Настоящее изобретение относится к производным соединений, таких как полисахариды, имеющие по крайней мере терминальные сиаловые звенья и предпочтительно состоящие из звеньев сиаловой кислоты, содержащих альдегидную группу для реакции с субстратами на восстанавливающем терминале и способам их получения. Производные пригодны для превращения в другие реакционно-активные производные и для конъюгации с такими субстратами, содержащими аминогруппу, как пептиды, белки, лекарственные препараты, системы для доставки лекарственных препаратов (например, липосомы), вирусы, клетки, например животные клетки, микроорганизмы, синтетические полимеры и т.д.
Полисиаловые кислоты (PSA) представляют собой природные неразветвленные полимеры сиаловой кислоты, вырабатываемые определенными штаммами бактерий и в определенных клетках у млекопитающих (Roth et al., 1993). Их можно получать с различной степенью полимеризации, от n = примерно 80 или более остатков сиаловой кислоты до n=2, неполным кислотным гидролизом или расщеплением нейраминидазами, или фракционированием природных вырабатываемых бактериями видов полимера. Состав различных полисиаловых кислот также изменяется таким образом, что существуют гомополимерные формы, т.е. альфа-2,8-связанная полисиаловая кислота, включающая капсульный полисахарид штамма К1 E. coli и В-группы менингококков, который также обнаруживают в эмбриональной форме молекулы клеточной адгезии нейрона (N-CAM). Также существуют гетерополимерные формы, такие как чередующаяся альфа-2,8 альфа-2,9 полисиаловая кислота штамма К92 E. coli и полисахариды С группы N. meningitidis. Сиаловую кислоту также можно обнаружить в чередующихся сополимерах с мономерами, отличающимися от сиаловой кислоты, таких как группа W135 или группа Y N. meningitidis. Полисиаловые кислоты обладают важным биологическим действием, включая уклонения патогенных бактерий от иммунной системы и системы комплемента и регуляцию глиоцетарной адгезивности незрелых нейронов в ходе эмбрионального развития (где полимер обладает антиадгезивным действием) [Muhlenhoff et. al., 1998; Rutishauser, 1989; Troy, 1990, 1992; Cho and Troy, 1994], хотя рецепторы полисиаловых кислот у млекопитающих не известны. Альфа-2,8-связанная полисиаловая кислота штамма К1 E. coli также известна как «коломиновая кислота» и ее (различной длины) используют в качестве примера настоящего изобретения.
Среди бактериальных полисахаридов альфа-2,8 связанная форма полисиаловой кислоты представляет собой единственную неиммуногенную (не вызывающую ни ответа Т-клеток, ни образования антител у млекопитающих, даже при конъюгации с иммуногенными носителями белками), что может отражаться на ее статусе как полимера млекопитающих (так же, как и бактериального). Более короткие формы полимера (до n=4) обнаруживают в ганглиозидах клеточной поверхности, которые широко распространены в организме и считаются эффективными для сообщения и поддержания иммунологической толерантности по отношению к полисиаловой кислоте. В последние годы биологические свойства полисиаловых кислот, в особенности биологические свойства альфа-2,8 связанной гомополимерной полисиаловой кислоты, были использованы для модификации фармакокинетических свойств белковых или низкомолекулярных молекул лекарственных веществ [Gregoriadis, 2001; Jain et al., 2003; US-A-5846951, WO-A-0187922]. Получение производных полисиаловой кислоты приводит к сильному улучшению с точки зрения периода полувыведения некоторого ряда терапевтических белков, включая каталазу и аспарагиназу [Fernandes and Gregoriadis, 1996 and 1997], а также делает возможным использование таких протеинов несмотря на ранее существовавшие антитела, возникшие как нежелательное (и в некоторых случаях неизбежное) следствие предшествующего воздействия терапевтического белка [Fernandes and Gregoriadis, 2001]. Во многих отношениях модифицированные свойства полисиалилированных белков сравнимы с белками, производными полиэтилен гликоля (PEG). Например, и в том и в другом случае увеличивается период полувыведения и белки и пептиды являются более стабильными по отношению к протеолитическому расщеплению, но сохранение биологической активности кажется большим у PSA по сравнению с PEG [Hreczuk-Hirst et al., 2002]. Также возникают вопросы об использовании PEG с терапевтическими агентами, которые следует вводить постоянно, поскольку PEG подвергается только очень медленному биологическому разрушению [Beranova et al., 2000] и формы с высокой молекулярной массой имеют тенденцию накапливаться в тканях [Bendele et al., 1998; Convers et al., 1997]. Было обнаружено, что полиэтиленгликолированные белки приводят к образованию анти-PEG антител, которые могут также влиять на время задержки конъюгата в кровотоке [Cheng et al., 1990]. Несмотря на то, что PEG исторически используют в качестве парентерально вводимого полимера, конъюгированного с лекарственными препаратами, требуется лучшее понимание его иммунотоксикологии, фармакологии и метаболизма [Hunter and Moghimi, 2002; Brocchini, 2003]. Также существуют сомнения по поводу использования PEG в терапевтических агентах, которые могут требовать высокой дозировки, поскольку аккумуляция PEG может приводить к токсичности. Следовательно, альфа-2,8-связанная полисиаловая кислота (PSA) предлагает привлекательную альтернативу PEG, представляя собой иммунологически невидимый биологически разрушаемый полимер, являющийся естественной частью человеческого организма, и который разрушается тканевыми нейроаминидазами до сиаловой кислоты, нетоксичного сахарида.
Группа заявителей описала в предшествующих научных публикациях и выданных патентах пользу от применения природных полисиаловых кислот для улучшения фармакокинетических свойств белковых терапевтических агентов [Gregoriadis, 2001; Fernandes and Gregoriadis, 1996, 1997, 2001; Gregoriadis et al., 1993, 1998, 2000; Hreczuk-Hirst et al., 2002; Mital, 2004; Jain et al., 2003, 2004; US-A-05846951; WO-A-0187922]. Теперь заявители описывают новые производные PSA, которые делают возможными новые композиции и способы получения PSA-производных белков (и других форм терапевтического агента). Эти новые вещества и способы в особенности применимы для получения PSA-производных терапевтических агентов, предназначенных для использования у человека и животных, в которых основную важность представляет собой химическое и молекулярное определение лекарственных объектов из-за требований безопасности, медицинской этики и распорядительных органов (Управление по контролю за продуктами и лекарствами США (FDA), EMEA).
Ранее были описаны способы для присоединения полисахаридов к терапевтическим агентам, таким как белки [Jennings and Lugowski, 1981; US-A-5846951; WO-A-0187922]. Некоторые из этих способов зависят от химического производного «не восстанавливающего» конца полимера для получения альдегидного остатка, реакционно-способного по отношению к белку (фиг.1). Это происходит потому что восстанавливающий конец PSA и других полисахаридов имеет низкую реакционную способность по отношению к белкам в мягких условиях, необходимых для сохранения белковой конформации и химической целостности PSA и белка в ходе конъюгации. Невостанавливающее терминальное звено сиаловой кислоты, поскольку оно содержит вицинальные диолы, может быть легко (и селективно) окислено периодатом с получением моноальдегидной формы, которая является в значительной степени более реакционно-активной по отношению к белкам и которая включает пригодный реакционно-способный элемент для присоединения белков посредством восстановительного аминирования и других химических реакций. Заявители ранее описывали это в US-A-5846951 и WO-A-0187922. Реакция проиллюстрирована на фиг.1, где
а) показывает окисление коломиновой кислоты (альфа-2,8 связанной полисиаловой кислоты E. coli) периодатом натрия с получением реакционно-способного в отношении белков альдегида на невосстанавливающем конце и
b) показывает селективное восстановление основания Шиффа цианборгидридом натрия с получением стабильной необратимой ковалентной связи с аминогруппой белка.
Из различных способов, которые были описаны для присоединения полисиаловых кислот к терапевтическим агентам [US-A-5846951; WO-A-0187922], ни один из них не является специально предназначенным для конъюгирования по восстанавливающему концу из-за его низкой реакционной способности по отношению к терапевтическим белкам. Хотя реакция теоретически пригодная, достижение приемлемых выходов конъюгата посредством реакции белков с полукеталем восстанавливающего конца PSA требует времени реакции, которое не представляется благоприятным для стабильности белка. Во-вторых, необходимы концентрации реагента (избытка полимера), которые могут быть недостижимыми или экономически невыгодными. Тем не менее, несмотря на неэффективность этой реакции, заявители заметили, что она приводит к побочным продуктам в ходе реакций конъюгации, предназначенных для получения конъюгатов с белком посредством введенного альдегида на (противоположном) невосстанавливающем конце полимера. Потенциал таких побочных продуктов очевиден в опубликованных исследованиях по каталазе, инсулину и аспарагиназе [Fernandes and Gregoriadis, 1996, 1997, 2001; Jain et al. 2003], в которых полукеталь природной (химически не модифицированной) формы полимера дает начало белковым конъюгатам с низкой степенью эффективности (менее 5% белка становится производными, см. далее ниже в примерах сравнения и таблице 1) в ходе восстановительного аминирования.
Реакционная активность восстанавливающего конца коломиновой кислоты, хотя и низкая по отношению к белковым мишеням, достаточна для того, чтобы причинять беспокойство при производстве химически определенных конъюгатов такого типа для того, чтобы быть предпочтительными с точки зрения распорядительных органов при терапевтическом использовании у человека и животных. В отличие от природного полимера коломиновой кислоты, который в слабой степени является монофункциональным, форма PSA, окисленная периодатом (имеющая альдегид на одном конце и полукеталь на другом), неизбежно приводит к получению комплекса продуктов, которые серьезно затрудняют задачу получения молекулярно определенных и фармацевтически приемлемых конъюгатов (фиг.2). Фиг.2а представляет собой принципиальную схему, показывающую образование побочных продуктов в ходе полисиалилирования (исходный способ). Фиг.2b представляет собой более подробную принципиальную схему, показывающую образование побочных продуктов в ходе полисиалилирования (исходный способ), конкретно
i) асимметрического димера;
ii) линейного полимера;
iii) разветвленного полимера и
iv) различных более сложных структур.
С первого взгляда казалось бы простым очистить ожидаемый реакционный продукт от различных побочных продуктов, описанных на фиг.2, однако не существует способов прямой очистки, поскольку физико-химические свойства ожидаемых форм (размер, заряд и т.д.) являются в значительной степени одинаковыми, в действительности почти идентичными по сравнению с намеченными формами продукта. Это исключило бы необходимость очистки ожидаемых типов из реакционной смеси такими способами как ионообменная хроматография и гельпроникающая хроматография (которые разделяют на основе заряда и размера соответственно) и исключило бы многие другие способы очистки. В настоящий момент заявители решили указанные проблемы разработкой нового способа конъюгации полисахаридов, содержащих группы сиаловой кислоты на восстанавливающем терминальном конце, с белками, в соответствии с чем низкая реакционная способность восстанавливающего конца может быть использована как благоприятное воздействие, которое исключает сложность получения продукта, описанную на Фиг 2(b) при использовании установленного способа (фиг.1) восстановительного аминирования белков с окисленной периодатом природной коломиновой кислотой.
Jennings and Lugovski в патенте США 4356170 описывают получение производных бактериальных полисахаридов с белками за счет активированного восстанавливающего терминального звена, включающее стадию предварительного восстановления и затем стадию окисления. Они предлагают этот подход в том случае, если терминальное восстанавливающее звено представляет собой N-ацетил маннозамин, глюкозу, глюкозамин, рамнозу и рибозу.
В Европейской заявке на патент EP-A-0454898 аминогруппу белка связывают с альдегидной группой, полученной восстановлением и частичным окислением восстанавливающего терминального остатка сахара глюкозаминогликана. Глюкозаминогликаны, обработанные таким образом, включают гиалуроновую кислоту, хондроитинсульфат, гепарин, гепаринсульфат и дерматансульфат. Ни одно из указанных соединений не содержит звена сиаловой кислоты на восстанавливающем терминальном конце.
В изобретении обеспечивается новый способ получения альдегидного производного сиаловой кислоты, в котором исходное вещество, содержащее звено сиаловой кислоты на своем восстанавливающем терминальном конце, подвергают последовательным стадиям:
a) восстановления до восстановленного раскрытого кольца восстановливающего терминального звена сиаловой кислоты с образованием вицинальной диольной группы; и
b) селективного окисления вицинальной диольной группы, полученной в стадии а) с получением альдегидной группы.
Исходное вещество предпочтительно представляет собой ди-, олиго-, или полисахарид, хотя изобретение может иметь применение и для других исходных веществ.
Исходное вещество, используемое в способе по изобретению, должно предпочтительно содержать звено сиаловой кислоты на восстанавливающем терминальном конце, присоединенное к соседнему звену посредством восьмого атома углерода. В стадии b) 6,7-диольную группу окисляют с образованием альдегида при 7 атоме углерода.
В альтернативном варианте осуществления, в котором звено сиаловой кислоты на восстанавливающем терминальном конце связано с соседним звеном посредством 9 углеродного атома, в стадии b) получают 7,8-диольную группу и окисляют с получением альдегида при 8 атоме углерода.
В способе по изобретению, в котором исходное вещество представляет собой ди-, олиго- или полисахарид, предпочтительно, чтобы исходное вещество имело на невосстанавливающем конце терминальную сахаридную группу, содержащую вицинальную диольную группу, и в котором исходное вещество подвергают предварительной стадии перед стадией а), селективного окисления вицинальной диольной группы до альдегида, в соответствии с чем в стадии а) альдегид также восстанавливают до образования гидроксигруппы, которая не является частью вицинальной диольной группы. Изобретение в особенности приемлемо в том случае, если терминальное звено восстанавливающего конца исходного вещества представляет собой звено сиаловой кислоты. В альтернативном варианте осуществления исходное вещество может иметь вицинальную диольную группу, которая остается в таком виде на невосстанавливающем терминальном сахаридном звене исходного вещества для стадии а). Она не будет модифицирована стадией восстановления, но будет окислена на стадии окисления с образованием альдегидной группы. Продукт будет бифункциональным и может проявлять полезные терапевтические активности из-за его способности сшивать субстраты по реакции обеих альдегидных групп с приемлемыми функциональными группами субстрата.
Согласно второму аспекту изобретения обеспечивают новый способ, в котором сиаловую кислоту исходного вещества, имеющего терминальную сиаловую кислоту на невосстанавливающем терминальном конце, подвергают следующим стадиям:
c) стадии селективного окисления невосстанавливающего терминального звена сиаловой кислоты в 7,8-вицинальную диольную группу для образования 7-альдегида; и
d) стадию восстановления 7-альдегидной группы до соответствующего спирта.
Этот аспект изобретения обеспечивает производные сиаловой кислоты, которые имеют пассивированный невосстанавливающий терминальный конец, делающим возможным активацию восстанавливающего терминального конца для последующей реакции. Активация может представлять собой восстановление/окисление, например, первого аспекта изобретения с необязательными последующими стадиями превращения альдегидной группы в другую группу, такими как аминирование с образованием амина. Могут быть разработаны другие стадии для активации восстанавливающего терминального конца.
Предпочтительно этот второй аспект изобретения представляет собой часть способа, в котором исходное вещество имеет восстанавливающее терминальное звено и впоследствии требует быть конъюгированным с другой молекулой посредством указанного звена. В таком способе восстанавливающее терминальное звено обычно активируют, например, реакцией, которая бы иным способом активировала часть невосстанавливающих терминальных звеньев сиаловой кислоты, в том случае, если она не приводит к пассивации. Такая реакция, например, представляет собой селективное окисление вицинального диольного фрагмента и ее проводят после стадии d).
В изобретении предпочтительное полисахаридное исходное вещество может содержать в молекуле звенья, отличные от сиаловой кислоты. Например, звенья сиаловой кислоты могут чередоваться с другими сахаридными звеньями. Предпочтительно однако, чтобы полисахарид содержал в значительной степени только звенья сиаловой кислоты. Предпочтительно они соединены 2→8 и/или 2→9.
Предпочтительно полисахаридное исходное вещество имеет по крайней мере 2, более предпочтительно по крайней мере 5, более предпочтительно по крайней мере 10, например по крайней мере 50 сахаридных звеньев. Например, полисахарид может включать по крайней мере 5 звеньев сиаловой кислоты.
Полисиаловую кислоту можно получать из любого источника, предпочтительно природного источника, такого как бактериальный источник, например E. coli K1 или K92, менингококков группы В, или даже коровьего молока, или N-CAM полимер сиаловой кислоты может быть гетерополимерным, таким как группа 135 или группа V N. meningitides. Полисиаловая кислота может быть в форме соли или свободной кислоты. Она может быть в гидролизованной форме, такой, чтобы молекулярная масса уменьшалась после выделения из бактериального источника. Полисиаловая кислота может быть веществом с широким диапазоном молекулярных масс, таким, чтобы полидисперсность составляла более 1,3, например составляла 2 или более. Предпочтительно полидисперсность молекулярной массы составляет менее 1,2, например составляет до 1,01.
Часть полисиаловых кислот, с широким диапазоном молекулярной массы, может быть фракционирована с низкими полидисперсностями, т.е. на фракции с различными значениями средней молекулярной массой. Фракционирование представляет собой предпочтительно анионообменную хроматографию при элюировании подходящим основным буфером. Заявители обнаружили пригодную анионообменную среду i) препаративную среду, такую как сильное ионообменное вещество на основе активированной агарозы, содержащей в боковых группах четвертичный аммониевый ион (т.е. сильное основание). Элюционный буфер не реакционно-активен и предпочтительно является летучим, так что желаемый продукт можно выделять из основания каждой фракции выпариванием. Пригодными примерами являются амины, такие как триэтаноламин. Выделение может представлять собой, например, лиофильную сушку. Способ фракционирования пригоден для полисиаловой кислоты в качестве исходного вещества так же, как и для ее производных. Технологический способ можно, таким образом, использовать до или после необходимых стадий способа по изобретению.
Заявители считают, что впервые ионообменную хроматографию применяют для получения фракций ионных полисахаридов с молекулярной массой более примерно 5 кДа, в особенности полисиаловой кислоты с такой молекулярной массой (MW). Согласно дополнительному аспекту настоящего изобретения обеспечивают способ для фракционирования части ионизируемого полисахарида с молекулярной массой (MW) выше чем 5 кДа при использовании ионообменной хроматографии с элюционным буфером основания или кислоты, которое предпочтительно является летучим. Предпочтительно полисахарид содержит группы карбоновых кислот и ионный обмен представляет собой анионный обмен. Предпочтительно элюционный буфер содержит амин, более предпочтительно триэтаноламин. Более предпочтительно полисахарид выделяют из фракции при помощи лиофильной сушки. Этот способ можно применять для фракционирования коломиновой кислоты (CA), содержащей другие реакционно-активные фрагменты (имид малеиновой кислоты или иодацетат и т.д.) или другие природные (например, декстрансульфат) или синтетические (например, полиглутаминовая кислота, полилизин в последнем случае при использовании катионообменной хроматографии) заряженные полимеры. Заявители полагают, что также впервые ионообменную хроматографию (IEC) используют для разделения ионных полисахаридов в сочетании с технологическими способами осаждения и/или способами ультрафильтрации. Способы IEC должны удалять также побочные продукты получения как эндотоксины, которые остаются в коммерчески доступных PSA и СА.
В предварительной стадии окисления и стадии с) селективное окисление следует предпочтительно проводить при таких условиях, чтобы в значительной степени не происходило расщепления в средней части основной углеводородной цепи длинноцепного (полимерного) исходного вещества, чтобы не происходило уменьшения молекулярной массы. Можно использовать ферменты, отвечающие за проведение этой стадии. Наиболее удобно, чтобы окисление представляло собой химическое окисление. Реакцию можно проводить с иммобилизированными реагентами, такими как перрутенат на основе полимера. Наиболее прямой способ проводят с растворенными реагентами. Окислитель представляет собой перрутенат или, предпочтительно, периодат. Окисление можно проводить периодатом при концентрации в диапазоне от 1 мМ до 1 М, при рН в диапазоне от 3 до 10, температуре в диапазоне от 0 до 60°С в диапазоне от 1 минуты до 48 часов.
В способе стадия а) представляет собой стадию, в которой звено сиаловой кислоты на восстанавливающем конце восстанавливают. Обычно звено на восстанавливающем конце исходного вещества находится в форме кетального кольца и восстановление в стадии а) раскрывает кольцо и восстанавливает кетон до спирта. Таким образом, гидроксильная группа при 6-углеродном атоме представляет собой часть вицинального диольного фрагмента.
При подходящих условиях восстановление (для стадий а) и d)) можно использовать водород с катализаторами или предпочтительно гидриды, такие как бор. Они могут быть иммобилизированными, как, например, Amberlite (торговая марка) - боргидрид на подложке. В качестве восстанавливающего агента предпочтительно используют гидриды щелочных металлов, такие как боргидрид натрия при концентрации в диапазоне от 1 мкМ до 0,1 М, при рН в диапазоне от 6,5 до 10, при температуре в диапазоне от 0 до 60°С в диапазоне от 1 минуты до 48 часов. Условия реакции выбирают таким образом, чтобы боковые карбоксильные группы исходного вещества не восстанавливались. Когда предварительная стадия окисления проведена, полученную альдегидную группу восстанавливают до спиртовой группы, не являющейся частью вицинальной диольной группы. Другими пригодными восстанавливающими агентами являются цианоборгидрид в кислых условиях, например цианоборгидрид на полимерной подложке или цианоборгидрид щелочного металла, L-аскорбиновая кислота, мета-бисульфит натрия, L-селектрид, триацетоксиборгидрид и т.д.
Между любой предварительной стадией окисления и стадией восстановления а) и после стадии b), и между стадией окисления с) и стадией восстановления d), и между стадией d) и любой последующей стадией окисления соответствующий интермедиат должен быть отделен от соответствующих окисляющих и восстанавливающих агентов соответственно перед последующей стадией. Если стадию проводят в жидкой фазе, отделение можно проводить обычными способами, такими как расходование избытка окисляющего агента при использовании этиленгликоля, диализ полисахарида и ультрафильтрация для концентрирования водного раствора. Смесь продуктов из стадии восстановления вновь можно разделять диализом и ультрафильтрацией. Можно предложить реакции, проводимые при использовании иммобилизованных окисляющих или восстанавливающих реагентов, приводящие к прямой изоляции продукта.
Стадию селективного окисления, стадию b) удобно проводить при условиях, сходных со стадией предварительного окисления, как описано выше. Таким же образом, окисляющий агент следует расходовать перед выделением продукта при использовании этиленгликоля. Затем продукт выделяют такими подходящими способами, как диализ и ультрафильтрация.
Способ по первому аспекту изобретения и предпочтительного варианта осуществления второго аспекта, который включает последующую стадию окисления после стадии d) для активирования восстановливающего терминального сахаридного звена, приводит к получению активированного производного, содержащего реакционно-активный альдегидный фрагмент, полученный из восстановливающего терминального конца. Предпочтительный способ, включающий окисление, затем восстановление, затем стадию окисления, приводит к получению активированного продукта, имеющего один реакционно-активный альдегидный фрагмент. Если не проводят предварительной стадии окисления и исходное вещество содержит невосстанавливющее терминальное звено, которое содержит вициальную диольную группу (например, сиаловую кислоту), продукт будет содержать альдегидную группу на каждом из терминальных концов, которые могут использоваться.
Альдегидные группы пригодны для конъюгации с субстратами, содержащими аминогруппу, или гидразиновыми соединениями. Способы, в которых активированный продукт из стадии окисления в последствие конъюгирует с субстратом, образуют дополнительный аспект изобретения. Предпочтительно за реакцией конъюгации, как описано в предшествующих публикациях заявителей, как описано выше, которая включает конъюгацию с амином с образованием основания Шиффа, предпочтительно следует восстановление с образованием вторичного аминового фрагмента. Способ в особенности ценен для белков, которые могут быть модифицированы, в которых аминогруппа представляет собой эпсилон-аминогруппу лизиновой группы или N-терминальную аминогруппу. Способ в особенности ценен для белков, которые могут быть модифицированы, или пептидных терапевтически активных агентов, таких как цитокины, гормоны роста, ферменты, гормоны, антитела или фрагменты. В качестве альтернативы способ может быть использован для получения производных систем доставки лекарств, таких как липосомы, например, реакцией альдегида с аминогруппой компонентов, образующих липосому. Другие системы доставки лекарств описаны в более ранней заявке заявителя US-A-5846951. Другие вещества, которые могут быть модифицированы, включают вирусы, микроорганизмы, клетки, включая клетки животных, и синтетические полимеры.
В качестве альтернативы субстрат может содержать гидразиновую группу, в таком случае продукт представляет собой гидразон. Его можно восстанавливать при желании для дополнительной стабильности до алкил гидразида.
В другом предпочтительном варианте осуществления за стадией окисления b) или последующей стадией окисления d) следует реакция одной или каждой из альдегидной групп с соединением посредством линкера, включающим аминогруппу или гидразидную группу и другую функциональную группу, пригодную для селективного получения производного белков или других терапевтически активных соединений, или систем доставки лекарств. Такой линкер может, например, включать соединение, содержащее заместитель функциональной группы для специфической реакции с сульфогидрильными группами и двухосновную органическую группу, соединяющими амин или гидразидную группу и функциональную группу. В реакции остатка альдегида с амино- или гидразидной группой образуется реакционно-активный конъюгат, пригодный для связывания с субстратом, содержащим тиольную (сульфогидрильную) группу. Такие конъюгаты обладают особой ценностью для селективного или сайт-специфического получения производных белков и пептидов.
Получение производных белков и систем доставки лекарств может приводить к увеличению периода полувыведения, улучшенной стабильности, пониженной иммуногенности и/или контролю растворимости и, следовательно, биологической доступности и фармакокинетических свойств или может улучшать растворимость активных веществ или вязкость растворов, содержащих производное активного вещества.
Согласно изобретению также обеспечивают новое соединение, которое представляет собой альдегидное производное ди-, олиго- или полисахарида, включающего фрагменты сиаловой кислоты, в которых терминальное звено восстанавливающего конца представляет собой OR-группу, в которой R выбирают из
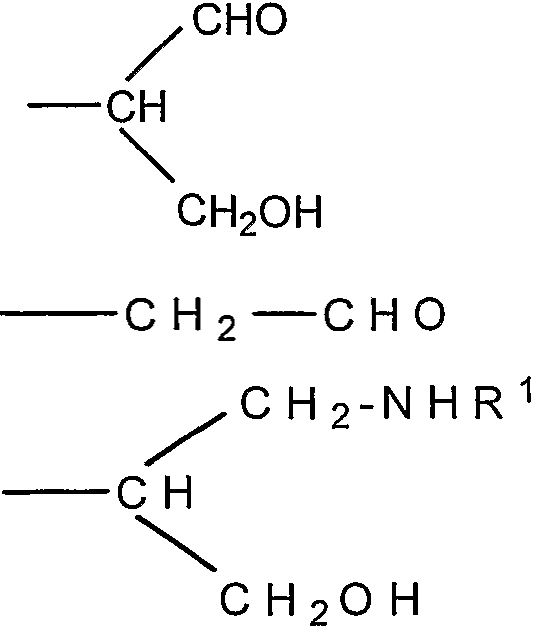
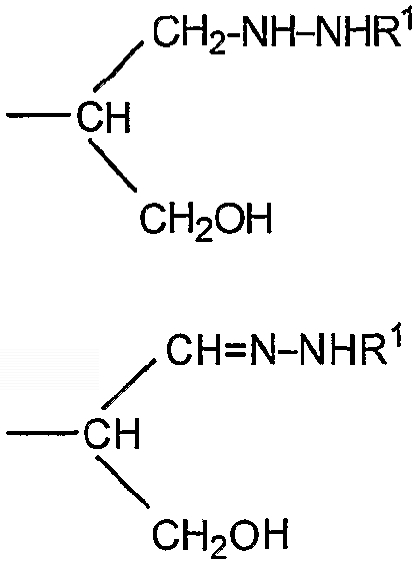
-CH2CH2NHR1, CH2CH=N-NHR1 и CH2CH2NHNHR1, в которых R1 представляет собой Н, С1-24 алкил, арил С2-6 алканоил или полипептид, или белок, связанный через N-терминальный конец или аминогруппу боковой цепи лизинового остатка, систему доставки лекарств, или органическую группу, содержащую функциональный заместитель, адаптированный для реакции с сульфогидрильной группой и, предпочтительно, чтобы терминальный фрагмент на невосстанавливающем конце был пассивирован.
Новое соединение может включать среднюю часть цепи из сахаридных звеньев между двумя терминальными звеньями. Звенья средней части цепи состоят исключительно из звеньев сиаловой кислоты или, в качестве альтернативы, могут включать другие сахаридные звенья в дополнение к терминальным звеньям, которые получают из звеньев сиаловой кислоты. Соединение может быть обычно получено, как описано выше в отношении первого аспекта изобретения.
Новое соединение может быть полисиалилированным субстратом, включающим по крайней мере одну группу полисиаловой кислоты (полисахаридную), конъюгированную с каждой молекулой субстрата конъюгация, включая связь вторичного амина, гидразона или алкилгидразида через восстанавливающий терминальный конец полисиаловой кислоты и в значительной степени не содержащая сшивок невосстанавливающего конца полисиаловой кислоты с другой молекулой или субстратом. Субстрат может представлять собой, например, биологически активное соединение, например фармацевтически активное соединение, в особенности пептидный или белковый терапевтический агент, или систему доставки лекарств. Такие активные вещества широко описаны ниже.
Новое соединение может иметь общую формулу I
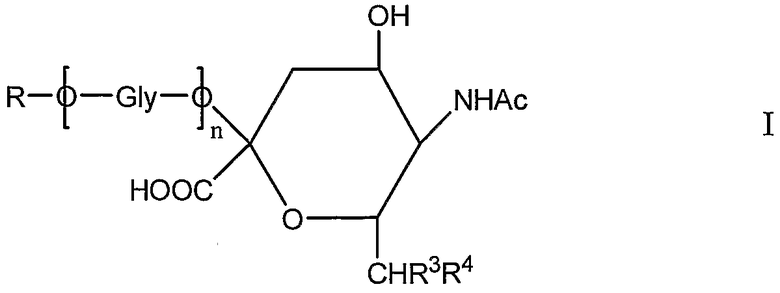
в котором R выбран из
-CH2CH2NHR1, CH2CH=N-NHR1 и CH2CH2NHNHR1, в которых R1 представляет собой Н, С1-24 алкил, арил С2-6 алканоил или полипептид, или белок, связанный через N-терминальный конец или γ-аминогруппу лизинового остатка, систему доставки лекарств, или органическую группу, содержащую функциональный заместитель, адаптированный для реакции с сульфогидрильной группой;
R3 и R4 выбраны из
i) R3 представляет собой Н и R4 представляет собой ОН;
ii) где R представляет собой СН(СН2ОН)СН2ОН или -СН2СНО, R3 и R4 вместе составляют =О;
iii) где R представляет собой CH(CH2OH)CH2NHR1 или -CH2CH2NHR1, R3 представляет собой H и R4 представляет собой -NHR1;
iv) где R представляет собой -CH(CH2OH)CH2NHNHR1 или -CH2CH2NHNHR1, R3 представляет собой H и R4 представляет собой -NHNHR1; или
v) -CHCH=N-NHR1, R3 и R4 вместе составляют =N-NHR1; Ас представляет собой ацетил,
n представляет собой 0 или более; и
GlyO представляет собой глюкозильную группу.
Если R представляет собой группу
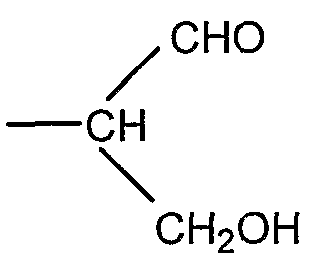
соединение общей формулы I представляет собой полисахарид, который является производным полисиаловой кислоты, имеющим альдегидную группу на восстанавливающем терминальном звене.
Если R представляет собой группу
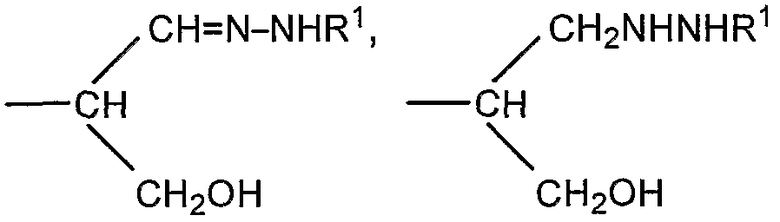
CH2CH=N-NHR1 или CH2CH2NHNHR1 соединение представляет собой конъюгат, образованный реакцией альдегидного производного полисиаловой кислоты с гидразидом R1NHNH2. Гидразид предпочтительно представляет собой ацильный гидразид (R1 содержит терминальную карбонильную группу).
Если R представляет собой группу
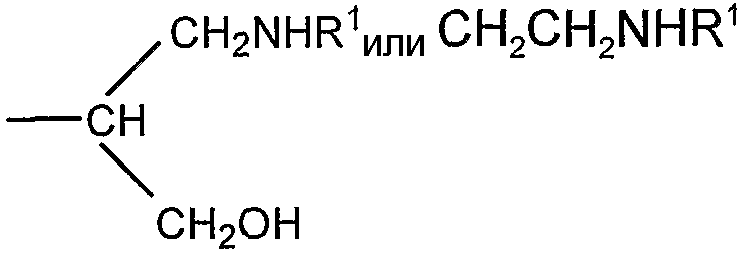
соединение представляет собой конъюгат, образованный реакцией альдегидного производного полисиаловой кислоты с первичной амино группой, содержащей соединение R1NH2.
R1 может представлять собой остаток пептида или белкового терапевтического агента, например антитела или фрагмента, фермента или другого биологически активного соединения, как описано выше. Группа R1 может включать линкерный фрагмент между активным соединением и полисиаловой кислотой.
В качестве альтернативы R1 может представлять собой остаток линкерного реагента, например, для получения производного полисиаловой кислоты, пригодного для конъюгации с группами, отличающимися от аминогрупп или гидразидов, на активных соединениях. Примеры представляют собой линкерные реагенты формулы
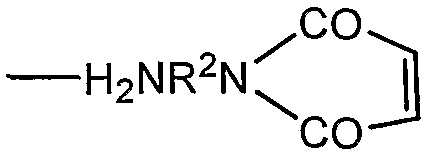
что представляет собой N-соединения имида малеиновой кислоты, в которых R2 представляет собой двухосновную органическую группу, например арилен олиго(алкокси)алкан, или, предпочтительно, алкандиильную группу, например С2-12-алкандиильную группу.
Настоящее изобретение наиболее применимо, если новое соединение является монофункциональным и пассивировано на терминальном звене невосстанавливающего конца. В таких соединениях R3 представляет собой Н и R4 представляет собой ОН. R может принимать любые значения, указанные выше. Глюкозильные группы, предпочтительно, включают звенья сиаловой кислоты и, более предпочтительно, состоят только из таких звеньев, связанных 2-8 или 2-9, например чередованием 2-8/2-9 друг с другом.
Изобретение дополнительно обеспечивает композиции, включающие новые соединения и разбавитель, а также фармацевтические композиции, включающие новые соединения, в которых R1 обладает биологической активностью, и фармацевтически приемлемый наполнитель. Фармацевтические композиции можно вводить перорально, внутривенно, внутрибрюшинно, внутримышечно, подкожно, интраназально, внутрикожно, местно или внутритрахеально.
Во втором аспекте изобретения обеспечивают новое соединение, которое представляет собой продукт способа, согласно второму способу аспекта, которое имеет общую формулу II
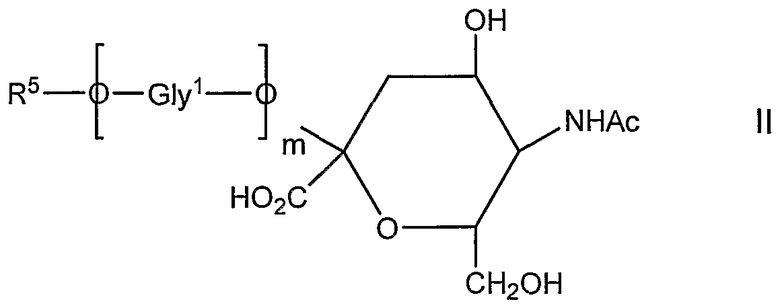
в которой
Ас представляет собой ацетил;
m составляет 0 или больше;
Gly1О представляет собой гликозил; и
R5 представляет собой органическую группу, предпочтительно восстановленную форму терминального восстанавливающего сахаридного звена, его окисленное производное, которое представляет собой альдегид или продукт реакции такого альдегида, который, например, представляет собой амин или гидразид.
Предпочтительно R5 выбирают из тех же групп, что и R выше. В качестве альтернативы R5 представляет собой группу III, соединенную через один углерод, 8 или 9, с
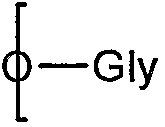
(посредством чего другой из углеродов, 8 или 9, замещен гидроксилом:
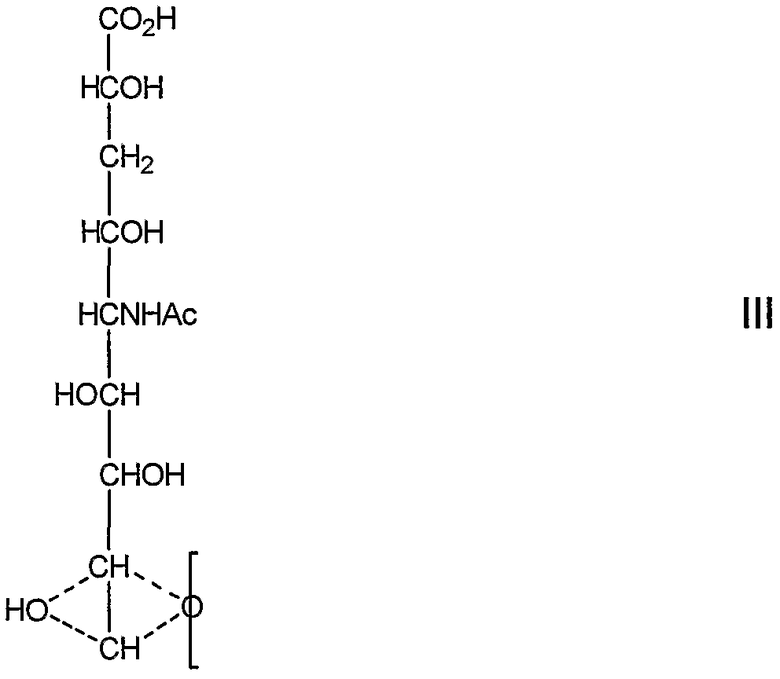
которое представляет собой продукт восстановления с открытием цикла восстанавливающей терминальной сиаловой кислоты.
Предпочтительно группы Gly1O включают звенья сиаловой кислоты, наиболее предпочтительно состоят из звеньев сиаловой кислоты. Значение m предпочтительно равно 2 или более, более предпочтительно составляет 5-1000, например 10-500, более предпочтительно от 10 до 50.
Новый способ в особенности ценен для создания монофункциональной полисиаловой кислоты (PSA). Это основывается на понимании таутомерного равновесия, восстанавливающего концевого кольца PSA, например, коломиновой кислоты (СА), которое описано на фиг.3. Восстанавливающий конец остатка сиаловой кислоты PSA самопроизвольно образует кетон с открытием кольца посредством таутомеризации (фиг.3). При динамическом равновесии между кольцевой и линейной структурами остатка сиаловой кислоты восстанавливающего конца в любой отдельно взятый момент кетонный фрагмент присутствует только у части молекул PSA. Однако, как было отмечено выше, здесь подчеркивается, что реакционная активность полукеталя восстанавливающего конца является недостаточной для практического использования присоединения PSA к белкам, исходя из чего ранее описанные способы не используют эту часть полимера для присоединения белков или других лекарств. Таким образом, как проиллюстрировано на фиг.3, в растворе терминальный остаток сиаловой кислоты на восстанавливающем конце полисиаловой кислоты находится в таутомерном равновесии. Форма с открытым кольцом, хотя и присутствует в небольшом избытке в равновесии, является слабо реакционно-активной по отношению к аминогруппам белков и приводит к образованию ковалентных аддуктов с белками в присутствие цианоборгидрида натрия.
В предпочтительном варианте осуществления изобретения, для лучшего достижения белковой конъюгации описанных продуктов с PSA, заявители создали химически модифицированную форму полисиаловой кислоты, которая является монофункциональной. Новая форма включает химические модификации обоих концов природной молекулы полисиаловой кислоты. В отличие от исходной формы реакции (фиг.1), в которой полимер становится конъюгированным преимущественно от 2 до 8 положении, с наиболее удаленным «восстанавливающим концом», у новой формы полимера прикрепление происходит исключительно в противоположной ориентации.
Новая предпочтительная монофункциональная форма полисиаловой кислоты или другого полисахаридного альдегидного производного представляет собой более пригодную для синтеза и производства фармацевтически приемлемого продукта, поскольку она исключает значительную сложность, которая в противном случае возникает при использовании полимерных форм с немодифицированными восстанавливающими концами (фиг.2). Получение новых форм полимера (фиг.4) включает селективное окисление, предпочтительно периодатом, как в предшествующих описаниях заявителей, для введения альдегидной функции на невосстанавливающем конце. В отличие от предшествующей технологии, показанной на фиг.1, этот альдегидный фрагмент затем разрушают восстановлением, например боргидридом. На другом конце полимера стадия восстановления боргидридом также одновременно блокирует кольцевую структуру восстанавливающего конца за счет восстановления полукеталя. Одновременное восстановление кетона до гидроксильного фрагмента вводит новую диольную функциональную группу, которая теперь подлежит селективному окислению во второй стадии окисления. Когда природный полимер был (успешно) окислен периодатом, восстановлен боргидридом и окислен второй раз периодатом, образуется новая форма полимера, которая действительно является монофункциональной, имеющей одну реакционно-активную группу (альдегид) только на восстанавливающем конце (фиг.3).
Реакционная активность белка (при восстановительном аминировании) различных интермедиатов, описанная в способе «двойного окисления» фиг.4, описана в таблице 2. В особенности, данные показывают, что интермедиат «CAOR» (коломиновая кислота - полисиаловая кислота - окисленная/редуцированная), полученная восстановлением боргидридом окисленного периодатом полимера, является инертной по отношению к белковым мишеням, обеспечивая, чтобы оба, альдегидный и полукетальный фрагмент разрушались при восстановлении боргидридом. Во втором цикле окисления периодатом «инертного по отношению к белку» интермедиата CAOR получают новое производное полисиаловой кислоты (CAORO), которое вновь является реакционно-активным по отношению к белкам (таблица 2) и, более того, действительно является монофункциональным по своей природе, обладая единственной альдегидной группой на «восстанавливающем конце» полимера и являясь реакционно-неактивным по отношению к белкам на другом конце. Монофункциональная PSA может приводить к исключительно единственным образом ориентированному присоединению к белкам крайним невосстанавливаемым концом и не способна приводить к непреднамеренно сшитым белкам (фиг.5). Эта новая реакционная схема (фиг.4), известная как способ «двойного окисления» позволяет элегантно избежать необходимости очистки ожидаемого продукта от различных непреднамеренно полученных продуктов (см. фиг.2), которых удается полностью избежать в представленной новой реакционной схеме.
Последующее представляет собой краткое описание чертежей.
Фиг.1а представляет собой реакционную схему, показывающую активирование по предшествующему уровню техники невосстанавливающего терминального звена сиаловой кислоты;
фиг.1b представляет собой реакционную схему, показывающую восстановительное аминирование по предшествующему уровню техники альдегидного фрагмента продукта реакционной схемы 1а при использовании фрагмента белок-амин;
фиг.2а представляет собой принципиальную схему, показывающую возможные побочные реакции, имеющие место в реакции фиг.1b, вовлекающие восстанавливающий конец;
фиг.2b схематически представляет возможные побочные продукты побочных реакций фиг.2а;
фиг.3 представляет собой реакционную схему, показывающую таутомерию между кеталем и формы с замкнутым кольцом восстанавливающего терминального звена сиаловой кислоты PSA;
фиг.4а представляет собой реакционную схему, показывающую предпочтительные реакции окисления восстановления окисления PSA;
фиг.4b дает представление о подходящих условиях для стадий схемы фиг.4 и объясняет аббревиатуры, используемые для исходных веществ, интермедиатов и конечных продуктов;
фиг.5 представляет собой принципиальную схему, сходную с фиг.2b, но показывающую продукты реакции фиг.4;
фиг.6 показывает результат анализа методом гельпроникающей хроматографии (GPC) продуктов примера 1;
фиг.7 показывает результаты денатурирующего электрофореза в полиакриламидном геле (SDS-PAGE) примера 2;
фиг.8 показывает фармакокинетику периода полувыведения конъюгатов, испытываемых in vivo на мышах в примере 3;
фиг.9 показывает результаты IEC для коломиновой кислоты (СА) 22,7 кДа в примере сравнения 2;
фиг.10 показывает исходные результаты электрофореза в полиакриламидном геле (PAGE) для СА 22,7 кДа в примере сравнения 2;
фиг.11 показывает исходные результаты электрофореза в полиакриламидном геле (PAGE) для нескольких веществ СА как загружаемых, так и отделенных фракций, как и в примере сравнения 2.2;
фиг.12 показывает GPC хроматограммы для 3 из фракций СА, выделенных как в примере сравнения 2.2;
фиг.13 показывает исходные результаты PAGE для двух из образцов, использованных на фиг.12, и других образцов СА и САО, как описано в примере сравнения 2.2;
фиг.14 показывает результаты ультрафильтрации СА 22,7 кДа, как описано в примере сравнения 2.4;
фиг.15 показывает SDS PAGE для примера 5;
фиг.16 показывает результаты SDS PAGE для фракционированных конъюгатов гормона роста GH-CA, полученных в примере 5; и
фиг.17 показывает результаты примера 7.
Изобретение дополнительно проиллюстрировано сопроводительными примерами.
Примеры
Материалы
Карбонат аммония, этиленгликоль, полиэтиленгликоль (8 кДа), цианоборгидрид натрия (>98% чистоты), мета-периодат натрия и маркеры молекулярной массы получали от Sigma Chemical Laboratory, UK. Используемая коломиновая кислота, линейные α-(2→8)-связанные полисиаловые кислоты E. coli (22,7 кДа в среднем, высокая полидисперсность 1,34, 39 кДа, полидисперсность 1,4; 11 кДа полидисперсность 1,27) были от Camida, Ireland, радиоактивный иодид (Na125I) приобретали от Amersham, UK. Другие вещества включали 2,4 динитрофенил гидразин (Aldrich Chemical Company, UK), трубчатый аппарат для диализа (пределы отсечения 3,5 кДа и 10 кДа; Medicell International Limited, UK), Sepharose SP HiTrap, PD-10 колонки (Pharmacia, UK), Трис-глицин полиакриламидные гели (4-20% и 16%), Трис-глицин додецилсульфат натриевый протекающий буфер и загружаемый буфер (Novex, UK). Деионизированную воду получали из водоочистной установки Elgastat Option 4 (Elga Limited, UK). Все используемые реагенты были аналитической степени чистоты. Для спектрофотометрических измерений при анализе белков или СА использовали спектрофотометр для прочтения планшетов (Dynex Technologies, UK). Беспородных мышей CD1 (в возрасте 8-9 недель; вес тела 29-35 г) приобретали от Charles River (UK) и акклиматизировали в течение по крайней мере одной недели перед использованием.
Способы
Определение белков и коломиновой кислоты
Количественную оценку полисиаловых кислот (как сиаловой кислоты) с резорциновым реагентом проводили резорциновым способом [Svennerholm, 1957], как описано в других публикациях [Gregoriadis et al., 1993; Fernandes and Gregoriadis, 1996, 1997]. Fab (белок) измеряли на основе BCA колориметрического способа.
Пример сравнения 1
Ковалентные PSA-белок конъюгаты получали восстановительным аминированием с цианоборгидридом натрия при использовании природной формы полисиаловой кислоты (коломиновая кислота, СА) из E. coli, посредством ее слабо реакционно-способного восстанавливающего конца. СА = коломиновая кислота; САО = окисленная коломиновая кислота как в Fernandes and Gregoriadis, 1996; Jain et al., 2003. Цианоборгидрид натрия (NaCNBH3) использовали в концентрации 4 мг мл-1.
Результаты представлены в таблице 1. Молярные соотношения в колонке 1 составляли соотношение исходного СА(О) к белку (n=3, ± стандартное отклонение).
Пример 1 Получение монофункциональной полисиаловой кислоты:
1а Активация коломиновой кислоты
Свежеприготовленный 0,1 М раствор метапериодата натрия (NaIO4) смешивали с СА (100 мг СА/мл NaIO4) при 20°С и реакционную смесь перемешивали магнитной мешалкой в течение 15 минут в темноте. Затем в реакционную смесь добавляли двукратный объем этиленгликоля для расходования избытка NaIO4 и смесь оставляли перемешиваться при 20°С еще в течение 30 минут. Окисленную коломиновую кислоту подвергали экстенсивному диализу (предел отсечения 3,5 КДа трубчатый аппарат для диализа) (24 часа) против 0,01% буфера карбоната аммония (рН 7,4) при 4°С. Ультрафильтрацию (свыше предела отсечения 3,5 кДа) использовали для концентрации раствора САО из трубчатого аппарата для диализа. Вслед за концентрированием до требуемого объема фильтрат лиофилизировали и хранили при -40°С до дальнейшего использования.
1b Восстановление коломиновой кислоты
Окисленную коломиновую кислоту (САО; 22,7 кДа) восстанавливали в присутствие боргидрида натрия. Свежеприготовленный 0,15 мМ раствор боргидрида натрия (NaBH4; в 0,1 М NaOH, разбавленном до рН 8-8,5 путем разбавления разбавленным раствором H2SO4) смешивали с САО (100 мг СА/мл) при 20°С и реакционную смесь перемешивали в течение 2 часов в темноте. За счет завершения реакции рН понижалась до 7. Окисленную/восстановленную коломиновую кислоту (CAOR) подвергали диализу (предел отсечения трубчатого аппарата для диализа молекулярная масса 3,5 КДа) против 0,01% раствора буфера карбоната аммония с рН (7) при 4°С. Для концентрации CAOR раствора из трубчатого аппарата для диализа использовали ультрацентрифугирование. Фильтрат лиофилизировали и хранили при 4°С до дальнейшего использования. Определение любого содержания альдегида определяли, как описано в «определении окисления СА».
1с Повторное окисление СА
После подтверждения отсутствия содержания альдегида в окисленной/восстановленной коломиновой кислоте (CAOR) ее вновь окисляли так же, как описано в активации коломиновой кислоты, за исключением того, что CAOR инкубировали с раствором периодата в течение более длительного времени (вплоть до 1 часа). Степень окисления CAORO продукта измеряли на лиофилизированном порошке, также полученном в этой стадии.
1d Определение состояния окисления СА и производных
Качественную оценку степени окисления коломиновой кислоты проводили с 2,4-динитрофенилгидразином (2,4-DNPH), что при взаимодействии с карбонильными соединениями приводило к плохо растворимым 2,4-динитрофенилгидразонам. Неокисленную (СА), окисленную (САО), восстановленную (CAOR) и повторно окисленную (CAORO) (5 мг каждой) добавляли к 2,4-DNPH реагенту (1,0 мл), растворы встряхивали и затем оставляли при 37°С до появления кристаллического осадка [Shriner et al., 1980]. Степень (количественную) окисления СА измеряли при помощи способа [Park and Johnson, 1949], основанного на восстановлении феррицианидных ионов в щелочном растворе до ферроцианида железа (персидский голубой), которое затем измеряли при 630 нм. В этом случае в качестве стандарта использовали глюкозу.
1е Гельпроникающая хроматография
Образцы коломиновой кислоты (СА, САО, CAOR и CAORO) растворяли в NaNO3 (0,2 М), CH3CN (10%; 5 мг/мл) и хроматографировали на двух GMPWXL колонках с определением на основе показателя преломления (ГПХ (GPC) система: VE1121 GPC помпа растворителя, VE3580RI детектор и сравнение при использовании программного обеспечения Trisec 3 (Viscoteck Europe Ltd). Образцы (5 мг/мл) фильтровали через 0,45 мкм нейлоновую мембрану и пропускали при 0,7 см/мин 0,2 М NaNO3 и CH3CN (10%) в качестве подвижной фазы.
Результаты
Коломиновая кислота (СА), полисиаловая кислота, представляет собой линейный альфа-2,8-связанный гомополимер остатков N-ацетилнейроаминовой кислоты (Neu5Ac) (фиг.1а). Однако периодат представляет собой сильный окисляющий агент и несмотря на то, что он является селективным [Fleury and Lang, 1932] по отношению к углеводородам, включающим гидроксильные группы при соседних атомах углерода, он может вызывать зависящее от времени расщепление внутренних остатков Neu5Ac. В связи с этим, в настоящей работе время окисления коломиновой кислоты ограничивали 15-60 минутами при использовании 100 мМ периодата при комнатной температуре [Lifely et al., 1981]. Более того, поскольку периодат разлагается при воздействии света с образованием более реакционно-способных соединений [Dyer, 1956], реакционные смеси хранили в темноте. Целостность внутренних альфа-2,8-связанных Neu5Ac остатков после обработки периодатом и боргидридом анализировали при использовании гельпроникающей хроматографии и хроматограммы, полученные для окисленных (CAO), окисленных восстановленных (CAOR), дважды окисленных (CAORO) веществ сравнивали с аналогичными для нативной СА. Было обнаружено (фиг.6), что окисленная (15 минут) (CAO) (6b), восстановленная (CAOR) (6с), дважды окисленная (1 час) (CAORO) (6d) и нативная (6а) СА обладают фактически идентичными профилями элюирования без свидетельств, что последовательные стадии окисления и восстановления привели к значительной фрагментации полимерной цепи. Небольшие пики отвечают буферным солям. Количественные измерения состояния окисления СА проводили восстановлением иона феррицианида в щелочном растворе до ферроцианида (персидский голубой) [Park and Johnson, 1949] при использовании глюкозы в качестве стандарта [результаты показаны в таблице 2]. В таблице 2 показано, что окисленная коломиновая кислота имеет больше чем стехиометрическое (>100%) количество восстанавливающего агента, т.е. 112% моль кажущегося содержания альдегида, включающего комбинированную восстанавливающую силу восстанавливающего конца полукеталя и введенного альдегида (с другого конца). У CAOR не наблюдалось реакционной активности, это показывает, что нейтрализация как альдегида, так и полукеталя САО была успешно осуществлена восстановлением боргидридом. После второго цикла окисления периодатом содержание альдегида в полимере сохранилось на 95% CAORO (при ошибке эксперимента 10%), указывая на успешное введение нового альдегидного фрагмента на восстанавливающем конце.
Результаты количественных исследований интермедиатов каломиновой кислоты в способе двойного окисления при использовании феррицианида (таблица 2) совпадали с результатами качественных опытов, проведенных с 2,4-динитрофенилгидразином, который давал светло-желтый осадок после 10 минут реакции при комнатной температуре с нативной СА и интенсивный оранжевый цвет - с формами полимера, содержащими альдегид.
Таблица 2: Степень окисления различных интермедиатов коломиновой кислоты в схеме реакции двойного окисления при использовании глюкозы в качестве стандарта (100%, 1 моль альдегида на моль глюкозы n=3±стандартное отклонение).
Пример 2 - Получение конъюгатов Fab-коломиновая кислота
Fab растворяли в 0,15 М фосфатно-солевого буферного раствора (PBS) (рН 7,4) и ковалентно связывали с различными коломиновыми кислотами (CA, CAO, CAOR и CAORO) посредством восстановительного аминирования в присутствии цианоборгидрида натрия (NaCNBH3). Коломиновую кислоту из каждой стадии синтеза (исходное вещество и продукты из каждого из примеров с 1а до с) вместе с Fab в молярных отношениях СА:Fab 100:1 вводили в реакцию в 0,15 М PBS (рН 7,4; 2 мл), содержащего цианоборгидрид натрия (4 мг/мл) в герметично закрытых сосудах при перемешивании магнитной мешалкой при 35±2°С в печи. Смеси затем подвергали осаждению сульфатом аммония ((NH4)2SO4) путем медленного добавления соли при непрерывном перемешивании для получения 70% мас./об. насыщения. Образцы, перемешиваемые в течение 1 часа при 4°С, центрифугировали в течение 15 минут (5000·g) и гранулы, содержащие полисиалилированный Fab, суспендировали в насыщенном растворе (NH4)2SO4 и вновь центрифугировали в течение 15 минут (5000·g). Выделенные осадки повторно растворяли в 1 мл 0,15 М Na-фосфатного буфера с добавлением 0,9% NaCl (рН 7,4; PBS) и активно диализировали (24 часа) при 4°С опять с тем же PBS. Диализаты затем исследовали на содержание сиаловой кислоты и Fab и выход конъюгата выражали в виде молярного соотношения СА:Fab. Контроль включал конъюгацию исходного белка способу в присутствии неокисленной СА или в отсутствии СА при описанных условиях. Перемешивание поддерживали на минимуме для того, чтобы избежать сопутствующую денатурацию белка. Полисиалилированный Fab дополнительно характеризовали гельпроникающей хроматографией, ионообменной хроматографией и денатурирующим электрофорезом в полиакриламидном геле (SDS-PAGE).
2b Ионообменная хроматография
Образцы (0,5 мл) в начальный момент времени (контроль) и через 48 часов из реакционных смесей подвергали ионообменной хроматографии (IEC) на катионообменной колонке Sepharose SP (1 мл; скорость потока 1 мл/мин; связывающий/промывающий буфер 50 мМ фосфата натрия, рН 4,0; элюционный буфер, 50 мМ фосфата натрия, рН 4,0, содержащий 1 М хорида натрия). Колонки промывали, элюировали и элюированные фракции анализировали на содержание СА и белка (Fab). Для обессоливания образцов перед использованием в колонке использовали PD-10 колонки.
2с SDS электрофорез в полиакриламидном геле
Электрофорез в полиакриламидном геле (SDS-PAGE) (MiniGel, вертикальная гелевая установка, модель VGT 1, энергоснабжение модель Consort E132; VWR, UK) использовали для определения изменений размера молекул Fab при полисиалилировании. SDS-PAGE Fab и его конъюгатов (с СА, САО, CAOR и CAORO) образцов в начальный момент времени (контроль) и через 48 часов из реакционных смесей, а также в качестве контроля способа (неокисленная СА) проводили при использовании 4-20% полиакриламидного геля. Образцы калибровали в широком диапазоне молекулярной массы маркеров.
В предшествующих экспериментах [Jain et. Al., 2003; Gregoriadis, 2001] с другими белками было обнаружено, что оптимальные выходы молекулярной конъюгации CA:Fab (полученного из иммуноглобулина (IgG) овцы) требуют температуры 35±2°C в 0,15 М PBS буфера при рН 6-9 в течение 48 часов. Иминовые образцы (основания Шиффа), образованные при этих условиях между альдегидом полимера и белком, успешно восстанавливали NaCNBH3 с получением стабильного вторичного амина [Fernandes and Gregoriadis, 1996; 1997]. Воздействие на белок окисленной периодатом природной СА приводит к образованию метастабильного аддукта присоединения основания Шиффа СА-белок (как описано для полисиалилирования каталазы) [Fernandes and Gregoriadis, 1996]. Подобным образом в реакции окисленной формы СА с Fab заявители сначала получают метастабильный аддукт присоединения основания Шиффа путем инкубации окисленного полимера с Fab в течении 48 часов при 37°С, который затем укрепляют селективным восстановлением (восстановительным аминированием) с NaCNBH3 (который восстанавливает иминовую структуру основания Шиффа, но не альдегидный фрагмент полимера). Для того, чтобы характеризовать реакционную способность по отношению к белку различных интермедиатов СА «способа двойного окисления», Fab подвергали восстановительному аминированию в присутствии природной СА (СА), окисленной СА (САО), окисленной восстановленной СА (CAOR) и «дважды окисленной» СА (CAORO). Для этих исследований использовали 22,7 кДа PSA при молярном соотношении СА: Fab (100:1). После 48 часов инкубации в присутствии NaCNBH3 конъюгаты Fab выделяли из реакционных смесей осаждением сульфатом аммония (как описано в Примерах) и результаты выражали в терминах молярных соотношений СА:Fab в полученных конъюгатах (таблица 3).
Синтез соединений Fab (белок) коломиновой кислоты
Из таблицы 3 очевидно, что если использовали природную неокисленную СА (в присутствии цианоборгидрида), наблюдали значительную конъюгацию, но на низком уровне (результатом которой являлось молярное соотношение СА:Fab 0,21:1) за счет реакции с полуацетальной группой СА на ее восстанавливающем конце.
Образование конъюгатов CA-Fab было дополнительно доказано совместным осаждением двух фрагментов при добавлении (NH4)2SO4 (СА сама по себе не осаждается в присутствие этой соли). Доказательство конъюгации также получали ионообменной хроматографией (IEC, не показано) и электрофорезом в полиакриламидном геле (SDS-PAGE; фиг.7).
Для ионообменной хроматографии полисиалилированный Fab, полученный осаждением (NH4)2SO4, повторно растворяли в буфере фосфата натрия (50 мМ, рН 4,0) и подвергали ионообменной хроматографии (IEC) при использовании Sepharose SP HiTrap колонки (катионообменная). В противоположность результатам, показывающим полное растворение СА (в промывочной воде) и Fab (в элюированных фракциях), и СА и Fab в реакционных образцах 48 часов совместно элюировали в фракциях промывочной воды, показывая присутствие СА-Fab конъюгата.
Фиг.7 описывает анализ антител на описанные выше Fab конъюгаты. Эти данные подтверждают, что распределения молекулярной массы двух конъюгатов являются чрезвычайно сходными (как ожидалось, поскольку побочные продукты, полученные из ассиметрично бифункциональной СА, составляют только небольшой процент от общей части молекул). Также из фиг.7 очевидно, что вне зависимости, получали ли конъюгаты Fab из асимметрической бифункциональной СА (т.е. окисленной периодатом природной СА) или из монофункциональной PSA, образовывались конъюгаты с широким диапазоном молекулярной массы, превышающей молекулярную массу не переведенного в производные Fab контроля. Это согласуется с известной полидисперсностью природного полимера, описанной в предыдущих публикациях заявителя. Фиг.7 также подтверждает, что восстановительное аминирование монофункциональной СА приводит к получению Fab конъюгата с выходом, сравнимым с выходом ранее описанного способа, основанного на окисленной периодатом природной СА (описанного на фиг.1). Также из фиг.7 очевидно, что только следовые количества непереведенного в производное Fab остаются в каждом образце конъюгата. Следовые количества оставшегося Fab удаляли из указанных конъюгатов ионообменной хроматографией перед исследованиями in vivo (пример 3 ниже).
Пример 3 - Исследования in vivo
В образцы Fab фрагмента овечьего иммуноглобулина (IgG) или в конъюгаты с САО или CAORO вводили радиоактивную метку 125I следующим образом:
10% по объему каждого из указанных образцов отбирали (˜100 мкл) и помещали в свежие IODO-gen пробирки. 20 мкл образца PBS, содержащего 200 мкКи 125I (в виде NaI), добавляли к белку или конъюгату и пробирки закрывали и оставляли инкубироваться при комнатной температуре в течение 10 минут. Содержимое пробирок затем переносили на 500 мкл центробежные фильтры (предел отсечения молекулярной массы 3,5 кДа) и образцы центрифугировали при 6500 об/мин в микроцентрифуге. Элюент отбрасывали и объем ретентата (выше мембраны) доводили до 500 мкл. Этот способ повторяли еще 5 раз, измеряли радиоактивность выше (белок) и ниже (свободный иод) мембраны для образца в 5 мкл при использовании счетчика Pakard Cobra Gamma. Если число квантов свободного 125I составляло менее 5% от числа квантов в конъюгированной фракции, то дополнительной очистки не проводили. Если свободный 125I составлял >5%, очистительный цикл повторяли и образцы анализировали повторно.
CD1 мышам (вес тела 29-35 г) вводили дозу 40 мкг (100 мкл объема в PBS) белка каждой мыши (˜1,6 мг/кг) внутривенно (через хвостовую вену) в виде одной инъекции и отбирали 50 мкл образцы крови (при использовании обработанных гепарином градуированных капилляров) через временные интервалы из другой хвостовой вены и помещали в 500 мкл PBS. Последнее кровотечение представляло собой обескровливание для получения достаточного количества квантов. Образцы затем центрифугировали при 3000 об/мин в течение 10 минут и полученную надосадочную жидкость удаляли и помещали в пробирки для счетчика гамма-квантов. Образцы подсчитывали вместе с представленными образцами инъецируемого белка в Pakard Cobra II автоматическом счетчике гамма-квантов. Зарегистрированное число квантов выражали как процент от исходной инъецируемой дозы.
Образцы меченных радиоактивным иодом Fab, CAO и CAORO Fab конъюгатов вводили в виде инъекции внутривенно мышам для контроля периода полувыведения из кровотока. На фиг.8 показаны фармакокинетики нативного Fab по сравнению с конъюгатами Fab-коломиновой кислоты, полученных исходным способом (при использовании САО) и нового способа двойного окисления (при использовании CAORO). Представленные результаты показывают, что CAO-Fab и CAORO-Fab приводят к выраженному и значительно более длительному времени удержания в кровотоке по сравнению с наблюдаемым в случае неизмененного Fab, приводя к увеличению в 6,28 раз и 5,28 раз (соответственно) AUC величин по сравнению с нативным Fab.
Пример 4 - Синтез конъюгата имида малеиновой кислоты
CAORO, синтезированную в примере 1с выше, вводили в реакцию с 5 молярными эквивалентами гидразида N-[β-малеимидпропионовой кислоты] в 0,1 М ацетате натрия в течение 2 часов при 37°С. Полученный гидразон осаждали в этаноле, повторно суспендировали в ацетате натрия и повторно осаждали в этаноле, повторно растворяли в воде и подвергали сухой заморозке. Продукт является пригодным для сайт-специфической конъюгации с тиоловыми группами фрагментов цистеина в белках и пептидах. Альдегидное производное монофункциональной полисиаловой кислоты можно также вводить в реакцию с линкерным соединением, содержащим гидразидный фрагмент и N-малеимидный фрагмент с образованием стабильного гидразона, содержащего активную малеимидную функциональность, пригодную для реакции с тиольной группой.
Пример сравнения 2 - Фракционирование коломиновой кислоты при использовании ионообменной хроматографии (СА, 22,7 кДа, полидисперсность 1,34)
Пример сравнения 2.1 Крупномасштабное фракционирование
XK50 колонку (Amersham Biosciences, UK) заполняли 900 мл Sepharose Q FF (Amersham Biosciences) и уравновешивали 3 объемами колонки промывающего буфера (20 мМ триэтаноламин; рН 7,4) при скорости потока 50 мл/мин. СА (25 грамм в 200 мл промывающего буфера) загружали в колонку при 50 мл в минуту через шприцевый вход. Вслед за этим промывали колонку 1,5 объемами (1350 мл) промывающего буфера.
Связанную СА элюировали 1,5 объемами колонки различных элюционных буферов (триэтаноламиновый буфер, 20 мМ рН 7,4 с NaCl в диапазоне от 0 мМ до 475 мМ с шагом 25 мМ) и, наконец, 1000 мМ NaCl в том же буфере для удаления остаточной А и других остатков (если какие-либо присутствуют).
Образцы концентрировали до 20 мл ультрафильтрацией при высоком давлении через 5кДа мембрану (Vivascience, UK). В этих образцах буфер заменяли на деионизированную воду при использовании повторяющейся ультрафильтрации при 4°С. Образцы анализировали на среднюю молекулярную массу и другие параметры гельпроникающей хроматографией (GPC) (как описано в примере 1е) и нативным электрофорезом в акриламидном геле (окрашенном альциановым синим).
Пример сравнения 2.2
Фракционирование в меньших масштабах
Следующие образцы фракционировали при использовании идентичной промывочной воды и системы градиентов в меньшем масштабе (матрица до 75 мл; 0,2-3 грамма коломиновой кислоты):
коломиновая кислота (СА, 22,7 кДа, полидисперсность 1,34; СА, 39 кДа, полидисперсность 1,4), коломиновая кислота-альдегид (САО, 22,7 кДа, полидисперсность 1,34), монофункциональная коломиновая кислота (CAORO, 22,7 кДа; полидисперсность 1,34); амин коломиновой кислоты (СА-NH2, 22,7 кДа, полидисперсность 1,34), малеимид коломиновой кислоты (САМ, как для примера 4 и молекулярная масса СА отслеживалась в течение всего процесса).
Узкие фракции СА, полученные при использовании описанной выше процедуры, окисляли 10 мМ периодата и анализировали гельпроникающей хроматографией (GPC) и нативным PAGE на изменение размера полимера.
Результаты
Ионообменная хроматография СА 22,7:
увеличение масштаба (матрица 75 мл, 3 г СА)
* не проводилось
Коломиновую кислоту и ее производные (22,7 кДа) успешно фракционировали в различные узкие фракции с полидисперсностью менее чем 1,1, со средней молекулярной массой до 46 кДа с различными % доли. Фиг.9 и 10; таблица 4 показывают результаты разделения 22,7 кДа веществ в масштабе 75 мл. Фиг.9 представляет собой результат GPC и фиг.10 представляет собой нативный PAGE.
Этот способ позволял осуществлять масштабирование от 1 мл до 900 мл матрицы с практически идентичным профилем фракционирования для каждого из масштабов (не все результаты показаны).
Фракционирование большего полимера (СА, 39 кДа, полидисперсность 1,4) приводило к получению образцов вплоть до 90 кДа. Этот способ может быть успешно использован для фракционирования даже больших партий полимера. Фиг.11 показывает результаты нативного PAGE для 3 подаваемых образцов СА и фракций, разделенных ионообменной хроматографией, анализированных в таблице 4. Результаты PAGE показывают, что ионообменные фракции имеют узкую полидисперсность. Это согласуется с данными GPC, показанными на фиг.12, которые показывают, результаты для 3 из фракций, отделенных от 22,7 кДа СА. Удерживаемые объемы показаны в таблице 5.
22,7 кДа вещества отделяли в больших масштабах. При использовании GPC анализировали фракции из ионного обмена. Выделяли следующие фракции, показанные в таблице 6.
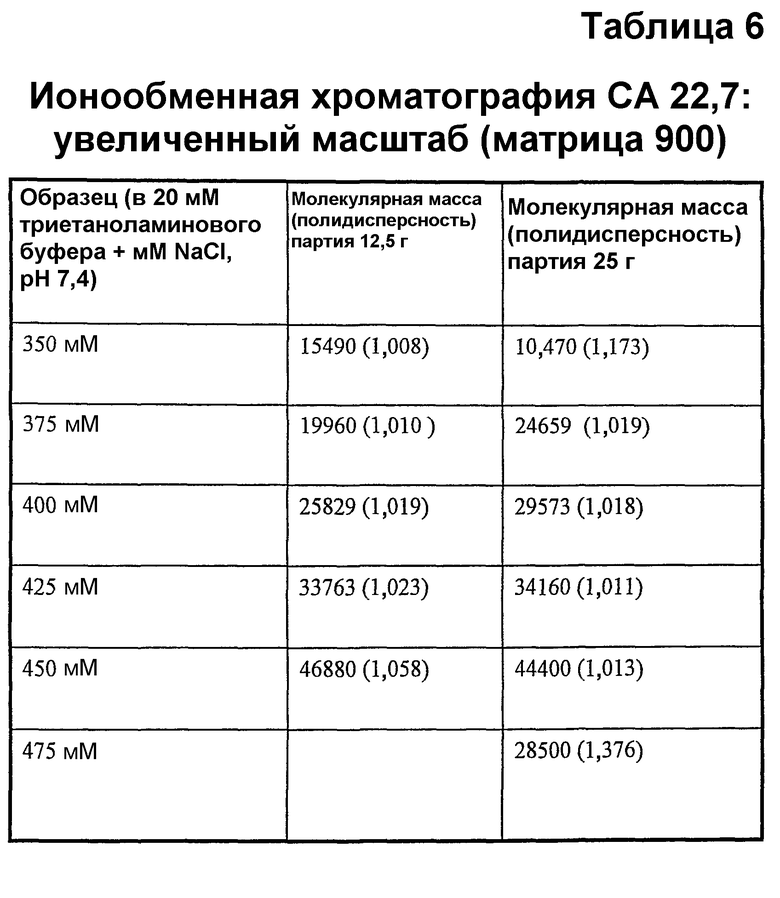
Все узкие фракции успешно окисляли 10 мМ периодатом и образцы, отобранные на различных стадиях способа получения и проанализированные GPC и нативным PAGE, не показали изменения молекулярной массы и полидисперсности. Данные для некоторых из образцов показаны на фиг.13.
2.3 Осаждение коломиновой кислоты
Дифференциальное осаждение этанолом использовали для осаждения коломиновой кислоты с разной длиной цепи.
Результаты
Дифференциальное осаждение этанолом показало, что меньшие СА требовали больше этанола (EtOH). Широко полидисперсные полимеры 22,7 кДа осаждали 70% EtOH, дающим выход >80% получаемого полимера. Концентрация EtOH 80% требовалась для осаждения >80% с более низкой молекулярной массой 6,5 кДа (полидисперсность <1,1). Этот способ также удаляет любые соли, загрязняющие продукт.
2.4 Фракционирование коломиновой кислоты фильтрацией
Образцы 22,7 кДа очищали ультрафильтрацией на мембранах с различными пределами отсечения (5, 10, 30, 50 и 100 кДа). Во всех случаях ретентат анализировали GPC и нативным PAGE.
Результаты
Образцы 22,7 кДа очищали ультрафильтрацией на мембранах с различными пределами отсечения, что показало уменьшение полидисперсности полимера и сдвиг по направлению к более высокой молекулярной массе с увеличением пределов отсечения мембраны (фиг.14).
Можно использовать также комбинированные способы и хроматографию ионных пар для фракционирования полимеров.
Пример 5 - Синтез конъюгатов гормона роста (GH)-коломиновой кислоты (с широкой и узкой дисперсностью)
Для получения конъюгатов GH использовали окисленную коломиновую кислоту (САО; 22,7 кДа) и окисленную коломиновую кислоту с узкой дисперсностью (NCAO; 22,7 кДа, полидисперсность = 1,09; 40,9 кДа, полидисперсность = 1,02), полученную в примере сравнения 2.2.
Получение конъюгатов гормона роста - коломиновой кислоты
Гормон роста растворяли в 0,15 М PBS (рН 7,4) и ковалентно связывали с различными коломиновыми кислотами (САО и NCAO). Различные СА (22,7 кДа, САО; 22,7 кДа & 40,9 кДа, NCA) по отдельности добавляли к GH (2 мг) в молярных соотношениях СА: GH (12,5:1), цианоборгидрид натрия добавляли до конечной концентрации 4 мг/мл. Реакционные смеси герметично закрывали и перемешивали при помощи магнитной мешалки в течение 24 часов при 35±2°С. Смеси затем осаждали сульфатом аммония ((NH4)2SO4) путем медленного добавления соли при непрерывном перемешивании для достижения 70% мас./об. насыщения, перемешивали 1 час при 4°С, затем центрифугировали (5000·g) в течение 15 минут, и гранулы повторно суспендировали в насыщенном растворе (NH4)2SO4 и вновь центрифугировали в течение 15 минут (5000·g). Выделенные осадки повторно растворяли в 1 мл PBS рН 7,4 и подвергали активному диализу (24 часа) при 4°С против того же буфера. Контроли включали конъюгацию нативного белка в присутствие неокисленной СА или в отсутствие СА. Встряхивание было сведено к минимуму, чтобы избежать сопутствующей денатурации белка. Полисиалилированный GH характеризовали SDS-PAGE. Полисиалилированный GH подвергали ионообменной хроматографии, как описано в примере сравнения 2, и фракции продукта подвергали SDS-PAGE.
Результаты
Результаты (фиг.15) показывают, что в контрольной ячейке (с GH) миграция образца представляла собой такую же, как и для свежего GH. В полосах конъюгатов существуют сдвиги, что обычно указывает на увеличение массы, свойственное полисиалилированому GH. Ширина полосы значительно сужалась в случае конъюгатов узкодисперсных полимеров по сравнению с конъюгатами широкодисперсных полимеров. Более того, GH конъюгаты (с широкодисперсным полимером) разделялись на различные образцы анионообменной хроматографией (фиг.16).
Пример 6 - Синтез конъюгатов инсулин-коломиновая кислота
Активированную полисиаловую кислоту (окисленная коломиновая кислота (САО)) и монофункциональную полисиаловую кислоту (окисленная восстановленная окисленная коломиновая кислота (CAORO)), полученную в примере 1, использовали для получения rh-инсулин конъюгатов.
Получение конъюгатов инсулин-коломиновая кислота
Инсулин растворяли в минимальном объеме 15 мМ HCl с последующим разбавлением 0,15 М PBS (рН 7,4) и ковалентно связывали с различными коломиновыми кислотами (СА, САО и монофункциональной CAORO). Коломиновую кислоту (22,7 кДа) вместе с инсулином (2 мг) в молярных соотношениях СА:инсулин (25:1) вводили в реакцию в течении 48 часов в 0,15 М PBS (рН 7,4; 2 мл), содержащем цианоборгидрид натрия (4 мг/мл) в герметично закрытых сосудах при перемешивании магнитной мешалкой при 35±2°С в инкубаторе. Смеси затем подвергали осаждению сульфатом аммония ((NH4)2SO4) путем медленного добавления соли при непрерывном перемешивании до достижении 70% мас./об. насыщения. Образцы перемешивали в течение 1 часа при 4°С, затем центрифугировали (5000·g) в течение 15 минут и гранулы суспендировали в насыщенном растворе (NH4)2SO4 и вновь центрифугировали в течение 15 минут (5000·g). Полученные осадки вновь растворяли в 1 мл 0,15 М натрий фосфатного буфера с добавлением 0,9% NaCl (рН 7,4; PBS) и подвергали экстенсивному диализу (24 часа) при 4°С против такого же PBS. Диализаты затем анализировали на содержание сиаловой кислоты и белка и выход конъюгации выражали в терминах молярного соотношения СА:инсулин (как в примере 1). Контроли включали конъюгацию нативного белка в присутствие неокисленной СА или в отсутствие СА при описанных условиях. Встряхивание было сведено к минимуму, чтобы избежать сопутствующей денатурации белка. Полисаилилированный инсулин дополнительно характеризовали ионообменной хроматографией и SDS-PAGE. Результаты выражали в терминах молярного соотношения СА:инсулин в полученных конъюгатах (таблица 7).
Синтез соединений инсулин (белок) коломиновой кислоты
Из таблицы 7 очевидно, что если использовали природную неокисленную СА (в присутствии цианоборгидрида), наблюдали значительную конъюгацию, но на низком уровне (результатом которой являлось молярное соотношение СА:инсулин 0,07:1) за счет реакции с полуацетальной группой СА на ее восстанавливающем конце.
Образование конъюгатов CA-инсулин дополнительно подтверждали совместным осаждением двух фрагментов при добавлении (NH4)2SO4 (СА сама по себе не осаждается в присутствие этой соли). Доказательство конъюгации также получали ионообменной хроматографией (IEC) и электрофорезом в полиакриламидном геле (SDS-PAGE).
Пример 7 - исследования in vivo
Инсулин и полисиалилированный инсулин, полученный в примере 6, исследовали на их способность понижать уровень глюкозы в крови у нормальных Т/О беспородных мышей (вес тела 22-24 грамм). Животных разделяли на группы по пять, вводили подкожно (s.c.) инсулин (0,3 единицы каждой мыши в 0,9% растворе хлорида натрия или тот же эквивалент белка в виде полисиалилированного инсулина) и измеряли уровень глюкозы в образцах крови через временные интервалы при использования набора для определения глюкозы (Accu-Chek Advantage, Roche, UK).
Результаты
Фармакологическую активность полисиалилированных инсулиновых компонентов сравнивали с активностью немодифицированного инсулина у нормальных мышей, которым вводили препарат подкожно и отбирали кровь через временные интервалы. Уровни глюкозы в крови мышей для 3 инсулинов показаны на фиг.17. Точки на графике показывают среднее для 5 образцов и планки погрешностей представляют s.e.m. величины. Результаты на фиг.17 явно показывают, что полисиалилированные инсулины (полученный исходным способом (при использовании САО) и новым способом двойного окисления (при использовании монофункциональной CAORO)) вызывают более продолжительное понижение уровней глюкозы в крови. Таким образом, тогда как уровни глюкозы, достигнувшие самого низкого значения через 0,75 часа, возвращаются к нормальному уровню через два часа после введения немодифицированного инсулина, уровни глюкозы у мышей, которым вводили полисиалилированный белок, хотя также достигали самого низкого значения через 0,75 часа, возвращались к нормальным величинам через 6 часов. Эти результаты показывают, что САО-инсулин и CAORO-инсулин приводят к получению заметного и значительно более длительного времени удержания в кровотоке по сравнению с немодифицированным инсулином, приводя к увеличению площади под кривой, по сравнению с нативным инсулином.
Литература
1. Bendele, A., Seely, J., Richey, C, Sennello, G., Shopp, G., Renal tubular vacuolation in animals treated with polyethylene-glycol conjugated proteins, Toxicological sciences, 42 (1998) 152-157.
2. Beranova, M., Wasserbauer, R., Vancurova, D., Stifter, M., Ocenaskova, J., Mora, M., Biomaterials, 11 (2000) 521-524.
3. Brocchini, S., Polymers in medicine: a game of chess. Drug Discovery Today, 8, (2003)111-112.
4. Cheng T, Wu, M., Wu,P., Chern, J, Roffer, SR., Accelerated clearance of polyethylene glycol modified proteins by anti-polyethylene glycol IgM. Bioconjugate chemistry, 10 (1999) 520-528.
5. Cho, J.W. and Troy, F. A., Polysialic acid engineering: Synthesis of polysialylated neoglycosphingolipid by using the polytransferase from neuroinvasive E.coliK1, Proceedings of National Academic Sciences, USA, 91 (1994) 11427-11431.
6. Convers, C. D., Lejeune, L., Shum, K., Gilbert, C., Shorr, R.G.L, Physiological effect of polyethylene glycol conjugation on stroma-free bovine hemoglobin in the conscious dog after partial exchange transfusion, Artificial organ, 21(1997) 369-378.
7. Dyer, J.R., Use of periodate oxidation in biochemical analysis, Methods of Biochemical Analysis, 3 (1956) 111-152.
8. Fernandes, A.I., Gregoriadis, G., Polysialylated asparaginase: preparation, activity and pharmacokinetics, Biochimica et Biophysica Acta, 1341 (1997)26-34.
9. Fernandes, A.I., Gregoriadis, G., Synthesis, characterization and properties of polysialylated catalase, Biochimica et Biophysica Acta, 1293 (1996) 92-96.
10. Fernandes, A.I., Gregoriadis, G., The effect of polysialylation on the immunogenicity and antigenicity of asparaginase: implications in its pharmacokinetics, International Journal of Pharmaceutics, 217 (2001) 215-224.
11. Fleury, P., Lange, J., Sur I'oxydation des acides alcools et des sucres par I'acid periodique, Comptes Rendus Academic Sciences, 195 (1932) 1395-1397.
12. Gregoriadis, G., Drug and vaccine delivery systems, in: PharmaTech, World Markets Research Centre Limited, London (2001) 172-176.
13. Gregoriadis, G., Fernandes, A., McCormack, B., Mital, M., Zhang, X, Polysialic acids: Potential for long circulating drug, protein, liposome and other microparticle constructs, in Gregoriadis, G and McCormack, B (Eds), Targeting of Drugs, Stealth Therapeutic Systems, Plenum Press, New York (1998) 193-205.
14. Gregoriadis, G., Fernandes, A., Mital, M., McCormack, B., Polysialic acids: potential in improving the stability and pharmacokinetics of proteins and other therapeutics, Cellular and Molecular Life Sciences, 57 (2000) 1964-1969.
15. Gregoriadis, G., McCormack, B., Wang, Z., Lifely, R., Polysialic acids: potential in drug delivery, FEBS Letters, 315 (1993) 271-276.
16. Hreczuk-Hirst, D., Jain, S., Genkin, D., Laing, P., Gregoriadis, G., Preparation and properties of polysialylated interferon-α-2b, AAPS Annual Meeting, 2002, Toronto, Canada, M1056.
17. Hunter, A. C, Moghimi, S. M., Therapeutic synthetic polymers: a game of Russian Roulette. Drug Discovery Today, 7 (2002) 998-1001.
18. Jain, S., Hirst, D. H., McCormack, B., Mital, M., Epenetos, A., Laing, P., Gregoriadis, G., Polysialylated insulin: synthesis, characterization and biological activity in vivo, Biochemica et. Biophysica Acta, 1622 (2003) 42-49.
19. Jain, S., Hirst, D.H., Laing, P., Gregoriadis, G., Polysialylation: The natural way to improve the stability and pharmacokinetics of protein and peptide drugs, Drug Delivery Systems and Sciences, 4(2) (2004) 3-9.
20. Jennings, H. J., Lugowski, C, Immunogenicity of groups A, B, and C meningococal polysaccharide tetanus toxoid conjugates, Journal of Immunology, 127(1981)1011-1018.
21. Lifely, R., Gilhert, A.S., Moreno, C.C., Sialic acid polysaccharide antigen of Neisseria meningitidis and Escherichia coli: esterification between adjacent residues, Carbohydrate Research, 94 (1981) 193-203.
22. Mital, M., Polysialic acids: a role for optimization of peptide and protein therapeutics, Ph.D. Thesis, University of London, 2004.
23. Muflenhoff, M., Ectehardt, M., Gerardy-Schohn, R., Polysialic acid: three-dimensional structure, biosynthesis and function, Current opinions in Structural Biology, 8(1998)558-564.
24. Park, J.T., Johnson, M.J., A submicrodetermination of glucose, Journal of Biological Chemistry, 181 (1949) 149-151.
25. Roth, J., Rutishauser, U., Troy, F.A. (Eds.), Polysialic acid: from microbes to man, Birkhauser Verlag, Basel, Advances in Life Sciences, 1993.
26. Rutishauser, U., Polysialic acid as regulator of cell interactions in: R.U. Morgoles and R.K. Margalis (eds.), Neurobiology of Glycoconjugates, pp 367-382, Plenum Press, New York, 1989.
27. Shriner, R. L, Fuson, R.D.C., Curtin, D.Y., Morill, T.C., The Systematic Identification of Organic Compounds, 6th ed., Wiley, New York, 1980.
28. Svennerholm, L, Quantitative estimation of sialic acid II: A colorimetric resorcinol-hydrochloric acid method, Biochimca et Biophysica Acta, 24 (1957)604-611.
29. Troy, F. A. Polysialylation of neural cell adhesion molecules, Trends in Glycoscience and Glycotechnology, 2 (1990) 430-449.
39. Troy, F.A., Polysialylation: From bacteria to brain, Glycobiology, 2 (1992) 1-23.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПОЛИСИАЛОВОЙ КИСЛОТЫ | 2004 |
|
RU2327703C2 |
| ГЛИКОПОЛИСИАЛИРОВАНИЕ БЕЛКОВ, НЕ ЯВЛЯЮЩИХСЯ БЕЛКАМИ СВЕРТЫВАНИЯ КРОВИ | 2014 |
|
RU2662807C2 |
| ГЛИКОПОЛИСИАЛИРОВАНИЕ БЕЛКОВ, НЕ ЯВЛЯЮЩИХСЯ БЕЛКАМИ СВЕРТЫВАНИЯ КРОВИ | 2010 |
|
RU2533619C2 |
| N-КОНЦЕВОЕ ПОЛИСИАЛИЛИРОВАНИЕ | 2007 |
|
RU2432175C2 |
| СПОСОБ СНИЖЕНИЯ СОДЕРЖАНИЯ ЭНДОТОКСИНА В ОБРАЗЦЕ, СОДЕРЖАЩЕМ ПОЛИСИАЛОВУЮ КИСЛОТУ И ЭНДОТОКСИН | 2008 |
|
RU2473567C2 |
| КОНЪЮГАТЫ БЕЛКОВ СВЕРТЫВАНИЯ КРОВИ | 2010 |
|
RU2595442C2 |
| КОНЪЮГАТЫ БЕЛКОВ СВЕРТЫВАНИЯ КРОВИ | 2010 |
|
RU2744370C2 |
| ЛЕКАРСТВЕННЫЙ ПРЕПАРАТ И СПОСОБ УЛУЧШЕНИЯ РЕОЛОГИЧЕСКИХ СВОЙСТВ МОКРОТЫ И ИНГАЛЯЦИОННОЕ ПРИМЕНЕНИЕ ТАКОГО ПРЕПАРАТА | 2009 |
|
RU2522846C2 |
| ОКСИНТОМОДУЛИН ЧЕЛОВЕКА, ЕГО ПРИМЕНЕНИЕ, ЛЕКАРСТВЕННЫЙ ПРЕПАРАТ НА ЕГО ОСНОВЕ И СПОСОБ ПРИМЕНЕНИЯ ПРЕПАРАТА ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ГИПЕРГЛИКЕМИИ | 2009 |
|
RU2524204C2 |
| Способ получения гибридного белка, состоящего из рекомбинантного белка аналога интерферона гамма, конъюгированного с олигосахаридом | 2016 |
|
RU2656140C2 |
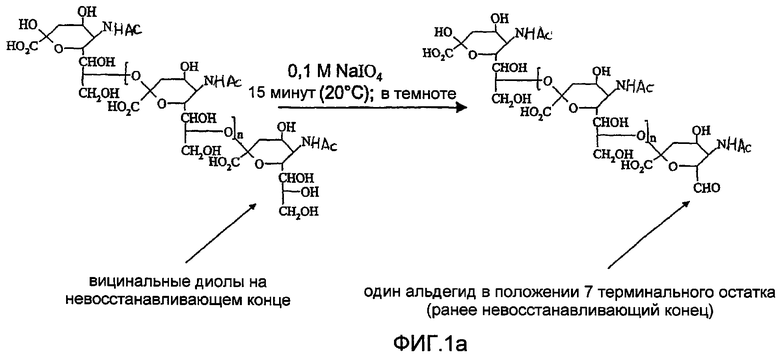
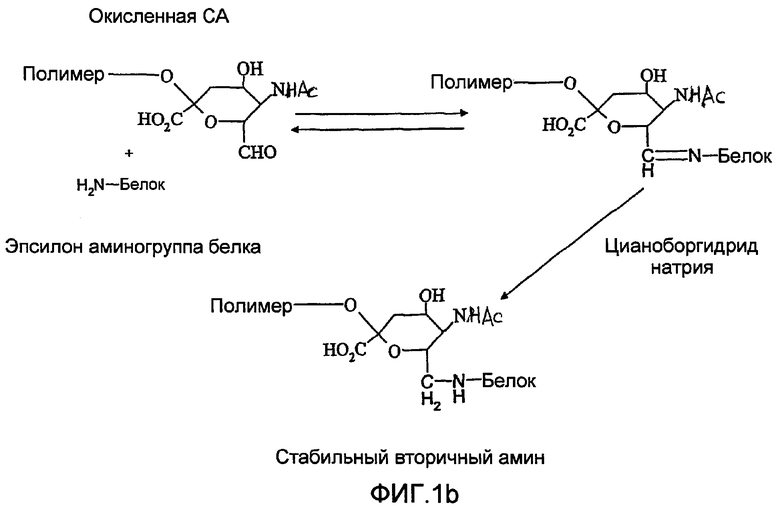
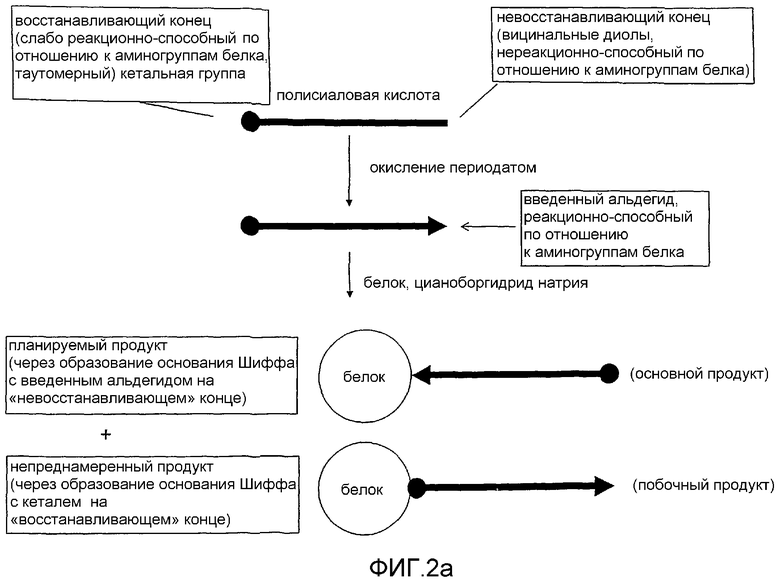
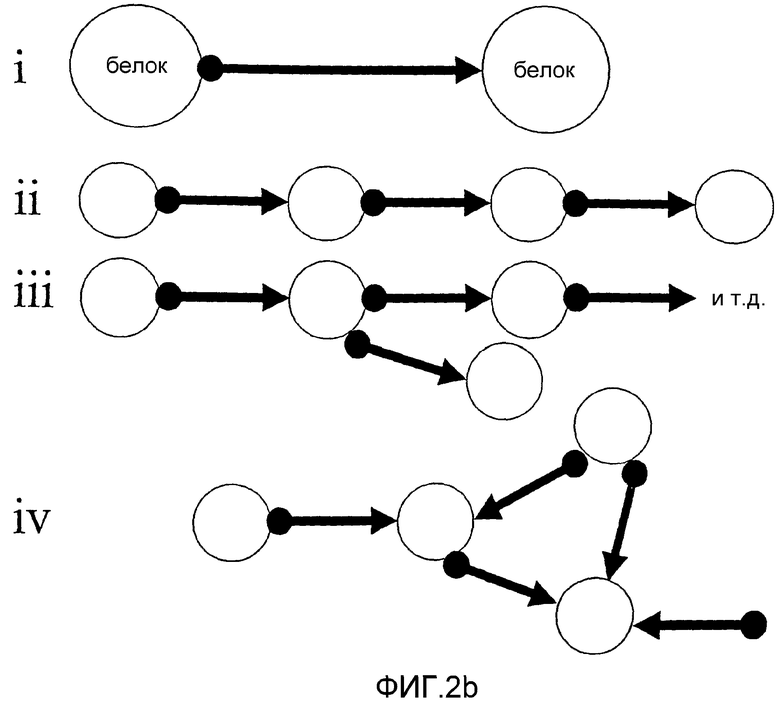
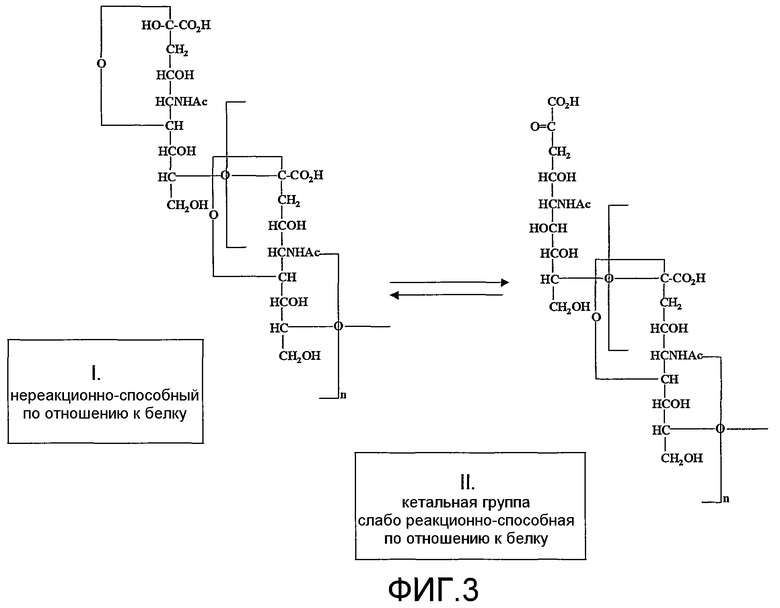
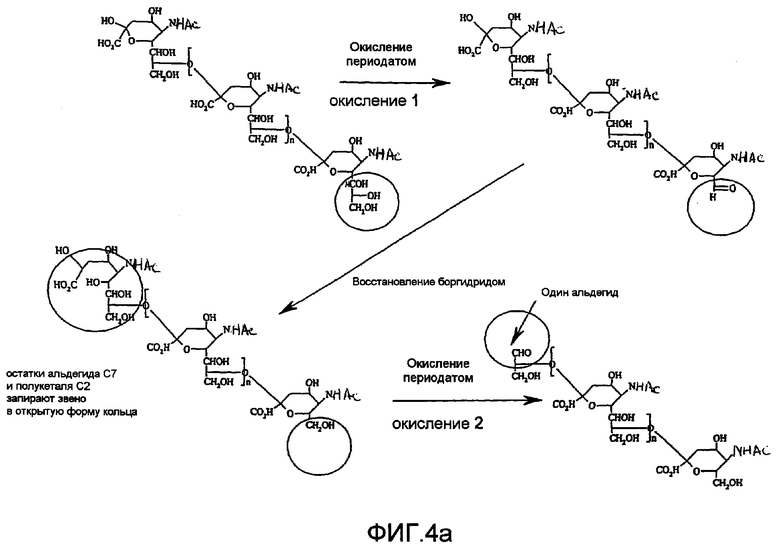
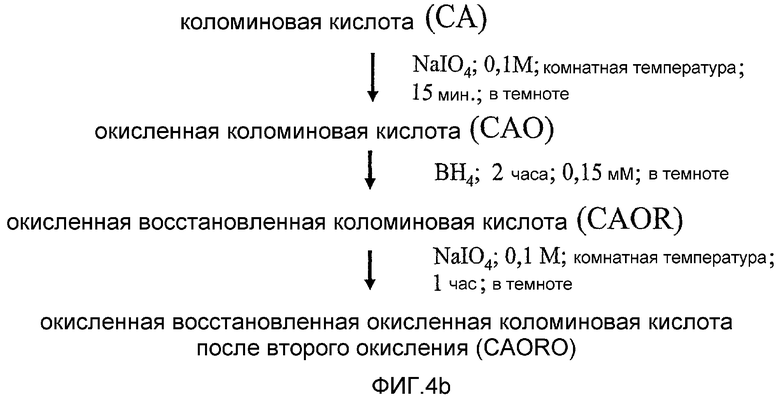

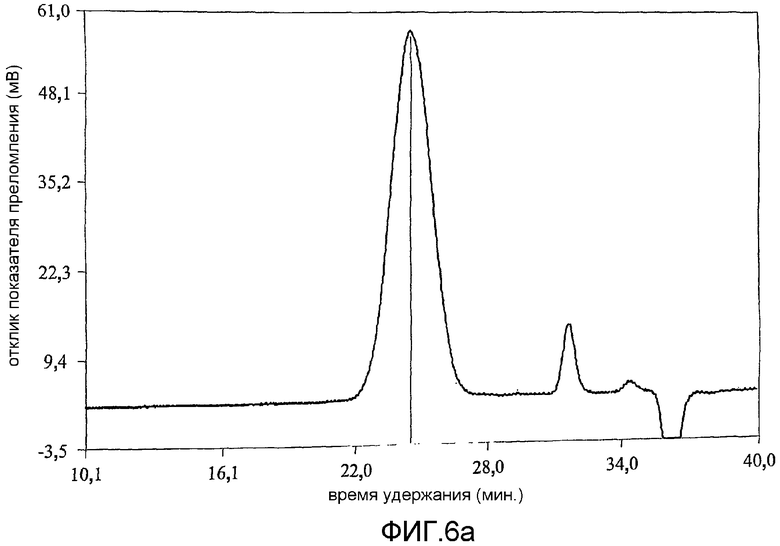
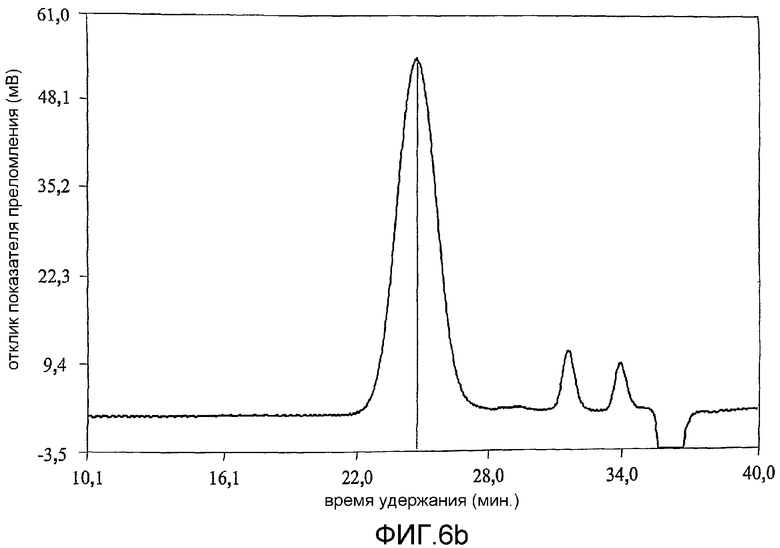
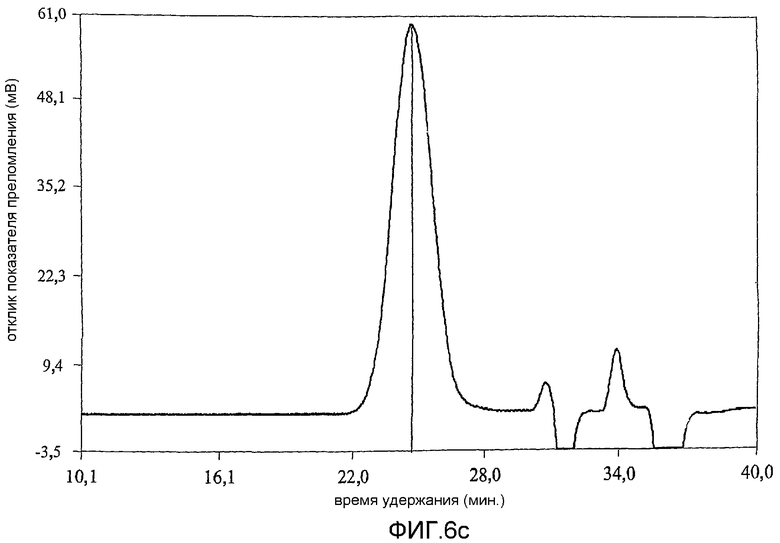
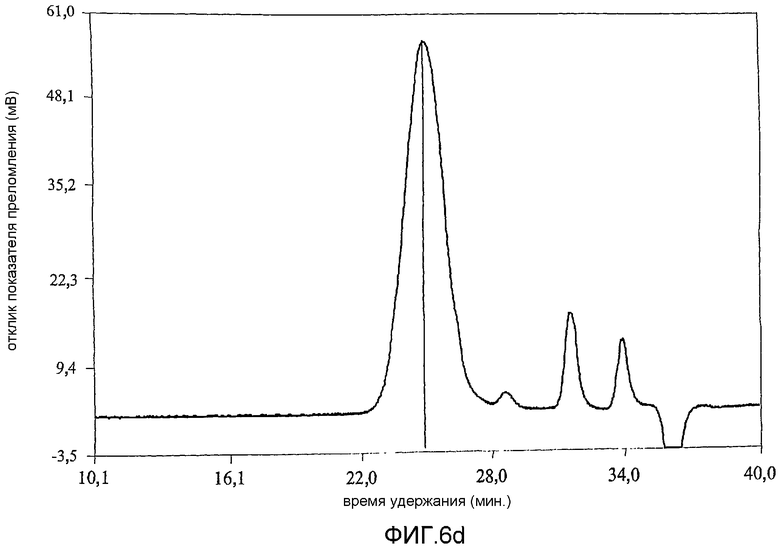
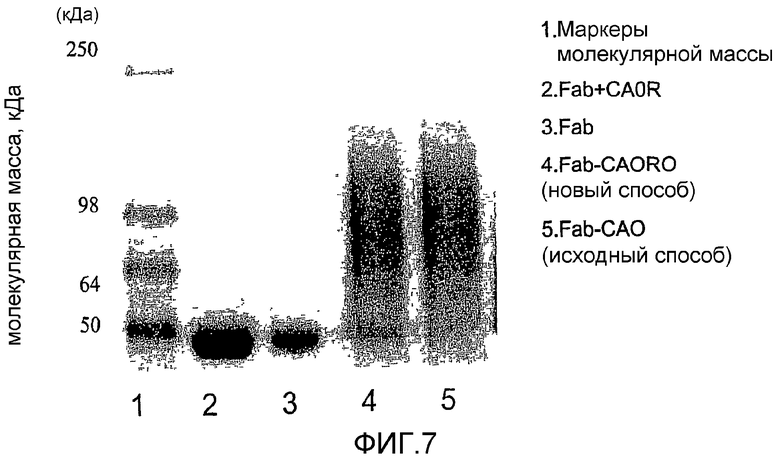
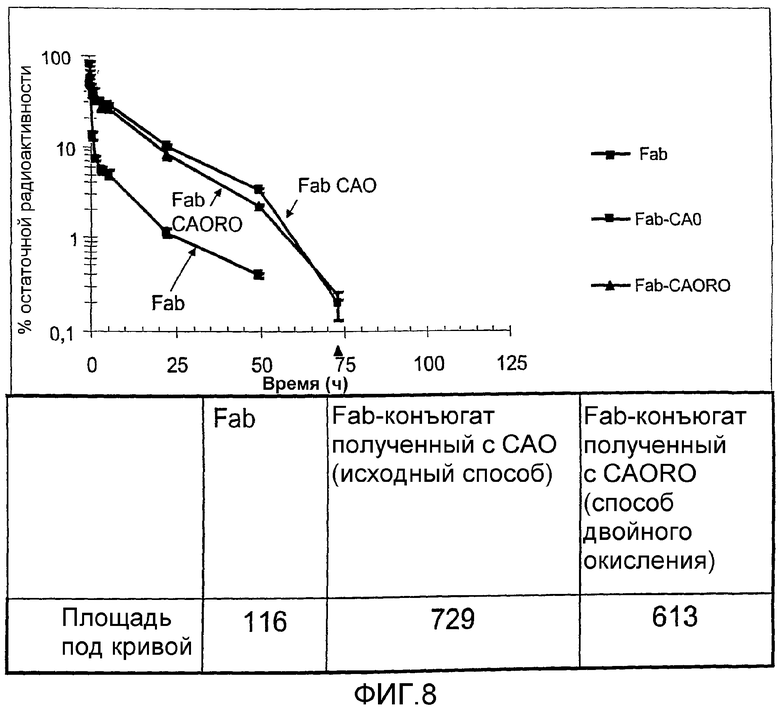
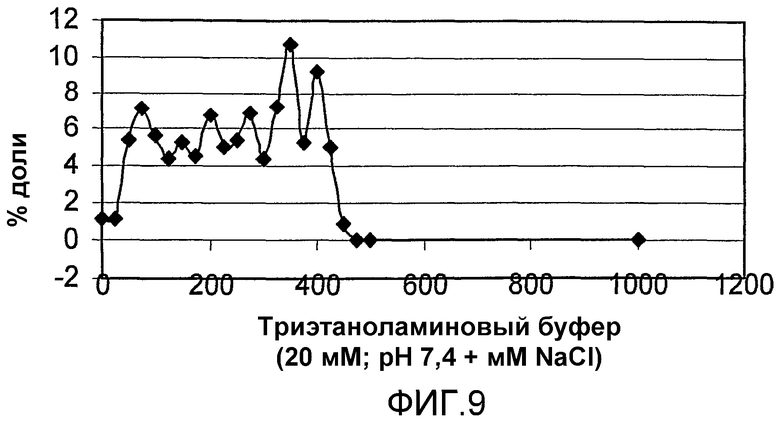
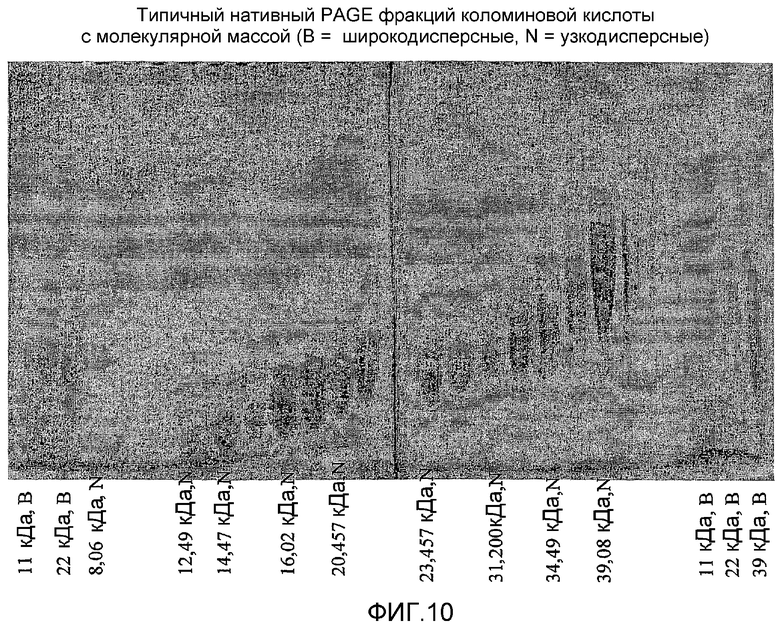
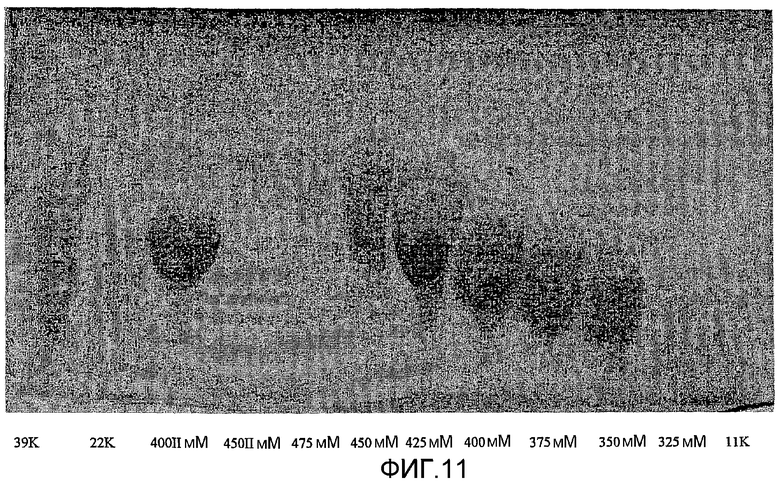


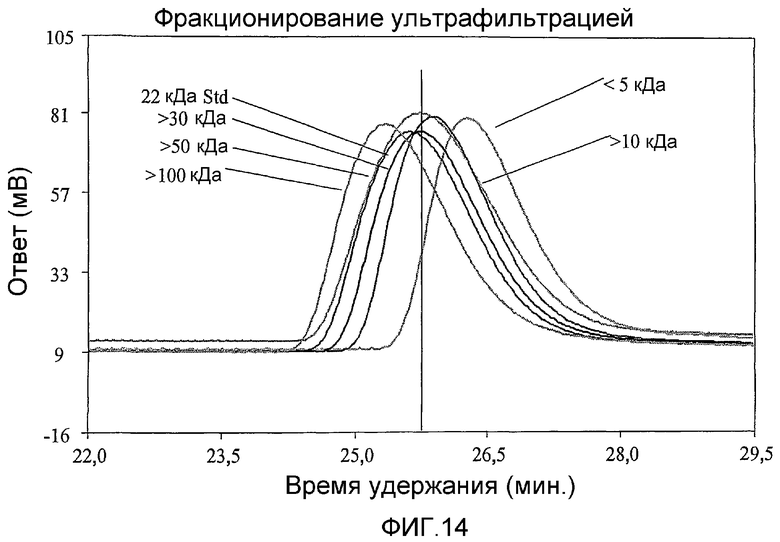
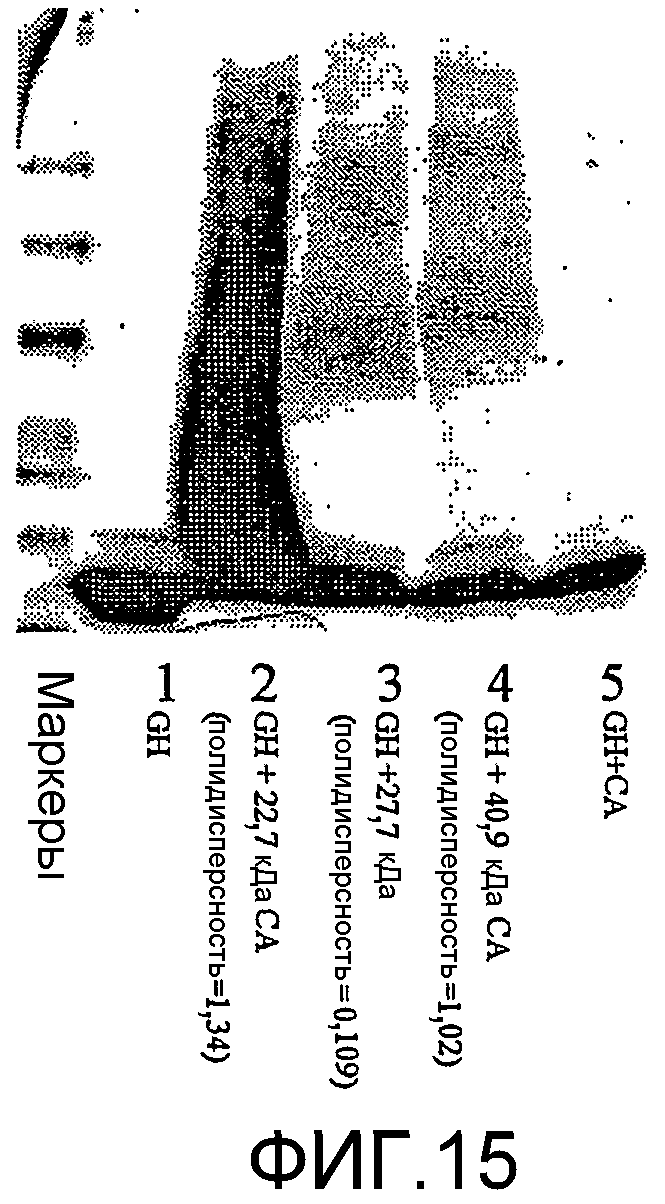
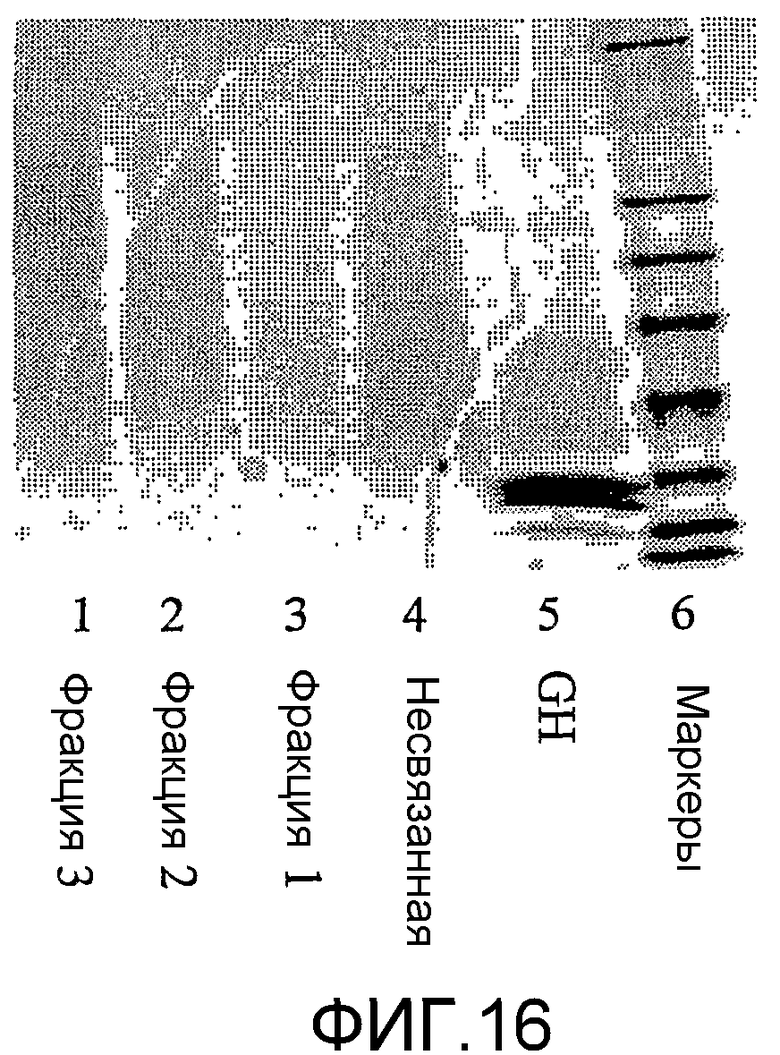
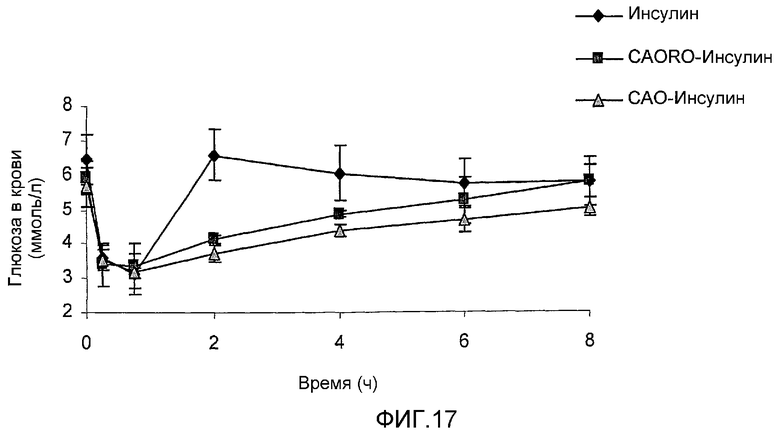
Изобретение относится к альдегидным производным и конъюгатам ди-, олиго- или полисахарида, имеющим общую формулу (I),к способам их получения и фармацевтической композиции, обладающей способностью длительное время удерживаться в кровотоке, на их основе.
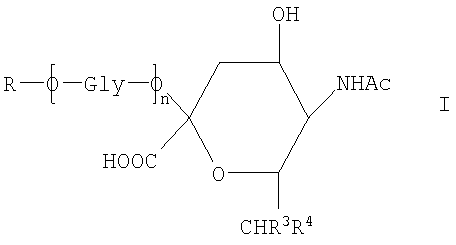
где R представляет собой -СН(СНО)СН2ОН, -СН2СНО, -CH(СН2NHR1)СН2ОН, -CH(CH2NHNHR1)CH2OH, -CH(CH=NNHR1)CH2OH, -CH2CH2NHR1, -CH2CH=N-NHR1, -CH2CH2NHNHR1; R1 представляет собой полипептид или белок; GlyO представляет собой звено сиаловой кислоты; R3 представляет собой Н; R4 представляет собой ОН; п равно 2 и более. 5 н. и 15 з.п. ф-лы, 7 табл., 22 ил.
a) предварительное селективное окисление вицинальной диольной группы до альдегида;
b) восстановление до восстановленного раскрытого кольца восстанавливающего терминального звена сиаловой кислоты, посредством чего получают вицинальную диольную группу и в которой альдегид, образованный в стадии а) также восстанавливают до гидроксильной группы, которая не является частью вицинальной диольной группы; и
c) селективное окисление вицинальной диольной группы, полученной в стадии b) с получением альдегидной группы.
а) предварительное селективное окисление вицинальной диольной группы до альдегида;
b) восстановление до восстановленного раскрытого кольца восстанавливающего терминального звена сиаловой кислоты, посредством чего получают вицинальную диольную группу и в которой альдегид, образованный в стадии а) также восстанавливают до гидроксильной группы, которая не является частью вицинальной диольной группы; и
с) селективное окисление вицинальной диольной группы, полученной в стадии b) с получением альдегидной группы и затем вводят в реакцию с субстратом, имеющим первичную аминогруппу или группу гидразида, выбранным из белка или пептида, с последующим, при необходимости, восстановлением полученного продукта.

где R представляет собой -СН(СНО)СН2OH, -СН2СНО, GlyO представляет собой звено сиаловой кислоты; R3 представляет собой Н; R4 представляет собой ОН; п равно 2 и более.
где R представляет собой -CH(CH2NHR1)CH2OH, -CH(CH2NHNHR1)CH2OH, -CH(CH=NNHR1)CH2OH, -CH2CH2NHR1, -CH2CH=N-NHR1, -CH2CH2NHNHR1, где R1 представляет собой полипептид или белок; GlyO представляет собой звено сиаловой кислоты; R3 представляет собой Н; R4 представляет собой ОН; п равно 2 и более.
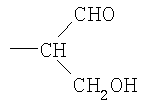 .
.
 .
.
| US 4356170 А, 26.10.1982 | |||
| US 5097020 А, 17.03.1992 | |||
| Устройство для определения опорных усилий стоящего человека | 1973 |
|
SU454898A1 |
| ПОЛИСАХАРИД NEISSERIA MENINGITIDIS С МОДИФИЦИРОВАННОЙ ГРУППОЙ B, КОНЪЮГИРОВАННЫЙ АНТИГЕН, ВАКЦИНА ПРОТИВ МЕНИНГИТА ГРУППЫ B И СПОСОБ ИНДУКЦИИ ОТВЕТА АНТИТЕЛ К МЕНИНГИТУ ГРУППЫ B | 1990 |
|
RU2105568C1 |
| SEN G | |||
| ЕТ AL | |||
| The specificity of the binding site of Achatinin, a sialic acid-binding lectin from Achatina fulica | |||
| Carbohydrate Research, 1995, vol.268, №1, pp.115-125. | |||

