Область изобретения
Данное изобретение касается новой фармацевтической комбинации для лечения рака, причем указанная комбинация проявляет синергический эффект. Фармацевтическая комбинация включает цитотоксическое противоопухолевое средство, выбранное из группы, включающей паклитаксел, доцетаксел, доксорубицин и гемцитабин или их фармацевтически приемлемую соль и, по меньшей мере, один ингибитор циклин-зависимой киназы (CDK), выбранный из соединений формулы I (как описано здесь), или фармацевтически приемлемую соль, или сольват его. Данное изобретение также касается способа лечения рака, который включает введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества указанной комбинации.
Предпосылки изобретения
Рак - это общий термин, используемый для описания болезней, при которых анормальные клетки делятся без контроля. Раковые клетки могут проникать в соседние ткани и могут распространяться по кровяному руслу и лимфатической системе в другие части организма. Существуют различные типы рака, такие как рак мочевого пузыря, рак молочной железы, рак толстой кишки, рак прямой кишки, рак головы и шеи, эндометриальный рак, рак почки (почечноклеточный), лейкемия, мелкоклеточный рак легкого, немелкоклеточный рак легкого, рак поджелудочной железы, рак простаты, рак щитовидной железы, рак кожи, Неходжкинская лимфома и меланома. В настоящее время существует много способов, приемлемых для лечения рака, чем когда-либо ранее, включая химиотерапию, радиацию, хирургию, гормональную терапию, иммунную терапию и генную терапию. Химиотерапию обычно используют для лечения многих типов рака. Наиболее широко используемые химиотерапевтические средства (противоопухолевые средства) включают паклитаксел, доцетаксел, доксорубицин, этопозид, карбоплатин, цисплатин, топотекан и гемцитабин. Эти и другие подобные противоопухолевые средства успешно использовались для лечения различных раков. Однако в свое время у некоторых пациентов, страдающих раком, как выяснили, развивается невосприимчивость к монотерапии, включающей применение таких стандартных противоопухолевых средств. Устойчивость или невосприимчивость к лекарственному средству представляет главное препятствие успешному лечению. Такую невосприимчивость часто рассматривают или как врожденную (то есть присутствующую в начале лечения), или приобретенную (то есть возникшую во время курсов химиотерапии). Исследование, связанное с воздействием на человеческие раковые клетки немелкоклеточного рака легкого (NCI-H460) постепенно возрастающих концентраций доксорубицина, подтвердило появление новой клеточной линии (NCI-H460/R), которая была невосприимчива к доксорубицину (96,2 раза) и кросс-невосприимчива к этопозиду, паклитакселу, винбластину и эпирубицину (J.Chemother., 2006 Feb; 18(1) 66-73). В другом исследовании сообщалось о распространенности in vitro невосприимчивости к химиотерапии в культурах опухолевых клеток немелкоклеточного рака легкого (NSCLC), чрезмерной невосприимчивости к лекарственному средству или промежуточной невосприимчивости к лекарственному средству к ряду противоопухолевых средств, включающих цисплатин, доксорубицин, этопозид, гемцитабин, навелбин, паклитаксел, таксотер и топотекан (Ann. Thorac. Surg. 2006 Feb;81(2):440-6; discussion 446-7). Гемцитабин рассматривали как наиболее клинически активное лекарственное средство для лечения рака поджелудочной железы, однако он не смог значительно улучшить состояние пациентов с раком поджелудочной железы по причине предсуществующей или приобретенной хемоневосприимчивости большинства опухолевых клеток к лекарственному средству (Oncogene 2003 May 22; 22(21): 3243-51). Другой наблюдаемой или распространенной при лечении рака проблемой является тяжелая токсичность, связанная с большинством противоопухолевых средств. Частота проявления тяжелых побочных эффектов, таких как кардиотоксичность в случае с такими лекарственными средствами, как доксорубицин, описана в J Egypt Natl Cane Inst. 2005 Dec 17(4) 291-300. Несмотря на частоту проявления невосприимчивости и тяжелую токсичность, связанную с традиционными противоопухолевыми средствами, например гемцитабином, паклитакселом, эти средства будут оставаться важными при лечении рака, потому что они обладают способностью уменьшать опухолевую массу. Для улучшения скорости реакции и предотвращения токсичности, связанной с традиционными противоопухолевыми средствами, оценивают новые терапевтические подходы. Один такой подход направлен на протокол, включающий объединение различных противораковых средств, обладающих различным биологическим механизмом (Jekunen et al., Br. J.Cancer, 69, 299-306 (1994); Yeh et al., Life Sciences, 54, 431-35 (1994)). Оптимальный комбинационный химиотерапевтический протокол может привести к усилению терапевтической эффективности, снижению токсичности реципиента и минимальной или отсроченной невосприимчивости к лекарственному средству. Когда комбинируют лекарственные средства с различной токсичностью, каждое лекарственное средство можно использовать в его оптимальной дозе, помогая минимизировать непереносимые побочные эффекты, как сообщили для комбинации капецитабина и доцетаксела в "Oncology" (Williston Park). 2002 Oct; 16:17-22. Некоторые из противоопухолевых средств, как выяснили, синергически эффективны, когда используются в комбинации с другими противораковыми средствами, чем когда используются как монотерапия. Например, циклофосфамид и 5-фторурацил действуют синергически в клетках овариальной светлоклеточной аденокарциномы, как сообщается в Cancer Lett. 2001 Jan 10; 162 (1):39-48. Комбинационную химиотерапию также можно успешно использовать для лечения рака в поздних стадиях, которые трудно лечить монотерапией, радиацией или хирургическим лечением, например, сообщают о комбинации паклитаксела и гемцитабина для лечения метастатического немелкоклеточного рака легкого (Cancer, 2006 Sep 1; 107(5): 1050-4).
В последнее время была опробована комбинация одного или более стандартных противоопухолевых средств, таких как паклитаксел, цисплатин и т.д., с молекулярно нацеленным противораковым средством для лечения рака для повышения норм ответа на лекарственное средство и для обращения к невосприимчивости к противоопухолевым средствам. Молекулярно нацеленные средства, например иматиниб мезилат, флавопиридол и т.д., модулируют белки, такие как киназы, чья активность является более специфично связанной с раковыми клетками. За долгий период исследования доказали, что члены семейства циклин-зависимой киназы (CDK) играют ключевые роли в различных клеточных процессах. На данный момент известно 11 членов семейства CDK, среди которых CDK1, 2, 3, 4 и 6, как известно, играют важные роли в клеточном цикле (Cyclins and cyclin-dependent kinases: theme and variations. Adv Cancer Res. 1995; 66:181 -212). CDK активированы формирующими нековалентными комплексами с циклинами, такими как циклины А-, В-, С-, D- (D1, D2 и D3) и Е-типов. Каждый изофермент этого семейства отвечает за конкретные аспекты (клеточная сигнализация, транскрипция и т.д.) клеточного цикла, и некоторые из CDK изоферментов специфичны к определенным видам тканей. Аберрантная экспрессия и надэкспрессия этих киназ проявляются при многих болезненных состояниях. Ряд соединений, обладающих потенциально полезными свойствами ингибитора CDK, разработаны и отражены в литературе. Флавопиридол является первым мощным ингибитором циклин-зависимых киназ (CDK), дошедшим до клинического испытания. Как обнаружили, флавопиридол синергически делает возможным цитотоксический ответ на традиционные противоопухолевые средства в различных линиях раковых клеток. Например, о последовательном лечении НСТ116 рака толстой кишки доцетакселом, флавопиридолом и 5-фторурацилом сообщается в Acta Pharmacol Sin. 2006 Oct; 27(10):1375-81. Также о комбинированном лечении доцетакселом и флавопиридолом раковых клеток легкого сообщается в Radiother Oncol. 2004 May; 71(2):213-21, а о лечении рака желудка - в Mol Cancer Ther. 2003 Jun; 2(6): 549-55.
Хотя доказано, что комбинации противораковых средств обладают значительным успехом в протоколах лечения рака, существуют еще некоторые неудовлетворенные потребности и возможности для улучшений в лекарствах для лечения онкологических заболеваний, которые с трудом поддаются лечению или которые показывают невосприимчивость к лечению традиционными противоопухолевыми средствами в качестве монотерапии. В частности, разработка нового комбинационного подхода к доставке известных противораковых средств, обладающих различным механизмом действия, будет представлять важное достижение в данной области. Хотя протокол, включающий комбинацию противораковых средств, обладающих различным механизмом действия, может работать в случае некоторых комбинаций, но он не может работать тем же образом в другой комбинации противораковых средств, и такая комбинация не всегда может стать комбинацией, обладающей благоприятными терапевтическими эффектами. Однако в данном изобретении определено, что новая фармацевтическая комбинация известных противораковых средств, включающих ингибитор циклин-зависимой киназы, выбранный из соединений, представленных формулой I (как описано здесь), и стандартного цитотоксического противоопухолевого средства для лечения различных онкологических заболеваний обеспечивает неожиданно большую эффективность, чем когда противораковые средства используются отдельно.
Краткое описание изобретения
В одном аспекте данное изобретение касается новой фармацевтической комбинации, включающей цитотоксическое противоопухолевое средство, выбранное из группы, включающей паклитаксел, доцетаксел, доксорубицин и гемцитабин или их фармацевтически приемлемую соль; и ингибитор циклин-зависимой киназы (CDK), выбранный из соединений формулы I (как описано здесь) или фармацевтически приемлемой соли, или сольвата их; причем указанная комбинация проявляет синергический эффект при лечении рака.
В другом аспекте данное изобретение касается фармацевтической комбинации, включающей цитотоксическое противоопухолевое средство, выбранное из группы, включающей паклитаксел, доцетаксел, доксорубицин и гемцитабин или их фармацевтически приемлемую соль; и ингибитор циклин-зависимой киназы (CDK), выбранный из соединений формулы I (как описано здесь) или фармацевтически приемлемой соли, или сольвата их, для одновременного или последовательного введения для лечения рака.
В следующем аспекте данное изобретение касается применения новой фармацевтической комбинации для лечения рака и для индуцирования клеточного апоптоза.
В следующем аспекте данное изобретение касается способа лечения рака, который включает введение пациенту, нуждающемуся в этом, терапевтически эффективного количества цитотоксического противоопухолевого средства, выбранного из группы, включающей паклитаксел, доцетаксел, доксорубицин и гемцитабин или их фармацевтически приемлемую соль; в комбинации с терапевтически эффективным количеством ингибитора циклин-зависимой киназы (CDK), выбранного из соединений формулы I (как описано здесь), или фармацевтически приемлемой соли, или сольвата их.
В еще одном аспекте данное изобретение касается применения новой комбинации для получения лекарства для лечения рака.
Другие аспекты и дополнительный объем применимости данного изобретения станет очевидным из следующего детального описания.
Краткое описание графических материалов
Фигура 1 показывает, что комбинация доксорубицина и соединения А при обработке клеток немелкоклеточного рака легкого Н-460 in vitro проявляет синергизм. График(и) А, В, С и D представляет(представляют) распределение клеточного цикла различных групп обработки, а именно контроль (в течение 96 часов), 200 нМ доксорубицина отдельно (в течение 24 часов), 800 нМ соединения А отдельно (в течение 72 часов) и комбинацию, включающую введение 200 нМ доксорубицина (в течение 24 часов), за которым следует 800 нМ соединения А (72 часа) соответственно.
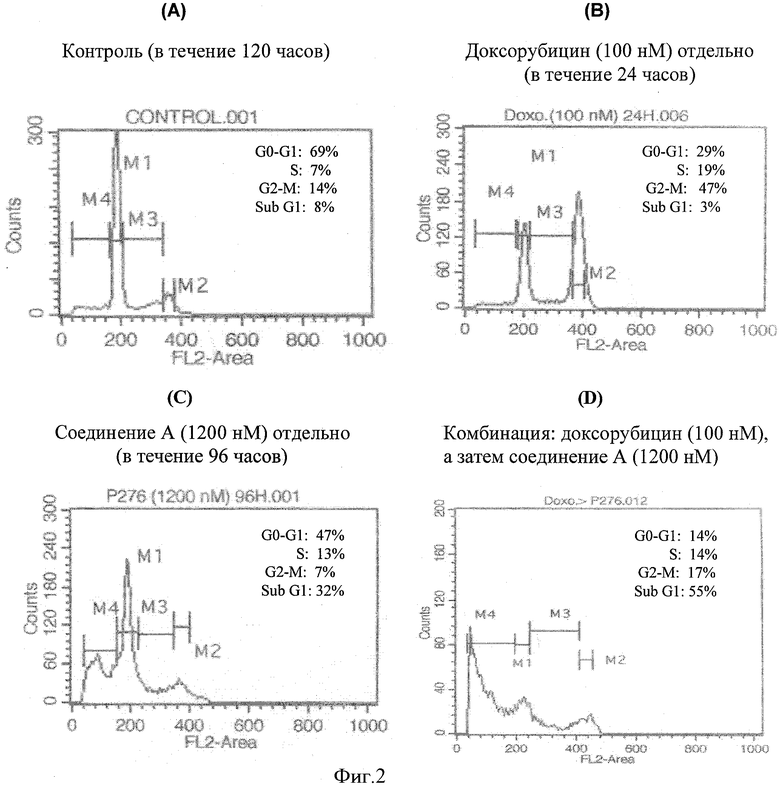
Фигура 2 показывает, что комбинация доксорубицина и соединения А при обработке клеток немелкоклеточного рака легкого Н-460 in vitro проявляет синергизм. График(и) А, В, С и D представляет(представляют) распределение клеточного цикла различных групп обработки, а именно контроль (в течение 120 часов), 100 нМ доксорубицина отдельно (в течение 24 часов), 1200 нМ соединения А отдельно (в течение 96 часов) и комбинацию, включающую введение 100 нМ доксорубицина (24 часа), за которым следует 1200 нМ соединения А (96 часов) соответственно.
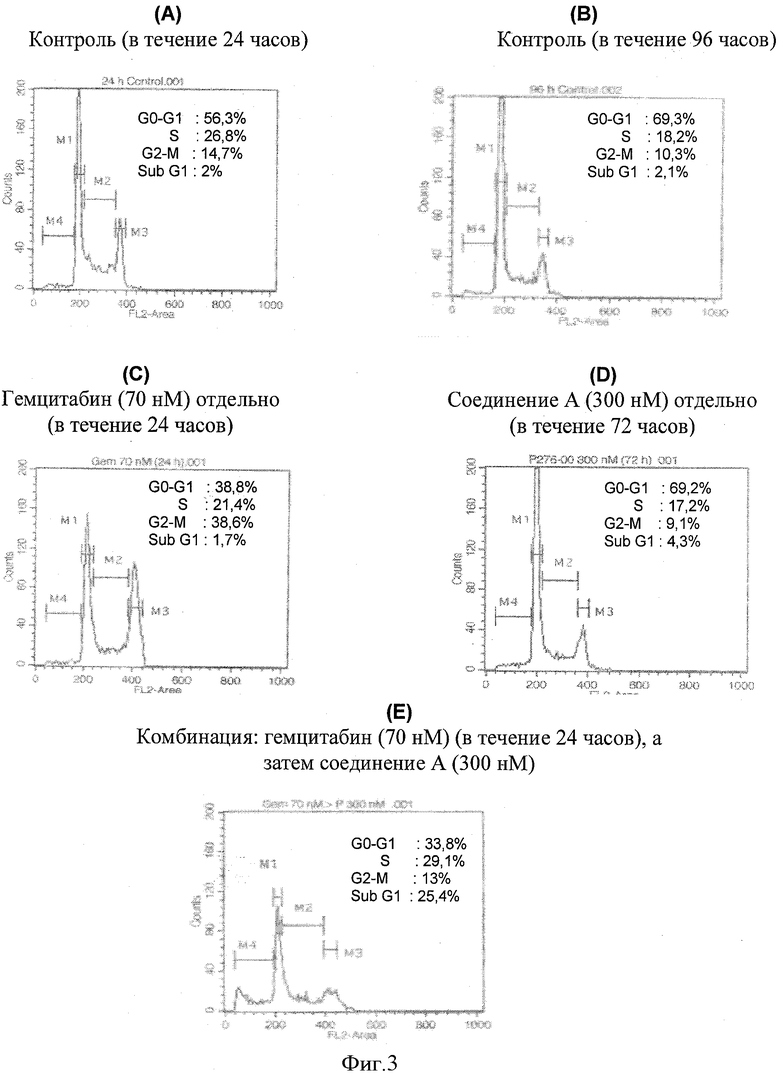
Фигура 3 демонстрирует, что применение комбинации гемцитабина и соединения А при обработке клеток поджелудочной железы (Panc-1) in vitro приводит к синергической активности. График(и) А, В, С и D представляет(представляют) распределение клеточного цикла различных групп обработки, а именно контроль (в течение 24 часов), контроль (в течение 96 часов), 70 нМ гемцитабина отдельно (в течение 24 часов), 300 нМ соединения А отдельно (в течение 72 часов) и комбинацию, включающую введение 70 нМ гемцитабина (24 часа), за которым следует 300 нМ соединения А (72 часа) соответственно.
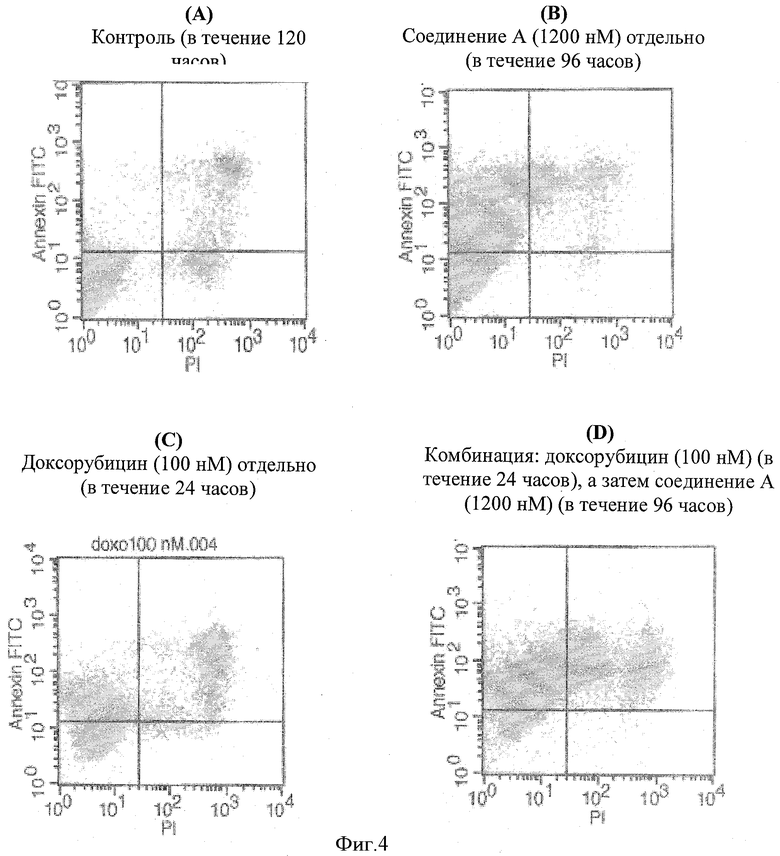
Фигура 4 демонстрирует определение раннего апоптоза при синергической комбинации доксорубицина, за которым следует соединение А в конце 120-часовой обработки, с помощью окрашивания аннексином V. График(и) А, В, С и D представляет(представляют) распределение клеток в четырех квадратах в различных группах обработки, а именно контроль (в течение 120 часов), 1200 нМ соединения А отдельно (в течение 96 часов), 100 нМ доксорубицина отдельно (в течение 24 часов) и комбинацию, включающую введение 100 нМ доксорубицина (24 часа), за которым следует 1200 нМ соединения А (96 часов) соответственно.
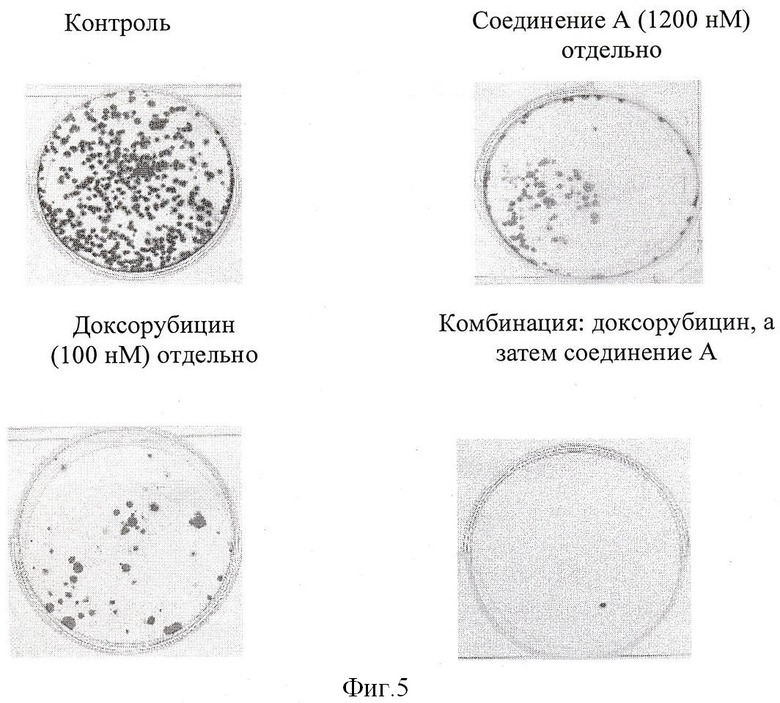
Фигура 5 показывает, что комбинация доксорубицина и соединения А при обработке клеток немелкоклеточного рака легкого Н-460 in vitro проявляет синергизм при тестировании в клоногенном испытании.
Фигура 6 показывает анализ вестерн-блоттинга различных белков, включенных в регуляцию клеточного цикла и апоптоз.
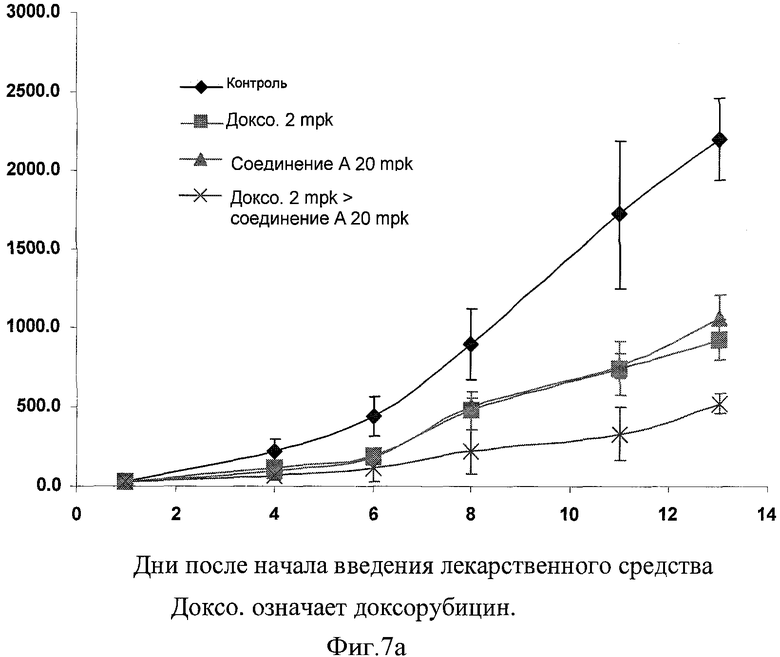
Фигура 7а иллюстрирует in vivo эффективность доксорубицина (2 мг на кг) по человеческим клеткам немелкоклеточной карциномы легкого (Н-460) и соединения А (20 мг на кг) комбинации на Н-460 ксенотрансплантатной модели.
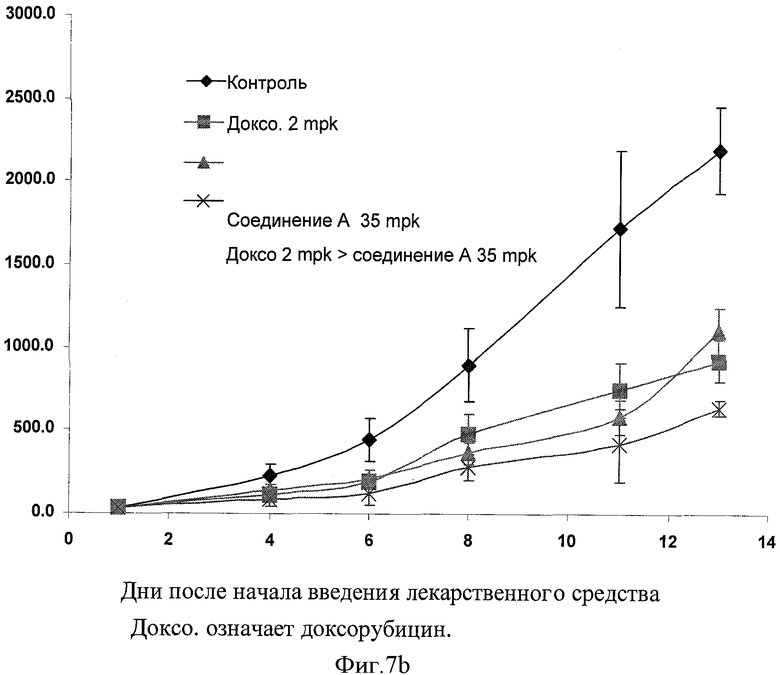
Фигура 7b иллюстрирует in vivo эффективность доксорубицина (2 мг на кг) и соединения А (35 мг на кг) комбинации на Н-460 ксенотрансплантатной модели.
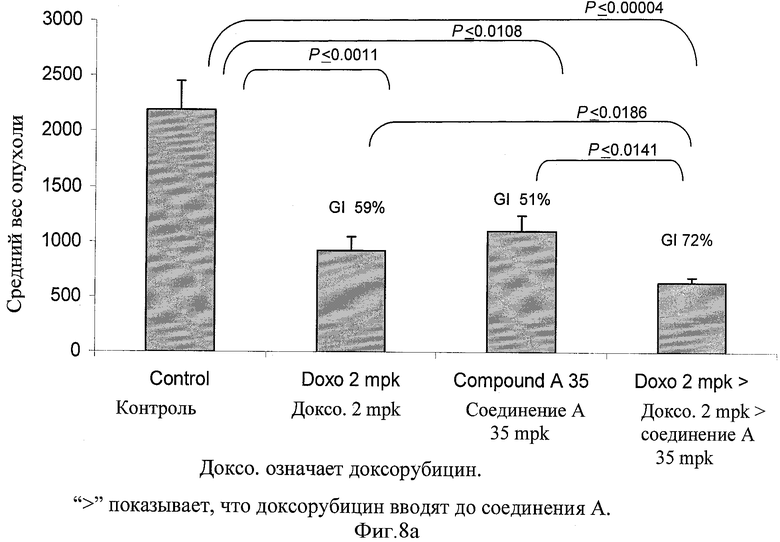
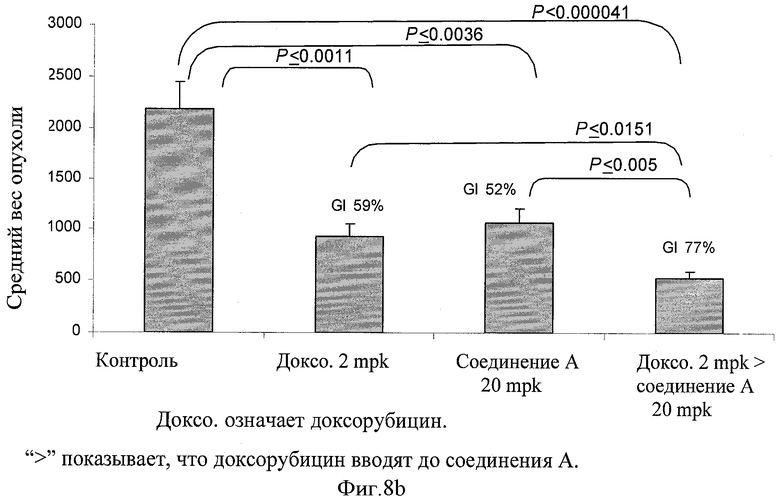
Фигуры 8а и 8b показывают средний вес опухоли в конце лечения и SE (столбцы) 8 опухолей от отдельной мыши в каждой группе в конце исследования. Процент ингибирования роста (GI) в конце обработки представлен для соответствующей группы на вершине каждого столбца. Парный t тест использовали для оценки статистической значимости различия между различными группами обработки. Статистически значимое различие считали присутствующим при Р<0,05.
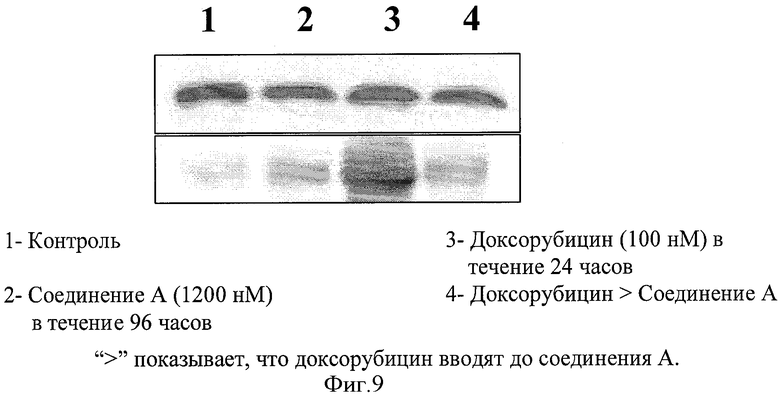
Фигура 9 показывает вестерн-блоттинг с помощью СОХ-2 антитела.
Детальное описание изобретения
В настоящее время установлено, что новая комбинация данного изобретения, которая включает традиционное цитотоксическое противоопухолевое средство, выбранное из группы, включающей паклитаксел, доцетаксел, доксорубицин и гемцитабин или их фармацевтически приемлемую соль, и ингибитор CDK, выбранный из соединений формулы I (как описано здесь), или фармацевтически приемлемой соли, или сольвата их, проявляет синергический эффект, когда применяется при лечении рака, особенно солидных опухолей.
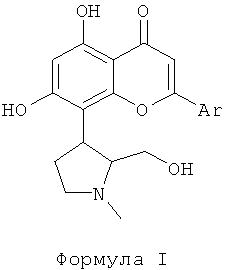
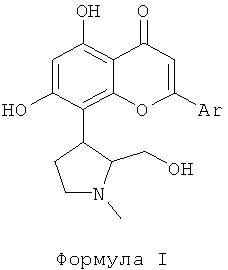
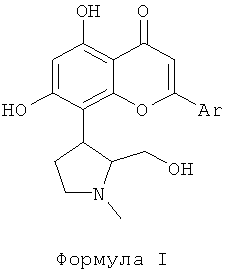
Ингибитор CDK, используемый в фармацевтической комбинации данного изобретения, выбран из соединений формулы I, как описано здесь ниже. Ингибиторы CDK, представленные следующей формулой I, раскрыты в патентной РСТ публикации WO 2004004632. Соединения формулы I являются перспективными ингибиторами CDK, которые могут ингибировать пролиферацию многих раковых клеток. Соединения формулы I, как используется в данном изобретении, эффективны против различных солидных и гематологических злокачественных новообразований. В данном изобретении отмечено, что объединение ингибиторов CDK формулы I с традиционным цитотоксическим противоопухолевым средством, выбранным из группы, включающей паклитаксел, доцетаксел, доксорубицин и гемцитабин, привели к усилению апоптоза или программированной клеточной смерти.
Ингибиторы CDK, используемые в данном изобретении, выбраны из соединений, представленных следующей формулой I
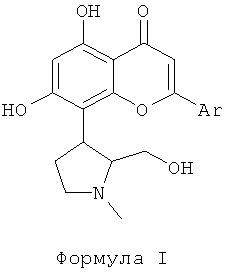
где Ar представляет собой фенильную группу, которая замещена или незамещена 1, 2 или 3 идентичными или различными заместителями, выбранными из: галогена, такого как хлор, бром, фтор или йод, нитро, циано, С1-С4-алкила, трифторметила, гидроксила, С1-С4-алкокси, карбокси, С1-С4-алкоксикарбонила, CONH2 и NR1R2;
где R1 и R2 каждый независимо выбран из водорода или С1-С4-алкила.
Получение соединений формулы I, которые могут быть в форме фармацевтически приемлемых солей и сольватов, и получение оральной и/или парентеральной фармацевтической композиции, включающей вышеуказанные соединения, раскрыты в патентной РСТ публикации WO 2004004632. Этот патент, который включен в данное описание путем ссылки, раскрывает, что ингибиторы CDK, представленные формулой I, проявляют значительную противораковую эффективность.
Как показано здесь выше, ингибиторы CDK формулы I можно использовать в форме их солей или сольватов. Предпочтительная соль соединений формулы I включает гидрохлоридную соль, соль метансульфоновой кислоты и соль трифторуксусной кислоты.
Специалист в данной области оценит, что соединения формулы I содержат, по меньшей мере, два хиральных центра. Таким образом, соединения формулы I существуют в форме двух различных оптических изомеров (то есть (+) или (-) энантиомеров). Все эти энантиомеры и смеси их, включая рацемические смеси, включены в объем данного изобретения. Энантиомеры соединения формулы I можно получить способами, раскрытыми в публикациях РСТ заявок WO 2004004632 и WO 2007148158, которые включены в данное описание путем ссылки во всей их полноте. Энантиомеры соединения формулы I также можно получить способами, хорошо известными в данной области, такими как хиральная HPLC (высокоэффективная жидкостная хроматография) и ферментативное разделение. Альтернативно, энантиомеры соединений формулы (I) можно синтезировать с помощью оптически активных исходных материалов. Таким образом, определение ингибитора CDK формулы I включает в себя все возможные стереоизомеры и их смеси. Определение формулы I включает рацемические формы и выделенные оптические изомеры, обладающие установленной активностью.
Традиционное цитотоксическое противоопухолевое средство(средства), используемое в новой фармацевтической комбинации данного изобретения, можно выбрать из группы, включающей паклитаксел, доцетаксел, доксорубицин, гемцитабин и аналогичные цитотоксические противоопухолевые средства, которые проявляют противораковую активность через сходный механизм действия.
Паклитаксел является натуральным дитерпеновым продуктом, выделенным из дерева тис тихий Taxus brevifolia (Rowinsky et. al., J.Natl. Cancer Inst, 82, 1247-1259 (1990)). Выделение паклитаксела и его структура раскрыты в J.Am. Chem. Soc. 93, 2325 (1971). Это противомикротрубочковое средство, которое обеспечивает систему микротрубочек из тубулиновых димеров и стабилизирует микротрубочки путем предотвращения деполимиризации. Паклитаксел одобрен для клинического применения для лечения овариального рака (Merkman et al.; Yale Journal Of Biology and Medicine, 64:583, 1991) и для лечения рака молочной железы (Holmes et al.; J.Nat. cancer Inst., 83; 1797, 1991), однако он также применим для лечения других онкологических заболеваний, например, его рассматривают как потенциальный кандидат для лечения рака головы и шеи (Forastire et. al., Sem. OncoL, 20: 56, 1990) и рака легкого (М.Ghaemmaghami et al.; Chest; 113; 86-91 (1998)). Паклитаксел раскрыт в патенте США №5670537, который включен в данное описание путем ссылки, для его идеи по применению или введению паклитаксела в лечении восприимчивых онкологических заболеваний. Паклитаксел коммерчески доступен как инъекционный раствор Таксол®. Применение паклитаксела как монотерапии обычно сопровождается нежелательными побочными эффектами, включающими реакции гиперчувствительности, гипотензию, брадикардию, гипертензию, тошноту и рвоту и реакции в месте инъекции.
Доцетаксел принадлежит таксановому семейству и является полусинтетическим производным паклитаксела. Доцетаксел показан главным образом для рака молочной железы и немелкоклеточного рака легкого. Его также применяют в лечении других онкологических заболеваний. Это соединение раскрыто в патенте США №4814470, который включен в данное описание путем ссылки, для его идеи синтеза и применения доцетаксела для лечения восприимчивых онкологических заболеваний. Доцетаксел тригидрат коммерчески доступен как инъекционный раствор Таксотер®. Все лечения на основе таксоидных производных, включая доцетаксел, могут показывать серьезные и болезненные токсичности, такие как миелосупрессия, нейтропения, гиперчувствительность, периферическая нейропатия и удержание жидкости среди прочих (Fumoleau et al., Bull. Cancer, (82)8: 629-636 (1995)).
Доксорубицин является дженерическим названием Адримицина® и коммерчески доступен в инъекционной форме. Доксорубицин был впервые выделен из ферментативного бульона Sreptomyces peucetius var caesius (патент США №3590028). Это цитотоксическое противоопухолевое средство связывается с нуклеиновыми кислотами, вероятно, с помощью специфичного интеркалирования плоского антрациклинового ядра с двойной спиралью ДНК, что приводит к анормальной клеточной репликации. Доксорубицин используют в лечении раковых заболеваний молочной железы, мочевого пузыря, печени, легкого, простаты, желудка и щитовидной железы; сарком костной и мягких тканей; лимфом и лейкемий и опухолей детского возраста. Применение доксорубицина обычно сопровождается некоторыми побочными эффектами, включая миелосупрессию, тошноту и рвоту, кожно-слизистые и сердечные эффекты.
Гемцитабин является дженерическим названием 2'-деокси-2',2'-дифторцитидина. Он коммерчески доступен как моногидрохлоридная соль и как β-изомер. Гемцитабин раскрыт в патентах США №№4808614 и 5464826, которые включены в данное описание путем ссылки, для его идеи синтеза и применения гемцитабина для лечения восприимчивых онкологических заболеваний. Коммерческий состав гемцитабина гидрохлорида как отдельного средства показан как терапия первого ряда для пациентов с локально запущенной или метастатической аденокарциномой поджелудочной железы или карциномой клеток легкого (NSCLC), и его обычно используют для пациентов, которых ранее лечили 5-фторурацилом.
Общие выражения, используемые выше и ниже, предпочтительно имеют в контексте этого описания следующие значения, если не определено иное. Формы единственного числа включают ссылку на множественное число, если контекст явно диктует иное.
Выражение "противоопухолевое средство" является синонимом "химиотерапевтическому средству" или "противораковому средству" и означает терапевтическое средство, которое действует путем ингибирования или предотвращения роста новообразований. Выражение "противоопухолевое средство" или "противораковой средство" обычно означает соединения, которые предотвращают размножение раковых клеток (то есть противопролиферативные средства). Обычно противоопухолевое средство(средства) относятся к двум классам, противопролиферативные цитотоксические и противопролиферативные цитостатические. Цитотоксические средства предотвращают размножение раковых клеток путем (1) препятствования способности клеток реплицировать ДНК и (2) индуцирования клеточной смерти и/или апоптоза в раковых клетках.
Противопролиферативные цитостатические средства действуют посредством модулирования, препятствования или ингибирования процессов клеточной сигнальной трансдукции, которые регулируют клеточную пролиферацию. В данном изобретении противоопухолевые средства, включенные в фармацевтическую комбинацию данного изобретения, являются цитотоксическими средствами и, следовательно, относятся к цитотоксическим противоопухолевым средствам.
Как используется здесь, выражение "синергический" означает, что эффект, достижимый способами и комбинациями данного изобретения, больше, чем сумма эффектов, полученных от применения цитотоксического противоопухолевого средства(средств) или его фармацевтически приемлемой соли и ингибитора CDK формулы I или фармацевтически приемлемой соли или сольвата его, отдельно. Преимущественно такой синергизм обеспечивает большую эффективность при тех же дозах и/или предотвращает или задерживает развитие мультиневосприимчивости к лекарственному средству.
Как используется здесь, выражение "терапевтически эффективное количество" означает количество химиотерапевтического средства, обеспечивающее максимальный апоптоз пролиферативных клеток при минимальной токсичности к непролиферативным клеткам.
Выражение "апоптоз" означает тип клеточной смерти, при котором серии молекулярных этапов в клетке ведут к ее смерти. Для организма это является нормальным способом организма избавляться от ненужных или анормальных клеток. Процесс апоптоза может быть заблокирован в раковых клетках. Также его называют программированной клеточной смертью (Dictionary of cancer terms. National Cancer Institute).
Как используется здесь, выражение "возрастающий апоптоз" определяется как повышение нормы программированной клеточной смерти, то есть у большего числа клеток индуцирован процесс смерти по сравнению с воздействием (контактом) либо цитотоксического противоопухолевого средства отдельно, либо ингибитора CDK отдельно.
Выражение "субъект", как используется здесь, означает животное, предпочтительно млекопитающее, наиболее предпочтительно человек, которое является объектом лечения, наблюдения или эксперимента.
В одном варианте осуществления данное изобретение касается новой фармацевтической комбинации для лечения рака, где указанная комбинация включает цитотоксическое противоопухолевое средство, выбранное из группы, включающей паклитаксел, доцетаксел, доксорубицин или гемцитабин или их фармацевтически приемлемую соль, и, по меньшей мере, один ингибитор циклин-зависимой киназы (CDK), выбранный из соединений формулы I (как описано здесь), или фармацевтически приемлемой соли, или сольвата их.
В одном варианте осуществления фармацевтическая комбинация, включающая ингибитор CDK формулы I и цитотоксические противоопухолевые средства, как описано здесь, не ограничена исключительно теми комбинациями, которые получены физическим объединением указанных ингредиентов, а также охватывает те, которые позволяют отдельное введение, которое может быть одновременным, последовательным или растягиваемым на период времени с тем, чтобы получить максимальную эффективность комбинации. Таким образом, фармацевтическую комбинацию можно вводить одновременно или растягивать на период времени для эффективного лечения рака.
Для цели данного изобретения ингибитор CDK, выбранный из соединений формулы I, можно вводить, например, до, после или одновременно с цитотоксическим противоопухолевым средством. В предпочтительном варианте осуществления данного изобретения цитотоксическое противоопухолевое средство или его фармацевтически приемлемую соль вводят до введения ингибитора CDK формулы I, или фармацевтически приемлемой соли, или сольвата его, в диапазоне дозировки, описанном ниже. Однако оптимальный способ и последовательность введения ингибитора CDK и цитотоксического противоопухолевого средства при данных условиях могут быть надлежащим образом выбранными специалистом в данной области с помощью следующих установленных методик и информации, содержащейся в данном описании.
В одном варианте осуществления составляющие, включенные в комбинацию, должны быть введены различными путями, потому что у них различные физические и химические характеристики. Например, ингибиторы CDK формулы I можно вводить либо орально, либо парентерально для образования и поддержания их оптимальных уровней в крови, тогда как цитотоксическое противоопухолевое средство(средства) можно вводить парентерально, внутривенным, подкожным или внутримышечным путем.
Для орального применения ингибиторы CDK формулы I можно вводить, например, в форме таблеток или капсул, порошков, дисперсных гранул, или крахмальных капсул или как водных растворов, или суспензий. В случае таблеток для орального применения носители, которые обычно используют, включают лактозу, кукурузный крахмал, магния карбонат, тальк и сахар и обычно добавляют смазывающие средства, такие как магния стеарат. Для орального введение в капсулированной форме применимые носители включают лактозу, кукурузный крахмал, магния карбонат, тальк и сахар.
Для внутримышечного, внутриперитонеального, подкожного и внутривенного применения обычно применяют стерильные растворы активного ингредиента (цитотоксического противоопухолевого средства(средств) или ингибитора CDK), а рН растворов должен быть надлежащим образом отрегулирован и буферизован.
В другом варианте осуществления данное изобретение касается способа лечения рака, который включает введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества указанной комбинации. Следовательно, в способе данного изобретения рак лечат у субъекта путем введения субъекту терапевтического количества цитотоксического противоопухолевого средства, эффективного для лечения рака, в комбинации с терапевтически эффективным количеством ингибитора CDK, выбранного из соединений формулы I, или фармацевтически приемлемой соли, или сольвата их, где возникает синергический эффект.
Как показано здесь ранее, активные ингредиенты, содержащиеся в фармацевтической композиции, можно вводить одновременно или последовательно.
Таким образом, согласно данному изобретению способ лечения рака включает введение субъекту, нуждающемуся в таком лечении, терапевтическое количество цитотоксического противоопухолевого средства одновременно с терапевтическим количеством ингибитора CDK, представленного соединениями формулы I.
В одном варианте осуществления способ лечения рака включает последовательное введение терапевтического количества цитотоксического противоопухолевого средства и терапевтического количества ингибитора CDK, представленного соединениями формулы I, субъекту, нуждающемуся в таком лечении.
В другом варианте осуществления способ лечения рака включает введение субъекту, нуждающемуся в таком лечении, терапевтического количества цитотоксического противоопухолевого средства до введения ингибитора CDK, представленного соединениями формулы I.
Способ и фармацевтическую комбинацию данного изобретения можно использовать в лечении рака, выбранного из группы, включающей рак молочной железы, рак легкого (включая мелко- и немелкоклеточный рак легкого и аденокарциному легкого), овариальный рак, рак поджелудочной железы (включая карциному экзокринной поджелудочной железы), рак желудка, колоректальный рак и гепатоцеллюлярный рак.
В предпочтительном варианте осуществления фармацевтическую комбинацию данного изобретения можно использовать в лечении рака, выбранного из немелкоклеточного рака легкого и рака поджелудочной железы.
Фактическая дозировка активных ингредиентов, содержащихся в комбинации, может варьировать в зависимости от потребностей пациента и серьезности состояния, подлежащего лечению. Определение подходящей дозировки для конкретной ситуации известно специалисту в данной области. Как правило, лечение начинают с меньших доз, которые ниже, чем оптимальная доза соединения. Затем дозу каждого ингредиента увеличивают на небольшие количества, пока не будет достигнуто оптимального эффекта при таких обстоятельствах. Однако количество каждого ингредиента в фармацевтической комбинации типично будет меньше, чем количество, которое произвело бы терапевтический эффект при отдельном введении. Для удобства полную дневную дозу можно делить и вводить порциями в течение дня, если нужно. В предпочтительном варианте осуществления цитотоксическое противоопухолевое средство или его фармацевтически приемлемую соль и ингибитор CDK, представленный соединениями формулы I, или фармацевтически приемлемой солью, или сольватом их, вводят последовательно в инъекционных формах, так что цитотоксическое противоопухолевое средство вводят в синергически эффективной дозе, изменяющейся от 10 мг до 1400 мг, предпочтительно изменяющейся от 15 мг до 1000 мг, а ингибитор CDK вводят в синергически эффективной дозе, изменяющейся от 5 мг до 750 мг, предпочтительно изменяющейся от 10 мг до 300 мг.
В одном предпочтительном варианте осуществления данного изобретения, когда цитотоксическим противоопухолевым средством является паклитаксел, его вводят в синергически эффективной дозе, изменяющейся от 30 мг до 300 мг.
В еще одном предпочтительном варианте осуществления данного изобретения, когда цитотоксическим противоопухолевым средством является доцетаксел, его вводят в синергически эффективной дозе, изменяющейся от 20 мг до 175 мг.
В еще одном предпочтительном варианте осуществления данного изобретения, когда цитотоксическим противоопухолевым средством является доксорубицин, его вводят в синергически эффективной дозе, изменяющейся от 17,5 мг до 75 мг.
В следующем предпочтительном варианте осуществления данного изобретения, когда цитотоксическим противоопухолевым средством является гемцитабин, его вводят в синергически эффективной дозе, изменяющейся от 70 мг до 1200 мг.
Комбинации, представленные данным изобретением, оценили некоторыми аналитическими системами и несколькими различными режимами введения in vitro. Детали эксперимента представлены здесь ниже. Представленные здесь данные четко указывают, что цитотоксическое противоопухолевое средство, когда комбинируется с ингибитором CDK формулы I, проявляет синергический эффект. Четко указано, что противораковые средства, когда используются в комбинации при лечении рака, усиливают апоптоз или цитотоксичность у пролиферативных клеток, чем когда клетки обработаны только ингибитором CDK формулы I отдельно или цитотоксическим противоопухолевым средством отдельно. Например, из данных, представленных в Таблицах 2-4, можно четко видеть, что ингибитор CDK, представленный соединением формулы I, обозначенным здесь как соединение А, синергически усилил цитотоксичность доксорубицина в анализе in vitro по отношению к клеткам немелкоклеточной карциномы легкого Н-460.
Представленное соединение, соединение А, используемое в фармакологических испытаниях, является (+)-trans-2-(2-хлор-фенил)-5,7-дигидрокси-8-(2-гидроксиметил-1-метил-пирролидин-3-ил)-хромен-4-оном гидрохлоридом и является одним из соединений, раскрытых в РСТ публикации патентной заявки WO 2004004632, включенной здесь путем ссылки.
В данном изобретении также созданы ксенотрансплантатные модели для расширенных in vitro наблюдений in vivo системы. В данном изобретении тестирована комбинация данного изобретения по ее in vivo эффективности с применением ксенотрансплантатных моделей немелкоклеточного рака легкого SCID (тяжелая комбинированная иммунная недостаточность) у самцов мышей. Было отмечено, что ингибитор CDK синергически усиливал эффективность доксорубицина, когда вводился в последовательной комбинации с доксорубицином. Как видно из графического представления на фигурах 7а и 7b, фармацевтическая комбинация данного изобретения проявила терапевтически синергическую активность на ксенотрансплантатных моделях немелкоклеточного рака легкого SCID мышей.
В параллельном in vitro исследовании, проведенном по данному изобретению, с применением комбинации, включающей традиционное цитотоксическое противоопухолевое средство, доксорубицин и другой известный ингибитор CDK, флавопиридол, в лечении человеческих линий клеток немелкоклеточной карциномы легкого Н-460, обнаружили, что комбинация доксорубицина и флавопиридола независимо от последовательности введения дает аддитивный эффект, а не синергизм (Таблица 16-А, В, С). Детали этого исследования продемонстрированы здесь ниже. Таким образом, нельзя с уверенностью сказать, что комбинация противораковых средств, обладающих различным механизмом действия, может всегда привести к благоприятным терапевтическим эффектам. Однако в данном изобретении четко продемонстрирована синергическая эффективность новой фармацевтической комбинации данного изобретения.
Синергический эффект комбинации данного изобретения, включающей цитотоксическое противоопухолевое средство и ингибитор CDK, будет объяснен более детально со ссылкой на его предпочтительные варианты осуществления. Следует отметить, что они представлены только как примеры и не предназначены ограничивать данное изобретение.
Фармакологические испытания
In vitro испытание цитотоксичности
Используемое испытание цитотоксичности представляло собой испытание MTS (3-(4,5-диметилтиазол-2-ил)-5-(3-карбоксиметоксифенил)-2-(4-сульфофенил)-2Н-тетразолиум, внутренняя соль). Человеческие клетки немелкоклеточной карциномы легкого Н-460 высеяли с плотностью 1500 клеток/лунка в 180 µл культуральной среды в 96-луночный планшет и инкубировали в течение ночи, чтобы позволить клеткам прилипнуть. Варьирующие концентрации лекарственных средств, содержащихся в комбинации, добавили в лунки и инкубировали в течение соответствующего периода времени в увлажненном 5% СО2 инкубаторе при 37°С в случае воздействия отдельного лекарственного средства. При обработке двумя лекарственными средствами традиционное цитотоксическое противоопухолевое средство (паклитаксел, доцетаксел и доксорубицин) вводили в течение 3 часов или 24 часов с последующим удалением среды и промыванием клеток один раз средой. После промывания клеток две различные концентрации соединения А добавили в лунки и планшеты инкубировали в течение 48, 72 или 96 часов в увлажненном 5% СО2 инкубаторе при 37°С. Контрольные лунки обработали наполнителем. В конце периода инкубации среду удалили из лунок и добавили 20 µл MTS (2 мг/мл в фосфатном буфере, рН 6-6,5 и простерилизовали посредством фильтрации) и 1 µл феназин метосульфата (PMS, 3 мМ в фосфатном буфере, рН 7,3 и простерилизовали посредством фильтрации) в каждую лунку и полный объем отрегулировали до 200 µл полной средой. Планшет инкубировали в течение 2-4 часов в увлажненном 5% СО2 инкубаторе при 37°С. Планшет считывали при 490 нМ на спектрофотометре (SpectraMax, Molecular Devices); процент цитотоксичности и IC50 рассчитали с помощью программного обеспечения SoftMax для SpectraMax.
Пример 1
Этот пример показывает синергический эффект комбинации доксорубицина и соединения А, где доксорубицин и соединение А вводили последовательно, так что доксорубицин вводили до соединения А. Человеческие клетки немелкоклеточной карциномы легкого Н-460 высеяли с плотностью 1500 клеток/лунка. Клетки сначала обработали одним из двух лекарственных средств отдельно, то есть доксорубицином или соединением А отдельно. Обработка доксорубицином длилась сначала 24 часа, за ней последовала обработка полной средой в течение 72 часов, тогда как в случае соединения А сначала 24 часа в полной среде, а затем соединением А в течение следующих 72 часов. Используемые концентрации доксорубицина составляли 100 нМ и 200 нМ, тогда как соединение А использовали при концентрации 800 нМ (IC30 концентрация после 48-часовой обработки). При исследовании комбинации клетки сначала обработали 200 нМ или 100 нМ доксорубицина в течение сначала 24 часов, а затем 800 нМ соединения А в течение 72 часов. После завершения обработки лекарственными средствами, то есть в конце 96 часов, планшеты подвергли испытанию MTS выживаемости, а процент цитотоксичности рассчитали по сравнению с контролем. Результаты представлены в следующей Таблице 1.
Пример 2
Этот пример показывает синергический эффект комбинации доксорубицина и соединения А, где доксорубицин и соединение А вводили последовательно, так что доксорубицин вводили до соединения А. В этом примере использовали соединение А при концентрации 1200 нМ. Человеческие клетки немелкоклеточной карциномы легкого Н-460 высеяли с плотностью 1500 клеток/лунка. Клетки сначала обработали одним из двух лекарственных средств отдельно, то есть доксорубицином или соединением А отдельно. Обрабатывали доксорубицином сначала в течение 24 часов, а затем полной средой в течение 72 часов, тогда как в случае соединения А сначала 24 часа в полной среде, а затем соединением А в течение следующих 72 часов. Концентрации используемого доксорубицина составили 200 нМ и 100 нМ, тогда как соединение А использовали при концентрации 1200 нМ (IC50 концентрация после 48-часовой обработки). В исследовании комбинации клетки сначала обработали 100 нМ или 200 нМ доксорубицина в течение 24 часов, а затем 1200 нМ соединения А в течение 72 часов. После завершения обработки лекарственными средствами, то есть в конце 96 часов, планшеты подвергли испытанию MTS выживаемости и процент цитотоксичности рассчитали по сравнению с контролем. Результаты представлены в следующей Таблице 2.
Пример 3
Этот пример показывает синергический эффект комбинации доксорубицина и соединения А, где доксорубицин и соединение А вводили последовательно, так что доксорубицин вводили до соединения А. В этом примере соединение А использовали при концентрации 1200 нМ, а период времени длился 96 часов. Человеческие клетки немелкоклеточной карциномы легкого Н-460 высеяли с плотностью 1500 клеток/лунка. Клетки сначала обработали одним из двух лекарственных средств отдельно, то есть доксорубицином или соединением А отдельно. Обработка доксорубицином длилась сначала в течение 24 часов, а затем полной средой в течение 96 часов, тогда как в случае соединения А сначала 24 часа в полной среде, а затем соединением А в течение следующих 96 часов. Использовали концентрации доксорубицина 100 нМ и 200 нМ, тогда как соединение А использовали при концентрации 1200 нМ (IC50 концентрация после 48-часовой обработки). В исследовании комбинации клетки сначала обработали 100 нМ или 200 нМ доксорубицина в течение 24 часов, а затем 1200 нМ соединения А в течение 96 часов. После завершения обработки лекарственными средствами, то есть в конце 120 часов, планшеты подвергли испытанию MTS выживаемости и процент цитотоксичности рассчитали по сравнению с контролем. Результаты представлены в следующей Таблице 3.
Пример 4
Этот пример показывает синергический эффект комбинации доксорубицина и соединения А, введенных одновременно в течение 120 часов. Человеческие клетки немелкоклеточной карциномы легкого Н-460 высеяли с плотностью 1500 клеток/лунка. Клетки сначала обработали одним из двух лекарственных средств отдельно, то есть доксорубицином или соединением А отдельно в течение 120 часов каждым. Использовали концентрации доксорубицина 30 нМ и 100 нМ, тогда как соединение А использовали при концентрации 800 нМ и 1200 нМ (~IC30 и ~IC50 концентрация после 48-часовой обработки). В исследовании этой комбинации 30 нМ или 100 нМ доксорубицина и 1200 нМ или 800 нМ соединения А соответственно добавили вместе в течение 120 часов. После завершения обработкой лекарственными средствами, то есть в конце 120 часов, планшеты подвергли испытанию MTS выживаемости и процент цитотоксичности рассчитали по сравнению с контролем. Результаты представлены в следующей Таблице 4.
Пример 5
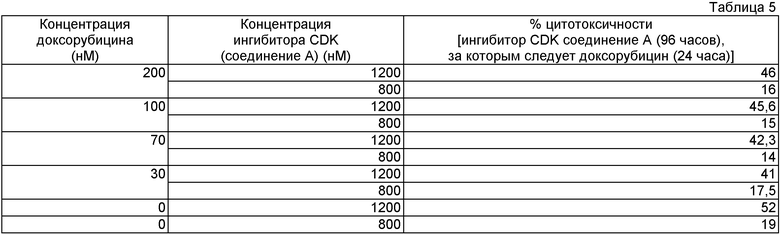
Этот пример показывает, что нет синергического эффекта, когда соединение А вводили перед цитотоксическим противоопухолевым средством доксорубицином. Человеческие клетки немелкоклеточной карциномы легкого Н-460 высеяли с плотностью 1500 клеток/лунка. Клетки сначала обработали одним из двух лекарственных средств отдельно, то есть доксорубицином или соединением А отдельно. Обработали соединением А сначала в течение 96 часов, а затем полной средой в течение 24 часов, тогда как в случае доксорубицина сначала 96 часов в полной среде, а затем доксорубицином в течение 24 часов. Использовали концентрации доксорубицина 30 нМ, 70 нМ, 100 нМ и 200 нМ, тогда как соединение А использовали при концентрации 800 нМ и 1200 нМ (~IC30 и -IC50 концентрация после 48-часовой обработки). В этом исследовании комбинации добавили 1200 нМ или 800 нМ соединения А в течение сначала 96 часов, а затем 30 нМ, 70 нМ, 100 нМ или 200 нМ доксорубицина в течение 24 часов. После завершения обработки лекарственными средствами, то есть в конце 120 часов, планшеты подвергли испытанию MTS выживаемости и процент цитотоксичности рассчитали по сравнению с контролем. Таблица 6 показывает, что процент цитотоксичности в этой комбинации ниже, чем цитотоксичность соединения А при введении отдельно. Поэтому этот эффект последовательности является антагонистическим, поскольку доксорубицин не потенцирует эффект первого лекарственного средства, которое является соединением А в этом случае. Результаты представлены в следующей Таблице 5.
Примеры 1-4 четко показывают, что ингибитор CDK синергически потенцирует эффект доксорубицина, когда ингибитор CDK вводят после или одновременно с цитотоксическим лекарственным средством. Пример 5 также показывает важность последовательного лечения. Лечение доксорубицином, за которым следует ингибитор CDK, оказывается синергическим, тогда как обратная последовательность не эффективна.
Пример 6
Этот пример показывает синергический эффект комбинации доцетаксела и соединения А, где доцетаксел и соединение А вводили последовательно, так что доцетаксел вводили до соединения А. Человеческие клетки немелкоклеточной карциномы легкого Н-460 высеяли с плотностью 3000 клеток/лунка. Клетки сначала обработали одним из двух лекарственных средств отдельно, то есть доцетакселом или соединением А отдельно. Обрабатывали доцетакселом в течение сначала 3 часов, а затем полной средой в течение 45 часов, тогда как в случае обработки соединением А сначала 3 часа полной средой, а затем соединением А в течение следующих 45 часов. Использовали концентрации доцетаксела 0,1 нМ и 3 нМ, тогда как соединение А использовали при концентрации 700 нМ (~IC30 концентрация после 48-часовой обработки). В этом исследовании комбинации клетки сначала обработали 0,1 нМ или 3 нМ доцетаксела в течение 3 часов, а затем 700 нМ соединения А в течение 45 часов. После завершения обработки лекарственными средствами, то есть в конце 48 часов, планшеты подвергли испытанию MTS выживаемости и процент цитотоксичности рассчитали по сравнению с контролем. Результаты представлены в следующей Таблице 6.
Пример 7
Этот пример показывает синергический эффект комбинации доцетаксела и соединения А, где доцетаксел и соединение А вводили последовательно, так что доцетаксел вводили до соединения А. Человеческие клетки немелкоклеточной карциномы легкого Н-460 высеяли с плотностью 3000 клеток/лунка. Клетки сначала обработали одним из двух лекарственных средств отдельно, то есть доцетакселом или соединением А отдельно. Обрабатывали доцетакселом в течение сначала 3 часов, а затем полной средой в течение 45 часов, тогда как в случае обработки соединением А сначала 3 часа полной средой, а затем соединением А в течение следующих 45 часов. Использовали концентрации доцетаксела 0,1 нМ и 3 нМ, тогда как соединение А использовали при концентрации 1000 нМ (~IC50 концентрация после 48-часовой обработки). В исследовании комбинации клетки сначала обработали 0,1 нМ или 3 нМ доцетаксела в течение 3 часов, а затем 1000 нМ соединения А в течение 45 часов. Планшеты подвергли испытанию MTS выживаемости и процент цитотоксичности рассчитали по сравнению с контролем. Результаты представлены в следующей Таблице 7.
Пример 8
Этот пример показывает синергический эффект комбинации паклитаксела и соединения А, где паклитаксел и соединение А вводили последовательно, так что паклитаксел вводили до соединения А. Человеческие клетки немелкоклеточной карциномы легкого Н-460 высеяли с плотностью 1500 клеток/лунка. Клетки сначала обработали одним из двух лекарственных средств отдельно, то есть паклитакселом или соединением А отдельно. Обрабатывали паклитакселом в течение сначала 3 часов, а затем полной средой в течение 45 часов, тогда как в случае обработки соединением А сначала 3 часа полной средой, а затем соединением А в течение следующих 45 часов. Концентрации используемого паклитаксела составляли 10 нМ, тогда как соединение А использовали при концентрации 700 нМ (~IC30 концентрация после 48-часовой обработки). В исследовании комбинации клетки сначала обработали 10 нМ паклитаксела в течение 3 часов, а затем 700 нМ соединения А в течение 45 часов. После завершения обработки лекарственными средствами, то есть в конце 48 часов, планшеты подвергли испытанию MTS выживаемости и процент цитотоксичности рассчитали по сравнению с контролем. Результаты представлены в следующей Таблице 8.
Пример 9
Этот пример показывает синергический эффект комбинации гемцитабина и соединения А, где гемцитабин и соединение А вводили последовательно, так что гемцитабин вводили до соединения А. Клетки из человеческой клеточной линии поджелудочной железы (Panc-1) высеяли с плотностью 1500 клеток/лунка. Клетки сначала обработали одним из двух лекарственных средств отдельно, то есть гемцитабином или соединением А отдельно. Обрабатывали гемцитабином в течение сначала 24 часов, а затем полной средой в течение 72 часов. Тогда как в случае обработки соединением А сначала 24 часа полной средой, а затем соединением А в течение следующих 72 часов. Гемцитабин использовали при концентрации 70 нМ, тогда как соединение А использовали при концентрации 300 нМ (~IC30 концентрация после 48-часовой обработки). В исследовании комбинации клетки сначала обработали 70 нМ гемцитабина в течение 24 часов, а затем 300 нМ соединения А в течение 72 часов. После завершения обработки лекарственными средствами, то есть в конце 96 часов, планшеты подвергли испытанию MTS выживаемости и процент цитотоксичности рассчитали по сравнению с контролем. Результаты представлены в следующей Таблице 9.
часа), за которым следует ингибитор CDK (соединение А) (72 часа)
Анализ распределения клеточного цикла и проточной цитометрии
Человеческую немелкоклеточную карциному легкого Н-460 высеяли в 25 мм3 колбы с культурой ткани. Через 24 часа клетки обработали соединением А отдельно в течение 72 часов или 96 часов и цитотоксическим противоопухолевым средством доксорубицином отдельно в течение 24 часов. Для исследования комбинации клетки обработали сначала цитотоксическим противоопухолевым средством доксорубицином в течение 24 часов, а затем соединением А в течение 72 часов или 96 часов после удаления цитотоксического противоопухолевого средства и промывания клеток дважды PBS (фосфатным буферным раствором). Контрольные клетки оставили необработанными в течение 96 часов или 120 часов. И отдельные, и прилипшие клетки собрали в различные точки времени. Клетки промыли дважды приблизительно 5 мл PBS с центрифугированием при 1000 rpm (оборотов в минуту) в течение 10 минут. Клетки повторно суспендировали в 500 µл PBS и фиксировали в 500 µл охлажденного до 0°С 70% этанола. Фиксированные клетки инкубировали при комнатной температуре в течение 30 минут, вращали при 1000 rpm в течение 10 минут. К клеточному осадку добавили 1 мл охлажденного 70% этанола и затем клеточный осадок держали в холоде до следующего анализа. Клетки промыли дважды PBS для удаления фиксажа и повторно суспендировали в 250 µл PBS. К ним добавили 50 µл пропидий йодида (4 мг/мл d PBS) и 12,5 µл РНКазы А (1 мг/мл). После инкубации при 37°С в течение 30 минут клетки анализировали с помощью проточной цитометрии.
Использовали проточный цитометр Becton Dickinson FACS Calibur согласно рекомендациям производителя. Использовали установку лазер на ионах аргона при 488 нм как источник возбуждения. Клетки с содержанием ДНК между 2n и 4n были определены как находящиеся в фазах G1, S и G2/M клеточного цикла, как определили уровнем красной флуоресценции. Клетки, обладающие содержанием ДНК менее чем 2n, обозначили как суб-G1 клетки. Число клеток в каждом отделении клеточного цикла выразили как процент полного числа присутствующих клеток.
Пример 10
Этот пример дает распределение клеточного цикла для различных обработок, как показано на фигуре 1. Около 1-2×106 клеток высеяли в колбу с культурой ткани для групп обработки. Протокол испытания был таким же, как упомянуто выше в "Анализ распределения клеточного цикла и проточная цитометрия". Клеточный цикл разделили на четыре части, которые представлены на фигуре 1 как M1, M2, М3 и М4. M1 соответствует G1 фазе, M2 - S фазе, М3 - G2-M фазе, а М4 - суб-G1 фазе, которая представляет клетки, подвергающиеся апоптозу. 96-часовый контроль, где не было обработки лекарственным средством, показал незначительный апоптоз только 2%, тогда как группа обработки любым лекарственным средством отдельно показала только 10% апоптоз и для соединения А, и для доксорубицина отдельно. Комбинация обоих лекарственных средств показала повышенный апоптоз 34%.
Пример 11
Этот пример дал распределение клеточного цикла для различных групп обработки, как показано на фигуре 2. Около 1-2×106 клеток высеяли в колбу с культурой ткани для групп обработки. Протокол испытания был таким же, как упомянуто выше в "Анализ распределения клеточного цикла и проточная цитометрия". Клеточный цикл разделили на четыре части, которые представлены на фигуре как M1, M2, М3 и М4. M1 соответствует G1 фазе, M2 - S фазе, М3 - G2-M фазе, а М4 -суб-G1 фазе, которая представляет клетки, подвергающиеся апоптозу. 120-часовый контроль, где не было обработки лекарственным средством, показал незначительный апоптоз только 8%, тогда как группа обработки любым лекарственным средством отдельно показала 32% и 3% апоптоз для соединения А и доксорубицина соответственно. Комбинация обоих лекарственных средств показала повышенный апоптоз 65%.
Пример 12
Этот пример дал распределение клеточного цикла для различных групп обработки, как показано на фигуре 3. Около 1-2х106 клеток высеяли в колбу с культурой ткани для групп обработки. Протокол испытания был таким же, как упомянуто выше в "Анализ распределения клеточного цикла и проточная цитометрия". Клеточный цикл разделили на четыре части, которые представлены на фигуре как M1, М2, М3 и М4. M1 соответствует G1 фазе, М2 - S фазе, М3 - G2-M фазе, а М4 - суб-G1 фазе, которая представляет клетки, подвергающиеся апоптозу. 96-часовый контроль, где не было обработки лекарственным средством, показал незначительный апоптоз только 2,1%, тогда как группа обработки любым лекарственным средством отдельно показала 4,3% и 1,7% апоптоз для соединения А и гемцитабина соответственно. Комбинация обоих лекарственных средств показала повышенный апоптоз 25,4%.
Пример 13
Окрашивание аннексином V-FITC (для определения раннего апоптоза)
Аннексии V-FITC (флуоресцинизотиоцианат) является чувствительной пробой для идентификации апоптических клеток. Во время раннего апоптоза мембранный фосфолипидный фосфотидил серии (PS) перемещается от внутреннего к внешнему листу плазматической мембраны, тем самым PS подвергается внешней клеточной окружающей среде. Аннексин V является 35-36 кДа кальций фосфолипид связывающим белком, который обладает высокой аффинностью к PS и связывается с клетками с открытым PS. Пропидий йодид (PI) представляет собой полярный краситель, который входит в клетки через неплотные мембраны и, следовательно, используется в сочетании с FITC для определения позднего апоптоза.
Человеческую немелкоклеточную карциному легкого Н-460 высеяли в 25 мм3 колбы с культурой ткани. Через 24 часа клетки обработали 1200 нМ соединения А или 100 нМ доксорубицина отдельно в течение 96 часов и 24 часов соответственно. Для исследования комбинации клетки обработали сначала 100 нМ цитотоксического противоопухолевого средства доксорубицина в течение 24 часов, а затем 1200 нМ соединения А в течение 96 часов после удаления цитотоксического противоопухолевого средства (доксорубицина) и промывания клеток один раз средой. Контрольные клетки оставили необработанными в течение 120 часов. Плавающие в среде клетки собрали и объединили с прилипшими клетками после сбора с трипсином в различные моменты времени. Клетки промыли дважды холодным PBS с центрифугированием при 1000 rpm в течение 10 минут. Клеточный осадок повторно суспендировали в IX связывающем буфере (10 мм HEPES (N-2-гидроксиэтилпиперазин-N-2-этансульфоновая кислота), рН 7,4, 140 мМ NaCl, 2,5 мМ CaCl2) при концентрации 1×106 клетки/мл. 100 µл раствора (1×105 клеток) окрасили аннексином V-FITC и пропидий йодидом. Клетки инкубировали в течение 15 минут при комнатной температуре в темноте и образец анализировали проточной цитометрией.
Использовали проточный цитометр Becton Dickinson FACS Calibur согласно рекомендациям производителя. Использовали установку лазер на ионах аргона при 488 нм как источник возбуждения. Фигура 4 показывает распределение клеток на четыре квадрата. Квадрат I, расположенный в нижнем левом углу (LL), показывает клетки, негативные по FITC и PI, что свидетельствует о том, что они являются здоровыми. Квадрат II, расположенный в нижнем правом углу (LR), показывает клетки, которые позитивны только по PI, что свидетельствует о том, что они полностью апоптические. Квадрат III в верхнем правом углу (UR) показывает клетки, которые позитивны и по аннексину, и PI, что свидетельствует о том, что они переходят от раннего апоптоза в поздний апоптоз. Квадрат IV в верхнем левом углу (UL) показывает, что клетки позитивны только по аннексину, что свидетельствует о том, что они находятся в раннем апоптозе.
Если клетки, даже после прекращения воздействия соединения, продолжают входить в апоптоз, они будут положительно окрашены аннексином. Клетки один раз в раннем апоптозе подвергнуты программированной клеточной смерти и в точке необратимости. Результаты указаны в Таблице 10. Было установлено, что самый высокий процент клеток в комбинации находятся или в раннем, или в раннем - позднем апоптозе по сравнению с любым лекарственным средством отдельно.
Пример 14
Клоногенное испытание
Человеческие клетки немелкоклеточной карциномы легкого (Н-460) высеяли с плотностью 750-1000 клеток на 35 мм планшет для культуры ткани. Инкубировали в течение ночи при 37°С для прикрепления клеток к планшету. Клетки обработали цитотоксическим противоопухолевым средством в течение 24 часов с последующим промыванием клеток и добавлением свежей среды, включающей соединение А, в течение 96 часов. В конце обработки среду снова заменили свежей полной средой, включающей 10% FCS, и инкубировали в течение 7-14 дней для формирования колоний. Как только на планшете появились видимые колонии, среду удалили, а колонии фиксировали смесью метанола и уксусной кислоты в соотношении 2:1 в течение 5 минут. Планшеты промыли водой и повторили процедуру фиксации. Планшеты высушили и колонии окрасили 0,1% кристаллическим фиолетовым красителем в течение 3-5 минут. Планшеты осторожно ополоснули водой, высушили и колонии сосчитали на Geldoc.
Клетки обработали 1200 нМ соединения А или 100 нМ доксорубицина отдельно в течение 96 часов и 24 часов соответственно или в комбинации 100 нМ доксорубицина, за которым следует 1200 нМ соединения А, в течение 96 часов. Фигура 5 показывает синергический эффект комбинации, поскольку только одна колония была видна в комбинации по сравнению с контролем или любым из лекарственных средств отдельно.
Эксперименты восстановления после лечения
Протокол испытания для обработки клеток соединением А и доксорубицином отдельно или в комбинации был таким же, как описано в анализе распределения клеточного цикла. После обработки лекарственным средством клеткам позволили восстановиться в свежей полной среде, включающей 10% FCS. Клетки при восстановлении анализировали с помощью FACS при 0-, 6-, 18-, 24- и 48-часовых моментах времени для обработки любым лекарственным средством отдельно и комбинацией. В следующих примерах восстановление клеток представлено процентом клеток, подвергающихся апоптозу.
Пример 15
Клетки обработали только цитотоксическим противоопухолевым средством доксорубицином в течение 24 часов с последующим удалением среды и замещением свежей полной средой. FACS анализ выполнили, как описание в способе, установленном для определения процента клеток, подвергающихся апоптозу во время периода восстановления после окончания обработки лекарственным средством. Апоптоз определяли в 0, 6, 18, 24 и 48 часов во время периода восстановления. В конце 24 часов обработки лекарственным средством процент апоптоза составлял 3%, который во время периода восстановления значительно не увеличился, указывая, что клетки в конечном итоге восстановились от обработки лекарственным средством. Результаты показаны в Таблице 11.
Пример 16
Испытание выполнили, как описано в протоколе. Клетки обработали только соединением А в течение 96 часов с последующим удалением среды и замещением свежей полной средой. FACS анализ выполнили, как описано в способе, позволяющем определить процент клеток, подвергающихся апоптозу во время периода восстановления после окончания обработки лекарственным средством. Апоптоз определили в 0, 6, 18, 24 и 48 часов во время периода восстановления. В конце 96 часов обработки лекарственным средством процент апоптоза составил 32%, который во время периода восстановления снизился от 24% до 19% в конце 48 часов восстановления, показывая, что клетки постепенно восстанавливаются при увеличении периода восстановления. Результаты показаны в Таблице 12.
Пример 17
Испытание выполнили, как описано в протоколе. Клетки обработали доксорубицином в течение 24 часов, а затем соединением А в течение 96 часов с последующим удалением среды и замещением свежей полной средой. FACS анализ выполнили, как описано в способе, позволяющем определить процент клеток, подвергающихся апоптозу во время периода восстановления после окончания обработки лекарственным средством. Апоптоз определяли в 0, 6, 18, 24 и 48 часов во время периода восстановления. В конце обработки лекарственным средством процент апоптоза составлял 55%. Во время периода восстановления дополнительные 32% вошли в апоптоз в конце 6 часов, которые повысились до 57% в конце 48 часов восстановления, показывая, что клетки не восстановились, а вместо этого продолжают входить в апоптоз во время периода восстановления. Результаты показаны в Таблице 13.
Пример 18
Анализ вестерн-блоттинга
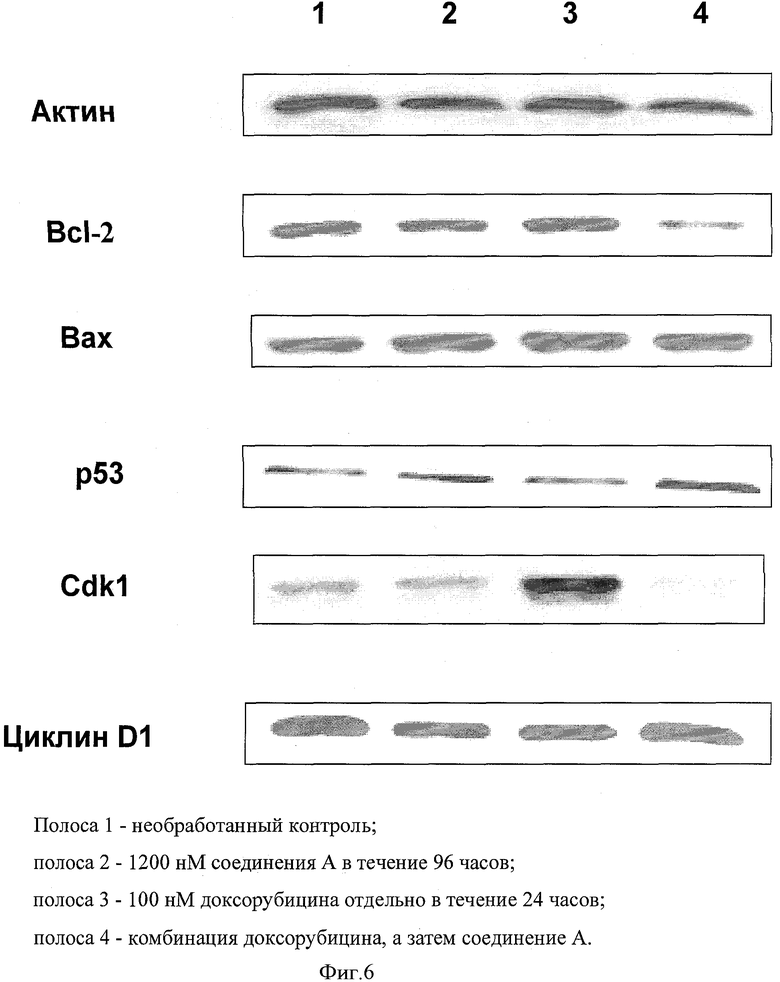
Человеческие клетки немелкоклеточной карциномы легкого (Н-460) были необработанными, то есть контрольными клетками, или обработанными 100 нМ доксорубицина отдельно в течение 24 часов или 1200 нМ соединения А отдельно в течение 96 часов. В обработке комбинацией клетки сначала обработали 100 нМ доксорубицина в течение 24 часов, а затем 1200 нМ соединения А в течение 96 часов. В конце периода обработки клетки лизировали и оценили содержание белка лизата с помощью реагента Брэдфорда. 40 µг белка загрузили в SDS-PAGE (электрофорез в полиакриламидном геле с додецилсульфатом натрия) и перенесли на PVDF (поливинилиденфторидную) мембрану. Мембраны испытали с помощью р53, Вах, Вс1-2, циклин Dl, Cdk1 и актин антител. Первичное антитело определяют вторичным антителом с пероксидазой хрена и подвергают реагентам west pico хемилюминесценции.
Фигура 6 показывает анализ вестерн-блоттинга различных белков, вовлеченных в регуляцию клеточного цикла и апоптоз. Равное количество белка загрузили в четыре полосы. Различные образцы, загруженные в лунки, описаны в подписях на фигуре. Результаты показывают, что противоапоптический белок Bcl-2 был существенно подавлен в обработке комбинацией по сравнению с любым лекарственным средством отдельно, что было почти эквивалентно контролю. Это коррелирует с повышенным апоптозом, наблюдаемым в обработке комбинацией в FACS анализе. Проапоптический белок Вах слегка активирован по отношению к контролю во всех обработанных образцах. Белок - супрессор опухолевого роста р53 был существенно активирован в обработке комбинацией по сравнению с контролем, но не так сильно, как в других двух группах обработки. Cdkl-B1 является инициатором митоза. Дерегулирование этого фермента ведет к онкогенезу. Таким образом, ингибирование Cdk1 будет ингибировать его активность и, следовательно, инициацию митоза и клеточной пролиферации. Фигура показывает, что доксорубицин существенно индуцирует уровни Cdkl, тогда как добавление соединения А понижает Cdkl до незначительных уровней, таким образом, предотвращая клетки от вхождения в митоз. Соединение А отдельно показало уровни, равные контролю. Уровни циклина D1 не показали существенного изменения в различных группах обработки. В обработке комбинацией уровни были эквивалентны контролю, тогда как с соединением А и доксорубицином отдельно наблюдалось незначительное снижение в уровнях.
Пример 19
Этот пример показывает in vivo тестирование эффективности комбинации доксорубицина и ингибитора CDK, соединения А, на немелкоклеточного легкого (Н-460) ксенотрансплантатной модели.
Для исследования использовали человеческую клеточную линию немелкоклеточной карциномы легкого (Н-460), полученную из американской коллекции типовых культур (АТСС). Доксорубицин и соединение А для i.p.(внутриперитонеального) введения получили путем растворения соединений в солевом растворе.
Использовали группу 36 самцов мышей с тяжелым комбинированным иммунодефицитом SCID, весом ~20 г возрастом 6-8 недель.
Человеческие клетки немелкоклеточной карциномы легкого (Н-460) выращивали на среде RPMI 1640, включающей 10% фетальной телячьей сыворотки в 5% СО2 инкубаторе при 37°С. Клетки осадили центрифугированием при 1000 rpm в течение 10 минут. Клетки повторно суспендировали в солевом растворе до получения количества 25×106 клеток на мл, 0,2 мл этой клеточной суспензии инъецировали подкожным (s.c.) путем SCID мышам. Мышей осматривали через день на предмет пальпируемой опухолевой массы. Как только размер опухоли достиг 5-7 мм в диаметре, животных рандомизировали на группы соответствующего лечения, как показано в следующей Таблице 14. Доксорубицин вводили один раз каждую неделю, а соединение А вводили один раз каждый день в течение 5 дней, как показано в Таблице 15. Сначала вводили дозу доксорубицина, а затем соединения А с интервалом 6 часов с последующим соединением А ежедневно периодом пять дней, что заключало в себе один цикл. После двухдневного перерыва должен был начинаться следующий цикл. Лечение включало всего 2 цикла. Ежедневно регистрировали вес тела. Размер опухоли и другие признаки токсичности регистрировали через день. Не наблюдалась значительная потеря веса или признаки заболеваемости. Вес опухоли (мг) оценивали по формуле для удлиненного элипсоида: {Длина (мм)×[ширина (мм)2]×0,5}, принимая удельный вес за единицу, а π за тройку. Рост опухоли у обработанных соединением животных рассчитали как Т/С (обработка/контроль)×100%, а процент ингибирования роста (GI%) составлял [100-Т/С%]. Результаты представлены графически на фигурах 7а, 7b, 8 и 9.
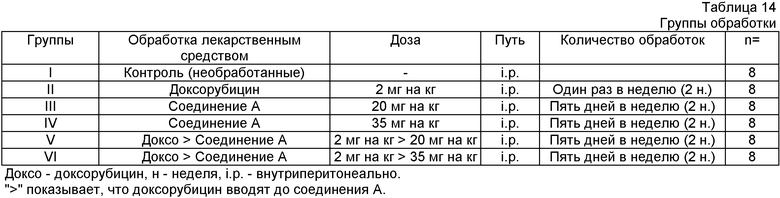
М, Т, W, Т и F: дни недели (Monday, Tuesday, Wednesday, Thursday и Friday)
">" показывает, что доксорубицин вводят до соединения А.
Пример 20
Анализ вестерн-блоттинга с помощью СОХ-2 антитела
Фигура 9 показывает анализ вестерн-блоттинга с помощью СОХ-2 антитела. Различные образцы, загруженные в лунки, показаны в подписях на фигуре. Результаты показывают, что:
- Контроль показал базальные уровни СОХ-2, которые являются низкими.
- Соединение А отдельно также показало низкие уровни СОХ-2.
- Доксорубицин сильно индуцировал СОХ-2, который отвечает за хемоневосприимчивость через путь NFκB сигнализации.
- Добавление соединения А после доксорубицина существенно подавляло СОХ-2.
Таким образом, опосредованное NFκB ингибирование СОХ-2 соединением А могло быть включено в супрессию роста опухоли и индуцированной доксорубицином хемоневосприимчивости в опухолевом ксенотрансплантате человеческой немелкоклеточной карциномы легкого (Н-460).
Пример 21
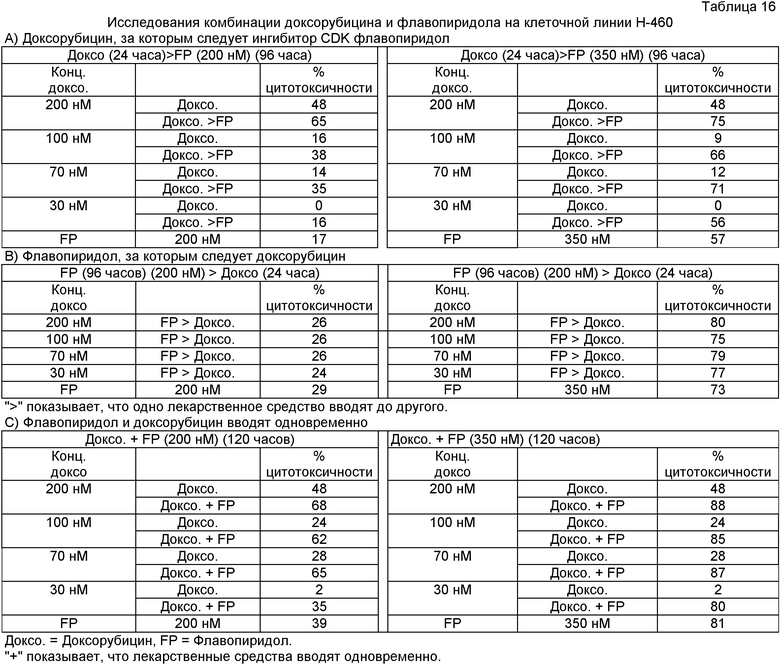
Исследования комбинации доксорубицина и флавопиридола на человеческой клеточной линии немелкоклеточной карциномы легкого (Н-460)
Этот пример показывает, что не было синергического эффекта, когда флавопиридол вводили после (Таблица 16А), перед. (Таблица 16В) или параллельно с (Таблица 16С) цитотоксическим противоопухолевым средством доксорубицином. Человеческие клетки немелкоклеточной карциномы легкого Н-460 высеяли с плотностью 1500 клеток/лунка. Согласно Таблице 16А клетки сначала обработали одним из лекарственных средств отдельно, то есть доксорубицином или флавопиридолом отдельно. Обрабатывали доксорубицином в течение сначала 24 часов, а затем полной средой в течение 96 часов, а в случае флавопиридола сначала 24 часа полной средой, а затем флавопиридолом в течение следующих 96 часов. Использовали концентрации доксорубицина 30 нМ, 70 нМ, 100 нМ и 200 нМ, а флавопиридол использовали при концентрации 200 нМ и 350 нМ (IC30 и IC50 концентрации соответственно после 48-часовой обработки). В исследовании комбинации клетки сначала обработали 30 нМ, 70 нМ, 100 нМ и 200 нМ доксорубицина в течение сначала 24 часов, а затем 200 нМ и 350 нМ флавопиридола в течение 96 часов. После завершения обработки лекарственным средством, то есть в конце 120 часов, планшеты подвергли испытанию MTS выживаемости и процент цитотоксичности рассчитали по сравнению с контролем. Результаты показаны в Таблице 16А.
Согласно Таблице 16В клетки сначала обработали одним из лекарственных средств отдельно, то есть доксорубицином или флавопиридолом отдельно. Обрабатывали флавопиридолом в течение сначала 96 часов, а затем полной средой в течение 24 часов, а в случае доксорубицина сначала 96 часов полной средой, а затем доксорубицином в течение 24 часов. Использовали концентрации доксорубицина 30 нМ, 70 нМ, 100 нМ и 200 нМ, а флавопиридол использовали при концентрации 200 нМ и 350 нМ (~IC30 и ~IC50 концентрация после 48-часовой обработки). В этом исследовании комбинации добавили 200 нМ или 350 нМ флавопиридола в течение сначала 96 часов, а затем 30 нМ, 70 нМ, 100 нМ или 200 нМ доксорубицина в течение 24 часов. После завершения обработки лекарственными средствами, то есть в конце 120 часов, планшеты подвергли испытанию MTS выживаемости и процент цитотоксичности рассчитали по сравнению с контролем. Результаты показаны в Таблице 16 В.
Таблица 16С не показывает синергический эффект комбинации доксорубицина и флавопиридола, введенных одновременно в течение 120 часов. Человеческие клетки немелкоклеточной карциномы легкого Н-460 высеяли с плотностью 1500 клеток/лунка. Клетки сначала обработали одним из лекарственных средств отдельно, то есть доксорубицином или флавопиридолом отдельно, в течение 120 часов каждым. Использовали концентрации доксорубицина 30 нМ, 70 нМ, 100 нМ и 200 нМ, а флавопиридол использовали при концентрации 200 нМ и 350 нМ (~IC30 и ~IC50 концентрация после 48-часовой обработки). В этом исследовании комбинации добавили 30 нМ, 70 нМ, 100 нМ или 200 нМ доксорубицина и 200 нМ или 350 нМ флавопиридола соответственно вместе в течение 120 часов. После завершения обработки лекарственными средствами, то есть в конце 120 часов, планшеты подвергли испытанию MTS выживаемости и процент цитотоксичности рассчитали по сравнению с контролем. Результаты представлены в следующей Таблице 16С.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМБИНИРОВАННАЯ ХИМИОТЕРАПИЯ | 2001 |
|
RU2284818C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ПРОТИВООПУХОЛЕВЫМ АЛКАЛОИДОМ | 2018 |
|
RU2757373C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ПРОТИВООПУХОЛЕВЫМ АЛКАЛОИДОМ | 2011 |
|
RU2743643C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ПРОТИВООПУХОЛЕВЫМ АЛКАЛОИДОМ | 2018 |
|
RU2767664C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ПРОТИВООПУХОЛЕВЫМ АЛКАЛОИДОМ | 2011 |
|
RU2605335C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМБИНАЦИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2018 |
|
RU2801665C2 |
| АКТИВАЦИЯ ПРОКАСПАЗЫ-3 С ПОМОЩЬЮ КОМБИНИРОВАННОЙ ТЕРАПИИ | 2013 |
|
RU2659936C2 |
| ЛЕЧЕНИЕ РАКА КОМБИНАЦИЕЙ ЛУЧЕВОЙ ТЕРАПИИ, НАНОЧАСТИЦ ОКСИДА ЦЕРИЯ И ХИМИОТЕРАПЕВТИЧЕСКОГО СРЕДСТВА | 2015 |
|
RU2704811C2 |
| КОМБИНИРОВАННАЯ ХИМИОТЕРАПИЯ | 2006 |
|
RU2429838C2 |
| КОМБИНИРОВАННАЯ ХИМИОТЕРАПИЯ | 2010 |
|
RU2587013C2 |

Предложены: фармацевтическая комбинация, включающая цитотоксическое противоопухолевое средство, выбранное из группы, включающей паклитаксел, доцетаксел, доксорубицин и гемцитабин или их фармацевтически приемлемую соль, и, по меньшей мере, один ингибитор циклин-зависимой киназы (CDK) формулы I, ее применение для лечения рака молочной железы, мелкоклеточного или немелкоклеточного рака легкого, овариального рака, рака поджелудочной железы, желудка, колоректального рака и гепатоцеллюлярного рака; вариант применения, когда цитотоксическое противоопухолевое средство и указанный ингибитор циклин-зависимой киназы (CDK) вводятся одновременно или последовательно. Показаны синергические эффекты заявленной комбинация при лечении рака. 3 н. и 16 з.п. ф-лы, 9 ил., 16 табл.
1. Фармацевтическая комбинация, адаптированная для лечения рака, выбранного из группы, включающей рак молочной железы, мелкоклеточный рак легкого, немелкоклеточный рак легкого, овариальный рак, рак поджелудочной железы, рак желудка, колоректальный рак и гепатоцеллюлярный рак, включающая цитотоксическое противоопухолевое средство, выбранное из группы, включающей паклитаксел, доцетаксел, доксорубицин и гемцитабин или их фармацевтически приемлемую соль; и ингибитор циклин-зависимой киназы (CDK), или энантиомер, или его фармацевтически приемлемую соль, или сольват, где указанный ингибитор циклин-зависимой киназы (CDK) представлен следующей формулой I:
где Аr представляет собой фенил, который незамещен или замещен 1, 2 или 3 идентичными или различными заместителями, выбранными из: галогена, выбранного из хлора, брома, фтора или йода, нитро, циано, С1-С4-алкила, трифторметила, гидроксила, С1-С4-алкокси, карбокси, С1-С4-алкоксикарбонила, CONH2 и NR1R2; где R1 и R2 каждый независимо выбран из водорода или C1-C4-алкила.
2. Фармацевтическая комбинация по п.1, где ингибитор циклин-зависимой киназы (CDK) является соединением формулы I, где фенильная группа замещена 1, 2 или 3 идентичными или разными заместителями, выбранными из: галогена, выбранного из хлора, брома, фтора или йода, С1-С4-алкила или трифторметила.
3. Фармацевтическая комбинация по п.2, где ингибитор циклин-зависимой киназы (CDK) является соединением формулы I, где фенильная группа замещена 1, 2 или 3 галогенами, выбранными из хлора, брома, фтора или йода.
4. Фармацевтическая комбинация по п.3, где ингибитор циклин-зависимой киназы (CDK) является соединением формулы I, где фенильная группа замещена хлором.
5. Фармацевтическая комбинация по п.2, где ингибитор циклин-зависимой киназы (CDK) является соединением формулы I, где фенильная группа замещена 1, 2 или 3 трифторметильными группами.
6. Фармацевтическая комбинация по п.1, где ингибитор циклин-зависимой киназы (CDK) представлен соединением формулы I, которое представляет собой (+)-trаns-2-(2-хлор-фенил)-5,7-дигидрокси-8-(2-гидроксиметил-1-метил-пирролидин-3-ил)-хромен-4-он или его фармацевтически приемлемую соль.
7. Фармацевтическая комбинация по п.1, где указанное цитотоксическое противоопухолевое средство является паклитакселом.
8. Фармацевтическая комбинация по п.1, где указанное цитотоксическое противоопухолевое средство является доцетакселом.
9. Фармацевтическая комбинация по п.1, где указанное цитотоксическое противоопухолевое средство является доксорубицином.
10. Фармацевтическая комбинация по п.1, где указанное цитотоксическое противоопухолевое средство является гемцитабином.
11. Фармацевтическая комбинация по п.1, где рак является немелкоклеточным раком легкого или раком поджелудочной железы.
12. Применение фармацевтической комбинации, включающей цитотоксическое противоопухолевое средство, выбранное из группы, включающей паклитаксел, доцетаксел, доксорубицин и гемцитабин или их фармацевтически приемлемую соль; и ингибитор циклин-зависимой киназы (CDK), или энантиомер, или его фармацевтически приемлемую соль, или сольват, где указанный ингибитор циклин-зависимой киназы (CDK) представлен следующей формулой I:
где Аr представляет собой фенил, который незамещен или замещен 1, 2 или 3 идентичными или различными заместителями, выбранными из: галогена, выбранного из хлора, брома, фтора или йода, нитро, циано, С1-С4-алкила, трифторметила, гидроксила, С1-С4-алкокси, карбокси, С1-С4-алкоксикарбонила, CONH2 и NR1R2; где R1 и R2 каждый независимо выбран из водорода или С1-С4-алкила; для лечения рака, выбранного из группы, включающей рак молочной железы, мелкоклеточный рак легкого, немелкоклеточный рак легкого, овариальный рак, рак поджелудочной железы, рак желудка, колоректальный рак и гепатоцеллюлярный рак.
13. Применение по п.12, где рак является немелкоклеточным раком легкого или раком поджелудочной железы.
14. Применение по п.12 или 13, где комбинация проявляет терапевтический синергизм.
15. Применение фармацевтической комбинации, включающей цитотоксическое противоопухолевое средство, выбранное из группы, включающей паклитаксел, доцетаксел, доксорубицин и гемцитабин или их фармацевтически приемлемую соль; и ингибитор циклин-зависимой киназы (CDK), или энантиомер, или его фармацевтически приемлемую соль, или сольват, где ингибитор циклин-зависимой киназы (CDK) представлен следующей формулой I:
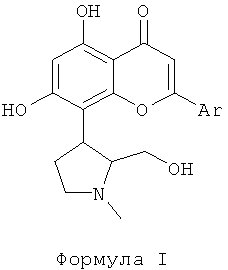
где Аr представляет собой фенил, который незамещен или замещен 1, 2 или 3 идентичными или различными заместителями, выбранными из: галогена, выбранного из хлора, брома, фтора или йода, нитро, циано, С1-С4-алкила, трифторметила, гидроксила, С1-С4-алкокси, карбокси, С1-С4-алкоксикарбонила, CONH2 и NR1R2; где R1 и R2 каждый независимо выбран из водорода или C1-C4-алкила; для лечения рака, выбранного из группы, включающей рак молочной железы, мелкоклеточный рак легкого, немелкоклеточный рак легкого, овариальный рак, рак поджелудочной железы, рак желудка, колоректальный рак и гепатоцеллюлярный рак, где указанное цитотоксическое противоопухолевое средство и указанный ингибитор циклин-зависимой киназы (CDK) вводятся одновременно или последовательно.
16. Применение по п.15, где цитотоксическое противоопухолевое средство или его фармацевтически приемлемая соль и ингибитор циклин-зависимой киназы (CDK), представленный соединениями формулы I, или энантиомером, или его фармацевтически приемлемой солью, или сольватом, вводятся одновременно.
17. Применение по п.15, где цитотоксическое противоопухолевое средство или его фармацевтически приемлемая соль и ингибитор циклин-зависимой киназы (CDK), представленный соединениями формулы I, или энантиомером, или его фармацевтически приемлемой солью, или сольватом, вводятся последовательно.
18. Применение по п.17, где цитотоксическое противоопухолевое средство или его фармацевтически приемлемая соль вводится до ингибитора циклин-зависимой киназы (CDK), представленного соединениями формулы I, или энантиомером, или его фармацевтически приемлемой солью, или сольватом.
19. Применение по любому одному из пп.15-18, где комбинация проявляет терапевтический синергизм.
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| ИНГИБИТОРЫ ЦИКЛИН-ЗАВИСИМЫХ КИНАЗ И ИХ ПРИМЕНЕНИЕ | 2003 |
|
RU2334746C2 |
| Д.Г.ГРЭХАМ-СМИТ, Дж.К.АРОНСОН | |||
| Оксфордский справочник по клинической фармакологии и фармакотерапии | |||
| - М.: Медицина, 2000, с.533-537 | |||
| Pennati M et al | |||
| Способ получения цветных позитивных отпечатков | 1926 |
|
SU6140A1 |

