Изобретение относится к способу получения производных морфинона, которые являются промежуточными соединениями для получения производных 14-гидроксиморфинона, которые, в свою очередь, используются для получения антагонистов опиатов-производных оксиморфона. Антагонисты опиатов являются важными лекарственными средствами для лечения и диагностики наркомании.
Наиболее широко применяемым способом производства антагонистов опиатов - производных оксиморфона, таких как, например, налтрексон, и налоксон является способ, описанный в The Organic Chemistry of Drug Synthesis. Vol. 1 John Wiley&Sons, New York, 1990, p. 289-291, включающий окисление тебаина с получением 14-гидроксикодеинона, его гидрирование, последовательное деалкилирование метильных групп у фенильного кольца и азота с обработкой полученного нороксиморфона соответствующим алкилгалогенидом. Основным недостатком данного способа получения является необходимость использования в качестве исходного сырья дорогостоящего тебаина, т.к. количество производимого тебаина лимитировано вследствие очень низкого его содержания в опиумной смоле.
В патентах США № 5922876 и № 6008355 раскрыты способы получения производных оксиморфона и оксикодона, включающие получение производных морфинона из морфина и кодеина прямым окислением по Сверну, а именно действием ДМСО с оксалилхлоридом в хлористом метилене при -78°С с получением технического продукта с последующей обработкой уксусным ангидридом и получением диенолацетатов, вторичным их окислением перекисью или надкислотой с получением производных 14-гидроксиморфинона, которые гидрируют с получением оксикодона и оксиморфона, которые далее деалкелируют по азоту с получением норпроизводных, которые алкилируют с получением антагонистов опиатов. Основным недостатком указанного способа является проведение окисления морфина и кодеина в производные морфинона в жестких условиях, что делает данный способ неудобным для использования его в промышленности.
Помимо способа получения производных морфинона окислением кодеина и морфина, который описан выше как одна из стадий способа получения производных оксиморфона и оксикодона, наиболее широко применимым может считаться способ получения данных продуктов из тебаина реакцией галогеноводорода в присутствии катализатора с последующим гидролизом, описанный в патенте США № 4052402. Недостатком этого способа также является использование дорогостоящего тебаина.
Производные морфинона также могут быть получены окислением морфина и кодеина различными сильными окислителями, однако селективность этого окисления низкая, причем в некоторых случаях окисление идет в сторону образования других продуктов (Beilstein, 27 II 136).
Задачей настоящего изобретения является разработка более простого, технологичного, с использованием более доступного и дешевого исходного сырья способа получения производных морфинона, производных 14-гидроксиморфинона и способ получения антагонистов опиатов - производных оксиморфона, которые являются в настоящее время основными средствами терапии опиатной зависимости и диагностики наркомании.
Авторами настоящего изобретения неожиданно было обнаружено, что использование таких сильных неспецифично окисляющих агентов, как соли хлорноватистой и бромноватистой кислот, в мягких условиях позволяет получить с высокими выходом и чистотой производные морфинона.
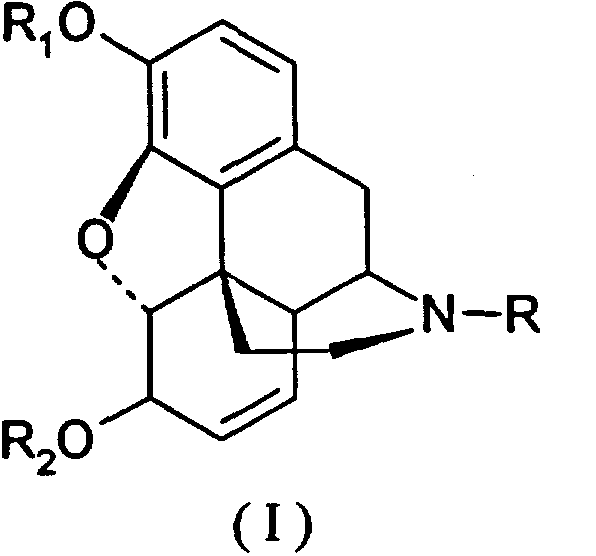
Таким образом, настоящее изобретение относится к способу получения производных морфинона, включающему окисление солями хлорноватистой и бромноватистой кислот производных морфина общей формулы I:
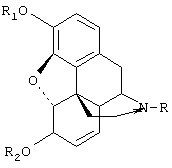
(I)
где К представляет низший алкил, С3-С6циклоалкил, бензил или замещенный бензил, циан; R1 представляет алкил, бензил или алкилкарбонил; R2 представляет водород, алкил, бензил или алкилкарбонил.
Окисление (по Схеме 1) предпочтительно ведут солями хлорноватистой и бромноватистой кислот, преимущественно полученными непосредственно перед окислением. Для окисления используются предпочтительно гипохлориты или гипобромиты натрия и калия.

Окисление проводят простым смешиванием водного раствора окислителя и раствора исходного вещества либо его суспензии в органическом растворителе. Предпочтительно в качестве растворителя используют ацетонитрил. Процесс проводят при температуре в интервале от 0 до 20°С в течение от 30 мин до 2 часов. Далее целевой продукт выделяют, используя известные методики.
В предпочтительном варианте осуществления изобретения соотношения органический растворитель - раствор окислителя выбирают таким образом, чтобы образовывалась двухфазная система, в которой целевой продукт находится в органической фазе, что облегчает его выделение. Предпочтительно указанное выше соотношение составляет 1:3.
Другим объектом настоящего изобретения является способ получения производных 14-гидроксиморфинона. Общая схема способа представлена на Схеме 2.

Производные морфина (I) окисляют гипохлоритом или гипобромитом с получением соединения (II) способом, представленным на Схеме 1. Далее производные морфинона (II) обрабатывают уксусным ангидридом с получением эфира енола (III), который окисляют далее перекисью водорода с получением производных 14-гидроксиморфинона. Вторую и третью стадии способа проводят по известным методикам, например, как описано в патенте США 6008355.
Еще одним объектом настоящего изобретения является способ получения производных оксиморфона по общей схеме, представленной на Схеме 3:

где R’ замещенный алкил или алкилкарбонил.
Первую стадию окисления производных морфина (I) в производные морфинона (II) проводят, как описано выше (Схема 1). Далее до производных 14-гидроксиморфинона (IV) процесс ведут, как показано на Схеме 2. Превращение производных 14-гидроксиморфинона (IV) в производные оксиморфона (V) производят гидрированием, далее (V) деалкелируют по азоту с получением норпроизводных (VI), которые алкилируют с получением антагонистов опиатов (VII), как показано ранее.
Ниже приведен ряд конкретных примеров воплощения изобретения, которые даны для его иллюстрации.
Примеры
Пример 1. Получение кодеинона (II; R=СН3, R1=СН3).
Готовят раствор окислителя, растворяя твердый гидроксид натрия (3,0 г) в 25 мл воды, и при охлаждении смесью лед/вода барботируют через этот раствор газообразный хлор, испаряя 2 мл жидкого хлора.
Отдельно готовят суспензию кодеина (5,0 г, 0,0168 моль) в ацетонитриле (8 мл). К суспензии добавляют раствор окислителя, поддерживая температуру реакционной массы 5-10°С. Реакционную массу выдерживают при этой температуре в течение часа и затем доводят температуру до комнатной. Отделяют органический слой. К органическому слою прибавляют хлористый метилен (25 мл) и промывают полученный раствор водой (2×25мл). Раствор продукта сушат сульфатом натрия и упаривают. После перекристаллизации из этилацетата получают 4,96 г целевого продукта (Тпл=184-185°С). Выход 94,3%.
Пример 2. Получение 14-гидроксикодеинона(IV; R=СН3, R1=СН3).
К кодеинону (4,96 г; 0,0158 моль), полученному по методике, приведенной в Примере 1, прибавляют уксусный ангидрид (13,6 г; 0,158 моль) и толуол (5 мл) и кипятят с обратным холодильником в течение 3 часов. Полученный раствор охлаждают, разбавляют хлористым метиленом и добавляют водный раствор гидрокарбоната натрия (27,72 г; 0,33 моль), поддерживая температуру реакционной массы 5-10°С, доводят температуру до комнатной, отделяют органический слой, промывают его водой (2×50 мл) и сушат сульфатом натрия. Затем раствор упаривают досуха и остаток растирают с 50 мл низкокипящего петролейного эфира и фильтруют. Полученный технический диенолацетат кодеинона представляет собой осадок коричневого цвета, который без дальнейшей очистки подвергают окислению, смешивая с муравьиной кислотой (3,7 г; 0,1 моль), раствором перекиси водорода (3,87 г 30%) и водой (10 мл) и выдерживают реакционную массу при 35-40°С в течение 5 часов. Реакционную массу доводят до рН 9 аммиачной водой и экстрагируют хлористым метиленом (2×25 мл). После упаривания получают 3,55 г целевого продукта. Выход 72,0%.
Пример 3. Получение налтрексона (VII; R’=циклопропилметил, R1=H).
К 14-гидроксикодеинону (3,55 г; 0,0114 моль), полученному по методике, описанной в примере 2, помещенному в автоклав, добавляют метанол (15 мл) и палладий на угле (5%) (0,5 г) и гидрируют при 20 ати в течение 24 часов при комнатной температуре. Реакционную массу фильтруют и упаривают. Остаток смешивают с 20 мл гексаметилдисилазана и кипятят с обратным холодильником в течение 2 часов. Избыток гексаметилдисилазана отгоняют в вакууме. Остаток перекристаллизовывают из гексана (25 мл). Полученное 14-триметилсилилокси соединение смешивают с раствором бромциана в хлороформе (10 мл 1M) и кипятят полученную смесь в течение 20 часов. Реакционную массу промывают водой, сушат сульфатом натрия и упаривают. К полученному цианпроизводному добавляют раствор уксусной кислоты (60 мл, 80%) и реакционную массу кипятят в течение 2 часов. Смесь подщелачивают 25% раствором аммиака до рН 9. Выпавший осадок отфильтровывают, добавляют хлорметилциклопропан (7,0 г), йодид калия (2,0 г) и этанол (100 мл) и реакционную массу нагревают до кипения. Реакционную массу охлаждают, подщелачивают аммиачной водой до рН 9, экстрагируют хлористым метиленом (2×25 мл) и сушат сульфатом натрия. Раствор охлаждают до 0-5°С и добавляют раствор трибромида фосфора в хлористом метилене (50 мл 1М), реакционную массу перемешивают при комнатной температуре, подщелачивают аммиачной водой до рН 9 и отделяют органический слой, промывают его водой (2×30 мл), сушат сульфатом натрия и упаривают в вакууме досуха. Получают 2,17 г целевого продукта в виде основания. Выход 56,0%.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ПОЛУЧЕНИЯ 14-ГИДРОКСИНОРМОРФИНОНОВ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1998 |
|
RU2183636C2 |
| ПРОИЗВОДНОЕ 14-ГИДРОКСИНОРМОРФИНОНА, ПРОИЗВОДНОЕ МОРФИНОНА, ПРОИЗВОДНОЕ МОРФИНА, СПОСОБЫ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО 14-ГИДРОКСИНОРМОРФИНОНА, ПРОИЗВОДНОЕ МОРФИНОНА, НОРОКСИМОРФОНА | 2002 |
|
RU2297419C2 |
| СПОСОБ ПОЛУЧЕНИЯ НАЛТРЕКСОНА | 2011 |
|
RU2448709C1 |
| Способ получения производных морфина | 1978 |
|
SU856381A3 |
| КОНТРОЛИРУЕМОЕ ВЫСВОБОЖДЕНИЕ ФЕНОЛЬНЫХ ОПИАТОВ | 2007 |
|
RU2469038C2 |
| СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ МОРФИНАН-6-ОНА | 2014 |
|
RU2652786C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ МОРФИНА | 2007 |
|
RU2426736C2 |
| Способ получения производных N-деметил-морфинов | 1985 |
|
SU1398776A3 |
| ПРОИЗВОДНЫЕ β-КАРБОЛИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 1999 |
|
RU2210571C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ОКСАДИАЗОЛАЛКИЛПУРИНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ КИСЛОТНО-АДДИТИВНЫХ СОЛЕЙ (ЕГО ВАРИАНТЫ) | 1987 |
|
RU2007404C1 |
Изобретение относится к способу получения производных морфинона, которые являются промежуточными соединениями для получения производных 14-гидроксиморфинона, которые, в свою очередь, используются для получения антагонистов опиатов - производных оксиморфона. Способ получения производных морфинона включает окисление солями хлорноватистой и бромноватистой кислот производных морфина общей формулы I:

где R - низший алкил, С3-С6циклоалкил, бензил или замещенный бензил, циан; R1 - алкил, бензил или алкилкарбонил; R2 - водород, алкил, бензил или алкилкарбонил. Окисление проводят солями хлорноватистой и бромноватистой кислот, преимущественно полученными непосредственно перед окислением. Для окисления используются предпочтительно гипохлориты или гипобромиты натрия и калия. Антагонисты опиатов являются важными лекарственными средствами для лечения и диагностики наркомании. 3 с. и 4 з.п. ф-лы.
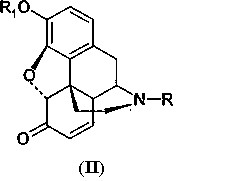
где R представляет низший алкил, необязательно замещенный С3-С6циклоалкилом, С3-С6-циклоалкил, бензил или замещенный бензил, циан;
R1 представляет алкил, бензил или алкилкарбонил,
включающий окисление солями хлорноватистой или бромноватистой кислоты производных морфина общей формулы (I):

где R и R1 принимают значения, определенные выше;
R2 представляет водород, алкил, бензил или алкилкарбонил.

где R и R1 принимают значения, определенные в п.1,
включающий окисление солями хлорноватистой или бромноватистой кислоты производных морфина общей формулы (I)
где R и R1 принимают значения, определенные выше,
R2 представляет водород, алкил, бензил или алкилкарбонил,
с получением производных морфинона (II)
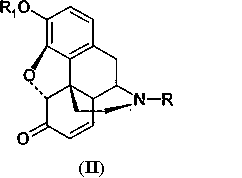
где R и R1 принимают значения, определенные в п.1,
обработку производных морфинона (II) уксусным ангидридом с получением эфира енола (III)
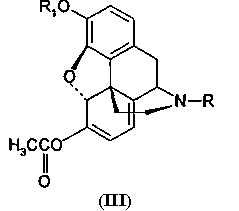
где R и R1 принимают значения, определенные выше,
окисление эфира енола (III) перекисью водорода с получением производных 14-гидроксиморфинона (IV).

где R’ - замещенный алкил или алкилкарбонил;
R и R1 принимают значения, определенные в п.1,
включающий окисление солями хлорноватистой или бромноватистой кислоты производных морфина общей формулы (I)
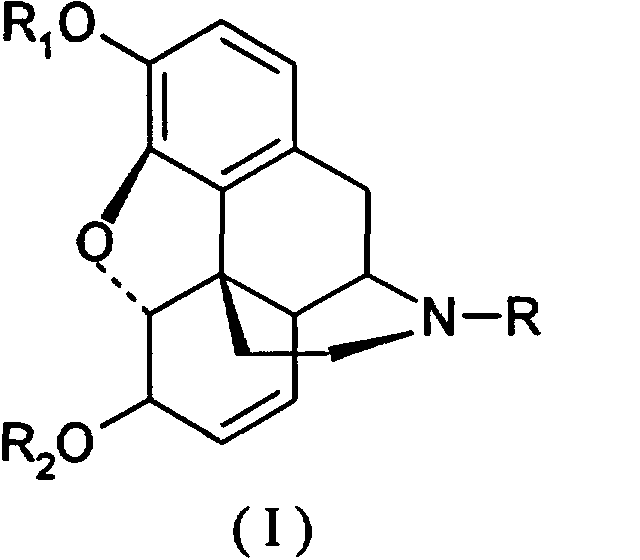
где R и R1 принимают значения, определенные выше;
R2 представляет водород, алкил, бензил или алкилкарбонил,
с получением производных морфинона (II)
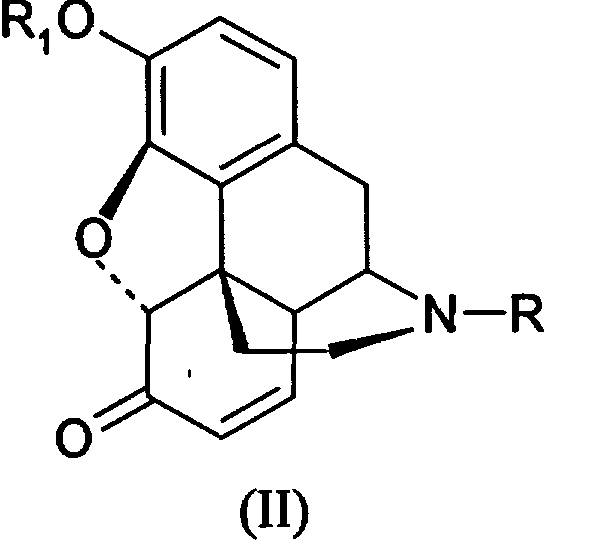
где R и R1 принимают значения, определенные в п.1,
обработку производных морфинона (II) уксусным ангидридом с получением эфира енола (III)

где R и R1 принимают значения, определенные выше,
окисление эфира енола (III) перекисью водорода с получением производных 14-гидроксиморфинона (IV)

где R и R1 принимают значения, определенные выше,
гидрирование производных 14-гидроксиморфинона (IV) с получением производных оксиморфона (V)

где R и R1 принимают значения, определенные выше,
деалкилирование по азоту производных оксиморфона (V) с получением норпроизводных (VI)

где R и R1 принимают значения, определенные выше,
алкилирование норпроизводных (VI) с получением производных оксиморфона (VII).
| US 6008355 A, 28.12.1999 | |||
| US 5922876 A, 13.07.1999 | |||
| US 4052402 A, 04.10.1977. |

