Тамоксифен (1-п-диметиламиноэтоксифенил-транс-1,2-дифенилбут-1-ен), представленный структурной формулой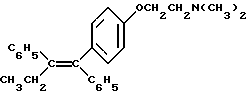
является широко известным антиэстрогенным соединением, которое применяется для лечения и профилактики рака молочной железы у млекопитающих. Смотри The Merk Index, 11th Ed., 1430 (1989). Известно, что, хотя тамоксифен действительно эффективен при лечении/профилактике данного заболевания, он вызывает некоторые гистеротрофные эффекты, которые могут быть вредны для пациенток, получающих лечение тамоксифеном. Таким образом, было бы выгодным, если бы было доступно фармацевтическое средство, которое не оказывало бы влияние на антинеопластические полезные свойства, которые обеспечивает тамоксифен, в то же время сводя к минимуму или элиминируя вредный гистеротрофный эффект.
Таким образом, настоящее изобретение относится к способу минимизации гистеротрофного эффекта тамоксифена и некоторых аналогов тамоксифена путем одновременного или последовательного введения некоторых нафтильных фармацевтических средств. Также предложены фармацевтические композиции.
Сущность изобретения.
Настоящее изобретение относится к способу минимизации гистеротрофного эффекта нестероидных антиэстрогенных соединений формулы II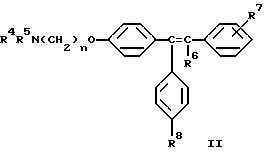
где либо R4 является H или низшим алкильным радикалом и R5 является низшим алкильным радикалом, либо R4 и R5 соединены вместе с соседним атомом азота с образованием гетероциклического радикала;
R6 является H или низшим алкильным радикалом;
R7 является H, галогено, OH, низшим алкильным радикалом или является бута-1,3-диенильным радикалом, который вместе с соседним бензольным кольцом образует нафтильный радикал;
R8 является H или OH; и
n равно 2;
или их фармацевтически приемлемых солей, когда указанное соединение формулы II вводят женщине для лечения или профилактики рака молочной железы, который заключается в одновременном или последовательном введении данной женщине соединения формулы I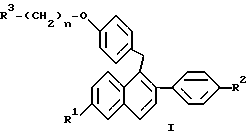
где R1 представляет -H, -OH, -O(C1-C4 алкил), -OCOC6H5, -OCO(C1-C6 алкил) или -OSO2(C4-C6 алкил);
R2 представляет -H, -OH, -O(C1-C4 алкил), -OCOC6H5, -OCO(C1-C6 алкил) или -OSO2(C4-C6 алкил);
n равен 2 или 3; и
R3 представляет 1-пиперидинил, 1-пирролидинил, метил-1-пирролидинил, диметил-1-пирролидинило, 4-морфолино, диметиламино, диэтиламино или 1-гексаметиленимино;
или его фармацевтически приемлемые соли.
Также описывается фармацевтические композицию, содержащие соединение формулы I и соединение формулы II совместно с фармацевтически приемлемым носителем, наполнителем или разбавителем.
Подробное описание изобретения.
Настоящее изобретение относится к открытию того, что отдельная группа фармацевтически активных нафтильных соединений (соединения формулы I) применима для минимизации гистеротрофного эффекта нестероидных антиэстрогенных соединений формулы II. Формулы I и II приведены ниже.
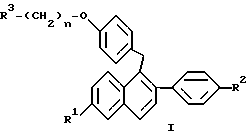
где R1 представляет -H, -OH, -O(C1-C4 алкил), -OCOC6H5, -OCO(C1-C6 алкил) или -OSO2(C4-C6 алкил);
R2 представляет -H, -OH, -O(C1-C4 алкил), -OCOC6H5, -OCO(C1-C6 алкил) или -OSO2(C4-C6 алкил);
n равен 2 или 3; и
R3 представляет 1-пиперидинил, 1-пирролидинил, метил-1-пирролидинил, диметил-1-пирролидинил, 4-морфолино, диметиламино, диэтиламино или 1-гексаметиленимино; или его фармацевтически приемлемая соль; и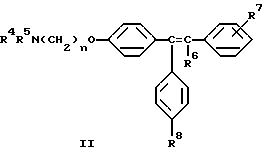
где либо R4 представляет H или низшим алкильным радикалом и R5 является низшим алкильным радикалом, либо R4 и R5 соединены вместе с соседним атомом азота с образованием гетероциклического радикала;
R6 является H или низшим алкильным радикалом;
R7 является H, галогено, OH, низшим алкильным радикалом или является бута-1,3-диенильным радикалом, который вместе с соседним бензольным кольцом образует нафтильный радикал;
R8 является H или OH; и
n равно 2;
или его фармацевтически приемлемая соль.
Описательные химические термины, применяемые для формул I и II, имеют свое обычное значение. Например, термин "галоген" включает бром, хлор, фтор и йод. Термин "низший алкил" или "C1-C4 алкил" относится к неразветвленным и разветвленным алифатическим радикалам, состоящим из 1-4 атомов углерода, включающим метил, этил, пропил, изопропил, н-бутил, изобутил, втор-бутил и трет-бутил. Кроме этого, термин "C1-C4 алкокси" относится к неразветвленным и разветвленным алифатическим эфирным радикалам, состоящим из 1-4 атомов углерода, таким как метокси, этокси, пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси и трет-бутокси.
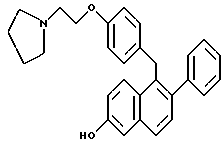
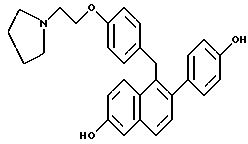
Соединения формулы I, в частности соединение, в котором каждый из R1 и R2, каждый являются -OH, и R3 является 1-пиперидинилом, являются ядерными регуляторными молекулами. Эти соединения связываются с рецепторами к эстрогену и являются полезными при лечении различных клинических симптомов, связанных с постклимактерическим синдромом, фиброидной болезнью матки, эндометриозом и пролиферацией гладкомышечных клеток аорты. Действительно, соединения формулы I блокируют действие эстрогена в некоторых клетках, но в других типах клеток соединения формулы I активируют те же гены, которые активируют и эстроген, и проявляют те же фармакологические свойства (например, препятствование остеопорозу, вызываемого дефицитом эстрогена, и снижение уровня сывороточного холестерина). По существу, на соединения формулы I можно ссылаться как на тканеселективные антиэстрогены, обладающие смешанными агонист-антагонистическими свойствами.
Хотя соединения формулы I и эстроген используют и конкурируют за одни и те же рецепторы, фармакологический эффект введения этих двух групп средств нельзя предсказать с уверенностью, и он индивидуален для каждого из них.
Соединения формулы I получают в соответствии со способами, приведенными ниже.
Исходный продукт для одного пути получения соединений формулы I по настоящему изобретению, соединения формулы VII, приведенной ниже, получают, по существу, как описано в Патенте США N 4230862, выданном 28 октября 1980 года, который включен здесь в качестве ссылки.
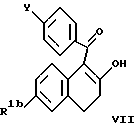
где R1b представляет -H или O(C1-C4 алкил); и
Y является метокси или R3-(CH2)n-O-, в котором R3 и n определены выше. Предпочтительно, R1b является метокси, Y является R3-(CH2)n-O-, R3 является 1-пиперидинилом и n равно 2.
Обычно легкодоступный тетралон или его соль формулы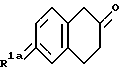
где R1a определяется выше, подвергают взаимодействию ацилирующим агентом, таким как фенилбензоат формулы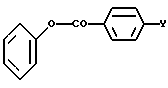
где Y определен выше. Реакцию обычно проводят в присутствии умеренно сильного основания, такого как амид натрия, и она протекает при комнатной температуре или ниже.
Что касается следующей стадии, по одному варианту селективное соединение формулы VII, после преобразования в производное енолфосфата in situ, подвергают взаимодействию в реакционных условиях Гриньяра с реактивом Гриньяра формулы
R2b-MgBr
где R2b обозначает -H или -O(C1-C4 алкил), с получением формулы IIIa, ниже, которые также известны в данной области (смотри, например, Патент США N 4230862, выше).
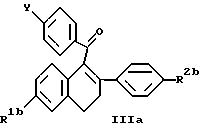
где R1b, R2b и Y определены выше, или его фармацевтически приемлемой соли.
Когда Y в соединении формулы III является R3-(CH2)n-O-, такие соединения могут быть восстановлены или подвергнуты снятию защиты, как описано ниже. Когда Y в соединении формулы III является метокси, используется сначала один из путей синтеза, показанных на схеме I (см. в конце описания, используется сначала. R1b, R2b, R3 и n на схеме I определены выше.
Каждую стадию путей синтеза A и B на схеме I проводят с помощью методов, хорошо известных специалисту в данной области.
Например, соединения формулы IIIc получают путем обработки соединений формулы IIIb гидрохлоридом пиридина при нагревании с обратным холодильником. Если в данных условиях R1b и/или R2b являются алкокси, данные группы будут деалкилированы до гидроксигрупп. Применение этого способа снимает необходимость стадии удаления защиты такой (их) алкоксигруппы на более поздней стадии, если это желательно.
Альтернативно, метоксигруппа Y в формуле IIIb может быть избирательно деметилирована путем обработки соединения эквивалентом тиоэтоксида натрия в инертном растворителе, таком как N,N-диметилформамид (ДМФ), в условиях умеренно повышенной температуры в пределах от около 80oC до около 100oC. За протеканием данной стадии можно следить с помощью обычных хроматографических методов, таких как тонкослойная хроматография (ТСХ).
Когда соединение формулы IIIc получено, можно осуществить его взаимодействие с соединением формулы
R3-(CH2)n-Q
где R3 определен выше и Q обозначает бром или предпочтительно хлор с получением соединений формулы IIId. Данная реакция изображена в виде последней стадии пути A на схеме I.
В обычных условиях алкилирования эта реакция будет протекать по каждой из гидроксигрупп, которые могут присутствовать в молекуле формулы IIIc. Однако, с путем проведения реакции в присутствии избытка мелкодисперсного порошкообразного карбоната калия, и используя от эквивалента до небольшого избытка реагента Q-(CH2)-R3, может быть достигнуто избирательное алкилирование по 4-гидроксибензоильной группе.
Для получения соединений формулы IIIe, как показано в пути B на схеме I, соединение формулы IIIc подвергают взаимодействию с избытком алкилирующего агента формулы
Z-(CH2)n-Z'
где каждый из Z и Z' представляют одинаковые или различные уходящие группы, в растворе щелочи.
Приемлемые уходящие группы включают, например, сульфонаты, такие как метансульфонат, 4-бромсульфонат, толуолсульфонат, этансульфонат, изопропансульфонат, 4-метоксибензолсульфонат, 4-нитробензолсульфонат, 2-хлорбензолсульфонат и тому подобное, галогены, такие как бром, хлор, иод и тому подобное, и другие родственные группы. Предпочтительным алкилирующим агентом является 1,2-дибромэтан, и на один эквивалент субстрата используется, по крайней мере, 2 эквивалента, предпочтительно более чем 2 эквивалента 1,2-дибромэтана.
Предпочтительный щелочной раствор для данной реакции алкилирования состоит из карбоната калия в инертном растворителе, таком как метилэтилкетон (МЭК) или ДМФ. В данном растворе 4-гидроксигруппа бензоильного радикала соединения формулы IIId существует в виде феноксид-иона, который замещает одну из уходящих групп алкилирующего агента.
Данная реакция протекает наилучшим образом, когда раствор щелочи, содержащий реактивы и реагенты, доводят до кипения и оставляют до полного протекания реакции. При использовании МЭК в качестве предпочтительного растворителя время протекания реакции меняется от около 6 часов до около 20 часов.
Затем реакционный продукт в данной стадии, соединение формулы IIIe, подвергают взаимодействию с 1-пиперидином, 1-пирролидином, метил-1-пирролидином, диметил-1-пирролидином, 4-морфолином, диметиламином, диэтиламином или 1-гексаметиленимином обычным способом с получением соединений формулы IIId. Предпочтительно, солянокислую соль пиперидина подвергают взаимодействию с соединением формулы IIIe в инертном растворителе, таком как безводный ДМФ, и нагревают до температуры в пределах от около 60oC до около 110oC. Когда смесь нагревают до предпочтительной температуры около 90oC, реакция протекает в течение лишь от около 30 минут до около 1 часа. Однако изменения в условиях протекания реакции будут влиять на время, которое требуется для того, чтобы реакция завершилась. Разумеется, что ход данной стадии реакции можно отслеживать с помощью обычных хроматографических методов.
Соединения формулы IIId представляют собой исходный продукт для одного способа получения фармацевтически активных соединений формулы Ia, как показано в схеме II (см. в конце описания).
где R1a, R2a, R3 и n определяются, как выше.
На схеме II соединение формулы IIId или его соль прибавляют к подходящему растворителю и подвергают взаимодействию с восстановителем, таким как, например, литийалюминийгидрид (ЛАГ). Хотя в данной реакции можно использовать свободное основание соединения формулы IIId, часто добавления кислоты, предпочтительно солянокислая соль, является более удобной.
Количество восстановителя, используемого в данной реакции, представляет собой количество, достаточное для восстановления карбонильной группы соединения формулы IIId с образованием карбинольных соединений формулы IV. Обычно используют произвольный избыток восстановителя на эквивалент субстрата.
Подходящие растворители включают любой растворитель или смесь растворителей, которые будут оставаться инертными в условиях восстановления. Подходящие растворители включают диэтиловый эфир, диоксан и тетрагидрофуран (ТГФ). Безводная форма данных растворителей является предпочтительной, а безводный ТГФ является особенно предпочтительным.
Температурой, используемой на данной стадии, является та, которая будет достаточной для полного протекания реакции восстановления. Температура окружающей среды в пределах от приблизительно 17oC до приблизительно 25oC обычно является достаточной.
Промежутком времени для данной стадии является промежуток, необходимый для протекания реакции. Обычно, данная реакция протекает в течение от около 1 часа до около 20 часов. Оптимальное время может быть определено с помощью слежения за ходом реакции посредством широко распространенных хроматографических методов.
Карбинольные продукты на данной стадии реакции (соединения формулы IV) выделяют, по существу, по способу, описанному в примере 7, ниже и применяют в способах, описанных здесь.
Когда карбинол формулы IV получен, такое соединение прибавляют в инертный растворитель, такой как, например, этилацетат, с последующим добавлением сильной протонной кислоты, такой как соляная кислота, и получением соединений формулы Ia. Данная реакция обычно протекает при температуре окружающей среды от около 17oC до около 25oC и обычно завершается в течение лишь от около нескольких минут до около 1 часа. Кристаллизацию конечного продукта проводят по стандартным методикам, по существу, как описано в примере 1, ниже.
Деалкилирование/снятие защиты концевых защищенных гидроксигрупп может быть проведено до получения соединений формулы IV, до получения соединений формулы Ia или после получения защищенных соединений формулы Ia, по способам, известным специалистам в данной области. Однако предпочтительным является деалкилирование защищенного соединения формулы Ia после его получения.
Реакция, показанная на схеме II, дает фармацевтически активные соединения формулы Ia, в которых каждый из R1a и R2a является водородом или C1-C4 алкокси. Предпочтительными соединениями формулы Ia являются такие, где R1a и R2a, каждый, представляет метокси или R1a и R2a, каждый, являются гидрокси, R3 является пиперидинилом и n равно 2. Данные предпочтительные соединения, причем последние особенно предпочтительны, так же, как и другие соединения формулы Ia, могут быть использованы в качестве фармацевтических средств или далее могут быть использованы для других соединений формулы I, которые также применимы для осуществления на практике способов по настоящему изобретению.
В качестве альтернативы реакциям, показанным на схеме II, для получения соединений формулы Ia по настоящему изобретению может быть использован одностадийный процесс, заключающийся в восстановлении кетона формулы V, приведенной ниже. Более конкретно, когда R1a и/или R2a обозначает -O(C1-C4 алкил), эти гидроксизащитные группы могут быть удалены до применения настоящего нового способа или, необязательно, могут быть удалены in situ после настоящего одностадийного процесса восстановления. В дополнение к этому продукт по данному процессу, который может содержать 1 или 2 незащищенных или защищенных гидроксильных радикала, можно необязательно перевести в соль по известным способам или как описывается здесь.
В данном способе соединение формулы V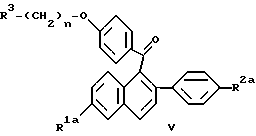
где R1a, R2a, R3 и n определены выше, или его соль подвергают взаимодействию с восстановителем, таким как литийалюминийгидрид или Red-Al® [бис(2-метоксиэтоксиалюминийгидрид)натрия] в присутствии растворителя с температурой кипения в интервале от около 150oC до около 200oC.
Соединения формулы V получают путем взаимодействия соединения формулы IIIb (как описано выше) с около 2 эквивалентами 2,3-дихлор-5,6-дициано-1,4-бензохинона (ДДХ) в присутствии инертного растворителя или смеси растворителей, таких как, например, диоксан, дихлорметан, толуол, дихлорэтан или бензол. Реакционную смесь обычно кипятят с обратным холодильником в течение около от 1 до 2 часов и затем оставляют перемешиваться при температуре окружающей среды в течение от около 36 до приблизительно 72 часов. Полученное соединение формулы VI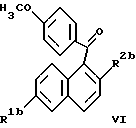
где R1b и R2b определены выше, затем деметилируют, как описано выше, и алкилируют соединением формулы
R3-(CH2)n-Q
где R3 определено выше, по вышеописанным способам.
В данной восстановительной реакции количеством восстановителя, используемого в данной реакции, является количество, достаточное для восстановления карбонильной группы соединения формулы V с получением соединения формулы Ia. Обычно используют произвольный избыток восстановителя на эквивалент субстрата.
Требованиями к растворителю, используемому в процессе, является наличие относительно высокой температуры кипения в интервале от около 150oC до около 200oC, какой обладают растворители, такие как, например, н-пропилбензол, диглим (1,1'-оксибис[2-метоксиэтан]) и анизол. Из них н-пропилбензол является предпочтительным растворителем для соединений формулы V, когда R1a и/или R2a обозначают -OCH3 и -C6H4-4'-O(C1-C4 алкил). Red-Al, используемый как в качестве растворителя, так и в качестве восстановителя, является предпочтительным, когда R1a обозначает -OH и/или R2a является -C6H4-4'-OH.
Температурой, используемой в данной реакции, является та, которая достаточна для протекания реакции восстановления до конца. Предпочтительно, реакционную смесь кипятят с обратным холодильником в течение от около 15 минут до около 6 часов, дают остыть до температуры окружающей среды и обрабатывают обычными методами [смотри, например, Fieser and Fieser, Reagents for Organic Synthesis, Том 1, стр. 584 (1968)] и, как описано далее здесь в примерах. Оптимальный промежуток времени для протекания данной реакции, от около 10 минут до приблизительно 1 часа, может быть определен с помощью слежения за ходом реакции по стандартным методикам.
Продукты формулы Ia по одностадийной реакции выделяют, по существу, как описано в примере 2, ниже. Предпочтительными соединениями формулы Ia по данной реакции являются такие же соединения, что и предпочтительные соединения формулы Ia, описанные выше, и они могут использоваться в качестве фармацевтически активных агентов в способах, описанных здесь, или они могут быть использованы для получения других соединений формулы I, которые также применимы в настоящих способах.
Например, когда R1a и/или R2a в соединении формулы I являются C1-C4 алкильными гидроксизащитными группами (таким образом, не являясь деалкилированными, как предложено в одном варианте схемы I), такие группы могут быть удалены обычными методами деалкилирования, как описано в примере 2, ниже, с получением особенно предпочтительного соединения формулы Ia.
Другие предпочтительные соединения формулы I получают путем замещения заново образованных R1a и/или R2a гидроксигрупп соединения формулы Ia радикалом формулы -O-CO-(C1-C6 алкил) или -O-SO2-(C4-C6 алкил) по хорошо известным методам. Смотри, например, Патент США N 4358593.
Например, когда желательной является -O-CO-(C1-C6 алкильная) группа, дигидроксисоединение формулы Ia подвергают взаимодействию с агентом, таким как ацилхлорид, бромид, цианид или азид или с подходящим ангидридом или смешанным ангидридом. Реакции удобно проводить в основном растворителе, таком как пиридин, лутидин, хинолин или изохинолин или в третичноаминном растворителе, таком как триэтиламин, трибутиламин, метилпиперидин и тому подобное. Реакция также может быть проведена в инертном растворителе, таком как этилацетат, диметилформамид, диметилсульфоксид, диоксан, диметоксиэтан, ацетонитрил, ацетон, метилэтилкетон и тому подобное, к которому добавлен, по крайней мере, один эквивалент акцептора кислоты (за исключениями, указанными ниже), такого как третичный амин. При желании могут быть использованы катализаторы ацилирования, такие как 4-диметиламинопиридин или 4-пирролидинопиридин. Смотри, например, Haslam et al., Tetrahedron, 36:2409-2433 (1980).
Реакции ацилирования, которые дают вышеуказанные концевые R1 и R2 группы соединения формулы I, приводят при умеренных температурах в интервале от приблизительно -25oC до приблизительно 100oC, зачастую в инертной атмосфере, такой как атмосфера газообразного азота. Однако обычно для протекания реакции достаточно температуры окружающей среды.
Такие ацилирования данных гидроксигрупп также могут быть осуществлены путем кислотно-катализируемых реакций, подходящих карбоновых кислот в инертных органических растворителях или при нагревании. Используются кислотные катализаторы, такие как серная кислота, полифосфорная кислота, метансульфоновая кислота и тому подобное.
Вышеуказанные R1 и/или R2 группы в соединениях формулы I могут быть также получены с помощью образования активного сложного эфира подходящей кислоты, такого как эфиры, образованные такими известными реагентами, как дициклогексилкарбодиимид, ацилимидазолы, нитрофенолы, пентахлорфенол, N-гидроксисукцинимид и 1-гидроксибензотриазол. Смотри, например, Bull. Chem. Soc. Japan, 38: 1979 (1965) и Chem. Ber., 788 и 2024 (1970).
Каждый из вышеуказанных методов, который приводит к -O-CO-(C1-C6 алкильным) радикалам, осуществляют в растворителях, как обсуждено выше. Те методы, при которых в ходе реакции не образуется кислотный продукт, разумеется, не требуют использования в реакционной смеси акцептора кислоты.
Когда желательным является соединение формулы I, в котором R1a и/или R2a группу соединения формулы Ia преобразуют в группу формулы -O-SO2(C4-C6 алкил), (20 мл) и реакционную смесь кипятят с обратным холодильником. Летучие компоненты выпаривают в вакууме и остаток очищают при помощи колоночной хроматографии на силикагеле (200 г) с использованием в качестве элюента дихлорметана и смеси этанола и 25% NH3 (водн.) (9:1) (градиент от 0% до 10%). Это дает 0,30 г 3-амино-4-оксо-2,3,4,5-тетрагидро-1H-нафто[2,1-b]азепина.
1H ЯМР (CDCl3) δ 2,05-2,15 (м, 1H); 2,64-2,75 (м, 1H); 2,95-3,05 (м, 1H); 3,44 (дд, 1H); 3,48-3,52 (м, 1H); 7,13 (д, 1H, J = 9 Гц); 7,49 (т, 1H, J = 9 Гц); 7,56 (т, 1H, J = 9 Гц); 7,60 (ушир.с, 1H); 7,75 (д, 1H, J = 9 Гц); 7,85 (д, 1H, J = 9 Гц); 8,07 (д, 1H, J = 9 Гц).
Трет-бутиловый эфир (1,1-диметил-2-(4-оксо-2,3,4,5-тетрагидро-1H- нафто[2,1-b]азепин-3-илкарбамоил)этил)карбаминовой кислоты.
К раствору 3-трет-бутилоксикарбониламино-3-метилбутановой кислоты (0,26 г, 1,2 ммоль) в ДМФА (15 мл) добавляли гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (0,24 г, 1,2 ммоль). Через 15 минут выдерживания при комнатной температуре добавляют 3-амино-4-оксо-2,3,4,5-тетрагидро-1H-нафто[2,1-b] азепин (0,25 г, 1,1 ммоль) и реакционную смесь перемешивают в течение 6 ч. Добавляют воду (80 мл) и раствор экстрагируют этилацетатом (30 мл). Органическую фазу промывают бикарбонатом натрия иодистоводородную, азотную, серную, фосфорную, гипофосфорную и тому подобное. Соли, являющиеся производными органических кислот, таких как алифатические моно- и дикарбоновые кислоты, фенилзамещенные алкановые кислоты, гидроксиалкановые и гидроксиалкандионовые кислоты, ароматические кислоты, алифатические и ароматические сульфоновые кислоты, также могут быть использованы. Такие фармацевтически приемлемые соли, таким образом, включают ацетата, фенилацетат, трифторацетат, акрилат, аскорбат, бензоат, хлорбензоат, динитробензоат, гидроксибензоат, метоксибензоат, метилбензоат, о-ацетоксибензоат, нафталин-2-бензоат, бромид, изобутират, фенилбутират, β- гидроксибутират, бутин-1,4-диоат, гексин-1,4-диоат, капрат, каприлат, хлорид, циннамат, цитрат, формиат, фумарат, гликолят, гептаноат, гиппурат, лактат, малат, малеат, гидроксималеат, малонат, манделат, мезилат, никотинат, изоникотинат, нитрат, оксалат, фталат, терефталат, фосфат, моногидрофосфат, дигидрофосфат, метафосфат, пирофосфат, пропиолат, пропионат, фенилпропионат, салицилат, себацинат, сукцинат, суберат, сульфат, бисульфат, пиросульфат, сульфит, бисульфит, сульфонат, бензолсульфонат, п-бромфенилсульфонат, хлорбензолсульфонат, этансульфонат, 2-гидроксиэтансульфонат, метансульфонат, нафталин-1-сульфонат, нафталин-2-сульфонат, п-толуолсульфонат, ксилолсульфонат, тартрат и тому подобное. Предпочтительной солью является солянокислая соль.
Фармацевтически приемлемые соли добавления кислот обычно получают путем взаимодействия соединения формулы I с эквимолярным или избыточным количеством кислоты. Реагенты обычно смешивают в общем растворителе, таком как диэтиловый эфир или этилацетат. Соль обычно осаждается из раствора в течение от около одного часа до 10 суток и может быть выделена путем фильтрования, или растворитель может быть удален с помощью широко распространенных способов.
Фармацевтически приемлемые соли обычно обладают улучшенными характеристиками растворимости по сравнению с соединением, производными которого они являются, и, таким образом, часто лучше подходят для приготовления в виде жидкостей или эмульсий.
Соединения формулы II, применяемые по способам и в фармацевтических композициях по настоящему изобретению, получают известными способами, такими как описаны в Патенте США N 4623600, который включен здесь в качестве ссылки. Фармацевтически приемлемые соли добавления кислот соединений формулы II получают вышеописанным способом.
Предпочтительное соединение формулы II, в котором каждый из R4 и R5 является метилом, R6 является этилом, каждый из R7 и R8 является H, и n равно 2, известно в данной области как тамоксифен. Тамоксифен и его аналоги формулы II являются антиэстрогенными соединениями, и тамоксифен преимущественно применяется для лечения рака молочной железы у женщин. В дополнение к данной хорошо известной активности в данной области также широко признано, что тамоксифен может вызывать определенные побочные эффекты, в частности рак эндометрия, который потенциально может угрожать жизни. [смотри, например, Fisher, B., et al., JNCI, 86(7): 527-537 (1994)].
С одной стороны, настоящее изобретение относится к способу минимизации гистеротрофного эффекта нестероидного антиэстрогенного соединения формулы II, в частности тамоксифена, путем введения соединения формулы I, в частности соединения, в котором каждый из R1 и R2 является -OH, и R3 является 1-пиперидинилом, женщине, подвергающейся лечению соединением формулы II для лечения или профилактики рака молочной железы. В данном контексте "гистеротрофный эффект" означает пролиферацию эпителиальных клеток матки, что часто может быть побочным эффектом введения тамоксифена женщинам. Представляется, что данный гистеротрофный эффект непосредственно вовлечен в развитие рака эндометрия.
Введение соединения формулы I, в частности соединения, в котором каждый из R1 и R2 является -OH, и R3 является 1-пиперидинилом, минимизирует гистеротрофный эффект одновременно или последовательно вводимого соединения формулы II, в частности тамоксифена, без влияния на действие соединения формулы II в отношении рака молочной железы. Термин "минимизирование" или его производные включает в себя частичное или полное ингибирование тамоксифен-индуцированного гистеротрофного действия на эпителиальные клетки матки.
Для лечения рака молочной железы у человека может вводиться тамоксифен или другое соединение формулы II, одно или в сочетании с другими химиотерапевтическими средствами и/или радиотерапией, в качестве вспомогательного средства к хирургическому вмешательству или, при определенных обстоятельствах, может рассматриваться на предмет использования в качестве химиосупрессивного/химиопрофилактического средства. Поскольку каждая из данных схем введения может представлять различные степени риска развития гистеротрофных побочных эффектов, лечащий врач может наилучшим образом решить, следует ли вводить соединение формулы I одновременно или последовательно по отношению к введению соединения формулы II.
При последовательном введении фармацевтические составы соединений формул I и II получают описанными здесь способами.
При одновременном введении соединения формулы I и формулы II могут быть получены в виде фармацевтических составов выше указанными известными способами и введены в виде раздельных доз. Альтернативно, они могут быть объединены с образованием фармацевтической композиции по настоящему изобретению, которая включает в себя эффективное количество соединения формулы I и эффективное количество соединения формулы II, предпочтительно соединение формулы I, в котором каждый из R1 и R2 является -OH, и R3 является пиперидинилом, и тамоксифен, соответственно, вместе с фармацевтически приемлемым носителем, наполнителем или разбавителем.
Как использовано выше и во всем данном описании, термин "эффективное количество" означает, что дозировка активного(ых) соединения(й) достаточна для обеспечения терапевтического лечения конкретного клинического симптома.
Термин "активное соединение", как используется во всем данном описании, относится к соединению формулы I или его фармацевтически приемлемой соли или сольвату и/или к соединению формулы II или его фармацевтически приемлемой соли.
Для терапевтического лечения конкретно симптома может вводиться соединение формулы I, как таковое, с соединением формулы II или без него, или может быть смешано и включено в состав фармацевтических композиций в виде стандартных дозированных формах для парентерального, чрескожного, ректального назального, внутривенного или преимущественно перорального введения. Такие фармацевтические композиции готовят способами, хорошо известными в данной области, и они содержат соединения формулы I, необязательно включая соединения формулы II. При приготовлении композиций по настоящему изобретению активные ингредиенты обычно смешивают с носителем, или разбавлены носителем, или заключены в носитель, который может быть в виде капсулы, саше, бумаги или другой емкости. Когда носитель выступает в роли разбавителя, он может быть твердым, полутвердым или жидким материалом, который действует как растворитель, наполнитель или среда для активного ингредиента. Таким образом, композиция может быть в виде таблеток, пилюль, порошков, лепешек, саше, крахмальных таблеток, эликсиров, эмульсий, растворов, сиропов, суспензий, мягких и твердых желатиновых капсул, стерильных растворов для инъекций и стерильно упакованных порошков.
Кроме того, соединения настоящей композиции, в частности соединения формулы I, хорошо подходят для получения препаратов в виде лекарственных форм с удерживаемым высвобождением и тому подобного. Составы могут быть изготовлены так, что они высвобождают активные ингредиенты только или предпочтительно в конкретном физиологическом местоположении, возможно, в течение некоторого периода времени. Покрытие, оболочки и защитные матрицы могут быть изготовлены, например, из полимерных веществ или восков.
Некоторые примеры подходящих носителей, наполнителей и разбавителей включают лактозу, декстрозу, сахарозу, сорбит, маннитол, крахмалы, аравийскую камедь, кальций фосфат, альгинаты, салицилат кальция, микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, трагакант, желатин, сироп, метилцеллюлозу, метил- и пропилгидроксибензоаты, тальк, стеарат магния, воду и минеральное масло. Композиции могут дополнительно содержать смазывающие агенты, увлажняющие агенты, эмульгаторы и суспенгаторы агенты, консерванты, подсластители или ароматизаторы. Композиции могут быть составлены так, чтобы обеспечивать быстрое, удерживаемое или отсроченное высвобождение активного(ых) ингредиента(ов) после введения пациенту способами, хорошо известными в данной области. Для перорального введения соединения, необязательно включая компонент второго соединения, может быть смешано с носителями и разбавителями, спрессовано в таблетки или заключено в желатиновые капсулы. Смеси могут быть по выбору растворены в жидкостях, таких как 10% водный раствор глюкозы, изотонический физиологический раствор, стерильная вода и тому подобное, и введены внутривенно или с помощью инъекций.
Композиции предпочтительно готовят в виде стандартной дозированной формы, причем каждая доза содержит от около 1 до около 500 мг и более часто от около 5 до около 300 мг активного(ых) ингредиента(ов). Термин "стандартная дозированная форма" относится к физически дискретным единицам, подходящим в качестве разовых доз для человека, причем каждая единица содержит заранее определенное количество активных ингредиентов, рассчитанное так, чтобы вызывать желательный терапевтический эффект в сочетании с требуемым фармацевтически приемлемым носителем. Под "фармацевтически приемлемым" понимают, что носитель, разбавитель или наполнитель должны быть с другими ингредиентами состава и не должен приносить вреда его реципиенту.
Соединения формулы I, сами по себе или в сочетании с фармацевтическим средством по настоящему изобретению, обычно вводятся в виде обычного препарата. Следующие примеры составов являются только иллюстративными и не должны трактоваться как ограничение объема настоящего изобретения.
Составы 1-5 см (в конце описания).
Конкретные составы 1-5 могут быть изменены в соответствии с предусмотренными обоснованными вариациями.
Таблеточные препараты готовят, используя нижеуказанные ингредиенты (см. в конце описания составы 6, 7).
Для формирования таблетки компоненты смешивают и спрессовывают.
Альтернативно, таблетки, каждая из которых содержит 25-1000 мг соединения формулы I, получают следующим образом (см. в конце описания состав 7).
Соединение формулы I, крахмал и целлюлозу пропускают через сито N 45 меш U. S. и тщательно перемешивают. Раствор поливинилпирролидона смешивают с полученными порошками, которые затем пропускают через сито N 14 меш U.S. Гранулы, полученные таким образом, сушат при 50-60oC и пропускают через сито N 18 меш U.S. Натрийкарбоксиметилкрахмал, стеарат магния и тальк, предварительно пропущенный через сито N 60 меш U.S., добавляют затем к гранулам, которые после перемешивания спрессовывают с помощью таблетирующего аппарата, что позволяет получить таблетки.
Суспензии, каждая из которых содержит 25-1000 мг лекарства в дозе 5 мл, получают следующим образом (см. в конце описания состав 8).
Активный ингредиент пропускают через сито N 45 меш U.S. и перемешивают с натрийкарбоксиметилцеллюлозой и сиропом с получением однородной пасты. Добавляют при перемешивании раствор бензойной кислоты, ароматизатор и краситель, разбавленные большим количеством воды. Затем добавляют воду в количестве, достаточном для достижения требуемого объема (см. в конце описания состав 9, 10).
Конкретная доза соединения формулы I, требуемая для минимизации гистеротрофного эффекта нестероидного антиэстрогенного соединения формулы II по данному изобретению, будет зависеть от тяжести состояния, пути введения и связанных с ними факторов, которые оцениваются лечащим врачом. Обычно приемлемые и эффективные суточные дозы соединения формулы I будут составлять от около 0,1 мг до около 1000 мг/сутки и, более типично, от около 50 мг до около 600 мг/сутки. Такие дозы вводят субъекту, нуждающемуся в лечении, ежедневно от одного до трех раз или чаще, как необходимо для эффективного лечения присутствующего симптома. Обычно предпочтительным является введение соединения формулы I в виде соли добавления кислоты, что и обычно используют при введении лекарственных средств, содержащих основную группу, такую как пиперидиновое кольцо. Также предпочтительно вводить такое соединение перорально.
Соединения формулы II, в частности тамоксифен, вводят для лечения рака молочной железы в дозах и в строках, которые согласуются с таковыми, хорошо известными в данной области. Однако предпочтительным является введение существенного избытка соединения формулы I по отношению к соединению формулы II.
Следующие примеры приведены для дальнейшей иллюстрации получения соединений по настоящему изобретению. Не следует думать, что объем изобретения ограничивается каким-либо следующим примером.
Данные ЯМР для следующих примеров получают на ЯМР-спектрометре GE 300 МГц и в качестве растворителя используют безводный d-6 ДМСО, если не указано иное.
Получение 1.
[3,4-Дигидро-2-(4-метоксифенил)-6-метоксинафталин-1-ил] (4- метоксифенил)метанон.
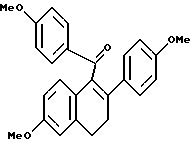
К суспензии гидрида натрия (12,75 г в 60% масляной дисперсии, предварительно промытой гексаном, 0,32 моль), перемешиваемой в тетрагидрофуране (ТГФ) (650 мл) при 0oC, добавляли раствор (3,4-дигидро-2-гидрокси-6-метокси-1-нафталинил)(4-метоксифенил)метанона (90,0 г, 0,29 ммоль; (Смотри, например, патент США N 4230862) и дифенилхлорфосфата (77,8 г, 0,29 ммоль) в ТГФ (750 мл). Скорость добавления была такой, чтобы температура реакции поддерживалась ниже 8oC. После перемешивания в течение 3 часов при 0oC по каплям добавляли 4-MeOC6H4MgBr (1,5 эквивалента 0,064 г/мл раствора в ТГФ) и полученной смеси дали постепенно нагреться до комнатной температуры. Через 12 часов раствор гасили добавлением холодного водного хлорида аммония. Органическую часть отделяли от смеси и водную часть экстрагировали этилацетатом. Объединенные органические экстракты сушили (сульфат натрия), фильтровали и концентрировали. К полученному маслу добавляли ацетонитрил (1 л) до образования осадка. Твердые продукты удаляли фильтрованием и фильтрат концентрировали, что давало масло, которое очищали методом флэш-хроматографии (силикагель, метиленхлорид). Желаемый продукт последовательно очищали кристаллизацией из метанола с получением 96,7 г (83%) указанного в заголовке соединения в виде желтого кристаллического твердого продукта: т.пл. = 172-173oC; 1H ЯМР (ДМСО-d6) δ 7,75 (д, J = 8,7 Гц, 2H), 7,16 (д, J = 8,6 Гц, 2H), 6,60-6,90 (сложный, 7H), 3,74 (с, 3H), 3,71 (с, 3H), 3,64 (с, 3H), 2,96 (м, 2H), 2,96 (м, 2H); МС (FD) m/e 400 (M+).
Получение 2.
[3,4-Дигидро-2-(4-метоксифенил)-6-метоксинафталин-1-ил] (4- гидроксифенил)метанон.
δ
К раствору литийэтантиола [полученного путем добавления n-BuLi (87,8 мл 1,6 М раствора в гексане, 140 ммоль) к раствору этантиола (12,1 мл, 164 ммоль), перемешиваемому при 0oC в этиловом эфире (400 мл) с последующим коротким перемешиванием и концентрированием], перемешиваемому в диметилформамиде (400 мл), добавляли продукт по получению 1 (46,7 г, 117 ммоль). Смесь затем нагревали по 100oC. Через 1 час реакцию концентрировали, полученное коричневое масло растворяли в хлороформе. Данный раствор экстрагировали добавлением водного хлорида аммония. Водную часть обрабатывали 1H соляной кислотой, пока не получали pH 5, и последовательно экстрагировали хлороформом. Объединенные органические экстракты промывали насыщенным раствором соли, сушили (сульфат натрия), фильтровали и концентрировали. Полученное коричневое масло очищали методом флэш-хроматографии (силикагель, градиент этилацетата/гексана), что давало 30,0 г (66%) указанного в заголовке продукта в виде желтого масла: 1H-ЯМР (300 МГц, CDCl3) 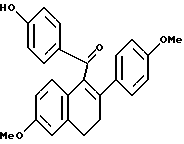 7,74 (м, 2H), 7,16 (м, 2H), 6,85 (д, J = 8,0 Гц, 1H), 6,77 (с, 1H), 6,65 (м, 5H), 6,11 (с, 1H), 3,78 (с, 3H), 3,69 (с, 3H), 3,00 (м, 2H), 2,77 (м, 2H); 13C-ЯМР (75 МГц, CDCl3) δ 201,1, 162,4, 159,7, 159,6, 137,5, 137,2, 134,6, 134,2, 133,3, 130,6, 129,6, 127,6, 127,2, 116,5, 114,7, 114,5, 112,3, 56,2, 56,0, 30,7, 29,6; Анал. расч. для: C 77,70; H 5,74. Найдено C 77,46; H 5,91. МС (FD) m/e 386 (M+); ИК (хлороформ) 3400,94, 1641,63, 1601,12 см-1.
7,74 (м, 2H), 7,16 (м, 2H), 6,85 (д, J = 8,0 Гц, 1H), 6,77 (с, 1H), 6,65 (м, 5H), 6,11 (с, 1H), 3,78 (с, 3H), 3,69 (с, 3H), 3,00 (м, 2H), 2,77 (м, 2H); 13C-ЯМР (75 МГц, CDCl3) δ 201,1, 162,4, 159,7, 159,6, 137,5, 137,2, 134,6, 134,2, 133,3, 130,6, 129,6, 127,6, 127,2, 116,5, 114,7, 114,5, 112,3, 56,2, 56,0, 30,7, 29,6; Анал. расч. для: C 77,70; H 5,74. Найдено C 77,46; H 5,91. МС (FD) m/e 386 (M+); ИК (хлороформ) 3400,94, 1641,63, 1601,12 см-1.
Получение 3.
[3,4-Дигидро-2-(4-метоксифенил)-6-метоксинафталин-1-ил] (4- [2-(1-пиперидинил)этокси]фенил]метанон.
δ
К раствору продукта по получению 2 (36 г, 93 ммоль), перемешиваемому в диметилформамиде (ДМФ; 1 л), добавляли йодид калия (30 мг, 0,18 ммоль) с последующим добавлением карбоната калия (64,2 г, 465 ммоль) и 1-(2-хлорэтил)пиперидин-моногидрохлорида (18,9 г, 102 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение ночи, затем концентрировали и полученное масло растворяли в хлороформе. Данный раствор тщательно промывали водой, насыщенным раствором соли, сушили (сульфат натрия), фильтровали и концентрировали. Полученное масло очищали методом флэш-хроматографии (силикагель, градиент метанола/хлороформа), что давало 43 г (93%) указанного в заголовке продукта в виде желтой пены: 1H-ЯМР (300 МГц, ДМСО-d6) 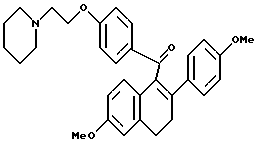 7,72 (д, J = 8,0 Гц, 1H), 7,15 (д, J = 10 Гц, 3H), 6,87 (д, J = 11 Гц, 3H), 6,72 (д, J = 8 Гц, 2H), 6,62 (с, 2H), 4,05 (м, 2H), 3,69 (с, 3H), 3,63 (с, 3H), 2,95 (м, 2H), 2,62 (м, 4H), 2,38 (м, 4H), 1,44 (м, 4H), 1,33 (м, 2H); 13C-ЯМР (75 МГц, ДМСО-d6) δ 197,2, 168,22, 168,18, 162,3, 158,4, 158,3, 136,4, 134,9, 133,0, 131,3, 129,6, 128,6, 125,9, 125,4, 114,4, 113,7, 113,6, 113,4, 111,5, 65,7, 62,5, 57,0, 55,0, 54,9, 54,1, 29,1, 28,0, 25,4, 23,7; Анал. расч. для: C 77,24; H 7,09; N 2,81. Получ: C 77,44; H 7,13; N 2,75. МС (FD) m/e 497 (M+); ИК (хлороформ) 1672,5 см-1.
7,72 (д, J = 8,0 Гц, 1H), 7,15 (д, J = 10 Гц, 3H), 6,87 (д, J = 11 Гц, 3H), 6,72 (д, J = 8 Гц, 2H), 6,62 (с, 2H), 4,05 (м, 2H), 3,69 (с, 3H), 3,63 (с, 3H), 2,95 (м, 2H), 2,62 (м, 4H), 2,38 (м, 4H), 1,44 (м, 4H), 1,33 (м, 2H); 13C-ЯМР (75 МГц, ДМСО-d6) δ 197,2, 168,22, 168,18, 162,3, 158,4, 158,3, 136,4, 134,9, 133,0, 131,3, 129,6, 128,6, 125,9, 125,4, 114,4, 113,7, 113,6, 113,4, 111,5, 65,7, 62,5, 57,0, 55,0, 54,9, 54,1, 29,1, 28,0, 25,4, 23,7; Анал. расч. для: C 77,24; H 7,09; N 2,81. Получ: C 77,44; H 7,13; N 2,75. МС (FD) m/e 497 (M+); ИК (хлороформ) 1672,5 см-1.
Пример 1.
[2-(4-Метоксифенил)-6-метоксинафталин-1-ил][4-[2-(1- пиперидинил)этокси] фенил]метангидрохлорид.
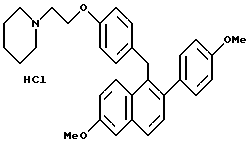
К суспензии литийалюминийгидрида (3,80 г, 94,8 ммоль), перемешиваемой при 0oC в сухом ТГФ (100 мл), медленно добавляли раствор продукта по получению 3 (23,6 г, 47,4 ммоль) в ТГФ (50 мл) в течение 45-минутного промежутка времени. Реакционную смесь оставляли перемешиваться при температуре окружающей среды в течение 14 часов, охлаждали до 0oC и осторожно гасили водой (5 мл). К данному раствору по каплям добавляли гидроксид натрия (15 мл 15% по массе водного раствора) с последующим добавлением воды (5 мл). Смесь перемешивали в течение 0,5 часа, фильтровали и твердые вещества тщательно промывали этилацетатом. Фильтрат затем концентрировали, что давало 21 г (89%) промежуточного продукта (карбинола) в виде белой пены, который затем использовали без дальнейшей очистки. К промежуточному продукту (23,6 г, 47,2 ммоль), перемешиваемому при температуре окружающей среды в этилацетате (100 мл), добавляли соляную кислоту [100 мл насыщенного этилацетатного раствора]. Осадок непосредственно образовывался в течение концентрирования смеси. Полученное твердое вещество перекристаллизовывали из метанола, что давало 19,4 г (79%) указанного в заголовке продукта в виде белого кристаллического твердого вещества: 1H-ЯМР (300 МГц, ДМСО-d6) δ 10,54 (ушир.с, 1H), 7,72-7,80 (сложный, 2H), 7,34-7,38 (сложный, 2H), 7,23 (д, J = 8,5 Гц, 2H), 7,08 (дд, J = 8,4, 2,3 Гц, 1H), 6,80-6,96 (сложный, 6H), 4,30 (ушир.с, 4H), 3,85 (с, 3H), 3,76 (с, 3H), 3,37-3,45 (сложный, 4H), 2,90-2,99 (м, 2H), 1,61-1,82 (сложный, 5H), 1,32-1,39 (м, 1H); МС (FD) m/e 481 (M+-соляная кислота); Анал. расч. для: C 74,19; H 7,00; N 2,70. Найдено: C 74,28; H 7,10; N 2,66.
Пример 2.
[2-(4-Гидроксифенил)-6-гидроксинафталин-1-ил] [4-[2-(1- пиперидинил)этокси]фенил]метангидрохлорид.
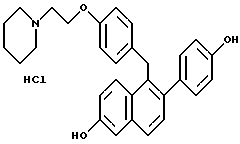
К раствору продукта по примеру 1 (5,0 г, 9,6 ммоль), перемешиваемому в 1,2-дихлорэтане (50 мл) при комнатной температуре, добавляли трихлорид бора (20 мл, 234 ммоль). Полученную темно-пурпурную реакцию оставляли перемешиваться при температуре окружающей среды в течение ночи, затем охлаждали до 0oC. Затем осторожно по каплям добавляли метанол (50 мл) в течение 2-часового промежутка времени (осторожно: выделение газа), причем в течение этого времени образовывался осадок. Твердое вещество отфильтровывали, промывали холодным метанолом и затем диэтиловым эфиром. Перекристаллизация из метанола давала продукт в виде белого порошка: 1H-ЯМР (300 МГц, ДМСО-d6) δ 10,38 (ушир. с, 0,5H), 9,74 (с, 1H), 9,52 (с, 1H), 7,61-7,68 (сложный, 2H), 7,28 (д, J = 8,3 Гц, 1H), 7,08-7,14 (сложный, 3H), 6,99 (дд, J = 9,1, 2,4 Гц, 1H), 6,75-6,91 (сложный, 6H), 4,28-4,31 (сложный, 4H), 3,34-3,45 (сложный, 4H), 2,95 (м, 1H), 1,63-1,75 (сложный, 5H), 1,35 (м, 1H); МС (FD) m/e 454 (M+-соляная кислота); Анал. расч. для: C 73,53; H 6,58; N 2,86. Найдено: C 73,48; H 6,57; N 3,01.
Пример 3.
[2-(4-Бензоилоксифенил)-6-бензоилоксинафталин-1-ил] [4-[2-(1- пиперидинил)этокси]фенил]метан.
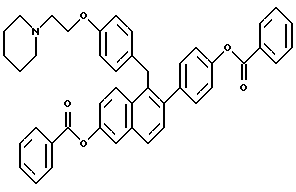
К суспензии продукта по примеру 2 (4,1 г, 8,4 ммоль), перемешиваемого в ТГФ (200 мл), добавляли N,N-диметиламинопиридин (10 мг, в качестве катализатора). Смесь охлаждали до 0oC и добавляли триэтиламин (8,5 г, 83,7 ммоль). Через 10 минут по каплям добавляли бензоилхлорид (4,7 г, 33,5 ммоль) и раствору давали перемешиваться в течение 60 часов. Осадок затем отфильтровывали и фильтрат концентрировали. Очистка данного продукта методом препаративной ВЭЖХ (хлороформ до 25% этилацетата в градиенте хлороформа) с последующей перекристаллизацией из метанола давала 3,78 г соединения заголовка в виде белого порошка: 1H-ЯМР (300 МГц, ДМСО-d6) δ 8,18 (арр т, J = 9,1 Гц, 4H), 7,91-8,05 (сложный, 3H), 7,75 (м, 1H), 7,61-7,69 (м сложный, 2H), 7,58 (д, J = 8,9 Гц, 1H), 7,42-7,50 (сложный, 3H), 7,38 (д, J = 8,3 Гц, 2H), 6,91 (д, J = 8,5 Гц, 2H), 6,80 (д, J = 8,5 Гц, 2H), 4,40 (с, 2H), 3,97 (т, J = 3,5 Гц, 2H), 2,60 (т, J = 3,3 Гц, 2H), 2,39 (сложный, 4H), 1,31-1,52 (сложный, 6H); МС (FD) m/e 661 (M+); Анал. расч. для: C 79,86; H 5,94; N 2,12. Найдено: C 79,59; H 6,05; N 1,96.
Пример 4.
[2-(4-Пивалоилоксифенил)-6-пивалоилоксинафталин-1-ил] [4-[2-(1- пиперидинил)этокси]фенил]метан.
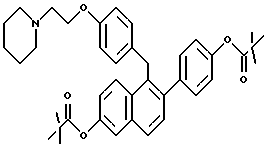
К суспензии продукта по примеру 2 (0,250 г, 0,510 ммоль), перемешиваемой в ТГФ (25 мл), добавляли N,N-диметиламинопиридин (2 мг) с последующим добавлением триэтиламина (0,78 мл, 5,6 ммоль) и триметилацетилхлорида (0,25 мл, 2,0 ммоль). Полученную смесь перемешивали при температуре окружающей среды в течение 2 часов, затем вливали в этилацетат/воду (100 мл, 1:1 по объему). Органический слой отделяли и водную часть экстрагировали добавлением этилацетата (50 мл). Объединенные органические экстракты промывали насыщенным водным хлоридом аммония (1х25 мл), насыщенным водным бикарбонатом натрия (2х25 мл) и насыщенным раствором соли (1х25 мл). Очистка методом радиальной хроматографии (силикагель, 2 мм, 10:8:1:1 этилацетат:гексаны:триэтиламин: этанол) давала 0,268 г соединения заголовка (85%) в виде густой маслянистой жидкости: ИК (хлороформ) 2977, 2939, 1746, 1510, 1176, 1146, 1122 см-1; 1H-ЯМР (300 МГц, CDCl3) δ 7,87-7,90 (д, 1H, J = 9,3 Гц), 7,75-7,78 (д, 1H, J = 8,6 Гц), 7,56-7,57 (д, 1H, J = 2,4 Гц), 7,43-7,46 (д, 1H, J = 8,4 Гц), 7,28-7,31 (м, 3H), 7,10-7,14 (дд, H1, J = 9,2 Гц, J = 2,4 Гц), 7,03-7,06 (м, 2H), 6,86-6,88 (д, 2Н, J=8,5 Гц), 6,71-6,74 (м, 2H), 4,34 (с, 2H), 4,10-4,15 (м, 2H), 2,79-2,83 (м, 2H), 2,52-2,57 (м, 4H), 1,65-1,68 (м, 4H), 1,45-1,51 (м, 2H), 1,39 (с, 9H), 1,36 (с, 9H); МС (FD) m/e 621 (M+).
Пример 5.
[2-(4-n-Бутилсульфонилоксифенил)-6-n-бутилсульфонилоксинафталин-1- ил] [4-[2-(1- пиперидинил)этокси]фенил]метан.
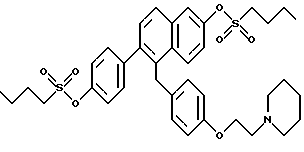
К суспензии продукта по примеру 2 (0,250 г, 0,510 ммоль), перемешиваемой в ТГФ (25 мл), добавляли по очереди N,N-диметиламинопиридин (2 мг), триэтиламин (0,78 мл, 5,6 ммоль) и бутансульфонилхлорид (0,26 мл, 2,04 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 2 часов, затем вливали в этилацетат/воду (100 мл, 1:1) и органический слой впоследствии отделяли. Водную часть экстрагировали добавлением этилацетата (50 мл) и объединенные органические слои промывали насыщенным водным хлоридом аммония (1х25 мл), затем насыщенным водным бикарбонатом натрия (2х25 мл) и насыщенным раствором соли (1х25 мл). Очистка методом радиальной хроматографии (силикагель, 2 мм, 10:8:1:1 этилацетат:гексаны:триэтиламин:метанол) давала 0,289 г (82%) соединения заголовка в виде густого сиропа: ИК (хлороформ) 3032, 2966, 2940, 2879, 1609, 1510, 1375, 1245, 1171, 1149, 1129, 870, 839 см-1; 1H-ЯМР (300 МГц, CDCl3) δ 7,92-7,95 (д, 1H, J = 9,3 Гц), 7,81-7,84 (д, 1H, J = 8,6 Гц), 7,77-7,78 (д, 1H, J = 2,5 Гц), 7,46-7,49 (д, 1H, J = 8,4 Гц), 7,24-7,34 (м, 5H), 6,86-6,88 (д, 2H, J = 8,6 Гц), 6,74-6,77 (д, 2H, J = 8,6 Гц), 4,33 (с, 2H), 4,05-4,09 (м, 2H), 3,25-3,32 (м, 4H), 2,72-2,81 (м, 2H), 2,48-2,52 (м, 4H), 1,93-2,06 (м, 4H), 1,44-1,61 (м, 10H), 0,96-1,01 (м, 3H); МС (FD) m/e 694 (M+).
Пример 6.
[2-(4-n-Гексилсульфонилоксифенил)-6-n-гексилсульфонилоксинафталин- 1-ил] [4-[2-(1-пиперидинил)этокси]фенил]метан.
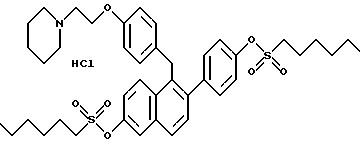
К раствору продукта по примеру 2 (0,49 г, 1,00 ммоль), перемешиваемому в ТГФ (200 мл) при температуре окружающей среды, последовательно добавляли N, N-диметилформамид (10 мг), триэтиламин (0,50 г, 5 ммоль) и гексилсульфонилхлорид (0,46 мл, 2,5 ммоль). Через 18 часов реакционную смесь концентрировали и полученную темную маслянистую жидкость делили между этилацетатом и насыщенным водным раствором бикарбоната натрия. Органический экстракт отделяли, сушили (сульфат натрия) и концентрировали. Неочищенный продукт растворяли в этилацетате и добавляли эфирную соляную кислоту (10 мл насыщенного раствора). Полученный осадок растирали с Et2O и сушили, что давало 1,2 г желательного продукта в виде густого смолистого твердого вещества: 1H-ЯМР (300 МГц, CDCl3) согласующийся со структурой; МС (FD) m/e 938 (M+-соляная кислота).
Получение 4.
[3,4-Дигидро-2-фенил-6-метоксинафталин-1-ил](4-гидроксифенил)метанон.
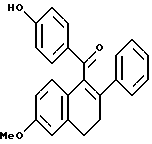
К раствору литийэтантиола [полученного путем добавления n-BuLi (63,7 мл 1,6 М раствора в гексане, 101,4 ммоль), к раствору этантиола (101,4 ммоль), перемешиваемому при 0oC в Et2O (400 мл), с последующим концентрированием], перемешиваемому в диметилформамиде (400 мл), добавляли (3,4-дигидро-6-метокси-2-фенил-1-нафталинил)(4-метоксифенил)метанон, полученный как описано в Jones, et al., J. Med. Chem., 53:931-938 (1992), выше, (30,0 г, 78,0 ммоль). Смесь затем нагревали до 85oC. После 0,5 часа смесь концентрировали и полученное коричневое твердое вещество растворяли в хлороформе. Данный раствор экстрагировали добавлением насыщенного водного хлорида аммония. Водную часть обрабатывали 1н. соляной кислотой, пока не получали pH 5, и впоследствии удаляли хлороформ. Объединенные органические экстракты промывали насыщенным раствором соли, сушили (сульфат натрия), фильтровали и концентрировали. Полученную коричневую жидкость очищали методом флэш-хроматографии (силикагель, градиент этилацетата/гексана), что давало 24,7 г (87%) желательного продукта в виде желтой пены: 1H-ЯМР (300 МГц, CDCl3) δ 7,74 (д, J = 8,6 Гц, 2H), 7,15-7,18 (м, 2H), 7,05-7,18 (м, 3H), 6,86 (д, J = 8,6 Гц, 1H), 6,78 (д, J = 2,7 Гц, 1H), 6,60-6,70 (м, 3H), 6,23 (ушир. с, 1H), 3,78 (с, 3H), 2,95-3,05 (м, 2H), 2,75-2,85 (м, 2H); Анал. расч. для: C 80,87; H 5,66. Найдено: C 80,66; H 5,48; МС (FD) m/e 354 (M+).
Получение 5.
[3,4-Дигидро-2-фенил-6-метоксинафталин-1-ил] [4-[2-(1- пиперидинил)этокси]фенил]метанон.
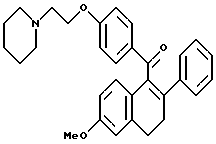
К раствору продукта по получению 4 (20,4 г, 57,0 ммоль), перемешиваемому в диметилформамиде (400 мл) при температуре окружающей среды, добавляли иодид калия (30 мг, 0,18 ммоль) с последующим добавлением карбоната калия (39,3 г, 285 ммоль) и 1-(2-хлорэтил)пиперидинмоногидрохлорида (11,6 г, 62,7 ммоль). Через 16 часов реакционную смесь концентрировали и полученное масло растворяли в хлороформе. Данный раствор тщательно промывали водой, насыщенным раствором соли, сушили (сульфат натрия), фильтровали и концентрировали. Полученное масло очищали методом флэш-хроматографии (силикагель, градиент метанола/хлороформа), что давало 25,1 г (94%) желательного продукта в виде коричневой маслянистой жидкости: 1H-ЯМР (300 МГц, CDCl3) δ 7,79 (д, J = 8,7 Гц, 2H), 7,20-7,33 (м, 2H), 7,04-7,20 (м, 3H), 6,88 (д, J = 8,5 Гц, 1H), 6,70-6,82 (м, 3H), 6,62 (м, 1H), 4,08 (т, J = 6,0 Гц, 2H), 3,70 (с, 3H), 3,03(т, J = 7,5 Гц, 2H), 2,70-2,90 (м, 4H), 2,40-2,60 (м, 4H), 1,55-1,65 (м, 4H), 1,40-1,52 (м, 2H); 13C-ЯМР (75 МГц, CDCl3) δ 198,33, 162,84, 158,97, 141,21, 136,71, 135,97, 137,78, 131,79, 130,44, 128,08, 127,48, 127,24, 126,59, 114,17, 113,80, 111,37, 66,15, 57,68, 55,23, 55,05, 29,73, 28,80, 25,89, 24,12; Анал. Расч. для: C 79,63; H 7,11; N 2,99. Получ: C 79,92; H 7,15; N 3,07. МС (FD) m/e 467 (M+).
Получение 6.
[3,4-Дигидро-2-фенил-6-метоксинафталин-1-ил] [4-[2-(1- пирролидинил)этокси]фенил]метанон.
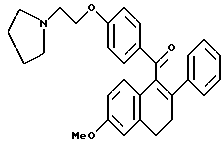
Реакция продукта по получению 4 (1,9 г, 5,3 ммоль), 1-(2-хлорэтил)пирролидинмоногидрохлорида (0,99 г, 5,8 ммоль) и карбоната калия (3,65 г, 29,1 ммоль) в диметилформамиде (50 мл) в соответствии с методикой по получению 5 давала 81% выход соединения заголовка в виде густой маслянистой жидкости: 1H-ЯМР (300 МГц, CDCl3) δ 7,79 (д, J = 7,8 Гц, 2H), 7,20-7,30 (м, 2H), 7,05-7,20 (м, 3H), 6,87 (д, J = 8,6 Гц, 1H), 6,73-6,84 (м, 3H), 6,60 (д, J = 8,6 Гц, 1H), 4,08 (т, J = 5,8 Гц, 2H), 3,78 (с, 3H), 3,00 (т, J = 8,0 Гц, 2H), 2,76-2,96 (м, 4H), 2,50-2,70 (м, 4H), 1,75-1,85 (м, 4H). МС (FD) m/e 453 (M+).
Пример 7.
[3,4-Дигидро-2-фенил-6-метоксинафталин-1-ил] [4-[2-(1- пирролидинил)этокси]фенил]метанол.
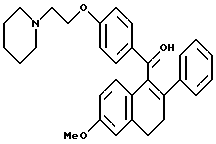
К суспензии литийалюминийгидрида (1,60 г, 42,8 ммоль), перемешиваемой при 0oC в сухом ТГФ (200 мл), добавляли раствор продукта по получению 5 (10,0 г, 21,4 ммоль) в ТГФ (125 мл) по каплям в течение 5-минутного промежутка времени. Реакционной смеси давали нагреться до температуры окружающей среды и впоследствии перемешивали в течение 1 часа. Раствор затем охлаждали до 0oC и осторожно гасили водой (1,6 мл). К данному раствору по каплям добавляли гидроксид натрия (4,8 мл 15% по массе водного раствора) с последующим добавлением воды (1,6 мл). После перемешивания в течение 30 минут смесь фильтровали и твердые вещества тщательно промывали ТГФ. Фильтрат затем концентрировали, что давало 8,7 г (87% желательного продукта в виде желтого масла, которое использовали без дальнейшей очистки: 1H-ЯМР (300 МГц, CDCl3) δ 7,20-7,45 (м, 7H), 6,82 (д, J = 8,3 Гц, 2H), 6,71 (с, 1H), 6,53 (м, 1H), 5,83 (ушир. с, 1H), 4,07 (т, J = 6,1 Гц, 2H), 3,75 (с, 3H), 2,91 (т, J = 6,1 Гц, 2H), 2,60-2,80 (м, 4H), 2,40-2,60 (м, 4H), 1,80-1,95 (м, 2H), 1,52-1,70 (м, 4H), 1,43 (с, 1H); МС (FD) m/e 469 (M+).
Пример 8.
[3,4-Дигидро-2-фенил-6-метоксинафталин-1-ил] [4-[2-(1- пирролидинил)этокси]фенил]метанол.
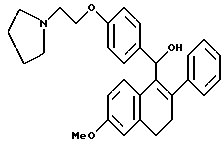
Реакция продукта по получению 4 (1,8 г, 4,0 ммоль), литийалюминийгидрида (0,31 г, 8,0 ммоль) в ТГФ (65 мл) в соответствии с получением продукта по примеру 7 давала 87% выход соединения заголовка в виде белой пены: 1H-ЯМР (300 МГц, CDCl3) δ 7,20-7,40 (м, 7H), 6,86 (д, J = 8,6 Гц, 2H), 6,71 (с, 1H), 6,51 (м, 1H), 5,83 (д, J = 4,9 Гц, 1H), 4,07 (т, J = 6,3 Гц, 2H), 3,75 (с, 3H), 2,82-2,95 (м, 4H), 2,55-2,73 (м, 6H), 2,27 (д, J = 3,8 Гц, 1H), 1,70-1,90 (м, 4H), 1,67 (с, 1H); MC (FD) m/e 455 (M+); HRMS FAB+ для C30H33NO3 расчетная 456,2539. Найдено 456,2531.
Пример 9.
[2-Фенил-6-метоксинафталин-1-ил] [4-[2-(1- пиперидинил)этокси]фенил]метангидрохлорид.
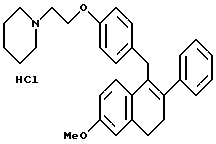
К раствору продукта по примеру 7 (8,7 г, 18,5 ммоль), перемешиваемому в этилацетате (100 мл), добавляли насыщенный раствор хлороводорода в этилацетате (250 мл). Через 0,5 мин полученный раствор концентрировали, что давало 8,0 г (89%) желательного продукта в виде белой пены, которую использовали без дальнейшей очистки: 1H-ЯМР (300 МГц, ДМСО-d6) δ 7,70-7,85 (м, 4H), 7,30-7,50 (м, 7H), 7,10 (с, 1H), 6,80-7,00 (м, 2H), 4,25-4,40 (м, 4H), 4,00-4,20 (ушир. с, 3H), 3,35-3,55 (м, 4H), 2,85-3,55 (м, 2H), 1,70-1,90 (м, 4H), 1,30-1,45 (м, 2H); Анал. расч. для: C 76,29; H 7,02; N 2,87. Найдено: C 76,56; H 7,18; N 2,91; МС (FD) m/e 452 (M+-соляная кислота).
Пример 10.
[2-Фенил-6-метоксинафталин-1-ил] [4-[2-(1- пирролидинил)этокси] фенил] метангидрохлорид.
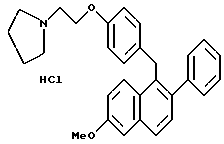
Реакция (1,57 г, 3,4 ммоль) с этилацетатом/соляной кислотой в соответствии с методикой примера 9 давала количественный выход указанного в заголовке продукта: 1H-ЯМР (300 МГц, ДМСО) δ 7,72-7,85 (м, 2H), 7,28-7,45 (м, 7H), 7,10 (м, 1H), 6,78-6,95 (м, 4H), 4,30 (м, 2H), 4,20-4,25 (м, 2H), 3,84 (с, 3H), 3,40-3,60 (м, 2H), 2,95-3,10 (м, 2H), 1,80-2,02 (м, 6H); МС (FD) m/e 437 (M+-соляная кислота); Анал. расч. для: C 76,01; H 6,80; N 2,95. Найдено: C 75,71; H 6,85; N 2,82.
Пример 11.
[2-Фенил-6-гидроксинафталин-1-ил] [4-[2-(1- пиперидинил)этокси] фенил] метан.
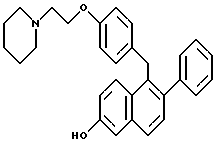
К раствору продукта по примеру 9 (4,0 г, 8,0 ммоль), перемешиваемому в 1,2-дихлорэтане (50 мл) при 0oC, добавляли трихлорид бора (10 мл, 117,0 ммоль). Полученный темно-пурпурный раствор растворяли при комнатной температуре в течение ночи в герметичной пробирке, затем охлаждали при 0oC. Осторожно по каплям добавляли метанол (50 мл) в течение 30-минутного промежутка времени (осторожно: выделение газа). Полученный раствор концентрировали и растворяли в этилацетате. Органический экстракт промывали насыщенным водным бикарбонатом натрия, насыщенным раствором соли, сушили (сульфат натрия), фильтровали и концентрировали. Полученную коричневую пену очищали методом флэш-хроматографии (силикагель, градиент метанола/хлороформа), что давало 2,7 г (63%) желательного продукта в виде белой пены: 1H-ЯМР (300 МГц, ДМСО) δ 9,72 (ушир. с, 1H), 7,62-7,80 (м, 2H), 7,22-7,50 (м, 6H), 7,10-7,22 (м, 2H), 7,00 (м, 1H), 6,80-6,90 (м, 2H), 6,78 (м, 1H), 4,23 (с, 2H), 3,85-4,10 (м, 2H), 2,50-2,75 (м, 2H), 2,25-2,50 (м, 4H), 1,25-1,56 (м, 6H); Анал. расч. для: C 82,35; H 7,14; N 3,20. Найдено: C 82,17; H 7,11; N 3,35; МС (FD) m/e 437 (M+); ИК (KBr) 2935,07; 2855,01, 1621,38, 1597,26 см-1.
Пример 12.
[2-Фенил-6-гидроксинафталин-1-ил] [4-[2-(1- пирролидинил)этокси] фенил] метанол.
Реакция продукта по примеру 10 (1,27 г, 2,7 ммоль) с трихлоридом бора (10 мл, 117 ммоль) в 1,2-дихлорэтане (30 мл) в соответствии с методикой примера 11 давало 32% выход желательного продукта в виде белого твердого вещества: ИК (KBr) 2932,17, 2876,23, 2815,47, 1620,41, 1597,26 см-1; 1H-ЯМР (300 МГц, CDCl3) δ 7,74 (д, J = 8,5 Гц, 1H), 7,61 (д, J = 8,5 Гц, 1H), 7,20-7,40 (м, 7H), 7,13 (с, 1H), 7,00 (м, 1H), 6,85 (д, J = 8,3 Гц, 2H), 6,66 (д, J = 8,3 Гц, 2H), 4,31 (с, 2H), 4,06 (т, J = 5,9 Гц, 2H), 2,95 (т, J = 5,8 Гц, 2H), 2,65-2,80 (м, 4H), 1,77-1,90 (м, 4H); МС (FD) m/e 424 (M+); Анал. расч. для: C 82,24; H 6,90; N 3,31. Найдено: C 82,01; H 6,84; N 3,37.
Пример 13.
[3,4-Дигидро-2-(4-метоксифенил)нафталин-1-ил] [4-[2-(1- пиперидинил)этокси]фенил]метанол.
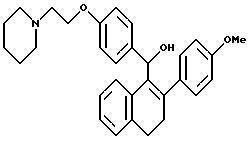
К суспензии [2-(4-метоксифенил)-3,4-дигидронафт-1-ил][4-[2-(1- пиперидинил)этокси] фенил] метанонмезилата [Jones, et al., J. Med. Chem. 35:931 (1992), выше] (2,00 г, 3,35 ммоль), перемешиваемой в ТГФ (100 мл) при температуре окружающей среды, медленно добавляли литийалюминийгидрид (1,0 г, 26 ммоль) в течение 20-минутного промежутка времени. Через 18 часов раствор концентрировали почти досуха, затем осторожно гасили водой (50 мл). Полученную смесь экстрагировали добавлением этилацетата (3х100 мл). Объединенные органические экстракты промывали водой, сушили (сульфат натрия) и концентрировали. Очистка методом жидкостной хроматографии (Waters Prep 500, силикагель, градиент от хлороформа до 25% хлороформ:метанол) давала 1,0 г желательного продукта в виде желто-коричневого аморфного порошка: 1H-ЯМР (300 МГц, CDCl3) согласующийся со структурой; МС (FD) m/e 469 (M+).
Пример 14.
[3,4-Дигидро-2-(4-метоксифенил)нафталин-1-ил] [4-[2-(1- пирролидинил)этокси]фенил]метанол.
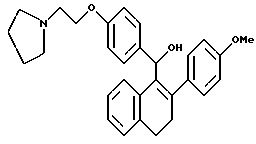
Реакция [2-(4-метоксифенил)-3,4-дигидронафт-1-ил] [4-[2-(1- пирролидинил)этокси] фенил[метанонмезилата [Jones, et al. , J. Med. Chem. 35:931 (1992), выше] (0,85 г, 1,9 ммоль) и литийалюминийгидрида (0,16 г, 4,0 ммоль) в ТГФ (150 мл) в соответствии с экспериментальной методикой опыта 13 давала желательное соединение в виде желто-коричневого аморфного твердого вещества: 1H-ЯМР (300 МГц, CDCl3) согласующийся со структурой; МС (FD) m/e 455 (M+); Анал. расч. для: C 79,20; H 7,26; N 3,08. Найдено: C 79,11; H 7,47; N 2,93.
Пример 15.
[2-(4-Метоксифенил)нафталин-1-ил] [4-[2-(1- пиперидинил)этокси] фенил] метангидрохлорид.
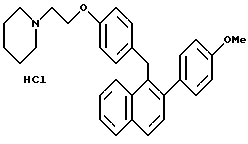
К раствору продукта по примеру 13 (1,90 г, 4,21 ммоль), перемешиваемому в метаноле (40 мл) при температуре окружающей среды, добавляли соляную кислоту в метаноле (10 мл насыщенного раствора). Через 48 часов реакционную смесь концентрировали и сушили. Растирание с эфиром с последующим фильтрованием и сушкой давало 580 мг желательного соединения в виде белого порошка: 1H-ЯМР (CDCl3, 300 МГц) согласующийся со структурой; МС (FD) m/e 451 (M+-соляная кислота).
Пример 16.
[2-(4-Метоксифенил)нафталин-1-ил] [4-[2-(1- пирролидинил)этокси] фенил] метангидрохлорид.
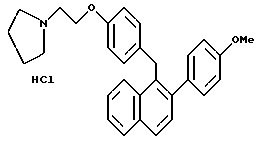
К раствору продукта по примеру 14 (2,0 г, 4,58 ммоль), перемешиваемому в метаноле (50 мл) при температуре окружающей среды, добавляли соляную кислоту в метаноле (10 мл насыщенного раствора). Реакционную смесь затем концентрировали до 20 мл и охлаждали до -20oC в течение нескольких часов. Фильтрование давало 0,62 г желательного продукта в виде белого порошка: 1H-ЯМР (CDCl3, 300 МГц) согласующийся со структурой; МС (FD) m/e 437 (M+-соляная кислота); Анал. расч. для: C 76,01; H 6,80; N 2,96. Получ: C 75,95; H 6,76; N 2,98.
Получение 7.
[3,4-Дигидро-2-(4-метоксифенил)-6-метоксинафталин-1-ил] [4-[2-(1- пирролидинил)этокси]фенил]метанон.
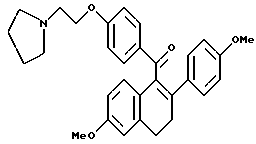
К раствору по получению 2 (2,0 г, 5,2 ммоль), перемешиваемому в диметилформамиде (50 мл), добавляли карбонат калия (3,6 г, 26 ммоль) и 1-(2-хлорэтил)пирролидин-моногидрохлорид (0,8 г, 5,7 ммоль). Реакционную смесь перемешивали в течение ночи при температуре окружающей среды и концентрировали. Полученное масло растворяли в хлороформе и полученный раствор тщательно промывали водой, насыщенным раствором соли, сушили (сульфат натрия), фильтровали и концентрировали. Полученное масло очищали методом флэш-хроматографии (силикагель, градиент метанола/хлороформа), что давало 2,25 г (90%) желательного продукта в виде коричневой маслянистой жидкости: 1H-ЯМР (300 МГц, CDCl3) δ 7,80 (д, J = 9,4 Гц, 2H), 7,18 (д, J = 6,8 Гц, 2H), 6,87 (д, J = 8,6 Гц, 2H), 6,65-6,85 (м, 4H), 6,60 (м, 1H), 4,09 (т, J = 5,8 Гц, 2H), 3,78 (с, 3H), 3,71 (с, 3H), 3,01 (т, J = 7,5 Гц, 2H), 2,88 (т, J = 5,8 Гц, 2H), 2,65-2,85 (м, 2H), 2,60-2,75 (м, 4H), 1,80-1,90 (м, 4H); МС (FD) m/e 483 (M+).
Пример 17.
[3,4-Дигидро-2-(4-метоксифенил)-6-метоксинафталин-1-ил][4-[2-(1- пирролидинил)этокси]фенил]метанол.
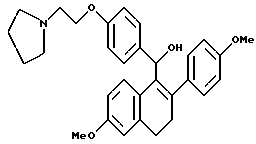
К суспензии литийалюминийгидрида (0,34 г, 8,80 ммоль), перемешиваемой при 80oC в ТГФ (40 мл), медленно добавляли раствор продукта по получению 7 (2,14 г, 4,4 ммоль) в ТГФ (25 мл) в течение 5-минутного промежутка времени. Реакционную смесь нагревали до температуры окружающей среды. Через 1 час смесь охлаждали до 0oC и осторожно гасили водой (0,4 мл). К данному раствору по каплям добавляли гидроксид натрия (1,2 мл 15% по массе водного раствора) с последующим добавлением воды (0,4 мл). После перемешивания в течение 30 минут смесь фильтровали и твердые вещества тщательно промывали ТГФ. Фильтрат концентрировали, что давало 1,60 г (75%) желательного продукта в виде белой пены, которую использовали без дальнейшей очистки: 1H-ЯМР (300 МГц, ДМСО) δ 7,40 (д, J = 8,2 Гц, 2H), 7,33 (д, J = 7,6 Гц, 1H), 7,16 (д, J = 8,1 Гц, 2H), 6,90 (д, J = 7,7 Гц, 2H), 6,75 (д, J = 7,8 Гц, 2H), 6,66 (с, 1H), 6,45 (д, J = 7,6 Гц, 1H), 5,69 (с, 1H), 5,64 (с, 1H), 3,95 (т, J = 5,5 Гц, 2H), 3,72 (с, 3H), 3,64 (с, 3H), 2,65-2,85 (м, 4H), 2,40-2,65 (м, 6H), 1,60-1,80 (м, 4H); МС (FD) m/e 485 (M+).
Пример 18.
[2-(4-Метоксифенил)-6-метоксинафталин-1-ил] [4-[2-(1- пирролидинил)этокси]фенил]метангидрохлорид.
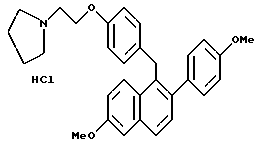
К раствору продукта по примеру 17 (1,61 г, 3,30 ммоль), перемешиваемому в этилацетате (50 мл) при температуре окружающей среды, добавляли насыщенный раствор хлороводорода в этилацетате (50 мл). Полученную смесь концентрировали, что давало 1,66 г (100%) желательного продукта в виде белой пены, которую использовали без дальнейшей очистки: 1H-ЯМР (300 МГц, ДМСО-d6) δ 7,70-7,80 (м, 2H), 7,30-7,40 (м, 2H), 7,20-7,30 (м, 2H), 7,05 (м, 1H), 6,80-7,00 (м, 6H), 4,29 (с, 2H), 4,20-4,25 (м, 2H), 3,84 (с, 3H), 3,75 (с, 3H), 3,42-3,75 (м, 4H), 3,00-3,15 (м, 2H), 1,80-2,00 (м, 4H); МС (FD) m/e 467 (M+-соляная кислота).
Пример 19.
[2-(4-Гидроксифенил)-6-гидроксинафталин-1-ил] [4-[2-(1- пирролидинил)этокси]фенил]метан.
К раствору продукта по примеру 18 (1,61 г, 2,60 ммоль) в 1,2-дихлорэтане (30 мл), перемешивая при 0oC, добавляли трихлорид бора (10 мл, 117 ммоль). Полученный темно-пурпурный раствор перемешивали в течение ночи при температуре окружающей в герметичной пробирке. После охлаждения раствора до 0oC осторожно добавляли метанол (25 мл) в течение 30-минутного промежутка времени (осторожно, выделение газа). Раствор впоследствии концентрировали и полученный продукт растворяли в 30% изопропаноле/хлороформе, затем промывали насыщенным бикарбонатом натрия, насыщенным раствором соли, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали методом радиальной хроматографии (градиент метанола/хлороформа), что давало 0,34 г (27%) желательного продукта в виде белой пены: 1H-ЯМР (300 МГц, ДМСО-d6) δ 9,45 (с, 1H), 9,36 (с, 1H), 7,72 (д, J = 8,8 Гц, 1H), 7,62 (д, J = 9,2 Гц, 1H), 7,28 (д, J = 8,7 Гц, 1H), 7,00-7,10 (м, 2H), 6,80-6,90 (м, 2H), 6,70-6,80 (м, 4H), 5,45 (с, 1H), 4,84 (с, 1H), 4,25 (с, 2H), 3,90-4,05 (м, 2H), 2,75-2,90 (м, 2H), 2,50-2,65 (м, 4H), 1,60-1,80 (м, 4H); 13C-ЯМР (75 МГц, ДМСО-d6) δ 203,32, 191,97, 188,16, 186,14, 185,95, 177,43, 173,46, 169,60, 167,74, 163,48, 162,30, 159,87, 158,14, 154,98, 152,43, 60,50, 56,25, 54,00, 45,05, 41,00, 37,50, 35,00, 30,05, 27,50, 26,00, 22,50, 20,00; Анал. расч. для: C 79,24; H 6,65; N 3,19. Получ: C 78,99; H 6,51; N 2,92. МС (FD) m/e 440 (M+); ИК (KBr) 3382,61, 2964,00, 1610,77, 1509,49 см-1.
Получение 8.
[3,4-Дигидро-2-(4-метоксифенил)-6-метоксинафталин-1-ил] [4-[2-(1- N,N-диметиламино)этокси]фенил]метанон.
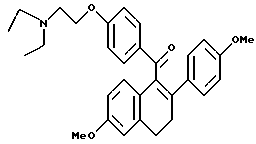
Реакция продукта по получению 2 (1,6 г, 4,1 ммоль) 2-диэтиламиноэтилхлоридгидрохлорида (0,8 г, 4,5 ммоль) и карбоната калия (2,3 г, 16,4 ммоль) в диэтилформамиде (50 мл) в соответствии с методикой по получению 3 давала 95% выход желательного продукта: 1H-ЯМР (300 МГц, CDCl3) δ 7,82 (д, J=8,8 Гц, 2Н), 7,20 (д, J = 8,7 Гц, 2H), 6,89 (д, J = 8,5 Гц, 1H), 6,65-6,80 (м, 5H), 6,62 (м, 1H), 4,03 (т, J = 6,3 Гц, 2H), 3,80 (с, 3H), 3,72 (с, 3H), 3,03 (т, J = 7,7 Гц, 2H), 2,75-2,90 (м, 4H), 2,61 (ABк, J = 7,2 Гц, Δν = 14,4 Гц, 4H), 1,06 (т, J = 7,2 Гц, 6H); МС (FD) m/e 485 (M+); Анал. расч. для: C 76,67; H 7,26; N 2,88. Получ: C 76,97; H 7,43; N 2,91.
Получение 9.
[3,4-Дигидро-2-(4-метоксифенил)-2,4-дигидро-6-метоксинафталин- 1-ил] [4-[3-(1-пиперидинил)пропокси]фенил]метанон.
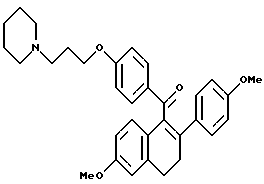
Реакция продукта по получению 2 (1,6 г, 4,1 ммоль), 1-(3-хлорпропил)пиперидингидрохлорида (0,9 г, 4,5 ммоль) и карбоната калия (2,3 г, 16,4 ммоль) в ДМФ (50 мл) в соответствии с методикой по получению 7 давала 95% выход желательного продукта: 1H-ЯМР (300 МГц, CDCl3) δ 7,80 (д, J = 8,7 Гц, 2H), 7,19 (д, J = 5,0 Гц, 2H), 6,86 (д, J = 8,4 Гц, 1H), 6,63-6,80 (м, 5H), 6,60 (м, 1H), 3,98 (т, J = 6,4 Гц, 2H), 3,78 (с, 3H), 3,70 (с, 3H), 3,00 (т, J = 7,7 Гц, 2H), 2,75-2,85 (м, 2H), 2,30-2,50 (м, 6H), 1,90-2,00 (м, 2H), 1,50-1,65 (м, 4H), 1,40-1,50 (м, 2H); МС (FD) m/e 511 (M+); Анал. расч. для: C 77,47; H 7,29; N 2,74. Получ: C 77,42; H 7,36; N 2,72.
Пример 20.
[3,4-Дигидро-2-(4-метоксифенил)-6-метоксинафталин-1-ил] [4-[2-(1- N,N-диэтиламино)этокси]фенил]метанол.
Реакция продукта по получению 8 (1,7 г, 3,4 ммоль) с литийалюминийгидридом (0,3 г, 6,8 ммоль) в ТГФ (80 мл) в соответствии с методикой примера 17 давала количественный выход желательного продукта: 1H-ЯМР (300 МГц, CDCl3) δ 7,33 (д, J = 8,5 Гц, 2H), 7,20-7,30 (м, 3H), 6,80-6,90 (м, 4H), 6,71 (с, 1H), 6,50 (м, 1H), 5,85 (д, J = 3,9 Гц, 1H), 4,01 (т, J = 6,4 Гц, 2H), 3,78 (с, 3H), 3,74 (с, 3H), 2,86 (ABк, J = 8,2 Гц, Δν = 14,7 Гц, 4H), 2,60-2,70 (м, 6H), 1,85 (м, 1H), 1,05 (т, J = 7,2 Гц, 6H); МС (FD) m/e 487 (M+).
Пример 21.
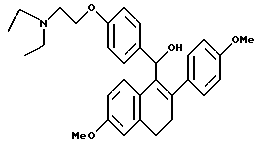
[3,4-Дигидро-2-(4-метоксифенил)-6-метоксинафталин-1-ил][4-[3-(1- пиперидинил)пропокси]фенил]метанол.
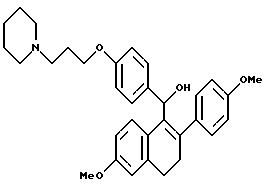
Реакция продукта по получению 9 (1,77 г, 3,50 ммоль) с литийалюминийгидридом (0,27 г, 7,00 ммоль) в ТГФ (50 мл) в соответствии с методикой по примеру 17 давала 97% выход желательного продукта: 1H-ЯМР (300 МГц, CDCl3) δ 7,32 (д, J = 8,4 Гц, 2H), 7,20-7,30 (м, 4H), 6,80-6,90 (м, 3H), 6,70 (с, 1H), 6,50 (м, 1H), 5,85 (с, 1H), 3,96 (т, J = 6,3 Гц, 2H), 3,78 (с, 3H), 3,74(с, 3H), 2,85-2,95 (м, 2H), 2,60-2,70 (м, 2H), 2,25-2,50 (м, 6H), 1,90-2,00 (м, 2H), 1,54-1,60 (м, 4H), 1,43 (с, 2H); МС (FD) m/e 514 (M+1).
Пример 22.
[2-(4-Метоксифенил)-6-метоксинафталин-1-ил] [4-[2-(1- N, N-диэтиламино)этокси]фенил]метангидрохлорид.
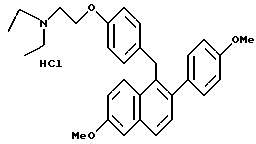
Реакция продукта по примеру 20 (1,6 г, 3,3 ммоль) с соляной кислотой (100 мл насыщенного раствора в этилацетате) в этилацетате (100 мл) в соответствии с методикой по примеру 18 давала 90% выход желательного продукта: ИК (KBr) 3416,37, 2935,07, 2835,72, 2575,30, 2437,37, 1624,27, 1608,84, 1510,45 см-1; 1H-ЯМР (300 МГц, CDCl3) δ 7,72 (т, J = 8,6 Гц, 2H), 7,15-7,30 (м, 4H), 7,05 (м, 1H), 6,85-6,95 (м, 3H), 6,72 (д, J = 8,6 Гц, 2H), 4,40-4,50 (м, 2H), 4,35 (с, 3H), 3,92 (с, 3H), 3,83 (с, 3H), 3,35-3,45 (м, 2H), 3,20-3,35 (м, 4H), 1,43 (т, J = 7,2 Гц, 6H); МС (FD) m/e 470 (M+ -соляная кислота); Анал. расч. для: C 73,57; H 7,17; N 2,77. Найдено: C 73,80; H 7,35; N 2,77.
Пример 23.
[2-(4-Метоксифенил)-6-метоксинафталин-1-ил][4-[3-(1- пиперидинил)пропокси]фенил]метангидрохлорид.
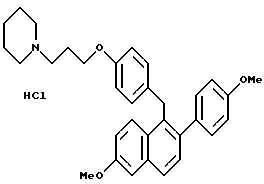
Реакция продукта по примеру 21 (1,5 г, 2,9 ммоль) с соляной кислотой (50 мл насыщенного этилацетатного раствора) в этилацетате (50 мл) в соответствии с методикой по примеру 18 давала 97% выход желательного продукта: 1H-ЯМР (300 МГц, CDCl3) δ 7,70-7,80 (м, 2H), 7,42 (д, J = 8,4 Гц, 1H), 7,15-7,30 (м, 3H), 7,05 (м, 1H), 6,85-6,95 (м, 4H), 6,69 (д, J = 8,6 Гц, 2H), 4,34 (с, 2H), 3,97-4,03 (м, 2H), 3,92 (с, 3H), 3,82 (с, 3H), 3,50-3,60 (м, 2H), 2,57-2,70 (м, 2H), 2,20-2,50 (м, 4H), 1,80-2,00 (м, 4H); МС (FD) m/e 495 (M+ - соляная кислота); Анал. расч. для: C 74,49; H 7,20; N 2,63. найдено: C 74,74; H 7,36; N 2,75.
Пример 24.
[2-(4-Гидроксифенил)-6-гидроксинафталин-1-ил] [4-[2-(1-N, N- диэтиламино)этокси]фенил]метан.
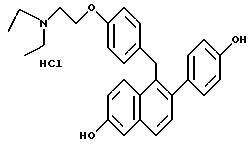
Реакция продукта по примеру 22 (1,32 г, 2,60 ммоль) с трихлоридом бора (10,0 мл, 117,0 ммоль) в 1,2-дихлорэтане (30 мл) в соответствии с методикой по примеру 19 давала 76% выход желательного продукта в виде белого порошка: ИК (KBr) 3356,57, 2973,65, 1734,23, 1704,33, 1610,77, 1509,49 см-1; 1H-ЯМР (300 МГц, ДМСО-d6) δ 9,62 (с, 1H), 9,43 (с, 1H), 7,56-7,70 (м, 2H), 7,24 (д, J = 8,4 Гц, 1H), 7,00-7,15 (м, 3H), 6,95 (м, 1H), 6,82 (д, J = 8,6 Гц, 2H), 6,65-6,78 (м, 4H), 4,23 (с, 2H), 4,00 (т, J = 6,4 Гц, 2H), 2,65-2,75 (м, 2H), 2,40-2,60 (м, 4H), 0,90 (т, J = 7,1 Гц, 6H); 13C-ЯМР (75 МГц, ДМСО-d6) δ 156,53, 156,45, 154,87, 136,65, 134,44, 133,49, 132,66, 132,28, 130,14, 128,90, 128,73, 126,93, 126,57, 125,18, 118,73, 115,01, 114,32, 109,43, 66,22, 51,43, 47,00, 39,00, 33,81, 11,87; МС (FD) m/e 442 (M+); HRMS (FАB+) для C29H31NO3 рассчитано 442,2382, получено 442,2381.
Получение 10.
[3,4-Дигидро-2-(4-метоксифенил)-6-метоксинафталин-1-ил] [4-[3-(1- бром)этокси]фенил]метанон.
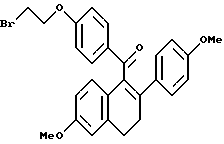
К раствору продукта по получению 2 (4,00 г, 10,0 ммоль), перемешиваемому в 2-бутаноле (100 мл) при температуре окружающей среды, добавляли карбонат калия (2,76 г, 20,0 ммоль) и 1,2-дибромэтан (17,2 мл, 100 ммоль). Данный раствор нагревали с обратным холодильником в течение ночи, затем фильтровали и концентрировали. Полученное коричневое масло очищали методом флэш-хроматографии (силикагель, 20% этилацетат/гексаны), что давало 4,40 г (89%) желательного продукта в виде коричневой маслянистой жидкости. 1H-ЯМР (300 МГц, CDCl3) δ 7,81 (д, J = 8,7 Гц, 2H), 7,18 (д, J = 8,7 Гц, 2H), 6,86 (д, J = 8,6 Гц, 1H), 6,76 (д, J = 6,1 Гц, 3H), 6,78 (д, J = 6,8 Гц, 2H), 6,60 (м, 1H), 4,26 (т, J = 6,1 Гц, 2H), 3,78 (с, 3H), 3,70 (с, 3H), 3,60 (т, J = 6,4 Гц, 2H), 3,01 (т, J = 7,7 Гц, 2H), 2,75-2,85 (м, 2H); Анал. расч. для: C 65,73; H 5,11. Найдено: C 65,96; H 5,28.
Получение 11.
[3,4-Дигидро-2-(4-метоксифенил)-6-метоксинафталин-1-ил][4-[3-(1- гексаметилениминил)этокси]фенил]метанон.
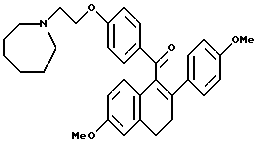
К продукту по получению 10 (2,1 г, 4,3 ммоль), перемешиваемому в диметилформамиде (50 мл) при температуре окружающей среды, добавляли карбонат калия (1,8 г, 13 ммоль) и гексаметиленимин (0,9 мл, 13 ммоль). Раствор впоследствии нагревали до 100oC. После перемешивания в течение ночи смесь концентрировали и полученное коричневое масло делили между хлороформом и водой. Органический экстракт промывали насыщенным раствором соли, сушили (сульфат натрия), фильтровали и концентрировали. Полученную желтую маслянистую жидкость очищали методом радиальной хроматографии (градиент этилацетата/гексана/метанола), что давало 0,95 г (43%) желательного продукта в виде желтого масла: 1H-ЯМР (300 МГц, CDCl3) δ 7,81 (д, J = 8,7 Гц, 2H), 7,21 (д, J = 6,9 Гц, 1H), 6,81 (д, J = 8,5 Гц, 1H), 6,60-6,85 (м, 7H), 4,00-4,50 (м, 2H), 3,80 (с, 3H), 3,72 (с, 3H), 2,85-3,10 (м, 4H), 2,70-2,85 (м, 6H), 1,50-1,80 (м, 8H); Анал. расч. для: C 77,47; H 7,29; N 2,74. найдено: C 77,25; H 7,16; N 2,71; МС (FD) m/e 511 (M+).
Пример 25.
[3,4-Дигидро-2-(4-метоксифенил)-6-метоксинафталин-1-ил] [4-[2-(1- гексаметилениминил)этокси]фенил]метанол.
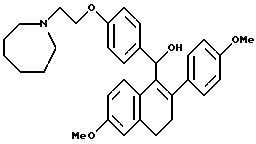
К суспензии литийалюминийгидрида (0,3 г, 7,2 ммоль), перемешиваемой при 0oC в ТГФ (40 мл), медленно добавляли раствор продукта по получению 11 (1,8 г, 3,6 ммоль) в ТГФ (25 мл) в течение 5-минутного промежутка времени. Реакционной смеси давали нагреться до температуры окружающей среды. Через 1 час смесь охлаждали до 0oC и осторожно гасили водой (0,4 мл). К данному раствору медленно добавляли гидроксид натрия (1,2 мл 15% по массе водного раствора) с последующим добавлением воды (0,4 мл). После перемешивания в течение 30 минут смесь фильтровали и твердые вещества тщательно промывали ТГФ, фильтрат концентрировали, что давало 1,71 г (93%) желательного продукта в виде белой пены, которую использовали без дальнейшей очистки: 1H-ЯМР (300 МГц, CDCl3) δ 7,34 (д, J = 8,5 Гц, 2H), 7,20-7,30 (м, 3H), 6,80-6,90 (м, 4H), 6,73 (с, 1H), 6,55 (м, 1H), 5,88 (с, 1H), 4,06 (т, J = 6,3 Гц, 2H), 3,81 (с, 3H), 3,76 (с, 3H), 2,85-3,00 (м, 4H), 2,75-2,85 (м, 4H), 2,63-2,75 (м, 2H), 2,95 (м, 1H), 1,60-1,75 (м, 8H); Анал. расч. для: C 77,16; H 7,65; N 2,73. Найдено: C 77,33; H 7,79; N 2,71. МС (FD) m/e 513 (M+).
Пример 26.
Солянокислая соль [2-(4-Метоксифенил)-6-метоксинафталин-1- ил][4-[2-(1-гексаметилениминил)этокси]фенил]метана.
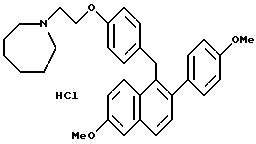
К раствору продукта по примеру 25 (1,7 г, 3,3 ммоль), перемешиваемому в этилацетате (100 мл) при температуре окружающей среды, добавляли соляную кислоту (100 мл насыщенного раствора в этилацетате). Полученную смесь концентрировали, что давало 1,66 г (94%) желательного продукта, который использовали без очистки: 1H-ЯМР (300 МГц, CDCl3) δ 7,48 (т, J = 8,9 Гц, 2H), 7,43 (д, J = 8,6 Гц, 1H), 7,20-7,35 (м, 3H), 7,10 (м, 1H), 6,85-7,00 (м, 4H), 6,75 (д, J = 8,6 Гц, 2H), 4,45-4,60 (м, 2H), 4,37 (с, 2H), 3,94 (с, 3H), 3,85 (с, 3H), 3,55-3,70 (м, 2H), 3,40-3,50 (м, 2H), 3,00-3,20 (м, 2H), 2,10-2,25 (м, 2H), 1,80-2,00 (м, 4H), 1,60-1,80 (м, 2H); 13C-ЯМР (75 МГц, ДМСО) δ 155,6, 137,15, 134,29, 134,19, 134,08, 132,29, 130,15, 129,01, 128,79, 127,28, 126,91, 125,95, 124,94, 118,63, 114,61, 113,70, 106,79, 62,42, 55,20, 55,13, 55,10, 54,85, 54,10, 33,77, 30,44, 26,05, 22,72; Анал. расч. для: C 74,49; H 7,20; N 2,63. Получ: C 74,73; H 7,16; N 2,62; МС (FD) m/e 495 (M+ - соляная кислота); ИК (KBr) 2234,10, 2862,73, 2835,72, 2448,94, 1624,27, 1608,84, 1511,42 см-1.
Пример 27.
[2-(4-Гидроксифенил)-6-гидроксинафталин-1-ил] [4-[2-(1- гексаметилениминил)этокси]фенил]метана.
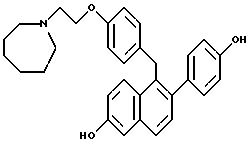
К раствору продукта по примеру 26 (1,3 г, 2,4 ммоль), перемешиваемому в 1,2-дихлорэтане (30 мл) при 0oC, добавляли трихлорид бора (10 мл, 117 ммоль). Полученный темно-пурпурный раствор перемешивали в течение ночи при температуре окружающей среды в герметичной пробирке, затем охлаждая до 0oC. Метанол (25 мл) медленно добавляли в течение 30-минутного промежутка времени (осторожно: выделение газа) и полученный раствор концентрировали. Неочищенный продукт растворяли в 20% метаноле/хлороформе и впоследствии промывали насыщенным бикарбонатом натрия и насыщенным раствором соли. Органический экстракт сушили (сульфат натрия), фильтровали и концентрировали. Полученную коричневую пену очищали методом радиальной хроматографии (градиент этилацетата/триэтиламина/метанола/гексана), что давало желто-коричневое твердое вещество. Данный продукт растворяли в этилацетате, затем промывали насыщенным бикарбонатом натрия. Органический экстракт концентрировали, что давало 0,60 г (54%) желательного продукта в виде белой пены: 1H-ЯМР (300 МГц, ДМСО-d6) δ 9,64 (с, 1H), 9,41 (с, 1H), 7,55-7,70 (м, 2H), 7,24 (д, J = 8,5 Гц, 1H), 7,00-7,10 (м, 3H), 6,95 (м, 1H), 6,81 (д, J = 8,6 Гц, 2H), 6,70-6,78 (м, 4H), 4,23 (с, 2H), 3,91 (т, J = 6,0 Гц, 2H), 2,70-2,80 (м, 2H), 2,55-2,70 (м, 4H), 1,40-1,60 (м, 8H); Анал. расч. для: C 79,63; H 7,11; N 2,99. Получ: C 79,35; H 6,87; N 2,75; МС (FD) m/e 468 (M+); ИК (KBr) 3362,35, 2926,39, 2855,98, 1734,23, 1704,33, 1610,77, 1509,49 см-1.
Получение 12.
[3,4-Дигидро-2-(4-метоксифенил)-3,4-дигидро-6-метоксинафталин- 1-ил] [4-[2-(1-морфолинил)этокси]фенил]метанон.
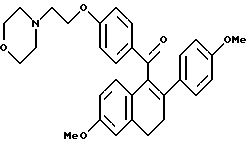
Реакция продукта по получению 10 (2,1 г, 4,3 ммоль) морфолина (1,13 мл, 12,9 ммоль) и карбоната калия (1,78 г, 12,9 ммоль) в ДМФ (50 мл) в соответствии с методикой по получению 11 давала 80% выход желательного продукта в виде густой маслянистой жидкости: 1H-ЯМР (300 МГц, CDCl3) δ 7,83 (д, J = 8,7 Гц, 2H), 7,60 (м, 1H), 7,20 (д, J = 8,8 Гц, 2H), 6,88 (д, J = 8,7 Гц, 1H), 6,65-6,80 (м, 5H), 4,05-4,20 (м, 2H), 3,80 (с, 3H), 3,73 (с, 3H), 3,70-3,80 (м, 4H), 2,90 (т, J = 7,9 Гц, 2H), 2,75-2,85 (м, 4H), 2,50-2,60 (м, 4H); МС (FD) m/e 499 (M+); анал. расч. для: C 74,53; H 6,66; N 2,80. Получ: C 74,75; H 6,58; N 2,83.
Получение 13.
[3,4-Дигидро-2-(4-метоксифенил)-6-метоксинафталин-1-ил] [4-[2-(1- (3,3-диметил)пирролидинил)этокси]фенил]метанон.
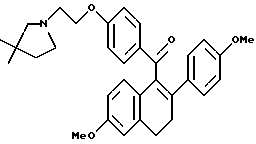
Реакция продукта по получению 10 (2,1 г, 4,3 ммоль), 3,3-диметилпирролидина (1,2 мл, 12 ммоль) и карбоната калия (1,8 г, 13 ммоль) в ДМФ (100 мл) в соответствии с методикой по получению 11 давала 60% выход желательного продукта в виде густой маслянистой жидкости: 1H-ЯМР (300 МГц, CDCl3) δ 7,80 (д, J = 8,7 Гц, 2H), 7,18 (д, J = 8,7 Гц, 2H), 6,87 (д, J = 8,6 Гц, 1H), 6,73-6,80 (м, 3H), 6,67 (д, J = 8,6 Гц, 2H), 6,60 (м, 1H), 4,05 (м, 2H), 3,78 (с, 3H), 3,71 (с, 3H), 2,89-3,05 (м, 2H), 2,73-2,86 (м, 4H), 2,64-2,75 (м, 2H), 2,04 (с, 2H), 1,60 (т, J = 6,9 Гц, 2H), 1,07 (с, 6H); МС (FD) m/e 511 (M+).
Пример 28.
[3,4-Дигидро-2-(4-метоксифенил)-6-метоксинафталин-1-ил][4-[2-(1- морфолинил)этокси]фенил]метанол.
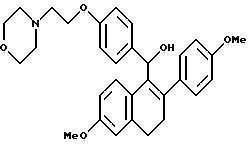
Реакция продукта по получению 12 (1,6 г, 3,2 ммоль) с литийалюминийгидридом (0,3 г, 7,2 ммоль) в ТГФ (65 мл) в соответствии с методикой примера 25 давала 98% выход желательного продукта в виде белой пены: 1H-ЯМР (300 МГц, CDCl3) δ 7,39 (д, J = 8,7 Гц, 2H), 7,20-7,30 (м, 4H), 6,80-7,00 (м, 3H), 6,73 (м, 1H), 6,55 (м, 1H), 5,86 (д, J = 4,2 Гц, 1H), 4,09 (т, J = 5,6 Гц, 2H), 3,80 (с, 3H), 3,70-3,80 (м, 4H), 3,76 (с, 3H), 2,85-3,00 (м, 2H), 2,75-2,85 (м, 2H), 2,65 (м, 1H), 2,55-2,65 (м, 4H), 1,05-1,10 (м, 2H); МС (FD) m/e 501 (M+); Анал. расч. для: C 74,23; H 7,03; N 2,79. Найдено: C 74,51; H 7,18; N 2,79.
Пример 29.
[3,4-Дигидро-2-(4-метоксифенил)-6-метоксинафталин-1-ил] [4-[2-(1- (3,3-диметил)пирролидинил)этокси]фенил]метанол.
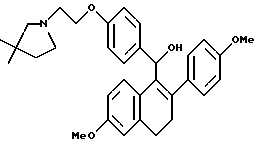
Реакция продукта по получению 13 (1,3 г, 2,5 ммоль) с литийалюминийгидридом (0,2 г, 5,0 ммоль) в ТГФ (65 мл) в соответствии с методикой примера 25 давала 98% выход желательного продукта в виде белой пены:: 1H-ЯМР (300 МГц, CDCl3) δ 7,33 (д, J = 8,6 Гц, 2H), 7,20-7,30 (м, 3H), 6,80-6,90 (м, 4H), 6,70 (м, 1H), 6,52 (м, 1H), 5,85 (с, 1H), 4,04 (т, J = 6,1 Гц, 2H), 3,79 (с, 3H), 3,74 (с, 3H), 2,80-2,95 (м, 4H), 2,60-2,75 (м, 4H), 2,42 (с, 2H), 2,20 (м, 1H), 1,85 (м, 1H), 1,61 (м, 1H), 1,08 (с, 6H); МС (FD) m/e 513 (M+); Анал. расч. для: C 77,16; H 7,65; N 2,73. Найдено: C 77,33; H 7,51; N 2,69.
Пример 30.
[2-(4-Метоксифенил)-6-метоксинафталин-1-ил] [4-[2-(1- морфолинил)этокси] фенил]метангидрохлорид.
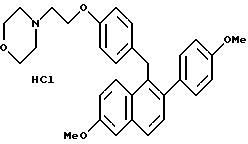
Реакция продукта по примеру 28 (1,58 г, 3,1 ммоль) с соляной кислотой (100 мл насыщенного раствора в этилацетате) в этилацетате (100 мл) в соответствии с методикой примера 26 давала 94% выход желательного продукта в виде белой пены: 1H-ЯМР (300 МГц, CDCl3) δ 7,70-7,85 (м, 2H), 7,44 (д, J = 8,4 Гц, 1H), 7,20-7,40 (м, 4H), 6,86-7,15 (м, 4H), 6,70-6,86 (м, 2H), 4,50-4,65 (м, 2H), 4,25-4,50 (м, 4H), 3,83-4,10 (м, 2H), 2,94 (с, 3H), 3,85 (с, 3H), 3,50-3,70 (м, 2H), 3,40-3,50 (м, 2H), 3,00-3,20 (м, 2H); МС (FD) m/e 483 (M+ - соляная кислота).
Пример 31.
[2-(4-Метоксифенил)-6-метоксинафталин-1-ил] [4-[2-(1-(3,3- диметил)пирролидинил)этокси]фенил]метангидрохлорид.
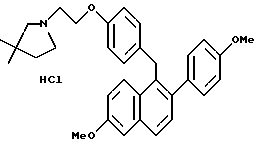
Реакция продукта по примеру 29 (1,2 г, 2,4 ммоль) с соляной кислотой (100 мл насыщенного раствора в этилацетате) в этилацетате (100 мл) в соответствии с методикой примера 26 давала 92% выход желательного продукта в виде белой пены: 1H-ЯМР (300 МГц, CDCl3) δ 7,29 (т, J = 9,3 Гц, 2H), 7,41 (д, J = 8,2 Гц, 1H), 7,15-7,30 (м, 3H), 7,19 (д, J = 6,8 Гц, 1H), 6,85-7,00 (м, 4H), 6,73 (д, J = 7,52 Гц, 2H), 4,48 (с, 2H), 4,35 (с, 2H), 3,93 (с, 3H), 3,83 (с, 3H), 3,60 (м, 1H), 3,15-3,50 (м, 2H), 3,15 (м, 1H), 2,76 (м, 1H), 2,05 (м, 1H), 1,85 (м, 1H), 1,75 (м, 1H), 1,33 (с, 3H), 1,33 (с, 3H); МС (FD) m/e 495 (M+ - соляная кислота); Анал. расч. для: C 74,49; H 7,20; N 2,63. Найдено: C 74,70; H 7,18; N 2,47.
Пример 32.
[2-(4-Гидроксифенил)-6-гидроксинафталин-1-ил] [4-[2-(1- морфолинил)этокси]фенил]метан.
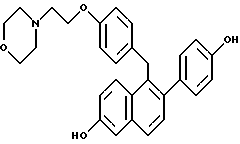
Реакция продукта по примеру 30 (1,28 г, 2,40 ммоль) с трихлоридом бора (10 мл, 117 ммоль) в 1,2-дихлорэтане (30 мл) в соответствии с методикой примера 27 давала 28% выход желательного продукта в виде белого твердого вещества: ИК (KBr) 3317,99, 2927,35, 2868,51, 1610,77, 1509,49 см-1; 1H-ЯМР (300 МГц, CDCl3) δ 7,75 (д, J = 9,3 Гц, 1H), 7,55 (м, 1H), 7,37 (д, J = 8,5 Гц, 1H), 7,10-7,20 (м, 2H), 6,65-7,05 (м, 8H), 5,50 (ушир. с, 2H), 4,32 (с, 2H), 4,00-4,20 (м, 2H), 3,70-3,80 (м, 4H), 2,70-2,85 (м, 2H), 2,50-2,70 (м, 4H); МС (FD) m/e 456 (M+); Анал. расч. для: C 76,46; H 6,42; N 3,07. Получ: C 76,75; H 6,44; N 3,02.
Пример 33.
[2-(4-Гидроксифенил)-6-гидроксинафталин-1-ил] [4-[2-(1-(3,3- диметил)пирролидинил)этокси]фенил]метан.
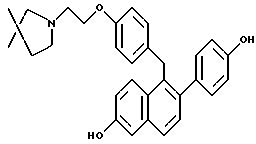
Реакция продукта по примеру 31 (1,2 г, 2,3 ммоль) с трихлоридом бора (10 мл, 117 ммоль) в 1,2-дихлорэтане (30 мл) в соответствии с методикой примера 27 давала 58% выход желательного продукта в виде белого твердого вещества: ИК (KBr) 3370,07, 2955,32, 2869,48, 1711,08, 1610,77, 1510,46 см-1; 1H-ЯМР (300 МГц, CDCl3) δ 7,71 (д, J = 9,2 Гц, 1H), 7,56 (д, J = 8,5 Гц, 1H), 7,32 (д, J = 8,4 Гц, 1H), 7,10-7,15 (м, 3H), 6,98 (м, 1H), 6,75-6,85 (м, 4H), 6,58 (д, J = 8,5 Гц, 2H), 4,28 (с, 2H), 4,11 (т, J = 7,70 Гц, 2H), 2,90 (т, J = 5,9 Гц, 2H), 2,82 (т, J = 6,7 Гц, 2H), 2,79 (т, J = 6,7 Гц, 2H), 1,66 (т, J = 6,9 Гц, 2H), 1,10 (с, 6H); МС (FD) m/e 468 (M+); Анал. расч. для: C 79,63; H 7,11; N 3,00. Получ: C 79,65; H 7,24; N 2,72.
Получение 14.
[2-(4-Метоксифенил)-6-метоксинафталин-1-ил][4-метоксифенил)метанон.
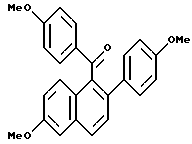
К 50 мл диоксана добавляли 6,0 г (15 ммоль) [3,4-дигидро-2-(4-метоксифенил)-6-метоксинафталин-1-ил] (4- метоксифенил)метанона и 7,3 г (32 ммоль) 2,3-дихлор-5,6-дициано-1,4-бензохинона. Смесь нагревали с обратным холодильником в течение 2 часов, затем оставляли перемешиваться при температуре окружающей среды в течение 60 часов. Смесь затем концентрировали досуха и остаток помещали в 500 мл метиленхлорида и трижды промывали 400 мл 2Н гидрохлорида натрия с последующим однократным промыванием 500 мл деионированной воды. Полученный органический слой отделяли, сушили над сульфатом натрия и растворитель удаляли под вакуумом. Полученный продукт затем очищали методом флэш-хроматографии (силикагель, градиент 20% этилацетат/гексана), что давало выход 4,75 г (80%)а соединения заголовка в виде белой пены: ЯМР QE300 МГЦ в CDCl3: (3,80 ч/м, с, 3H), (4,00 ч/с, с, 3H), (6,75 ч/м, д, 2H), (7,20 ч/м, дд, 1H), (7,30 ч/м, дс, 1H), (7,40 ч/м, д, 2H), (7,60 ч/м, д, 1H), (7,75 ч/м, д, 2H), (7,95 ч/м, д, 1H). МС (FD) me/e 398 (M+); Анал. расч. для: C 78,37; H 5,57. Получ: C 78,55; H 5,78.
Пример 34.
[2-(4-Метоксифенил)-6-метоксинафталин-1-ил][4-[2-(1- пиперидинил)этокси] фенил]метангидрохлорид.
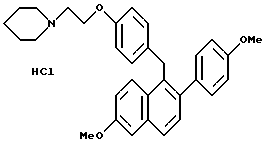
К 20 мл пропилбензола добавляли 240 мг (6,01 ммоль) 95% литийалюминийгидрида и 240 мг (0,484 ммоль) соединения по получению 14. Смесь нагревали с обратным холодильником в течение 35 минут и давали остыть до температуры окружающей среды. К смеси осторожно добавляли 1 мл деионированной воды с последующим добавлением 3 мл 15% гидроксида натрия/деионированной воды (по массе) и затем еще 1 мл деионированной воды. Смесь перемешивали в течение 15 минут при температуре окружающей среды и осадок удаляли с помощью вакуум-фильтра. Маточный раствор разбавляли метиленхлоридом (100 мл), однократно промывали насыщенным раствором соли, сушили над сульфатом натрия и сушили на роторном испарителе досуха. Коричневое клейкое вещество очищали по методу радиальной хроматографии на 4 мм пластинке и 19:1 метиленхлориде:метаноле в качестве элюента, что давало соединение заголовка. ЯМР QE300 МГц в CDCl3: (1,55 ч/м, м, 2H), (1,75 ч/м, сложный, 4H), (2,60 ч/м, сложный, 4H), (2,85 ч/м, т, 2H), (3,95 ч/м, с, 3H), (4,05 ч/м, с, 3H), (4,20 ч/м, т, 2H), (4,45 ч/м, с, 2H), (6,85 ч/м, д, 2H), (7,00 ч/м, сложный, 4H), (7,15 ч/м, дд, 1H), (7,25 ч/м, дс, 1H), (7,35 ч/м, д, 2H), (7,50 ч/м, д, 1H), ), (7,80 ч/м, д, 1H), (7,90 ч/м, д, 1H), МС (FD) me/e 481 (M+).
Пример 35.
[2-(4-Гидрофенил)-6-гидроксинафталин-1-ил] [4-[2-(1- пиперидинил)этокси] фенил]метан.
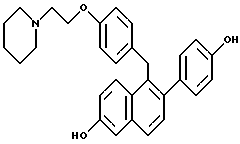
К суспензии лишенного защиты продукта по получению 12, где такое лишение защиты осуществляют по стандартным методикам, как описано здесь, (0,51 г, 1,00 ммоль), перемешиваемому в n-пропилбензоле, добавляют Red-Al® (0,87 г, 6,00 ммоль) и смесь нагревают с обратным холодильником. Через 3 часа раствор охлаждают до температуры окружающей среды и осторожно гасят 1,0 Н соляной кислотой. Полученную двухфазную смесь экстрагируют добавлением этилацетата и объединенные органические экстракты промывают насыщенным
водным бикарбонатом, насыщенным раствором соли, сушат (MgSO4), фильтруют и концентрируют. Очистка сырого продукта методом радиальной хроматографии (силикагель, этилацетат/гексаны/метанол/триэтиламин 2,5/2,5/0,7/0,3) дает продукт заголовка.
Методика проведения испытаний.
Общая методика получения.
В примерах, иллюстрирующих методы, использовали модель постклимактерического типа, в которой определяли ответ матки на различные курсы лечения.
Из Charles River Laboratories (Portage, MI) получили 75 старых женских особей крыс Sprague Dawley (весом от 200 до 250 г). Животные подвергались двусторонней овариэктомии (ОВЭ) В Charles River Laboratories и были отправлены через одну неделю. По получении они были помещены в металлические подвесные клетки и имели ad libitum доступ к пище (содержание кальция 0,5%) и воде в течение одной недели. Температура в помещении поддерживалась на 22,2 ± 1,7oC с минимальной относительной влажностью 40%. Световой режим в помещении состоял из 12 светлых часов и 12 темных часов. Экспериментальные группы составляли от 5 до 6 крыс.
Отбор тканей по схемам введения лекарственного вещества. По прошествии однонедельного акклиматизационного периода (таким образом, через две недели после ОВЭ) начали ежедневное введение доз испытываемого соединения. Испытываемые соединения вводили подкожно в виде суспензии в 20% β- гидроксициклодекстрине. Животным дозы вводились перорально в течение 4 дней. В ходе осуществления схемы введения лекарственного вещества животных взвешивали и анестезировали смесью кетамина:ксилазина (2:1, по объему) и образец крови отбирали с помощью кардиальной пункции. Животных умерщвляли с помощью удушения CO2, матку удаляли через срединный разрез и определяли сырой вес матки.
Подавление стимуляции тамоксифеном матки у крыс с помощью соединения формулы I.
Данные, представленные в таблице 1, показывают сравнительные результаты по крысам после овариэктомии (получавшим лечение только 20% циклодекстрином), крысам, получавшим лечение 0,01, 0,1, 1,0 и 10,0 мг/кг тамоксифена, и крысам, получавшим лечение теми же дозами тамоксифена плюс 0,1, 1,0 и 10,0 мг/кг соединения формулы I, в котором R1 и R2 являются -OH, и R3 является 1-пиперидинилом ("соединение формулы I" в таблице 1).
Эти данные показывают, что соединение формулы I, вводимое в дозе 10 мг/кг совместно с тамоксифеном, существенно подавляет эффект стимуляции матки тамоксифена, особенно будучи введенным совместно с более высокими терапевтическими дозами тамоксифена.



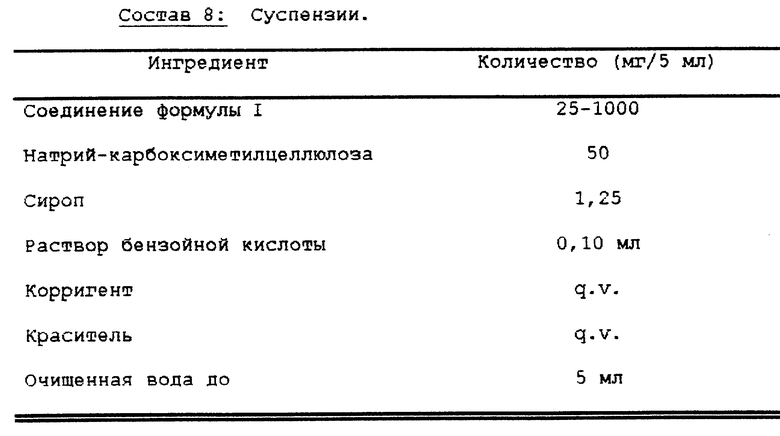



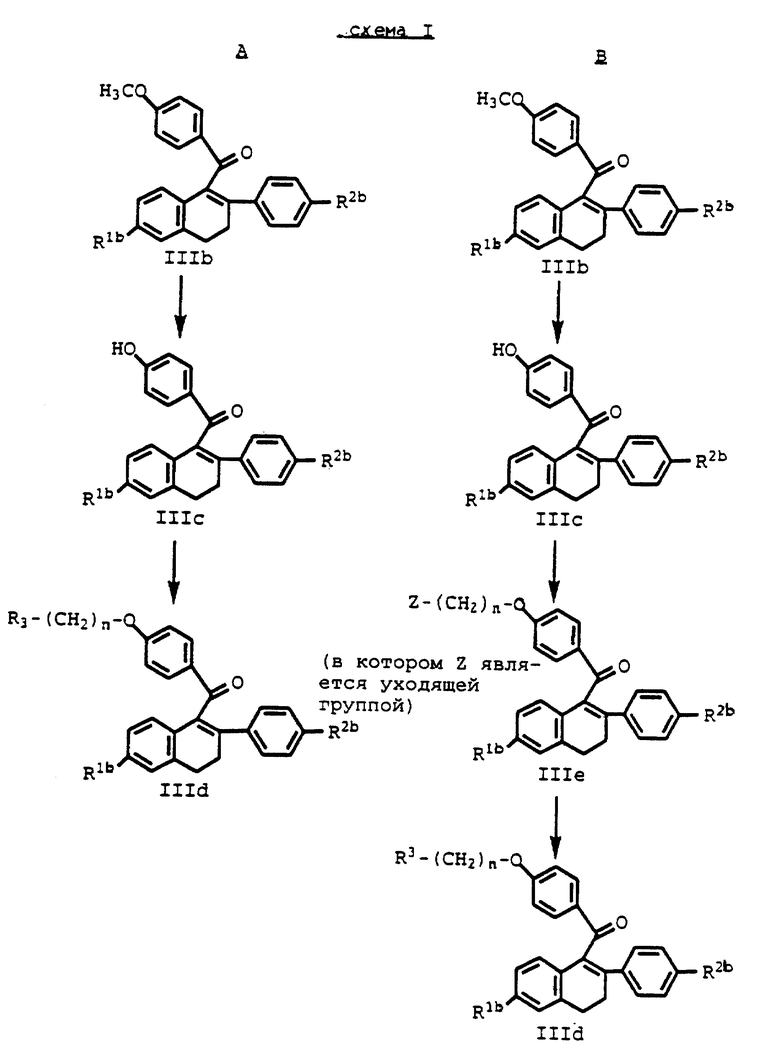
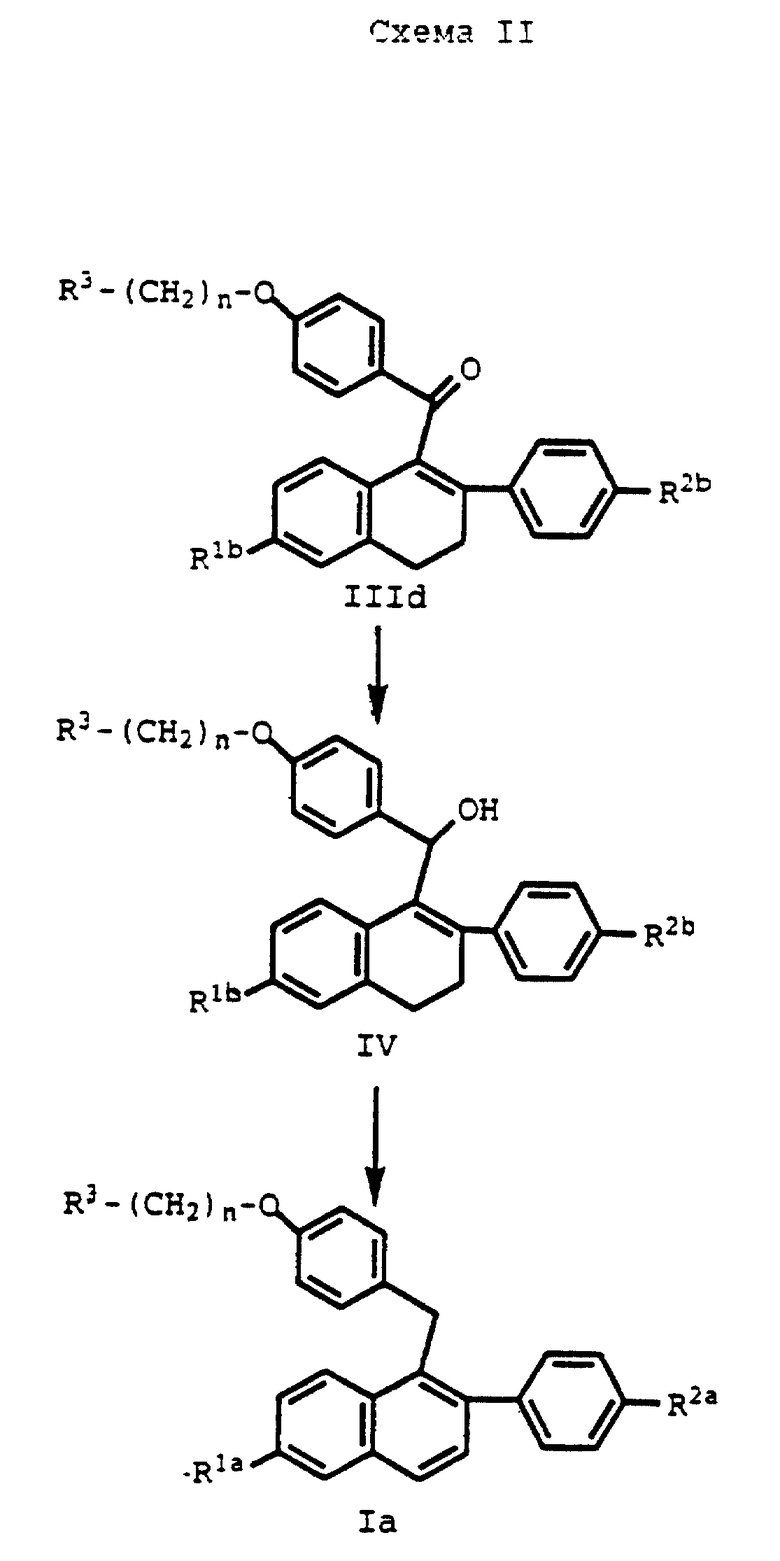
Изобретение относится к способу минимизации гистеротрофного эффекта нестероидных антиэстрогенных соединений формулы II, где значения радикалов R4, R5, R6, R7, R8 и n указаны в формуле изобретения, или его фармацевтически приемлемой соли, путем одновременного или последовательного введения соединения формулы I, где значения радикалов R1, R2, R3 и n указаны в формуле изобретения, или его фармацевтически приемлемой соли, и к фармацевтической композиции для лечения или профилактики рака молочной железы. Фармацевтическая композиция не оказывает влияние на антинеопластические полезные свойства, в то же время сводя к минимуму или элиминируя вредный гистеротрофный эффект. 2 с. и 9 з.п.ф-лы, 1 табл.
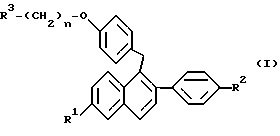
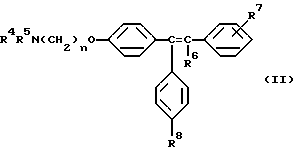
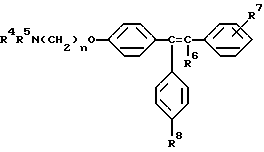
где либо R4 является H или низшим алкильным радикалом и R5 является низшим алкильным радикалом, либо R4 и R5 соединены с соседним атомом азота с образованием гетероциклического радикала;
R6 является H или низшим алкильным радикалом;
R7 обозначает H, галоген, OH, низший алкильный радикал или является бута-1,3-диенильным радикалом, который вместе с соседним бензольным кольцом образует нафтильный радикал;
R8 является H или OH;
n равно 2,
или его фармацевтически приемлемой соли, когда указанное соединение формулы II вводят женщине для лечения или профилактики рака молочной железы, заключающийся в одновременном или последовательном введении указанной женщине соединения формулы I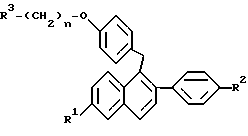
где R1 представляет -H, -OH, -O(C1-C4 алкил), -OCOC6H5, -OCO(C1-C6 алкил) или -OSO2(C4-C6 алкил);
R2 представляет -H, -OH, -O(C1-C4 алкил), -OCOC6H5, -OCO(C1-C6 алкил) или -OSO2(C4-C6 алкил);
n равно 2 или 3;
R3 обозначает 1-пиперидинил, 1-пирролидинил, метил-1-пирролидинил, диметил-1-пирролидинил, 4-морфолино, диметиламино, диэтиламино или 1-гексаметиленимино;
или его фармацевтически приемлемой соли.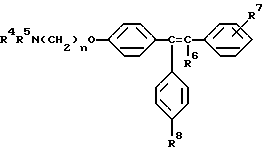
где либо R4 является H или низшим алкильным радикалом, и R5 является низшим алкильным радикалом, либо R4 и R5 соединены с соседним атомом азота с образованием гетероциклического радикала;
R6 является H или низшим алкильным радикалом;
R7 представляет H, галоген, OH, низший алкильный радикал или является бута-1,3-диенильным радикалом, который вместе с соседним бензольным кольцом образует нафтильный радикал;
R8 является H или OH; и
n равно 2,
или его фармацевтически приемлемую соль, когда для лечения или предотвращения рака молочной железы женщине вводят указанный первый компонент, и эффективное количество второго компонента, который представляет собой соединение формулы I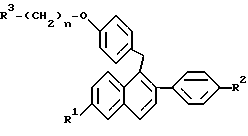
где R1 обозначает -H, -OH, -O(C1-C4 алкил), -OCOC6H5, -OCO(C1-C6 алкил) или -OSO2(C4-C6 алкил);
R2 является -H, -OH, -O(C1-C4 алкил), -OCOC6H5, -OCO(C1-C6 алкил) или -OSO2(C4-C6 алкил);
n равно 2 или 3; и
R3 представляет 1-пиперидинил, 1-пирролидинил, метил-1-пирролидинил, диметил-1-пирролидинил, 4-морфолино, диметиламино, диэтиламино или 1-гексаметиленимино,
или его фармацевтически приемлемую соль и указанный второй компонент вводят для минимизации гистеротрофного эффекта, индуцированного соединением формулы II, вместе с фармацевтически приемлемым носителем, наполнителем или разбавителем.
| US 4230862 A, 28.10.1980 | |||
| МАШКОВСКИЙ М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, 1993, ч | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| ЭЛЕКТРОМЕХАНИЧЕСКИЙ СНЕГООЧИСТИТЕЛЬ ДЛЯ ЖЕЛЕЗНЫХ ДОРОГ | 1922 |
|
SU549A1 |
