Изобретение относится к производным рапамицина, способу их получения, их применению в качестве фармацевтических средств и фармацевтическим композициям, содержащим их.
Рапамицин является известным [McAlpine, J.B., et al., J. Antibiotics (1991), 44, 668; Schreiber, S. L, et al., J. Am. Chem. Soc. (1991), 113. 7433; патент US N 3929992] макролидным антибиотиком, продуцируемым Streptomyces hygroscopicus, имеющим структуру, отображенную в формуле A:
(Предложено много различных схем для обозначения атомов, входящих в формулу рапамицина. Чтобы избежать путаницы, при упоминании здесь конкретных производных рапамицина названия приводятся относительно рапамицина, использующего схему обозначений формулы A). Рапамицин является сильнодействующим иммунодепрессантом, и было также показано, что он обладает противоопухолевой и противогрибковой активностью. Его применение в качестве фармацевтического средства, однако, ограничено из-за его очень низкой и непостоянной биологической доступности. Кроме того, рапамицин нерастворим и лишен стабильности, что затрудняет получение стабильных галеновых композиций.
Известно много различных производных рапамицина. Некоторые 40-O-замещенные рапамицины описаны, например, в патенте US 5258389 и Международной заявке WO 94/09010 (O-алкилрапамицины); в Международной заявке WO 92/05179 (эфиры карбоновых кислот), в патентах US 5118677 (амидные эфиры), US 5118678 (карбаматы), US 5100833 (фторированные эфиры), US 5151413 (ацетали) и US 5120842 (силильные эфиры).
Неожиданно было открыто, что некоторые производные рапамицина обладают улучшенным фармакологическим профилем по сравнению с рапамицином, а также показывают более высокую стабильность.
В соответствии с изобретением предлагается соединение формулы I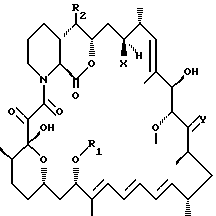 I
I
где R1 представляет собой алкил, алкенил, алкинил, гидроксиалкил, гидроксиалкенил, гидроксиалкинил, бензил, алкоксибензил или хлорбензил, R2 выбран из формулы II или формулы III:

где R3 выбран из H, алкил, алкенил, алкинил, арил, тиоалкил, арилалкил, гидроксиарилалкил, гидроксиарил, гидроксиалкил, дигидроксиалкил, гидроксиалкоксиалкил, гидроксиалкиларилалкил, дигидроксиалкиларилалкил, алкоксиалкил, алкилкарбонилоксиалкил, аминоалкил, алкиламиноалкил, алкоксикарбониламиноалкил, алкилкарбониламиноалкил, арилсульфонамидоалкил, аллил, дигидроксиалкилаллил, диоксоланилаллил, карбалкоксиалкил и алкилсилил групп; R4 представляет собой H, метил или вместе с R3 образует C2-6 алкилен; R5 представляет собой R6O-CH2-, где R6 выбран из H, алкил, алкенил, алкинил, арил, алкилкарбонил, арилкарбонил, гетероарилкарбонил, гидроксиалкилкарбонил, аминоалкилкарбонил, формил, тиоалкил, арилалкил, гидроксиарилалкил, гидроксиарил, гидроксиалкил, дигидроксиалкил, гидроксиалкоксиалкил, гидроксиалкиларилалкил, дигидроксиалкиларилалкил, алкоксиалкил, алкилкарбонилоксиалкил, аминоалкил, алкиламиноалкил, алкоксикарбониламиноалкил, алкилкарбониламиноалкил, арилсульфонамидоалкил, аллил, дигидроксиалкилаллил, диоксоланилаллил и карбалкоксиалкил групп: R7CO-, где R7 выбран из H, алкил, гидрокси-, алкокси-, арилокси-, амино-, алкиламино-групп, остатка аминокислоты или N,N- дизамещенной амино-группы, в которой заместители (а) выбраны из алкил, арил или арилалкил групп, или (б) образуют гетероциклическую структуру: R8NCH-, где R8 представляет собой алкил, арил, амино-, алкиламино-, ариламино-, гидрокси-, алкокси- или арилсульфониламино- группу; -O-CH-O; или замещенный диоксиметилин;
Y представляет собой О, (H, ОН) и (H, OR9), где R9 выбран из C1-4алкил, алкилкарбонил, арилкарбонил, гетероарилкарбонил, гидроксиалкилкарбонил, аминоалкилкарбонил, формил или арил групп; и
X представляет собой OH или H:
где "алк" или "алкил" относится к C1-10 алифатическому заместителю, возможно прерванному окси-связью; и "ар" или "арил" относится к моноциклическому, возможно гетероциклическому, возможно замещенному, C4-14 ароматическому заместителю,
при условии, что когда X представляет собой ОН, R1 представляет собой алкил и R2 представляет собой остаток формулы II, тогда R3 является другим, чем Н.
Любая группировка "алк" или "алкил", упомянутые выше, может быть разветвленной, линейной или циклической; предпочтительно это C1-8 алифатический заместитель, возможно прерванный окси-связью, более предпочтительно не прерванный.
Примеры группировки "ар" или "арил", упомянутые выше и возможно замещенные, могут включать в себя, например, фенил, бензил, толил, пиридил и т.п.
В случае когда R1 является хлорбензилом или алкоксибензилом, заместитель предпочтительно располагается в орто- положении.
В случае когда R7CO- является N,N-дизамещенным карбамоилом, он может быть например N-метил-N-(2-пиридин-2-ил-этил)-карбамоилом, (4-метил- пиперазин-1-ил)-карбонилом или (морфолин-4-ил)-карбонилом.
В случае когда R5 представляет собой замещенный диоксиметилин, он может быть например O,O-(алкилен)-диоксиметилином, т.е. таким, в котором 2 атома кислорода связаны алкиленовой группой.
В соединениях формулы I предпочтительны следующие значения либо индивидуально либо в комбинации или субкомбинации:
1. X является OH, a R1 является C3-10алкинилом или C3-10гидроксиалкинилом, предпочтительно C3-10алк-2-инилом или C3-10гидроксиалк-2-инилом, более предпочтительно C3-6алк-2-инилом;
2. X является H, a R1 является C1-10алкилом, C3-10алк-2-енилом, C3-10гидроксиалк-2-енилом, C3-10алк-2-инилом, C3-10гидроксиалк-2-инилом или C1. C1-10алкокси C1-10алкилом, предпочтительно C1-6алкилом или C3-6алк-2-инилом, более предпочтительно C1-4алкилом, наиболее предпочтительно метилом;
3. C3-6алкинил в качестве R1 является 2-пропинилом или пент-2-инилом, предпочтительно пент-2-инилом;
4. Y является O, (H, OH) или (H, C1-4алкоксилом), предпочтительно O;
5. R2 является остатком формулы II;
6. В остатке формулы II R3 является H, C1-6гидроксиалкилом, гидрокси-C1-6алкокси-C1-6алкилом, (C1-6алкил)карбониламино-C1-6алкилом, C1-6алкокси-C1-6алкоксилом или амино-C1-6алкилом, предпочтительно H, гидроксиэтилом, гидрокси пропилом, гидроксиэтоксиэтилом, метоксиэтилом или ацетиламиноэтилом; особенно H, когда X является H или когда X является ОН и R1 является алкинилом:
7. В остатке формулы II R4 является метилом.
8. R2 является остатком формулы III, где R5 является R6OCH2-, где R6 выбран из групп: H, C1-6алкил, C3-6алк-2-енил, C3-6алк-2-инил, арил, C1-6алкилкарбонил, арилкарбонил, гидроксиC1-6алкил, C1-6алкокси-C1-6алкил или аминоC1-6алкил; R7CO-, где R7 выбран из H, гидрокси-, C1-6алкокси-, амино-, C1-6алкиламино-групп, остатка аминокислоты или N,N-дизамещенной амино-группы, в которой заместители (а) выбраны из C1-6алкила или арила, или (б) образуют гетероциклическую структуру; R8NCH-, где R8 представляет собой ал кил, арил, амино-, алкиламино-, ариламино-, гидрокси-, алкокси- или арилсульфониламино- группу; - O-CH-O-; или замещенный диоксиметилин.
Предпочтительными соединениями являются соединения формулы Ia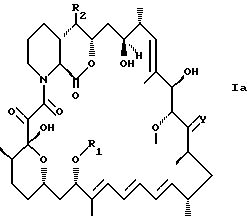
где R1, R2 и Y такие, как определено выше, предпочтительно имеют любое из предпочтительных значений, приведенных выше в пп. 1 и 3-8; и формулы Iб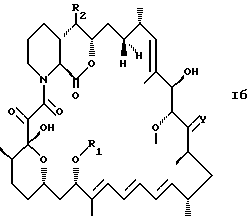
где R1, R2 и Y такие, как определено выше, предпочтительно имеют любое из предпочтительных значений, приведенных выше в пп. 2-8.
Особенно предпочтительные соединения включают в себя
(1) 32-деоксорапамицин;
(2) 16-O-пент-2-инил-32-деоксорапамицин;
(3) 16-O-пент-2-инил-32-деоксо-40-O-(2-гидроксиэтил)-рапамицин;
(4) 16-O-пент-2-инил-32(S)-дигидрорапамицин;
(5) 16-O-пент-2-инил-32(S)-дигидро-40-O-(2-гидроксиэтил)-рапамицин;
(6) 32(S)-дигидро-40-O-(2-метокси)этилрапамицин;
(7) 32(S)-дигидро-40-O-(2-гидроксиэтил)-рапамицин.
Соединения формулы I проявляют изомерию и, соответственно, далее будут существовать изомерные формы. Следует понимать, что настоящее изобретение охватывает соединения формулы I, индивидуальные изомеры формулы I'
где R1, R2, Y и X такие, как определено выше, а также их изомерные смеси.
Индивидуальные изомеры можно разделить способами, известными из уровня техники.
В настоящем изобретении также предлагается способ производства соединений формулы I, при котором
а) чтобы получить соединение формулы I, где X представляет собой H, восстановительно удаляют карбонил в положении 32 соединения формулы IVa
где R1, R2, и Y такие, как определено выше, в защищенной или незащищенной форме,
и, где требуется, удаляют присутствующие защитные группы: или
б) чтобы получить соединение формулы I, где X представляет собой ОН, стереоселективно восстанавливают карбонил в положении 32 соединения формулы IVa, как определено выше: или
в) превращают соединение формулы I, где R1 представляет собой алкил, чтобы обеспечить соединение формулы I, где R1 другой, чем алкил.
В стадии а) способа соединение формулы IVa находится предпочтительно в защищенной форме, т.е. оно может содержать защитные группы на функциональных группах, которые не участвуют в реакции, например ОН в положении 28 и возможно в положении 40 в случаев когда R2 представляет собой остаток формулы II, или в положении 39 в случае, когда R2 представляет собой остаток формулы III.
Восстановление а) до 32-деоксо соединения формулы I можно проводить в две стадии:
(1) подвергают взаимодействию соединения формулы IVa предпочтительно в защищенной форме с гидридом, например гидридом диизобутилалюминия или предпочтительно гидридом литий три-трет-бутоксиалюминия, с получением соответствующего 32- дигидросоединения. Другие способы и реагенты, известные из уровня техники для восстановления кетонов, могут быть использованы для получения 32-дигидро-соединения из соответствующего кетона. Они включают в себя, например, гидрирование, восстановление металлами, восстановление гидридами металлов [Comprehensive Organic Transformations, R.C. Larock, VCH Publishers Inc. , New York, 1989, pp. 527-535, sections 7.1.1- 7.1.4] и способами асимметрического восстановления для кетонов [Comprehensive Organic Transformations, R.C. Larock, VCH Publishers Inc., New York, 1989, pp. 540-547, sections 7.1.15]. После стадии восстановления (1) следует
(2) превращают 32-дигидросоединение в соответствующее 32-гало-производное, например 32-бромо- или (предпочтительно) 32-йодо-производное, которое затем восстанавливают, например гидридом, в желаемое 32-деоксо производное и, когда требуется, снимают защиту полученного соединения. Далее, реагенты, такие которые применяются для восстановления галидов, могут быть использованы, и они включают в себя например низковалентные металлы (литий, натрий, магний и цинк) и гидриды металлов (гидриды алюминия, боргидриды, силаны, гидриды меди) [Comprehensive Organic Transformations, R.C. Larock, VCH Publishers Inc., New York, 1989, pp. 18-20, sections 1.5.1. и 1.5.2.].
Альтернативно, восстановление галида может быть достигнуто с использованием водорода или источника водорода (т.е. муравьиной кислоты или ее соли) в присутствии подходящего металлического катализатора (т.е. никеля Ренея, металлического палладия или палладиевых комплексов, комплексов родия или рутения) [Comprehensive Organic Transformations, R.C. Larock, VCH Publishers Inc., New York, 1989, pp. 20-24, section 1.5/3.]. Кроме того, могут быть использованы известные [Comprehensive Organic Transformations, R.C. Larock, VCH Publishers Inc. , New York, 1989, pp. 27-31, sections 1.9.1.-1.9.4] способы, применяемые для превращения спирта в соответствующее деоксисоединение. Эти способы включают в себя например прямое восстановление или восстановление промежуточного фосфористого соединения, сульфоната, тиокарбоната, тиокарбамата или ксантата.
Подходящие гидрокси защитные группы и способы их образования и удаления известны [Protective Groups in Organic Synthesis, second ed. T.W. Greene and P.G.M. Wuts, John Wiley & Sons, New York, 1991, Chapter 2 и ссылки в ней] из уровня Техники. Предпочтительными ОН защитными группами являются, например, триорганосилил-группы, такие как три(C1-6)алкилсилил (например, триметилсилил, триэтилсилил), триизопропилсилил, изопропилдиметилсилил, трет-бутилдиметилсилил, триарилсилил (например, трифенилсилил) или триаралкилсилил (например, трибензилсилил). Снятие защиты может быть проведено в умеренно кислотных условиях.
Стадию восстановления (1) можно эффективно проводить при низкой температуре, например от -10 до -80oC.
На стадии (2) 32-дигидросоединение, возможно в защищенной форме, предпочтительно 32(R)-диастереоизомер, превращают в сложный эфир, предпочтительно сульфонат, например мезилат, тозилат, нозилат или трифлат, с последующим замещением подходящим галидом, например, йодидом или бромидом натрия, йодидом или бромидом тетрабутиламмония, предпочтительно в присутствии основания, например амина. 32(R)-диастереоизомер может быть выделен из смеси в соответствии с известными методами разделения, например хроматографией.
Гидриды, подходящие для восстановления 32-галосоединения, включают в себя например гидриды радикалов, такие как гидрид трибутилолова, или трис-(триметилсилил)-силан. Восстановление можно также проводить в отсутствии или присутствии радикального инициатора, например 2,2'-азобисизобутиронитрила или предпочтительно Et3B, обычно при температуре от 0 до 80oC.
После стадии (1) или (2) восстановления может быть добавлен, если требуется, оксидант, такой как ацетат меди, чтобы заново окислить селективно в карбонил продукты нежелательного побочного восстановления, которое может происходить например в положении 9.
Альтернативно, 32-дигидропроизводное можно непосредственно превратить в галид способами, известными из уровня техники, например с использованием трифенилфосфина в комбинации с N-бром- или N-йод- сукцинимидом, карбонтетрабромидом или -тетрайодидом, 1,2-дибромтетрахлороэтаном, 2,4,5-трибром- или трийодимидазолом, йодом, 1,2-дийодэтаном, или с использованием тионилбромида или метилтрифеноксифосфоний йодидом.
Восстановление карбонила в положении 32 в 32-деоксо производное можно также осуществлять через образование тозилгидразона с последующей обработкой бораном, например катехолбораном, или через образование дитиана с последующим подходящим восстановлением, например никелем Ренея или гидридом, например трибутилолово гидридом. Можно применять другие известные способы трансформации кетона в соответствующий алкан; такие способы включают в себя например прямое восстановление [Comprehensive Organic Transformations, R.C. Larock, VCH Publishers Inc., New York, 1989, pp. 35-37, section 1.12.1.] или восстановление через гидразоны [Comprehensive Organic Transformations, R.C. Larock, VCH Publishers Inc., New York, 1989, pp. 37-38, section 1.12.2.] и через производные серы и селена [Comprehensive Organic Transformations, R.C. Larock, VCH Publishers Inc., New York, 1989, pp. 34-35, section 1.11.].
Стадию б) восстановления до 32(S)-дигидросоединения формулы I проводят при селективных условиях. Предпочтительно используют восстанавливающий агент, который в значительной степени способствует восстановлению до 32(S), например триэтилборгидрид натрия. Восстановление можно проводить при низкой температуре, например от -50 до -80oC, в инертном растворителе, например в тетрагидрофуране (ТГФ), диэтиловом эфире, глиме, диглиме или метил-трет-бутиловом эфире. Отделение 32(S)-дигидросоединения от получаемого небольшого количества 32(R)-дигидросоединения можно осуществлять способами, известными из уровня техники, например колоночной хроматографией, хроматографией с обращенной фазой.
При необходимости гидроксигруппы в положении 28 и возможно в положении 40 можно защитить до восстановления и снять защиту после, например как описано выше. Предпочтительно осуществлять стадию б) восстановления без OH-защиты.
Стадию в) превращения можно проводить в соответствии со способами, известными из уровня техники. Например, соединение формулы I, где R1 представляет собой алкил, предпочтительно метил, можно подвергнуть взаимодействию с соединением Rx-OH, где Rx представляет собой алкинил или гидроксиалкинил, чтобы получить соединение формулы I, где R1 представляет собой алкинил или гидроксиалкинил. Реакцию можно проводить в апротонном растворителе, например дихлорметане, толуоле, ацетонитриле или ТГФ, в кислотных условиях.
Предпочтительно восстановление в положении 32, в частности стадию б) восстановления, проводят на соединении формулы IVa, где R1 уже имеет желаемое значение, например R1 представляет собой алкинил, таким образом избегая последующего превращения после восстановления. Соединение формулы IVa, где R1 представляет собой алкинил или гидроксиалкинил, применяемое в качестве исходного материала, можно получить с использованием соединения Rx-OH, как описано выше.
Соединения, применяемые в качестве исходных материалов, можно получить аналогично способам, известным [USP 5258389, WO 94/09010, WO 95/16691, USP 5120842 и т.д.] из уровня техники и применяемым в технике.
Следующие примеры иллюстрируют изобретение. Все температуры приведены в oC. Использованы следующие сокращения:
ТГФ = тетрагидрофуран
ТЭС = триэтилсилил
MS = масс-спектрометрия
FAB = бомбардировка быстрыми атомами
Пример 1: 32-деоксорапамицин (R1 = CH3; R2 = II, где R3 = H и R4 = CH3; X = H; Y = О)
К перемешиваемому, охлажденному (-78o) раствору 26,1 г (22,85 ммоль) 28,40-бис-О-ТЭС-рапамицина в 260 мл ТГФ добавляют 50,3 мл (50,3 моль) 1М раствора гидрида литий-три-трет-бутоксиалюминия в ТГФ. Полученной смеси дают возможность нагреться до -15o в течение 2 часов. Затем охлаждающую баню заменяют на ледяную, доводя температуру до 0o, и при этой температуре продолжают перемешивание в течение 1 часа. Реакционную смесь выливают в разделительную воронку, содержащую 750 мл этилацетата и 400 мл охлажденного на льду 2н. водного раствора лимонной кислоты, и быстро встряхивают. Водный слой отделяют и дважды экстрагируют холодным этилацетатом. Объединенный органический раствор промывают охлажденным на льду 2н. водным раствором лимонной кислоты, водой, насыщенным водным раствором бикарбоната натрия и дважды насыщенным рассолом, затем сушат над безводным карбонатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток, содержащий смесь 32(R)-дигидро- 28,40-бис-О-ТЭС-рапамицина и (32R)9,32-бис-дигидро-28,40-бис-O- ТЭС-рапамицина, растворяют без дополнительной очистки в 260 мл метанола. Этот раствор охлаждают до 0o и обрабатывают 6,85 г (34,31 ммоль) ацетата меди. После перемешивания в течение 1 часа полученную суспензию разбавляют метил-трет-бутиловым эфиром и дважды промывают водой и дважды - насыщенным рассолом. Водные слои снова экстрагируют метил-трет-бутиловым эфиром. Объединенный органический раствор сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток очищают хроматографией на силикагеле (60:40 гексан/метил-трет-бутиловый эфир), получая чистый 32(R)-дигидро-28,40-бис-O-ТЭС-рапамицин в виде белого твердого вещества.
1H ЯМР (CDCl3) 4:1 смесь ротамеров, химические сдвиги в скобках относительно минорного ротамера δ 0,72 (1H, dd, H-38ax), 1,63 (1,60) (3H, s, C17-CH3), 1,66 (1,69) (3H, s, C29-CH3), 1,77 и 1,81 (H-33), 2,46 (1H, m, H-31), 2,82 (2,91) (1H, m, H-25), 2,91 (1H, m, H-39), 3,13 (3H, s, C16-OCH3), 3,26 (3H, s, C27-OCH3), 3,41 (1H, m, H-40), 3,43 (3H, s, C39-OCH3), 3,62 (1H, m, H-32), 3,75 (3,57) (1H, d, H-27), 4,10 (1H, d, H-28), 4,81 (1H, широкий s, C10-OH), 5,05 (1H, d, Н-34), 5,27 (1H, d, H-30), 5,36 (1H, d, Н-2), 5,69 (1H, dd, Н-22), 6,03 (5,96) (1H, d, H-18), 6,15 (1H, dd, H-21), 6,33 (1H, dd, H-20), 6,40 (1H, dd, H-19).
MS (FAB, Lil матрица) m/z 1150 ([M + Li]+) (отн. интенсивность 100)
К перемешиваемому, охлажденному (-15o) раствору 20,69 г (18/10 ммоль) 32(R)-дигидро-28,40-бис-O-ТЭС-рапамицина и 7,55 мл (54,27 ммоль) триэтиламина в 200 мл метиленхлорида добавляют 2,10 мл (27,02 ммоль) метансульфонилхлорида. Смесь перемешивают в течение 20 минут, затем разбавляют этилацетатом и добавляют насыщенный водный бикарбонат натрия. Слои разделяют и водный слой экстрагируют трижды этилацетатом. Объединенную органическую фазу промывают насыщенным водным бикарбонатом натрия и рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток может быть очищен колоночной хроматографией на силикагеле (80:20 гексан/этилацетат) с получением чистого 32(R)-дигидро-32-O-мезил-28,40-бис-O-ТЭС- рапамицина в виде белого твердого вещества, но на практике неочищенный продукт используют в последующих стадиях без дополнительной очистки.
1H ЯМР (CDCl3) δ 0,77 (1H, dd, H-38ax), 1,67 (3H, s, C17-CH3), 1,72 (3H, s, C29-CH3), 2,77 (1H, М, H-25), 2,92 (1 H, m, H-39), 3,03 (3H, s, C16-OCH3), 3,17 (3H, s, C27-OCH3), 3,21 (3H, s, C39-OCH3), 3,42 (1H, m, H-40), 3,45 (3H, s, CH3SO3), 3,91 (1H, d, H-27), 4,10 (1H, d, H-28), 4,72 (1H, m, H-32), 4,94 (1H, s, C10-OH), 5,12 (1H, m, H-34), 5,25 (1H, d, H-30), 5,43 (1H, d, H-2), 5,88 (1H, dd, H-22), 6,03 (1H, d, H-18), 6,18 (1H, dd, H-21), 6,37 (1H, dd, H-20), 6,44 (1H, dd, H-19) MS (FAB, Lil матрица) m/z 1228 ([M + Li] +) (отн. интенсивность 68), 1132 ([(M - CH3SO3H) + Li]+) (отн. интенсивность 100)
Смесь 22,35 г (18,30 ммоль) 32(R)-дигидро-32-O-мезил-28,40- бис-O-ТЭС-рапамицина, 27,50 г (183,33 ммоль) йодида натрия и 6,3 мл (36,68 ммоль) диизопропилэтиламина в 400 мл ТГФ нагревают до образования флегмы в течение 6 часов, затем дают ей возможность остыть до комнатной температуры. Полученную смесь разбавляют этилацетатом и обрабатывают 38,4%-ным водным раствором бисульфита натрия. Разделяют слои. Органическую фазу трижды промывают насыщенным водным бикарбонатом натрия и один раз насыщенным рассолом. Затем сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (83 : 17 гексан/этилацетат), получая чистый 32(S)-деоксо-32-йодо-28,40-6ис-O-ТЭС-рапамицин.
1H ЯМР (CDCl3) 1,5:1 смесь ротамеров, химические сдвиги в скобках относительно минорного ротамера δ 0,73 (1H, dd, H-38ax), 1,68 (1,66) (6H, s, C17-CH3 и C29-CH3), 2,72 (1H, m, H-25), 2,91 (2H, m, H-32 и H-39), 3,15 (3H, s C16-OCH3), 3,30 (3,31) (3H, s, C27-OCH3), 3,43 (3,41) (3H, s, C39-OCH3), 3,77 (3,91) (1H, d, H- 27), 4,21 (4,25) (1H, d, H-28), 4,51 (1H, s, C10-OH), 5,45 (5,48) (1H, d, H-30), 5,60 (5,79) (1H, dd, H-22), 6,02 (5,85) (1H, d, H-18)
MS (FAB, Lil матрица) m/z 1260 ([M + Li]+) (отн. интенсивность 100)
К перемешиваемому, охлажденному (0o) раствору 16,79 г (13,19 ммоль) 32(S)-деоксо-32-йодо-28,40-бис-O-ТЭС- рапамицина в 190 мл толуола добавляют 7 мл (26,38 ммоль) трибутилолово гидрида, затем 1,3 мл (1,30 ммоль) 1М раствора триэтилборана в гексане. Эту смесь перемешивают в течение 30 минут и нейтрализуют насыщенным водным хлоридом аммония. Слои разделяют, и водный слой дважды экстрагируют этилацетатом. Объединенные органические слои промывают водой, насыщенным водным бикарбонатом натрия, водой и трижды насыщенным рассолом, затем сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Осадок очищают колоночной хроматографией на силикагеле (75 : 25 гексан/метил-трет-бутиловый эфир), получая чистый 32-деоксо-28,40-бис-O-ТЭС-рапамицин в виде белого твердого вещества.
1H ЯМР (CDCl3) 2,5:1 смесь ротамеров, химические сдвиги в скобках относительно минорного ротамера δ 0,73 (1H, dd, H-38ax), 1,62 (1,57) (3H, s, C17-CH3), 1,68 (1,72) (3H, s, C29-CH3), 2,77 (2,91) (1H, m, H-25), 2,91 (1H, m, H-39), 3,15 (3H, s, C16-OCH3), 3,27 (3,25) (3H, s, C27-OCH3), 3,43 (3,45) (3H, s, C39-OCH3), 3,70 (3,67) (1H, d, H-27), 4,11 (4,07) (1H, d, H-28), 4,57 (1H, широкий s, C10-ОH), 4,87 (4,67) (1H, d, H-34), 5,19 (5,08) (1H, d, H-30), 5,32 (1H, d, H-2), 5,60 (5,66) (1H, dd, H-22), 6,01 (5,92) (1H, d, H-18), 6,17 (1H, dd, H-21), 6,30 (1H, dd, H-20), 6,40 (1h, dd, H-19) MS (FAB, Lil матрица) m/z 1134 ([M + Li]+) (отн. интенсивность 100)
К перемешиваемому, охлажденному (-15oC) раствору 10,73 г (9,52 ммоль) 32-деоксо-28,40-бис-O-ТЭС-рапамицина в 85 мл метанола добавляют по каплям 9,5 мл 2н. водного раствора серной кислоты. По завершении процедуры добавления реакционную смесь нагревают до 0oС и перемешивают в течение 1,5 часов, затем разбавляют этилацетатом и нейтрализуют насыщенным бикарбонатом натрия. Слои разделяют и водный слой экстрагируют тремя порциями этилацетата. Объединенную органическую фазу трижды промывают насыщенным бикарбонатом натрия и рассолом, затем сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток растворяют в диэтиловом эфире, после чего выкристаллизовывается желаемый 32-деоксорапамицин (бесцветные кристаллы).
1H ЯМР (CDCl3) 3:1 смесь ротамеров, химические сдвиги в скобках относительно минорного ротамера δ 0,70 (1H, dd, H-38ax), 1,14 и 1,32 (H-32), (1,56 (H-33), 1,65 (1,62) (3H, s, C17-CH3), 1,68 (1,70) (3H, s, C29-CH3), 2,31 (2H, m, H-23 и H-31), 2,82 (2,95) (1H, m, H-25), 2,95 (1H, m, H-39), 3,14 (3H, s, C16-OCH3), 3,32 (3H, s, C27-OCH3), 3,38 (1H, m, H-40), 3,43 (3,41) (3H, s, C39-OCH3), 3,61 (1H, d, H-27), 4,12 (1H, d, H-28), 4,80 (4,71) (1H, d, H-34), 5,22 (1H, d, H-30), 5,31 (1H, d, H-2), 5,56 (1H, dd, H-22), 5,95 (5,87) (1H, d, H-18), 6,16 (1H, dd, H-21), 6,36 (1H, dd, H-20), 6,41 (1H, dd, H- 19)
MS (FAB, Lil матрица) m/z 906 ([M + U]/) (отн. интенсивность 100)
Пример 2: 16-пент-2-инилокси-32(5)-дигидрорапамицин (R1 = пент-2-инил; R2 = II, где R3 = H и R4 = CH3; X = OH; Y = O)
К перемешиваемому, охлажденному (0o) раствору 970 мг (1,06 ммоль) 32(S)-дигидрорапамицина и 1,39 мл (15,00 ммоль) 2-пентин-1-ола в 20 мл метиленхлорида добавляют 0,50 мл (6,50 ммоль) трифторуксусной кислоты. Смесь перемешивают при 0o в течение 3 часов и нейтрализуют насыщенным водным бикарбонатом натрия. Слои разделяют, и водный слой экстрагируют тремя порциями этилацетата. Объединенный органический раствор промывают насыщенным рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Неочищенную смесь очищают колоночной хроматографией на силикагеле (20 : 80 гексан-этилацетат), затем HPLC (жидкостной хроматографией высокого разрешения) с обращенной фазой (обращенная фаза RP18, 81:19 метанол-вода), получая титульное соединение в виде белого аморфного твердого вещества.
1H ЯМР (CDCl3) 2,5:1 смесь ротамеров, химические сдвиги в скобках относительно минорного ротамера δ 0,71 (1H, dd, H-38ax), 1,13 (1,05) (3H, t, CH3CH2CCCH2O), 1,67 (3H, s, 17-CH3), 1,69 (3H, s, 29-CH3), 2,21 (2H, qt, CH3CH2CCCH2O), 2,96 (1H, m, H-39), 3,33 (3,37) (3H, s, 27-OCH3), 3,41 (3,39) (3H, s, 39-OCH3), 3,78 (1H, dt, CH3CH2CCCHHO), 4,0 (1H, dt, CH3CH2CCCHHO), 5,52 (5,71) (1H, dd, H- 22), 5,98 (5,83) (1H, d, H-18), 6,15 (1H, m, H-21), 6,30 (1H, dd, H-20), 6,40 (1H, dd, H-19)
MS (FAB) m/z 974 ([M + Li]+)
Пример 3: 16-пент-2-инилокси-32(8)-дигидрорапамицин
(Альтернативный способ)
Рапамицин подвергают взаимодействию с 2-пентин-1-олом способом, аналогичным способу Примера 2, с получением 16-пент-2- инилокси-рапамицина.
К перемешиваемому, охлажденному (-77o) раствору 17,5 г (18,1 ммоль) 16-деметокси-16-пент-2-инилоксирапамицина в 180 мл ТГФ добавляют 21,7 мл (21,7 ммоль) 1М раствора триэтилборгидрида натрия в ТГФ. После 1 часа при -77oС реакцию гасят и нейтрализуют 10%-ным водным раствором лимонной кислоты. Затем реакционной смеси дают возможность нагреться до комнатной температуры, и большую часть ТГФ удаляют путем выпаривания при пониженном давлении. Полученный раствор дважды экстрагируют этилацетатом, органические фазы объединяют и сушат над сульфатом натрия. После выпаривания растворителя неочищенный продукт хроматографируют на силикагеле, элюируя смесью гексан/ацетон 7/3. Окончательную очистку проводят способом препаративной HPLC (RP-18, 76: 24 метанол: вода), получая титульное соединение в виде белого аморфного твердого вещества. Спектральные данные идентичны данным для продукта, полученного другим способом.
Пример 4: 32(8)-дигидро-40-O-(2-метокси)этилрапамицин (R1=CH3; R2=II, где R3=2-метоксиэтил и R4 = CH3; X=OH; Y=O)
К перемешиваемому, охлажденному (0o) раствору 2,17 г (2,00 ммоль) 40-O-(2-метокси)этил-28-O-ТЭС-рапамицина в 20 мл ТГФ добавляют по каплям 4,4 мл (4,4 ммоль) 1М раствора L-Selectide® в ТГФ. Полученный желтый раствор перемешивают в течение трех часов при 0o, и избыток гидридного реагента гасят добавлением 2 мл MeOH. Раствор разбавляют метил-трет-бутиловым эфиром и добавляют насыщенный водный раствор соли Рошеля. Смеси дают возможность нагреться до комнатной температуры и перемешивают в течение 15 минут. Слои разделяют, и органический раствор промывают холодной 1н. HCl, насыщенным рассолом, 1н. бикарбонатом натрия и снова рабсолом. Водные промывки обратно экстрагируют метил-трет-бутиловым эфиром. Объединенные органические слои сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении, получая неочищенную смесь 32S и 32R изомеров 32-дигидро-40-O-(2-метокси)этил-28-O-ТЭС рапамицина.
Полученный по способу, описанному выше, неочищенный продукт растворяют в 20 мл ацетонитрила и охлаждают до 0o. К полученному раствору добавляют 2 мл комплекса HF пиридин. Перемешивание продолжают в течение 1 часа, затем добавляют 1н бикарбонат натрия. Эту смесь трижды экстрагируют метил-трет-бутиловым эфиром. Объединенный органический раствор промывают 1н бикарбонатом натрия и рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Очистку проводят при помощи HPLC с обращенной фазой (RP 18, 5 мкм, 50:50-100:0 ацетонитрил-вода в течение 60 минут), получая 32(S)-дигидро-40-O-(2-метокси)этилрапамицин и 32(R)-дигидро-40-O-(2-метокси)этилрапамицин как побочный продукт.
32(S)-дигидро-40-O-(2-метокси)этилрапамицин: 1H ЯМР (CDCl3) 2:1 смесь ротамеров, химические сдвиги в скобках относительно минорного ротамера δ 0,77 (1H, dd, H-38ax), 1,67 (6H, s, C17-CH3 и C29-CH3), 2,50 (1H, m, H- 31), 3,01 (1H, m, H-25), 3,12 (2H, m, H-39 и H-40), 3,14 (3,15) (3H, s, OCH3), 3,28 (1H, m, H-32), 3,36 (3,34) (3H, s, OCH3), 3,39 (3,38) (3H, s, OCH3), 3,48 (3,46) (3H, s, OCH3), 3,55 и 3,75 (4H, 2m, OCH2CH2O), 3,84 (1H, m, H-14), 4,12 (4,16) (1 H, d, H-28), 4,73 (1H, s, C10-OH), 5,03 (1H, m, H-34)
MS (FAB) m/z 980 ([M + Li]+)
Пример 5: (32S)-дигидро-40-O-(2-гидрокси)этилрапамицин (R1=CH3; R2=II, где R3 = -CH2CH2OH и R4 = CH3; X = OH; Y = O)
Титульное соединение получают по способу, описанному в Примере 4, но с использованием подходящего исходного материала.
(32S)-дигидро-40-O-(2-гидрокси)этилрапамицин: 1H ЯМР (CDCl3) 1,7:1 смесь ротамеров, химические сдвиги в скобках относительно минорного ротамера δ 0,76 (1H, dd, H-38ax), 2,50 (1H, m, H-31), 3,10 (1H, m, H- 39), 3,13 (3,14) (3H, s, C16-OCH3), 3,20 (1H, m, H-40), 3,28 (1H, m, H-32), 3,36 (3,38) (3H, s, C27-OCH3), 3,45 (3,43) (3,41) (3H, s, C39-OCH3), 3,50 (1H, d, H-27), 3,58 и 3,70 (4H, m, OCH2CH2OH), 4,12 (4,16) (1H, d, H-28), 5,06 (1H, m, H-34), 5,60 (1H, dd, H-22), 5,99 (1H, d, H-18), 6,17 (1H, dd, H-21), 6,33 (1H, dd, H-20), 6,42 (1H, dd, H-19)
MS (FAB, Lil матрица) m/z 966 ([M + Li]+) (отн. интенсивность 100)
Пример 6: 16-пент-2-инилокси-32-деоксорапамицин (R1 = пент-2-инил; R2= ll, где R3=H и R4 = CH3; X=H; Y=O)
Титульное соединение получают по способу, описанному в примерах 1 и 2 или 3, но с использованием подходящих исходных материалов.
1H ЯМР (CDCl3) δ 0,76 (1H, dd, H-38ax), 1,23 (3H, t, CH3CH2CCCH2O), 2,21 (2H, ddq, CH3CH2CCCH2O), 2,78 (1H, m, H-25), 2,94 (1H, m, H-39), 3,31 (3H, s, C27-OCH3), 3,42 (3H, s, C39-OCH3), 3,62 (1H, d, H-27), 3,78 (1H, ddd, CH3CH2CCCH2O),
4,02 (1H, ddd, CH3CH2CCCH2O), 4,12(1H, d, H-28), 4,79 (1H, m, H-34), 5,20 (1H, d, H-30), 5,28 (1H, широкий d, H-2), 5,50 (1H, dd, H-22), 5,97 (1H, d, H-18), 6,14 (1H, dd, H-21), 6,30 (1H, dd, H-20), 6,38 (1H, dd, H-19)
MS (FAB, Lil матрица) m/z 958 ([M + Li]+) (отн. интенсивность 100)
Соединения формулы I проявляют фармацевтическую активность и, следовательно, применимы в качестве фармацевтических средств.
В частности, соединения формулы I обладают иммуносупрессорной и противопролиферативной активностью, как продемонстрировано в следующих in vitro и in vivo тест-методах:
1. Реакция смешанной культуры лимфоцитов (MLR)
Изначально реакция смешанной культуры лимфоцитов была разработана в связи с аллотрансплантатами для оценки тканевой совместимости между потенциальными донорами органа и реципиентами, и является одной из самых лучших моделей, разработанных для оценки иммунной реакции in vitro. Модель MLR на мышах [Т. Meo в "Immonological Methods", L. Lefkovits and В. Pernis, Eds., Academic Press, N.Y., pp. 227-239 (1979)] применяют для демонстрации иммуносупрессорного воздействия соединений формулы I. Клетки селезенки (2 • 105/лунку) мышей линии Balb/c (самки, 8-10 недель) инкубируют на микротитрационных планшетах в течение 5 дней вместе с 0,5 • 106 клетками селезенки мышей линии СВА (самки, 8-10 недель), облученными (2000 рад) или обработанными митомицином C. Облученные аллогенные клетки индуцируют пролиферативный ответ в Balb/c клетках селезенки, который можно измерить введением в ДHК меченного предшественника. Поскольку стимуляторные клетки облучены (или обработаны митомицином C), они не реагируют в виде пролиферации на Balb/c клетки, но сохраняют свои антигенные свойства. Антипролиферативный эффект соединений формулы I на Balb/c клетки измеряют при различных разведениях и подсчитывают концентрацию, дающую 50%-ное ингибирование пролиферации клеток (IC50). Ингибиторную способность тест-образца можно сравнить с таковой рапамицина и выразить в виде относительной IC50 (т.е. IC50 тест-образца/IC50 рапамицина). Обнаружено, что соединения примеров 1 и 2 обладают в этом тесте относительной IC50 равной 0,3 и 0,08, соответственно.
2. IL-6 обусловленная пролиферация (IL-6 PROL)
Способность соединений формулы I препятствовать сигнальным путям, ассоциированным с фактором роста, оценивают, применяя интерлейкин- 6 (IL- 6) зависимую линию мышиных гибридомных клеток. Анализ проводят на 96-луночных микротитрационных планшетах. 5000 клеток на лунку культивируют в среде без сыворотки [М. H. Schreier и R. Tees в Immunological Methods, 1. Lefkovits and В. Pernis, Eds., Academic Press 1981, Vol. II, pp. 263-275], обогащенной 1 нг рекомбинантного IL-6/мл. После 66-часовой инкубации в отсутствии или присутствии тест-образца клетки подвергают мечению 1 мкКюри (3-H)-тимидина/лунку на еще 6 часов, собирают и подсчитывают методом жидкостной сцинтилляции. Встраивание (3-H)-тимидина в ДHК коррелирует с увеличением числа клеток и, таким образом, служит инструментом для измерения клеточной пролиферации. Серия разведений тест-образца позволяет подсчитать концентрацию 50%-ного ингибирования клеточной пролиферации (IC50). Ингибиторную способность тест-образца можно сравнить с таковой рапамицина и выразить в виде относительной IC50 (т.е. IC50 тест-образца/IC50 рапамицина). Обнаружено, что соединения Примеров 1 и 2 обладают в этом тесте относительной 1050 равной 0,2 и 0,09, соответственно.
3. Анализ связывания макрофилина (МВА)
Известно, что как рапамицин, так и структурно родственный иммунодепрессант FK-506 оба связывают in vivo макрофилин-12. (также известный как FK-506 связывающий белок или FKBP-12), и представляется, что это связывание связано с иммуносупрессорной активностью этих соединений. Анализ на конкурентное связывание показал, что соединения формулы I также обладают сильным связывающим сродством к макрофилину-12.
В этом анализе FK-506, связанный с BSA (бычьим сывороточным альбумином), применяют для фиксирования в микротитрационных лунках. Рекомбинантный человеческий макрофилин-12 с биотином (biot-MAP) связывается в присутствии или отсутствии тест-образца с иммобилизированным FK-506. После промывки (чтобы удалить неспецифически связанный макрофилин) связанный biot-MAP оценивают путем инкубации со стрептавидин-алкалин фосфатазным конъюгатом с последующей промывкой и последующим добавлением п-нитрофенилфосфата в качестве субстрата. Оптическую плотность (OD) считывают при 405 нм. Связывание тест-образца с biot-MAP выражается в виде снижения количества biot-MAP, связанного с FK-506, и, таким образом, в снижении значений OD405. Серия разведений тест-образца позволяет определить концентрацию, приводящую к 50% ингибированию связывания biot-MAP с иммобилизированным FK-506 (IC50). Ингибиторную способность тест-образца сравнивают с IC50 свободного FK-506, взятого в качестве стандарта, и выражают в виде относительной IC50 (т.е. IC50 тест-образца/IC50 FK-506). Обнаружено, что соединения Примеров 1, 2 и 5 обладают в этом тесте относительной IC50 равной 1, 2,8 и 2,25, соответственно.
4. Локализованная реакция "трансплантат против хозяина" (GvH)
In vivo эффективность соединений формулы I доказывают на подходящей животной модели [Ford et al., TRANSPLANTATION 10 (1970) 258]. Клетки селезенки (1 • 107) 6-недельных самок крысы линии Wistar/Furth (WF) инъецируют подкожно в день 0 в левые задние лапы самкам крыс линии (F344 x WF)F1 весом около 100 г. Животных обрабатывают 4 дня подряд и на 7 день удаляют и взвешивают подколенные лимфатические узлы. Разницу в весе между двумя лимфатическими узлами принимают в качестве параметра для оценки реакции.
5. Реакция почечного аллотрансплантата у крысы
Одну почку из крысы-донора линии DA(RT1a) или Brown-Norway (BN) (RT1n) трансплантируют на почечный сосуд унилатерально (левая сторона) нефректомированной крысы-реципиента (Lewis RT11), применяя анастомоз типа "конец-в-конец". Мочеточниковый анастомоз тоже "конец-в-конец". Обработку препаратом начинают в день трансплантации и продолжают 14 дней. Контралатеральную нефректомию производят через семь дней после трансплантации, оставляя реципиенту только донорскую почку. Выживание трансплантированных реципиентов берут в качестве параметра для функционального трансплантата.
6. Экспериментально индуцируемый аутоиммунный энцефаломиелит (EAE) у крыс
Оценивают эффективность соединений формулы 1 при ЕДЕ [Levine & Wenk, AMER. J. PATH 41 (1965), 61: McFarlin et al., J. IMMUNOL. 113 (1974), 712; Borel, TRANSPLANT. & CLIN. IMMUNOL. (1981), 3]. EAE является широко распространенной моделью рассеянного склероза. Самок крыс линии Wistar инъецируют в заднюю лапу смесью жидкости из спинного мозга быка и полным адъювантом Фрейнда. Симптомы заболевания (паралич хвоста и обеих задних ног) обычно развиваются за период в 16 дней. Регистрируется число заболевших животных, а также время наступления заболевания.
7. Артрит, вызванный адъювантом Фрейнда
Эффективность против экспериментально вызванного артрита показывают при помощи процедуры, описанной, например, в Winter & Nuss, ARTHRITIS & RHEUMATISM 9 (1966) 394: Billingham & Davies, HANDBOOK OF EXPERIMENTAL PHARMACOL (Vane & Ferreira Eds, Springer-Verlag, Berlin) 50/11 (1979) 108-144. Крыс линий OFA и Wistar (самцы и самки 150 г весом) инъецируют внутрикожно в основании хвоста или в заднюю лапу 0,1 мл минерального масла, содержащего 0,6 мг лиофилизированного штамма Mycobacterium smegmatis, убитого нагреванием. В модели развивающегося артрита обработку препаратом начинают непосредственно после инъекции адъюванта (дни 1-18); в модели установившегося артрита обработку препаратом начинают на 14 день, когда хорошо разовьется вторичное воспаление (дни 14-20). К концу эксперимента опухоль сустава измеряют при помощи микроциркуля. Дозой агента, приводящей к 50%-ому эффекту (ED50), служит пероральная доза, мг/кг, уменьшающая опухоль (первичную или вторичную) на половину по сравнению с контрольной.
8. Противоопухолевая и MDR (реакция элиминации макрофагов) активность
Демонстрируется противоопухолевая активность соединений формулы 1 и их способность усиливать работу противоопухолевых агентов путем смягчения резистентности к множеству лекарств, например, путем введения противоопухолевого агента, например, колхицина или этопозида в клетки с резистентностью к множеству лекарственных препаратов и клетки, чувствительные к лекарственным препаратам in vitro или животным, имеющим опухоли или инфекции, резистентные к множеству лекарственных препаратов или чувствительные к лекарственным препаратам, с или без одновременным введением тестируемых соединений формулы I и с введением только соединения формулы I.
Такое in vitro тестирование проводят с применением любой подходящей линии клеток, резистентной к лекарственным препаратам, и контрольной (родительской) линии клеток, полученной, например, как описано у Ling et al. , J. Cell. Physiot. 83, 103-116 (1974) и Bech-Hansen et al., J. Cell. Physiol. 88, 23-32 (1976). Конкретными отобранными клонами являются линия с резистентностью к множеству лекарственных препаратов (например, колхицин-резистентная) линия CHR (субклон C5S3.2) и родительская, чувствительная линия AUX B1 (субклон AB1 S11).
Показывают in vivo противоопухолевую и анти-MDR активность, например, у мышей, инъекцированных опухолевыми клетками с резистентностью к множеству лекарственных препаратов и чувствительностью к лекарственным препаратам. Сублинии карциномы асцитов Эрлиха (EA), резистентные к лекарственным веществам DR, VC, AM, ET, TE или CC, развиваются при последовательном переносе клеток EA на последующие поколения BALB/c мышей-хозяина в соответствии со способами, описанными в Slater et al., J. Clin. Invest, 70 1131 (1982).
Эквивалентные результаты можно получить, применяя соединения формулы I в тест-моделях сравнимой структуры, например in vitro, или применяя экспериментальных животных, инфицированных вирусными штаммами, резистентными или чувствительными к лекарственным препаратам, резистентными к антибиотикам (например, к пенициллину) и чувствительными бактериальными штаммами, резистентными к противогрибковым препаратам и чувствительными грибковыми штаммами, а также протозойными штаммами, резистентными или чувствительными к лекарственным препаратам, например плазмодиальными штаммами, например естественно возникающими субштаммами Plasmodium falciparum, проявляющим приобретенную хемотерапевтическую антималярийную резистентность.
9. Ингибирование Mip (усилитель инфективности макрофагов) и Mip-подобных факторов
Дополнительно, соединения формулы I связывают и блокируют целый ряд Mip (усилитель инфективности макрофагов) и Mip-подобных факторов, которые структурно схожи с макрофилином. Mip и Mip- подобные факторы являются вирулентными факторами, продуцируемыми целым рядом патогенов, включая представителей родов Chlamidia, например Chlamidia trachomatis; Neisseria, например Neisseria meningitis; и Legionella, например Legionella pneumophiliai а также облигатными паразитами отряда Rickettsiales. Эти факторы играют ключевую роль в возникновении внутриклеточной инфекции. Эффективность соединений формулы I в уменьшении инфективности патогенов, которые продуцируют Mip или Mip-подобные факторы, можно показать путем сравнения инфективности патогенов в культуре клеток в присутствии и отсутствии макролидов, например с применением способов, описанных в Lundemose, et al., Mol. Microbiol. (1993) 7: 777.
10. Хроническое отторжение аллотоансплантата
Почку самки крысы линии DA (RTI1a) ортотопически трансплантируют самке крысы- реципиента линии Lewis (RT11). Всего трансплантируют 24 животных. Всех животных подвергают обработке циклоспорином A, 7,5 мг/кг/день перорально, в течение 14 дней, начиная со дня трансплантации, для предотвращения острого клеточного отторжения. Контралатеральную нефректомию не проводят. Каждую экспериментальную группу обрабатывают отличающейся дозой соединения формулы 1 или плацебо (шесть животных).
Начиная с 53-64 дня после трансплантации, животные-реципиенты получают перорально в течение следующих 69-72 дней соединение формулы I или плацебо. Через 14 дней после трансплантации животные подвергаются оценке состояния трансплантата при помощи магнитной резонансной сцинтиграфии (MRI) с перфузионным измерением почек (с сравнением трансплантированной и родной контралатеральной почки). Эту процедуру повторяют на 53-64 день после трансплантации и к концу эксперимента. Затем животные подвергаются аутопсии. Определяют и статистически анализируют параметры отторжения, такие как счет MRI, уровень относительной перфузии трансплантированной почки и гистологическая оценка почечного аллотрансплантата по клеточному отторжению и изменению сосудов. В данной модели аллотрансплантата почки крысы введение соединения формулы I, например соединения примера 1 или 2 в дозе 0,5-2,5 мг/кг, приводит к снижению всех вышеперечисленных параметров отторжения.
11. Ангиопластика
Баллонную катетеризацию проводят на 0 день, в частности как описано Powell et al., (1989). Под изофлуорановой (Isofluorane) анестезией вводят катетер Fogarty 2F в левую общую сонную артерию через внешний каротидный и вздутый сосуд (растяжение = 20 мкл H2O). Вздутый баллон трижды протягивают по всей длине общей сонной артерии, последние два раза чуть проворачивая для получения равномерной деэндотелиализации. Затем катетер удаляют, вокруг внешнего каротидного сосуда помещают лигатуру для предотвращения кровотечения и животных оставляют в покое для восстановления.
Для исследования используют 2 группы крыс линии RoRo (400 г, возраст - примерно 24 недели): одна контрольная группа и одна группа, получающая исследуемое соединение. Крысы являются полностью рандомизированными во время всей обработки, экспериментальных процедур и анализа.
Тестируемое соединение вводят перорально (через желудочный зонд), начиная за 3 дня до баллонного повреждения (день -3) до конца эксперимента, на 14 день после баллонного повреждения (день +14). Крыс держат в отдельных клетках и дают пищу и воду ad libidum. Затем крыс анестезируют изофлуораном (Isofluorane), перфузионный катетер вставляют через левый желудочек и закрепляют в дуге аорты, а аспирационную канюлю вставляют в правый желудочек. Животных перфузируют при перфузионном давлении 150 мм рт.ст., сначала в течение 1 минуты 0,1 М солевым буферным раствором (физиологический раствор с фосфатным буфером, PBS, pH 7,4) а затем в течение 15 минут 2,5%-ным глутаральдегидом в фосфатном буфере (pH 7,4). Перфузионное давление составляет 150 мм рт. ст. на конце канюли (≈ 100 мм рт.ст. в сонной артерии, как определено в предварительном эксперименте, вводя канюлю, присоединенную к датчику давления во внешнем каротидном сосуде). Затем сонные артерии вырезают, отделяют от окружающей ткани и помещают в 0,1 М какодилатный буфер (pH 7,4), содержащий 7% сахарозы и инкубируют в течение ночи при 4oC. На следующий день сосуды помещают в 0,05%-ный KMnO4 в 0,1 М какодилате и встряхивают в течение 1 часа при комнатной температуре. Затем ткани дегидратируют в серии разной концентрации этанола: 2х10 мин в 75%, 2х10 мин в 85%, 3х10 мин в 95% и 3х10 мин в 100% этаноле. Затем дегидратированные сосуды фиксируют в Technovit 7100 в соответствии с рекомендациями производителей. Фиксирующую среду оставляют полимеризоваться на ночь в эксикаторе в атмосфере аргона, поскольку обнаружено, что кислород тормозит нормальный процесс отверждения блоков.
При помощи ножа из твердого металла на роторном микротоме нарезают срезы по 1-2 мкм толщиной из средней части каждого сосуда и прокрашивают в течение 2 минут красителем Гимза. Таким образом из каждого сосуда получают около 5 срезов и поперечные срезы средней области, неоинтимы и просвета оценивают морфометрически при помощи системы анализа изображения (MCID, Торонто, Канада). В этом анализе соединения формулы 1, например соединение Примера 1 или 2, подавляют миоинтимальную пролиферацию при введении перорально при суточной дозе 0,5-2,5 мг/кг.
Соединения формулы I также применимы в анализах по качественному или количественному определению макрофилин-связывающих соединений, например в конкурентных анализах для диагностических или скрининговых целей. Таким образом, в другом практическом воплощении в изобретении предлагается применение соединений формулы I в качестве инструментов скрининга для определения присутствия макрофилин- связывающих соединений в тест-растворе, например крови, сыворотке крови или тест-бульоне. Предпочтительно соединение формулы I иммобилизуют в микротитрационных лунках, а затем дают ему возможность связаться в присутствии или отсутствии тест-раствора с меченным макрофилином-12 (FKBP-12). Альтернативно, FKBP-12 иммобилизуют в микротитрационных лунках, а затем дают возможность связаться в присутствии или отсутствии тест-раствора с меченным соединением формулы I, например флюоро-, ферментно- или радиомеченным, например соединением формулы I, где R1 содержит меченную группу. Планшеты промывают и измеряют количество связанного меченного соединения. Количество макрофилин- связывающего вещества в тест-растворе приблизительно обратно пропорционально количеству связанного меченного соединения. Для количественного анализа стандартную кривую связывания строят с использованием известных концентраций макрофилин-связывающего соединения.
Соединения формулы I, следовательно, применимы при следующих состояниях:
а) Лечение и профилактика острого и хронического отторжения трансплантированного органа или ткани, например, при лечении реципиентов, например сердца, легкого, комбинации сердца и легкого, печени, почки, поджелудочной железы, кожных и роговичных трансплантатов. Они также показаны для профилактики заболевания "трансплантат-против-хозяина", например такого, которое является следствием пересадки костного мозга.
б) Лечение и профилактика трансплантатных васкулопатий, например атеросклероза.
в) Лечение и профилактика клеточной пролиферации гладкой мускулатуры и миграции, приводящей к утолщению интимы сосудов, обструкции кровеносных сосудов, обструктивного коронарного атеросклероза, рестеноза.
г) Лечение и профилактика аутоиммунного заболевания и воспалительных состояний, в частности воспалительных состояний с этиологией, включая аутоиммунный компонент, таких как артрит (например, ревматоидный артрит, хронический прогрессирующий артрит и деформирующий артрит) и ревматические заболевания. Специфические аутоиммунные заболевания, для которых могут быть применимы соединения формулы I, включают в себя аутоиммунные гематологические расстройства (включая, например, гемолитическую анемию, гипопластическую анемию, анемию эритроцитов и идиопатическую тромбоцитопению), системный волчанковый эритематоз, полихондрит, склеродому, грануломатоз Вегенера, дерматомиозит, хронический активный гепатит, миастению, псориаз, синдром Стивена-Джонсона, идиопатический синдром мальабсорбции, аутоиммунную воспалительную болезнь кишечника (включая, например, язвенный колит и гранулематозную болезнь), эндокринную офтальмопатию, болезнь Грейвса, саркоидоз, рассеянный склероз, первичный биллиарный цирроз печени, ювенильный диабет (диабет типа I), увеит (передний и задний), сухой и мокрый кератоконъюнктивиты, интерстициальный фиброз легкого, псориазный артрит, гломерулонефрит (с и без нефротического синдрома, например включая идиопатический нефротический синдром или нефропатию с минимальными изменениями) и ювенильный дерматомиозит.
д) Лечение и профилактика астмы.
е) Лечение резистентности к множеству лекарственных препаратов (MDR). Соединения формулы I подавляют P-гликопротеины (Pgp), которые являются мембранными транспортными молекулами, ассоциированными с MDR. MDR, в частности, проблематична для пациентов, страдающих раком и СПИДом, которые не могут реагировать на традиционную хемотерапию из-за того, что лекарственное соединение вытесняется из клеток белками Pgp. Соединения формулы I, следовательно, применимы для усиления эффективности других хемотерапевтических агентов при лечении и контроле состояний резистентности к множеству лекарственных препаратов, таких как рак с резистентностью к множеству лекарственных препаратов или СПИД с резистентностью к множеству лекарственных препаратов.
ж) Лечение пролиферативных заболеваний, например опухолей, гиперпролиферативных кожных болезней и т.п.
з) Лечение грибковых инфекций.
и) Лечение и профилактика воспалений, особенно при потенциации действия стероидов.
к) Лечение и профилактика инфекций, особенно инфекции патогенами, имеющими Mip и Mip-подобные факторы.
Требуемая доза при вышеперечисленных показаниях будет, конечно, варьироваться, например в зависимости от состояния, показанного для лечения (например, типа болезни или природы резистентности), желаемого эффекта и способа введения. В общем, однако, удовлетворительные результаты получены при пероральном введении в дозах порядка от 0,05 до 5 или вплоть до 10 мг/кг/день, например от 0,1 до 2 или вплоть до 7,5 мг/кг/день, вводимых за один раз или, разделенных на дозы для введения 2-4 раза в день: или при введении парентерально, например внутривенно, например с помощью внутривенной капельницы или инфузии, в дозах от 0,01 до 2,5 вплоть до 5 мг/кг/день, например от 0,05 до 0,1 вплоть до 1,0 мг/кг/день. Подходящие суточные дозы для пациентов, таким образом, составляют порядка 500 мг перорально, например порядка от 5 до 100 мг перорально, или порядка от 0,5 до 125 вплоть до 250 мг внутривенно, например порядка от 2,5 до 50 мг внутривенно.
Иначе и даже предпочтительно, дозировку подбирают для каждого пациента специально, чтобы обеспечить предопределенные уровни в крови, например как определено при помощи радиоиммуноанализа (RIA). Таким образом, дозировка для пациента может быть подобрана так, чтобы достичь регулярных текущих уровней в крови, как измерено методом RIA, и составлять значения порядка от 50 или 150 вплоть до 500 или 1000 нг/мл, т.е. аналогично методам дозирования, ныне применяемым для иммуносуппрессорной терапии циклоспоринами.
Соединения формулы I можно вводить как активный ингредиент отдельно или вместе с другими лекарствами. Например, при иммуносуппрессорных применениях, таких как профилактика и лечение заболевания "трансплантат-против-хозяина", отторжения трансплантата или аутоиммунного заболевания, соединения формулы I можно применять в комбинации с циклоспоринами или аскомицинами или их иммуносуппрессивными аналогами, например, циклоспорином A, циклоспорином G, FK-506 и др. ; кортикостероидами; циклофосфамидом; азатиоприном; метотрексатом; бреквинаром; лефлуномидом; мизорибином: микофенольновой кислотой; мофетил микофенолятом; иммуносуппрессивными моноклональными антителами, например моноклональными антителами к лейкоцитарным рецепторам, например, MHC, CD2, CD3, CD4, CD7, CD25, CD28, CTLA4, B7, CD45 или CD58 или их лигандам: или другими иммуномодуляторными соединениями. Для противовоспалительных применений соединения формулы I можно применять вместе с противовоспалительными агентами, например кортикостероидами. Для противоинфекционных применений соединения формулы I можно применять в комбинации с другими противоинфекционными агентами, например противовирусными лекарствами или антибиотиками.
Соединения формулы I вводят традиционными способами, в частности энтерально, например перорально, например в виде растворов для питья, таблеток или капсул, или парентерально, например в виде инъекционных растворов или суспензий. Подходящие формы стандартной дозы для перорального введения содержат, например, от 1 до 5 мг соединения формулы I, обычно 1-10 мг. Фармацевтические композиции, содержащие соединения формулы I, можно производить традиционным способом, например аналогично тому, как производят фармацевтические композиции, содержащие рапамицин, например как описано в ЕРА 0041795.
Предпочтительно фармацевтические композиции содержат соединение формулы I и среду носителя, которая содержит гидрофильную фазу, липофильную фазу и поверхностно-активное вещество. Они могут быть в виде эмульсии или микроэмульсионного преконцентрата. Такие эмульсии или микроэмульсионные преконцентраты описаны, например, в Патентной заявке UK 2278780 A. Предпочтительно липофильная фаза составляет 10-85 мас.% от среды носителя: поверхностно-активное вещество составляет 5-80 мас.% от среды носителя; гидрофильная фаза составляет 10-50 мас. % от среды носителя. Соединение формулы I предпочтительно присутствует в количестве 2-15 мас.%. Особенно предпочтительная фармацевтическая композиция включает в себя микроэмульсионный преконцентрат в качестве среды носителя, который содержит:
1) продукт реакции касторового масла и окиси этилена,
2) продукт транэстерификации растительного масла и глицерина, содержащий главным образом линолевую кислоту или олеиновую кислоту, моно-, ди- и триглицериды или полиоксиалкилированное растительное масло,
3) 1,2-пропиленгликоль, и
4) этанол.
В соответствии с вышеупомянутым в настоящем изобретении также предлагается:
A. Соединение формулы I для применения в качестве фармацевтического средства, например при профилактике или лечении вышеуказанных заболеваний.
Б. Фармацевтическая композиция, содержащая соединение формулы I вместе с его фармацевтически приемлемым разбавителем или носителем.
В. Способ профилактики или лечения заболеваний, как указано выше, у субъекта, нуждающегося в таком лечении, при котором на вышеуказанный субъект воздействуют эффективным количеством соединения формулы I.
Г. Набор или упаковку для применения при иммуносуппрессии, воспалении или инфекциях, как указано выше, включающий в себя фармацевтическую композицию, содержащую соединение формулы I и фармацевтическую композицию, содержащую либо иммунодепрессант либо иммуномодуляторное лекарственное средство или противовоспалительный агент или противоинфекционный агент.
Неожиданно было обнаружено, что соединения формулы I, где X = OH, т.е. 32(S)-дигидросоединения, обладают улучшенной активностью, например в анализах, описанных выше, и являются более стабильными, чем соответствующие энантиомеры, т.е. 32(R)-дигидросоединения, например как следует из теста:
Тестируемые соединения инкубируют в крысиной сыворотке и их связывающее сродство к FKBP-12 измеряют в МВД анализе после инкубации с различным временем. С уменьшением сродства возрастает номинальная IC50. Снижение сродства в общем приписывают нестабильности соединения в крысиной сыворотке.
Описываются новые производные рапамицина формулы I, где R1 представляет собой С1-С10алкил, С3-С10алкинил, С3-С10гидроксиалкинил, R2 выбирается из формулы II где R3 выбирается из Н, С1-С6алкил, гидроксиС1-С6алкил или С1-С6-алкоксиС1-С6алкил; R4 представляет собой Н или метил; Х представляет собой ОН или Н, при условии, что когда Х представляет собой ОН, R1 представляет собой C1-С6алкил и R2 представляет собой остаток формулы II, тогда R3 не является Н. Соединения формулы I обладают иммуносупрессорной активностью. Описывается такая фармацевтическая композиция на их основе. 2 с. и 4 з.п.ф-лы.
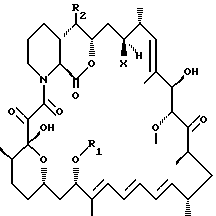

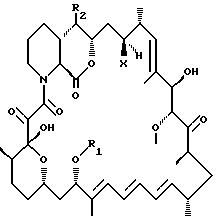
где R1 представляет собой С1-С10алкил, C3-C10алкинил, С3-С10гидроксиалкинил;
R2 остаток формулы II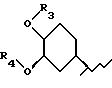
где R3 - Н, С1-С6алкил, гидроксиС1-С6алкил или С1-С6-алкоксиС1-С6алкил; R4 представляет собой Н или метил; а
Х представляет собой ОН или Н,
при условии, что когда Х представляет собой ОН, R1 представляет собой С1-С6алкил и R2 представляет собой остаток формулы II, тогда R3 не является Н.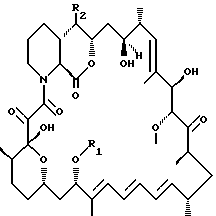
где R1 представляет собой С3-10алк-2-инил или С3-10гидроксиалк-2-инил;
R2 представляет собой остаток формулы II, как определено в п.1.
где R1 представляет собой С1-10алкил, С3-10алк-2-инил или С3-10гидроксиалк-2-инил;
R2 представляет собой остаток формулы II как определено в п.1.
Приоритет по признакам:
09.06.95 - по п.2 формулы изобретения;
06.07.95 - по п.3 формулы изобретения;
09.06.95 и 06.07.95 - по п.1 формулы изобретения.
