Изобретение относится к новым α-(1-пиперазинил)-ацетамидопроизводным аренкарбоновой кислоты, пригодным для лечения диабета.
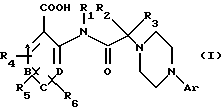
Объектом настоящего изобретения являются, таким образом, соединения общей формулы (I)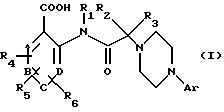
в которой Ar выбран из моно-, би- или трициклической арильной группы, имеющей от 6 до 14 атомов углерода,
гетероароматической группы, выбранной из пиридила, пиримидинила, пирролила, фурила, тиенила, хинолила, индолила, бензотиенила, бензофурила, бензопиранил, бензотиопиранила, дибензофурила, карбазолила и бензотиазинила,
причем остаток Аr может иметь от 1 до 3 заместителей, выбранных из группы, включающей С1-С8алкил, (С3-С8)циклоалкил(С1-С6)алкил, C1-С8алкокси,
(C3-C8)циклоалкилокси(C1-C6)aлкил, (С3-С8)циклоалкил(С1-С6)алкокси(С1-С6)алкил, (С3-С8)циклоалкилокси,
(С3-C8)-циклоалкил(С1-С6)алкокси, (С1-С6)алкокси(С1-С6)алкил, С6-С14арил, С6-С14гетероарил, (С6-С14)гетероарил (С1-С6)алкил, (С6-С14)арил(С1-С6)алкил,
(C6-C14)apил (C1-C6)aлкил(C6-C14)aрил, С6-С14арилокси, (С6-С14)арилокси(С1-С6)алкил, (С6-С14)арил(С1-С6)алкилокси, (С6-С14)-арил(С1-С6)алкилокси(С1-С6)алкил, галоид, трифторметил, трифторметокси, циано, гидрокси, нитро, амино, карбокси, (C1-С6)алкоксикарбонил, карбамоил, (С1-С8)алкилтио, (C1-С8)алкилсульфинил, (С1-С8)алкилсульфонил, сульфоамино, (C1-C8)aлкилcyльфoнилaминo, сульфамоил и (С1-С8)алкилкарбониламино, или два из этих заместителей образуют метилендиоксигруппу, за исключением из значений для остатка Аr 4-карбоксифенильной и замещенной 4-карбоксифенильной группы,
R1, R2 и R3 независимо друг от друга выбраны из группы, включающей атом водорода,
С1-С8алкил, (С1-С6)алкокси (С1-С6)алкил, циклоалкил, содержащий от 3 до 8 атомов углерода, (С3-С8)циклоалкил (С1-С6)алкил, (С3-С8)циклоалкилокси (С1-С6)алкил, (С3-С8)циклоалкил (С1-С6)алкокси (С1-С6)алкил, С6-С14арил,
С6-С14гетероарил, (С6-С14)-гетероарил (С1-С6)алкил, (С6-С14)арил (C1-С6)алкил, (С6-С14)арил (С1-С6)алкил (С6-С14)арил, (С6-С14)арил (С1-С6)алкокси (С1-С6)алкил и (С6-С14)арилокси (С1-С6)алкил,
A, В, С и D представляют собой группы =СН-, причем один или два из этих остатков могут представлять собой также атом азота,
R4, R5 и R6 независимо друг от друга выбраны из группы, включающей: атом водорода,
С1-С6алкил, (С3-С8)циклоалкил(С1-С6)алкил, С1-С8алкокси, (С3-С8)циклоалкилокси(С1-С6)алкил, (С3-С8)циклоалкилокси, (С3-С8)циклоалкил(С1-С6)алкокси, (С3-С8)циклоалкил(С1-С6)алкокси(С1-С6)алкил, (С1-С6)алкокси(С1-С6)алкил,
С6-С14арил, (С6-С14)арил(С1-С6)алкил, (С6-С14)-арил(С1-С6)алкил(С6-С14)арил, (С6-С14)арилокси, (С6-С14)арилокси(С1-С6)алкил, (С6-С14)арил(С1-С6)алкокси, (С6-С14)арил(С1-С6)алкилокси(С1-С6)алкил, галоид, трифторметил, трифторметокси, циано, карбокси, гидрокси, нитро, амино, (С1-С6)алкоксикарбонил, карбамоил, (С1-С6)алкилтио, (С1-С8)алкилсульфинил, (C1-C8) алкилсульфонил, сульфоамино, (С1-С8)алкилсульфониламино, сульфамоил и (С1-С8)алкилкарбониламино,
при этом две из этих групп могут образовать метилендиоксигруппу или фенильное кольцо, сконденсированное с кольцом, к которому они присоединены, при этом различные арильные группы сами могут быть замещены 1-3 заместителями, выбранными из группы, включающей С1-С8алкил, С1-С8алкокси, галоид, трифторметил, трифторметокси, гидрокси, нитро и амино, а также их сольваты и фармацевтически приемлемые соли.
В качестве примера арильной группы можно назвать фенил, α-нафтил, β-нафтил или флуоренил.
С1-С8алкильные группы могут быть линейными или разветвленными. В качестве примера таких групп можно назвать метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил и пентил.
С1-С8алкоксигруппы также могут быть линейными или разветвленными. В качестве примера таких групп можно назвать метокси, этокси, пропокси, изопропокси, бутокси и изобутокси.
Галоиды могут быть выбраны из фтора, хлора, брома и иода. Гетероарильные группы в качестве значений для R1, R2 и R3 могут представлять собой, в частности, те же группы, что и гетероарильные группы, указанные в качестве значений для Аr.
Изобретение также относится к таутомерным формам и энатиомерам, диастереоизомерам и эпимерам соединений общей формулы (I).
Соединения общей формулы (I) имеют функциональную карбоксигруппу и могут образовывать соли с основаниями.
Примеры солей, образованных соединениями общей формулы (I) с основаниями, включают фармакологически приемлемые соли, такие как натриевые соли, калиевые соли, кальциевые соли и другие соли того же типа.
Соединения общей формулы (I) могут быть переведены в солевую форму путем взаимодействия с аминами с получением фармацевтически приемлемых солей. Так, например, соединения общей формулы (I) могут образовывать соли с глюкаминами, N-метилглюкамином, N, N-диметилглюкамином, этаноламином, морфолином, N-метилморфолином или лизином.
Соединения общей формулы (I) содержат основные атомы азота и могут образовывать моно- или двухосновные соли с неорганическими или органическими кислотами. Примеры солей соединений общей формулы (I) с кислотами включают (но не ограничиваясь только ими) такие фармацевтически приемлемые соли, как гидрохлорид, гидробромид, сульфат, сукцинат, малеат, фумарат, малат или тартрат, и сульфонаты, такие как метансульфонат, бензолсульфонат или толуолсульфонат.
Изобретение также относится к способу получения соединений общей формулы (I). Этот способ по изобретению предусматривает взаимодействие ароматического амина общей формулы (II):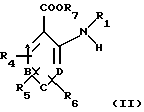
в которой А, В, С, D, R1, R4, R5 и R6 имеют вышеуказанные значения, a R7 означает атом водорода, С1-С6алкильную или бензильную группу, с галогенангидридом галогензамещенной карбоновой ксилоты общей формулы (III):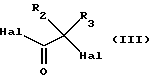
в которой R2 и R3 имеют вышеуказанные значения, a Hal обозначает атом хлора или брома, с получением соединения общей формулы (IV):
в которой А, В, С, D, R1, R2, R3, R4, R5, R6, R7 и Hаl имеют вышеуказанные значения, и взаимодействие соединения общей формулы (IV) с соединением общей формулы (V):
в которой Аr имеет вышеуказанное значение, в присутствии основного агента, такого как триэтиламин, с получением соединения общей формулы (VI):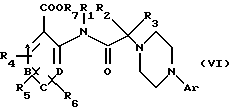
в которой Аr, А, В, С, D, R1, R2, R3, R4, R5, R6 и R7 имеют вышеуказанные значения.
В том случае, если R7 обозначает алкильную группу, соединение общей формулы (VI) может быть подвергнуто гидролизу с использованием обычных кислот или оснований с получением соединения общей формулы (I).
В том случае, если R7 обозначает бензильную группу, соединение общей формулы (VI) может быть подвергнуто гидрогенолизу в присутствии катализатора, такого как палладий на активированном угле, с получением соединения общей формулы (I).
Соединения формул (II) и (V) известны или могут быть получены известными методами.
Так, в частности, соединения формулы (II) описаны в Organic Preparation and Procedures International, 13, 189, 1981.
Соединения формулы (V) могут быть получены по методу, описанному у R. Ratouis и др. (J. Med. Chem., 8, 104, 1965) или у Prelog и др. (Collection Czechoslov. Chem. Communications, 6, 211, 1934).
Например, соединение (VI), в котором R7 обозначает алкильную группу, может быть подвергнуто гидролизу в присутствии основного агента, такого как разбавленный раствор гидроксида натрия.
Энантиомеры соединений формулы (I) могут быть выделены путем последовательной перекристаллизации соли кислоты (I) с оптически активным основанием в растворителях, таких как ацетон, этилацетат или изопропанол, с последующим вытеснением из соли в оптически активную кислоту неорганической или органической кислотой обычным методом.
Благодаря их гипогликемическому действию и нетоксичности в активных дозах соединения по изобретению могут применяться при лечении диабета, в частности инсулиннезависимого диабета.
Таким образом, еще одним объектом настоящего изобретения являются фармацевтические композиции, содержащие эффективное количество соединения по изобретению.
Фармацевтические композиции по изобретению могут быть представлены в форме, предназначенной для парентерального, орального, ректального, чрескожного введения и введения через слизистую.
Они могут быть представлены в форме инъецируемых растворов или суспензий либо в форме лекарственных препаратов в соответствующих емкостях, содержащих несколько доз лекарственного средства, в форме таблеток без покрытия или с покрытием, таблеток с сахарным покрытием, капсул, в том числе твердых желатиновых капсул, пилюль, крахмальных облаток, порошков, суппозиториев или ректальных капсул, растворов или суспензий для чрескожного применения в полярном растворителе или в форме средств для применения через слизистую.
Приемлемыми наполнителями для таких применений являются производные целлюлозы или микрокристаллической целлюлозы, карбонаты щелочно-земельных металлов, фосфат магния, крахмалы, модифицированные крахмалы или лактоза для твердых форм.
Предпочтительными наполнителями для ректального введения являются масло какао или стеараты полиэтиленгликоля.
Наиболее предпочтительными наполнителями для парентерального введения являются вода, водные растворы, физиологический раствор или изотонические растворы.
Доза может варьироваться в широких пределах в зависимости от терапевтических показаний и пути введения лекарственного средства, а также от возраста и веса пациента.
В приведенных ниже примерах проиллюстрировано получение соединений формулы (I) и промежуточных соединений формул (II) и (IV).
А. Пример получения соединения формулы (II)
Получение метил-2-циклогексилметиламино-5-метоксибензоата
17,6 г метил-5-метоксиантранилата, 11,8 мл циклогексанкарбоксальдегида и 2 г 10%-ного палладия на активированном угле (50% воды) вводят в 200 мл метанола в 1-литровом аппарате гидрогенизации. В этом аппарате создают атмосферу азота и его содержимое перемешивают при комнатной температуре в течение 3 ч. После этого добавляют 300 мл дихлорметана, палладий на активированном угле отделяют путем фильтрации и полученный фильтрат концентрируют под вакуумом. Полученное масло кристаллизуют из смеси этанола (200 мл) и воды (50 мл), получая 25,4 г желтого твердого вещества с tпл 58-60oС.
ИК (КВг): 1683 см-1 (С=O), 1528-1 (С=O).
1H-ЯМР (СDСl3, 200 МГц): δ част./млн 1,06-1,64 (1Н, m, циклогексил), 2,93 (2Н, t, СН2), 3,68 (3Н, s, ОСН3), 3,78 (3Н, s, ОСН3), 6,56 (1Н, d, протон фенила), 6,96 (1Н, dd, протон фенила), 7,34 (2Н, d+s, протон фенила + NH).
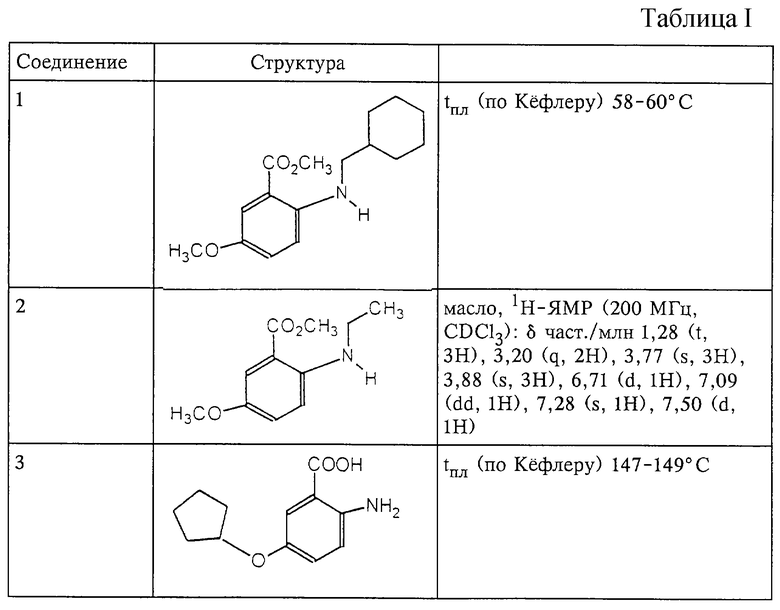
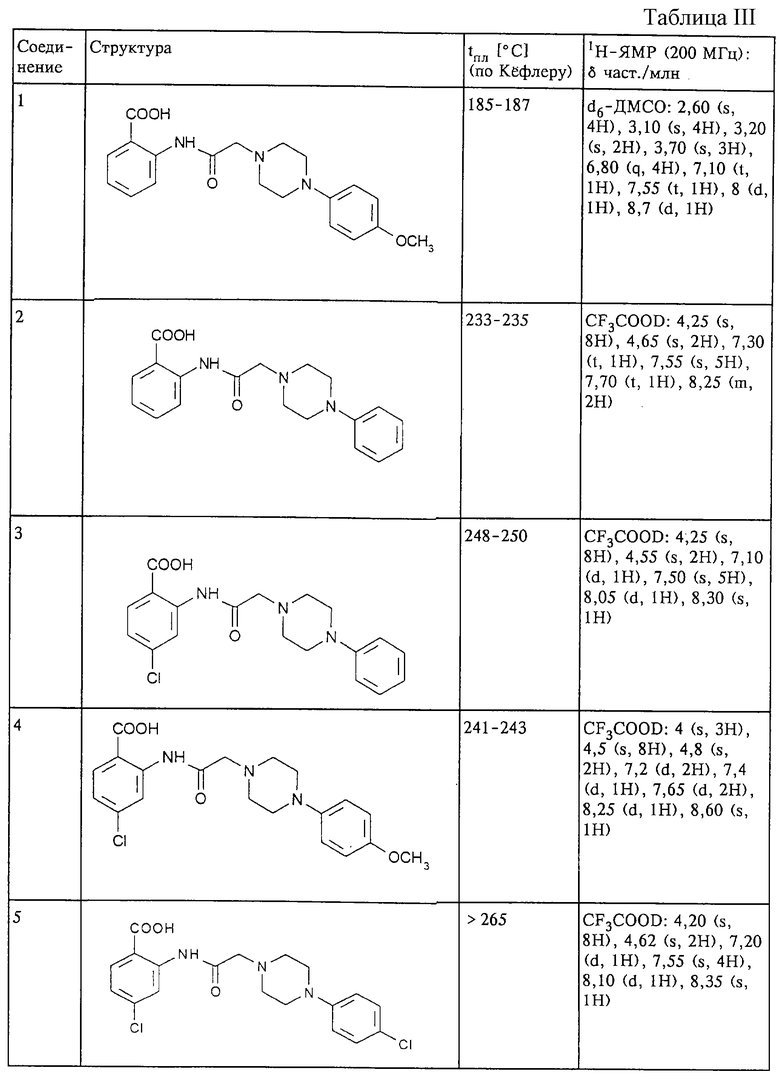
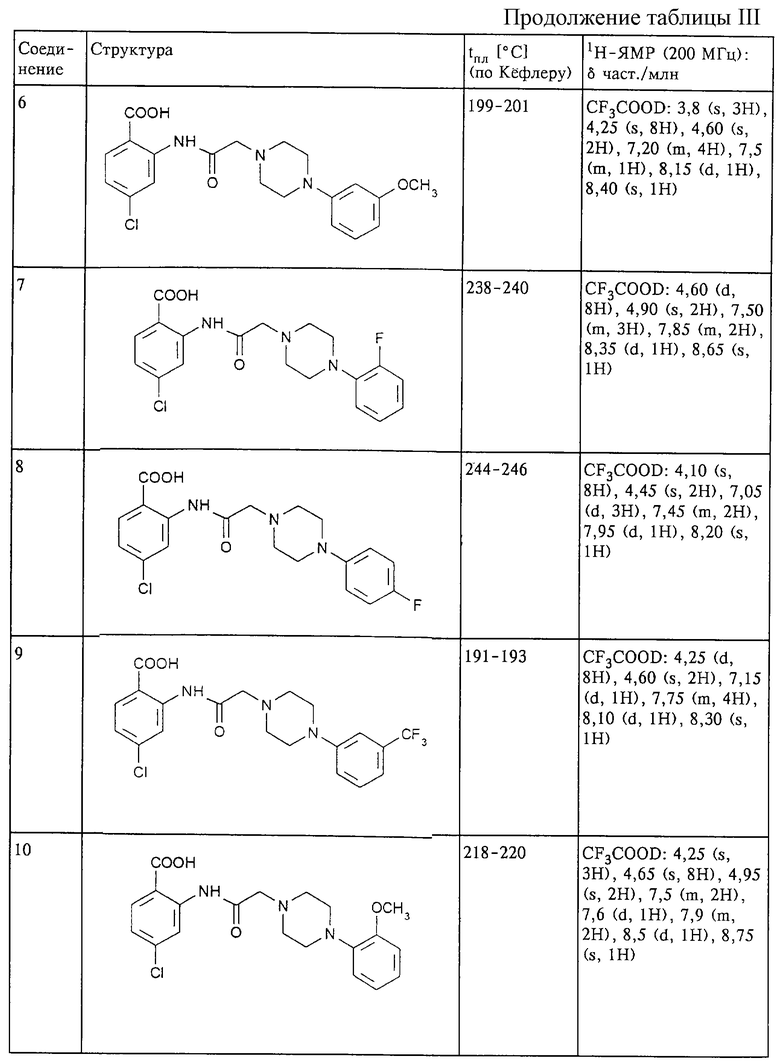
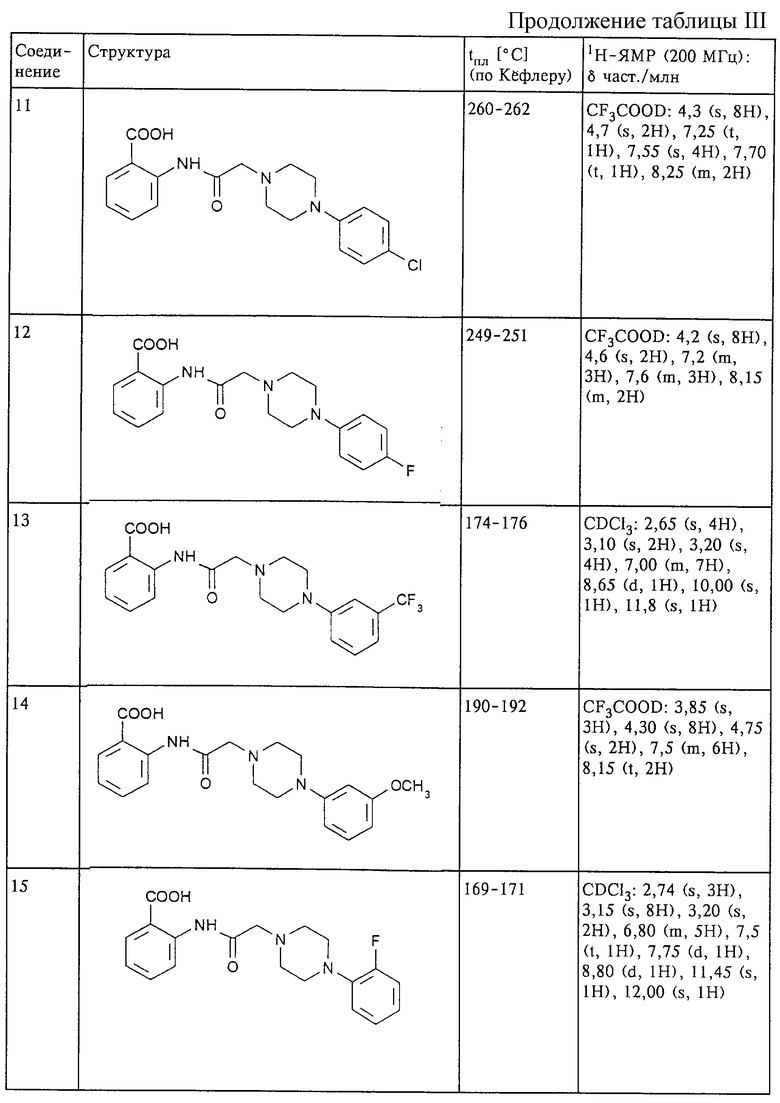
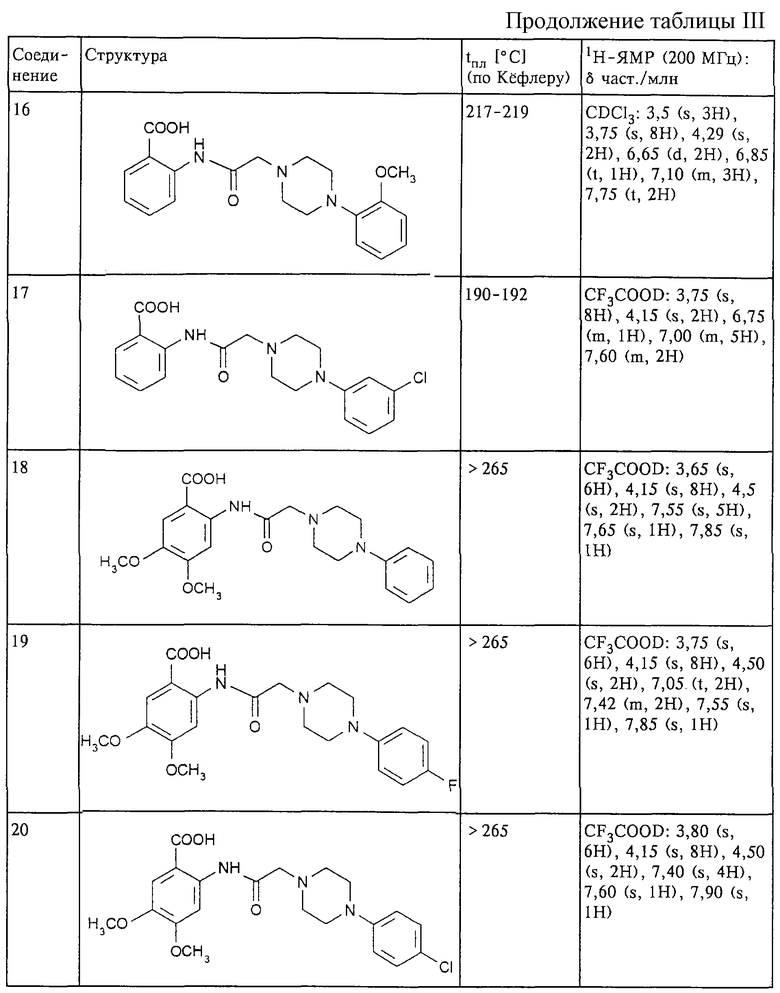
Структура и характеристики соединений формулы (II) приведены в таблице I.
Б. Пример получения соединения формулы (IV)
Получение 4-хлор-2-(хлорацетамидо)бензойной кислоты
25,5 мл хлорацетилхлорида добавляют по каплям при перемешивании к 50 г 2-амино-4-хлорбензойной кислоты в 600 мл диоксана, поддерживая температуру реакционной смеси на уровне 20oС. Перемешивание продолжают в течение 2 ч при комнатной температуре и после этого добавляют 1200 мл воды. Целевой продукт осаждают, смесь перемешивают в течение 1 ч, фильтруют и затем полученное твердое вещество промывают водой. После сушки получают 60,7 г 4-хлор-2-(хлорацетамидо)бензойной кислоты с tпл 194-196oС.
ИК: 1676 см-1 (С=O).
1H-ЯМР (d6-ДМСО, 200 МГц): δ част./млн 4,30 (2Н, s, СН2), 7,1 (1Н, d, протон фенила), 7,7 (1Н, d, протон фенила), 8,5 (1Н, s, протон фенила), 11,75 (1Н, s, NH), 13,90 (1Н, широкий s, COOH).
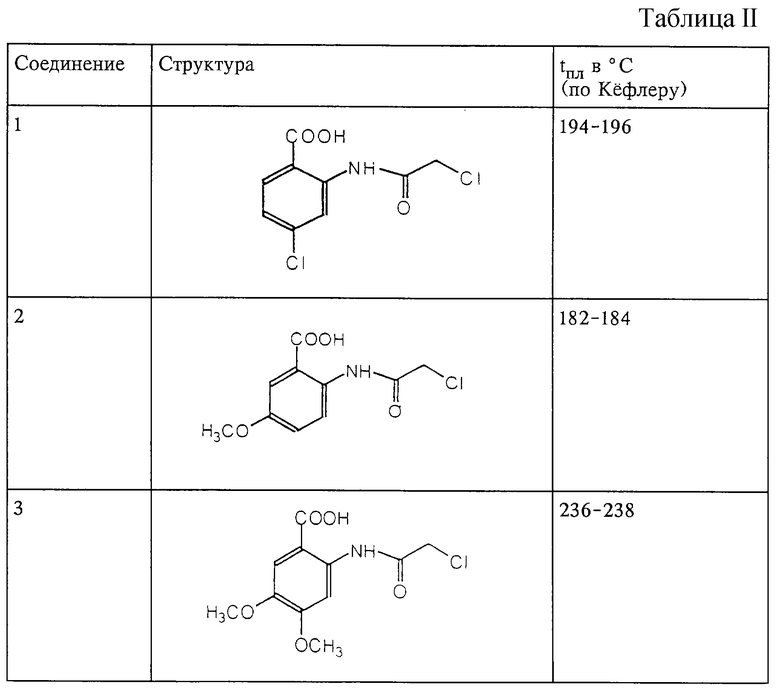
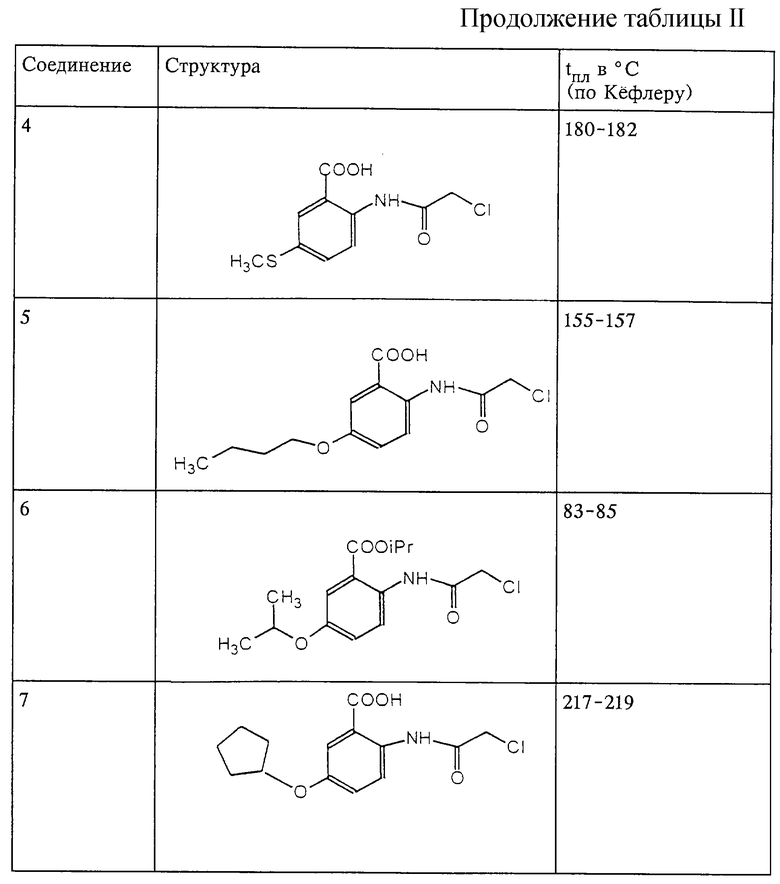
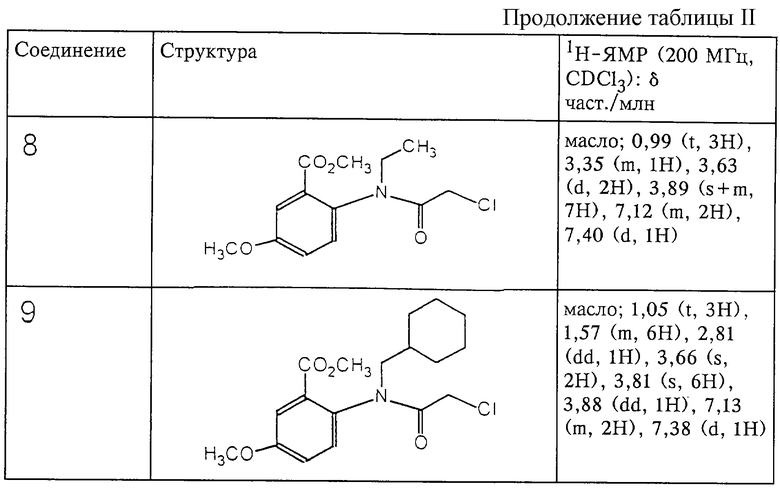
Структура и характеристики соединений формулы (IV) приведены в таблице II.
В. Пример получения соединения формулы (II)
Получение 4-хлор-2- {[4-(2-метоксифенил)-1-пипразинил]-ацетамидо} бензойной кислоты
15 г 4-хлор-2-(хлорацетамидо) бензойной кислоты добавляют при перемешивании и комнатной температуре к 11,6 г 1-(2-метоксифенил)пиперазина и 17 мл триэтиламина в 120 мл диметилформамида (ДМФ). Реакционную смесь продолжают перемешивать в течение 48 ч при комнатной температуре и затем в нее добавляют 500 мл воды. Экстрагируют дихлорметаном (3 раза порциями по 300 мл). Растворитель выпаривают под вакуумом и полученное твердое вещество снова растворяют в 300 мл 2н. водного раствора гидроксида натрия. Раствор промывают диэтиловым эфиром (3 раза порциями по 300 мл) и водную фазу затем подкисляют уксусной кислотой. Твердое вещество кристаллизуют и после фильтрации получают 22,5 г сырого продукта. После перекристаллизации из диоксана получают 21,1 г 4-хлор-2-{ [4-(2-метоксифенил)-1-пиперазинил] ацетамидо} бензойной кислоты в виде белого твердого вещества с tпл 218-220oС.
ИК: 1699 см-1 (С=O), 1673 см-1 (С=O).
1H-ЯМР (СF3СООВ): δ част./млн 4,25 (3Н, s, ОСН3), 4,65 (8Н, (широкий) s, 4 СН2), 4,95 (2Н, s, СН2), 7,5 (2Н, m, протоны фенила), 7,6 (1Н, d, протон фенила), 7,90 (2Н, m, протоны фенила), 8,50 (1Н, d, протон фенила), 8,75 (1Н, s, протон фенила).
Г. Другой вариант получения соединения формулы (I)
Получение 2- {[4-(4-фторфенил)-1-пиперазинил]ацетамидо} -4,5-(метилендиокси) бензойной кислоты
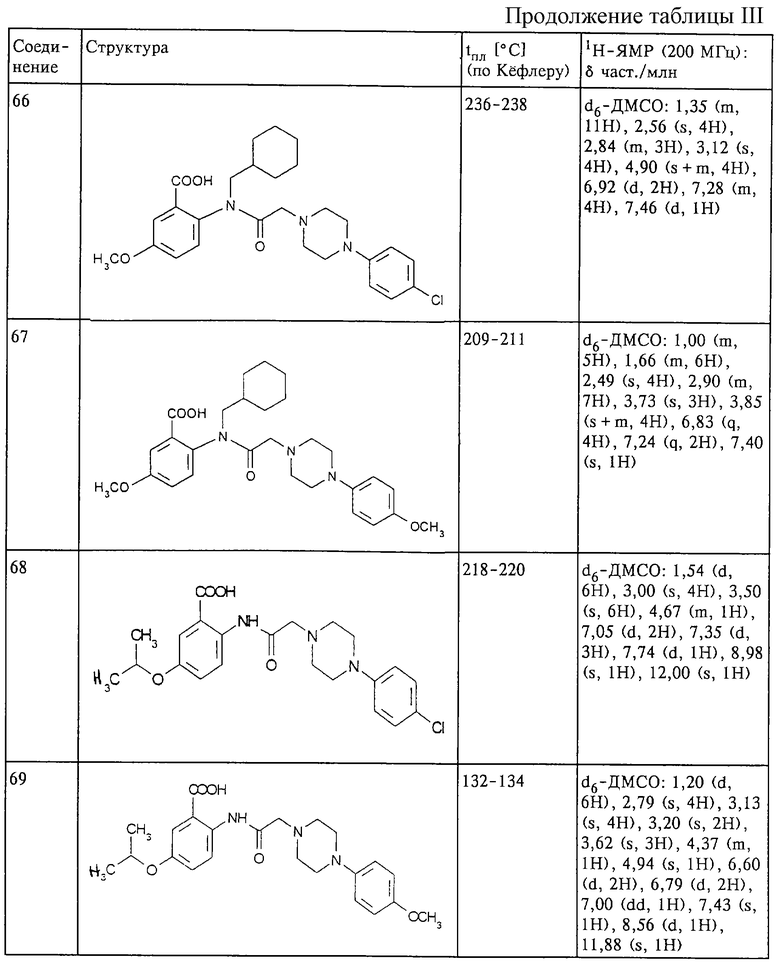
15 г 2-(хлорацетамидо)-4,5-(метилендиокси)бензойной кислоты добавляют при перемешивании и комнатной температуре к 10,5 г 1-(4- фторфенил)пиперазина и 16,2 мл триэтиламина в 150 мл ДМФ. Реакционную смесь продолжают перемешивать в течение 48 ч при комнатной температуре. Добавляют 3,5 мл уксусной кислоты и медленно добавляют 150 мл воды. Кислота кристаллизуется, и ее разбавляют 300 мл воды. Смесь перемешивают в течение 30 мин, фильтруют и полученное твердое вещество промывают водой. После перекристаллизации из смеси диоксана с ДМФ получают 14,9 г 2- {[4-(4-фторфенил) -1-пипразинил] ацетамидо} -4,5- (метилендиокси) бензойной кислоты с tпл 254-256oС.
ИК (КВг): 1654 см-1 (С=O).
1H-ЯМР (СF3СООD, 200 МГц): δ част./млн 4,40 (8Н, s, пиперазинил), 4,67 (2Н, s, СН2), 6,05 (2Н, s, О-СН2-О), 7,30 (2Н, t, протон фенила), 7,65 (3Н, m, протон фенила), 7,90 (1Н, s, протон фенила).
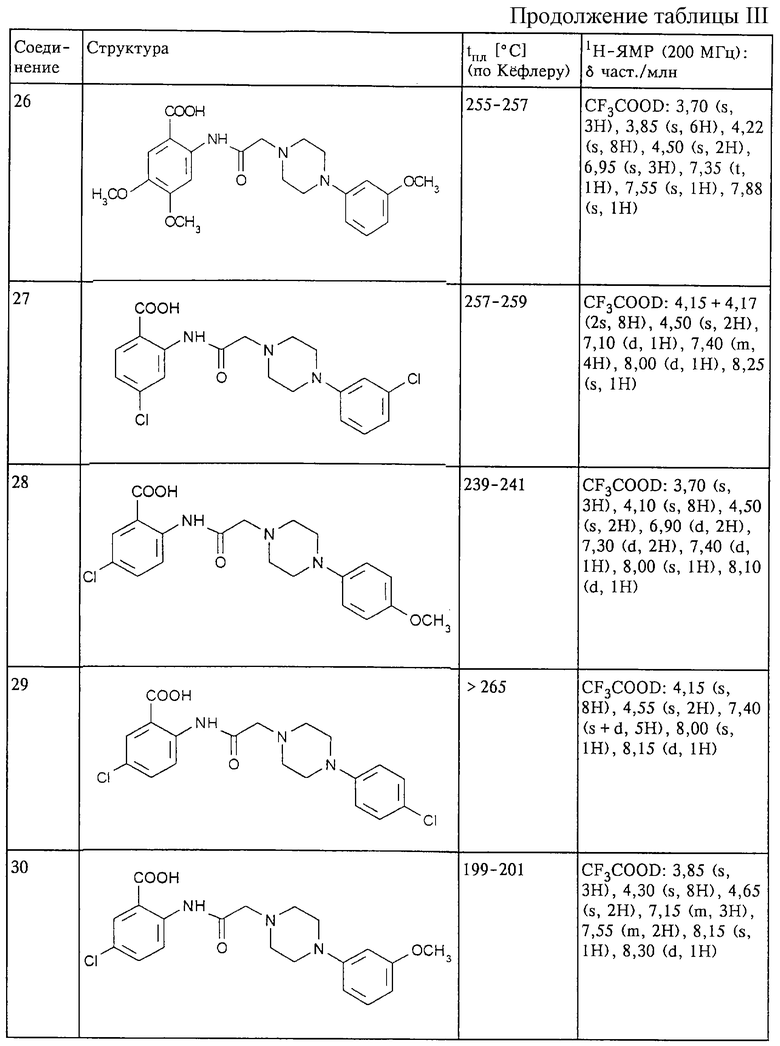
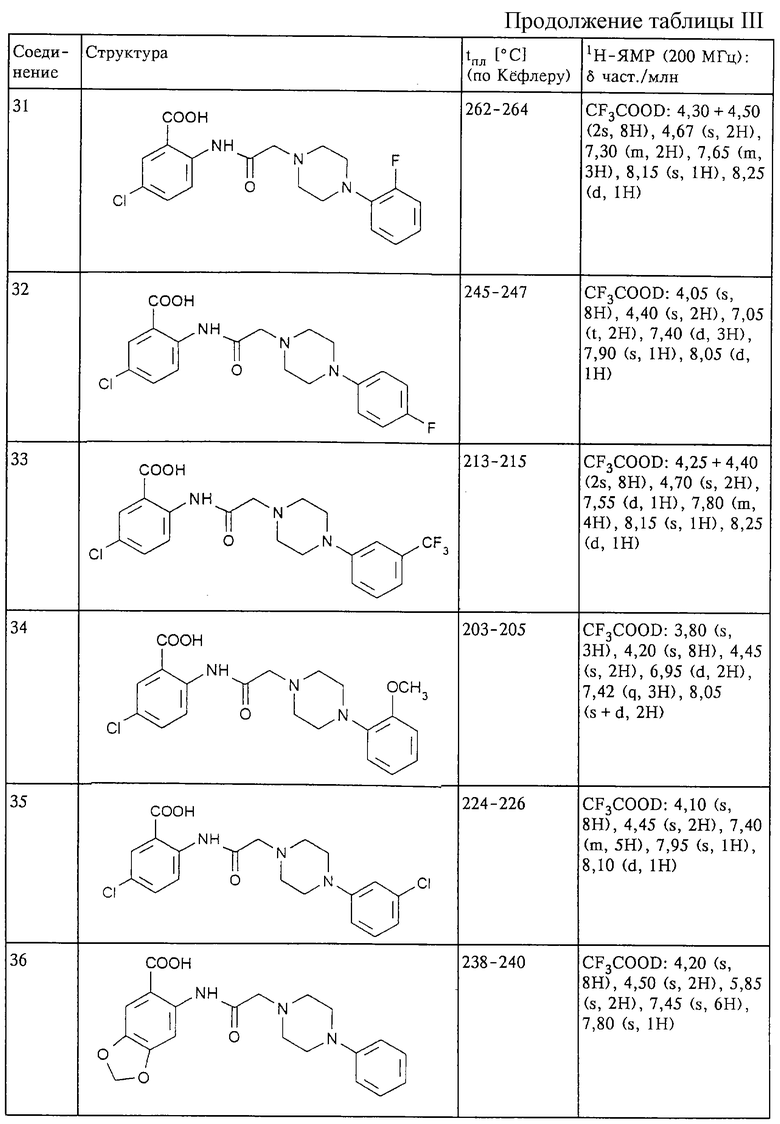
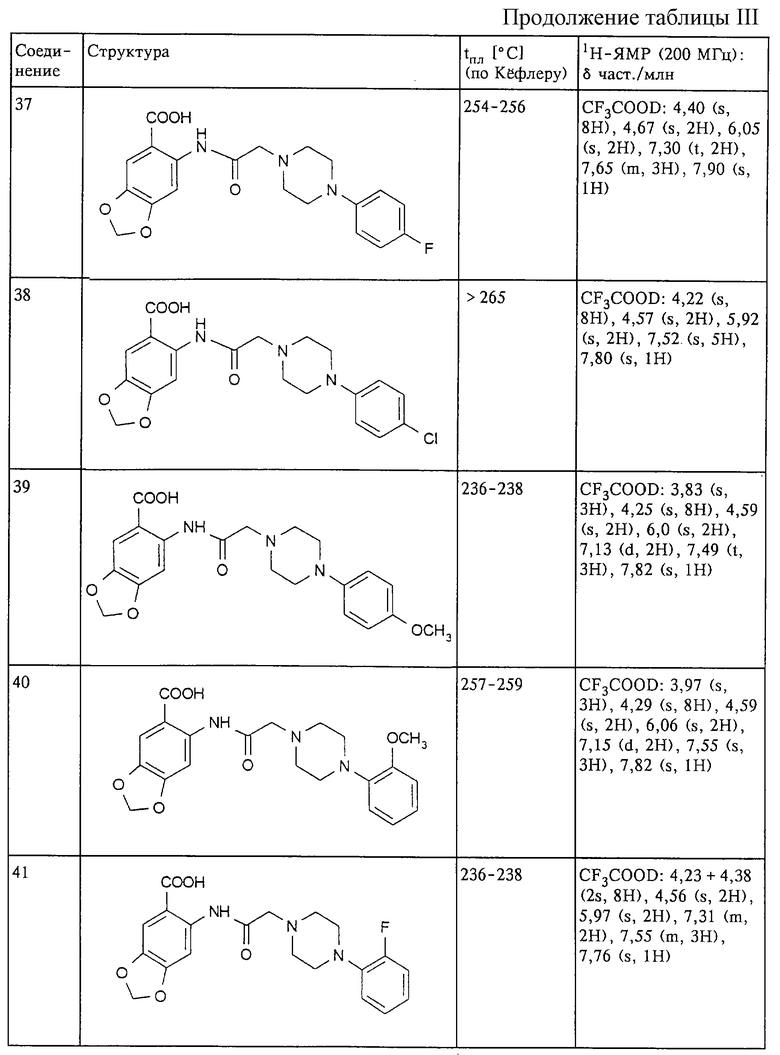
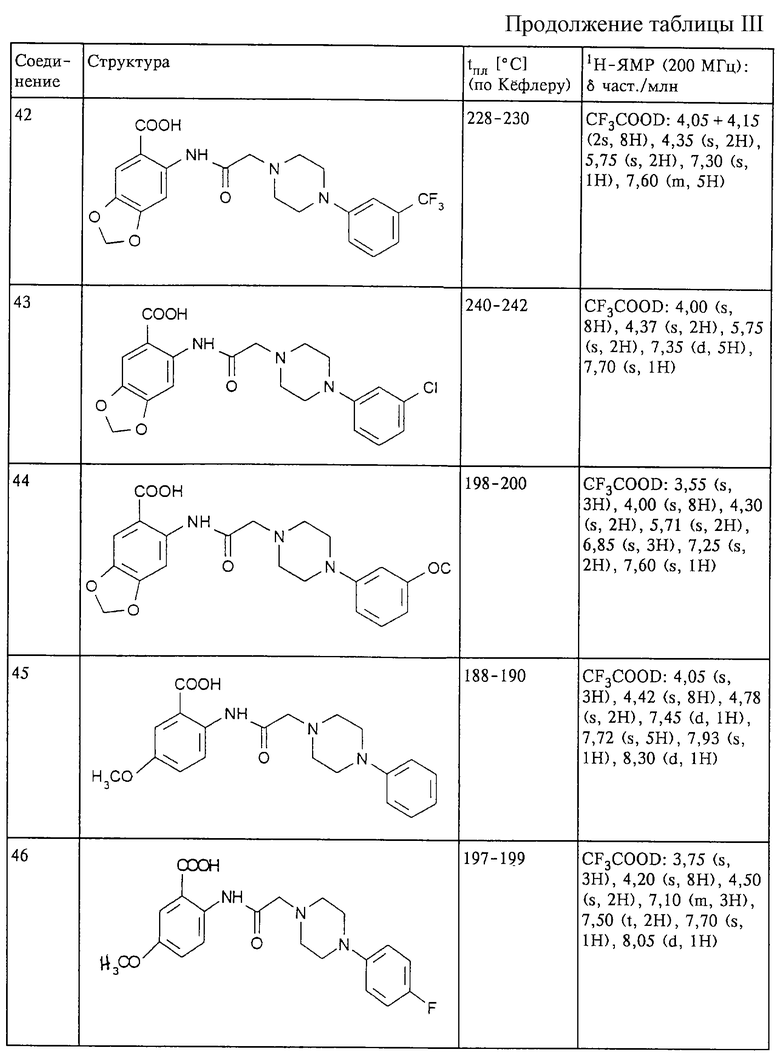
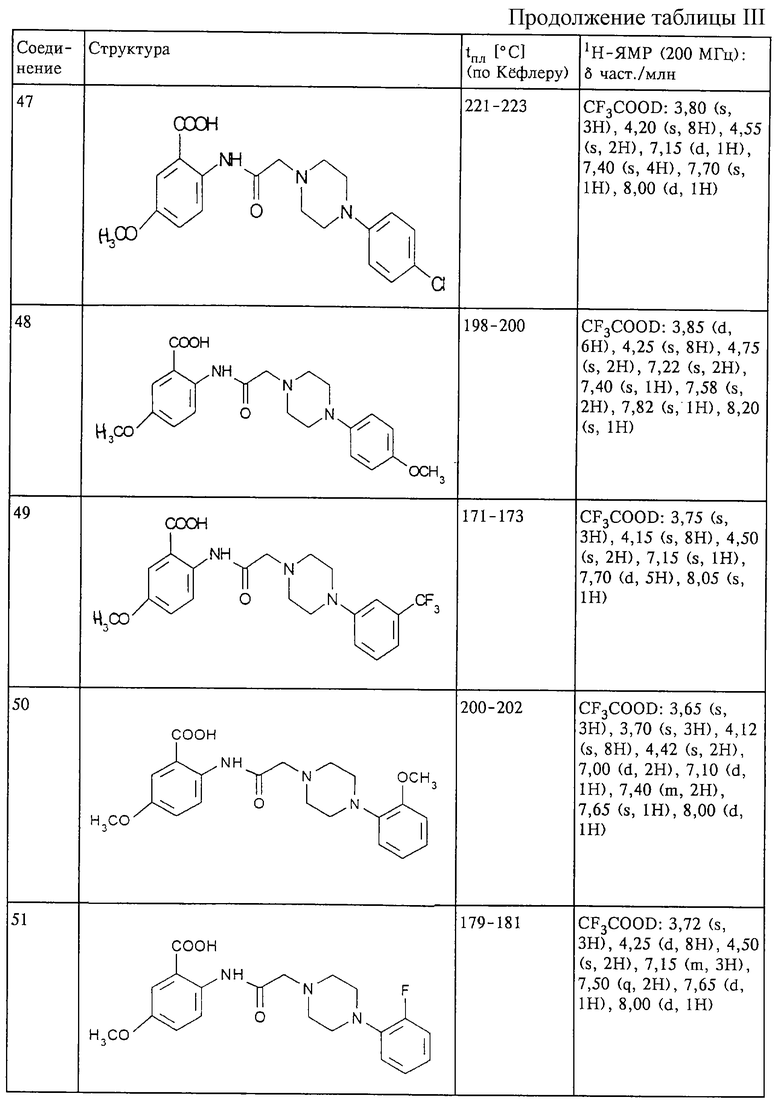
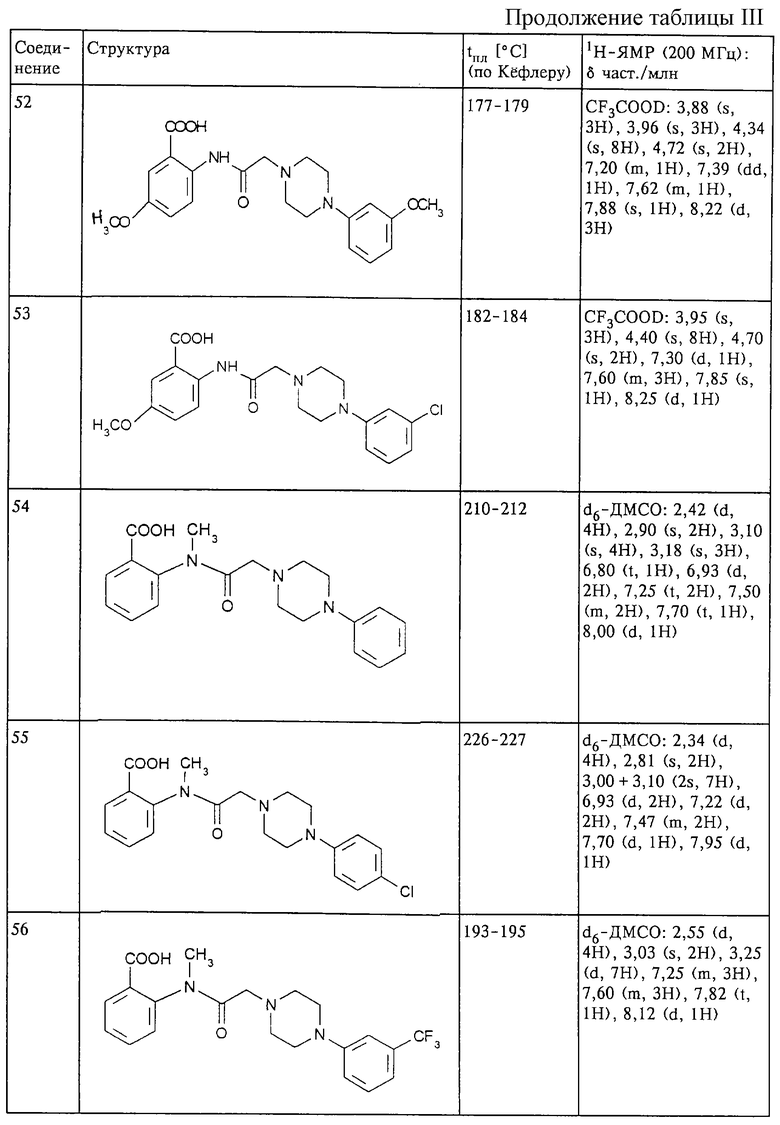
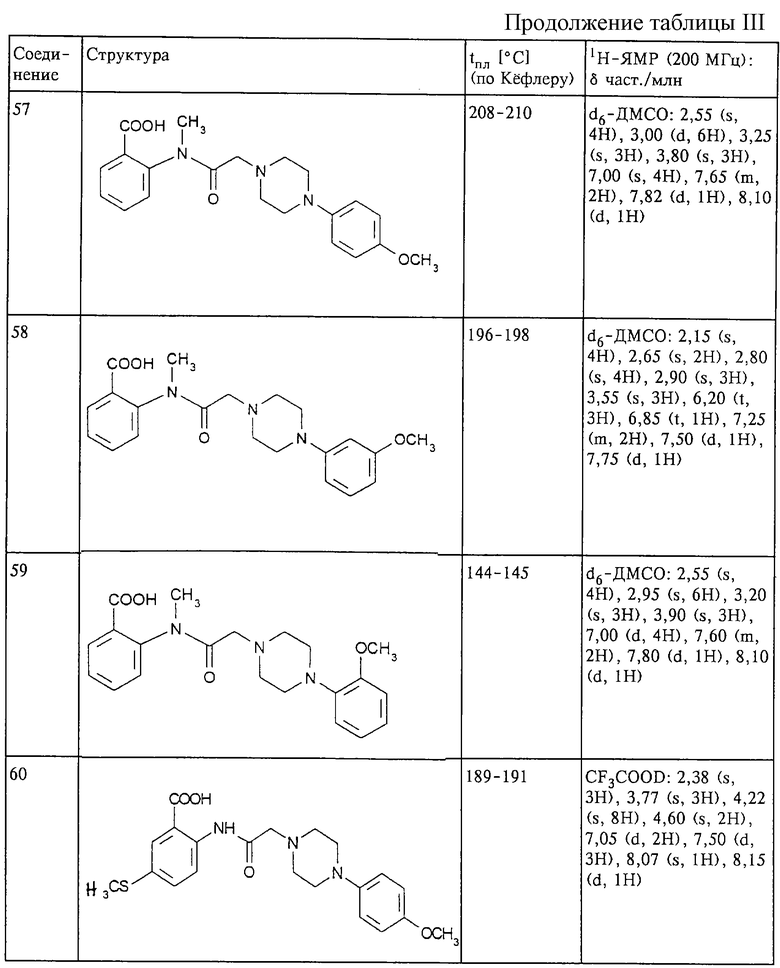
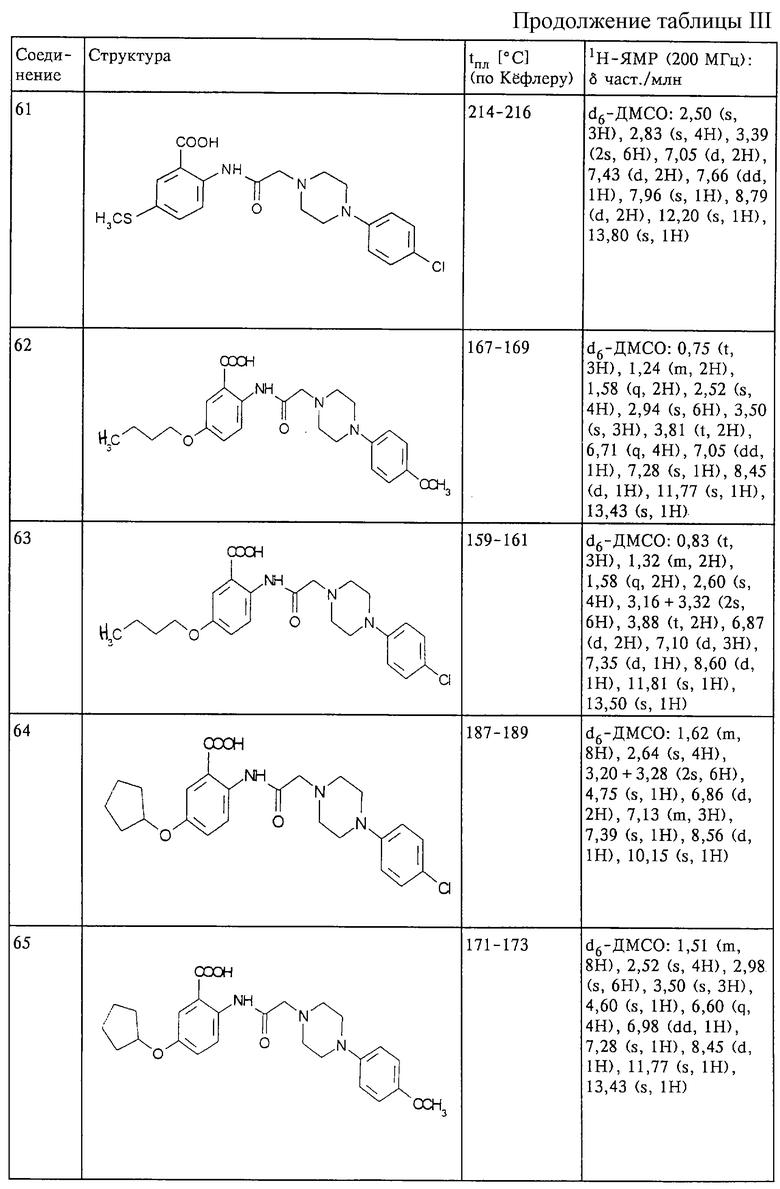
Структура и характеристики соединений формулы (I) приведены в таблице III.
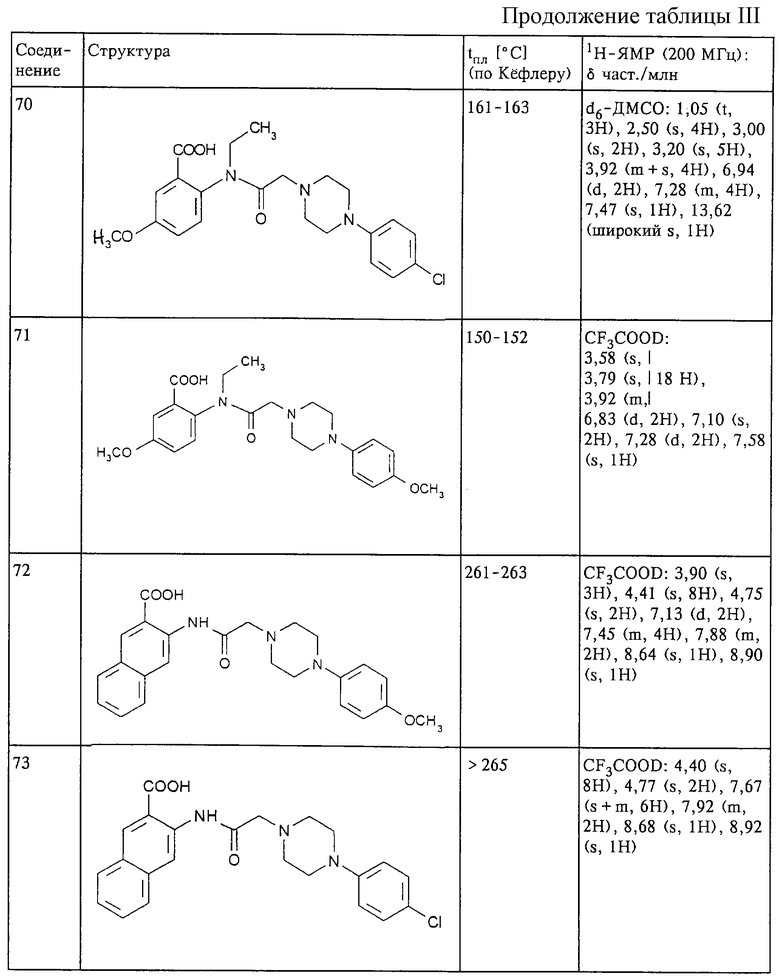
Ниже представлены результаты фармакологических исследований. Исследование антидиабетической активности на крысах линии NOSTZ
Анитидиабетическую активность соединений формулы (I) при оральном введении определяли на экспериментальной модели инсулиннезависимого диабета, вызванного у крысы стрептозотоцином.
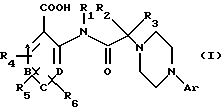
Инсулиннезависимый диабет моделируют на крысах путем инъекции стрептозотоцина новорожденным (в день появления на свет).
Возраст больных диабетом крыс, на которых проводились исследования, составлял восемь недель. Начиная со дня их появления на свет и до дня эксперимента животных содержали в виварии при температуре в пределах от 21 до 22oС и при фиксированном цикле день/ночь (продолжительность световой фазы с 7 до 19 ч; продолжительность темновой фазы с 19 до 7 ч). Животных содержали на поддерживающей диете, при этом вода и корм предоставлялись по желанию, но за 2 ч перед опытом корм убирали (постабсорбционное состояние).
Крысам орально вводили в течение дня тестируемый продукт. Через два часа после последнего введения и по истечении 30 мин после анастезии животных пентабарбиталом натрия (нембутал) из кончика хвоста брали образцы крови в количестве 300 мкл.
Полученные основные результаты представлены в таблице IV. Эти результаты свидетельствуют о том, что соединения формулы (I) эффективно снижают гликемию у животных, больных диабетом.
Приведенные результаты выражены в процентах изменения гликемии в день Д4 (4 дня лечения) в сравнении с первым днем Д0 (до лечения).


Изобретение относится к новым α-(1-пиперазинил)ацетамидопроизводным аренкарбоновой кислоты общей формулы I, в которой Ar выбран из моно-, би- или трициклической арильной группы, имеющей от 6 до 14 атомов углерода, возможно замещенный С1-С6 алкокси, галоидом или CF3; R1, R2, R3 независимо выбраны из Н, С1-С8 алкила, С3-С8 циклоалкил (С1-С6) алкила; А, В,С и D представляют= СН-; R4, R5 и R6 независимо Н, С1-8 алкокси, С3-С8 циклоалкокси, галоид, С1-С6 алкилтио или два из этих радикалов могут образовать фенил, сконденсированный с кольцом, к которому они присоединены; или их сольфатам, фармацевтически приемлемым солям. Соединения I пригодны для лечения инсулиннезависимого диабета. 6 с. и 1 з.п. ф-лы, 4 табл.
в которой Аr выбран из моно-, би- или трициклической арильной группы, имеющей от 6 до 14 атомов углерода, необязательно имеющей от одного до трех заместителей, выбранных из группы, включающей (С1-С6)алкокси, галоид и трифторметил;
R1, R2 и R3 независимо друг от друга выбраны из группы, включающей атом водорода, (С1-С8)алкил и (С3-С8)циклоалкил(С1-С6)алкил;
А, В, С и D представляют собой группу =СН-;
R4, R5 и R6 независимо друг от друга выбраны из группы, включающей атом водорода, (C1-C8)aлкокси, (С3-С8)циклоалкокси, галоид и (C1-С6)алкилтио, при этом две из этих групп могут образовать фенильное кольцо, сконденсированное с кольцом, к которому они присоединены,
а также их сольваты и фармацевтически приемлемые соли.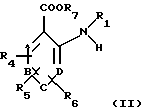
в которой А, В, С, D, R1, R4, R5 и R6 имеют вышеуказанные значения;
R7 выбран из группы, включающей атом водорода, С1-С6алкил и бензил, с галоидангидридом галогензамещенной карбоновой кислоты формулы (III)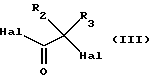
в которой R2 и R8 имеют вышеуказанные значения;
Hal обозначает хлор или бром,
с получением соединения формулы (IV)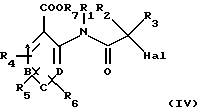
в которой А, В, С, D, R1, R2, R3, R4, R5, R6, R7 и Hаl имеют вышеуказанные значения,
и взаимодействие соединения формулы (IV) с соединением формулы (V)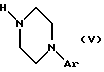
в которой Аr имеет вышеуказанное значение,
в присутствии основного агента с получением соединения формулы (VI)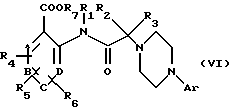
в которой Аr, А, В, С, D, R1, R2, R3, R4, R5, R6 и R7 имеют вышеуказанные значения,
и в случае, если R7 обозначает алкил, гидролиз этого соединения с получением соединения формулы (I), а в случае, если R7 обозначает бензил, гидрогенолиз этого соединения с получением соединения формулы (I).
| Способ обработки заполнителя | 1977 |
|
SU638568A1 |
| Спутник-конструктор - учебно-демонстрационная модель | 2017 |
|
RU2693722C2 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| ТРИНУС Ф.П | |||
| Фармакотерапевтический справочник | |||
| - Киев: Здоровья, 1989, с.261. | |||

