Изобретение относится к органической химии, конкретно к новым химическим соединениям α- полифторалкил-α-нитроалкиламинам и их N-ацилпроизводным общей формулы 1
где а) RF=CF3; C2F5; C3F7; C4F9; C6F13; H(CF2CF2)n, где n=1,2,3;
R=H, алкил C1-C4, Ph;
R1=H; C(O)R3, где R3=H, алкил C1-C6, Ph;
R2=H;
б) RF=C2F5; C3F7; C4F9; H(CF2CF2);n, где n=1,2,3;
R1=H•HCl;
R2=H.
которые могут быть использованы как промежуточные продукты в синтезе биологически активных соединений.
Указанные соединения могут быть использованы, в частности, для получения фторсодержащих аналогов α- аминокислот путем превращения нитрометильной группы в карбоксильную аналогично тому, как это описано в известной работе /S. Sabelle, D.Luset, T.Le Gall, Ch. Mioskowski. "Enantioselective synthesis of α-aminoacids from nitroalkenes". Tetrahedron Lett. 1998, v. 39.P. 2111-2114/. Фторсодержащие α-аминоксилоты и их аналоги представляют значительный интерес как вещества, обладающие биологической активностью. /"Соединения фтора. Синтез и применение". Под.ред. Н. Исикава. М.; Мир. 1990. С. 306-337/. С этой точки зрения заявляемые соединения могут рассматриваться как предшественники фторсодержащих α-аминокислот, что и определяет возможные аспекты их практического использования.
Ближайшим аналогом по строению является гидрохлорид 1,1,1-трифтор-2-амино-3-нитропропана формулы (2), который применяется в органическом синтезе для получения 1,1,1-трифтор-2-диазо-3-нитропропана /Айзикович А.Я., Базыль И. Т. "1,1,1-Трифтор-2-диазо-3-нитропропансинтез и реакции с енаминами" ЖОрХ 1987. Т. 23. N 6. С. 1331-1332/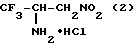
Способ получения соединения формулы (2) состоит во взаимодействии свежеприготовленной натриевой соли нитрометана в тетрагидрофуране с трифторацетонитрилом в присутствии хлористого триэтилбензиламмония. Образующийся α- нитроенамин выделяют обработкой реакционной массы раствором H2SO4 в тетрагидрофуране с последующей перегонкой в вакууме. Затем продукт растворяют в бензоле и добавляют NaBH4, а также каталитическое количество бромистого тетрабутилламония. Перемешивают 6 ч при 40-50oC, фильтруют и в фильтрат пропускают HCl до прекращения выделения гидрохлорида амина. Остаток фильтруют и кристаллизуют из этанола. Выход 59%, т.пл. 106-107oC /Айзикович А.Я., Базыль И.Т. 1,1,1-Трифтор-2-диазо-3-нитропропан - синтез и реакции с енаминами. ЖОрХ 1987. Т. 23 N 6. С. 1331-1332/.
Задачей изобретения является синтез новой группы соединений, которые могут служить исходными продуктами для получения веществ с потенциальной биологической активностью, в частности фторсодержащих аналогов α- аминокислот.
Поставленная задача достигается структурой новых соединений, а именно наличием полифторалкильного заместителя, нитрометильной и аминогрупп, а также заместителей у атома азота аминогруппы.
Поставленная задача достигается также тем, что нитрил полифторкарбоновой кислоты подвергают взаимодействию с натриевой солью нитроалкана в 1,4-диоксане, а затем подвергают взаимодействию с боргидридом натрия и уксусной кислотой в бензоле с выделением конечных продуктов известными методами, при этом предпочтительно соотношение боргидрид натрия-уксусная кислота составляет 1: 0.75-1. N-Производные аминов были получены взаимодействием гидрохлоридов аминов общей формулы (1) с хлорангидридами карбоновых кислот в присутствии основных агентов.
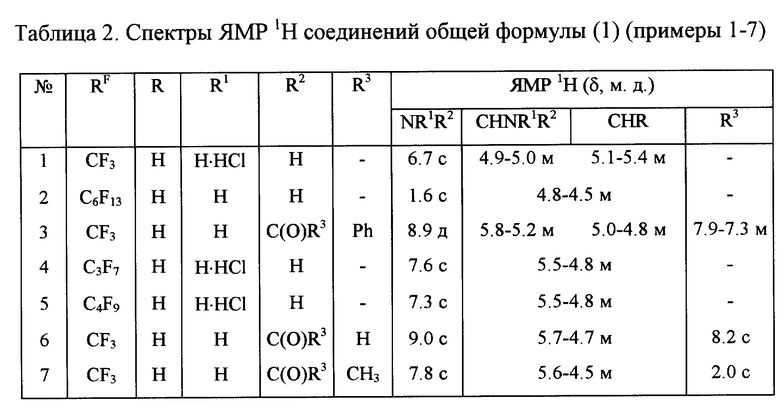
Все соединения являются кристаллическими веществами, устойчивыми в течение длительного срока. Их структура однозначно установлена методами элементного анализа и ЯМР 1H и 19F-спектроскопии (таблицы 1, 2).
Пример 1. Получение гидрохлорида 1,1,1-Трифтор-2-амино-3-нитропропана.
В суспензию 8.3 г (0.1 моль) натриевой соли нитрометана в 100 мл сухого 1.4-диоксана 30-40 мин пропускали трифторацетонитрил, получаемый одновременно из 20.0 г (0.15 моль) амида трифторуксусной кислоты и 21.3 г (0.15 моль) P2O5. Затем реакционную смесь перемешивали в течение 0.5 ч в присутствии 0,1 г (3 • 10-4 моль) тетрабутиламмония бромистого, после чего медленно добавляли 2.7 мл (0.05 моль) концентрированной серной кислоты, охлаждая реакционную смесь на водяной бане со льдом. Выпавший осадок сульфата натрия отфильтровывали, растворитель упаривали в вакууме при 15-20 мм рт.ст. Остаток, представляющий собой свободный енамин, растворяли в 60 мл сухого бензола и медленно при перемешивании добавляли его к суспензии 3.7 г (0.1 моль) NaBH4 в 60 мл сухого бензола. После добавления всего количества раствора енамина реакционную массу перемешивали 30 мин, затем медленно добавляли по каплям раствор 6.0 мл (0.1 моль) уксусной кислоты в 50 мл сухого бензола, перемешивали еще 30 мин при комнатной температуре и 1.5-2 ч при 50-60oC, охлаждали до комнатной температуры, осадок отфильтровывали, а через фильтрат барботировали сухой HCl. Выпавший гидрохлорид отфильтровывали и перекристаллизовывали из этанола. Выход и т.пл. приведены в таблице 1, а спектр ПМР - в таблице 2.
Пример 2. Получение 1,1,1,2,2,3,3,4,4,5,5,6,6-тридекафтор-7- амино-8-нитрооктана.
К суспензии 8.3 г (0.1 моль) натриевой соли нитрометана в 200 мл 1,4-диоксана при перемешивании прикапывали 34.5 г (0.1 моль) нитрила перфторгептановой кислоты в течение 3 ч в присутствии 0.1 г (3 • 10-4 моль) тетрабутиламония бромистого. После добавления всего количества нитрила реакционную смесь перемешивали 30 мин, после чего медленно добавляли 2.7 мл (0.05 моль) концентрированной серной кислоты, охлаждая реакционную смесь на водяной бане со льдом. Выпавший осадок сульфата натрия отфильтровывали, фильтрат выливали в большое количество воды, выпавший осадок, представляющий свободный енамин, отфильтровывали, сушили, растворяли в 100 мл сухого бензола и медленно при перемешивании добавляли его к суспензии 3.7 г (0.1 моль) NaBH4 в 100 мл сухого бензола. После добавления всего количества раствора енамина реакционную массу кипятили 6 ч, затем, не прекращая нагревание, медленно добавляли по каплям раствор 6.0 мл (0.1 моль) уксусной кислоты в 50 мл сухого бензола, перемешивали еще 30 мин при комнатной температуре и 1.5-2 ч при 50-60oC, вновь охлаждали до комнатной температуры, осадок отфильтровывали, а через фильтрат барботировали сухой HCl. Выпавший осадок свободного амина отфильтровывали и перекристаллизовывали из этанола. Выход и т.пл. приведены в табл. 1, а спектр ПМР - в табл. 2.
Пример 3. Получение 1,1,1-трифтор-2-(N-бензоил)амино-3-нитропропана.
Смесь 8.1 г (0.05 моль) гидрохлорида 1,1,1-трифтор-2-амино-3-нитропропана и 7.0 (0.05 моль) бензоилхлорида кипятили в течение 4 часов в 50 мл бензола в присутствии 7.9 г (0.1 моль) пиридина, после чего растворитель упаривали в вакууме при 10-15 мм рт.ст., а остаток промывали насыщенным раствором NaCl. Выпавший осадок отфильтровывали, промывали холодным эфиром, растворяли в хлороформе, добавляли гексан, выпавший осадок отфильтровывали. Выход и т.пл. приведены в таблице 1, а спектр ПМР - в таблице 2.
Соединения (4-7), физико-химические константы которых приведены в табл. 1 и 2, были получены аналогичными методами.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПЕРФТОРАЛКИЛЗАМЕЩЕННЫЕ N,N'-ЭТИЛЕНБИС-β-АМИНОВИНИЛКЕТОНАТЫ НИКЕЛЯ, ПАЛЛАДИЯ И МЕДИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1996 |
|
RU2101275C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПЛЕНОК НА ОСНОВЕ СЛОЖНЫХ ОКСИДОВ | 1992 |
|
RU2048619C1 |
| МАТЕРИАЛ ДЛЯ ИЗГОТОВЛЕНИЯ ВНЕ- И ВНУТРИГЛАЗНОГО ТРАНСПЛАНТАТА | 1997 |
|
RU2132701C1 |
| СПОСОБ ИДЕНТИФИКАЦИИ ОБЪЕКТОВ | 1991 |
|
RU2012430C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПЛЕНОК НА ОСНОВЕ СЛОЖНЫХ ОКСИДОВ | 1992 |
|
RU2048618C1 |
| МАГНИТНАЯ СИСТЕМА | 1998 |
|
RU2138871C1 |
| УСТРОЙСТВО ДЛЯ ИДЕНТИФИКАЦИИ ОБЪЕКТОВ | 1996 |
|
RU2123176C1 |
| КОГЕРЕНТНЫЙ СУПЕРГЕТЕРОДИННЫЙ СПЕКТРОМЕТР ЭЛЕКТРОННОГО ПАРАМАГНИТНОГО РЕЗОНАНСА | 1990 |
|
SU1739751A1 |
| СПОСОБ ПОЛУЧЕНИЯ ПЛЕНОК НА ОСНОВЕ СЛОЖНЫХ ОКСИДОВ | 1992 |
|
RU2048617C1 |
| СПОСОБ КОНТРОЛЯ КАЧЕСТВА ЦВЕТОВОГО ЗРЕНИЯ | 1994 |
|
RU2108056C1 |

Описываются α-полифторалкил-α-нитроалкиламины и их производные, которые могут быть использованы в качестве реагентов для синтеза соединений, обладающих потенциальной биологической активностью, общей формулы
RFCHN(R1R2)CHRNO2,
где RF = CF3, C2F5, C3F7, C4F9, C6F13, H(CF2CF2)n, где n = 1,2,3; R = H, алкил C1 - C4, Ph; R1 = H, H•HCl; C(O)R3, где R3 = H, алкил C1-C6, Ph; R2 = H; R1+R2 = фталил,
а также способ их получения, отличающийся тем, что нитрилы полифторкарбоновых кислот подвергают взаимодействию с натриевыми солями нитроалканов в 1,4 - диоксане, а затем подвергают взаимодействию с боргидридом натрия и уксусной кислотой в бензоле в соотношении боргидрид натрия: уксусная кислота 1: 0,75-1 с выделением продуктов известными методами. 2 с. и 1 з.п. ф-лы, 2 табл.
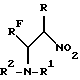
где а) RF = CF3; C2F5; C3F7; C4F9; C6F13; H(CF2CF2)n, где n = 1, 2, 3;
R = Н, алкил С1-С4, Ph;
R1 = Н, C(O)R3, где R3 = Н, алкил С1-С6, Ph;
R2 = Н,
или
или б) RF = C2F5; C3F7; C4F9; C6F13; H(CF2CF2)n, где n = 1, 2, 3;
R1= H•HCl,
R2= H.
где а) RF = CF3; C2F5; C3F7; C4F9; C6F13; H(CF2CF2)n, где n = 1, 2, 3;
R = Н, алкил С1-С4, Ph;
R1 = Н, C(O)R3, где R3 = H, алкил С1-С6, Ph;
R2 = Н,
или
или б) RF = C2F5; C3F7; C4F9; C6F13; H(CF2CF2)n, где n = 1, 2, 3;
R1= H•HCl
R2= H,
отличающийся тем, что нитрил соответствующей полифторкарбоновой кислоты подвергают взаимодействию с натриевой солью соответствующего нитроалкана в 1,4-диоксане, а затем подвергают взаимодействию с боргидридом натрия и уксусной кислотой в бензоле с последующей при необходимости обработкой полученного соединения хлористым водородом с получением гидрохлорида соединения формулы I, который при необходимости может быть обработан хлорангидридом соответствующей карбоновой кислоты в присутствии основного агента, и выделением конечного продукта.
| Новиков С.С | |||
| и др | |||
| Химия алифатических и алициклических нитросоединений | |||
| - М.: Химия, 1974, с | |||
| Приспособление для автоматического тартания | 1922 |
|
SU416A1 |
| Айзикович А.Я | |||
| и др | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| ЖОрХ | |||
| Кузнечная нефтяная печь с форсункой | 1917 |
|
SU1987A1 |
| Прибор для равномерного смешения зерна и одновременного отбирания нескольких одинаковых по объему проб | 1921 |
|
SU23A1 |
| Нагревательные методические колодцы с разделением пламени | 1920 |
|
SU1331A1 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
