Настоящее изобретение относится к способу очистки соединений нороксиморфона. Данное изобретение относится также к способу получения чистых соединений нороксиморфона, в особенности налтрексона и налоксона, в особенности чистого налтрексона.
Нороксиморфон имеет химическое название 7,8-дигидро-14-гидроксинорморфинон или α,β-дигидро-14-гидроксинорморфинон и соответствует формуле
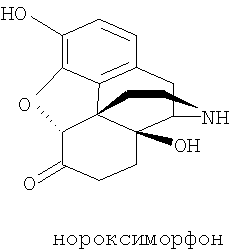
Соединения нороксиморфона и их получение описаны, например, в немецком патенте DE 2727805. Выбранным производным нороксиморфона является соединение, известное как налтрексон, которое соответствует следующей химической формуле:
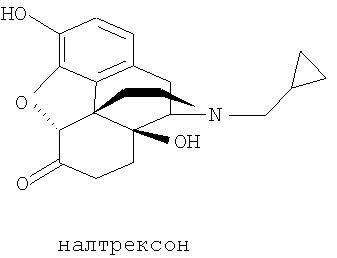
Налтрексон, его производные и соли, например налтрексона гидрохлорид, N-метилналтрексона бромид (метилналтрексон) или налтрексона метобромид, являются известными фармацевтически активными соединениями, которые применяют, в частности, для снижения психологической зависимости при злоупотреблении наркотиками. Налтрексона метобромид применяют, например, в качестве антагониста мю-рецепторов для профилактики побочных действий наркотиков. Налоксон (CAS №465-65-5), имеющий аллильный радикал в качестве заместителя у атома азота, обладает сходной фармацевтической активностью. Поскольку эти соединения являются производными морфина, их синтезируют из предшественников из класса морфиноподобных алкалоидов дикого мака. Поскольку полный синтез сложных природных веществ этого класса очень труден, исходные материалы для синтеза соединений нороксиморфона получают из растений путем экстракции. Однако экстракция из растений, в данном случае из растений мака, не позволяет получить индивидуальное соединение, а дает смесь многих структурно близких соединений. Многие из этих экстрагированных соединений токсичны или из них образуются токсичные соединения в ходе дальнейших химических превращений, например при последующем синтезе с образованием оксиморфона, нороксиморфона и налтрексона. Особенно проблемными примесями оказались α,β-ненасыщенные соединения, например соединения с формулами (Ia), (Ib) и (Ic).
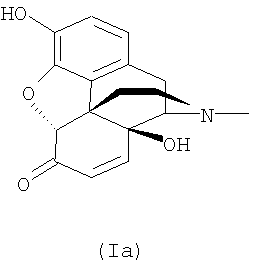
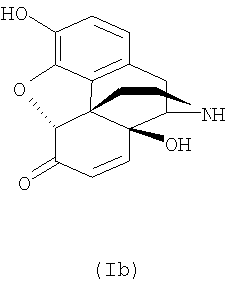
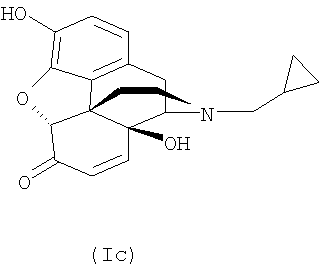
В смеси, полученной экстракцией растительного материала, среди примесей могут присутствовать также возможные предшественники этих соединений, например соответствующие α-замещенные и/или β-замещенные спирты, которые, в свою очередь, могут образовывать α,β-ненасыщенные соединения, например соединения с формулами (Ia), (Ib) и (Ic). Кроме того, при получении налтрексона из указанных растительных экстрактов могут образовываться токсичные α,β-ненасыщенные соединения, которые могут обладать мутагенной, тератогенной и/или канцерогенной активностью. Поэтому предельные уровни этих соединений в налтрексоне и производных налтрексона были снижены до 100 частей на млн. и в некоторых случаях до 10 частей на млн. Однако эти требования трудно выполнить в отношении продуктов, синтезируемых из сырья, полученного путем экстракции растительного материала известными способами.
Было обнаружено, что требования в отношении указанного выше предельного уровня 10 частей на млн. можно выполнить и обеспечить даже более низкое содержание вышеупомянутых α,β-ненасыщенных соединений, если растительный экстракт, содержащий, кроме соединения нороксиморфона, соответствующее α,β-ненасыщенное соединение и другие примеси, или продукт последующей стадии синтеза выбранного соединения нороксиморфона (а) подвергнуть реакции, в результате которой гидроксильные группы, присутствующие в этой смеси, превращаются в отщепляемые группы, (b) эти отщепляемые группы, при необходимости, можно снова удалить и затем (с) подвергнуть полученную смесь избирательному гидрированию.
Стадии (а) и (b), в том числе возможное выделение продуктов реакции, проводят предпочтительно в неводной среде, предпочтительно также в среде, не содержащей спирта. Предпочтение отдается удалению отщепляемых групп перед гидрированием. Гидрирование, т.е. стадию (с), можно проводить в присутствии апротонных растворителей и, в мягких условиях, в присутствии протонных растворителей, таких как вода и спирты. После гидрирования все оставшиеся отщепляемые группы можно удалить с помощью гидролиза.
В результате такой конверсии присутствующих в смеси гидроксильных групп в отщепляемые группы [стадия (а)] и, при необходимости, последующего удаления этих отщепляемых групп [стадия (b)] все нормируемые примеси, присутствующие в исходном материале в количестве порядка 1000 частей на млн., удаляются при гидрировании [стадия (с)] в такой степени, что их уже невозможно обнаружить с помощью анализа методом высокоэффективной жидкостной хроматографии (ВЭЖХ).
Особенно удивительно то, что в результате предварительной обработки исходного продукта, т.е. растительного экстракта, проведенной в соответствии с изобретением, гидрирование действует настолько избирательно, что все нормируемые побочные продукты удаляются практически полностью, а нужные гидроксильные группы снова образуются из отщепляемых групп в соединениях нороксиморфона, при этом имеющаяся кетогруппа не гидрируется, не удаляется и не превращается в гидроксильную группу. Такую высокую степень чистоты невозможно получить путем простого гидрирования исходной смеси. Предполагается, что возможные предшественники соединения нороксиморфона, например соответствующие α-замещенные и/или β-замещенные спирты, присутствующие в качестве примеси в смеси, полученной при экстракции из растений, изменяются в результате реакций согласно изобретению на стадии (а) или стадиях (а) и (b) в такой степени, что они или последующие продукты этих реакций (например, продукты элиминирования) превращаются в метиленовые группы в результате гидрирования на стадии (с). Однако данное изобретение не ограничивается этим объяснением.
Согласно изобретению возможно также, например, при получении нороксиморфона или при его дальнейшей переработке в налтрексон или налоксон и их соли, подвергать обработке согласно изобретению исходную смесь или любой промежуточный или конечный продукт, т.е. налтрексон или налоксон, предпочтительно исходную смесь или промежуточный продукт, на стадии (а) и стадии (b) и затем подвергать их гидрированию.
Исходная смесь состоит в основном из оксиморфона
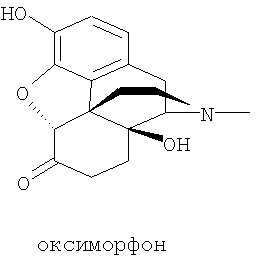
полученного из природных веществ тебаина или орипавина, экстрагированных из растительных материалов.
Данное изобретение относится к способу очистки растительных экстрактов, которые состоят, главным образом, из соединений нороксиморфона согласно формуле (II) и включают, в качестве примесей, α,β-ненасыщенные соединения нороксиморфона и другие соединения нороксиморфона
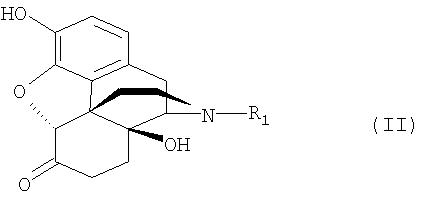
где R1 - водород или, при необходимости, замещенный фенилом или хлором алкил с 1-8 атомами углерода, алкенил с 2-4 атомами углерода или сама по себе известная, отделяемая отщепляемая группа,
отличающемуся тем, что (а) растительный экстракт или продукт последующей стадии синтеза выбранного соединения нороксиморфона подвергают реакции, в которой присутствующие в смеси гидроксильные группы превращаются в отщепляемые группы формулы -OR2, где R2 - введенный радикал отщепляемой группы, (b) эти отщепляемые группы, при необходимости, можно опять удалить, после чего (с) полученную смесь подвергают избирательному гидрированию, в результате которого образуется насыщенная связь в α,β-положении примесей соединений нороксиморфона и все оставшиеся отщепляемые группы превращаются в гидроксильную группу, после чего может быть (d) выделено чистое соединение нороксиморфона.
Настоящее изобретение относится также к соединениям оксиморфона формулы (II), очищенным по способу согласно изобретению или к смеси таких соединений, а также к фармацевтическим составам, содержащим соединение как таковое.
R1 - предпочтительно водород, алкил с 1-8 атомами углерода, алкенил с 2-4 атомами углерода или отщепляемая группа, предпочтительно алкил с 1-6 атомами углерода, аллил или водород, предпочтительно алкил с 1-6 атомами углерода или водород.
R1 в качестве отщепляемой группы является предпочтительно (С1-С4)-алкилоксикарбонилом [(С1-С6)-алкил-О-С(О)-] или фенилоксикарбонилом [фенил-О-С(О)-], предпочтительно этилоксикарбонилом, изобутилкарбонилом или трет-бутилкарбонилом (Вос), циклогексилоксикарбонилом, предпочтительно этилоксикарбонилом или трет-бутилкарбонилом (Boc). Способ введения этого радикала путем реакции соединения общей формулы (II) (где R1 - водород или замещаемый радикал), например, с ангидридом Вос (Вос-О-Вос) {[(СН3)3С-О-С(О)]2-О} или карбаматом Вос [(СН3)3С-О-С(О)-N(С1-4-алкил)2] сам по себе известен. Такие радикалы и их введение у атомов азота сами по себе известны.
Если соединение по формуле (II) является конечным продуктом, R1 в нем является предпочтительно метиленциклопропилом (-СН2-С3Н5) или аллилом (-СН2-СН=СН2). Предпочтение отдается гидрированию соединения или смеси соединений, в которых R1 не является ни метиленциклопропилом, ни аллилом, и предпочтительный конечный продукт получают из гидрированного продукта.
Определение "которые состоят, главным образом, из соединений нороксиморфона согласно формуле (II) и включают, в качестве примесей, α,β-ненасыщенные соединения нороксиморфона и другие соединения, являющиеся примесями нороксиморфона" означает, что растительные экстракты в сухом состоянии содержат всего, по меньшей мере, около 70% по массе, предпочтительно, по меньшей мере, около 80% по массе и предпочтительно, по меньшей мере, около 90% по массе соединения нороксиморфона, отношение соединения нороксиморфона формулы (II) к загрязняющим соединениям нороксиморфона составляет от 99,800 до 99,999% по массе соединений нороксиморфона формулы (II), от около 0,200 до 0,001% по массе загрязняющих соединений, и все сухие вещества, имеющееся в экстракте, составляют в сумме 100% по массе.
В отщепляемой группе формулы -OR2, -OR2 предпочтительно представляет собой эфирную группу, например формильный эфирный радикал [R2=НС(О)-], ацетильный эфирный радикал [R2=СН3С(О)-, метилкарбонил], трихлорацетильный эфирный радикал [R2=CCl3C(О)-], трифторацетильный эфирный радикал [R2=CF3C(О)-, трифторметилкарбонил], бензоильный эфирный радикал [R2=С6Н5С(О)-], при необходимости, также замещенные бензильные эфирные группы, или эфиры сульфоновых кислот, в которых R2 предпочтительно является метилсульфонилом, бензилсульфонилом или п-толуолсульфонилом. Или же -OR2 может быть остатком эфира угольной кислоты, в котором R2 - (С1-С8)-алкилоксикарбонил или фенилоксикарбонил, предпочтительно этилоксикарбонил, изобутилкарбонил или трет-бутилкарбонил (Boc), циклогексилоксикарбонил, предпочтительно этилоксикарбонил или трет-бутилкарбонил (Boc).
Способ образования эфирного остатка, например, в случае введения ацетила или трет-бутилкарбонила (Boc), путем реакции соединения общей формулы (II) с уксусным ангидридом или ацетилхлоридом или ангидридом Вос (Вос-О-Вос) {[(СН3)3С-О-С(О)]2-О} сам по себе известен. Ацетил и Вос являются здесь представителями других соединений, реагирующих таким же образом, т.е. соединений, в которых метильный или трет-бутильный радикал заменен на другой радикал с такой же реакционной способностью. Отшепляемые группы обычно удаляются в ходе реакции, например, на стадии (b) или при гидрировании, но, если это необходимо в конкретном случае, их можно удалить также после гидрирования способом, который сам по себе известен.
Предпочтение отдается введению отщепляемой группы или получению производных с помощью реакций с хлоридами кислот или ангидридами кислот, например уксусным ангидридом, ацетилхлоридом, трифторуксусным ангидридом, метансульфонилхлоридом, метансульфонилангидридом, толуолсульфонилхлоридом и родственными соединениями, которые сами по себе известны.
Превращение R1 в отщепляемую группу, если R1 представляет собой алкильную группу, известно из литературы для аналогичных реакций и не требует более подробного описания.
Предпочтение отдается проведению дальнейшей конверсии реакционной смеси, полученной на стадии (а), в безводной среде, предпочтительно также в бесспиртовой среде, поскольку в присутствии воды могут образовываться примеси, в особенности спирты в α- или β-положении, в результате присоединения воды и, возможно, спирта к α,β-ненасыщенным соединениям. Это относится к подготовительной стадии (а) и к удалению отщепляемых групп на стадии (b), в том числе к возможному выделению продуктов реакции. Гидрирование на стадии (с) можно проводить в мягких условиях в водных и/или спиртовых растворителях. Для такой обработки в безводных и предпочтительно бесспиртовых средах подходят, в частности, апротонные растворители, например трет-бутиловые эфиры.
Отщепляемая группа на стадии (а) вводится путем обработки реакционной смеси алкилирующим агентом, как описано выше, при необходимости с нагреванием. Последующее добавление органических растворителей, например МТБЭ, (метил-трет-бутилового эфира), приводит к осаждению продукта.
Способом удаления отщепляемых групп на стадии (b) предпочтительно является нагревание продукта реакции стадии (а) в неводных растворителях, если необходимо, более нескольких часов, предпочтительно в апротонных растворителях, таких как ТГФ (тетрагидрофуран), диоксан, этилацетат, МТБЭ (метил-трет-бутиловый эфир), ДМФ (диметилформамид), ДМСО (диметисульфонамид) и т.п., необязательно с добавлением основания, такого как калия трет-бутоксид или гидроксид лития, в апротонные растворители, например в ТГФ, диоксан или этилацетат. Затем продукт предпочтительно осаждается путем добавления апротонного растворителя.
Условия гидрирования сами по себе известны и приведены, например, в европейском патенте ЕР 0158476, международных заявках WO 99/02529, WO 95/32973 или WO 91/05768. В соответствии с изобретением предпочтение отдается условиям гидрирования, при которых для гидрирования на стадии (с) применяют элементарный водород и/или режимы или соединения, приводящие к образованию элементарного водорода in situ. В соответствии с изобретением, в данном случае, предпочтение отдается условиям гидрирования, при которых для гидрирования на стадии (с) в качестве источников водорода используют элементарный водород, циклогексен и/или циклогексадиен (который реагирует с выделением водорода и образованием бензола), и/или формиат аммония (который разлагается с выделением водорода и образованием диоксида углерода и аммиака) или в растворителе из класса полярных органических растворителей, при необходимости с добавлением воды для солюбилизации, например, катализаторов гидрирования. Такие катализаторы гидрирования описаны ниже. Реакции гидрогенизации с переносом водорода обычно проводят при нормальном давлении и сами по себе известны.
Особое предпочтение отдается каталитическому гидрированию с использованием благородного металла в качестве катализатора в гетерогенной или гомогенной форме. Такие катализаторы на основе благородного металла предпочтительно выбирают из соединений группы переходных металлов периодической таблицы элементов, в особенности выбирают из металлов VIII группы периодической таблицы, их соединений и комплексов, в особенности рутения (Ru) и осмия (Os), кобальта (Со), родия (Rh) и иридия (Ir), никеля (Ni), палладия (Pd) и платины (Pt). Предпочтение отдается родию, палладию и платине, в особенности палладию. Эти металлы используют как катализаторы гидрирования в способе, который сам по себе известен. Таким образом, можно проводить гидрирование в гетерогенной форме, когда катализаторы наносят на подложку, предпочтительно на активированный уголь или глинозем или на другую подложку, саму по себе известную, предпочтительно на активированный уголь.
Соединения этих металлов можно предпочтительно также использовать как гомогенные катализаторы, предпочтительно соединения палладия. Примерами таких соединений палладия являются соединения Pd(0), сами по себе известные, такие как тетракис(трифенилфосфин)палладий, и соответствующие комплексы с такими лигандами, как три(2-толуил)фосфин, три(2-фурил)фосфины, три(трет-бутил)фосфин, или бидентатные лиганды dppm [1,1-бис(дифенилфосфинометан)], dppe [1,2-бис(дифенилфосфино)этан] и родственные соединения, и комплекс трис(дибензилиденацетон)дипалладий-хлороформ, и соединения Pd(II), предпочтительно PdCl2, Pd(dppe)Cl2, Pd(OAc)2, Pd(dppe)(OAc)2, π-аллил-Pd комплексы, предпочтительно димер π-аллилпалладия хлорида. Предпочтение отдается соединениям Pd(0). Эти соединения, соли и комплексы сами по себе известны и описаны в литературе.
Эти катализаторы используют в каталитических количествах, предпочтительно в количествах 0,0005-0,01% по массе благородного металла, предпочтительно около 0,001-0,005% по массе благородного металла, по отношению к общей массе реагента. Однако указанный верхний предел не является критическим. Поэтому можно также использовать большие количества катализаторов, например эквимолярные количества по массе исходного продукта. Однако обычно в этом нет необходимости.
Гидрирование предпочтительно проводят газообразным водородом предпочтительно в инертном растворителе, например в органических кислотах, предпочтительно в ледяной уксусной кислоте, муравьиной кислоте, пропионовой кислоте или смеси этих соединений; в спиртах, предпочтительно в метаноле, этаноле, изопропиловом спирте, н-бутаноле или в смеси этих соединений; в нитрилах, предпочтительно в ацетонитриле и/или пропионитриле; в кетонах, предпочтительно в ацетоне и/или 2-бутаноне; в эфирах, таких как этилацетат, в полярных апротонных растворителях, предпочтительно в диметилформамиде (ДМФ) или диметилсульфонамиде (ДМСО), при необходимости с добавлением воды. Предпочтение отдается протонным растворителям, в особенности метанолу, этанолу, изопропиловому спирту, н-бутанолу, или апротонным полярным растворителям, предпочтительно ацетону, ДМФ, ацетонитрилу, при необходимости, в смеси с 1-99% по массе воды и предпочтительно в присутствии органической кислоты, например уксусной кислоты, трифторуксусной кислоты, пропионовой кислоты, муравьиной кислоты, предпочтительно уксусной кислоты, предпочтительно в концентрации от 0,1% по массе до 99% по массе. Гидрирование предпочтительно проводят при температуре в интервале от 0°С до 150°С, предпочтительно в интервале от 20°С до 100°С, предпочтительно в интервале от нормального давления до 100 бар, предпочтительно в интервале от нормального давления до 10 бар.
Вместо водорода можно также использовать соединения, которые выделяют водород в реакции in situ, например в реакции переноса водорода с формиатом аммония, циклогексеном и/или циклогексадиеном. В этом случае водород удаляется в предшествующей реакции с катализом реагента.
Настоящее изобретение относится также к способу получения чистого нороксиморфона из растительных экстрактов, которые состоят, главным образом, из нороксиморфона и содержат загрязняющие соединения нороксиморфона, отличающемуся тем, что оксиморфон указанной выше формулы (II), в которой R1 - метил, вначале загружают в виде растительного экстракта и (а) растительный экстракт подвергают реакции, в результате которой присутствующие в смеси гидроксильные группы превращаются в отщепляемые группы формулы -OR2, в которой R2 представляет собой введенный радикал отщепляемой группы, такой как те, что описаны ранее для R1, предпочтительно ацил, предпочтительно ацетил;
(а1) N-метильную группу [соответствующая определению R1 для соединения формулы (II)] удаляют и заменяют отщепляемую группу R3, в которой R3 - отщепляемая группа, такая как те, что описаны ранее для R1, предпочтительно алкилоксикарбонил, предпочтительно этилоксикарбонил или Boc, предпочтительно этилоксикарбонил;
(а2) отщепляемые группы R2 и R3 могут быть удалены из продукта реакции, полученного на стадиях (а) и (а1);
(b) по меньшей мере, один из продуктов, полученных на стадиях (а), (а1) и/или (а2), предпочтительно один из продуктов, полученных на стадиях (а1) или (а2), предпочтительно на стадии (а2), подвергают реакции избирательного гидрирования, как описано выше, и
(c) при необходимости, выделяют чистое соединение нороксиморфона.
Продукт, полученный на стадии (а2), может быть подвергнут дальнейшей обработке, предпочтительно с образованием налтрексона или налоксона, или соли этих соединений, или четвертичного производного этих соединений, предпочтительно гидрохлорида, гидробромида, метохлорида или метобромида, предпочтительно соответствующих солей или четвертичных производных налтрексона.
Избирательное гидрирование также удаляет отщепляемые группы, но, при необходимости, это может быть сделано отдельно на стадии (а2) и/или если необходимо, в заключение, после гидрирования.
На стадии (а) оксиморфон предпочтительно этерифицируют уксусным ангидридом с образованием метил-трет-бутилового эфира (МТБЭ), обрабатывают в безводных условиях и выделяют с получением диацетилоксиморфона (R2=ацетил).
На стадии (а1) предпочтение отдается превращению с помощью этилхлорформиата в апротонном растворителе, предпочтительно ацетонитриле, деметилированию в щелочных условиях, например с K2СО3, с выделением соединения оксиморфона, в котором R3 - этоксикарбонил, или конечное соединение является соответствующим этоксикарбаматом диацетилоксиморфона.
На стадии (а2) отщепляемые группы R2 и R3 удаляют из продукта реакции, полученного на стадиях (а) и (а1). С этой целью продукт реакции стадии (а) или (а1) нагревают в неводных растворителях, предпочтительно в апротонных растворителях, таких как ТГФ, диоксан, этилацетат, МТБЭ, ДМФ, ДМСО и т.п., если необходимо свыше нескольких часов, если необходимо с добавлением основания, такого как калия трет-бутоксид или гидроксид лития, в апротонных растворителях, например ТГФ, диоксане, этилацетате. Этот продукт затем предпочтительно осаждают, добавляя апротонный растворитель.
На стадии (b) выделенный продукт, например диацетилоксиморфона этоксикарбамат, предпочтительно растворяют в ледяной уксусной кислоте и подвергают гидрированию, вводя газообразный водород в указанных выше условиях, с использованием палладия на активированном угле в качестве катализатора. Впоследствии оставшиеся отщепляемые группы R4 и R5 удаляют, добавляя в реакционную смесь 40%-ную серную кислоту с образованием нороксиморфона сульфата, который можно, при необходимости, выделить. Добавление основания, например, путем добавления раствора аммиака в смеси этанол/вода, позволяет нейтрализовать и подготовить реакционную смесь для выделения свободного нороксиморфона. Свободный нороксиморфон нерастворим в смеси вода/этанол при слабощелочном рН, предпочтительно рН 8-10, и выпадает в виде кристаллического осадка при доведении рН, что позволяет отделить его фильтрованием. При анализе с помощью ВЭЖХ α,β-ненасыщенные соединения не обнаруживают в выделенном нороксиморфоне. Полученный таким образом нороксиморфон можно подвергнуть дальнейшей обработке, предпочтительно с образованием высокочистого налтрексона или налоксона (CAS №465-65-5) или с образованием солей или четвертичных производных. Предпочтительными солями являются гидрохлориды и гидробромиды. Предпочтительными четвертичными производными являются соединения налтрексон метобромид (известный как метилналтрексон) или налоксон метобромид [известный как метилналоксон (CAS №73232-50-5)]. Предпочтение отдается налтрексона гидрохлориду или гидробромиду и налтрексона метобромиду.
Нороксиморфон, полученный в соответствии с изобретением, можно подвергнуть обработке, например, с образованием высокочистого налтрексона или высокочистого налоксона, или с образованием высокочистой соли или четвертичного производного этих соединений.
В этой связи данное изобретение относится к способу получения высокочистых солей и четвертичных производных налтрексона и налоксона, в которых содержание предельных олефиновых примесей ниже предела обнаружения, предпочтительно солей налтрексона, путем проведения реакции исходного материала, содержащего нороксиморфон, с соответствующим алкилирующим агентом, т.е. с циклопропилметил-бромидом (для налтрексона) или аллилбромидом (для налоксона), и реакции продукта, содержащего налтрексон или налоксон, с кислотой, предпочтительно с разбавленной соляной кислотой или бромистоводородной кислотой, с образованием соответствующей соли, в описанном случае с образованием гидрохлорида или гидробромида; или с дальнейшей реакцией с алкилирующим агентом, предпочтительно метилбромидом, с получением налтрексона метобромида или налоксона метобромида, отличающемуся тем, что, по меньшей мере, исходный материал или продукт стадий (а) или (b), полученный в качестве промежуточного продукта или конечного продукта, предпочтительно продукт стадий (а) или (b), предпочтительно стадии (b), полученный в качестве промежуточного продукта, подвергают реакции гидрирования, как описано выше. Следующие примеры иллюстрируют изобретение.
Пример 1 [получение диацетилоксиморфона (ДАОМ), введение отщепляемой группы с прямым удалением отщепляемой группы]
20 г оксиморфона суспендируют в смеси 10 г трет-бутилметилового эфира и 21 г уксусного ангидрида (3,24 экв.) при комнатной температуре. Реакционный раствор нагревают с обратным холодильником в течение 5 часов. Затем охлаждают и добавляют 70 г трет-бутилметилового эфира. Нагревают суспензию еще раз до температуры флегмы, затем охлаждают до 0-4°С и перемешивают до полного осаждения. Продукт отфильтровывают с отсасыванием, промывают трет-бутилметиловым эфиром и высушивают до постоянного веса при 90°С при пониженном давлении. Выход 23 г (91% в расчете на использованный оксиморфон); чистота по ВЭЖХ 98%. Продукт содержит следы (около 1000 частей на млн.) α,β-ненасыщенного соединения; 3,8,14-триацетилоксиморфон не обнаруживают.
Пример 2 [получение диацетилоксиморфона (ДАОМ), содержащего следы 3,8,14-триацетилоксиморфона; введение отщепляемой группы]
20 г оксиморфона суспендируют в смеси 10 г трет-бутилметилового эфира и 21 г уксусного ангидрида (3,24 экв.) при комнатной температуре. Реакционный раствор нагревают при температуре не выше 30-40°С в течение 48 часов. Затем охлаждают и добавляют 70 г трет-бутилметилового эфира. Эту смесь охлаждают до 0-4°С и перемешивают до полного осаждения. Продукт отфильтровывают с отсасыванием, промывают трет-бутилметиловым эфиром и высушивают до постоянного веса при температуре 30°С при пониженном давлении. Выход 26,8 г (91% в расчете на использованный оксиморфон); чистота по ВЭЖХ 98%. Продукт содержит следы 3,8,14-триацетилоксиморфона.
Пример 3 [получение диацетилоксиморфона карбамата] Суспендируют 30 г диацетилоксиморфона с 66 г этилхлорформиата (8 экв.) и гетерогенного основания (1 экв. карбоната калия) в органическом растворителе (74 г ацетонитрила) и нагревают при повышенной температуре (65-68°С) в течение нескольких часов (24-28 часов). После окончания реакции отгоняют ацетонитрил и этилхлорформиат при пониженном давлении. Добавляют к осадку 73 г ацетонитрила. Затем отфильтровывают гетерогенное основание (KHCO3/K2CO3) при комнатной температуре. Отгоняют ацетонитрил при пониженном давлении и добавляют 60 г трет-бутилметилового эфира для полного осаждения. После нагревания до температуры флегмы охлаждают смесь до 0-5°С и продолжают перемешивать, затем отфильтровывают твердый осадок с отсасыванием и промывают сначала трет-бутилметиловым эфиром, затем водой. Высушивают бесцветный продукт при пониженном давлении и при 80°С до постоянного веса. По данным ВЭЖХ продукт содержит >1000 частей на млн. α,β-ненасыщенных соединений. Выход 29 г (86% в расчете на использованный диацетилоксиморфон). Чистота по ВЭЖХ >95%.
Пример 4 [конверсия 3,14-диацетилоксиморфона (ДАОМ), содержащего следы 3,8,14-триацетилоксиморфона, до 3,14-диацетилоксиморфона (ДАОМ), не содержащего 3,8,14-триацетилоксиморфона; удаление отщепляемой группы]
Суспендируют 20 г диацетилоксиморфона со следами 3,8,14-триацетилоксиморфона в смеси 20 г трет-бутилметилового эфира и 3-5 г уксусной кислоты при комнатной температуре. Нагревают реакционный раствор при температуре 70°С в течение 10-15 часов. Затем охлаждают и добавляют 70 г трет-бутилметилового эфира. Смесь охлаждают до 0-4°С и перемешивают до полного осаждения. Продукт отфильтровывают с отсасыванием, промывают трет-бутилметиловым эфиром и высушивают до постоянного веса при температуре 30°С при пониженном давлении. Выход 15,7 г (91% в расчете на использованный диацетилоксиморфон); чистота по ВЭЖХ 98%, продукт содержит около 1000 частей на млн. α,β-ненасыщенного соединения, 3,8,14-триацетилоксиморфон не обнаруживается.
Пример 5 [гидрирование с отщепляемой группой]
Растворяют 20 г диацетилоксиморфона со следами 3,8,14-триацетилоксиморфона, полученного, как в примере 2, в 60 г ледяной уксусной кислоты при комнатной температуре. Добавляют 0,6 г увлажненного водой палладия на активированном угле (10% Pd по сухому веществу, содержание воды около 50%). Затем вводят газообразный водород при температуре продукта 50-60°С и давлении 2,7 бар. После гидрирования отфильтровывают катализатор и концентрируют раствор до половины объема при пониженном давлении. Затем добавляют МТБЭ и охлаждают смесь до 0-4°С. Продукт отфильтровывают с отсасыванием, промывают МТБЭ и высушивают при температуре 70°С при пониженном давлении. Чистота 98%; α,β-ненасыщенные соединения и 3,8,14-триацетилоксиморфон не обнаруживаются, выход 85% (диацетилоксиморфона в расчете на использованный диацетилоксиморфон).
Пример 6 [гидрирование с удаленными отщепляемыми группами; получение нороксиморфона]
Растворяют 30 г диацетилоксиморфона карбамата, полученного, как в примере 3, в 60 г ледяной уксусной кислоты при комнатной температуре. Добавляют 0,6 г увлажненного водой палладия на активированном угле (10% Pd по сухому веществу, содержание воды около 50%). Затем вводят газообразный водород при температуре продукта 50-60°С и давлении 2,7 бар. После гидрирования отфильтровывают катализатор и концентрируют раствор до половины объема при пониженном давлении. Добавляют тройной объем 40% серной кислоты к концентрированному раствору в ледяной уксусной кислоте. Кипятят карбамат с обратным холодильником для образования свободного амина. В ходе этого процесса продукт выпадает в виде сульфатной соли. Отфильтровывают образовавшуюся соль и промывают небольшим количеством охлажденного этанола. Растворяют полученный твердый осадок в смеси вода/этанол и доводят рН раствора до 9 водным раствором аммиака. При этом рН выпадает свободный нороксиморфон, который нужно отфильтровать. При анализе методом ВЭЖХ α,β-ненасыщенные побочные продукты не обнаруживают. Выход 70-75% (в расчете на использованный диацетилоксиморфонкарбамат).
Чистота по ВЭЖХ >98%, α, β-ненасыщенное соединение и 3,8,14-триацетилоксиморфон не обнаруживают.
Пример 7 [получение налтрексона гидрохлорид]
(A) 100 г нороксиморфона, полученного как в примере 6, суспендируют в 165 г N-метилпирролидона с 37 г карбоната натрия, после чего добавляют 56 г бромметилциклопропана. Суспензию нагревают до 70°С в течение 3 часов и охлаждают до комнатной температуры. Продукт кристаллизуют с помощью 800 г воды, внося затравку кристаллов налтрексона. При добавлении всего количества воды значение рН доводят до 9,5 путем добавления гидроксида натрия. Продукт выделяют, промывают водой и сушат под вакуумом. Выход 113 г сырого налтрексона, чистота более чем 95% (чистота >95%).
(B) 102 г сырого налтрексона, полученного, как в пункте (А), нагревают в колбе с обратным холодильником вместе с 800 г этилацетата и 7 г активированного угля. Смесь фильтруют при 75°С. Фильтрат еще раз смешивают с активированным углем и нагревают в колбе с обратным холодильником. После фильтрации активированного угля часть этилацетата выпаривают при атмосферном давлении. В полученный раствор добавляют затравку кристаллов налтрексона и продукт кристаллизуют охлаждением раствора до 0°С. Кристаллизованный налтрексон отфильтровывают, промывают некоторым количеством этилацетата и сушат под вакуумом. Выход 77 г кристаллического налтрексона; чистота более чем 99% (чистота >99%).
(C) 100 г кристаллического налтрексона, полученного, как в пункте (В), нагревают в 300 г воды и 34 г концентрированной соляной кислоты до температуры кипения. Прозрачный раствор медленно охлаждают до 2°С и добавляют затравку налтрексон · HCl. Продукт выделяют, промывают водой и сушат под вакуумом. Выход 99 г налтрексон · HCl, чистота более чем 99,8% (чистота >99,8%).
Пример 8 [получение налоксона гидрохлорида]
(А) 80 г нороксиморфона, полученного, как в примере 6, суспендируют в 140 г диметилацетамида, после чего добавляют 36 г аллилбромида и 35 г диизопропилэтиламина. Смесь перемешивают в течение 6 часов при 25°С. Затем 128 г ацетона добавляют и медленно добавляют 300 г воды, после чего осаждается основание налоксона. Суспензию далее перемешивают в течение 1 часа при 25°С, затем охлаждают до 0°С и далее перемешивают при данной температуре в течение 4 часов. Продукт отфильтровывают, промывают смесью 80 г ацетона и 80 г воды и сушат под вакуумом при 60°С. Выход 81,5 г основания налоксона, чистота более чем 99% (чистота >99%).
(В) 30 г основания налоксона, полученного, как в пункте (А), нагревают вместе с 2 г активированного угля и смесью 240 г изопропанола и 25 г ацетонитрила при 45°С. Смесь затем фильтруют. Далее добавляют смесь 15 г воды и 12 г концентрированной соляной кислоты. В раствор добавляют затравку налоксон · HCl и выдерживают в течение 60 минут при 45°С, в результате чего продукты начинают кристаллизоваться. Суспензию продукта далее медленно охлаждают до 0°С. Перемешивание продолжают при данной температуре в течение 3 часов. Продукт, налоксон · HCl, отфильтровывают, промывают 20 г изопропанола и сушат под вакуумом при 40°С в течение 17 часов. Выход 31,0 г; чистота более чем 99,8% (чистота >99,8%).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ МОРФИНАН-6-ОНА | 2014 |
|
RU2652786C2 |
| СПОСОБ ПОЛУЧЕНИЯ НАЛТРЕКСОНА | 2011 |
|
RU2448709C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ МОРФИНОНА, СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 14-ГИДРОКСИМОРФИНОНА, И СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ОКСИМОРФОНА | 2002 |
|
RU2236412C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ 14-ГИДРОКСИНОРМОРФИНОНОВ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1998 |
|
RU2183636C2 |
| ПРОИЗВОДНЫЕ НИТРОСУЛЬФОБЕНЗАМИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ ДЛЯ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ СУЛЬФОНИЛМОЧЕВИНЫ И ИХ ПРЕДШЕСТВЕННИКОВ | 2000 |
|
RU2359963C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГИДРИРОВАННОГО НИТРИЛЬНОГО КАУЧУКА, ОБЛАДАЮЩЕГО НИЗКОЙ МОЛЕКУЛЯРНОЙ МАССОЙ | 2010 |
|
RU2548681C9 |
| СПОСОБ ПОЛУЧЕНИЯ ОКСИМОРФОНА | 2014 |
|
RU2637934C2 |
| КАТАЛИЗАТОРЫ НА ОСНОВЕ РУТЕНИЯ ДЛЯ МЕТАТЕЗИСА НИТРИЛЬНЫХ КАУЧУКОВ | 2010 |
|
RU2566501C9 |
| ЦИКЛОПЕНТАДИЕНИЛЬНЫЕ ПРОИЗВОДНЫЕ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1996 |
|
RU2159758C2 |
| ДИФОСФИНЫ И МЕТАЛЛОКОМПЛЕКСЫ | 2006 |
|
RU2408600C2 |
Изобретение относится к способу очистки растительных экстрактов, которые состоят, главным образом, из соединений нороксиморфона и содержат в качестве примесей α,β-ненасыщенные соединения нороксиморфона, путем (а) преобразования растительного экстракта или продукта последующей стадии в синтезе выбранного соединения нороксиморфона в результате реакции, превращающей присутствующие в смеси гидроксильные группы в отщепляемые группы формулы -OR2, в которой R2 представляет собой введенный радикал отщепляемой группы, (b) эти отщепляемые группы, при необходимости, могут быть снова удалены, после чего (с) полученную смесь подвергают избирательному гидрированию так, что образуется насыщенная связь в α,β-положении ненасыщенных соединений нороксиморфона, и все оставшиеся отщепляемые группы превращают в гидроксильную группу, после чего (d) чистое соединение нороксиморфона выделяют; подвергают переработке нороксиморфона, очищенного таким образом, в налтрексон или налоксон, или соль этих соединений, или четвертичное производное этих соединений, которые являются известными фармацевтически активными соединениями, применяемыми, в частности, для снижения психологической зависимости и при злоупотреблении наркотиками. 2 н. и 19 з.п. ф-лы.
1. Способ очистки растительных экстрактов, которые состоят, в основном, из соединений нороксиморфона формулы (II) и которые содержат в качестве примесей α,β-ненасыщенные соединения нороксиморфона и другие загрязняющие соединения нороксиморфона:
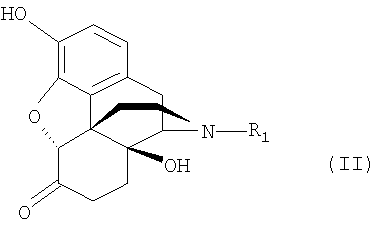
где R1 - водород, или, при необходимости, замещенный фенилом или хлором алкил с 1-8 атомами углерода, алкенил с 2-4 атомами углерода, или сама по себе известная отделяемая отщепляемая группа, предпочтительно карбаматная группа, отличающийся тем, что (а) растительный экстракт или продукт последующей стадии синтеза выбранного соединения нороксиморфона преобразуют по реакции, превращающей гидроксильные группы, присутствующие в смеси, в отщепляемые группы формулы -OR2, где R2 - введенный радикал отщепляемой группы, (b) эти отщепляемые группы, при необходимости, снова удаляют, затем (с) полученную смесь подвергают избирательному гидрированию, в результате которого образуется насыщенная связь в α,β-положении примесей соединений нороксиморфона и все оставшиеся отщепляемые группы превращаются в гидроксильную группу, после чего, при необходимости, (d) выделяют чистое соединение нороксиморфона.
2. Способ по п.1, отличающийся тем, что отщепляемые группы удаляют перед гидрированием.
3. Способ по п.1, отличающийся тем, что стадию (а) и стадию (b), включающие возможное выделение продуктов реакции, проводят в безводной среде, предпочтительно в среде, не содержащей спирта.
4. Способ по п.1, отличающийся тем, что гидрирование [стадию (с)] проводят в присутствии апротонных или протонных растворителей, предпочтительно в воде и спиртах.
5. Способ по п.1, отличающийся тем, что отщепляемые группы, оставшиеся после гидрирования [стадия (с)], удаляют с помощью гидролиза.
6. Способ по п.1, отличающийся тем, что R1 представляет собой водород, алкил с 1-8 атомами углерода, алкенил с 2-4 атомами углерода или отщепляемую группу; предпочтительно алкил с 1-6 атомами углерода, аллил или водород, предпочтительно алкил с 1-6 атомами углерода или водород, и, если соединение формулы (II) является конечным продуктом, R1 является предпочтительно метиленциклопропилом или аллилом.
7. Способ по п.1, отличающийся тем, что отщепляемая группа R1 представляет собой (С1-С4)-алкилоксикарбонил или фенилоксикарбонил, предпочтительно этилоксикарбонил, изобутилкарбонил, или трет-бутилкарбонил (Boc), циклогексил-оксикарбонил, предпочтительно этилоксикарбонил или трет-бутилкарбонил (Boc).
8. Способ по п.1, отличающийся тем, что отщепляемая группа формулы -OR2 является эфирным остатком, предпочтительно формильным эфирным радикалом, ацетильным эфирным радикалом, трихлорацетильным эфирным радикалом, трифторацетильным эфирным радикалом, бензоильным эфирным радикалом, возможно замещенными бензильными эфирными группами, т.е. тем, что R2 в радикале -OR2 является формилкарбонилом, метилкарбонилом, трихлорметилкарбонилом, трифторметилкарбонилом, фенилкарбонилом, при необходимости, замещенным фенилкарбонилом, или -OR2 является эфирным остатком сульфоновой кислоты, в котором R2 - метилсульфонил, бензилсульфонил или п-толуолсульфонил.
9. Способ по п.1, отличающийся тем, что -OR2 является эфирным остатком карбоновой кислоты, в котором R2 является (С1-С8)-алкилоксикарбонилом или фенилоксикарбонилом; предпочтительно этилоксикарбонилом, изобутилкарбонилом или трет-бутилкарбонилом (Boc), циклогексилоксикарбонилом, предпочтительно этилоксикарбонилом или трет-бутилкарбонилом (Boc).
10. Способ по п.1, отличающийся тем, что растительный экстракт, в котором R1 не является ни метиленциклопропилом, ни аллилом, гидрируют и из этого гидрированного продукта получают соединение, в котором R1 является метиленциклопропилом или аллилом.
11. Способ по п.1, отличающийся тем, что растительный экстракт в сухом виде содержит, по меньшей мере, около 70% по массе, предпочтительно, по меньшей мере, 80% по массе и предпочтительно, по меньшей мере, около 90% по массе соединений нороксиморфона, отношение соединений нороксиморфона формулы (II) к загрязняющим соединениям нороксиморфона находится в интервале от 99,800 до 99,999% по массе соединений нороксиморфона формулы (II) от около 0,200 до 0,001% по массе загрязняющих соединений, и все сухие вещества, имеющиеся в экстракте, составляют в сумме 100% по массе.
12. Способ п.1, отличающийся тем, что для гидрирования используют элементарный водород и/или соединения, образующие элементарный водород in situ.
13. Способ по п.12, отличающийся тем, что для гидрирования используют элементарный водород, циклогексен, циклогексадиен и/или формиат аммония в растворителе, выбранном из класса полярных органических растворителей и воды, и в присутствии катализаторов гидрирования.
14. Способ по п.1, отличающийся тем, что гидрирование проводят водородом с использованием благородного металла в качестве катализатора в гетерогенной или гомогенной форме, который предпочтительно выбирают из соединений группы переходных металлов Периодической таблицы элементов, предпочтительно из металлов VIII группы Периодической таблицы, их соединений и комплексов, предпочтительно из рутения, осмия, кобальта, родия, иридия, никеля, палладия, платины, предпочтительно из родия, палладия и платины, предпочтительно палладия.
15. Способ по п.14, отличающийся тем, что гидрирование проводят в гетерогенной форме, при этом катализаторы наносят на подложку, предпочтительно на активированный уголь или глинозем или на другую, саму по себе известную подложку, предпочтительно на активированный уголь.
16. Способ по п.14, отличающийся тем, что для гидрирования используют гомогенные катализаторы, предпочтительно соединения палладия, предпочтительно сами по себе известные соединения Pd(0).
17. Способ по п.16, отличающийся тем, что в качестве катализатора используют тетракис(трифенилфосфин)палладий и соответствующие комплексы с такими лигандами, как три-(2-толуил)фосфин, три-(2-фурил)фосфины, три-(трет-бутил)фосфин, или бидентатные лиганды dppm [1,1-бис(дифенилфосфинометан)], dppe [1,2-бис(дифенилфосфино)этан] и родственные соединения, комплекс трис-(дибензилиденацетон)дипалладий-хлороформ, соединения Pd(II), предпочтительно PdCl2, Pd(dppe)Cl2, Pd(OAc)2, Pd(dppe)(OAc)2, π-аллил-Pd комплексы, предпочтительно димер π-аллилпалладия хлорида.
18. Способ по п.14, отличающийся тем, что эти катализаторы используют в каталитических количествах, предпочтительно в количествах 0,0005-0,01% по массе благородного металла, предпочтительно около 0,001-0,005% по массе благородного металла, по отношению к массе неочищенного реагента.
19. Способ по п.1, отличающийся тем, что гидрирование проводят с помощью газообразного водорода в инертном растворителе, предпочтительно в органическом протонном или апротонном полярном растворителе, при необходимости, в смеси с водой, 1-99% по массе, предпочтительно в присутствии органической кислоты, в концентрации предпочтительно от 0,1 до 99% по массе и при температуре в интервале предпочтительно от 0 до 150°С, предпочтительно в интервале от 20 до 100°С, и предпочтительно в интервале от нормального давления до 100 бар, предпочтительно в интервале от нормального давления до 10 бар.
20. Способ по п.12, отличающийся тем, что вместо водорода используют соединения, которые выделяют водород в реакции in situ, предпочтительно в форме гидрирования с переносом водорода с формиатом аммония, циклогексеном или циклогексадиеном.
21. Способ получения чистого нороксиморфона из растительных экстрактов, которые состоят, главным образом, из нороксиморфона и содержат загрязняющие соединения нороксиморфона, отличающийся тем, что оксиморфон формулы (II) по п.1, где R1 - метил, вначале загружают в виде растительного экстракта и (а) растительный экстракт подвергают реакции, в результате которой гидроксильные группы, присутствующие в смеси, превращаются в отщепляемые группы формулы -OR2, в которой R2 представляет собой введенный радикал отщепляемой группы по пп.1, 8 и 9, предпочтительно ацил, предпочтительно ацетил;
(а1) N-метильную группу удаляют и заменяют на отщепляемую группу R3, в которой R3 - введенный радикал отщепляемой группы по пп.1, 8 и 9, предпочтительно алкилоксикарбонил, предпочтительно этилоксикарбонил или Boc, предпочтительно этилоксикарбонил;
(а2) отщепляемые группы R2 и R3, при необходимости, удаляют из продукта реакции, полученного на стадиях (а) и (а1);
(b) по меньшей мере, один из продуктов, полученных на стадиях (а), (а1) и/или (а2), предпочтительно один из продуктов, полученных на стадиях (а1) или (а2), предпочтительно на стадии (а2), подвергают реакции избирательного гидрирования, как описано выше, и
(c) при необходимости, чистое соединение нороксиморфона выделяют.
| 0 |
|
SU156478A1 | |
| WO 9902529 А1, 21.01.1999 | |||
| WO 9532973 А1, 07.12.1995 | |||
| WO 9105768 А1, 02.05.1991 | |||
| АМИНОПИРАЗИНОВЫЕ СОЕДИНЕНИЯ СО СВОЙСТВАМИ АНТАГОНИСТА A2A | 2015 |
|
RU2727805C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ МОРФИНОНА, СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 14-ГИДРОКСИМОРФИНОНА, И СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ОКСИМОРФОНА | 2002 |
|
RU2236412C2 |

