Настоящее изобретение относится к новому применению агониста рецептора сфингозин-1-фосфата (С1Ф), особенно в лечении рака.
Агонисты С1Ф-рецептора являются средствами, ускоряющими хоминг лимфоцитов (ХЛ), которые вызывают лимфопению, являющуюся результатом перераспределения, предпочтительно обратимого, лимфоцитов из кровообращения во вторичную лимфатическую ткань, не оказывая при этом генерализованного иммунодепрессивного действия. Секвестируются “необученные” клетки; стимулируется миграция Т-клеток CD4 и CD8 и В-клеток из крови в лимфоузлы (ЛУ) и пейеровы бляшки (ПБ), что приводит, например, к ингибированию инфильтрации клеток в трансплантированные органы.
Агонисты С1Ф-рецептора являются типичными аналогами сфингозина, такими как 2-замещенные производных 2-аминопропан-1,3-диола или 2-аминопропанола, например, соединение, включающее группу формулы Х
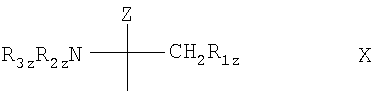
где Z означает Н; C1-С6алкил; С2-С6алкенил; С2-С6алкинил; фенил; фенил, замещенный ОН-группой; C1-С6алкил с 1-3 заместителями, выбранными из группы, состоящей из галогена, С3-С8циклоалкила, фенила и ОН-замещенного фенила; или CH2-R4z, где R4z означает ОН, ацилоксигруппу или остаток формулы (а)
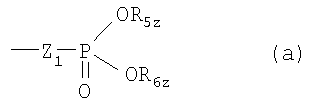
где Z1 означает простую связь или О, предпочтительно О; каждый из R5z и R6z, независимо, означает Н или С1-С4алкил, необязательно замещенный 1, 2 или 3 атомами галогена;
R1z означает ОН, ацилоксигруппу или остаток формулы (а); и каждый из R2z и R3z, независимо, означает Н, С1-С4алкил или ацил.
Группа формулы Х является функциональной группой, присоединенной в качестве концевой группы к части молекулы, которая может быть гидрофильной или липофильной и включать один или несколько алифатических, алициклических, ароматических и/или гетероциклических остатков; при этом молекула, в которой по крайней мере одна из групп Z или R1z является или включает в себя остаток формулы (а), действуя как агонист одного или нескольких рецепторов сфингозин-1-фосфата.
Агонисты С1Ф-рецептора являются соединениями, которые сигнализируют как агонисты у одного или нескольких рецепторов сфингозин-1-фосфата, например, с С1Ф1 по С1Ф8. Связывание агониста с С1Ф-рецептором может, например, привести к диссоциации внутриклеточных гетеротримерных G-белков на Gα-GTP и Gβγ-GTP и/или к повышенному фосфорилированию агонистсвязанного рецептора и активации нижерасположенных сигнальных метаболических путей/киназ. Способность к связыванию агонистов рецептора С1Ф можно измерить, как описано в представленном ниже параграфе И.
Подходящими агонистами С1Ф-рецептора являются, например:
- соединения, описанные в ЕР 627406 A1, например, соединение формулы I

где R1 означает прямую или разветвленную (C12-C22) углеродную цепь - которая может иметь в цепи связь или гетероатом, выбранные из двойной связи, тройной связи, О, S, NR6, где R6 означает Н, алкил, аралкил, ацил или алкоксикарбонил, и карбонил, и/или
- которая может иметь в качестве заместителя алкоксигруппу, алкенилоксигруппу, алкинилоксигруппу, аралкилоксигруппу, ацил, алкиламиногруппу, алкилтиогруппу, ациламиногруппу, алкоксикарбонил, алкоксикарбониламиногруппу, ацилоксигруппу, алкилкарбамоил, нитрогруппу, галоген, аминогруппу, гидроксииминогруппу, гидроксигруппу или -карбоксигруппу;
или R1 означает
- фенилалкил, в котором алкил означает прямую или разветвленную (С6-С20) углеродную цепь; или
- фенилалкил, в котором алкил означает прямую или разветвленную (С1-С30) углеродную цепь, где указанный фенилалкил имеет в качестве заместителей
- прямую или разветвленную (С6-С20) углеродную цепь, необязательно замещенную галогеном,
- прямую или разветвленную (С6-С20) алкокси цепь, необязательно замещенную галогеном,
- прямую или разветвленную (С6-С20) алкенилоксигруппу,
- фенилалкоксигруппу, галофенилалкоксигруппу, фенилалкоксиалкил, феноксиалкоксигруппу или феноксиалкил,
- циклоалкилалкил, замещенный С6-С20алкилом,
- гетероарилалкил, замещенный C6-C20алкилом,
- гетероциклический С6-С20алкил или
- гетероциклический алкил, замещенный С2-С20алкилом,
и где
алкильная часть может иметь
- в углеродной цепи связь или гетероатом, выбранные из двойной связи, тройной связи, О, S, сульфинила, сульфонила, или NR6, где R6 соответствует указанному выше, и
- в качестве заместителя алкоксигруппу, алкенилоксигруппу, алкинилоксигруппу, аралкилоксигруппу, ацил, алкиламиногруппу, алкилтиогруппу, ациламиногруппу, алкоксикарбонил, алкоксикарбониламиногруппу, ацилоксигруппу, алкилкарбамоил, нитрогруппу, галоген, аминогруппу, гидроксигруппу или карбоксигруппу, и каждый из R2, R3, R4 и R5 независимо означает Н, C1-C4алкил или ацил; или фармацевтически приемлемая соль этого соединения;
- соединения, описанные в ЕР 1002792 А1, например, соединение формулы II
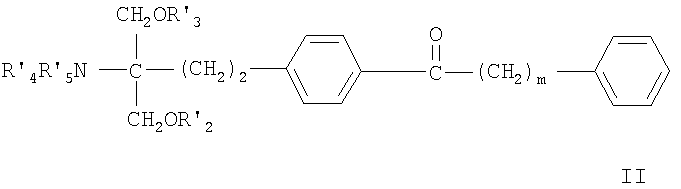
где m означает от 1 до 9 и каждый из R'2, R'3, R'4 и R'5 независимо означает Н, алкил или ацил, или фармацевтически приемлемую соль этих соединений;
- соединения, описанные в ЕР 0778263 А1, например, соединение формулы III

где W означает Н; C1-C6алкил, С2-С6алкенил или С2-С6алкинил; незамещенный или ОН-замещенный фенил; R''4O(CH2)n; или C1-C6алкил, имеющий от 1 до 3-х заместителей, выбранных из группы, состоящей из галогена, С3-С8циклоалкила, фенила и фенила, замещенного ОН;
Х означает Н или незамещенный или замещенный алкил с прямой цепью, имеющий число атомов углерода р, или незамещенную или замещенную алкоксигруппу с прямой цепью, имеющую число (р-1) атомов углерода, например, с 1-3 заместителями, выбранными из группы, содержащей C1-C6алкил, ОН, C1-С6алкоксигруппу, ацилоксигруппу, аминогруппу, C1-C6алкиламиногруппу, ациламиногруппу, оксогруппу, галоС1-С6алкил, галоген, незамещенный фенил и фенил с 1-3 заместителями, выбранными из группы, состоящей из С1-С6алкила, ОН, C1-C6алкоксигруппы, ацила, ацилоксигруппы, аминогруппы, C1-C6алкиламиногруппы, ациламиногруппы, галоC1-С6алкила и галогена; Y означает Н, C1-C6алкил, ОН,
C1-C6алкоксигруппу, ацил, ацилоксигруппу, аминогруппу, C1-C6алкиламиногруппу, ациламиногруппу, галоС1-С6алкил или галоген; Z2 означает простую связь или алкилен с прямой цепью, имеющий число атомов углерода q;
Каждое из р и q независимо означают целое число от 1 до 20, при условии, что 6≤p+q≤23, m' означает 1, 2 или 3, n означает 2 или 3, каждый из R''1, R''2, R''3 и R''4 независимо означает Н, C1-C4алкил или ацил;
или фармацевтически приемлемая соль этих соединений;
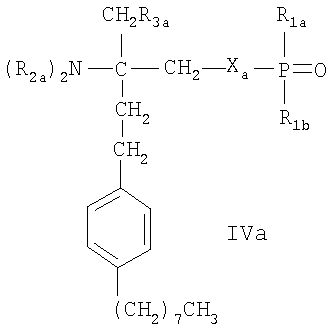
- соединения, описанные в WO 02/18395, например, соединения формулы IVa или IVb
 или
или 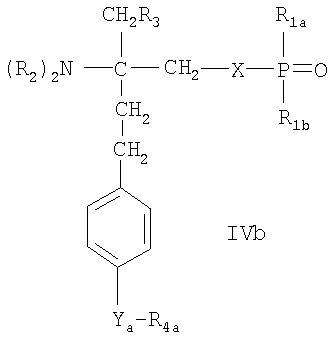
где Ха означает О, S, NR1s или группу -(СН2)na-, которая является незамещенной или имеет в качестве заместителей от 1 до 4 атомов галогена; nа означает 1 или 2, R1s означает Н или (C1-C4)алкил, незамещенный или замещенный галогеном;
R1a означает Н, ОН, (C1-C4)алкил или O(C1-C4)алкил, где алкил является незамещенным или имеет в качестве заместителей от 1 до 3 атомов галогена; R1b означает Н, ОН или
(С1-С4)алкил, который является незамещенным или замещен галогеном; каждый из R2a, независимо, выбран из Н или (C1-C4)алкила, незамещенного или замещенного галогеном; R3a означает Н, ОН, галоген или O(C1-C4)алкил, где алкил является незамещенным или замещенным галогеном; и R3b означает Н, ОН, галоген,
(C1-C4)алкил, где алкил является незамещенным или замещенным гидроксигруппой, или O(C1-C4)алкил, где алкил является незамещенным или замещен галогеном; Ya означает -СН2-, -С(O)-, -СН(ОН)-, -C(=NOH)-, О или S, и R4a означает (С4-С14)алкил или (С4-С14)алкенил; или фармацевтически приемлемые соли этих соединений;
- соединения, описанные в WO 02/076995, например, соединение формулы V
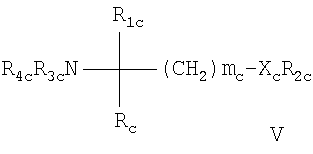
где mc означает 1, 2 или 3; Xc означает О или прямую связь; R1c означает Н;
C1-C6алкил, необязательно замещенный ОН, ацилом, галогеном, С3-С10циклоалкилом, фенилом или гидроксифениленом; С2-С6алкенил; C2-C6алкинил; или фенил, необязательно замещенный ОН; R2c означает
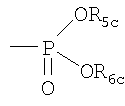
где R5c означает H или C1-C4алкил, необязательно замещенный 1, 2 или 3 атомами галогена, и R6с означает Н или C1-C4алкил, необязательно замещенный галогеном;
каждый из R3с и R4c независимо означает Н, C1-C4алкил, необязательно замещенный галогеном, или ацил, и Rc означает С13-С20алкил, который может необязательно иметь в цепи атом кислорода и который может необязательно быть замещенным нитрогруппой, галогеном, аминогруппой, гидроксигруппой или карбоксигруппой; или остаток формулы (а)
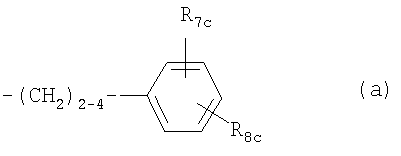
где R7c означает Н, C1-C4алкил или C1-C4алкоксигруппу, и R8c означает замещенный С1-С20алканоил, фенилС1-С14алкил, где C1-C14алкил необязательно замещен галогеном или ОН, циклоалкилС1-С14алкоксигруппу или фенилС1-14алкоксигруппу, где циклоалкильное или фенильное кольцо необязательно замещено галогеном, C1-C4алкилом и/или C1-C4алкоксигруппой, фенил-C1-C14алкоксиС1-С14алкил, феноксиС1-С14алкоксигруппу или феноксиС1-С4алкил, Rc также означает остаток формулы (а), где R8c означает C1-С14алкоксигруппу, когда R1c означает C1-C4алкил, С2-С6алкенил или C2-C6алкинил;
или соединение формулы VI
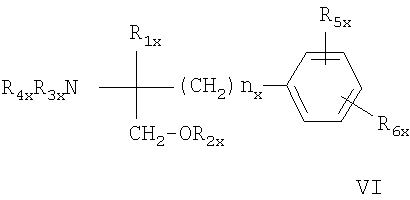
где nx означает 2, 3 или 4; R1x означает Н; C1-C6алкил, необязательно замещенный ОН, ацилом, галогеном, циклоалкилом, фенилом или гидроксифениленом; С2-С6алкенил; С2-С6алкинил; или фенил, необязательно замещенный ОН; R2х означает Н; C1-C4алкил или ацил, каждый из R3х и R4x, независимо означает Н, C1-C4алкил необязательно замещенный галогеном или ацилом, R5x означает Н, С1-С4алкил или С1-С4алкоксигруппу, и R6x означает C1-С20алканоил, замещенный циклоалкилом;
циклоалкилС1-С14алкоксигруппу, где циклоалкильное кольцо необязательно замещено галогеном, C1-C4алкилом и/или С1-С4алкоксигруппой; фенилС1-С14алкоксигруппу, где фенильное кольцо необязательно замещено галогеном, С1-С4алкилом и/или С1-С4алкоксигруппой, R6x также означает С4-С14алкоксигруппу, когда R6x означает С2-С4алкил, замещенный ОН, или пентилоксигруппу или гексилоксигруппу, когда R1x означает С1-С4алкил, при условии, что R6x не означает фенилбутиленоксигруппу, когда или R5x означает Н, или R1x означает метил; или фармацевтически приемлемая соль этих соединений;
- соединения, описанные в WO 02/06268 AI, например, соединение формулы VII
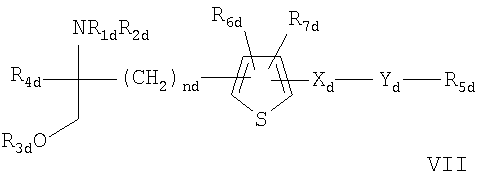
где каждый из R1d и R2d независимо означает Н или аминозащитную группу;
R3d означает водород или гидроксизащитную группу; R4d означает низший алкил;
nd означает целое число от 1 до 6; Хd означает этилен, винилен, этинилен, группу, имеющую формулу -D-СН2- (где D означает карбонил, -СН(ОН)-, О, S или N), арил или арил, содержащий до трех заместителей, выбранных из группы а, указанной ниже; Yd означает простую связь, C1-С10алкилен, C1-С10алкилен, содержащий до трех заместителей, выбранных из групп а и б, C1-С10алкилен, содержащий атомы О и S в середине или в конце углеродной цепи, или C1-С10алкилен, содержащий атомы О и S в середине или в конце углеродной цепи, которая содержит до трех заместителей, выбранных из групп а и б;
R5d означает водород, циклоалкил, арил, гетероцикл, циклоалкил, имеющий до трех заместителей, выбранных из групп а и б, арил, содержащий до трех заместителей, выбранных из групп а и б, или гетероцикл, содержащий до трех заместителей, выбранных из групп а и б; и каждый из R6d и R7d независимо означает Н или заместитель, выбранный из группы а;
<группа а> означает галоген, низший алкил, галогенсодержащий низший алкил, низшую алкоксигруппу, низшую алкилтиогруппу, карбоксил, низший алкоксикарбонил, гидроксигруппу, низший алифатический ацил, аминогруппу, низшую моноалкиламиногруппу, низшую диалкиламиногруппу, низшую алифатическую ациламиногруппу, цианогруппу или нитрогруппу;
<группа б> означает циклоалкил, арил, гетероцикл, каждый из которых необязательно имеет до трех заместителей, выбранных из группы а;
при условии, что когда R5d означает водород, Yd означает либо простую связь, либо линейный С1-С10алкилен; или фармацевтически приемлемые соли или сложные эфиры этих соединений;
- соединения, описанные в JP 14316985 (JP 2002316985), например, соединение формулы VIII
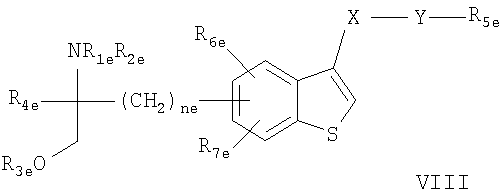
где R1e, R2e, R3е, R4e, R5e, R6е, R7е, ne, Хе И Ye описаны В JP 14316985; или фармацевтически приемлемые соли или сложные эфиры этих соединений.
- Соединения, описанные в WO 03/29184 и WO 03/29205, например, соединения формулы IX

где Xf означает О или S; R1f, R2f, R3f, и nf, как описано в WO 03/29184 и 03/29205, например, 2-амино-2-[4-(3-бензилоксифенокси)-2-хлорфенил]пропил-1,3-пропандиол или 2-амино-2-[4-(бензилоксифенилтио)-2-хлорфенил]пропил-1,3-пропандиол.
В каждом случае, где приводятся цитаты из заявок на получение патента, они включаются в настоящее изобретение в виде ссылок.
Ацил может быть остатком Ry-CO-, где Ry означает С1-С6алкил, С3-С6циклоалкил, фенил или фенил-С1-С4алкил. Если не оговорено иначе, алкил, алкоксигруппа, алкенил или алкинил могут быть прямыми или разветвленными.
Когда в соединениях формулы I углеродная цепь, такая как R1, замещена, предпочтительными заместителями являются галоген, нитрогруппа, аминогруппа, гидроксигруппа или карбоксигруппа. В случае, когда углеродную цепь прерывает фенилен, у которого необязательно имеются заместители, углеродная цепь предпочтительно является незамещенной. Когда фениленовая часть является замещенной, она предпочтительно замещена галогеном, нитрогруппой, аминогруппой, метоксигруппой, гидроксигруппой или карбоксигруппой.
Предпочтительными соединениями формулы I являются такие соединения, у которых R1 означает С13-С20алкил, необязательно замещенный нитрогруппой, галогеном, аминогруппой, гидроксигруппой или карбоксигруппой. Более предпочтительными являются соединения, у которых R1 означает фенилалкил, замещенный С6-С14алкильной цепью, необязательно замещенной галогеном, и алкильная часть означает C1-С6алкил, необязательно замещенный гидроксигруппой. Более предпочтительно, когда R1 означает фенил-С1-С6алкил, замещенный по фенилу прямой или разветвленной, предпочтительно прямой, С6-С14алкильной цепью. С6-С14алкильная цепь может быть в орто-, мета- или пара-, предпочтительно в пара-положении.
Предпочтительно, когда каждый от R2 до R5 означает Н.
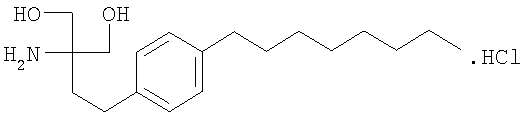
Предпочтительным соединением формулы I является 2-амино-2-тетрадецил-1,3-пропандиол. Особенно предпочтительным агонистом С1Ф-рецептора формулы I является соединение FTY720, т.е. 2-амино-2-[2-(4-октилфенил)этил]пропан-1,3-диол в свободной форме или в форме фармацевтически приемлемой соли (далее упоминаемое как “соединение А”), например, в форме гидрохлорида
Предпочтительным соединением формулы II является такое соединение, где каждый от R'2 до R'5 означает Н и m означает 4, т.е. 2-амино-2-{2-[4-(1-оксо-5-фенилпентил)фенил]этил} пропан-1,3-диол, в свободной форме или в форме фармацевтически приемлемой соли (далее упоминаемое как “соединение Б”), например, в форме гидрохлорида.
Предпочтительным соединением формулы III является соединение, где W означает СН3; каждый от R''1 до R''3 означает Н; Z2 означает этилен; Х означает гептилоксигруппу и Y означает Н; т.е. 2-амино-4-(4-гептилоксифенил)2-метилбутанол. Данное соединение может быть в свободной форме или в форме фармацевтически приемлемой соли (далее упоминаемое как “соединение В”), например, в форме гидрохлорида. Особенно предпочтителен R-энантиомер.
Предпочтительным соединением формулы IVa является соединение FTY720-фосфат (R2a означает Н, R3а означает ОН, Ха означает О, R1a и R1b означают ОН). Предпочтительным соединением формулы IVb является соединение В-фосфат (R2a означает Н, R3а означает ОН, Ха означает О, R1a и R1b означают ОН, Ya означает О, R4a означает гептил). Предпочтительным соединением формулы V является соединение Б-фосфат.
Предпочтительным соединением формулы V является сложный эфир фосфорной кислоты и моно[(R)-2-амино-2-метил-4-(4-пентилоксифенил)бутила].
Предпочтительным соединением формулы VIII является (2R)-2-амино-4-[3-(4-циклогексилоксибутил)бензо[b]тиен-6-ил]-2-метилбутан-1-ол.
В тех случаях, когда соединения формул с I по IX имеют в молекуле один или несколько ассиметричных центров, предметом настоящего изобретения также являются различные оптические изомеры, рацематы, диастереоизомеры и их смеси. Соединения формул III или IVb в тех случаях, когда углеродный атом, несущий аминогруппу, является ассиметричным, предпочтительно имеют R-конфигурацию по этому углеродному атому.
В качестве примеров фармацевтически приемлемых солей соединений формул от I до IX можно привести их соли неорганических кислот, такие как гидрохлорид, гидробромид и сульфат; соли органических кислот, такие как ацетат, фумарат, малеат, бензоат, цитрат, малат, метансульфонат, бензолсульфонат; или, в ряде случаев, соли, содержащие такие металлы, как, например, натрий, калий, кальций и алюминий; соли аминов, например, триэтиламин; а также соли двухосновных аминокислот, таких как лизин. В способах настоящего изобретения включены гидратные и сольватные формы соединений и солей.
Установлено, что агонисты С1Ф-рецептора благодаря обнаруженной активности, например, хоминга лимфоцитов, например, как описано в ЕР 627406 А1 или US 6004565, эффективны, например, в качестве иммунодепрессантов, например, в лечении острого отторжения аллотрансплантата. В настоящее время известно, что агонисты С1Ф-рецептора имеют ценные свойства, благодаря которым они могут использоваться в химиотерапии рака, особенно солидных опухолей, особенно прогрессирующих солидных опухолей. Тем не менее все еще существует необходимость расширения арсенала средств лечения солидных форм рака, особенно в тех случаях, когда лечение противораковыми соединениями не приводит к регрессии или стабилизации заболевания.
На основе полученных результатов в настоящем изобретении представлены:
1.1. Способ лечения солидных опухолей у субъекта, нуждающегося в таком лечении, который заключается в введении упомянутому субъекту терапевтически эффективного количества агониста С1Ф-рецептора, содержащего группу формулы X, или его фармацевтически приемлемой соли.
1.2. Способ подавления роста солидных опухолей у субъекта, нуждающегося в таком лечении, который включает введение упомянутому субъекту терапевтически эффективного количества агониста С1Ф-рецептора, содержащего группу формулы X, или его фармацевтически приемлемой соли.
1.3. Способ индукции регрессии опухоли, например редукции опухолевого образования, у субъекта, нуждающегося в таком способе; способ включает введение упомянутому субъекту терапевтически эффективного количества агониста С1Ф-рецептора, включающего группу формулы X, или его фармацевтически приемлемой соли.
1.4. Способ лечения инвазивности солидной опухоли или симптомов, связанных с таким опухолевым ростом, у субъекта, нуждающегося в таком лечении, который включает введение упомянутому субъекту терапевтически эффективного количества агониста С1Ф-рецептора, включающего группу формулы X, или его фармацевтически приемлемой соли.
1.5. Способ предотвращения метастазирования опухолей, или предотвращения, или ингибирования роста микрометастаз применительно к субъекту, нуждающемуся в таком способе; способ заключается в введение упомянутому субъекту терапевтически эффективного количества агониста С1Ф-рецептора, включающего группу формулы X, или его фармацевтически приемлемой соли.
1.6. Способ ингибирования или регулирования неконтролируемого ангиогенеза, например, ангиогенеза, опосредованного сфингозин-1-фосфатом (С1Ф), применительно к субъекту, нуждающемуся в таком способе; способ заключается в введение упомянутому субъекту терапевтически эффективного количества агониста С1Ф-рецептора, включающего группу формулы X, или его фармацевтически приемлемой соли.
1.7. Способ предупреждения или лечения заболеваний, связанных с процессом неоангиогенеза или с неконтролируемым ангиогенезом, применительно к субъекту, который нуждается в этом; способ заключается в введение упомянутому субъекту терапевтически эффективного количества агониста С1Ф-рецептора, включающего группу формулы X, или его фармацевтически приемлемой соли.
Под понятием “солидные опухоли” подразумеваются опухоли и/или метастазы (любой локализации), не относящиеся к лимфатическому раку, например, опухоли мозга и другие опухоли центральной нервной системы (например, опухоли оболочек мозга, спинного мозга, краниальных нервов и других частей центральной нервной системы, например, глиобластома или бластома костного мозга); рак головы и/или шеи; опухоли молочной железы; опухоли кровеносной системы (например, опухоли сердца, средостения и плевры, и других внутригрудных органов, опухоли сосудов и опухоли ткани, ассоциированной с сосудами); опухоли выделительной системы (например, опухоли почки, почечной лоханки, мочеточника, мочевого пузыря, а также опухоли другой и неопределенной локализации органов мочевой системы); опухоли желудочно-кишечного тракта (например, опухоли пищевода, желудка, тонкой кишки, толстой кишки, сигмовидной кишки, прямой кишки, ануса и анального канала), опухоли, затрагивающие печень и внутрипеченочные желчные протоки, желчный пузырь, опухоли другой и неопределенной локализации отделов желчного тракта, опухоли поджелудочной железы и другие опухоли пищеварительной системы); опухоли, затрагивающие ротовую полость (губу, язык, десну, дно полости рта, небо и другие части рта, околоушную железу и другие части слюнных желез, миндалины, ротоглотку, носоглотку, грушевидный карман, гортаноглотку и другие участки губы, ротовой полости и глотки); опухоли репродуктивной системы (например, вульвы, влагалища, шейки матки, тела матки, матки, яичника и других частей, связанных с женскими половыми органами, плаценты, пениса, предстательной железы, яичка и других частей, связанных с мужскими половыми органами); опухоли дыхательного тракта (например, носовой полости и среднего уха, околоносовых пазух, гортани, трахеи, бронха и легкого, например, мелкоклеточный рак легкого и немелкоклеточный рак легкого); опухоли костной системы (например, кости и суставного хряща конечностей, костного суставного хряща и других частей); опухоли кожи (например, злокачественная меланома кожи, немеланомный рак кожи, базалиома кожи, плоскоклеточная карцинома кожи, мезотелиома, саркома Капоши); и опухоли, вовлекающие другие ткани, включая периферические нервы и вегетативную нервную систему, соединительную и мягкую ткань, забрюшинное пространство и брюшину, глаз и придатки, щитовидную железу, надпочечник и другие эндокринные железы и родственные структуры, вторичная и неопределенная злокачественная неоплазма лимфатических узлов, вторичная злокачественная неоплазма респираторной и пищеварительной систем и вторичная злокачественная неоплазма другой локализации.
В описании настоящего изобретения при упоминании опухоли, опухолевого заболевания, карциномы или рака также подразумевается как альтернатива или как дополнение метастазирование в пораженном органе или ткани независимо от локализации опухоли и/или метастаза.
В том случае, когда агонистом С1Ф-рецептора является соединение формулы I, например, соединение А или соединения формул IVa или IVb, в одном из вариантов осуществления настоящего изобретения в нем используют способы 1.1, 1.2, 1.3 или 1.4 для лечения солидной опухоли, не являющейся опухолью молочной железы, предстательной железы, мочевого пузыря, почки и легкого.
В сериях дальнейших специфических или альтернативных вариантов осуществления настоящего изобретения в нем также представлены:
1.8. Способ повышения активности химиотерапевтического средства или преодоления устойчивости к химиотерапевтическому средству, который применяется к субъекту, нуждающемуся в нем, и заключается в введении указанному субъекту терапевтически эффективного количества агониста С1Ф-рецептора, например, агониста С1Ф-рецептора, включающего группу формулы X, или его фармацевтически приемлемой соли параллельно или последовательно с упомянутым химиотерапевтическим средством.
1.9. Способ по п.1.8, в котором химиотерапевтическое средство является ингибитором метаболических путей сигнальной трансдукции, направленных либо против клеток хозяина, либо против процессов, вовлеченных в формирование опухоли и/или процесс метастазирования, либо используемых опухолевыми клетками для пролиферации, выживания, дифференциации или развития лекарственной устойчивости.
1.10. Способ, соответствующий способу, указанному выше, в котором агонист С1Ф-рецептора вводят с интервалами.
В дальнейших сериях специфических или альтернативных вариантов осуществления настоящего изобретения в нем также представлены:
2.1. Агонист С1Ф-рецептора, включающий группу формулы X, или его фармацевтически приемлемая соль, для использования в одном из способов, указанных выше под п.п.1.1-1.4, предпочтительно для солидной опухоли, за исключением опухолей молочной железы, предстательной железы, мочевого пузыря, почки или легкого, когда агонист С1Ф-рецептора является соединением формулы I, например, соединением А, или соединением формулы IVa или IVb.
2.2. Агонист С1Ф-рецептора, например, агонист С1Ф-рецептора, включающий группу формулы X, или его фармацевтически приемлемая соль, для применения по одному из способов, указанных выше под п.п.1.5-1.10, или ниже под п.7.
3.1. Агонист С1Ф-рецептора, содержащий группу формулы X, или его фармацевтически приемлемая соль для использования в приготовлении фармацевтической композиции для применения в одном из способов, указанных выше под п.п.1.1-1.4; использование композиции предпочтительно предназначено в случае солидной опухоли, но не опухоли молочной железы, предстательной железы, мочевого пузыря, почки или легкого, когда агонист С1Ф-рецептора является соединением формулы I, например, соединением А, или соединением формулы IVa или IVb.
3.2. Агонист С1Ф-рецептора, например, агонист С1Ф-рецептора, содержащий группу формулы X, или его фармацевтически приемлемая соль, для использования в приготовлении фармацевтической композиции для применения в одном из способов, указанных выше под п.п.1.5-1.10, или ниже под п.7.
4.1. Фармацевтическая композиция для использования в одном из способов, указанных выше под п.п.1.1-1.4, содержащая агонист С1Ф-рецептора, включающий группу формулы X, или его фармацевтически приемлемую соль вместе с одним или несколькими фармацевтически приемлемыми растворителями или носителями; предпочтительно композиция предназначена для солидной опухоли, но не опухоли молочной железы, предстательной железы, мочевого пузыря, почки или легкого, когда агонист С1Ф-рецептора является соединением формулы I, например, соединением А, или соединением формулы IVa или IVb.
4.2. Фармацевтическая композиция для использования в одном из способов, как указано выше под п.п.1.5-1.10, или ниже под п.7, содержащая агонист С1Ф-рецептора, например, агонист С1Ф-рецептора, содержащий группу формулы X, или его фармацевтически приемлемую соль вместе с одним или несколькими фармацевтически приемлемыми растворителями или носителями.
5.1. Фармацевтическая комбинация, включающая а) первое средство, которое является агонистом С1Ф-рецептора, например, агонистом С1Ф-рецептора, включающим группу формулы X, или его фармацевтически приемлемую соль, и б) сопутствующее средство, которое является химиотерапевтическим средством, например, таким как указано ниже.
5.2. Фармацевтическия комбинация, включающая а) первое средство, которое является агонистом С1Ф-рецептора, например, агонистом С1Ф-рецептора, включающим группу формулы X, или его фармацевтически приемлемую соль, и б) сопутствующее средство, которое является химиотерапевтическим средством, выбранным из соединений, упомянутых ниже в разделе XI, для создания синергидного терапевтического эффекта.
6. Способ, как указано выше, включающий совместное введение, например, одновременное или последовательное, терапевтически эффективного количества агониста С1Ф-рецептора, например, агониста С1Ф-рецептора, включающего группу формулы X, или его фармацевтически приемлемой соли, и второй лекарственной субстанции, являющейся химиотерапевтическим средством, например, таким как указано в настоящем патенте.
7. Способ лечения лимфопролиферативного или миелопролиферативного заболевания, например, лечения инвазивности опухоли или симптомов, связанных с таким опухолевым ростом, у субъекта, который в этом нуждается; способ включает совместное введение упомянутому субъекту, например, одновременно или последовательно, агониста С1Ф-рецептора, например, агониста С1Ф-рецептора, включающего группу формулы X, или его фармацевтически приемлемой соли, и второй лекарственной субстанции, являющейся химиотерапевтическим средством, например, таким как указано в настоящем патенте.
Под “лимфатическим раком” подразумеваются, например, опухоли кровеносной и лимфатической систем (например, болезнь Ходжкина, неходжкинская лимфома, лимфома Беркитта, СПИД-ассоциированные лимфомы, злокачественные иммунопролиферативные заболевания, множественная миелома и злокачественные плазмоклеточные неоплазмы, лимфоидный лейкоз, острый или хронический миелоидный лейкоз, острый или хронический лимфолейкоз, моноцитарный лейкоз, другие лейкозы определенного клеточного типа, лейкоз неопределенного клеточного типа, другие и неопределенные злокачественные неоплазмы лимфоидной, кроветворной и родственных тканей, например, диффузная крупноклеточная лимфома, Т-клеточная лимфома или кожная Т-клеточная лимфома). Миелоидный рак включает, например, острый или хронический миелоидный лейкоз.
Под понятием “химиотерапевтическое средство” подразумевается главным образом любое химиотерапевтическое средство, иное чем агонист С1Ф-рецептора. Понятие включает, но ими не ограничивается:
i. ингибитор ароматазы,
ii. антиэстроген, антиандроген (особенно в случае рака предстательной железы) или агонист гонадорелина,
iii. ингибитор топоизомеразы I или ингибитор топоизомеразы II,
iv. средство, активное в отношении микротрубочек, алкилирующее средство, противоопухолевый антиметаболит или соединение платины,
v. соединение, связывающее/понижающее активность протеин- или липидкиназы или активность протеин- или липидфосфатазы, другое антиангиогенное соединение или соединение, которое индуцирует процессы клеточной дифференциации,
vi. рецептор брадикинина 1 или антагонист ангиотензина II,
vii. ингибитор циклооксигеназы, дифосфонат, ингибитор гистондезацетилазы, ингибитор гепараназы (предотвращает гепарансульфатную деградацию), например, продукт PI-88, модификатор биологического ответа, предпочтительно лимфокин или интерфероны, например, интерферон γ, ингибитор многих мишеней, или ингибитор, который блокирует метаболические пути антиапоптоза,
viii. ингибитор Ras-онкогенных изоформ, например, H-Ras, K-Ras или N-Ras, или ингибитор фарнезилтрансферазы, например, L-744832 или DK8G557,
ix. ингибитор теломеразы, например, теломестатин,
х. ингибитор протеазы, ингибитор металлопротеиназы матрикса, ингибитор метионинаминопептидазы, например, бенгамид или его производное, или ингибитор протеосом, например, PS-341, и/или
xi. Ингибитор mTOR.
Понятие “ингибитор ароматазы”, как оно используется в настоящем изобретении, относится к соединению, которое ингибирует образование эстрогена, например, превращение субстратов андростендиона и тестостерона в эстрон и эстрадиол соответственно. Понятие включает, но не ограничивается ими, стероиды, особенно атаместан, экземестан и форместан и, в частности, нестероидные соединения, особенно аминоглутетимид, роглетимид, пиридоглутетимид, трилостан, тестолактон, кетоконазол, ворозол, фадрозол, анастрозол и летрозол. Экземестан можно вводить, например, в форме коммерческого продукта AROMASINТМ. Форместан можно вводить, например, в форме коммерческого продукта LENTARONТМ. Фадрозол можно вводить, например, в форме коммерческого продукта AFEMAТМ. Анастрозол можно вводить, например, в форме коммерческого продукта ARIMIDEXТМ. Летрозол можно вводить, например, в форме коммерческого продукта FEMARAТМ или
FEMARТМ. Аминоглутетимид можно вводить, например, в форме коммерческого продукта ORIMETENТМ. Комбинация настоящего изобретения, включающая химиотерапевтическое средство, которое является ингибитором ароматазы, особенно полезна при лечении опухолей, имеющих рецепторы гормонов, например опухолей молочной железы.
Понятие “антиэстроген”, как оно используется в настоящем изобретении, относится к соединению, которое противодействует влиянию эстрогенов на уровень рецепторов эстрогенов. Понятие включает тамоксифен, фулвестрант, ралоксифен и гидрохлорид ралоксифена, но не ограничивается этими соединениями. Тамоксифен можно вводить, например, в форме коммерческого продукта NOLVADEXТМ. Гидрохлорид ралоксифена можно вводить, например, в форме коммерческого продукта EVISTAТМ. Фулвестрант можно приготовить, как описано в US 4659516, или можно вводить, например, в форме комерческого продукта FASLODEXТМ. Комбинация настоящего изобретения, включающая в качестве химиотерапевтического средства антиэстроген, особенно полезна при лечении, например, опухолей, имеющих рецепторы эстрогенов, например опухолей молочной железы.
В настоящем изобретении понятие “антиандроген” относится к соединениям, способным ингибировать биологическое действие андрогенных гормонов; к антиандрогенам относится, но им не ограничивается, бикалутамид (продукт
CASODEXТМ), который можно приготовить, например, как описано в US 4636505.
Понятие “агонист гонадорелина”, как оно используется в настоящем изобретении, включает абареликс, гозерелин и ацетат гозерелина, но не ограничивается этими соединениями. Гозерелин описан в US 4100274, его можно вводить, например, в форме коммерческого продукта под торговой маркой ZOLADEXТМ. Абареликс можно приготовить, например, как указано в US 5843901.
Понятие “ингибитор топоизомеразы I” в настоящем изобретении включает, но не ограничивается ими, топотекан, иринотекан, 9-нитрокамптотецин и макромолекулярный камптотециновый конъюгат PNU-166148 (соединение А1 в WO 99/17804). Иринотекан можно вводить, например, в форме коммерческого продукта CAMPTOSARТМ. Топотекан можно вводить, например, в форме коммерческого продукта HYCAMTINТМ.
Понятие “ингибитор топоизомеразы II'', как оно используется в настоящем изобретении, включает, но не ограничивается ими, антрациклины, такие как доксорубицин (включая липосомную форму, например, продукт CAELYXТМ), даунорубицин, эпирубицин, идарубицин и неморубицин, антрахиноны митоксантрон и лозоксантрон, подофиллотоксины этопозид и тенипозид. Этопозид можно вводить, например, в форме коммерческого продукта ETOPOPHOSТМ. Тенипозид можно вводить, например, в форме коммерческого продукта VM 26-BRISTOLТМ. Доксорубицин можно вводить, например, в форме коммерческого продукта ADRIBLASTINТМ. Эпирубицин можно вводить, например, в форме коммерческого продукта FARMORUBICINТМ. Идарубицин можно вводить, например, в форме коммерческого продукта ZAVEDOSТМ. Митоксантрон можно вводить, например, в форме коммерческого продукта NOVANTRONТМ.
Понятие “средство, активное в отношении микротрубочек” относится к средствам, стабилизирующим микротрубочки и дестабилизирующим микротрубочки; понятие включает, но не ограничивается ими, таксаны, например, паклитаксел и доцетаксел, алкалоиды барвинка, например винбластин, особенно сульфат винбластина, винкристин, особенно сульфат винкристина, винорельбин, дискодермолиды и эпотилоны и их производные, например, эпотилон В или его производное. Паклитаксел можно вводить, например, в форме коммерческого продукта TAXOLТМ. Доцетаксел можно вводить, например, в форме коммерческого продукта
TAXOTEREТМ. Сульфат винбластина можно вводить, например, в форме коммерческого продукта VINBLASTIN R.P.ТМ. Сульфат винкристина можно вводить, например, в форме коммерческого продукта FARMISTINТМ. Дискодермолид можно получить, например, как описано в US 5010099.
Понятие “алкилирующее средство”, как оно используется в настоящем изобретении, включает, но не ограничивается ими, бусульфан, хлорамбуцил, циклофосфамид, ифосфамид, мелфалан или нитрозомочевину (BCNU или продукт GliadelТМ). Циклофосфамид можно вводить, например, в форме коммерческого продукта CYCLOSTINТМ. Ифосфамид можно вводить, например, в форме коммерческого продукта HOLOXANТМ.
Понятие “противоопухолевый антиметаболит” включает, но не ограничивается ими, 5-фторурацил, капецитабин, гемцитабин, цитарабин, флударабин, тиогуанин, метотрексат и эдатрексат.Капецитабин можно вводить, например, в форме коммерческого продукта XELODAТМ. Гемцитабин можно вводить, например, в форме коммерческого продукта GEMZARТМ.
Понятие “соединение платины”, как оно используется в настоящем изобретении, включает, но не ограничивается ими, карбоплатин, цисплаптин и оксалиплатин. Карбоплатин можно вводить, например, в форме коммерческого продукта CARBOPLATТМ. Оксалиплатин можно вводить, например, в форме коммерческого продукта ELOXATINТМ.
Понятие “соединения, связывающие/понижающие активность протеин- или липидкиназ, или другие антиангиогенные соединения”, как оно используется в настоящем изобретении, включает, но не ограничивается ими, ингибиторы протеинтирозинкиназы и/или серин- или треонинкиназы или ингибиторы липидкиназы, например, соединения, связывающие, понижающие или ингибирующие активность представителей семейства факторов роста эпидермиса рецепторных тирозинкиназ (EGFR, ErbB2, ErbB3, ErbB4, в виде гомо- или гетеродимеров); представителей семейства факторов роста эндотелия сосудов рецепторных тирозинкиназ (VEGFR); рецепторы тромбоцитарного фактора роста (PDGFR); рецепторы факторов роста фибробластов (FGFR); рецептор 1 инсулиноподобного фактора роста (IGF-1R); представителей семейства Trk-рецепторных тирозинкиназ; представителей семейства Ах1-рецепторных тирозинкиназ; Ret-рецепторную тирозинкиназу; Kit/SCFR-рецепторную тирозинкиназу; представителей Ab1-семейства и продукты слияния их генов (например, BCR-Ab1); представителей протеинкиназы С (РКС) и Raf семейства серин/треониновых киназ; представителей МЕК, SRC, JAK, FAK, PDK или PI(3) семейства киназ или семейства киназ, близких киназе PI(3); и/или членов семейства циклинзависимых киназ (CDK). Понятие также включает антиангиогенные соединения, имеющие другой механизм активности, например, не связанный с ингибированием протеин- или липидкиназ.
Соединениями, которые связывают, понижают или ингибируют активность VEGFR, являются, главным образом, соединения, белки или антитела, которые ингибируют VEGF-рецепторную тирозинкиназу, ингибируют VEGF-рецептор или связываются с VEGF. В частности, это соединения, белки или моноклональные антитела, которые в общих чертах или подробно описываются в WO 98/35958, например, 1-(4-хлоранилино)-4-(4-пиридилметил)фталазин или его фармацевтически приемлемая соль, например, сукцинат, в WO 00/27820, например, производное амида 7У-арил(тио)антраниловой кислоты, например, 2-[(4-пиридил)метил]амино-N-[3-метокси-5-трифторметил)фенил]бензамид или 2-[(1-оксидо-4-пиридил)метил]амино-N-[3- трифторметилфенил]бензамид, или в WO 00/09495, WO 00/59509, WO 98/11223, WO 00/27819 и в ЕР 0769947; соединения, которые описаны М. Prewett и др. в Cancer Research, 59, 1999, сс.5209-5218; F.Yuan и др. в Proc. Nail. Acad. Sci. USA, 93, 1996, сс.14765-14770; Z. Zhu и др. в Cancer Res., 58, 1998, cc.3209-3214; J. Mordenti и др. в Toxicologic Pathology, 27, 1999, сс.14-21; в WO 00/37502 и WO 94/10202; продукт AngiostatinТМ, описанный M.S. O'Reilly и др. в Cell, 79, 1994, сс.315-328; продукт EndostatinТМ, описанный M.S. O'Reilly и др. в Cell, 88, 1997, cc.277-285; амидные производные антраниловой кислоты; ZD4190; ZD6474; SU5416; SU6668; или антитела к VEGF, или антитела к рецептору VEGF, например, RhuMab.
Под антителами подразумевают интактные моноклональные антитела, поликлональные антитела, полиспецифические антитела, образованные, по крайней мере, из 2 интактных антител, и фрагменты антител такой длины, которая обеспечивает желаемую биологическую активность.
Соединения, которые связывают, понижают или ингибируют активность семейства рецепторов факторов роста эпидермиса, являются, главным образом, соединениями, белками или антителами, которые ингибируют представителей семейства EGF-рецепторных тирозинкиназ, например, EGF-рецептора, ErbB2, ErbB3 и ErbB4, связывают EGF или EGF-родственные лиганды, или которые обладают двойственным ингибирующим воздействием по отношению к ErbB- и VEGF-рецепторной киназе. В частности, это такие соединения, протеины или моноклональные антитела, которые в общих чертах или подробно описываются в WO 97/02266, например, соединение из примера 39, или в ЕР 0564409, WO 99/03854, ЕР 0520722, ЕР 0566226, ЕР 0787722, ЕР 0837063, US 5747498, WO 98/10767, WO 97/30034, WO 97/49688, WO 97/38983 и, особенно, в WO 96/30347 (например, соединение, известное как СР 358774), WO 96/33980 (например, соединение ZD 1839) и WO 95/03283 (например, соединение ZM105180) или РСТ/ЕР 02/08780; например, трастузумаб (продукт HerpetinR), цетуксимаб, Иресса, OSI-774, CI-1033, ЕКВ-569, GW-2016, Е1.1, Е2.4, Е2.5, Е6.2. Е6.4, Е2.11, Е6.3 или Е7.6.3.
Соединениями, которые связывают, понижают или ингибируют активность PDGFR, являются, главным образом, соединения, которые ингибируют PDGF-рецептор, например, производное N-фенил-2-пиримидинамина, например иматиниб.
Соединениями, которые связывают, понижают или ингибируют активность представителей семейства с-Ab1 и продуктов слияния их генов, являются, например, производное N-фенил-2-пиримидинамина, например, иматиниб; PD180970; AG957; или NSC 680410.
Соединения, которые связывают, понижают или ингибируют активность представителей семейств протеинкиназы С, Raf, MEK, SRC, JAK, FAK и PDK, или представителей семейства PI(3)-киназы или родственной PI(3)-киназы, и/или представителей семейства циклинзависимых киназ (CDK), являются, главным образом, такими соединениями, как производные стауроспорина, которые описываются в ЕР 0296110, например, мидостаурин. Примеры других соединений включают, например, UCN-01, сафингол, BAY 43-9006, Бриостатин 1, Перифозин; UO 126; Илмофозин; RO 318220 и RO 320432; GO 6976, Изис 3521; или LY333531/LY379196.
Другими антиангиогенными соединениями являются, например, талидомид (THALOMID) и TNP-470.
Соединения, которые связывают, понижают или ингибируют активность протеин- или липидфосфатазы - это, например, ингибиторы фосфатазы 1, фосфатазы 2А, PTEN или CDC25, например, окадаевая кислота или ее производное.
Соединениями, которые индуцируют процессы клеточной дифференциации, являются, например, ретиноевая кислота, α-, γ- или δ-токоферол или α-, γ- или δ-токотриенол.
Понятие “ингибитор циклооксигеназы”, как оно используется в настоящем изобретении, включает, но не ограничивается ими, например, целекоксиб (продукт CelebrexR, рофекоксиб (продукт VioxxR), эторикоксиб, вальдекоксиб или 5-алкил-2-ариламинофенилуксусную кислоту, например, 5-метил-2-(2'-хлор-6'-фторанилино)фенилуксусную кислоту.
Понятие “ингибитор гистондезацетилазы”, как оно используется в настоящем изобретении, включает MS-27-275, SAHA, пироксамид, FR-901228 и вальпроевую кислоту, но не ограничивается ими.
Понятие “дифосфонаты”, как оно используется в настоящем изобретении, включает этридоновую, клодроновую, тилудроновую, памидроновую, алендроновую, ибандроновую, ризедроновую и золедроновую кислоту, но не ограничивается этими соединениями. Этридоновую кислоту можно вводить, например, в форме коммерческого продукта DIDRONELТМ. Клодроновую кислоту можно вводить, например, в форме коммерческого продукта BONEFOSТМ. Тилудроновую кислоту можно вводить, например, в форме коммерческого продукта SKELIDТМ. Памидроновую кислоту можно вводить, например, в форме коммерческого продукта AREDIAТМ. Алендроновую кислоту можно вводить, например, в форме коммерческого продукта FOSAMAXТМ. Ибандроновую кислоту можно вводить, например, в форме коммерческого продукта BONDRANATТМ. Ризедроновую кислоту можно вводить, например, в форме коммерческого продукта ACTONELТМ. Золедроновую кислоту можно вводить, например, в форме коммерческого продукта ZOMETAТМ.
Понятие “ингибитор металлопротеиназы матрикса”, как оно используется в настоящем изобретении, включает, но не ограничивается ими, коллагеновые пептидомиметические и непептидомиметические ингибиторы, производные тетрациклина, например, гидроксамат пептидомиметического ингибитора батимастата и его биодоступный при пероральном введении аналог маримастат, приномастат, BMS-279251, BAY 12-9566, ТАА211 или AAJ996.
Понятие “mTOR-ингибитор”, как оно используется в настоящем изобретении, означает рапамицин (сиролимус) или его производное, но не ограничивается этими соединениями. Рапамицин - известный макролидный антибиотик, образуемый Streptomyces hygroscopicus. Приемлемые производные рапамицина включают, например, соединения формулы А
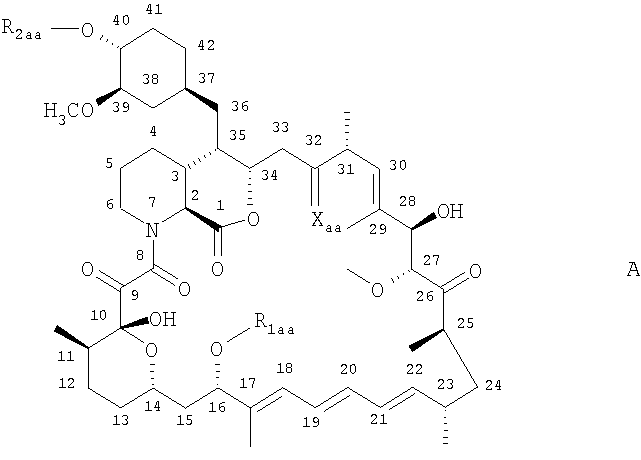
где
R1aa означает СН3 или С3-С6алкинил
R2aa означает Н или -СН2-СН2-ОН, 3-гидрокси-2-(гидроксиметил)-2-метилпропаноил или тетразолил, и
Хаа означает =O, (Н, Н) или (Н, ОН)
при условии, что R2aa не является Н, когда Хаа означает =O и R1aa означает СН3;
или его пролекарство, когда R2aa означает -СН2-СН2-ОН, например, его физиологически гидролизуемый простой эфир.
Соединения формулы А описываются, например, в WO 94/09010, WO 95/16691, WO 96/41807, US 5362718 или WO 99/15530, в настоящее изобретение они включаются в виде ссылок. Их можно приготовить согласно описанию или аналогично способам, описанным в этих ссылках.
Предпочтительными производными рапамицина являются 32-дезоксорапамицин, 16-пент-2-инилокси-32-дезоксорапамицин, 16-пент-2-инилокси-32(S)дигидрорапамицин, 16-пент-2-инилокси-32(S)-дигидро-40-орто-(2-гидроксиэтил)рапамицин и, более предпочтительно, 40-орто-(2-гидроксиэтил)рапамицин. Другими примерами производных рапамицина являются, например, CCI779 или 40-[3-гидроксиметил)-2-метилпропаноат]рапамицин или его фармацевтически приемлемая соль, как описано в US 5362718, АВТ578 или 40-(тетразолил)рапамицин, особенно 40-эпи-(тетразолил)-рапамицин, например, описанные в WO 99/15530, или рапалоги, описанные в WO 98/02441 и W001/14387, например, АР23573.
В каждом случае, когда упоминаются заявки на патенты или научные публикации, касающиеся сущности соединений, они включаются в настоящее изобретение в виде ссылок. Подобным образом включаются также фармацевтически приемлемые соли этих соединений, соответствующие рацематы, диастереоизомеры, энантиомеры, таутомеры и соответствующие кристаллические модификации описанных выше соединений, если таковые представлены, например, сольваты, гидраты и полиморфы, которые описаны в цитируемых публикациях. Соединения, которые в настоящем изобретении используются в качестве активных ингредиентов комбинаций, могут быть получены и введены, как описано в цитируемых документах. Также в рамках настоящего изобретения представлена комбинация, содержащая более двух разных активных ингредиентов, как изложено выше; например, фармацевтическая комбинация в рамках настоящего изобретения могла бы включать три или более активных ингредиента. Более того, и первое средство, и сопутствующее средство не являются одним и тем же ингредиентом.
Применение агонистов С1Ф, например, агонистов С1Ф, включающих группу формулы X, в лечении солидных опухолей, которые детально изложены в настоящем изобретении выше, можно продемонстрировать в испытаниях на животных и в клинических исследованиях, например, в соответствии со способами, описанными ниже.
A. In vitro
A.1. Противоопухолевая активность
Используют линию клеток рака молочной железы мыши, первоначально выделенную из карциномы молочной железы, например, линию JygMC(A). Перед началом исследования количество клеток доводят до титра 5×105 для посева в планшет со свежей питательной средой. Клетки инкубируют в свежей среде, содержащей 2,5 мМ тимидина, без фетальной телячьей сыворотки (ФТС) в течение 12 ч и затем дважды промывают физиологическим раствором с фосфатным буфером (ФСБ) с последующим добавлением свежей среды, содержащей 10% ФТС, и дополнительно инкубируют еще в течение 12 ч. После этого клетки инкубируют в свежей среде без ФТС, содержащей 2,5 мМ тимидина, в течение 12 ч. Для отделения клеток их дважды смывают ФСБ и вновь высевают в планшеты со свежей средой, содержащей 10% ФТС. После синхронизации клетки инкубируют без/с различными концентрациями соединения формулы I в течение 3, 6, 9, 12, 18 или 24 ч. После обработки 0,2% раствором ЭДТА клетки собирают, фиксируют 70% раствором этанола, охлажденным до температуры таяния льда, гидролизуют с РНКазой А (тип 1-А: фирма Sigma Chem. Co.), взятой в количестве 250 мкг/мл, при 37°С в течение 30 мин и окрашивают иодидом пропидиума в концентрации 10 мг/мл в течение 20 мин. После инкубационного периода количество клеток определяют двумя способами: подсчетом клеток в счетчике Coulter и SRB-колориметрическим анализом. При данных условиях агонист С1Ф, например соединение Б в форме гидрохлорида, ингибирует пролиферацию опухолевых клеток в диапазоне концентраций 10-12 - 10-6 М.
А.2. Анализ С1Ф-опосредованного образования трубочек в эндотелиальных клетках пупочной вены человека (ЭКПВЧ)
Для анализа образования трубочек используют клетки ЭКПВЧ из 2-8 пассажа; при этом до сбора клеток их плотность никогда не превышает 70%. Клетки готовят для анализа, промывая сбалансированным для герпеса физиологическим раствором (HBSS, фирма Clonetics) с последующей обработкой раствором трипсин/ЭДТА (0,25 мг/мл, фирма Clonetics). После отделения примерно 90% клеток от планшета добавляют равный объем трипсин-нейтрализующего раствора (TNS, фирма Clonetics), затем клетки собирают в конические пробирки, содержащие, по крайней мере, 10 мл сред ЕВМ-2 (фирма Clonetics)+0,1% бычьего сывороточного альбумина (БСА) (фирма Sigma). Клетки центрифугируют при 1000 об./мин в течение 5 мин, супернатант удаляют и добавляют 5 мл свежей среды ЕВМ-2+0,1% БСА. Клетки подсчитывают с помощью гематоцитометра и объем клеточной суспензии доводят до конечной концентрации 500000 клеток/мл. В конические пробирки помещают по 100 нмоль изучаемых соединений и по 1 мл раствора коклюшного токсина (КТ) в концентрации 10 нг/мл, затем вносят по 1 мл клеточной суспензии в каждую пробирку. После этого пробирки инкубируют в течение получаса при 37°С в атмосфере 5% CO2. Миграционный анализ проводят, используя 24-многолуночные вставные планшеты Fluoro-Blok, покрытые фибронектином (размер пор 8 мкм, фирма Falcon #351147), вместо отдельных вставок в 24-луночный планшет. Клетки и исследуемые соединения подготавливают и предварительно инкубируют, как описано выше, затем по 100 мкл добавляют в каждую соответствующую лунку вставного планшета. По 300 мкл среды ЕВМ-2+2% десорбированного древесного угля, без С1Ф, вносят в лунки, помеченные “нет стимуляции (-)”, и по 300 мкл среды, содержащей С1Ф (500 нмоль), вносят в лунки, помеченные “стимуляция (+)”. После этого планшет инкубируют в течение 4 ч при 37 С в атмосфере 5% СО2.
Кальцеин AM, 50 мкг/ампулу (фирма Molecular Probes #C3100) готовят следующим образом: сначала добавляют в ампулу 20 мкл ДМСО, затем 12,5 мл HBSS (на планшет) нагревают до 37°С и 150 мкл добавляют в ампулу; содержимое ампулы переносят обратно в оставшийся HBSS до получения конечной концентрации 4 мкг/мл кальцеина AM.
Планшет Fluoro-blok вынимают из термостата, отделяют верхний вставной планшет и постукивают по нему, чтобы удалить излишек среды, прилипший к вставкам. Затем вставной планшет переносят на свежий 24-луночный планшет, содержащий по 500 мкл в лунке кальцеина AM, в концентрации 4 мкг/мл. После этого планшет инкубируют в течение 11/2 ч при 37°С в атмосфере 5% CO2.
После инкубации планшет считывают на приборе Cytofluor II при волне возбуждения 485 нм и волне эмиссии 530 нм. Покрытие Fluoro-Blok во вставках позволяет подсчитывать только те клетки, которые мигрируют на дно. Для анализа данные заносят в программу Excel, построение диаграмм осуществляют с использованием программы SigmaPlot, для критериев значимости (проверка по критерию Стьюдента) используют программу SigmaStat (фиг.7).
Образование трубочек определяют путем подсчета количества точек ветвления (два независимых соединяющихся тяжа) в трех независимых полях зрения при 4-кратном увеличении. Результаты описывают следующим образом:
Данные результаты показывают уникальную способность соединения FTY720-фосфат или соединения В-фосфат действовать в качестве агониста ангиогенеза как такового и, что удивительно, в качестве антагониста С1Ф-опосредованного ангиогенеза. Предпочтительной формой для соединения В-фосфата является рацемат или R-энантиомер. КТ используют в качестве контроля ингибирования Giα (EDG-1)-опосредованной активности.
Б. In vivo
Б.1. Противоопухолевая активность
Противоопухолевую активность выражают отношением O/К% (среднее увеличение опухолевых масс у обработанных животных к среднему увеличению опухолевых масс у контрольных животных, умноженное на 100).
Аликвотные пробы раковых клеток (1×107), например, клеток меланомы человека А375, прививают мышам линии BALB/c-пи/пи. Когда опухоли достигнут размера примерно 10×10 мм, животных произвольно делят на четыре подгруппы и начинают лечить соединением формулы I. После двухнедельного лечения животных забивают, а опухоли и ткани готовят для морфологического и молекулярного анализа. Размер опухолей определяют с помощью циркуля. В данном анализе агонист С1Ф, например соединения Б или В (в форме гидрохлорида), замедляет рост опухолей при введении в дозе от 0,5 до 5 мг/кг; контролем является физиологический раствор: например, соединение В-HCl при 5-кратном введении в течение недели приводит к значению О/К - 30%.
Б.2. Комбинация с ингибитором VEGF-R-протеинтирозинкиназы
Голых мышей с привитыми опухолями молочных желез человека MDA-MB-435 лечат в течение двух недель ингибитором VEGF-R-протеинтирозинкиназы, например, сукцинатом 1-(4-хлоранилино)-4-(4-пиридилметил)фталазина, в дозе 100 мг/кг перорально при 5-кратном введении в течение недели, агонистом С1Ф-рецептора, например, соединением В (гидрохлоридом), в дозе 2,5 мг/кг внутривенно при 5-кратном введении в течение недели, или комбинацией из обоих соединений. Противоопухолевый эффект выражают отношением О/К%, как указано выше. Комбинация соединения В-HCl с сукцинатом 1-(4-хлоранилино)-4-(4-пиридилметил)фталазина оказывает большее противоопухолевое действие (О/К% 27) по сравнению с одним из средств (соединением В-HCl, О/К 66%; сукцинатом 1-(4-хлоранилино)-4-(4-пиридилметил)фталазина, O/К% 91). Хорошие противоопухолевые ответы получают также, когда голым мышам прививают клетки меланомы человека A3 75 и лечат сходным образом с помощью той же комбинации: комбинированное лечение приводит к показателю O/К% 15, в то время как лечение с помощью только одного из средств - к показателям O/К% 35 и 44 соответственно.
Б.3. Антиангиогенная активность
Пористые камеры, содержащие (i) сфингозин-1-фосфат (5 мкмоль/камеру) или (ii) VEGF человека (1 мкг/камеру) в 0,5 мл 0,8% (мас./объем) агара (содержащего гепарин, 20 ед./мл) вводят мышам подкожно в бок. С1Ф или VEGF индуцируют рост васкуляризированной ткани вокруг камеры. Данный ответ является дозозависимым и характеризуется количественно измерением веса и кровяного наполнения ткани. Мышей обрабатывают один раз в сутки (i) перорально соединением А (0,3, 3, 30 или 50 мг/кг), или (ii) внутривенно R-энантиомером соединения В (2,5 мг/кг), или (iii) внутривенно S-энантиомером соединения В (2,5 мг/кг), или (iv) перорально, или внутривенно раствором (5% глюкоза, 10 мл/кг), начиная за 4-6 ч до введения камер и продолжая в течение 4-х суток. Животных забивают для измерения васкуляризированных тканей через 24 ч после введения последней дозы. Определяют вес и кровяное наполнение васкуляризированных тканей вокруг камеры. Животные, которых лечили соединением А, или R- или S-энантиомером соединения В, показывают снижение веса и/или кровяного наполнения васкуляризированных тканей по сравнению с аналогичными показателями у животных, которые получали только один растворитель.
В. Клиническое исследование
B.1. Исследование клинической эффективности агонистов С1Ф-рецептора, например, соединений формул I, II или III, например, соединений А, Б или В
В исследовании участвуют 20 больных с прогрессирующими солидными опухолями в поздней стадии, резистентными или трудно поддающимися стандартной терапии; они получают упомянутое соединение в дозе, которая подбирается путем постепенного повышения дозы. Проводят еженедельное врачебное и лабораторное обследование общего клинического состояния больных. Изменение состояния опухоли и метастаз оценивают каждые 2 месяца путем рентгенологического исследования. Первоначально больные получают лечение в течение 2-х месяцев. После этого лечение продолжают до тех пор, пока заболевание не перестает прогрессировать и пока больные удовлетворительно переносят лекарство.
Основные переменные для оценки результата лечения: безопасность (побочные эффекты), стандартные биохимические показатели сыворотки и форменных элементов крови, размеры опухоли по оценке методом компьютерной томографии (КТ) или магнитно-резонансной интроскопии (МРИ).
В.2. Комбинированное лечение
Подходящими клиническими исследованиями являются, например, открытые нерандомизированные с повышением доз исследования пациентов с запущенными солидными опухолями. Такие исследования показывают, в частности, синергизм активных ингредиентов в комбинации, предлагаемой в настоящем изобретении. Благоприятные эффекты на пролиферативные заболевания можно установить непосредственно по результатам этих исследований или по изменениям в ходе исследования, которые по существу известны любому специалисту в данной области. Такие исследования, в частности, применимы для сравнения эффектов монотерапии, в которой используются активные ингредиенты, и эффектов от применения комбинации настоящего изобретения. Предпочтительно дозу средства (а) повышают до тех пор, пока не будет достигнута максимально переносимая доза; сопутствующее средство (б) вводят в фиксированной дозе. Альтернативно средство (а) вводят в фиксированной дозе, а дозу сопутствующего средства (б) повышают. Каждый больной получает дозы средства (а) либо ежедневно, либо с перерывами. Эффективность лечения в таких исследованиях может быть установлена, например, после 12, 18 или 24 недель путем рентгенологической оценки опухолей с интервалом 6 недель.
Альтернативой является двойной слепой метод с плацебо контролем; его можно использовать для того, чтобы подтвердить преимущества упомянутой в настоящем изобретении комбинации.
Суточные дозы, необходимые при варианте осуществления способа настоящего изобретения, в нем, когда используется только один агонист С1Ф-рецептора, варьируют в зависимости, например, от соединения, которое используется, организма, способа введения и тяжести заболевания, лечение которого проводится. Предпочтительная суточная доза находится в диапазоне от 0,1 до 100 мг; ее вводят в качестве однократной дозы или делят на несколько доз. Приемлемыми суточными дозами для больных являются дозы, например, порядка 0,1-50 мг, прием пероральный. Агонист С1Ф-рецептора может быть введен любым стандартным способом, особенно энтерально, например, перорально, например, в форме таблеток, капсул или микстур, через нос, посредством ингаляций или парентерально, например, в форме растворов или суспензий для инъекций. Приемлемые формы унифицированной дозировки для перорального введения включают примерно от 0,1 до 30 мг, обычно от 0,25 до 30 мг агониста рецептора С1Ф, вместе с одним или несколькими фармацевтически приемлемыми растворителями или носителями. Для того чтобы ингибировать ангиогенез, важно подобрать достаточно высокую дозу агониста рецептора С1Ф, поскольку низкие концентрации агонистов С1Ф-рецептора стимулируют ангиогенез. Приемлемую дозу для обеспечения антиангиогенного действия, когда больному вводится С1Ф-агонист, можно подобрать путем повышения концентрации и дозы, как описано выше для А, Б и В.
Комбинацию настоящего изобретения можно также применять в комбинации с хирургическим вмешательством, слабо выраженной продолжительной гипертермией целого организма и/или лучевой терапией.
Введение фармацевтической комбинации настоящего изобретения приводит к положительному эффекту, например, синергетическому терапевтическому эффекту, например, в отношении замедления, купирования или реверсирования новообразований, распространения или роста метастаз, или большей длительности опухолевого ответа, или подавления ангиогенеза; применение фармацевтической комбинации может также привести и к другим ценным эффектам, например, к уменьшению побочных эффектов, улучшенному качеству жизни или уменьшенной смертности и болезненности по сравнению с монотерапией, при которой применяется только один из фармацевтически активных ингредиентов, используемых в комбинации настоящего изобретения. Особенно это касается лечения рака, который не поддается лечению другими химиотерапевтическими средствами, известными как противораковые.
Другое преимущество заключается в том, что в комбинации настоящего изобретения можно использовать более низкие дозы активных ингредиентов, дозы могут быть меньше и применяться реже или более низкие дозы можно использовать для того, чтобы уменьшить степень побочных эффектов в ходе сдерживания роста опухолевого образования. Это находится в соответствии с пожеланиями и потребностями пациентов.
В соответствии с одним из вариантов осуществления настоящего изобретения в нем предпочтительная фармацевтическая комбинация включает:
а) соединения формул I, II, III, IVa, IVb, V или VI, например, соединение А, Б или В, и
б) одно или несколько соединений в качестве сопутствующих средств, таких как указанные выше в параграфах (ii), (iii), (iv), (v), (vii) или (xi), например, карбоплатин, цисплатин, паклитаксел, доцетаксел, гемцитабин, доксорубицин, соединение, которое связывает, понижает или ингибирует активность представителей семейства факторов роста эндотелия сосудов рецепторных тирозинкиназ (VEGFR) или рецепторов тромбоцитарного фактора роста (PDGFR), или дифосфонатов или mTOR-ингибитора.
Другой вариант осуществления настоящего изобретения относится к использованию агониста С1Ф-рецептора (а) в комбинации с химиотерапевтическим средством (б) в лечении лимфатического или миелоидного рака, например, как описано выше. Комбинация может включать в качестве дополнительного сопутствующего средства (б), например, бусульфан, цитарабин, 6-тиогуанин, флударабин, гидроксимочевину, прокарбазин, блеомицин или метотрексат. В качестве сопутствующих средств (б), например, для использования в лечении лимфатического рака, предпочтительными являются ингибиторы топоизомеразы II, например, даунорубицин или, особенно, соединения, которые связывают, понижают или ингибируют активность PDGFR или представителей семейства с-Ab1 и продуктов слияния их генов, например иматиниб.
Понятия “совместное введение”, или “комбинированное введение”, или подобные понятия, как они используются в настоящем изобретении, означают введение выбранных терапевтических средств одному больному и подразумевают также применение таких режимов лечения, при которых средства не обязательно вводятся одним и тем же способом или в одно и то же время.
Одной из задач настоящего изобретения было представить фармацевтическую композицию, включающую количество комбинации настоящего изобретения, которое при совместном применении терапевтически эффективно в отношении пролиферирующего злокачественного заболевания. В этой композиции первое средство (а) и сопутствующее средство (б) могут вводиться вместе, одно за другим, или раздельно в форме одной комбинированной унифицированной дозы, или в виде двух унифицированных дозовых форм. Форма унифицированной дозы также может быть фиксированной комбинацией.
Фармацевтические композиции согласно настоящему изобретению можно приготовить способом, известным per se. Они пригодны для энтерального, например, перорального или ректального, и парентерального введения млекопитающим (теплокровным животным) включая людей. Композиции содержат терапевтически эффективное количество, по крайней мере, одного фармакологически активного компонента комбинации, например, как указано выше, или в комбинации с одним или несколькими фармацевтически приемлемыми носителями или растворителями, особенно пригодными для энтерального или парентерального применения.
Приемлемые фармацевтические композиции содержат, например, активный ингредиент (или активные ингредиенты) приблизительно в количестве от 0,1% до 99,9%, предпочтительно, от 1% до 60%. Фармацевтические препараты для комбинированной терапии для энтерального или парентерального введения являются, например, формами унифицированных доз, таких как таблетки с сахарным покрытием, таблетки, капсулы, суппозитории или ампулы. Если не указано иначе, их готовят известным per se способом, например, с помощью таких обычных процессов, как перемешивание, гранулирование, покрытие сахарной оболочкой, растворение или лиофилизация.
Ценным является то, что содержание единицы смешанной фармацевтической композиции состоит из отдельных доз каждого составляющего компонента комбинации, взятых в количестве, которое в отдельности недостаточно для проявления активности; необходимое эффективное количество может быть достигнуто введением множества дозовых единиц.
В частности, терапевтически эффективное количество каждого компонента комбинации настоящего изобретения можно вводить одновременно или последовательно в любом порядке, компоненты можно вводить по отдельности или в виде фиксированной комбинации. Например, способ сдерживания прогрессирования или лечения пролиферирующего злокачественного заболевания согласно настоящему изобретению может включать: (i) введение первого средства (а) в свободной форме или в форме фармацевтически приемлемой соли и (ii) введение сопутствующего средства (б) в свободной форме или в форме фармацевтически приемлемой соли; введение осуществляется одновременно или последовательно в любом порядке, в количествах, терапевтически эффективных при совместном применении, предпочтительно в количествах, оказывающих синергетический эффект, например, в суточных или прерывистых дозировках, соответствующих количествам, описанным в настоящем изобретении. Отдельные компоненты комбинации настоящего изобретения можно вводить по отдельности в разное время на протяжении курса лечения или одновременно в виде раздельных форм или в виде единой комбинированной формы. Понятие “введение” также включает в себя использование пролекарства как компонента комбинации, которое in vivo превращается в компонент комбинации. Настоящее изобретение поэтому следует понимать как охватывающее все перечисленные режимы одновременного или чередующегося лечения, и понятие “введение” следует интерпретировать соответствующим образом.
Эффективная дозировка каждого из компонентов комбинации настоящего изобретения может варьировать в зависимости от того, используется ли индивидуальное соединение или фармацевтическая композиция, а также в зависимости от способа введения, заболевания, лечение которого проводится, и его тяжести. Поэтому режим дозировки комбинации настоящего изобретения выбирается с учетом различных факторов, включая способ введения, функцию почек и печени больного. Рядовой врач или ветеринар может легко определить и прописать эффективное количество отдельных активных ингредиентов, необходимое для предупреждения, противодействия или сдерживания развития заболевания. Оптимальным является выбор такой концентрации активных ингредиентов, которая попадает в допустимый диапазон концентраций, обеспечивающих эффективность лечения при отсутствии токсичности; такой выбор основывается на кинетической способности активных ингредиентов достигать мишеней.
Суточные дозировки первого средства или компонента (а) будут, конечно, варьировать в зависимости от различных факторов, например, от выбранного соединения, особенности заболевания, лечение которого проводится, и желаемого эффекта. В целом удовлетворительные результаты достигаются при введении агониста С1Ф-рецептора, например, соединения А, Б или В в суточных дозах порядка приблизительно 0,1-100 мг, в виде однократной дозы или в виде нескольких раздельных доз. Агонист С1Ф-рецептора можно вводить любым традиционным способом, в особенности энтерально, например, перорально, например, в форме таблеток, капсул, микстур, или парентерально, например, в форме растворов или суспензий для инъекций. Приемлемые формы разовых доз для перорального введения включают приблизительно от 0, 1 до 30 мг компонента (а), например, от 0,1 до 25 мг, совместно с одним или несколькими фармацевтически приемлемыми растворителями или носителями.
Фадрозол можно вводить человеку перорально в диапазоне доз, варьирующих приблизительно от 0,5 до 10 мг/сутки, предпочтительно от 1 до 2,5 мг/сутки. Экземестан можно вводить человеку перорально в диапазоне доз, варьирующих приблизительно от 5 до 200 мг/сутки, предпочтительно от 10 до 25 мг/сутки. Этот препарат можно также вводить парентерально, приблизительно от 50 до 500 мг/сутки, предпочтительно от 100 до 250 мг/сутки. Если это лекарство будет вводиться в отдельной фармацевтической композиции, его можно вводить в форме, описанной в GB 2177700. Форместан можно вводить человеку парентерально в диапазоне доз, варьирующих приблизительно от 100 до 500 мг/сутки, предпочтительно от 250 до 300 мг/сутки. Анастрозол можно вводить человеку перорально в диапазоне доз, варьирующих приблизительно от 0,25 до 20 мг/сутки, предпочтительно от 0,5-2,5 мг/сутки. Аминоглутемид можно вводить человеку в диапазоне доз, варьирующих приблизительно от 200 до 500 мг/сутки.
Цитрат тамоксифена можно вводить человеку в диапазоне доз, варьирующих приблизительно от 10 до 40 мг/сутки.
Винбластин можно вводить человеку в диапазоне доз, варьирующих приблизительно от 1,5 до 10 мг/м2/сутки. Сульфат винкристина можно вводить человеку парентерально в диапазоне доз, варьирующих приблизительно от 0,025 до 0,05 мг/кг массы тела в неделю. Винорельбин можно вводить человеку в диапазоне доз, варьирующих приблизительно от 10 до 50 мг/м2/сутки.
Фосфат этопозида можно вводить человеку в диапазоне доз, варьирующих приблизительно от 25 до 115 мг/м2/сутки, например, 56,8 или 113,6 мг/м2/сутки.
Тенипозид можно вводить человеку в диапазоне доз, варьирующих приблизительно от 75 до 150 мг, приблизительно каждые две недели. Доксорубицин можно вводить человеку в диапазоне доз, варьирующих приблизительно от 10 до 100 мг/м2/сутки, например, 25 или 50 мг/м2/сутки.
Эпирубицин можно вводить человеку в диапазоне доз, варьирующих приблизительно от 10 до 200 мг/м2/сутки. Идарубицин можно вводить человеку в диапазоне доз, варьирующих приблизительно от 0,5 до 50 мг/м2/сутки.
Митоксантрон можно вводить человеку в диапазоне доз, варьирующих приблизительно от 2,5 до 25 мг/м2/сутки.
Паклитаксел можно вводить человеку в диапазоне доз, варьирующих приблизительно от 50 до 300 мг/м2/сутки. Доцетаксел можно вводить человеку в диапазоне доз, варьирующих приблизительно от 25 до 100 мг/м2/сутки.
Циклофосфамид можно вводить человеку в диапазоне доз, варьирующих приблизительно от 50 до 1500 мг/м2/сутки. Мелфалан можно вводить человеку в диапазоне доз, варьирующих приблизительно от 0,5 до 10 мг/м2/сутки.
5-Фторурацил можно вводить человеку в диапазоне доз, варьирующих приблизительно от 50 до 1000 мг/м2/сутки, например, 500 мг/м2/сутки.
Капецитабин можно вводить человеку в диапазоне доз, варьирующих приблизительно от 10 до 1000 мг/м2/сутки. Гемцитабин гидрохлорид можно вводить человеку в дозе примерно, равной или большей чем 1000 мг/м2 в неделю. Метотрексат можно вводить человеку в диапазоне доз, варьирующих приблизительно от 5 до 500 мг/м2 в сутки.
Топотекан можно вводить человеку в диапазоне доз, варьирующих приблизительно от 1 до 5 мг/м2/сутки. Иринотекан можно вводить человеку в диапазоне доз, варьирующих приблизительно от 50 до 350 мг/м2/сутки.
Карбоплатин можно вводить человеку в диапазоне доз, варьирующих приблизительно от 200 до 400 мг/м2 приблизительно каждые четыре недели. Цисплатин можно вводить человеку в диапазоне доз, варьирующих приблизительно от 25 до 75 мг/м2 приблизительно каждые три недели. Оксалиплатин можно вводить человеку в диапазоне доз, варьирующих приблизительно от 50 до 85 мг/м2 каждые две недели.
Иматиниб можно вводить человеку в диапазоне доз, варьирующих приблизительно от 2,5 до 850 мг в сутки, более предпочтительно от 5 до 600 мг/сутки и еще более предпочтительно от 20 до 300 мг/сутки.
Алендроновую кислоту можно вводить человеку в диапазоне доз, варьирующих приблизительно от 5 до 10 мг/сутки. Клодроновую кислоту можно вводить человеку, например, в диапазоне доз, варьирующих приблизительно от 750 до 1500 мг/сутки. Этридоновую кислоту можно вводить человеку в диапазоне доз, варьирующих приблизительно от 20,0 до 400 мг/сутки. Ибандроновую кислоту можно вводить человеку в диапазоне доз, варьирующих приблизительно от 1 до 4 мг каждые три - четыре недели. Ризедроновую кислоту можно вводить человеку в диапазоне доз, варьирующих приблизительно от 20 до 30 мг/сутки. Памидроновую кислоту можно вводить человеку в диапазоне доз, варьирующих приблизительно от 15 до 90 мг каждые три - четыре недели. Тилудроновую кислоту можно вводить человеку в диапазоне доз, варьирующих приблизительно от 200 до 400 мг/сутки.
Трастузумаб можно вводить человеку в диапазоне доз, варьирующих приблизительно от 1 до 4 мг/м2 в неделю.
Бикалутамид можно вводить человеку в диапазоне доз, варьирующих приблизительно от 25 до 50 мг/м2/сутки.
1-(4-хлоранилино)-4-(4-пиридилметил)фталазин или его соль, например, сукцинат, можно вводить человеку в диапазоне доз приблизительно от 50 до 1500, более предпочтительно от 100 до 750 и наиболее предпочтительно от 250 до 500 мг/сутки.
Рапамицин или его производное, например, 40-орто-(2-гидроксиэтил)-рапамицин, можно вводить в диапазоне доз, варьирующих приблизительно от 0,1 до 25 мг.
Пример состава: мягкие капсулы
Агонисты С1Ф-рецептора, например, агонист С1Ф-рецептора, включающий группу формулы X, хорошо переносятся пациентами в дозировках, которые необходимы для применения в соответствии с настоящим изобретением. Например, острая LD50 для соединения А>10 мг/кг при пероральном ведении крысам и обезьянам.
С другой стороны, настоящее изобретение относится к использованию С1Ф-агонистов в качестве проангиогенных лекарственных средств. В последнее время считается общепризнанным, что индукция неоангиогенеза является отличной мишенью при ряде заболеваний (например, таких как миокардиальный ангиогенез, заживление раны или диабетическая сосудистая дисфункция/ васкулопатия).
Как описано выше, агонисты С1Ф-рецептора в высоких концентрациях (2 мкМ или выше, например, 2-5 мкМ или около 5 мкМ) проявляют антиангиогенные эффекты, и агонисты С1Ф-рецептора могут ингибировать VEGF-индуцированный ангиогенез. В противоположность этому низкие концентрации (0,1-1 мкМ, например, 0,1-0,5 мкМ или 0,5-1 мкМ) С1Ф-агонистов усиливают ангиогенез и способны потенцировать VEGF-опосредованный ангиогенез. Таким образом, С1Ф-агонисты могут проявлять двухфазные эффекты в отношении ангиогенеза.
Соответственно настоящее изобретение также предусматривает:
8. Использование С1Ф-агониста, например, С1Ф-агониста, содержащего группу формулы X, например, соединение А или соединение А-фосфат, в индукции процесса неоангиогенеза, например, в качестве проангиогенного агента, например, в тех случаях, где показано стимулирование ангиогенеза;
9. Способ приготовления лекарственного средства для лечения или предупреждения заболеваний, связанных с ингибированием процесса неоангиогенеза, например, заболеваний, связанных с антиангиогенными факторами, например, в тех случаях, где показано стимулирование ангиогенеза, например, в заживлении раны или в лечении инфаркта миокарда или диабетической сосудистой дисфункции/васкулопатии; способ включает использование агониста С1Ф-рецептора, например, С1Ф-агониста, включающего группу формулы X, например, соединение А или соединение А-фосфат в качестве активного ингредиента;
10. Способ лечения или предупреждения заболеваний, связанных с ингибированием процесса неоангиогенеза, например, заболеваний, связанных с антиангиогенными факторами, например, в тех случаях, где показано стимулирование ангиогенеза, например, в таких случаях, как заживление раны или лечение инфаркта миокарда или диабетической сосудистой дисфункции/васкулопатии; способ включает введение субъекту, который нуждается в таком лечении, эффективного количества агониста С1Ф-рецептора, например, С1Ф-агониста, включающего группу формулы X, например, соединения А или соединения А-фосфат.
С1Ф-агонисты, приемлемые для стимулирования ангиогенеза, включают соединения, которые указаны выше в качестве средств для лечения рака, например, С1Ф-агонисты, включающие группу формулы X, или соединения, соответствующие формулам I-IX, или их фармацевтически приемлемые соли или сложные эфиры. Предпочтительным С1Ф-агонистом является соединение А-фосфат. Можно использовать только один С1Ф-агонист, или комбинацию с одним или несколькими другими средствами, которые стимулируют ангиогенез, например, VEGF.
Для того чтобы стимулировать ангиогенез, важно подобрать в достаточной мере низкую дозу агониста С1Ф-рецептора, поскольку высокие концентрации агонистов С1Ф-рецептора ингибируют ангиогенез. Приемлемую дозу для того, чтобы обеспечить проангиогенное действие, когда С1Ф-агонист вводится больному, можно подобрать на основании исследований повышения концентраций и доз, как описано выше, п.п.А, Б и В.
Описание фигур
Фиг.1 показывает, что соединение А-фосфат существенно стимулирует образование капилляроподобной сети колоколообразным дозозависимым способом с максимальной активностью в примерной дозе 0,5 мкМ.
Фиг.2 показывает, что ни соединение А-фосфат, ни соединение А в концентрациях от 0,5 до 1 мкМ не ослабляют VEGF-опосредованное ремоделирование, но лучше взаимодействуют с полипептидным фактором роста.
Фиг.3 показывает, что образование трубочек, стимулируемое как соединением А-фосфат, так и С1Ф, практически полностью ингибируется коклюшным токсином (КТ, 50 нг/мл), ингибитором гетеротримерных G-белков αi/o-типа. Это можно объяснить возможным вовлечением сигнальных событий, опосредованных EDG-1 (С1Ф1)-рецептором, в стимулируемые соединением А-фосфат биологические ответы.
Фиг.4 показывает, что сфингозин в концентрации 1 мкМ, который сам, по-видимому, является менее эффективным, чем С1Ф, ослабляет способность как С1Ф, так и соединения А-фосфат индуцировать капилляроподобные структуры без ингибирующего воздействия на VEGF-индуцированное образование трубочек. В этом отношении сфингозин ведет себя иначе, чем соединение А. Эти данные указывают на то, что равновесие между сфингозином и С1Ф, по-видимому, является крайне важным для активации эндотелиальных клеток/ангиогенеза, что наиболее вероятно осуществляется через представителей семейства EDG-рецепторов. Существенно, что в высоких концентрациях сфингозин и соединение А (2-5 мкМ) ингибируют образование трубочек, инициированное VEGF.
Фиг.5 показывает, что обработка ЭКПВЧ соединением А-фосфат в концентрации 0,5 мкМ может вызвать временную активацию ERK1/2 с пиком фосфорилирования/ активации на 10-й минуте и возвратом к исходному уровню на 20-й минуте.
Фиг.6. Исследовали способность соединения А, соединения А-фосфат, сфингозина и С1Ф также индуцировать тканевой фактор на ЭКПВЧ. Полученные данные показывают, что ни одно из этих соединений, примененное само по себе, или в комбинации с другими соединениями, не повышают активности тканевого фактора, как показано на Фиг.6. Соединение А и соединение А-фосфат могут несколько усилить содержание VEGF-индуцируемого тканевого фактора, но не ФНО-α-индуцируемого тканевого фактора.
Фиг.7 показывает действие соединения В в анализе С1Ф-опосредованного образования трубочек ЭКПВЧ.
Список сокращений:
БСА: бычий сывороточный альбумин
ECGS: совокупность факторов роста эндотелиальных клеток
С: сфингозин
JNK1/2: c-jun-N-концевая киназа 1/2
Эквиваленты ТФ: эквиваленты тканевого фактора
EGR-1/NFAT: белок 1 ответного начального роста / ядерный фактор активированных Т-клеток
А 1 Ф: соединение А-фосфат (FTY720-фосфат)
ПХЛ: повышенная хемилюминесценция
ФСБ: фосфатно-солевой буфер
Роль агонистов С1Ф-рецептора, например С1Ф-агонистов, содержащих группу формулы X, в стимулировании ангиогенеза можно продемонстрировать на примере описанных ниже методов.
Г. Культура клеток и материалы
Эндотелиальные клетки пупочной вены человека (ЭКПВЧ) культивируют при температуре 37°С в атмосфере 5% СО2 в среде M199 с добавлением 20% SCS (фирма HyClone, Логан, Юта, США), 1 ед./мл гепарина, 50 мкг/мл ECGS, 2 мМ глутамина, 100 ед./мл пенициллина и 0,1 мг/мл стрептомицина, В эксперименте используют клетки до пятого пассажа. Используют клетки, инкубированные в условиях кратковременного голодания, для чего ЭКПВЧ культивируют в течение 5 ч в среде M199 с пониженным содержанием SCS (1%). Рекомбинантный VEGF165 человека получают от фирмы PromoCell (Гейдельберг, Германия). Поликлональнае антитела к фосфоспецифической ERK1/2, р38 киназе, антитела к нефосфо-ERK1/2 и хемилюминесцентный реагент LumiGLO (фирма New England Biolabs, Беверли, Массачусетс), поликлональные антитела к 1кВ (фирма Santa Cruz Biotechnology, Санта-Круз, Калифорния). Конъюгаты ослиного антикроличьего иммуноглобулина G (IgG) и овечьего антимышиного IgG с пероксидазой приобретены у фирмы Amersham LIFE SCIENCE (Amersham Place, Англия). Блоттинг-мембраны Immobilon P - продукция фирмы Millipore (Бедфорд, Массачусетс, США). Сфингозин получен от фирмы Sigma Chemical Co.; С1Ф - от фирмы Biomol. Исходный раствор соединения А-фосфат готовят следующим образом. Соединение А-фосфат растворяют в метаноле, содержащем следы концентрированной HCl (0,5 мг соединения А-фосфат в 500 мкл метанола, содержащего 2 мкл HCl). Из полученного раствора упаривают под вакуумом растворитель и полученный осадок растворяют (вариант 1) в 0,1% растворе обезжиренного БСА в стерильной деионизированной воде (500 мкл) или (вариант 2) в 0,5% растворе Тритона Х-100 в деионизированной воде. Полученные исходные растворы (2,5 мМ) обрабатывают ультразвуком и хранят при температуре 4°С.
Анализ коагуляции
Клетки высевают в 6-луночные планшеты при плотности 80-90% и выращивают в течение ночи. Клетки собирают из планшет и анализируют активность тканевого фактора по методике, описанной М. Clauss в J. Biol. Chem., 271, 1996, ее. 17629-17634; D. Mechtcheriakova в Blood, 93, 1999, сс.3811-3823. Вкратце анализ проводят следующим образом: клетки, после индукции в течение 4 ч с VEGF (1,5 нМ), ФНО-α ((100 ед./мл), С (0,5-2 мкМ), С1Ф (0,5-2 мкМ), соединением А (0,5-2 мкМ) и соединением А-фосфат (0,5-2 мкМ), дважды промывают, после чего собирают в 1 мл коагулирующего буфера (12 мМ ацетата натрия, 7 мМ диэтилбарбитата и 130 мМ хлорида натрия; рН 7,4). 50 мкл ресуспендированных клеток смешивают с 50 мкл цитратной плазмы; время коагуляции определяют после рекальцификации в 50 мкл раствора CaCl3 (концентрация 20 мМ) при температуре 37°С. Эквиваленты ТФ определяют, используя стандартную кривую, полученную по тромбопластину из мозга кролика.
Д. Вестерн-блот анализ
После различных обработок клетки дважды промывают холодным ФСБ, лизируют в 100 мкл буфера Лэммли, собирают и прогревают в течение 5 мин при температуре 95°С. Полученные в итоге лизаты клеток разделяют путем денатурирующего электрофореза в полиакриламидном геле в присутствии додецилсульфата натрия (SDS-ПААГ) и переносят на Immobilon Р-мембрану. Мембрану блокируют с помощью ФСБ, содержащего 0,1% твин-20 и 3% снятого молока, в течение 30 мин и инкубируют при комнатной температуре в течение 1 ч с первичным антителом, разведенным в блокирующем буфере. Полученную мембрану промывают три раза в течение 5 мин ФСБ, содержащим 0,1% твин-20, и инкубируют с конъюгатом вторичного антитела с пероксидазой в течение 1 ч при комнатной температуре. После промывания мембрану инкубируют в течение 1 мин с реагентом ECL (реагент для электрогенерированной хемилюминесценции) и экспонируют пленку в течение времени, достаточного для получения результата. Для повторного исследования с другим антителом мембрану дважды промывают в ФСБ, десорбируют в течение 30 мин при 55°С с десорбирующим буфером (62,5 мМ трис-HCl, рН 6,8, 2% SDS, 100 мМ 2-меркаптоэтанол) и промывают ФСБ три раза в течение 5 мин при комнатной температуре. После каждого иммунологического анализа мембраны хранят влажными, завернутыми в саран (SaranWrap), при 4°С.
Анализ ангиогенеза на матригеле in vitro
Морфогенез эндотелиальных клеток, заключающийся в образовании капилляроподобных структур, проводят на матрице матригеля с редуцированным количеством фактора роста (фирма BD Bioscience) в соответствии с инструкцией по применению. Вкратце анализ проводят следующим образом: ЭКПВЧ обрабатывают трипсином, ресуспендируют в бессывороточной среде M199, содержащей соевый ингибитор трипсина (1 мг/мл, фирма Sigma). После центрифугирования клетки ресуспендируют в бессывороточной среде, доводя до плотности 0,5×105 клеток/мл, и клеточную суспензию высевают в 96-луночные планшеты для культур клеток (фирма Costar, Corning Incorporated), в которые предварительно вносят по 50 мкл матригеля без/с различными стимуляторами: VEGF в концентрации 1,5 нМ, С1Ф в концентрации 0,1-2 мкМ, С в концентрации 0,5-2 мкМ, соединение А в концентрации 0,5-2 мкМ и соединение А-фосфат в концентрации 0,1-2 мкМ. Через 8 ч клетки на матригеле фиксируют 3% раствором формальдегида в ФСБ и хранят при температуре 4°С. Результаты оценивают количественно по изображениям, сделанным с помощью микроскопа “Nikon Diaphot”, оснащенным охлаждаемой камерой с многозарядным устройством (фирма Kappa GmbH, Gleichen, Германия) путем прямого подсчета точек ветвления в двух полях зрения микроскопа по каждой лунке в двукратной повторности.
Е. Анализ in vitro на матригеле формирования трубочек (капилляроподобных структур) в результате морфогенеза эндотелиальных клеток вследствие индукции соединением А-фосфат и возможное участие в этом процессе Gi-опосредованного сигнального метаболического пути (или путей)
Эффект соединения А и соединения А-фосфат на морфогенетическую дифференциацию эндотелиальных клеток определяют с использованием анализа ангиогенеза in vitro на матригеле. Морфогенез эндотелиальных клеток представляет собой сложный процесс, который требует взаимодействий клетки с внеклеточным матриксом, сопровождаемый реконструкцией матрикса, индуцированной миграцией, межклеточными взаимодействиями, и периваскулярным протеолизом. Как показано на фиг.1, соединение А-фосфат в большой степени может активировать образование капилляроподобной сети колоколообразным дозозависимым способом, показывающим максимальную активность вблизи концентрации 0,5 мкМ. Количество точек ветвления в поле зрения микроскопа, которое отражает индукционную потенцию стимула, сравнимо для соединения А-фосфат и С1Ф и может значительно превышать эффекты, инициируемые VEGF. Само по себе соединение А в концентрации 0,5-1 мкМ имеет слабое по сравнению с соединением А-фосфат, но последовательно повышающееся действие. Ни соединение А-фосфат, ни соединение А в концентрации 0,5-1 мкМ не ослабляют реконструкцию, опосредованную VEGF, но даже скорее взаимодействуют с полипептидным фактором роста (см., например, фиг.2). Более того, образование трубочек, стимулируемое соединением А-фосфат или С1Ф, полностью ингибируется коклюшным токсином (КТ, 50 нг/мл), ингибитором гетеротримерных G-белков αi/о-типа. Это можно интерпретировать как возможное вовлечение EDG-1 (С1Ф1) рецепторопосредованных сигнальных действий в стимулируемые соединением А-фосфат биологические ответы (см., например, фиг.3). Сфингозин в концентрации 1 мкМ, который сам, по-видимому, является менее активным, чем С1Ф, ослабляет способность и С1Ф, и соединения А-фосфат индуцировать капилляроподобные структуры без проявления ингибирующего действия на образование трубочек, индуцируемое VEGF (см., например, фиг.4). В этом отношении сфингозин ведет себя иначе, чем соединение А. Эти данные указывают на то, что равновесие между сфингозином и С1Ф, по-видимому, является крайне важным для активации эндотелиальных клеток/ангиогенеза, наиболее вероятно, через семейство EDG-рецепторов. Существенно то, что высокие концентрации сфингозина и соединения А (2-5 мкМ) ингибируют VEGF-инициируемое образование трубочек. Эти данные говорят о двухфазном дозозависимом воздействии соединения А и соединения А-фосфат на ангиогенез in vitro.
Ж. Активация ERK1/2 МАР-киназ соединением А-фосфат
Сигнальная трансдукция посредством МАР-киназ играет ключевую роль в осуществлении различных функций эндотелиальных клеток. Обработка ЭКПВЧ соединением А-фосфат в концентрации 0,5 мкМ может вызвать временную активацию ERK1/2, с пиком фосфорилирования/активации на 10-й минуте и возвратом к исходному уровню на 20-й минуте (см. фиг.5). При этом в ЭКПВЧ не обнаруживают активации соединением А-фосфат р38-киназы и JNK1/2. Более того, соединение А-фосфат может инициировать активацию ERK1/2 дозозависимым способом, показывая наибольшую активность при концентрации 2 мкМ. Эти данные противоположны результатам анализа образования трубочек, где соединение А-фосфат в концентрации 2 мкМ может проявлять меньшую активность, чем в концентрации 0,5 мкМ. Ни соединение А, ни сфингозин не способны индуцировать активацию МАР-киназы в эндотелиальных клетках в кинетическом диапазоне обработки от 5 минут до 60 минут. Чтобы оценить возможную роль воспалительной/NFκB-зависимой программы в биологических ответах эндотелиальных клеток, стимулируемых соединением А-фосфат, повторно исследуют мембраны с антителами к IκВ. Обработка соединением А-фосфат не влияет на уровень IκВ. Более того, обработка эндотелиальных клеток соединением А-фосфат вероятно не индуцирует экспрессию Е-селектина как NFκB-зависимого вторичного ответного гена. Таким образом, эти данные убедительно указывают на то, что подача сигнала соединением А-фосфат не вовлекает NFκB-активацию - главный каскад в острой воспалительной реакции в эндотелиальных клетках.
З. Соединения А и А-фосфат не индуцируют экспрессии тканевого фактора на эндотелиальных клетках
Важной отличительной особенностью как классического индуктора воспаления ФНО-α, так и главного ангиогенного фактора роста VEGF, на эндотелиальных клетках является их способность к усилению тканевого фактора. Исследовали способность соединения А, соединения А-фосфат, сфингозина и С1Ф также индуцировать тканевой фактор на ЭКПВЧ. Полученные данные показывают, что ни одно из этих соединений, ни в одиночку, ни в комбинациях, не может повышать активность тканевого фактора (см., например, фиг.6). Соединение А и соединение А-фосфат могут в небольшой степени усиливать тканевой фактор, индуцированный VEGF, но не ФНО-α. Вместе полученные данные указывают на то, что соединение А, соединение А-фосфат, сфингозин и С1Ф по механизму действия отличаются от ангиогенного VEGF и индуцирующего воспаление ФНО-α.
И. Способность агонистов С1Ф-рецептора к связыванию с отдельными С1Ф-рецепторами человека можно установить в следующих испытаниях:
Временная трансфекция С1Ф-рецепторов человека в клетки НЕК293
Проводят клонирование EDG-рецепторов и Gi-белков, смешивают равные количества 4-х кДНК: EDG-рецептора, Gi-α, Gi-β и Ci-γ; полученную смесь используют для трансфекции монослойных культур клеток НЕК293, для чего применяют метод преципитации фосфатом кальция (M.Wigler и др. в Cell., 11, 1977, с.223; DS. Im и др. в Mol. PharmacoL, 57, 2000, с.753). Вкратце метод заключается в следующем: смесь ДНК, содержащую 25 мкг ДНК и 0,25 М CaCl, добавляют к раствору 2 мМ Na2HPO4 в HEPES-буфере. Субконфлюэнтные монослои клеток НЕК293 протравливают 25 мМ хлорохином и затем осадок ДНК наносят на клетки. Спустя 4 ч монослои отмывают физиологическим раствором с фосфатным буфером и добавляют питательную среду (90% 1:1 модифицированная по Dulbecco основная среда (DMEM):F-12+10% фетальная телячья сыворотка). Спустя 48-72 ч после добавления ДНК клетки собирают, смывая их НМЕ-буфером (в мМ: 20 HEPES, 5MgCl2, 1 ЭДТА, рН 7,4), содержащем 10% сахарозы, на льду, и гомогенизируют с помощью гомогенизатора Даунса. После центрифугирования при 800 g супернатант разводят НМЕ без сахарозы и центрифугируют при 100000 g в течение 1 ч. Образовавшийся осадок повторно гомогенизируют и центрифугируют в течение еще одного часа при 100000 g. Полученный грубый осадок мембран ресуспендируют в НМЕ с сахарозой, делят на аликвоты и быстро замораживают путем погружения в жидкий азот. Мембраны хранят при температуре -70°С. Концентрацию белка определяют спектроскопическим анализом белков по Bradford.
Анализ связывания ГТФγS с использованием С1Ф-рецепторных / НЕК293 мембранных препаратов
Исследования связывания ГТФγS проводят по методике, описанной DS. Im и др. в Mol. PharmacoL, 57, 2000, с.753. Лигандопосредованное связывание ГТФγ8 с G-белками оценивают в ГТФ-связывающем буфере (в мМ: 50 HEPES, 100 NaCl, 10 MgCl2, pH 7,5), используя 25 мкг препарата мембран клеток НЕК293, подвергнутых временной трансфекции. Лиганд добавляют к мембранам в присутствии 10 мкМ ГДФ и 0,1 нМ [35S] ГТФγS (1200 Ки/ммоль) и инкубируют при 30°С в течение 30 мин. Связанный ГТФγS отделяют от несвязанного, используя сборник фирмы Brandel (Гейтерсберг, Мэриленд, США) и считают с помощью жидкостного сцинтилляционного счетчика.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРИМЕНЕНИЕ СРЕДСТВ, СВЯЗЫВАЮЩИХ EDG-РЕЦЕПТОР, В ЛЕЧЕНИИ РАКОВОГО ЗАБОЛЕВАНИЯ | 2008 |
|
RU2426555C2 |
| МОДУЛЯТОРЫ РЕЦЕПТОРА S1P ДЛЯ ЛЕЧЕНИЯ РАССЕЯННОГО СКЛЕРОЗА | 2007 |
|
RU2569732C9 |
| МОДУЛЯТОРЫ РЕЦЕПТОРА S1P ДЛЯ ЛЕЧЕНИЯ РАССЕЯННОГО СКЛЕРОЗА | 2007 |
|
RU2617502C2 |
| МОДУЛЯТОРЫ РЕЦЕПТОРА S1P ДЛЯ ЛЕЧЕНИЯ РАССЕЯННОГО СКЛЕРОЗА | 2007 |
|
RU2495664C2 |
| ПРОИЗВОДНЫЕ ИЗОТИОМОЧЕВИНЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ, ИХ ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2005 |
|
RU2376297C2 |
| ПРИМЕНЕНИЕ ПРОИЗВОДНОГО РАПАМИЦИНА | 2011 |
|
RU2483727C1 |
| ИНГИБИРОВАНИЕ ИЛИ ПРОФИЛАКТИКА НАРУШЕННОЙ РЕГУЛЯЦИИ АНГИОГЕНЕЗА С ПОМОЩЬЮ ПРОИЗВОДНЫХ РАПАМИЦИНА И ФАРМАЦЕВТИЧЕСКИЕ КОМБИНАЦИИ, СОДЕРЖАЩИЕ ПРОИЗВОДНОЕ РАПАМИЦИНА | 2006 |
|
RU2445093C2 |
| ЛЕЧЕНИЕ РАКА | 2005 |
|
RU2325906C2 |
| НОВЫЕ СОЕДИНЕНИЯ | 2016 |
|
RU2747645C2 |
| ПРИМЕНЕНИЕ ПРОИЗВОДНОГО РАПАМИЦИНА | 2013 |
|
RU2665138C2 |
Изобретение относится к медицине, в частности к онкологии, и касается создания лекарственного средства для контроля ангиогенеза. Для этого предлагается применять агонист рецептора сфингозин-1-фосфата, который представляет собой 2-амино-2-[2-(4-октилфенил)этил]пропан-1,3 диол, или его гидрохлорид, или фосфат. Настоящее изобретение также может включать комбинацию этих веществ с химиотерапевтическим средством. Изобретение обеспечивает регуляцию ангиогенеза, в том числе ингибирование неконтролируемого неоангиогенеза, в частности при лечении солидных опухолей. 8 з.п. ф-лы, 7 ил.
1. Применение агониста рецептора сфингозин-1-фосфата, которым является 2-амино-2-[2-(4-октилфенил)этил]пропан-1,3 диол в свободной форме или форме фармацевтически приемлемой соли для приготовления лекарственного средства для контроля ангиогенеза.
2. Применение агониста рецептора сфингозин-1-фосфата по п.1 в форме его гидрохлорида.
3. Применение агониста рецептора сфингозин-1-фосфата по п.1, где агонист рецептора сфингозин-1-фосфата является соединением формулы IVa
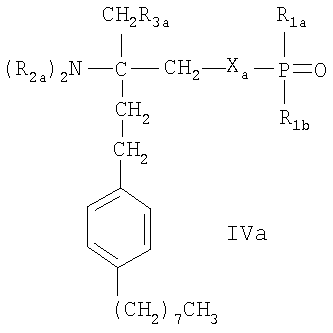
где R2a означает водород, R3a означает ОН, X означает О, каждый из R1a и R1b означает ОН или его фармацевтически приемлемая соль.
4. Применение агониста рецептора сфингозин-1-фосфата по пп.1, 2 или 3 в концентрации более чем 2 мкМ для приготовления лекарственного средства для ингибирования нарушенного ангиогенеза.
5. Применение агониста рецептора сфингозин-1-фосфата по п.3, где концентрация находится между 2 мкМ и 5 мкМ.
6. Применение агониста рецептора сфингозин-1-фосфата по пп.1, 2 или 3 в концентрации 0,1-1,0 мкМ для приготовления лекарственного средства для усиления ангиогенеза.
7. Применение агониста рецептора сфингозин-1-фосфата по пп.1, 2 или 3 для приготовления лекарственного средства для предупреждения или лечения болезней, опосредованных процессом неоангиогенеза или связанных с нарушением регуляции ангиогенеза.
8. Применение по пп.1, 2 или 3, где лекарственное средство дополнительно включает химиотерапевтическое средство.
9. Применение по п.7, где химиотерапевтическое средство выбирают из
i) ингибитора ароматазы,
ii) антиэстрогена, антиандрогена или агониста гонадорелина,
iii) ингибитора топоизомеразы I или ингибитора топоизомеразы II,
iv) средства, активного в отношении микротрубочек, алкилирующего средства, противоопухолевого антиметаболита или соединения платины,
v) соединения, связывающего/понижающего активность протеин- или липидкиназы или активность протеин- или липидфосфатазы, другого антиангиогенного соединения или соединения, которое индуцирует процессы клеточной дифференциации,
vi) рецептора брадикинина 1 или антагониста ангиотензина II,
vii) ингибитора циклооксигеназы, дифосфоната, ингибитора гистондезацетилазы, ингибитора гепараназы, модификатора биологического ответа, ингибитора многих мишеней, или ингибитора, который блокирует антиапоптозные метаболические пути,
viii) ингибитора Ras-онкогенных изоформ,
ix) ингибитора теломеразы,
x) ингибитора протеазы, ингибитора металлопротеиназы матрикса, ингибитора метионинаминопептидазы, или ингибитора протеосом, и/или
xi) mTOR-ингибитора.
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| RU 2000101856 А, 10.11.2001 | |||
| Металлический водоудерживающий щит висячей системы | 1922 |
|
SU1999A1 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| МАШКОВСКИЙ М.Д | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| - M.: ООО «Новая Волна», изд | |||
| С.Б | |||
| Дивов, 2001, т.2, с.422-423, см | |||
| Самоцентрирующийся лабиринтовый сальник | 1925 |
|
SU423A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Н.И.Переводчиковой | |||
| - М.: Медицина, 1986, с.65-69. | |||

