировали в вакууме. Остаток разбавляли с помощью EtOAc (40 мл) и промывали насыщенным раствором NaHCO3 (25 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме и получали 6-хлор-2,5-дифторпиридин-3-амин Х-2 (0,28 г) в виде бледно-коричневого твердого вещества. Это соединение использовали в следующей реакции без дополнительной очистки.
Выход: 42%.
ЖХМС в щелочной среде; методика 2 (ЭР-): 163 (М-Н)-, чистота 23%.
А.3. Синтез 6-хлор-5-фтор-2-метоксипиридин-3-амина Х-3:

Стадия 1: Синтез 2-хлор-3-фтор-6-метокси-5-нитропиридина Х-3а:
К раствору 2-хлор-3,6-дифтор-5-нитропиридина Х-2с (0,67 г, 3,44 ммоля) в МеОН (10 мл) при -40°С по каплям добавляли NaOMe (0,82 мл, 3,79 ммоля) и реакционную смесь перемешивали при такой же температуре в течение 20 мин. За протеканием реакции следили с помощью ТСХ. После завершения реакции реакционную смесь выливали в охлажденный льдом 1 н. раствор HCl (10 мл) и экстрагировали гексаном (2×15 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме и получали 2-хлор-3-фтор-6-метокси-5-нитропиридин Х-3а (0,40 г) в виде желтого твердого вещества. Это соединение использовали в следующей реакции без дополнительной очистки.
Выход: 56%.
1H ЯМР (400 МГц, ДМСО-d6) δ 4,03 (s, 3Н), 8,82 (d, J = 7,83 Гц, 1Н).
Стадия 2: Синтез 6-хлор-5-фтор-2-метоксипиридин-3-амина Х-3:
При перемешивании к раствору 2-хлор-3-фтор-6-метокси-5-нитропиридина Х-3а (0,20 г, 0,97 ммоля) в уксусной кислоте (4 мл) при 0°С добавляли железо (0,22 г, 3,87 ммоля). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь выливали в охлажденный льдом насыщенный раствор NaHCO3 (25 мл). Реакционную смесь фильтровали через слой целита, промывали с помощью EtOAc (2×15 мл) и водный слой экстрагировали с помощью EtOAc (2×15 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное неочищенное вещество очищали с помощью колоночной хроматографии (диоксид кремния, 100-200 меш, 4% EtOAc в гексане) и получали 6-хлор-5-фтор-2-метоксипиридин-3-амин Х-3 (0,11 г, 63%) в виде почти белого твердого вещества.
Выход: 63%.
ЖХМС в щелочной среде, методика 2 (ЭР+): 177 (М+Н)+, чистота 98%.
1H ЯМР (400 МГц, ДМСО-d6) δ 3,84 (s, 3Н), 5,49 (brs, 2H), 6,90 (d, J = 9,78 Гц, 1H).
A.4. Синтез 6-хлор-2-фтор-5-метоксипиридин-3-амина Х-4:

Стадия 1: Синтез 3-бром-2-фтор-5-метоксипиридина Х-4а:
К раствору 5-бром-6-фторпиридин-3-ола (0,80 г, 4,17 ммоля) и NaH (0,33 г, 8,33 ммоля) в ДМФ (15 мл) при 0°С по каплям добавляли CH3I (0,31 мл, 5,00 ммоля). Реакционную смесь перемешивали при комнатной температуре в течение 4 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь выливали в холодную Н2О (20 мл) и экстрагировали с помощью EtOAc (2×30 мл). Органический слой отделяли, промывали рассолом (20 мл), сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное неочищенное вещество очищали с помощью колоночной хроматографии с использованием CombiFlash (30% EtOAc в гексане) и получали 3-бром-2-фтор-5-метоксипиридин Х-4а (0,80 г) в виде бледно-желтого твердого вещества.
Выход: 93%.
ЖХМС в щелочной среде, методика 2 (ЭР+): 206 (М+Н)+, чистота 99%.
1H ЯМР (400 МГц, ДМСО-d6) δ 3,85 (s, 3Н), 7,92 (t, J = 2,20 Гц, 1Н), 7,99 (dd, J = 7,34, 2,20 Гц, 1Н).
Стадия 2: Синтез 3-бром-2-фтор-5-метокси-1-оксидопиридин-1-ия X-4b:
К раствору 3-бром-2-фтор-5-метоксипиридина Х-4а (0,30 г, 1,35 ммоля) в ДХМ (15 мл) при 0°С добавляли гидропероксид мочевины (0,38 г, 4,05 ммоля) и реакционную смесь перемешивали в течение 10 мин. При 0°С по каплям добавляли ангидрид трифторуксусной кислоты (0,96 мл, 6,76 ммоля) и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь выливали в охлажденную льдом Н2О (15 мл), подщелачивали насыщенным раствором NaHCO3 (15 мл) до рН 8 и экстрагировали с помощью ДХМ (2×15 мл). Органический слой отделяли, промывали рассолом (10 мл), сушили над безводным Na2SO4 и концентрировали в вакууме и получали 3-бром-2-фтор-5-метокси-1-оксидопиридин-1-ий X-4b (0,16 г, 37%) в виде почти белого твердого вещества. Это соединение использовали в следующей реакции без дополнительной очистки.
Выход: 93%.
ЖХМС в щелочной среде, методика 2 (ЭР+): 222 (М+Н)+, чистота 69%.
Стадия 3: Синтез 5-бром-2-хлор-6-фтор-3-метоксипиридина Х4-с:
К раствору 3-бром-2-фтор-5-метокси-1-оксидопиридин-1-ия X-4b (0,40 г, 1,45 ммоля) в ДХМ (10 мл) добавляли POCl3 (0,35 мл, 3,88 ммоля), затем при 0°С добавляли ДМФ (0,1 мл) и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. За протеканием реакции следили с помощью ТСХ. После завершения реакции реакционную смесь концентрировали в вакууме. Остаток выливали в охлажденную льдом Н2О (15 мл), подщелачивали насыщенным раствором NaHCO3 (15 мл) до рН 8 и экстрагировали с помощью EtOAc (2×15 мл). Органический слой отделяли, промывали рассолом (15 мл), сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное неочищенное вещество очищали с помощью колоночной хроматографии с использованием CombiFlash (30% EtOAc в гексане) и получали 5-бром-2-хлор-6-фтор-3-метоксипиридин Х4-с (0,26 г) в виде бледно-желтого твердого вещества.
Выход: 74%.
1H ЯМР (400 МГц, ДМСО-d6) δ 3,93 (s, 3Н), 8,16 (d, J = 6,85 Гц, 1Н).
Стадия 4: Синтез N-(6-хлор-2-фтор-5-метокси-3-пиридил)-1,1-дифенилметанимина X4-d:
К раствору 5-бром-2-хлор-6-фтор-3-метоксипиридина Х4-с (0,25 г, 1,04 ммоля), бензофенонимина (0,21 г, 1,14 ммоля) в диоксане (15 мл) добавляли Cs2CO3 (1,02 г, 3,12 ммоля) и Xantphos (0,12 г, 0,21 ммоля). Реакционную смесь продували аргоном в течение 20 мин, затем добавляли Pd2(dba)3 (0,10 г, 0,10 ммоля). Реакционную смесь нагревали при 100°С в течение 16 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь разбавляли с помощью EtOAc (15 мл), фильтровали через слой целита, промывали с помощью EtOAc (2×15 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное неочищенное вещество очищали с помощью колоночной хроматографии с использованием CombiFlash (30% EtOAc в гексане) и получали N-(6-хлор-2-фтор-5-метокси-3-пиридил)-1,1-дифенилметанимин X4-d (0,25 г, 50%) в виде почти белого твердого вещества.
Выход: 93%.
ЖХМС в щелочной среде, методика 2 (ЭР+): 341 (М+Н)+, чистота 71%.
Стадия 5: Синтез 6-хлор-2-фтор-5-метоксипиридин-3-амина Х-4:
К раствору N-(6-хлор-2-фтор-5-метокси-3-пиридил)-1,1-дифенилметанимина X4-d (0,24 г, 0,51 ммоля) в МеОН (15 мл) при 0°С добавляли 1 н. раствор HCl (0,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь выливали в H2O (15 мл) и экстрагировали с помощью ДХМ (2×20 мл).
Органический слой отделяли, промывали рассолом (20 мл), сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное неочищенное вещество очищали с помощью колоночной хроматографии с использованием CombiFlash (30% EtOAc в гексане) и получали 6-хлор-2-фтор-5-метоксипиридин-3-амин Х-4 (0,06 г) в виде почти белого твердого вещества.
Выход: 62%.
ЖХМС в щелочной среде, методика 2 (ЭР+): 177 (М+Н)+, чистота 93%.
1H ЯМР (400 МГц, ДМСО-d6) δ 3,79 (s, 3Н), 5,64 (s, 2H), 6,99 (d, J = 8,80 Гц, 1Н).
A.5. Синтез 2,5-дифтор-6-метоксипиридин-3-амина Х-5:

Стадия 1: Синтез 2,3,6-трифтор-5-нитропиридина Х-5а:
При перемешивании к раствору 2,3,6-трифторпиридина (2,00 г, 15,0 ммоля) в дымящей HNO3 (12,5 мл, 301 ммоль) при 0°С по каплям добавляли концентрированную H2SO4 (12,0 мл, 225 ммолей). Реакционную смесь нагревали при 60°С в течение 1 ч. За протеканием реакции следили с помощью ТСХ. После завершения реакции реакционную смесь выливали на дробленый лед (40 мл) и экстрагировали гексаном (2×30 мл). Органический слой отделяли, промывали насыщенным раствором NaHCO3 (40 мл), сушили над безводным Na2SO4 и концентрировали в вакууме и получали 2,3,6-трифтор-5-нитропиридин Х-5а (1,20 г) в виде желтого масла. Это соединение использовали в следующей реакции без дополнительной очистки.
Выход: 45%.
1H ЯМР (400 МГц, ДМСО-d6) δ 9,20-9,26 (m, 2H).
Стадия 2: Синтез 2,5-дифтор-6-метокси-3-нитропиридина X-5b:
При перемешивании к раствору 2,3,6-трифтор-5-нитропиридина Х-5а (0,20 г, 1,12 ммоля) в МеОН (10 мл) при -78°С по каплям добавляли NaOMe (0,93 мл, 4,32 ммоля) и реакционную смесь перемешивали при такой же температуре в течение 2 ч. За протеканием реакции следили с помощью ТСХ. После завершения реакции реакцию при -78°С останавливали насыщенным раствором HCl (10 мл) и смесь экстрагировали гексаном (2×15 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме и получали 2,5-дифтор-6-метокси-3-нитропиридин Х-5b (0,136 г) в виде бледно-желтого твердого вещества. Это соединение использовали в следующей реакции без дополнительной очистки.
Выход: 64%.
1H ЯМР (400 МГц, ДМСО-d6) δ 4,06 (s, 3Н), 8,76 (dd, J = 8,80, 7,34 Гц, 1Н).
Стадия 3: Синтез 2,5-дифтор-6-метоксипиридин-3-амина Х-5:
При перемешивании к раствору 2,5-дифтор-6-метокси-3-нитропиридина X-5b (0,13 г, 0,68 ммоля) в уксусной кислоте (4 мл) при 0°С порциями добавляли железо (0,15 г, 2,74 ммоля). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакцию останавливали насыщенным раствором NaHCO3 (25 мл) и смесь экстрагировали с помощью EtOAc (2×25 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме и получали 2,5-дифтор-6-метоксипиридин-3-амин Х-5 (0,104 г, 94%) в виде коричневого твердого вещества. Это соединение использовали в следующей реакции без дополнительной очистки.
Выход: 62%.
ЖХМС в щелочной среде, методика 2 (ЭР+): 161 (М+Н)+, чистота 99%.
1H ЯМР (400 МГц, ДМСО-d6) δ 3,77 (s, 3Н), 5,04 (brs, 2H), 7,17 (dd, J = 10,76, 8,31, 1Н).
A.6. Синтез 5-фтор-2,6-диметоксипиридин-3-амина Х-6:

Стадия 1: Синтез 3-фтор-2,6-диметокси-5-нитропиридина Х-6а:
При перемешивании к раствору 2,3,6-трифтор-5-нитропиридина Х-5а (0,30 г; 1,68 ммоля) в МеОН (4 мл) при -40°С по каплям добавляли NaOMe (0,36 мл; 1,68 ммоля) и реакционную смесь перемешивали при такой же температуре в течение 2 ч. За протеканием реакции следили с помощью ТСХ. После завершения реакции реакцию при 0°С останавливали 2 н. раствором HCl (6 мл) и смесь экстрагировали гексаном (2×10 мл). Органический слой отделяли, сушили над безводным Na2SO4 и выпаривали в вакууме и получали 3-фтор-2,6-диметокси-5-нитропиридин Х-6а (0,32 г, 94%) в виде бледно-желтого твердого вещества. Это соединение использовали в следующей реакции без дополнительной очистки.
Выход: 94%.
1H ЯМР (400 МГц, ДМСО-d6) δ 4,06 (s, 3Н), 4,09 (s, 3H), 8,52 (d, J = 9,29 Гц, 1Н).
Стадия 2: Синтез 5-фтор-2,6-диметоксипиридин-3-амина Х-6:
При перемешивании к раствору 3-фтор-2,6-диметокси-5-нитропиридина X-6а (0,25 г, 1,24 ммоля) в уксусной кислоте (8 мл) при 0°С порциями добавляли железо (0,28 г, 4,95 ммоля). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь выливали в охлажденный льдом насыщенный раствор NaHCO3 (25 мл) и экстрагировали с помощью EtOAc (2×20 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме и получали 5-фтор-5-фтор-2,6-диметоксипиридин-3-амин Х-6 (0,19 г) в виде коричневого твердого вещества. Это соединение использовали в следующей реакции без дополнительной очистки.
Выход: 89%.
ЖХМС в щелочной среде, методика 2 (ЭР+): 173 (М+Н)+, чистота 99%.
1H ЯМР (400 МГц, ДМСО-d6) δ 3,82 (s, 3Н), 3,85 (s, 3Н), 4,58 (brs, 2H), 6,92 (d, J = 11,25 Гц, 1Н).
А.7. Синтез 2,5-дифтор-6-метилпиридин-3-амина Х-7:

К раствору 6-хлор-2,5-дифторпиридин-3-амина Х-2 (0,24 г, 1,38 ммоля) в диоксане (12 мл) при комнатной температуре добавляли метилбороновую кислоту (0,25 г, 4,15 ммоля) и раствор Cs2CO3 (1,13 г, 3,46 ммоля) в Н2О (4 мл) и реакционную смесь продували аргоном в течение 20 мин. Добавляли PdCl2(dppf) (0,10 г, 0,14 ммоля) и реакционную смесь продували аргоном в течение 10 мин. Реакционную смесь нагревали при 120°С в течение 6 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь охлаждали до комнатной температуры, фильтровали через слой целита, промывали с помощью EtOAc (2×60 мл) и фильтрат концентрировали в вакууме. Остаток разбавляли с помощью Н2О (60 мл) и экстрагировали с помощью EtOAc (3×40 мл). Органический слой отделяли, промывали рассолом (70 мл), сушили над безводным Na2SO4 и концентрировали в вакууме и получали 2,5-дифтор-6-метилпиридин-3-амин Х-7 (0,32 г) в виде бледно-коричневой жидкости. Это соединение использовали в следующей реакции без дополнительной очистки.
Выход: 51%.
ЖХМС в щелочной среде, методика 2 (ЭР+): 145 (М+Н)+, чистота 31%.
А.8. Синтез 6-(дифторметокси)-5-фтор-2-метоксипиридин-3-амина Х-8:

Стадия 1: Синтез 3-фтор-6-метокси-5-нитропиридин-2-ола Х-8а:
К раствору 2-хлор-3-фтор-6-метокси-5-нитропиридина Х-3а (0,90 г, 4,36 ммоля) в Н2О (6 мл) добавляли KOH (0,61 г, 10,9 ммоля) и реакционную смесь нагревали при 80°С в течение 16 ч. За протеканием реакции следили с помощью ТСХ. После завершения реакции реакционную смесь разбавляли с помощью Н2О (100 мл) и экстрагировали с помощью EtOAc (3×80 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме и получали 3-фтор-6-метокси-5-нитропиридин-2-ол Х-8а (0,30 г, неочищенный) в виде бледно-желтого твердого вещества. Это соединение использовали в следующей реакции без дополнительной очистки.
1H ЯМР (400 МГц, ДМСО-d6) δ 3,99 (s, 3Н), 8,42 (d, J = 9,60 Гц, 1H), 12,12 (s, 1H).
Стадия 2: Синтез 2-(дифторметокси)-3-фтор-6-метокси-5-нитропиридина X-8b:
К раствору 3-фтор-6-метокси-5-нитропиридин-2-ола Х-8а (0,29 г, 1,54 ммоля) в CH3CN (4 мл) при 40°С добавляли раствор KOH (0,87 г, 15,4 ммоля) в Н2О (1 мл) и бромдифторметилдиэтилфосфонат (2,74 мл, 15,4 ммоля) и реакционную смесь перемешивали при такой же температуре в течение 4 ч. За протеканием реакции следили с помощью ТСХ. После завершения реакции реакционную смесь разбавляли с помощью Н2О (50 мл) и экстрагировали с помощью EtOAc (3×40 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное неочищенное вещество очищали с помощью флэш-хроматографии (от 2 до 5% EtOAc в гексане) и получали 2-(дифторметокси)-3-фтор-6-метокси-5-нитропиридин Х-8b (0,24 г) в виде бледно-желтой жидкости. Это соединение использовали в следующей реакции без дополнительной очистки.
Выход: 65%.
1H ЯМР (400 МГц, ДМСО-d6) δ 4,04 (s, 3Н), 7,90 (t, J = 70,8 Гц, 1H), 8,80 (d, J = 9,60 Гц, 1Н).
Стадия 3: Синтез 6-(дифторметокси)-5-фтор-2-метоксипиридин-3-амина X-8:
К раствору 2-(дифторметокси)-3-фтор-6-метокси-5-нитропиридина Х-8b (0,23 г, 0,97 ммоля) в СН3СООН (8 мл) при 0°С медленно добавляли Fe (0,27 г, 4,83 ммоля) и реакционную смесь перемешивали при комнатной температуре в течение 4 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь фильтровали через слой целита, промывали с помощью EtOAc (80 мл) и фильтрат концентрировали в вакууме. Остаток выливали в насыщенный водный раствор NaHCO3 (80 мл) и экстрагировали с помощью EtOAc (2×70 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме и получали 6-(дифторметокси)-5-фтор-2-метоксипиридин-3-амин Х-8 (0,18 г) в виде коричневой жидкости. Это соединение использовали в следующей реакции без дополнительной очистки.
Выход: 77%.
ЖХМС в щелочной среде, методика 2 (ЭР-): 207 (М-Н)-, чистота 85%.
1H ЯМР (400 МГц, ДМСО-d6) δ 3,84 (s, 3Н), 5,18 (brs, 2H), 6,95 (d, J = 10,8 Гц, 1H), 7,39 (t, J = 74 Гц, 1Н).
A.9. Синтез 5-бром-3-метоксипиразин-2-амина Х-9:

Стадия 1: Синтез 3,5-дибромпиразин-2-амина Х-9а:
К раствору пиразин-2-амина (0,50 г, 5,26 ммоля) в ДМСО (10 мл) и Н2О (0,3 мл) при температуре ниже 15°С в течение 10 мин порциями добавляли NBS (1,97 г, 11,0 ммоля). Реакционную смесь перемешивали при отсутствии света при комнатной температуре в течение 5 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. Реакционную смесь выливали в охлажденную льдом Н2О (60 мл) и экстрагировали с помощью EtOAc (2×70 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное неочищенное вещество очищали с помощью колоночной хроматографии (диоксид кремния, 100-200 меш, 5% EtOAc в гексане) и получали 3,5-дибромпиразин-2-амин Х-9а (0,612 г) в виде почти белого твердого вещества.
Выход: 46%.
ЖХМС в щелочной среде, методика 2 (ЭР+): 252 (М+Н)+, чистота 100%.
1H ЯМР (400 МГц, ДМСО-d6) δ 6,97 (brs, 2H), 8,13 (s, 1H).
Стадия 2: Синтез 5-бром-3-метоксипиразин-2-амина Х-9:
Раствор 3,5-дибромпиразин-2-амина Х-9а (0,60 г, 2,37 ммоля) и NaOMe (0,15 г, 2,78 ммоля) в МеОН (15 мл) кипятили с обратным холодильником в течение 1 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь охлаждали до комнатной температуры. Осадившееся твердое вещество очищали с помощью колоночной хроматографии (диоксид кремния, 100-200 меш, 10% EtOAc в гексане) и получали 5-бром-3-метоксипиразин-2-амин Х-9 (0,295 г) в виде белого твердого вещества.
Выход: 61%.
ЖХМС в щелочной среде, методика 2 (ЭР+): 204 (М+Н)+, чистота 100%.
1H ЯМР (400 МГц, ДМСО-d6) δ 3,87 (s, 3Н), 6,52 (brs, 2H), 7,57 (s, 1H).
A. 10. Синтез 5-хлор-3-метоксипиразин-2-амина Х-10:
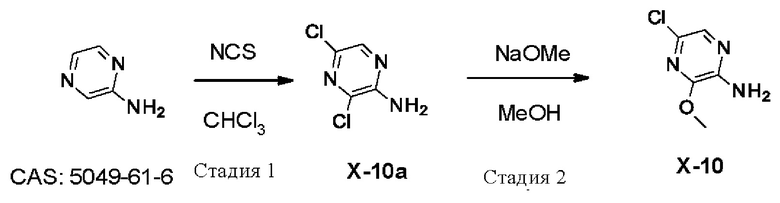
Стадия 1: Синтез 3,5-дихлорпиразин-2-амина Х-10а:
При перемешивании к раствору пиразин-2-амина (2,00 г, 21,0 ммоля) в CHCl3 (25 мл) порциями добавляли NCS (3,65 г, 27,3 ммоля) и реакционную смесь перемешивали при комнатной температуре в течение 6 ч. За протеканием реакции следили с помощью ТСХ. После завершения реакции реакционную смесь выливали в охлажденную льдом Н2О (20 мл) и экстрагировали с помощью EtOAc (2×40 мл). Органический слой отделяли, промывали рассолом (25 мл), сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное неочищенное вещество очищали с помощью хроматографии с использованием CombiFlash (20% EtOAc в гексане) и получали 3,5-дихлорпиразин-2-амин Х-10а (1,50 г) в виде почти белого твердого вещества.
Выход: 37%.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,02 (brs, 2Н), 8,06 (s, 1H).
Стадия 2: Синтез 5-хлор-3-метоксипиразин-2-амина Х-10:
При перемешивании к раствору 3,5-дихлорпиразин-2-амина Х-10а (0,80 г, 4,15 ммоля) в МеОН (20 мл) при комнатной температуре добавляли NaOMe (0,90 г, 16,6 ммоля). Реакционную смесь нагревали при 70°С в течение 16 ч. За протеканием реакции следили с помощью ТСХ. После завершения реакции реакционную смесь концентрировали в вакууме. Остаток разбавляли с помощью Н2О (15 мл) и экстрагировали с помощью EtOAc (3×25 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное неочищенное вещество очищали с помощью хроматографии с использованием CombiFlash (20% EtOAc в гексане) и получали 5-хлор-3-метоксипиразин-2-амин Х-10 (0,53 г) в виде почти белого твердого вещества.
Выход: 69%.
1H ЯМР (400 МГц, ДМСО-d6) δ 3,89 (s, 3Н), 6,52 (brs, 2H), 7,53 (s, 1H).
A. 11. Синтез 5-фтор-6-(2-фторэтокси)-2-метоксипиридин-3-амина Х-11:
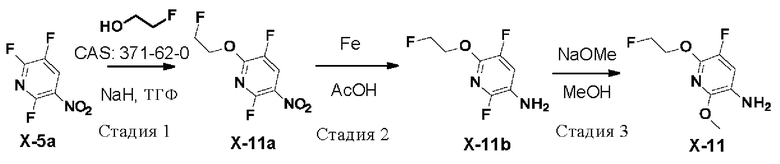
Стадия 1: Синтез 2,5-дифтор-6-(2-фторэтокси)-3-нитропиридина Х-11а:
При перемешивании к раствору 2-фторэтанола (1,19 г, 18,5 ммоля) в ТГФ (30 мл) при 0°С добавляли NaH (0,81 г, 20,2 ммоля) и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь охлаждали до -78°С, затем при такой же температуре медленно добавляли 2,3,6-трифтор-5-нитропиридин Х-5а (3,00 г, 16,8 ммоля) и реакционную смесь перемешивали при -78°С в течение 2 ч. За протеканием реакции следили с помощью ТСХ. После завершения реакции реакцию останавливали охлажденной льдом Н2О (50 мл) и смесь экстрагировали с помощью EtOAc (2×100 мл).
Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме и получали 2,5-дифтор-6-(2-фторэтокси)-3-нитропиридин Х-11а (3,10 г) в виде коричневой смолообразной жидкости. Это соединение использовали в следующей реакции без дополнительной очистки.
Выход: 83%.
1H ЯМР (400 МГц, ДМСО-d6) δ 4,64-4,69 (m, 1Н) 4,73-4,76 (m, 2H) 4,86-4,89 (m, 1H) 8,79-8,83 (m, 1Н).
Стадия 2: Синтез 2,5-дифтор-6-(2-фторэтокси)пиридин-3-амина Х-11b:
К раствору 2,5-дифтор-6-(2-фторэтокси)-3-нитропиридина Х-11a (2,70 г, 12,2 ммоля) в СН3СООН (25 мл) при 0°С добавляли Fe (6,79 г, 122 ммоля) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь фильтровали через слой целита и промывали с помощью Et2O (500 мл) и фильтрат концентрировали в вакууме. Остаток выливали в насыщенный водный раствор NaHCO3 (380 мл) и экстрагировали с помощью Et2O (2×500 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме и получали 2,5-дифтор-6-(2-фторэтокси)пиридин-3-амин Х-11b (2,00 г) в виде коричневого твердого вещества. Это соединение использовали в следующей реакции без дополнительной очистки.
Выход: 70%.
ЖХМС в щелочной среде, методика 2 (ЭР+): 193 (М+Н)+ чистота 82%.
Стадия 3: Синтез 5-фтор-6-(2-фторэтокси)-2-метоксипиридин-3-амина X-11:
К раствору 2,5-дифтор-6-(2-фторэтокси)пиридин-3-амина Х-11b (1,00 г, 4,27 ммоля) в МеОН (10 мл) при 0°С медленно добавляли NaOMe (25% раствор в МеОН, 1,85 мл, 8,55 ммоля) и реакционную смесь нагревали при 100°С в течение 16 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакцию останавливали охлажденным льдом 1 н. водным раствором HCl (50 мл) и смесь экстрагировали гексаном (2×500 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное неочищенное вещество очищали с помощью препаративной ВЭЖХ и получали 5-фтор-6-(2-фторэтокси)-2-метоксипиридин-3-амин Х-11 (0,46 г) в виде коричневого твердого вещества.
Выход: 50%.
ЖХМС в щелочной среде, методика 2 (ЭР+): 205 (М+Н)+, чистота 97%.
1H ЯМР (400 МГц, ДМСО-d6) δ 3,83 (s, 3Н) 4,41-4,43 (m, 1H) 4,48-4,50 (m, 1H) 4,65 (brs, 3Н) 4,76-4,78 (m, 1H) 6,90-6,98 (m, 1H).
A. 12. Синтез 6-(2-фторэтокси)-2-метоксипиридин-3-амина Х-12:

Стадия 1: Синтез 2,6-дифтор-3-нитропиридина Х-12а:
К раствору 2,6-дифторпиридина (5,00 г, 43,4 ммоля) в концентрированной HNO3 (36,3 мл, 869 ммолей) при 0°С медленно добавляли концентрированную H2SO4 (34,7 мл, 652 ммоля) и реакционную смесь нагревали при 60°С в течение 3 ч. За протеканием реакции следили с помощью ТСХ. После завершения реакции реакционную смесь охлаждали до комнатной температуры, выливали на дробленый лед (120 мл) и экстрагировали с помощью ДХМ (2×100 мл). Органический слой отделяли, промывали насыщенным водным раствором NaHCO3 (120 мл), сушили над безводным Na2SO4 и концентрировали в вакууме и получали 2,6-дифтор-3-нитропиридин Х-12а (3,20 г, неочищенный) в виде желтого масла. Это соединение использовали в следующей реакции без дополнительной очистки.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,47 (dd, J = 8,80, 2,45 Гц, 1H) 8,91-9,00 (m, 1H).
Стадия 2: Синтез 6-фтор-2-метокси-3-нитропиридина X-12b и 2-фтор-6-метокси-3-нитропиридина Х-12с:
К раствору 2,6-дифтор-3-нитропиридина Х-12а (2,90 г, 18,1 ммоля) в ТГФ (25 мл) при -78°С медленно добавляли NaOMe (25% раствор в МеОН, 4,31 мл, 19,9 ммоля) и реакционную смесь перемешивали при такой же температуре в течение 1 ч. За протеканием реакции следили с помощью ТСХ. После завершения реакции реакцию останавливали охлажденной льдом Н2О (60 мл) и смесь экстрагировали с помощью Et2O (2×100 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме и получали 6-фтор-2-метокси-3-нитропиридин X-12b и 2-фтор-6-метокси-3-нитропиридин Х-12с (2,51 г, смесь двух региоизомеров) в виде коричневой жидкости. Это соединение использовали в следующей реакции без дополнительной очистки.
1Н ЯМР (400 МГц, ДМСО-d6, 1Н ЯМР показал наличие смеси двух региоизомеров) δ 3,97 (s, 3Н), 4,02 (s, 3H) 6,97-7,00 (m, 2H) 8,58-8,71 (m, 2H).
Стадия 3: Синтез 6-(2-фторэтокси)-2-метокси-3-нитропиридина X-12d и 2-(2-фторэтокси)-6-метокси-3-нитропиридина Х-12е:
К раствору 2-фторэтанола (1,12 г, 17,5 ммоля) в ДМФ (40 мл) добавляли CS2CO3 (9,51 г, 29,2 ммоля) и смесь 6-фтор-2-метокси-3-нитропиридина X-12b и 2-фтор-6-метокси-3-нитропиридина Х-12c (2,51 г, 14,6 ммоля, смесь двух региоизомеров). Реакционную смесь нагревали при 100°С в течение 16 ч. За протеканием реакции следили с помощью ТСХ. После завершения реакции реакционную смесь разбавляли холодной Н2О (80 мл) и экстрагировали с помощью EtOAc (2×80 мл). Органический слой отделяли, промывали холодным Н2О (2×50 мл), сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное неочищенное вещество очищали с помощью колоночной хроматографии (диоксид кремния, 100-200 меш, от 5 до 12% EtOAc в гексане) и повторно очищали с помощью препаративной ВЭЖХ и получали 6-(2-фторэтокси)-2-метокси-3-нитропиридин X-12d (0,98 г) в виде почти белого твердого вещества и 2-(2-фторэтокси)-6-метокси-3-нитропиридин Х-12е (1,10 г) в виде почти белого твердого вещества.
6-(2-Фторэтокси)-2-метокси-3-нитропиридин X-12d:
Выход: 31%.
1H ЯМР (400 МГц, CDCl3) δ 4,12 (s, 3Н) 4,63 (t, J = 4 Гц, 1H) 4,69-4,74 (m, 2H) 4,84 (t, J = 4 Гц, 1H) 6,47 (d, J = 8,8 Гц, 1H) 8,39 (d, J = 8,8 Гц, 1H).
2-(2-Фторэтокси)-6-метокси-3-нитропиридин Х-12е:
Выход: 35%.
1H ЯМР (400 МГц, CDCl3) δ 4,01 (s, 3Н) 4,75-4,78 (m, 2H) 4,80-4,84 (m, 1H) 4,85-4,91 (m, 1H) 6,43 (d, J = 8,8 Гц, 1H) 8,37 (d, J = 8,8 Гц, 1H).
Стадия 4: Синтез 6-(2-фторэтокси)-2-метоксипиридин-3-амина Х-12:
К раствору 6-(2-фторэтокси)-2-метокси-3-нитропиридина X-12d (0,97 г, 4,49 ммоля) в СН3СООН (15 мл) медленно добавляли Fe (1,25 г, 22,4 ммоля) и реакционную смесь перемешивали при комнатной температуре в течение 6 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. Реакционную смесь фильтровали через слой целита, промывали с помощью EtOAc (80 мл) и фильтрат концентрировали в вакууме. Остаток разбавляли насыщенным раствором NaHCO3 (150 мл) и экстрагировали с помощью EtOAc (2×100 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное неочищенное вещество очищали с помощью флэш-хроматографии (от 10 до 20% EtOAc в гексане) и получали 6-(2-фторэтокси)-2-метоксипиридин-3-амин Х-12 (0,67 г) в виде бледно-коричневой жидкости.
Выход: 79%.
ЖХМС в щелочной среде, методика 2 (ЭР+): 187 (М+Н)+, чистота 97%.
1H ЯМР (400 МГц, ДМСО-d6) δ 3,84 (s, 3Н) 4,28-4,34 (m, 1H) 4,36 (s, 2H) 4,38-4,41 (m, 1H) 4,62-4,66 (m, 1H) 4,73-4,80 (m, 1H) 6,20 (d, J = 8,31 Гц, 1H) 6,96 (d, J = 7,83 Гц, 1H).
A. 13. Синтез 2,5-дифтор-6-метоксипиридин-3-амина Х-13:

Стадия 1: Синтез 2-(2,2-дифторэтокси)-3,6-дифтор-5-нитропиридина Х-13а:
При перемешивании к раствору 2,2-дифторэтанола (0,79 г, 11,2 ммоля) в ТГФ (20 мл) при 0°С добавляли NaH (1,35 г, 33,7 ммоля) и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь охлаждали до -78°С, затем при такой же температуре медленно добавляли 2,3,6-трифтор-5-нитропиридин Х-5а (2,00 г, 11,2 ммоля) и реакционную смесь перемешивали при -78°С в течение 2 ч. За протеканием реакции следили с помощью ТСХ. После завершения реакции реакцию останавливали охлажденной льдом Н2О (50 мл) и смесь экстрагировали с помощью EtOAc (2×100 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме. Неочищенное вещество очищали с помощью колоночной хроматографии (диоксид кремния, 100-200 меш, 2% EtOAc в гексане) и получали 2-(2,2-дифторэтокси)-3,6-дифтор-5-нитропиридин Х-13а (0,86 г) в виде коричневой жидкости.
Выход: 32%.
1H ЯМР (400 МГц, ДМСО-d6) δ 4,78 (td, J = 14,92, 2,93 Гц, 2 Н), 6,32-6,62 (m, 1 H), 8,87 (dd, J = 8,80, 7,34 Гц, 1 Н).
Стадия 2: Синтез 6-(2,2-дифторэтокси)-2,5-дифторпиридин-3-амина Х-13:
К раствору 2-(2,2-дифторэтокси)-3,6-дифтор-5-нитропиридин Х-13а (0,85 г, 3,5 ммоля) в СН3СООН (17 мл) при 0°С добавляли Fe (1,98 г, 35 ммолей) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь фильтровали через слой целита и промывали с помощью Et2O (500 мл) и фильтрат концентрировали в вакууме. Остаток выливали в насыщенный водный раствор NaHCO3 (380 мл) и экстрагировали с помощью Et2O (2×500 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме. Неочищенное вещество промывали пентаном и получали 6-(2,2-дифторэтокси)-2,5-дифторпиридин-3-амин Х-13 (0,51 г) в виде коричневого твердого вещества. Это соединение использовали в следующей реакции без дополнительной очистки.
Выход: 67%.
ЖХМС в щелочной среде, методика 2 (ЭР+): 211 (М+Н)+, чистота 99%.
1H ЯМР (400 МГц, ДМСО-d6) δ 4,43 (td, J = 14,92, 3,42 Гц, 2 Н), 5,20 (s, 2 H), 6,20-6,50 (m, 1 H), 7,21 (dd, J = 10,76, 8,31 Гц, 1 Н).
А.14. Синтез 6-(дифторметокси)-2-метоксипиридин-3-амина Х-14:
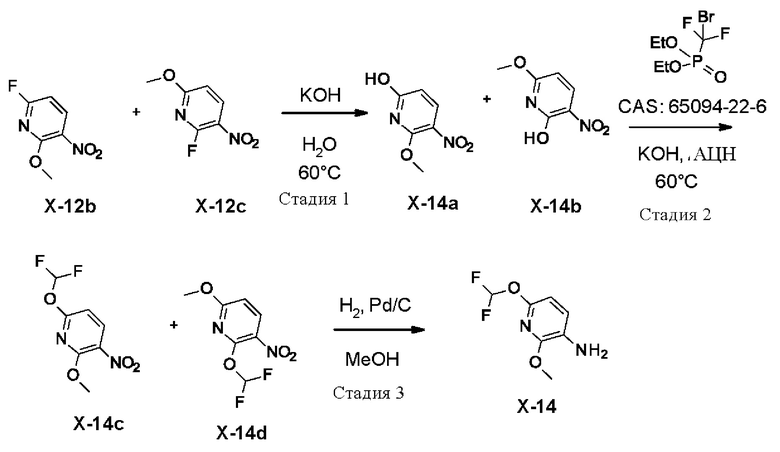
Стадия 1: Синтез 6-метокси-5-нитропиридин-2-ола Х-14а и 6-метокси-3-нитропиридин-2-ола Х-14b:
К раствору смеси 6-фтор-2-метокси-3-нитропиридина Х-12b и 2-фтор-6-метокси-3-нитропиридина Х-12с (0,60 г, 3,5 ммоля, смесь двух региоизомеров) в воде (20 мл) добавляли KOH (0,78 г, 13,9 ммоля). Реакционную смесь нагревали при 60°С в течение 3 ч. За протеканием реакции следили с помощью ТСХ. После завершения реакции реакционную смесь охлаждали до 0°С и подкисляли 1 н. раствором HCl (6 мл) до рН 4-5. Осадившееся твердое вещество отфильтровывали и сушили в вакууме и получали смесь 6-метокси-5-нитропиридин-2-ола Х-14а и 6-метокси-3-нитропиридин-2-ола Х-14b (0,45 г, смесь двух региоизомеров) в виде желтого твердого вещества.
Выход: 29%.
ЖХМС в щелочной среде, методика 2 (ЭР+): 171 (М+Н)+, чистота 99% (смесь состава 40/60).
Стадия 2: Синтез 6-(дифторметокси)-2-метокси-3-нитропиридина Х-14с и 2-(дифторметокси)-6-метокси-3-нитропиридина X-14d:
К раствору смеси 6-метокси-5-нитропиридин-2-ола Х-14а и 6-метокси-3-нитропиридин-2-ола Х-14b (1,2 г, 5,06 ммоля, смесь двух региоизомеров) в СН3СН (32 мл) и воде (8 мл) добавляли KOH (1,42 г, 25,3 ммоля) и бромдифторметилдиэтилфосфонат (6,75 г, 25,3 ммоля) и реакционную смесь перемешивали при 60°С в течение 4 ч. За протеканием реакции следили с помощью ТСХ. После завершения реакции реакционную смесь разбавляли с помощью Н2О (60 мл) и экстрагировали с помощью EtOAc (3×40 мл). Органический слой отделяли, промывали рассолом (2×50 мл) и сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное неочищенное вещество очищали с помощью флэш-хроматографии (2% EtOAc в гексане) и получали 6-(дифторметокси)-2-метокси-3-нитропиридин Х-14с (0,24 г) и 2-(дифторметокси)-6-метокси-3-нитропиридин X-14d (0,07 г) в виде почти белых твердых веществ.
6-(Дифторметокси)-2-метокси-3-нитропиридин Х-14с:
Выход: 22%.
1H ЯМР (400 МГц, ДМСО-d6) δ 4,12 (s, 3H), 6,60 (d, J = 8,80 Гц, 1Н), 7,41 (t, J = 72 Гц, 1 Н), 8,47 (d, J = 8,80 Гц, 1Н).
2-(Дифторметокси)-6-метокси-3-нитропиридин X-14d:
Выход: 7%.
1H ЯМР (400 МГц, ДМСО-d6) δ 4,03 (s, 3Н), 6,65 (d, J = 9,29 Гц, 1 Н), 7,51 (t, J = 72 Гц, 1 Н), 8,41 (d, J = 9,29 Гц, 1 Н).
Стадия 3: Синтез 6-(дифторметокси)-2-метоксипиридин-3-амина Х-14:
К раствору 6-(2-фторэтокси)-2-метокси-3-нитропиридина X-12d (50 мг, 0,22 ммоля) в МеОН (3 мл) добавляли Pd/C (10 мг) и реакционную смесь перемешивали при повышенном давлении водорода, при комнатной температуре в течение 2 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь фильтровали через целит, промывали с помощью МеОН (2×30 мл) и фильтрат концентрировали в вакууме и получали 6-(дифторметокси)-2-метоксипиридин-3-амин Х-14 (30 мг) в виде коричневой жидкости. Это соединение использовали в следующей реакции без дополнительной очистки.
Выход: 68%.
ЖХМС в щелочной среде, методика 2 (ЭР+): 191 (М+Н)+, чистота 97%.
А.15. Синтез 6-хлор-4-метоксипиридин-3-амина Х-15:
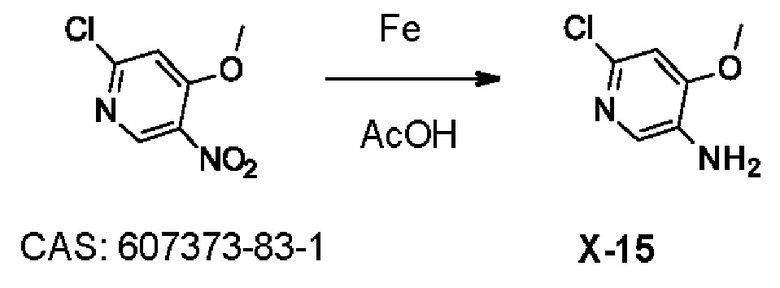
К раствору 2-хлор-4-метокси-5-нитропиридина (2,0 г, 10,6 ммоля) в СН3СООН (15 мл) при 0°С добавляли Fe (2,96 г, 53 ммоля) и реакционную смесь перемешивали при комнатной температуре в течение 3 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь фильтровали через слой целита и промывали с помощью EtOAc (2×30 мл) и фильтрат концентрировали в вакууме. Остаток выливали в насыщенный водный раствор NaHCO3 (100 мл) и экстрагировали с помощью EtOAc (2×60 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме. Неочищенное вещество промывали эфиром и получали 6-хлор-4-метоксипиридин-3-амин Х-15 (0,95 г) в виде почти белого твердого вещества.
Выход: 55%.
ЖХМС в щелочной среде, методика 2 (ЭР+): 159 (М+Н)+, чистота 100%.
1H ЯМР (400 МГц, ДМСО-d6) δ 3,85 (s, 3H), 5,01 (s, 2 H), 6,88 (s, 1 H), 7,60 (s, 1H).
А.16. Синтез 6-(2,2-дифторэтокси)-2-метоксипиридин-3-амина Х-16 и 2-(2,2-дифторэтокси)-6-метоксипиридин-3-амина Х-17:

Стадия 1: Синтез 6-(2,2-дифторэтокси)-2-метокси-3-нитропиридина Х-16а и 2-(2,2-дифторэтокси)-6-метокси-3-нитропиридина Х-17а:
К раствору смеси 6-фтор-2-метокси-3-нитропиридина Х-12b и 2-фтор-6-метокси-3-нитропиридина Х-12с (4,00 г, 23,2 ммоля, смесь двух региоизомеров) в ДМФ (40 мл) при 0°С добавляли Cs2CO3 (15,1 г, 46,5 ммоля) и 2-фторэтанол (1,79 г, 27,9 ммоля). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. За протеканием реакции следили с помощью ТСХ. После завершения реакции реакцию останавливали охлажденной льдом Н2О (50 мл) и смесь экстрагировали с помощью EtOAc (2×300 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме и получали 6-(2,2-дифторэтокси)-2-метокси-3-нитропиридин Х-16а и 2-(2,2-дифторэтокси)-6-метокси-3-нитропиридин Х-17а (4,20 г, неочищенные, смесь двух региоизомеров) в виде коричневой смолообразной жидкости.
1Н ЯМР (400 МГц, ДМСО-d6, 1Н ЯМР показал наличие смеси двух региоизомеров) δ 3,99 (s, 3Н) 4,69-4,76 (m, 2H) 6,64-6,68 (m, 1H) 8,46 (d, J = 3,91 Гц, 1H) 8,48 (d, J = 3,91 Гц, 1Н).
Стадия 2: Синтез 6-(2,2-дифторэтокси)-2-метоксипиридин-3-амина Х-16 и 2-(2,2-дифторэтокси)-6-метоксипиридин-3-амина Х-17:
К раствору 6-(2,2-дифторэтокси)-2-метокси-3-нитропиридина Х-16а и 2-(2,2-дифторэтокси)-6-метокси-3-нитропиридина Х-17а (0,50 г, 2,14 ммоля, смесь двух региоизомеров) в СН3СООН (10 мл) при 0°С медленно добавляли железо (1,19 г, 21,4 ммоля) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. За протеканием реакции следили с помощью ТСХ. Реакционную смесь фильтровали через слой целита®, промывали с помощью EtOAc (500 мл) и фильтрат концентрировали в вакууме. Остаток выливали в насыщенный водный раствор NaHCO3 (380 мл) и экстрагировали с помощью EtOAc (2×500 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме и получали 6-(2,2-дифторэтокси)-2-метоксипиридин-3-амин Х-16 и 2-(2,2-дифторэтокси)-6-метоксипиридин-3-амин Х-17 (0,32 г, неочищенные, смесь двух региоизомеров) в виде коричневого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6, 1H ЯМР показал наличие смеси двух региоизомеров) δ 3,99 (s, 3Н) 4,69-4,76 (m, 2H) 6,64-6,68 (m, 1H) 8,46 (d, J = 3,91 Гц, 1H) 8,48 (d, J = 3,91 Гц, 1Н). (Протоны NH2 не видны).
А.17. Синтез 6-хлор-4-метоксипиридин-3-амина Х-15:
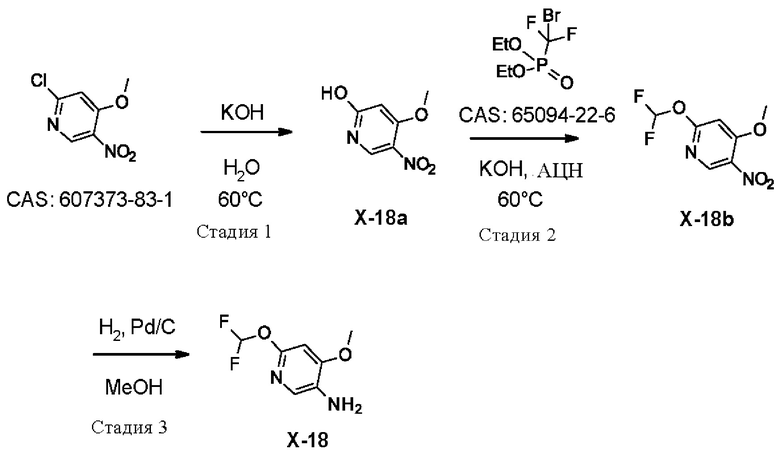
Стадия 1: Синтез 4-метокси-5-нитропиридин-2-ола Х- 18а:
К раствору 2-хлор-4-метокси-5-нитропиридина (1,00 г, 5,30 ммоля) в Н2О (25 мл) добавляли KOH (1,49 г, 26,5 ммоля) и реакционную смесь нагревали при 60°С в течение 3 ч. За протеканием реакции следили с помощью ТСХ. После завершения реакции реакционную смесь охлаждали до комнатной температуры, выливали в охлажденную льдом Н2О (100 мл), при 0°С подкисляли 1 н. раствором HCl (8 мл) до рН 4 и экстрагировали с помощью EtOAc (3×70 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме и получали 4-метокси-5-нитропиридин-2-ол Х-18а (0,71 г) в виде бледно-желтого твердого вещества. Это соединение использовали в следующей реакции без дополнительной очистки.
Выход: 61%.
ЖХМС в щелочной среде, методика 2 (ЭР+): 171 (М+Н)+, чистота 77%.
1H ЯМР (400 МГц, ДМСО-d6) δ 3,83 (s, 3Н) 5,87 (s, 1H) 8,48 (s, 1H) 12,17 (brs, 1H).
Стадия 2: Синтез 2-(дифторметокси)-4-метокси-5-нитропиридина X-18b:
К раствору 4-метокси-5-нитропиридин-2-ола Х-18а (0,60 г, 2,73 ммоля) в СН3СН (20 мл) и H2O (5 мл) при комнатной температуре медленно добавляли KOH (0,77 г, 13,6 ммоля) и бромдифторметилдиэтилфосфонат (3,64 г, 13,6 ммоля) и реакционную смесь нагревали при 60°С в течение 4 ч. За протеканием реакции следили с помощью ТСХ. После завершения реакции реакционную смесь разбавляли с помощью Н2О (60 мл) и экстрагировали с помощью EtOAc (3×40 мл). Органический слой отделяли, промывали рассолом (2×50 мл), сушили над безводным Na2SO4 и концентрировали в вакууме. Реакцию повторяли с использованием 2,70 г и вещества, полученные при проведении 2 реакций, объединяли и растворяли в ДХМ (150 мл) и полученное неочищенное вещество очищали с помощью колоночной хроматографии (диоксид кремния, 100-200 меш, 10% EtOAc в гексане) и получали 2-(дифторметокси)-4-метокси-5-нитропиридин X-18b (1,55 г, 36%) в виде бледно-желтой жидкости.
Выход: 36%.
ЖХМС в щелочной среде, методика 2 (ЭР+): 221 (М+Н)+, чистота 82%.
1H ЯМР (400 МГц, CDCl3) δ 4,05 (s, 3H) 6,53 (s, 1H) 7,52 (t, J = 72 Гц, 1Н) 8,76 (s, 1H).
Стадия 3: Синтез 6-(дифторметокси)-4-метоксипиридин-3-амина Х-18:
К раствору 2-(дифторметокси)-4-метокси-5-нитропиридина Х-18b (1,50 г, 5,62 ммоля) в МеОН (50 мл) при комнатной температуре добавляли 20% Pd/C (влажность 50%, 0,18 г) и реакционную смесь перемешивали при повышенном давлении водорода, при комнатной температуре в течение 4 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь фильтровали через слой целита®, промывали с помощью МеОН (2×60 мл) и фильтрат концентрировали в вакууме. Полученное неочищенное вещество очищали с помощью колоночной хроматографии (диоксид кремния, 100-200 меш, 30% EtOAc в гексане) и получали 6-(дифторметокси)-4-метоксипиридин-3-амин Х-18 (0,805 г, 75%) в виде белого твердого вещества.
Выход: 36%.
ЖХМС в щелочной среде, методика 2 (ЭР+): 191 (М+Н)+, чистота 96%.
1H ЯМР (400 МГц, ДМСО-d6) δ 3,85 (s, 3H) 4,72 (s, 2H) 6,56 (s, 1H) 7,44 (s, 1H) 7,49 (t, J = 74 Гц, 1H).
A. 18. Синтез 6-циклопропил-2,5-дифторпиридин-3-амина Х-19:

К раствору 6-хлор-2,5-дифторпиридин-3-амина Х-2 (0,25 г, 1,52 ммоля) в диоксане (8 мл) при комнатной температуре добавляли циклопропилбороновую кислоту (0,27 г, 3,18 ммоля) и раствор Cs2CO3 (1,24 г, 3,80 ммоля) в Н2О (2 мл) и реакционную смесь продували аргоном в течение 10 мин. Добавляли PdCl2(dppf) (0,11 г, 0,15 ммоля) и реакционную смесь нагревали при 120°С в течение 18 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь охлаждали до комнатной температуры, фильтровали через слой целита, промывали с помощью EtOAc (2×60 мл) и фильтрат концентрировали в вакууме. Остаток разбавляли с помощью H2O (60 мл) и экстрагировали с помощью EtOAc (3×40 мл). Органический слой отделяли, промывали рассолом (70 мл), сушили над безводным Na2SO4 и концентрировали в вакууме Полученное неочищенное вещество очищали с помощью колоночной хроматографии (диоксид кремния, 100-200 меш, 8% EtOAc в гексане) и получали 6-циклопропил-2,5-дифторпиридин-3-амин Х-19 (0,10 г) в виде бесцветной жидкости.
Выход: 37%.
ЖХМС в щелочной среде, методика 2 (ЭР+): 171 (М+Н)+, чистота 95%.
А.19. Синтез 6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-амина Х-20:

К раствору 6-(2,2-дифторэтокси)-2,5-дифторпиридин-3-амина Х-13 (1,00 г, 4,76 ммоля) в ТГФ (15 мл) при 0°С медленно добавляли NaOMe (25% в МеОН, 1,13 г, 5,23 ммоля) и реакционную смесь нагревали при 80°С в течение 16 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакцию останавливали охлажденной льдом H2O (50 мл) и смесь экстрагировали с помощью EtOAc (2×100 мл). Органический слой отделяли, сушили над безводным Na2S04 и концентрировали в вакууме. Полученное неочищенное вещество очищали с помощью колоночной хроматографии (диоксид кремния, 100-200 меш, от 5 до 10% EtOAc в гексане) и получали 6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-амин Х-20 (0,55 г) в виде коричневой жидкости.
Выход: 50%.
ЖХМС в щелочной среде, методика 2 (ЭР+): 223 (М+Н)+, чистота 91%.
1H ЯМР (400 МГц, ДМСО-d6) δ 3,85 (s, 3Н) 4,50 (td, J = 14,89, 3,69 Гц, 2Н) 4,74 (s, 2H) 6,23-6,54 (m, 1H) 6,96 (d, J = 11,32 Гц, 1Н).
А.20. Синтез 6-(дифторметокси)-5-фтор-2-метоксипиридин-3-амина Х-21:

Стадия 1: Синтез 2-(2-(дифторметокси)этокси)-3-фтор-6-метокси-5-нитропиридина Х-21а:
К раствору 3-фтор-6-метокси-5-нитропиридин-2-ола Х-8а (0,10 г, 0,53 ммоля) в ДМФ (4 мл) при комнатной температуре добавляли K2CO3 (0,22 г, 1,59 ммоля) и 1-бром-2-(дифторметокси)этан (0,09 г, 0,53 ммоля) и реакционную смесь нагревали в микроволновой печи при 90°С в течение 2 ч. За протеканием реакции следили с помощью ТСХ. После завершения реакции реакционную смесь охлаждали до комнатной температуры, выливали в Н2О (50 мл) и экстрагировали с помощью EtOAc (3×40 мл). Органический слой отделяли, промывали рассолом (2×50 мл), сушили над безводным Na2SO4 и концентрировали в вакууме и получали 2-(2-(дифторметокси)этокси)-3-фтор-6-метокси-5-нитропиридин Х-21а (0,09 г) в виде коричневой жидкости. Это соединение использовали в следующей реакции без дополнительной очистки.
Выход: 64%.
1H ЯМР (400 МГц, ДМСО-d6) δ 4,05 (s, 3Н) 4,22-4,27 (m, 2H) 4,70-4,76 (m, 2H) 6,75 (t, J = 74 Гц, 1Н) 8,57 (d, J = 9,78 Гц, 1Н).
Стадия 2: Синтез 6-(2-(дифторметокси)этокси)-5-фтор-2-метоксипиридин-3-амина Х-21:
К раствору 2-(2-(дифторметокси)этокси)-3-фтор-6-метокси-5-нитропиридина Х-21а (0,09 г, 0,32 ммоля) в СН3СООН (2 мл) при 0°С медленно добавляли железо (0,18 г, 3,19 ммоля) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь фильтровали через слой целита, промывали с помощью EtOAc (50 мл) и фильтрат концентрировали в вакууме. Остаток выливали в насыщенный водный раствор NaHCO3 (10 мл) и экстрагировали с помощью EtOAc (2×50 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное неочищенное вещество очищали путем промывки пентаном (3×70 мл) и получали 6-(2-(дифторметокси)этокси)-5-фтор-2-метоксипиридин-3-амин Х-21 (0,08 г, 77%) в виде коричневого твердого вещества. Это соединение использовали в следующей реакции без дополнительной очистки.
МС (ИЭР) m/е [М+Н]+/ВУ (время удерживания)/%: 253,00/1,72/77,7%.
Выход: 77%.
ЖХМС в щелочной среде, методика 2 (ЭР+): 253 (М+Н)+, чистота 78%.
А.21. Синтез 6-хлор-4-метоксипиридин-3-амина Х-22:

К раствору 6-хлор-5-фтор-2-метоксипиридин-3-амина Х-3 (2,0 г, 11,33 ммоля) в МеОН (38 мл) в атмосфере аргона в течение 5 мин добавляли Pd/C (20%, 0,43 г) и реакционную смесь перемешивали в атмосфере водорода при комнатной температуре в течение 16 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь фильтровали через слой целита и промывали с помощью EtOAc (3×100 мл). Фильтрат концентрировали в вакууме. Полученное неочищенное вещество очищали с помощью колоночной хроматографии (диоксид кремния, 100-200 меш, от 4 до 10% EtOAc в гексане) и получали 5-фтор-2-метоксипиридин-3-амин Х-22 (0,48 г) в виде коричневого твердого вещества.
Выход: 30%.
ЖХМС в щелочной среде, методика 2 (ЭР+): 143 (М+Н)+, чистота 96%.
1H ЯМР (400 МГц, ДМСО-d6) δ 3,83 (s, 3Н) 5,32 (br s, 2H) 6,72 (dd, J = 2,8, 9,6 Гц, 1H) 7,24 (d, J = 2,8 Гц, 1Н).
А.22. Синтез 6-циклопропил-5-фтор-2-метоксипиридин-3-амина Х-23:

К раствору 6-циклопропил-2,5-дифторпиридин-3-амина Х-19 (1,50 г, 8,75 ммоля) в МеОН (20 мл) при комнатной температуре добавляли NaOMe (25% в МеОН, 3,78 мл, 17,5 ммоля) и реакционную смесь нагревали при 100°С в течение 24 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь концентрировали в вакууме. Остаток разбавляли с помощью Н2О (20 мл) и экстрагировали с помощью EtOAc (3×25 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное неочищенное вещество очищали с помощью хроматографии с использованием CombiFlash (20% EtOAc в гексане) и получали 6-циклопропил-5-фтор-2-метоксипиридин-3-амин Х-23 (1,10 г) в виде коричневого масла.
Выход: 69%.
ЖХМС в щелочной среде, методика 2 (ЭР+): 183 (М+Н)+, чистота 99%.
1H ЯМР (400 МГц, ДМСО-d6) δ 0,76-0,86 (m, 4Н) 1,94-2,03 (m, 1H) 3,75 (s, 3H) 4,94 (s, 2H) 6,68 (d, J = 10,76 Гц, 1Н).
В. Синтез промежуточных продуктов формулы XI
B.1. Синтез 6-(дифторметил)-1Н-пирроло[2,3-b]пиридина XI-1:
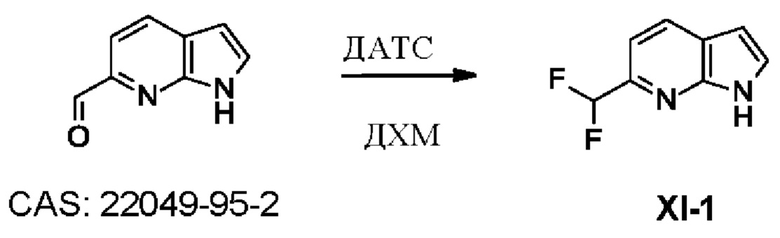
К раствору 1Н-пирроло[2,3-b]пиридин-6-карбальдегида (196 мг, 1,26 ммоля) в дихлорметане (4 мл) при 0°С добавляли диэтиламинотрифторид серы (260 мкл, 1,91 ммоля). Реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь выливали в смесь льда и NaHCO3 и 3 раза экстрагировали с помощью ДХМ. Органическую фазу сушили над Na2SO4 и растворители выпаривали и получали 6-(дифторметил)-1Н-пирроло[2,3-b]пиридин XI-1 (96 мг) в виде коричневого твердого вещества.
Выход: 45%.
ЖХМС в щелочной среде, методика 1 (ЭР+): 169 (М+Н)+, чистота 82%.
В.2. Синтез 6-(дифторметокси)-1Н-индола XI-2:

Стадия 1: Синтез трет-бутил-6-((трет-бутоксикарбонил)окси)-1Н-индол-1-карбоксилата XI-2а:
К раствору lH-индол-6-ола (5,00 г, 37,6 ммоля) в CH3CN (50 мл) добавляли ди-трет-бутилдикарбонат (25,9 мл, 113 мл), ДМАП (2,29 г, 18,8 ммоля) и триэтиламин (15,7 ммоля, 113 ммоля). Реакционную смесь перемешивали при 25°С в течение 16 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь концентрировали в вакууме. Полученное неочищенное вещество очищали с помощью колоночной хроматографии (диоксид кремния, 100-200 меш, 20% EtOAc в гексане) и получали трет-бутил-6-((трет-бутоксикарбонил)окси)-1Н-индол-1-карбоксилат XI-2а (10,0 г) в виде бледно-желтой жидкости.
Выход: 80%.
ЖХМС в щелочной среде, методика 2 (ЭР-): 332 (М-Н)-, чистота 99%.
Стадия 2: Синтез трет-бутил-6-гидрокси-1Н-индол-1-карбоксилата XI-2b:
К раствору трет-бутил-6-((трет-бутоксикарбонил)окси)-1Н-индол-1-карбоксилата XI-2а (9,90 г, 29,6 ммоля) в ДХМ (100 мл) добавляли морфолин (51,8 мл, 592 ммоля) и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. За протеканием реакции следили с помощью ТСХ. После завершения реакции реакционную смесь разбавляли с помощью Н2О (200 мл) и экстрагировали с помощью EtOAc (3×100 мл). Органический слой отделяли, промывали рассолом (2×100 мл), сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное неочищенное вещество очищали с помощью колоночной хроматографии (диоксид кремния, 100-200 меш, 15% EtOAc в гексане) и получали трет-бутил-6-гидрокси-1Н-индол-1-карбоксилат XI-2b (6,70 г) в виде бесцветного масла.
Выход: 97%.
1H ЯМР (400 МГц, ДМСО-d6) δ 1,61 (s, 9H) 6,55 (d, J = 3,94 Гц, 1Н) 6,71 (dd, J = 8,37, 1,97 Гц, 1Н) 7,36 (d, J = 8,86 Гц, 1Н) 7,43 (d, J = 3,45 Гц, 1Н) 7,51 (s, 1Н) 9,41 (s, 1Н).
Стадия 3: Синтез трет-бутил-6-(дифторметокси)-1Н-индол-1-карбоксилата XI-2c:
К раствору трет-бутил-6-гидрокси-1Н-индол-1-карбоксилата XI-2b (2,00 г, 8,57 ммоля) в CH3CN (20 мл) и H2O (20 мл) при -78°С медленно добавляли KOH (9,62 г, 171 ммоль) и бромдифторметилдиэтилфосфонат (3,05 мл, 17,1 ммоля). Через 15 мин реакционную смесь перемешивали при 0°С в течение 3 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь разбавляли с помощью Н2О (200 мл) и экстрагировали с помощью EtOAc (2×200 мл). Органический слой отделяли, промывали рассолом (2×30 мл), сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное неочищенное вещество очищали с помощью колоночной хроматографии (диоксид кремния, 100-200 меш, 15% EtOAc в гексане) и получали трет-бутил-6-(дифторметокси)-1Н-индол-1-карбоксилат XI-2c (0,68 г) в виде желтого масла.
Выход: 21%.
ЖХМС в щелочной среде, методика 2 (ЭР-): 282 (М-Н)-, чистота 74%.
1H ЯМР (400 МГц, ДМСО-d6) δ 1,63 (s, 9H) 6,73 (d, J = 3,91 Гц, 1Н) 7,09 (dd, J = 8,80, 1,47 Гц, 1Н) 7,23 (t, J = 76 Гц, 1Н) 7,66 (d, J = 8,31 Гц, 1Н) 7,69 (d, J = 3,42 Гц, 1Н) 7,86 (s, 1Н).
Стадия 4: Синтез 6-(дифторметокси)-1Н-индола XI-2:
К раствору трет-бутил-6-(дифторметокси)-1Н-индол-1-карбоксилата XI-2 с (0,67 г, 1,76 ммоля) в ДХМ (25 мл) при 0°С добавляли ТФК (трифторуксусная кислота, 40 мл) и реакционную смесь перемешивали при такой же температуре в течение 5 мин, затем при комнатной температуре в течение 1 ч. За протеканием реакции следили с помощью ТСХ. После завершения реакции реакционную смесь концентрировали в вакууме. Остаток разбавляли с помощью Н2О (100 мл), насыщенным раствором NaHCO3 (50 мл) и экстрагировали с помощью EtOAc (2×200 мл). Органический слой отделяли, сушили над безводным NaHSO4 и концентрировали в вакууме и получали 6-(дифторметокси)-1Н-индол XI-2 (0,31 г) в виде коричневого масла.
Выход: 76%.
ЖХМС в щелочной среде, методика 2 (ЭР-): 182 (М-Н)-, чистота 79%.
1H ЯМР (400 МГц, ДМСО-d6) δ 6,42-6,44 (m, 1Н) 6,83 (dd, J = 8,56, 1,71 Гц, 1Н) 7,14 (t, J = 74 Гц, 1Н) 7,18 (s, 1Н) 7,37 (t, J = 2,45 Гц, 1Н) 7,54 (d, J = 8,80 Гц, 1Н) 11,17 (brs, 1Н).
В.3. Синтез 6-хлор-7-фтор-1Н-индола XI-3:

К раствору 1-хлор-2-фтор-3-нитробензола (2,50 г, 14,2 ммоля) в ТГФ (50 мл) при -78°С добавляли винилмагнийбромид (5,61 г, 42,7 ммоля) и реакционную смесь перемешивали при такой же температуре в течение 1 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакцию останавливали насыщенным раствором NH4Cl (100 мл), смесь разбавляли с помощью Н2О (400 мл) и экстрагировали с помощью EtOAc (500 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное неочищенное вещество очищали с помощью колоночной хроматографии (диоксид кремния, 100-200 меш, 5% EtOAc в гексане) и получали 6-хлор-7-фтор-1Н-индол XI-3 (0,60 г) в виде красной жидкости.
Выход: 17%.
ЖХМС в щелочной среде, методика 2 (ЭР-): 168,00 (М-Н)-, чистота 66%.
В.4. Синтез 1-(бензолсульфонил)-6-бензилоксипирроло[2,3-b]пиридина XI-4:
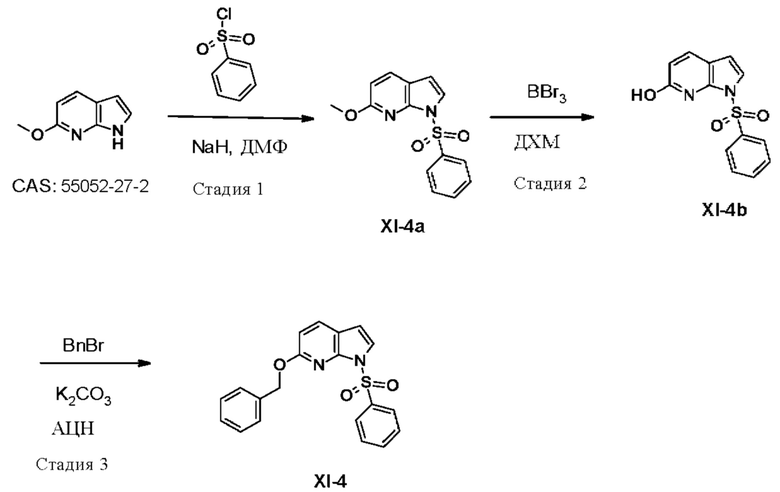
Стадия 1: Синтез 1-(бензолсульфонил)-6-метоксипирроло[2,3-b]пиридина Х1-4а:
Раствор 6-метокси-1Н-пирроло[2,3-b]пиридина (998 мг, 5,4 ммоля) в 10 мл ДМФ обрабатывали гидридом натрия (60% в парафине, 238 мг, 6 ммолей) и перемешивали при комнатной температуре в течение 1 ч. Затем добавляли хлорангидрид бензолсульфоновой кислоты (0,8 мл, 6,5 ммоля). Реакционную смесь перемешивали при комнатной температуре в течение 18 ч, добавляли воду (100 мл) и суспензию экстрагировали этилацетатом (3×30 мл). Объединенные органические экстракты сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученное вещество очищали с помощью колоночной хроматографии на силикагеле (с использованием смеси петролейный эфир:этилацетат, 80:20). Собранные фракции выпаривали и получали 980 мг 1-(бензолсульфонил)-6-метоксипирроло[2,3-b]пиридина XI-4а в виде белого порошкообразного вещества.
Выход: 63%.
ЖХМС в нейтральной среде, методика 3 (ЭР+): 289 (М+Н)+, чистота 100%.
1Н ЯМР (600 МГц, ДМСО-d6) δ 3,89 (s, 3Н), 6,71 (dd, J=6,2, 2,3 Гц, 2Н), 7,67-7,61 (m, 3Н), 7,75-7,70 (m, 1Н), 7,91 (d, J=8,5 Гц, 1H), 8,13 (dd, J=8,5, 1,3 Гц, 2Н).
Стадия 2: Синтез 1-(бензолсульфонил)пирроло[2,3-b]пиридин-6-ола XI-4b:
К раствору 1-(бензолсульфонил)-6-метоксипирроло[2,3-b]пиридина XI-4а (800 мг, 2,7 ммоля) в дихлорметане (35 мл) при 0°С добавляли 1,0 М раствор трибромида бора в дихлорметане (5 мл, 5 ммолей), затем нагревали до комнатной температуры и перемешивали при такой же температуре в течение 95 ч. Реакционную смесь гидролизовали путем добавления насыщенного раствора NaHCO3 (40 мл). Добавляли воду и водную фазу экстрагировали этилацетатом (3×35 мл). Объединенные органические экстракты сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученное вещество очищали с помощью колоночной хроматографии на силикагеле (с использованием смеси петролейный эфир:этилацетат, 80:20). Собранные фракции выпаривали и получали 580 мг 1-(бензолсульфонил)пирроло[2,3-b]пиридин-6-ола XI-4b в виде белого порошкообразного вещества. Выход: 79%.
ЖХМС в нейтральной среде, методика 3 (ЭР+): 275 (М+Н)+, чистота 93%.
1Н ЯМР (600 МГц, ДМСО-d6) δ 6,58 (d, J=8,4 Гц, 1H), 6,66 (d, J=4,0 Гц, 1Н), 7,55 (d, J=4,0 Гц, 1H), 7,64-7,58 (m, 2Н), 7,75-7,69 (m, 1Н), 7,84 (d, J=8,4 Гц, 1Н), 8,20-8,14 (m, 2Н), 10,92 (s, 1Н).
Стадия 3: Синтез 1-(бензолсульфонил)-6-бензилоксипирроло[2,3-b]пиридина XI-4:
Смесь 1-(бензолсульфонил)пирроло[2,3-b]пиридин-6-ола XI-4b (767 мг, 2,8 ммоля), бензилбромида (0,29 мл, 205 ммолей, 0,89 экв.) и карбоната калия (967,2 мг, 7 ммолей, 2,5 экв.) в сухом ацетонитриле (20 мл) нагревали в атмосфере аргона при 50°С в течение 22 ч. После охлаждения реакционную смесь фильтровали для удаления непрореагировавшего карбоната калия и тщательно промывали этилацетатом (100 мл). После выпаривания органического растворителя получали 1-(бензолсульфонил)-6-бензилоксипирроло[2,3-b]пиридин XI-4 в виде белого твердого вещества (600 мг). Это соединение использовали в следующей реакции без дополнительной очистки.
Выход: 59%.
ЖХМС в нейтральной среде, методика 3 (ЭР+): 365 (М+Н)+, неочищенный.
С. Синтез промежуточных продуктов формулы XII
С.1. Синтез 6-хлор-1Н-индол-3-сульфонилхлорида XII-1:

Стадия 1: Синтез 6-хлор-1Н-индол-3-сульфоновой кислоты XII-1а:
К раствору 6-хлориндола (1,00 г, 6,62 ммоля) в пиридине (10 мл) добавляли комплекс пиридин-триоксид серы (1,57 г, 9,93 ммоля) и реакционную смесь кипятили с обратным холодильником в течение 16 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь разбавляли с помощью Н2О (100 мл) и экстрагировали с помощью Et2O (250 мл). Водный слой отделяли и концентрировали в вакууме. Полученное неочищенное вещество выпаривали с толуолом и получали 6-хлор-1H-индол-3-сульфоновую кислоту XII-1а (2,30 г, неочищенная) в виде коричневого полужидкого вещества. Это соединение использовали в следующей реакции без дополнительной очистки.
ЖХМС в щелочной среде, методика 2 (ЭР-): 230 (М-Н)-, чистота 98%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 6,98-7,04 (m, 1H), 7,12-7,26 (m, 1H), 7,44 (s, 1H), 7,69-7,75 (m, 1H), 11,13 (brs, 1Н).
Стадия 2: Синтез 6-хлор-1Н-индол-3-сульфонилхлорида XII-1:
К раствору 6-хлор-1Н-индол-3-сульфоновой кислоты XII-1а (2,00 г, 6,45 ммоля) в сульфолане (5 мл) и CH3CN (5 мл) при 0°С по каплям добавляли POCl3 (1,30 мл, 14,2 ммоля) и реакционную смесь нагревали при 70°С в течение 3 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакцию останавливали охлажденной льдом Н2О (100 мл) и смесь экстрагировали с помощью EtOAc (2×50 мл). Органический слой отделяли, промывали рассолом (50 мл), сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное неочищенное вещество очищали с помощью колоночной хроматографии (диоксид кремния, 100-200 меш, 30% EtOAc в гексане) и получали 6-хлор-1Н-индол-3-сульфонилхлорид XII-1 (1,00 г) в виде светло-розового твердого вещества.
Выход: 62%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 7,32 (dd, J=8,56, 1,22 Гц, 1H), 7,71 (s, 1Н), 8,03 (d, J=8,80 Гц, 1H), 8,45 (d, J=2,93 Гц, 1H), 12,38 (brs, 1H).
С.2. Синтез 6-бром-1Н-индол-3-сульфонилхлорида XII-2:
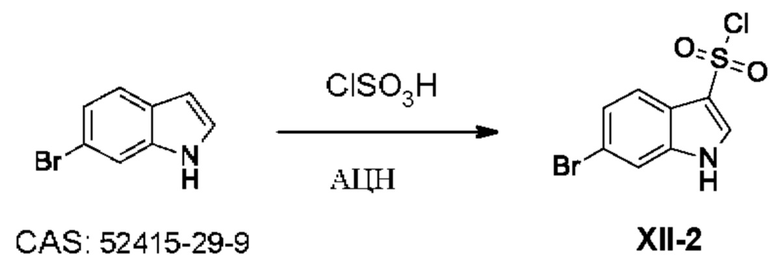
К раствору 6-бром-1H-индола (5 г, 25,5 ммоля) в CH3CN (60 мл) при 0°С добавляли ClSO3H (1 мл) и реакционную смесь перемешивали при комнатной температуре в течение 12 ч. За протеканием реакции следили с помощью ТСХ. После завершения реакции реакционную смесь выливали в охлажденную льдом Н2О (200 мл) и перемешивали в течение 30 мин. Осадившееся твердое вещество отфильтровывали и сушили в вакууме и получали 6-бром-1H-индол-3-сульфонилхлорид XII-2 (5 г) в виде коричневого твердого вещества. Это соединение использовали в следующей реакции без дополнительной очистки.
Выход: 66%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 7,38-7,48 (m, 1H) 7,85 (s, 1Н) 7,97 (d, J=8,37 Гц, 1Н) 8,44 (d, J=3,45 Гц, 1H) 12,55 (brs, 1H).
С.3. Синтез 1-(бензолсульфонил)-6-хлориндол-3-сульфонилхлорида XII-3:
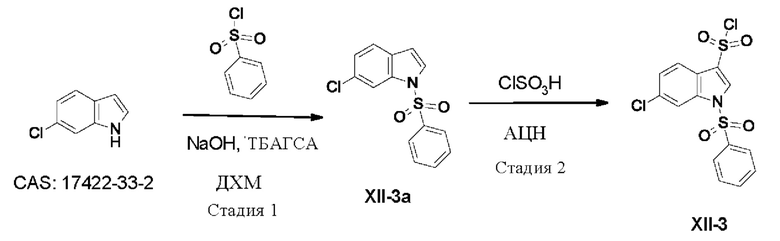
Стадия 1: Синтез 1-(бензолсульфонил)-6-хлориндола XII-3а:
Суспензию тонкоизмельченного гидроксида натрия (24,5 г, 613 ммолей) в дихлорметане (300 мл) перемешивали в бане со льдом и одной порцией добавляли 6-хлориндол (30 г, 197 ммолей), затем тетрабутиламмонийгидросульфат (1,75 г, 5,15 ммоля). Затем в течение 20 мин по каплям добавляли бензолсульфонилхлорид (2,2 мл, 218 ммолей) и реакционную смесь перемешивали при 0°С в течение 1 ч. Затем баню со льдом удаляли и смесь перемешивали при комнатной температуре в течение еще 1 ч. После завершения реакции по данным ЖХ/МС реакционную смесь фильтровали через слой целита и последний промывали с помощью ДХМ, объединенные фильтрат и промывочные растворы выпаривали досуха. Продукт растирали с эфиром, отфильтровывали, промывали небольшим количеством эфира, затем гексаном и сушили, фильтрат концентрировали и получали вторую порцию вещества, всего получали 50,54 г 1-(бензолсульфонил)-6-хлориндола XII-3а в виде светло-коричневого твердого вещества.
Выход: 88%.
1Н ЯМР (400 МГц, CDCl3) δ 8,04 (dd, J=1,8, 0,9 Гц, 1Н), 7,91 (t, J=1,4 Гц, 1H), 7,89 (t, J=1,8 Гц, 1H), 7,67-7,54 (m, 2H), 7,53-7,48 (m, 2H), 7,48-7,42 (m, 1H), 7,23 (dd, J=8,4, 1,9 Гц, 1H), 6,65 (dd, J=3,7, 0,9 Гц, 1H).
Стадия 2: Синтез 1-(бензолсульфонил)-6-хлориндол-3-сульфонилхлорида XII-3:
Раствор 1-(бензолсульфонил)-6-хлориндола XII-3а (50 г, 171,4 ммоля) в ацетонитриле (500 мл) перемешивали в бане со льдом и в течение 20 мин по каплям добавляли хлорсульфоновую кислоту (100,8 г, 856,8 ммоля) и реакционную смесь перемешивали при комнатной температуре в течение 5 дней. Затем ее при перемешивании в течение 20 мин медленно выливали в воду со льдом (2,2 л), фильтровали, несколько раз промывали водой и сушили с отсасыванием и получали 63,77 г 1-(бензолсульфонил)-6-хлориндол-3-сульфонилхлорида XII-3 в виде светло-коричневого твердого вещества.
Выход: 95%.
1Н ЯМР (400 МГц, CDCl3) δ 8,36 (s, 1Н), 8,07 (d, J=1,8 Гц, 1H), 8,04 (t, J=1,3 Гц, 1Н), 8,02 (d, J=1,5 Гц, 1Н), 7,91 (d, J=8,6 Гц, 1H), 7,79-7,70 (m, 1H), 7,68-7,59 (m, 2Н), 7,47 (dd, J=8,6, 1,8 Гц, 1Н).
С.4. Синтез 1-(бензолсульфонил)-6-хлорпирроло[2,3-b]пиридин-3-сульфонилхлорида XII-4:
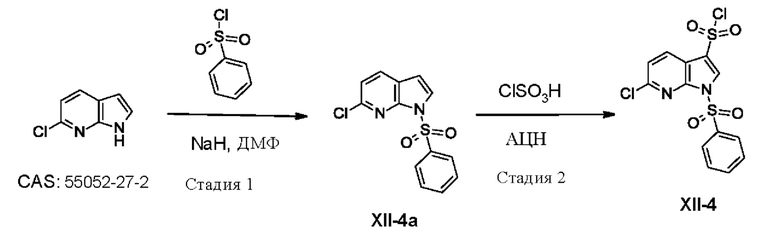
Стадия 1: Синтез 1-(бензолсульфонил)-6-хлорпирроло[2,3-b]пиридина XII-4а:
К раствору 6-хлор-1Н-пирроло[2,3-b]пиридина (1,37 г, 8,97 ммоля) в ДМФ (100 мл) добавляли гидрид натрия (60% в парафине, 1 г, 41 ммоль). Раствор перемешивали в течение 30 мин, при этом ему давали нагреться от 0°С до КТ. Затем по каплям добавляли хлорангидрид бензолсульфоновой кислоты (1,5 мл, 11,8 ммоля). Суспензию перемешивали при комнатной температуре в течение 3 ч и гидролизовали водой со льдом. Полученное твердое вещество отфильтровывали при пониженном давлении, тщательно промывали водой (75 мл) и в заключение петролейным эфиром (15 мл). Полученное вещество сушили при 60°С и очищали с помощью колоночной хроматографии (элюент: чистый дихлорметан) и получали 856 мг 1-(бензолсульфонил)-6-хлорпирроло[2,3-b]пиридина XII-4а в виде коричневатого твердого вещества.
Выход: 32%.
Стадия 2: Синтез 1-(бензолсульфонил)-6-хлорпирроло[2,3-b]пиридин-3-сульфонилхлорида XII-4:
Полученный 1-(бензолсульфонил)-6-хлорпирроло[2,3-b]пиридин XII-4а (150 мг, 0,51 ммоля) растворяли в ацетонитриле (5 мл) и по каплям обрабатывали хлорсульфоновой кислотой (2 мл, 2,91 ммоля). Смесь кипятили с обратным холодильником в течение 3 ч, охлаждали до комнатной температуры, гидролизовали водой со льдом (50 мл) и нейтрализовывали насыщенным раствором гидрокарбоната натрия. Неочищенный продукт экстрагировали дихлорметаном (3 раза по 50 мл). Объединенные органические экстракты сушили над MgS04, фильтровали и концентрировали. Полученное вещество очищали с помощью колоночной хроматографии (элюент: чистый дихлорметан) и получали 163 мг 1-(бензолсульфонил)-6-хлорпирроло[2,3-b]пиридин-3-сульфонилхлорида XII-4 в виде желтоватого твердого вещества.
Выход: 81%.
1Н ЯМР (600 МГц, CDCl3) δ: 8,48 (s, 1Н), 8,32 (d, J=7,8 Гц, 2H), 8,18 (d, J=8,3 Гц, 1H), 7,71 (t, J=7,5 Гц, 1H), 7,60 (t, J=7,9 Гц, 2H), 7,41 (d, J=8,4 Гц, 1H).
C.5. Синтез 1-(бензолсульфонил)-6-(дифторметил)пирроло[2,3-b]пиридин-3-сульфонилхлорида XII-5:

Стадия 1: Синтез 1-(бензолсульфонил)-6-(дифторметил)пирроло[2,3-b]пиридина XII-5а:
Суспензию гидроксида натрия (76 мг, 1,88 ммоля) в дихлорметане (1 мл) перемешивали в бане со льдом и добавляли 6-(дифторметил)-1Н-пирроло[2,3-b]пиридин XI-14 (125 мг, 0,74 ммоля), затем тетрабутиламмонийгидросульфат (7,5 г, 0,022 ммоля). Затем по каплям добавляли бензолсульфонилхлорид (105 мкл, 0,81 ммоля) и реакционную смесь перемешивали при комнатной температуре в течение ночи. После завершения реакции смесь фильтровали через слой целита и последний промывали с помощью ДХМ, объединенные фильтрат и промывочные растворы выпаривали досуха. Неочищенный продукт очищали с помощью хроматографии (SiO2, элюирование дихлорметаном) и получали 1-(бензолсульфонил)-6-(дифторметил)пирроло[2,3-b] пиридин XII-5а (200 мг) в виде светло-коричневого твердого вещества.
Выход: 70%.
ЖХМС в щелочной среде, методика 1 (ЭР+): 309 (М+Н)+, чистота 100%.
Стадия 2: Синтез 1-(бензолсульфонил)-6-(дифторметил)пирроло[2,3-b]пиридин-3-сульфонилхлорида XII-5:
Раствор 1-(бензолсульфонил)-6-(дифторметил)пирроло[2,3-b]пиридина XII-3а (76 мг, 0,24 ммоля) в ацетонитриле (10 мл) перемешивали в бане со льдом и по каплям добавляли хлорсульфоновую кислоту (54 мкл, 0,78 ммоля) и реакционную смесь перемешивали при 50°С в течение 4 дней. Затем добавляли оксихлорид фосфора (100 мкл, 1,06 ммоля) и реакционную смесь нагревали при 70°С в течение ночи. Затем после охлаждения ее медленно выливали в воду со льдом и экстрагировали хлороформом (3×). Органические слои сушили над сульфатом магния и выпаривали досуха и получали 1-(бензолсульфонил)-6-хлориндол-3-сульфонилхлорид XII-5 (100 мг) в виде твердого вещества. Неочищенный продукт использовали в следующей реакции без дополнительной очистки.
Выход: 95%.
ЖХМС в щелочной среде, методика 1 (ЭР): 387 (М-Н)- (соответствует массе сульфоновой кислоты), чистота 88%.
С.6. Синтез 1-(бензолсульфонил)-6-(дифторметил)индол-3-сульфонилхлорида XII-6:

Стадия 1: Синтез 1-(бензолсульфонил)индол-6-карбальдегида XII-6а:
При перемешивании к суспензии тонкоизмельченного гидроксида натрия (8,26 г, 206,7 ммоля) в дихлорметане (130 мл), предварительно охлажденной в верхней части бани со льдом, одной порцией добавляли 1H-индол-6-карбальдегид (10,0 г, 68,89 ммоля), затем тетрабутиламмонийгидросульфат (1,754 г, 5,17 ммоля). Перемешивание продолжали в течение еще 10 мин, затем в течение 20 мин по каплям добавляли раствор бензолсульфонилхлорида (9,67 мл, 75,78 ммоля, 1,1 экв.) в дихлорметане (20 мл) и реакционную смесь перемешивали при 0°С в течение 1 ч. Охлаждающую баню удаляли и смесь перемешивали при температуре окружающей среды в течение еще 1 ч. Реакционную смесь фильтровали через слой кизельгура, промывая осадок на фильтре дихлорметаном (2×100 мл), и фильтрат концентрировали в вакууме. Затем остаток растирали с диэтиловым эфиром (100 мл) и твердое вещество собирали фильтрованием, промывая осадок на фильтре диэтиловым эфиром (2×50 мл). Затем твердое вещество сушили в вакууме и получали 17,5 г искомого соединения (содержащего примесь тетрабутиламмонийгидросульфата, ~8% мас./мас). Твердое вещество растворяли в этилацетате (350 мл) и раствор промывали водой (150 мл) и рассолом (100 мл), сушили над безводным сульфатом натрия, фильтровали и растворитель выпаривали в вакууме и получали 1-(бензолсульфонил)индол-6-карбальдегид XII-6а (15,29 г) в виде темно-бежевого твердого вещества.
Выход: 70%.
ЖХМС в кислой среде, методика 4 (ЭР+): 286 (М+Н)+, чистота 84%.
1H ЯМР (400 МГц, CDCl3) δ 6,74 (dd, J=3,6, 0,7 Гц, 1Н), 7,52-7,44 (m, 2Н), 7,60-7,53 (m, 1H), 7,66 (d, J=8,2 Гц, 1H), 7,81-7,75 (m, 2Н), 7,98-7,88 (m, 2Н), 8,54-8,45 (m, 1H), 10,09 (s, 1H).
Стадия 2: Синтез 1-(бензолсульфонил)-6-(дифторметил)индола XII-6b:
При перемешивании к раствору 1-(бензолсульфонил)индол-6-карбальдегида XII-6а (3,58 г, 12,55 ммоля) в дихлорметане (55 мл) по каплям добавляли диэтиламинотрифторид серы (7,5 мл, 56,77 ммоля). Перемешивание продолжали при температуре окружающей среды в течение 21 ч. Реакцию останавливали насыщенным водным раствором гидрокарбоната натрия (100 мл) и затем смесь экстрагировали дихлорметаном (2×150 мл). Объединенные органические слои промывали рассолом (100 мл), сушили над безводным сульфатом натрия, фильтровали и растворитель выпаривали в вакууме. Остаток очищали с помощью флэш-хроматографии (колонка KP-SIL, 340 г) в градиентном режиме с использованием этилацетата в гептане (от 5 до 30%) и получали 1-(бензолсульфонил)-6-(дифторметил)индол XII-6b (2,91 г) в виде почти белого твердого вещества.
Выход: 75%.
ЖХМС в кислой среде, методика 4 (ЭР+): 308 (М+Н)+, чистота 100%.
1Н ЯМР (400 МГц, CDCl3) δ 6,70 (dd, J=3,7, 0,7 Гц, 1H), 6,76 (t, J=56,5 Гц, 1H), 7,39 (dd, J=8,2, 0,8 Гц, 1Н), 7,50-7,42 (m, 2H), 7,59-7,51 (m, 1H), 7,64-7,59 (m, 1H), 7,66 (d, J=3,7 Гц, 1H), 7,93-7,85 (m, 2H), 8,17 (d, J=0,8 Гц, 1H).
Стадия 3: Синтез 1-(бензолсульфонил)-6-(дифторметил)индол-3-сульфонилхлорида XII-6:
При перемешивании к раствору 1-(бензолсульфонил)-6-(дифторметил)индола XII-6b (5,7 г, 18,55 ммоля) в ацетонитриле (57 мл), предварительно охлажденному в верхней части бани со льдом, в течение 20 мин по каплям добавляли хлорсульфоновую кислоту (10,8 г, 92,74 ммоля) и реакционную смесь перемешивали при температуре окружающей среды в течение 3 дней. Реакционную смесь при перемешивании в течение 20 мин медленно выливали в воду со льдом (220 мл). Осадившееся твердое вещество собирали фильтрованием, промывая осадок на фильтре охлажденной льдом водой (3×25 мл). Затем осадок на фильтре сушили в токе азота в течение 1 ч, промывали циклогексаном (25 мл) и сушили в токе азота в течение еще 2 ч и получали 1-(бензолсульфонил)-6-(дифторметил)индол-3-сульфонилхлорид XII-6 (7,51 г) в виде почти белого твердого вещества.
Выход: 99%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 7,20 (t, J=55,9 Гц, 1Н), 7,51 (d, J=8,3 Гц, 1Н), 7,75-7,57 (m, 3Н), 7,76 (s, 1Н), 7,91 (d, J=8,2 Гц, 1H), 8,11-8,01 (m, 2Н), 8,14 (s, 1Н).
С.7. Синтез 6-хлор-1Н-пирроло[2,3-b]пиридин-3-сульфонилхлорида XII-7:

Смесь 6-хлор-1Н-пирроло[2,3-b]пиридина (0,50 г, 3,28 ммоля) и ClSO3H (10 мл) нагревали при 90°С в течение 16 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакцию останавливали охлажденной льдом Н2О (30 мл), смесь фильтровали, промывали с помощью Н2О (30 мл) и сушили в вакууме и получали 6-хлор-1Н-пирроло[2,3-b]пиридин-3-сульфонилхлорид XII-7 (0,45 г) в виде почти белого твердого вещества. Это соединение использовали в следующей реакции без дополнительной очистки.
Выход: 55%.
ЖХМС в щелочной среде, методика 2 (ЭР-): 230 (М-Н)- (соответствует сульфоновой кислоте), чистота 98%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 7,16 (d, J=7,98 Гц, 1H) 7,50 (d, J=2,99 Гц, 1Н) 8,08 (d, J=8,48 Гц, 1Н) 11,88 (brs, 1H).
С.8. Синтез 6-(дифторметокси)-1Н-индол-3-сульфонилхлорида XII-8:

Стадия 1: Синтез 6-(дифторметокси)-1Н-индол-3-сульфоновой кислоты XII-8а:
К раствору 6-(дифторметокси)-1Н-индола XI-2 (0,30 г, 1,30 ммоля) в CH3CN (15 мл) при 0°С медленно добавляли ClSO3H (0,13 мл, 1,95 ммоля) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь выливали в охлажденную льдом Н2О (50 мл) и экстрагировали с помощью EtOAc (100 мл). Органический слой отделяли, промывали рассолом (20 мл) и концентрировали в вакууме и получали 6-(дифторметокси)-1H-индол-3-сульфоновую кислоту XII-8а (0,33 г, неочищенная) в виде коричневого полужидкого вещества.
ЖХМС в щелочной среде, методика 2 (ЭР-): 262 (М-Н)-, чистота 75%.
Стадия 2: Синтез 6-(дифторметокси)-1Н-индол-3-сульфонилхлорида XII-8:
К раствору 6-(дифторметокси)-1Н-индол-3-сульфоновой кислоты XII-8а (0,15 г, 0,43 ммоля) в ДХМ (5 мл) при 0°С добавляли оксалилхлорид (0,15 мл, 1,70 ммоля), затем добавляли ДМФ (0,007 мл, 0,09 ммоля) и реакционную смесь перемешивали при комнатной температуре в течение 3 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь концентрировали в вакууме и получали 6-(дифторметокси)-1Н-индол-3-сульфонилхлорид XII-8 (0,13 г, неочищенный) в виде коричневого полужидкого вещества. Это соединение использовали в следующей реакции без дополнительной очистки.
ЖХМС в щелочной среде, методика 2 (ЭР-): 262 (М-Н)- (соответствует сульфоновой кислоте), чистота 80%.
С.9. Синтез 1-(бензолсульфонил)-6-хлор-7-фториндол-3-сульфонилхлорида XII-9:

Стадия 1: Синтез 1-(бензолсульфонил)-6-хлор-7-фториндола XII-9а:
При перемешивании к суспензии тонкоизмельченного гидроксида натрия (3,54 г, 0,088 моля) в дихлорметане (60 мл), предварительно охлажденной в верхней части бани со льдом, одной порцией добавляли 6-хлор-7-фтор-1H-индол XI-3 (5 г, 0,029 моля), затем тетрабутиламмонийгидросульфат (0,501 г, 0,001 моля). Перемешивание продолжали в течение еще 10 мин, затем в течение 20 мин по каплям добавляли раствор бензолсульфонилхлорида (4,2 мл, 0,033 моля) в дихлорметане (15 мл) и реакционную смесь перемешивали при 0°С в течение 1 ч. Баню со льдом удаляли и смесь перемешивали при температуре окружающей среды в течение еще 1 ч. Реакционную смесь фильтровали через слой кизельгура, промывая осадок на фильтре дихлорметаном (2×50 мл). Фильтрат промывали водой (4×50 мл) и рассолом (50 мл), сушили над безводным сульфатом натрия, фильтровали и растворитель выпаривали в вакууме и получали 1-(бензолсульфонил)-6-хлор-7-фториндол XII-9а (8,57 г) в виде темно-бежевого твердого вещества. Выход: 90%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 6,95 (dd, J=3,7, 2,3 Гц, 1H), 7,37 (dd, J=8,4, 6,2 Гц, 1H), 7,48 (d, J=8,6 Гц, 1H), 7,66 (tt, J=6,9, 1,9 Гц, 2Н), 7,80-7,71 (m, 1H), 7,98-7,91 (m, 2Н), 7,99 (d, J=3,7 Гц, 1H).
Стадия 2: Синтез 1-(бензолсульфонил)-6-хлор-7-фториндол-3-сульфонилхлорида XII-9:
При перемешивании к раствору 1-(бензолсульфонил)-6-хлор-7-фториндола XII-9а (8,50 г, 0,027 моля) в ацетонитриле (85 мл), предварительно охлажденному в верхней части бани со льдом, в течение 20 мин по каплям добавляли хлорсульфоновую кислоту (9,12 мл, 0,137 моля) и реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч. Реакционную смесь при перемешивании в течение 20 мин медленно выливали в воду со льдом (340 мл). Осадившееся твердое вещество собирали фильтрованием, промывая осадок на фильтре охлажденной льдом водой (3×50 мл) и циклогексаном (50 мл). Осадок на фильтре затем сушили в токе азота в течение 2 ч и затем в вакуумном сушильном шкафу при 40°С в течение 16 ч и получали 1-(бензолсульфонил)-6-хлор-7-фториндол-3-сульфонилхлорид XII-9 (7,82 г) в виде светло-розового твердого вещества.
Выход: 66%.
ЖХМС в кислой среде, методика 4 (ЭР+): 388 (М+Н)+, чистота 95%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 7,44 (dd, J=8,5, 6,3 Гц, 1H), 7,72-7,61 (m, 3Н), 7,80-7,72 (m, 1H), 7,81 (s, 1Н), 8,05-7,98 (m, 2Н).
С.10. Синтез 1-(бензолсульфонил)-6-метилиндол-3-сульфонилхлорида XII-10:
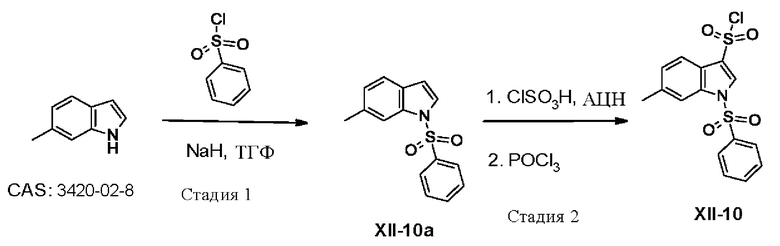
Стадия 1: Синтез 1-(бензолсульфонил)-6-метилиндола XII-10а:
К раствору 6-метил-1Н-индола (1 г, 7,39 ммоля) в ТГФ (20 мл) при 0°С добавляли гидрид натрия (60% в парафине, 0,35 г, 8,9 ммоля). Раствор перемешивали в течение 30 мин, при этом ему давали нагреться от 0°С до КТ. Затем по каплям добавляли хлорангидрид бензолсульфоновой кислоты (1,1 мл, 8,9 ммоля). Реакционную смесь перемешивали при комнатной температуре в течение ночи и гидролизовали водой. Затем ее экстрагировали с помощью EtOAc. Органический слой отделяли, промывали рассолом, сушили над MgSO4 и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии (элюент: 25-40% AcOEt в гептане) и получали 1,97 г 1-(бензолсульфонил)-6-метилиндол XII-10а в виде бесцветного масла.
Выход: 70%.
ЖХМС в щелочной среде, методика 1 (ЭР-): 270 (М-Н)-, чистота 71%.
Стадия 2: Синтез 1-(бензолсульфонил)-6-метилиндол-3-сульфонилхлорида XII-10:
Полученный 1-(бензолсульфонил)-6-метилиндол XII-10а (0,9 г, 3,15 ммоля) разбавляли ацетонитрилом (9 мл) и по каплям обрабатывали хлорсульфоновой кислотой (0,32 мл, 4,72 ммоля). Через 2 ч добавляли оксихлорид фосфора (0,65 мл, 6,93 ммоля) и реакционную смесь нагревали при 70°С в течение ночи. После охлаждения до комнатной температуры и разбавления хлороформом органический слой отделяли и промывали водой, затем рассолом. Объединенные органические экстракты сушили над MgSO4, фильтровали и концентрировали в вакууме и получали 1,4 г 1-(бензолсульфонил)-6-метилиндол-3-сульфонилхлорид XII-10 в виде коричневатого твердого вещества. Это соединение использовали в следующей реакции без дополнительной очистки. ЖХМС в щелочной среде, методика 1 (ЭР-): 419 (М-Н)-, до проведения анализа реакцию в аликвоте смеси останавливали морфолином.
С.11. Синтез 1-(бензолсульфонил)-6-метоксииндол-3-сульфонилхлорида XII-11:

Стадия 1: Синтез 1-(бензолсульфонил)-6-метоксииндола XII-11а:
К раствору 6-метоксииндола (2,5 г, 17 ммолей) в ДМФ (50 мл) при 0°С добавляли гидрид натрия (60% в парафине, 1,7 г, 71 ммоль). Суспензию перемешивали в течение 30 мин, затем нагревали до комнатной температуры. Затем раствор при перемешивании по каплям обрабатывали бензолсульфонилхлоридом (2,8 мл, 3,70 г, 22 ммоля). После перемешивания при комнатной температуре в течение 2,5 ч к реакционной смеси при энергичном перемешивании добавляли охлажденную льдом воду. Полученный осадок отфильтровывали при пониженном давлении, тщательно промывали водой (100 мл) и затем петролейным эфиром (10 мл). После сушки при 60°С получали 1-(бензолсульфонил)-6-метоксииндол XII-11а в виде бесцветного твердого вещества (3,2 г).
Выход: 65%.
1Н ЯМР (600 МГц, CDCl3) δ: 7,87-7,81 (m, 2Н), 7,53-7,48 (m, 2Н), 7,45-7,39 (m, 3Н), 7,36 (d, J=8,5 Гц, 1Н), 6,84 (dd, J=8,6/2,3 Гц, 1H), 6,56 (dd, J=3,7/0,9 Гц, 1H), 3,85 (s, 3Н).
Стадия 2: Синтез 1-(бензолсульфонил)-6-метоксииндол-3-сульфонилхлорида XII-11:
Раствор 1-(бензолсульфонил)-6-метоксииндола XII-11а (500 мг, 1,74 ммоля) в дихлорметане (15 мл) обрабатывали комплексом SO3⋅ДМФ (1,2 г, 7,8 ммоля) и перемешивали при комнатной температуре в течение 2 ч (за протеканием реакции следили с помощью ТСХ). Ожидаемую промежуточную индолсульфоновую кислоту не выделяли. Затем добавляли тионилхлорид (1 мл, 14 ммолей) и смесь перемешивали при комнатной температуре в течение 16 ч. Смесь гидролизовали насыщенным раствором NaHCO3 (50 мл) и экстрагировали дихлорметаном (3 раза по 50 мл). Объединенные органические экстракты сушили над MgSO4, фильтровали и концентрировали путем выпаривания в вакууме. Остаток очищали с помощью колоночной хроматографии (силикагель 60, элюент: дихлорметан/петролейный эфир = 1:1) и получали 1-(бензолсульфонил)-6-метоксииндол-3-сульфонилхлорид XII-11 в виде бесцветного твердого вещества (504 мг).
Выход: 75%.
1H ЯМР (600 МГц, CDCl3) δ: 8,23 (s, 1Н), 8,00-7,93 (m, 2Н), 7,80 (d, J=8,8 Гц, 1Н), 7,70-7,63 (m, 1Н), 7,59-7,53 (m, 2Н), 7,47 (d, J=2,2 Гц, 1H), 7,06 (dd, J=8,8, 2,2 Гц, 1H), 3,89 (s, 3Н).
С.12. Синтез 1Н-пирроло[3,2-11]хинолин-3-сульфонилхлорида XII-12:

Стадия 1: Синтез 1H-пирроло[3,2-h]хинолин-3-сульфоновой кислоты XII-12а:
К раствору 1Н-пирроло[3,2-h]хинолина (400 мг, 2,3 ммоля) в пиридине (6 мл) при 0°С добавляли комплекс пиридин-триоксид серы (1,2 г, 3,5 ммоля). Затем реакционную смесь при перемешивании нагревали при 120°С в течение 2 ч, охлаждали до комнатной температуры и выпаривали досуха. Бежевое твердое вещество растворяли в воде и водную фазу промывали хлороформом (3×). При выдерживании в водной фракции образовывался осадок и его отфильтровывали, промывали водой и сушили в вакууме при 35°С и получали 470 мг 1H-пирроло[3,2-Н]хинолин-3-сульфоновой кислоты XII-12а в виде бежевого твердого вещества.
Выход: 79%.
ЖХМС в щелочной среде, методика 1 (ЭР+): 249 (М+Н)+, чистота 100%.
Стадия 2: Синтез 1Н-пирроло[3,2-b]хинолин-3-сульфонилхлорида XII-12:
К раствору 1H-пирроло[3,2-h]хинолин-3-сульфоновой кислоты XII-12а (855 мг, 3,44 ммоля) в ацетонитриле (8,5 мл), охлажденному до 0°С, в атмосфере аргона по каплям добавляли оксихлорид фосфора (1,06 г, 6,88 ммоля). Затем реакционную смесь при перемешивании нагревали при 70°С в течение ночи. После охлаждения до комнатной температуры при энергичном перемешивании осторожно добавляли воду со льдом. Осаждалось твердое вещество и его отфильтровывали, промывали водой и сушили в вакууме при 35°С и получали 284 мг 1Н-пирроло[3,2-h]хинолин-3-сульфонилхлорида XII-12 в виде бежевого твердого вещества.
Выход: 27%.
ЖХМС в щелочной среде, методика 1 (ЭР+): 275 (М+Н)+, до проведения анализа реакцию в аликвоте смеси останавливали этиламином.
С.13. Синтез 1Н-бензо[g]индол-3-сульфонилхлорида XII-13:

Стадия 1: Синтез 1Н-бензо[g]индол-3-сульфоновой кислоты XII-13а:
К раствору 1Н-бензо[g]индола (1 г, 5,8 ммоля) в пиридине (16 мл) при 0°С добавляли комплекс пиридин-триоксид серы (1,38 г, 8,7 ммоля). Затем реакционную смесь при перемешивании нагревали при 125°С в течение 5 ч, охлаждали до комнатной температуры и выпаривали досуха. Коричневое масло разбавляли водой. При выдерживании образовывался осадок и его отфильтровывали, промывали водой и сушили в вакууме при 35°С и получали 1,4 г 1Н-бензо[g]индол-3-сульфоновой кислоты XII-13а в виде бежевого твердого вещества.
Выход: 98%.
ЖХМС в щелочной среде, методика 1 (ЭР-): 246 (М-Н)-.
Стадия 2: Синтез 1Н-бензо[g]индол-3-сульфонилхлорида XII-13:
К раствору 1Н-бензо[g]индол-3-сульфоновой кислоты XII-13а (100 мг, 0,4 ммоля) в ацетонитриле (1 мл), охлажденному до 0°С, в атмосфере аргона по каплям добавляли оксихлорид фосфора (76 мкл, 0,8 ммоля). Затем реакционную смесь нагревали при 70°С в течение 1 ч. После охлаждения до комнатной температуры при энергичном перемешивании осторожно добавляли воду со льдом. Осаждалось твердое вещество и его отфильтровывали, промывали водой и сушили в вакууме при 35°С и получали 65 мг 1Н-бензо[g]индол-3-сульфонилхлорида XII-13 в виде коричневого твердого вещества.
Выход: 60%.
ЖХМС в щелочной среде, методика 1 (ЭР-): 246 (М-Н)- (соответствует сульфоновой кислоте).
С.14. Синтез 5-бром-6-хлор-1Н-пирроло[2,3-b]пиридин-3-сульфонилхлорида XII-14:

К раствору 5-бром-6-хлор-1Н-пирроло[2,3-b]пиридина (0,50 г, 2,16 ммоля) в CH3CN (10 мл) при 0°С добавляли ClSO3H (5 мл) и реакционную смесь нагревали при 80°С в течение 12 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь выливали в смесь лед-H2O (150 мл), осадок отфильтровывали и сушили в вакууме и получали 5-бром-6-хлор-1Н-пирроло[2,3-b]пиридин-3-сульфонилхлорид (0,605 г, неочищенный) в виде коричневого твердого вещества. Это соединение использовали в следующей реакции без дополнительной очистки. ЖХМС в щелочной среде, методика 2 (ЭР-): 309 (М-Н)- (соответствует сульфоновой кислоте), чистота 97%.
С.15. Синтез 1-(бензолсульфонил)-6-бензилоксипирроло[2,3-b]пиридин-3-сульфонилхлорида XII-15:

Раствор 1-(бензолсульфонил)-6-бензилоксипирроло[2,3-b]пиридина XI-4 (570 мг, 2 ммоля) в дихлорметане обрабатывали комплексом триоксид серы/ДМФ (1224 мг, 8 ммолей). Смесь перемешивали при комнатной температуре в течение 0,5 ч (анализ с помощью ТСХ показывал отсутствие исходного вещества и полное превращение в ожидаемую сульфоновую кислоту, элюент: чистый дихлорметан). Затем добавляли тионилхлорид (1,4 мл, 20 ммолей) и образовавшуюся суспензию перемешивали при комнатной температуре в течение 22 ч. За реакцией в полученном прозрачный растворе следили с помощью ТСХ (наблюдали одно пятно, соответствующее ожидаемому продукту, элюент: петролейный эфир:этилацетат, 80:20). Смесь гидролизовали насыщенным водным раствором NaHCO3 (75 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические экстракты сушили над MgSO4, фильтровали и концентрировали при пониженном давлении и получали 920 мг 1-(бензолсульфонил)-6-бензилоксипирроло[2,3-b]пиридин-3-сульфонилхлорида XII-15. Это соединение использовали в следующей реакции без дополнительной очистки.
Выход: 64%.
1Н ЯМР (600 МГц, ДМСО-d6) δ 5,42 (s, 2Н), 6,82 (d, J=8,5 Гц, 1Н), 7,42-7,31 (m, 3Н), 7,49 (d, J=9,1 Гц, 3Н), 7,55 (t, J=7,9 Гц, 2Н), 7,70 (t, J=7,5 Гц, 1Н), 8,01-7,96 (m, 1H), 8,07 (dd, J=8,5, 1,1 Гц, 2Н).
ПРИМЕРЫ СОЕДИНЕНИЙ
D. Синтез соединений общей формулы I
Все соединения, предлагаемые в настоящем изобретении, специально раскрытые в настоящем изобретении, обозначены, как "I-х", где любой "х" обозначает число, определяющее отдельные соединения. Соответственно, соединения примеров обозначены, как I-1, I-2, I-3 и т.п. Обозначения используют независимо от того, что любое соединение может быть также описано субродовой формулой, предлагаемой в настоящем изобретении, например, формулой II, III или IV и т.п.
D.1. Методика А. Синтез 6-хлор-Х-(2,5-дифторпиридин-3-ил)-1Н-индол-3-сульфонамида I-1:

К раствору соединения XII-1 (0,50 г, 1,97 ммоля) в пиридине (10 мл) добавляли соединение Х-1 (0,18 г, 1,25 ммоля) и ДМАП (0,012 г, 0,09 ммоля). Реакционную смесь нагревали при 80°С в течение 16 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь концентрировали в вакууме. Остаток разбавляли с помощью Н2О (100 мл), 1 н. раствором HCl (50 мл) и экстрагировали с помощью EtOAc (100 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное неочищенное вещество очищали с помощью колоночной хроматографии (диоксид кремния, 100-200 меш, 40% EtOAc в гексане) и получали 6-хлор-N-(2,5-дифторпиридин-3-ил)-1Н-индол-3-сульфонамид 1-1 (0,05 г) в виде почти белого твердого вещества.
Выход: 7%.
ЖХМС в щелочной среде, методика 1 (ЭР-): 342 (М-Н)-, чистота 97%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 7,25 (dd, J=8,31, 1,47 Гц, 1H), 7,54 (s, 1Н), 7,71-7,81 (m, 2Н), 7,89 (s, 1Н), 8,16 (d, J=2,93 Гц, 1H), 10,73 (brs, 1Н), 12,22 (brs, 1Н).
Соединения, приведенные ниже в таблице 4, можно синтезировать по методике, аналогичной методике А.


6-Хлор-N-(6-хлор-2,5-дифторпиридин-3-ил)-1Н-индол-3-сульфонамид I-2
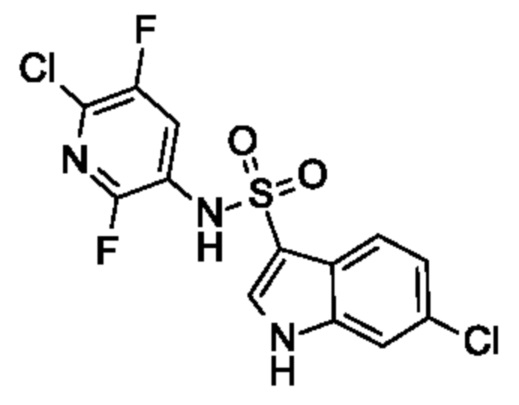
ЖХМС в щелочной среде, методика 2 (ЭР+): 378 (М+Н)+, чистота 98%.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,25 (d, J=8,31 Гц, 1H), 7,54 (s, 1Н), 7,75-7,84 (m, 1Н), 7,94 (t, J=7,58 Гц, 1 Н), 8,16 (brs, 1H), 10,86 (brs, 1H), 12,22 (brs, 1Н).
6-Хлор-N-(6-хлор-5-фтор-2-метоксипиридин-3-ил)-1H-индол-3-сульфонамид I-3
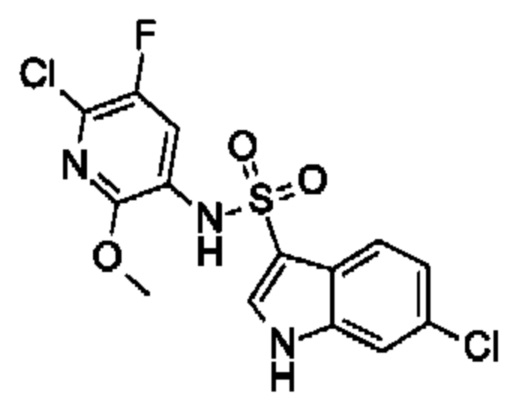
ЖХМС в щелочной среде, методика 2 (ЭР-): 388 (М-Н)-, чистота 99%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,53 (s, 3Н), 7,23 (dd, J=8,56, 1,71 Гц, 1Н), 7,52 (d, J=1,96 Гц, 1H), 7,71 (d, J=9,29 Гц, 1H), 7,82 (d, J=8,80 Гц, 1H), 8,08 (d, J=2,93 Гц, 1H), 10,14 (brs, 1Н), 12,12 (brs, 1H).
6-Хлор-N-(6-хлор-2-фтор-5-метоксипиридин-3-ил)-1H-индол-3-сульфонамид I-4
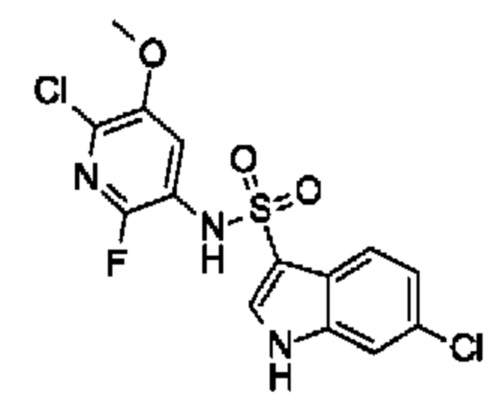
ЖХМС в щелочной среде, методика 2 (ЭР-): 388 (М-Н)-, чистота 94%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,82 (s, 3Н), 7,22-7,28 (m, 1Н), 7,51-7,59 (m, 2H), 7,76 (d, J=8,80 Гц, 1H), 8,06 (d, J=2,93 Гц, 1H), 10,51 (s, 1H), 12,17 (brs, 1H).
6-Хлор-N-(2,5-дифтор-6-метоксипиридин-3-ил)-1Н-индол-3-сульфонамид I-5

ЖХМС в щелочной среде, методика 2 (ЭР-): 372 (М-Н)-, чистота 99%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,79 (s, 3Н), 7,19 (d, J=8,80 Гц, 1H), 7,55-7,60 (m, 2Н), 7,61-7,65 (m, 1Н), 7,84 (s, 1Н), 10,02 (s, 1Н), 12,09 (brs, 1H).
6-Хлор-Х-(5-фтор-2,6-диметоксипиридин-3-ил)-1H-индол-3-сульфонамид I-6

ЖХМС в щелочной среде, методика 2 (ЭР-): 384 (М-Н)-, чистота 99%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,24 (s, 3Н), 3,82 (s, 3Н), 7,19 (dd, J=8,56, 1,71 Гц, 1Н), 7,47 (d, J=10,27 Гц, 1Н), 7,51 (d, J=1,47 Гц, 1H), 7,68 (d, J=8,31 Гц, 1H), 7,78 (s, 1Н), 9,44 (s, 1Н), 11,95 (brs, 1H).
6-Хлор-N-(2,5-дифтор-6-метилпиридин-3-ил)-1Н-индол-3-сульфонамид I-7
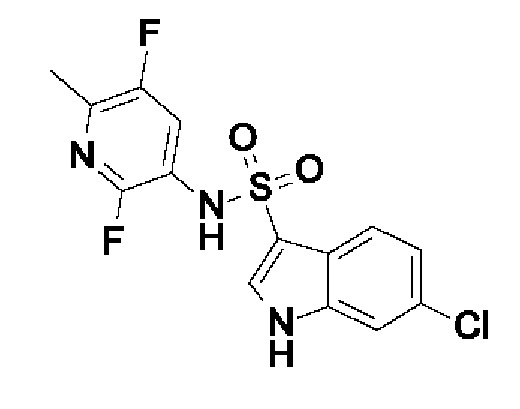
ЖХМС в щелочной среде, методика 2 (ЭР-): 356 (М-Н)-, чистота 99%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 2,24 (d, J=2,8 Гц, 3Н), 7,24 (dd, J=8,8 Гц, 1,6 Гц, 1Н), 7,53 (d, J=2,4 Гц, 1Н), 7,64 (t, J=8,8 Гц, 1H), 7,77 (d, J=8,8 Гц, 1Н), 8,05 (d, J=2,4 Гц, 1Н), 10,50 (s, 1H), 12,15 (brs, 1H).
6-Хлор-N-(2-хлор-6-метоксипиридин-3-ил)-1H-индол-3-сульфонамид I-8

ЖХМС в нейтральной среде, методика 3 (ЭР+): 372 (М+Н)+, чистота 95%.
1Н ЯМР (500 МГц, ДМСО-d6) δ 3,75 (s, 3Н), 6,77 (d, J=8,51 Гц, 1Н), 7,18 (dd, J=1,89, 8,51 Гц, 1Н), 7,48-7,56 (m, 2Н), 7,65 (d, J=8,51 Гц, 1H), 7,77 (d, J=1,89 Гц, 1Н), 10,90 (s, 1H), 12,10 (brs, 1Н).
6-Хлор-N-[6-(дифторметокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-индол-3-сульфонамид I-9
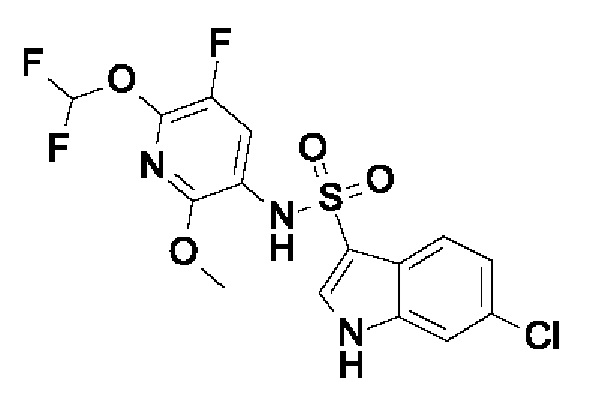
ЖХМС в щелочной среде, методика 2 (ЭР): 420 (М-Н)-, чистота 96%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,39 (s, 3Н), 7,22 (dd, J=1,2, 8,4 Гц, 1Н), 7,52 (d, 3=1,6 Гц, 1Н), 7,58 (t, J=72,8 Гц, 1Н), 7,70-7,77 (m, 2Н), 7,94 (d, J=2,4 Гц, 1Н), 9,85 (brs, 1Н), 12,06 (brs, 1H).
N-(5-Бром-3-метоксипиразин-2-ил)-6-хлор-1Н-индол-3-сульфонамид I-10
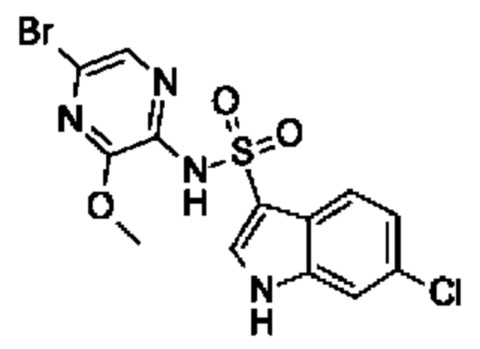
ЖХМС в щелочной среде, методика 2 (ЭР+): 417 (М+Н)+, чистота 98%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,84 (s, 3Н), 7,21 (dd, J=8,37, 1,97 Гц, 1H), 7,54 (d, J=1,48 Гц, 1H), 7,82 (s, 1H), 7,89 (d, J=8,37 Гц, 1Н), 8,09 (s, 1H), 10,93 (brs, 1H), 12,14 (brs, 1H).
6-Хлор-N-(5-хлор-3-метоксипиразин-2-ил)-1H-индол-3-сульфонамид I-11

ЖХМС в щелочной среде, методика 2 (ЭР+): 373 (М+Н)+, чистота 99%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,89 (s, 3Н), 7,24 (dd, J=8,56, 1,71 Гц, 1Н), 7,53 (d, J=l,47 Гц, 1H), 7,82 (s, 1H), 7,95 (d, J=8,31 Гц, 1Н), 8,14 (d, J=2,94 Гц, 1Н), 10,97 (s, 1Н), 12,15 (brs, 1H).
6-Хлор-N-[5-фтор-6-(2-фторэтокси)-2-метоксипиридин-3-ил]-1Н-индол-3-сульфонамид I-12

ЖХМС в щелочной среде, методика 2 (ЭР-): 416 (М-Н)-, чистота 99%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,24 (s, 3Н), 4,42-4,46 (m, 1Н), 4,50-4,55 (m, 1Н), 4,62-4,66 (m, 1H), 4,75 4,78 (m, 1H), 7,20 (dd, J=8,80, 1,96 Гц, 1H), 7,49-7,54 (m, 2Н), 7,68 (d, J=8,31 Гц, 1Н), 7,81 (d, J=2,45 Гц, 1H), 9,48 (s, 1Н), 11,97 (brs, 1H).
6-Хлор-N-[6-(2-фторэтокси)-2-метоксипиридин-3-ил]-1Н-индол-3-сульфонамид I-13

ЖХМС в щелочной среде, методика 2 (ЭР+): 400 (М+Н)+, чистота 97%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,24 (s, 3Н), 4,31-4,36 (m, 1Н), 4,40-4,43 (m, 1Н), 4,60-4,63 (m, 1H), 4,71-4,76 (m, 1H), 6,34 (d, J=8,31 Гц, 1Н), 7,17 (dd, J=8,56, 1,71 Гц, 1H), 7,44 (d, J=8,31 Гц, 1H), 7,51 (s, 1H), 7,65 (d, J=8,31 Гц, 1Н), 7,72 (s, 1H), 9,21 (s, 1Н), 11,92 (brs, 1H).
6-Хлор-N-(6-хлор-4-метоксипиридин-3-ил)-1Н-индол-3-сульфонамид I-14

ЖХМС в щелочной среде, методика 2 (ЭР+): 372 (М+Н)+, чистота 98%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 7,01 (s, 1Н), 7,21 (d, J=8,31 Гц, 1Н), 7,52 (s, 1Н), 7,72 (d, J=8,31 Гц, 1Н), 7,86 (s, 1Н), 8,04 (s, 1Н), 9,68 (br s, 1Н), 12,04 (br s, 1Н) (сигналы 3Н объединены с пиком растворителя).
6-Хлор-N-(2,5-дифтор-6-метоксипиридин-3-ил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид I-33
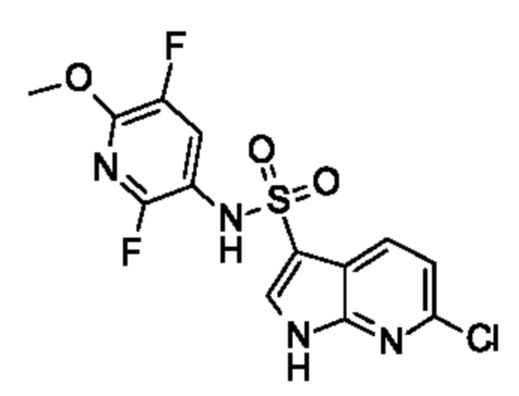
ЖХМС в щелочной среде, методика 1 (ЭР-): 373 (М-Н)-, чистота 99%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,84 (s, 3Н), 7,37 (d, J=8,3 Гц, 1Н), 7,70 (dd, J=9,9, 7,4 Гц, 1Н), 8,05 (d, J=2,7 Гц, 1Н), 8,10 (d, J=8,3 Гц, 1H), 10,12 (s, 1Н), 12,83 (s, 1Н).
6-Хлор-N-(5-фтор-2,6-диметоксипиридин-3-ил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид I-34

ЖХМС в щелочной среде, методика 1 (ЭР-): 385 (М-Н)-, чистота 99%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,24 (s, 3Н), 3,85 (s, 3Н), 7,36 (d, J=8,3 Гц, 1H), 7,53 (d, J=10,4 Гц, 1Н), 7,95 (s, 1H), 8,11 (d, J=8,4 Гц, 1H), 9,59 (s, 1Н), 12,69 (s, 1Н).
6-Хлор-N-(6-хлор-2-метоксипиридин-3-ил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид I-35
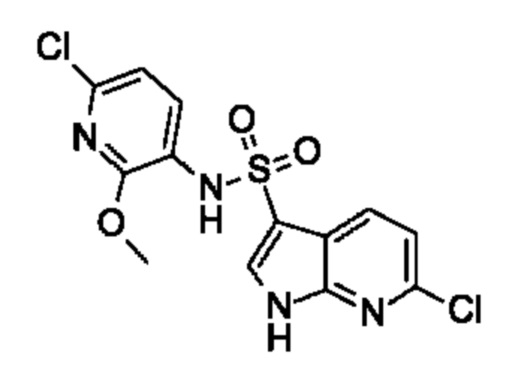
ЖХМС в щелочной среде, методика 1 (ЭР-): 371 (М-Н)-, чистота 99%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,44 (s, 3Н), 7,03 (d, J=8,0 Гц, 1Н), 7,37 (d, J=8,3 Гц, 1H), 7,63 (d, J=8,0 Гц, 1H), 8,05 (s, 1H), 8,19 (d, J=8,3 Гц, 1H), 9,88 (s, 1Н), 12,78 (s, 1Н).
6-Хлор-N-[5-фтор-6-(2-фторэтокси)-2-метоксипиридин-3-ил]-1Н-пирроло[2,3-b]пиридин-3-сульфонамид I-36

ЖХМС в щелочной среде, методика 1 (ЭР-): 417 (М-Н)-, чистота 99%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,24 (d, J=1,4 Гц, 3Н), 4,45 (t, J=4,4 Гц, 1Н), 4,53 (t, J=4,1 Гц, 1H), 4,68-4,62 (m, 1H), 4,80-4,74 (m, 1Н), 7,35 (dd, J=8,3, 1,4 Гц, 1H), 7,56 (dd, J=10,4, 1,4 Гц, 1H), 7,96 (d, J=1,4 Гц, 1H), 8,10 (dd, J=8,4, 1,4 Гц, 1H), 9,8 (s, 1Н), 12,6 (s, 1H).
6-Хлор-N-[6-(дифторметокси)-2-метокси-3-пиридил]-1Н-пирроло[2,3-b]пиридин-3-сульфонамид I-37

ЖХМС в щелочной среде, методика 1 (ЭР-): 403 (М-Н)-, чистота 98%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,34 (s, 3Н), 6,58 (d, J=8,2 Гц, 1Н), 7,36 (d, J=8,3 Гц, 1H), 7,58 (t, 1H), 7,65 (d, J=8,2 Гц, 1H), 7,96 (s, 1H), 8,13 (d, J=8,3 Гц, 1Н), 9,68 (s, 1Н), 12,72 (s, 1H).
6-Хлор-N-[6-(2,2-дифторэтокси)-2-метоксипиридин-3-ил]-1Н-индол-3-сульфонамид I-38

ЖХМС в щелочной среде, методика 2 (ЭР+): 418 (М+Н)+, чистота 94%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,28 (s, 3Н) 4,43 (td, J=14,92, 3,91 Гц, 2Н) 6,17-6,46 (m, 1H) 6,39 (d, J=8,31 Гц, 1Н) 7,18 (dd, J=8,56, 1,71 Гц, 1Н) 7,46 (d, J=8,31 Гц, 1Н) 7,51 (d, J=1,96 Гц, 1H) 7,66 (d, J=8,80 Гц, 1H) 7,74 (s, 1H) 9,28 (brs, 1H) 11,93 (brs, 1H).
6-Хлор-N-[2-(2,2-дифторэтокси)-6-метоксипиридин-3-ил]-1Н-индол-3-сульфонамид I-39

ЖХМС в щелочной среде, методика 2 (ЭР+): 418 (М+Н)+, чистота 96%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,74 (s, 3Н) 4,05 (td, J=14,31, 4,16 Гц, 2Н) 5,61-5,95 (m, 1H) 6,39 (d, J=8,31 Гц, 1Н) 7,12-7,21 (m, 1Н) 7,48 (d, J=8,31 Гц, 1H) 7,52 (s, 1Н) 7,65 (d, J=8,80 Гц, 1Н) 7,77 (s, 1H) 9,36 (brs, 1Н) 11,95 (brs, 1H).
6-Хлор-N-[6-(дифторметокси)-4-метоксипиридин-3-ил]-1Н-индол-3-сульфонамид I-40

ЖХМС в щелочной среде, методика 2 (ЭР+): 404 (М+Н)+, чистота 99%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,28 (s, 3Н) 6,56 (s, 1H) 7,20 (dd, 3=2,61, 1,72 Гц, 1Н) 7,52 (d, J=l,48 Гц, 1H) 7,63 (t, J=74 Гц, 1H) 7,69 (d, J=8,37 Гц, 1Н) 7,79 (d, J=2,46 Гц, 1Н) 7,85 (s, 1H) 9,49 (s, 1H) 11,99 (brs, 1Н).
6-Хлор-N-(6-циклопропил-2,5-дифторпиридин-3-ил)- Ш-индол-3-сульфонамид I-41

ЖХМС в щелочной среде, методика 2 (ЭР+): 384 (М+Н)+, чистота 97%.
1H ЯМР (400 МГц, ДМСО-d6) δ 0,77-0,79 (m, 2Н) 0,93-1,00 (m, 2Н) 2,11-2,14 (m, 1Н) 7,22 (d, J=8,31 Гц, 1H) 7,53 (s, 1Н) 7,59-7,65 (m, 1Н) 7,73 (d, J=8,31 Гц, 1H) 8,04 (d, J=2,93 Гц, 1H) 10,40 (s, 1H) 12,16 (brs, 1H).
6-Хлор-N-[6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-индол-3-сульфонамид I-42

ЖХМС в щелочной среде, методика 2 (ЭР+): 436 (М+Н)+, чистота 94%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,30 (s, 3Н) 4,54 (td, J=14,92, 3,42 Гц, 2Н) 6,20-6,51 (m, 1H) 7,20 (dd, J=8,31, 1,96 Гц, 1Н) 7,52 (d, J=l,96 Гц, 1Н) 7,55 (d, J=10,27 Гц, 1H) 7,70 (d, J=8,31 Гц, 1H) 7,83 (s, 1H) 9,55 (brs, 1H) 11,99 (brs, 1H).
6-Хлор-N-[6-[2-(дифторметокси)этокси]-5-фтор-2-метоксипиридин-3-ил]-1Н-индол-3-сульфонамид I-43

ЖХМС в щелочной среде, методика 2 (ЭР+): 466 (М+Н)+, чистота 95%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,23 (s, 3Н) 4,09-4,15 (m, 2Н) 4,37-4,44 (m, 2Н) 6,68 (t, J=76 Гц, 1H) 7,18 (dd, J=8,37, 1,48 Гц, 1Н) 7,45-7,51 (m, 2Н) 7,66 (d, J=8,86 Гц, 1Н) 7,80 (s, 1H) 9,54 (brs, 1Н) 11,96 (brs, 1H).
6-Хлор-N-[6-(2,2-дифторэтокси)-2,5-дифторпиридин-3-ил]-1Н-пирроло[2,3-b]пиридин-3-сульфонамид I-44

ЖХМС в щелочной среде, методика 1 (ЭР-): 423 (М-Н)-, чистота 96%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 4,52 (td, J=15,0, 3,3 Гц, 2Н), 6,36 (tt, J=54,1, 3,3 Гц, 1H), 7,37 (d, J=8,3 Гц, 1H), 7,79 (dd, J=9,8, 7,4 Гц, 1Н), 8,21-7,92 (m, 2Н), 10,24 (s, 1Н), 12,85 (s, 1H).
6-Хлор-N-[6-(2,2-дифторэтокси)-2-метоксипиридин-3-ил]-1Н-пирроло[2,3-b]пиридин-3-сульфонамид I-45

ЖХМС в щелочной среде, методика 1 (ЭР-): 417 (М-Н)-, чистота 99%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,28 (s, 3Н), 4,45 (td, J=14,9, 3,6 Гц, 2Н), 6,51-6,12 (m, 2Н), 7,34 (d, J=8,3 Гц, 1Н), 7,51 (d, J=8,2 Гц, 1H), 7,90 (s, 1Н), 8,08 (d, J=8,3 Гц, 1Н), 12,66 (s, 1Н), 9,47 (s, 1H).
6-Хлор-N-[6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-пирроло[2,3-b]пиридин-3-сульфонамид I-46

ЖХМС в щелочной среде, методика 1 (ЭР-): 435 (М-Н)-, чистота 97%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,28 (s, 3Н), 4,56 (td, J=14,8, 3,5 Гц, 2Н), 6,37 (t, 1Н), 7,37 (d, J=8,4 Гц, 1Н), 7,62 (d, J=10,2 Гц, 1H), 7,99 (s, 1Н), 8,13 (d, J=8,3 Гц, 1Н), 9,71 (s, 1H), 12,72 (s, 1H).
6-Хлор-N-(6-циклопропил-2,5-дифторпиридин-3-ил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид I-47
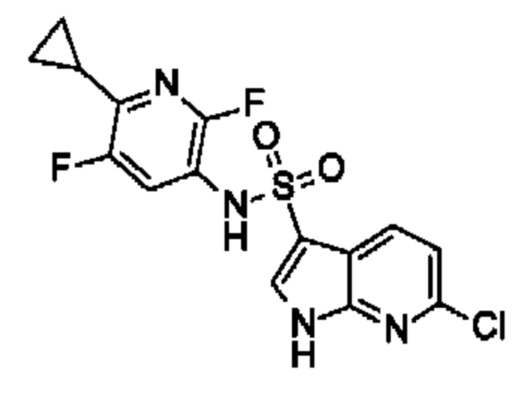
ЖХМС в щелочной среде, методика 1 (ЭР-): 383 (М-Н)-, чистота 95%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 1,12-0,64 (m, 4Н), 2,24-2,02 (m, 1H), 7,38 (d, J=8,4 Гц, 1Н), 7,66 (dd, J=9,5, 7,5 Гц, 1Н), 8,25-8,11 (m, 2Н), 10,46 (s, 1H), 12,89 (s, 1Н).
6-Хлор-N-[6-[2-(дифторметокси)этокси]-5-фтор-2-метоксипиридин-3-ил]-1Н-пирроло[2,3-b]пиридин-3-сульфонамид I-48
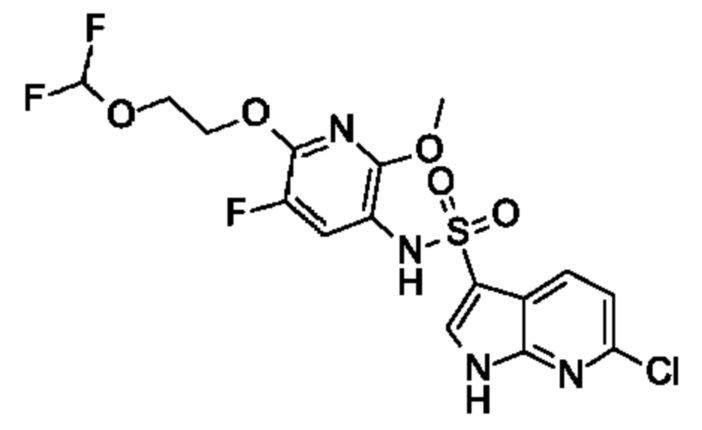
ЖХМС в щелочной среде, методика 1 (ЭР-): 465 (М-Н)-, чистота 94%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,24 (s, 3Н), 4,30 (d, J=122,2 Гц, 4Н), 6,71 (s, 1Н), 8,25-7,18 (m, 4Н), 9,65 (s, 1Н), 12,63 (s, 1Н).
6-Хлор-N-(5-фтор-2-метоксипиридин-3-ил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид I-49
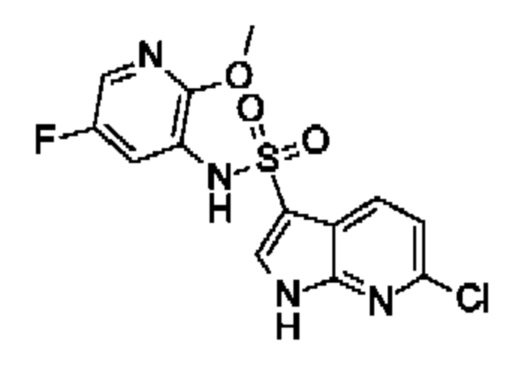
ЖХМС в щелочной среде, методика 1 (ЭР-): 355 (М-Н)-, чистота 99%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,51 (s, 3Н), 7,38 (d, J=8,3 Гц, 1Н), 7,57 (dd, J=9,4, 2,8 Гц, 1Н), 7,85 (d, J=2,8 Гц, 1Н), 8,31-8,16 (m, 2Н), 10,07 (s, 1H), 12,83 (s, 1Н).
6-Хлор-N-(6-циклопропил-5-фтор-2-метоксипиридин-3-ил)-1Н-индол-3-сульфонамид I-50

ЖХМС в щелочной среде, методика 2 (ЭР+): 396 (М+Н)+, чистота 99%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 0,81-0,87 (m, 2Н) 0,89-0,94 (m, 2Н) 2,08-2,10 (m, 1Н) 3,36 (s, 3Н) 7,20 (dd, J=8,80, 1,47 Гц, 1H) 7,41 (d, J=10,27 Гц, 1H) 7,51 (s, 1H) 7,75 (d, J=8,80 Гц, 1H) 7,95 (d, J=2,45 Гц, 1Н) 9,68 (s, 1H) 12,04 (brs, 1Н).
N-[6-(2,2-Дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-6-(дифторметокси)-1H-индол-3-сульфонамид I-51

ЖХМС в щелочной среде, методика 2 (ЭР+): 468 (М+Н)+, чистота 99%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,29 (s, 3Н) 4,50-4,60 (m, 2Н) 6,22-6,52 (m, 1H) 7,00-7,05 (m, 1H) 7,20 (t, J=74, 1Н) 7,25 (s, 1H) 7,55 (d, J=10,34 Гц, 1H) 7,71 (d, J=8,86 Гц, 1H) 7,83 (d, J=2,46 Гц, 1H) 9,54 (s, 1H) 11,95 (brs, 1H).
N-[6-(Дифторметокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-пирроло[3,2-п]хинолин-3-сульфонамид I-68

ЖХМС в щелочной среде, методика 1 (ЭР-): 437 (М-Н)-, чистота 96%.
1Н ЯМР (400 МГц, ДМСО-d6) 5 3,32 (s, 3Н), замаскирован пиком растворителя, 8,04-7,30 (m, 6Н), 8,45 (d, J=8,2 Гц, 1H), 8,92 (d, J=4,5 Гц, 1Н), 9,91 (s, 1H), 13,17 (s, 1Н).
N-[6-(2,2-Дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-пирроло[3,2-h]хинолин-3-сульфонамид I-69

ЖХМС в щелочной среде, методика 1 (ЭР-): 451 (М-Н)-, чистота 94%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,25 (s, 3Н), 4,51 (td, J=14,8, 3,5 Гц, 2Н), 6,33 (t, J=3,5 Гц, 1Н), 7,71-7,54 (m, 3Н), 7,80 (s, 1Н), 7,89 (d, J=8,7 Гц, 1Н), 8,45 (dd, J=8,3, 1,6 Гц, 1Н), 8,92 (dd, J=4,3, 1,6 Гц, 1H), 9,63 (s, 1Н), 13,10 (s, 1H).
N-(2,6-Диметоксипиридин-3-ил)-1H-бензо[g]индол-3-сульфонамид I-70

ЖХМС в нейтральной среде, методика 3 (ЭР+): 384 (М+Н)+, чистота 98%.
1Н ЯМР (500 МГц, ДМСО-d6) δ 3,21 (s, 3Н), 3,70 (s, 3Н), 6,28 (d, J=8,3 Гц, 1H), 7,52-7,38 (m, 2Н), 7,65-7,54 (m, 2Н), 7,83-7,69 (m, 2Н), 8,01-7,86 (m, 1Н), 8,45-8,32 (m, 1H), 9,13 (s, 1Н), 12,71 (s, 1Н).
5-Бром-6-Хлор-N-[6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-пирроло[2,3-b]пиридин-3-сульфонамид I-71

ЖХМС в щелочной среде, методика 2 (ЭР+): 515 (М+Н)+, чистота 94%.
1H ЯМР (400 МГц, ДМСО-d6) δ 3,29 (s, 3Н) 4,55 (td, J=14,80, 3,18 Гц, 2Н) 6,22-6,54 (m, 1H) 7,64 (d, J=10,27 Гц, 1Н) 8,08 (s, 1Н) 8,47 (s, 1H) 9,80 (brs, 1H) 12,90 (brs, 1Н).
D.2. Синтез 6-Хлор-N-(6-метоксипиридин-3-ил)-1Н-индол-3-сульфонамида I-15:
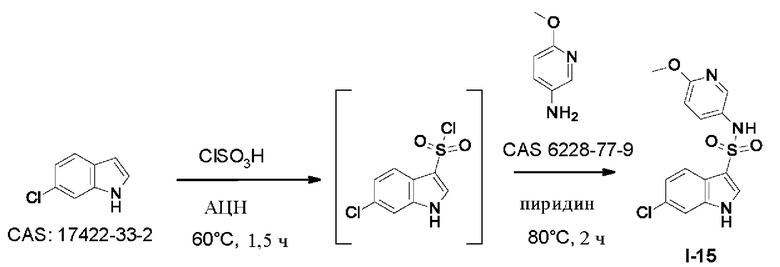
В сосуде раствор 6-хлориндола (630 мг, 4,1 ммоля) в ацетонитриле (25,2 мл) перемешивали в бане со льдом и по каплям добавляли хлорсульфоновую кислоту (714 мкл, 10,7 ммоля) и реакционную смесь перемешивали в течение 30 мин. Баню со льдом удаляли и реакционную смесь нагревали при 60°С в течение 1,5 ч. После охлаждения до комнатной температуры добавляли пиридин (54,6 мл) и раствор становился желтым. Во второй герметизированный сосуд помещали 6-метоксипиридин-3-амин (37,2 мг, 0,3 ммоля) и добавляли аликвоту полученного выше раствора (2,8 мл, 0,15 ммоля). Реакционную смесь перемешивали при 80°С в течение 2 ч, затем выпаривали в центробежном испарителе. Остаток очищали с помощью хроматографии с обращенной фазой в щелочной среде и с МС-детектированием и получали 21,8 мг 6-Хлор-N-(6-метоксипиридин-3-ил)-1Н-индол-3-сульфонамида I-15.
Выход: 42%.
ЖХМС в щелочной среде, методика 1 (ЭР-): 336 (М-Н)-, чистота 100%.
D.3. Методика В. Синтез N-[6-(дифторметокси)-5-фтор-2-метоксипиридин-3-ил]-6-(дифторметил)-1H-индол-3-сульфонамида I-16:
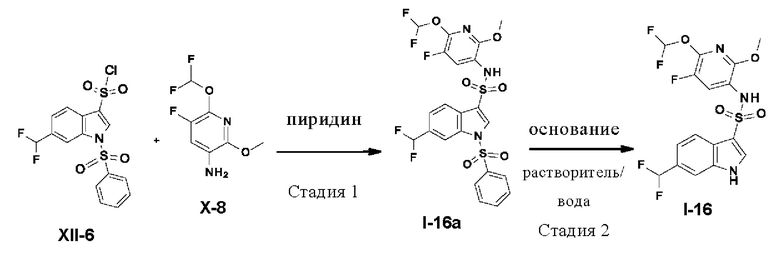
Стадия 1: Синтез 1-(бензолсульфонил)-N-[6-(дифторметокси)-5-фтор-2-метокси-3-пиридил]-6-(дифторметил)индол-3-сульфонамида I-16а:
В герметизированном сосуде в атмосфере аргона 6-(дифторметокси)-5-фтор-2-метоксипиридин-3-амин Х-8 (150 мг, 0,49 ммоля) растворяли в пиридине (3 мл). При 0°С добавляли 1-(бензолсульфонил)-6-(дифторметил)индол-3-сульфонилхлорид XII-6 (290 мг, 0,71 ммоля), затем перемешивали при комнатной температуре в течение ночи. Реакционную смесь выливали в воду и экстрагировали этилацетатом (трижды). Органические фазы сушили над MgSO4 и выпаривали. Остаток очищали с помощью флэш-хроматографии на диоксиде кремния (при элюировании в градиентном режиме с использованием ДХМ и метанола, от 100/0 до 95/5) и получали 310 мг 1 -(бензолсульфонил)-N-[6-(дифторметокси)-5-фтор-2-метокси-3-пиридил]-6-(дифторметил)индол-3-сульфонамида I-16а в виде белого твердого вещества.
Выход: 75%.
ЖХМС в щелочной среде, методика 1 (ЭР-): 576 (М-Н)-, чистота 100%.
Стадия 2: Синтез N-[6-(дифторметокси)-5-фтор-2-метоксипиридин-3-ил]-6-(дифторметил)-1H-индол-3-сульфонамида I-16:
В герметизированной пробирке 1-(бензолсульфонил)-N-[6-(дифторметокси)-5-фтор-2-метокси-3-пиридил]-6-(дифторметил)индол-3-сульфонамид I-16а (310 мг, 0,54 ммоля) растворяли в ТГФ (4 мл). Добавляли тетрабутиламмонийфторид (1,3 мл, 1М раствор в воде, 1,3 ммоля) и реакционную смесь перемешивали при 90°С в течение ночи. Реакционную смесь выливали в воду и экстрагировали этилацетатом (трижды). Органические фазы сушили над MgSO4 и выпаривали. Остаток очищали с помощью флэш-хроматографии на диоксиде кремния (при элюировании смесью ДХМ/метанол, 95/5) и получали 170 мг N-[6-(дифторметокси)-5-фтор-2-метоксипиридин-3-ил]-6-(дифторметил)-1Н-индол-3-сульфонамида I-16 в виде ярко-желтого твердого вещества.
Выход: 58%.
ЖХМС в щелочной среде, методика 1 (ЭР-): 436 (М-Н)-, чистота 97%.
1Н ЯМР (400 МГц, ДМСО-d6 δ 3,37 (s, 3Н), 7,12 (t, J=56,0 Гц, 1Н), 7,80-7,32 (m, 4Н), 7,89 (d, J=8,3 Гц, 1Н), 8,04 (s, 1Н), 9,84 (s, 1Н), 12,22 (s, 1Н).
Соединения, приведенные ниже в таблице 5 можно синтезировать по методикам, аналогичным методике В.


6-Хлор-N-(5-хлор-2-метоксипиридин-3-ил)-1Н-индол-3-сульфонамид I-17
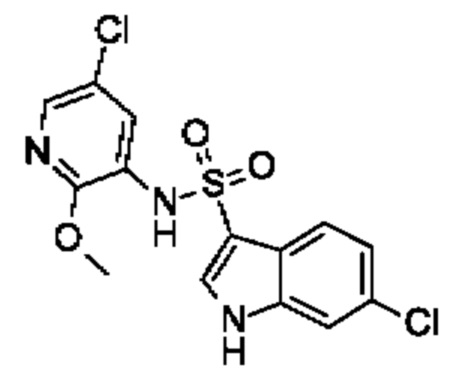
ЖХМС в щелочной среде, методика 1 (ЭР-): 370 (М-Н)-, чистота 100%.
N-(5-Бромпиразин-2-ил)-6-хлор-1Н-индол-3-сульфонамид I-18

ЖХМС в щелочной среде, методика 1 (ЭР-): 385 (М-Н)-, чистота 98%.
6-Хлор-N-(6-цианопиридин-3-ил)-1Н-индол-3-сульфонамид I-19
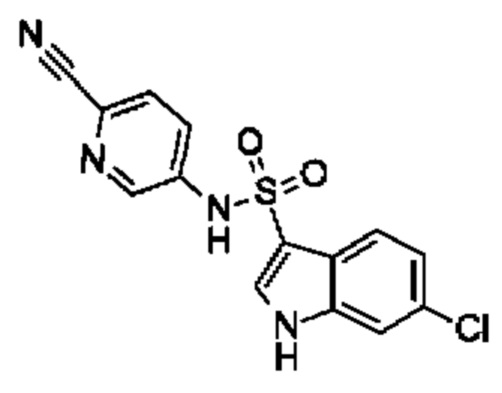
ЖХМС в щелочной среде, методика 1 (ЭР-): 331 (М-Н), чистота 98%.
N-(6-Бромпиридин-3-ил)-6-хлор-1Н-индол-3-сульфонамид I-20

ЖХМС в щелочной среде, методика 1 (ЭР-): 384 (М-Н)-, чистота 98%.
6-Хлор-N-(6-йодпиридин-3-ил)-1Н-индол-3-сульфонамид I-21

ЖХМС в щелочной среде, методика 1 (ЭР-): 432 (М-Н)-, чистота 100%.
6-Хлор-N-[6-(дифторметокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-пирроло[2,3-b]пиридин-3-сульфонамид I-22

ЖХМС в щелочной среде, методика 1 (ЭР-): 421 (М-Н)-, чистота 97%.
1Н ЯМР (400 МГц, ДМСО-d6 δ 3,37 (s, J=l,2 Гц, 3Н), 7,37 (d, J=8,4, 1,2 Гц, 1Н), 7,77-7,40 (t, 1Н), 7,77 (d, J=4,l Гц, 1Н), 8,09 (s, J=2,l Гц, 1Н), 8,18 (d, J=8,3, 1,2 Гц, 1Н), 9,95 (s, 1Н), 12,77 (s, 1Н).
6-Хлор-N-(6-хлор-5-фторпиридин-3-ил)-1Н-индол-3-сульфонамид I-23
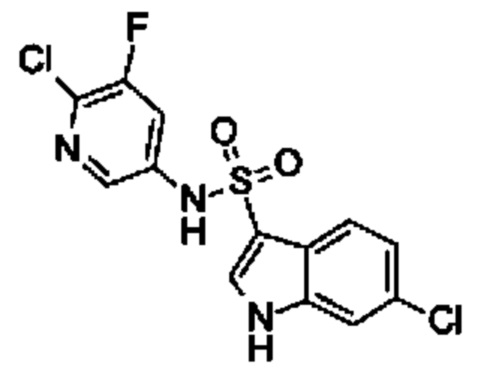
ЖХМС в нейтральной среде, методика 3 (ЭР+): 360 (М+Н)+, чистота 99%.
1Н ЯМР (500 МГц, ДМСО-d6) δ 7,25 (dd, J=l,93, 8,53 Гц, 1Н), 7,52 (d, J=l,83 Гц, 1Н), 7,54 (dd, J=2,38, 10,09 Гц, 1Н), 7,78 (d, J=8,62 Гц, 1Н), 7,97 (d, J=2,38 Гц, 1Н), 8,22 (d, J=2,93 Гц, 1Н), 10,97 (br s, 1Н),12,22 (br s, 1Н).
6-Хлор-N-(б-хлорпиридин-3-ил)-1Н-индол-3-сульфонамид I-24
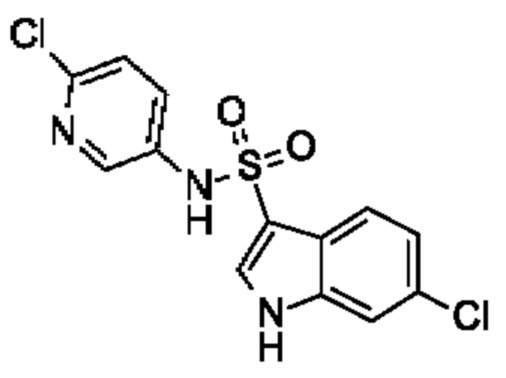
ЖХМС в нейтральной среде, методика 3 (ЭР+): 342 (М+Н)+, чистота 95%.
1Н ЯМР (500 МГц, ДМСО-d6) δ 7,23 (dd, J=l,83, 8,62 Гц, 1Н), 7,33 (d, J=8,62 Гц, 1Н), 7,46-7,57 (m, 2Н), 7,76 (d, J=8,44 Гц, 1Н), 8,01-8,14 (m, 2Н), 10,62 (br s, 1Н), 12,15 (br s, 1H).
6-Хлор-N-(6-хлор-4-фторпиридин-3-ил)-1Н-индол-3-сульфонамид I-25
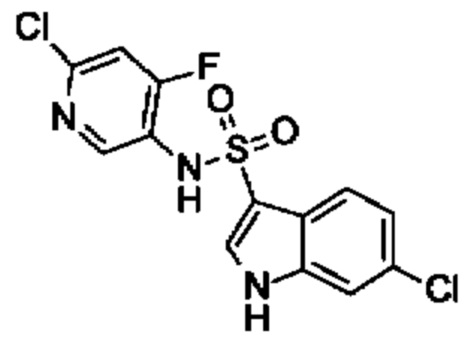
ЖХМС в нейтральной среде, методика 3 (ЭР+): 360 (М+Н)+, чистота 97%.
1Н ЯМР (500 МГц, ДМСО-d6 δ 7,22 (dd, J=l,93, 8,53 Гц, 1Н), 7,53 (d, J=l,83 Гц, 1Н), 7,54 (d, J=9,54 Гц, 1Н), 7,69 (d, J=8,62 Гц, 1Н), 7,96 (d, J=2,75 Гц, 1Н), 8,22 (d, J=9,90 Гц, 1Н), 10,35 (br. s., 1Н), 12,14 (br. s., 1Н).
6-Хлор-N-(4,6-дихлорпиридин-3-ил)-1Н-индол-3-сульфонамид I-26

ЖХМС в нейтральной среде, методика 3 (ЭР+): 376 (М+Н)+, чистота 96%.
1Н ЯМР (500 МГц, ДМСО-d6) δ 7,19 (d, J=8,4 Гц, 1Н), 7,65 (d, J=8,6 Гц, 1Н), 7,53 (s, 1Н), 7,68 (s, 1Н), 7,90 (d, J=4,0 Гц, 1Н), 8,22 (s, 1Н), 10,17 (s, 1Н), 12,13 (s, 1Н).
N-(6-Хлор-2-метоксипиридин-3-ил)-6-(дифторметил)-1Н-индол-3-сульфонамид 1-27
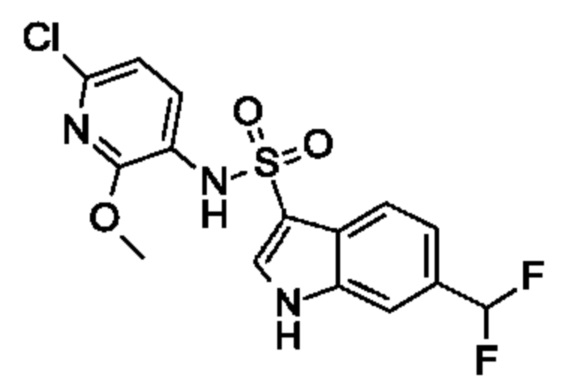
ЖХМС в щелочной среде, методика 1 (ЭР-): 386 (М-Н)-, чистота 99%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 7,26-6,95 (m, 2Н), 7,40-7,33 (m, 1Н), 3,43 (s, 3Н), 7,61 (d, J=8,l Гц, 1Н), 7,69 (d, J=l,5 Гц, 1Н), 7,90 (dd, J=8,4, 0,8 Гц, 1Н), 8,00 (s, 1Н), 9,78 (s, 1Н), 12,21 (s, 1Н).
N-(6-Хлор-2-метоксипиридин-3-ил)-6-(дифторметил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид I-28

ЖХМС в щелочной среде, методика 1 (ЭР-): 387 (М-Н)-, чистота 97%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,40 (s, 3Н), 7,27-6,63 (m, 2Н), 7,77-7,35 (m, 2Н), 8,27 (d, J=73,6 Гц, 2Н), 9,90 (s, 1Н), 12,89 (s, 1Н).
N-(6-Хлор-5-фтор-2-метоксипиридин-3-ил)-6-(дифторметил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид I-29
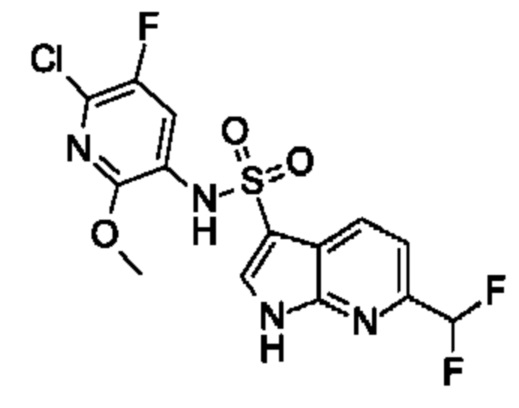
ЖХМС в щелочной среде, методика 1 (ЭР-): 405 (М-Н)-, чистота 97%.
N-[6-(2,2-Дифторэтокси)-2,5-дифтор-3-пиридил]-6-(дифторметил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид I-30
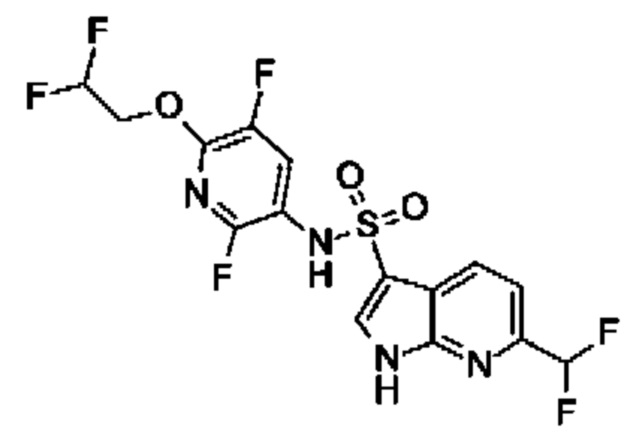
ЖХМС в щелочной среде, методика 1 (ЭР-): 439 (М-Н)-, чистота 99%.
N-[6-(Дифторметокси)-5-фтор-2-метоксипиридин-3-ил]-6-(дифторметил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид I-31

ЖХМС в щелочной среде, методика 1 (ЭР-): 437 (М-Н)-, чистота 99%.
N-[6-(2,2-Дифторэтокси)-2,5-дифторпиридин-3-ил]-6-(дифторметил)-1Н-индол-3-сульфонамид I-52

ЖХМС в щелочной среде, методика 1 (ЭР-): 438 (М-Н)-, чистота 96%.
1Н ЯМР (400 МГц, ДМСО-d6 δ 4,51 (td, J=15,l, 3,3 Гц, 2Н), 6,53-6,17 (m, 1Н), 7,14 (t, J=56,l Гц, 1Н), 7,38 (d, J=8,4 Гц, 1Н), 7,80-7,65 (m, 2Н), 7,83 (d, J=8,3 Гц, 1Н), 8,04 (d, J=2,9 Гц, 1Н), 10,17 (s, 1Н), 12,31 (s, 1Н).
N-[6-(2,2-Дифторэтокси)-2-метоксипиридин-3-ил]-6-(дифторметил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид I-53

ЖХМС в щелочной среде, методика 1 (ЭР-): 433 (М-Н)-, чистота 100%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,21 (s, 3Н), 4,43 (td, J=14,9, 3,6 Гц, 2Н), 6,48-6,13 (m, 2Н), 7,10 (d, J=55,l Гц, 1Н), 7,53 (dd, J=14,9, 8,2 Гц, 2Н), 8,03 (s, 1Н), 8,21 (d, J=8,2 Гц, 1Н), 9,46 (s, 1Н), 12,77 (s, 1Н).
N-[6-(2,2-Дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-6-(дифторметил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид I-54

ЖХМС в щелочной среде, методика 1 (ЭР-): 451 (М-Н-), чистота 95%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,23 (s, 3Н), 4,54 (td, J=14,8, 3,5 Гц, 2Н), 6,36 (tt, J=54,5, 3,6 Гц, 1Н), 7,04 (t, J=55,l Гц, 1Н), 7,60 (dd, J=17,0, 9,2 Гц, 2Н), 8,13 (s, 1Н), 8,27 (d, J=8,2 Гц, 1Н), 9,72 (s, 1Н), 12,83 (s, 1Н).
6-(Дифторметил)-N-[5-фтор-6-(2-фторэтокси)-2-метоксипиридин-3-ил]-1Н-пирроло[2,3-b]пиридин-3-сульфонамид I-55

ЖХМС в щелочной среде, методика 1 (ЭР-): 433 (М-Н)-, чистота 99%.
6-(Дифторметил)-N-(5-фтор-2,6-диметоксипиридин-3-ил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид I-56

ЖХМС в щелочной среде, методика 1 (ЭР-): 401 (М-Н)-, чистота 100%.
6-(Дифторметил)-N-(2,5-дифтор-6-метилпиридин-3-ил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид I-57

ЖХМС в щелочной среде, методика 1 (ЭР-): 373 (М-Н)-, чистота 98%.
N-[6-[2-(Дифторметокси)этокси]-5-фтор-2-метоксипиридин-3-ил]-6-(дифторметил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид I-58

ЖХМС в щелочной среде, методика 1 (ЭР-): 481 (М-Н)-, чистота 99%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,18 (s, 3Н), 4,28 (d, J=120,0 Гц, 4Н), 7,24-6,35 (m, 2Н), 7,57 (d, J=9,2 Гц, 2Н), 8,11 (s, 1Н), 8,24 (d, J=8,l Гц, 1Н), 9,66 (s, 1Н), 12,81 (s, 1Н).
6-Хлор-N-[6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-7-фтор-1Н-индол-3-сульфонамид I-59

ЖХМС в щелочной среде, методика 1 (ЭР-): 452 (М-Н)-, чистота 95%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,31 (s, 3Н), 4,78-4,42 (m, 2Н), 6,60-6,09 (m, 1Н), 7,30 (t, J=7,6 Гц, 1Н), 7,58 (dt, J=22,5, 9,7 Гц, 2Н), 7,92 (s, 1Н), 9,76 (s, 1Н), 12,7 (s, 1Н).
N-[6-(Дифторметокси)-5-фтор-2-метоксипиридин-3-ил]-6-метил-1Н-индол-3-сульфонамид I-60
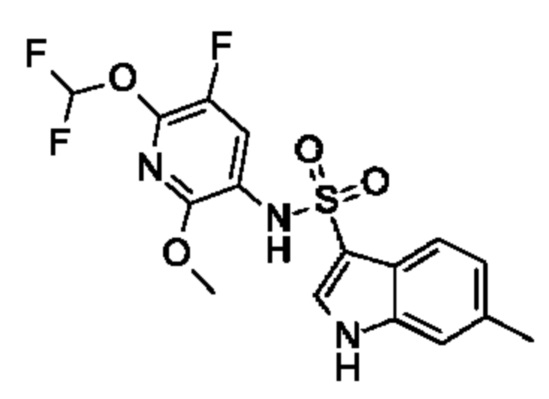
ЖХМС в щелочной среде, методика 1 (ЭР-): 400 (М-Н)-, чистота 98%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 2,39 (s, 3Н), 3,45 (s, 3Н), 7,00 (d, J=8,l Гц, 1Н), 7,25 (s, 1Н), 7,74-7,56 (m, 3Н), 7,84 (d, J=2,9 Гц, 1Н), 9,71 (s, 1Н), 11,82 (s, 1Н).
6-Хлор-N-(2,6-диметоксипиридин-3-ил)-1Н-индол-3-сульфонамид I-61
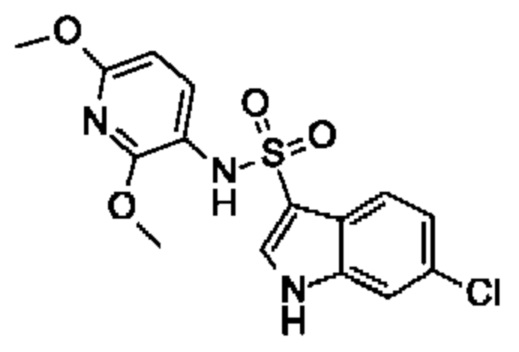
ЖХМС в нейтральной среде, методика 3 (ЭР+): 368 (М+Н)+, чистота 99%.
1Н ЯМР (500 МГц, ДМСО-d6) δ 3,24 (s, 3Н), 3,73 (s, 3Н), 6,28 (d, J=8,3 Гц, 1Н), 7,17 (dd, J=8,6, 1,9 Гц, 1Н), 7,41 (d, J=8,3 Гц, 1Н), 7,51 (d, J=l,9 Гц, 1Н), 7,66 (d, J=8,6 Гц, 1Н), 7,69 (s, 1Н), 9,16 (s, 1Н), 11,89 (s, 1Н).
N-[6-(2,2-Дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-6-(дифторметил)-1Н-индол-3-сульфонамид I-62

ЖХМС в щелочной среде, методика 1 (ЭР-): 450 (М-Н)-, чистота 99%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,25 (s, 3Н), 4,52 (td, J=14,8, 3,6 Гц, 2Н), 6,35 (tt, J=54,5, 3,5 Гц, 1Н), 7,12 (t, J=56,l Гц, 1Н), 7,86-7,29 (m, 4Н), 7,95 (d, J=2,2 Гц, 1Н), 9,56 (s, 1Н), 12,17 (s, 1Н).
N-[6-(Дифторметокси)-5-фтор-2-метоксипиридин-3-ил]-6-метокси-1Н-индол-3-сульфонамид I-63
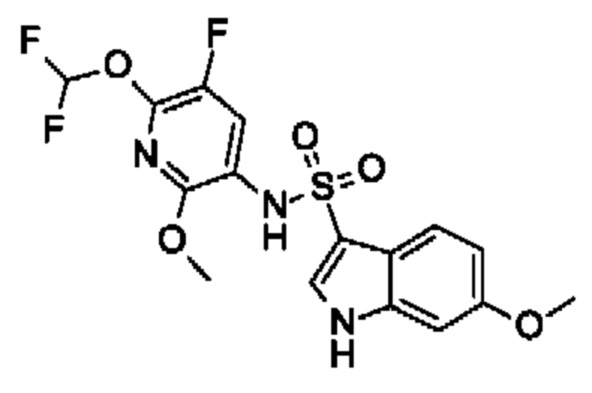
ЖХМС в щелочной среде, методика 1 (ЭР-): 414 (М-Н)-, чистота 95%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,45 (s, 3Н), 3,77 (s, 3Н), 6,87-6,76 (m, 1Н), 6,93 (s, 1Н), 7,76-7,33 (m, 3Н), 7,78 (d, J=2,9 Гц, 1Н), 9,73 (s, 1Н), 11,74 (s, 1Н).
N-[6-(2,2-Дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-6-метокси-1Н-индол-3-сульфонамид I-64

ЖХМС в щелочной среде, методика 1 (ЭР-): 430 (М-Н)-, чистота 95%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,36 (s, 3Н), 3,77 (s, 3Н), 4,54 (td, J=14,9, 3,6 Гц, 2Н), 6,36 (t, J=3,5 Гц, 1Н), 6,80 (dd, J=8,8, 2,3 Гц, 1Н), 6,93 (d, J=2,3 Гц, 1Н), 7,53 (dd, J=19,8, 9,6 Гц, 2Н), 7,66 (d, J=2,8 Гц, 1Н), 9,42 (s, 1Н), 11,67 (s, 1Н).
6-Хлор-N-[6-(2,2-дифторэтокси)-2,5-дифторпиридин-3-ил]-1Н-индол-3-сульфонамид I-67

ЖХМС в щелочной среде, методика 1 (ЭР-): 422 (М-Н)-, чистота 99%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 4,49 (t, J=14,8 Гц, 2Н), 6,34 (t, J=54,4 Гц, 1Н), 7,20 (d, J=8,6 Гц, 1Н), 7,52 (s, 1Н), 7,70 (d, J=8,5 Гц, 2Н), 7,88 (s, 1Н), 10,14 (s, 1Н), 12,03 (s, 1Н).
N-(6-Хлор-5-фтор-2-метоксипиридин-3-ил)-6-фенилметокси-1Н-пирроло[2,3-b]пиридин-3-сульфонамид I-72

ЖХМС в нейтральной среде, методика 3 (ЭР+): 463 (М+Н)+, чистота 96%.
1Н ЯМР (500 МГц, ДМСО-d6) δ 3,56 (s, 3Н), 5,36 (s, 2Н), 6,79 (d, J=8,6 Гц, 1Н), 7,30 (t, J=7,3 Гц, 1Н), 7,36 (t, J=7,3 Гц, 2Н), 7,45 (d, J=6,9 Гц, 2Н), 7,70 (d, J=9,2 Гц, 1Н), 7,90 (d, J=2,7 Гц, 1Н), 8,11 (d, J=8,5 Гц, 1Н), 10,07 (s, 1Н), 12,40 (s, 1Н).
D.4. Синтез 6-хлор-N-(6-хлор-5-фтор-2-метоксипиридин-3-ил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамида I-32:

В герметизированном сосуде 1-(бензолсульфонил)-6-хлорпирроло[2,3-b]пиридин-3-сульфонилхлорид XII-4 (100 мг, 0,26 ммоля) растворяли в пиридине (4 мл). Добавляли 6-хлор-5-фтор-2-метоксипиридин-3-амин Х-3 (67 мг, 0,38 ммоля), затем перемешивали при 70°С в течение 2 ч. Реакционную смесь выливали в воду и экстрагировали этилацетатом (дважды). Органические фазы сушили над MgSO4 и выпаривали. Остаток очищали с помощью препаративной ЖХМС в щелочной среде, методика 1, и получали 11 мг 6-хлор-N-(6-хлор-5-фтор-2-метоксипиридин-3-ил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид I-32 в виде бледно-желтого твердого вещества.
Выход: 11%.
ЖХМС в щелочной среде, методика 1 (ЭР-): 389 (М-Н)-, чистота 96%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 3,63 (s, 3Н), 7,39 (d, J=8,4 Гц, 1Н), 7,98 (d, J=9,0 Гц, 1Н), 8,29 (dd, J=18,4, 5,6 Гц, 2Н), 11,17 (s, 1Н), 12,87 (s, 1Н).
D.5. Синтез 6-циано-N-[6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-индол-3-сульфонамида I-65:

Стадия 1: Синтез 6-бром-N-(6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил)-1Н-индол-3-сульфонамида I-65 а:
К раствору 6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-амина Х-20 (0,30 г, 1,34 ммоля) в пиридине (8 мл) при 0°С в течение 10 мин порциями добавляли 6-бром-1Н-индол-3-сульфонилхлорид XII-2 (1,58 г, 5,35 ммоля), затем при 0°С добавляли ДМАП (0,02 г, 0,13 ммоля) и реакционную смесь нагревали при 90°С в течение 20 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь концентрировали в вакууме. Остаток растирали с 2 н. раствором HCl (10 мл), разбавляли с помощью Н2О (70 мл) и экстрагировали с помощью EtOAc (3×35 мл). Органический слой отделяли, промывали рассолом (2×60 мл), сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное неочищенное вещество очищали с помощью колоночной хроматографии (диоксид кремния, 100-200 меш, 30% EtOAc в гексане) и получали 6-бром-N-(6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил)-1Н-индол-3-сульфонамид I-65а (0,42 г) в виде почти белого твердого вещества.
Выход: 62%.
ЖХМС в щелочной среде, методика 2 (ЭР+): 481 (М+Н)+, чистота 95%.
Стадия 2: Синтез 6-циано-N-[6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-индол-3-сульфонамида I-65:
К раствору 6-бром-N-(6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил)-1Н-индол-3-сульфонамида I-65а (0,20 г, 0,40 ммоля) в ДМФ (6 мл) добавляли Zn(CN)2 (0,14 г, 1,19 ммоля) и реакционную смесь продували аргоном в течение 15 мин. Добавляли Pd2(dba)3 (0,02 г, 0,02 ммоля) и 1,1'-бис(дифенилфосфанил)ферроцен (0,02 г, 0,04 ммоля) и реакционную смесь нагревали в микроволновой печи при 110°С в течение 3 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь разбавляли с помощью Н2О (80 мл) и экстрагировали с помощью EtOAc (3×40 мл). Органический слой отделяли, промывали рассолом (2×60 мл), сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное неочищенное вещество очищали с помощью колоночной хроматографии (диоксид кремния, 100-200 меш, 35% EtOAc в гексане) и получали 6-циано-N-[6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-индол-3-сульфонамид1-65 (0,077 г, 45%) в виде почти белого твердого вещества.
Выход: 45%.
ЖХМС в щелочной среде, методика 2 (ЭР-): 425 (М-Н)-, чистота 99%.
1Н ЯМР (400 МГц, ДМСОd6) δ 3,21 (s, 3Н) 4,53 (td, 1=14,67, 3,42 Гц, 2Н) 6,20-6,50 (m, 1Н) 7,54 (dd, J=8,31, 0,98 Гц, 1Н) 7,59 (d, J=10,27 Гц, 1Н) 7,86 (d, J=8,31 Гц, 1Н) 7,99 (s, 1Н) 8,04 (s, 1Н) 9,68 (brs, 1Н) 12,41 (brs, 1Н).
D.6. Синтез 6-циано-N-[6-(дифторметокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-индол-3-сульфонамида I-66:

Стадия 1: Синтез 6-бром-N-(6-(дифторметокси)-5-фтор-2-метоксипиридин-3-ил)-1Н-индол-3-сульфонамида I-66а:
К раствору 6-(дифторметокси)-5-фтор-2-метоксипиридин-3-амина Х-8 (0,10 г, 0,47 ммоля) в пиридине (2 мл) при 0°С в течение 20 мин порциями добавляли 6-бром-1Н-индол-3-сульфонилхлорид XII-2 (0,44 г, 1,50 ммоля), затем при такой же температуре добавляли ДМАП (0,006 г, 0,05 ммоля) и реакционную смесь нагревали при 100°С в течение 18 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь концентрировали в вакууме. Остаток растирали с 2 н. раствором HCl (5 мл), разбавляли с помощью Н2О (10 мл) и экстрагировали с помощью EtOAc (3×25 мл). Органический слой отделяли, промывали рассолом (2×30 мл), сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное неочищенное вещество очищали с помощью колоночной хроматографии (диоксид кремния, 100-200 меш, 40% EtOAc в гексане) и получали 6-бром-N-(6-(дифторметокси)-5-фтор-2-метоксипиридин-3-ил)-1Н-индол-3-сульфонамид I-66а (0,15 г) в виде почти белого твердого вещества.
Выход: 63%.
ЖХМС в щелочной среде, методика 2 (ЭР-): 46 (М-Н)-, чистота 92%.
Стадия 2: Синтез 6-циано-N-[6-(дифторметокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-индол-3-сульфонамида I-66:
К раствору 6-бром-N-(6-(дифторметокси)-5-фтор-2-метоксипиридин-3-ил)-1Н-индол-3-сульфонамида I-66а (0,09 г, 0,18 ммоля) в ДМФ (2 мл) добавляли CuCN (0,03 г, 0,36 ммоля) и реакционную смесь продували аргоном в течение 20 мин, затем добавляли Pd(PPh3)4 (0,02 г, 0,02 ммоля). Реакционную смесь продували аргоном в течение 5 мин и нагревали при 110°С в течение 16 ч. За протеканием реакции следили с помощью ТСХ и ЖХМС. После завершения реакции реакционную смесь разбавляли с помощью Н2О (20 мл) и EtOAc (20 мл), фильтровали через слой целита. Органический слой отделяли и водный слой экстрагировали с помощью EtOAc (3×15 мл). Объединенные органические слои промывали рассолом (2×20 мл), сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное неочищенное вещество очищали с помощью колоночной хроматографии (диоксид кремния, 100-200 меш, 50% EtOAc в гексане) и получали 6-циано-N-[6-(дифторметокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-индол-3-сульфонамид I-66 (0,025 г) в виде почти белого твердого вещества.
Выход: 33%.
ЖХМС в щелочной среде, методика 2 (ЭР-): 411 (М-Н)-, чистота 96%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 7,53-7,55 (m, 1Н) 7,55 (t, J=72 Гц, 1Н) 7,73-7,77 (m, 1Н) 7,91 (d, J=8,31 Гц, 1Н) 7,99 (s, 1Н) 8,13 (s, 1Н) 9,94 (brs, 1Н) 12,45 (brs, 1Н) (сигналы 3Н объединены с пиком растворителя).
Проводили исследования соединений примеров и их активности по данным исследований Са2+ и цАМФ дополнительно приведены в представленной ниже таблице 6.
В. БИОЛОГИЯ/ФАРМАКОЛОГИЯ
B-I. Культуры клеток
Рекомбинантная линия клеток GPR17:
Клетки Flp-In T-REx СНО, стабильно экспрессирующие рецептор GPR17 (СНО hGPR17), полученные из лаборатории Evi Kostenis (Bonn University, Germany) выращивали при 37°C в увлажненной атмосфере, содержащей 5% СО2. Клетки выращивали в МДСИ (модифицированная по методике Дульбекко среда Игла) с добавлением питательной смеси F-12 и с добавлением гигромицина В (500 мкг/мл) и бластицидина (30 мкг/мл). Экспрессирование из локуса Flp-In индуцировали путем обработки доксициклином (1 мкг/мл) за 16-20 ч до проведения исследований.
Первичные олигодендроциты:
Клетки-предшественники первичных олигодендроцитов (КПО) выделяли из переднего мозга детены1Ней крыс Wistar в день 0-2 после рождения. Образцы головного мозга отбирали механически с помощью 1Нприца и двух ра3Ных полых игл (сначала 1,2×40 и затем 0,60×30). Не содержащую комков суспензию клеток фильтровали через устройство для просеивания клеток с отверстиями размером 70 мкм и помещали в матрац для клеточных культур площадью 75 см2 и с покрытием из поли-D-лизина со средой МДСИ с добавлением 10% (об./об.) термически инактивированной фетальной телячьей сыворотки, пенициллина (100 Ед./мл) и стрептомицина (0,1 мг/мл), причем каждый второй день проводили замену среды. После выдерживания при 37°С в увлажненной атмосфере, содержащей 5% СО2, в течение 8-11 дней смешанные культуры встряхивали при 240 об/мин в течение 14-24 ч для отделения КПО от астроцитов и микроглии. Для дополнительного обогащения с помощью КПО суспензию помещали в не содержащие покрытия чашки Петри на 45 мин. Затем КПО высевали в планшеты с покрытием из поли-L-орнитина и выдерживали при 37°С в увлажненной атмосфере, содержащей 5% СО2, в среде для пролиферации Neurobasal с добавлением 2% (об./об.) В27, 2 мМ GlutaMAX, 100 Ед./мл пенициллина, 0,1 мг/мл стрептомицина, 10 нг/мл PDGF-AA (рецептор тромбоцитарного фактора роста) и 10 нг/мл щелочного раствора FGF (фактор роста фибробластов), проводя замену среды через день.
В-II: Функциональные исследования GPR17 in vitro
В-II-1: Функциональное исследование мобилизации кальция
GPR17 представляет собой рецептор, связанный с белком G. Активация GPR17 запускает передачу сигнала белка G типа Gq, что приводит к выделению кальция (Са2+) из депо - эндоплазматического ретикулума в цитозоль, количество можно определить с помощью красителя Calcium 5, флуоресцентного индикаторного красителя для определения концентрации Са2+ в цитозоле. Соединения, предлагаемые в настоящем изобретении, испытывали с использованием исследования Са2+ или исследования для цАМФ GPR17, дополнительно описанного ниже. Некоторые типичные соединения примеров исследовали с использованием обоих исследований активности, как это указано в приведенной ниже таблице 6.
Описание исследования Са2+:
Клетки СНО hGPR17 оттаивали и высевали в черные 384-луночные планшеты с прозрачным дном при плотности, равной 20000 клеток/лунка. Клетки инкубировали при 37°С в увлажненной атмосфере, содержащей 5% СО2, в течение ночи. Через 16-22 ч после высевания к клеткам СНО hGPR17 в течение 60 мин добавляли краситель Calcium 5, флуоресцентный индикаторный краситель для определения концентрации Са2+ в цитозоле, в соответствии с инструкциями изготовителя. Зависимость интенсивности флуоресценции от концентрации Са2+ в цитозоле в течение времени определяли при комнатной температуре в помощью устройства для считывания FLIPR Tetra. Клетки сначала инкубировали при комнатной температуре в течение 30 мин в буфере ССРХ (сбалансированный солевой раствор Хенкса) - Hepes (N-2-гидроксиэтилпиперазин-N-2-этансульфоновая кислота), обладающем значением рН, равным 7,4, содержащем исследуемые соединения при увеличивающейся концентрации (обычно от 10-11 до 10-6 М). Затем к клеткам добавляли 50 нМ MDL29,951, агониста GPR17. Определяли ингибирующее воздействие исследуемых соединений при ра3Ных концентрациях и определяли значения pIC50. Все инкубирования повторяли дважды и результаты сопоставляли с зависимостью концентрация-эффект для эталонных соединений - агониста и антагониста GPR17. Анализ и аппроксимацию зависимости проводили с помощью программного обеспечения ActivityBase ХЕ с использованием 4-параметрического логистического уравнения XLfit: у=А+((В-A)/(l+((C/x)^D))), где А, В, С и D означают минимальное значение у, максимальное значение у, IC50 и угол наклона соответственно.
Результаты исследования Са2+:
По данным исследования мобилизации Са2+ соединения, приведенные в примерах, обычно обладают значениями pIC50, большими или равными 6,5; более предпочтительно большими или равными 7,5 и еще более предпочтительно большими или равными 8,5. Активности исследованных соединений примеров представлены в таблице 6 в приведенном ниже разделе В-IIB. Диапазоны активностей А, В и С означают следующие значения pIC50, полученные в исследовании Са2+: "A": pIC50 6,5≤х<7,5, "В": pIC50 7,5≤х<8,5, "С":pIC50≥8,5.
В-IIB. Функциональное исследование накопления цАМФ
Активация GPR17 также может обеспечить передачу сигнала белка G типа Gi, что приводит к уменьшению концентрации внутриклеточного циклического аденозинмонофосфата (цАМФ). Изменение концентрации внутриклеточного цАМФ можно определить с использованием набора для динамического исследования цАМФ с помощью ОФРВ, выпускающегося фирмой CisBio (Codolet, France). В исследовании использовали методику однородной флуоресценции с разре1Нением по времени (ОФРВ) и оно основано на определении конкурентности между нативным цАМФ, продуцируемым клетками, и цАМФ, меченым с помощью красителя d2. Связывание метки определяли с использованием антител к цАМФ, меченных криптатом.
Описание исследования цАМФ
Клетки СНО hGPR17 разводили в ЗФФ (забуференный фосфатом физиологический раствор), содержащем ЭДТК (этилендиаминтетрауксусная кислота), и помещали в черные 384-луночные планшеты при плотности, равной 5000 клеток/лунка. Клетки сначала инкубировали при комнатной температуре в течение 30 мин в буфере ССРХ - Hepes (рН 7,4), содержащем разбавитель или исследуемые соединения - антагонисты/обратные агонисты GPR17 при ра3Ных концентрациях. Затем к разбавителю и к исследуемому соединению - антагонисту/обратному агонисту GPR17 при каждой концентрации добавляли агонист GPR17, MDL29,951, в соответствии с зависимостью доза-ответ (обычно от 10-5 до 10-10 М) при конечном объеме буфера ССРХ - Hepes (рН 7,4), содержащего 1% ДМСО, 5 мкМ форсколина и 0,1 мМ ИБМК (8-метоксиметил-3-изобутил-1-метилксантин), равном 20 мкл. После инкубирования при комнатной температуре в течение 60 мин реакцию останавливали и клетки лизировали путем добавления реагента для детектирования d2 и содержащего криптат реагента в 10 мкл буфера для лизиса в соответствии с инструкциями изготовителя. После инкубирования в течение 60 мин определяли изменение концентраций цАМФ в соответствии с инструкциями изготовителя с использованием устройства для считывания планшетов Envision при возбуждении лазером. Все инкубирования повторяли дважды. Результаты анализировали с помощью программного обеспечения GraphPad Prism с использованием 4-параметрического логистического уравнения для определения значений рЕС50 для MDL29,951 при отсутствии и в присутствии исследуемых соединений - антагонистов/обратных агонистов GPR17. Строили зависимость отношения доз (ОД) от концентрации антагониста и с помощью анализа по методике Шильда получали рассчитанное сродство pA2 для соединений - антагонистов/обратных агонистов GPR17.
Результаты исследования цАМФ:
По данным исследования цАМФ соединения, приведенные в примерах, обычно обладают значениями рА2, большими или равными 6,5; предпочтительно большими или равными 7,5; более предпочтительно большими или равными 8,5. Активности исследованных соединений примеров приведены в представленной ниже таблице. Диапазоны активностей А, В и С означают следующие значения рА2, полученные в исследовании цАМФ: "А": рА2 6,5≤х<7,5, "В": рА2 7,5≤х<8,5, "С": рА2≥8,5.
В представленной ниже таблице 6 приведены значения pIC50 и рА2 для соединений примеров, изученных с помощью исследований Са2+ и цАМФ. Отсутствие результата в столбце со значениями рА2 или Са2+ означает, что соответствующее соединение не изучали с использованием соответствующего исследования или что результат еще не получен.
В-IIC: Исследование созревания/миелинизации олигодендроцитов
Воздействие негативных модуляторов GPR17 на созревание/миелинизацию первичных олигодендроцитов можно определить с помощью проводимых in vitro иммунологических исследований с использований антител к основному миелиновому белку (ОМБ), использующихся в качестве маркера наличия зрелых олигодендроцитов.
Описание исследования ОМБ, вестерн-блоттинг/олигодендроцит/миелинизация
После выдерживания в среде для пролиферации в течение 3-4 дней первичные КПО высевали в 12-луночные планшеты для выращивания культур тканей при плотности, равной 25000 клеток/см2, и среду заменяли на не содержащую фактора роста среду Neurobasal для индуцирования самопроизвольной дифференциации in vitro и экспрессии белка GPR17. Для конечной дифференциации и количественного анализа экспрессии белка через 24-48 ч в не содержащую фактора роста среду добавляли 0,20 нг/мл трийодтиронина (ТЗ) и 10 нг/мл цилиарного нейротрофического фактора вместе с исследуемыми соединениями - антагонистами/обратными агонистами GPR17 при концентрации, равной 1 мкМ, или с разбавителем и выдерживали в течение еще 3 дней. После обработки соединением клетки дважды промывали охлажденным льдом ЗФФ и лизировали в охлажденном льдом буфере для лизиса (25 мМ Tris (трис(гидроксиметиламинометан)), рН 7,4, 150 мМ NaCl, 1 мМ ЭДТК, 1% Triton Х-100, 1% IGEPAL), содержащем смесь ингибитора протеазы. Лизаты перемешивали с вращением при 4°С в течение 20 мин и центрифугировали при скорости, равной 15000×g, при 4°С в течение 10 мин. Концентрацию белка определяли с помощью анализа содержания белка Pierce ВСА в соответствии с инструкциями изготовителя. 7,5-15 мкг Белка разделяли с помощью электрофореза на полиакриламидном геле с использованием 10% ДСН (додецилсульфат натрия) и переносили на нитроцеллюло3Ную мембрану путем электроблоттинга. После промывки мембраны блокировали с помощью Roti-Block при комнатной температуре в течение 1 ч и инкубировали при 4°С в течение ночи в Roti-Block с добавлением антител к ОМБ (1:5000, LifeSpan Biosciences). Мембраны 3 раза промывали с помощью ЗФФ, содержащего 0,1% Tween, и затем инкубировали при комнатной температуре в течение 1 ч в Roti-Block с добавлением конъюгированных с пероксидазой хрена козьих антимышиных антител IgG. Иммунореактивные белки визуализировали с помощью хемилюминесценции с использованием реагента для детектирования с помощью вестерн-блоттинга Amersham Biosciences ECL Prime и определяли количество с помощью денситометрии с использованием программного обеспечения Gelscan. Для нормировки на одинаковое содержание и перенос белка мембраны повторно исследовали с использованием антител к β-актину (1:2500, BioLegend; вторичные козьи антикроличьи антитела IgG HRP (ABIN)). Количество экспрессированного ОМБ в присутствии исследуемых соединений сопоставляли с количеством экспрессированного ОМБ при контрольных условиях.
Результаты исследования добавления 1 мкМ соединения I-22 (6-хлор-N-[6-(дифторметокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-пирроло[2,3-b]пиридин-3-сульфонамида) на экспрессирование миелина представлены на фиг. (n4, среднее и СО (стандартное отклонение)).
Описание исследования ОМБ, планшеты с волоконным покрытием/созревание/миенилизация олигодендроцита
Активность соединений, предлагаемых в настоящем изобретении, также можно определить с помощью исследования с использованием планшетов с волоконным покрытием следующим образом:
КПО высевали в 96-луночные план1Неты с волоконным покрытием Mimetix Aligned (Electrospining company) при плотности, равной 16000-22000 клеток/см2 После выдерживания в среде для пролиферации в течение 2 дней и не содержащей фактора роста среде Neurobasal в течение 2 дней для индуцирования самопроизвольной дифференциации in vitro и экспрессии белка GPR17 добавляли разбавитель или исследуемые соединения - антагонисты/обратные агонисты при концентрации, равной 1 мкМ, в среде для конечной дифференциации с добавлением 0,20 нг/мл трийодтиронина и 10 нг/мл цилиарного нейротрофического фактора и выдерживали в течение 6 дней, причем через 3 дня проводили замену среды. Затем клетки фиксировали в 4% параформальдегиде, затем промывали с помощью ЗФФ, обеспечивали проницаемость мембран с помощью 0,1% TritonX-100 в ЗФФ и блокировали с помощью 10% козьей сыворотки и 1% бычьего сывороточного альбумина в забуференном фосфатом физиологическом растворе. Антитела к ОМБ разводили в блокирующем буфере (1:2000) и инкубировали при 37°С в течение 1 ч. Клетки повторно промывали с помощью ЗФФ и инкубировали с конъюгированными с Су2 вторичными антимы1Ниными антителами IgG (Millipore, 1:500) в течение 1 ч. После промывки с помощью ЗФФ клетки окрашивали с помощью 0,2 мкг/мл ДАФИ (4,6-диамидино-2-фенилиндол, повторно промывали и закрепляли с помощью Mowiol. Флуоресцентные изображения получали с помощью микроскопа Zeiss AxioObserver.Z1, снабженного системой для визуализации ApoTome и объективом Plan-Apochromat 20×/0,8, с использованием фильтра для eGFP (возбуждение: 470/40 нм; испускание: 525/50 нм) и фильтра для ДАФИ (возбуждение: 365 нм; испускание: 445/50 нм). Получали изображения по меньшей мере для 15 случайно выбранных областей для контрольных образцов (среда для конечной дифференциации с добавлением 0,1% ДМСО) и для исследуемых соединений с использованием одинаковых условий, обеспеченных с помощью программного обеспечения Zeiss ZEN2.3. Изменения количеств миелинированных волокон приводили для групп волокон, обладающих разной диной (от 0 до 40 мкм, от 41 до 60 мкм, от 61 до 80, от 81 до 100, от 101 до 120 и >120 мкм) при отсутствии или в присутствии негативного модулятора GPR17.
| название | год | авторы | номер документа |
|---|---|---|---|
| (АЗА)ИНДОЛ-, БЕНЗОТИОФЕН- И БЕНЗОФУРАН-3-СУЛЬФОНАМИДЫ | 2017 |
|
RU2767904C2 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2700004C1 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛОВ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2689777C1 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2679914C9 |
| ПРОИЗВОДНЫЕ ИМИДАЗОПИРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2695664C1 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2015 |
|
RU2733400C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОПИРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2678305C1 |
| КОНДЕНСИРОВАННЫЕ БИЦИКЛИЧЕСКИЕ ГЕТЕРОАРОМАТИЧЕСКИЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2685234C1 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОИМИДАЗОПИРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2697090C1 |
| ПРОИЗВОДНЫЕ ИМИДАЗОПИРИДАЗИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2679609C1 |

Группа изобретений относится к фармацевтической химии и включает соединение формулы I, его фармацевтически приемлемые соли, способ лечения рассеянного склероза и терапевтическую композицию. В формуле I X1 обозначает N или C(R7), R2 и R4 оба обозначают водород, R5 обозначает водород или галоген, R6 выбран из группы, включающей галоген, цианогруппу, бензилоксигруппу, C1-С3-алкоксигруппу и С1-С3-алкил, где каждый из следующих: алкил и алкоксигруппа, может содержать один - три галогена, или R6 вместе с R7 и с атомами С, к которым они присоединены, образуют 5- или 6-членное ароматическое или неароматическое кольцо, которое может содержать 1 или 2 образующих кольцо N-атома, где указанное кольцо является незамещенным, R7, если он содержится, выбран из группы, включающей водород и галоген, или R7 вместе с R6 образуют кольцо, как это описано выше, R8 выбран из группы, включающей водород, галоген, метоксигруппу, этоксигруппу, фторметоксигруппу и фторэтоксигруппу, R10 выбран из группы, включающей водород, цианогруппу, галоген, С3-С5-циклоалкил, С1-С4-алкоксигруппу и С1-С4-алкил, где каждый из следующих: алкил и алкоксигруппа, может содержать один - три заместителя, выбранных из группы, включающей галоген и фтор(С1-С3)алкоксигруппу, R11 выбран из группы, включающей водород, фтор и метоксигруппу, Х2 обозначает N или C(R12), R12 выбран из группы, включающей водород, метоксигруппу и галоген. Технический результат - соединение формулы I, обладающее ингибирующей активностью в отношении GPR17, используемое для лечения рассеянного склероза. 4 н. и 19 з.п. ф-лы, 1 ил., 6 табл.
 I
I
1. Соединение формулы I

в которой X1 обозначает N или C(R7),
R2 и R4 оба обозначают водород,
R5 обозначает водород или галоген,
R6 выбран из группы, включающей галоген, цианогруппу, бензилоксигруппу, C1-С3-алкоксигруппу и С1-С3-алкил, где каждый из следующих: алкил и алкоксигруппа, может содержать один - три галогена, или
R6 вместе с R7 и с атомами С, к которым они присоединены, образуют 5- или 6-членное ароматическое или неароматическое кольцо, которое может содержать 1 или 2 образующих кольцо N-атома, где указанное кольцо является незамещенным,
R7, если он содержится, выбран из группы, включающей водород и галоген, или
R7 вместе с R6 образуют кольцо, как это описано выше,
R8 выбран из группы, включающей водород, галоген, метоксигруппу, этоксигруппу, фторметоксигруппу и фторэтоксигруппу,
R10 выбран из группы, включающей водород, цианогруппу, галоген, С3-С5-циклоалкил, С1-С4-алкоксигруппу и С1-С4-алкил, где каждый из следующих: алкил и алкоксигруппа, может содержать один - три заместителя, выбранных из группы, включающей галоген и фтор(С1-С3)алкоксигруппу,
R11 выбран из группы, включающей водород, фтор и метоксигруппу,
Х2 обозначает N или C(R12),
R12 выбран из группы, включающей водород, метоксигруппу и галоген,
и его фармацевтически приемлемые соли.
2. Соединение по п. 1, в котором
X1 обозначает N или C(R7),
R2 обозначает водород,
R4 обозначает водород,
R5 обозначает водород или галоген,
R6 выбран из группы, включающей галоген, цианогруппу, бензилоксигруппу, С1-С2-алкоксигруппу и С1-С2-алкил, где каждый из следующих: алкил и алкоксигруппа, может содержать один или два галогена, или
R6 вместе с R7 и с атомами С, к которым они присоединены, образуют пиридильное кольцо таким образом, что пиридил вместе с бициклической кольцевой системой, с которой он аннелирован, образуют 1Н-пирроло[3,2-h]хинолин,
R7, если он содержится, выбран из группы, включающей водород и галоген, или
R7 вместе с R6 образуют кольцо, как это описано выше,
R8 выбран из группы, включающей водород, галоген, метоксигруппу, этоксигруппу, фторметоксигруппу и фторэтоксигруппу,
R10 выбран из группы, включающей водород, цианогруппу, галоген, С3-С5-циклоалкил, С1-С4-алкоксигруппу и С1-С4-алкил, где каждый из следующих: алкоксигруппа, может содержать один - три заместителя, выбранных из группы, включающей галоген и фтор(С1-С3)алкоксигруппу,
R11 выбран из группы, включающей водород, фтор и метоксигруппу,
Х2 обозначает N или C(R12),
R12 выбран из группы, включающей водород, метоксигруппу и галоген,
и его фармацевтически приемлемые соли.
3. Соединение по любому из предыдущих пунктов, в котором R2, R4 и R5 все обозначают водород и R6 выбран из группы, включающей галоген, цианогруппу, фторметоксигруппу и фторметил.
4. Соединение по любому из предыдущих пунктов, в котором R8 выбран из группы, включающей фтор и метоксигруппу.
5. Соединение по любому из предыдущих пунктов, в котором R10 выбран из группы, включающей галоген, циклопропил и С1-С2-алкоксигруппу, где алкоксигруппа необязательно может содержать один - три заместителя, выбранных из группы, включающей фтор и фтор(С1-С2)алкоксигруппу.
6. Соединение по любому из предыдущих пунктов, в котором R10 выбран из группы, включающей галоген, метоксигруппу, этоксигруппу, фторметоксигруппу, фторэтоксигруппу и фторметоксиэтоксигруппу.
7. Соединение по любому из предыдущих пунктов, в котором R11 обозначает водород или фтор, предпочтительно фтор.
8. Соединение по любому из предыдущих пунктов, в котором
R2, R4 и R5 все обозначают водород,
R6 выбран из группы, включающей хлор, цианогруппу, метил, метоксигруппу, фторметоксигруппу и фторметил,
X1 обозначает N или C(R7),
R7, если он содержится, выбран из группы, включающей водород и фтор,
R8 выбран из группы, включающей фтор и метоксигруппу,
R10 выбран из группы, включающей хлор, бром, циклопропил и С1-С2-алкоксигруппу, где алкоксигруппа замещена с помощью вплоть до 3 атомов фтора или содержит один заместитель, выбранный из группы, включающей фторметоксигруппу и фторэтоксигруппу,
R11 обозначает водород или фтор,
Х2 обозначает N или C(R12),
R12, если он содержится, обозначает водород или фтор, предпочтительно водород,
и его фармацевтически приемлемые соли.
9. Соединение по любому из пп. 1-8, в котором Х2 обозначает N, таким образом, описывающееся формулой II, в которой все заместители являются такими, как описано в предыдущих пунктах, и его фармацевтически приемлемые соли, 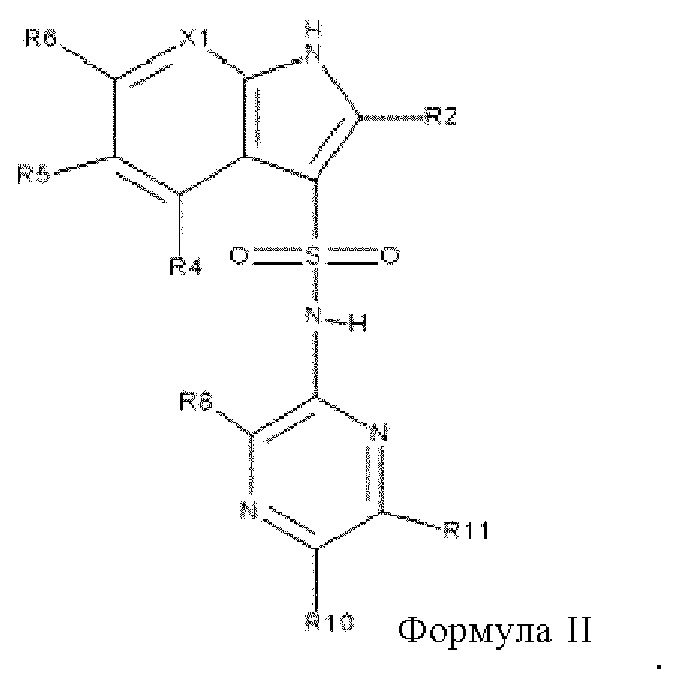
10. Соединение по п. 9, в котором
X1 обозначает N или C(R7)
R2, R4 и R5 все обозначают водород,
R6 обозначает хлор или фторметил,
R7, если он содержится, выбран из группы, включающей водород и фтор,
R8 выбран из группы, включающей фтор и метоксигруппу,
R10 выбран из группы, включающей хлор, бром, метоксигруппу, фторметоксигруппу, фторэтоксигруппу, фторметоксиэтоксигруппу и фторэтоксиметоксигруппу,
R11 обозначает фтор,
Х2 обозначает N
и R12, если он содержится, обозначает водород.
11. Соединение по любому из пп. 1-8, в котором Х2 обозначает C(R12), таким образом, описывающееся формулой III
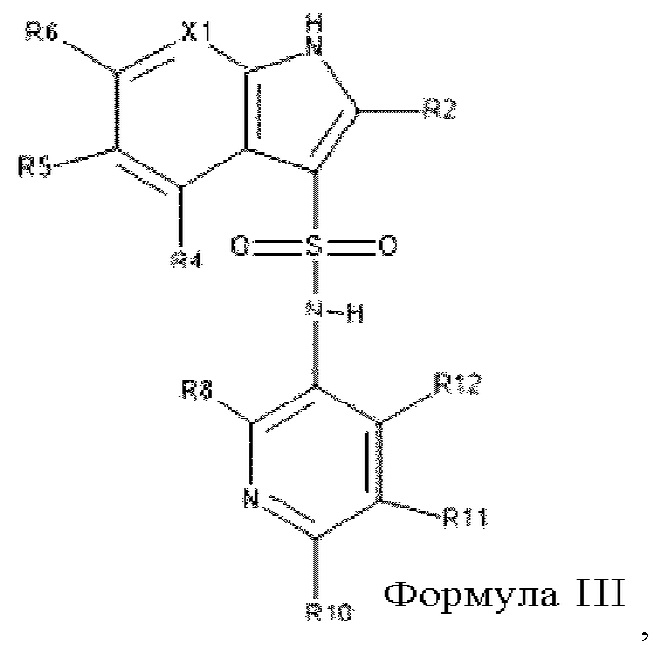
в которой все заместители являются такими, как описано в пп. 1-8, и его фармацевтически приемлемые соли.
12. Соединение по п. 11, в котором
R2, R4 и R5 все обозначают водород,
R6 выбран из группы, включающей галоген, цианогруппу, метил, изопропил, метоксигруппу, фторметоксигруппу, фторметил и бензилоксигруппу,
X1 обозначает N или C(R7),
R7, если он содержится, выбран из группы, включающей водород и галоген,
R8 выбран из группы, включающей водород, фтор, С1-С2-алкоксигруппу и фтор(C1-С2)алкоксигруппу,
R10 выбран из группы, включающей галоген, циклопропил, С1-С2-алкил и С1-С2-алкоксигруппу, где алкоксигруппа, может необязательно содержать один - три заместителя, выбранных из группы, включающей фтор и фтор(С1-С2)алкоксигруппу,
R11 выбран из группы, включающей водород, метоксигруппу и фтор,
R12 выбран из группы, включающей водород и фтор,
и его фармацевтически приемлемые соли.
13. Соединение по любому из пп. 1-8, в котором X1 обозначает C(R7), таким образом, описывающееся формулой IV

в которой все заместители являются такими, как описано в пп. 1-8, и его фармацевтически приемлемые соли.
14. Соединение по п. 13, в котором
R2, R4 и R5 все обозначают водород,
R6 выбран из группы, включающей галоген, цианогруппу, метил, изопропил, метоксигруппу, фторметоксигруппу, фторметил и бензилоксигруппу,
R7 выбран из группы, включающей водород, галоген, фторметоксигруппу и фторметил,
Х2 обозначает N или C(R12),
R8 выбран из группы, включающей водород, фтор, С1-С2-алкоксигруппу и фтор(С1-С2)алкоксигруппу,
R10 выбран из группы, включающей галоген, циклопропил, С1-С2-алкил и С1-С2-алкоксигруппу, где алкоксигруппа, может необязательно содержать один - три заместителя, выбранных из группы, включающей фтор и фтор(С1-С2)алкоксигруппу,
R11 выбран из группы, включающей водород, метоксигруппу и фтор,
R12, если он содержится, выбран из группы, включающей водород, метоксигруппу и фтор,
и его фармацевтически приемлемые соли.
15. Соединение по любому из пп. 1-8, в котором X1 обозначает N, таким образом, описывающееся формулой V

в которой все заместители являются такими, как описано в пп. 1-8, и его фармацевтически приемлемые соли.
16. Соединение по п. 15, в котором
R2, R4 и R5 все обозначают водород,
R6 выбран из группы, включающей галоген, цианогруппу, метил, изопропил, метоксигруппу, фторметоксигруппу, фторметил и бензилоксигруппу,
Х2 обозначает N или C(R12),
R8 выбран из группы, включающей водород, фтор, С1-С2-алкоксигруппу и фтор(С1-С2)алкоксигруппу,
R10 выбран из группы, включающей галоген, циклопропил, С1-С2-алкил и С1-С2-алкоксигруппу, где алкоксигруппа, может необязательно содержать один – три заместителя, выбранных из группы, включающей фтор и фтор(С1-С2)алкоксигруппу,
R11 выбран из группы, включающей водород, метоксигруппу и фтор, предпочтительно из группы, включающей водород и фтор,
R12, если он содержится, выбран из группы, включающей водород, метоксигруппу и фтор.
17. Соединение по п. 1, описывающееся формулой VI

в которой n равно 0,
Х3 обозначает СН или N,
R2 обозначает водород,
R4 обозначает водород,
R5 обозначает водород или галоген,
Х2 обозначает N или C(R12),
R8 выбран из группы, включающей водород, галоген, метоксигруппу, этоксигруппу, фторметоксигруппу и фторэтоксигруппу,
R10 выбран из группы, включающей водород, цианогруппу, галоген, С3-С5-циклоалкил, C1-С4-алкоксигруппу и C1-С4-алкил, где алкоксигруппа, может содержать один – три заместителя, выбранных из группы, включающей галоген и фтор(C1-С3)алкоксигруппу,
R11 выбран из группы, включающей водород, фтор и метоксигруппу,
R12 выбран из группы, включающей водород, метоксигруппу и галоген,
и его фармацевтически приемлемые соли.
18. Соединение по п. 17, в котором
n равно 0,
Х3 обозначает N или СН,
R2 и R4 оба обозначают водород,
R5 обозначает водород или галоген, предпочтительно водород,
R8 выбран из группы, включающей водород, фтор и метоксигруппу,
R10 выбран из группы, включающей галоген, циклопропил и С1-С2-алкоксигруппу, где алкоксигруппа необязательно может содержать один - три заместителя, выбранных из группы, включающей фтор и фтор(С1-С2)алкоксигруппу,
R11 обозначает водород или фтор,
Х2 обозначает N или C(R12) и
R12, если он содержится, выбран из группы, включающей водород, метоксигруппу и фтор,
и его фармацевтически приемлемые соли.
19. Соединение, выбранное из группы, включающей
6-хлор-N-[6-(2,2-дифторэтокси)-2,5-дифторпиридин-3-ил]-1H-индол-3-сульфонамид,
N-[6-(дифторметокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-пирроло[3,2-h]хинолин-3-сульфонамид,
N-[6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-пирроло[3,2-h]хинолин-3-сульфонамид,
5-бром-6-хлор-N-[6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
6-хлор-N-[6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-7-фтор-1H-индол-3-сульфонамид,
6-циано-N-[6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-1H-индол-3-сульфонамид,
N-[6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-6-(дифторметокси)-1Н-индол-3-сульфонамид,
N-[6-[2-(дифторметокси)этокси]-5-фтор-2-метоксипиридин-3-ил]-6-(дифторметил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
6-хлор-N-[6-[2-(дифторметокси)этокси]-5-фтор-2-метоксипиридин-3-ил]-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
N-[6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-6-(дифторметил)-1H-индол-3-сульфонамид,
6-хлор-N-(6-циклопропил-5-фтор-2-метоксипиридин-3-ил)-1H-индол-3-сульфонамид,
6-хлор-N-(5-фтор-2-метоксипиридин-3-ил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
N-[6-(дифторметокси)-5-фтор-2-метоксипиридин-3-ил]-6-метокси-1H-индол-3-сульфонамид,
N-[6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-6-метокси-1H-индол-3-сульфонамид,
6-хлор-N-[6-[2-(дифторметокси)этокси]-5-фтор-2-метоксипиридин-3-ил]-1Н-индол-3-сульфонамид,
N-[6-(дифторметокси)-5-фтор-2-метоксипиридин-3-ил]-6-метил-1Н-индол-3-сульфонамид,
6-циано-N-[6-(дифторметокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-индол-3-сульфонамид,
6-(дифторметил)-N-(2,5-дифтор-6-метилпиридин-3-ил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
6-(дифторметил)-N-(5-фтор-2,6-диметоксипиридин-3-ил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
6-(дифторметил)-N-[5-фтор-6-(2-фторэтокси)-2-метоксипиридин-3-ил]-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
6-хлор-N-(6-циклопропил-2,5-дифторпиридин-3-ил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
6-хлор-N-[6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-индол-3-сульфонамид,
6-хлор-N-(6-циклопропил-2,5-дифторпиридин-3-ил)-1Н-индол-3-сульфонамид,
N-[6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-6-(дифторметил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
6-хлор-N-[6-(2,2-дифторэтокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
N-[6-(2,2-дифторэтокси)-2-метоксипиридин-3-ил]-6-(дифторметил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
6-хлор-N-[6-(2,2-дифторэтокси)-2-метоксипиридин-3-ил]-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
N-[6-(2,2-дифторэтокси)-2,5-дифторпиридин-3-ил]-6-(дифторметил)-1Н-индол-3-сульфонамид,
6-хлор-N-[6-(дифторметокси)-4-метоксипиридин-3-ил]-1Н-индол-3-сульфонамид,
6-хлор-N-[6-(2,2-дифторэтокси)-2-метоксипиридин-3-ил]-1Н-индол-3-сульфонамид,
6-хлор-N-[2-(2,2-дифторэтокси)-6-метоксипиридин-3-ил]-1Н-индол-3-сульфонамид,
N-[6-(дифторметокси)-5-фтор-2-метоксипиридин-3-ил]-6-(дифторметил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
6-хлор-N-[6-(дифторметокси)-2-метокси-3-пиридил]-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
N-(6-хлор-5-фтор-2-метоксипиридин-3-ил)-6-(дифторметил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
6-хлор-N-[6-(2-фторэтокси)-2-метоксипиридин-3-ил]-1Н-индол-3-сульфонамид,
6-хлор-N-[5-фтор-6-(2-фторэтокси)-2-метоксипиридин-3-ил]-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
6-хлор-N-[5-фтор-6-(2-фторэтокси)-2-метоксипиридин-3-ил]-1Н-индол-3-сульфонамид,
N-[6-(дифторметокси)-5-фтор-2-метоксипиридин-3-ил]-6-(дифторметил)-1Н-индол-3-сульфонамид,
6-хлор-N-[6-(дифторметокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
6-хлор-N-(5-хлор-3-метоксипиразин-2-ил)-1Н-индол-3-сульфонамид,
6-хлор-N-[6-(дифторметокси)-5-фтор-2-метоксипиридин-3-ил]-1Н-индол-3-сульфонамид,
N-(5-бром-3-метоксипиразин-2-ил)-6-хлор-1Н-индол-3-сульфонамид,
6-хлор-N-(2,5-дифтор-6-метилпиридин-3-ил)-1Н-индол-3-сульфонамид,
6-хлор-N-(5-фтор-2,6-диметоксипиридин-3-ил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
6-хлор-N-(6-хлор-5-фтор-2-метоксипиридин-3-ил)-1Н-пирроло[2,3-b]пиридин-3-сульфонамид,
6-хлор-N-(5-фтор-2,6-диметоксипиридин-3-ил)-1Н-индол-3-сульфонамид,
6-хлор-N-(2,5-дифтор-6-метоксипиридин-3-ил)-1Н-индол-3-сульфонамид,
6-хлор-N-(6-хлор-5-фтор-2-метоксипиридин-3-ил)-1Н-индол-3-сульфонамид,
6-хлор-N-(6-хлор-2,5-дифторпиридин-3-ил)-1Н-индол-3-сульфонамид,
6-хлор-N-(6-йодпиридин-3-ил)-1Н-индол-3-сульфонамид,
6-хлор-N-(6-хлор-4-фторпиридин-3-ил)-1Н-индол-3-сульфонамид,
и его фармацевтически приемлемые соли.
20. Соединение по любому из предыдущих пунктов для применения в терапии в качестве обратного агониста рецептора GPR17.
21. Соединение по любому из предыдущих пунктов для применения для лечения или предупреждения рассеянного склероза.
22. Способ лечения или предупреждения рассеянного склероза, включающий введение нуждающемуся в нем пациенту соединения по любому из пп. 1-19 в терапевтически эффективном количестве.
23. Терапевтическая композиция, обладающая ингибирующей активностью в отношении GPR17, содержащая эффективное количество соединения по любому из пп. 1-19 и фармацевтически приемлемый носитель.
| WO 2012059869 A1, 10.05.2012 | |||
| БЕЗОПАСНЫЕ КАЧЕЛИ | 2014 |
|
RU2567698C2 |
| WO 2006045476 A2, 04.05.2006 | |||
| Saravanan Konda Mani et al., Identification of novel GPR17- agonists by structural bioinformatics and signaling activation | |||
| International journal of biological macromolecules, 2017, vol.106, pp.901-907 | |||
| RU 2006109108 A, 20.11.2007. | |||
