Область изобретения
Настоящее изобретение относится к терапиям лечения рака, в особенности к синергическому применению двух или более противораковых агентов, имеющих антипролиферативную цитотоксическую и цитостатическую активности.
Уровень техники
Химиотерапию, представляющую собой систематическое введение противоопухолевых агентов, которые проходят сквозь организм через циркулирующую систему крови, перед и часто вместе с хирургическим вмешательством и/или лучевой терапией, широко используют на протяжении многих лет при лечении различных раковых образований. К сожалению, доступные химиотерапевтические лекарственные препараты часто оказываются вредными для пациентов, потому что убивают множество здоровых клеток и, таким образом, вызывают серьезные побочные эффекты, что приводит к необходимости ограничения доз, которые могут быть введены пациентам.
В особенности, раковые опухоли трудно лечить, потому что они содержат как пролиферативные, так и непролиферативные раковые клетки. Поскольку раковая опухоль растет, васкулярное развитие часто не может успевать за быстрым размножением популяции злокачественных клеток. Следовательно, твердые массы раковых опухолей обычно демонстрируют аномальную сеть кровеносных сосудов, которые в отличие от сосудов в нормальных тканях не могут обеспечить адекватное питание злокачественных раковых клеток, для оптимального роста. В большинстве злокачественных твердых опухолей непролиферативные опухолевые клетки составляют большую часть от общей популяции раковых клеток. Более того, по мере того, как опухоль увеличивается в размере, пропорция непролиферативных опухолевых клеток в соотношении также увеличивается. Поскольку наиболее распространено то, что противораковые агенты поражают пролиферативные клетки, популяцию непролиферативных опухолевых клеткок определяют как главный фактор, влияющий на неудачу при проведении лучевой или химиотерапии, когда их используют по отдельности или вместе при лечении опухолевой болезни.
Как упомянуто выше, по мере того, как опухоль увеличивается в размере, она обычно становится все более невосприимчивой к большинству курсов химиотерапии. Соответственно множество курсов, направленных на уничтожение опухоли, включают стадию уменьшения ее в объеме для снижения массы перед введением противоопухолевых агентов.
Однако уменьшение в объеме не всегда приводит к разрушению опухоли, даже когда оно протекает совместно с применением сильных противоопухолевых агентов. Таким образом, существует потребность в разработке новых способов лечения, которые нацелены как на пролиферативные, так и непролиферативные раковые клетки для лечения злокачественных опухолей.
WO 98/54966 раскрывает комбинацию терапий, применяющих противоопухолевый агент или лучевую терапию совместно с ингибитором пренил-протеиновой трансферазы, который может быть полезньм при лечении рака. Однако WO 98/54966 не раскрывает применение соединений формулы I по настоящему изобретению.
US 6011029 раскрывает соединения формулы I по настоящему изобретению и способы их применения в качестве противораковых агентов. Кроме того, патент вообще раскрывает, что соединения формулы I могут быть пригодны в комбинации с другими терапиями рака. Однако US 6011029 не раскрывает никакую конкретную комбинацию способов лечения, которая действует синергически в качестве противораковых способов лечения.
Таким образом, объектом настоящего изобретения является обеспечение синергического способа лечения рака.
Также объектом настоящего изобретения является создание фармацевтической композиции для синергического лечения рака.
Эти и другие объекты настоящего изобретения будут более понятны из описания, которое представлено ниже.
Сущность изобретения
Настоящее изобретение предусматривает синергический способ лечении рака, который включает введение такому виду как млекопитающее, нуждающемуся в этом, синергически терапевтически эффективного количества (1) по крайней мере одного агента, выбранного из группы, состоящей из антипролиферативных цитотоксических агентов и антипролиферативных цитостатических агентов, паклитаксел [1S-[1R*,3R*(E),7R*,10S*,11R*,12R*,16S*]]-7-11-дигидрокси-8,8,10, 12, 16-пентаметил-3-[1-метил-2-(2-метил-4-тиазолил)этенил]-4-аза-17 оксабицикло [14.1.0] гептадекан-5,9-дион, СРТ-11, гемцитабин, 4-(3 бромфениламино)-6,7-бис (метокси)хиназолин, трастузумаб, 4-(3-хлор-4-фторфениламино)-7-метокси -6-(3-(4-морфофенил)пропокси)хиназолин, тамоксифен и N-[5-[[[5-(1,1-диметилэтил)-2-оксазолил]метил]тио]-2-тиазолил]-4-пиперидинкарбоксамид, а также соединения формулы (I)
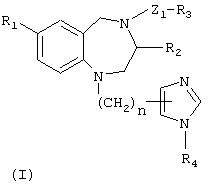
или его фармацевтически приемлемой соли, где
R1 представляет собой Br или CN;
R2 представляет собой необязательно замещенный бензил;
R3 представляет собой необязательно замещенный низший алкил, необязательно замещенный фенил, необязательно замещенный 2-тиенил или необязательно замещенный 1-пиперидинил;
R4 представляет собой водород или метил;
Z1 представляет собой CO, SO2 или SO2N(R5)-; и
n имеет значение 1; а
R5 представляет собой необязательно замещенный низший алкил или необязательно замещенный фенил;
при условии, что противораковый агент вводят одновременно с или до введения соединения формулы I.
В предпочтительном воплощении изобретения цитотоксический или цитостатический агент вводят до введения соединения формулы I. В другом воплощении изобретения цитотоксический или цитостатический агент вводят одновременно с соединением формулы I.
Краткое описание чертежей
Фиг.1 показывает, что в НСТ-116 модели твердой карциномы прямой кишки человека, развитой в лишенных шерсти мышах, большинство опухолевых клеток находятся в непролиферативной (G0) фазе роста. Непролиферативные клетки, присутствующие в НСТ-116 твердом новообразовании карциномы прямой кишки человека, развитом подкожно, идентифицируют путем введения пролонгированной BrdUrd метки (24 часа) при длительном вливании. Только 20% от общей популяции опухолевых клеток, рассасываемых из твердого новообразования, окрашены точно BrdUrd, которые селективно метят пролиферативные клетки.
Фиг.2 показывает селективное поражение непролиферативных НСТ-116 опухолевых клеток in vitro соединением 2. Опухолевые клетки в экспоненциальном росте (день 2 несливающихся и высоко пролиферативных клеток) были в >44 раза менее чувствительными к соединению 2 (IC90=0.3 мкм) по сравнению с опухолевыми клетками в стационарной фазе роста (день 8 высоко сливающихся и непролиферативных клеток).
Фиг.3 показывает селективное поражение непролиферативных НСТ-116 опухолевых клеток in vitro соединением 6. Опухолевые клетки в экспоненциальном росте (день 2 несливающихся и высоко пролиферативных клеток) были в ˜67 раз менее чувствительными к соединению 6 (IC90=1.04 μМ) по сравнению с клетками новообразования в стационарной фазе роста (день 8 высоко сливающихся и нелролиферативных клеток).
Фиг.4 показывает селективное поражение непролиферативных НСТ-116 опухолевых клеток in vitro соединением 4. Опухолевые клетки в экспоненциальном росте (день 2 несливающихся и высоко пролиферативных клеток) были в ˜91.2 раз менее чувствительным к соединению 4 (IC90=1.05 мкм) по сравнению с клетками новообразования в стационарной фазе роста (день 8 высоко сливающихся и непролиферативных клеток).
Фиг.5 показывает, что в отличие от соединения 2 и близких к нему соединений, антипролиферативные агенты, такие как паклитаксел, селективно поражают размножающиеся НСТ-116 опухолевые клетки in vitro. Раковые клетки в экспоненциальном росте (день 2 несливающихся и высоко пролиферативных клеток) были ≫10 раз более чувствительным к паклитакселю (IC90=17.8 μМ) по сравнению с клетками новообразования в стационарной фазе роста (день 8 высоко сливающихся и непролиферативных клеток).
Фиг.6 показывает, что эпотилоны, другой класс противоопухолевых антипролиферативных агентов, селективно поражает пролиферативные клетки. В этом примере НСТ-116 раковые клетки в экспоненциальном росте (день 2 несливающихся и высоко пролиферативных клеток) были ≫83 раз более чувствительным к эпотилону В (IC90=1.3 нМ) по сравнению с НСТ-116 клетками в стационарной фазе роста (день 8 высоко сливающихся и непролиферативных клеток).
Фиг.7 показывает результаты комбинированной химиотерапии с соединением 2 и паклитакселем в клеточной линии карциномы прямой кишки человека, НСТ-116. Эти данные показывают, что применение комбинированного лечения соединением 2 и паклитакселем для лечения НСТ-116 раковых клеток in vitro приводит к заметно синергической противораковой активности. Последовательность, при которой вводят два агента, как было показано, является важной для определения того, будет ли проявляться синергизм. Паклитаксел вводят сначала в течение 20 часов с последующим введением соединения 2 (0.33 мкм), период в течение последующих 20 часов является явно синергическим, как при одновременном лечении двумя агентами при установленных концентрациях в течение 20 часов. Наоборот, комбинирование соединения 2 (0.3 μМ) с последующим введением паклитакселя, как было обнаружено, является антагонистическим.
Фиг.8 показывает комбинированную химиотерапию с соединением 2 и соединением 1 в клеточной линии карциномы прямой кишки человека НСТ-116. Эти данные показывают, что применение комбинированного лечения соединением 2 и соединением 1 для лечения НСТ-116 раковых клеток in vitro приводит к заметной синергической противораковой активности. Соединение 1 вводят сначала в течение 20 часов с последующим введением соединения 2 (1 мкм) в течение последующих 20 часов периода лечения.
Фиг.9 и 10 дополнительно показывают синергизм комбинированной химиотерапии с использованием соединения 2 и соединения 1. Синергизм получают в области концентраций соединения 1 и соединения 2 и, как оказалось, он не зависит от конкретной концентрации каждого агента, используемого в комбинации. В случае использования соединения 2 концентрации 1 мкм (фиг.8), 0.33 мкм (фиг.9) и 0.11 мкм (фиг.10) все приводят к синергическому взаимодействию с соединением 1 в различных концентрациях. В этих экспериментах соединение 1 вводят сначала в течение 20 часов с последующим введением соединения 2 в течение последующих 20 часов периода лечения.
Фиг.11 показывает синергизм in vivo в ксенотрансплантантах опухолей человека (НСТ-116 карциномы прямой кишки человека), развитых в лишенных шерсти мышах, полученный путем следующей комбинированной химиотерапии, использующей соединение 2 и паклитаксел. В этом эксперименте соединение 2 вводят пролонгированным внутривенным вливанием (24 часа) в дозе 125 мг/кг. Паклитаксел вводят внутрибрюшинно в дозе 24 мг/кг в конце периода вливания соединения 2 (считается одновременным введением).
Фиг.12 показывает синергизм in vivo в паклитаксел-резистентном ксенотрансплантанте опухоли человека (Pat-7 карцинома яичников человека), развитом в лишенных шерсти мышах, с последующей комбинированной химиотерапией при использовании соединения 2 и паклитаксел. Паклитаксел и соединение 2 вводят одновременно; паклитаксел путем внутривенного введения и соединение 2 путем внутрибрюшинного введения. Приведенные данные отражают максимальные толерантные режимы: паклитаксел (36 мг/кг, внутривенно, q3dx3); соединение 2 (350 мг/кг, внутрибрюшинно, q3dx3).
Фиг.13 показывает терапевтический синергизм in vivo в лекарственно-резистентном ксенотрансплантанте опухоли человека (HCTVM46 карциномы прямой кишки человека), развитом в лишенных шерсти мышах с последующей комбинированной химиотерапией, при использовании соединения 2 и соединения 1. Соединение 1 вводят внутривенно за 24 часа до введения соединения 2 внутрибрюшинно. Приведенные данные отражают максимальные толерантные режимы: соединение 1 отдельно (15 мг/кг, q4dx3), соединение 2 отдельно (400 мг/кг, q4dx3), комбинированно (соединение 1 при 6 мг/кг с последующим введением соединения 2 при 400 мг/кг).
Фиг.14 показывает схематичную зависимость комбинирования соединения 1 и соединения 2 in vivo против лекарственно-резистентного ксенотрансплантанта опухоли человека (HC7VM46 карцинома прямой кишки человека), развитого в лишенных шерсти мышах. В противоположность другим схемам, описанными выше, введение соединения 2 за один день до соединения 1 не приводит к терапевтическому синергизму. Приведенные данные отражают максимальные толерантные режимы: соединение 1 отдельно (10 мг/кг, внутривенно, q4dx3), соединение 2 отдельно (400 мг/кг, внутрибрюшинно, q4dx3), комбинированно (соединение 2 при 300 мг/кг с последующим введением соединения 1 при 10 мг/кг).
Фиг.15 показывает, что комбинированная химиотерапия с соединением 2 и СРТ-11 приводит к синергической противоопухолевой активности в обширной (300-500 мг) карциноме прямой кишки человека НСТ116, развитой в лишенных шерсти мышах. СРТ-11 вводят за 1 час до соединения 2. СРТ-11 вводят внутривенно при или около его MTD в 30 мг/кг/инъекцию. Соединение 2 вводят двумя различными уровнями доз: 60 и 80 мг/кг/ инъекцию, внутривенно.
Фиг.16 (А) показывает, что комбинированная химиотерапия с гемцитабином (Gem) плюс соединение 2 вызывает повышение ингибирования роста новообразования карциномы прямой кишки человека НТ-29. Фиг. 16 (В) показывает, что синергическая противоопухолевая активность, являющаяся результатом комбинированной терапии гемцитабина и соединения 2, также имеет место, когда регрессию опухоли используют как конечную точку канцирогенного эффекта. Gem вводят за 1 час до соединения 2. Gem вводят внутривенно при двух уровнях доз, 24 и 36 мг/кг/инъекцию, Q2D×4 (MTD=36 мг/кг/инъекцию.). Соединение 2 вводят при двух различных уровнях доз: 60 и 80 мг/кг/инъекцию, внутривенно.
Фиг.17 (А) показывает, что комбинированная химиотерапия с паклитакселем (Ptxl) плюс соединение 2 вызывает синергическую противоопухолевую активность с точки зрения роста опухоли, против карциномы прямой кишки человека НСТ116. Фиг. 17 (В) показывает, что синергическая противоопухолевая активность, являющаяся результатом комбинированной терапии паклитакселя и соединения 2, также имеет место, когда регрессия опухоли и скорость лечения используют как конечные точки отклика канцирогенного эффекта. Паклитаксел вводят за 3 часа до соединения 2. Паклитаксел вводят внутривенно при 20 мг/кг/инъекцию, Q7D×4. Соединение 2 вводят двумя различными уровнями доз: 40 и 80 мг/кг/инъекцию, внутривенно.
Фиг.18A-18G показывают противораковое действие различных противоопухолевых агентов, используемых отдельно или в комбинации. Результаты показывают, что соединение 2 и Her-1 ингибитор (соединение 8) при использовании в комбинации вызывают синергическое цитотоксическое действие in vitro против Her-1 активированной SAL-2 раковой клеточной линии (сравнить фиг.18В с фиг.18D). Фиг.18А: клоногенная выживаемость SAL-2 клеточной линии под воздействием паклитакселя в течение 20 часов при указанных концентрациях. Фиг.18В, клоногенная выживаемость SAL-2 клеточной линии под воздействием соединения 2 в течение 20 часов при указанных концентрациях. Фиг.18 С: антагонистическое взаимодействие между паклитакселем и Her-1 ингибитором, соединение 8. SAL-2 клетки сначала обрабатывают соединением 8 в течение 20 часов до дополнительного воздействия паклитакселем в течение дополнительных 20 часов. Фиг.18D: синергическое взаимодействие между соединением 2 и Her-1 ингибитором, соединение 8. SAL-2 клетки сначала обрабатывают соединением 9 в течение 20 часов до дополнительного воздействия соединением 2 в течение дополнительных 20 часов. Соединение 2 повышает противоопухолевую активность Her-1 (EGFR) ингибитора Iressa® в Her-1 экспрессированной А431 модели ксенотрансплантанта карциномы чешуйчатой клетки человека в лишенных шерсти мышах. Фиг.18Е: Комбинированное действие Iressa® (200 мг/кг/прием, РО, QlD×11) и соединения 2 (60 мг/кг/инъекцию, внутривенно, Q2D×5). Iressa® терапию проводят за 3 дня до начала лечения соединением 2. Фиг.18F: Комбинированное действие Iressa® (200 мг/кг/прием, РО, Q1D×11) и соединения 2 (80 мг/кг/инъекцию, внутривенно, Q2D×5). Фиг.18G: Комбинированное действие Iressa® (200 мг/кг/прием, РО, Q1D×11) и паклитакселя (24 мг/кг/инъекцию, внутривенно, Q2D×5).
Фиг.19 показывает, что комбинированное лечение соединением 2 и герцептином приводит к синергической антипролиферативной активности против ВТ474 клеточной линии карциномы молочной железы человека. Лечение Herceptin® проводят за 2 дня до соединения 2, как показано на чертеже.
Фиг.20 Соединение 2 усиливает противоопухолевую активность Тамоксифена в MCF-7 эстрогензависимой ксенотрансплантантной моделе карциномы груди человека в лишенных шерсти мышах. Тамоксифен вводят РО, Q2D×14. Соединение 2 вводят РО в течение 2 курсов, Q1D×10, как проиллюстрированно на чертеже.
Фиг.21 Соединение 2 усиливает противоопухолевую активность хирургической кастрации в андрогензависимой ксенотрансплантантной модели карциномы простаты человека MDA-PCa-2b в лишенных шерсти мышах. Хирургическую кастрацию осуществляют на 21 день после имплантации опухоли. Соединение 2 и терапию паклитакселя начинают спустя 3 дня после хирургической кастрации. Соединение 2 вводят РО, Q1D×10. Паклитаксел вводят IV, Q2D×5.
Фиг.22 Соединение 2 повышает противоопухолевую активность ингибитора андрогенового рецептора Casodex® против андрогензависимой, Casodex®-чувствительной карциномы простаты человека на ксенотрансплантантной моделе MDA-РСа-2b-AI в лишенных шерсти мышах. Casodex® вводят РО, Q1D×10. Терапию Соединения 2 начинают спустя 3 дня после введения Casodex®. Соединение 2 вводят РО, QlD×10.
Фиг.23 показывает, что комбинация ингибитора CDK (CDKI), Соединения 9, с Соединением 2 обеспечивает последовательно-зависимое, синергическое цитотоксическое воздействие на А2780 раковые клетки яичников in vitro. 4-часовое лечение с помощью 1.5 мкМ Соединения 9 (неэффективная доза) объединяют с 20 часовым лечением в повышающихся концентрациях соединения 2. Образование колоний оценивают на 10 день. Panel А. Обработка CDKI предшествует Соединению 2. Panel В, обработка Соединением 2 предшествует CDKI.
Детальное описание изобретения
Преимущественно настоящее изобретение представляет собой способ синергического лечения рака, который включает введение синергически терапевтически эффективного количества (1) по крайней мере одного агента, выбранного из группы, состоящей из антипролиферативного цитотоксического агента и антипролиферативного цитостатического агента, и (2) соединения формулы I, такому виду как млекопитающее, предпочтительно человеку, нуждающемуся в этом.
Неожиданно было найдено, что применение (1) по крайней мере одного антипролиферативного цитотоксического агента и/или антипролиферативного цитостатического агента и (2) соединения формулы I, когда их используют в комбинации, обеспечивает синергический способ лечения рака. Как его используют здесь, термин "синергический" означает, что эффект, достигнутый с помощью способов и композиций настоящего изобретения, оказывается выше, чем сумма эффектов, полученных от способов и композиций, содержащих цитотоксический или цитостатический агент или агенты и соединение формулы I настоящего изобретения по отдельности и в количествах, используемых в предложенных способах и композициях. Преимущество такого синергизма между активными инградиентами позволяет использовать меньшие дозы одного или обоих активных ингредиентов, позволяет использовать более низкие дозы противоопухолевых агентов или агента, лучевой терапии, обеспечивает большую эффективность при одних и тех же дозах и/или предотвращает или замедляет наращивание мультилекарственной резистентности.
Кроме того, дополнительные преимущества по сравнению с раскрытыми ранее способами заключаются в способности настоящей комбинации соединений формулы I и по крайней мере одного агента, выбранного из группы, состоящей из цитостатического агента и цитотоксического агента(ов) к индивидуальному варьированию в зависимости от природы раковых клеток, которые должны быть обработаны. Также предвидится, что терапевтический эффект от настоящей композиции может быть достигнут с меньшими количествами цитотоксического или цитостатического агента(ов) и соединения формулы I, которые потребуются, если такие противоопухолевые агенты и соединения формулы I будут вводиться по одному. Такой подход исключает любые не базирующиеся на механизме вредные токсические эффекты, которые могут появиться от введения определенного количества противоопухолевого агента или агентов, или соединения формулы I, или одной лучевой терапии, достаточных для достижения того же самого терапевтического эффекта. Настоящие композиции достигают синергический терапевтический эффект и демонстрируют неожиданное терапевтическое преимущество над эффективностью любого из компонентов соединений или способов, которые используют по одному.
Степень селективности двух или более противоопухолевых агентов, которые составляют способ по настоящему изобретению, обеспечивает терапевтические преимущества над ранее раскрытыми способами использования одного противоопухолевого агента для лечения рака. В частности, использование двух или более независимых фармацевтически активных компонентов, которые обладают комплементарными, по существу неперекрывающимися активностями, позволяет врачу, использующему настоящий способ лечения, независимо и точно варьировать активность комбинации без необходимости синтезировать единственное лекарственное средство, имеющее определенный фармацевтический профиль. В дополнение, такие комбинации должны эффективно поражать как пролиферативные, так и непролиферативные клетки.
Антипролиферативный цитотоксический агент(ы), который включает лучевую терапию, может быть применен одновременно с или перед соединением формулы I. В предпочтительном воплощении настоящего изобретения антипролиферативный цитотоксический агент(ы) вводят и/или лучевую терапию осуществляют до применения соединения формулы I. Как используют здесь, термин "одновременный" или "одновременно" означает, что антипролиферативный цитотоксический агент(ы) или лучевую терапию и соединение формулы I вводят в течение 24 часов, предпочтительно 12 часов, более предпочтительно 6 часов и более предпочтительно 3 часа или менее, каждого из остальных.
В дополнение к антипролиферативному цитотоксическому агенту(ам) и лучевой терапии, описанных выше, агенты, которые заставляют клетки становиться "непролиферативными" или "неактивными", которые определяют здесь как "антипролиферативные цитостатические агенты" или "неактивные агенты," могут необязательно быть введены пациенту, нуждающемуся в этом. Антипролиферативные цитостатические агенты могут быть введены одновременно или последовательно с соединением формулы I или лучевой терапией или цитотоксическим агентом(ами).
Настоящее изобретение предусматривает способы синергического лечения разнообразных раковых образований, включая в том числе, но не ограничиваясь ими, следующие: карцинома, в том числе мочевого пузыря (в том числе быстротекущего и метастатического рака мочевого пузыря), молочной железы, толстой или прямой кишки (в том числе проктологического рака), почки, печени, легкого (в том числе маленьких и немаленьких клеток рака легкого и аденокарценомы легкого), яичников, простаты, яичка, мочеполового тракта, лимфатической системы, гортани, поджелудочной железы (в том числе внешнесекреторная карцинома поджелудочной железы), пищевода, желудка, желчного пузыря, шеи, щитовидной железы и кожи (в том числе сквамозная карцинома клетки);
кроветворные опухоли лимфоидного происхождения, в том числе лейкемия, острая лимфоцитарная лейкемия, острая лимфобластная лейкемия, лимфома В-клетки, лимфома Т-клетки, лимфома Ходкинса (Hodgkins), лимфома не-Hodgkins, лимфома опасной клетки, гистоцитарная лимфома и лимфома Бюркета (Burketts лимфома);
кроветворные опухоли миелоидного происхождения, в том числе острая и хроническая миеломная лейкемии, миелодисплазийный синдром, миелоидная лейкемия и промиеломоноцитная лейкемия;
опухоли центральной и периферийной нервной системы, в том числе астроцитома, нейробластома, глиома и шваномасс;
опухоли мезенхимального происхождения в том числе фиброкарцинома, рабдомиокарцинома, и остеокарцинома и другие опухоли, в том числе меланома, остеокарцинома, пигментолсан, кератоактантома, симинома, фолликулярный рак щитовидной железы и тератосаркинома.
Более предпочтительно изобретение используют для лечения быстротекущих или метастатических раковых образований мочевого пузыря, рака поджелудочной железы, рака предстательной железы, рака небольших клеток легкого, проктологического рака и рака молочной железы.
В предпочтительном воплощении настоящего изобретения способ обеспечивает синергическое лечение раковых опухолей. Преимущественно синергический способ по настоящему изобретению подавляет развитие опухолей, подавляет бремя опухоли или вызывает регрессию опухоли в организме млекопитающего.
Как используют здесь, определение "лучевая терапия" включает, но не ограничивается, рентгеновским излучением или гамма-излучением, которое поступает либо из внешнего применяемого источника, такого как лазер, или путем имплантации небольших источников излучения. Лучевая терапия может также рассматриваться как антипролиферативный цитотоксический агент.
Как используют здесь, определение "противоопухолевый агент" представляет собой синоним "химиотерапевтического агента" и относится к соединениям, которые предотвращают размножение раковых клеток (т.е. антипролиферативные агенты). Вообщем, агент(ы) по настоящему изобретению попадают в два класса, антипролиферативные цитотоксический и антипролиферативные цитостатические. Цитотоксические агенты предотвращают раковые клетки от размножения за счет того, что (1) лишают клетку способности репродуцировать ДНК и (2) вызывают некроз клетки и/или апоптоз раковых клеток. Антипролиферативные цитостатические или неактивные агенты действуют через модуляцию, предотвращение или ингибирование процессов импульсной модулярной трансдукции клетки, которая регулирует размножение клетки. Большинство химиотерапевтических агентов являются цитотоксическими, которые поражают пролиферативные клетки.
Классы соединений, которые могут быть использованы в качестве антипролиферативных цитотоксических агентов включают следующие:
Алкилирующие агенты (в том числе без ограничения азотные иприты, производные этиленимина, алкилсульфонаты, нитрозомочевины и триазены): урациловый иприт, хлорметин, циклофосфамид (Cytoxan®), ифосфамид, мелфалан, хлорамбуцил, пипоброман, триэтиленмеламин, триэтилентиофосфорамин, бузулфан, кармустин, ломустин, стрептозоцин, дакарбазин и темозоломид.
Антиметаболиты (в том числе без ограничесния антагонисты фолиевой кислоты, аналоги пиримидина, аналоги пурина и ингибиторы аденозин деаминазы): метотрексат, 5-фторурацил, флоксуридин, цитарабин, 6-меркаптопурин, 6-тиогуанин, фосфат флударабина, пентостатин и гемцитабин.
Природные продукты и их производные (например, винка алкалоиды, противоопухолевые антибиотики, ферменты, лимфокины и эпиподофиллотоксины): винбластин, винкристин, виндезин, блеомицин, дактиномицин, даунорубицин, доксорубицин, эпирубицин, идарубицин, Ара-С, паклитаксел (паклитаксел имеет коммерческое название Taxol®), митрамицин, деоксоформицин, митомицин-С, L-аспаргиназа, интерфероны (особенно IFN-a), этопозид и тенипозид.
Другие антипролиферативные цитотоксические агенты представляют собой навелбен, СРТ-11, анастразол, летразол, капецитабин, релоксафин, циклофосфамид, ифозамид и дролоксафин.
Агенты, поражающие микроканальцы, препятствуют клеточному делению и хорошо известны из уровня техники благодаря их антипролиферативной цитотоксической активности. Агенты, поражающие микроканальцы, пригодные по изобретению, включают без ограничения аллоколхицин (NSC 406042), Халихлондрин В (NSC 609395), колхицин (NSC 757), производные колхицина (например, NSC 33410), доластатин 10 (NSC 376128), маитанзин (NSC 153858), ризоксин (NSC 332598), паклитаксел (Taxol®, NSC 125973), производные Taxol® (например, производные (например, NSC 608832), тиоколхицин NSC 361792), тритилцистеин (NSC 83265), винбластин сульфат (NSC 49842), винкристин сульфат (NSC 67574), природные и синтетические эпотилоны, включающие в том числе без ограничения эпотилон А, эпотилон В и дискодермолид (see Service, (1996) Science, 274:2009), эстрамустин, нокодазол, МАР4 и тому подобное. Примеры таких агентов также описаны в научной и патентной литературе, см, например, Bulinski (1997) J. Cell Sci. 110:3055 3064; Panda (1997) Proc. Nat. Acad. Sci. USA 94:10560-10564; Muhlradt (1997) Cancer Res. 57:3344-3346; Nicolaou (1997) Nature 387:268-272; Vasquez (1997) Mol. Biol. Cell. 8:973-985; Panda (1996) J. Biol. Chem 271:29807-29812.
Термин "паклитаксел", как его используют здесь, относится к коммерчески доступному лекарственному средству Taxol® (NSC номер: 125973). Taxol® ингибирует репликацию эукариотической клетки путем усиления полимеризации тубилиновых остатков в стабилизированных пучках микроканальцев, которые не способны организоваться в подходящие структуры из-за митоза. Из многих пригодных химиотерапевтических лекарственных средств паклитаксел вызывает интерес благодаря его эффективности в клинических испытаниях против устойчивых к лекарственным средствам опухолям, включая опухоли яичников и молочной железы (Hawkins (1992) Oncology, 6: 17-23, Horwitz (1992) Trends Pharmacol. Sci. 13: 134-146, Rowinsky (1990) J. Natl. Cane. Inst. 82: 1247-1259).
Особенно предпочтительные антипролиферативные цитотоксические агенты представляют собой соединения с активностью, аналогичной активности паклитакселя. Они включают без ограничения паклитаксел и производные паклитакселя (паклитакселподобные соединения) и аналоги. Паклитаксел и его производные являются коммерчески доступными. Кроме того, способы получения паклитакселя и производных паклитакселя, а также аналогов хорошо известны специалистам в данной области (см., например, US Patent Nos: 5,569,729; 5,565,478; 5,530,020; 5,527,924; 5,508,447; 5,489,589; 5,488,116; 5,484,809; 5,478,854; 5,478,736; 5,475,120; 5,468,769; 5,461,169; 5,440,057; 5,422,364; 5,411,984; 5,405,972 и 5,296,506).
Таким образом, антипролиферативные цитотоксические агенты, которые пригодны для применения в способах и композициях настоящего изобретение, включают без ограничения агенты, стабилизирующие микроканальцы, такие как паклитаксел (также известный как Taxol®), доцетаксел (также известный как Taxotere®), 7-O-метилтиометилпаклитаксел (раскрытый в US 5,646,176), 4-дезацетил-4-метилкарбонатпаклитаксел, 3'-трет-бутил-3'-N-трет-бутилоксикарбонил-4-децетил-3'-дефенил-3'-N-дебензоил-4-O-метоксикарбонилпаклитаксел (раскрытый в USSN 60/179,965 filed on February 3, 2000, и пример 17 здесь), С-4 метилкарбонатпаклитаксел (раскрытый в WO 94/14787), эпотилон А, эпотилон В, эпотилон С, эпотилон D, дезоксиэпотилон А, дезоксиэпотилон В [1S-[1R*,3R*(E),7R*,10S*,11R*,12R*,16S*]]-7-11-дигидрокси-8,8,10, 12, 16-пентаметил-3-[1-метил-2-(2-метил-4-тиазолил)этенил]-4-аза-17 оксабицикло [14.1.0]гептадекан-5,9-дион (раскрытый в WO 99/02514), [1S-[1R*,3R*(Е),7R*,10S*,11R*,12R*,16S*]]-3-[2-[2-(аминометил)-4-тиазолил]-1-метилэтенил]-7,11-дигидрокси-8,8,10,12,16-пентаметил-4-17-оксабицикло[14.1.0]-гептадекан-5,9-дион (раскрытый в USSN 09/506,481 filed on February 17, 2000 и примеры 7 и 8, приведенные здесь) и его производные, а также агенты, разрушающие микроканальцы.
Также пригодными являются цитотоксические агенты, такие как эпидофиллотоксин; противоопухолевый энзим; ингибитор топоизомеразы; прокарбазин; митоксантрон; координационные комплексы платины, такие как цис-платин и карбоплатин; биологически чувствительные модификаторы; ингибиторы роста; антигормональные терапевтические агенты; лейкворин; тегафур и плазмотопоитические факторы роста.
Кроме того, антипролиферативные цитотоксические агенты включают мелфалан, гексаметилмеламин, тиотепу, цитарабин, идатрексат, триметрексат, дакарбазин, L-аспарагиназу, камтофецин, топотекан, бикалутамид, флутамид, лейпролид, производные пиридобензоиндола, интерфероны и интерлейкины. Предпочтительные классы антипролиферативных цитотоксических агентов представляют собой EGFR ингибиторы, Her-2 ингибиторы, CDK ингибиторы и Herceptin® (трастузумаб). Некоторые особенно предпочтительные антипролиферативные цитостатические агенты представляют собой паклитаксел, цис-платин, карбоплатин, эпотилоны, гемцитабин, СРТ-11,5-фторурацил, тегафур, лейковорин и EGFR ингибиторы, такие как Iressa® (ZD 1839,4-(3-хлор-4-фторфениламино)-7-метокси-6-(3-(4-морфолинил)пропокси)хиназолин и OSI-774 (4-(3-этинилфениламино)-6,7-бис(2-метоксиэтокси)хиназолин.
В одном воплощении настоящего изобретения пролиферативные раковые клетки оказываются непролиферативными перед или во время лечения в соответствии с настоящим изобретением при обработке цитостатическим агентом. Как используют здесь, "цитостатический агент" представляет собой синоним "неактивного агента" и относится к любым агентам, снижающим скорость деления клетки или роста опухоли, так что клетки становятся непролиферативными или, таким образом, что их поведение приближается к поведению непролиферативных клеток. Типичные антипролиферативные цитостатические или "неактивные" агенты по изобретению включают без ограничения гормоны и стероиды (в том числе синтетические аналоги): 17α-Этинилэстрадиол, Диэтилстилбестрол, Тестостерон, Преднизон, Флуоксиместерон, Дромостанолон пропионат, Тестолактон, Мегестролацетат, Метилпреднизолон, Метилтестостерон, Преднизолон, Триамцинолон, Хлортрианизен, Гидроксипрогестерон, Аминоглутетимид, Эстрамустин, Медроксипрогестеронацетат, Лейпролид, Флутамид, Торемифен, Золадекс.
Также пригодны для применения в качестве цитостатических агентов, агенты, предотвращающие развитие кровеносных сосудов, такие как матричные ингибиторы маталлопротеиназы и другие VEGF ингибиторы, такие как анти-VEG антитела и небольшие молекулы, такие как ZD6474 и SU6668, которые также включены. Анти-Нег2 антитела из Genetech могут быть также использованы. Пригодный EGFR ингибитор представляет собой ЕКВ-569 (необратимый ингибитор). Также включено Imclone антитело С225, иммуноспецифическое для EGFR, и src ингибиторы.
Кроме того, пригодными для применения в качестве антипролиферативного цитостатического агента является Казодекс (Casodex®) (бикалутамид, Astra Zeneca), который оказывается андрогензависимым карциномным непролиферативным. Еще один пример цитостатического агента представляет собой антиэстрогенный Тамоксифен, который ингибирует пролиферацию или рост эстрогензависимого рака молочной железы. Ингибиторы трансдукции клеточных пролиферативных импульсов представляют собой цитостатические агенты. Примерами являются эпидермальные ингибиторы фактора роста, Her-2 ингибиторы, ингибиторы МЕК-1 киназы, ингибиторы МАРК киназы, PI3 ингибиторы, Src ингибиторы киназы и ингибиторы PDGF.
Как указывалось, цитостатические агенты также содержат агенты, предотвращающие развитие кровеносных сосудов, а также антиваскулярные агенты, которые путем прерывания потока крови в твердых опухолях делают раковые клетки неактивными из-за отсутствия питания. Кастрация, которая также делает андрогензависимую карциному непролиферативной, также может быть использована. Голодание другими средствами, отличными от хирургического разрушения потока крови, является другим примером цитостатического агента. Особенно предпочтительный класс антиваскулярных цитостатических агентов относится к комбретастатинам. Другие примеры цитостатических агентов включают ингибиторы MET киназы, ингибиторы MAP киназы, ингибиторы нерецепторных и рецепторных тирозинкиназ, интегринсигнальные ингибиторы и ингибиторы рецепторов инсулинподобных факторов роста.
В предпочтительных воплощениях изобретения способ включает введение комбинации двух или более противоопухолевых агентов. Например, данные, приведенные в описании, показывают, что ксенотрансплантанты рака простаты человека MDA-PCa-2b оказываются неактивными в случае хирургической кастрации, произведенной в организме животных перед лечением их с помощью соединения формулы I.
Способы безопасного и эффективного введения большинства из этих химиотерапевтических агентов хорошо известны специалисту в этой области. Кроме того, их введение описано в стандартной литературе. Например, введение большинства химиотерапевтических агентов описано в "Physicians' Desk Reference" (PDR), например, 1996 edition (Medical Economics Company, Montvale, NJ 07645-1742, USA), которая включена в описание в качестве ссылки.
Предпочтительными соединениями формулы I являются те, в которых
R1 представляет собой Br или CN;
R2 представляет собой необязательно замещенный бензил;
R3 представляет собой необязательно замещенный низший алкил, необязательно замещенный фенил, необязательно замещенный 2-тиенил или необязательно замещенный 1-пиперидинил;
R4 представляет собой водород или метил;
Z1 представляет собой CO, SO2 или SO2N(R5)-;
R5 представляет собой необязательно замещенный низший алкил или необязательно замещенный фенил;
и имеет значение 1.
Более предпочтительными соединениями формулы I для применения в способах и композициях для настоящего изобретения являются те, в которых
R1 представляет собой CN;
R2 представляет собой необязательно замещенный бензил;
R3 представляет собой необязательно замещенный низший алкил, необязательно замещенный фенил, необязательно замещенный 2-тиенил или необязательно замещенный 1-пиперидинил;
R4 представляет собой водород или метил;
Z представляет собой СО или SO2;
n имеет значение 1.
Наиболее предпочтительными соединениями формулы I для применения в настоящем изобретении являются те, в которых
R1 представляет собой CN;
R2 представляет собой бензил;
R3 представляет собой н-пропил, н-бутил, 3-метоксипропил, 2-тиенил, 5-бром-2-тиенил, фенил, 4-метоксифенил или 1-пиперидинил;
R4 представляет собой водород;
Z представляет собой SO2;
n имеет значение 1.
Соединения формулы I, которые особенно пригодны для способов и композиций настоящего изобретения, включают
(R)-2,3,4,5-тетрагидро-1-(1H-имидазол-4-илметил)-3-(фенилметил)-4-(2-тиенилсульфонил)-1Н-1,4-бензодиазепин-7-карбонитрил;
(R)-7-циано-2,3,4,5-тетрагидро-1-(1Н-имидазол-4-илметил)-4-(1Н-имидазол-4-илметил)-4-)1-оксобутил)-3-(фенилметил)-1Н-1,4-бензодиазепин;
(R)-4-[(5-бром-2-тиенил)сульфонил]-7-циано-2,3,4,5-тетрагидро-1-(1Н-имидазолил илметил)-3-(фенилметил)-1Н-1,4-бензодиазепин;
(R)-7-циано-2,3,4,5-тетрагидро-1-(1Н-имидазол-4-илметил)-4-[(4-метоксифенил)сульфонил]-3-(фенилметил)-1Н-1,4-бензодиазепин;
(R)-7-циано-2,3,4,5-тетрагидро-1-(1Н-имидазол-4-илметил)-3-(фенилметил)-4-(фенилсульфонил)-1Н-1,4-бензодиазепин;
(R)-7-циано-2,3,4,5-тетрагидро-1-(1Н-имидазол-4-илметил)-3-(фенилметил)-4-(пропилсульфонил)-1Н-1,4-бензодиазепин;
(R)-4-(бутилсульфонил)-7-циано-2,3,4,5-тетрагидро-1-(1Н-имидазол-4-илметил)-3-(фенилметил)-1Н-1,4-бензодиазепин;
(R)-7-циано-2,3,4,5-тетрагидро-1-(1Н-имидазол-4-илметил)-3-(фенилметил)-4-(1-пиперидинилсульфонил)-1Н-1,4-бензодиазепин;
(R)-4-(3-метоксипропилсульфонил)-7-циано-2,3,4,5-тетрагидро-1-(1H-имидазол-4-илметил)-3-(фенилметил)-1Н-1,4-бензодиазепин; и их фармацевтически приемлемые соли.
Соль метансульфокислоты и (R)-2,3,4,5-тетрагидро-1-(1Н-имидазол-4-илметил)-3-(фенилметил)-4-(2-тиенилсульфонил)-1Н-1,4-бензодиазепин-7-карбонитрила особенно предпочтительна для применения в способах и композицих по настоящему изобретению.
В предпочтительном воплощении настоящего изобретения антипролиферативный цитотоксический агент выбирают из группы, состоящей из паклитакселя, доцетаксела, 7-O-метилтиометилпаклитакселя, 4-дезацетил-4-метилкарбонатпаклитакселя, 3'-трет-бутил-3'-N-трет-бутилоксикарбонил-4-деацетил-3'-дефенил-3'-N-дебензоил-4-O-метоксикарбонил-паклитакселя, С-4 метилкарбонатпаклитакселя, эпотилона А, эпотилона В, эпотилона С, эпотилона D, дезоксиэпотилона А, дезоксиэпотилона В [1S-[1R*,3R*(E),7R*,10S*,11R*,12R*,16S*]]7,1'1-дигидрокси-8,8,10,12,16-пентаметил-3-[1-метил-2-(2-метил-4-тиазолил)этенил]-4-аза-17-оксабицикло[14.1.0]гептадекан-5,9-диона и [1S-[1R*,3R*(E),7R*,10S*, 11R*,12R*,16S*]]-3-[2-[2-(аминометил)-4-тиазолил]-1-метилэтенил]-7, 11-дигидрокси-8,8,10,12,16-пентаметил-4,17-диоксабицикло[14.1.0]гептадекан-5,9-диона и соединения формулы I представляет собой (R)-2,3,4,5-тетрагидро-1-(1Н-имидазол-4-илметил)-3-(фенилметил)-4-(2-тиенилсульфонил)-1Н-1,4-бензодиазепин-7-карбонитрил или его фармацевтически приемлемую соль.
В другом предпочтительном воплощении изобретения цитотоксический агент представляет собой паклитаксел и соединение формулы I, которое представляет собой (R)-2,3,4,5-тетрагидро-1-(1Н-имидазол-4-илметил)-3-(фенилметил)-4-(2-тиенилсульфонил)-1Н-1,4-бензодиазепин-7-карбонитрил или его фармацевтически приемлемую соль.
В другом предпочтительном воплощении настоящего изобретения цитотоксический агент представляет собой S-[1R*,3R*(E),7R*,10S*,11R, 12R*,16S*]]7,11-дигидрокси-8,8,10,12,16-пентаметил-3-[1-метил-2-(2-метил-4-тиазолил)этенил]-4-аза-17-оксабицикло-[14.1.0]гептадекан-5,9-дион и соединение формулы I, которое представляет собой (R)-2,3,4,5-тетрагидро-1-(1Н-имидазол-4-илметил)-3-(фенилметил)-4-(2-тиенилсульфонил)-1Н-1,4-бензодиазепин-7-карбонитрил или его фармацевтически приемлемую соль.
Способы, в которых применяют цитостатический агент, выбранный из группы, состоящей из Iressa®, Herceptin®, Тамоксифена и хирургической или химической кастрации, особенно предпочтительны для применения в комбинациях способов по настоящему изобретению.
Когда описывают соединения по настоящему изобретению, термин "низший алкил" или "низший алк" (как часть другой группы) означает незамещенную алкильную группу, состоящую из 1 до 6, предпочтительно от 1 до 4 атомов углерода.
Термин "аралкил" относится к арильной группе, связанной непосредственно через низшию алкильную группу. Предпочтительно аралкильная группа представляет собой бензил.
Термин "арил" относится к моноциклическим или бициклическим ароматическим углеводородным группам, имеющим от 6 до 12 атомов углерода в кольце. Типичным арилом в данном случае является фенильная, нафтинильная и бифенильная группы.
Термин "гетероцикло" относится к полностью насыщенной или ненасыщенной, ароматической или неароматической циклической группе, которая представляет собой от 4 до 7-членную моноциклическую, от 7 до 11-членную бициклическую или от 10 до 15-членную трициклическую кольцевую систему, которая имеет по крайней мере один гетероатом в по крайней мере одном углеродсодержащем кольце. Каждое кольцо гетероциклической группы, содержащей гетероатом, может иметь 1, 2, 3 или 4 гетероатомов, выбранных из азота, кислорода и серы, где азотный и серный гетероатомы могут также необязательно быть окисленными и гетероатом азота может также необязательно быть кватернизирован. Гетероциклогруппа может быть связана с любым гетероатомом или углеродным атомом.
Типичные моноциклические гетероциклогруппы включают пирролидинил, пирролил, индолил, пиразолил, оксетанил, пиразолинил, имидазолил, имидазолинил, имидазолидинил, оксазолил, оксазолидинил, изоксазолинил, изоксазолил, тиазолил, тиадиазолил, тиазолидинил, изотиазолил, изотиазолидинил, фурил, тетрагидрофурил, тиенил, оксадиазолил, пиперидинил, пиперазинил, 2-оксопиперазинил, 2-оксопиперидинил, 2-оксопирролидинил, 2-оксазепинил, азепинил, 4-пиперидонил, пиридил, N-оксопиридил, пиразинил, пиримидинил, пиридазинил, тетрагидротиопиранил, тетрагидропиранил, морфолинил, тиаморфолинил, тиаморфолинил сульфоксид, тетрагидротиопиранилсульфон, тиаморфолинилсульфон, 1,3-диоксолан, тетрагидро-1,1-диоксотиенил, диоксанил, изотиазолидинил, тиетанил, тииранил, триазинил, триазолил и тому подобное.
Примеры бициклических гетероциклогрупп включают бензотиазолил, бензоксазолил, бензотиенил, хинолинил, хинолинил-N-оксид, тетрагидроизохинолинил, изохинолинил, бензимидазолил, бензопиранил, индолизинил, бензофурил, хромонил, кумаринил, циннолинил, хиноксалинил, индазолил, пирролопиридил, фуропиридинил (такие как фуро[2,3-с]пиридинил, фуро[3,1-b]пиридинил или фуро[2,3-b]пиридинил), дигидроизоиндолил, дигидрохиназолинил (такие как 3,4-дигидро-4-оксохиназолинил), бензизотиазолил, бензизоксазолил, бензодиазинил, бензофуразанил, бензотиопиранил, бензотриазолил, бензпиразолил, дигидробензофурил, дигидробензотиенил, дигидробензотиопиранил, дигидробензотиопиранил сульфон, дигидробензопиранил, индолинил, изохроманил, изоиндолинил, нафтиридинил, фталазинил, пиперонил, пуринил, пиридопиридил, хиназолинил, тетрагидрохинолинил, тиенофурил, тиенопиридил, тиенотиенил и тому подобное.
Когда группа относится к тем группам, которые являются необязательно замещенными, она может быть замещенной от одного до пяти, предпочтительно от одного до трех, заместителей таких как F, Cl, Br, I, трифторметил, трифторметокси, гидрокси, низший алкокси, циклоалкокси, гетероциклоокси, оксо, низший алканоил, арилокси, низший алканоилокси, амино, низший алкиламино, ариламино, аралкиламино, циклоалкиламино, гетероциклоамино, дизамещенные амины, в которых два аминозаместителя независимо выбраны из низшего алкила, арила или аралкила, низшего алканоиламино, ароиламино, аралканоиламино, замещенного низшего алканоиламино, замещенного ариламино, замещенного аралкиланоиламино, тиола, низшего алкилтио, арилтио, аралкилтио. циклоалкилтио, гетероциклотио, низшего алкилтионо, арилтионо, аралкилтионо, низшего алкилсульфонила, арилсульфонила, аралкилсульфонила, сульфонамида (например, SO2NH2), замещенного сульфонамида, нитро, циано, карбокси, карбамаила (например, CONH2), замещенного карбамаила (например, CONH-низший алкил, CONH-арил, CONH-аралкил или случаев, когда два заместителя, находящихся на атомах азота, независимо выбраны из низшего алкила, арила или аралкила), низшего алкоксикарбонила, арила, замещенного арила, гуандино и гетероциклов (например, индолила, имидазолила, фурила, тиенила, тиазолила, пирролидила, пиридила, пиримидила и тому подобное). При этом, как было отмечено выше, если заместитель дополнительно замещен, то он замещается F, Cl, Br, I, необязательно замещенным низшим алкилом, гидрокси, необязательно замещенным низшим алкокси, необязательно замещенньм арилом или необязательно замещенным аралкилом.
Все стреоизомеры соединений формулы I настоящего изобретения включены либо в виде их смеси, или в виде их чистой или существенно частой форме. Определения соединений формулы I охватывает все возможные стреоизомеры и их смеси. Определение формулы I в особенности охватывает рацемические формы и выделенные оптические изомеры, имеющие определенную активность.
Соединения формулы I могут быть получены в соответствии со способами, описанными в US 6,011,029. Все описания и патенты, приведенные здесь, включены как ссылки.
Соединения формулы I пригодны в различных формах фармацевтически приемлемых солей. Термин "фармацевтически приемлемая соль" относится к такой форме соли, которая будет пригодна для химика фармацевта, т.е. к тем, которые существенно нетоксичны и которые обеспечивают желаемые фармакокинетические свойства, приятный вкус, абсорбцию, распределение, метаболизм или экскрецию. Другие факторы, более практичные по природе, но также важные при выборе представляют собой стоимость сырого материала, легкость кристаллизации, выход, стабильность, гигроскопичность и текучесть полученной основы лекарственного средства. Соответственно фармацевтические композиции могут быть получены из активных ингредиентов или их фармацевтически приемлемых солей с фармацевтически приемлемым носителем.
Фармацевтически приемлемые соли соединений формулы I, цитотоксические агенты и цитостатический агенты, которые пригодны для применения в способах и композициях настоящего изобретения включают без ограничения соли, сформированные с различными органическими и неорганическими кислотами, такими как соляная кислота, гидроксиметансульфоновая кислота, бромистоводородная кислота, метансульфоновая кислота, серная кислота, уксусная кислота, трифторуксусная кислота, малеиновая кислота, бензоилсульфоновая кислота, толуолсульфоновая кислота, сульфаминовая кислота, гликолевая кислота, стеариновая кислота, молочная кислота, яблочная кислота, памовая кислота, сульфаниловая кислота, 2-ацетоксибензойная кислота, фумаровая кислота, толуолсульфоновая кислота, метансульфоновая кислота, этандисульфоновая кислота, щавелевая кислота, изэтионовая кислота и включает различные другие фармацевтически приемлемые соли, такие как, например, нитраты, фосфаты, бораты, тартраты, цитраты, сукцинаты, бензоаты, аскорбаты, салицилаты и тому подобное. Катионы, такие как ионы четвертичного аммония, включены как фармацевтически приемлемые противоионы для анионных частиц.
Предпочтительные соли соединений формулы I включают соли соляной кислоты, соли метансульфоновой кислоты и соли трифторуксусной кислоты с солями метансульфоновой кислоты, являющимися более предпочтительными. В дополнение фармацевтически приемлемые соли соединений формулы I могут быть сформированы со щелочными металлами, такими как натрий, калий и литий; щелочноземельными металлами, такими как кальций и магний; органическими основаними, такими как дициклогексиламин, трибутиламин и пиридин; и аминокислотами, такими как аргинин, лизин и тому подобное.
Фармацевтически приемлемые соли по настоящему изобретению могут быть синтезированы удобными химическими способами. Как правило, соли получают реакцией свободного основания или кислоты со стехометрическими количествами или избытком желаемой солеобразующей неорганической или органической кислоты или основания в пригодном растворителе или комбинации растворителей.
Настоящее изобретение также охватывает фармацевтические композиции, пригодные для лечения рака, пригодные для введения терапевтически эффективного количества комбинаций по настоящему изобретению, с или без фармацевтически приемлемыми носителями или растворителями. Синергические фармацевтические композиции по настоящему изобретению содержат необязательный антипролиферативный цитотоксический агент или агенты, необязательный неактивный агент, соединение формулы I и фармацевтически приемлемый носитель. Способы обуславливают применение цитотоксического и/или цитостатического агента в комбинации с соединением формулы I. Композиции по настоящему изобретению могут дополнительно содержать один или более фармацевтически приемлемый(ых) дополнительный(ых) компонент(ы), такие как квасцы, стабилизаторы, антимикробные агенты, буферы, красители, отдушки, присадки и тому подобное. Противоопухолевые агенты, необязательно цитостатические агенты (если это химическое соединение), соединения формулы I и композиции по настоящему изобретению могут быть введены орально или парентерально, включая внутривенные, внутримышечные, внутрибрюшные, подкожные, ректальные и местные пути введения.
Для орального применения противоопухолевые агенты, цитостатические агенты, соединения формулы I и композиции настоящего изобретение могут быть введены, например, в форме таблеток или капсул, порошков, дисперсионных гранул или облаток или как водные растворы или суспензии. В случае таблеток для орального применения, носители, которые обычно используют, включают лактозу, кукурузный крахмал, карбонат магния, тальк, сахар и смазывающие агенты, такие как стеарат магния, которые обычно добавляют. Для орального введения в форме капсулы пригодные носители включают лактозу, кукурузный крахмал, карбонат магния, тальк и сахар. Когда используют водные суспензии для орального введения, обычно добавляют эмульгированные и/или суспендированные агенты. В дополнение могут быть введены подсластители и/или приправы для оральной композиции. Для внутримышечного, внутрибрюшного, подкожного и внутривенного применения обычно используют стерильные растворы активных ингредиентов и рН растворов должна быть соответственно отрегулированной и буферизирована. Для внутривенного применения общая концентрация раствора(ов) должна контролироваться, чтобы получить изотоники. В предпочтительном воплощении настоящего изобретения соединения формулы I или их фармацевтически приемлемые соли формируют с сульфобутилэфиром-7-β-циклодекстрина или 2-гидроксипропил-β-циклодекстрином для внутривенного введения.
Для приготовления суппозиторов в соответствии с изобретением сначала расплавляют воски с низкой температурой плавления, такие как смесь глицеридов жирной кислоты или масло какао, после чего диспергируют гомогенно в воск активный ингредиент, например, путем перемешивания. Жидкие приготовления включают растворы, суспензиии, эмульсии. Расплавленную гомогенную смесь затем выливают в соответствующего размера формы и оставляют остывать, пока они затвердевают. Аэрозольные приготовления, пригодные для ингаляции, могут включать растворы и твердые вещества в виде порошка, который может быть в комбинации с фармацевтически приемлемым носителем, таким как инертный сжатый газ.
Также оказываются включенными приготовления, которые затем подвергаются превращениям за короткое время перед применением, в жидкие формы для либо орального, либо парентерального введения. Такие жидкие формы включают растворы, суспензии и эмульсии.
Соединения формулы I, такие как цитотоксические и цитостатические агенты, описанные здесь, могут также быть нанесены трансдермально. Трансдермальные композиции могут находится в форме кремов, лосьенов, аэрозолей и/или эмульсий и могут включать трансдермальные пластыри, матрицы или резервуарного типа, как это принято для выполнения задач, поставленных в этой области.
Комбинации настоящего изобретения могут также быть использованы вместе с другими хорошо известными терапиями, которые выбирают, исходя из их полезности с точки зрения той болезни, которую лечат.
Если составы содержат фиксированную дозу, активные ингредиенты в комбинациях композиций настоящего изобретения применяют в пределах интервалов доз, описанных ниже. Альтернативно цитотоксические агенты, цитостатические агенты и соединения формулы I могут быть введены раздельно в интервалах доз, описанных ниже. В предпочтительном воплощении настоящего изобретения противоопухолевый агент вводят в дозовом интервале, описанными ниже до введения соединения формулы I в дозовом интервале, описанным ниже.
В таблице 1 приведены предпочтительные химиотерапевтические комбинации и типичные дозы для применения в способах настоящего изобретения в том случае, когда указано "Соединение формулы I", любой из вариантов соединений формулы I рассмотрен для применения в химиотерапевтических комбинациях. Предпочтительно применяют Соединение 2.
В приведенной выше таблице 1 "5FU" обозначает 5-фторурацил, "Лейковорин" может быть использован как кальций леукворин, "UFT представляет собой 1:4 в мольном соотношении тегафур: урацил, и "Эпотилон" представляет собой предпочтительно соединение, описанное в WO 99/02514 или WO 00/50423, оба включены в описание как ссылки.
Поскольку таблица 1 раскрывает типичные интервалы доз соединений формулы I и конкретных противоопухолевых агентов по изобретению, при составлении фармацевтических композиций по изобретению клиницист может применять предпочтительные дозы, которые диктуются состоянием пациента, который подвергается лечению. Например, Соединение 2, соединение формулы I, может предпочтительно быть введено при дозе, лежащей в интервале от около 25-500 мг/м2, каждые три недели в течение такого времени, которое требуется для лечения. Предпочтительные дозы для цисплатина находятся примерно в интервале от около 75-120 мг/м2 для введения в течение трех недель. Предпочтительные дозы для карбоплатина находятся внутри интервала от около 200-600 мг/м2 или AUC около 0.5-8 мг/мл × минута; более предпочтительные дозы AUC составляют около 4-6 мг/мл × минута. Когда в применяемом способе используют лучевая терапия, предпочтительные дозы находятся в предпочтительном интервале от около 200-6000 cGY. Предпочтительные дозы для СРТ-11 находятся в предпочтительном интервале от около 100-125 мг/м2 один раз в неделю. Предпочтительные дозы для паклитакселя составляют примерно 130-225 мг/м2 каждые 21 дня. Предпочтительные дозы для гемцитабина находятся в предпочтительном интервале от около 80-1500 мг/м2, которые вводятся еженедельно. Предпочтительно UFT используют в пределах интервала от около 300-400 мг/м2 каждый день, когда его введение комбинируют с лейковорином. Предпочтительные дозы для лейковорина составляют примерно 10-600 мг/м2, вводимые еженедельно.
Фактически применяемая доза может варьироваться в зависимости от требования пациента и тяжести больного, которого лечат. Определение подходящей дозы для конкретной ситуации остается за специалистом. Как правило, лечение начинают с небольших доз, которые ниже оптимальной дозы для соединения. Затем дозу увеличивают небольшими количествами, пока не достигнут оптимального эффекта в сложившихся обстоятельствах. Для удобства общая дневная доза может быть разделена и вводиться порциями в течение дня по желанию. Может также применяться прерывистая терапия (например, одна неделя из трех или три недели из четырех) может также быть использована.
Определенные виды опухолей могут эффективно лечиться соединениями формулы I и множеством противораковых агентов. Такие тройные и четверные комбинации могут обеспечивать большую эффективность. В случаях использования таких тройных и четверных комбинациях могут применяться дозы, определенные выше. Другие такие комбинации в приведенной Таблице 1 могут таким образом включать "Соединение 2" в комбинации с (1) митоксантроном + преднизоном; (2) доксорубицином + таксаном; или (3) герцептином + таксаном. 5-FU может быть заменен на UFT в любой из этих комбинаций.
Когда применяют способы или композиции по настоящему изобретению, могут вводиться при желании другие агенты, использумые для модуляции роста опухоли или метастазов в клинических условиях, такие как антиэметики.
Также могут быть проведены клинические испытания, в которых применяют комбинацию химиотерапевтических способов по изобретению. В особенности предпочтительно при таких курсах введение путем примерно 3-часового вливания Taxol ® (135 мг/м2), а затем примерно 1-часовое вливание соединения 2 (50 мг/м2) с при трехнедельных интервалах. В другом курсе Taxol® (80 мг/м2) вливают примерно один час, а затем проводят вливание соединения 2 (80 мг/м2). Этот курс лечения протекает в течение недели. Другой курс заключается во введении тройной комбинации, включающей 3-часовое вливание Taxol® (135 мг/м2), а затем примерно в течение двадцати минут вливание карбоплатина (AUC==6), при этом Taxol® и карбоплатин оказываются примененными каждые три недели. В этом курсе соединение 2 вводят пациентам еженедельно в виде примерно одночасового вливания при 80 мг/м2.
Настоящее изобретение охватывает способ синергического лечения рака, в котором цитотоксический агент и/или цитостатический агент, а также соединение формулы I применяют одновременно или последовательно. Таким образом, в то время как фармацевтическая композиция, содержащая противоопухолевый(е) агент(ы) и соединение формулы I, могут быть успешно введена на протяжении одного конкретного курса лечения, предварительное введение цитотоксического или цитостатического агента(ов) могут быть успешно проведено во время другого курса лечения. Должно быть понятно, что настоящая комбинация противоопухолевого (ых) агента (ов) и соединения формулы I может быть использована совместно с другими методами для лечения рака (предпочтительно раковых опухолей) в том числе, но не ограничиваясь ими, лучевой терапией и хирургией. Кроме того, должно быть понятно, что цитостатический агент, как и любой другой, может быть применен последовательно или одновременно с любой или всеми другими синергическими терапиями.
Комбинации по настоящему изобретению могут также быть совместно введены с другими хорошо известными терапевтическими агентами, которые выбирают благодаря их особенной пригодности против заболеваний, которые лечат. Комбинации по настоящему изобретению могут быть альтернативно использованы последовательно с известными фармацевтически приемлемым(ми) агентом(ами), когда мультикомбинационный состав является неприемлемым.
Химиотерапевтический(е) агент(ы) и/или лучевая терапия могут применяться согласно методикам хорошо известным из уровня техники. Для специалиста в этой области является очевидным, что введение химиотерапевтического(их) агента(ов) и/или проведение лучевой терапии могут варьироваться в зависимости от заболевания, которое лечат и от изученного воздействия химиотерапевтического(их) агента(ов) и/или лучевой терапии на указанное заболевание. Также в соответствии со знаниями клиницистов терапевтические курсы (например, дозировка, время применения) могут варьироваться в зависимости от наблюдаемого эффекта от введения терапевтических агентов (т.е. противоопухолевого агента(ов) или лучевой терапии) на пациента и зависимости от наблюдаемой реакции со стороны болезни на введение терапевтических агентов.
В способах по настоящему изобретению соединение формулы I вводят одновременно или последовательно с цитотоксическим агентом(ами), и/или с лучевой терапией, и/или цитостатическим агентом. Так, не представляется необходимым, что химиотерапевтический(е) агент(ы) и соединение формулы I или лучевая терапия и соединение формулы I должны вводиться одновременно или по существу одновременно. Преимущество от одновременного или по существу одновременного введения полностью определяется клиницистом.
Также в общем соединение формулы I, необязательный цитостатический агент, и необязательный цитотоксический агент(ы) не обязательно должны быть введены в одной и той же фармацевтической композиции и могут из-за разных физических и химических свойств вводиться разными путями. Например, соединение формулы I может быть введено орально, для того чтобы способствовать и поддерживать хорошие уровни крови, в то время как химиотерапевтический(е) агент(ы) могут быть введены внутривенно. Определение вида и целесообразности введения, где это возможно, в одной и той же фармацевтической композиции является компетенцией клинициста. Первоначальное введение может быть осуществлено согласно известным специалистам курсов лечения и затем основываться на наблюдаемых эффектах, дозировки, пути введения и время введения могут изменяться специалистом в этой области.
Конкретный выбор соединения формулы I и цитотоксического агента(ов), и/или лучевой терапии, и/или цитостатического агента будет зависеть от диагноза больного, которого лечат, и от развития заболевания, а также соответствующего курса лечения. Соединение формулы I, и/или цитотоксический агент(ы), и/или цитостатический агент, и/или лучевая терапия могут быть применены совместно (например, одновременно, по существу одновременно или во время одного того же курса лечения) или последовательно в зависимости от природы пролиферативного заболевания, состояния пациента, и конкретного выбора химиотерапевтического(их) агента(ов), и/или цитостатического агента, и/или лучевой терапии, которые должны быть введены вместе (т.е. во время одного курса лечения) с соединением формулы I.
Если соединение формулы I, и цитотоксический агент(ы), и/или цитостатический агент не вводят одновременно или по существу одновременно, тогда первоначальный порядок введения соединения формулы I и химиотерапевтического(их) агента(ов), и/или цитостатического агента, и/или лучевой терапии могут варьироваться. Так, например, соединение формулы I может быть введено сначала вслед за введением цитотоксического (их) агента(ов) и/или лучевой терапии; или цитотоксический агент(ы) и/или лучевая терапия могут быть применены сначала с последующим введением соединения формулы I. Это альтернативное введение может повторяться на протяжении одного курса лечения. Определение порядка введения и число повторений введения каждого терапевтического агента во время курса лечения полностью находится в компетенции специалиста после проведенной оценки заболевания, которое лечат, и состояния пациента. Например, цитостатический(е) агент(ы) и/или лучевая терапия могут быть введены вначале, особенно если применяют цитотоксический агент. Лечение затем продолжают с применением соединения формулы I и необязательно с последующим применением цитостатического агента, пока курс лечения не будет завершен.
Так, в соответствии с опытом и знаниями практикующий клиницист может изменить на протяжение курса введение каждого компонента (терапевтического агента), т.е. соединение формулы I, цитостатический агент, цитотоксический(е) агент(ы) или лучевую терапию в соответствии с индивидуальными потребностями пациента в лечебном процессе.
Опытный клиницист при определении того, является ли лечение эффективным при вводимых дозах, будет учитывать общее состояние пациента, также как и более определенные признаки, стихание симптомов, относящихся к заболеванию, ингибирование роста опухоли, заметное сокращение новообразования или ингибирование метастазов. Размер опухоли может быть измерен стандартными методами, такими как радиологическое измерение, например CAT или MRI сканирование, и успешное измерение может быть использовано для вынесения суждения о том, что рост опухоли замедляется или нет или даже происходит обратное развитие. Стихание зависимых от заболевания симптомов, таких как боль и улучшение общего состояния, могут также оказать помощь при вынесении суждения об эффективности лечения.
Чтобы облегчить дальнейшее понимание изобретения, приведены следующие примеры, в основном, с целью иллюстрировать его более детально. Область изобретения не должна ограничиваться приведенными примерами, но она охватывает тот предмет изобретения, который включен в формулу изобретения.
Эксперимент.
Соединения:
Следующие обозначения используют в примерах для исследуемых соединений:
Соединение 1: [1S-[1R*,3R*(E),7R*10S*,11R*,12R*16S*]]-7,11-дигидрокси-8,8,10,12,16-пентаметил-3-[1-метил-2-(2-метил-4-тиазолил)этенил]-4-аза-17-оксабицикло[14.1.0]гептадекан-5,9-дион
Соединение 2: (R)-2,3,4,5-тетрагидро-1-(1Н-имидазол-4-илметил)-3-(фенилметил)-4-(2-тиенилсульфонил)-1Н-1,4-бензодиазепин-7-карбонитрил, гидрохлоридная соль
Соединение 3: (R)-4-(бутилсульфонил)-7-циано-2,3,4,5-тетрагидро-1-(1Н-имидазол-4-илметил)-3-(фенилметил)-1Н-1,4-бензодиазепин, гидрохлоридная соль
Соединение 4: (R)-4-(3-метоксипропилсульфонил)-7-циано-2,3,4,5-тетрагидро-1-(1Н-имидазол-4-илметил)-3-(фенилметил)-1Н-1,4-бензодиазепин, гидрохлоридная соль
Соединение 5: (R)-4-[(5-бром-2-тиенил)сульфонил]-7-циано-2,3,4,5-тетрагидро-1-(1Н-имидазол-4-илметил)-3-(фенилметил)-1Н-1,4-бензодиазепин, гидрохлоридная соль
Соединение 6: (R)-7-циано-2,3,4,5-тетрагидро-1-(1Н-имидазол-4-илметил)-3-(фенилметил)-4-(1-пиперидинилсульфонил)-1Н-1,4-бензодиазепин, гидрохлоридная соль
Соединение 7: (R)-7-циано-2,3,4,5-тетрагидро-1-(1Н-имидазол-4-илметил)-4-[(4-метоксифенил)сульфонил]-3-(фенилметил)-1Н-1,4-бензодиазепин, гидрохлоридная соль
Соединение 8: 4-(3-бромфениламино)-6,7-бис(метокси) хиназолин
Соединение 9: N-[5-[[[5-(1,1-диметилэтил)-2-оксазолил]метил]тио]-2-тиазолил]-4-пиперидинкарбоксамид.
Введение лекарственного средства:
Для введения соединения 1 (эпотилона) грызунам используют два различных наполнителя: (1) этанол/вода (1:9, об./об.) и (2) Cremophor®/этанол/вода (1:1:8, об./об.). Соединение 1 сначала растворяют в этаноле или смеси Cremophor*/этанол (50:50). Окончательное разведение до требуемой степени в дозе осуществляют менее, чем за 1 час до введения препарата. Для парентерального введения (IV) разбавление осуществляют в воде так, что дозированные растворы содержат определенную композицию наполнителей, описанную выше. Для орального применения (РО) разбавление осуществляют с 0.25 М натрийфосфатным буфером (рН 8.0) при соотношении 30/70, об./об. Паклитаксел растворяют в 50/50 смеси этанола и Cremophor* и хранят при 4°С; окончательное разбавление паклитакселя осуществляют непосредственно перед введением препарата с NaCl 0.9%. Объем всех впрыскиваемых соединений составляет 0.01 мл/г мышей и 0.005 мл/г крыс.
Испытания на образование колоний клоногенных клеток:
Способность соединений уничтожать клоногенные опухолевые клетки (клетки, которые способны делиться неограниченно с образованием колоний) in vitro оценивалась клоногенными испытаниями. В конце 16 часов лекарственного воздействия монослойную клеточную культуру диссоциируют обработкой клеток 0.05% трипсином в течение 5 минут при 37°С. Клетки ресуспендируют в полной среде (содержащей 10% FBS), рассчитанной с помощью Coulter Channelyzer, разбавляют и помещают в питательную среду с 5-кратным разбавлением в пластиковой чашке Петри для тканевой культуры. Клетки инкубируют во влажной атмосфере в течение 10 дней при 37°С. Колонии клеток окрашивают кристаллическим фиолетовым и колонии с >50 клетками на колонию записывают. Была определена концентрация, требуемая для уничтожения клоногенных НСТ-116 клеток карциномы прямой кишки человека на 90% (т.е. IC90).
BrdUrd метка асинхронно растущих опухолей:
BrdUrd растворяют в стерильном фосфат-буферном соляном растворе (PBS), рН 7.4, и вводят пролонгированным вливанием (24 часа) через хвостовую вену мышам в дозе 100 mpk. Мышей с новообразованием умерщвляют в конце периода вливания и опухоли подвергают BrdUrd/DNA анализу.
Распределение опухоли и окрашивание клеток:
Опухоли вырезают и измельчают ножницами, а затем диссоциируют, используя смесь ферментов, состоящую из 0.025% коллагеназы (Sigma Chemical Co., St Louis, МО), 0.05% проназы (Calbiochem, LaJolla, CA) и 0.04% ДНазы (Sigma) в течение 1 часа при 37°С. После удаления остатков пропусканием клеточной суспензии через 70 мкм нейлоновые сетки клетки промывают в PBS, собирают и ресуспендируют в 75% метаноле. Осевшие клетки хранят в холодильнике при 4°С до анализа.
В день испытания клетки осаждают в 75% метаноле, промывают один раз PBS, ресуспендируют в пепсине в 2N HCI (0.2 мг/мл) и инкубируют в течение 20 минут при 37°С. Клетки затем промывают дважды 1 мл PBS, содержащим 0.5% эмбриональной бычьей сыворотки (FBS) и 0.5% Tween 80. После двух промываний клетки ресуспендируют в 1 мл PBS, содержащим 2% FBS, и инкубируют при комнатной температуре в течение 20 минут. Клетки затем встряхивают и комочки ресуспендируют в 100 мкм анти-BrdUrd-FITC (10 μг/мл) (Boeringer Mannheim). После инкубационного периода в 45 минут клетки промывают в 1 мл PBS, содержащем 0.5% FBS и 0.5% Tween 80. Промытые клетки ресуспендируют в 1 мл РНазы (1 мг/мл) и инкубируют в течение 30 минут при комнатной температуре. Затем добавляют пропидиййодид (Sigma) в концентрации 10 мкм/мл в PBS.
In vivo противоопухолевые тесты
Ксенотрансплантанты опухоли человека определяют в Balb/c nu/nu лишенных шерсти мышах. Опухоли размножают как подкожные трансплантанты в подходящей мышиной линии, используя фрагменты новообразования, полученные из донорских мышей.
Требуемое количество животных, необходимых для определения заметной реакции (6-10), суммируют в начале эксперимента и каждому вводят подкожного имплантант фрагмента новообразования (˜50 мг) с 13-мерным троакаром. Для лечения опухолей ранней стадии животных опять объединяют до распределения на различные курсы лечения и контрольные группы. Для лечения животных с развитой фазой заболевания опухоли оставляют расти до предопределенного размера (опухоли вне области исключаются) и животных поровну распределяют на различные курсы лечения и контрольные группы. Лечение каждого животного основано на индивидуальном весе тела. Обрабатываемых животных контролируют ежедневно в течение курса лечения относительно токсичности/смертности. Каждую группу животных взвешивают до начала лечения (Wt1) и затем после последней лечебной дозы (Wt2). Различие в весе тела (Wt2-Wt1) дает величину токсичности лечения.
Реакцию опухоли определяют измерением ее кронциркулем дважды в неделю, пока опухоли не достигнут предопределенного "конечного" размера в 0.5 или 1.0 г. Вес новообразования (мг) вычисляют по формуле
Вес опухоли=(длина × ширина 2) + 2
Максимум толерантной дозы (MTD) определяют как уровень дозы, непосредственно выше которой немедленно проявляется избыточная токсичность (т.е. более, чем одна смерть). MTD часто эквивалентна оптимальной дозе (OD). Активность описана при OD. Обрабатываемые мыши, умершие до того, как их опухоли достигали конечного размера, считались умершими от токсичности лекарства. Ни одной контрольной мыши не умерло до того, как опухоли были меньше требуемого размера. Лечение групп с более чем одной смертью, вызванной лекарственной токсичностью, считалось имеющим избыточную токсичность лечения и их данные не включены в число соединений с противоопухолевой эффективностью.
Реакцию опухоли в конечной точке выражают с точки зрения замедления ее роста (Т-С величина), определенном как различие во времени (дни), требуемом для лечения опухолей (Т) для достижения предопределенного конечного размера по сравнению со временем контрольной группы (С).
Для определения степени уничтожения опухолевых клеток сначала вычисляют время удвоения объема опухоли (TVDT) по формуле
TVDT=Среднее время (дни) для достижения контрольной опухоли конечного размера - Среднее время (дни) для достижения контрольной опухоли половины конечного размера И, Log уничтоженных клеток (LCK)=Т-С÷(3.32 × TVDT)
Новообразование определяют как "вылеченное", когда не было определено заболевания в конце времени изучения (день >75 дней после имплантации опухоли); интервал между окончанием изучения и окончанием лекарственного лечения всегда был в 10 раз выше времени удвоения объема у любого отдельного типа новообразования.
Группа для курса лечения и для контроля обычно включает восемь мышей. Статистические анализы данных по реакции проводят, используя общий тест Gehan Wilcoxon.
ПРИМЕР 1
Опухоли, содержащие пролиферативные и непролиферативные клеточные популяции
В процессе роста опухоли развитие системы сосудов часто не может выдержать темп быстрой пролиферации злокачественной клеточной популяции. Следовательно, твердая масса новообразования обычно вызывает нарушение сети кровеносных сосудов, которые, в отличие от сосудов в нормальных тканях, непременно обеспечивают адекватную питательную поддержку клеткам новообразования для оптимального роста. Твердые опухоли, следовательно, содержат пролиферативные и непролиферативные опухолевые клетки. В более твердой опухоли непролиферативные опухолевые клетки составляют большинство всех клеточных популяций новообразования. Распространенные противораковые агенты влияют на биохимические процессы пролиферативных клеток.
Непролиферативные клеточные популяции новообразования и/или стационарные клетки только минимально подвергаются воздействию таких агентов. Следовательно, главный составляющий фактор неуспеха лучевой терапии или химиотерапии в лечении опухолевых заболеваний заключается в неспособности этих соединений воздействовать на неподвижную(неактивную) клеточную популяцию новообразования. В ксенотрансплантанте НСТ116 карциномы прямой кишки человека, используя пролонгированный (24 часа) инфузионный BrdUrd режим, было установлено, что пролиферативная клеточная фракция в различных опухолях in vivo составляет только небольшую фракцию от общей клеточной популяции. В 200-300 мг НСТ-116 новообразования только около 20% клеточной популяции содержит пролиферативные клетки, в то время как оставшиеся 80% составляют непролиферативные клетки (фиг.1). Также был исследован ряд других опухолей и в каждом случае непролиферативные клетки составляют большинство клеток, присутствующих в опухолях (таблица 2).
Ограниченное в питании микроокружение твердого новообразования может быть воспроизведено в клеточной культуре in vitro путем контролирования условий роста среды культуры. Следовательно, при уменьшении питательной поддержки (день 8 культуры) в клетках опухоли может быть вызвано непролиферативное или неподвижное (стационарная фаза, около 90% непролиферативных) состояние, тогда как при оптимальных условиях питания (день 3 культуры) практически все опухолевые клетки размножаются экспоненциально (log фаза, около 90% пролиферативных).
ПРИМЕР 2
Композиции по изобретению, селективно уничтожающие непролиферативные опухолевые клетки
Способность соединения 2 уничтожать клоногенные опухолевые клетки (клетки, которые способны неограниченно делиться с образованием колоний) in vitro оценивают клоногенными испытаниями. В конце 16 часов лекарственного воздействия монослойные клеточные культуры диссоциируют обработкой клеток 0.05% трипсином в течение 5 минут при 37°С. Клетки ресуспендируют в полной среде (содержащей 10% PBS), рассчитанной с помощью Coulter Channelyzer, разбавляют и помещают в питательную среду с 5-кратным разбавлением в пластиковой чашке Петри для тканевой культуры. Клетки инкубируют во влажной атмосфере в течение 10 дней при 37°С. Колонии клеток окрашивают кристаллическим фиолетовым и колонии с >50 клетками на колонию записывают. Определяют концентрацию, необходимую для уничтожения клоногенных НСТ-116 клеток карциномы прямой кишки человека на 90% (т.е. IC90).
Соединение 2 и его аналоги селективно уничтожают клетки непролиферативной (стационарной) НСТ116 карциномы прямой кишки человека по сравнению с пролиферативными (Log) клетками (фиг.2-4 и таблица 3). Степень (отношение) различий в чувствительности между пролиферативной и непролиферативной популяцией находится в пределах 2.7-181.
Большинство противораковых лекарственных средств влияют на синтез ДНК или ее функции и, следовательно, поражают пролиферативные клетки, в то время как остающиеся неактивные или неподвижные клетки остаются неповрежденными, пока такие клетки не делятся вскоре под воздействием лекарственного средства. Следовательно, эффективность противораковых средств часто ограничена указанной стойкой фракцией опухолевых клеток. Например, в прямо противоположно соединению 2 обычно используемые раковые агенты, такие как паклитаксел, эпотилон В и 5-фторурацил, являются предпочтительно цитотоксическими по отношению к пролиферативным клеткам опухоли и существенно неэффективными против непролиферативных клеток (фиг.5 и 6 и таблица 4).
ПРИМЕР 3
Антипролиферативные агенты в комбинации с соединениями по изобретению действуют синергично для уничтожения опухолевых клеток in vitro
Как описано выше, механистические изучения строго предполагают, что соединение 2 является селективно цитотоксическим по отношению к непролиферативным опухолевым клеткам. Эта уникальная особенность повышает перспективу синергической комбинированной терапии с существующими противораковыми средствами, которые исходно поражают пролиферативные опухолевые клетки. Например, паклитаксел высоко цитотоксичен в log фазе НСТ-116 клеток in vitro с IC90 величиной, составляющей 17.8 нМ (фиг.5). Однако против непролиферативной стационарной клеточной популяции он полностью нетоксичен при концентрациях таких высоких, как 162 нМ (фиг.5). Такое же сильное различие наблюдается с эпотилоном В (фиг.6) с IС90 1.3 и >108 нМ для пролиферативных и непролиферативных клеток соответственно (таблица 3). In vitro, комбинирование паклитакселя с соединением 2 приводит к синергической цитотоксичности, такой, что комбинация вызывает большее дополнительное (аддитивное) уничтожение клеток (фиг.7). Более того, было обнаружено, что для того, чтобы наблюдать эту синергическую комбинацию, два агента должны применяться в особой последовательности. Синергизм наблюдался, когда клетки обрабатывали двумя агентами одновременно или когда паклитаксел предшествовал соединению 2. Когда соединение 2 вводят до паклитакселя за 24 часа, наблюдался антагонизм. Подобный синергизм и зависимость от последовательности наблюдался при комбинировании соединения 2 и соединения 1 (фиг.8-10).
ПРИМЕР 4
Антипролиферативные агенты в комбинации с соединениями по изобретению действуют синергично для уничтожения опухолевых клеток в опухолевых ксенотрансплантатах человека
Комбинированную терапию с инфузионным соединением 2 плюс внутривенно болюсом паклитакселя осуществляют, используя sc HCT-116 модель опухоли (фиг.11). В то время как индивидуальные агенты, вводимые отдельно, являются неактивными (0.4 и 0.6 LCK для соединения 2 и паклитакселя соответственно), комбининация (соединение 2, 125 mpk 24 ч внутривенно вливанием плюс паклитаксел, 24 мг/кг внутривенно) приводит к получению 1.8 LCK синергического действия.
Комбинированная химиотерапия с соединением 2 и паклитакселем, вводимым с помощью болюса, также демонстрирует синергизм в Pat-7 клетках карциномы яичников человека, полученных от пациента, больного раком, выработавшего резистентность к TAXOL®. Паклитаксел и соединение 2, когда их дают в соответственно максимальных толерантных дозах, приводят к менее чем 1 log уничтожению клеток (LCK) как ответной реакции опухоли в этой модели резистентного новообразования, в случае лечения одним агентом. Однако комбинирование соединения 2 и паклитакселя в максимуме толерантных доз обеспечивает терапевтический синергизм и значительную противоопухолевую активность 1.7 LCK (фиг.12). Синергическую противоопухолевую активность также наблюдают в НСТ116 ксенотрансплантанте карциномы прямой кишки человека (фиг.13). Соединение 2 при уровнях доз, которые приводят к таким системным воздействиям, которые наблюдаются у пациентов (20-80 мкм·час), как было показано, проявляют синергизм с противоопухолевым действием паклитакселя.
Терапевтический синергизм также ясно проявляется при комбинировании соединения 1 и соединения 2 in vivo в ксенотрансплантантах карциномы прямой кишки человека HCT/VM46 с мультирезистентиостью к лекарственным препаратам. Оба соединения 1 и соединения 2 имеют умеренную противоопухолевую активность в этой модели при одиночном введении агента при лечении (фиг.13). Оба агента вызывают более чем 1 LCK ответную реакцию опухоли (1.6 и 1.1 LCK соответственно), но не вызывают ее лечения. Однако, когда два агента вводят в комбинации (соединение 1 на 24 часа позднее соединения 2), наблюдается значительное улучшение противоопухолевой активности. Замечено высокое усиление в замедлении роста опухоли (3.7 LCK), в том числе повышение лечащих эффектов наблюдалось у 3 из 7 мышей (фиг.13).
Была показана зависимость от последовательности комбинирования. При введении соединения 2 за 24 часа до соединения 1 терапевтический синергизм не наблюдался (фиг.14), комбинация действовала так, как будто вводят одно соединение 1.
ПРИМЕР 5
Соединения по изобретению повышают противоопухолевую активность in vivo ингибитора топоизомеразы I CPT-11 против твердой карциномы человека
СРТ-11 является антипролиферативным цитотоксическим противораковым агентом, который особенно взаимодействует с ферментом топоизомеразы I, который уменьшает торсионное напряжение в ДНК путем стимулирования реверсивных однонитевых промежутков. Цитотоксичность СРТ-11 обусловлена повреждением двухцепочечной ДНК в процессе синтеза ДНК, когда ферменты репликации взаимодействуют с третичным комплексом, образованным топоизомеразой 1-ДНК, и либо СРТ-11 или его активным метаболитом SN-38. Следовательно, СРТ-11 поражает селективно пролиферативные клетки, которые подвергаются активному синтезу ДНК. В этом примере показано, что комбинированное лечение мышей, несущих обширный (300-500 мг) НСТ 116 ксенотрансплантант карциномы прямой кишки человека с помощью СРТ-11 за 1 час до соединения 2, приводит к синергической противоопухолевой активности, которая намного превосходит активность каждого агента, введенного по отдельности (фиг.15А и 15В). В этом испытании СРТ-11 вводят внутривенно при или около MTD 30 мг/кг/инъекцию. Соединение 2 вводят двумя различными уровнями доз: 60 и 80 мг/кг/инъекцию, внутривенно. Эти дозы соединения 2 выбраны, потому что они создают лекарственные воздействия, которые приблизительно равны клинически доступному воздействию. Как показано на фиг.15А и 15В, оба уровня доз соединения 2 при их комбинировании с СРТ-11 вызывают значительно большую противоопухолевую активность, чем каждый из агентов отдельно. Более того, комбинирование вызывает значительно большие скорости частичной и полной регрессии опухоли, чем один агент отдельно.
Противоопухолевая активность комбинированной химиотерапии с соединением 2 и СРТ-11 против обширной НСТ116 карциномы прямой кишки человека
1Режим лекарственного лечения был внутривенным, Q2D × 5. СРТ-22 вводят за 1 час до соединения 2
2Опухоли находились в пределах 300-500 (средние ˜200 мг) во время инициации лечения
3LCK = Log уничтожения клеток
4PR = Частичная ответная реакция, уменьшение размера опухоли на 50% или более.
5CR = Полная ответная реакция, определенная как уничтожение при лечении всех легко определяемых в размерах опухолях. Новообразование определено как неизмеримое, когда оно составляет менее 35 мг. Полная реакция отличается от вылечивания тем, что могут оставаться микроскопические или оккультные опухоли, и они могут привести к последующей прогрессии опухоли.
ПРИМЕР 6
Соединения по изобретению повышают противоопухолевую активность in vivo гемцитабина (GEMZAR) против твердых опухолей человека
Гемцитабин является антиметаболитом типа антипролиферативного цитотоксического химиотерапевтического агента, который проявляет специфичность клеточной фазы. Агент в первую очередь уничтожает клетки, подвергающиеся синтезу ДНК (S-фаза) и также блокирует прохождение клеток через границу G1/S-фазы. Следовательно, гемцитабин является селективно токсичным против клеток, которые активно делятся или пролиферируют. Показано, что комбинирование гемцитабина с соединением 2 (гемцитабин вводят за 1 час до соединения 2) повышает противоопухолевую активность против обширных НТ-29 ксенотрансплантантов карциномы прямой кишки человека в лишенных шерсти мышах (фиг.16 А и 16В). Существенно повышенная противоопухолевая активность наблюдалась не только относительно замедления роста (LCK), но, что более важно, также, что она приводила к явному улучшению в скорости ответной реакции (регрессия опухоли). Следовательно, комбинирование соединения 2 с гемцитабином приводит к 100% PR у всех излечиваемых мышей, в то время как отдельный агент соединения 2 приводит к 12.5% PR и отдельный агент гемцитабин приводит к 0 и 25% скорости отклика соответственно при дозе 24 и 6 мг/кг/инъекцию (фиг.16В).
ПРИМЕР 7
Паклитаксел в комбинации с соединениями по изобретению действует синергично при уничтожении ракового ксенотрансплантанта прямой кишки человека
Паклитаксел уничтожает опухолевые клетки, как только они вступают в митоническую фазу цикла деления клетки. Следовательно, паклитаксел является селективно токсичным против пролиферативных клеток (фиг.5). Показано, что комбинация соединения 2 с паклитакселем вызывает синергическую противоопухолевую активность против модели карциномы прямой кишки человека, НСТ116, в лишенных шерсти мышах. Фиг.17А и 17В показывают, что внутривенное введение 40 или 80 mpk соединения 2 через три часа после внутривенного введения паклитакселя (20 mpk, неделя × 4) вызывает сильное повышение противоопухолевого действия по сравнению с одиночным агентом; степень терапевтического усиления явно превосходит степень, которую получают при использовании двух агентов, просто вызывающих дополнительную противоопухолевую активность. Это синергическое взаимодействие может быть рассмотрено не только относительно замедления роста опухоли, но также в отношении клинически релевантных измерений ответной реакции опухоли, а именно частичной и полной регрессии опухоли (или отклика PR и CR соответственно) и ее вылечивания. Данные приведены в таблице 6 и фиг.17С.
Противоопухолевая комбинированная химиотерапия с соединением 2 и паклитакселем против обширной НСТ116 карциномы прямой кишки человека
Соединение 2
1Режим лекарственного лечения - внутривенно, Q7D × 4. Паклитаксел вводят за 3 часа до соединения 2
2Опухоли находились в пределах 130-300 (средняя ˜200 мг) во время лечебного воздействия
3LCK = Log уничтоженных клеток
4PR = Частичная ответная реакция, уменьшение размера опухоли на 50% или более
5CR = Полная ответная реакция, определенная как уничтожение при лечении всех легко распознаваемых опухолей. Новообразование определяют как неизмеримое, когда оно составляет менее 35 мг. Полная ответная реакция отличается от вылечивания тем, что могут оставаться микроскопические или оккультные опухоли, и они могут приводить к последующей прогрессии опухоли
6Лечение - нет данных о продолжении заболевания на день 77 после имплантирования опухоли, что определялось при вскрытии.
ПРИМЕР 8
Her-1 ингибиторы (Соединение 8) в комбинации с соединениями по изобретению действуют синергично для уничтожения SAL-2 раковых клеток in vitro и А431 чешуйчатых клеток карциномы in vivo
Рецептор эпидермального фактора роста (EGF, или Her-1) играет роль во многих карциномах эпителиальных и чешуйчатых клетках человека. Поражение EGF с его рецепторами в клетках опухоли приводит к активизации протеинтирозинкиназной активности и автофосфорилированию тирозина. Это, с другой стороны, приводит к активации каскада биохимических ответных реакций, которые включены в сигнальную трансдукцию митогенных (пролиферативных) раковых клеток. Ингибирование EGF связывания рецептора, как было показано на чувствительных клетках, приводит к ингибированию клеточного размножения, задержке клеточного цикла и апоптозу. Следовательно, EGF или Her-1 ингибиторы селективно поражают пролиферативные клетки и при нарушении митогенной сигнальной трансдукции делают раковые клетки непролиферативными. Исходя из селективности соединений по настоящему изобретению, которые поражают непролиферативные клетки, была выдвинута гипотеза, что можно использовать преимущество этого особого способа действия Her-1 ингибиторов, в котором при введении раковых клеток в непролиферативное состояние будет "повышаться чувствительность" их к проапоптозной активности соединений 2 и родственным соединениям по настоящему изобретению. Эта гипотеза была доказана как in vitro, так и in vivo экспериментами. Для in vitro опытов, для того чтобы определить терапевтическую эффективность комбинации Her-1 ингибитора с соединением 2, были проведены эксперименты с использованием SAL-2 опухолевых клеток, которые системно сверхэкспрессируют активированную форму Her-1 рецепторов. Для этой цели используют Her-1 ингибитор, соединение 8. Известно, что паклитаксел используют в качестве примера агентов, которые поражают только пролиферативные клетки и в соответствии с такой гипотезой он не будет хорошо работать в настоящих экспериментальных условиях, т.е. последовательного лекарственного воздействия: Her-1 ингибитор сначала вызывает стаз клетки, далее вводят соединение 2 или паклитаксел. Данные показывают, что экспоненциальный рост SAL2 раковых клеток является относительно резистентным по отношению к соединению 2 и паклитакселю при введении каждого агента по отдельности, как показано на фиг.18A-18G. Последовательное комбинирование Her-1 ингибитора '832, а затем паклитаксел является антагонистичным, что предсказано нашей гипотезой (фиг.18С). Наоборот, последовательность комбинирования Her-1 ингибитора (соединение 8) с последующим введением соединения 2 является синергической (фиг.18D).
Для демонстрации эффективности объединения Her-1 ингибитора с соединениями настоящего изобретения in vivo применяют Her-1 ингибитор Iressa® в комбинации с соединением 2 против карциномы чешуйчатой клетки человека, А431, которая, как известно, сверхэкспрессирует Her-1 рецептор. Как показано на фиг.18Е и F, два различных уровня доз соединения 2 при использовании в комбинации с Iressa® вызывают повышение противоопухолевой активности по сравнению с введением каждого агента по отдельности. Следует заметить, что Iressa вводят в режиме ежедневного лечения, Q1D × 11, лечение которым начинают за 3 дня до начала лечения соединением 2: Соединение 2 вводят по Q2D × 5 схеме. Важно, этот режим последовательного лекарственного лечения (т.е. сначала Iressa®) плохо работает с паклитакселем (фиг.18G). Комбинирование Iressa® и паклитакселя вызывает противоопухолевое действие, которое не является лучшим, чем любой из агентов, использующийся отдельно.
ПРИМЕР 9
Комбинирование соединения 2 с Her-2, направленным на антитело Herceptin, оказывает синергичные ингибиторные действия на рост Her-2 экспрессированных раковых клеток молочной железы человека
HER-2 (или с-еrbВ2) протоонкоген кодирует протеин трансмембранного рецептора в 185 kDa, который структурно напоминает рецептор эпидермального фактора роста, или Her-1. Сверхэкспрессия HER2 протеина наблюдается в 25%-30% исходных раковых образованиях молочной железы. Herceptin® является рекомбинантным ДНК-производным моноклональным антителом человека, который селективно связывается с внеклеточным доменом Her-2 протеина. Herceptin®, как было показано в in vitro испытаниях и на животных, вызывает пролиферацию опухолевых клеток человека, которые сверхэкспрессируют Her-2. Следовательно, в соответствии с гипотезой, предложенной в настоящем изобретении, Herceptin® является примером класса агентов, который действует путем перевода раковых клеток в непролиферативные, таким образом повышая чувствительность опухолевых клеток к противораковому действию соединений настоящего изобретения, которые селективно поражают непролиферативные клетки. Следовательно, в этом примере предварительное лечение раковой клеточной линии молочной железы человека ВТ-474, которая сверхэкспрессирует Her-2, за 2 дня до лечения соединением 2, вызывает синергическое антипролиферативное действие на рост раковых клеток (фиг.19).
ПРИМЕР 10
Тамоксифен в комбинации с соединениями по изобретению действует синергично при уничтожении раковых клеток в ксенотрансплантатах эстрогензависимого рака молочной железы человека
Большинство раковых образований молочной железы человека нуждаются в эстрогене при росте и прогрессировании опухоли (эстрогензависимые). При лечении эстрогензависимых раковых образований молочной железы антиэстрогенами, такими как тамоксифен, как было показано, подавляется клеточное размножение опухоли in vitro и in vivo. Для определения эффективности объединения противораковых агентов по изобретению в модели рака молочной железы человека развивают ксенотрансплантат MCF-7 в лишенных шерсти мышах и обрабатывают их, как описано далее. Комбинированная химиотерапия при использовании соединения 2 и тамоксифена приводит к повышению противоопухолевой активности. Действительно, ремиссия опухоли наблюдается в группе от 3 до 8 мышей в модели ксенотрансплантанта MCF-7 рака молочной железы человека. Фиг.20 показывает, что когда соединение 2 вводят как единственный агент, оказывается очень небольшой эффект на рост опухоли. Тамоксифен в случае использования его одного оказывается значительно более активным в этой модели, приблизительно он так эффективен, как удаление эстрадиальных (estradial) гранул, но тем не менее после лечения с помощью тамоксифена или удаления гранул, все опухоли возобновляют рост и никакое лечение не наблюдается. В противоположность этому, 40 mpk тамоксифена в комбинации с соединением 2 (ро, 100 mpk, qdx×10, два курса лечения) приводят к разкому снижению среднего веса опухоли в дополнение к обозначенному выше лечению.
ПРИМЕР 11
Андрогенотсекающая терапия (хирургическая или химическая) усиливает чувствительность андрогензависимых опухолей простаты человека к соединению 2, вызывая непролиферативное состояние в опухолевых клетках (отсутствие активности)
Как было упомянуто ранее, клетки оказываются непролиферативными или "неактивными" под воздействием различных биологических условий. Один из приемлемых подходов к лечению опухоли простаты человека заключается в химической кастрации. Поскольку настоящее изобретение не строится ни на какой конкретной теории или механизме, можно определить, что в андрогензависимом раке простаты гормональное удаление действует как агент предварительной дезактивации путем удаления критического стимула роста в отношении пролиферации опухоли. Эффективность противоопухолевых агентов по настоящему изобретению была таким образом определена в моделях ксенотрансплантата рака простаты человека.
В андрогензависимой sc MDA-Pca-2b модели ксенотрансплантата карциномы простаты человека лечение с помощью одного соединения 2 (400 mpk, po, qld×10) имеет незначительный эффект на рост опухоли, как показано на фиг.21. Андрогенудаляющая терапия (хирургическая кастрация) также снижает скорость роста опухоли, но не вызывает регрессии. Как показывают данные (фиг.21), введение соединения 2 в течение 3 дней после кастрации приводит к быстрой регрессии опухоли.
Другой способ, при котором андрогензависимые клетки карциномы простаты оказываются непролиферативными, заключается в применении химических антиандрогенных агентов, таких как Casodex®. Действительно, в настоящее время известно, что некоторые карциномы простаты остаются чувствительными к Casodex® терапии даже после того, как они стали значительно андрогеннезависимыми и наоборот. Этот феномен по гипотезе является результатом специфических изменений, происходящих в андрогеновом рецепторе, который дифференциально воздействует на поражение андроген и антиандрогенового рецептора. Для исследования противоопухолевой эффективности объединенных Casodex® и соединения 2 была использована Casodex® чувствительная, но андрогеннезависимая модель простаты человека, например модель Рса-2b-AI. Как показано на фиг.22, объединенные эффекты Casodex® и соединения 2 были значительно выше на этой модели опухоли, чем эффекты воздействия каждого из агентов, при использовании их в отдельности, когда оба вводили при максимально переносимых (толерантных) дозах и режимах (600 mpk, po, q1D×10 соединение 2; 150 mpk, po, q1D×4 для Casodex® соответственно). Таким образом, по определению, комбинация Casodex® и соединения 2 обеспечивает синергическую противоопухолевую активность, которая превышает активность каждого из агентов, вводимых по одному.
ПРИМЕР 12
Соединение 2 в комбинации с CDK ингибитором (Соединением 9), создающее синергический эффект разрушения клеток в строго зависимой последовательности (фиг.23)
Митотическое деление клеток млекопитающих требует скоординированных действий семейства серин/треонин протеиновых киназ, известных как циклинзависимые киназы (CDKs). Ингибиторы CDKs, особенно CDK2, как было показано, подавляют пролиферацию клеток и задерживают клетки на G1/S границе клеточного цикла. Чтобы определить эффективность комбинированной химиотерапии с CDKs ингибитором (CDKI) и соединением настоящего изобретения, было проведено исследование, в котором экспотенциально пролиферированные клетки A2780s карциномы яичников человека, были обработаны в течение периода в 4 часа 1.5 мкм селективным CDK2 ингибитором. Соединение 9 (неэффективная доза) было объединено с обработкой в течение 20 часов при повышающейся концентрации соединением 2. Были осуществлены две последовательные обработки: (1) соединение 9 в течение 4 часов, а затем соединение 2 в течение 20 часов или (2)а соединение 2 в течение 20 часов, а затем соединение 9 в течение 4 часов. Как показано на фиг.23, синергическое разрушение клеток наблюдалось, когда CDKI вводят первым, в то же время эффект отсутствовал, когда сначала вводили соединение 2. Это подтверждает гипотезу, что за счет введения CDKI первым, клетки оказываются непролиферативнными, что в свою очередь делает их чувствительными по отношению к цитотоксическому действию соединения 2.
ПРИМЕР 13
Превращение Эпотилона В в Эпотилон F
 (i) 1.98 г (3.90 ммоль) Эпотилона В помещают в атмосферу аргона и растворяют в 60 мл сухого CH2Cl2. К полученному раствору добавляют 0.720 г mCPBA (4.17 ммоль, 1.07 эквивалентов). Смесь подвергают перемешиванию при 25°С в течение 5.5 часов. Затем реакционную массу гасят 60 мл NaHCO3 и экстрагируют 3×75 мл CHCl3. Органическую фазу промывают последовательно 100 мл воды, 70 мл 5% Na2SO3(вод.) и затем 70 мл рассола. Органическую фазу затем сушат над Na2SO4. Сырой продукт хроматографируют, используя элюирование на силикагеле с 2% МеОН в CHCl3, чтобы получить 0.976 г N-оксида (48%) в виде белого пушистого твердого вещества.
(i) 1.98 г (3.90 ммоль) Эпотилона В помещают в атмосферу аргона и растворяют в 60 мл сухого CH2Cl2. К полученному раствору добавляют 0.720 г mCPBA (4.17 ммоль, 1.07 эквивалентов). Смесь подвергают перемешиванию при 25°С в течение 5.5 часов. Затем реакционную массу гасят 60 мл NaHCO3 и экстрагируют 3×75 мл CHCl3. Органическую фазу промывают последовательно 100 мл воды, 70 мл 5% Na2SO3(вод.) и затем 70 мл рассола. Органическую фазу затем сушат над Na2SO4. Сырой продукт хроматографируют, используя элюирование на силикагеле с 2% МеОН в CHCl3, чтобы получить 0.976 г N-оксида (48%) в виде белого пушистого твердого вещества.
(ii) В разгерметезированную пробирку в атмосфере аргона добавляют 0.976 г N-оксида (1.86 ммоль), растворенного в 35 мл сухого CH2Cl2, 2,6-лутидин (1.73 мл, 14.88 ммоль, 8 эквивалентов) и (CF3CO)2O (1.84 мл, 13.02 ммоль, 7 эквивалентов). Пробирку герметизируют и нагревают до 70°С в течение 25 минут. Смеси дают возможность охладиться и растворитель удаляют под током аргона с последующей концентрацией до нескольких мл под вакуумом и образованием темно-желтого раствора. Реакционную смесь разбавляют 25 мл MeOH и добавляют 2.9 мл 28% NH4OH (вод.). Смесь нагревают до 45°С в течение 20 минут, затем охлаждают до комнатной температуры. Сырой продукт концентрируют на роторном испарителе и хроматографируют, используя элюирование на силикагеле 4% MeOH в CHCl3, чтобы получить 0.815 г Эпотилона F (84%).
ПРИМЕР 14
Получение [1S-[1R*,3R*,(E),7R*,10S*,11R*,12R*,16S*]]-3-[2-[2-(Азидометил)-4-тиазолил]-1-метилэтенил1-7,11-дигидрокси-8,8,10,12,16-пентаметил-4,17-диоксабицикло[14.1.0]гептадекан-5,9-дион

В перемешиваемый раствор эпотилона F из примера 13, приведенного выше (957 мг, 1.83 ммоль), в 20.0 мл тетрагидрофурана при 0°С в атмосфере аргона добавляют 0.47 мл дифенилфосфорилазида (604 мг, 2.19 ммоль, 1.2 эквивалентов). Смесь перемешивают в течение приблизительно 3 минут. Затем добавляют 1,8-диазабицикло[5.4.0]ундец-7-ен (0.27 мл, 278 мг, 1.83 ммоль, 1 эквивалент) и смесь перемешивают при 0°С. Через 2 часа смесь нагревают до 25°С и перемешивают в течение 20 часов. Реакционную смесь разбавляют 150 мл этилацетатом и промывают 50 мл Н2О. Водный слой экстрагируют 35 мл этилацетата. Объединенные органические слои сушат над Na2SO4 и концентрируют под вакуумом. Сырое вещество хроматографируют, используя элюирование на силикагеле с 50% этилацетатом в гексане, чтобы получить 913 мг (91%) 21-азидоэпотилона В в виде прозрачного, бесцветного масла. MS (ESI+): 549.3 (М+Н)+; 1H-ЯМР (300 мГц, CDCl3); δ=6.59 (расширенный синглет, 17-H), 7.04 (с, 19-Н), 4.63 (с, 21-Н2) HRMS (DCl); С27Н4N4O6S: [M+] вычислено: 549.2747, найдено: 549.2768.
ПРИМЕР 15
Получение[1S-[1R*,3R*(Е),7R*,10S*,11R*,12R*,16S*]]-3-[2-[2-(Аминометил)-4-тиазолил]-1-метилэтенил]-7,11-дигидрокси-8,8,10,12,16-пентаметил-4,17-диоксабицикло[14.1.0]гептадекан-5,9-дион
Катализатор Линдлара, 18.0 мг, суспендируют в 500 мкл этанола в атмосфере Н2 и насыщают им. Затем добавляют 15.9 мг (29.0 мкмоль) 21-азидоэпотилона В из примера 14, приведенного выше, растворенного в смеси этанол-метанола. После перемешивания в течение 30 минут при комнатной температуре суспензию фильтруют через кизельгур и промывают этилацетатом. Растворитель удаляют из органической фазы и сушат в высоком вакууме. Очистку сырого продукта осуществляют через PSC (растворитель: СН2Cl2 /метанол 90:10), после чего получают 12.3 мг (81%) 21-аминоэпотилона В и 1 мг (6%) эдукта.
1Н-ЯМР (300 мГц, CDCl3); δ=6.58 (расширенный синглет, 17-Н), 7.05 (с, 19-Н), 4.15 (с, 21-Н2); HRMS (DCl); C27H42N2O6S: [М+H+] вычислено: 522.2764, найдено: 522.2772.
ПРИМЕР 16
Получение [1S-[1R*,3R*(E),7R*,10S*,11R*,12R*,16S*]]-3-[2[2-(Аминометил)-4-тиазолил]-1-метилэтенил]-7,11-дигидрокси-8,8,10,12,16-пентаметил-4,17-диоксабицикло[14.1.0]гептадекан-5,9-диод (альтернативный метод)
В перемешиваемый раствор 21-азидоэпотилона В (пример 14) (1.070 г, 1.950 ммоль) в 30.0 мл тетрагидрофурана в атмосфере аргона добавляют 0.22 мл триметилфосфина (0.163 г, 2.145 ммоль, 1.1 эквивалента). Затем добавляют Н2О (5.5 мл) и смеси дают возможность перемешиваться при 25°С. Через 3 часа, когда азид полностью вступил в реакцию, добавляют 3 мл 28% водного раствора NH4OH, чтобы завершить превращение фосфорнилимина в амин. После перемешивания при 25°С в течение 1 часа растворители удаляют под вакуумом. Сырое вещество хроматографируют, используя элюирование на силикагеле 1% Et3N, 2.5% МеОН в CHCl3, чтобы получить 924 мг (91%) 21-аминоэпотилона В в виде белого твердого вещества. MS (ESI+): 523.3 (М+Н)+.
ПРИМЕР 17
Получение(+)-цис-4-трет-Бутил-1-трет-бутилоксикарбонил-3-триэтилсилилоксиазетидин-2-она
Триметилацетальдегид (20.3 мл, 1.25 экв.) добавляют к перемешиваемой суспензии n-анизидина (18.4 г, 0.150 моль) и сухого Na2SO4 (150 г) в безводном дихлорметане (250 мл) при комнатной температуре. Через 2 часа полученную массу фильтруют и сухой остаток промывают дополнительно безводным дихлорметаном. Растворитель удаляют из фильтрата и кристаллический остаток растворяют в безводном дихлорметане (750 мл), после чего помещают в атмосферу азота. Добавляют триэтиламин (48.0 мл, 2.3 экв.) и реакцию охлаждают до -78°С. Затем добавляют по каплям бензилоксиацетилхлорид (27.2 мл, 1.15 экв.) и реакции дают возможность нагреться до комнатной температуры. Через 24 часа ее промывают 0.5 М HCl (дважды) насыщенным водным раствором NaHCO3, рассолом и сушат (Na2SO4). Растворитель удаляют и остаток хроматографируют на колонке с силикагелем (градиент элюирования 20% дихлорметана в гексане, содержащий от 0 до 20% EtOAc), чтобы получить (+)-цис-4-трет-бутил-3-бензилокси-1-п-метоксибензилазетидинон в виде кристаллического вещества.
(46.9 г, 92%): 1Н ЯМР (CDCl3) δ 1.09 (с, 9Н), 3.81 (с, 3Н), 4.15 (д, 1Н, J=5.5 Гц), 4.77 (д, 1Н, J=11.9 Гц), 4.81 (д, 1Н, J=5.5 Гц), 5.03 (д, 1Н, J=11.9 Гц), 6.87-7.43 (м, 9 Гц); LRMS (ESI) 340 ([М+Н)+. Раствор цериевого нитрата аммония (60.4 г, 3.6 экв.) в 900 мл воды добавляют к хорошо перемешиваемому раствору азетидинона (10.38 г, 30.6 ммоль) в ацетонитриле (600 мл) на ледяной бане в течение более 1 часа. Реакционную массу затем экстрагируют EtOAc (дважды) и объединенные органические экстракты промывают насыщенным водным раствором NaHCO3 (дважды), 20% водным раствором NaHSO3, насыщенным водным раствором NaHCO3 и рассолом. После сушки (Na2SO4) растворители удаляют и остаток хроматографируют на колонке с силикагелем (градиент элюирования с порцией гексана, содержащий от 10 до 40% EtOAc), чтобы получить 5.64 г слегка загрязненного (±)-цис-3-бензилокси-4-трет-бутилазетидин-2-она: 1H ЯМР (CDCl3) δ 1.04 (с, 9Н), 3.51 (д, 1Н, J=5.2 Гц), 4.71 (м, 2Н), 4.96 (д, 1Н, J=11.9 Гц), 6.10 (расширенный синглет, 1Н), 7.35 (м, 5Н). Суспензию данного вещества (5.54 г, 23.8 ммоль) и 2.5 г 10% Pd на угле в абсолютном EtOH (100 мл) гидрируют 34 psi (237,8 кПа) Н2 аппарат Парра) в течение 23 часов. Вводят дополнительно 2 г Pd катализатора и гидрирование продолжают еще 17 часов при 50 psi (349,8 кПа)Н2. Катализатор удаляют фильтрированием и растворитель удаляют из фильтрата, при этом остается неочищенный (±)-цис-3-гидрокси-4-(трет-бутил)-азетидин-2-он: 1Н ЯМР (CDCl3+1 капля D2O) δ 1.05 (с, 9Н), 3.48 (д, 1Н, J=5.0 Гц), 4.98 (д, 1Н, J=5.0 Гц). Это вещество растворяют в сухом N,N-диметилформамиде (40 мл), после чего добавляют имидазол (3.24 г, 2 экв.), триэтилсилилхлорид (4.0 мл, 1 экв.). Через 10 минут реакционную смесь разделяют между водой, смесью EtOAc и гексаном (1:1). Органическую фазу промывают водой (дважды), рассолом и затем сушат (Na2SO4). Растворители удаляют и остаток хроматографируют на колонке с силикагелем (градиент элюирования от 20 до 25% EtOAc в гексане), что дает (±)-цис-4-трет-бутил-3-триэтилсилилоксиазетидин-2-он (3.86 г): 1H ЯМР (CDCl3) δ 0.70 (м, 6Н), 0.98 (м, 18Н),3.39 (д, 1Н, J=5.0 Гц),4.88 (дд, 1Н, J=2.1,5.0 Гц), 6.08 (расширенный синглет, 1Н). Раствор указанного азетидинона (2.04 г, 7.92 ммоль), диизопропилэтиламина (1.66 мл, 1.2 экв.), ди-трет-бутилдикарбоната (1.90 г, 1.1 экв.) и п-диметиламинопиридина (194 мг, 0.2 экв.) в сухом дихлорметане (24 мл) перемешивают при комнатной температуре в течение 3 часов. Реакционную смесь разбавляют дихлорметаном, промывают рассолом и сушат (Na2SO4). Удаление растворителя, которое следует за хроматографией на колонке с силикагелем (градиент элюирования от 0 до 20% EtOAc в гексане), дает 2.71 г (96%) названного соединения в виде масла: 1H ЯМР (CDCl3) δ 0.70 (м, 6Н), 1.00 (м, 9Н), 1.09 (с, 9Н), 1.53 (с, 9Н), 3.90 (д, 1Н, J=6.5 Гц), 4.93 (д, 1Н, J=6.5 Гц).
ПРИМЕР 18
Получение производного баккатина А
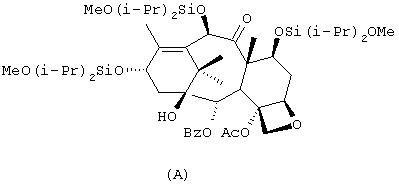
К раствору 10-дезацетилбаккатина (47.4 г, 87 ммоль) в безводном N,N-диметилформамиде (ДМФА) (500 мл) при температуре окружающей среды добавляют имидазол (47 г, 691 ммоль). Раствор перемешивают в течение 10-15 минут, пока он не станет прозрачным. Затем к реакционной смеси добавляют по каплям диизопропилдихлорсилан (58 мл, 322 ммоль). Реакционную смесь перемешивают в течение 16 часов при температуре окружающей среды. К раствору добавляют дополнительное количество диизопропилдихлорсилана (6 мл) и полученную реакционную смесь вновь перемешивают в течение 60 минут. HPLC на этой точке реакции показывает ее завершение. К смеси добавляют метанол (36 мл) и раствор перемешивают в течение 60 минут. Реакцию останавливают и разбавляют смесью трет-бутилметилкетона (ТВМЕ) (500 мл) и воды (200 мл). Слои отделяют и органическую фазу промывают рассолом (250 мл), сушат (сульфат натрия) и упаривают, чтобы получить трисилилированное производное баккатина А (91 г, >100% выход) в виде белого аморфного соединения, которое используют на следующей стадии без дополнительно очистки. LRMS(ESI)M+ вычислено: C50H84O13Si3:977. найдено: 977.
ПРИМЕР 19
Получение производного баккатина В
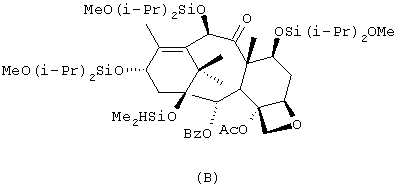
(В) К раствору производного баккатина А (90 г, 92 ммоль) в ДМФА (500 мл) при °С добавляют имидазол (22 г, 320 ммоль). Затем при 0°С добавляют по каплям диметилхлорсилан (35 мл, 320 ммоль). В этот момент наблюдают выпадение соединения в виде осадка. Реакционную смесь (взвесь) перемешивают в течение 0.5 часов при 0°С.
Сухой остаток фильтруют и промывают холодным ДМФА (3×150 мл). После высушивания на воздухе сухой остаток повторно растворяют в ТВМЕ (700 мл) и раствор промывают водой (3×200 мл), рассолом (250 мл) и сушат (сульфат натрия). Раствор фильтруют через небольшую силикагелевую подушку. Удаление растворителя под вакуумом дает В с 77% выходом (709).
LRMS(ESI)M+ вычислено: для C50H90O13S14 1035. найдено: 1035.
ПРИМЕР 20
Получение производного баккатина С
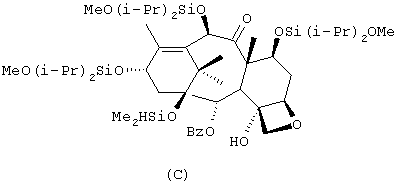
В перемешиваемый раствор В (66.3 г, 64 ммоль) в толуоле (680 мл) при -34°С добавляют по каплям Red-Al® (50 мл, 160 ммоль, 65% по массе раствор натрий бис(2-метоксиэтокси)алюминий гидрида в толуоле) в течение 10 минут. Реакционную смесь нагревают до -25°С и перемешивают в течение 1.5 часов. К реакционной смеси добавляют по каплям метанол (62 мл), поддерживая внутреннюю температуру между -20 и -25°С. Раствор разбавляют ТВМЕ (500 мл), а затем 1N раствором гидроксида натрия (60 мл) и рассолом (60 мл). Затем его перемешивают в течение 30 минут. К смеси добавляют кизельгур (12 г), перемешивают в течение 10 минут и фильтруют через подушку из кизельгура. Слои отделяют. Органический слой промывают водой, рассолом и сушат (сульфат натрия). После этого раствор пропускают через небольшую подушку из силикагеля перед удалением растворителя. Соединение получают с 97% выходом (62 г) в виде белого твердого вещества. LRMS(ESD) M+ вычислено: для C50H88O12S14:993. найдено: 993.
ПРИМЕР 21
Получение производного баккатина D

В атмосфере аргона к раствору производного баккатина С (62 г, 62 ммоль) в безводном тетрагидрофуране (ТГФ) (600 мл) при -60°С добавляют бис(триметилсилил)амид лития (125 мл, 125 ммоль, 1М раствор в ТГФ) по каплям. Раствор перемешивают в течение 15 минут, а затем добавляют метилхлорформиат (9 мл, 116 ммоль); внутренняя температура раствора остается -60°С. Реакцию медленно нагревают до 0°С и смесь перемешивают в течение 3 часов. После завершения реакции добавляют насыщенный раствор хлорида аммония (300 мл). Реакционную смесь экстрагируют ТВМЕ (100 мл). Органические слои промывают насыщенным раствором хлорида аммония (200 мл), водой (200 мл), рассолом (200 мл), сушат (сульфат натрия) и упаривают, что обеспечивает D в виде масла (67 г, >100%). Сырое вещество используют на следующей стадии без дополнительно очистки. LRMS(ESI)M+ вычислено: для С52 Н90O14Si4: 1051. найдено: 1051.
ПРИМЕР 22
Получение производного баккатина Е

К раствору производного баккатина D (62 г, 59 ммоль) в сухом ТГФ (260 мл) добавляют комплекс триэтиламин•фтористоводородная кислота (56 мл, 344 ммоль) при температуре окружающей среды. Реакционную массу перемешивают в течение 3 часов. Затем ее разбавляют этилацетатом (350 мл) и промывают водой (200 мл), рассолом (200 мл), сушат (сульфат натрия) и упаривают, чтобы получить Е (43 г, >100% сырого продукта, выход). Повторное суспендирование сырого соединения в смеси горячего этилацетата (350 мл) и гексана (50 мл) дает чистый Е с 90% выходом. LRMS(ESI)M+ вычислено: для С29Н36O11: 560. найдено: 560.
ПРИМЕР 23
Получение производного баккатина F
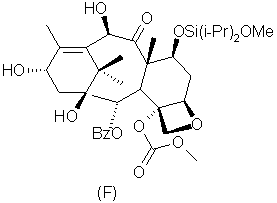
В перемешиваемый раствор производного баккатина Е (32 г, 57 ммоль) и имидазола (11.7 г, 172 ммоль в ДМФА (220 мл)) при -65°С добавляют диизопропилдихлорсилан (26.8 мл) в атмосфере аргона. Температуру реакционной смеси поддерживают при -60°С и смесь перемешивают в течение 2 часов. После завершения реакции (HPLC) добавляют раствор имидазола в метаноле (11.7 г имидазол, растворенный в 35 мл метанола) и раствор перемешивают при 0°С в течение 30 минут. Смесь экстрагируют ТВМЕ (500 мл). Органическую фазу промывают водой (4×150 мл), сушат (сульфат натрия) и испаряют, чтобы получить неочищенный F (45 г). Сырое вещество дополнительно растворяют в ацетонитриле (150 мл) и раствор промывают гексаном (3×100 мл). Удаление ацетонитрила дает чистый F в виде белого твердого вещества (34 г, 84% выход).
LRMS(ESI)M+ вычислено: для С36Н52O12Si: 704. найдено: 704.
ПРИМЕР 24
Получение 4-деацетил-7-[бисизопропил(метокси)]силилокси-4-метоксикарбонил-баккатина
К раствору производного баккатина F (33.2 г, 47 ммоль) в ДМФА (200 мл) добавляют по каплям бис(триметилсилил)амид лития (61.2 мл, 61.2 ммоль, 1М раствор в ТГФ) при -43°С. Реакционную смесь перемешивают в течение 15 минут, а затем добавляют уксусный ангидрид (5.8 мл, 63 ммоль). Реакционную массу вновь перемешивают в течение 30 минут при -40°С. Добавляют уксусную кислоту (3.6 мл), после чего удаляют охлаждающую баню. Реакционную смесь экстрагируют ТВМЕ (300 мл). Органический слой отделяют и промывают водой (3×150 мл), рассолом (150 мл), сушат (сульфат натрия) и упаривают, чтобы получить сырой продукт. Очистки указанного соединения достигают кристаллизацией из смеси ТГФ:гептан (1:6). При очистке 40 г обеспечивается 21 г перекристаллизованного названного продукта (60% выход). LRMS(ESI)M+ вычислено: для C38H54O13Si: 746. найдено: 746.
ПРИМЕР 25
Получение 3'-трет-Бутил-3'-N-трет-бутилоксикарбонил-4-деацетил-3'-дефенил-3'-N-дебензоил-4-O-метоксикарбонил-паклитаксел
Раствор (±)-цис-4-трет-бутил-1-(трет-бутилоксикарбонил)-3-триэтилсилилоксиазетидин-2-она (2.71 г, 5 экв.) и 4-деацетил-7-[бисизопропил(метокси)]силилокси-4-метоксикарбонилбаккатина (1.13 г, 1.52 ммоль) в сухом ТГФ (100 мл) под N2 охлаждают до -50°С и добавляют раствор бис(триметилсилил)амида лития (1.97 мл, 1.3 экв., 1.0 М в ТГФ). Через 5 минут полученную смесь помещают на баню, что дает возможность поддерживать температуру от -35 до -30°С в течение 20 часов, а затем -25°С в течение 24 часов. Реакционную массу затем гасят насыщенным водным раствором MH4Cl и экстрагируют смесью EtOAc и гексана (1:1). Органические экстракты промывают рассолом и сушат (Na2SO4). Растворители удаляют и остаток хроматографируют (радиальная хроматография на пластине с 6 мм силикагеля; градиент элюирования от 5 до 20% EtOAc в гексане), чтобы получить 1.55 г 3'-трет-бутил-3'-N-трет-бутилоксикарбонил-7-[бисизопропил(метокси)]силилокси-4-деацетил-3'-дефенил-3'-N-дебензоил-4-O-метоксикарбонил-2'-триэтилсилилоксипаклитаксел в виде смеси 2',3'-диастереоизомеров. Полученную смесь растворяют в сухом ТГФ (60 мл) и добавляют триэтиламинтригидрофторид (0.92 мл 4 экв.). Через 22 часа при комнатной температуре реакционную смесь нейтрализуют насыщенным водным раствором NaHCO3 и затем экстрагируют EtOAc. Органические экстракты промывают рассолом, сушат (Na2SO4) и растворители удаляют. Остаток хроматографируют (радиальная хроматография; 2 мм пластина с силикагелем; градиент элюирования от 10 до 50% EtOAc в гексане), чтобы получить (в зависимости от элюирования): 210 мг (18%) 2'S,3'R-3'-трет-бутил-3-'N-трет-бутилоксикарбонил-4-деацетил-3'-дефенил-3'-N-дебензоил-4-O-метоксикарбонилпаклитаксел {1Н ЯМР (CDCl3) δ 1.04 (с, 9Н), 1.13 (с, 3Н), 1.20 (с, 3Н), 1.37 (с, 9Н), 1.65 (с, 1Н), 1.66 (с, 3Н), 1.84-1.93 (м, 2Н), 2.17 (с, 3Н), 2.25 (с, 3Н), 2,55 (м, 3Н), 3.00 (д, 1Н, J=6.5 Гц), 3.74 (д, 1Н, J=10.8 Гц), 3.79 (д, 1Н, J=6.9 Гц), 3.92 (с, 3Н), 4.16 (д, 1Н, J=8.5 Гц), 4.33 (д, 1Н, J=8.5 Гц), 4.42 (м, 1Н), 4.54 (д, 1Н, J=6.5 Гц) 4.87 (д, 1Н, J=10.6 Гц), 5.01 (д, 1Н, J=7.7 Гц), 5.68 (д, 1Н, J=7.0 Гц), 5.76 (м, 1Н), 6.32 (с, 1Н), 7.44 -8.05 (м, 5Н); LRMS (ESI) 846 [(М+Н)+]} и 668 мг (56%) названного соединения {1Н ЯМР (CDCl3) δ 1.07 (с, 9Н), 1.14 (с, 3Н), 1.24 (с, 3Н), 1.33 (с, 9Н), 1.66 (с, 4Н), 2.23 (с, 3Н), 2.38-2.59 (м, 4Н), 3.11 (д, - 1Н, J=5.8 Гц), 3.77 (д, 1Н, J=11.1 Гц), 3.82 (д, 1Н, J=7.0 Гц), 3.96 (с, 3Н), 4.20 (д, 1Н, J=8.6 Гц), 4.33 (д, - 1Н, J=8.6 Гц), 4.39 (м, 1Н), 4.53 (д, 1Н, J=5.4 Гц) 4.88 (д, 1Н, J=10.6 Гц), 4.98 (д, 1Н, J=7.9 Гц), 5.69 (д, 1Н, J=7.1 Гц), 6.03 (м, 1Н), 6.28 (с, 1Н), 7.40-8.11 (м, 5Н); LRMS (ESI) 846 [(M+H)+]}.
Настоящее изобретение не ограничивается воплощениями, описанными выше, и может варьироваться и модифицироваться без выхода за рамки, определенные пунктами формулы.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПЛЕКС ИНГИБИТОРА RAS-ФАРНЕЗИЛТРАНСФЕРАЗЫ, КОМПОЗИЦИЯ ИНГИБИТОРА RAS-ФАРНЕЗИЛТРАНСФЕРАЗЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1999 |
|
RU2230062C2 |
| СИНЕРГИЧЕСКАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМБИНАЦИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2007 |
|
RU2438664C2 |
| ВИДЫ КОМБИНИРОВАННОЙ ТЕРАПИИ ДЛЯ ЛЕЧЕНИЯ РАКА | 2016 |
|
RU2727474C2 |
| N,N'-(АЛКАНДИИЛ)БИС[ЛАБДА-7(9),13,14-ТРИЕН-4-КАРБОКСАМИДЫ], ОБЛАДАЮЩИЕ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 2017 |
|
RU2654201C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМБИНАЦИЯ ДЛЯ ЛЕЧЕНИЯ И/ИЛИ ХЕМОСЕНСИБИЛИЗАЦИИ ОПУХОЛЕЙ, УСТОЙЧИВЫХ К ПРОТИВОРАКОВЫМ СРЕДСТВАМ | 2007 |
|
RU2396974C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ПРОТИВООПУХОЛЕВЫМ АЛКАЛОИДОМ | 2018 |
|
RU2757373C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ПРОТИВООПУХОЛЕВЫМ АЛКАЛОИДОМ | 2011 |
|
RU2743643C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ПРОТИВООПУХОЛЕВЫМ АЛКАЛОИДОМ | 2018 |
|
RU2767664C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ПРОТИВООПУХОЛЕВЫМ АЛКАЛОИДОМ | 2011 |
|
RU2605335C2 |
| СПОСОБ ПРОФИЛАКТИКИ И/ИЛИ ЛЕЧЕНИЯ РАКОВЫХ ЗАБОЛЕВАНИЙ | 2006 |
|
RU2480201C2 |
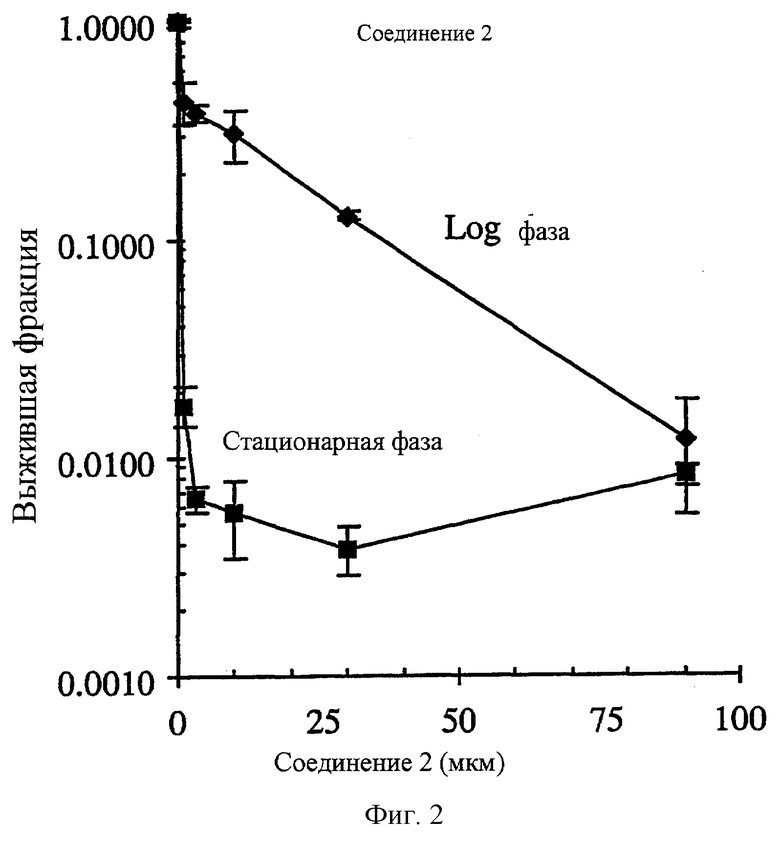
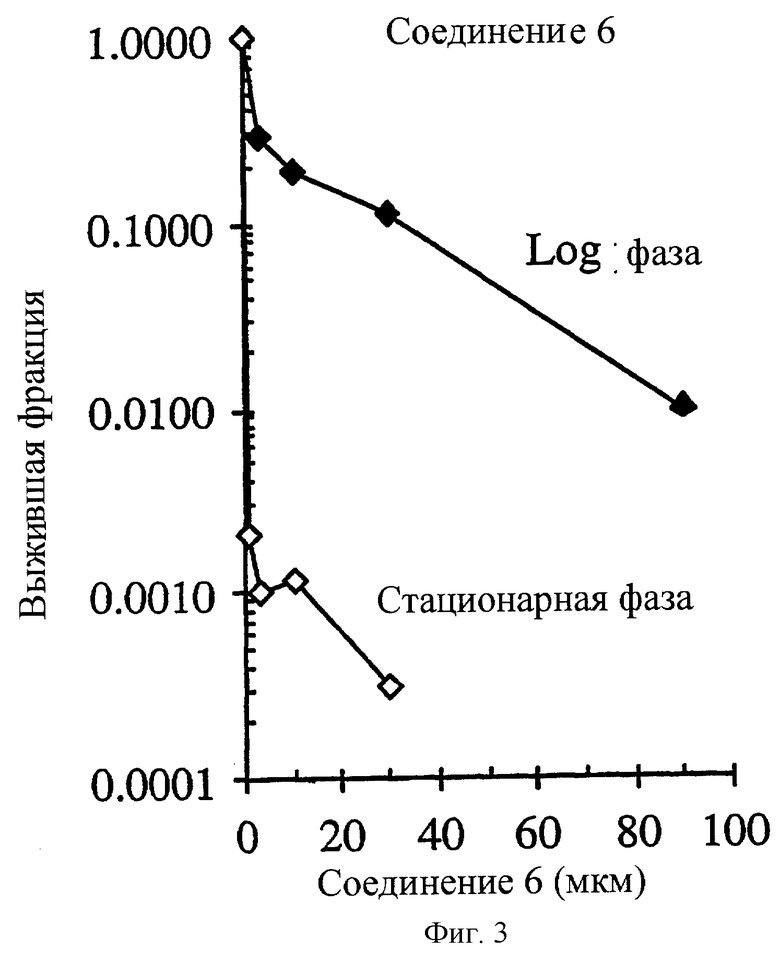
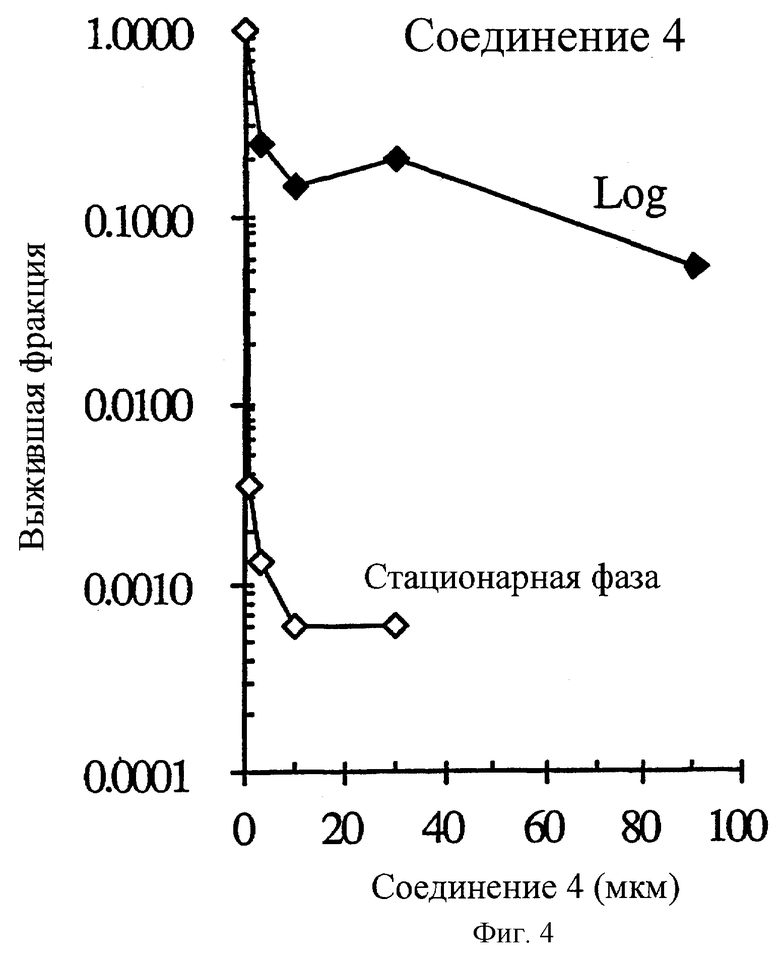
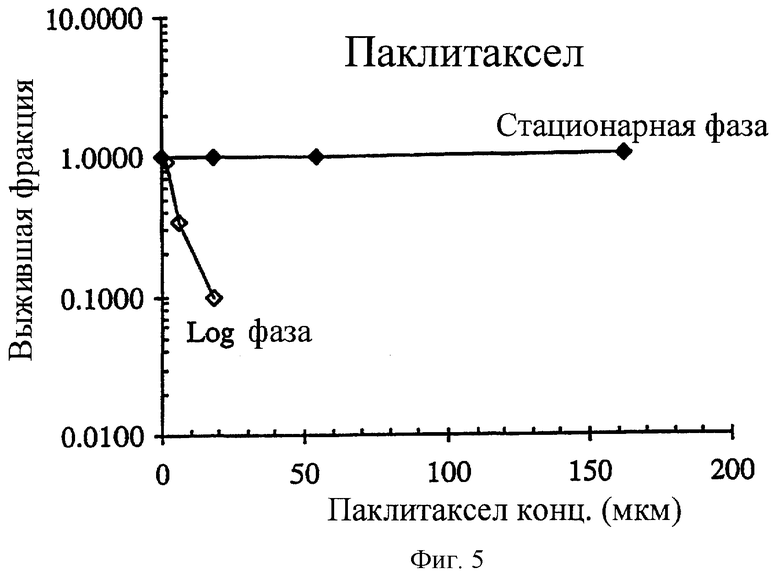
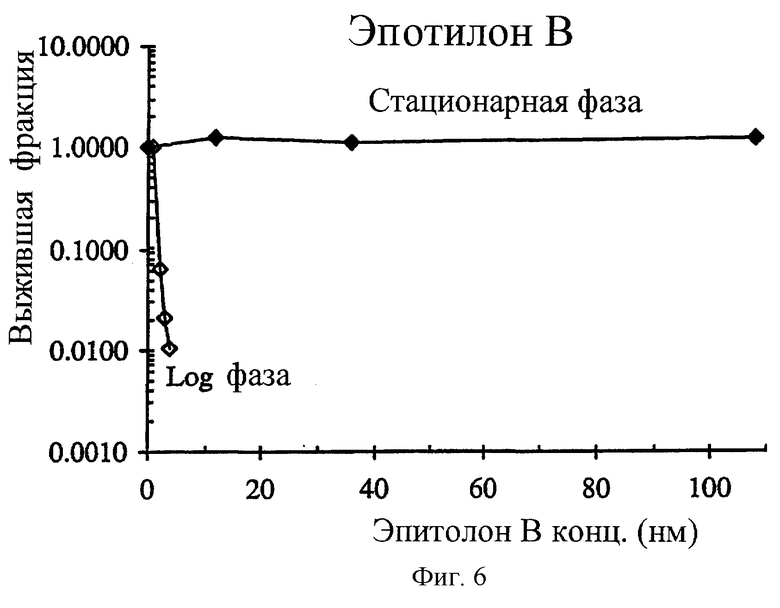
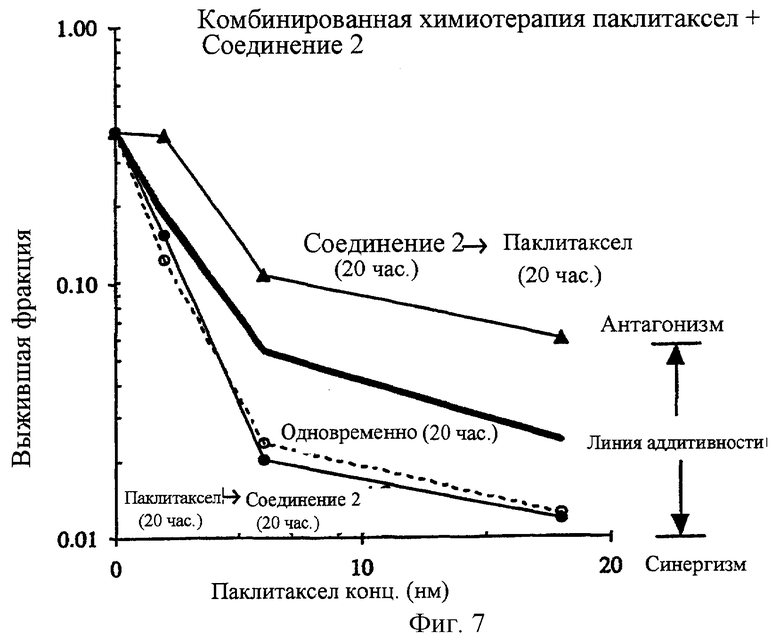
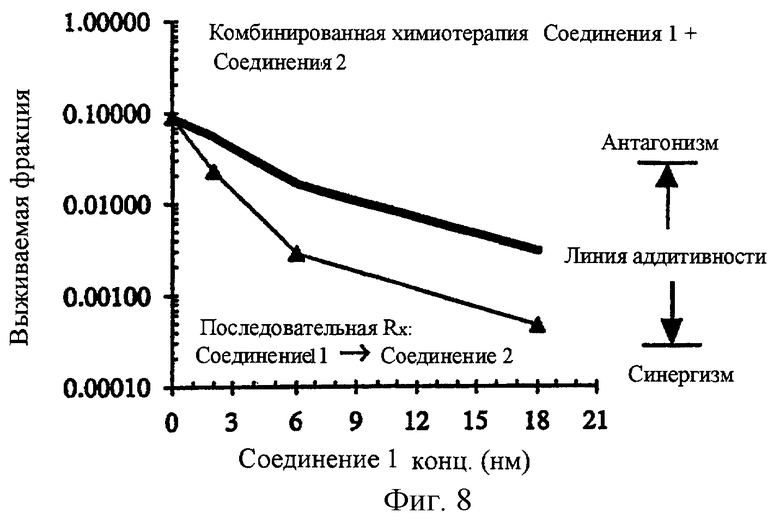



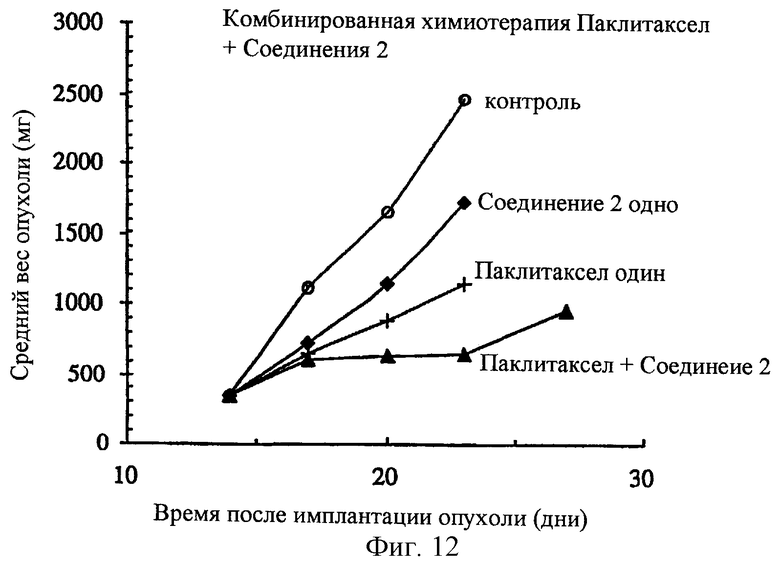
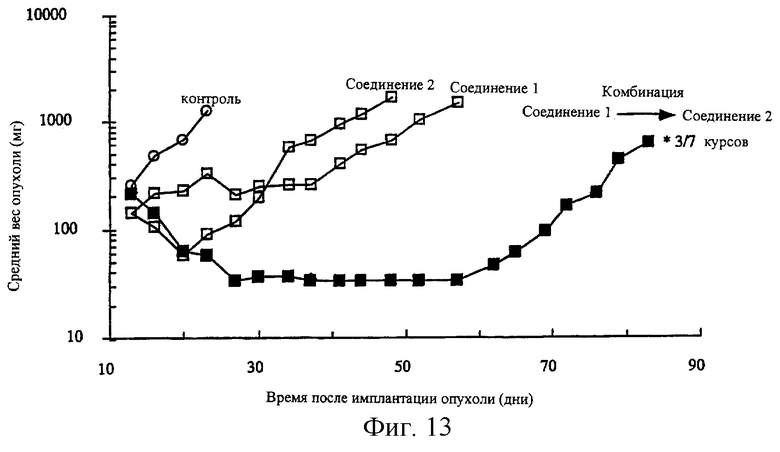
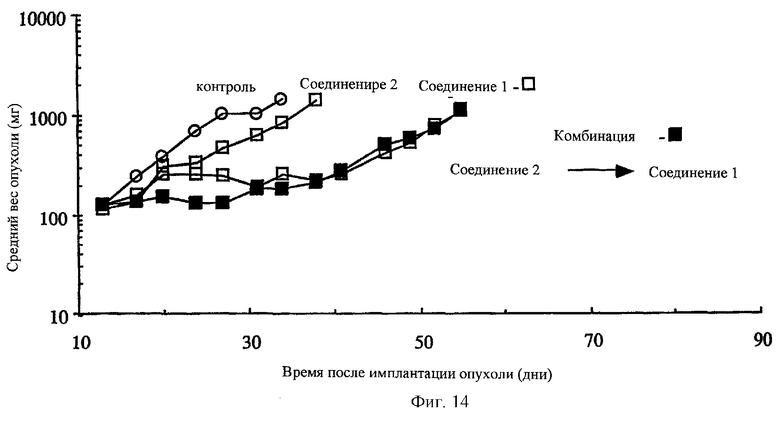
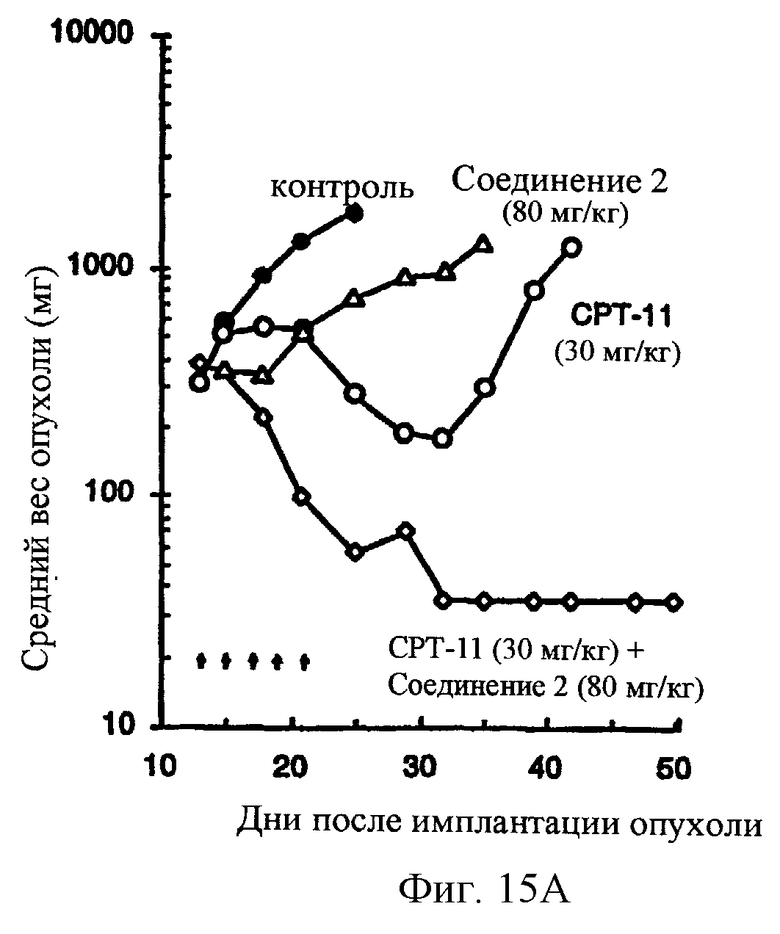
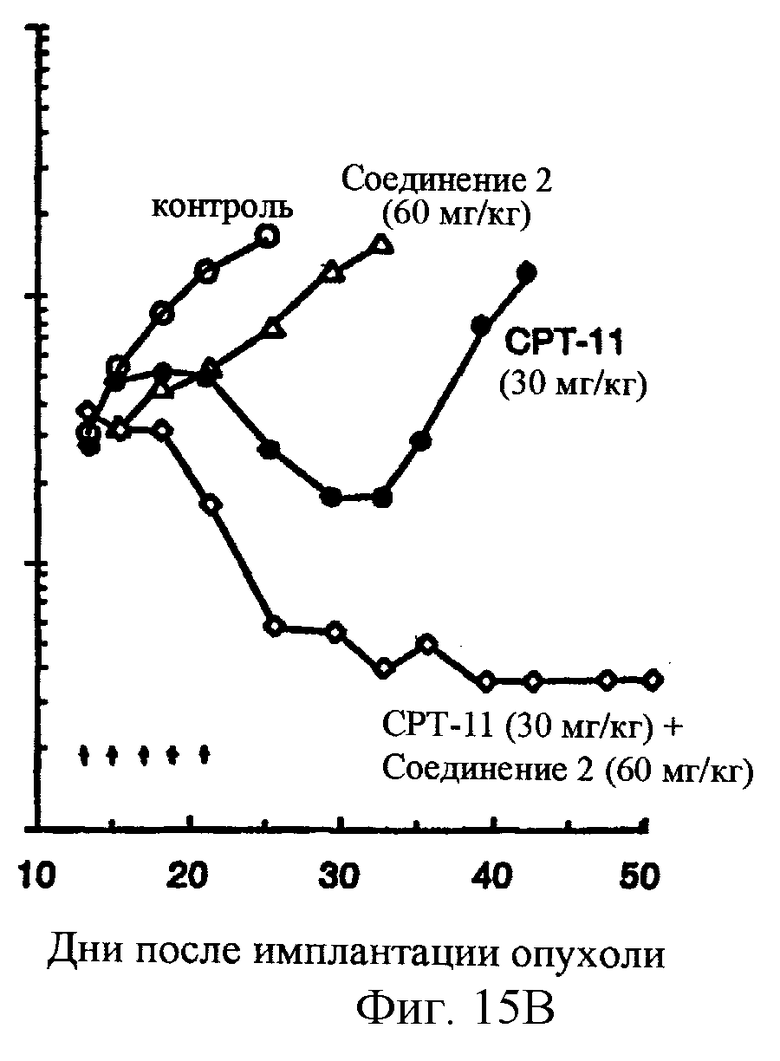
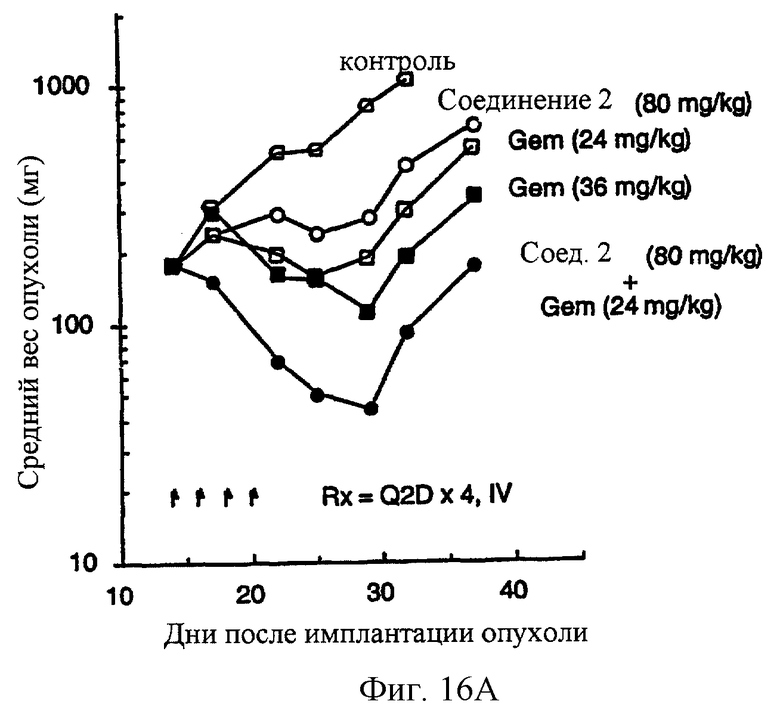
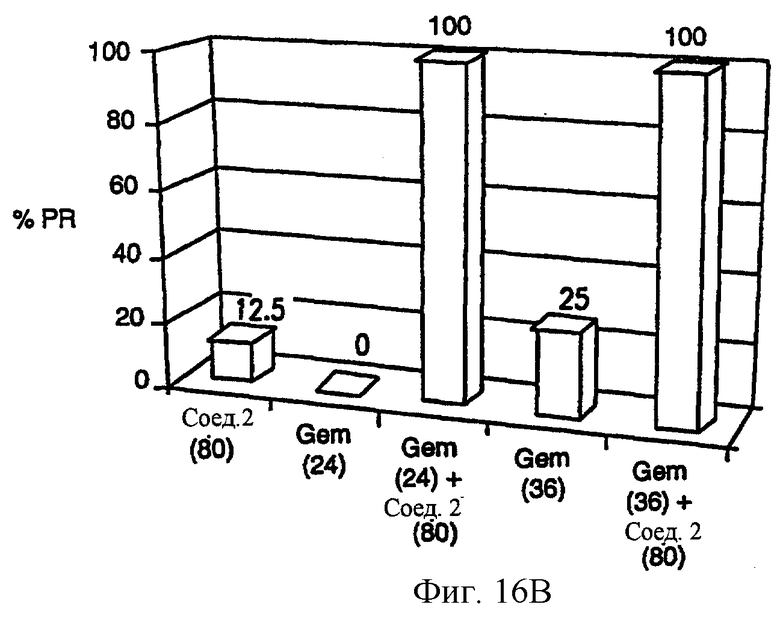


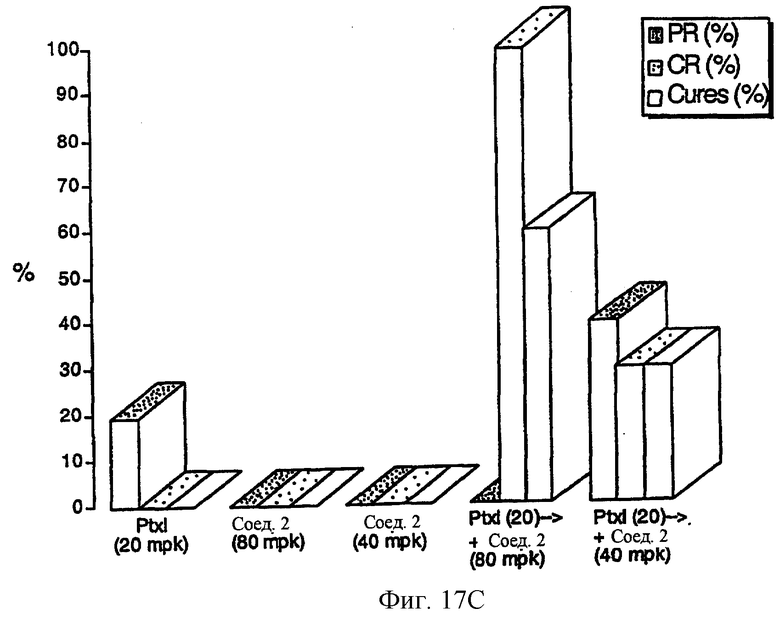
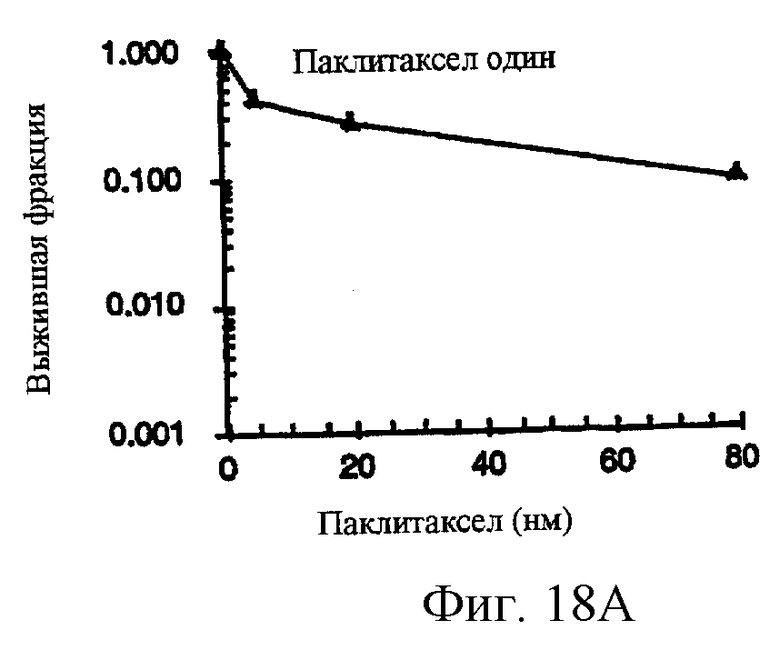
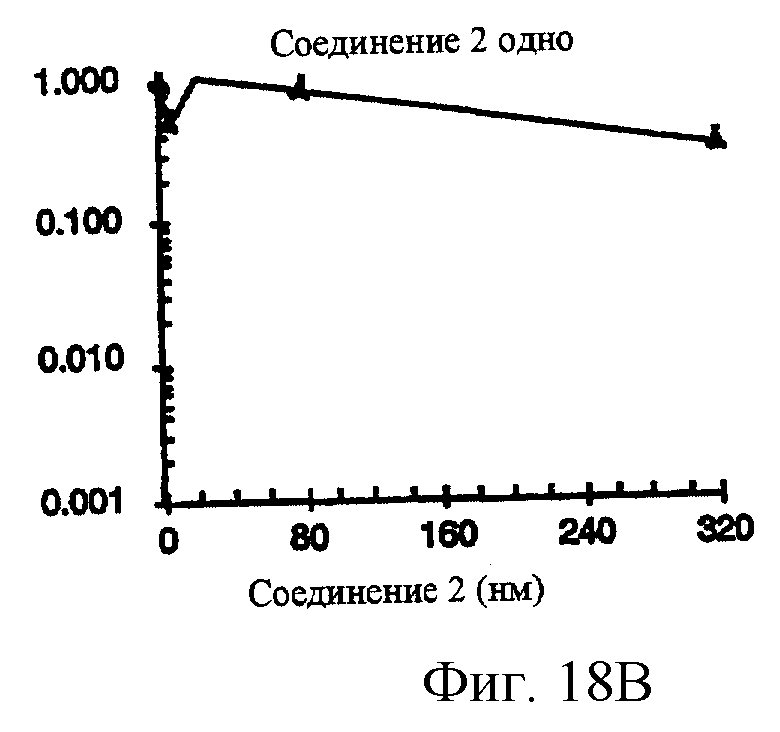
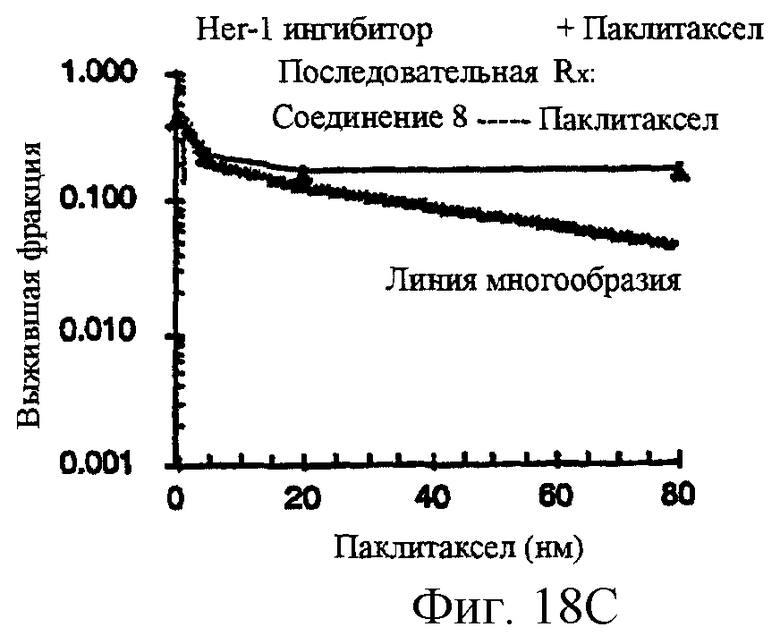
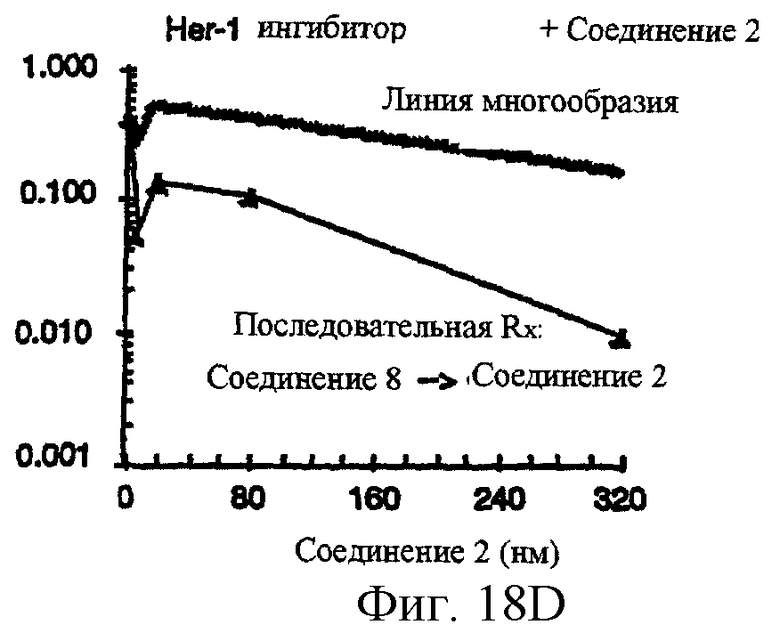
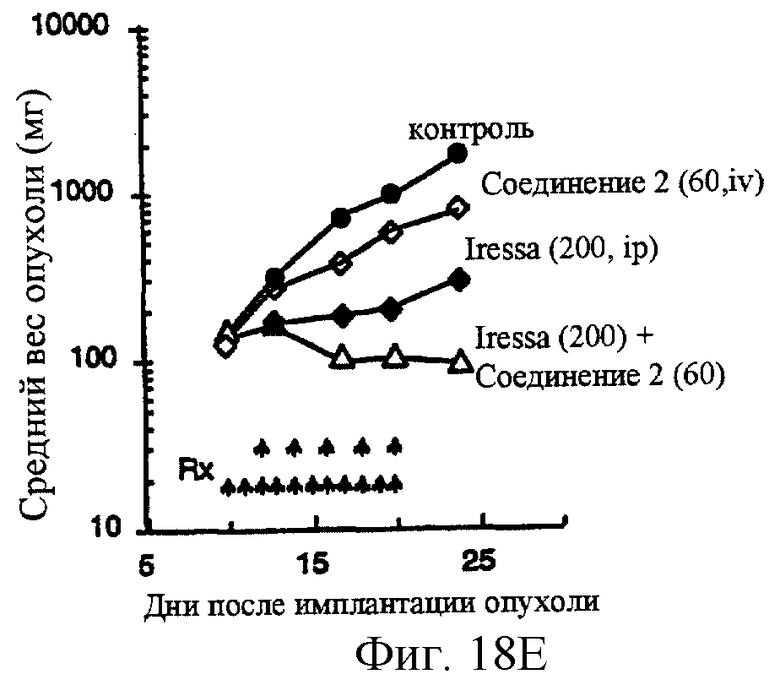
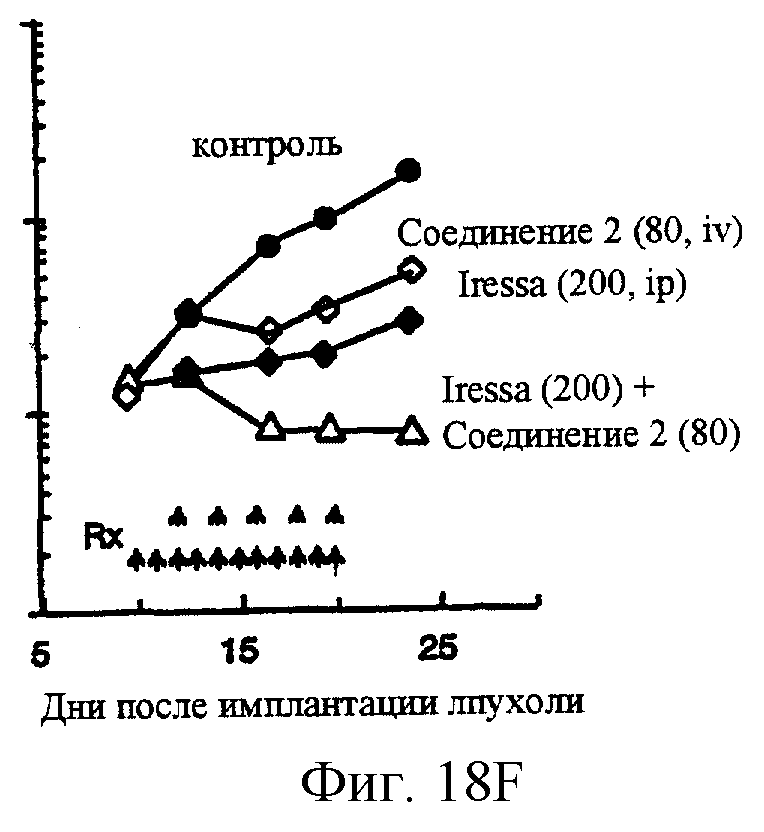

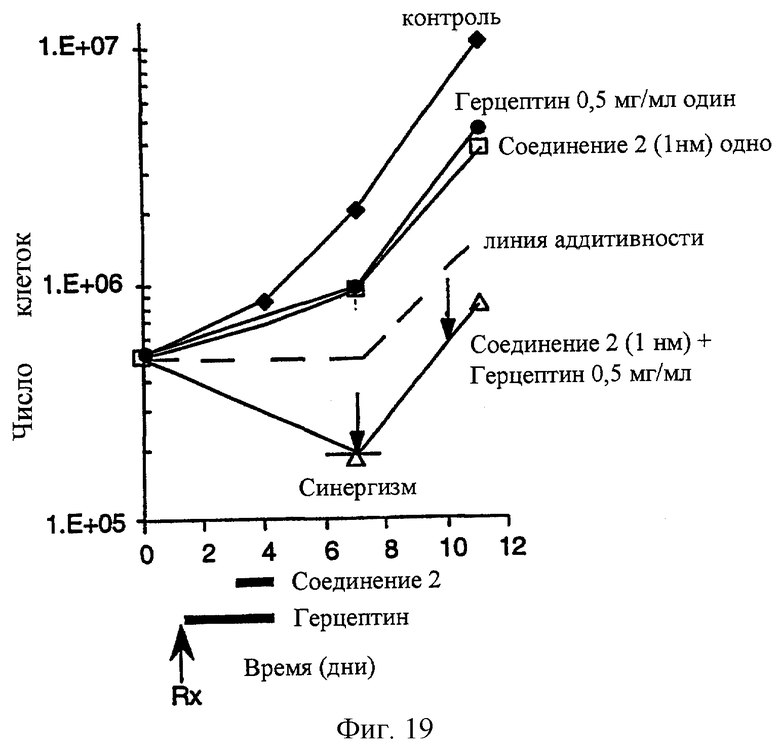
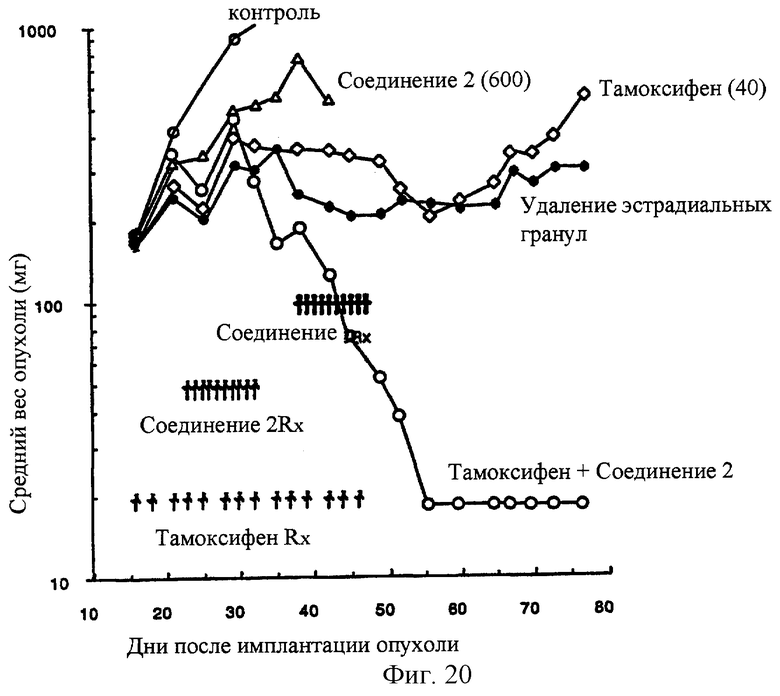
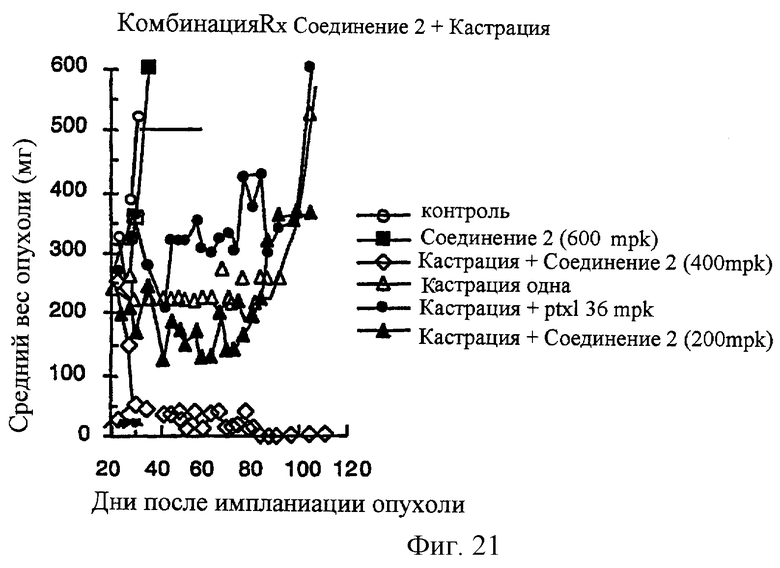
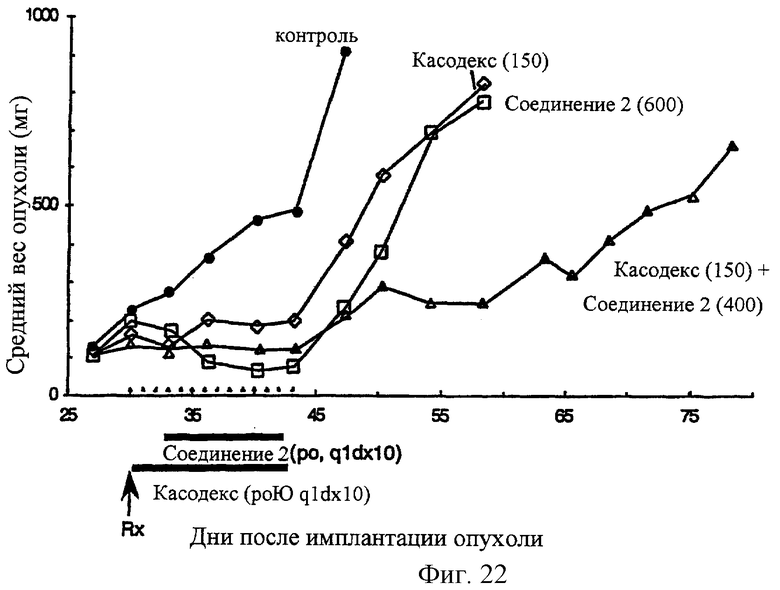
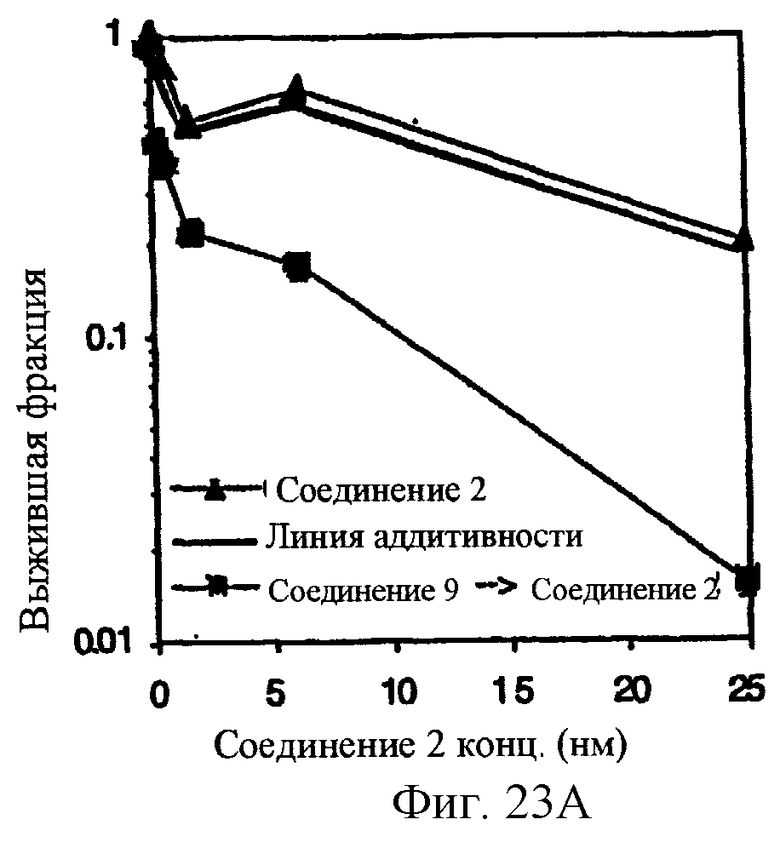
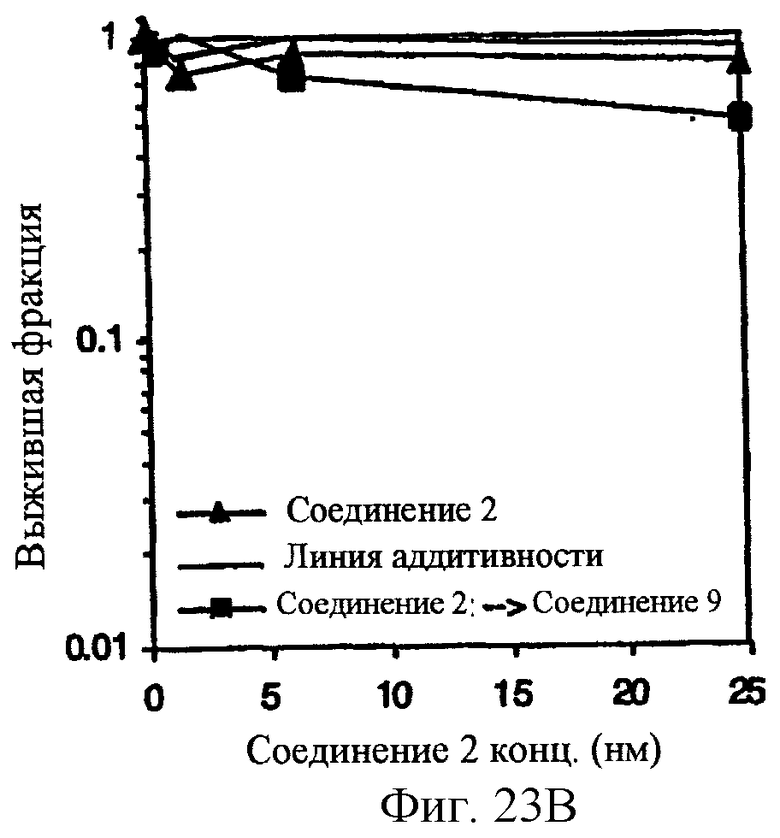
Изобретение относится к области медицины, к лечению рака. Способ включает введение млекопитающему, нуждающемуся в этом, синергически терапевтически эффективного количества (1) агента, выбранного из группы, состоящей из цитотоксических агентов и цитостатических агентов и (2) соединения формулы (I) или его фармацевтически приемлемой соли. Способ обеспечивает синергический противоопухолевый эффект при использовании меньших доз одного или обоих активных ингредиентов, предотвращает или замедляет развитие мультилекарственной резистентности опухоли с обеспечением поражения как пролиферативных, так и непролиферативных опухолевых клеток. 6 з.п. ф-лы, 6 табл., 23 ил.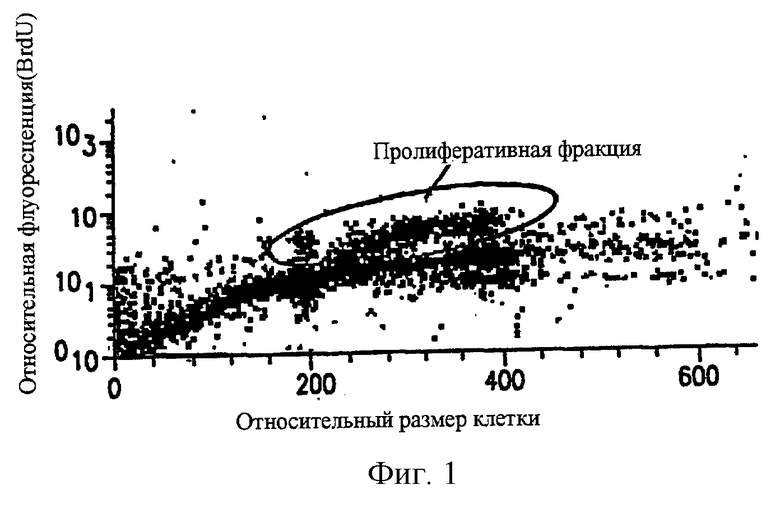
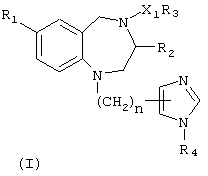
или его фармацевтически приемлемой соли, где
R1 представляет собой Br или CN;
R2 представляет собой необязательно замещенный бензил;
R3 представляет собой необязательно замещенный низший алкил, необязательно замещенный фенил, необязательно замещенный 2-тиенил или необязательно замещенный 1-пиперидинил;
R4 представляет собой водород или метил;
Z1 представляет собой СО, SO2, или S02N(R5)- и
n имеет значение 1, а
R5 представляет собой необязательно замещенный низший алкил или необязательно замещенный фенил,
при условии, что противораковый агент вводят одновременно с или до введения соединения формулы I.
R1 представляет собой CN;
R2 представляет собой необязательно замещенный бензил;
R3 представляет собой необязательно замещенный низший алкил, необязательно замещенный фенил, необязательно замещенный 2-тиенил или необязательно замещенный 1-пиперидинил;
R4 представляет собой водород или метил;
Z1 представляет собой СО или SO2 и
n имеет значение 1.
R1 представляет собой CN;
R2 представляет собой бензил;
R3 представляет собой н-пропил, н-бутил, 3-метоксипропил, 2-тиенил, 5-бром-2-тиенил, фенил, 4-метоксифенил или 1-пиперидинил;
R4 представляет собой водород;
Z1 представляет собой SO2 и
n имеет значение 1.
(R)-7-циано-2,3,4,5-тетрагидро-1-(1Н-имидазол-4-илметил)-4-(1Н-имидазол-4-илметил)-4-)1-оксобутил)-3-(фенилметил)-1Н-1,4-бензодиазепин;
(R)-4-[(5-бром-2-тиенил)сульфонил]-7-циано-2,3,4,5-тетрагидро-1-(1H-имидазолил илметил)-3-(фенилметил)-1Н-1,4-бензодиазепин;
(R)-7-циано-2,3,4,5-тетрагидро-1-(1Н-имидазол-4-илметил)-4-[(4-метоксифенил)сульфонил]-3-(фенилметил)-1Н-1,4-бензодиазепин;
(R)-7-циано-2,3,4,5-тетрагидро-1-(1Н-имидазол-4-илметил)-3-(фенилметил)-4-(фенилсульфонил)-1Н-1,4-бензодиазепин;
(R)7-циано-2,3,4,5-тетрагидро-1-(1Н-имидазол-4-илметил)-3-(фенилметил)-4-(пропилсульфонил)-1Н-1,4-бензодиазепин;
(R)-4-(бутилсульфонил)-7-циано-2,3,4,5-тетрагидро-1-(1Н-имидазол-4-илметил)-3-(фенилметил)-1Н-1,4-бензодиазепин;
(R)-7-циано-2,3,4,5-тетрагидро-1-(1Н-имидазол-4-илметил)-3-(фенилметил)-4-(1-пиперидинилсульфонил)-1Н-1,4-бензодиазепин;
R)-4-(3-метоксипропилсульфонил)-7-циано-2,3,4,5-тетрагидро-1-(1Н-имидазол-4-илметил)-3-(фенилметил)-1Н-1,4-бензодиазепин; и их фармацевтически приемлемых солей.
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| MICCOLI I | |||
| Potentiation of lonidamide and diazepam, two agents acting on mitochondria, in human glioblastoma treatment//J | |||
| Natl | |||
| Cancer Inst | |||
| Способ и аппарат для получения гидразобензола или его гомологов | 1922 |
|
SU1998A1 |
| PubMed на сайте http;//www.pubmed.com | |||
| БЕРТРАМ Г.КАТЦУНГ | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| М.: Бином - СПб: Невский | |||

