Изобретение относится к новым химическим соединениям с ценными свойствами, в частности к соединениям общей формулы (I)
где
A - линейный алкил с 1 - 8 атомами углерода,
B - галоген,
D - группа формулы -CH2OH или -CO-R1, где R1 означает водород или гидроксигруппу,
R - остаток формулы -CO-R2, -CO-NHR3 или
где
R2 означает гидроксил или линейный или разветвленный алкоксил с 1 - 8 атомами углерода, а R3 - остаток формулы -SO2R5 или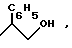
где
R5 означает фенил, который может быть замещен галогеном или линейным алкилом с 1 - 6 атомами углерода,
R4 - водород или трифенилметильную группу, или их соли.
В общем имеются в виду соли с органическими или неорганическими основаниями или кислотами.
В рамках настоящего изобретения предпочитают физиологически переносимые соли
Физиологически переносимыми солями имидазолилзамещенных производных циклогексана могут представлять собой соли предлагаемых соединений с минеральными кислотами, карбоновыми кислотами или сульфокислотами. Особенно предпочтительными являются, например, соли с хлористоводородной кислотой, бромистоводородной кислотой, серной кислотой, фосфорной кислотой, метансульфокислотой, этансульфокислотой, толуолсульфокислотой, бензолсульфокислотой, нафталиндисульфокислотой, уксусной кислотой, пропионовой кислотой, молочной кислотой, винной кислотой, лимонной кислотой, фумаровой кислотой, малеиновой кислотой или бензойной кислотой.
Физиологически переносимыми солями могут быть также соли с металлами или аммониевые соли тех предлагаемых соединений, которые содержат свободную карбоксильную группу. Особенно предпочтительными являются натриевые, калиевые, магниевые или кальциевые соли, а также аммониевые соли, которые образованы с аммиаком или органическими аминами, например этиламином, ди- или триэталмином, дициклогексиламином, диметиламиноэтанолом, аргинином, лизином или этилендиамином.
В рамках настоящего изобретения предлагаемые соединения могут иметься в разных стереоизомерных формах, при этом предлагаемые соединения имеются или в виде изображения и зеркального изображения (энантиомеры), или не в виде изображения и зеркального изображения (диастереомеры). Изобретение относится как к энантиомерам, так и к диастереомерам, а также к их соответствующим смесям. Как рацемические формы, так и диастереомеры можно разделять известными приемами на чистые стереоизомерные формы (E.L.Eliel, Stereochemistry of Carbon Compounds, McGraw Hill, 1962).
Новые соединения общей формулы (I) можно получать, например, за счет того, что соединение общей формулы (II)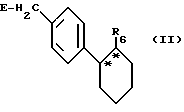
где
E - обычная удаляемая группа, например, хлор, бром, иод, тозилат или мезилат, предпочтительно бром,
R6 - линейный или разветвленный алкоксикарбонил с 1 - 4 атомами углерода в алкоксильной части или группа трифенилметил-тетразолил-1-ила,
подвергают взаимодействию с соединением общей формулы (III)
где
A, B и D имеют указанное выше значение,
в инертных растворителях, при необходимости в присутствии основания или в среде защитного газа до получения соединения общей формулы (IV)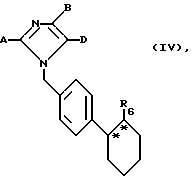
где
A, B, D и R6 имеют указанное выше значение,
и в случае кислот (R = CO2H) омыляют сложные эфиры, а в случае амидов и сульфамидов, полученных из соответствующих карбоновых кислот, амидируют после предварительного активирования с помощью соединений общей формулы
H-NHR3 (V),
где R3 имеет указанное выше значение,
при необходимости в присутствии основания и/или вспомогательного средства, например водоудаляющего средства, в инертных растворителях, и в случае свободных тетразолов, отщепляют тритильную группу кислотами, предпочтительно трифторуксусной кислотой или соляной кислотой в диоксане, и при необходимости вводят заместители A, B и D обычными способами, например, восстановлением, окислением, алкилированием или гидролизом, или переводят в другие группы, при необходимости разделяют изомеры и в случае получения солей обрабатывают соответствующими основаниями или кислотами.
Описанный способ может быть пояснен схемой, представленной в конце текста.
В качестве растворителя при осуществлении вышеуказанного способа можно использовать инертные органические растворители, не изменяющиеся в условиях реакций. К ним относятся простые эфиры как, например, простой диэтиловый эфир, диоксан, тетрагидрофуран, диметиловый эфир гликоля, углеводороды, например, бензол, толуол, ксилол, гексан, циклогексан, нефтяные фракции, галогенированные углеводороды как, например, дихлорметан, трихлорметан, тетрахлорметан, дихлорэтилен, трихлорэтилен или хлорбензол, или эфир уксусной кислоты, триэтиламин, пиридин, диметилсульфоксид, диметилформамид, триамид гексаметилфосфорной кислоты, ацетонитрил, ацетон или нитрометан. Кроме того, можно также использовать смеси указанных растворителей. Особенно предпочтительно используют диметилформамид и тетрагидрофуран.
В качестве оснований пригодны известные органические и неорганические основания. Предпочтительно используют гидроокиси щелочных металлов как, например, гидроокись натрия или калия, гидроокиси щелочноземельных металлов как, например, гидроокись бария, карбонаты щелочных металлов как, например, карбонат натрия или калия, или карбонаты щелочноземельных металлов как, например, карбонат кальция, или алкоголяты щелочных или щелочноземельных металлов как, например, метанолат или этанолат натрия или калия или трет.бутилат калия, или органические амины [триалкил(C1-C6)амины], например, триэтиламин, или гетероциклы, например, 1,4-диазабицикло[2.2.2]октан, 1,8-диазабицикло[5.4.0] ундек-7-ен, пиридин, диаминопиридин, метилпиперидин или морфолин. Можно использовать в качестве оснований щелочные металлы, например, натрий, или их гидриды, например, гидрат натрия. Предпочтительными являются гидрид натрия, карбонат калия, триэтиламин, пиридин и трет.бутилат калия.
Обычно основание используют в количестве 0,05 -10 моль, предпочтительно 1 - 2 моль, в пересчете на 1 моль соединения формулы (III).
Способ обычно проводят при температуре от -30 до +100oC, предпочтительно от -10 до +60oC, и при атмосферном давлении. Однако способ можно также осуществлять при повышенном или пониженном давлении (например, 0,5 - 5 бар).
В качестве оснований для омыления пригодны обычные неорганические основания. К ним предпочтительно принадлежат гидроокиси щелочных или щелочноземельных металлов, такие как, например, гидроокись лития, натрия, калия или бария, или карбонаты щелочных металлов, такие как, например, карбонаты натрия, калия или бикарбонат натрия, или алкоголяты щелочных металлов, как, например, метанолат натрия, этанолат натрия, метанолат калия, этанолат калия или трет. бутанолат калия. Особенно предпочтительно используют гидроокись лития, натрия или гироокись калия.
В качестве растворителей пригодны для гидролиза вода или обычно используемые для гидролиза органические растворители. К ним предпочтительно принадлежат спирты, такие как метанол, этанол, пропанол, изопропанол, бутанол или трет.бутанол, или простые эфиры, как, например, тетрагидрофуран или диоксан, или диметилформамид, или диметилсульфоксид. Особенно предпочтительно используют спирты, как, например, метанол, этанол, пропанол или изопропанол. Кроме того, можно также использовать смеси упомянутых растворителей.
Гидролиз предпочтительно осуществляют кислотами, как, например, трифторуксусной кислотой, уксусной кислотой, хлористоводородной кислотой, смесью хлористоводородной кислоты с диоксаном, бромистоводородной кислотой, метансульфокислотой, серной кислотой или надхлорной кислотой, предпочтительно трифторуксусной кислотой или хлористоводородной кислотой в диоксане.
Гидролиз обычно осуществляют при температуре 0 - 100 oC, предпочтительно при 20 - 80oC.
Обычно гидролиз проводят при атмосферном давлении. Однако его можно также осуществлять при пониженном или повышенном давлении (например, при давлении 0,5 - 5 бар).
При осуществлении омыления основания обычно используют в количестве от 1 до 3 моль, предпочтительно от 1 до 1,5 моль, в пересчете на 1 моль сложного эфира. Особенно предпочтительно используют молярное количество реагентов,
При проведении реакции сначала получают карбоксилаты предлагаемых соединений в качестве промежуточных продуктов, которые можно выделять. Предлагаемые кислоты получают путем обработки карбоксилатов обычными неорганическими кислотами. К ним предпочтительно принадлежат минеральные кислоты, как, например, хлористоводородная кислота, бромистоводородная кислота, серная кислота, фосфорная кислота или трифторукусная кислота. При этом при получении карбоновых кислот целесообразным оказалось подкисление щелочной реакционной смеси омыления на второй стадии без выделения карбоксилатов. Затем кислоты можно выделять известными приемами.
Амидирование и сульфонамидирование соединений общей формулы (IV) обычно осуществляют в одном из вышеуказанных растворителей, предпочтительно в тетрагидрофуране или дихлорметане.
Амидирование и сульфонамидирование могут протекать, в случае необходимости, через стадию активирования галогенангидридов кислоты или смешанных ангидридов, которые можно получать путем взаимодействия с тионилхлоридом, трихлоридом фосфора, пентахлоридом фосфора, трибромидом фосфора, оксалилхлоридом или метансульфохлоридом.
Амидирование и сульфонамидирование обычно проводят при температуре от -50 до +8-oC, предпочтительно от -30 до +20oC, и атмосферном давлении.
Кроме вышеупомянутых оснований в качестве основания пригодны также предпочтительно триэтиламин и/или диметиламинопиридин, 1,5-диазабицикло[3.4.0] ундецен-5 и 1,4-диазабицикло[2.2.2]октан.
Основание используют в количестве 0,5 - 10 моль, предпочтительно 1 - 5 моль, в пересчет на 1 моль соединений общей формулы (IV).
В качестве связывающего кислоту агента для амидирования можно использовать карбонаты щелочных или щелочноземельных металлов, как, например, карбонат натрия, карбонат калия, гидроокислы щелочных или щелочноземельных металлов, как, например, гидроокись натрия или калия, или органические основания, как, например, пиридин, триэтиламин, N-метилпиперидин или бициклические амидины, как, например, 1,5-диазабицикло[3.4.0] -нонен-5 или 1,5-диазабицикло[3.4.0]ундецен-5. Предпочтительно используют триэтиламин.
В качестве агента дегидратации пригодны карбодиимиды, как, например, диизопропилкарбодиимид, дициклогексилкарбодиимид, гидрохлорид N-(3-диметиламинопропил)-N'-этилкарбодиимида, карбонильные соединения, как, например, карбонилдиимидазол, 1,2-оксазолиевые соединения, как, например, 3-сульфонат-2-этил-5-фенил-1,2-оксазолия, ангидрид пропанфосфорной кислоты, изобутилхлороформат, гексилфторфосфат бензотриазолилокси-трис-(диметиламино)фосфония, амид сложного дифенилового эфира фосфоновой кислоты или хлорангиндрид метансульфокислоты, в случае необходимости, в присутствии основания, как, например, триэтиламина, N-этилморфолина, N-метилпиперидина, дициклогексилкарбодиимида и N-оксисукцинимида (J.C.Sheehan, S.L.LEdis, J. Am. Chem. Soc. 95, 875 (1973); F.E.Frerman et al., J. Biol. Chem. 225, 507 (1982) и N.B.Benoton, K.Kluroda, Int. Pept. Prot. Res. 13, 403 (1979), 17, 187 (1981)).
Связывающие кислоту агенты и агенты дегидратации обычно используют в количестве 0,5 - 3 моль, предпочтительно 1 - 1,5 моль, в пересчете на 1 моль соответствующей карбоновой кислоты.
Получение производных заместителей A, B и D происходит обычно известными из литературы способами, причем ниже поясняется восстановление альдегидов или алкоксикарбонильных соединений до спиртов (а), окисление альдегидов до карбоновых кислот (б) и алкилирование (в):
а) Восстановление алкоксикарбонильных соединений или альдегидов до соответствующих спиртов обычно проводят гидридами, как, например, алюмогидридом лития или боргидридом натрия, предпочтительно алюмогидродом лития, в среде инертных растворителей, как, например, простых эфиров, углеводородов или спиртов или в среде их смесей, предпочтительно в среде простых эфиров, как, например, простого диэтилового эфира, тетргидрофурана или диоксана, или спиртов, как, например, этанола, в случае альдегидов, предпочтительно боргидридом натрия в этаноле, при температуре 0 - 150oC, предпочтительно 20 - 100oC, при атмосферном давлении.
б) Окисление альдегидов до карбоновых кислот обычно проводят в одном из вышеприведенных растворителей, предпочтительно трет.бутаноле, с перманганатом калия в присутствии гидрофосфата натрия и сульфита натрия при температуре от -30 до +20oC, предпочтительно от -20 до +20oC и атмосферном давлении.
в) Алкилирование обычно проводят в среде одного из вышеприведенных растворителей, алкилирующим агентом, таким, как, например, алкилгалогенид с 1 - 8 атомами углерода, сложный эфир сульфокислоты, незамещенные или замещенные диалкилсульфаты с 1 - 6 атомами углерода или диарилсульфаты с 1 - 10 атомами углерода, предпочтительно метилйодид, сложный эфир п-толуолсульфокислоты или диметилсульфат.
Производные цилкогексана общей формулы (II) являются новыми и могут быть получены тем, что соединения общей формулы (VI)
сначала переводят гидрированием с палладием на угле в одном из вышеприведенных растворителей, например, метаноле, в среде водорода в соединения общей формулы (VII) ,
,
и во второй стадии, если R ≠ тетразолил, этерифицируют обычными способами, и если R = тетразолил, подвергают взаимодействию с хлорсульфонилизоцианатом в дихлорметане до получения соответствующих цианосоединений, затем вводят тетразолильную группу с помощью смеси азида натрия и триэтиламмонийхлорида в присутствии одного из вышеприведенных оснований, предпочтительно N,N-диметилформамида, в среде азота, дальнейшим взаимодействием с трифенилметилхлоридом в присутствии одного из вышеприведенных растворителей или оснований, предпочтительно дихлорметана и триэтиламина, вводят трифенилметильную группу и затем проводят бромирование по метиленовой группе, при необходимости в присутствии катализатора.
Восстановление двойной связи проводят при температуре 0 - 40oC, предпочтительно при 20oC и давлении 1 бар.
Этерификацию проводят в одном из вышеприведенных растворителей, предпочтительно толуоле и тетрагидрофуране, после описанного выше предварительного активирования соответствующей карбоновой кислоты, предпочтительно через хлорангидриды карбоновой кислоты, и последующего взаимодействия с соответствующим алкоголятом при температуре 0 - 60oC, предпочтительно при 10 - 35oC и атмосферном давлении.
Превращение до цианосединений и тетразолильных соединений проводят обычно при температуре кипения растворителей и атмосферном давлении.
Введение трифенилметильной группы в тетразольное кольцо происходит обычно при 0oC.
Бромирование проводят предпочтительно N-бромсукцинимидом, в среде одного из вышеприведенных растворителей, предпочтительно четыреххлористого углерода, при температуре 40 - 100oC, предпочтительно при 60 - 90oC и атмосферном давлении.
В качестве катализатора для бромирования пригодны, например, азобисизобутиронитрил, перекись дибензоила, предпочтительно азобисизобутиронитрил, причем катализатор используют в количестве 0,01 - 0,1 моль, предпочтительно 0,01 - 0,05 моль, в пересчете на 1 моль соединения общей формулы (VII).
Соединения общей формулы также являются новыми и могут быть получены, например, тем, что соединения общей формулы (VIII)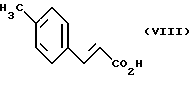 ,
,
где
R1 имеет указанное выше значение,
подвергают взаимодействию в одном из вышеуказанных растворителей, предпочтительно толуоле, с 1,3-бутадиеном в присутствии гидрохинона при температуре 180 - 230oC, предпочтительно при 200oC и давлении примерно 20 бар (Eur. J. Med. Chem. 11, 493 (1976)).
Соединения общей формулы (VIII) известны или могут быть получены известными способами (Organikum, VEB Deutscher Verlag der Wissenschaften, Berlin 1977, с. 572).
Соединения общих формул (IV) и (VII) являются новыми и могут быть получены, например, одним из вышеописанных способов.
Соединения общей формулы (III) известны (ЕП N 324377, Beilstein 25, 163; 23, 45; пат. США N 4355040) или могут быть получены известными способами.
Амины общей формулы (V) являются известными или могут быть получены известным способом (Beilstein 11/104, Р.В.Вицгерт, Успехи химии 32, 3 (1963); РЖХ 32, 1 (1969); Beilstein 4, 87).
Соединения общей формулы (I) согласно изобретению обладают неожиданным ценным фармакологическим спектром действия.
Предлагаемые соединения имеют специфическое антагонистическое действие в отношении ангиотензина II, так как они тормозят связь ангиотензина II с рецепторами. Они подавляют сосудосуживающие и стимулирующие секрецию альдостерона эффекты ангиотензина II. Кроме того, они ингибируют пролиферацию гладких мышечных клеток.
Поэтому их можно использовать в лекарствах для лечения повышенного артериального давления и артериосклероза. Кроме того, их можно использовать для лечения коронарных заболеваний сердца, недостаточности сердца, нарушения мозговой деятельности, ишемии мозговых заболеваний, нарушения периферического кровообращения, нарушения функций почки и надпочечника, бронхоспазмические и васкулярно вызванных заболеваний дыхательных путей, ретенции натрия и отеков.
Исследования задержки индуцированного агонистами сокращения.
Самок и самцов кроликов умерщвляли ударом в затылок и обескровливали, в некоторых случаях наркотизировали нембуталом (около 60 - 80 мг/кг внутривенно) и умерщвляли путем открытия грудной клетки. Грудную аорту вынимали, прилегающую к ней соединительную ткань удаляли, аорту подразделяли на кольцевые участки шириной 1,5 мм, каждый из которых при начальной нагрузке около 3,5 г подавали в 10 мл ванны, содержащей аэрированный карбогеном питательный раствор Кребса/Хензелайта, имеющий температуру 37oC, следующего состава: 119 ммоль/л хлористого натрия; 2,5 ммоль/л дигидрата хлористого кальция; 1,2 ммоль/л дигидрогенфосфата калия; 10 ммоль/л глюкозы; 4,8 ммоль/л хлористого калия; 1,4 ммоль/л семигидрата сульфата магния и 25 ммоль/л бикарбоната натрия.
Сокращения определяли изометрически при помощи ячеек марки Статам УС2 через мостиковые усилители, сигналы преобразовывали в цифровые величины при помощи аналого-цифрового преобразователя и оценивали. Кривые по дозам агониста и действию составляли ежечасно. При этом каждую кривую составляли на основании 3 - 4 отдельных концентраций исследуемого соединения, которое вводили в ванну в промежутках 4 мин. После окончания составления кривой и последующих циклов промывки (16 раз в течение около 5 с/мин вышеупомянутым питательным раствором) проводилась 28-минутная фаза покоя или инкубации, за которую, как правило, сокращения снова достигали исходного значения.
Каждая третья кривая использовалась в качестве базовой величины для оценки каждого исследуемого в дальнейших опытах соединения, которое при составлении дальнейших кривых подавалось в ванны в повышенных дозах, начиная с проведения периода инкубации. При этом каждый кольцевой сегмент аорты стимулировался тем же агонистом.
Агонисты и их стандартные концентрации (объем аппликации на отдельную дачу = 100 мкл)
KCl 22,7; 32,7; 42,7; 52,7 ммоль/л
1-норадренали 3•10-9; 3•10-8; 3•10-7; 3•10-6 г/мл
Серотонин 10-8; 10-7; 10-6; 10-5 г/мл
В-НТ 920 10-7; 10-6; 10-5 г/мл
метоксамин 10-7; 10-6; 10-5 г/мл
ангиотензин II 3•10-9; 10-8; 3•10-8; 10-7 г/мл
Для определения КТ50 (концентрации, при которой исследуемое соединение вызывает 50%-ное торможение) исходили из эффекта, получаемого при третьей субмаксимальной концентрации агониста.
Предлагаемые соединения тормозят в зависимости от дозы индуцируемое ангиотензином II сокращение изолированной аорты кролика. Индуцированное деполяризацией калия или другими агонистами сокращение вообще не тормозилось или только слабо тормозилось при наличии высоких концентраций. Результаты опыта приведены в табл. 1.
Определение антигипертенсивной активности у ненаркотизированных гипертенсивных крыс.
Оральная антигипертенсивная активность предлагаемых соединений исследовалась у ненаркотизированных крыс при помощи хирургически индуцированного одностороннего стеноза артерий почки. Для этой цели правую артерию почки сужали при помощи серебряной скобки шириной в просвете 0,18 мм. При таком виде гипертонии активность ренина плазмы крови повышена в течение первых шести недель после вмешательства. Артериальное кровяное давление этих животных бескровно измеряли при помощи "хвостовой манжеты" в определенных временных промежутках после дачи исследуемого соединения. Исследуемые соединения в виде тилозной суспензии давали в различных дозах внутрижелудочно (орально) при помощи соответствующего зонда. Предлагаемые соединения снижали артериальное кровяное давление гипертенсивных крыс в клинически релевантных дозах.
Кроме того, в зависимости от концентрации предлагаемые соединения тормозили специфическую связь радиоактивного ангиотензина II.
Взаимодействие предлагаемых соединений с рецептором ангиотензина II на мембранных фракциях коры надпочечника (крупного рогатого скота).
Свежую кору надпочечника крупного рогатого скота тщательно очищали от мозгового вещества и оболочки, после чего в 0,32 М раствора сахарозы при помощи мешалки типа Ультра-турракс (фирмы Янке унд Кункель, г. Штауфен, ДЕ) измельчали до крупного мембранного гомогената, который на двух стадиях центрифугирования очищали с получением мембранных фракций. Исследования по определению связывания с рецептором проводили на частично очищенных мембранных фракциях с применением радиоактивного ангиотензина II. Анализируемая среда объемом 0,25 мл содержала частично очищенные мембраны (50 - 80 мкг), 3H-ангиотензина II (3 - 5 нМ), буферный раствор (50 мМ Трис, pH 7,2), 5 мМ MgCl2, а также исследуемые соединения. После 60-минутной инкубации при комнатной температуре несвязанную радиоактивность проб отделяли при помощи влажных стекловолоконистых фильтров, а связанную радиоактивность спектрофотометрически определяли в сцинтиляционной среде после промывки протеина холодным буферным раствором (50 мМ Трис/HCl, pH 7,4, 5%-ного полиэтиленгликоля с молекулярным весом 6000). Данные обрабатывали при помощи компьютерных программ для получения значений Ki или KT50 (Ki: исправленные для используемой радиоактивности значения KT50; KT50 - концентрация, при которой исследуемое соединение вызывает 50%-ное торможение специфической связи радиолиганда).
Результаты опыта приведены в табл. 2.
Исследования торможения пролиферации гладких мышечных клеток предлагаемыми соединениями.
С целью определения антипролифераторного действия соединений использовались гладкие мышечные клетки, полученные из аорт крыс или свиней при помощи технически Медиа-Эксплантат (R. Ross. J. Cell. Biol. 50, стр. 172, 1971). Клетки высевали в пригодных чашках, содержащих, как правило, 96 углублений, и культивировали в среде 199, содержащей 7,5% телячьей эмбриональной сыворотки, 7,5% сыворотки новорожденного теленка, 2 мМ L-глутамина и 15 мМ буфера HEPES, pH 7,4 в атмосфере, содержащей 5% CO2, при температуре 37oC в течение 2 - 3 дней. Затем клетки синхронизировали путем удаления сыворотки на 2 - 3 дня и затем культивировали в присутствии ангиотензина II, сыворотки и других необходимых факторов. Одновременно добавляли исследуемые соединения. Через 16 - 20 ч добавляли 1 мк Ci3H-тимидина и через дальнейшие 4 ч определяли встроение этого вещества в ДНК клеток, осажденных путем обработки трихлоруксусной кислотой.
Для определения значений по КТ50 рассчитывали концентрацию исследуемого соединения, которая при последовательном разведении обеспечивает полумаксимальную задержку включения тимидина, вызываемого 1%-ной телячьей эмбриональной сывороткой.
Результаты опыта приведены в табл. 3.
Новое активное вещество можно переводить известными приемами в обычные препараты, такие как, например, таблетки, драже, пилюли, грануляты, аэрозоли, сиропы, эмульсии, суспензии и растворы с использованием инертных, нетоксичных, фармацевтически пригодных носителей или растворителей. При этом активное соединение должно иметься в терапевтически эффективной концентрации, например, в концентрации около 0,5 - 90% от веса соответствующей композиции, т.е. в количестве, достаточном для обеспечения указанной ниже дозировки.
Препараты получают, например, путем смешивания активного вещества с растворителями и/или носителями, в случае необходимости при использовании эмульгаторов и/или диспергаторов, причем, например, в случае использования воды в качестве разбавителя органические растворители можно использовать в качестве вспомогательных растворителей.
Аппликацию осуществляют обычным образом, предпочтительно орально или парентерально, в частности чрезъязычно или внутривенно.
В случае парентерального применения можно использовать растворы активного вещества с применением пригодных жидких носителей.
Для достижения эффективных результатов при внутривенной аппликации целесообразно использовать активное вещество в количестве около 0,001 - 1 мг/кг, предпочтительно около 0,01 - 0,5 мг/кг веса тела, а при оральной аппликации дозировка составляет около 0,01 - 20 мг/кг, предпочтительно 0,1 - 10 мг/кг веса тела.
Но в случае необходимости может быть целесообразным применение активного вещества и в других количествах, а именно в зависимости от веса тела или вида аппликации, от индивидуального поведения к лекарству, вида препарата, момента или промежутка, когда осуществляют его аппликацию. Так, например, в некоторых случаях может быть достаточным использование активного вещества в количестве, меньшем указанного минимального количества, в то время как в других случаях активное вещество следует использовать в количестве, большем указанного максимального количества. В случае аппликации большего количества рекомендуется распределять его по нескольким дневным дозам.
В нижеследующих примерах использовались для определения Rf (тонкослойная хроматография) следующие растворители:
А - дихлорметан : метанол - = 5 : 1
Б - дихлорметан : метанол - = 3 : 1
В - дихлорметан : метанол - = 20 : 1
Г - дихлорметан : метанол - = 10 : 1
Д - петролейный эфир : этилацетат - = 10 : 1
Е - петролейный эфир : этилацетат - = 5 : 1
Ж - петролейный эфир : этилацетат - = 2 : 1
З - петролейный эфир : этилацетат - = 1 : 1
И - дихлорметан : метанол - = 50 : 1
Нижеследующие примеры поясняют получение исходных соединений.
Пример 1. Транс-6-(4-толил)-циклогекс-3-ен-1-карбоновая кислота.
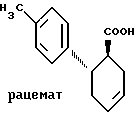 .
.
Известным способом (Eur. J. Med. Chem. 11, 493, 1976) 275 г (1,695 моль) 3-(4-толил)акриловой кислоты подвергают взаимодействию с 580 мл 1,3-бутадиена (количество определено в конденсированном виде) в 480 мл толуола с добавлением 3 г гидрохинона в течение 22 ч примерно при 200oC и 20 бар. Смесь разбавляют толуолом и экстрагируют 0,5 м. водного натрового щелока. Затем подкисляют водные фазы 1 м. соляной кислотой и экстрагируют диэтиловым эфиром. Эфирные растворы сушат над сульфатом натрия, упаривают и опять растворяют в толуоле. После кипячения в течение 15 мин в присутствии 5 г активированного угля фильтруют в горячем виде и упаривают растворитель до приблизительно 120 - 160 мл; при 0 -4oC выкристаллизовывается 124 г (573 моль) продукта. Фильтрат еще немного сгущают и опять охлаждают для дополнительной кристаллизации. При повторении этой операции выпадает в общей сложности еще 42 г (194 ммоль) продукта. Rf=0,39 (Г).
Пример II. Транс-2-(4-толил)-циклогексан-1-карбоновая кислота .
.
155 г (717 ммоль) соединения по примеру 1 растворяют в 1 л метанола и обрабатывают водородом на 10 г палладия (10% на животном угле) при температуре 20oC и давлении 1 бар. Спустя 16 ч отфильтровывают катализатор и упаривают растворитель в глубоком вакууме. Выход: 153 г (701 ммоль). Rf=0,38 (Г).
Пример III. Трет.бутиловый эфир транс-2-(4-толил)-циклогексан-1-карбоновой кислоты.
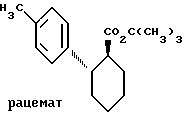 .
.
Способ А.
45,8 г (184 ммоль) соединения по примеру II растворяют в 600 мл толуола и подвергают взаимодействию с 49,5 мл (387 ммоль) оксалилхлорида при кипячении с обратным холодильником. Спустя 2 ч отгоняют растворитель вместе с избытком реагента, для чего сырой хлорангидрид карбоновой кислоты при необходимости должен повторно смешиваться с толуолом, после чего еще раз отгоняют. Полученный таким образом продукт растворяют в 500 мл тетрагидрофурана, перемешивают с 24,8 г (221 ммоль) трет. бутанолата калия при 0oC и продолжают перемешивать 20 ч при 20oC. Затем прибавляют воду и диэтиловый эфир и несколько раз экстрагируют. Органическую фазу сушат над сульфатом натрия, упаривают и остаток очищают хроматографией на силикагеле марки 60 (продукт фирмы Мерк, DE; элюент B). Выход: 39,6 (130 ммоль). Rf=0,47 (Д).
Способ Б.
20,0 г (91,6 ммоль) соединения по примеру II в 7 мл концентрированной серной кислоты суспендируют в 100 мл диэтилового эфира и смешивают при -30oC с 80 мл (713 ммоль) изобутена в автоклаве. Смесь нагревают в закрытом автоклаве до 20oC и реакцию осуществляют в течение 20 ч. Затем охлаждают опять до -30oC, открывают автоклав и при перемешивании реакционную смесь подают в смесь 300 мл 3 м. натриевой щелочи и 400 мл диэтилового эфира при 20oC. Водную фазу экстрагируют диэтиловым эфиром, органический раствор сушат над сульфатом натрия и упаривают. Выход: 23,3 г (84,9 ммоль).
Пример IV. Трет. бутиловый эфир транс-2-(4-бромметилфенил)- циклогексан-1-карбоновой кислоты.

11,70 г (42,6 ммоль) соединения по примеру III подвергают взаимодействию с 7,59 г (42,6 ммоль) N-бросукциниамида и 1,4 г азобисизобутиронитрила при кипячении с обратным холодильником в 100 мл тетрахлорметана. Спустя 4 ч смесь охлаждают, отфильтровывают выпавший сукцинимид и упаривают фильтрат. Выход: 14,2 г (40,2 ммоль), Rf = 0,48 (Д).
Пример V. Транс-2-(4-толил)-циклогексан-1-карбонитрил.
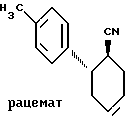
100,0 г (458,0 ммоль) соединения по примеру II в 1 л дихлорметана подвергают взаимодействию с 84,3 г (595,5 г) хлорсульфонилизоцианата в 100 мл дихлорметана при кипячении в течение часа (Organic Synthesis 50, 18 (1970)). Затем к охлаждающейся реакционной смеси прикапывают 72 мл (938,9 ммоль) N, N-диметилформамида и перемешивают 18 ч. Выливают на 350 г льда, после таяния льда разделяют фазы и упаривают и остаток перегоняют. Получают 57,8 г (290,2 ммоль) продукта. Т. кип.: 122 - 131oC (0,2 мбар). Rf = 0,81 (дихлорметан).
Пример VI. 5-[транс-2-(4-толил)-циклогекс-1-ил]тетразол.
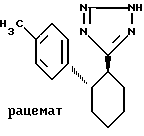 .
.
15,34 г (69,6 ммоль) соединения по примеру V в 230 л безводного формамида подвергают взаимодействию с 22,6 г (348 ммоль) азида натрия и 47,9 г (348 ммоль) хлорида триэтиламмония при кипячении в атмосфере азота; спустя 20 ч выливают после охлаждения в диэтиловый эфир и 1 М серную кислоту и экстрагируют затем 10%-ным натровым щелоком. Водную фазу при 0oC доводят до pH 1,5 с помощью 1 м. соляной кислоты и отфильтровывают выпавший осадок, промывают водой и сушат в высоком вакууме над пятиокисью фосфора и гидроокисью натрия. Выход: 11,2 г (46,2 ммоль). Rf = 0,23 (B).
Пример VII. 5-[транс-2-(4-толил)-циклогекс-1-ил]-2-трифенилметил-тетразол.
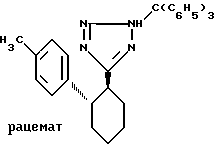 .
.
11,0 г (45,7 ммоль) соединения по примеру VI в 170 мл дихлорметана подвергают взаимодействию с 13,4 г (48,2 ммоль) трифенилметилхлорида и 7,57 г (54,6 ммоль) триэтиламина при 0oC. Перемешивают около 20 ч, причем смесь нагревается до комнатной температуры. Экстрагируют диэтиловым эфиром и водной лимонной кислотой. Органические фазы сушат сульфатом натрия и упаривают. Выход: 22,1 г (45,5 ммоль). Rf = 0,67 (E).
Пример VIII. 5-[транс-2-(4-бромметилфенил)-циклогекс-1-ил]-2-трифенилметил-тетразол.
 .
.
22,1 г (45,5 ммоль) соединения по примеру VII в 300 мл дихлорметана подвергают взаимодействию с 8,1 г (45,5 ммоль) N-бромсукцинимида и 0,3 г азобисизобутиронитрила при кипячении с обратным холодильником в течение 3 ч. Затем охлаждают до комнатной температуры и потом до 0oC и отфильтровывают осадок. Фильтрат упаривают и получают сырой продукт (26,2 г), который без дальнейшей очистки подвергают дальнейшей обработке. Rf = 0,47 (D).
Нижеследующие примеры поясняют получение имидазолилзамещенных производных циклогексана формулы (I).
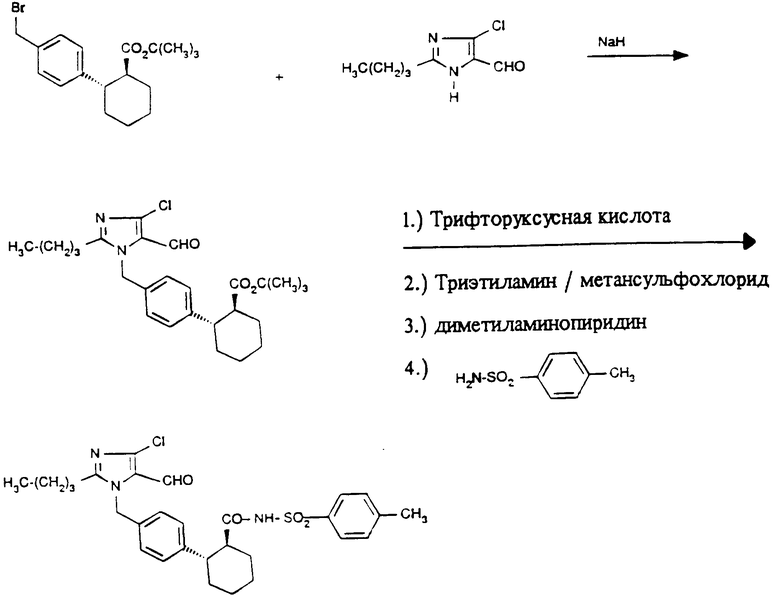
Пример 1. Трет.бутиловый эфир транс-2-[4-(2-бутил-4-хлор-5-формил-имидазол-1-ил-метил)фенил]- циклогексан-1-карбоновой кислоты.
 .
.
8,16 г (43,7 ммоль) 2-бутил-4-хлор-5-формил-имидазола (ЕП N 324377) перемешивают в 10 мл диметилформамида с 1,32 г (43,7 ммоль) гидрида натрия (80%-ный, стабилизированный парафином) до окончания выделения водорода при температуре около 0oC. Затем прикапывают раствор 18,4 г (437, ммоль) соединения по примеру VI в 100 мл диметилформамида и перемешивают 20 ч при 20oC. Для обработки добавляют воду и экстрагируют эфиром. Органические фазы сушат над сульфатом натрия и упаривают. Полученный остаток очищают хроматографией на силикагеле 60 (Мерк, растворитель (D)). Выход: 7,81 г (17,0 ммоль), Rf = 0,67 (E).
Пример 2. Транс-2-[4-(2-бутил-4-хлор-5-формил-имидазол- 1-ил-метил)фенил]-циклогексан-1-карбоновая кислота.

2,7 г (5,2 ммоль) соединения по примеру 1 подвергают взаимодействию в 40 мл диоксана с 16 мл концентрированной соляной кислоты. После проведения реакции в течение 18 ч при 20oC разбавляют эфиром, смешивают с 1 м. водного натриевого щелока и экстрагируют в длительной воронке при встряхивании. Водную щелочную фазу (pH 13 - 14) освобождают в вакууме от органического растворителя и устанавливают pH 2 при 0oC с помощью 2 М соляной кислоты. Выпавший осадок отфильтровывают, промывают водой и сушат в глубоком вакууме над гидроокисью натрия и пятиокисью фосфора. Выход: 2,0 г (5,0 ммоль), Rf = 0,28 (B).
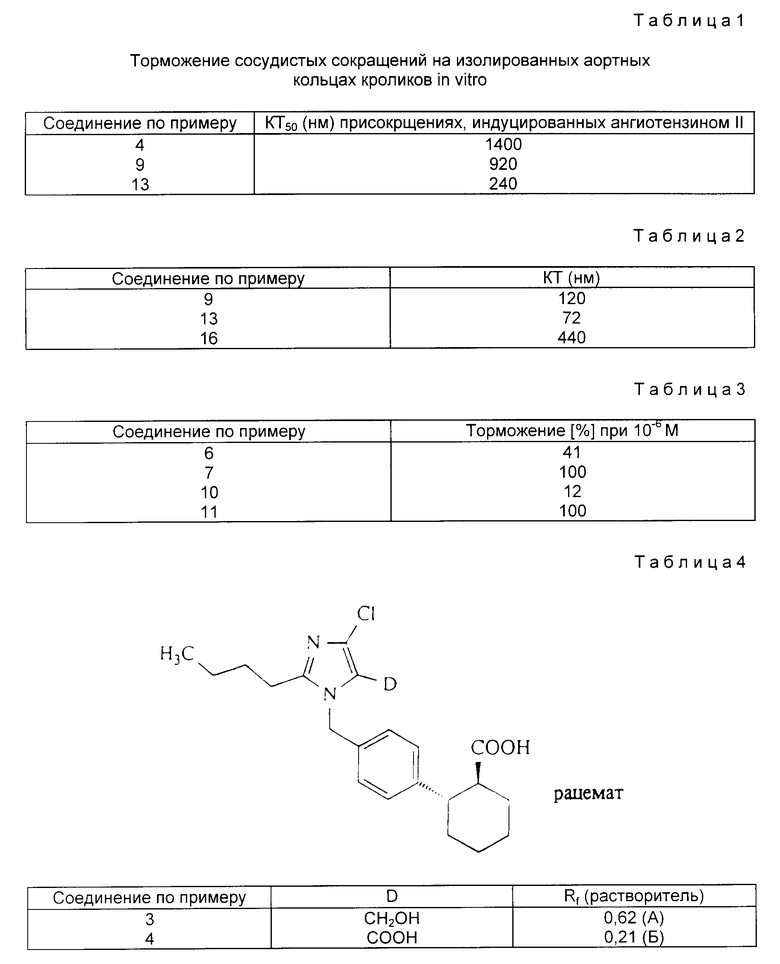
Аналогично примеру 2 получают приведенные в табл.4 соединения.
Пример 5. N-(4-толилсульфонил)амид транс-2-[4-(2-бутил-4-хлор-5-формил-имидазол-1-ил-метил)фенил]- циклогексан-1-карбоновой кислоты.
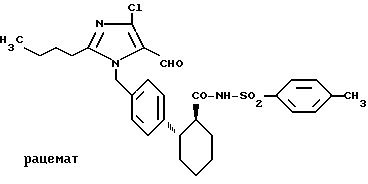 .
.
0,7 г (1,5 ммоль) соединения по примеру 2 подвергают взаимодействию в 30 мл тетрагидрофурана при -20oC с 0,127 мл (1,65 ммоль) хлорангидрида метансульфокислоты и 0,91 мл (6,6 ммоль) триэтиламина. Спустя 2 ч прибавляют при этой температуре 0,73 г (6,0 ммоль) 4-(N,N-диметиламино)пиридина и 0,31 г (1,8 ммоль) 4-толуолсульфамида и перемешивают 24 ч при 20oC. Затем реакционную смесь выливают в 1 М соляной кислоты и многократно экстрагируют эфиром. Органические фазы сушат сульфатом натрия, упаривают и полученный остаток очищают на силикагеле 60 (Мерк, растворитель Ж). Выход: 0,72 г (1,3 ммоль), Rf = 0,72 (B).
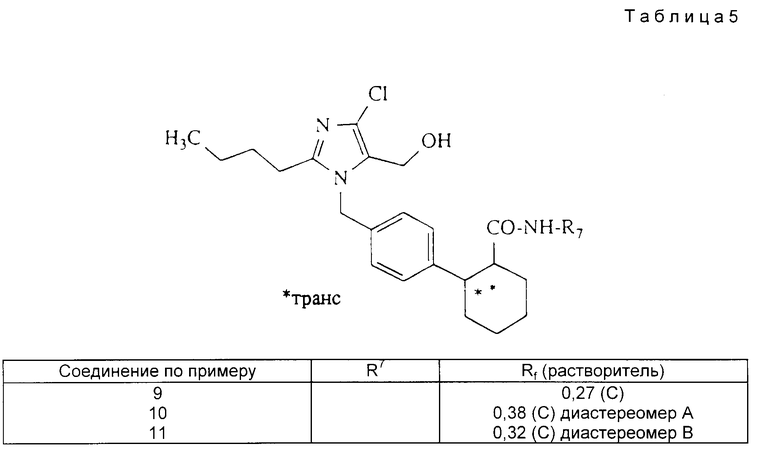
Примеры 6 и 7. (S)-фенилглициноламид[1,2-транс]-2-[4-(2- бутил-4-хлор-5-формил-имидазол-1-ил-метил)-фенил]-циклогексан-1- карбоновой кислоты.
 .
.
1,3 (2,8 ммоль) соединения по примеру 2 подвергают взаимодействию при -30oC в 30 мл тетрагидрофурана с 0,78 мл (5,6 ммоль) триэтиламина и 0,235 мл (3,1 ммоль) хлорангидрида метансульфокислоты. Спустя 30 мин при -30oC прикапывают раствор 459 мг (3,3 ммоль) (S)-фенилглицинола и 0,34 г (2,8 ммоль) 4-(N, N-диметиламино) пиридина в 10 мл тетрагидрофурана и смесь перемешивают при нагревании до 20oC в течение 24 ч. Выливают в 1 М. соляную кислоту и экстрагируют многократно эфиром. Органические фазы сушат сульфатом натрия, упаривают и остаток разделяют хроматографией (силикагель 60, Мерк).
Выход: 186 мг (0,36 ммоль) пример 6 (диастереомер A); 591 мг пример 6/7 (смесь диастереомеров A + B); 230 мг (0,44 ммоль) пример 7 (диастереомер B); Rf = 0,32 (3) пример 6; Rf = 0,17 (3) пример 7.
Пример 8. Трет. бутиловый эфир транс-2-[4-(2-бутил-4- хлор-5-гидроксиметил-имидазол-1-ил-этил)фенил]-циклогексан- 1-карбоновой кислоты.
 .
.
1 г (1,9 ммоль) соединения по примеру 1 растворяют в 10 мл этанола и подвергают взаимодействию при 20oC с 74,2 мг (2,0 ммоль) бораната натрия. Спустя 1 ч прибавляют воду и экстрагируют эфиром. Органическую фазу сушат сульфатом натрия и упаривают. Выход: 0,97 г (1,85 ммоль), Rf = 0,53 (3).
Аналогично примеру 8 получают приведенные в табл. 5 соединения.
Пример 12. Трет.бутиловый эфир транс-2-[4-(2-бутил-4-хлор- 5-карбокси-имидазол-1-ил-метил)фенил]-циклогексан-1-карбоновой кислоты.
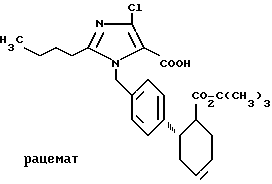 .
.
11,0 г (1,9 ммоль) соединения по примеру 1 растворяют в 9 мл трет. бутанола и подвергают взаимодействию при 20oC с 7,7 мл 1,25 м. водного раствора гидрофосфата натрия и 11,5 мл 1 м. водного раствора перманганата калия. Через 10 мин останавливают реакцию добавлением насыщенного водного раствора сульфата натрия, устанавливают pH 3,51 M соляной кислотой и экстрагируют этилацетатом. После отгонки растворителя вносят в диэтиловый эфир и экстрагируют 2 M водного натрового щелока. Водную фазу освобождают в вакууме от остатков растворителя и при 0oC устанавливают pH 11 добавлением соляной кислоты, выпавший осадок отфильтровывают, промывают водой и сушат в глубоком вакууме над смесью гидроокиси натрия и пятиокиси фосфора. Выход: 120 мг (0,2 ммоль). Rf = 0,28 (Г). Эфирная фаза содержит после экстракции щелочью 81% исходного продукта.
Пример 13. N-(4-толиосульфонил)амид транс-2-[4-(2- бутил-4-хлор-5-карбокси-имидазол-1-ил-метил)фенил]-циклогексан- 1-карбоновой кислоты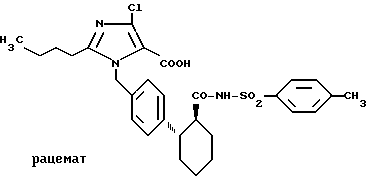 .
.
Аналогично примеру 12 получают целевое соединение. Rf = 0,11 (B).
Пример 14. 5-[транс-2-(4-{2-бутил-4-хлор-5-формил-имидазол-1- ил-метил} -фенил]-циклогекс-1-ил]-2-трифенилметил-тетразол.
Аналогично примеру 1 получают целевое соединение .
.
Rf = 0,72 (Ж).
Пример 15. 5[транс-2-(4{2-бутил-4-хлор-5-гидроксиметил- имидазол-1-ил-метил}фенил)-циклогекс-1-ил]-2-трифенилметил- тетразол.
Аналогично примеру 8 получают целевое соединение .
.
Rf = 0,23 (Ж).
Пример 16. 5-{ транс-2[4-(2-бутил-4-хлор-5-формил-имидазол- 1-ил-метил)-фенил]-циклогекс-1-ил}-тетразол.
 .
.
0,2 г (0,3 ммоль) соединения по примеру 14 подвергают взаимодействию в 2 мл тетрагидрофурана с 1 мл воды и 1 мл трифторуксусной кислоты. Спустя 2 ч при комнатной температуре выливают в смесь диэтилового эфира с водой и устанавливают pH 13 добавлением 10%-ного натрового щелока. Водную фазу подкисляют при 0oC 1 М соляной кислотой до pH 2, при этом выпадает продукт. Отфильтровывают и сушат в глубоком вакууме над пятиокисью фосфора и гидроокисью натрия. Получают 0,1 г (0,2 ммоль) продукта. Rf = 0,10 (И).
Пример 17. 5-{ транс-2-(4[2-бутил-4-хлор-5-гидроксиметил- имидазол-1-ил-метил)фенил]-циклогекс-1-ил}-тетразол.
Аналогично примеру 16 получают целевое соединение .
.
Rf = 0,41 (Г).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ЗАМЕЩЕННОЙ ГЕТЕРОЦИКЛОМ ФЕНИЛ-ЦИКЛОГЕКСАН-КАРБОНОВОЙ КИСЛОТЫ, СМЕСЬ ИХ ИЗОМЕРОВ ИЛИ ОТДЕЛЬНЫЕ ИЗОМЕРЫ И ИХ СОЛИ | 1994 |
|
RU2125990C1 |
| ПРОИЗВОДНЫЕ ЗАМЕЩЕННОЙ ГЕТЕРОЦИКЛОМ ФЕНИЛ-ЦИКЛОГЕКСАН-КАРБОНОВОЙ КИСЛОТЫ, СМЕСЬ ИХ ИЗОМЕРОВ ИЛИ ОТДЕЛЬНЫЕ ИЗОМЕРЫ И ИХ СОЛИ | 1994 |
|
RU2119480C1 |
| ПРОИЗВОДНЫЕ ПИРИДОНБИФЕНИЛА И ИХ СОЛИ | 1993 |
|
RU2118956C1 |
| ПРОИЗВОДНЫЕ АМИДА ФЕНИЛЦИКЛОГЕКСИЛКАРБОНОВОЙ КИСЛОТЫ, СМЕСЬ ИХ ИЗОМЕРОВ ИЛИ ОТДЕЛЬНЫЕ ИЗОМЕРЫ И ИХ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ С АНТИАРТЕРИОСКЛЕРОТИЧЕСКОЙ И АНТИРЕСТЕНОЗНОЙ АКТИВНОСТЬЮ | 1996 |
|
RU2158261C2 |
| ПРОИЗВОДНЫЕ 2-ФЕНИЛИНДОЛА, СМЕСЬ ИХ ИЗОМЕРОВ, ИЛИ ОТДЕЛЬНЫЕ ИЗОМЕРЫ, ИЛИ ИХ ФИЗИОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОЯВЛЯЮЩАЯ АНТИПРОЛИФЕРАТИВНУЮ АКТИВНОСТЬ | 1996 |
|
RU2162842C2 |
| ПРОИЗВОДНЫЕ ЦИКЛОАЛКАНО-ИНДОЛА И АЗАИНДОЛА, СМЕСЬ ИХ ИЗОМЕРОВ ИЛИ ОТДЕЛЬНЫЕ ИЗОМЕРЫ И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ПРОИЗВОДНЫЕ КАРБОНОВОЙ КИСЛОТЫ В КАЧЕСТВЕ ИСХОДНЫХ СОЕДИНЕНИЙ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИНГИБИРУЮЩАЯ ВЫСВОБОЖДЕНИЕ АССОЦИИРОВАННЫХ С АПОЛИПОПРОТЕИНОМ В-100 ЛИПОПРОТЕИНОВ | 1995 |
|
RU2157803C2 |
| ИЗОБУТИЛЗАМЕЩЕННЫЕ АМИДЫ МЕТАНСУЛЬФОНИЛХИНОЛИЛМЕТОКСИФЕНИЛЦИКЛОАЛКИЛУКСУСНОЙ КИСЛОТЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1993 |
|
RU2103261C1 |
| ПРОИЗВОДНЫЕ ХИНОЛИН-2-ИЛ-МЕТОКСИБЕНЗИЛГИДРОКСИМОЧЕВИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И 4-(ХИНОЛИН-2-ИЛ-МЕТОКСИ)ФЕНИЛ-ЦИКЛОАЛКИЛКЕТОН В КАЧЕСТВЕ ИСХОДНОГО СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ХИНОЛИН-2-ИЛ-МЕТОКСИБЕНЗИЛГИДРОКСИМОЧЕВИНЫ | 1992 |
|
RU2048466C1 |
| ПРОИЗВОДНЫЕ БИФЕНИЛПИРИДОНА И ИХ СОЛИ | 1992 |
|
RU2100350C1 |
| ПРОИЗВОДНЫЕ БЕНЗОФУРАНИЛАЛКАН-КАРБОНОВОЙ КИСЛОТЫ, ИЛИ СМЕСЬ ИХ ИЗОМЕРОВ, ИЛИ ОТДЕЛЬНЫЕ ИЗОМЕРЫ, ИЛИ ИХ СОЛИ | 1994 |
|
RU2125564C1 |
Объектом изобретения являются имидазолилзамещенные производные циклогексана общей формулы (I)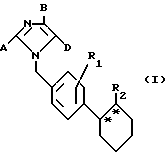
где A - линейный или разветвленный алкил или алкенил с 1 - 8 атомами углерода каждый или циклоалкил с 3 - 8 атомами углерода; B - водород, галоген или перфторалкил с 1 - 5 атомами углерода; D - группа формулы -CH2OR3 или -CO-R4, где R3 - водород или линейный или разветвленный алкил с 1 - 8 атомами углерода, R4 - водород, гидроксигруппа или линейный или разветвленный алкоксил с 1 - 8 атомами углерода, R1 - водород, галоген, нитрогруппа, трифторметильная группа, линейный или разветвленный алкил, алкоксил или алкоксикарбонил с 1 - 6 атомами углерода каждый, цианогруппа или карбоксил, R2 - остаток формул -CO-R5, -CO-NR6R7 или
где R5 - гидроксил или линейный или разветвленный алкоксил с 1 - 8 атомами углерода, R6 - водород, линейный или разветвленный алкил с 1 - 6 атомами углерода, R7 - остаток формул -SO2R9 или
где R9 - линейный или разветвленный алкил с 1 -8 атомами углерода, который может быть замещен фенилом или толилом, или фенил, котрый может быть замещен галогеном или линейным или разветвленным алкилом с 1 - 6 атомами углерода, R10 - водород, линейный или разветвленный алкил с 1 - 6 атомами углерода или защищающая гидроксилгруппа, R8 - водород, линейный или разветвленный алкил с 1 - 4 атомами углерода или трифенилметильная группа,
и их соли. Другими объектами изобретения являются способ получения соединений формулы (I) и их солей, содержащее их лекарственное средство и способ его получения, а также исходные соединения и способ их получения. 1 з.п. ф-лы, 5 табл.

где А - линейный С1 - С8-алкил;
В - галоген; D - группа формулы -СН2ОН или -СО - R1, где R1 - водород или гидроксигруппа;
R - остаток формулы -СО - R2, -СО - NHR3 или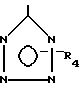
где R2 - гидроксил или линейный или разветвленный С1 - С8-алкоксил;
R3 - остаток формулы
- SO2R5
или
где R5 - фенил, который может быть замещен галогеном или линейным С1 - С6-алкилом;
R4 - водород или трифенилметильная группа,
или их соли.
| SU, патент, 9999666, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
