Изобретение относится к лекарственному средству и способу его получения из органов животных, в частности к выделению биологически активного вещества из хрящей (грудинно-реберного сочленения) и получению лекарственной формы для парентерального введения, которая может использоваться в медицине как средство, нормализующее функции хрящевой ткани.
Известны способы получения биологически активных веществ и лекарственных средств из животного сырья (а.с. SU №1218521, 1994; а.с. SU №1227198, 1986; патент РФ 1298879, 1993; патент РФ №1448443, 1994; патент РФ №1522486, 1993; патент РФ №2075944, 1997; патент РФ №2161501, 2001; патент US №4341765; патент GB 1161896, 1969; патент FR 2583982, 1987).
Известно использование пептидов в фармацевтических композициях, включающих эти пептиды для нормализации функции хрящевой ткани (патент US №20020177554, 2002, А61К 038/17), являющийся наиболее близким аналогом для предлагаемого средства, нормализующего функции хрящевой ткани.
Известны способы получения комплексных пептидных препаратов, обладающих тканеспецифическим действием (патент РФ №944191, 1994; патент РФ №1122606, 1993; патент РФ №1417244, 1993; патент РФ №2104702, 1998).
Известен способ получения указанных препаратов (Морозов В.Г., Хавинсон В.Х. Пептидные биорегуляторы. СПб.:, Наука, 1996. 74 с.), включающий получение биологически активного вещества из органов и тканей животных путем экстракции 3%-ным раствором уксусной кислоты с добавлением хлористого цинка и обработкой надосадочной жидкости ацетоном, являющийся наиболее близким аналогом-прототипом для предлагаемого способа получения средства, нормализующего функции хрящевой ткани. К недостаткам указанного способа следует отнести извлечение в процессе экстракции из ткани большого количества (более 70%) веществ непептидной природы - балластных компонентов, которые, являясь примесями, не определяют биологическую активность выделенного активного вещества, а также его низкий выход.
Настоящим изобретением поставлена и решена задача разработки способа получения средства, нормализующего функции хрящевой ткани, в виде лекарственной формы для парентерального введения, выделенного из хрящей (грудинно-реберного сочленения) телят не старше 12-месячного возраста или свиней, техническим результатом которого является оптимальная технология выделения пептидного комплекса с содержанием низкомолекулярной фракции от 70 до 90% с молекулярной массой входящих в него пептидных компонентов в пределах от 75 до 846 Да и получение водного раствора экстракта с концентрацией полипептидов 2,5÷2,9 мг/мл, позволяющая не только очистить получаемый продукт от примесей, но и увеличить его выход.
Экспериментальная проверка показала, что предлагаемый способ обеспечивает фармацевтическую стабильность, максимальную биологическую ценность и терапевтическую эффективность средства, нормализующего функции хрящевой ткани, выполненного в виде лекарственной формы для парентерального введения, что подтверждено результатами экспериментов, приводимыми в примерах, иллюстрирующих изобретение.
Другим аспектом изобретения является средство, нормализующее функции хрящевой ткани, в виде лекарственной формы для парентерального введения, представляющее собой пептидный комплекс с содержанием низкомолекулярной фракции от 70 до 90%, с молекулярной массой входящих в него пептидных компонентов в пределах от 75 до 846 Да, с концентрацией полипептидов 2,5÷2,9 мг/мл, технический результат которого заключается в том, что выделенное вещество отличается от известных веществ, полученных ранее из животного сырья, по молекулярной массе (от 500 до 1500 Да) входящих в него пептидных компонентов (патенты РФ №№1298879, 2104702, 2163129), а также по его нетоксичности и апирогенности за счет полной очистки от примесей.
В ходе исследований в экспериментах на животных выявлено, что полученный водный раствор средства в концентрации полипептидов 2,5÷2,9 мг/мл является терапевтически эффективной дозой, что позволяет считать показанным использование средства при различных видах патологии опорно-двигательного аппарата. Это подтверждается приводимыми ниже примерами.
В процессе проведения исследований была установлена важность следующих стадий способа получения средства, нормализующего функции хрящевой ткани, выполненного в виде лекарственной формы для парентерального введения (далее - препарат):
- повторное осаждение образовавшегося гомогенизированного осадка двукратными объемами ацетона не менее двух раз и последующее промывание на нутч-фильтре двукратными объемами ацетона до получения осадка светло-серого цвета, что свидетельствует о полной очистке осадка от примесей, таких как липидные, белковые, фосфолипидные и другие;
- получение водного раствора экстракта в концентрации полипептидов 2,5÷2,9 мг/мл; его центрифугирование, фильтрование с последующей ультрафильтрационной очисткой на установке при противодавлении не более 1,0 кгс/см2 через материалы с задерживающей способностью 15000 Да и добавление в ультрафильтрат гликокола до конечной концентрации 10÷20 мг/мл при рН 5,6÷6,6;
- стерилизующая фильтрация, ампулирование и автоклавирование в течение 8 минут при температуре 120°С и атмосферном давлении 1,1 кгс/см2, что обеспечивает стабильность препарата при сохранении терапевтической эффективности.
Режим и время автоклавирования были установлены экспериментально.
Указанный технический результат достигается тем, что средство, нормализующее функции хрящевой ткани, выполнено в виде лекарственной формы для парентерального введения и представляет собой пептидный комплекс с содержанием низкомолекулярной фракции от 70 до 90%, с молекулярной массой входящих в него пептидных компонентов в пределах 75 до 846 Да, с концентрацией полипептидов 2,5÷2,9 мг/мл и получено из хрящей (грудинно-реберного сочленения) телят не старше 12-месячного возраста или свиней путем экстракции уксусной кислотой в присутствии хлористого цинка.
Указанный технический результат достигается также тем, что предлагаемый способ получения средства, нормализующего функции хрящевой ткани, характеризуется тем, что хрящи (грудинно-реберное сочленение) телят не старше 12-месячного возраста или свиней замораживают при температуре не менее минус 40°С, выдерживают при температуре минус 20÷22°С в течение не менее двух месяцев, затем измельчают, добавляют 3% раствор уксусной кислоты в объемном соотношении 1:5 при температуре 20±5°С, экстракцию проводят при постоянном перемешивании, после получения однородной взвеси в нее добавляют 1% раствор хлористого цинка в объемном соотношении 50:1, охлаждают при постоянном перемешивании до температуры 7÷16°С, затем перемешивают по 1 ч через каждые 4 ч отстаивания в течение 48 ч, экстракт отделяют от балластных веществ сепарированием, к экстракту добавляют ацетон в объемном соотношении 1:5, выдерживают при температуре 3÷5°С в течение 4 ч, образовавшийся гомогенизированный осадок повторно осаждают ацетоном не менее 2-х раз, затем осадок, содержащий активное вещество, промывают на нутч-фильтре двукратными объемами охлажденного до температуры 7÷16°С ацетона до получения осадка светло-серого цвета, протирают через металлическое сито, высушивают, растворяют в дистиллированной воде при комнатной температуре и постоянном перемешивании до концентрации полипептидов 2,5÷2,9 мг/мл, раствор центрифугируют, фильтруют, подвергают ультрафильтрационной очистке на установке при противодавлении не более 1,0 кгс/см2 через материалы с задерживающей способностью 15000 Да, в ультрафильтрат добавляют гликокол до его конечной концентрации 10÷20 мг/мл при рН 5,6÷6,6, раствор подвергают стерилизующей фильтрации под давлением не более 2,0 кгс/см2, разливают в ампулы по 2 мл и автоклавируют в течение 8 минут при температуре 120°С и атмосферном давлении 1,1 кгс/см2.
Сущность изобретения поясняется чертежом и таблицей.
На чертеже изображено влияние препарата на развитие эксплантатов хрящевой ткани.
В таблице показано влияние препарата на морфологические и биохимические показатели периферической крови морских свинок при изучении токсичности.
Изобретение иллюстрируется следующими примерами: пример 1 - способ получения препарата; пример 2, подтверждающий биологическую активность препарата; пример 3 - изучение токсичности препарата; пример 4, подтверждающий терапевтическую эффективность препарата, выполненного в виде лекарственной формы для парентерального введения.
Пример 1. Способ получения препарата
В качестве сырья используются хрящи (грудинно-реберное сочленение) телят (не старше 12-месячного возраста) или свиней, которые замораживают при температуре не менее минус 40°С и выдерживают при температуре минус 20÷22°С в течение не менее двух месяцев.
В реактор для экстракции перекачивают 300 л 3% раствора уксусной кислоты и охлаждают до температуры (20±5)°С, затем в реактор при постоянном перемешивании загружают 60 кг измельченного до получения гомогенной массы сырья. Экстракцию проводят при постоянном перемешивании, после получения однородной взвеси добавляют в нее 1% раствор хлористого цинка в объемном соотношении 50:1, охлаждают при постоянном перемешивании до температуры плюс 7÷16°С, затем перемешивают по 1 ч через каждые 4 ч отстаивания в течение 48 ч.
Экстракт отделяют от балластных веществ сепарированием при (5000±500) об/мин в течение 1 ч. К экстракту добавляют ацетон в объемном соотношении 1:5. Выдерживают при температуре плюс 3÷5°С в течение 4 ч. Образовавшийся осадок гомогенизируют и повторно осаждают ацетоном не менее двух раз. Затем осадок, содержащий активное вещество, промывают на нутч-фильтре двукратными объемами охлажденного до температуры 7÷16°С ацетона до получения осадка светло-серого цвета. Промытый осадок протирают через металлическое сито, выкладывают тонким слоем в эмалированные кюветы, закрывают двойным слоем ткани хлопчатобумажной и высушивают при периодическом помешивании в вытяжном шкафу до полного удаления запаха ацетона.
Выход активного вещества (порошка биологически активного пептидного комплекса, выделенного из хрящей) составляет 30 г на 1 кг исходного сырья.
Полученный порошок растворяют в дистиллированной воде при постоянном перемешивании при комнатной температуре в течение 40 мин до концентрации пептидов 2,5÷2,9 мг/мл. Полученный раствор центрифугируют при (3000±200) об/мин в течение (20±5) мин. Центрифугат фильтруют через фильтр типа АР-15 или аналогичный.
Фильтрат подвергают ультрафильтрационной очистке на установке для ультрафильтрации при противодавлении не более 1,0 кгс/см2 через материалы с задерживающей способностью 15000 Да.
В ультрафильтрат добавляют расчетную навеску гликокола, до его конечной концентрации 10÷20 мг/мл, перемешивают до полного растворения гликокола при сохранении рН 5,6÷6,6.
Раствор подвергают стерилизующей фильтрации, которую проводят под давлением, не превышающим 2,0, кгс/см2.
Полученный раствор разливают в ампулы по 2,0 мл и автоклавируют, причем ампулы с препаратом подвергают стерилизации в течение 8 минут при температуре 120°С и атмосферном давлении не более 1,1 кгс/см2.
Препарат представляет собой бесцветный, прозрачный раствор и содержит пептидный комплекс с концентрацией полипептидов 2,5÷2,9 мг/мл.
Тестирование на отсутствие высокомолекулярных белковых компонентов осуществляют путем добавления к содержимому 1 ампулы препарата 1 мл 10%-ного раствора трихлоруксусной кислоты. Прозрачность раствора свидетельствует об отсутствии высокомолекулярных белковых компонентов.
Для определения в препарате пептидных связей к его раствору добавляют биуретовый реактив. Окрашивание раствора в фиолетовый цвет свидетельствует об имеющихся в препарате пептидных связях.
С целью определения в препарате полипептидов и их фракций используют методы ультрафиолетовой спектрофотометрии, гель-хроматографии, масс-спектрометрии, высокоэффективной жидкостной хроматографии и электрофореза в полиакриламидном геле.
Ультрафиолетовый спектр раствора препарата снимают в области длин волн от 250 до 350 нм: максимум поглощения отмечается при длине волны 270±5 нм.
Молекулярную массу полипептидов, входящих в препарат, определяют следующими методами:
методом гель-хроматографии на сефадексах G-25 и G-50 ("Pharmacia", Швеция). Для калибровки колонки 1,6×60 см используют Peptide Molecuar Weight Kit MS III ("Serva", Германия);
методом масс-спектрометрии. Спектры получают на времяпролетном масс-спектрометре Voyager DE Biospectrometry с лазерной десорбцией и ионизацией с помощью матрицы (MALDI-TOF) при относительной интенсивности азотного лазера 2300÷2400, ускоряющем напряжении 25000 В, времени задержки 90 нс и давлении в вакуумной камере 2,6×106 тор;
методом электрофореза в 15%-ном полиакриламидном геле в сравнении со стандартным набором маркерных белков.
Перечисленными выше методами установлено, что в состав препарата входят полипептиды с молекулярной массой 75 до 846 Да.
С помощью обращенно-фазовой высокоэффективной жидкостной хроматографии в градиенте ацетонитрила (сорбент "Lichrosorb C18", колонка 2×62 мм) установлено, что в состав препарата входят преимущественно низкомолекулярные фракции - от 70 до 90%, а высокомолекулярные компоненты в препарате отсутствуют.
Пирогенность препарата определяют на кроликах общепринятым методом (ГФ XI, вып.2, с.183) при тест-дозе препарата 0,25 мг на 1 кг массы животного в 1,0 мл изотонического 0,9% раствора натрия хлорида для инъекций. Показано, что препарат является апирогенным.
Таким образом, вышеизложенным способом получен препарат - лекарственная форма для парентерального введения, представляющая собой пептидный комплекс с содержанием низкомолекулярной фракции от 70 до 90%, с молекулярной массой входящих в него пептидных компонентов в пределах от 75 до 846 Да, с концентрацией полипептидов 2,5÷2,9 мг/мл.
Пример 2. Влияние препарата на развитие эксплантатов хрящевой ткани
Эксперименты проведены на 47 фрагментах хрящевой ткани проксимальной головки бедренной кости крыс линии "Wistar" с массой тела 150÷200 г. Питательная среда для культивирования эксплантатов состояла из 35% раствора Игла, 25% фетальной сыворотки теленка, 35% раствора Хенкса, 5% куриного эмбрионального экстракта, в среду добавляли глюкозу (0,6%), инсулин (0,5 ед./мл), пенициллин (100 ед./мл), глютамин (2 мМ). Фрагменты хрящевой ткани помещали в эту среду и культивировали на коллагеновой подложке в чашках Петри в термостате при температуре 36,7°С в течение 2 суток. В экспериментальную среду добавляли препарат в концентрациях 1, 10, 50, 100, 200 и 400 нг/мл. Критерием биологической активности служил индекс площади (ИП) - соотношение площади всего эксплантата вместе с зоной роста к исходящей площади фрагмента хрящевой ткани. Значения ИП выражали в процентах, контрольное значение ИП принималось за 100%.
На чертеже показано влияние препарата на развитие эксплантатов хрящевой ткани.
Установлено, что через 1 сутки культивирования происходило распластывание эксплантатов на коллагеновой подложке и начиналось выселение пролиферирующих и мигрирующих клеток по периферии эксплантата. На 3-и сутки культивирования при концентрации препарата 50 нг/мл наблюдалось достоверное повышение ИП эксплантатов на 31% по сравнению с контрольными значениями ИП. При исследовании эксплантатов хрящевой ткани на более длительных сроках культивирования (7 дней) было выявлено аналогичное стимулирующее действие препарата в той же концентрации.
Таким образом, в отношении хрящевой ткани препарат оказывал тканеспецифическое действие, проявляющееся в стимуляции роста эксплантатов.
Пример 3. Изучение токсичности препарата
Общетоксическое действие препарата исследовали в соответствии с требованиями "Руководства по экспериментальному (доклиническому) изучению новых фармакологических веществ" (2000): острой токсичности при однократном введении препарата, а также подострой и хронической токсичности при длительном введении препарата.
Исследование по изучению острой токсичности проведено на 72 белых беспородных мышах-самцах с массой тела 20÷23 г. Животные были рандомизированно разделены на 6 равных групп. Препарат вводили животным однократно внутримышечно в дозах 35 мг/кг, 50 мг/кг, 100 мг/кг, 150 мг/кг, 200 мг/кг в 0,25 мл стерильного 0,9% раствора NaCl. Животным контрольной группы в том же объеме вводили 0,9% раствор NaCl.
Исследования по изучению подострой токсичности проведено на 60 белых беспородных крысах-самцах с массой тела 150÷180 г. Ежедневно однократно животным подопытных групп вводили препарат внутримышечно в течение 90 дней в дозах 1 мг/кг, 10 мг/кг, 100 мг/кг в 0,5 мл стерильного 0,9% раствора NaCl. Животным контрольной группы вводили в том же объеме стерильный 0,9% раствор NaCl. До введения препарата, на 30, 60 и 90 сутки после начала введения препарата у животных исследовали морфологический состав и свойства периферической крови. При завершении эксперимента исследовали биохимические и коагулологические показатели крови.
Исследования по изучению хронической токсичности проводили в течение 6 месяцев, исходя из длительности рекомендуемого клинического назначения препарата на 84 морских свинках-самцах массой 250÷280 г. Животные подопытных групп получали ежедневно однократно внутримышечно препарат в течение 6 мес в дозах 1 мг/кг, 10 мг/кг, 100 мг/кг в 0,5 мл стерильного 0,9% раствора NaCl. В контрольной группе животным вводили по аналогичной схеме стерильный 0,9% раствор NaCl в том же объеме. У животных в периферической крови общепринятыми методами определяли: количество эритроцитов, гемоглобина, ретикулоцитов, тромбоцитов, лейкоцитов, лейкоцитарную формулу, скорость оседания эритроцитов (СОЭ), резистентность эритроцитов. Наряду с этим определяли содержание в сыворотке крови общего белка по методу Лоури, калия и натрия методом плазменной спектрофотометрии. После завершения эксперимента проводили патоморфологическое исследование головного и спинного мозга, спинномозговых ганглиев, щитовидной железы, паращитовидных желез, надпочечников, семенников, гипофиза, сердца, легких, аорты, печени, почки, мочевого пузыря, поджелудочной железы, желудка, тонкой кишки, толстой кишки, тимуса, селезенки, лимфатических узлов, костного мозга.
При изучении острой токсичности установлено, что однократное введение исследуемого препарата животным в дозе, превышающей терапевтическую, рекомендованную для клинического применения более чем в 5000 раз, не вызывает токсических реакций, что свидетельствует о большой терапевтической широте препарата.
Изучение подострой и хронической токсичности препарата свидетельствует об отсутствии побочных эффектов при длительном применении препарата в дозах, превышающих терапевтическую в 300÷3000 раз. При исследовании влияния препарата на морфологический состав и биохимические показатели периферической крови морских свинок через 3 и 6 месяцев после начала введения препарата достоверного изменения показателей не выявлено (таблица).
При оценке общего состояния животных, морфологических и биохимических показателей периферической крови, морфологического состояния внутренних органов, состояния сердечно-сосудистой и дыхательной систем, функции печени и почек патологические изменения в организме не обнаружены.
Следовательно, препарат, полученный предлагаемым способом, при длительном введении животным не обладает токсическими свойствами, препятствующими дальнейшему его применению в качестве лекарственного средства, выполненного в виде лекарственной формы для парентерального введения.
Пример 4. Эффективность применения препарата у больных остеохондрозом поясничного отдела позвоночника
Исследование проведено с участием 33 больных остеохондрозом поясничного отдела позвоночника в возрасте 42÷59 лет. Больные часто отмечали появление болей в нижней части спины с иррадиацией по ходу седалищного нерва, значительно усиливающихся при изменении положения тела, ходьбе, физической нагрузке. Все больные ранее длительное время получали анальгетики и противовоспалительные средства, применение которых вызывало кратковременный терапевтический эффект, требующий увеличения дозы препаратов на курс лечения и продолжительного их приема.
15 пациентам основной группы вводили препарат в дозе 5 мг в 2 мл физиологического раствора внутримышечно однократно ежедневно в течение 20 дней. Контрольная группа включала 18 пациентов, получавших инъекции физиологического раствора по аналогичной схеме.
Эффективность применения препарата у больных оценивали по динамике клинических показателей и данных рентгенологического исследования.
Необходимо отметить, что рентгенологические симптомы дегенеративно-дистрофических заболеваний позвоночника являются не только объективными диагностическими критериями стадии развития патологического процесса, но и имеют большую прогностическую значимость при проводимой лекарственной терапии.
Установлено, что применение препарата у больных остеохондрозом поясничного отдела позвоночника на фоне комплексной терапии способствовало снижению болевого синдрома в 67,4% случаев. Подобная динамика была наиболее характерна для лиц среднего возраста. Прогрессирующее с возрастом течение заболевания, сопровождающееся характерными рентгенологическими симптомами (сужение просвета между смежными телами позвонков за счет уплощения дегенеративно измененных межпозвонковых дисков, образование передних и задних остеофитов тел позвонков, наличие артрозных изменений в задних и латеральных межпозвонковых суставах в виде сужения щелей, неровности контуров, развития остеофитов по краям суставных концов, изменения конфигурации межпозвонковых отверстий), способствует развитию спондилеза и спондилоартроза и формированию нейродистрофических и нейрососудистых синдромов. В этих случаях применение препарата у больных сглаживало болевую симптоматику при нагрузке на позвоночник и нижние конечности. Улучшение клинической картины течения заболевания коррелировало с улучшением объективных критериев по ренгтгенограммам.
Таким образом, результаты проведенного исследования подтверждают нормализующее действие препарата в терапевтически эффективной дозе 5 мг (2,5 мг/мл) на функцию хрящевой ткани и свидетельствуют о лечебной эффективности препарата и целесообразности его применения в комплексном лечении дегенеративно-дистрофических заболеваний позвоночника в дозе 5 мг внутримышечно однократно ежедневно в течение 20 дней.
| название | год | авторы | номер документа |
|---|---|---|---|
| СРЕДСТВО, НОРМАЛИЗУЮЩЕЕ ФУНКЦИИ ПОЧЕК, И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2006 |
|
RU2302868C1 |
| СРЕДСТВО, НОРМАЛИЗУЮЩЕЕ ФУНКЦИИ ЩИТОВИДНОЙ ЖЕЛЕЗЫ, И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2006 |
|
RU2302875C1 |
| СРЕДСТВО, НОРМАЛИЗУЮЩЕЕ ТОНУС МОЧЕВОГО ПУЗЫРЯ, И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2006 |
|
RU2302867C1 |
| СРЕДСТВО, НОРМАЛИЗУЮЩЕЕ РЕПРОДУКТИВНУЮ ФУНКЦИЮ У МУЖЧИН, И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2006 |
|
RU2302874C1 |
| СРЕДСТВО, НОРМАЛИЗУЮЩЕЕ ФУНКЦИИ ГОЛОВНОГО МОЗГА, И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2006 |
|
RU2302871C1 |
| СРЕДСТВО, НОРМАЛИЗУЮЩЕЕ РЕПРОДУКТИВНУЮ ФУНКЦИЮ У ЖЕНЩИН, И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2006 |
|
RU2303454C1 |
| СРЕДСТВО, НОРМАЛИЗУЮЩЕЕ ФУНКЦИИ КРОВЕНОСНЫХ СОСУДОВ, И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2006 |
|
RU2301072C1 |
| ГЕПАТОПРОТЕКТОРНОЕ СРЕДСТВО И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2006 |
|
RU2301071C1 |
| СРЕДСТВО, ОБЛАДАЮЩЕЕ ГЕРОПРОТЕКТОРНОЙ АКТИВНОСТЬЮ, И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2006 |
|
RU2302870C1 |
| ПЕПТИД, НОРМАЛИЗУЮЩИЙ МЕТАБОЛИЗМ В КОСТНОЙ И ХРЯЩЕВОЙ ТКАНЯХ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ И СПОСОБ ЕЕ ПРИМЕНЕНИЯ | 2006 |
|
RU2299741C1 |
Изобретение относится к выделению биологически активного вещества из хрящей животных и получению лекарственной формы для парентерального введения, которая может использоваться в медицине как средство, нормализующее функции хрящевой ткани. Средство выполнено в виде лекарственной формы для парентерального введения и представляет собой пептидный комплекс с содержанием низкомолекулярной фракции от 70 до 90%, с молекулярной массой входящих в него пептидных компонентов в пределах 75 до 846 Да, с концентрацией полипептидов 2,5÷2,9 мг/мл и получено из хрящей телят не старше 12-месячного возраста или свиней путем экстракции уксусной кислотой в присутствии хлористого цинка. Предлагаемый способ получения средства заключается в том, что хрящи телят не старше 12-месячного возраста или свиней замораживают при температуре не менее минус 40°С, выдерживают при температуре минус 20÷22°С в течение не менее двух месяцев, затем измельчают, добавляют 3% раствор уксусной кислоты в объемном соотношении 1:5 при температуре 20÷5°С, экстракцию проводят при постоянном перемешивании, после получения однородной взвеси в нее добавляют 1% раствор хлористого цинка в объемном соотношении 50:1, охлаждают при постоянном перемешивании до температуры 7÷16°С, затем перемешивают по 1 ч через каждые 4 ч отстаивания в течение 48 ч, экстракт отделяют от балластных веществ сепарированием, к экстракту добавляют ацетон в объемном соотношении 1:5, выдерживают при температуре 3÷5°С в течение 4 ч, образовавшийся гомогенизированный осадок повторно осаждают ацетоном не менее 2-х раз, затем осадок, содержащий активное вещество, промывают на нутч-фильтре двукратными объемами охлажденного до температуры 7÷16°С ацетона до получения осадка светло-серого цвета, протирают через металлическое сито, высушивают, растворяют в дистиллированной воде при комнатной температуре и постоянном перемешивании до концентрации полипептидов 2,5÷2,9 мг/мл, раствор центрифугируют, фильтруют, подвергают ультрафильтрационной очистке на установке при противодавлении не более 1,0 кгс/см2 через материалы с задерживающей способностью 15000 Да, в ультрафильтрат добавляют гликокол до его конечной концентрации 10÷20 мг/мл при рН 5,6÷6,6, раствор подвергают стерилизующей фильтрации под давлением не более 2,0 кгс/см2, разливают в ампулы по 2 мл и автоклавируют в течение 8 минут при температуре 120°С и атмосферном давлении 1,1 кгс/см2. Изобретение обеспечивает оптимальную технологию выделения пептидного комплекса с содержанием низкомолекулярной фракции от 70 до 90%, с молекулярной массой входящих в него пептидных компонентов в пределах от 75 до 846 Да и получение водного раствора экстракта с концентрацией полипептидов 2,5÷2,9 мг/мл, позволяющую не только очистить получаемый продукт от примесей, но и увеличить его выход; а также то, что выделенное вещество отличается от известных веществ, полученных ранее из животного сырья, по молекулярной массе входящих в него пептидных компонентов, а также по его нетоксичности и апирогенности за счет полной очистки от примесей. 2 н.п. ф-лы, 1 табл., 1 ил.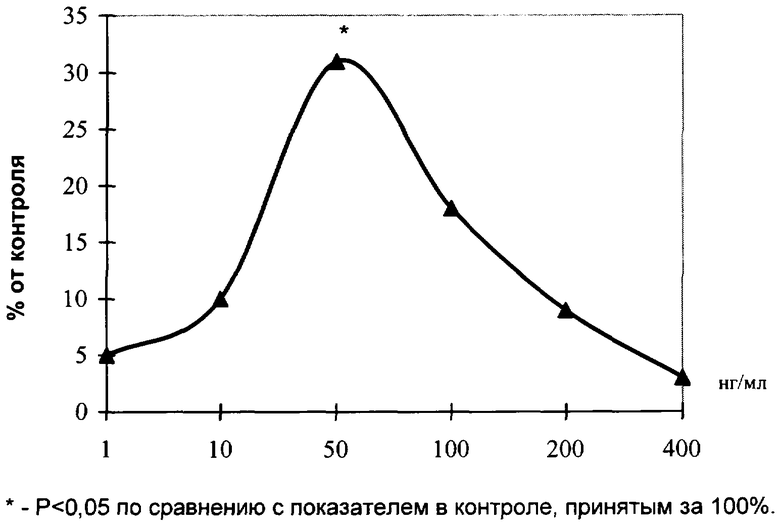
| МОРОЗОВ В.Г | |||
| и др | |||
| Пептидные биорегуляторы, СПб, Наука, 1996, с.74 | |||
| US 2002177554 A1, 28.11.2002 | |||
| НОВЫЕ ПЕПТИДЫ, ЯВЛЯЮЩИЕСЯ ПРОИЗВОДНЫМИ АУТОАНТИГЕНА, ПРЕДНАЗНАЧЕННЫЕ ДЛЯ ИСПОЛЬЗОВАНИЯ В ИММУНОТЕРАПИИ АУТОИММУННЫХ ЗАБОЛЕВАНИЙ | 1995 |
|
RU2178797C2 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| JP 2003137807 A1, 14.05.2003. | |||

