Родство с предшествующими заявками.
Данная заявка является частичным продолжением заявки, серийный номер 08/633275, поданной 16 апреля 1996 г.
Предпосылки создания изобретения.
Настоящее изобретение относится к новому способу получения катализатора, пригодного для синтеза ненасыщенных сложных эфиров путем газофазной реакции. В частности, настоящее изобретение касается нового способа получения катализатора, пригодного для газофазного образования винилацетата в результате взаимодействия этилена, кислорода и уксусной кислоты.
Известно в данной области техники получение винилацетата путем осуществления взаимодействия этилена, кислорода и уксусной кислоты в газовой фазе в присутствии катализатора, содержащего палладий, золото, и ацетата щелочного металла на некоторых носителях, таких как диоксид кремния. Такие каталитические системы могут проявлять высокую активность. К сожалению, результаты применения таких золотопалладиевых катализаторов неустойчивы. Эта неустойчивость, видимо, основана в некоторой степени на картине (профиле) распределения компонентов катализатора, нанесенных на носитель и связанных с ним. Например, при использовании известных винилацетатных каталитических систем, содержащих пористый носитель с палладием и золотом, металлические компоненты, осажденные во внутренних (центральных) областях носителя, или около них, не всегда обеспечивают существенное содействие механизмам реакции, поскольку реагенты не способны легко диффундировать в центральные (внутренние) области пористой структуры катализатора. Еще важнее то, что продукты каталитического синтеза, образованные во внутренней части катализатора, должны диффундировать из внутренней части наружу, опять вступая в контакт с активной фазой в наружной области катализатора. Следовательно, эти образованные во внутренней части продукты подвергаются дальнейшей реакции и часто превращаются в ненужные побочные продукты. Наиболее эффективные реакции происходят тогда, когда каталитический металл нанесен в виде тонкой оболочки на поверхностные области катализатора, что облегчает диффузию реагентов и продуктов и обеспечивает хороший выход продукта и пониженное содержание побочных продуктов.
Уже выдано много разных патентов, основанных на желании более равномерно распределить и закрепить такие каталитические компоненты, как золото и палладий, в тонком слое на поверхности носителя, чтобы создать винилацетатный катализатор, обладающий высокой производительностью, хорошей избирательностью и длительным сроком службы. Примеры таких патентов включают патенты США N 4087622, N 4048096, N 3822308, N 3775342 и патент Великобритании N 1521652.
Основной способ получения винилацетатного катализатора, содержащего палладий и золото, нанесенные на носитель, включает в себя (1) пропитку носителя водными растворами водорастворимых соединений палладия и золота, (2) осаждение не растворимых в воде соединений палладия и золота на носителе путем контактирования пропитанного носителя катализатора с раствором соединений, способных взаимодействовать с водорастворимыми соединениями палладия и золота с образованием не растворимых в воде соединений благородных металлов, (3) промывку обработанного носителя водой для удаления анионов, высвободившихся из первоначально пропитанных соединений палладия и золота во время осаждения, и (4) преобразование не растворимых в воде соединений палладия и золота в свободный металл путем обработки восстанавливающим агентом. Окончательная обработка обычно включает (5) пропитку восстановленного катализатора водным раствором ацетата щелочного металла и (6) высушивание конечного каталитического продукта.
Попытки обеспечить равномерное распределение палладия и золота на носителе включали выполнение вышеупомянутых стадий и/или использование материалов носителей, имеющих различные конкретно указанные размеры пор. Особенно полезные усовершенствования в производстве высокоактивных катализаторов для получения винилацетата описаны в совместно переуступленных патентах США N 5314858 и N 5332710, которые оба включены в данное описание путем ссылки. В этих двух патентах описаны способы улучшения распределения палладия и золота на носителе путем выполнения стадии (2) осаждения и "фиксирование" (закрепление) водорастворимых соединений благородных металлов на носителе в виде соединений, не растворимых в воде. В патенте США N 5314858 фиксирование благородных металлов на носителе обеспечивают, используя две отдельные стадии осаждения, чтобы избежать использования больших избытков "фиксирующего" соединения. В патенте США N 5332710 описано фиксирование благородных металлов путем вращения пропитанных носителей при погружении пропитанных носителей в реакционный раствор, по крайней мере, в начальном периоде осаждения. Как было обнаружено, такой метод погружения с вращением дает катализаторы, в которых осажденные на носителе металлы более равномерно распределены в тонком слое на поверхности носителя.
Попытки повышения каталитической активности, обычно оцениваемой по объемной производительности (STY), включали использование носителей с конкретным размером пор или конкретными формами. Носители катализатора, пригодные для получения сложных виниловых эфиров, обычно состоят из диоксида кремния, оксида алюминия, силикатов алюминия или шпинелей. Диоксид кремния является предпочтительным носителем, потому что он порист и является нейтральным носителем для нанесения благородных металлов. Обычно носители имеют форму сфер, таблеток или цилиндров. Часто используют сферические носители, имеющие диаметр в диапазоне 4-8 мм. Когда каталитическая активность повышается, то является предпочтительным для производства ненасыщенного сложного эфира в промышленном масштабе увеличивать объем проходящего через катализатор газообразного сырьевого материала, состоящего из олефина, органической карбоновой кислоты и кислорода. На объемный поток газа через катализатор влияют форма и пористость катализатора. Одна причина увеличения объема газообразного сырьевого материала, проходящего через катализатор, заключается в предотвращении образования участков перегрева на активном катализаторе. Поскольку реакции образования ненасыщенных сложных эфиров являются экзотермическими, то повышение каталитической активности может вызвать чрезмерный нагрев некоторых участков катализатора. Неэффективное распределение тепла на катализаторе вызывает нежелательные побочные реакции, такие, как образование диоксида углерода, что приводит к снижению избирательности по отношению к образованию ненасыщенного сложного эфира, такого, как винилацетат.
Другой проблемой, связанной с повышением активности катализаторов образования сложных виниловых эфиров, является образование тяжелых фракций в процессе синтеза сложных виниловых эфиров. Тяжелые фракции представляют собой остатки из побочных продуктов, содержащие высокомолекулярные органические соединения, образованные в процессе синтеза ненасыщенного сложного эфира. Такие тяжелые фракции включают (но не ограничиваются ими) этилидендиацетат, 1,1-диацетоксиэтилен, цис- и транс-диацетоксиэтилен, этиленгликольдиацетат, винилацетоксиацетат, винилацетоксиуксусную кислоту, этиленгликольмоноацетат и циклопропанкарбоновую кислоту. Синтез ненасыщенного сложного эфира, такой, как синтез винилацетата, часто может давать избирательность по тяжелым фракциям до примерно 2% от реагирующего этилена. Тяжелые фракции можно легко удалить и отделить от целевого продукта путем перегонки со сбором остатков, содержащих тяжелые фракции, и выбросом их в отвал или сжиганием. Однако такой метод выброса тяжелых фракций не практикуется. Тяжелые фракции считаются токсичными, и их сжигание может вызвать образование и выделение токсических продуктов в окружающую среду. Законы и инструкции по борьбе с загрязнением во многих частях мира строго ограничивают возможность складирования токсичных твердых отходов в отвалах или их сжигания. Поэтому всякое уменьшение образования тяжелых фракций во время синтеза ненасыщенного сложного эфира является весьма желательным.
Каталитическая активность катализатора синтеза сложного винилового эфира уже была повышена путем увеличения относительного количества золота, нанесенного на носитель. Было найдено, что увеличенное содержание золота в катализаторе синтеза сложного винилового эфира повышает срок службы катализатора и снижает избирательность по CO2 и образование тяжелых фракций.
Хотя существуют катализаторы и способы получения катализаторов с улучшенным равномерным распределением таких металлов, как палладий и золото, на носителе катализатора и с высокой каталитической активностью, все еще имеется потребность в способе получения катализаторов синтеза винилацетата, имеющих более равномерное распределение палладия и золота на поверхности носителя, и, в частности, в увеличении золотого содержания носителя. Существующие способы нанесения золота на носитель не всегда гарантируют содержание золота в готовом катализаторе из-за значительного выщелачивания или стирания золота, или солей золота, с катализатора в процессе его получения.
Таким образом, задачей настоящего изобретения является создание способа получения катализатора синтеза сложных виниловых эфиров, имеющего повышенное количество металлического золота.
Другой задачей настоящего изобретения является создание способа получения катализатора синтеза сложных виниловых эфиров, имеющего повышенную избирательность по сложному виниловому эфиру, такому, как винилацетат.
Еще одной задачей настоящего изобретения является создание способа получения катализатора синтеза винилацетата, имеющего (катализатора) пониженную избирательность по диоксиду углерода и этилацетату.
И еще одной задачей настоящего изобретения является создание катализатора синтеза сложных виниловых эфиров с высокой каталитической активностью.
Другие задачи и преимущества настоящего изобретения изложены в следующем далее подробном описании и будут очевидны для специалиста в данной области техники при практическом осуществлении настоящего изобретения.
Краткое изложение сущности изобретения.
Теперь найдено, что высокоактивные катализаторы на носителе, содержащие палладий и золото, полезны для получения сложных виниловых эфиров из этилена, низших карбоновых кислот с 2-4 углеродными атомами и кислорода в газовой фазе при повышенной температуре и при нормальном или повышенном давлении, можно получить путем нанесения золота на катализатор в две стадии. Обычно золотой компонент катализатора синтеза сложных виниловых эфиров наносили на носители в одну единственную стадию пропитки и фиксировали (закрепляли) на них. Например, золото наносят путем пропитки на носитель катализатора в виде раствора водорастворимой соли или кислоты одновременно с водорастворимой солью металлического палладия или, в соответствии с другим вариантом, золото наносят на стадии, отдельной от нанесения палладия. Впитанные водорастворимые соединения палладия и золота затем фиксируют путем образования не растворимых в воде соединений палладия и золота с помощью щелочного фиксирующего раствора, после чего не растворимые в воде соединения палладия и золота восстанавливают до металлов палладия и золота. К сожалению, как было указано выше, нанесение золота на носитель в одну стадию часто приводит к потере золота с поверхности носителя из-за выщелачивания или стирания во время приготовления катализатора. Поэтому не всегда можно обеспечить высокую каталитическую активность и высокую избирательность по винилацетату.
Чтобы устранить указанные проблемы, в соответствии с настоящим изобретением усовершенствованный катализатор синтеза сложных виниловых эфиров получают путем (1) одновременной или последовательной пропитки носителя катализатора водными растворами водорастворимой соли палладия и первого количества водорастворимого соединения золота, таких, как хлорид натрий палладия и хлорид золота (III), (2) фиксирования благородных металлов на носителе осаждением не растворимых в воде соединений палладия и золота путем обработки пропитанных носителей реакционноспособным основным (щелочным) раствором, таким, как водный раствор гидроксида натрия, который взаимодействует с соединениями палладия и золота с образованием гидроксидов палладия и золота на поверхности носителя, (3) промывки водой для удаления хлорид-иона (или другого аниона) и (4) восстановления всех гидроксидов благородных металлов до свободных палладия и золота, причем усовершенствование содержит, (5) пропитки носителя вторым количеством водорастворимого соединения золота после фиксирования первого количества водорастворимого соединения золота и (6) фиксирования второго количества водорастворимого соединения золота. Первое и второе количества золота могут быть восстановлены либо после каждой из соответственных стадий фиксирования, либо все золото может быть восстановлено на конечной стадии восстановления после раздельного фиксирования первого и второго количеств золота. Было обнаружено, что использование катализаторов, полученных описанным способом, обеспечивает сохранение каталитической активности по отношению к образованию сложных виниловых эфиров, таких, как винилацетат, путем осуществления взаимодействия этилена, низшей карбоновой кислоты и кислорода в газовой фазе и существенное уменьшение побочной реакции образования диоксида углерода.
Подробное описание изобретения.
В усовершенствованном способе получения катализатора для использования в синтезе ненасыщенных сложных эфиров золото наносят на носитель катализатора в две отдельные стадии.
Материал носителя для катализатора по настоящему изобретению может иметь самую разнообразную геометрическую форму. Например, носитель может быть в форме сфер, таблеток или цилиндров. Геометрические размеры носителя могут, как правило, находиться в диапазоне 1-8 мм. Наиболее подходящей геометрической формой является, в частности, сферическая форма в виде, например, сфер диаметром в диапазоне 4-8 мм.
Удельная площадь поверхности носителя может изменяться в широких пределах. Например, подходящими являются носители, имеющие площадь внутренней поверхности примерно 50-300 м2/г и, в частности, примерно 100-200 м2/г (при измерении методом БЭТ, т.е. методом Брунауэра-Эммета-Теллера).
Примеры материалов носителей, которые могут быть использованы, включают диоксид кремния, оксид алюминия, силикаты алюминия и шпинели. Предпочтительным материалом является диоксид кремния.
В соответствии со способом по настоящему изобретению носитель катализатора сначала пропитывают водным раствором, содержащим водорастворимое соединение палладия и первое количество водорастворимого соединения золота. Можно также использовать последовательно отдельные растворы соединений палладия и золота, но такой способ обработки менее удобен. Примерами подходящих водорастворимых соединений палладия являются хлорид палладия (II), хлорид натрий-палладия (II), нитрат палладия (II) или сульфат палладия (II), а в качестве водорастворимых соединений золота могут быть использованы хлорид золота (III) или золото (III)-кислота. Золото (III)-кислота и хлорид натрий- палладия (II) являются предпочтительными из-за их хорошей растворимости в воде. Важен объем раствора, используемого для пропитки носителя благородными металлами. Для эффективного нанесения объем пропитывающего раствора должен составлять от примерно 95 до примерно 100% безводной абсорбционной способности носителя катализатора и предпочтительно равен примерно 98-99%. Такой метод пропитки характеризуют как метод "начального увлажнения".
После пропитки носителя водорастворимым соединением палладия и первым количеством водорастворимого соединения золота эти водорастворимые соединения благородных металлов затем фиксируют на носителе в виде не растворимых в воде соединений палладия и золота. Фиксирующий раствор содержит щелочной раствор, например водный раствор, содержащий гидроксиды щелочных металлов, бикарбонаты щелочных металлов и/или карбонаты щелочных металлов. Является особенно предпочтительным использовать водные растворы гидроксида натрия и гидроксида калия. Количество используемого щелочного соединения должно обеспечивать отношение щелочного металла к аниону от водорастворимых соединений благородных металлов от примерно 1:1 до примерно 2:1, предпочтительно от примерно 1,2:1 до примерно 1,8:1. Путем обработки щелочным раствором водорастворимые соединения благородных металлов преобразуют в не растворимые в воде соединения, видимо, являющиеся гидроксидами и/или оксидами, по крайней мере, в случае, когда щелочной раствор является раствором гидроксида натрия или гидроксида калия.
Пропитку носителя на первой стадии фиксирования золота предпочтительно осуществляют способом "погружения с вращением", который описан в патенте США N 5332710, выданном 26 июля 1994 г. на имя Nicolau et al., все описание которого включено в данное описание путем ссылки. В этом способе пропитанный носитель погружают в щелочной фиксирующий раствор и галтуют или вращают в нем в течение начальных стадий осаждения не растворимых в воде соединений благородных металлов. Вращение или галтовку носителей в щелочном фиксирующем растворе предпочтительно проводят в течение, по крайней мере, около 0,5 часа при начальной обработке, а наиболее предпочтительно в течение, по крайней мере, около 2,5 часов. Обработку погружением с вращением можно продолжать до примерно 4 часов, после чего обработанному носителю можно дать постоять в фиксирующем растворе, чтобы гарантировать полное осаждение соединений благородных металлов.
Можно использовать любой тип вращательного или галтовочного оборудования, поскольку нет необходимости в использовании конкретного устройства. Но что важно, так это степень вращательного движения. Так, достаточным предпочтительно является такое вращение, которое обеспечивает равномерное контактирование всех поверхностей пропитанных носителей с щелочным фиксирующим раствором. Вращение предпочтительно не должно быть настолько резким, чтобы происходило фактически истирание не растворимых в воде соединений благородных металлов до полного стирания их с поверхности носителя. С другой стороны, было обнаружено, что некоторая небольшая степень истирания не растворимых в воде соединений благородных металлов фактически обеспечивает более равномерное распределение не растворимых в воде соединений благородных металлов на поверхности носителя. Частота вращения предпочтительно составляет от примерно 1 до примерно 10 об/мин и, возможно, даже выше, в зависимости от конкретного используемого носителя и от количества благородного металла, осаждаемого на носителе. Используемую частоту вращения можно регулировать, причем она может также зависеть от используемого устройства, размера и формы носителя, типа носителя, дозировки металла и т.д., но предпочтительно она соответствует описанным выше требованиям недопущения фактически стирания не растворимых в воде соединений с поверхности носителя, хотя небольшая степень истирания может быть выгодной.
Другим методом фиксирования благородных металлов на носителе является метод "начального увлажнения", при котором, как было описано выше, определенный объем фиксирующего раствора, например водного раствора гидроксида щелочного металла, равный поглощающей способности носителя в сухом состоянии, выливают на пористые носители, уже пропитанные водорастворимыми соединениями благородных металлов. Обработанным носителям дают постоять до завершения осаждения. При выполнении фиксирования методом начального увлажнения пропитанные носители подвергают перед фиксированием водным раствором гидроксида металла воздушной сушке.
В соответствии с другим вариантом стадия фиксирования может быть разделена на, по крайней мере, два отдельных этапа обработки щелочным фиксирующим раствором. Такой способ фиксирования раскрыт в патенте США N 5314858, выданном 24 мая 1994 г. на имя Colling, полное описание которого включено в данное описание путем ссылки. При каждой отдельной обработке количество щелочного реакционноспособного соединения не превышает того, которое равно молярному количеству, необходимому для реагирования со всем количеством соединения благородного металла, присутствующего на носителе в виде водорастворимого соединения. Реакционноспособное соединение используют без избытка. Предпочтительно количество реакционноспособного соединения, используемого на каждом этапе фиксирования, меньше, чем молярное количество, необходимое для реагирования со всем водорастворимым соединением благородного металла. Каждый этап фиксирования проводят путем пропитки высушенного пропитанного носителя щелочным фиксирующим раствором в количестве, примерно равном поглощающей способности носителя в сухом состоянии. Количество щелочного соединения, содержащегося в растворе, должно обеспечивать отношение щелочного металла к аниону от водорастворимых соединений благородных металлов от примерно 0,7 до 1:1 (молярное) на первом этапе и от примерно 0,2 до 0,9:1 (молярное) на втором этапе. Предпочтительно общее количество щелочного металла относительно аниона находится в пределах от примерно 1,2 до примерно 1,6:1 (молярное) для всей стадии фиксирования. После обработки на первом этапе фиксирования обработанным носителям дают постоять в течение достаточного периода времени, чтобы обеспечить возможность осаждения не растворимых в воде соединений благородных металлов. Период времени можно изменять, но обычно он составляет от примерно 2 часов до примерно 8 часов перед повторной обработкой носителей второй частью щелочного фиксирующего раствора. После обработки на втором этапе фиксирования обработанным носителям опять дают постоять, по крайней мере, еще 2 часа, предпочтительно, по крайней мере, около 4 часов, а возможно, до завершения осаждения в течение до примерно 16 часов.
Обработка на втором этапе фиксирования может быть одинаковой с обработкой на первом этапе, причем обработанные и частично зафиксированные носители пропитывают фиксирующим раствором с требуемой концентрацией щелочи и при общем объеме раствора, также эквивалентном безводной поглощательной способности носителя. Носитель можно также пропитывать на втором этапе фиксирования описанным выше способом "погружения с вращением". При этом способе уже один раз фиксированные носители погружают в щелочной фиксирующий раствор или галтуют или вращают в нем в течение начальных этапов осаждения не растворимых в воде соединений благородных металлов, как было описано ранее.
После того как уже зафиксировано первое количество водорастворимого золота, второе количество золота может быть нанесено пропиткой и зафиксировано так же, как описано выше в отношении впитывания и фиксирования первого количества золота на носителе. При этом для нанесения второго количества золота можно использовать любые соли золота, описанные выше для первого этапа нанесения золота. Аналогичным образом, для осаждения на носителе не растворимого соединения золота можно использовать любой из описанных выше методов фиксирования, таких, как метод "погружения с вращением", метод начального увлажнения и метод двойного фиксирования. Предпочтительно второе количество золота фиксируют на носителе методом "начального увлажнения" путем пропитки носителя раствором второго количества водорастворимого соединения золота и водным раствором щелочного фиксирующего агента или путем обработки носителя, пропитанного вторым количеством водорастворимого соединения золота, водным щелочным фиксирующим раствором и обеспечения пропитанному носителю возможности постоять в течение до примерно 16 часов или более, чтобы гарантировать осаждение не растворимых в воде соединений золота. Объем фиксирующего раствора равен сухой поглощающей способности носителя, а количество используемого щелочного соединения превышает молярное количество, необходимое для реагирования со всем количеством впитанных водорастворимых соединений золота.
После фиксирования соединений благородных металлов на носителе носитель промывают деионизированной водой для удаления анионов, таких, как хлорид-ионы, которые все еще находятся на носителе и высвобождаются из пропитывающих растворов. Промывку проводят до полного удаления анионов с носителя. Чтобы гарантировать по существу полное удаление анионов, таких, как хлорид-ион, с катализатора, производят тестирование промывных вод нитратом серебра, до тех пор, пока тест нитратом серебра не станет отрицательным, т.е. не будет образования хлорида серебра. После вымывания ионов из катализатора его высушивают при температурах, не превышающих примерно 150oC, в инертной атмосфере, такой, как непрерывный поток азота. Промывку и сушку можно проводить после того, как на носителе уже зафиксированы палладий и первое количество золота, а также после того, как нанесено и зафиксировано второе количество золота, или же промывку и сушку проводят, когда все благородные металлы уже зафиксированы на носителе, т.е. после фиксирования второго количества золота.
Зафиксированные материалы затем обрабатывают восстанавливающим агентом, чтобы преобразовать присутствующие соли и соединения благородных металлов на носителе в металлическую форму. Восстановление может быть осуществлено в жидкой фазе, например, гидратом водного гидразина, или, предпочтительно, в газовой фазе, например, водородом или углеводородами, например этиленом. Если восстановление осуществляют раствором гидрата гидразина, реакцию предпочтительно проводят при нормальной температуре. Когда восстановление осуществляют в газовой фазе, реакцию целесообразно проводить при повышенной температуре, например при примерно 100-200oC в случае восстановления этиленом. Восстанавливающий агент целесообразно использовать в избытке, чтобы быть уверенным, что все соли и соединения благородных металлов преобразованы в металлическую форму. При использовании гидразина массовое отношение гидразина к драгоценным металлам находится в пределах от примерно 10:1 до примерно 15: 1, а предпочтительно составляет примерно 12:1. После восстановления не растворимых в воде соединений палладия и золота носитель сушат в инертной атмосфере при примерно 150oC. При использовании стадий промывки и сушки восстановление благородных металлов может быть осуществлено после каждого этапа фиксирования или после того, как все благородные металлы зафиксированы на носителе. Кроме того, восстановление может быть проведено либо до, либо после промывки катализатора для удаления анионов.
Относительное количество водорастворимого соединения золота, наносимого на каждой стадии пропитки, не имеет важного значения. От примерно 1/2 до примерно 3/4 общего количества золота на готовом катализаторе наносят на первой стадии пропитки, а остальное - на второй. Предпочтительно количество соединений палладия и золота должно быть таким, чтобы обеспечить от примерно 3 до примерно 8 г палладия и от примерно 1,5 до примерно 14 г золота на литр готового катализатора. Катализаторы, содержащие более высокие или более низкие количества благородных металлов по сравнению с указанными выше, могут быть полезными в получении винилацетата путем осуществления взаимодействия этилена, кислорода и уксусной кислоты в паровой фазе, если они получены новым способом, раскрытым в данном описании. При этом отношение золото/палладий в готовом катализаторе может находиться в пределах от примерно 0,2:1 до примерно 2:1, предпочтительно от примерно 0,4:1 до примерно 1,5:1.
В зависимости от применения, для которого предназначен катализатор, полученный способом по настоящему изобретению, катализатор может также быть снабжен традиционными добавками. Например, полезным является добавление ацетатов щелочных металлов, когда катализатор предназначен для получения ненасыщенных сложных эфиров из олефинов, кислорода и органических кислот. В таком случае, например, катализатор может быть пропитан водным раствором ацетата калия, ацетата натрия, ацетата лития, ацетата рубидия или ацетата цезия и затем промыт и высушен.
Катализаторы по настоящему изобретению могут быть применены с особой выгодой для получения винилацетата из этилена, кислорода и уксусной кислоты в газовой фазе. Особенно подходящими для этой цели являются такие катализаторы по настоящему изобретению, которые содержат диоксид кремния в качестве материала носителя и добавки ацетатов щелочных металлов. При получении винилацетата такие катализаторы также отличаются высокой активностью и избирательностью по винилацетату и длительным сроком службы.
Когда винилацетат получают с применением катализаторов по настоящему изобретению, поток газа, содержащий этилен, кислород или воздух и уксусную кислоту, пропускают поверх катализатора. Состав газового потока можно изменять в широких пределах с учетом ограничений в отношении взрывоопасности. Например, молярное отношение этилена к кислороду может находиться в пределах от примерно 80:20 до примерно 98:2 и молярное отношение уксусной кислоты к этилену - в пределах от примерно 100:1 до примерно 1:100, а содержание газообразного ацетата щелочного металла может составлять примерно 2-200 м.д. (миллионных долей) относительно используемой уксусной кислоты. Газовый поток может также содержать другие, инертные газы, такие, как азот, диоксид углерода и/или насыщенные углеводороды. Температуры реакции используют повышенные, предпочтительно находящиеся в диапазоне примерно 100-250oC, предпочтительно 130-200oC. Давление можно использовать несколько пониженное, нормальное или повышенное, предпочтительно избыточное давление до примерно 20 атм.
Катализаторы по настоящему изобретению при использовании для получения винилацетата показывают повышенную объемную производительность, пониженную избирательность по диоксиду углерода и этилацетату, а также уменьшенное образование тяжелых фракций.
Следующие далее примеры предназначены для дальнейшей иллюстрации настоящего изобретения и не должны рассматриваться как ограничивающие объем настоящего изобретения.
Примеры I, II и III.
Катализаторы получали на сферических носителях на основе диоксида кремния, которые были поставлены ф. Sud Chemie с диаметрами около 5 мм. Носители были разделены на три порции (примеры I, II и III) по 250 см3 каждая. Катализаторы в примере I получали стандартным способом (описан ниже). Катализаторы в примере II получали таким же способом, как и катализаторы в примере I, за исключением того, что на катализатор наносили дополнительное количество золота. Количество золота было примерно равно количеству палладия, нанесенного на катализатор. Катализаторы примера III содержали такие же количества палладия и золота, как в примере II, но были получены двухстадийным способом нанесения золота по настоящему изобретению.
Все носители пропитывали водными растворами, содержавшими тетрахлорат натрий-палладия и тетрахлораурат натрия. Используемый объем раствора был эквивалентен количеству раствора, которое носители были способны поглотить (метод начального увлажнения). В примере I носители пропитывали достаточным количеством водорастворимых солей палладия и золота, чтобы полученные катализаторы имели примерно 7 г/л металлического палладия и примерно 4 г/л металлического золота. В примерах II и III носители пропитывали достаточным количеством водорастворимых солей палладия и золота, чтобы каждый катализатор имел примерно 7 г/л металлического палладия и примерно 7 г/л металлического золота. В примере III во время первой пропитки наносили 4 г/л золота.
После пропитки носители помещали в роторный испаритель (без вакуума) и обрабатывали 283 см3 50%-ного (мас./мас.) водного раствора гидроксида натрия для фиксирования водорастворимых солей палладия и золота на носителях в виде не растворимых в воде гидроксидов палладия и золота. Использованное количество гидроксида натрия составляло примерно 120% стехиометрического эквивалента, необходимого для преобразования солей металлов в их гидроксиды металлов. Носители немедленно вращали при примерно 5 об/мин в течение примерно 2,5 часов. Температуру в процессе погружения с вращением поддерживали на уровне примерно 70oC путем вращения в горячей водяной бане.
Фиксированные носители сливали и помещали в 500 мл градуированные цилиндры с погружными трубками и промывали в течение 5 часов деионизированной водой до тех пор, пока тестирование промывных вод нитратом серебра не стало отрицательным, т.е. не было образования хлорида серебра. Затем промытые носители помещали в 500 мл круглодонные колбы и ставили в печь под непрерывным потоком азота с высушиванием в течение ночи, т.е. примерно 16 часов. Температуру печи поддерживали на уровне примерно 150oC.
Затем зафиксированные не растворимые в воде соединения палладия и золота на высушенных носителях восстанавливали до металлического палладия и металлического золота парофазным методом с образованием катализаторов. Поверх носителей в печи пропускали смесь 5%-ного этилена в азоте в течение примерно 5 часов при температуре около 150oC. Расход газовой смеси составлял примерно 0,5 стандартных кубических футов в час (0,142 м3/час) при атмосферном давлении. После восстановления катализаторы извлекали из печи и давали им остыть до комнатной температуры.
Катализаторы в примерах I и II пропитывали водным раствором примерно 10 г ацетата калия (концентрация около 40 г/л) методом начального увлажнения и высушивали в сушилке с псевдоожиженным слоем в течение примерно 1 часа при примерно 150oC.
После восстановления в этилене, как описано выше, катализаторы примера III дополнительно пропитывали раствором тетрахлораурата натрия и 1,65 г 50%-ного (мас./мас.) водного фиксирующего раствора гидроксида натрия, чтобы катализаторы имели дополнительные 3 г/л золота, а всего примерно 7 г/л золота на готовых катализаторах. Количество использованного гидроксида натрия соответствовало примерно 180% стехиометрических эквивалентов, необходимых для преобразования солей золота в металлическое золото. Катализаторам давали постоять в растворе всю ночь, т.е. в течение примерно 16 часов. Затем катализаторы помещали в 500 мл градуированный цилиндр с погружной трубкой и промывали 5 часов деионизированной водой до получения отрицательного теста промывных вод нитратом серебра.
Промытые катализаторы помещали в 500 мл круглодонной колбе в печь при примерно 150oC под непрерывным потоком азота на всю ночь. Поверх промытых и высушенных катализаторов пропускали смесь 5%-ного этилена в азоте в течение примерно 5 часов при примерно 150oC для восстановления солей золота в металлическое золото.
После охлаждения катализаторов до комнатной температуры их пропитывали водным раствором примерно 10 г ацетата калия (концентрация около 40 г/л). Давали катализаторам постоять в течение примерно 15 минут и затем высушивали их в сушилке с псевдоожиженным слоем при примерно 100oC в течение примерно 1 часа.
Пробу (30 см3) катализатора помещали в трубчатый реактор с поршнеобразным вводом пробы длиной 3 фута (0,914 м) и внутренним диаметром 0,75 дюйма (19 мм). Трубку снабжали концентрической термопарой 0,125 дюйма (3,175 мм). Через трубку пропускали уксусную кислоту, этилен, кислород и азот при температуре, обеспечивавшей преобразование в продукт примерно 45% кислорода. Продукты анализировали автономно (off-line) путем парофазной хромотографии.
Результаты анализа продуктов представлены в таблице 1. Результаты показывают, что катализаторы, полученные двухстадийным способом нанесения золота, проявляют более низкую (5,4%) избирательность по диоксиду углерода, более низкую (0,08%) избирательность по этилацетату и повышенную (611) объемную производительность (STY) по сравнению с более высокими избирательностями по диоксиду углерода и по этилацетату и более низкой объемной производительностью (STY) катализаторов, полученных обычным способом. Кроме того, катализаторы, полученные двухстадийным способом нанесения золота, имеют более низкую (0,97%) избирательность по тяжелым фракциям по сравнению с 1,26% тяжелых фракций при использовании катализаторов, полученных обычным способом с таким же количеством золота, как у катализаторов, полученных двухстадийным способом. Таким образом, двухстадийный способ нанесения золота по настоящему изобретению дает усовершенствованные катализаторы синтеза винилацетата.
Примерно 60 мл каждого типа катализатора, полученного так, как описано выше, помещали в отдельные корзины из хромоникелевой стали. Температуру в каждой корзине измеряли посредством термопары вверху и внизу каждой корзины. Каждую корзину помещали в реактор Берти и держали при температуре, которая обеспечивала примерно 45%-ную конверсию кислорода, посредством электронагревательной сетки. Через каждую корзину заставляли перемещаться под давлением примерно 12 атм газовую смесь, состоявшую из примерно 50 нормальных литров (при измерении при нормальных условиях, т.е. при нормальных температуре и давлении) этилена, примерно 10 нормальных литров кислорода, примерно 49 нормальных литров азота и примерно 50 г уксусной кислоты. Анализ продуктов проводили путем неавтономного (он-лайнового) газового хроматографического анализа в сочетании с автономным (офф-лайновым) анализом жидких продуктов путем конденсации продуктного потока при примерно 10oC для обеспечения оптимального анализа конечных продуктов.
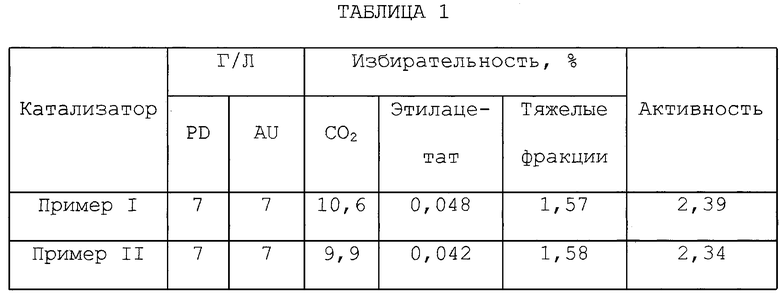
Результаты испытаний представлены в таблице 2. Испытания показывают явное снижение избирательности по таким побочным продуктам, как диоксид углерода и этилацетат (9,9 против 10,6 для избирательности по диоксиду углерода и 0,042 против 0,048 для избирательности по этилацетату), по сравнению в этом испытании с таким же катализатором, но полученным известным способом. Применение катализатора по настоящему изобретению на количество тяжелых фракций не влияло, а на активность катализатора влияло лишь незначительно.

Изобретение относится к способу получения катализатора для синтеза ненасыщенных сложных эфиров путем газофазной реакции, в частности, для газофазного образования винилацетата в результате взаимодействия этилена, кислорода и уксусной кислоты. В способе получения катализатора носитель катализатора пропитывают водорастворимыми соединениями палладия и золота с последующим фиксированием (закреплением) и затем восстановлением зафиксированных соединений палладия и золота до металлических палладия и золота, после чего носитель пропитывают вторым количеством водорастворимого соединения золота. Второе количество соединения золота фиксируют, после чего восстанавливают до металлического золота. Затем катализатор пропитывают ацетатом щелочного металла, таким как ацетат калия. Технический результат - катализаторы при использовании для получения винилацетата показывают повышенную объемную производительность, пониженную избирательность по диоксиду углерода и этилацетату, уменьшенное образование тяжелых фракций. 28 з.п.ф-лы, 2 табл.
| US 5314858 A, 24.05.1994 | |||
| US 5332710 A, 26.07.1994 | |||
| Катализатор для получения винилацетата и способ его приготовления | 1977 |
|
SU694054A3 |
| Способ обработки зубчатых колес | 1976 |
|
SU685451A1 |

