Эта заявка является частичным продолжением заявки с серийным N 07/781039, поданной 21 октября 1991 г., которая также частичное продолжение заявки с серийным N 07/748116, поданной 21 августа 1991 г.
Настоящее изобретение относится к гекса- и октагидробензо[f]хинолинонам, фармацевтическим препаратам, содержащим эти соединения и их использованию в качестве стероидных ингибиторов 5 α -редуктазы.
Общеизвестно, что некоторые нежелательные физиологические условия, такие как доброкачественный избыточный рост предстательной железы, обычное облысение мужчин, обыкновенные угри, себоррея, андрогенное облысение, гирсутизм и рак предстательной железы диктуются андрогеном и зависят от 5 α -дигидротестостерона (ДГТ).
Фермент 5 α -редуктаза является посредником в преобразовании тестостерона в более сильнодействующий локально андроген ДГТ, то есть на сам орган. Было установлено, что ингибиторы 5 α -редуктазы блокируют образование ДГТ и улучшают нежелательные физиологические состояния. Недавно были описаны два изофермента 5 α -редуктазы в организме человека (обозначены как тип 1 и 2). Andersson et al. , Рroc. Natl. Acad. Sci U.S.A., 87. 3640-3644 (1990); Andersson et al., Nature, 354, 159-161 (1991). Кроме некоторых структурных различий два фермента демонстрируют различия в отношении их биохимических свойств, особенностей экспрессии, генетики и фармакологии, Andersson et al., Nature, 354, 159-161 (1991); Jenkius, et al., Journal of Clinical Investigation, 89, 293-300 (1992). В настоящее время серьезно исследуется роль, которую играют два изофермента 5 α -редуктазы в действии андрогена. Эти изоферменты обычно описываются как 5 α -редуктаза 1 или 2 или 5- α -редуктаза типа 1 или типа 2.
Полезными соединениями для торможения 5 α -редуктазы являются производные стероида, такие как азастероиды (Rasmusson, et al., J.Med.Chem., 29 (11), 2298-2315 (1986); и производные бензоиламинофенокси-бутановой кислоты, такие, которые описаны в ЕПВ 291245.
Некоторые соединения бензо[f]хинолинона известны, См., например, Cannon, et al. , Synthesis, 6, 494-496 (1986); Kiguchi, et al., Heterocycles, 18, (Special Jssue), 217-220 (1982); Cannon, et al., J. Med. Chem., 22 (4), 341-347 (1979); Cannon, et al., J. Med. Chem., 23 (1), 1-5 (1980); Ninomiya, et al., J. Med. Chem. Soc. Perkin Traus 1, 12, 2911-2917 (1984); and Horri, et al. , Chem. Pharm. Bull., 16, (4), 668-671 (1968). Эти ссылки в основном касаются синтеза и допаминергической оценки раскрытых в них соединений. Ссылки не предполагают новых гекса- и октагидробензо[f]хинолинонов настоящего изобретения или того, что такие соединения будут полезны в качестве стероидных ингибиторов 5α- редуктазы.
Соответственно, одной из целей изобретения является получение новых гекса- и октагидробензо[f] хинолинонов, которые представляют собой сильнодействующие селективные стероидные ингибиторы 5 α -редуктазы, полезные в лечении доброкачественных образований предстательной железы, облысения у мужчин, угрей, себорреи, андрогенного облысения, гирсутизма и рака предстательной железы.
Другая цель изобретения - дать терапевтические композиции для лечения упомянутых заболеваний.
Еще одна цель изобретения - методы лечения этих заболеваний.
Другие цели, признаки и преимущества будут очевидны специалистам из следующего описания и формулы изобретения.
Настоящее изобретение обеспечивает получение новых гекса- и октагидробензо[f]хинолин-3-онов, которые являются эффективными стероидными ингибиторами 5 α -редуктазы.
Более конкретно, изобретение относится к соединениям, имеющим формулу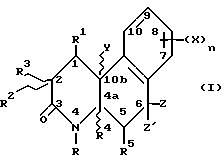
где R - водород, C1-C4 алкил, незамещенный или замещенный фен(C1-C4)алкил;
Z и Z' - независимо выбраны из водорода и C1-C4 алкила или один из Z и Z' объединяется с R5, чтобы образовать связь углерод-углерод;
Y - водород или метил или объединяется с R1, чтобы образовать связь углерод-углерод;
R1 - водород или объединяется с одним из Y или R, чтобы образовать связь углерод-углерод;
R2 - водород или C1-C4 алкил;
R3 - водород или объединяется с R1, чтобы образовать связь углерод-углерод;
R4 - водород или объединяется с R5, чтобы образовать связь углерод-углерод;
R5 - водород или объединяется с одним из Z или Z', чтобы образовать связь углерод-углерод;
n - 1 или 2;
X - водород, галоген, NO2, циано, CF3, C1-C6 алкил, C1-C6 алкокси, карбокси, C1-C6 алкоксикарбонил, амино, C1-C4 алкиламино, C1-C4 диалкиламино, амидо, C1-C4 алкиламидо, C1-C4 диалкиламидо, меркапто, C1-C6 алкилтио, C1-C6 алкилсульфинил, C1-C6 алкилсульфонил, или группа -A-R6, где A - C1-C6 алкилен, C2-C6 алкенилен или C2-C6 алкинилен; и R6 - галоген, гидрокси, CF3, C1-C6 алкокси, карбокси, C1-C6 алкоксикарбонил, амино, C1-C4 алкиламино, C1-C4 диалкиламино, амидо, C1-C4 алкиламидо, C1-C4 диалкиламидо, C1-C4 алкилсульфониламино, аминосульфонил или C1-C4 алкиламиносульфонил,
или их фармацевтически приемлемая соль; при условии, что
(a) по меньшей мере один из R1 и R5 - водород;
(b) если R - водород, метил, этил или бензил, X - не водород или метокси; и
(c) если R -метил, R2 - не метил.
Это изобретение также предусматривает фармацевтические препараты, которые включают соединение вышеуказанной формулы I или его фармацевтически приемлемую соль вместе с фармацевтически приемлемым наполнителем, разбавителем или носителем.
Раскрывается также способ торможения 5α- редуктазы. Более конкретно, предложены методы лечения множества заболеваний, которые связаны с активностью 5α- редуктазы у млекопитающих. Среди этих заболеваний - доброкачественные образования предстательной железы, облысение у мужчин, угри, себоррея, андрогенное облысение, гирсутизм и рак предстательной железы. Эти методы используют соединение формулы I или его фармацевтически приемлемую соль. Хотя соединения настоящего изобретения тормозят действие обоих изоферментов 5α- редуктазы, они демонстрируют большую селективность как ингибиторы 5α- редуктазы типа 1.
Еще один аспект изобретения - класс новых промежуточных соединений, полезных в получении соединений этого изобретения, а также способ получения достаточно оптически чистых активных соединений настоящего изобретения.
Промежуточные соединения имеют формулу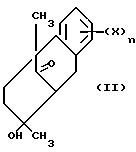
где X - водород, галоген, NO2, циано, CF3, C1-C6 алкил, C1-C6 алкокси, карбокси, C1-C6 алкоксикарбонил, амино, C1-C4 алкиламино, C1-C4 диалкиламино, амидо, C1-C4 алкиламидо, C1-C4 диалкиламидо, меркапто, C1-C6 алкилтио, С1-C6 алкилсульфинил, C1-C6 алкилсульфонил, или группа-A-R6, где A - C1-C6 алкилен, C2-C6 алкенилен или C2-C6 алкинилен; и R6 - галоген, гидрокси, CF3 C1-C6 алкокси, карбокси, C1-C6 алкоксикарбонил, амино, C1-C4 алкиламино, C1-C4 диалкиламино, амидо, C1-C4 алкиламидо, C1-C4 диалкиламидо, C1-C4 алкилсульфониламино, аминосульфонил или C1-C4 алкиламиносульфонил; и n - 1 или 2; или их фармацевтически приемлемая соль.
Аспект способа изобретения, который использует промежуточное соединение формулы II - способ для получения фактически оптически чистого активного соединения с формулой
где R - водород, C1-C4 алкил, незамещенный или замещенный фен (C1-C4)алкил;
Z и Z' - независимо выбраны из водорода и C1-C4 алкила;
Y - метил;
R2 - водород или C1-C4 алкил;
n- 1 или 2;
X - водород, галоген, NO2, циано, CF3, C1-C6 алкил, C1-C6 алкокси, карбокси, C1-C6 алкокси-карбонил, амино, C1-C4 алкиламино, C1-C4 диалкиламино, амидо, C1-C4 алкиламидо, C1-C4 диалкиламидо, меркапто, C1-C6 алкилтио, C1-C6 алкилсульфинил, C1-C6 алкилсульфонил, или группа -A-R6, где A - C1-C6 алкилен или C2-C6 алкенилен или C2-C6 алкинилен; и R6 - галоген, гидрокси, CF3, C1-C6 алкокси, карбокси, C1-C6 алкоксикарбонил, амино, C1-C4 алкиламино, C1-C4 диалкиламино, амидо, C1-C4 алкиламидо, C1-C4 диалкиламидо, C1-C4 алкилсульфониламино, аминосульфонил или C1-C4 алкиламиносульфонил,
или его фармацевтически приемлемой соли; который включает
a) реакцию 1-метил-2-тетралона с оптически активным амином для получения соответствующего 1-метиленамина; и
b) реакцию 1-метиленамина с α, β - ненасыщенным карбониловым соединением для получения соответствующего метанобензоциклооктан-4-она; и
c) реакцию метанобензоциклооктан-4-она с кислотным или основным катализатором для получения соответствующего 2,3,4,4a, 9,10-гексагидро-4a-метил-фенантрен-2-она, и
d) окислительное расщепление указанного фенантрен-2-она для получения соответствующего 3-[1-метил-1-(2-кето-1,2,3,4-тетрагидронафтил)]пропионовой кислоты; и
e) реакцию указанной пропионовой кислоты с аммиаком или первичным амином для получения соответствующего 10b-метил-1,2,3,4,6,10b-гексагидробензо[f] хинолин-3-она;
f) восстановление указанного гексагидробензо[f]хинолин-3-она для получения соответствующего октагидробензо[f]хинолин-3-она, как указано выше.
Используемый здесь термин "алкил" означает алкиловый радикал с прямой или разветвленной цепью, имеющий указанное число атомов углерода, такие алкильные группы включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, вторичный бутил, т-бутил и, если указано, высшие гомологи и изомеры, такие как н-пентил, н-гексил, 2-метилпентил и прочие.
Термин "алкилен" означает двухвалентный алкиловый радикал с прямой цепью с указанным числом атомов углерода, такой как метилен, 1,2-этандиил, 1,3-пропандиил, 1,4-бутандиил, 1,5-пентандиил, 1,6-гександиил. Таким же образом "алкенилен" означает двухвалентную ненасыщенную углеводородную группу с прямой цепью, имеющую указанное число атомов углерода и одну двойную связь углерод-углерод, такую как винилен, 1-пропилен-1,3-диил, 2-пропилен-1,3-диил, 2-бутен-1,4-диил, 1-бутен-1,4-диил и прочие. "Алкинилен" означает двухвалентную углеводородную группу с прямой цепью, имеющую указанное число атомов углерода и одну тройную связь углерод-углерод, такую как 1,2-ацетилендиил, 1-пропин-1,3-диил, 2-бутин-1,4-диил и другие.
Термин "фен(C1-C4)алкил" означает алкиловый радикал с прямой или разветвленной цепью с 1-4 углеродами, однозамещенный незамещенным или замещенным фенильным кольцом, в котором заместители одинаковые или разные галоген, C1-C4 алкил, C1-C4 алкокси, амино, C1-C4 алкиламино или C1-C4 диалкиламино. Типичные фен(C1-C4)алкильные группы включают бензил, 2-фенет -1-ил, 3-фенпроп-1-ил, 4-фенбут-1-ил, 1-фенет-1-ил, 2-фенпроп- -1-ил, 2-(4-галофенил)ет-1-ил, 4-галобензил и прочее.
Термин "алкокси" означает любой из метокси, этокси, н-пропокси, изопропокси и другие. Термин "галоген" и "гало" означает любой из фторо, хлоро, бромо и йодо. Термин "алкилтио" означает любой из метилтио, этилтио, н-пропилтио, изопропилтио и другие.
Термин "амидо" означает аминокарбонильную (-C(O)NH2) группу. Термин "алкиламино" означает группу - NH(C1-C4 акил) и термин "алкиламидо" означает группу -C(O)NH (C1-C4 алкил). Там, где указан заместитель "C1-C4 диалкиламино"[(-N) C1-C4 алкил)2] или "C1-C4 диалкиламидо" /-C(O/N)C1-C4 алкил)2/, каждая алкильная группа независимо имеет 1-4 атомов углерода.
Термин "алкилсульфонил" означает группу S(O) (алкил), где алкильная группа имеет указанное число атомов углерода. Таким же образом термин "алкилсульфонил" означает группу - SO2 (алкил), где алкильная группа имеет указанное число атомов углерода. Термин "алкилсульфониламино" означает группу - NHSO2, (C1-C4 алкил). Термин "аминосульфонил" означает группу - SO2NH2 и термин "алкиламиносульфонил" означает группу - SO2NH (C1-C4 алкил).
Октагидробензо[f] хинолинонами настоящего изобретения являются те соединения формулы I, в которых R1, R3, R4 и R5 - водород. Соответственно, гексагидробензо[f]хинолонами настоящего изобретения являются соединения формулы I, имеющие на два протона меньше, чем описано в определениях для формулы I.
Соединения настоящего изобретения имеют по меньшей мере один асимметричный углерод, представленный атомом углерода со звездочкой в формуле Ia ниже.
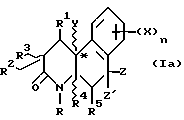
Соединения настоящего изобретения также существуют как индивидуальные цис-d- и цис-l-стереоизомеры, а также как транс-d- и трас-l-стереоизомеры и смеси таких изомеров. Две дис и две транс конфигурации показаны ниже в формуле 1b-1e.
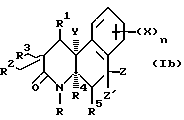
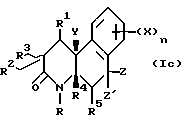
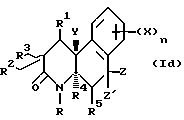
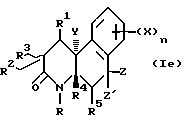
Соответственно, соединения настоящего изобретения включают не только смеси двух или нескольких таких индивидуальных изомеров, но также индивидуальный изомер.
Кроме того, существуют другие диастереомеры в зависимости от R2, Z и Z' заместителей. Соединения настоящего изобретения включают смеси двух или нескольких диастереомеров и индивидуальные изомеры.
Следующие соединения иллюстрируют соединения, охваченные объемом формулы I:
цис-dl-8-бромо-4-метил-1,2,3,4,4a, 5,6,10b-октагидробензо[f] хинолин-3-он;
транс-dl-8-бромо-4-метил-1,2,3,4,4a, 5,6,10b-октагидробензо[f] хинолин-3-он;
транс-dl-8-бромо-1,2,3,4,4a,5,6,10b-октагидробензо[f] хинолин-3-он;
транс-dl-8-йодо-4-метил-1,2,3,4,4a, 5,6,10b-октагидробензо[f] хинолин-3-он;
транс-dl-8,9-дихлоро-1,2,3,4,4a,5,6,10b-октагидробензо[f] хинолин-3-он;
транс-dl-8,9-дихлоро-4-метил-1,2,3,4,4a, 5,6,10b-окгагидробензо [f]хинолин-3-он;
транс-dl-8-хлоро-4-метил-1,2,3,4,4a, 5,6,10b-октагидробензо[f] хинолин-3-он;
цис-dl-8-хлоро-4-метил-1,2,3,4,4a, 5,6,10b-октагидробензо[f] хинолин-3-он;
транс-dl-4,8-диметил-1,2,3,4,4a,5,6,10b-октагидробензо[f] хинолин-он;
цис-dl-4,8-диметил-1,2,3,4,4a,5,6,10b-октагидробензо[f] хинолин-он;
транс-dl-8-фторо, 4-метил-1,2,3,4,4a, 5,6,10b-окгагидробензо[f] хинолин-3-он;
цис-dl-8-фторо-4-метил-1,2,3,4,4a, 5,6,10b-октагидробензо[f] хинолин-3-он;
транс-dl-4-метил-1,2,3,4,4a,5,6,10b-октагидробензо[f] хинолин-3-он;
цис-dl-4-метил-1,2,3,4,4a,5,6,10b-октагидробензо[f] хинолин-3-он;
цис-dl-1,2,3,4,4a,5,6,10b-окгагидробензо[f]хинолин-3-он;
транс-dl-8-фторо-1,2,3,4,4a,5,6,10b-октагидробензо[f] хинолин-3-он;
транс-dl-8-этоксикарбонилэтендиил-4-метил-1,2,3,4,4a, 5,6,10b-октагидробензо[f]хинолин-3-он;
транс-dl-8-хлоро-1,2,3,4,4a,5,6,10b-октагидробензо[f] хинолин-3-он;
транс-dl-8-этокси-1,2,3,4,4a,5,6,10b-октагидробензо[f] хинолин-3-он;
транс-dl-8-метокси-4-метил-1,2,3,4,4a, 5,6,10a-октагидробензо[f] хинолин-3-он;
транс-dl-8-метил-1,2,3,4,4a,5,6,10b-октагидробензо[f] хинолин-3-он;
транс-dl-1,2,3,4,4a, 5,6,10b-октагидробензо[f] хинолин-3-он;
транс-dl-8-этоксикарбонилэтандиил-4-метил-1,2,3,4,4a, 5,6,10b-октагидробензо[f]хинолин-3-он;
транс-dl-8-метоксикарбонилэтендиил-4-метил-1,2,3,4,4a,5,6, 10b-октагидробензо[f]хинолин-3-он;
транс-dl-8-карбоксиэтендиил-4-метил-1,2,5,4,4a, 5,6,10b-октагидробензо [f]хинолин-3-он;
транс-dl-8-т-бутиламинокарбонилэтендиил-4-метил-1,2,3,4,4a, 5 6,10b-октагидробензо[f]хинолин-3-он;
транс-dl-8-хлоро-2-( α -метил)-4-метил-1,2,3,4,4a,5,6, 10b-октагидробензо[f]хинолин-3-он;
транс-dl-8-хлоро-2-( β метил)-4-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-он;
транс-dl-8-бромо-6,6-диметил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-он;
транс-dl-8-бромо-4,6,6-триметил-1,2,3,4,4a,5,6, 10b-октагидробензо[f]хинолин-3-он;
цис-dl-8-бромо-4,6,6-триметил-1,2,3,4,4a, 5,6,10b-октагидробензо[f] хинолин-3-он;
транс-dl-8-т-бутил-1,2,3,4,4a,5,6,10b-октагидробензо[f] хинолин-3-он;
транс-dl-8-т-бутил-4-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-он;
транс-dl-8-фторо-4, 10b-диметил-1,2,3,4,4a,5,6, 10b-октагидробензо[f]хинолин-3-он;
цис-dl-8-фторо-4,10b-диметил-1,2,3,4,4a, 5,6,10b-октагидробензо[f] хинолин-3-он;
транс-dl-8-хлоро-4,10b-диметил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-он;
цис-dl-8-хлоро-4,10b-диметил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-он;
транс-dl-10b-метил-1,2,3,4,4a,5,6,10b-октагидробензо[f] хинолин-3-он;
транс-dl-4,10b-диметил-1,2,3,4,4a, 5,6,10b-октагидробензо[f] хинолин-3-он;
транс-dl-3-хлоро-10b-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-он;
цис-dl-8-хлоро-10b-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-он;
транс-dl-9-нитро-1,2,3,4,4a,5,6,10b-октагидробензо[f] хинолин-3-он;
транс-dl-9-нитро-4-метил-1,2,3,4,4a, 5,6,10b-октагидробензо[f] хинолин-3-он;
транс-dl-9-амино-4-метил-1,2,3,4,4a, 5,6,10b-октагидробензо[f] хинолин-3-он;
транс-dl-9-хлоро-4-метил-1,2,3,4,4a, 5,6,10b-октагидробензо[f] хинолин-3-он;
транс-dl-8-хлоро-3,4,4a,5,6,10b-гексагидробензо[f] хинолин-3-он;
dl-8-хлоро-2,3,4,4a,5,6-гексагидробензо[f]хинолин-3-он;
транс-dl-8-бромо-4-метил-3,4,4a,5,6,10b-гексагидробензо[f] хинолин-3-он;
транс-dl-8-хлоро, 4-метил-3,4,4a,5,6,10b-гексагидробензо[f] хинолин-3-он;
dl-8-хлоро-4-метил-2,3,4,4a,5,6-гексагидробензо[f] хинолин-3-он;
транс-dl-8-хлоро-2-( α -метил)-4-метил-1,2,3,4,4a, 10b-гексагидробензо[f]хинолин-3-он;
транс-dl-8-т-бутиламинокарбонилэтандиил-4-метил-1,2,3,4,4a, 5,6,10b-октагидробензо[f]хинолин-3-он;
транс-dl-8-фенил-4-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-он;
транс-dl-8-винил-4-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-он;
транс-dl-8-этоксикарбонил-4-метил-1,2,3,4,4a, 5,6,10b октагидробензо[f] хинолин-3-он.
Предпочтительными соединениями настоящего изобретения являются соединения формулы I, в которых:
R - водород или C1-C4 алкил;
Z и Z' - независимо водород или метил;
Y - водород или метил и находится в транс-конфигурации по отношению к водороду 4a положения;
R1, R3, R4 и R5 - водород;
R2 - водород или метил;
n - 1 или 2;
X - галоген, CF3, C1-C6 алкил, C1-C4 алкокси или -A-R6, где A - C1-C4 алкилен и R6 - C1-C4 алкоксикарбонил; или их фармацевтически приемлемая соль; при условии, что
(b) если R - водород, метил или этил, X - не водород или метокси; и
(c) если R - метил; R2 - не метил.
Наиболее предпочтительными соединениями настоящего изобретения являются те с формулой I, в которых
R - водород или метил;
Z и Z' - оба водород или метил;
Y - водород или метил и находится в транс-конфигурации по отношению к водороду 4a положения;
R1, R3, R4 и R5 -водород;
R2- водород или метил;
n - 1 или 2;
X - галоген, CF3, или C1-C4 алкил, или их фармацевтически приемлемая соль; при условии, что
(c) если R - метил, R2 - не метил.
Как упоминалось выше, изобретение включает фармацевтически приемлемые соли соединений, определенных упомянутой формулой. Хотя по сути нейтральное, конкретное соединение этого изобретения может иметь достаточно кислотную, достаточно основную или обе функциональные группы и соответственно реагировать с любым из ряда нетоксичных неорганических оснований и нетоксичных неорганических и органических кислот, чтобы образовать фармацевтически приемлемую соль. Кислотами, обычно используемыми для получения солей кислотного присоединения, являются неорганические кислоты, такие как хлористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, фосфорная кислота и прочие, и органические кислоты, такие как n-толуол сульфо, метан- сульфокислота, щавелевая кислота, n-бромо-фенил-сульфокислота, угольная кислота, янтарная кислота, лимонная кислота, бензойная, уксусная и прочие кислоты. Таким образом, примерами таких фармацевтически приемлемых солей являются сульфат, пиросульфат, бисульфат, сульфит, бисульфит, фосфат, моногидрогенфосфат, дигидрогенфосфат, метафосфат, пирофосфат, хлорид, бромид, йодид, ацетат, пропионат, деканоат, каприлат, акрилат, формат, изобутират, капроат, гептаноат, пропиолат, оксалат, малонат, сукцинат, суберат, себакат, фумарат, малеат, бутин-1,4-диоат, гексин-1,6-диоат, бензоат, хлорбензоат, метилбензоат, динитробензоат, гидроксибензоат, метоксибензоат, фталат, сульфонат, ксиленсульфонат, фенилацетат, фенилпропионат, фенилбутират, цитрат, лактат, гамма-гидроксибутират, гликоллят, тартрат, метансульфонат, пропансульфонат, нафталин-1-сульфонат, нафталин-2-сульфонат, манделят и прочие. Препочтительными фармацевтически приемлемыми солями кислотного присоединения являются те, которые формируются с минеральными кислотами, такими как хлористоводородная кислота и бромистоводородная кислота, и те, которые формируются с органическими кислотами, такими как малеиновая кислота и метансульфокислота.
Соли кислотного присоединения включают те, которые получают от нетоксичных неорганических оснований, таких как гидроксидов, карбонатов, бикарбонатов щелочных или щелочноземельных металлов или аммония и другие. Такие основания, полезные в получении солей этого изобретения, включают гидроксид натрия, гидроксид калия, гидроксид аммония, карбонат калия. Наиболее предпочтительны соли калия и натрия. Соединения настоящего изобретения получают с использованием известных специалистам процедур. Эти соединения настоящего изобретения, где Y - водород, предпочтительно синтезируются по схеме 1 (см. в конце текста).
Если X, n и R - как определены выше для формулы I, R-EAA - электрофильный алкилирующий агент и Ra и Rb - независимо выбраны из водорода и C1-C4 алкила или могут быть взяты вместе с атомом азота, чтобы получить 5-7 членную гетероциклическую группу, которая может также включать атом кислорода, при условии, что оба Ra и Rb не могут быть водородом в одно и то же время.
Как показано на синтетической схеме 1, Δ 4a-10b гексагидробензо[f]хинолиноны представляют собой промежуточные соединения, которые при восстановлении двойной связи дают соединения этого изобретения и/или соединения, полезные в качестве промежуточных соединений для получения соединений этого изобретения.
Гексагидробензохинолиноны получаются из незамещенного или замещенного в кольце фенацетилхлорида. Фенацетилхлорид коммерчески доступен или получается хорошо известными специалистам процедурами. Обычно, соответствующе замещенная фенилуксусная кислота реагирует с тионилхлоридом, фосфористым трихлоридом, оксалилхлоридом или фосфористым пентахлоридом, предпочтительно тионилхлоридом, при условиях, хорошо известных специалистам, чтобы получить соответствующий фенацетилхлорид.
Реакцией ациляции Friedel-crafts фенацетилхлорида с этиленом в присутствии кислотного катализатора Льюиса и в инертном или почти инертном растворителе или смеси растворителей достигается замыкание кольца в получении 2-тетралона. Подходящие кислотные катализаторы Льюиса включают AlBr3, AlCl3, CaCl3, FeCl3, SbCl5, ZrCl4, SnCl4, BCl3, BF3, SbCl3 и прочие, предпочтительно AlCl3. Растворители, полезные для этой реакции, включают карбон дисульфид, метиленхлорид, нитрометан, 1,2-дихлорэтан, нитробензол и другие, предпочтительно метиленхлорид. Активация фенацетилхлорида с кислотой Льюиса проводится при температурах от -78oC до 25oC.
Добавление этилена экзотермично по характеру и при использовании стандартных процедур охлаждения применяются температуры от -78oC до около 30oC.
Затем продукт реакции 2-тетралона аминируется первичным или вторичным амином, предпочтительно пирролидином, в инертном или почти инертном растворителе или смеси растворителей, чтобы получить соответствующий энамин. В случае первичного амина это может сопровождаться таутомером имина. Реакция завершается удалением воды, что может делаться при повышенных температурах от 80 до 110oC с использованием подходящего азеотропа растворителя или при комнатной температуре использованием подходящего обезвоживающего агента, такого как молекулярные сита или сульфат магния. Подходящими растворителями являются апротонные органические растворители, такие как бензол, толуол, ТГФ, CH2Cl2 и этилацетат.
Продукт реакции энамина затем реагирует с акриламидом в присутствии кислоты и в присутствии или отсутствии инертного или почти инертного растворителя или смеси растворителей, чтобы получить гексагидро-2-(1Н)-бензо[f] хинолинон. Кислоты в этой реакции включают сильные органические или минеральные кислоты, предпочтительно п-толуол сульфокислоту (пТСК). Хотя реакция может проводиться в растворителе, желательно его не использовать. Реакция проводится при температурах от 90oC до около 130oC.
Затем гексагидро-2(1Н)-бензо[f] хинолинон может восстанавливаться до соответствующих октагидробензо[f] хинолинонов настоящего изобретения. Далее октагидробензо[f]хинолиноны могут N-алкилироваться для получения последующих соединений настоящего изобретения.
Как вариант, гексагидро-2(1Н)-бензо[f]хинолиноны вначале могут N-алкилироваться, а затем восстанавливаться до соответствующих N-алкил-октагидробензо[f]хинолин(3)-онов настоящего изобретения.
Восстановление проводится реакцией гексагидробензо[f] хинолинона или N-алкил-гексагидробензо[f] хинолинона с соответствующим восстановительным агентом в инертном или почти инертном растворителе или смеси растворителей. Подходящие восстановительные агенты включают гидрогенизацию над металлическим катализатором и реагенты переноса гидрида, такие как формат аммония над катализатором металла, предпочтительно триэтилсилан/трифторуксусная кислота. Подходящие растворители включают инертные или почти инертные органические растворители, предпочтительно метиленхлорид. Используются температуры от 0oC до 69oC, предпочтительно около 25oC.
N-алкилирование проводится реакцией гексагидробензо[f] хинолинона или октагидробензо[f] хинолинона с электрофильным алкилирующим агентом, R-EAA, где R - как определено выше для формулы 1, в присутствии основания в инертном или почти инертном растворителе или смеси растворителей. Для этой реакции EAA - предпочтительно йодо. Основание - обычно гидрид металла, амид или алкоксид металла, предпочтительно гидрид натрия. Обычно эта реакция проводится при температурах от -30oC до температуры нагревания с обратным холодильником растворителя.
Эти соединения настоящего изобретения, где Y - метил, предпочтительно синтезируются по схеме 2 (см. в конце текста), где X, n и R - как определены выше для формулы I, и Ra и Rb и R-EAA - как определены выше для схемы 1.
Как показано на схеме 2, 4a-метил-гексагидробензо[f] хинолин-2(1Н)-он является промежуточным соединением, который восстанавливается для получения соединений настоящего изобретения и/или соединений, полезных в качестве промежуточных соединений для получения соединений этого изобретения.
Энамин готовится процедурами, показанными на схемах 1 и 2 и описанными выше для синтетической схемы 1. Энамин алкилируется реакцией с электрофильным алкилирующим агентом, предпочтительно метилйодидом в инертном или почти инертном растворителе или смеси растворителей. Температуры для этой реакции обычно от 0oC до 60oC. Реакционная смесь затем подвергается гидролизу с водной кислотой, предпочтительно смесью ацетата натрия, этилацетата и уксусной кислоты. Для получения 1-метил-2-тетралона в этой реакции используются температуры от 0oC до около 30oC.
Далее 1-метил-2-тетралон реагирует, как показано на схеме 2 с использованием реагентов и процедур, описанных выше для схемы 1, с тем, чтобы получить соединения настоящего изобретения, где Y - метил.
Эти соединения настоящего изобретения, где Z, Z' или оба - C1-C4 алкил, готовятся в соответствии с процедурами по схеме 1 и схеме 2, за исключением того, что в замыкании кольца Friedel-crafts используется реакция фенацетилхлорида, соответствующего алкена, а не этилена; показанного на обеих схемах 1 и 2. Примеры подходящих алкенов для использования в этой реакции включают пропилен, 1-бутен, изо-бутилен, 3,3-диметил-1-бутен, 2-пентен, 4-метил-2-пентен, 3-метил-1-бутен, 2-метил-2-бутен, 2,3-ди-метил-2-бутен и другие.
Эти соединения формулы I, где R2 - C1-C4 алкил, готовятся из соединений, полученных по схемам 1 и 2, предпочтительно, где R - C1-C4 алкил, незамещенный или замещенный фен (C1-C4) алкил, как показано на следующей схеме 3 реакции: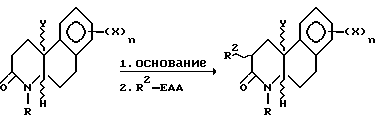
где Y, X, n, R и R2 - как описаны выше для формулы I, за исключением того, что R - не водород, и R2-EAA электрофильный алкилирующий агент, где R2 - как определено выше для схемы 1. R-(алкил или феналкил) соединение реагирует с основанием, таким как амид металла или алкоксид металла, предпочтительно гексаметилдисилазид калия, в инертном или почти инертном растворителе для смеси растворителей при температуре от -78oC до около 25oC. Затем алкилирование осуществляется добавлением соответствующего электрофильного алкилирующего агента, предпочтительно C1-C4 алкилйодида, чтобы получить 2-(C1-C4 алкил) соединения формулы I.
Для этих соединений формулы I, где R - H, атом азота 4- положения сначала блокируется подходящей амино защитной группой, такой как т-бутоксикарбонил или бензилоксикарбонил, и затем реагирует, как показано выше, на схеме 3. После алкилирования в 2-положении атом азота 4-положения - не защищен. Реакции защиты и разблокирования защиты проводятся при стандартных условиях для таких реакций.
Альтернативный метод получения этих соединений настоящего изобретения, где Y - метил, показан ниже на схеме 4 (см. в конце текста), где R, X и n - как определены выше для формулы I.
Другой аспект этого изобретения относится к конкретному способу получения оптически активных изомеров соединений этого изобретения. Как показано выше на схеме 4, 1-метил-2-тетралон реагирует с хиральным амином, таким как 1-фенилэтиламин в инертном или почти инертном растворителе или смеси растворителей для получения соответствующего энамина. Если используется первичный хиральный амин, энамин можно получить посредством таутомера имина. Реакция проводится удалением воды, что можно сделать при повышенных температурах от 80oC до 110oC с использованием подходящего азеотропа растворителя или при комнатной температуре использованием подходящего обезвоживающего агента, такого как молекулярные сита или сульфат магния.
Энамин затем реагирует с подходящим альфа, бета-ненасыщенным карбониловым соединением, предпочтительно метилвинилкетоном в реакции присоединения michael с последующим гидролизом мягкой водной кислотой, чтобы получить 5, 6, 7, 8, 9, 10-гексагидро-8-гидрокси-5,8-диметил-5,9-метанобензо- циклооктан-11-он. Эта реакция проводится в эфирном растворителе, таком как ТГФ, диоксан или другие в инертной атмосфере, такой как аргон или азот при температуре от 10oC до 50oC, предпочтительно при комнатной температуре. Альфа, бета-ненасыщенное карбониловое соединение в этой реакции используется от стехеометрических количеств реактантов до избытка с предпочтением избытка. Подходящие кислоты включают органические карбоновые кислоты и хлорную кислоту, предпочтительно уксусную кислоту.
Метанобензоциклооктан-II-он обрабатывается кислотным или основным катализатором, предпочтительно этоксидом натрия или калия в протонном растворителе, предпочтительно этаноле при нагревании с обратным холодильником для получения 2,3,4,4a,9,10-гексагидро-4a-метил-фенантрен-2-она.
Фенантрен-2-он окислительно расщепляется подходящим окислительным агентом, таким как озон, KMnO4, CrO3 или предпочтительно тетраокисью рутения в инертном или почти инертном растворителе или смеси растворителей от -78oC до около 100oC, предпочтительно от -10oC до около 10oC, чтобы получить 6-[1-метил-1-/2-оксо-1,2,3,4-тетрагидронафтил/] пропионовую кислоту. Обычно растворитель будет инертным или смесью растворителей, а предпочтительно смесью из 2 частей тетрахлорметана, 3 частей ацетонитрила и 2 частей воды.
Пропионовая кислота реагирует с аммиаком или первичным амином (NHR, где R - как определено выше для формулы I) в инертном или почти инертном растворителе или смеси растворителей, предпочтительно 2-пропаноле при температуре от 95oC до 200oC, предпочтительно от 165oC до 180oC, чтобы получить 10b-метил-1,2,3,4,6,10b-гексагидробензо[f] хинолин-3-он настоящего изобретения. Предпочтительно эта реакция проводится при относительном отсутствии окисляющих агентов, таких как воздух, в герметичном реакторе или в чем-либо в этом роде.
Гексагидробензо[f]хинолинон можно восстановить до соответствующего октагидросоединения настоящего изобретения практически теми же процедурами, которые описаны выше на схемах 1 и 2.
Следуя процедурам на схеме 4 и описанным выше, получают практически чистые оптически активные изомеры соединений этого изобретения, где Y - метил.
Асимметричный синтез отдельных энантиомерных соединений формулы I или их предшественников проводится реакцией энамина формулы
Схема 5
с производным акрилоила формулы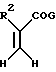
где Y, X, R2 и n - как определены выше для формулы I; C - уходящая группа, такая как хлор, бром, фтор, йод, толуолсульфонат, метансульфонат и симметричные или асимметричные ангидриды; и Rc - 1-фенэтил. 1-Фенэтил заместитель в последующем расщепляется с использованием трифторуксусной кислоты. -COG- радикал акрилоил производного является активированной формой -COOH, который можно активировать другими способами, такими как активными эфирами, смешанными ангидридами и другими.
Условия способа для проведения синтеза отдельного изомера на схемах чрезвычайно мягкие. В большинстве случаев было выявлено, что хороших выходов можно достичь в краткие периоды времени при температурах диапазона окружающей среды. Например, используются температуры от 0oC до 150oC и время реакции в пределах от нескольких минут до нескольких часов, максимум, является достаточным. Среда реакции - двухфазная смесь подходящего органического растворителя и водного раствора мягкого основания. Полезные растворители включают, например, галоалканы, эфиры, включающие тетрагидрофуран, и нитрилы, включающие ацетонитрил. Предпочтительными мягкими основаниями являются карбонаты и бикарбонаты щелочных металлов; более высокоосновные реагенты, такие как гидроксиды щелочных и щелочноземельных металлов и другие могут использоваться в некоторых случаях, но бикарбонаты предпочтительнее. При желании процесс может проводиться без основания.
Продукты этого синтеза легко изолируются обычными этапами обработки. Использование этого способа обеспечивает особо чистый синтез форм продукта реакции с одним изомером.
Надо понимать, что продукты настоящего способа могут использоваться как таковые, учитывая их биологическую активность, или их можно использовать как промежуточные соединения в дополнительных способах, чтобы получить активные соединения в объеме формулы I.
Гексагидробензо[f] хинолин-3-оны формулы I, имеющие Δ1 или Δ5 двойную связь углерод-углерод, готовятся из соответствующих октагидробензо[f]хинолин-3-онов реакциями присоединения/отщепления. Октагидробензо[f]хинолин-3-он реагирует с серой или селено электрофилом в присутствии основания в апротонном растворителе. Основание представляет собой обычно гидрид металла или амид металла, предпочтительно гидрид металла, такой как гидрид натрия. Хотя на каждый эквивалент октагидробензо[f]хинолина добавляется обычно один эквивалент основания, для тех соединений, где R - водород, добавляется второй эквивалент основания. Температуры для этой реакции от 20oC до температуры нагревания с обратным холодильником растворителя. Реагентом присоединения является сера или селено электрофил и проводится при температуре от -50oC до -100oC. Подходящие электрофилы серы подобны группам серы, используемым в нуцелофильном замещении и известны специалистам, Patai, "The Chemistry of the Thiol Group", Wiley, New York (1974); Reid, "Organic Chemistry of Bivalent Sulfur", Chemical publishing company, New York (1958, 1963); Kharasch, "Organic Sulfur Compounds", Perganon, New York (1961).
Подходящие селено соединения включают фенилселеннилхлорид, фенилселеннилбромид, N -(фенилселено)фталимид, дифенил диселенид, бензолселениновый ангидрид и селеноксиды. Специфические условия для определенного селено реагента хорошо известны, Clive, Tetrahedron, 34, 1049-1132 (1978) Aldrichimica Acta, 11, 43-49 (1978); и Miyoshi, et al., Tetrahedron Lett., 23, 4813 (1982).
Реакция отщепления обычно проводится при окислительных условиях в апротонном растворителе. March, "Advanced Organic Chemistry 3, изд, стр. 912-914, Wiley-Inter-science, N.Y.(1985).
Δ10b соединения настоящего изобретения получаются перегруппировкой (изомеризацией) из соответствующих Δ1 соединений, если Y - водород. Эта реакция проводится в апротонном растворителе в присутствии кислотного или основного катализатора при условиях, хорошо известных специалистам.
Δ4a соединения настоящего изобретения готовятся как промежуточные соединения процедурами, описанными выше на схеме 4, и изолируются, а не восстанавливаются до соответствующего октагидробензо[f]хинолин-3-она.
Оптически активные изомеры рацематов изобретения также считаются частью этого изобретения. Такие оптически активные изомеры можно получить из их соответствующих оптически активных предшественников процедурами, описанными выше или расщеплением рацемических смесей. Это расщепление можно проводить дериватизацией хиральным реагентом с последующей хроматографией или повторной кристаллизацией. Удаление хирального посредника стандартными методами позволяет получить по сути оптически чистые изомеры соединений настоящего изобретения или их предшественников. Дальнейшие подробности относительно расщепления можно получить из Jacques, et al., Enantiomers, Racemates and Resolutions, John Wiley & Sons, 1981.
Другой аспект настоящего изобретения и предпочтительный способ расщепления рацематов этих соединений формулы 1, где R - водород или C1-C4 алкил; Z и Z' - независимо выбраны из водорода и C1-C4 алкила; Y - водород; R1, R2, R3, R4 и R5 - все водород; n - 1 или 2; и X - водород, галоген, NO2, CF3, C1-C6 алкил, C1-C4 алкокси, амино, C1-C4 алкиламино, C1-C4 диалкиламино, меркапто или C1-C6-алкилтио;
на их компонентные оптические изомеры, включающий этапы:
(a) контактирования раствора метанола рацемата с сильной кислотой, чтобы получить 1-(2-метоксикарбонилэтил)-2-(амино)- 1,2,3,4-тетрагидронафталин;
(b) контактирования указанного тетрагидронафталина из (a) с раствором метанола оптически активной ди-п-толуоилвинокаменной кислоты, чтобы получить соответствующую соль тетрагидронафталина; и
(c) обработки указанной соли из (b) основанием, чтобы получить оптически активный изомер.
Как еще один аспект настоящего изобретения в дополнение к способу расщепления рацемических смесей этих соединений формулы I предусмотрено получение ди-п-толуоил-(D)- и (L) солей винокаменной кислоты 1-(2-метоксикарбонилэтил)-2-(амино или C1-C4 алкиламино)-1,2,3,4-тетрагидронафталина.
Это расщепление осуществляется растворением рацемической смеси оптически активных изомеров, как определено выше, в метаноле и контактированием указанного раствора с сильной кислотой, чтобы получить промежуточное соединение 1-(2-метоксикарбонилэтил)-2-(амино или C1-C4 алкиламино)-1,2,3,4-тетрагидронафталина. Подходящие сильные кислоты включают неорганические кислоты, такие как хлористоводородная кислота, азотная кислота, фосфорная, серная, бромистоводородная, йодистоводородная кислоты и другие, а также органические кислоты, такие как ароматические сульфокислоты и другие. Неорганические кислоты предпочтительны и наиболее предпочтительна серная кислота. Затем промежуточное тетрагидронафталина контактируется с раствором метанола оптически активной ди-п-толуоилвинокаменной кислоты, чтобы получить соответствующую соль тетрагидронафталина. Там, где желателен (+) энантиомер, используется (-)-ди-п-толуоил-L-винокаменная кислота. Соответственно, там, где желателен (-) изомер, используется (+)-ди-п-толуоил-D-винокаменная кислота.
Полученную соль можно отделить от смеси обычными способами. Например, отделенную соль можно обработать в водной среде основанием, чтобы сформировать свободный амин, который можно экстрагировать из водной фазы водонесмешиваемым растворителем. Свободный амин можно нагреть до 35-120oC для рециркуляции и получить нужный октагидробензо[f] хинолинон, в зависимости от используемого экстрагирующего растворителя.
Подходящими основаниями для использования в упомянутом способе являются обычно слабые основания, предпочтительно карбонат или бикарбонат калия или натрия, а лучше бикарбонат натрия. Подходящие водонесмешиваемые растворители включают метиленхлорид, толуол, этилацетат, метилтрет-бутиловый эфир и диэтиловый эфир, лучше метиленхлорид.
Специалист знает, что селективная кристаллизация одного диастереомера из органического раствора также проводится концентрацией. Относительно низкая концентрация дает чистый диастереомер более высокой чистоты, но с низким выходом, а более высокая концентрация рацемата и расщепляющего агента обычно дает более высокие выходы твердого вещества во многом за счет оптической чистоты.
Соединения, используемые в качестве первоначальных исходных материалов в синтезе соединений этого изобретения, хорошо известны и легко синтезируются стандартными процедурами, известными специалистам.
Фармацевтически приемлемые соли изобретения обычно формируются реакцией октагидробензо[f] хинолинона этого изобретения, который обладает подходящей кислотной или основной функциональностью, с эквимолярным или избыточным количеством кислоты или основания. Реагенты обычно объединяются в растворитель, такой как диэтиловый эфир или бензол для солей кислотного присоединения, или воду или спирты для солей основного присоединения, и соль обычно выпадает из раствора в течение от 1 часа до 10 дней, и может быть изолирована фильтрацией или другими известными средствами.
Кроме того, некоторые из соединений настоящего изобретения могут формировать сольваты с водой или обычными органическими растворителями. Такие сольваты включены как соединения этого изобретения.
Следующие примеры далее иллюстрируют соединения настоящего изобретения и способы их синтеза. Примеры никоим образом не ограничивают объем изобретения и не должны быть так истолкованы.
Пример 1
Получение цис-dl и транс-dl-8-бромо-4-метил-1,2,3,4, 4a,5,6,10b-октагидробензо[f]хинолин-3-она
A. 4-Бромофенилацетилхлорид
В 250 мл колбу с круглым дном, снабженную магнитной мешалкой, добавлялась 4-бромофенилуксусная кислота (100,0 г; 0,465 M) и 100 мл тионилхлорида (163,1 г; 1,37 моль). Полученная суспензия перемешивалась при комнатной температуре 22,5 часа. Избыток тионилхлорида выпаривался в вакууме для получения 108,5 г соединения A в виде жидкости коричневого цвета.
B. 6-Бромо-2-тетралон
К холодной (-78oC; сухой лед/изопропаноловая ванна) суспензии AlCl3 (125 г; 0,94 моль) в 1,400 мл CH2Cl2 был добавлен кислотный хлорид, полученный на этапе A (108,5 г; 0,47 моль), растворенный в 400 мл сухого CH2Cl2 с перемешиванием в течение 1 часа. Ванна сухой лед/изопропанол удалялась и раствор согревался до -10oC. В колбу пропускался пузырьками этилен с энергичным взбалтыванием. Реакция согревалась изотермически до 20oC и добавление этилена прекращалось. Смесь перемешивалась при комнатной температуре 3 часа, затем охлаждалась до 0oC и добавлялся лед до тех пор, пока не наблюдалось экзотерма. Реакционная смесь разбавлялась 1 л холодной ледяной воды и перемешивалась до растворения всех твердых веществ. Полученные слои разделялись и органический слой промывался дважды порциями в 1 л 1 NHCl и затем единожды 1 л насыщенной Na2HCO4. Органический слой высушивался над Na2SO4 и концентрировался в вакууме до получения бледно-желтого кристаллического вещества.
6-Бромо-2-тетралоновые кристаллы забирались минимальным количеством эфира. Осторожно добавлялся гексан по начала помутнения раствора. Смесь охлаждалась 4 часа, фильтровалась и промывалась холодным гексаном для получения 75,6 г соединения B в виде бледно-желтого кристаллического вещества (выход 71%) с точкой плавления 71-73oC.
C. 2-Пирролидинил-6-бромо-3,4-дигидронафталин
В 250 мл колбу с круглым дном добавлялось 5,00 г (22,21 мМ) 6-бромотетралона, полученного на этапе B; 70 мл сухого толуола и 3,1 г (3,7 мл) пирролидина. Колба снабжалась ловушкой Dean-Stark, конденсатором, входной трубкой для азота и магнитной мешалкой и реакция проводилась нагреванием с обратным холодильником 4 часа. Растворитель выпаривался в вакууме для получения 6,0 г (97,4%) искомого соединения в виде коричневого кристаллического материала, который использовался без дальнейшей очистки.
D. 8-Бромо-1,2,3,4,5,6-гексагидробензо[f]хинолин-3-он
Энамин (2,15 г, 7,73 мМ) из этапа C, акриламид (1,10 г, 15,46 мМ) и 100 мг п-толуол сульфокислоты (пТСК) тщательно смешивались в ступке с пестиком. Смесь переносилась в 250 мл колбу с круглым дном, снабженную магнитной мешалкой и входом для азота. С использованием бани из минерального масла смесь нагревалась до 89oC, при этом перемешанная смесь становилась черной и плавилась. Температура поддерживалась постоянной при 89oC 1,5 часа. Далее она поднималась до 130oC и выдерживалась такой 0,5 часа. Масляная баня удалялась и осторожно добавлялось 60 мл воды. Полученный темно-серый материал тщательно перемешивался шпателем и для фильтрации добавлялось 80 мл воды. После фильтрации оставались кристаллы коричневого цвета. Кристаллы забирались в CHCl3 и добавлялся активированный уголь. Эта смесь перемешивалась 15 минут, фильтровалась и выпаривалась в вакууме. Остаток забирался в минимальное количество этилацетата с помощью паровой бани и переносился в колбу Erlenmeyer, снабженную магнитной перемешивающей пластиной и погруженную в ванну сухой лед/ацетон с перемешиванием, чтобы получить искомое соединение в виде кристаллического вещества белого цвета (точка плавления 215-217 разложение). 1й сбор 940 мг; 2й сбор 175 мг (выход 55%).
E. 8-Бромо-4-метил-1,2,3,4,5,6-гексагидробензо[f]хинолинон-3-он
Следуя процедурам, описанным выше, было получено 5,17 г 8-бромо-1,2,3,4,5,6-гексагидробензо[f] хинолин-3-она. Гексагидробензохинолинон (5,17 г, 19,6 мМ) растворялся в 60 мл сухого диэтилового эфира в 250-милилитровой колбе с круглым дном. К раствору добавлялось 1,2 г гидрида натрия (60% дисперсия в минеральном масле). Колба прилаживалась к конденсатору нагревания с обратным холодильником с перемешивающей пластиной и смесь нагревалась с обратным холодильником 2 часа. Затем она охлаждалась до комнатной температуры и добавлялось 7,35 мл метилйодида. После добавления реакционная смесь нагревалась с обратным холодильником еще 3 часа. После охлаждения она быстро остужалась осторожным добавлением 5 мл воды. Затем смесь концентрировалась в вакууме с получением бледных кристаллов, которые забирались в смесь этилацетат/вода и полученные слои разделялись. Органический слой дважды промывался водой и единожды рассолом и затем высушивался над MgSO4 и выпаривался в вакууме для получения 5,22 г кристаллического вещества желтого цвета. Вещества рекристаллизовалось из ацетона для получения 3,55 г (62%) искомого соединения в виде вещества бледно-желтого цвета. Точка плавления 126-128oC.
F. 8-Бромо-4-метил-1,2,3,4,4a,5,6,10b-октагидробензо[f] хинолин-3-он
К раствору гексагидробензохинолинона, полученного выше в Этапе E (1,17 г; 4,0 мМ), в 10 мл сухого дихлорметана был добавлен триэтилсилан (1,37 г; 11,8 мМ). Полученная смесь перемешивалась 10 минут при комнатной температуре. Реакционная смесь охлаждалась в ледяной ванне и добавлялась трифторуксусная кислота (5 мл). Полученная смесь перемешивалась при комнатной температуре 4 дня. Реакционная смесь концентрировалась в вакууме. Масляный остаток забирался в CH2Cl2 и промывался насыщенным NaHCO3. Органический слой высушивался над сульфатом натрия и концентрировался в вакууме для получения масла оранжевого цвета. Мгновенная хроматография на SiO2 (элюирование 0,5% метанолом/CH2Cl2) дала 1,14 г масла светло-коричневого цвета. Протонная ЯМР спектроскопия выявила соотношение транс:цис как 3,2:1.
G. Цис-dl и транс-dl-8-бромо-4-метил-1,2,3,4,4a, 5,6,10b-октагидробензо[f]хинолин-3-он
Цис и транс изомеры разделялись ВЭЖХ на SiO2 (в гексане с увеличиващимся градиентом этилацетата). Транс изомер (пример 1A) выделился первым в количестве 631 мг; и цис изомер (пример 1B) вторым в количестве 192 мг. Транс изомер рекристаллизовался из диэтил эфиро/гексанов для получения 176 мг; точка плавления 103-104,5oC.
Элементарный анализ:
Транс (1A)
Рассчитано: C 57,16 H 5,48. N 4,76.
Получено: C 57,57; H 5,53; N 4,64.
Цис (1B)
Рассчитано: C 57,16; H 5,48; N 4,76.
Получено: C 57,46; H 5,67; N 4,59.
Пример 2
Получение транс-dl-8-бромо-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-она
Соединение 8-бромо-1,2,3,4,5,6-гексагидробензо[f]хинолин-3-она было получено в соответствии с процедурами, описанными в примере 1, этапах A, B, C и D.
Искомое соединение было получено в соответствии с процедурой, описанной выше в примере 1, этапе в количестве 84 мг белого кристаллического материала (выход 29% с последующей рекристаллизацией из этилацетата; точка плавления 252-254oC разл.
Элементарный анализ
Рассчитано: C 55,73; H 5,04; N 5,00.
Получено: C 55,46; H 5,07; N 4,89.
Пример 3
Получение транс-dl-8-йодо-4-метил-1,2,3,4,4a,5,6, 10b-октагидробензо[f] -хинолин-3-она
Соединение транс-dl-8-бромо-4-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f] хинолин-3-она было получено в соответствии с процедурами, описанными в примере 1, этапах A, B, C, D, E, F и G.
К перемешанному раствору транс изомера (475 мг; 1,614 мМ) в 3,5 мл сухого диоксана был добавлен гексаметилдитин и 54 мг (3 моль%/ тетра-кис/трифенилфосфин)палладия. Реакционная смесь нагревалась с обратным холодильником 2,5 часа, охлаждалась до комнатной температуры, фильтровалась через (Celite) и концентрировалась до получения масла светло-желтого цвета. Материал далее концентрировался в высоком вакууме всю ночь при комнатной температуре, чтобы получить 677 мг соответствующего 8-триметилтин соединения в виде масла бледно-желтого цвета, которое использовалось без дальнейшей очистки.
К холодному раствору соединения 8-триметилтина, полученного выше, в 5,0 мл CH2Cl2 по каплям добавлялось 1,6 мл 1,0 М йодинмонохлорида. Реакционная смесь согревалась до комнатной температуры на протяжении 1,5 часов. Смесь остужалась 1 мл воды, фильтровалась, и летучие вещества выпаривались в вакууме, чтобы получить маслянистый материал черного цвета. Этот материал был подвергнут хроматографии на -SiO2 (с элюированием 5% изопропанолом/ CH2Cl2) с получением желтого кристаллического вещества, которое рекристаллизовалось из этилацетат/гексана для получения 141 мг искомого соединения (выход 86%) в виде белого кристаллического материала; точка плавления: 103-104,5oC.
Элементарный анализ:
Рассчитано: C 49,28; H 4,73; N 4,11.
Получено: C 49,48; H 4,72; N 3,96.
Пример 4
Получение транс-dl-8,9-дихлоро-1,2,3,4,4a, 5,6,10b октагидробензо[f]хинолин-3-она
Искомое соединение было получено с использованием 3,4-дихлорфенилуксусной кислоты в качестве исходного материала по процедуре, описанной выше в примере 1, этапах A, B, C, D и F в количестве 567 мг в виде белого кристаллического материала. Точка плавления 267-268oC, разложение.
Элементарный анализ:
Рассчитано: C 57,80; H 4,85; N 5,18.
Получено: C 58,22; H 5,04; N 5,18
Пример 5
Получение транс-dl-8,9-дихлоро-4-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-она
Следуя процедурам, описанным выше в примере 1, этапах A, B, C, D и F с последующей рекристаллизацией из этилацетата был получен транс-dl-8,9-дихлоро-1,2,3,4,4a, 5,6,10b-октагидробензо[f] хинолин-3-он с использованием 3,4-дихлорфенилуксусной кислоты в качестве исходного материала.
Искомое соединение было получено из транс-dl-8,9-дихлороктагидробензо[f] хинолинона по процедурам, описанным в примере 1, этапе E в количестве 117 мг (выход 35%) в виде твердого вещества бежевого цвета. Точка плавления 168-169oC.
Элементарный анализ:
Рассчитано: C 59,17; H 5,32; N 4,93.
Получено: C 59,45; H 5,08; N 4,83.
Пример 6
Получение транс-dl-8-хлоро-4-метил-1,2,3,4,4a,5,6,10b-октагидробензо[f] хинолин-3-она
Искомое соединение вместе с цис-dl-изомером (пример 7) было получено по процедурам, описанным в примере 1, этапах A, B, C, D, E и F с использованием п-хлорфенилуксусной кислоты в качестве исходного материала. Отделение по примеру 1, этап C дало 500 мг искомого соединения. Точка плавления 82oC.
Элементарный анализ:
Рассчитано: C 67,33; H 6,46; N 5,61.
Получено: C 67,60; H 6,63; N 5,67.
Пример 7
Получение цис-dl-8-хлоро-4-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f] хинолин-3-она
Искомое соединение вместе с транс-dl-изомером (пример 6) было получено по процедурам примера 1, этапы A, B, C, D, E и F с использованием п-хлорфенилуксусной кислоты в качестве исходного материала. Отделение по примеру 1, этап C дало 200 мг искомого соединения в виде масла.
Элементарный анализ:
Рассчитано: C 67,33; H 6,46; N 5,61.
Получено: C 67,57; H 6,82; N 5,70.
Пример 8
Получение транс-dl-4,8-диметил-1,2,3,4,4a,5,6, 10b-октагидробензо[f]хинолин-3-она
Искомое соединение вместе с цис-dl-изомером (пример 9) было получено по процедурам примера 1, этапы A, B, C, D, E и F с использованием п-хлорфенилуксусной кислоты в качестве исходного материала. Отделение по примеру 1, этап C дало 400 мг искомого соединения. Точка плавления 115-116oC.
Элементарный анализ:
Рассчитано: C 78,56; H 8,35; N 6,11.
Получено: C 78,79; H 8,32; N 6,11.
Пример 9
Получение цис-dl-4,8-диметил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-она
Искомое соединение вместе с транс-dl-изомером (пример 8) было получено в соответствии с процедурами, описанными в примере 1, этапах A, B, C, D, E и F с использованием п-толилуксусной кислоты в качестве исходного материала. Отделение по примеру 1, этап C дало 290 мг искомого соединения. Точка плавления 78oC.
Элементарный анализ:
Рассчитано: C 78,56; H 8,35; N 6,11.
Получено: C 78,26; H 8,56; N 5,87.
Пример 10
Получение транс-dl-8-фторо-4-метил-1,2,3,4,4a,5,6, 10b-октагидробензо[f] хинолин-3-она
Искомое соединение вместе с цис-dl-изомером (пример 11) было получено по процедурам примера 1, этапы A, B, C, D, E и F с использованием п-фторофенилуксусной кислоты в качестве исходного материала. Отделение по примеру 1, этап C дало 244 мг искомого соединения. Точка плавления 108-109oC.
Элементарный анализ:
Рассчитано: C 72,80; H 6,91; N 6,00.
Получено: C 72,07; H 6,89; N 6,09.
Пример 11
Получение цис-dl-8-фторо-4-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f] хинолин-3-она
Искомое соединение вместе с транс-dl-изомером (пример 10) было получено по процедурам, описанным в примере 1, этапах A, B, C, D, E и F с использованием п-фторофенилуксусной кислоты в качестве исходного материала. Отделение по примеру 1, этап C дало 130 мг искомого соединения. Точка плавления 136-137oC.
Элементарный анализ:
Рассчитано: C 72,08; H 6,91; N 6,00.
Получено: C 72,30; H 7,04; N 6,06.
Пример 12
Получение транс-dl-4-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-она
Искомое соединение вместе с цис-dl-изомером (пример 13) было получено по процедурам примера 1, этапы A, B, C, D, E и F с использованием фенилуксусной кислоты в качестве исходного материала. Отделение по примеру 1, этап C дало 200 мг искомого соединения. Точка плавления 128-129oC
Элементарный анализ:
Рассчитано: C 78,10; H 7,96; N 6,51.
Получено: C 77,87; H 7,85; N 6,46.
Пример 13
Получение цис-dl-4-метил-1,2,3,4,4a,5,6, 10b-октагидробензо[f]хинолин-3-она
Искомое соединение вместе с транс-dl-изомером (пример 12) было получено по процедурам, описанным в примере 1, этапах A, B, C, D, E и F с использованием фенилуксусной кислоты в качестве исходного материала. Отделение по примеру 1, этап C дало искомое соединение. Точка плавления 129-130oC
Элементарный анализ:
Рассчитано: C 78,10; H 7,96; N 6,51.
Получено: C 78,32; H 7,04; N 6,58.
Пример 14
Получение цис-dl-1,2,3,4,4a,5,6, 10b-октагидробензо[f]хинолин-3-она
Соединение 1,2,3,4,5,6-гексагидробензо[f] хинолин-3-он было получено по процедурам, описанным в примере 1, этапах A, B, C, и D с использованием фенилуксусной кислоты в качестве исходного материала.
К 94 мл уксусной кислоты добавлялся 1,2,3,4,5,6-гексагидробензо [f]хинолин-3-он (3 г; 15 мМ) и 3 г 5% палладия на активированном угле. Смесь выстаивалась при комнатной температуре три дня при первоначальном давлении водорода 60 фунт/дюйм2 (4,21 кг/см2). Катализатор удалялся фильтрацией. Фильтрат разбавлялся этилацетатом и делался основным насыщенным NaHCO3. Полученные слои разделялись и органический слой высушивался над MgSO4 и концентрировался до получения 1,4 г (выход 46%) искомого соединения. Точка плавления 178-179oC.
Элементарный анализ:
Рассчитано: C 77,58; H 7,51; N 6,96.
Получено: C 77,88; H 7,52; N 7,05.
Пример 15
Получение транс-dl-8-фторо-1,2,3,4,4a,5,6, 10b-октагидробензо[f]хинолин-3-она
Искомое соединение было получено по процедурам примера 1, этапы A, B, C, D, E, F и G с использованием п-фторофенилуксусной кислоты в качестве исходного материала для получения 14,2 мг искомого соединения. Точка плавления 262-263oC.
Элементарный анализ:
Рассчитано: C 71,21; H 6,44; N 6,39.
Получено: C 71,17; H 6,48; N 6,29.
Пример 16
Получение транс-dl-8-этоксикарбонил-этендиил-4-метил- 1,2,3,4,4a, 5,6,10b-октагидробензо[f]хинолин-3-она
Соединение транс-dl-8-бромо-4-метил-1,2,3,4,4a, 5,6,10b- октагидробензо[f]хинолин-3-он было получено по процедурам, описанным в примере 1, этапах A, B, C, D, E, F и G. Это соединение (1,52 г, 5,17 мМ) и ацетат палладия (11) (12 мг, 0,052 мМ), три-(о-толил)фосфин (64 мг, 0,28 мМ), этилакрилат (674 мг, 6,46 мМ) и триэтиламин (2,8 мл) соединялись в толстостенной трубке, снабженной магнитной пластиной, покрытой тефлоном и перемешивались. Реакционная смесь нагревалась до 100oC в герметичной трубке и выдерживалась там всю ночь. После охлаждения добавлялась IN HCl и твердое вещество зеленого цвета аккуратно перемешивалось шпателем. Твердые вещества собирались фильтрацией и растворялись в этаноле с нагреванием. Раствор фильтровался через кизельгур (Celite®) и промывался несколько раз этанолом. Летучие вещества выпаривались в вакууме до получения твердого остатка желтого цвета. Рекристаллизация остатка из смеси этилацетат/гексаны дала 1,24 г искомого соединения в виде рыхлого материала желтого цвета (88% выход). Точка плавления 115,5-116,5oC.
Масс-спектр с высоким разрешением: 313,1659 C19H43NO3.
Пример 17
Получение транс-dl-8-хлоро-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-она
Искомое соединение было получено в соответствии с процедурами, описанными в примере 1, этапах A, B, C, D, F и G с использованием п-хлорофенилуксусной кислоты в качестве исходного материала. Точка плавления 231-232oC.
Элементарный анализ:
Рассчитано: C 66,24; H 5,97; N 5,94.
Получено: C 66,44; H 6,17; N 6,06.
Пример 18
Получение транс-dl-8-метокси-1,2,3,4,4a,5,6, 10b-октагидробензо[f]хинолин-3-она
Искомое соединение было получено по процедурам примера 1, этапы A, B, C, D и F с последующей рекристаллизацией из этилацетата с использованием п-метоксифенилуксусной кислоты в качестве исходного материала для получения 198 мг (38%) высоко кристаллического материала белого цвета. Точка плавления 216-217oC.
Пример 19
Получение транс-dl-8-метокси-4-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-она
Искомое соединение было получено по процедурам, описанным в примере 1, этапах A, B, C, D, F, рекристаллизация из этилацетата и затем E использование п-метоксифенилуксусной кислоты в качестве исходного материала дало 38 мг желтого порошка. Точка плавления 102-103oC.
Элементарный анализ:
Рассчитано: C 72,20; H 7,40; N 6,05.
Получено: C 72,61; H 7,59; N 5,94.
Пример 20
Получение транс-dl-8-метил-1,2,3,4,4a,5,6, 10b-октагидробензо[f]хинолин-3-она
Искомое соединение было получено по процедурам, описанным в примере 1, этапах A, B, C, D, F и G, с использованием п-толилуксусной кислоты в качестве исходного материала. Точка плавления 226-227oC.
Элементарный анализ:
Рассчитано: C 78,10; H 7,96; N 6,51.
Получено: C 78,39; H 8,19; N 6,27.
Пример 21
Получение транс-dl-1,2,3,4,4a,5,6,10b-октагидробензо[f] хинолин-3-она
Искомое соединение было получено по процедурам, описанным в примере 1, этапах A, B, C, D и F с использованием фенилуксусной кислоты в качестве исходного материала для получения 327 мг (выход 30%) после четырех рекристаллизаций из этилацетата. Точка плавления 227-228oC.
Элементарный анализ:
Рассчитано: C 77,58; H 7,58; N 6,96.
Получено: C 77,29; H 7,74; N 6,99.
Пример 22
Получение транс-dl-8-этоксикарбонилэтандиил-4-метил-1,2,3, 4,4a, 5,6,10b-октагидробензо[f]хинолин-3-она
Соединение транс-dl-8-этоксикарбонилэтендиил-4-метил- 1,2,3,4,4a, 5,6,10b-октагидробензо[f]хинолин-3-он было получено по процедурам, описанным в примере 16. Это соединение соединялось (424 мг, 1,35 мМ) с 50 мг 5% палладия на активированном угле в 50 мл этанола в герметичном реакторе при комнатной температуре и первоначальном давления 60 фут/дюйм2 (4,21 кг/см2). После четырех часов катализатор удалялся фильтрацией. Фильтрат концентрировался в вакууме. Остаток подвергался ВЭЖХ хроматографии на SiO2 и элюирование 5% метанолом, CH2Cl2 дало 308 мг (72%) искомого соединения в виде светло-желтого масла, которое кристаллизовалось при выстаивании. Точка плавления 86-88oC. Масс-спектр высокого разрешения: 315.1840 C19H25NO3.
Пример 23
Получение транс-dl-8-метоксикарбонил-этендиил-4-метил-1,2,3, 4,4a, 5,6,10b-октагидробензо[f]хинолин-3-она
Искомое соединение было получено в соответствии с процедурами, описанными в примере 16, за исключением того, что вместо этилакрилата использовался метилакрилат для получения 1,18 г (выход 94%). Температура плавления 172-174oC.
Элементарный анализ:
Рассчитано: C 72,22; H 7,07; N 4,68.
Получено: C 71,97; H 7,87; N 4,72.
Пример 24
Получение, транс-dl-8-карбоксиэтендиил-4-метил-1,2,3,4,5,6, 10b-октагидробензо[f]хинолин-3-она
Соединение транс-dl-8-этоксикарбонилэтендиил-4-метил-1, 2,3,4,4a, 5,6,10b-октагидробензо[f] хинолин-3-он было получено в соответствии с процедурами, описанными в примере 16. К раствору КОН (435 мг; 7,77 мМ) в смеси метанола и воды 3:1 (об/об) добавлялся этиловый эфир (1,22 г, 3,89 мМ). Реакционная смесь нагревалась с обратным холодильником с перемешиванием 1 час. Метанол удалялся в вакууме и остальная смесь окислялась 5N HCl. Полученный белый осадок собирался фильтрацией и промывался водой. Рекристаллизация из этанола дала 741 мг (выход 67%) искомого соединения в виде белого кристаллического вещества. Температура плавления 311oC разл.
Элементарный анализ:
Рассчитано: C 71,56; H 6,71; N 4,91.
Получено: C 71,82; H 6,57; N 4,88.
Пример 25
Получение транс-dl-8-т-бутиламинокарбонилэтендиил-4-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-она
A. Транс-dl-8-(2-тиопиридилкарбонилэтендиил)-4-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-он
Соединение транс-dl-8-карбоксиэтендиил-4-метил-1,2,3,4, 4a,5,6,10b-октагидробензо[f]хинолин-3-он было получено по процедурам, описанным в примере 24. Суспензия этой кислоты (1,99 г, 6,97 мМ), трифенилфосфин (3,66 г, 13,9 мМ) и 2,2'-дитиодипиридин (3,07 г, 13,95 мМ) в 30 мл безводного толуола перемешивались при комнатной температуре всю ночь. Реакционная смесь фильтровалась и осадок промывался 100 мл диэтилового эфира и высушивался до получения 2,2 г соединения A в виде бледно-желтого вещества (81%).
B. Транс-dl-8-т-бутиламинокарбонилэтендиил-4-метил-1, 2,3,4,4a, 5,6,10b-октагидробензо[f]хинолин-3-он
К перемешанной суспензии тиопиридил-эфира, полученного на этапе A, выше (440 мг, 1,13 мМ) в сухом ТГФ (11,0 мл) добавлялся трет-бутиламин (95 мл, 9,04 мМ). Реакционная смесь перемешивалась при комнатной температуре 24 часа. Смесь фильтровалась и твердые вещества промывались гексанами до получения 256 мг (выход 66,5%) искомого соединения. Температура плавления 243-245oC разложение.
Элементарный анализ:
Рассчитано: C 74,08; H 8,29; N 8,23.
Получено: C 74,21; H 8,39; N 8,11.
Пример 26
Получение транс-dl-8-хлоро-2 -(α и β)- метил-4-метил-1, 2,3,4,4a, 5,6,10b-октагидробензо[f]хинолин-3-он
Соединение транс-dl-8-хлоро-4-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f] хинолин-3-он было получено в соответствии с процедурами, описанными в примере 6.
К холодному (-78oC; сухой лед/изопропанол) перемешанному раствору транс-dl-8-хлоро-4-метил-1,2,3,4,4a, 5,6,10b-октагидробензо[f] хинолин-3-она (759 мг, 2,88 мМ) в 45 мл сухого ТГФ добавлялось 17,6 мл 0,5 М гексметилдизилазида калия (1,1 эквивалент, 8,81 мМ) в толуоле по каплям. После добавления реакционная смесь перемешивалась в холоде еще 45 минут. Избыток (5,0 эквив/метилйодида (2,5 мл) добавлялся к реакционной смеси. Охлаждающая ванна убиралась и реакционная смесь выстаивалась до комнатной температуры 2 часа. Реакция остужалась осторожным добавлением воды и смесь переносилась в разделительную воронку. К смеси добавлялся этилацетат и 1N HCl и слои разделялись. Органический слой промывался 1N HCl, единожды насыщенным NaHCO3 и затем рассолом. Органический материал высушивался над сульфатом магния и выпаривался в вакууме для получения 2,11 искомого соединения в виде вещества желтого цвета.
α- и β- изомеры отделялись ВЭЖХ на силикагеле с использованием 0-75% этилацетат/толуол (об/об) градиента для получения 414 мг α- изомера (пример 26A, температура плавления 166-167oC) в виде твердого вещества белого цвета и 199 мг β- изомера (пример 26B, точка плавления 82-83oC) в виде бесцветного твердого вещества.
Элементарный анализ:
α- изомер
Рассчитано: C 68,30; H 6,88; N 5,31.
Получено: C 68,09; H 6,93; N 5,20.
β- изомер
Рассчитано: C 68,30; H 6,88; N 5,31.
Получено: C 68,05; H 6,68; N 5,55.
Пример 27
Получение транс-dl-8-бромо-6,6-диметил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-она
4,4-Диметил-6-бромо-2-тетралон был получен в соответствии с процедурами, описанными в примере 1, этапах A и B, за исключением того, что на этапе B вместо этилена использовался изобутилен и что полученная 1:1 смесь 4,4-диметил и 3,3-диметил региоизомерических тетралонов разделялась на силикагеле с использованием 7,5% этилацетат/гексаны градиента (об/об) для получения желаемого 4,4-диметил-6-бромо-2-тетралона. Искомое соединение было получено из этого тетралона по процедурам, описанным в примере 1, этапах C, D и F и затем рекристаллизация из этилацетата дала 109,3 мг вещества белого цвета. Температура плавления 281-282oC.
Элементарный анализ:
Рассчитано: C 58,45; H 5,89; N 4,54.
Получено: C 58,68; H 5,77; N 4,44.
Пример 28
Получение транс-dl-8-бромо-4,6,6-триметил-1,2,3,4,4a,5,6, 10b-октагидробензо[f]хинолин-3-она
Смесь транс- и цис-dl-8-бромо-6,6-диметил-1,2,3,4,4a, 5,6,10b-октагидробензо[f]хинолин-3-она была получена концентрацией фильтратов рекристаллизаций в этилацетате транс-dl- 8-бромо-6,6-диметил-1,2,3,4,4a,5,8,10b-октагидробензо[f] хинолин-3-она (пример 27). Искомое соединение готовилось из этого материала вместе с цис-dl-изомером (пример 29) с использованием процедур, описанных в примере 1, этапах E и C для получения 67,2 мг твердого вещества белого цвета. Температура плавления 133-136oC.
Элементарный анализ:
Рассчитано: C 59,64; H 6,26; N 4,35.
Получено: C 59,50; H 6,21; N 4,55.
Пример 29
Получение цис-dl-8-бромо-4,6,6-триметил-1,2,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-она
Смесь транс- и цис-dl-8-бромо-6,6-триметил-1,2,3,4,4a,5,6, 10b-октагидробензо[f] хинолин-3-она была получена концентрацией фильтратов рекристаллизаций в этилацетате транс-dl-8- бромо-6,6-диметил-1,2,3,4,4a,5,6,10b-октагидробензо[f] хинолин-3-она (пример 27). Искомое соединение было получено из этого материала вместе с транс-dl-изомером (пример 28) с использованием процедур, описанных в примере 1, этапах E и C для получения 67,2 мг твердого вещества белого цвета. Температура плавления 177-180oC.
Элементарный анализ:
Рассчитано: C 59,64; H 6,26; N 4,35.
Получено: C 59,85; H 6,16; N 4,28.
Пример 30
Получение транс-dl-8-т-бутил-1,2,3,4,4a, 5,6,10b- октагидробензо[f]хинолин-3-она
A. 6-трет-бутил-2-нафтол
2- литровая колба с круглым дном загружалась свежерасплавленным хлоридом цинка (45,0 г), β- нафтолом (150,0 г, 1,04 моль) и гексанами (450 мл). Смесь энергично перемешивалась при добавлении т-бутилхлорида (150,0 г, 1,62 моль) по каплям в течение 30 минут. Когда реакционная смесь постепенно нагревалась с обратным холодильником, раствор не был получен. Реакционная смесь охлаждалась до комнатной температуры и добавлялось 100 мл CH2 Cl2. Реакционная смесь нагревалась с обратным холодильником всю ночь, охлаждалась и концентрировалась в вакууме до получения вещества белого цвета. Твердое вещество нагревалось с обратным холодильником с 1800 мл 10% NaOH фильтровалось, и выстаивалось до охлаждения. Осажденная натриевая соль белого цвета собиралась фильтрацией. Вещество, собранное фильтрацией, перемешивалось с избытком 5,0 М HCl и полученный фенол собирался фильтрацией и промывался 2 л воды. Рекристаллизация из гептана дала 30,67 г соединения A в виде твердого вещества белого цвета.
B. 6-т-бутил-2-метоксинафталин
В 2-х литровую колбу с круглым дном добавлялся 6-т-бутил-2-нафтол (30,67 г, 0,153 мМ) и 550 мл 15% КОН в воде. Раствор перемешивался при добавлении по каплям в течение 30 минут диметилсульфата (6,0 эквив.). После завершения добавления смесь перемешивалась 2 часа. Твердые вещества собирались на фильтрате и промывались водой для получения 28,97 г (выход 88%) соединения B.
C. 6-т-бутил-2-тетралон
К перемешанному раствору 6-т-бутил-2-метоксинафталина (28,97 г, 0,135 мМ) в 350 мл безводного этанола добавлялись шарики натрия (36 г, 11,5 эквив. ) 2 часа со скоростью, чтобы поддержать нагревание с обратным холодильником. Вязкая реакционная смесь перемешивалась до тех пор, пока весь натрий не растворился. Смесь охлаждалась и осторожно добавлялось 140 мл воды. Добавлялась концентрированная HCl и реакционная смесь нагревалась с обратным холодильником 30 минут. После охлаждения реакционная смесь фильтровалась и водный слой экстрагировался трижды толуолом. Выпаривание летучих веществ в вакууме дало 28,1 г красного вязкого масла. Масло забиралось в 300 мл диэтилового эфира и перемешивалось с 50 мл насыщенного водного NaHCO3 всю ночь. Полученный белый осадок собирался фильтрацией и промывался несколько раз гексанами. Этот материал частично растворялся в 500 мл H2О добавлялось 200 мл диэтилового эфира. Смесь энергично перемешивалась и добавлялось 300 мл насыщенного водного Na2CO3. Смесь перемешивалась 1 час, слои разделялись и водный слой экстрагировался трижды диэтиловым эфиром. Органические слои соединялись, промывались рассолом, высушивались над сульфатом магния и концентрировались в вакууме для получения 5,74 г соединения C в виде оранжевого масла, которое медленно кристаллизовалось при выстаивании.
D. 6-т-бутил-2-пирролидинил-3,4-дигидронафталин
К перемешанному раствору 6-т-бутил-2-тетралона (5,74 н; 28,37 мМ) в 100 мл толуола добавлялось 1,5 эквив. пирролидина (3,56 мл; 42,56 мМ). Добавлялась 100 мг порция п-толуолсульфокислоты и смесь нагревалась с обратным холодильником. Вода, уходящая во время реакции, собиралась ловушкой Dean Stark. После 3,5 часов концентрация летучих веществ в вакууме дала 7,31 г соединения в виде твердого вещества пурпурного цвета.
E. 8-т-бутил-1,2,3,4,5,6-гексагидробензо[f]хинолин-3-он
К 6-т-бутил-2-пирролидинил-3,4-дигидронафталину (7,25 г, 28,37 мМ) было добавлено 3,0 эквив. акриламина (6,05 г; 85,11 мМ). Реакционная смесь перемешивалась при 89oC всю ночь. Затем температура возрастала до 130oC и выдерживалась такой 20 минут. Осторожно добавлялась вода (100 мл) и реакционная смесь охлаждалась до комнатной температуры. Полученное твердое вещество растиралось с водой в порошок и собиралось на фильтре для получения вещества коричневого цвета. Твердое вещество рекристаллизовалось дважды из диметилформамида (ДМФ) H2O для получения нужного соединения. Температура плавления 265-268oC разл.
F. Транс-dl-8-т-бутил-1,2,3,4,4a,5,6, 10b-октагидробензо[f]хинолин-3-он
К холодному (0oC) перемешанному раствору 8-т-бутил-1, 2,3,4,5,6-гексагидробензо[f]хинолин-3-она (4,00 г, 15,66 мМ) и триэтилсилана (7,29 г, 62,66 мМ) в 90 мл добавлялось 45 мл трифторуксусной кислоты. Охлаждающая ванна убиралась и смесь перемешивалась при комнатной температуре 24 часа. Реакционная смесь осторожно выливалась в насыщенный NaHCO3, встряхивалась и слои разделялись. Органический слой промывался однократно NaHCO3, высушивался над сульфатом натрия и концентрировался в вакууме для получения 5,86 г твердого вещества коричневого цвета. Рекристаллизация из этилацетата дала искомое соединение (2,5 г, выход 62%) в виде кристаллического материала бежевого цвета. Температура плавления более 280oC.
Элементарный анализ:
Рассчитано: C 79,00; H 9,01; N 5,44.
Получено: C 79,36; H 9,16; N 5,49.
Пример 31
Получение транс-dl-8-т-бутил-4-метил-1,2,3,4,5,6- 10b-октагидробензо[f] хинолин-3-она
Искомое соединение было получено в соответствии с процедурами, описанными в примере 30, этапах A, B, C, D, E и F и затем N-метилировано в соответствии с процедурами, описанными в примере 1, этапе E с использованием 1,2-диметоксиэтана как растворителя в количестве 1,14 (выход 73%) твердого вещества. Температура плавления 183-184oC.
Элементарный анализ:
Рассчитано: C 79,66; Н 9,29; N 5,16.
Получено: C 80,08; H 9,31; N 4,99.
Пример 32
Получение транс-dl-8-фторо-4,10b-диметил-1,2,3,4,4a, 5,6,10b-октагидробензо[f]хинолин-3-она
A. 6-фторо-2-пирролидинил-3,4-дигидронафталин.
Искомое соединение было получено в соответствии с процедурами, описанными в примере 1, этапах A, B и C, с использованием в качестве исходного материала п-фторфенилуксусной кислоты.
B. 6-фторо-1-метил-2-пирролидинил-3,4-дигидронафталин
К 6-фторо-2-пирролидинил-3,4-дигидронафталину (13 г, 60,8 мМ) в 200 мл сухого тетрагидрофурана (ТГФ) добавлялся метилйодид (30 мл, 482 мМ), и смесь нагревалась с обратным холодильником 2 часа. Реакционная смесь выстаивалась до охлаждения с перемешиванием, и в это время происходила кристаллизация. Твердые вещества собирались фильтрацией для получения соединения B.
C. К 6-фторо-1-метил-2-пирролидинил-3,4-дигипронафталину, полученному выше в этапе B, в 1700 мл этилацетата, добавлялся ацетат натрия (10,2 г, 124,4 мМ), уксусная кислота (10,2 мл, 178,2 мМ) и 102 мл воды. Реакционная смесь перемешивалась при комнатной температуре 4 часа. Слои разделялись, и органический слой промывался последовательно рассолом, 5% NaHCO3 и снова рассолом. Органический слой высушивался над MgSO4 и концентрировался до получения 7,9 г соединения C в виде темного масла оранжево-красного цвета (выход 60%),
D. 8-фторо-10b-метил-1,2,3,4,6, 10b-гексагидробензо[f]хинолин-3-он
К 6-фторо-3-метил-2-тетралону (7,06 г, 39,6 мМ) в круглодонной колбе добавлялась п-толуолсульфокислота (1,23 г, 6,5 мМ) и смесь перемешивалась при комнатной температуре в азоте 15 минут. Добавлялся акриламид (5,62 г, 79,2 мМ), и реакционная смесь нагревалась до 88-90oC в азоте в течение трех дней. Смесь разбавлялась этилацетатом и водой, и перемешивалась при комнатной температуре 1 час. Полученные слои разделялись. Органический слой трижды промывался водой, высушивался над MgSO4 и концентрировался до получения вязкого масла. Сырой продукт кристаллизовался из этилацетата до получения 1,59 г (выход 17%) соединения D. Температура плавления 202oC.
Элементарный анализ:
Рассчитано: C 72,71; H 6,10; N 6,06.
Получено: C 72,45; H 6,14; N 6,03.
E. 8-фторо-4,10b-диметил-1,2,3,4,6, 10b-гексагидробензо[f]хинолин-3-он
8-фторо-10b-метил-1,2,3,4,6, 10b-гексагидробензо[f]хинолин-3-он (1,38 г, 6 мМ) добавлялся к суспензии NaH (475 мг; 20 мМ) в глиме (glyme) (15 мл). Смесь нагревалась с обратным холодильником 1,5 часа и быстро охлаждалась до комнатной температуры. Добавлялся метилйодид (15 мл), и смесь нагревалась с обратным холодильником в течение 4 часов, затем выстаивалась до охлаждения до комнатной температуры. После добавления воды смесь подвергалась концентрации почти досуха. Остаток разделялся между этилацетатом и водой. Органический слой трижды промывался водой, высушивался над MgSO4 и концентрировался под вакуумом. Рекристаллизация из гексана давала 737 мг (выход 56%) соединения E. Температура плавления 110-111oC.
Элементарный анализ:
Рассчитано: C 73,45; H 6,57; N 5,71.
Получено: C 73,72; H 6,84; N 5,86.
F. 8-фторо-4,10b-диметил-1,2,3,4,4a,5,6, 10b-октагидробензо[f]хинолин-3-он
Триэтилоилан (1 мл; 6,12 мМ) добавлялся к 8-фторо-4,10b- диметил-1,2,3,4,6,10b-гексагидробензо[f]хинолин-3-ону (500 мг; 2,04 мМ) в CH2Cl2 (15 мл) при комнатной температуре. Реакционная смесь охлаждалась до 0oC, и в нее добавлялась трифторуксуcная кислота (2,6 мл). После перемешивания при комнатной температуре в течение 4 дней реакционная смесь разбавлялась CH2Cl2, и обрабатывалась насыщенным NaHCO3. Полученные слои разделялись, и органический слой промывался насыщенным NaHCO3, высушивался над MgSO4 и концентрировался под вакуумом до получения масла желтого цвета.
G. Транс-dl-8-фторо-4,10b-диметил-1,2,3,4,4a,5,6, 10b-октагидробензо[f] хинолин-3-он
Смесь, полученная выше, в этапе F, разделялась с помощью колонной хроматографии на SiO2 (элюирование этилацетат/гексанами 9:1 (об:об)). Соответствующие фракции, содержащие желаемый продукт, выпаривались почти до сухости и разбавлялись гексанами. Полученные кристаллы собирались фильтрацией для получения 190 кг соединения G. Температура плавления 130-131oC.
Элементарный анализ:
Рассчитано: C 72,85; H 7,34; N 5,66.
Получено: C 72,71; H 7,48; N 5,73.
Пример 33
Получение цис-dl-8-фторо-4,10b-диметил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-она
Искомое соединение было получено наряду с транс-dl-изомером в соответствии с процедурами, описанными в примере 32, этапах A-F.
Смесь, полученная в примере 32, этапе F, разделялась колонной хроматографией на SiO2, (элюирование этилацетатом/гексанами 9:1 (об:об:)). Соответствующие фракции, содержащие желаемый продукт, выпаривались почти досуха и разбавлялись гексанами. Полученные кристаллы собирались фильтрацией для получения искомого соединения.
Пример 34
Получение транс-dl-8-хлоро-4,10b-диметил-1,2,3,4,4a,5,6, 10b-октагидробензо[f]хинолин-3-она
Искомое соединение было получено наряду с цис-dl-изомером (пример 35) в соответствии с процедурами, описанными в примере 32, этапах A, B, C, D, E и F, с использованием п-хлорфенилуксусной кислоты в качестве исходного материала. Колонная хроматография в соответствии с примером 32, этапом G, дала 523 мг искомого соединения. Температура плавления 94oC.
Элементарный анализ:
Рассчитано: C 68,30; H 6,88; N 5,31.
Получено: C 68,51; H 6,67; N 5,36.
Пример 35
Получение цис-dl-8-хлоро-4,10b-диметил-1,2,3,4,4a, 5,6,10b-октагидробензо[f]хинолин-3-она
Искомое соединение было получено наряду с транс-dl-изомером (пример 34) в соответствии с процедурами, описанными в примере 32, этапах A, B, C, D, E и F, с использованием п-хлорфенилуксусной кислоты в качестве исходного материала. Колонная хроматография в соответствии с примером 32, этапом G, дала 145 мг искомого соединения.
Элементарный анализ:
Рассчитано: C 68,30; H 6,88; N 5,31.
Получено: C 68,09; H 6,76; N 5,11.
Пример 36
Получение транс-dl-10b-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-она
A. 10b-метил-1,2,3,4,6, 10b-гексагидробензо[f]хинолин-3-он
Следуя процедурам, описанным в примере 32, этапах A, B, C и D, за исключением использования фенилуксусной кислоты в качестве исходного материала и использованием колонной хроматографии на SiO2 вместо рекристаллизации из этилацетата, было получено искомое соединение в виде кристаллического белого вещества. Температура плавления 123-124oC.
Элементарный анализ:
Рассчитано: C 78,84; H 7,09; N 6,57.
Получено: C 78,82; H 6,95; N 6,58.
B. Транс-dl-10b-метил-1,2,3,4,6, 10b-октагидробензо[f]хинолин-3-он
К раствору 10b-метил-1,2,3,4,6, 10b-гексагидробензо[f]хинолин-3-она (500 мг, 2,3 мМ) в 50 мл уксусной кислоты добавлялось 500 мг 5% палладия на углероде. Реакционная смесь перемешивалась в течение ночи при комнатной температуре под первоначальным водородным давлением в 60 фунт/кв.дюйм (4,22 кг/кв. см). Реакционная смесь фильтровалась и концентрировалась досуха. Остаток распределялся между этилацетатом и водой. Органический слой дважды промывался насыщенным NaHCO3 и водой, а затем высушивался над MgSO4 и концентрировался под вакуумом. Остаток растирался в порошок с эфиром до получения 180 мг (выход 36%) соединения B. Температура плавления 178-179oC.
Элементарный анализ:
Рассчитано: C 78,00; H 7,96; N 6,51.
Получено: C 78,38; H 8,02; N 6,36.
Пример 37
Получение транс-dl-4,10b-диметил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f] хинолин-3-он
Следуя процедурам, описанным в примере 36, был получен транс- dl-10b-метил-1,2,3,4,4a, 5,6,10b-октагидробензо[f] хинолин-3-он. К 4 мл глима добавлялся NaH (31 мг; 1,26 мМ) и транс-dl-10b-метил- 1,2,3,4,4a,5,6,10b-октагидробензо[f] хинолин-3-он (130 мг; 0,6 мМ). Реакционная смесь нагревалась с обратным холодильником 1,5 часа. После охлаждения до комнатной температуры добавлялось 10 мл oC метилйодида, и реакционная смесь нагревалась с обратным холодильником 3 часа. Добавлялась вода, и смесь концентрировалась почти досуха. Остаток распределялся между этилацетатом/водой. Органический слой трижды промывался водой, высушивался над MgSO4 и концентрировался под вакуумом. Остаток растирался с петролейным эфиром до получения 60 мг (выход 44%) искомого соединения. Температура плавления 93oC.
Элементарный анализ:
Рассчитано: C 78,56; H 8,35; N 6,11.
Получено: C 78,29; H 8,16; N 6,03.
Пример 38
Получение транс-dl-8-хлоро-10b-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-она
Следуя процедурам, описанным в примере 32, этапах A, B, C, D, E и F, за исключением использования п-хлорфенилуксусной кислоты в качестве исходного материала, было получено соединение 8-хлоро-10b-метил-1,2,3,4,4a,5,6,10b-октагидробензо[f]хинолин-3-он в виде смеси цис- и транс-изомеров.
Смесь очищалась ВЭЖХ (обратная фаза, CN) элюированием ТГФ: изооктаном (48% ТГФ по объему) до получения 32 мг искомого соединения.
Элементарный анализ:
Рассчитано: C 67,33; H 6,46; N 5,61.
Получено: C 67,53; H 6,35; N 5,73.
Пример 39
Получение цис-dl-8-хлоро-10b-метил-1,2,3,4,4a,5,6, 10b-октагидробензо[f] хинолин-3-она
Искомое соединение было получено наряду с транс-dl-изомером (пример 38) в соответствии с процедурами, описанными в примере 38. Цис-dl-изомер был получен также очисткой с помощью ВЭЖХ и с использованием процедур, описанных в примере 38, сопровождаемых растиранием с диэтиловым эфиром.
Пример 40
Получение транс- и цис- R(-) 8-хлоро-10b-метил-1,2,3,4,4a,5, 6,10b-октагидробензо[f]хинолин-3-она
A. 6-хлоро-1-метил-2-тетралон
Следуя процедурам, описанным в примере 32, этапах A, B и C, за исключением использования п-хлорфенилуксусной кислоты в качестве исходного материала, было получено соединение A.
B. 1-метил -2-(α- метилбензиламино)-6-хлоро-1,2-дидегидротетралин
К 500 мл толуола добавлялся 6-хлорометил-2-тетралон (50,0 г, 0,256 М) и (R)-(+)-1-фенилэтиламин (35 мл, 0,27 М). Смесь нагревалась с обратным холодильником 4 часа с азеотропным удалением воды. Растворитель удалялся под вакуумом до получения 79 г соединения B наряду с его иминовым таутомером в виде масла желтого цвета, которое использовалось без дальнейшей очистки.
C. (5S)-5,6,7,8,9,10-гексагидро-8-гидрокси-2-хлоро-5,8- диметил-5,9-метанобензоциклооктен-11-он
К перемешанному раствору 1-метил -2-(α- метилбензиламино)- 6-хлоро-1,2-дидегидротетралина (79 г, 0,25 М) в 500 мл ТГФ добавлялся метилвинилкетон (23 мл; 0,28 М). Раствор перемешивался при температуре окружающей среды в атмосфере аргона, в темноте, в течение 96 часов. Добавлялась водная уксусная кислота (20%, 500 мл), и смесь перемешивалась еще 2 часа. Реакционная смесь затем распределялась между этилацетатом и водой. Органическая фаза промывалась насыщенным NaCO3 и высушивалась над Na2SO4. Растворитель удалялся под вакуумом для получения 82 г соединения C в виде масла коричневого цвета, которое использовалось без дальнейшей очистки.
D. (R) (+(8-хлоро-10b-метил-1,2,3,5,6,10b-гексагидро-фенантрен-3-он
К перемешанному раствору этоксида натрия, полученного из натрия (6,5 г), в этаноле (500 мл) добавлялось искомое соединение (82 г) из этапа C. Раствор нагревался при 50oC 3 часа в атмосфере азота. Раствор охлаждался до температуры окружающей среды и распределялся между диэтиловым эфиром и водой. Органическая фаза промывалась рассолом и высушивалась над Na2SO4 и концентрировалась под вакуумом. Остаток очищался хроматографией на SiO2 (элюирование 25% этилацетатом в гексане) для получения 34 г соединения в виде коричневого масла, затвердевавшего со временем.
E. (R) 3-[1-(1-метил-6-хлоро-2-тетралон)]пропановая кислота
К перемешанной смеси RuCl3 • nH2O (620 мг, 2,99 мМ) в смеси растворителей (200 мл) из 2 частей 4-х хлористого углерода, 3 частей ацетонитрила и 2 частей воды добавлялась йодная кислота (20,45 г, 89,7 мМ). Смесь охлаждалась до 0oC, и перемешивалась 15 минут. К смеси медленно добавлялось искомое соединение из этапа D (3,7 г, 14,95 мМ) в ацетонитриле, и смесь перемешивалась при 0oC 3 часа, добавлялся 2-пропанол (20 мл), и смесь перемешивалась в течение 1 часа. Затем реакционная смесь распределялась между этилацетатом и водой, и водная фаза трижды экстрагировалась этилацетатом. Объединенные органические слои фильтровались через кизельгур, и осадок на фильтре промывался этилацетатом. Раствор концентрировался в вакууме до получения 1,74 г соединения E, которое использовалось без дальнейшей очистки.
F. (R) (+)-8-хлоро-10b-метил-1,2,3,4,6, 10b-гексагидробензо[f]хинолин-3-он
К перемешанному раствору искомого соединения из этапа E (1,74 г, 6,457 мМ) в 15 мл 2-пропанола добавлялся аммиак (1 мл), и трубчатый реактор герметизировался. Реакционная смесь нагревалась до 180oC за 15 минут. Смесь охлаждалась до комнатной температуры и концентрировалась под вакуумом до получения стекловидного твердого вещества коричневого цвета. Твердое вещество помещалось в этилацет, фильтровалось через SiO2, элюировалось этилацетатом до получения соединения в виде светлого твердого вещества. Образец кристаллизовался из диэтилового эфира/гексана для получения 219 мг соединения F. Температура плавления 61-63oC.
Элементарный анализ:
Рассчитано: C 67,88; H 5,70; N 5,65.
Получено: C 67,54; H 5,65; N 5,44.
Оптическое вращение: 589 нм = - 40,32o /C=1, метанол 365 нм = -185,48o.
G. 8-Хлоро-10b-метил-1,2,3,4,4a,5,6, 10b-октагидробензо[f]хинолин-3-он
К перемешанному раствору искомого соединения из этапа F (776 мг, 3,1 мМ) и триэтилсилана (4,95 мл, 31 мМ) в CH2Cl2 при 0oC добавлялась трифторуксусная кислота (4,82 мл; 6,26 мМ). Раствор медленно подогревался до комнатной температуры и перемешивался 48 часов. Реакционная смесь разбавлялась этилацетатом, нейтрализовалась Na2CO3, экстрагировалась этилацетатом и концентрировалась под вакуумом до получения 790 мг неочищенного продукта, включающего соединение G.
H. Транс-8-хлоро-10b-метил-1,2,3,4,4a, 5,6, 10-октагидробензо[f]хинолин-3-он
Хроматография на SiO2 (этилацетат в качестве элюента) неочищенного продукта на этапе G давала 341 мг соединения H. Температура плавления 135-137oC.
Элементарный анализ:
Рассчитано: C 67,33; H 6,46; N 5,61.
Получено: C 67,41; H 6,55; N 5,36.
Оптическое вращение: 589 нм = +113,86o. (C = 1, CHCl3) 365 нм = +371,29o.
Пример 41
Получение цис-8-хлоро-10b-метил-1,2,3,4,4a, 5,6, 10-октагидробензо[f] хинолин-3-он
Искомое соединение было получено наряду с транс-изомером (пример 40) с помощью процедур, описанных в примере 40, этапах A-G. Хроматография на SiO2 (этилацетат в качестве элюента) неочищенного продукта из примера 40, этапа G, давала 91 мг искомого соединения. Температура плавления 178- 181oC.
Элементарный анализ:
Рассчитано: C 67,33; H 6,46; N 5,61.
Получено: C 66,73; H 6,73; N 5,36.
Оптическое вращение: 589 нм = +199,10o
(C = 1, CHCl3) 365 нм = +660,63o.
Пример 42
Получение транс-4-этил-10b-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f] хинолин-3-она
Следуя процедурам, описанным в примере 40, этапах A, B, C, D, E и F, используя фенилуксусную кислоту в качестве исходного материала и используя этиламин вместо аммиака, а также диглим вместо этиленгликоля в этапе 7, было получено соединение 4-этил-10b-метил-1,2,3,4,6,10b-гексагидробензо[f] хинолин-3-он. Этот гексагидробензо[f]хинолин-3-он подвергался гидрогенизации с помощью процедур, описанных в примере 22, за исключением того, что реакция проводилась при 70oC свыше 7 часов до получения сырой реакционной смеси. Реакционная смесь фильтровалась, и растворители выпаривались под вакуумом до получения стекловидного твердого остатка. Неочищенный продукт очищался хроматографией на SiO2 /градиент элюирования от 100% гексанов до 100% CHCl3/ для получения 92 мг искомого соединения в виде бесцветного масла.
Элементарный анализ:
Рассчитано: C 78,97; H 8,70; N 5,76.
Получено: C 79,07; H 8,90; N 5,56.
Пример 43
Получение транс-4-н-бутил-10b-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-она
Следуя процедурам, описанным в примере 40, этапах A, B, C, D, E и F, с использованием фенилуксусной кислоты в качестве исходного материала и в этапе F с использованием н-бутиламина вместо аммиака и диметоксиэтана вместо этиленгликоля, было получено соединение 4-н-бутил-10b-метил-1,2,3,4, 6,10b-гексагидробензо[f] хинолин-3-он. Этот гексагидробензо[f]хинолин-3-он подвергался гидрогенизации с помощью процедур, описанных в примере 22, за исключением того, что реакция проводилась при 60oC свыше 7 часов и осуществлялась в соответствии с процедурами, описанными в примере 42 для получения 61 мг искомого соединения в виде бесцветного масла.
Элементарный анализ:
Рассчитано: C 77,10; H 9,35; N 5,00.
Получено: C 77,44; H 9,28; N 4,95.
Пример 44
Получение транс-4-(4-метоксибензил-8-хлоро-10b-метил-1,2, 3,4,4a, 5,6,10b-октагидробензо[f]хинолин-3-она
A. (R) 3-[1-(1-метил-6-хлоро-2-тетралон)]пропановая кислота
Следуя процедурам, описанным в примере 40, этапах A, B, C, D, E, с использованием п-хлорофенилуксусной кислоты в качестве исходного материала, было получено соединение 3-[(метил-6-хлоро-2-тетралон)]-пропановая кислота.
B. 4-(4-метоксибензил)-8-хлоро-10b-метил-1,2,3,4,6, 10b-гексагидробензо[f]хинолин-3-он
К 40 мл диметоксиэтана добавлялись 3-[1-(1-метил-6- хлоро-2-тетралон)] -пропановая кислота (2 г) и п-метоксибензиламин (5 мл) в герметично закрытом трубчатом реакторе. Раствор грелся при 120oC в течение ночи. После выстаивания реакционной смеси для охлаждения до комнатной температуры растворитель удалялся под вакуумом. Остаток растворялся в CHCl3, промывался последовательно 1N HCl, водой, насыщенным NaHCO3 и рассолом. Органическая база высушивалась над Na2SO4 и концентрировалась под вакуумом. Остаток подвергался хроматографии на SiO2 (градиент элюирования от 100% гексанов до 100% этилацетата) для получения 394 мг соединения B в виде масла.
C. Транс-4-(4-метоксибензил)-8-хлоро-10b-метил-1,2,3,4,4a, 5,6,10b-октагидробензо[f]хинолин-3-он
К перемешанному раствору искомого соединения из этапа B (693 мг) в CH2Cl2 (2,5 мл) в атмосфере азота добавлялся триэтилсилан (1,4 мл). Реакционная смесь перемешивалась 15 минут. К раствору добавлялось 1,5 мл трифторуксусной кислоты, и реакционная смесь перемешивалась при комнатной температуре в течение ночи. Реакционная смесь распределялась между CHCl3 и насыщенным NaHCO3. Органическая фаза высушивалась над NaSO4, и растворитель удалялся вакуумно. Остаток очищался хроматографией на SiO2, (25% этилацетат в гексане в качестве элюента). Продуктосодержащие фракции выпаривались под вакуумом до получения соединения C в виде пены (459 мг). Температура плавления 55-60oC.
Элементарный анализ:
Рассчитано: (+1/2 М H2O) C 69,74; H 6,65; N 3,70.
Получено: C 70,27; H 6,49; N 3,68.
Пример 45
Получение транс-4-метил-10b-метил-1,2,3,4,4a,5,6, 10b-октагидробензо[f] хинолин-3-она
Следуя процедурам, описанным в примере 40, этапах A, B, C, D, E и F, с использованием фенилуксусной кислоты в качестве исходного материала и в этапе F с использованием метиламина вместо аммиака и диглима вместо этиленгликоля, было получено соединение 4-метил-10b-метил-1,2,3,4,6,10b-гексагидробензо[f] хинолин-3-она. Этот гексагидробензо[f]хинолин-3-он подвергался гидрогенизации с помощью процедур, описанных в примере 22, за исключением того, что реакция проводилась при 60oC свыше 7 часов. Реакционная смесь фильтровалась, и растворитель выпаривался под вакуумом. Остаток очищался хроматографией на SiO2 (CHCl3 в качестве элюента), сопровождающейся рекристаллизацией из этилацетата/гексанов для получения 154 мг искомого соединения. Температура 111- 113oC.
Элементарный анализ:
Рассчитано: C 78,56; H 8,56; N 6,11.
Получено: C 78,33; H 8,62; N 6,14.
Пример 46
Получение транс-dl-9-нитро-1,2,3,4,4a,5,6, 10b-октагидробензо[f]хинолин-3-она
Соединение транс-dl-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-он было получено в соответствии с процедурами, описанными в примере 21. К этому соединению (8,0 г; 39,75 мМ) в 320 мл смеси 1:1 (об:об) ледяной уксусной кислоты и концентрированной серной кислоты при 0oC добавлялось 1,4 эквивалента 90% дымящей азотной кислоты в количестве, которое не позволяло бы температуре подняться выше 10oC. Смесь перемешивалась 30 минут при 0oC и затем выливалась на лед. Полученное твердое вещество собиралось фильтрацией и рекристаллизовалось из смеси ДМФ/вода до выхода 5,40 г (55%) искомого соединения в виде твердого вещества желтого цвета. Температура плавления > 300oC.
Элементарный анализ:
Рассчитано: C 63,40; H 5,73; N 11,38.
Получено: C 63,61; H 5,97; N 11,39.
Пример 47
Получение транс-dl-9-нитро-4-метил-1,2,3,4,4a,5,6, 10b-октагидробензо[f] хинолин-3-она
Соединение транс-dl-4-метил-1,2,3,4,4a, 5,6,10b- октагидробензо[f]хинолин-3-он было получено в соответствии с процедурами, описанными в примере 12. Искомое соединение было получено в соответствии с процедурой, описанной в примере 46 с использованием транс-dl-4-метил-1,2,3,4,4a,5,6,10b-октагидробензо[f]хинолин-3-она в качестве исходного материала, за исключением того, что реакция охлаждалась водой, и продукт рекристаллизовался из этилацетата/гексанов. Точка плавления 172-172,5oC.
Элементарный анализ:
Рассчитано: C 64,60; H 6,20; N 10,76.
Получено: C 64,80; H 6,34; N 10,85.
Пример 48
Получение транс-dl-9-амино-4-метил-1,2,3,4,4a,5,6, 10b-октагидробензо[f] хинолин-3-она
Соединение транс-dl-9-нитро-4-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f] хинолин-3-он было получено в соответствии с процедурой, описанной в примере 47. Это соединение (700 мг, 2,69 ммоль) растворяли в 100 мл этанола и добавляли 100 мг 10% палладия на угле. Реакционною смесь гидрировали в течение 1 часа под давлением водорода 2,95 кг/см (42 д/кв.дюйм). Смесь отфильтровывали и фильтрат концентрировали в вакууме с получением светлого рыжевато-коричневого твердого вещества, которое перекристаллизировали из этилацетат/гексана с получением 308 мг целевого соединения в виде твердого белого вещества. Температура плавления 213-214,5oC.
Элементарный анализ:
Рассчитано: C 73,01; H 7,88; N 12,16.
Получено: C 73,22; H 8,02; N 12,20.
Пример 49
Получение транс-dl-9-хлоро-4-метил-1,2,3,4,4a,5,6, 10b-октагидробензо[f] хинолин-3-она
Соединение транс-dl-9-амино-4-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f] хинолин-3-он было получено в соответствии с процедурами, описанными в примере 48. При 0oC к раствору этого соединения (74 мг; 0,321 мМ) в 0,3 мл концентрированной HCl добавлялся нитрит натрия (23 мг; 0,324 мМ) в 0,15 мл воды. Реакционная смесь перемешивалась при 0oC 30 минут, а затем была добавлена при температуре 0oC к раствору хлорида меди (1) (35 мг; 0,353 мМ) в 0,2 мл концентрированной HCl. Реакционная смесь выстаивалась до подогрева до комнатной температуры свыше 2,5 часов, затем 30 минут нагревалась до 60oC. После охлаждения смесь распределялась между CHCl3 и насыщенным NaCl. Органический слой высушивался над Na2SO4 и концентрировался в вакууме до получения 65 мг твердого материала. Искомое соединение было получено в виде твердого вещества белого цвета после мгновенной хроматографии на SiO2 (10% изопропанол/этилацетат). Температура плавления 228-230oC.
Элементарный анализ:
Рассчитано: C 67,33; H 6,46; N 5,61.
Получено: C 67,19; H 6,57; N 5,53.
Пример 50
Получение транс-dl-8-хлоро-3,4,4a, 5,6, 10b-гексагидробензо[f]хинолин-3-она и транс-dl-8-хлоро-2,3,4,4a,5,6-гексагидробензо[f]хинолин-3-она
A. Транс-dl-4-т-бутилоксикарбонил-3-хлоро-1,2,3,4,4a,5,6, 10b-октагидробензо[f]хинолин-3-он
Соединение транс-dl-3-хлоро-1,2,3,4,4a,5,6, 10b-октагидробензо[f]хинолин-3-он было получено в соответствии с процедурами, описанными в примере 17. К этому соединению (1,56 г; 6,62 мМ) в 40 мл безводного 1,2-диметоксиэтана (1,2-ДМЭ) добавлялось 380 мг гидрида натрия в виде 60% дисперсии в минеральном масле. Смесь нагревалась с обратным холодильником 1 час в атмосфере азота, затем охлаждалась до комнатной температуры. Добавлялся раствор ди-т-бутил дикарбоната (1,74 г; 7,94 мМ) в 10 мл безводного 1,2-ДМЭ, и смесь нагревалась с обратным холодильником еще 1 час. Смесь затем охлаждалась, и к ней осторожно добавлялась вода, а потом диэтиловый эфир. Слои разделялись, и водный слой экстрагировался диэтиловым эфиром. Объединенные органические слои промывались насыщенным NaCl, высушивались над Na2SO4 и концентрировались до получения полутвердого вещества оранжевого цвета, которое очищалось колонной хроматографией (SiO2 1:1 этилацетат/гексаны) для выхода 1,2 г (54%) соединения A в виде твердого вещества белого цвета.
B. Транс-dl-4-т-бутилоксикарбонил-2-фенилселено-8-хлоро -1,2,3,4,4a, 5,6,10b-октагидробензо[f]хинолин-3-он
К защитному лактаму, полученному в этапе A (1,2 г; 3,58 мМ) в 20 мл безводного ТГФ при - 78oC по каплям добавлялся 0,5 М раствор гексаметилдисилазида (14,3 мл, 7,16 мМ) в толуоле. После выстаивания в течение 1 часа при -78oC добавлялся раствор фенилселенилхлорида (755 мг; 3,94 мМ) в 5 мл безводного ТГФ. Реакционная смесь выстаивалась до подогрева до комнатной температуры и перемешивалась 2 часа, после чего охлаждалась 5 мл насыщенного NH4Cl. Смесь распределялась между насыщенным NH4Cl и этилацетатом. Органические слои высушивались над Nа2SO4 и концентрировались в вакууме до получения 2,197 г соединения B в виде масла оранжевого цвета.
C. Транс-dl-4-т-бутилоксикарбонил-8-хлоро-3,4,4a, 5,6,10b-гексагидробензо[f] хинолин-3-он и транс-dl-4-т-бутилоксикарбонил-8-хлоро-2,3,4,4a,5, 6-гексагидробензо[f]хинолин-3-он
К раствору селенида, полученного выше в этапе B (2,197 г, 4,48 мМ) в 20 мл ТГФ при 0oC и содержащему буфер от избытка твердого NaHCO3, добавлялось 1,2 эквивалента 30% пероксида водорода (610 мг) в 4 мл ТГФ. По прошествии 30 минут реакционная смесь распределялась между водой и этилацетатом. Органический слой промывался насыщенным NaHCO3, высушивался над Nа2SO4 и концентрировался в вакууме до получения 645 мг смеси олефинов. Отделение Δ1,2 олефина от Δ10B,1 олефина проводилось с помощью колонной хроматографии над смесью 2:1 гексанов/этилацетата. Олефин Δ1,2 был получен в виде 293 мг твердого вещества белого цвета (пример 50A). Более полярный изомер олефина Δ10B,1 был получен в виде 149 мг твердого вещества белого цвета (пример 50B).
D. Транс-dl-8-хлоро-3,4,4a, 5,6, 10b-гексагидробензо[f] хинолин-3-он и dl-8-хлоро-2,3,4,4a,5,6-гексагидробензо[f]хинолин-3-он
К раствору защитного олефина Δ1,2, полученного в этапе C (290 мг, 0,869 мМ), в 20 мл CH2Cl2 добавлялась трифторуксусная кислота (0,14 мл, 1,74 мМ). Реакционная смесь перемешивалась при комнатной температуре один час и затем распределялась между CH2Cl2 и насыщенным NaHCO3. Органический слой высушивался над Na2SO4 и концентрировался в вакууме. Полученный сырой продукт рекристаллизовался из этилацетата для получения 142 мг (70%) белого кристаллического транс-dl-8-хлоро-3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-она (пример 50A). Температура плавления 227-228oC.
Элементарный анализ:
Рассчитано: C 66.81; H 5.18; N 5.99.
Получено: C 67.09; H 5.18; N 6.22.
Защитный олефин Δ1,10B (149 мг, 0.446 мМ) обрабатывался способом, описанным выше для защитного олефина Δ1,2, с тем исключением, что неочищенный продукт очищался колонной хроматографией на SiO2 (5% изопропанол) CHCl3 для получения 65 мг (62%) dl-8-хлоро-2,3,4,4а,5,6-гексагидробензо[-f] хинолин-3-она в виде твердого вещества белого цвета.
Температура плавления 241-243oC.
Элементарный анализ:
Рассчитано: C 66.81; H 5.18; N 5.99.
Получено C 67.06; H 5.35; N 5.93.
Пример 51
Получение транс-dl-8-бромо-4-метил-3,4,4a,5,6, 10b-гексагидробензо[f]хинолин-3-она
Транс-dl-8-бромо-4-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-он был получен в соответствии с процедурами, описанными в примере 1. Это соединение было превращено в искомое соединение в соответствии с процедурами, описанными в примере 50, этапах B и C, сопровождаемыми рекристаллизацией из этилацетата/гексанов. Температура плавления 136-138oC.
Элементарный анализ:
Рассчитано: C 57,55; H 4,83; N 4,79.
Получено: C 57,81; H 4,74; N 4,81.
Пример 52
Получение транс-dl-8-хлоро-4-метил-3,4,4a, 5,6, 10b-гексагидробензо[f] хинолин-3-она
Транс-dl-8-хлоро-4-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-он был получен в соответствии с процедурами, описанными в примере 6. Это соединение было превращено в искомое соединение в соответствии с процедурами, описанными в примере 50, этапе B и C, сопровождаемым колонной хроматографией на SiO2 (этилацетат). Температура плавления 124-125oC.
Элементарный анализ:
Рассчитано: C 67,88; H 5,70; N 5,65.
Получено: C 67,77; H 5,77; N 5,38.
Пример 53
Получение dl-8-хлоро-4-метил-2,3,4,4a,5,6, -гексагидробензо[f]хинолин-3-она
Транс-dl-8-хлоро-4-метил-3,4,4a, 5,6, 10b-гексагидробензо[f]хинолин-3-он был получен в соответствии с процедурами, описанными в примере 52. К этому соединению (250 мг, 1 мМ) в 30 мл безводного ТГФ добавлялось 800 мг (6,8 мМ) пиридин гидрохлорида. Реакционная смесь перемешивалась при комнатной температуре в течение 7 дней и затем распределялась между этилацетатом и 1N HCl. Органический слой промывался 1N HCl, водой, затем высушивался над MgSO4 и концентрировался в вакууме до объема 10 мл. Искомое соединение было кристаллизовано из этого раствора и собиралось фильтрацией. Температура плавления 99oC.
Элементарный анализ:
Рассчитано: C 68,16; H 5,31; N 5,67.
Получено: C 67,95; H 4,48; N 5,64.
Пример 54
Получение транс-dl-8-хлоро-2-a-метил-4-метил-1,2,3,4,4a, 10b-гексагидробензо[f]хинолин-3-она
Смесь транс-dl-8-хлоро-2-(a и b)-метил-4-метил-1,2,3, 4,4a,5,6,10b-октагидробензо[f] хинолин-3-онов была получена в соответствии с процедурой, описанной в примере 26. К раствору этой смеси (759 мг, 2,88 мМ) при -78oC в 15 мл безводного ТГФ в присутствии 2,0 эквивалентов гексаметилфосфориктриамила (ГМФА) добавлялось 11,6 мл 0,5 М раствора гексаметилдисилазида калия (5,76 мМ) в толуоле. Реакционная смесь перемешивалась при -78oC 90 минут, затем добавлялся раствор ферилселенилхлорида (607 мг, 3,17 мМ) в 5 мл безводного ТГФ. Раствор выстаивался для подогрева до комнатной температуры свыше 2 часов, охлаждался насыщенным раствором NH4Cl, и смесь экстрагировалась этилацетатом. Объединенные органические слои промывались насыщенным NH4Cl, высушивались над Na2SO4 и концентрировались в вакууме до получения 1,42 г масла коричневого цвета. Неочищенный продукт очищался колонной хроматографией на SiO2 (этилацетат) для получения 252 мг 6-фенилселенида. К раствору селенида в 10 мл ТГФ добавлялось 100 мг NaHCO3 и приблизительно 1,1 эквивалент 3-хлорпероксибензойной кислоты (м-ХПБК) (142 мг 80-85% м-ХПБК, 658-700 мМ). По прошествии 15 минут реакционная смесь распределялась между этилацетатом и водой. Водный слой экстрагировался этилацетатом, и объединенные органические слои промывались насыщенным раствором NaHCO3, высушивались над Na2SO4 и концентрировались в вакууме. Неочищенное масло, полученное таким образом, очищалось мгновенной хроматографией на SiO2 (70% этилацетат/гексаны) для выхода 114 мг искомого соединения в виде твердого вещества белого цвета.
1Н-ЯРМ анализ показал, что образовался Δ5,6 олефин и что 2-метиловая группа находится в альфа-положении. Температура плавления 130-132oC.
Элементарный анализ:
Рассчитано: C 68,83; H 6,16; N 5,35.
Получено: C 68,35; H 6,15; N 4,92.
Пример 55
Получение транс-dl-8-т-бутиламинокарбонилэтандиил- 4-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-она
Соединение транс-dl-8-т-бутиламинокарбонилэтендиил-4-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-он было получено в соответствии с процедурами, описанными в примере 25. К раствору этого соединения (122 мг, 0,358 мМ) в 150 мл безводного этанола добавлялось 15 мг 10% палладия на углероде. Смесь подвергалась гидрогенизации при комнатной температуре 6 часов под первоначальным водородным давлением в 40 фут/кв.дюйм (2,81 кг/кв.см). Катализатор удалялся фильтрацией через кизельгур (Celite®), и фильтрат концентрировался до получения твердого вещества белого цвета. Протонная ЯМР-спектроскопия показала, что реакция еще не завершилась и поэтому материал был снова помещен в те же реакционные условия на дополнительные 6 часов. Очистка полученного твердого вещества проводилась с использованием колонной хромагографии на SiO2 (20% изопропано/этилацетат) для получения искомого соединения в виде твердого вещества белого цвета. Точка плавления 178-179oC.
Элементарный анализ:
Рассчитано: C 73,65; H 8,83; N 8,18.
Получено: C 73,21; H 8,69; N 8,32.
Пример 56
Получение транс-dl-8-фенил-4-метил-1,2,3,4,4a,5,6, 10b-октагидробензо[f] хинолин-3-она
Соединение транс-dl-8-бромо-4-метил-1,2,3,4,4a, 5,6,10b- октагидробензо[f] хинолин-3-он было получено в соответствии с процедурами, описанными в примере 1. К смеси этого соединения (160 мг, 0,54 мМ) и тетракистрифенилфосфина палладия (0) (19 мг, 0,02 мМ) в 1,2 мл толуола в атмосфере азота добавлялось 0,6 мл 2 М раствора водного Na2CO3, а затем фенилборная кислота (80 мг, 0,653 мМ). Реакционная смесь грелась 18 часов при 80oC. Смесь выстаивалась до охлаждения и затем распределялась между 75 мл CH2Cl2 (75 мл) и 25 мл 2 М водного Na2CO3 добавлением 2 мл концентрированного NH4OH. Органический слой высушивался над MgSO4 и концентрировался до получения плотного твердого вещества. Искомое соединение (106 мг, 67%) в виде кристаллического твердого вещества белого цвета было получено после проведения мгновенной хроматографии на SiO2 (5% изопропанол/ CHCl3) и растирания продукта с гексаном. Температура плавления 186,5-137,5oC.
Элементарный анализ:
Рассчитано: C 82,44; H 7,26; N 4,81.
Получено: C 82,38; H 7,12; N 5,08.
Пример 57
Получение транс-dl-8-винил-4-метил-1,2,3,4,4a,5,6, 10b-октагидробензо[f] хинолин-3-она
Соединение транс-dl-8-бромо-4-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f] хинолин-3-он было получено в соответствии с процедурами, описанными в примере 1. В трубку с затвором помещались 8-бромобензохинолинон (1,00 г, 3,4 мМ), ацетат палладия (11) (7,6 мг, 0,034 мМ), три-о-толилфосфин (41 мг, 0,136 мМ), винилтрибутилтин (1,24 мл, 4,25 мМ) и 5 мл безводного диоксана. В течение 15 минут через смесь пропускался аргон. Реакционная трубка закупоривалась и нагревалась при перемешивании при 100oC 18 часов. Реакционная смесь охлаждалась и фильтровалась через кизельгур (Celite®) . Фильтрат концентрировался в вакууме, и полученный материал анализировался с помощью капиллярной газ-хроматографии (Г.Х.), которая обнаружила в нем смесь 4,7 к 1 искомого продукта к исходному материалу. Колонная хроматография этой смеси на SiO2 (7% метанол/CHCl3) дала искомое соединение в виде 212 мг твердого вещества белого цвета. Температура плавления 89-90oC.
Элементарный анализ:
Рассчитано: C 79,63; H 7,94; N 5,80.
Получено: C 79,39; H 7,98; N 5,56.
Пример 58
Получение транс-dl-8-этоксикарбонил-4-метил-1,2,3,4,4a,5,6, 10b-октагидробензо[f]хинолин-3-она
Соединение транс-dl-8-йодо-4-метил-1,2,3,4,4a,5,6, 10b-октагидробензо[f] хинолин-3-он было получено в соответствии с процедурами, описанными в примере 3. К раствору йодида (1,0034 г, 2,94 мМ) в 280 мл этанола добавлялись тетракистрифенилфосфин палладий (11) хлорид (160 мг) и 1,0 мл триэтиламина. Смесь 30 минут продувалась азотом, а затем монооксидом углерода. Реакционная смесь снабжалась CO баллоном и нагревалась с обратным холодильником 48 часов. После фильтрования реакционной смеси через кизельгур (Celite®), фильтрат концентрировался в вакууме до получения вязкого масла оранжевого цвета. Протонная ЯМР-спектроскопия выявила, что реакция не завершилась. Масло помещалось в условия, описанные выше, на дополнительные 48 часов. Капиллярная газ-хроматография показала, что реакция завершилась на 85%. Полученный материал очищался колонной хроматографией на SiO2, (10% метанол/CHCl3), а затем ВЭЖХ на CN колонне обратной фазы для получения 122 мг искомого соединения. Температура плавления 140-140.5oC.
Элементарный анализ:
Рассчитано: C 71,06; H 7,37; N 4,87.
Получено: C 70,97; H 7,17; N 4,60.
Пример 59
Получение (R) (+)-транс-4-метил-3-хлоро-10b-метил-1,2,3,4,4a, 5,6,10b-октагидробензо[f]хинолин-3-она
Следуя процедурам, описанным в примере 40, этапах A, B, C, D, E и F, с использованием п-хлорфенилуксусной кислоты в качестве исходного материала и в этапе F с использованием метиламина вместо аммиака и 2-пропанола вместо этиленгликоля, было получено соединение (R) (+)-4-метил-8-хлоро-10b-метил-1,2,3,4,6, 10b-гексагидробензо[f] хинолин-3-он. Этот гексагидробензо[f]хинолин был восстановлен в соответствии с процедурой, описанной в примере 40. этапе G. Неочищенный продукт очищался хроматографией на SiO2 (этилацетат в качестве элюента) для получения 5,6 г искомого соединения в виде светлого масла, затвердевавшего со временем. Температура плавления 60-61oC.
Элементарный анализ:
Рассчитано: C 68,30; H 6,88; N 5,31.
Получено: C 68,14; H 6,94; N 5,27.
Оптическое вращение: 589 нм= +76,16o
(C = 1, CHCl3)
Пример 60
Получение (R) (+)-транс-4-метил-8-хлоро-10b-метил-3,4,4a,5,6, 10b-гексагидробензо[f]хинолин-3-она
Следуя процедурам, описанным в примере 59, было получено соединение (R) (+)-транс-4-метил-8-хлоро-10b-метил-1,2,3,4, 4a, 5,6,10b-октагидробензо[f] хинолин-3-он в виде смеси (4:1 транс:цис) с его цис-изомером. Перемешанный, охлажденный (-78oC) раствор 1,4 г (5,56 мМ) этой смеси в ТГФ (25 мл) обрабатывался гексаметилдисилазидом калия (12,2 мл; 6,12 мМ), его раствором в толуоле. Раствор перемешивался 30 минут и обрабатывался раствором фенилселенилхлорида (1,17 г, 6,117 мМ) в 5 мл ТГФ. Раствор нагревался до температуры окружающей среды свыше 1 часа и затем охлаждался 20 мл насыщенного раствора NH4Cl. Смесь распределялась между этилацетатом и водой, и водная фаза высушивалась над Na2SO4 и концентрировалась до получения диастереометрической смеси фенилселенидов. Неочищенные фенилселениды растворялись в 10 мл этилацетата и охлаждались до 0oC. К раствору добавлялся насыщенный раствор NaHCO3 (2 мл), сопровождаемый 0,4 мл 30% H2O2. Смесь подогревалась до температуры окружающей среды и перемешивалась 2 часа. Смесь экстрагировалась этилацетатом, и остаток подвергался хроматографии на SiO2 (этилацетат в качестве элюента) для получения 611 мг искомого соединения в виде твердого воскообразного вещества. Температура плавления 45-48oC.
Элементарный анализ:
Рассчитано: C 68,83; H 6,16; N 5,35.
Получено: C 69,26; H 6,48; N 5,08.
Оптическое вращение: 589 нм = +87,38o
(C = 1, CHCl3)
Пример 61
Получение (R) (+)-транс-8-хлоро-10b-метил-3,4,4a, 5,6, 10b-гексагидробензо[f]хинолин-3-она
С помощью процедур, описанных в примере 40, этапах A, B, C, D, Е, F и G, было получено соединение (R) (+)-8-хлоро-10b-метил-1,2,3,4,4a,5,6, 10-октагидробензо[f] хинолин-3-он в виде смеси (4:1) транс- и цис-изомеров. Смесь (1,02 г; 4,07 мМ) растворялась в 15 мл диокоана и обрабатывалась дихлордицианохиноном (1,02 г; 4,45 мМ), сопровождаемым бис (триметилсилил)трифторацетамидом (4,8 мл; 17,9 мМ). Раствор перемешивался при температуре окружающей среды 4 часа, а затем нагревался до 100oC в течение 14 часов. Реакционная смесь охлаждалась до температуры окружающей среды и распределялась между этилацетатом и водой. Органическая фаза концентрировалась в вакууме, и остаток подвергался хроматографии на SiO2 (этилацетат в качестве элюента) для получения 670 мг искомого соединения в виде масла. Образец кристаллизовался из диэтилового эфира для получения 60 мг искомого соединения в виде светлого твердого вещества. Температура плавления 151-154oC.
Элементарный анализ:
Рассчитано: +1/2 М H2O C 65,50; H 5,89; N 5,46.
Получено: C 65,67; H 6,06; N 5,40.
Оптическое вращение: 589 нм = +51,33o
(C = 1, CHCl3) 365 нм = -85,55o
Пример 62
Получение транс-dl-8-трифторометил-4-метил-1,2,3,4,4a,5,6, 10b-октагидробензо[f]хинолин-3-она
Транс-dl-8-бромо-4-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-он был получен в соответствии с процедурами, описанными в примере 1. Смесь бромида (1,2 г, 4,1 мМ), трифторацетата натрия (2,22 г, 16,3 мМ) и йодида меди (1,55 г, 8,1 мМ) в 25 мл N-метил-2-пирролидинона нагревалась при 180oC 18 часов в атмосфере аргона. После охлаждения смесь фильтровалась через силикагель, а силикагелевая пробка промывалась этилацетатом. Объединенные органические слои два раза промывались водой и один раз рассолом, высушивались над MgSO4 и выпаривались в вакууме до получения 1,14 г темного масла, кристаллизующегося во времени. Неочищенный продукт очищался с помощью мгновенной хроматографии на силикагеле (этилацетат), сопровождаемой хроматографией обратной фазы (1:1 вода/ацетонитрил) для получения 312 мг твердого вещества белого цвета (выход 27%).
Масс-спектр с высоким разрешением. Рассчитано для C15H13F3NO (283,118480); получено 283,119620.
Пример 63
Получение (+)-(4aR)-(10bR)-8-хлоро-1,2,3,4,4a,5,6, 10b-октагидробензо[f] хинолин-3-она
A. (S)-(-)-8-хлоро-4-( α -метилбензил)-1,2,3,4,5,6,-гексагидробензо[f] хинолин-3-он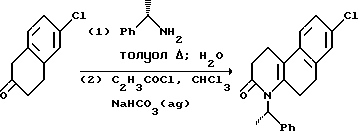
К перемешанному раствору 6-хлоро-2-тетралона (1,0 экв., 100,0 мМ, 18,06 г) в толуоле (300 мл) добавлялся (S)-(-)-α- метилбензиламин (1,0 экв., 100,0 мМ, 12,9 мл). Смесь нагревалась с обратным холодильником 3 часа с азеотропным удалением воды (Dean-Stark). Охлажденная реакционная смесь концентрировалась in vacuo до получения енамина, (S)-6-хлоро -2-(α-метилбензил)-3,4- дигидронафталина в виде масла пурпурного цвета. Масло помещалось в хлороформ (150 мл) и к нему добавлялся насыщенный водный бикарбонат натрия. Тщательно перемешанная смесь обрабатывалась акрилоилхлоридом (1,05 экв; 105,0 мМ, 8,5 мл), добавляемым по каплям в течение более 5 минут. Реакционная смесь помешивалась при температуре окружающей среды 20 минут, затем разбавлялась хлороформом, и слои разделялись. Водный слой экстрагировался хлороформом (1x). Объединенные органические слои промывались рассолом (1x), высушивались (Na2SO4) и концентрировались in vacuo до получения 37,9 г темного масла. Неочищенное масло подвергалось ВЭЖХ на силикагеле (градиент элюирования гексанами увеличивается до 25% этилацетата/гексанов) для получения 19,03 г масла пурпурного цвета, которое в дальнейшем подвергалось очистке мгновенной хроматографией на силикагеле (элюирование 20% этилацетатом/гексанами) для получения 17,96 г /53% выход/ искомого соединения в виде коричневого стекловидного вещества, которое оказалось гомогенным согласно ВЭЖХ (обратная фаза, C18, подвижная фаза: 60% ацетонитрил/ 0,5% водный буфер из ацетата аммония), тонкослойной хроматографии (силикагель, Rf = ~ 0,5, с 40% этилацетатом/гексанами), и 300 МГц
1Н ЯМР анализу. FDMS: m/e = 337. α[D] 589 = - 24,75 (C=1,0, CHCl3).
B. (-)-(4aR)-(10bR)-8-хлоро -4-(S-α- метилбензил)- 1,2,3,4,4a,5,6,10b-октагидробензо[f]хинолин-3-он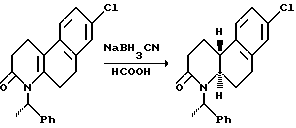
Перемешанный раствор (12,0 г; 35,4 мМ) продукта из этапа А в 96% муравьиной кислоте (175 мл) обрабатывался твердым цианоборогидридом натрия (2,0 экв. , 70,98 мМ, 4,46 г) каждый час в течение 5 часов (всего 10.экв., 354,9 мМ, 22,3 г). Смесь перемешивалась в течение ночи (14 часов) при температуре окружающей среды. Реакционная смесь концентрировалась до получения пасты светло-желтого цвета, которая помещалась в дихлорметан и промывалась 1N гидроксидом натрия (1x). Водный слой обратно экстрагировался дихлорметаном (2x). Объединенные органические слои промывались рассолом (1x), высушивались (Na2SO4) и концентрировались in vacuo до получения 12,0 г бесцветной пены. Предварительная ВЭЖХ остатка на силикагеле (градиент элюирования дихлорметаном - 1% этилацетат/дихлорметан) и соединенных фракций, которые были более чем на 90% диастереомерически чисты, дала 1,63 г искомого соединения в виде кристаллического твердого вещества. ВЭЖХ анализ: обратная фаза, C18, 230 нм, подвижная фаза; 60% ацетонитрил /40% 0,5% буфера ацетата аммония). Рекристаллизация твердого вещества из этилацетата дала искомое соединение в диастереомерически чистой форме в виде бесцветных игл (более чем 99: 1 диастереомерической чистоты, согласно ВЭЖХ анализу). Фракции, обогащенные искомым соединением, соединялись и концентрировались in vacuo до получения 3,0 г бесцветной пены. Фракциональная кристаллизация этого материала из этилацетата (3 кристаллизации) дала дополнительные 370 мг чистого искомого соединения, 2,50 г, выход 21%. Температура плавления 176-177oC. FDMS: m\e = 339. α[D]589 = -126,63 (C = 1,0, CHCl3). Полная конфигурация искомого соединения была выявлена
Теоретическое C 74,21; H 6,52; N 4,12.
Полученное С 74,07; Н 6,59; N 4,04.
C. (+)-(4aR)-(10bR)-8-хлоро-1,2,3,4,4a, 5,6,10b-октагидробензо[f]хинолин-3-он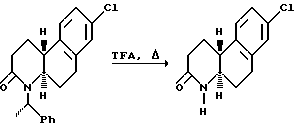
Смесь продукта из этапа B (1,0 экв., 4,09 мМ, 1,39 г) и трифторуксусной кислоты (35 мл) помещалась в круглодонную колбу. Перемешанная смесь нагревалась с обратным холодильником 2 часа. Охлажденная реакционная смесь концентрировалась in vacuo, помещалась в дихлорметан и промывалась насыщенным водным раствором бикарбоната натрия (1x). Слои разделялись, и водный слой обратно экстрагировался дихлорметаном (1x). Объединенные органические слои промывались рассолом (1х), высушивались (Na2SO4) и концентрировались in vacuo до получения твердого вещества белого цвета. Рекристаллизация из этилацетата дала 0,81 г, выход 84% искомого соединения в виде бесцветных игл. Температура плавления 241,5-242oC. FDMS: m\e = 235. α[D]589 = 32,61 (C = 1,0, ТГФ)
Теоретическое C 66,24; H 5,99; N 5,94.
Полученное С 66,35; Н 6,10; N 5,83.
Пример 64
(-)-(4AR)-(10bR)-8-хлоро-1-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f] хинолин-3-он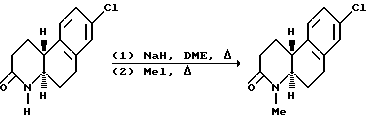
К перемешанному раствору продукта из этапа C примера 63 (1,0 экв., 0,288 мМ, 98 мг) в сухом 1,2-диметоксиэтане (5 мл) добавлялась 60% (вес/вес) дисперсия гидрида натрия (26 мг) в масле. Смесь нагревалась с обратным холодильником в атмосфере азота с перемешиванием в течение 1 часа, охлаждалась и обрабатывалась метилйодидом (5,0 экв., 1,44 мМ, 0,09 мл). Смесь нагревалась с обратным холодильником еще 1,5 часа. Охлажденная реакционная смесь охлаждалась водой (1 мл) и экстрагировалась дихлорметаном (3x). Объединенные органические слои промывались рассолом (1x), высушивались (Na2SO4) и концентрировались in vacuo до получения масла оранжевого цвета. Предварительная тонкослойная хроматография на силикагеле (2 мм тарелка, с этилацетатом) дала 50 мг, выход 69% бесцветного твердого вещества. Температура плавления 71.5-73oC. α[D]589 = 74.80 (C = 1.0, CHCl3) HRMS (FAB+) Рассчитано для C14H17NOCl: 250.1003. Получено 250.0999.
Пример 65
Получение (+)-(4aS)-(10bS)-8-хлоро-4-(метил)-1,2,3,4,4a, 5, 6,10b-октагидробензо[f]хинолин-3-она
A. (+)-(4aS)-(10bS)-8-хлоро -4-(S-α- метилбензил)- 1,2,3,4,4a,5,6,10b-октагидробензо[f]хинолин-3-он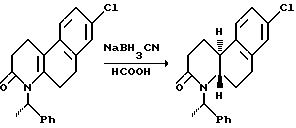
Перемешанный раствор продукта из этапа A, примера 63, в 96% муравьиной кислоте (20 мл) обрабатывался цианоборогидридом натрия по порциям более 8 часов (30 экв. , 88,8 мМ, 6,68 г) при температуре окружающей среды. Смесь затем нагревалась при 50oC 2 часа. Охлажденная реакционная смесь концентрировалась in vacuo до получения бесцветной пены. Остаток помещался в дихлорметан и промывался водой (1x). Водный слой обратно экстрагировался дихлорметаном (2x). Объединенные органические вытяжки промывались 1N гидроксидом натрия (1x), рассолом (1x), высушивались (Na2SO4) и концентрировались in vacuo до получения бесцветной пены. Жидкостная хроматография среднего давления на силикагеле (элюирование) 10% этилацетатом/дихлорметаном) дала 201 мг продукта из этапа B примера 63. Дальнейшее элюирование дало 129 мг искомого соединения в виде бесцветного твердого вещества, выход 13%. ВЭЖХ анализ этого материала (обратная фаза, C18, 230 нм, 60% ацетонитрил/ 40% 0,5% водного буфера ацетата аммония) выявил, что материал был более чем на 99% диастереомерически чистым.
B. (+)-(4aS)-(10bS)-8-хлоро-4-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-он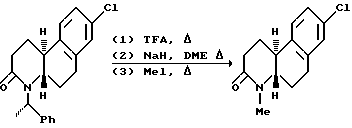
Перемешанный раствор продукта из этапа A (1,0 экв., 0,359 мМ, 122 мг) в трифторуксусной кислоте (6 мл) нагревался с обратным холодильником 1,5 часа. Охлажденная реакционная смесь концентрировалась in vacuo до выхода твердого вещества желтого цвета, который помещался в дихлорметан и промывался насыщенным раствором карбоната натрия (1x). Водный слой обратно экстрагировался дихлорметаном (1x). Объединенные органические вытяжки промывались рассолом (1x), высушивались (Na2SO4) и концентрировались in vacuo до получения (-)-(4aS)-(10bS)-8-хлоро-1,2,3,4,4a,5,6, 10b-октагидробензо[f]хинолин-3-она в виде светло-желтого твердого вещества. Остаток растворялся в сухом 1,2-диметоксиэтане (5 мл) и обрабатывался гидридом натрия (19 мг, 60% дисперсия в минеральном масле, промытая 3x пентаном). Смесь нагревалась с обратным холодильником 1,5 часа, охлаждалась и обрабатывалась метилйодидом (5,0 экв., 1,80 мМ, 0,2 мл). Реакционная смесь нагревалась еще 1,5 часа. Охлажденная реакционная смесь охлаждалась водой и экстрагировалась дихлорметаном (3x). Объединенные органические вытяжки промывались рассолом (1x), высушивались (Na2SO4) и концентрировались in vacuo до получения масла оранжевого цвета. Предварительная тонкослойная хроматография на силикагеле (2 мм тарелка, с этилацетатом) дала искомое соединение (56 мг, 62% выход) в виде бесцветного твердого вещества. Температура плавления 71,5-73oC. α[D]589 = = + 79,00 (C = 1,0CHCl3). HRMS(FAB+): Рассчитано для C14H17NOCl: 250,0999. Получено 250,1002.
В примерах 66-69 используются следующие сокращения "система A ВЭЖХ": 40% ацетонитрил в воде и 0,5% ацетат аммония на Waters Nova-pak C-8® при 220 нм, при 2,00 мл/мин, 25oC. "Система B ВЭЖХ": 50% ацетонитрил в воде и 0,5% ацетат аммония на Dupont Zorbax ®C-18 при 220 нм, при 2,00 мл/мин, 25oC. "Система C ВЭЖХ": 10% изопропиловый спирт в гексане на Chiralсel OD® при 254 нм, при 1,00 мл/мин и 25oC.
"Система D ВЭЖХ": 10% изопропиловый спирт в гексане на Chiralсel OD® при 220 нм, при 2,00 мл/мин и 40oC.
Пример 66.
Получение (+)-(4aR)-(10bR)-4-метил-8-хлоро-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-она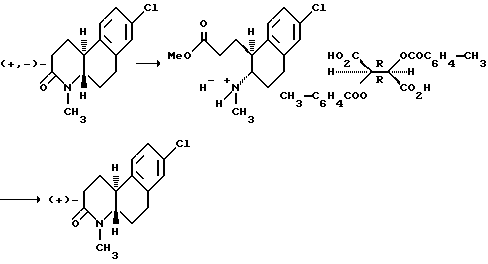
A. Соль ди-п-толуоилвинной кислоты 1-(2-метоксикарбонилэтил)- 2-(метиламино-6-хлоро)-1,2,3,4 -тетрагидронафталина.
Раствор транс (d, l)-4-метил-8-хлоро-1,2,3,4,4a,5,6, 10b-октагидробензо[f] хинолин-3-она (1,5 г), полученного в основном по примеру 6, в метаноле (30 мл) с концентрированной серной кислотой (3,5 г) нагревался с обратным холодильником 144 часа. Раствор концентрировался приблизительно до 20 мл и разбавлялся эфиром (50 мл), водой (100 мл) и насыщенным бикарбонатом натрия (20 мл, pH 9). Фазы разделялась, и водная фаза далее экстрагировалась этилацетатом (100 мл) и метиленхлоридом (100 мл). Объединенные органические фазы высушивались с помощью 4-A молекулярных сит и добавлялись к раствору моногидрата (-)-п-толуоил-L-винной кислоты (ДТВК) (2,00 г) в метаноле (10 мл). Все летучие соединения удалялись под вакуумом, а полученная пена растворялась приблизительно в 40 мл метанола. Твердые вещества отфильтровывались и высушивались с получением 1,01 г (теоретически 50%) соли, смешанной с 6,3% негидролизованного транс-dl-8-хлоро-4-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-она. Температура плавления 126-130oC.
ТСХ (силикагель с толуолом-этилацетатом-уксусной кислотой- метанолом 7: 4:1:4: Rf = 0,34)
ВЭЖХ (система A): tr (мин): 0,48 (43% ДТВК), tr = 1,33 (53% свободного аминоэстера), tr = 3,36 (3,15%, рацемат).
ВЭЖХ (система B): tr (мин) 1,06 (48,1%, ДТВК), 2,77 (1,2%, неизвестно), 3,73 (46,1%, свободный аминоэстер), 5,54 (4,5%, (искомое соединение).
1H ЯМР (CDCl3): δ 5,43 (с, 2H), 3,57 (с, 3H), 2,38 (с, 6H), УФ (метанол): λ(ε): 276 (30,100).
ИК (CHCl3): 1723 см-1.
B. (+)-(4aR)-(10bR)-4-метил-8-хлоро-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-он
Раствор вышеупомянутой соли ДТBК (100 мг) перемешивался с толуолом (5 мл), водой (10 мл) и бикарбонатом натрия (50 мл) 20 минут при 25oC. Толуоловый слой отделялся, высушивался (сульфатом натрия), нагревался (90-100oC, 18 часов) и выпаривался с получением искомого соединения.
1H ЯМР (бензол- d6): 6,99 (dxd, 1H), 6,83 (d, 1H), 6,57 (d, 1H), 2,67 (3H, с), 2,40 (dxt, 2H).
1H ЯМР (бензол- d6 с 2 экз. R-(-)-2,2,2-трифторо-1-(9-антрил)этанол (ТФАЭ): d 2,14 (3H, с), синглет метила в верхней части на 2,10 для 5-6% (-) энантиомера.
ВЭЖХ (система A): tr (мин) 3,36 (искомое соединение).
ВЭЖХ (система B): tr (мин) 5,68 (искомое соединение).
ВЭЖХ (система D): tr (мин) 13,85 (92%, искомое соединение), 15,8 (8%, (-) энантиомер).
1H ЯМР (CDCl3): 3,07 (3H, с).
Пример 67
Получение (+)-(4aR)-(10bR)-4-метил-8-хлоро-1,2,3,4,4a,5,6, 10b-октагидробензо[f]хинолин-3-он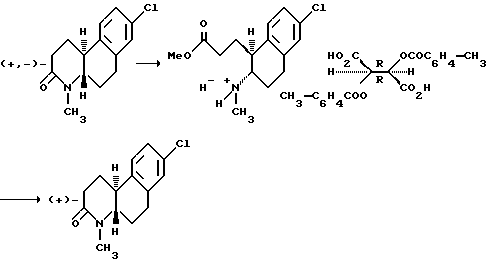
A. Ди-п-толуилвиннокислотная соль 1-(2-метоксикарбонилэтил)- 2-(метиламино)-6-хлоро-1,2,3,4-тетрагидронафталина
Раствор транс-d,l-8-хлоро-4-метил-1,2,3,4,4a,5,6, 10b-октагидробензо[f] хинолин-3-она (2,3 г) в 0,85 N соляной кислоте в метаноле нагревался 168 часов, после чего добавлялись концентрированная серная кислота (3,00 мл) и дополнительное количество метанола (100 мл). По прошествии 96 часов смесь концентрировалась и обрабатывалась твердым бикарбонатом натрия (20 г). Смесь, содержащая 1-(2-метоксикарбонилэтил)-2- (метиламино)-6-хлоро-1,2,3,4-тетрагидронафталин, распределялась между этилацетатом (100 мл) и водой (100 мл). Фазы разделялись, и водный слой экстрагировался 250 мл метиленхлорида. Органические фазы соединялись и фильтровались через 3-A молекулярные сита (15 г) в раствор 5,9 г (-)-п-толуол-L-винной кислоты (моногидрата) в метаноле. Все летучие компоненты удалялись под вакуумом, и белая пена сушилась в вакууме 30 часов до получения стереомерической соли (6,1 г, 99%; содержащей 5,0% негидролизованного транс-dl-8-хлоро-4-метил-1,2,3,4,4a,5,6, 10b-октагидробензо[f]хинолин-3-она, температура плавления 126-130oC.
Вышеупомянутая пена (соль DTBK) (5,69 г) вываривалась в метаноле (43oC, 32 мл), и полученный раствор охлаждался до 25oC. Смесь фильтровалась и сушилась в вакууме с получением 2,92 г соли DTBK. Рекристаллизация этой соли из 30 мл метанола дала 2,12 г чистой по строению соли (47%, 94% теоретически). ВЭЖХ (система A): 0,48 (46%, DTBK), 1,28 (54%, аминоэстер), других посторонних веществ не обнаружено.
B (+)-(4aR)-(10bR)-4-метил-8-хлоро-1,2,3,4,4a,5,6, 10b-октагидробензо[f] хинолин-3-он
Вышеупомянутая соль (1,50 г) перемешивалась (25oC, 10 минут) с толуолом (30 мл), насыщенным водным бикарбонатом натрия (10 мл) и водой (40 мл). Фазы разделялись, и толуоловый слой высушивался (3-A молекулярные сита). Полученный раствор нагревался (16 часов, 92oC) и выпаривался до получения 0,55 г (+)-(4aR)-(10bR)- 4-метил-8-хлоро-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-она, смешанного с 5-7% энантиомера (-)-(4aR)-(10bR)-8-хлоро-4-метил-1,2,3,4,4a,5,6, 10b-октагидробензо[f]хинолин-3-она.
ВЭЖХ (система A): 3,36 (97,9%, искомое соединение).
ВЭЖХ (система B): 5,76 (99,9%, искомое соединение).
ВЭЖХ (система C): 93% искомого соединения и 7% (-) энантиомера.
УФ (метанол) 204 (24100).
Пример 68
Получение (-)-(4aR)-(10bR)-8-хлоро-4-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-она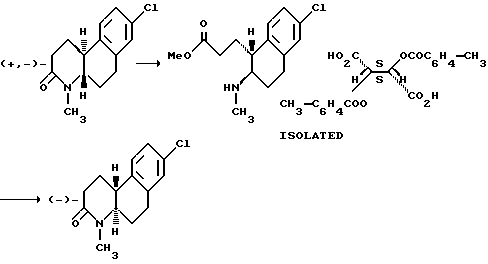
A. Ди-п-толуоилвиннокислая соль 1-(2-метоксикарбонилэтил-2-(метиламино)-6-хлоро-1,2,3,4- тетрагидронафталина
Раствор 90,0 г транс-dl-8-хлоро-4-метил-1,2,3,4,4a,5,6, 10b-октагидробензо[f] хинолин-3-она (приблизительно 95% чистоты согласно ВЭЖХ анализу), который готовился в основном в соответствии с процедурами примера 6, перемешивался при нагревании с обратным холодильником в азоте в безводном метаноле (4 л) и серной кислоте (98%, 200 мл) в течение 160 часов (до тех пор, пока не оставалось < 1,5% транс-dl-8-хлоро-4-метил-1,2,3,4,4a, 5,6,10b-октагидробензо[f] хинолин-3-она, согласно ВЭЖХ). Раствор концентрировался в вакууме и экстрагировался при 10-15oC с использованием метиленхлорида, бикарбоната натрия (700 г) и воды (2 л). Обработка органических вытяжек (+)-п-толуоил-L-винной кислоты (DTBK) (моногидрата) (производное от ненатуральной винной кислоты/ S,S изомер), 132,9 г в метаноле (700 мл) и три кристаллизации дали 48,2 г очищенной соли (температура плавления 125-130oC, годная для аналитического стандарта).
ВЭЖХ (система A): 0.48 (DTBK), 1,28 (аминоэстер).
УФ (метанол): 204 (51200), 239 (31400), 270 (2500).
B. (-)-(4aR)-(10bR)-8-хлоро-4-метил-1,2,3,4,4a, 5,6, 10b-октагидробензо[f]хинолин-3-он
Соль DTBK, вышеупомянутая, (500 мг) перемешивалась с 10 мл толуола, 5 мл воды и 5 мл насыщенного водного бикарбоната натрия (25oC). Водный слой отделялся и снова экстрагировался толуолом (5 мл). Объединенные толуоловые вытяжки промывались один раз насыщенным бикарбонатом натрия (5 мл), сушились (4-A молекулярные сита) и нагревались при 95-105oC (18 часов). Толуол удалялся в потоке азота при 40oC, и полученное масло растиралось с гексаном-эфиром до получения соединения B.
ТСХ (силикагель с толуолом-этилацетатом-уксусной кислотой, 7:4:1): Rf = 0,68.
ВЭЖХ (система A): 3,25 мин (> 99%, искомое соединение).
ВЭЖХ (система C): 31,12 мин (> 99% искомого соединения), 28 мин (< 1%) + (энантиомер).
1H ЯМР (CDCl3): 3,22 (dxd).
ИК (CHCl3): 1620 см-1.
УФ (метанол): 205 (20800).
Пример 69
Получение ди-п-толуоилвиннокислой соли 1-(2-метоксикарбонилэтил)-2-(метиламин)-6-хлоро-1,2,3, 4-тетрагидронафталина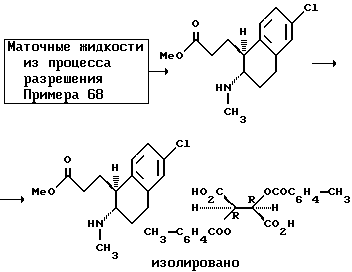
Начальные фильтраты растворения-выделения и маточная жидкость от первой рекристаллизации из примера 68, описанного выше, соединялись и выпаривались при 25oC в вакууме до получения пены белого цвета (190,4 г). Эта соль (180,4 г) эстрагировалась холодным метиленхлоридом, водой, бикарбонатом натрия, как описывалось в примере 68. Обработка вытяжек (-)-п-толуоил-L-винной кислотой -моногидратом (92,7 г) в метаноле (450 мл) и три кристаллизации дали чистую соль DTBK (57,0 г). Температура плавления 126-130oC.
ВЭЖХ (система A): 0,48 мин (47%, DTBK), 1,28 (53%, аминоэфир).
ИК (CHCl3): 1720 см-1.
УФ (метанол): 204 (46200), 239 (28000), 270 (4200).
Следуя процедурам, описанным выше, специалист может получить соединения формулы I.
Как указывалось ранее, соединения настоящего изобретения полезны в торможении преобразования тестостерона в 5α- дигидро тестостерон (ДГТ), и более конкретно, изофермент вида 1. Поэтому другим вариантом настоящего изобретения является способ торможения 5α- редуктазы назначением млекопитающему, нуждающемуся в торможении 5α- редуктазы, дозы (эффективного количества) соединения по формуле I или его фармацевтически приемлемой соли. Соединения настоящего изобретения полезны в чистом виде или в комбинации с другими ингибиторами 5α- редуктазы, в частности ингибиторами фермента типа 2, такими как финастерид, 3-карбоксистероиды, описанными в Holt et al., J. Med. Chem. 33, 943-950 (1990), и указанные здесь как ссылка, и соединения, описанные в ЕП 0291245, также данные здесь как ссылка.
Термин "эффективное количество", используемый здесь, означает количество соединения настоящего изобретения, которое способно тормозить преобразование тестостерона в 5α- дигидротестостерон, который катализуется ферментом 5α- редуктаза, и, в частности, тормозит 5α- редуктазу; более конкретно, изофермент типа 1. Торможение 5α- редуктазы настоящего способа включает как медицинское терапевтическое, так и/или профилактическое лечение. Определенная доза соединения, назначенные в соответствии с этим изобретением для достижения терапевтических и/или профилактических эффектов, будет, конечно определяться конкретными обстоятельствами случая, включающими, например, самосоединение, вид назначения и состояние организма. Типичная ежедневная доза содержит нетоксичный уровень дозировки от 0,01 мг/кг до 50 мг/кг веса тела активного соединения этого изобретения. Предпочтительные дозировки составляют от 0,05 до 20 мг/кг и идеально от 0,1 до 10 мг/кг.
Многие физиологические функции связаны с чрезмерной выработкой 5α- дигидротестостерона. Как таковые, соединения этого изобретения обладают способностью лечить у млекопитающих множество расстройств, связанных с 5α- дигидротестостероном, включающих доброкачественные разрастания предстательной железы (или гипертрофию), облысение у мужчин, угри, себоррею, андрогенное облысение, гирсутизм и рак предстательной железы. Поэтому настоящее изобретение также предусматривает методы лечения указанных расстройств дозировками, указанными выше, для торможения катализного преобразования 5α- редуктазы тестостерона в 5α- дигидротестостерон. Соединения настоящего изобретения используются самостоятельно или в комбинации с другими ингибиторами 5α- редуктазы, такими как финастерил, 3-карбоксистероиды и соединениями в ЕП 0291245, в таких методах лечения.
Соединения могут назначаться в разных видах применения, включающих оральное, ректальное кожное, подкожное, внутривенное, внутримышечное, через нос и наружное применение при облысении у мужчин, угрях и гирсутизме. Обычно эти соединения формуются в препараты по назначения. Поэтому другой аспект изобретения заключается в получении фармацевтического препарата, включающего эффективное количество соединения формулы I или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, разбавитель или наполнитель. Активный ингредиент таких препаратов содержит от 0,1% до 99,9% по весу препарата. "Фармацевтически приемлемый" означает носитель, разбавитель или наполнитель, которые совместимы с другими ингредиентами препарата и не вредны для реципиента. Эти фармацевтические препараты готовятся известными процедурами с известными ингредиентами. В приготовлении композиций настоящего изобретения активный ингредиент обычно смешивается с носителем, или разбавляется носителем, или заключается в носитель, который может быть в виде капсулы, пакетика, бумажного или другого контейнера. Если носитель служит разбавителем, он может быть твердым, полутвердым или жидким материалом, действующим как носитель, наполнитель или среда для активного ингредиента. Таким образом, композиция может быть в виде таблеток, пилюль, порошков, пакетиков, эликсиров, суспензий, эмульсий, растворов, сиропов, аэрозолей (в твердой или жидкой среде), мягких и твердых желатиновых капсул, шариков, стерильных растворов для инъекций, стерильных упакованных порошков, облаток, лепешек и прочее. Типичные препараты для наружного применения - мази, кремы, гели и лосьоны, содержащие, например, до 10 вес.% активного соединения.
Следующие примеры препаратов являются только иллюстративными и не ограничивают объем изобретения никоим образом. "Активный ингредиент" означает соединение по формуле I или его фармацевтически приемлемую соль.
Препарат 1
Твердые желатиновые капсулы готовятся с использованием следующих ингредиентов:
Ингредиенты - Количество (мг/капсулу)
Активный ингредиент - 250
Крахмал, высушенный - 200
Стеарат магния - 10
Всего - 460 мг
Препарат 2
Готовится таблетка с использованием следующих ингредиентов:
Ингредиенты - Количество (мг/капсулу)
Активный ингредиент - 250
Целлюлоза микрокристаллическая - 400
Диоксид кремния, расплавленный - 10
Стеариновая кислота - 5
Всего - 665 мг
Компоненты смешиваются и компрессией формуются таблетки, каждая весом 665 мг.
Препарат 3
Готовится аэрозольный раствор, содержащий следующие компоненты:
Ингредиенты - Вес
Активный ингредиент - 0,25
Этанол - 25,75
Реактивное вещество 22
(Хлордифторметан) - 70,00
Всего - 100,00
Активное соединение смешивается с метанолом и смесь добавляется к части реактивного вещества 22, охлаждается до -30oC и переносится в наполнительное устройство. Требуемое количество затем подается в контейнер из нержавеющей стали и разбавляется небольшим количеством реактивного вещества. Далее к контейнеру крепится клапан.
Препарат 4
Таблетки, каждая содержащая 60 мг активного ингредиента, готовятся следующим образом:
Активный ингредиент - 60 мг
Крахмал - 45 мл
Микрокристаллическая целлюлоза - 35 мг
Поливинилпирролидон (в виде 10% раствора в воде) - 4 мг
Натрий карбоксиметил крахмал - 4,5 мг
Стеарат магния - 0,5 мг
Тальк - 1 мг
Всего - 150 мг
Активный ингредиент, крахмал и целлюлоза пропускаются через сито 45 меш U. S. и тщательно смешиваются. Водный раствор, содержащий поливинил-пирролидон, смешивается с остальным порошком и смесь затем пропускается через сито N 14 меш U.S. Полученнае гранулы высушиваются при 50oC и пропускаются через сито и 18 меш U.S. Натрий карбоксиметил крахмал, стеарат магния и тальк, предварительно пропущенный через сито N 60 U.S., добавляются затем к гранулам, которые после смешивания прессуются на таблетирующей машине для получения таблеток весом 150 мг каждая.
Препарат 5
Капсулы, каждая, содержащая 80 мг активного ингредиента, готовятся следующим образом:
Активный ингредиент - 80 мг
Крахмал - 59 мг
Микрокристаллическая целлюлоза - 59 мг
Стеарат магния - 2 мг
Всего - 200 мг
Активный ингредиент, целлюлоза, крахмал и стеарат магния смешиваются, пропускаются через сито N 45 U.S. и заполняются в твердые желатиновые капсулы в количестве по 200 мг.
Препарат 6
Шарики, каждый содержащий 225 мг активного ингредиента, готовятся следующим образом:
Активный ингредиент - 225 мг
Глицериды насыщенной жирной кислоты - 2,000 мг
Всего - 2,225 мг
Активный ингредиент пропускается через U.S. сито N 60 и суспензируется в глицеридах насыщенной жирной кислоты, ранее расплавленных с необходимым минимальным теплом. Затем смесь выливается в форму для шариков емкостью 2 г и остывает.
Препарат 7
Суспензии, каждая содержащая 50 мг активного ингредиента на 5 миллилитровую дозу, готовятся следующим образом:
Активный ингредиент - 50 мг
Натрийкарбоксиметилцеллюлоза - 50 мг
Сироп - 1,25 мг
Раствор бензойной кислоты - 0,10 мл
Отдушка - q.v.
Краситель - q.v.
Очищенная вода до общего количества - 5 мл
Активный ингредиент пропускается через U.S. сито N 45 меш и смешивается с натрийкарбоксиметилцеллюлозой и сиропом для получения гладкой пасты. Раствор бензойной кислоты, отдушка и краситель разбавляются частью воды и добавляются к пасте с перемешиванием. Затем добавляется достаточное количество воды для получения нужного объема.
Препарат 8
Препарат для внутривенного применения может готовиться следующим образом:
Активный ингредиент - 100 мг
Изотонический физиологический раствор - 1,000 мл
Раствор вышеуказанных ингредиентов обычно назначается внутривенно со скоростью вливания 1 мл в минуту.
Мази обычно готовятся с использованием либо (1) масляного основания, т. е. состоящего из масел или углеводородов, таких как белый петролат или минеральное масло, либо (2) абсорбентного основания, т.е. состоящего из безводного вещества или вещества, которые могут абсорбировать воду, например безводного ланолина. Обычно после формирования основания, масляного или абсорбентного, активный ингредиент добавляется в количестве, необходимом для желаемой концентрации.
Кремы представляют собой масло/водные эмульсии. Они состоят из масляной фазы (внутренняя фаза), включающей обычно фиксированные масла, углеводороды и прочее, такие как воски, петролат, минеральное масло и другие вещества, и водной фазы (непрерывной фазы), включающей воду и любые водорастворимые вещества, такие как добавленные соли. Две фазы стабилизируются использованием эмульгирующего агента, например ПАВ, такого как натрий лаурилсульфат; гидрофильных коллоидов, таких как аравийские коллоидальные глины и другие. При формировании эмульсии активный ингредиент обычно добавляется в количестве, чтобы получить нужную концентрацию.
Гели включают основу, выбранную из масляной основы, воды или эмульсионно-суспензионную основу, такую как описана выше. К основе добавляется желирующий агент, который образует матрицу в основе, увеличивая ее вязкость. Примерами желирующих агентов являются гидроксипропилцеллюлоза, полимеры акриловой кислоты и другие. Обычно активный ингредиент добавляется к препарату в нужной концентрации до добавления желирующего агента.
Количество активного ингредиента, включенного в препарат этого изобретения не критичен; концентрация должна быть только в пределах, достаточных, чтобы позволить готовое применение препарата к области назначения в количестве, обеспечивающем нужное количество активного ингредиента.
Для демонстрации способности соединений настоящего изобретения в ингибировании 5α- редуктазы проводились следующие эксперименты.
Клеточная культура: активность 5α- редуктазы измерялась с использованием Hs 68 фибробластов кожи половых органов человека, которые были первоначально получены от American Type Culture Collection (Rockvill, MD). Клетки выращивались в среде Dulbecco's Modified Eagle's Medium (дМЕМ) плюс 10% очищенную околоплодную коровью сыворотку, которая была дополнена амфотерицином (0,25 мг/мл) и гентамицином (25,0 мг/мл) (Gldco, Grand Island, NY).
Сыворотка очищалась от эндогенных стероидов инкубацией с углем, покрытым декстраном до его добавления к среде. Клетки выдерживались при 37oC в атмосфере из 95% воздуха, 5% CO2 и каждые 7-10 дней подвергались контакту с раствором трипсин-EDTA (ЭДТК) (0,025% трипсина, 0,265 мМ EDTA). До анализа HS 68 клетки собирались и помещались в чашки Falcon с 6 капиллярами (Becton Dickinson Labware, Lincoln, Рark) с плотностью 6•104 клеток на капилляр. Клетки выращивались 4-5 дней до тех пор, пока они не достигали приблизительно 80% слияния.
Метод I анализа. Субстрат готовился растворением немаркированного тестостерона (Sigma Chemical Co, St Louis, MO) в абсолютном этаноле с последующим добавлением [7-3H/N/] -тестостерона (23,3 Ci) ммоль, New England Nucleаr, Boston. Раствор стероида доводился до сухости под потоком азота и затем вновь восстанавливался в среде.
Метод II анализа. Субстратом, используемым для этого метода был [14C]-тестостерон (50 мCi/ммоль) New England Nucleer. Аликвота субстрата доводилась до сухости под потоком азота. После добавления 30 л этанола тестостерон растворялся в соответствующем объеме среды.
Подготовка образца. Испытуемые соединения помещались в абсолютной этанол для достижения желаемой концентрации. Последующие разбавления испытуемых соединений средой осуществлялись Biomek100 Automated Laboratory Workstation (Beckman Instruments, polo Alto, CA). Существующая среда в капиллярах образца высасывалась и заполнялась свежей средой. Испытуемое соединение затем добавлялось в капилляры с последующим добавлением 0,5 мл субстрата. Объем инкубационной смеси поддерживался 2,0 мл. Окончательная концентрация субстрата была 12 мМ. Концентрация испытуемых соединений колебалась от 0,001 до 150 мМ. Дополнительные три капилляра (фон), содержащие среду и субстрат, но не клетки, также включались в расчет на неферментный метаболизм субстрата. Чашки возвращались в инкубатор и инкубировались 4 часа.
В конце инкубации среда собиралась и переносилась в экстракционную трубку, содержащую 5 мл толуол-этанола (9:1), к которому добавлялось 20-250 г каждого из немаркированных стероидов (эстриол, эстрадиол, эстрон, 5α-андростан-3α, 17β-диол, 5α-андростан-3β, 17β-диол, 4-андростен-3,17-дион, 5α- андростан-3,17-дион, тестостерон и 5α- дигидротестостерон Steraloids, Inc., Wieton, NH). В случае метода I анализа экстракционная трубка также содержала 1,000 и 10,000 dpm [4-14C]-дигидротестостерона (50-60 мCi /ммоль) [4-14C] -тестостерона (50 мCi/ммоль) (New England Nuclear, Boston, Ma) соответственно. [14C] -стероиды были включены как восстановительные стандарты до восполнения процедурных потерь. Небольшое количество NaCl также добавлялось к экстракционным трубкам для воспрепятствования ценообразованию. Образцы подвергались вращению 30 секунд и затем центрифугировались 10 минут при 500 x г. Органическая фаза собиралась и образцы высушивались, повторно растворялись в дихлорметане-метаноле (9:1) и анализировались тонкослойной хроматографией с использованием одного из методов, описанных ниже.
Хроматография Метод I (двухразмерная).
Экстрагированные образцы подавались на силикагелевые 60F254, толщиной 0,25 мм тарелки тонкослойной хроматографии (EM Science, Cincinnatl, OH). Тарелки наращивались в первом размере системой растворителей, содержащей дихлорметан-этилацетат-метанолуксусную кислоту (160:38:1,5:0,5, Mallinckrodt Inc. , Paris, KY). Тарелки удалялись из резервуаров и высушивались до того, как они наращивались во втором размере в системе растворителей, содержащей дихлорметан-метанол-гидроксид аммония (180:19:1, Mallinckrodt Inc., Paris, KY).
Хроматография Метод II (одноразмерная).
Экстрагированные образцы подавались на силикагелевые 60F254, толщиной 0,25 мм тарелки тонкослойной хроматографии (EM, Science, Cincinnati, OH). Тарелки наращивались в системе растворителя, содержащей либо циклогексан-этилацетат (1: 1, Mallinckrodt Inc., Paris, KY), либо хлороформ-этилацетат (3: 1, Mallinckrodt Inc. , Paris, KY). Обе эти системы растворителя давали адекватное разделение и позволяли лучшие результаты при сравнении с двухразмерной системой, описанной выше.
Тарелки вначале просматривались под 254 мм УФ светом и отмечались видимые пятна. Затем на них распылялся примулин Aldrich Chеmical Co., Milwaukee, WI) (0,001% в ацетоне-воде (4:1) в соответствии с методом Wright, R.S.,"A reagent for the non-olestructive localization of steroids and some other lopophilic materials on silica gel thin-layer chromatograms," J. Chramatogr., 59; 220-221 (1971), который позволял идентификацию дополнительных стероидов при 365 мм УФ свете. Образцы, полученные с использованием метода II анализа анализировались непосредственно с использованием Ambis Radioanalitic Jmaging System (Ambis Systems, Inc., San Diego., CA). В случае образцов по методу I анализа пятна соскребались с тарелки пипеткой Пастера со стекловатой, заложенной в нее, соединенной с вакуумной линией. Стероиды элюировались прямо в сцинтилляционные ампулы добавлением 0,2 мл дихлорметана с последующими двумя промывами 2,0 мл метанола. Органический растворитель выпаривался и добавлялись 10,0 мл сцинтилляционной жидкости (Ready Organic, Beckman Instments, Inc. , Fullerton, CA). Образцы анализировались спектрометрией жидкой сцинцилляции.
После удаления среды из экстракции клетки промывались фосфатным буферным раствором (ФБР, pH 7,4) и затем собирались в трипсин-EDTK раствор (0,025% трипсин, 0,265 мМ EDTK). Клетки собирались и центрифугировались при 1400 x г на 5 минут. Всплывающая липкость декантировалась и клетки повторно суспендировались в PBS. Аликвота клеточной суспензии просчитывалась в Coulter Electronics, Ltd., Luton Beds, England. Остальные клетки обрабатывались звуком и определялся протеин в соответствии с методом Bradford, M.M., "A rapid and sensitive method for protein quantitation of microgvaam guantities of protein utilizing the principle of protein-dye bibing", Anal. Biohem., 72; 248-254 (1976). Производилась коррекция для процедурных потерь и данные выражались в качестве процентного ингибирования, основанного или на концентрации стероида в пикомолях на мг/протеин, или пикомолях/105 клетках.
Результаты оценки даны в таблице. Процентное ингибирование используется на шкале 0-100%, где 0 равен нулевой активности и 100 равно полному ингибированию.
Следует учесть, что описание и примеры, изложенные здесь, даются как иллюстрация, а не как ограничение и что можно ввести различные модификации и изменения, не выходя за рамки духа и объема настоящего изобретения, определенного прилагаемой формулой.
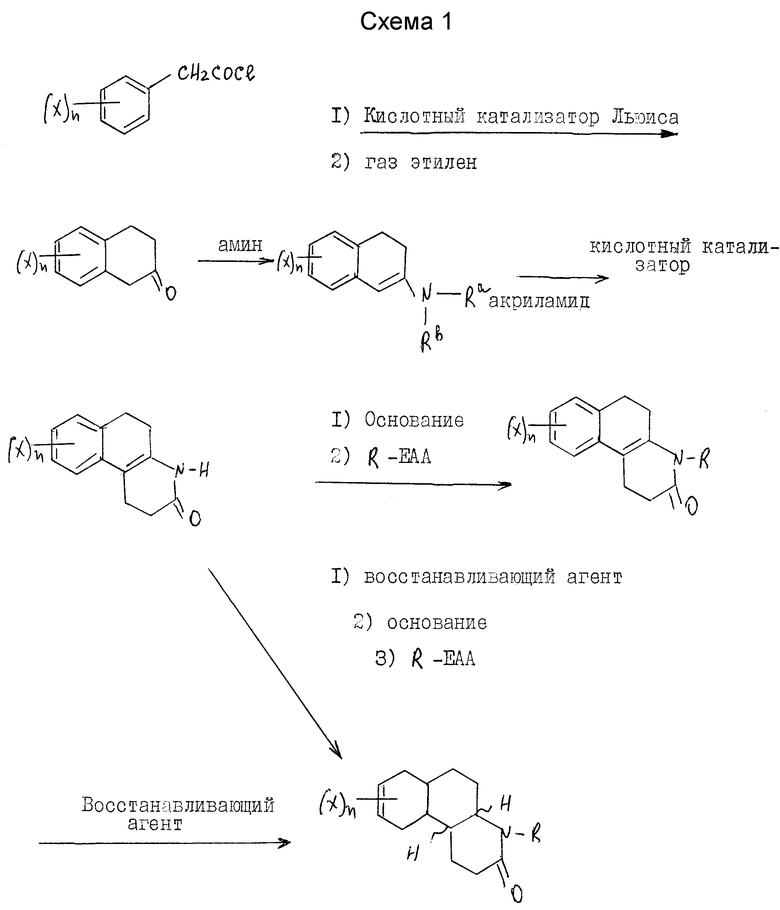

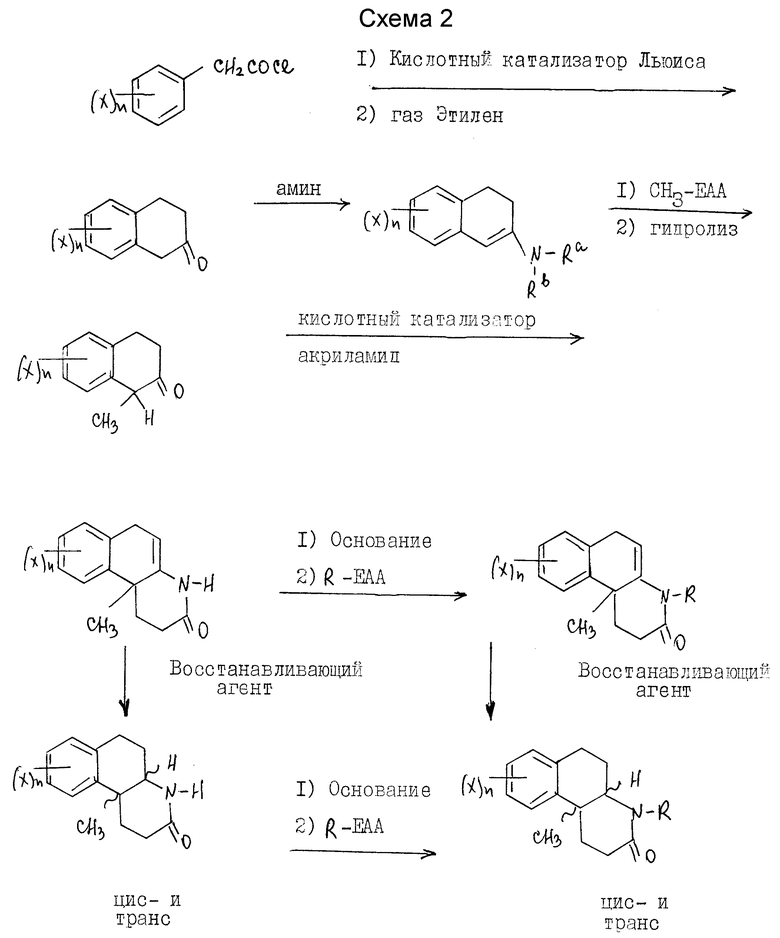
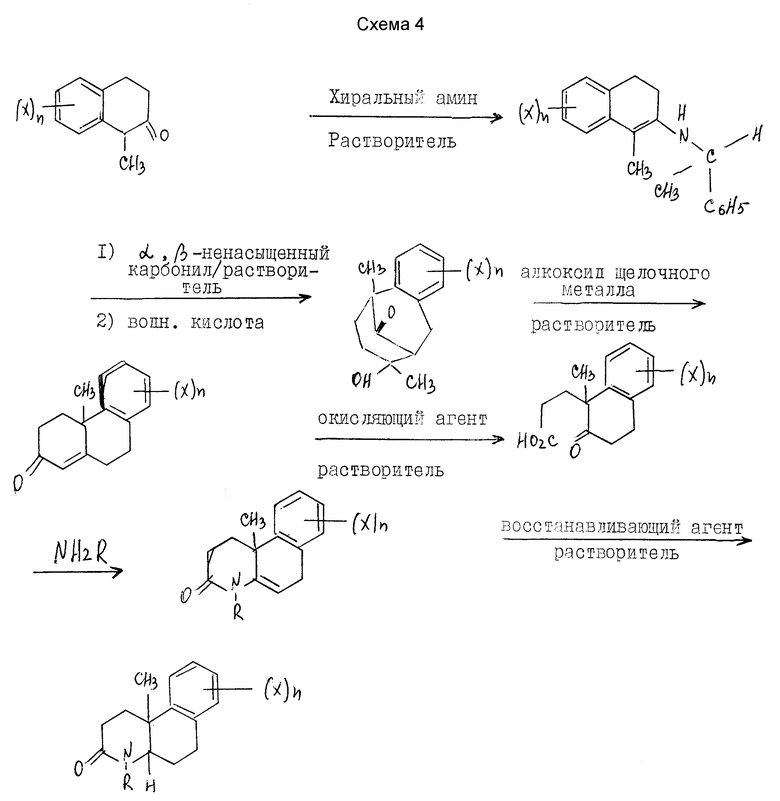
Бензо[f]хинолинон формулы I, где R - Н, алкил или феналкил; Z и Z' - Н, алкил или один из Z и Z' с R5 образуют связь углерод-углерод; Y - Н или метил или с R1 образует связь углерод-углерод; R1 - Н или с одним из Y или R3 образует связь углерод-углерод; R2 - Н или С1-4алкил; R3 - Н или с R1 образует связь углерод-углерод; R4 - Н или с R5 образует связь углерод-углерод; R5 - Н или с одним из Z или Z' образует связь углерод-углерод; n - 1 или 2; Х - Н, галоген, NO2, циано, СF3, С1-6алкил, алкокси, карбокси, алкоксикарбонил, амино, амидо или группа -А O- R6, где А - С1-6алкилен, С2-6алкенил или С2-6алкинилен; R6 -галоген, гидрокси, СF3, алкокси, карбокси, алкоксикарбонил, амино, алкиламино, амидо, алкиламидо или его фармацевтически приемлемая соль. Соединения 1 представляют собой сильнодействующие селективные стероидные ингибиторы 5α-редуктазы, полезные в лечении доброкачественных образований предстательной железы, облысения у мужчин, угрей, себореи. 7 с. и 12 з.п. ф-лы, 1 табл. 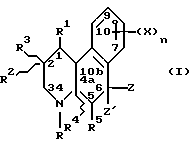 (I)
(I)

где R - водород, C1 - C4 алкил, незамещенный или замещенный фен(C1-C4)алкил;
Z и Z1 - независимо выбраны из водорода- и C1-C4 алкила или один из Z и Z1 с R5 образуют связь углерод-углерод;
Y - водород или метил или с R1 образует связь углерод-углерод;
R1 - водород или с одним из Y или R3 образует связь углерод-углерод;
R2 - водород или C1 - C4 алкил;
R3 - водород или с R1 образует связь углерод-углерод;
R4 - водород или с R5 образует связь углерод-углерод;
R5 - водород или с одним из Z или Z1 образует связь углерод-углерод;
n - 1 или 2;
Х - водород, галоген, NO2, циано, CF3, C1 - C6 алкил, C1 - C6 алкокси, карбокси, C1 - C6 алкоксикарбонил, амино, амидо, C1 - C4 алкиламидо, или группа -А-R6, где А - C1 - C6 алкилен, C2 - C6 алкенилен или C2 - C6 алкинилен; R6 - галоген, гидрокси, CF3, C1 - C6 алкокси, карбокси, C1 - C6 алкоксикарбонил, амино, C1 - C4 алкиламино, амидо, C1 - C4 алкиламидо,
или его фармацевтически приемлемая соль; при условии, что по меньшей мере один из R1 и К5 - водород; если R - водород, метил, этил или бензил, Х - не водород или метокси, и, если R - метил, R2 - не метил.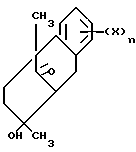
где Х - водород, галоген, NO2, циано, CF3, C1 - C6 алкил, C1 - C6 алкокси, карбокси, C1 - C6 алкоксикарбонил, амино, амидо, C1 - C4 алкиламидо, или группа -A-R6 , где А - C1 - C6 алкилен, C2 - C6 алкенилен или C2 - C6 алкинилен;
R6 - галоген, гидрокси, CF3, C1 - C6 алкокси, карбокси C1 - C6 алкоксикарбонил, амино, C1 - C4 алкиламино, амидо, C1 - C4 алкиламидо;
n - 1 иди 2;
или его фармацевтически приемлемая соль.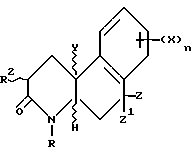
где R - водород, C1 - C4 алкил, незамещенный или замещенный фен(C1 - C4)алкил;
Z и Z1 - независимо выбраны из водорода и C1 - C4 алкила;
Y - водород или метил;
R2 - водород или C1 - C4 алкил;
n - 1 или 2;
Х - водород, галоген, NO2, циано, CF3, C1 - C6 алкил, C1 - C6 алкокси, карбокси, C1 - C6 алкоксикарбонил, амино, амидо, C1 - C4 алкиламидо или группа -A-R6, где A - C1 - C6 алкилен или C2 - C6 алкенилен или C2 - C6 алкинилен; и R6 - галоген, гидрокси, CF3, C1 - C6 алкокси, карбокси, C1 - C6 алкоксикарбонил, амино, C1 - C4 алкиламино, амидо, C1 - C4 алкиламидо,
или его фармацевтически приемлемой соли, отличающийся тем, что осуществляют
реакцию 1-метил-2-тетралона с оптически активным амином для получения соответствующего 1-метиленамина;
реакцию 1-метиленамина с α, β -ненасыщенным карбонильным соединением для получения соответствующего метанобензоциклооктан-4-она;
реакцию метанобензоциклооктан-4-она с алкоксидом щелочного металла для получения соответствующего 2,3,4,4а,9,10-гексагидро-4-а-метил-фенантрен-2-она;
окислительное расщепление указанного фенатрен-2-она для получения соответствующего 3-[1-метил-1(2-кето 1,2,3,4-тетрагидронафтил)]пропионовой кислоты;
реакцию указанной пропионовой кислоты с аммиаком или первичным амином для получения соответствующего 10b-метил-1,2,3,4,6,10b-гексагидробензо[f] хинолин-3-она;
восстановление указанного гексагидробензо[f]хинолин-3-она для получения соответствующего октагидробензо[f]хинолин-3-она.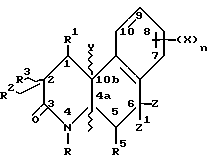
где R - водород или C1 - C4 алкил,
Z и Z1 - независимо выбраны из водорода и C1 - C4 алкила;
Y - водород;
R1 , R2, R3, R4 и R5 - водород;
n - 1 или 2;
Х - водород, галоген, NO2, CF3, C1 - C6 алкил, C1 - C4 алкокси или амино.
на их компонентные оптические изомеры, отличающийся тем, что осуществляют стадии:
контактирование раствора метанола рацемата с сильной кислотой для получения 1-(2-метоксикарбонилэтил)-2-(амино)-1,2,3,4-тетрагидронафталина;
контактирование указанного тетрагидронафталина с раствором метанола оптически активной ди-п-толуоилвинной кислоты для получения соответствующей соли тетрагидронафталина и обработку указанной соли основанием для получением одного оптически активного изомера.
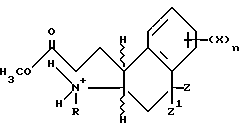
Приоритет по признакам.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| J.Med | |||
| Chem., 23(1) 1-5, 1980 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Hott et al | |||
| J.Med | |||
| Способ сопряжения брусьев в срубах | 1921 |
|
SU33A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Сатоскар Р.С., Бандаркар С.Д., Фармакология и фармакотерамия | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| - М.: Медицина, 1986, с.340-347. | |||

