Область изобретения
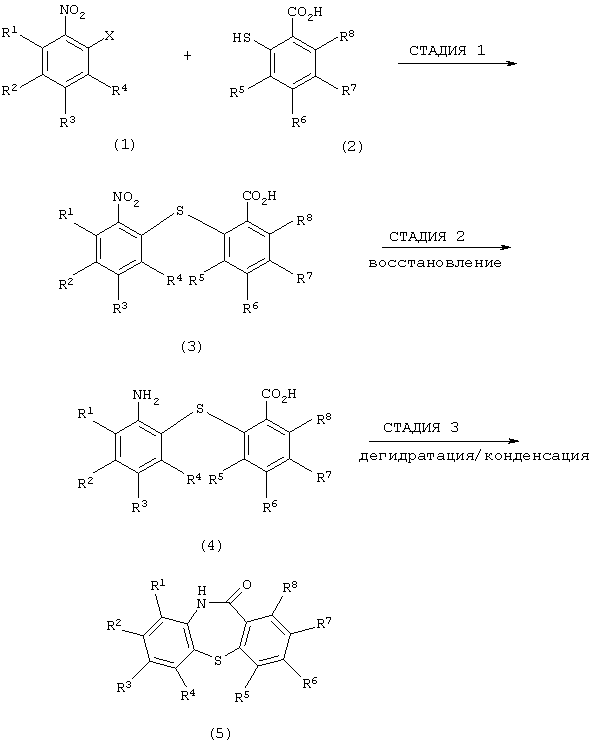
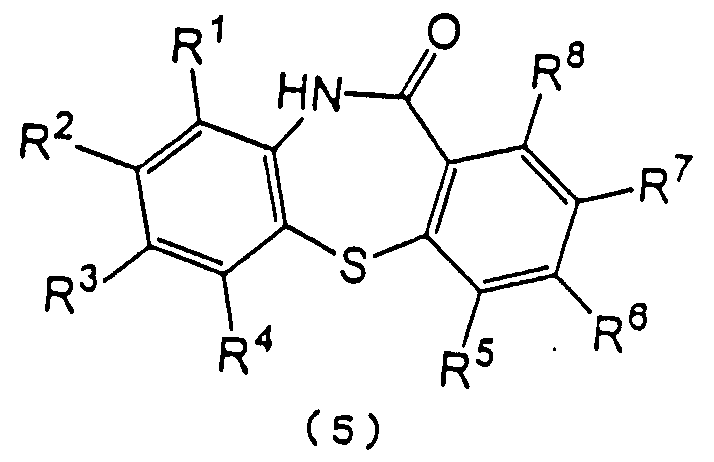
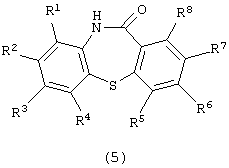
Настоящее изобретение относится к способу получения производного дибензотиазепина, представляющего интерес в качестве промежуточного соединения для получения фармацевтических препаратов. В частности, изобретение относится к способу получения производного дибензотиазепина следующей формулы (5):
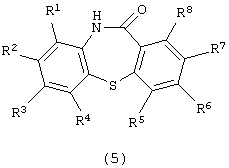
(в которой R1, R2, R3, R4, R5, R6, R7 и R8 являются одинаковыми или отличаются друг от друга и представляют атом водорода, алкильную группу, алкоксигруппу, алкилкарбонильную группу, арильную группу, арилоксигруппу или арилкарбонильную группу, причем каждая группа необязательно замещена), которое представляет интерес в качестве промежуточного соединения для получения 11-[4-(2-(2-гидроксиэтокси)этил]-1-пиперазинилдибензотиазепина и его производных, которые, как известно, являются эффективными в качестве антипсихотических фармацевтических препаратов.
Уровень техники
В заявке ЕР 0282236-А1 описано, что производные дибензотиазепина вышеприведенной формулы (5) могут быть переработаны для получения производного 11-[4-(2-(2-гидроксиэтокси)этил]-1-пиперазинилдибензотиазепина, которое представляет интерес в качестве антипсихотического фармацевтического препарата. Более подробно, дибензо[b,f] [1,4]тиазепин-11-он, который является представительным соединением производных дибензотиазепина формулы (5), вводят в реакцию с оксихлоридом фосфора для получения производного 11-хлордибензотиазепина; и к производному 11-хлордибензотиазепина добавляют пиперазин для получения производного 11-пиперазинилдибензотиазепина, которое затем реагирует с 2-хлорэтоксиэтанолом в щелочных условиях, образуя целевой 11-[4-(2-(2-гидроксиэтокси)этил]-1-пиперазинилдибензотиазепин.
Кроме того, в ЕР 0282236-А1 описано, что дибензо[b,f]-тиазепин-11-он получают из фенил-2-(фенилтио)фенилкарбамата или его аналогов циклизацией в присутствии полифосфорной кислоты.
В Helv. Chim. Acta, vol.42, pp.1263 (1959) описано, что производное дибензотиазепина может быть получено стадиями, включающими нагревание производного метилтиосалицилата с 2-галогенированным производным нитробензола в присутствии натрия для получения 2-нитро-2’-карбоксидифенилсульфидного производного, которое затем восстанавливают, используя катализатор никель Ренея, для получения 2-амино-2’-карбоксидифенилсульфидного производного, которое, наконец, нагревают для получения производного дибензотиазепина.
В Org. Prep. Proced. Int., pp. 287 (1974) описано, что производное дибензотиазепина может быть получено стадиями, включающими нагревание производного эфира тиосалициловой кислоты и производного 2-йоднитробензола в присутствии метилата натрия и меди, обработку полученного соединения последовательно щелочным раствором и кислотным раствором для получения 2-нитро-2’-карбоксидифенилсульфидного производного, восстановление производного сульфатом трехвалентного железа в водном растворе аммиака для получения 2-амино-2’-карбоксидифенилсульфидного производного и нагревание полученного производного при пониженном давлении.
В заявке WO 92/19607 описано, что производное дибензотиазепина формулы (5) может быть получено стадиями реакции 2-аминотиофенола с 2-фторбензонитрилом для получения 2-(2-аминофенилтио)бензонитрила, гидролиза полученного соединения для получения 2-(2-карбоксифенилтио)анилина и, наконец, циклизации анилинового производного.
Как описано выше, известны различные способы получения производного дибензотиазепина формулы (5). Однако известные способы имеют различные недостатки, такие как низкий выход, высокотемпературные условия реакции, использование труднодоступных исходных соединений и/или осложненная последующая переработка. Эти недостатки, вполне естественно, неблагоприятны для промышленного получения целевого производного дибензотиазепина.
Описание изобретения
Целью настоящего изобретения является способ промышленного получения производного дибензотиазепина формулы (5), т.е. способ получения производного дибензотиазепина с хорошим выходом без сложной последующей переработки с применением легкодоступного сырья.
В результате серьезных исследований найден новый способ получения производного дибензотиазепина формулы (5) с хорошим выходом, простым осуществлением с применением легкодоступного производного нитробензола, а также легкодоступного производного тиосалициловой кислоты.
Изобретение представляет способ получения производного дибензотиазепина следующей формулы (5):
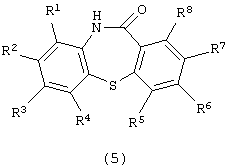
в которой каждый из R1, R2, R3, R4, R5, R6, R7 и R8 независимо представляет атом водорода, алкильную группу, алкоксигруппу, алкилкарбонильную группу, арильную группу, арилоксигруппу или арилкарбонильную группу, причем каждая группа необязательно замещена, который включает стадии:
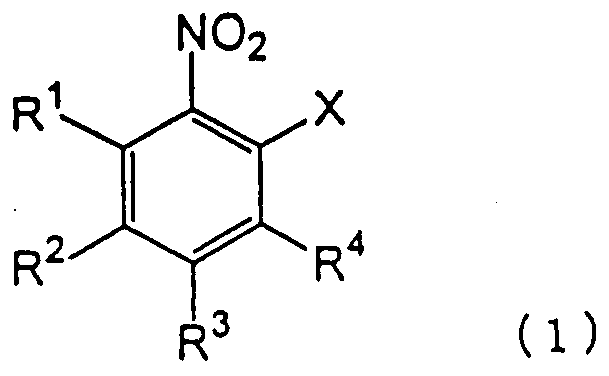
взаимодействия производного нитробензола следующей формулы (1):
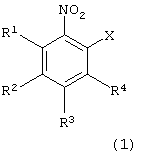
в которой каждый из R1, R2, R3 и R4 имеет указанное выше значение, и Х представляет атом галогена, с производным тиосалициловой кислоты следующей формулы (2):
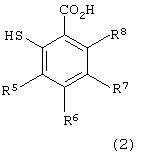
в которой каждый из R5, R6, R7 и R8 имеет указанное выше значение,
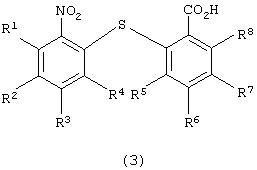
для получения 2-нитро-2’-карбоксидифенилсульфидного производного следующей формулы (3):
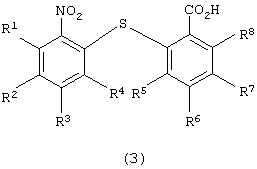
в которой каждый из R1, R2, R3, R4, R5, R6, R7 и R8 имеет значения, указанные выше;
восстановления полученного 2-нитро-2’-карбоксидифенилсульфидного производного для получения 2-амино-2’-карбоксидифенилсульфидного производного следующей формулы (4):
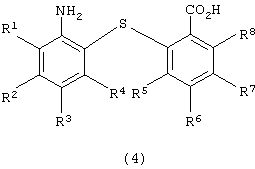
в которой каждый из R1, R2, R3, R4, R5, R6, R7 и R8 имеет значения, указанные выше;
и проведения реакции дегидратации-конденсации полученного 2-амино-2’-карбоксидифенилсульфидного производного.
Далее изобретение представляет способ получения производного дибензотиазепина формулы (5):
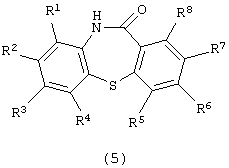
в которой каждый из R1, R2, R3, R4, R5, R6, R7 и R8 имеет значения, указанные выше,
который включает стадии:
восстановления 2-нитро-2'-карбоксидифенилсульфидного производного следующей формулы (3):
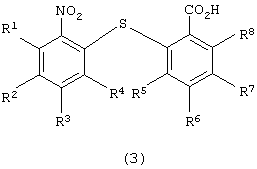
в которой каждый из R1, R2, R3, R4, R5, R6, R7 и R8 независимо представляет атом водорода, алкильную группу, алкоксигруппу, алкилкарбонильную группу, арильную группу, арилоксигруппу или арилкарбонильную группу, причем каждая группа необязательно замещена,
для получения 2-амино-2’-карбоксидифенилсульфидного производного следующей формулы (4):
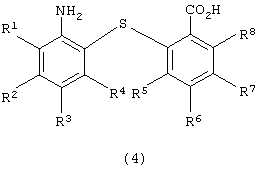
в которой каждый из R1, R2, R3, R4, R5, R6, R7 и R8 имеет значения, указанные выше;
и проведения реакции дегидратации-конденсации полученного 2-амино-2’-карбоксидифенилсульфидного производного.
Настоящее изобретение далее представляет 2-нитро-2'-карбоксидифенилсульфидное производное формулы (3).
Стадии способа получения производного дибензотиазепинового производного формулы (5) по изобретению проиллюстрирована следующей схемой:
Предпочтительное осуществление изобретения
В формулах соединений, вовлеченных в способ по изобретению, "алкильная группа, возможно имеющая заместитель", представленная символами от R1 до R8, означает алкильную группу с прямой или разветвленной цепью с числом атомов углерода от 1 до 10, не имеющую заместителей, или алкильную группу с прямой или разветвленной цепью с числом атомов углерода от 1 до 10, имеющую заместитель.
Вышеуказанная "алкильная группа с прямой или разветвленной цепью с числом атомов углерода от 1 до 10, не имеющая заместителей", предпочтительно представляет алкильную группу с прямой или разветвленной цепью с числом атомов углерода от 1 до 8, более предпочтительно от 1 до 5 атомов углерода. Примеры алкильных групп включают метил, этил, пропил (включая изомеры), бутил (включая изомеры), пентил (включая изомеры), гексил (включая изомеры), гептил (включая изомеры), нонил (включая изомеры) и децил (включая изомеры). Предпочтительными являются метил, этил, пропил (включая изомеры), бутил (включая изомеры), пентил (включая изомеры), гексил (включая изомеры) и октил (включая изомеры). Наиболее предпочтительными являются метил, этил, пропил (включая изомеры), бутил (включая изомеры) и пентил (включая изомеры).
Примеры алкильной части вышеуказанной "алкильной группы с прямой или разветвленной цепью с числом атомов углерода от 1 до 10, имеющей заместитель", включают алкильные группы, описанные в вышеприведенной формуле (1).
Заместитель вышеупомянутой "алкильной группы с прямой или разветвленной цепью с числом атомов углерода от 1 до 10, имеющей заместитель", может быть присоединен в любом положении алкильной части. Примеры заместителей включают прямые или разветвленные алкоксигруппы, имеющие от 1 до 10 атомов углерода, такие как метокси, этокси, пропокси (включая изомеры), бутокси (включая изомеры), пентилокси (включая изомеры), гексилокси (включая изомеры), гептилокси (включая изомеры), октилокси (включая изомеры), нонилокси (включая изомеры) и децилокси (включая изомеры); алкилкарбонильные группы, которые имеют от 2 до 6 атомов углерода и содержат прямую или разветвленную алкильную группу, имеющую от 1 до 5 атомов углерода, такие как ацетил, пропионил (включая изомеры), бутаноил (включая изомеры) и пентаноил (включая изомеры); фенилкарбонильные группы, которые могут иметь заместитель; и фенил, который может иметь заместитель.
"Фенилкарбонильная группа, которая может быть замещенной", означает фенилкарбонильную группу, не имеющую заместителя, или фенилкарбонильную группу, имеющую заместитель. "Фенильная группа, которая может быть замещенной", означает фенильную группу, не имеющую заместителя, или фенильную группу, имеющую заместитель. Заместителем в фенилкарбонильной группе или фенильной группе может быть фенил, фенилкарбонил, одна из вышеупомянутых алкильных, алкокси и алкилкарбонильных групп.
В изобретении "алкоксигруппа, возможно имеющая заместитель", представленная символами от R1 до R8 формул (2), (3), (4) и (5), означает алкоксигруппу, имеющую от 1 до 10 атомов углерода и содержащую прямую или разветвленную алкильную часть, которая не имеет заместителя и имеет от 1 до 10 атомов углерода, или алкоксигруппу, имеющую от 1 до 10 атомов углерода и содержащую прямую или разветвленную алкильную часть, которая имеет заместитель и имеет от 1 до 10 атомов углерода.
Примеры "алкоксигруппы, имеющей от 1 до 10 атомов углерода и содержащей прямую или разветвленную алкильную часть, которая не имеет заместителя и имеет от 1 до 10 атомов углерода", включают те, которые описаны выше. Примеры "алкоксигруппы, имеющей от 1 до 10 атомов углерода и содержащей прямую или разветвленную алкильную часть, которая имеет заместитель и имеет от 1 до 10 атомов углерода", включают вышеупомянутые алкильные группы, алкилкарбонильную группу, имеющую от 2 до 6 атомов углерода, фенилкарбонильную группу, которая может иметь заместитель, и фенил, который может иметь заместитель.
"Алкилкарбонильная группа, возможно имеющая заместитель", для символов от R1 до R8 в каждой формуле способа получения производного дибензотиазепина по изобретению означает алкилкарбонильную группу, имеющую от 2 до 11 атомов углерода и содержащую прямую или разветвленную алкильную часть, которая не имеет заместителя и имеет от 1 до 10 атомов углерода, или алкилкарбонильную группу, имеющую от 2 до 11 атомов углерода и содержащую прямую или разветвленную алкильную часть, которая имеет заместитель и имеет от 1 до 10 атомов углерода.
Примеры алкильных частей "алкилкарбонильной группы, имеющей от 2 до 11 атомов углерода и содержащей прямую или разветвленную алкильную часть, которая не имеет заместителя и имеет от 1 до 10 атомов углерода", включают те, которые описаны выше. Примеры заместителей "алкилкарбонильной группы, имеющей от 2 до 11 атомов углерода и содержащей прямую или разветвленную алкильную часть, которая имеет заместитель и имеет от 1 до 10 атомов углерода", включают те, которые описаны выше.
"Арильная группа, возможно имеющая заместитель", для символов от R1 до R8 в каждой формуле способа получения производного дибензотиазепина по изобретению означает арильную группу, не имеющую заместителя, или арильную группу, имеющую заместитель.
Примеры "арильной группы, не имеющей заместителя" включают фенил, нафтил и анторил. Предпочтительными являются фенил и нафтил. Наиболее предпочтительным является фенил. Примеры заместителей для "арильной группы, имеющей заместитель", включают те, которые были описаны выше для алкильных групп.
"Арилоксигруппа, возможно имеющая заместитель", для символов от R1 до R8 в каждой формуле способа получения производного дибензотиазепина по изобретению означает арилоксигруппу, имеющую арильную часть, не имеющую заместителя, или арилоксигруппу, имеющую арильную часть, имеющую заместитель.
Примеры арильных групп "арилоксигруппы, имеющей арильную часть, не имеющую заместителя", включают арильные группы, описанные выше. Примеры заместителей для "арилоксигруппы, имеющей арильную часть, имеющую заместитель" включают заместители, описанные выше для алкильных групп.
"Арилкарбонильная группа, возможно имеющая заместитель", для символов от R1 до R8 в каждой формуле способа получения производного дибензотиазепина по изобретению означает арилкарбонильную группу, имеющую арильную часть, не имеющую заместителя, или арилкарбонильную группу, имеющую арильную часть, имеющую заместитель.
Примеры арильных групп "арилкарбонильной группы, имеющей арильную часть, не имеющую заместителя", включают арильные группы, описанные выше. Примеры заместителей для "арилкарбонильной группы, имеющей арильную часть, имеющую заместитель", включают заместители, описанные выше для алкильных групп.
Группы от R1 до R8 могут быть одинаковыми или отличаться друг от друга, и каждая предпочтительно представляет атом водорода, алкильную группу, алкоксигруппу, алкилкарбонильную группу, арильную группу, арилоксигруппу или арилкарбонильную группу. Наиболее предпочтительными являются атом водорода, алкильная группа, алкоксигруппа и алкилкарбонильная группа.
Атом галогена для Х в формуле (1) может представлять фтор, хлор, бром или йод. Предпочтительными являются фтор, хлор и бром.
Каждая из стадий способа получения производных дибензотиазепина по изобретению описана здесь ниже более подробно.
На первой стадии способа получения производных дибензотиазепина по изобретению производное нитробензола формулы (1) и производное тиосалициловой кислоты формулы (2) вводят в реакцию в растворителе, предпочтительно в присутствии основания, для получения 2-нитро-2'-карбоксидифенилсульфидного производного формулы (3).
Примеры производных нитробензола формулы (1), используемых на первой стадии, включают 2-хлорнитробензол, 2-бромнитробензол, 2-фторнитробензол, 2-йоднитробензол, 2-хлор-5-метоксинитробензол, 2-бром-5-метоксинитробензол, 2-фтор-5-метоксинитробензол, 2-йод-5-метоксинитробензол, 2-хлор-5-метилнитробензол, 2-бром-5-метилнитробензол, 2-фтор-5-метилнитробензол, 2-йод-5-метилнитробензол, 2-хлор-5-фенилнитробензол, 2-бром-5-фенилнитробензол, 2-фтор-5-фенилнитробензол, 2-йод-5-фенил-нитробензол, 2-хлор-5-ацетилнитробензол, 2-бром-5-ацетилнитробензол, 2-фтор-5-ацетилнитробензол и 2-йод-5-ацетилнитробензол. Предпочтительными являются 2-хлорнитробензол и 2-бромнитробензол.
Примеры производных тиосалициловой кислоты формулы (2), используемых на первой стадии, включают тиосалициловую кислоту, 5-метокситиосалициловую кислоту, 5-метилтиосалициловую кислоту, 5-фенилтиосалициловую кислоту и 5-ацетилтиосалициловую кислоту. Предпочтительными являются тиосалициловая кислота и 5-метокситиосалициловая кислота.
Производное нитробензола формулы (1) обычно применяют в количестве от 0,7 до 10 моль, предпочтительно от 1,0 до 5 моль на один моль тиосалициловой кислоты формулы (2).
Вышеупомянутую первую стадию обычно проводят в растворителе. Специфические требования к растворителю отсутствуют, если только растворители не участвуют в реакции. Примеры растворителей включают воду; амидные растворители, такие как N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон и диметилимидазолидон; алифатические спирты, такие как метанол, этанол, н-пропанол, изопропанол и н-бутанол; кетоны, такие как ацетон, метилэтилкетон и метилизобутилкетон; и нитрилы, такие как ацетонитрил и бензонитрил. Предпочтительными являются вода, амиды и алифатические спирты.
Растворитель на первой стадии предпочтительно применяют таким образом, чтобы массовое отношение количества нитробензола формулы (1) к количеству растворителя было в интервале от 0,05 до 1,0, более предпочтительно от 0,1 до 0,8.
Реакцию на первой стадии обычно проводят при температуре не выше, чем температура кипения применяемого растворителя, предпочтительно при температуре от 0 до 150°С, более предпочтительно от 20 до 100°С. Время реакции на первой стадии сильно зависит от температуры реакции, но обычно реакция полностью завершается в пределах 20 часов.
Реакцию на первой стадии обычно проводят в присутствии основания. Примеры предпочтительных оснований включают карбонат калия, карбонат натрия, карбонат лития, гидроксид натрия, гидроксид калия, гидроксид лития и метилат натрия. Наиболее предпочтительными являются карбонат калия, карбонат натрия, гидроксид натрия, гидроксид калия и метилат натрия. Основание обычно применяют в количестве, соответствующем количеству от 1 до 10 молей, предпочтительно от 1,5 до 5 молей, на один моль общего количества исходных соединений.
В реакцию на первой стадии могут быть введены отличные от оснований добавки для ускорения реакции. Примеры добавок включают йодид калия и N,N-диметиламинопиридин. Добавки могут применяться в количестве от 0,0005 до 0,5 моль (моль добавки на моль производного нитробензола), предпочтительно от 0,001 до 0,1 моль на один моль производного нитробензола формулы (1).
Химическая структура 2-нитро-2’-карбоксидифенилсульфидного производного формулы (3), полученного на первой стадии по изобретению, зависит от химической структуры производного нитробензола формулы (1), а также от химической структуры производного тиосалициловой кислоты формулы (2). Примеры 2-нитро-2’-карбоксидифенилсульфидных производных включают 2-нитро-2’-карбоксидифенилсульфид, 2-нитро-4-метокси-2’-карбоксидифенилсульфид, 2-нитро-4-метил-2’-карбоксидифенилсульфид, 2-нитро-4-фенил-2’-карбоксидифенилсульфид, 2-нитро-4-ацетил-2’-карбоксидифенилсульфид и 2-нитро-2’-карбокси-4’-метоксидифенилсульфид. Предпочтительными являются 2-нитро-2’-карбоксидифенилсульфид и 2-нитро-2’-карбокси-4’-метоксидифенилсульфид.
2-Нитро-2’-карбоксидифенилсульфидное производное формулы (3), полученное на первой стадии, может быть выделено комбинацией обычной процедуры промывания и обычной процедуры выделения, такой как комбинация добавления кислоты для того, чтобы сделать реакционную смесь кислой, и фильтрации осажденного кристаллического продукта для получения сырого продукта, или комбинация добавления воды и экстрактивного растворителя (органического растворителя) к реакционной смеси и добавления кислоты для того, чтобы сделать кислой водную фазу реакционной смеси. Другим способом сырой продукт может быть выделен путем помещения части органического растворителя под пониженное давление. Полученный таким образом сырой продукт может быть применен как таковой на следующей стадии. Сырой продукт может быть дополнительно очищен, если требуется, колоночной хроматографией или перекристаллизацией. Способ очистки может быть выбран в зависимости от каждого очищаемого соединения. Предпочтительно применяемой кислотой является хлористо-водородная кислота, серная кислота, фосфорная кислота или уксусная кислота.
На второй стадии способа по изобретению 2-нитро-2’-карбоксидифенилсульфидное производное формулы (3) восстанавливают, чтобы получить 2-амино-2’-карбоксидифенилсульфидное производное формулы (4).
Выбор методики восстановления, проводимого на второй стадии, не ограничен, и могут быть использованы известные методики восстановления нитрогруппы. Предпочтительными являются способ с применением никеля Ренея (обозначенный далее как реакция (А)), способ с применением соли трехвалентного железа (обозначенный далее как реакция (В)), и способ с применением палладия, платины или их соединений (обозначенный далее как реакция (С)). В методике восстановления в качестве источника водорода применяют газообразный водород.
Реакция (А): Способ с использованием никеля Ренея
В способе может быть использован никель Ренея в количестве от 1,0 до 80 мас.% (в расчете на никель), предпочтительно от 5,0 до 40 мас.% на количество 2-нитpo-2’-кapбoкcи-дифенилсульфидного производного формулы (3). Примеры никелей Ренея, используемых в реакции, включают 10-60%-ный Ni-Al сплав и сплав, содержащий Cr и Mo. Может быть также использован стабилизированный никель. Усовершенствования метода катализа никелем Ренея слабо влияют на выход. Известный метод W-6 ("Raney Catalyst", Kubomatsu Teruo and Komatsu Shinichiro, pp.55, выпущена Kawaken Finechemical Co., Ltd, 10.05.1971) дает, пожалуй, наилучшие результаты. Достаточно эффективными могут быть другие усовершенствованные методики. В случае использования способа катализа никелем Ренея реакцию обычно проводят в присутствии газообразного водорода под давлением. Соответственно, реакцию обычно проводят в автоклаве. Давление газообразного водорода предпочтительно является настолько высоким, насколько возможно. Обычно давление газообразного водорода находится в интервале от 5 до 100 атм. Реакцию можно проводить при атмосферном давлении. В этом случае реакцию проводят в токе газообразного водорода.
Специфические ограничения на применяемые в реакции (А) растворители отсутствуют при условии, что растворители не участвуют в реакции. Примеры растворителей включают алифатические спирты, такие как метанол, этанол, н-пропанол, изопропанол и н-бутанол. Объем растворителя выбирают так, чтобы объем 2-нитро-2’-карбоксидифенилсульфидного производного формулы (3) составлял от 0,05 до 0,6 объема, предпочтительно от 0,1 до 0,6 объема на один объем растворителя (объем 2-нитро-2’-карбоксидифенилсульфидного производного формулы/объем растворителя).
Реакцию (А) можно проводить при температуре вплоть до температуры кипения растворителя. Реакцию обычно проводят при температуре от 20 до 200°С, предпочтительно от 25 до 150°С. Продолжительность реакции зависит от температуры и давления газообразного водорода. Обычно реакция полностью завершается в пределах 20 часов.
После завершения реакции (А) полученное восстановлением 2-амино-2’-карбоксидифенилсульфидное производное формулы (4) может быть выделено с помощью обычной комбинации операции промывания и операции выделения, такой как комбинация фильтрации реакционной смеси и концентрирования фильтрата при пониженном давлении. Полученный вышеуказанным образом продукт как таковой может быть использован на следующей стадии. Если требуется, продукт может быть очищен колоночной хроматографией или перекристаллизацией. Методика очистки может быть выбрана в зависимости от очищаемого продукта.
Реакция (В): Метод с использованием соли трехвалентного железа
Примеры солей трехвалентного железа, используемых в реакции, включают сульфат железа и хлорид железа. Данные соли могут быть использованы в форме гидрата или безводной форме. Предпочтительными являются 7 гидраты сульфата железа, безводные соли железа, 4 гидраты солей железа и n гидраты солей железа. Соль может быть использована в объеме от 0,1 до 30 (в расчете на атом железа), предпочтительно от 0,5 до 10 на один объем 2-нитро-2’-карбоксидифенилсульфидного производного формулы (3).
В качестве растворителя для реакции (В) обычно используют смесь воды и водного аммиака. Водный аммиак может быть получен, используя концентрированный водный аммиак (концентрация аммиака: от 25 до 28 мас.%). Могут быть также использованы водный аммиак более низкой концентрации или аммиачная вода, при условии, что содержание аммиака достаточно. Вода может быть использована так, чтобы объем 2-нитро-2’-карбоксидифенилсульфидного производного формулы (3) составил бы от 0,01 до 0,4 эквивалента на один объем воды (объем 2-нитро-2’-карбоксидифенилсульфидного производного/объем воды), предпочтительно от 0,02 до 0,2 эквивалента (то же, что и выше). Объем аммиака выбирают так, чтобы объем 2-нитро-2’-карбоксидифенилсульфидного производного формулы (3) составил бы от 0,005 до 0,5 эквивалента, предпочтительно от 0,01 до 0,5 эквивалента, на один объем аммиака (объем 2-нитро-2’-карбоксидифенилсульфидного производного/объем аммиака).
Реакцию (В) можно проводить при температуре вплоть до температуры кипения растворителя. Реакцию обычно проводят при температуре от 20 до 100°С, предпочтительно от 40 до 90°С. Продолжительность реакции зависит от температуры. Обычно реакция полностью завершается в пределах 2 часов.
После завершения реакции (В) полученное восстановлением 2-амино-2’-карбоксидифенилсульфидное производное формулы (4) может быть выделено обычной комбинацией операции промывки и операции выделения. Например, реакционную смесь фильтруют, и кислоту (например, хлористо-водородную кислоту, серную кислоту, фосфорную кислоту и уксусную кислоту) добавляют к фильтрату, смещая тем самым его рН в сторону кислотности. Полученный фильтрат концентрируют при пониженном давлении, чтобы получить сырое соединение. Полученный вышеописанным образом продукт как таковой может быть использован на следующей стадии. Если требуется, продукт может быть очищен колоночной хроматографией или перекристаллизацией. Методика очистки может быть выбрана в зависимости от очищаемого продукта.
Реакция (С): Способ с применением палладия или платины (или их соединений)
Реакцию можно проводить в присутствии восстанавливающего катализатора (т.е. катализатора гидрирования), выбранного из группы, включающей палладий (Pd), платину (Pt), соединения палладия и соединения платины. Восстанавливающий катализатор может быть нанесен на носитель, такой как углерод (С) или сульфат бария. Предпочтительными являются Pd/C, Pd/сульфат бария и оксид платины. Наиболее предпочтительным является Pd/C.
Восстанавливающий катализатор, включающий палладий или платину, может быть использован в количестве, соответствующем от 0,01 до 30 мас.% (в расчете на металлические палладий или платину), предпочтительно от 0,05 до 10 мас.%, от количества производного 2-нитро-2’-карбоксидисульфида формулы (3). Если катализатор нанесен на носитель, количество нанесенного катализатора может составлять от 1 до 10 мас.% (в расчете на металлические палладий или платину) от количества носителя. Если применяют катализатор Pd/C, может быть применен сухой катализатор с содержанием воды не более 5%, а также влажный катализатор, содержащий водный компонент в больших количествах. Влажный катализатор может содержать от 10 до 70 мас.% воды (количество воды на общее количество катализатора и носителя).
Если в реакции (С) в качестве восстанавливающего катализатора используют оксид платины, его предпочтительно применяют в количестве от 0,1 до 50 мас.%, предпочтительно от 1 до 30 мас.%, от количества производного 2-нитpo-2’-карбоксидифенилсульфида формулы (3).
Реакцию (С) обычно проводят в присутствии газообразного водорода под давлением. Соответственно, реакцию обычно проводят в автоклаве. Давление газообразного водорода предпочтительно является настолько высоким, насколько возможно. Обычно давление газообразного водорода находится в интервале от 5 до 100 атм. Реакцию можно проводить при атмосферном давлении. В этом случае восстановление (гидрирование) проводят в токе газообразного водорода.
Реакцию (С) обычно проводят в растворителе. Специфические ограничения на используемые в реакции (С) растворители отсутствуют при условии, что растворители не участвуют в реакции. Примеры растворителей включают алифатические спирты, такие как метанол, этанол, н-пропанол, изопропанол и н-бутанол, и амидные растворители, такие как N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон и диметилимидазолидон. Предпочтительными являются алифатические спирты. Растворитель предпочтительно используют в количестве от 2 до 70%, более предпочтительно от 5 до 50%, от количества производного 2-нитро-2’-карбоксидифенилсульфида формулы (3).
Реакцию (С) обычно проводят при температуре от 10 до 200°С, предпочтительно от 20 до 150°С. Продолжительность реакции зависит от температуры и давления газообразного водорода, но обычно она не превышает 30 часов.
Производное 2-амино-2’-карбоксидифенилсульфида формулы (4), полученное реакцией (С) (гидрирование), может быть выделено обычной комбинацией операции промывки и операции выделения, такой как комбинация фильтрации реакционной смеси и концентрирования фильтрата при пониженном давлении. Полученный вышеуказанным образом продукт может быть применен как таковой на следующей стадии. Если требуется, продукт может быть очищен колоночной хроматографией или перекристаллизацией. Методика очистки может быть выбрана в зависимости от очищаемого продукта.
Химическая структура производного 2-амино-2’-карбоксидифенилсульфида формулы (4), полученного на второй стадии (стадия восстановления), зависит от химической структуры производного 2-нитpo-2’-карбоксидифенилсульфида формулы (3), используемого на второй стадии в качестве исходного вещества. Примеры производных 2-амино-2’-карбоксидифенилсульфида формулы (4) включают 2-амино-2’-карбоксидифенилсульфид, 2-амино-4-метокси-2’-карбоксидифенилсульфид, 2-амино-4-метил-2’-карбоксидифенилсульфид, 2-амино-4-фенил-2’-карбоксидифенилсульфид, 2-амино-4-ацетил-2’-карбоксидифенилсульфид и 2-амино-2’-карбокси-4’-метоксидифенилсульфид. Предпочтительными являются 2-амино-2’-карбоксидифенилсульфид и 2-амино-2’-карбокси-4’-метоксидифенилсульфид.
На третьей стадии способа по изобретению производное 2-амино-2’-карбоксидифенилсульфида формулы (4) конденсируют дегидратацией для получения производного дибензотиазепина формулы (5).
Реакцию третьей стадии можно проводить без растворителя. Однако реакцию предпочтительно проводят в гидрофобном органическом растворителе, который не участвует в реакции. Примеры органических растворителей включают ароматические углеводороды, такие как толуол, ксилол, кумол и бензол; галогенированные ароматические углеводороды, такие как хлорбензол, 1,2-дихлорбензол, 1,3-дихлорбензол, 1,4-дихлорбензол, бромбензол, 1,2-дибромбензол, 1,3-дибромбензол, 1,4-дибромбензол; циклические алифатические углеводороды, такие как циклогексан, циклогептан и циклооктан; и алифатические сложные эфиры, такие как этилацетат, бутилацетат, метилбутират, этилбутират и бутилбутират. Предпочтительными являются толуол, ксилол, кумол и 1,2-дихлорбензол.
Специфические ограничения в отношении количества растворителя, используемого на третьей стадии, отсутствуют. Предпочтительно, однако, чтобы растворитель применялся в таком количестве, чтобы получить отношение массового количества производного 2-амино-2’-карбоксидифенилсульфида к объемному количеству растворителя (мас.%/об.) не менее 3%, предпочтительно в интервале от 4 до 40%. Реакцию третьей стадии можно проводить в аппарате Дина-Старка для осуществления азеотропной дегидратации (для дефлегмации с удалением образующейся в ходе реакции воды) так, чтобы увеличить скорость реакции и степень конверсии. Специфические ограничения в отношении температуры реакции третьей стадии отсутствуют при условии, что температура ниже температуры кипения используемого растворителя. Предпочтительной является температура от 100 до 200°С.
Химическая структура производного дибензотиазепина формулы (5), полученного на третьей стадии, зависит от химической структуры производного 2-амино-2’-карбоксидифенилсульфида формулы (4). Примеры производных дибензотиазепина формулы (5) включают дибензо[b,f][1,4]тиазепин-11-он, 8-метилдибензо[b,f][1,4]тиазепин-11-он, 8-фенилдибензо[b,f][1,4]-тиазепин-11-он, 8-метоксидибензо[b,f][1,4]тиазепин-11-он и 2-метоксидибензо[b,f][1,4]тиазепин-11-он. Предпочтительными являются дибензо-[b,f][1,4]тиазепин-11-он и 2-метоксидибензо[b,f][1,4] тиазепин-11-он.
Производное дибензотиазепина формулы (5), полученное на третьей стадии, может быть легко выделено охлаждением реакционной смеси для осаждения кристаллического продукта производного дибензотиазепина. Затем осажденный кристаллический продукт собирают фильтрацией, получая производное дибензотиазепина высокой чистоты. Если требуется дополнительная очистка, может быть использована перекристаллизация или колоночная хроматография. В другом случае реакционную смесь подщелачивают добавлением водного щелочного раствора, и затем, до осаждения полученного продукта, удаляют водную часть. Затем охлаждают оставшуюся органическую часть для осаждения кристаллического продукта производного дибензотиазепина. Водный щелочной раствор может быть получен с использованием бикарбоната натрия, карбоната натрия, карбоната калия, гидроксида натрия или гидроксида калия. Щелочное соединение в щелочном растворе предпочтительно находится в концентрации от 0,5 до 30 мас.%. Ограничения в отношении количества щелочного раствора отсутствуют, но щелочной раствор предпочтительно используют в количестве от 0,05 до 0,4 массовых частей на массовую часть продукта третьей стадии (т.е. производного дибензотиазепина формулы (5)).
Предпочтительные варианты осуществления изобретения описаны ниже.
1) Производное нитробензола формулы (1) представляет 2-хлорнитробензол или 2-бромнитробензол.
2) Производное тиосалициловой кислоты формулы (2) представляет тиосалициловую кислоту или 5-метокситиосалициловую кислоту.
3) На первой стадии способа получения производного дибензотиазепина по изобретению используют такие основания как карбонат калия, гидроксид натрия или метилат натрия.
4) Производное 2-нитро-2’-карбоксидифенилсульфида формулы (3) представляет 2-нитpo-2’-карбоксидифенилсульфид или 2-нитро-2’-карбокси-4’-метоксидифенилсульфид.
5) На первой стадии способа получения производного дибензотиазепина по изобретению в качестве растворителя в реакции используют N,N-диметилформамид или метанол.
6) При восстановлении на второй стадии процесса получения производного дибензотиазепина по изобретению в качестве восстанавливающего агента используют никель Ренея, а в качестве растворителя используют метанол или н-бутанол.
7) При восстановлении на второй стадии способа получения производного дибензотиазепина по изобретению в качестве восстанавливающего агента используют гидрат сульфата трехвалентного железа, а в качестве растворителя используют водный раствор аммиака.
8) Восстановление на второй стадии способа получения производного дибензотиазепина по изобретению проводят в присутствии любого катализатора, выбранного из Pd/C, Pd/сульфата бария, и оксида платины, используя в качестве растворителя метанол или этанол.
9) Производное 2-aминo-2’-кapбoкcидифeнилcyльфидa формулы (4) представляет 2-амино-2’-карбоксидифенилсульфид или 2-амино-2’-карбокси-4’-метоксидифенилсульфид или 2-метокси-дибензо[b,f][1,4]тиазепин-11-он.
10) Производное дибензотиазепина формулы (5) представляет дибензо[b,f][1,4]тиазепин-11-он или 2-метоксидибензо[b,f]-[1,4]тиазепин-11-он.
11) На первой стадии производным нитробензола формулы (1) является 2-хлорнитробензол или 2-бромнитробензол; производным тиосалициловой кислоты формулы (2) является тиосалициловая кислота или 5-метокситиосалициловая кислота; основанием является карбонат калия; растворителем является N,N-диметилформамид; и получаемое производное 2-нитpo-2’-кapбoкcидифенилсульфида формулы (3) представляет собой 2-нитpo-2’-карбоксидифенилсульфид или 2-нитpo-2’-карбокси-4’-метоксидифенилсульфид.
12) На второй стадии 2-нитро-2’-карбоксидифенилсульфид или 2-нитро-2’-карбокси-4’-метоксидифенилсульфид восстанавливают газообразным водородом в присутствии платины, палладия или их соединений для получения соответственно 2-aминo-2’-карбоксидифенилсульфида или 2-амино-2’-карбокси-4’-метоксидифенилсульфида.
13) На третьей стадии 2-амино-2’-карбоксидифенилсульфид или 2-амино-2’-карбокси-4’-метоксидифенилсульфид превращают соответственно в дибензо[b,f][1,4]тиазепин-11-он или 2-метоксидибензо[b,f][1,4]тиазепин-11-он.
Изобретение описано далее с помощью следующих неограничивающих примеров.
Пример 1
В 120 мл N,N-диметилформамида растворяли 94,5 г (0,60 моль) 2-хлорнитробензола и 159,0 г (1,15 моль) карбоната калия. К полученному раствору в N,N-диметилформамиде по каплям добавляли раствор 77,1 г (0,50 моль) тиосалициловой кислоты в 120 мл N,N-диметилформамида. Затем полученную смесь перемешивали при 70°С в течение 6 часов для проведения реакции. К реакционной смеси добавляли 800 мл воды и 700 мл этилацетата. Водную часть отделяли и подкисляли добавлением 400 г льда и 194 мл концентрированной хлористо-водородной кислоты. Кислый раствор перемешивали при комнатной температуре в течение одного часа. Осажденный кристаллический продукт собирали фильтрацией и сушили, получая 134,0 г (0,49 моль) 2-нитро-2’-карбоксидифенилсульфида в виде желтого порошка. Выход в расчете на тиосалициловую кислоту составлял 98%.
1H-ЯМР (ДМСО-d6): δ 7,1-8,3 (м, 8Н), 13,1-13,5 (шир., 1Н).
Пример 2
В 120 мл N,N-диметилформамида растворяли 94,5 г (0,60 моль) 2-хлорнитробензола и 159,0 г (1,15 моль) карбоната калия. К полученному раствору в N,N-диметилформамиде по каплям добавляли раствор 77,1 г (0,50 моль) тиосалициловой кислоты в 120 мл N,N-диметилформамида. Затем полученную смесь перемешивали при 70°С в течение 6 часов для проведения реакции. Водную часть отделяли и подкисляли добавлением 200 мл воды и 194 мл концентрированной хлористо-водородной кислоты. Кислый раствор перемешивали при комнатной температуре в течение одного часа. Осажденный кристаллический продукт собирали фильтрацией и сушили, получая 123,0 г (0,45 моль) 2-нитро-2’-карбоксидифенилсульфида в виде желтого порошка. Выход в расчете на тиосалициловую кислоту составлял 90%.
Пример 3
Повторяли методику примера 1 за исключением того, что использовали 121,2 г (0,60 моль) 2-бромнитробензола вместо 2-хлорнитробензола, получая 134,0 г (0,49 моль) 2-нитро-2’-карбоксидифенилсульфида. Выход в расчете на тиосалициловую кислоту составлял 98%.
Пример 4
Повторяли методику примера 1 за исключением того, что использовали 93,8 г (0,50 моль) 5-метокситиосалициловой кислоты вместо тиосалициловой кислоты, получая 137,3 г (0,45 моль) 2-нитро-2’-карбокси-4’-метоксидифенилсульфида. Выход в расчете на 5-метокситиосалициловую кислоту составлял 90%. Температура плавления: 185-187°С.
Пример 5
Повторяли методику примера 1 за исключением того, что использовали метанол вместо N,N-диметилформамида, получая 131,3 г (0,48 моль) 2-нитро-2’-карбоксидифенилсульфида. Выход в расчете на тиосалициловую кислоту составлял 96%.
Пример 6
Повторяли методику примера 5 за исключением того, что использовали 46,0 г (1,15 моль) гидроксида натрия вместо карбоната калия, получая 130,0 г (0,47 моль) 2-нитpo-2’-карбоксидифенилсульфида. Выход в расчете на тиосалициловую кислоту составлял 94%.
Пример 7
Повторяли методику примера 5 за исключением того, что использовали 62,1 г (1,15 моль) метилата натрия вместо карбоната калия и реакцию проводили в течение 5 часов, получая 131,8 г (0,48 моль) 2-нитро-2’-карбоксидифенилсульфида. Выход в расчете на тиосалициловую кислоту составлял 96%.
Пример 8
Повторяли методику примера 7 за исключением того, что для ускорения реакции к реакционной смеси добавляли 3,9 г (0,02 моль) йодида калия, получая 133,8 г (0,49 моль) 2-нитро-2’-карбоксидифенилсульфида. Выход в расчете на тиосалициловую кислоту составлял 97%.
Пример 9
В 300-миллилитровый автоклав помещали никель Ренея (50%-ный сплав, содержание Ni 4 г), 13,8 г (0,05 моль) 2-нитро-2’-карбоксидифенилсульфида, полученного в примере 1, и 100 мл метанола. Смесь перемешивали при комнатной температуре в течение 5 часов при давлении водорода 20 атм. Реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении, получая 11,3 г (0,046 моль) 2-aминo-2’-карбоксидифенилсульфида в виде бесцветного порошкообразного продукта. Выход в расчете на 2-нитро-2’-карбоксидифенилсульфид составлял 92%.
1H-ЯМР (ДМСО-d6): δ 5,0-5,9 (шир., 2Н), 6,5-8,1 (м, 8Н), 12,8-13,5 (шир., 1Н).
Пример 10
В 50 мл н-бутанола суспендировали никель Ренея (50%-ный сплав, содержание Ni 1 г) и 4,0 г (14,5 ммоль) 2-нитро-2’-карбоксидифенилсульфида, полученного в примере 1. Полученную в н-бутаноле суспензию перемешивали при 100°С в течение 15 часов при продувке водородом. Реакционную смесь фильтровали, и фильтрат концентрировали при пониженном давлении, получая 3,24 г (13,2 ммоль) 2-амино-2’-карбоксидифенилсульфида в виде бесцветного порошкообразного продукта. Выход в расчете на 2-нитро-2’-карбоксидифенилсульфид составлял 91%.
Пример 11
В 40 мл концентрированного водного раствора аммиака (концентрация аммиака: 28 мас.%) растворяли 2,75 г (10,0 ммоль) 2-нитро-2’-карбоксидифенилсульфида, полученного в примере 1. К полученной водно-аммиачной смеси по каплям добавляли раствор 21,6 г (77,8 ммоль) 7 гидрата сульфата трехвалентного железа в 70 мл воды. Полученную смесь нагревали при 80°С в течение 10 минут для проведения реакции. Реакционную смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали до 30 мл при пониженном давлении, и к концентрату добавляли 70 мл этилацетата и 2 мл уксусной кислоты. Отделенную органическую часть сушили над безводным сульфатом магния и фильтровали, чтобы отделить осушающий агент. Фильтрат концентрировали при пониженном давлении, получая 2,33 г (9,50 ммоль) 2-aминo-2’-карбоксидифенилсульфида в виде бесцветного порошкообразного продукта. Выход в расчете на 2-нитро-2’-карбоксидифенилсульфид составлял 95%.
Пример 12
Повторяли методику примера 10 за исключением того, что использовали 15,2 г (0,05 моль) 2-нитpo-2’-карбокси-4’-метоксидифенилсульфида вместо 2-нитро-2’-карбоксидифенилсульфида, получая 12,7 г (0,046 моль) 2-aминo-2’-кapбoкcи-4’-метоксидифенилсульфида в виде бесцветного порошкообразного продукта. Выход в расчете на 2-нитpo-2’-кapбoкcи-4’-мeтoкcидифенилсульфид составлял 92%. Температура плавления: 150-151°С.
Пример 13
В 300-миллилитровый автоклав помещали 1,37 г Pd (5 мас.%)/С, 13,7 г (0,05 моль) 2-нитpo-2’-карбоксидифенилсульфида, полученного в примере 1, и 95 мл метанола. Смесь перемешивали при 25°С в течение 6 часов при давлении водорода 10 атм для осуществления реакции гидрирования. Реакционную смесь фильтровали, и фильтрат концентрировали при пониженном давлении, получая 11,7 г (0,048 моль) 2-амино-2’-карбоксидифенилсульфида в виде бесцветного порошкообразного продукта. Выход в расчете на 2-нитро-2’-карбоксидифенилсульфид составлял 95%. Температура плавления: 150-151°С.
Пример 14
Повторяли методику примера 13 за исключением того, что температура и время реакции были изменены соответственно на 50°С и 4 часа, получая 12,0 г (0,049 моль) 2-aминo-2’-карбоксидифенилсульфида. Выход в расчете на 2-нитpo-2’-карбоксидифенилсульфид составлял 98%.
Пример 15
Повторяли методику примера 14 за исключением того, что вместо 1,37 г Pd (5 мас.%)/С использовали 2,91 г Pd (5 мас.%)/С (содержание воды: 52,9 мас.%), получая 11,9 г (0,049 моль) 2-aминo-2’-кapбoкcидифeнилcyльфидa. Выход в расчете на 2-нитро-2’-карбоксидифенилсульфид составлял 97%.
Пример 16
Повторяли методику примера 14 за исключением изменения количества метанола и времени реакции соответственно на 50 мл и 6 часов, получая 11,9 г (0,049 моль) 2-aминo-2’-карбоксидифенилсульфида. Выход в расчете на 2-нитро-2’-карбоксидифенилсульфид составлял 97%.
Пример 17
Повторяли методику примера 14 за исключением изменения количества метанола и времени реакции соответственно на 180 мл и 6 часов, получая 11,2 г (0,046 моль) 2-амино-2’-карбоксидифенилсульфида. Выход в расчете на 2-нитро-2’-карбоксидифенилсульфид составлял 91%.
Пример 18
Повторяли методику примера 14 за исключением того, что метанол был заменен этанолом, получая 11,2 г (0,046 моль) 2-амино-2’-карбоксидифенилсульфида. Выход в расчете на 2-нитро-2’-карбоксидифенилсульфид составлял 92%.
Пример 19
Повторяли методику примера 14 за исключением того, что вместо 1,37 г Pd (5 мас.%)/С использовали 640 мг оксида платины (PtO2), получая 10,8 г (0,044 моль) 2-амино-2’-карбоксидифенилсульфида. Выход в расчете на 2-нитро-2’-карбоксидифенилсульфид составлял 88%.
Пример 20
Повторяли методику примера 14 за исключением того, что применяли 15,2 г (0,05 моль) 2-нитро-2’-карбокси-4’-метоксидифенилсульфида, полученного в примере 4, получая 12,7 г (0,046 моль) 2-амино-2’-карбокси-4’-диметоксидифенилсульфида. Выход в расчете на 2-нитро-2’-карбокси-4’-диметоксидифенилсульфид составлял 92%.
Пример 21
В 300 мл толуола растворяли 25,4 г (0,10 моль) 2-амино-2’-карбоксидифенилсульфида. Для осуществления реакции полученный толуольный раствор кипятили с обратным холодильником в течение 20 часов. Реакционную смесь охлаждали до комнатной температуры, и выпавший в осадок кристаллический продукт собирали фильтрацией. Собранный продукт сушили, получая 15,7 г (0,069 моль) дибензо[b,f][1,4]тиазепин-11-она в виде бесцветных игл. Выход в расчете на 2-амино-2’-карбоксидифенилсульфид составлял 69%. Температура плавления: 259-260°С.
1H-ЯМР (ДМСО-d6): δ 7,05-7,80 (м, 8Н), 10,7 (с, 1Н).
Пример 22
В 300 мл толуола растворяли 25,4 г (0,10 моль) 2-амино-2’-карбоксидифенилсульфида. Для осуществления реакции полученный толуольный раствор кипятили с дефлегмированием в аппарате Дина-Старка в течение 20 часов с азеотропной дегидратацией. Реакционную смесь охлаждали до комнатной температуры, и выпавший в осадок кристаллический продукт собирали фильтрацией. Собранный продукт сушили, получая 18,2 г (0,080 моль) дибензо[b,f][1,4]тиазепин-11-она в виде бесцветных игл. Выход в расчете на 2-амино-2’-карбоксидифенилсульфид составлял 80%.
Пример 23
Повторяли методику примера 22 за исключением того, что в качестве растворителя в реакции использовали ксилол, и время реакции составляло 15 часов, получая 22,3 г (0,098 моль) дибензо[b,f][1,4]тиазепин-11-она в виде бесцветных игл. Выход в расчете на 2-амино-2’-карбоксидифенилсульфид составлял 98%.
Пример 24
Повторяли методику примера 22 за исключением того, что в качестве растворителя в реакции использовали кумол, и время реакции составляло 10 часов, получая 22,3 г (0,098 моль) дибензо[b,f][1,4]тиазепин-11-она в виде бесцветных игл. Выход в расчете на 2-aминo-2’-карбоксидифенилсульфид составлял 98%.
Пример 25
В 300 мл ксилола растворяли 25,4 г (0,10 моль) 2-амино-2’-карбоксидифенилсульфида, полученного в примере 14. Для осуществления реакции полученный ксилольный раствор кипятили с дефлегмированием в аппарате Дина-Старка в течение 15 часов с азеотропной дегидратацией. Реакционную смесь охлаждали до 75°С. Охлажденную смесь перемешивали при 75°С в течение 30 минут после добавления 240 мл насыщенного водного раствора бикарбоната натрия. Затем выпавший в осадок кристаллический продукт собирали фильтрацией. Собранный продукт сушили, получая 21,5 г (0,095 моль) дибензо[b,f][1,4]тиазепин-11-она в виде бесцветных игл. Выход в расчете на 2-амино-2’-карбоксидифенилсульфид составлял 95%.
Пример 26
Повторяли методику примера 25 за исключением того, что вместо насыщенного водного раствора бикарбоната натрия использовали 1N водный раствор гидроксида натрия, получая 21,1 г (0,093 моль) дибензо[b,f][1,4]тиазепин-11-она в виде бесцветных игл. Выход в расчете на 2-амино-2’-карбоксидифенилсульфид составлял 93%.
Пример 27
Повторяли методику примера 25 за исключением того, что в качестве растворителя в реакции использовали кумол, и время реакции составляло 10 часов, получая 22,0 г (0,097 моль) дибензо[b,f][1,4]тиазепин-11-она в виде бесцветных игл. Выход в расчете на 2-амино-2’-карбоксидифенилсульфид составлял 97%.
Пример 28
Повторяли методику примера 23 за исключением того, что использовали 27,5 г (0,10 моль) 2-амино-2’-карбокси-4’-метоксидифенилсульфида, полученного в примере 12, получая 23,6 г (0,092 моль) 2-метоксидибензо[b,f][1,4]тиазепин-11-она в виде бесцветных игл. Выход в расчете на 2-aминo-4-мeтoкcи-2’-карбоксидифенилсульфид составлял 92%. Температура плавления: 220-223°С.
Промышленное применение
Производное дибензотиазепина, представленное формулой (5) и представляющее интерес как промежуточное соединение для получения лекарственных препаратов, может быть легко получено с высоким выходом простыми методами согласно способу получения производного дибензотиазепина по настоящему изобретению, который включает стадии реакции производного нитробензола с производным тиосалициловой кислоты для получения производного 2-нитpo-2’-карбоксидифенилсульфида, восстановления продукта для получения производного 2-амино-2’-карбоксидифенилсульфида, и проведения реакции дегидратации-конденсации полученного продукта.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 2-ОКСО-1-ПИРРОЛИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2001 |
|
RU2292336C2 |
| ПРОИЗВОДНЫЕ 2-ОКСО-1-ПИРРОЛИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ | 2001 |
|
RU2355680C2 |
| КОМПОЗИЦИЯ, СПОСОБ И ПРИМЕНЕНИЕ | 2019 |
|
RU2802209C2 |
| НОВЫЕ ЗАМЕЩЕННЫЕ СУЛЬФАМАТНЫЕ ПРОТИВОСУДОРОЖНЫЕ ПРОИЗВОДНЫЕ | 2003 |
|
RU2328502C2 |
| СПОСОБ НИТРОВАНИЯ ФЕНОЛЬНЫХ СОЕДИНЕНИЙ | 2002 |
|
RU2318797C2 |
| ПРОИЗВОДНЫЕ ХИНОКСАЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И СРЕДСТВО С КВИСКВАЛАТ-АНТАГОНИСТИЧЕСКИМ ДЕЙСТВИЕМ НА ИХ ОСНОВЕ | 1992 |
|
RU2117663C1 |
| АЗОТСОДЕРЖАЩЕЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2011 |
|
RU2533708C2 |
| СПОСОБ РЕГИОСЕЛЕКТИВНОГО СИНТЕЗА ПРОИЗВОДНЫХ 1-АЛКИЛ-3-ГАЛОГЕНАЛКИЛПИРАЗОЛ-4-КАРБОНОВОЙ КИСЛОТЫ | 2009 |
|
RU2498977C9 |
| ПРОИЗВОДНЫЕ НУКЛЕОЗИДОВ ПИРИМИДИНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2085557C1 |
| Полимерное соединение и его применение в фотовольтаических устройствах | 2013 |
|
RU2640810C2 |
Изобретение относится к способу получения производного дибензотиазепина, представляющего интерес в качестве промежуточного соединения для получения фармацевтических препаратов. Описывается способ получения производного дибензотиазепина следующей формулы (5):
в которой каждый из R1, R2, R3, R4, R5, R6, R7 и R8 независимо представляет атом водорода, алкильную группу и алкоксигруппу, который включает стадии: взаимодействия производного нитробензола формулы (1):
в которой каждый из R1, R2, R3 и R4 имеет указанное выше значение, и Х представляет атом галогена, с производным тиосалициловой кислоты следующей формулы (2):
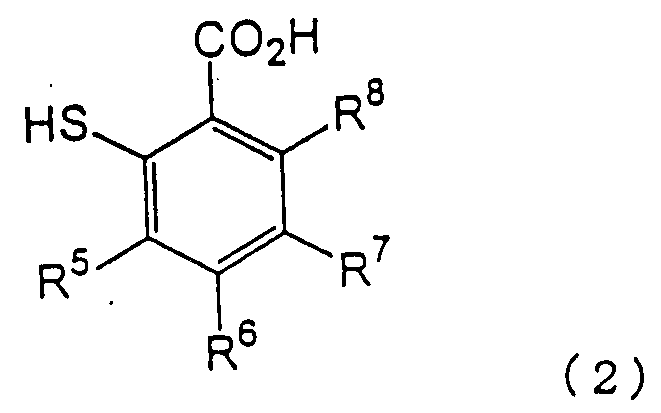
в которой каждый из R5, R6, R7 и R8 имеет указанное выше значение, с получением 2-нитро-2’-карбоксидифенилсульфидного производного следующей формулы (3):
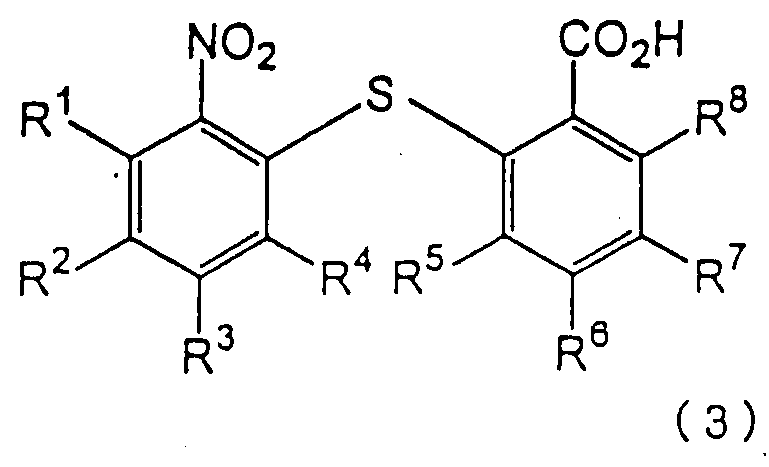
в которой каждый из R1, R2, R3, R4, R5, R6, R7 и R8 имеет значения, указанные выше; восстановления полученного производного 2-нитро-2’-карбоксидифенилсульфидного с получением 2-амино-2’-карбокси-дифенилсульфидного производного следующей формулы (4):
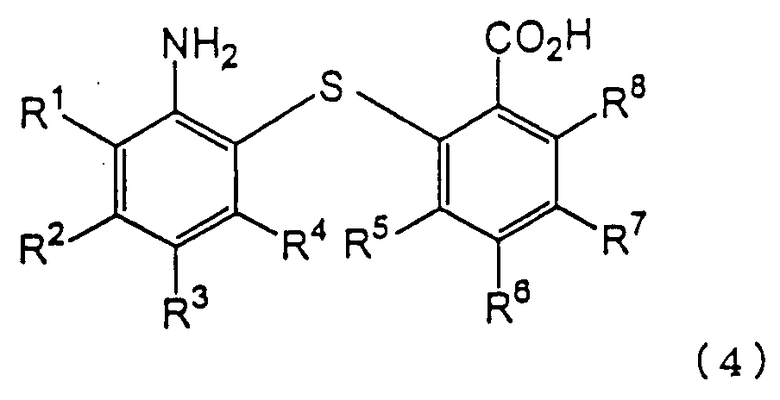
в которой каждый из R1, R2, R3, R4, R5, R6, R7 и R8 имеет значения, указанные выше; и реакции дегидратации-конденсации полученного производного 2-амино-2’-карбоксидифенилсульфида. Также описывается способ-аналог и способ получения промежуточного соединения. Технический результат - изложен способ промышленного получения производного дибензотиазепина с хорошим выходом без сложной последующей переработки с применением легкодоступного сырья. 3 н. и 9 з.п. ф-лы.
в которой каждый из R1 - R8 независимо представляет атом водорода, алкильную группу и алкоксигруппу,
который включает стадии взаимодействия производного нитробензола формулы (1)
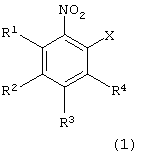
в которой каждый из R1 - R4 имеет указанное выше значение;
Х представляет атом галогена,
с производным тиосалициловой кислоты формулы (2)
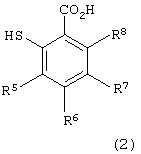
в которой каждый из R5 - R8 имеет указанное выше значение,
с получением производного 2-нитро-2’-карбоксидифенилсульфида формулы (3)
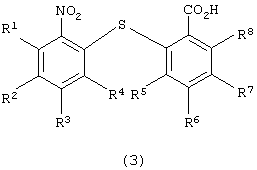
в которой каждый из R1 - R8 имеет значения, указанные выше,
восстановления полученного производного 2-нитро-2’-карбоксидифенилсульфида с получением производного 2-амино-2’-карбоксидифенилсульфида формулы (4)
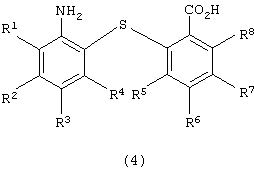
в которой каждый из R1 - R8 имеет значения, указанные выше,
и реакции дегидратации-конденсации полученного производного 2-амино-2’-карбоксидифенилсульфида.
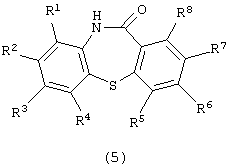
в которой каждый из R1 - R8независимо представляет атом водорода, алкильную группу и алкоксигруппу,
который включает стадии восстановления производного 2-нитро-2’-карбоксидифенилсульфида формулы (3)
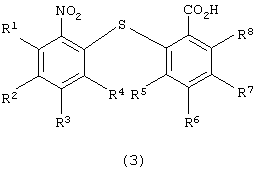
в которой каждый из R1 - R8 имеет значения, указанные выше,
с получением производного 2-амино-2’-карбоксидифенилсульфида формулы (4)
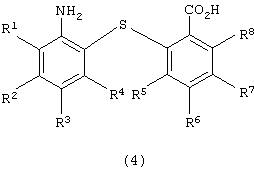
в которой каждый из R1 - R8имеет значения, указанные выше,
и реакции дегидратации-конденсации полученного производного 2-амино-2’-карбоксидифенилсульфида.
где R1 - R8 независимо представляют атом водорода, алкильную группу и алкоксигруппу,
который включает стадии взаимодействия производного нитробензола формулы (1)
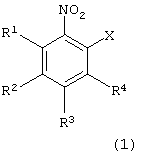
в которой каждый из R1 - R4 имеет указанное выше значение;
Х представляет атом галогена,
с производным тиосалициловой кислоты формулы (2)
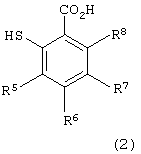
в которой каждый из R5 - R8 имеет указанное выше значение.
| J | |||
| HETEROCYCL | |||
| CHEM | |||
| Сплав для отливки колец для сальниковых набивок | 1922 |
|
SU1975A1 |
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| Автоматический огнетушитель | 0 |
|
SU92A1 |

