Настоящая заявка претендует на приоритет предварительной заявки США № 60/919323, внесенной в реестр 20 марта 2007, озаглавленной "4'-O-замещенные изоиндолиновые производные и содержащие их композиции и способы их применения", Ruchelman et al., которая вводится в настоящее изобретение полностью с помощью ссылки.
1. ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ НАСТОЯЩЕЕ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к 4'-O-замещенным изоиндолиновым производным. Также настоящее изобретение относится к фармацевтическим композициям, содержащим данные соединения, и способам лечения, предотвращения и менеджмента различных расстройств, применяя данные соединения и композиции.
2. УРОВЕНЬ ТЕХНИКИ
2.1. ПАТОБИОЛОГИЯ РАКА И ДРУГИХ ЗАБОЛЕВАНИЙ
Рак характеризуется, главным образом, увеличением числа патологических клеток, образованных из данной нормальной ткани, проникновением настоящих патологических клеток в соседние ткани, или распространением через лимфу или кровь раковых клеток к региональным лимфоузлам и к удаленным областям (метастаз). Клинические данные и молекулярно-биологические исследования показывают, что рак является многостадийным процессом, который начинается с незначительных предраковых изменений, которые могут при определенных условиях развиться до неоплазии. Опухолевое патологическое изменение может развиваться клонально и обнаруживать повышенную способность к проникновению, росту, метастазу и гетерогенизации, особенно при условиях, в которых опухолевые клетки ускользают от иммунологического надзора хозяина. Roitt, I., Brostoff, J and Kale, D., Immunology, 17,1-17,12 (3rd ed., Mosby, St. Louis, Mo., 1993).
Существует огромное разнообразие видов рака, которые подробно описывают в медицинской литературе. Примеры включает рак легких, толстой кишки, прямой кишки, предстательной железы, молочной железы, мозга и кишечника. Частота случаев возникновения рака продолжает расти по мере старения населения в целом, при возникновении новых видов рака, и по мере роста численности уязвимого населения (например, людей, зараженных СПИДом или подвергаемых избыточному действию солнечных лучей). Однако варианты лечения рака ограничены. Например, в случае рака крови (например, множественной миеломы) являются пригодными несколько вариантов лечения, особенно когда стандартная химиотерапия является неэффективной и не может быть выбрана трансплантация костного мозга. Следовательно, существует огромная необходимость в новых способах и композициях, которые можно применять для лечения пациентов с раком.
Многие виды рака связаны с образованием новых кровеносных сосудов, процессом, известным как ангиогенез. Выяснено несколько механизмов, участвующих в ангиогенезе, вызванном опухолью. Главное направление данных механизмов заключается в секреции опухолевыми клетками цитокинов с ангиогенными свойствами. Примеры данных цитокинов включают кислотный и основный фактор роста фибробластов (a,b-FGF), ангиогенин, фактор роста эндотелия сосудов (VEGF) и TNF-α. Альтернативно, опухолевые клетки могут высвобождать ангиогенные пептиды посредством производства протеаз и последующим разрушением внеклеточного матрикса, где хранятся некоторые цитокины (например, b-FGF). Ангиогенез может также быть вызван косвенно посредством стимуляции воспалительных клеток (в частности, макрофагов) и их последующим высвобождением ангиогенных цитокинов (например, TNF-α, b-FGF).
Ряд других заболеваний и расстройств также связан с, или характеризуется, неблагоприятным ангиогенезом. Например, усиленный или неконтролируемый ангиогенез участвует в ряде заболеваний и медицинских состояний, включая, но не ограничиваясь, глазные неоваскулярные заболевания, хороидальные неоваскулярные заболевания, неоваскулярные заболевания чувствительной оболочки глазного яблока, покраснение (неоваскуляризация углов), вирусные заболевания, генетические заболевания, воспалительные заболевания, аллергенные заболевания и аутоиммунные заболевания. Примеры данных заболеваний и состояний включают, но не ограничиваются: диабетическую ретинопатию; ретинопатию недоношенных; роговичное отторжение ткани; неоваскулярную глаукому; ретролентальную фиброплазию; артрит; и пролиферативную витреоретинопатию.
Соответственно, соединения, которые могут контролировать ангиогенез или ингибировать производство определенных цитокинов, включая TNF-α, могут быть пригодны для лечения и предотвращения различных заболеваний и состояний.
2.2. СПОСОБЫ ЛЕЧЕНИЯ РАКА
Современная терапия рака может включать хирургическую операцию, химиотерапию, гормональную терапию и/или лучевую терапию для того, чтобы уничтожить у пациента опухолевые клетки (смотри, например, Stockdale, 1998, Medicine, vol. 3, Rubenstein and Federman, eds., Chapter 12, Section IV). В последнее время, терапия рака может также включать биологическую терапию или иммунотерапию. Все из этих подходов имеют ряд недостатков для пациента. Хирургическая операция, например, может быть противопоказана из-за нездоровья пациента или может быть нежелательна для пациента.
Кроме того, хирургическая операция может удалить опухолевую ткань не полностью. Лучевая терапия является эффективной, только когда опухолевая ткань обладает более высокой чувствительностью к облучению, чем нормальная ткань. Лучевая терапия может также вызывать серьезные побочные эффекты. Гормональную терапию редко применяют отдельно. Хотя гормональная терапия может быть эффективной, ее часто применяют для того, чтобы предотвратить или препятствовать рецидиву рака после того, как с помощью других видов лечения удалено большинство раковых клеток. Виды биологической терапии и иммунотерапии количественно ограничены и могут вызывать побочные эффекты, такие как высыпание или опухание, симптомы, подобные симптомам при гриппе, включая повышение температуры, озноб и усталость, проблемы с пищеварительным трактом или аллергические реакции.
Что касается химиотерапии, существует множество химиотерапевтических средств, пригодных для лечения рака. Большинство противораковых химиотерапевтических средств действуют, ингибируя синтез ДНК, или прямо или косвенно ингибируя биосинтез предшественников дезоксирибонуклеотидтрифосфатов для того, чтобы предотвратить репликацию ДНК и сопутствующее ей деление клетки. Gilman et al., Goodman and Gilman's: The Pharmacological Basis of Therapeutics, Tenth Ed. (McGraw Hill, New York).
Несмотря на наличие множества химиотерапевтических средств, химиотерапия имеет много недостатков. Stockdale, Medicine, vol. 3, Rubenstein and Federman, eds., ch. 12, sect. 10, 1998. Почти все химиотерапевтические средства являются токсичными, и химиотерапия вызывает сильные, и очень опасные побочные эффекты, включая сильную тошноту, подавление деятельности костного мозга и подавление иммунной системы. Кроме того, даже при введении комбинаций химиотерапевтических средств, многие опухолевые клетки являются устойчивыми или развивают устойчивость к химиотерапевтическим средствам. Действительно, оказывается, что клетки, устойчивые к конкретным химиотерапевтическим средствам, применяемым в лечебном протоколе, являются устойчивыми к другим лекарствам, даже если данные средства действуют по механизму, отличному от механизма лекарств, применяемых при индивидуальном лечении. Данное явление называют плеотропная лекарственная резистентность или мультирезистентность. Из-за лекарственной устойчивости, многие виды рака оказываются или становятся резистентными для стандартных химиотерапевтических лечебных протоколов.
Другие заболевания или состояния, связанные с или характеризующиеся неблагоприятным ангиогенезом, могут также быть сложными для лечения. Однако предполагается, что некоторые соединения, такие как протамин, гепаин и стероиды, являются пригодными для лечения определенных конкретных заболеваний. Taylor et al., Nature 297:307 (1982); Folkman et al., Science 221:719 (1983); и патент США № 5001116 и 4994443.
Тем не менее, существует большая необходимость в безопасных и эффективных способах лечения, предотвращения и менеджемента рака и других заболеваний и состояний, включая заболевания, которые являются резистентными по отношению к стандартным способам лечения, таким как хирургическая операция, лучевая терапия, химиотерапия и гормональная терапия, при снижении или избегании токсичности и/или побочных действий, связанных с общепринятыми терапиями.
3. СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к 4'-O-замещенным изоиндолиновым соединениям и их фармацевтически приемлемым солям, сольватам (например, гидратам), пролекарствам, клатратам или стереоизомерам.
Также настоящее изобретение относится к способам лечения и менеджмента различных заболеваний или расстройств. Настоящие способы включают введение нуждающемуся в таком лечении пациенту терапевтически эффективного количества соединения, относящегося к настоящему изобретению, или его фармацевтически приемлемой соли, сольвата, пролекарства, клатрата или стереоизомера.
Кроме того, настоящее изобретение относится к способам предотвращения различных заболеваний и расстройств, которые включают введение нуждающемуся в таком предотвращении пациенту профилактически эффективного количества соединения, относящегося к настоящему изобретению, или его фармацевтически приемлемой соли, сольвата, пролекарства, клатрата или стереоизомера.
Также настоящее изобретение относится к фармацевтическим композициям, единичным стандартным лекарственным формам, режимам дозировки и наборам, которые содержат соединение, относящееся к настоящему изобретению, или его фармацевтически приемлемую соль, сольват, пролекарство, клатрат или стереоизомер.
4. ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В одном варианте осуществления настоящее изобретение относится к изоиндолиновым соединениям и их фармацевтически приемлемым солям, сольватам, пролекарствам, клатратам и стереоизомерам.
В другом варианте осуществления настоящее изобретение относится к способам лечения, менеджмента и предотвращения различных заболеваний и расстройств, которые включают введение нуждающемуся в таком лечении или предотвращении пациенту терапевтически или профилактически эффективного количества соединения, относящегося к настоящему изобретению или его фармацевтически приемлемой соли, сольвату, пролекарству, клатрату или стереоизомеру. Примеры заболеваний и расстройств описаны в настоящем изобретении.
В других вариантах осуществления соединение, относящееся к настоящему изобретению, или его фармацевтически приемлемую соль, сольват, пролекарство, клатрат или стереоизомер вводят в комбинации с другим лекарственным средством ("вторым активным средством") или лечением. Вторые активные средства включают малые молекулы и большие молекулы (например, белки и антитела), примеры которых относятся к настоящему изобретению, также как стволовые клетки. Способы или терапии, которые можно применять в комбинации с введением соединения, относящегося к настоящему изобретению, включают, но не ограничиваются, хирургическую операцию, переливание крови, иммунотерапию, биологическую терапию, лучевую терапию и другие виды терапий на основе нелекарственных средств, в настоящее время применяемые для того, чтобы лечить, предотвращать или контролировать различные расстройства, описанные в данном изобретении.
Настоящее изобретение также относится к фармацевтическим композициям (например, единичным стандартным лекарственным формам), которые можно применять в способах, относящихся к настоящему изобретению. В одном варианте осуществления фармацевтические композиции содержат соединение, относящееся к настоящему изобретению, или его фармацевтически приемлемую соль, сольват, пролекарство, клатрат или стереоизомер и, не обязательно, дополнительное активное средство.
4.1. СОЕДИНЕНИЯ
В одном варианте осуществления соединения, относящиеся к настоящему изобретению, для применения в фармацевтических композициях и способах имеют формулу I:
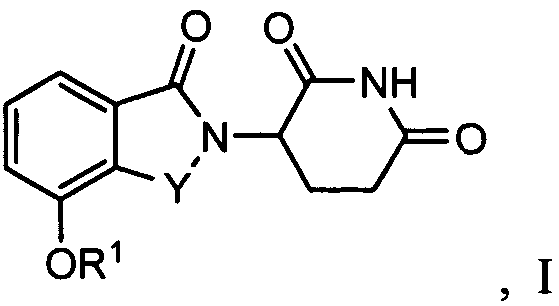
а также их фармацевтически приемлемые соли, сольваты, пролекарства, клатраты или стереоизомеры, где Y представляет C=O или CH2, и R1 представляет водород, алкил, алкенил, алкинил, арил, аралкил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, гетероарил, гетероарилалкил, ариламинокарбонил, алкилкарбонил, алкиламинокарбонил, диалкиламинокарбонил, алкоксикарбонил, циклоалкилкарбонил, гетероарилкарбонил или гетероциклилкарбонил; где R1 не обязательно замещен одним или более, в определенных вариантах осуществления, 1, 2, 3 или 4 заместителями, представляющими одну, две или три группы, выбранные из алкокси, галогена, алкила, карбокси, алкиламинокарбонила, алкоксикарбонила, нитро, амина, нитрила, галогеноалкила, гидрокси и алкилсульфонила.
В одном варианте осуществления Y представляет C=O. В другом варианте осуществления Y представляет CH2.
В определенных вариантах осуществления R1 представляет алкил, алкенил, алкинил, арил, аралкил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, гетероарил или гетероарилалкил, не обязательно, замещенный одним или более, в одном варианте осуществления, одной, двумя или тремя группами, выбранными из алкокси, галогена, алкила и алкилсульфонила. В одном варианте осуществления R1 представляет арил, аралкил или гетероарилалкил. В определенных вариантах осуществления арильное или гетероарильное кольцо в группе R1 представляет 5- или 6-членное моноциклическое кольцо. В определенных вариантах осуществления гетероарильное кольцо в группе R1 представляет 5- или 6-членное моноциклическое кольцо, содержащее 1-3 гетероатома, выбранные из O, N и S. В определенных вариантах осуществления арильное или гетероарильное кольцо в группе R1 представляет бициклическое кольцо. В определенных вариантах осуществления гетероарильное кольцо содержит 1-3 гетероатома, выбранные из O, N и S и присоединенные к алкильной группе через гетероатом в кольце. В определенных вариантах осуществления гетероарильное кольцо присоединено к алкильной группе через атом углерода в кольце.
В одном варианте осуществления R1 представляет фенил, бензил, нафтилметил, хинолилметил, бензофурилметил, бензотиенилметил, фурилметил или тиенилметил, не обязательно, замещенный одной или более, в одном варианте осуществления, одной, двумя или тремя группами, выбранными из алкокси, галогено, алкила и алкилсульфонила. В одном варианте осуществления R1 не обязательно замещают одним или двумя заместителями, выбранными из метокси, хлора, брома, фтора, метила и метилсульфонила.
В других вариантах осуществления R1 представляет 2-метоксифенил, бензил, 3-хлорбензил, 4-хлорбензил, 3,4-дихлорбензил, 3,5-дихлорбензил, 3-фторбензил, 3-бромбензил, 3-метилбензил, 4-метилсульфонилбензил, 3-метоксибензил, нафтилметил, 3-хинолилметил, 2-хинолилметил, 2-бензофурилметил, 2-бензотиенилметил, 3-хлортиен-2-илметил, 4-фторбензотиен-2-илметил, 2-фурилметил, 5-хлортиен-2-илметил или 1-нафт-2-илэтил.
В одном варианте осуществления R1 представляет гетероциклил. В определенных вариантах осуществления гетероциклильное кольцо в группе R1 представляет 5- или 6-членное моноциклическое кольцо, содержащее 1-3 гетероатома, выбранные из O, N и S. В определенных вариантах осуществления гетероциклильное кольцо в группе R1 представляет пиперидинил или тетрагидропиранил.
В определенных вариантах осуществления соединения имеют формулу II:
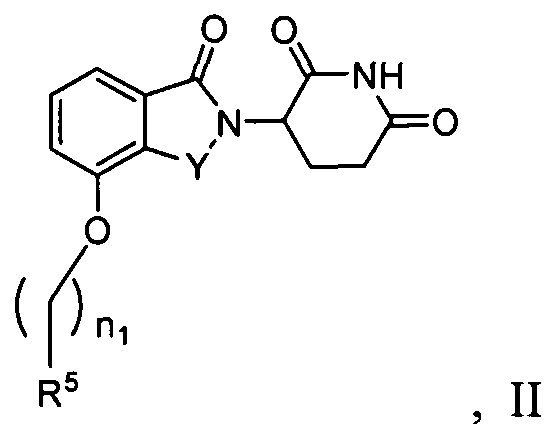
где Y представляет C=O или CH2, и R5 представляет арил или гетероарил, не обязательно замещенный одной, двумя или тремя группами, выбранными из алкила, галогена, алкокси, карбокси, алкиламинокарбонила, алкоксикарбонила, нитро, амина, нитрила, галогеноалкила, гидрокси и алкилсульфонила; n1 равно 0-5, и другие переменные представляют группы, как описано в другом месте настоящего изобретения.
В одном варианте осуществления Y представляет C=O. В другом варианте осуществления Y представляет CH2.
В одном варианте осуществления m равно 0 или 1. В определенных вариантах осуществления R5 выбирают из фенила, нафтила, фурила, тиенила, бензофурила, бензотиенила и хинолила, не обязательно замещенного одной или двумя группами, выбранными из метила, метокси, хлора, фтора, брома и метилсульфонила. В других вариантах осуществления R5 представляет фенил, 3-хлорфенил, 4-хлорфенил, 3,4-дихлорфенил, 3,5-дихлорфенил, 3-фторфенил, 3-бромфенил, 3-метилфенил, 4-метилсульфонилфенил, 3-метоксифенил, нафтил, 3-хинолил, 2-хинолил, 2-бензофурил, 2-бензотиенил, 3-хлортиен-2-ил, 4-фторбензотиен-2-ил, 2-фурил, 5-хлортиен-2-ил или 1-нафт-2-ил.
В одном варианте осуществления n1 равно 0 или 1. В определенных вариантах осуществления R5 выбирают из фенила, бензила, нафтила, фурила, тиенила, бензофурила, бензотиенила и хинолила, не обязательно замещенного одной или двумя группами, выбранными из метила, метокси, хлора, фтора, брома и метилсульфонила.
В одном варианте осуществления соединения имеют формулу III
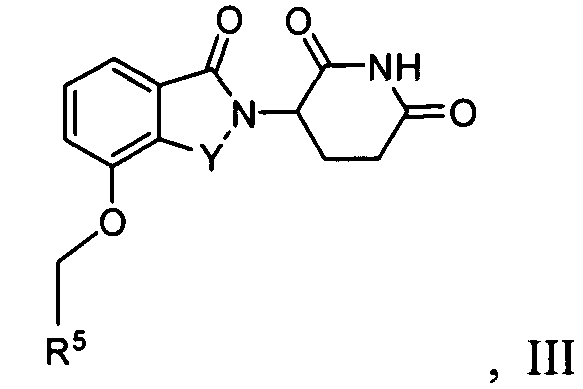
где переменные представляют группы, как описано в другом месте настоящего изобретения.
В одном варианте осуществления Y представляет C=O. В другом варианте осуществления Y представляет CH2.
В одном варианте осуществления R5 представляет
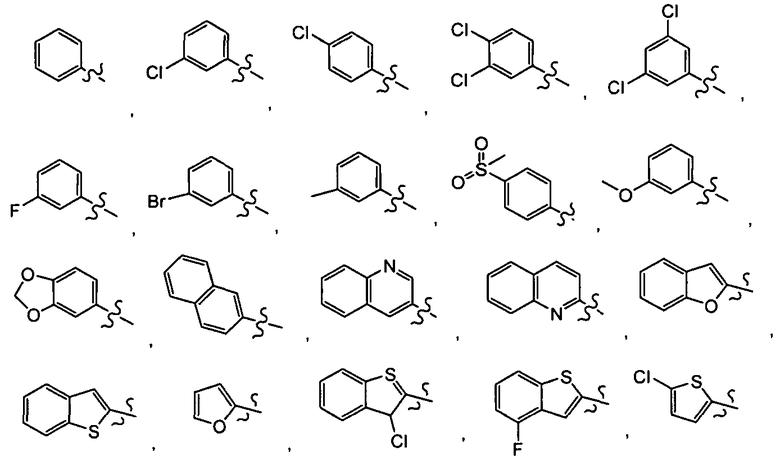
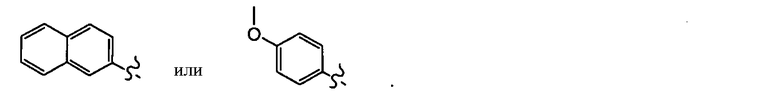
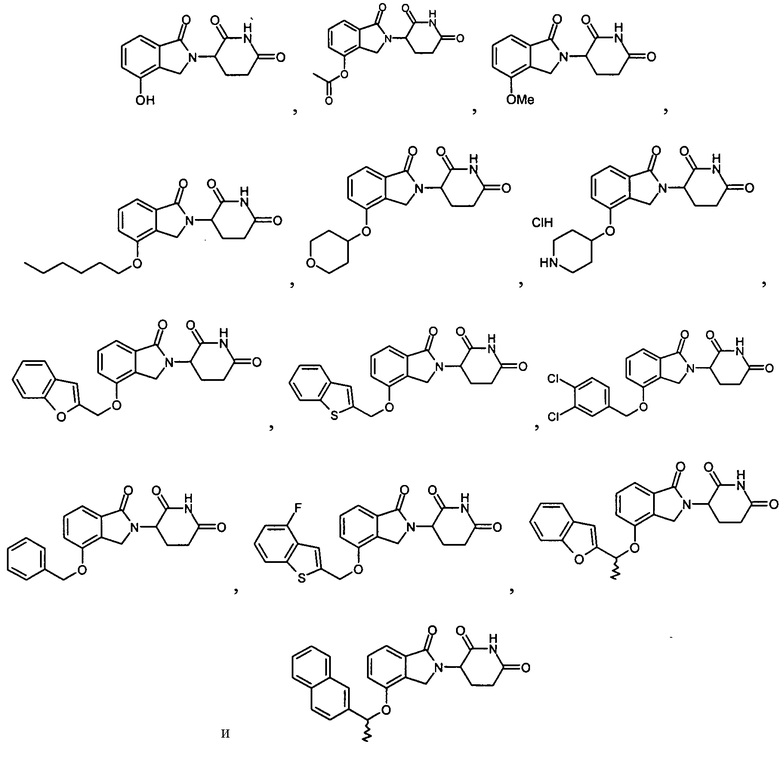
Примеры включают, но не ограничиваются, группы, перечисленные ниже или их фармацевтически приемлемую соль, сольват (например, гидрат), пролекарство, клатрат или стереоизомер:
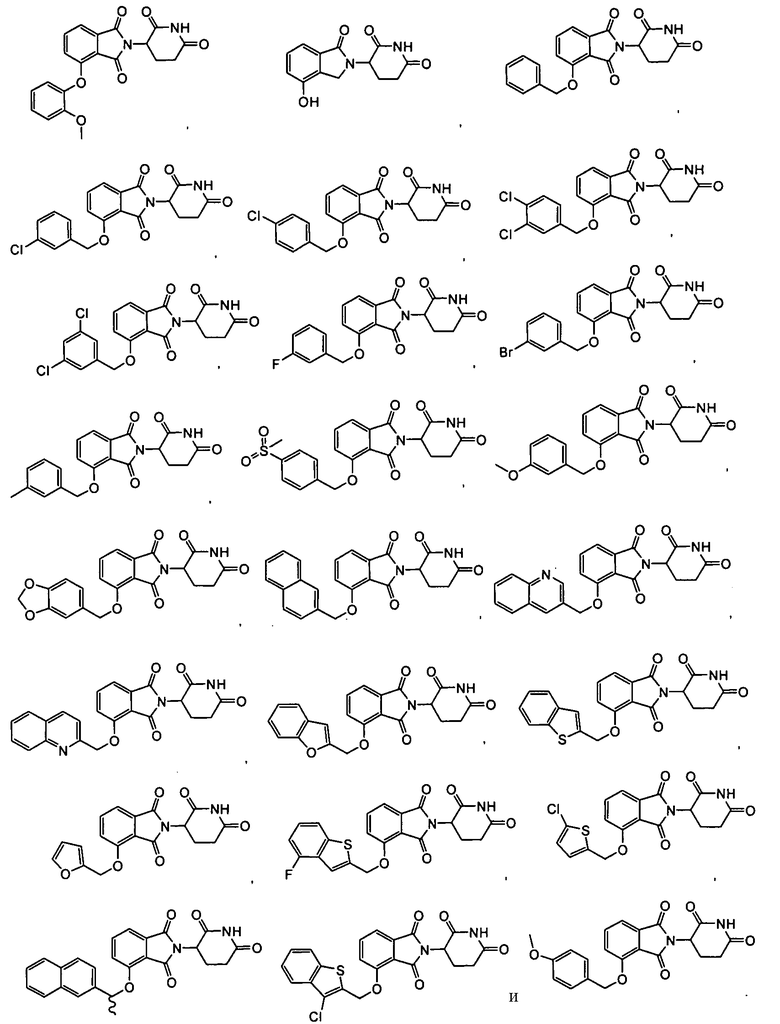
В определенных вариантах осуществления соединение представляет:
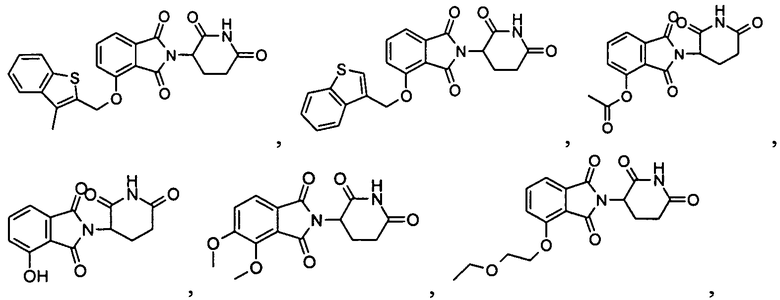
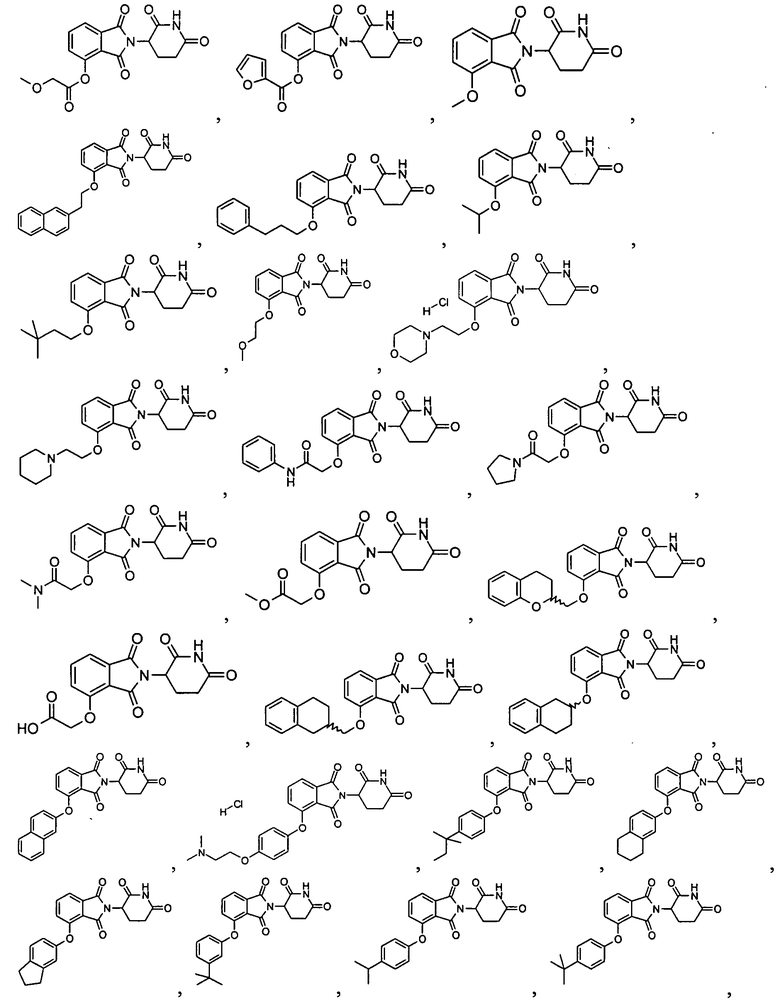
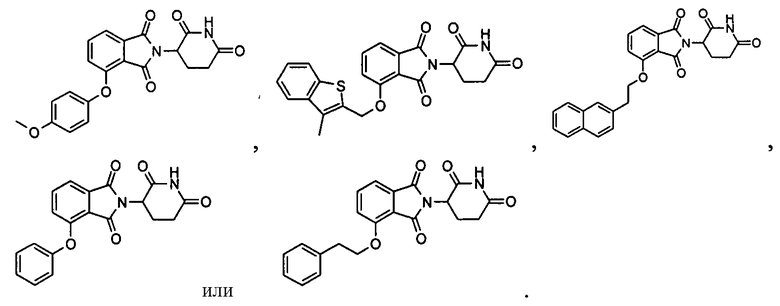
В одном варианте осуществления соединение выбирают из
Как применяют в настоящем изобретении и если не указано особо, термин "фармацевтически приемлемая соль" относится к соли, полученной из фармацевтически приемлемой нетоксичной кислоты, включая неорганические кислоты и органические кислоты. Подходящие нетоксичные кислоты включают неорганические и органические кислоты, такие как, но не ограничиваясь, уксусную, альгиновую, аминобензойную, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, муравьиную, фумаровую, фуранкарбоновую, глюконовую, глутаминовую, глюкуроновую, галактуроновую, глицидную, бромоводородную, хлороводородную, изэтионовую, молочную, малеиновую, яблочную, миндальную, метансульфоновую, слизевую, азотную, памовую, пантотеновую, фенилуксусную, пропионовую, фосфорную, салициловую, стеариновую, янтарную, сульфаниловую, серную, винную кислоту, п-толуолсульфокислоту и подобные. В одном варианте осуществления подходящими кислотами являются хлороводородная, бромоводородная, фосфорная и серная кислоты.
Как применяют в настоящем изобретении и если не указано особо, термин "сольват" относится к соединению, которое, кроме того, содержит стехиометрическое или нестехиометрическое количество растворителя, связанного нековалентными внутримолекулярными силами. Когда растворителем является вода, сольват представляет гидрат.
Как применяют в настоящем изобретении и если не указано особо, термин "пролекарство" относится к производному соединения, которое может гидролизоваться, окисляться или в других случаях реагировать в биологических условиях (in vitro или in vivo) для того, чтобы дать соединение. Примеры пролекарств включают, но не ограничиваются, соединения, которые содержат биогидролизуемые группы, такие как биогидролизуемые амиды, биогидролизуемые эфиры, биогидролизуемые карбаматы, биогидролизуемые карбонаты, биогидролизуемые уреиды и биогидролизуемые фосфатные аналоги. Другие примеры пролекарств включают соединения, которые содержат -NO, -NO2, -ONO, или -ONO2 группы. Пролекарства можно обычно получить, применяя хорошо известные способы, такие как способы, описанные в Burger's Medicinal Chemistry and Drug Discovery, 172-178, 949-982 (Manfred E. Wolff ed., 5th ed. 1995), и Design of Prodrugs (H. Bundgaard ed., Elselvier, New York 1985).
Как применяют в настоящем изобретении и если не указано особо, термины "биогидролизуемый карбамат," "биогидролизуемый карбонат", "биогидролизуемые уреид" и "биогидролизуемый фосфат" относятся к карбамату, карбонату, уреиду и фосфату, соответственно, соединения, которые или: 1) не препятствуют биологической активности соединения, но могут придавать полезные свойства при предпочтительных для данного соединения условиях in vivo, такие как абсорбция, продолжительность действия или начало действия; или 2) являются биологически неактивным, но превращаются in vivo в биологически активное соединение. Примеры биогидролизуемых карбаматов включают, но не ограничиваются, карбаматы, которые содержат низшую алкиламино, замещенную этилендиамино, аминокислотную, гидроксиалкиламино, гетероциклическую и гетероароматическую амино и полиэфирную аминогруппы.
Как применяют в настоящем изобретении и если не указано особо, термин "стереоизомер" включает все энантиомерно/стереомерно чистые и энантиомерно/стереомерно обогащенные соединения, относящиеся к настоящему изобретению.
Как применяют в настоящем изобретении и если не указано особо, термин "стереомерно чистая" относится к композиции, которая содержит один стереоизомер соединения и в значительной степени свободна от других стереоизомеров настоящего соединения. Например, стереомерно чистая композиция соединения, имеющего один хиральный центр, будет в значительной степени свободна от противоположного энантиомера соединения. Стереомерно чистая композиция соединения, имеющего два хиральных центра, будет в значительной степени свободна от других диастереомеров соединения. Типичное стереомерно чистое соединение содержит больше чем, приблизительно, 80% по весу одного стереоизомера соединения и меньше чем, приблизительно, 20% по весу других стереоизомеров соединения, больше чем, приблизительно 90% по весу одного стереоизомера соединения и меньше чем, приблизительно, 10% по весу других стереоизомеров соединения, больше чем, приблизительно 95% по весу одного стереоизомера соединения и меньше чем, приблизительно, 5% по весу других стереоизомеров соединения, больше чем, приблизительно 97% по весу одного стереоизомера соединения и меньше чем, приблизительно, 3% по весу других стереоизомеров соединения, больше чем, приблизительно 98% по весу одного стереоизомера соединения и меньше чем, приблизительно, 2% по весу других стереоизомеров соединения или больше чем, приблизительно 99% по весу одного стереоизомера соединения и меньше чем, приблизительно, 1% по весу других стереоизомеров соединения.
Как применяют в настоящем изобретении и если не указано особо, термин "стереомерно обогащенная" относится к композиции, которая содержит больше чем, приблизительно 55% по весу одного стереоизомера соединения, больше чем, приблизительно 60% по весу одного стереоизомера соединения, больше чем приблизительно 70% по весу, или больше чем, приблизительно 80% по весу одного стереоизомера соединения.
Как применяют в настоящем изобретении и если не указано особо, термин "энантиомерно чистая" относится к стереомерно чистой композиции соединения, имеющего один хиральный центр. Аналогично, термин "энантиомерно обогащенная" относится к стереомерно обогащенной композиции соединения, имеющего один хиральный центр.
Как применяют в настоящем изобретении и если не указано особо, термин "алкил" относится к насыщенному углеводороду с нормальной неразветвленной или разветвленной цепью, имеющему ряд атомов углерода, как указано в настоящем изобретении. В некоторых вариантах осуществления алкильные группы имеют 1-15, 1-10, 1-6 или 1-3 атомов углерода. Примеры насыщенных алкилов с нормальной неразветвленной цепью включают -метил, -этил, -н-пропил, -н-бутил, -н-пентил и -н-гексил; тогда как насыщенные алкилы с разветвленной цепью включают -изопропил, -сек-бутил, -изобутил, -трет-бутил, -изопентил, 2-метилбутил, 3-метилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилбутил и подобные. Термин "алкил" также включает циклоалкил.
Как применяют в настоящем изобретении, алкенил относится к углеводороду с нормальной неразветвленной цепью или разветвленной цепью, содержащему одну или более двойных связей. Примеры алкенильных углеродных цепей содержат от 2 до 20 атомов углерода, и в определенных вариантах осуществления, содержат 1-8 двойных связей, и алкенильные углеродные цепи из 2-16 атомов углерода, в определенных вариантах осуществления, содержат 1-5 двойных связей.
Как применяют в настоящем изобретении, алкинил относится к углеводороду с нормальной неразветвленной цепью или разветвленной цепью, содержащему одну или более тройных связей. Алкинильные углеродные цепи 2-20 атомов углерода, в определенных вариантах осуществления, содержат 1-8 тройных связей, и алкинильные углеродные цепи 2-16 атомов углерода, в определенных вариантах осуществления, содержат 1-5 тройных связей. Примеры алкенильных и алкинильных групп в настоящем изобретении включают, но не ограничиваются, этен, пропен, бутен, пентен, ацетилен и гексин. Как применяют в настоящем изобретении, низший алкил, низший алкенил и низший алкинил относятся к углеродным цепям, имеющим от, приблизительно, 1 или, приблизительно, 2 атомов углерода до, приблизительно, 6 атомов углерода.
Как применяют в настоящем изобретении и если не указано особо, термин "циклоалкил" относится к виду алкила, который является циклическим и содержит 3-15, 3-9, 3-6 или 3-5 атомов углерода, без чередующихся или резонирующих двойных связей между атомами углерода. Он может содержать 1-4 кольца. Примеры незамещенных циклоалкилов включают, но не ограничиваются, циклопропил, циклобутил, циклопентил, циклогексил и адамантил. Циклоалкил можно заместить одним или более заместителями. В некоторых вариантах осуществления циклоалкил может быть циклоалкилом, конденсированным с арильной или гетероарильной группами.
Как применяют в настоящем изобретении и если не указано особо, термин "гетероциклоалкил" относится к циклоалкилу, в котором один или более атомов углерода замещают гетероатомами, такими как, но не ограничиваясь, N, S и O. В некоторых вариантах осуществления гетероциклоалкильная группа содержит 2-14, 2-8, 2-7, 2-5 или 2-4 атомов углерода. В некоторых вариантах осуществления гетероциклоалкил может быть гетероциклоалкилом, конденсированным с арильной или гетероарильной группами.
Как применяют в настоящем изобретении, термин "арил" относится к карбоциклическому ароматическому кольцу, содержащему 5-14 атомов в кольце. Все атомы в кольце карбоциклической арильной группы являются атомами углерода. Арильные кольцевые структуры включают соединения, имеющие одну или более кольцевых структур, такие как моно-, би- или трициклические соединения, также как бензоконденсированные карбоциклические частицы, такие как 5,6,7,8-тетрагидронафтил и подобные. Конкретно, арильная группа может быть моно-, би- или трициклическим кольцом. Примеры арильных групп включают фенил, антраценил, флуоренил, инденил, азуленил, фенантренил и нафтил.
Как применяют в настоящем изобретении, "гетероарил" относится к моноциклической или мультициклической ароматической кольцевой системе, в определенных вариантах осуществления, от, приблизительно, 5 до, приблизительно, 15 членов, где один или более, в одном варианте осуществления 1-3, атомов в кольцевой система представляют гетероатом, т.е., элемент, отличный от углерода, включая, но не ограничиваясь, азот, кислород или серу. Гетероарильную группу можно, не обязательно, конденсировать с бензольным кольцом. Гетероарильные группы включают, но не ограничиваются, фурил, имидазолил, индолинил, пирролидинил, пиримидинил, тетразолил, тиенил, пиридил, пирролил, N-метилпирролил, хинолинил и изохинолинил.
Как применяют в настоящем изобретении, "гетероциклил" относится к моноциклической или мультициклической неароматической кольцевой системе, в одном варианте осуществления состоящей из 3-10 членов, в другом варианте осуществления состоящей из 4-7 членов, в следующем варианте осуществления состоящей из 5-6 членов, где один или более, в определенных вариантах осуществления 1-3, атомов в кольцевой системе представляют гетероатом, т.е., элемент, отличный от углерода, включая но не ограничиваясь, азот кислород или серу. В вариантах осуществления, где гетероатом (гетероатомы) представляет (представляют) азот, азот, не обязательно, замещают алкилом, алкенилом, алкинилом, арилом, гетероарилом, аралкилом, гетероаралкилом, циклоалкилом, гетероциклилом, циклоалкилалкилом, гетероциклилалкилом, ацилом, гуанидино, или азот можно кватернизировать для того, чтобы получить аммонийную группу, где заместители выбирают, как описано выше.
Как применяют в настоящем изобретении, "аралкил" относится к алкильной группе, в которой один из атомов водорода алкила замещают арильной группой.
Как применяют в настоящем изобретении, "гетероаралкил" относится к алкильной группе, в которой один из атомов водорода алкила замещают гетероарильной группой.
Как применяют в настоящем изобретении, "алкиламинокарбонил" относится к C(O)NHR, в которой R представляет алкил, включая низший алкил. Как применяют в настоящем изобретении, "диалкиламинокарбонил" относится к C(O)NR'R, в которой R' и R независимо представляют алкил, включая низший алкил; "карбоксамид" относится к группам формулы -NR'COR, в которой R' и R независимо представляют алкил, включая низший алкил.
Как применяют в настоящем изобретении, "ариламинокарбонил" относится к -C(O)NHR, в которой R представляет арил, включая низший арил, такой как фенил.
Как применяют в настоящем изобретении, "галогено", "галоген" или "галоид" относится к F, Cl, Br или I.
Когда число любого данного заместителя не указано (например, "галогеноалкил"), может присутствовать один или более заместителей. Например, "галогеноалкил" может содержать один или более одинаковых или различных галогенов.
Следует отметить, что если имеется разница между изображенной структурой и названием, данным этой структуре, изображенная структура является предпочтительней. Кроме того, если стереохимия структуры или части структуры не показана, например, жирными или пунктирными линиями, структуру или часть структуры следует рассматривать, как включающую все ее стереоизомеры.
4.2. СПОСОБЫ ЛЕЧЕНИЯ, ПРЕДОТВРАЩЕНИЯ И МЕНЕДЖМЕНТА
Относящимися к настоящему изобретению являются способы лечения, предотвращения и/или менеджемента различных заболеваний или расстройств, применяя соединение, относящееся к настоящему изобретению или его фармацевтически приемлемую соль, сольват (например, гидрат), пролекарство, клатрат или стереоизомер.
Примеры заболеваний или расстройств включают, но не ограничиваются, рак, расстройства, связанные с ангиогенезом, боль, включая, но не ограничиваясь, комплексный региональный синдром боли ("CRPS"), дегенерацию желтого пятна ("MD") и связанные с ним синдромы, кожные заболевания, легочные заболевания, расстройства, связанные с асбестом, паразитарные заболевания, состояния, связанные с иммунодефицитом, расстройства ЦНС, повреждения ЦНС, атеросклероз и связанные с ним расстройства, дисфункциональный сон и связанные с ним расстройства, гемоглобинопатию и связанные с ней расстройства (например, анемию), расстройства, связанные с TNFα, и другие различные заболевания и расстройства.
Как применяют в настоящем изобретении и если не указано особо, термины "лечить" и "лечение" относятся к ликвидации или уменьшению интенсивности заболевания, или расстройства, или одного или более симптомов, связанных с заболеванием или расстройством. В определенных вариантах осуществления термины относятся к снижению до минимума распространения или нарастания выраженности заболевания или расстройства, полученному в результате введения одного или более профилактических или терапевтических средств субъекту с данным заболеванием или расстройством.
Как применяют в настоящем изобретении, если не указано особо, термин "предотвращение" относится к лечению, применяя соединение, или введению соединения, относящегося к настоящему изобретению, с или без другого дополнительного активного соединения, перед возникновением симптомов, особенно пациентам с повышенным риском возникновения рака и/или других расстройств, описанных в данном изобретении. Термин "предотвращение" включает ингибирование или ослабление симптома конкретного заболевания. В частности, пациенты с семейным анамнезом заболевания являются кандидатами для профилактических режимов в определенных вариантах осуществления. Кроме того, пациенты, у которых уже проявлялись симптомы, также являются потенциальными кандидатами для предотвращения. В этой связи термин "предотвращение" можно взаимозаменяемо применять с термином "профилактическое лечение".
Как применяют в настоящем изобретении и если не указано особо, термин "менеджмент" относится к предотвращению или замедлению развития, распространения или нарастания выраженности заболевания, или расстройства, или одного или более его симптомов. В определенных случаях, положительное действие, которое субъект получает от профилактического или терапевтического средства, не приводит в результате к вылечиванию заболевания или расстройства.
Как применяют в настоящем изобретении и если не указано особо, "терапевтически эффективным количеством" соединения является количество, достаточное для того, чтобы получить терапевтический эффект при лечении или менеджементе заболевания или расстройства, или замедлить развитие, или снизить к минимуму проявление одного или более симптомов, связанных с заболеванием или расстройством. Терапевтически эффективное количество соединения относится к количеству терапевтического средства, отдельного или в комбинации с другими терапиями, которое оказывает терапевтический эффект при лечении или менеджементе заболевания или расстройства. Термин "терапевтически эффективное количество" может включать количество, которое улучшает общую терапию, ослабляет или помогает избежать симптомов или причин заболевания или расстройства, или увеличивает терапевтическую эффективность другого терапевтического средства.
Как применяют в настоящем изобретении и если не указано особо, "профилактически эффективным количеством" соединения является количество, достаточное для того, чтобы подавить или ослабить симптомы заболевания или для того, чтобы предотвратить рецидив заболевания. Профилактически эффективное количество соединения относится к количеству терапевтического средства, отдельного или в комбинации с другими средствами, которое приводит к профилактическому эффекту при подавлении или ослаблении симптомов заболевания или рецидива заболевания. Термин "профилактически эффективное количество" может включать количество, которое улучшает общую профилактику и увеличивает профилактическую эффективность другого профилактического средства.
Примеры рака и предраковых состояний включают, но не ограничиваются, примеры, описанные в патентах США № 6281230 и 5635517 Muller et al., в различных патентных публикациях США Zeldis, включая публикацию № 2004/0220144A1, внесенную в реестр 4 ноября 2004 (Treatment of Myelodysplastic Syndrom); 2004/0029832A1, внесенную в реестр 12 февраля 2004 (Treatment of Various Types of Cancer); и 2004/0087546, внесенную в реестр 6 мая 2004 (Treatment of Myeloproliferative Diseases). Примеры также включают примеры, описанные в WO 2004/103274, внесенной в реестр 2 декабря 2004. Все данные документы вводятся в настоящее изобретение полностью с помощью ссылки.
Конкретные примеры рака включают, но не ограничиваются, рак кожи, такой как меланома; лимфоузлов; молочной железы; шейки матки; желудочно-кишечного тракта; легких; яичника; предстательной железы; толстой кишки; прямой кишки; рта; мозга; головы и шеи; глотки; яичек; почек; поджелудочной железы; кости; селезенки; печени; мочевого пузыря; гортани; носовых каналов; и рак, связанный со СПИДом. Соединения также являются пригодными для лечения рака крови и костного мозга, такого как множественная миелома и острая и хроническая лейкемия, например лимфобластная, миелогенная, лимфоцитарная и миелоцитарная лейкемия. Соединения, относящиеся к настоящему изобретению, можно применять для лечения, предотвращения или менеджемента или первичной, или метастатической опухоли.
Другие конкретные виды рака включают, но не ограничиваются, запущенную опухоль, амилоидоз, нейробластому, менингиому, гемангиоперицитому, множественный метастаз в головном мозге, мультиформы глиобластомы, глиобластому, глиому ствола мозга, раковую опухоль мозга с неблагоприятным прогнозом, злокачественную глиому, повторную злокачественную глиому, анапластическую астроцитому, анапластическую олигодендроглиому, нейроиндокринную опухоль, ректальную аденокарциному, колоректальный рак стадии C и D по Дюку, нерезектабельную колоректальную карциному, метастатическую гепатоцеллюлярную карцинома, саркому Капоши, кариотипическую острую миелоидную лейкемию, хроническую лимфоцитарную лейкемию (CLL), лимфому Ходжкина, неходжкинскую лимфому, кожную T-клеточную лимфому, кожную B-клеточную лимфому, диффузную B-крупноклеточную лимфому, фолликулярную лимфому низкой степени злокачественности, метастатическую меланому (локализованную меланому, включая, но не ограничиваясь, глазную меланому), злокачественную мезотелиому, синдром мезотелиомы со злокачественным плевральным выпотом, перитонеальную карциному, папиллярную серозную карциному, гинекологическую саркому, саркому мягких тканей, склеродерму, кожный васкулит, гистиоцитоз клеток Лангенгарта, лейомиосаркому, прогрессирующую оссифицирующую фибродисплазию, устойчивый к действию гормонов рак предстательной железы, саркому с высоким риском возникновения после удаления мягких тканей, нерезектабельную гепатоцеллюлярную карциному, болезнь Вальденстрема, тлеющую миелому, вялотекущую миелому, рак фаллопиевой трубы, андроген-независимый рак предстательной железы, андроген-зависимый неметастатический рак стадии 4 предстательной железы, нечувствительный к гармонам рак предстательной железы, нечувствительный к химиотерапии рак предстательной железы, папиллярную тироидную карциному, фоккулярную тироидную карциному, меддулярную тироидную карциному и лейомиому. В конкретном варианте осуществления рак является метастатическим. В другом варианте осуществления рак является резистентным или невосприимчивым к химиотерапии или облучению.
В одном варианте осуществления относящимися к настоящему изобретению являются способы лечения, предотвращения или менеджемента различных форм лейкемии, такой как хроническая лимфоцитарная лейкемия, хроническая миелоцитарная лейкемия, острая лимфобластная лейкемия, острая миелогенная лейкемия и острая миелобластная лейкемия, включая лейкемию, которая является рецидивной, резистентной или невосприимчивой, как описано в публикации США № 2006/0030594, внесенной в реестр 9 февраля 2006, которая введена в настоящее изобретение полностью с помощью ссылки.
Термин "лейкемия" относится к злокачественным новообразованиям кроветворящих тканей. Лейкемия включает, но не ограничивается, хроническую лимфоцитарную лейкемию, хроническую миелоцитарную лейкемию, острую лимфобластную лейкемию, острую миелогенную лейкемию и острую миелобластную лейкемию. Лейкемия может быть рецидивной, резистентной или невосприимчивой к общепринятому лечению. Термин "рецидивная" относится к ситуации, когда у пациентов, у которых была ремиссия лейкемии после лечения, появляются лейкозные клетки в костный мозг и уменьшается количество нормальных кровяных клеток. Термин "резистентная или невосприимчивая" относится к обстоятельствам, когда у пациентов, даже после интенсивного лечения, имеются остаточные лейкозные клетки в костном мозге.
В другом варианте осуществления относящимися к настоящему изобретению являются способы лечения, предотвращения или менеджемента различных типов лимфом, включая неходжкинскую лимфому (NHL). Термин "лимфома" относится к гетерогенной группе новообразований, возникающих в ретикулоэндотелиальной и лимфатической системах. "NHL" относится к раковой моноклональной пролиферации лимфоидных клеток в областях иммуной системы, включая лимфоузлы, костный мозг, селезенку, печень и желудочно-кишечный тракт. Примеры NHL включают, но не ограничиваются, лимфому коры головного мозга (MCL), лимфоцитарную лимфому промежуточной дифференциации, промежуточную лимфоцитарную лимфому (ILL), диффузную низкодифференцированную лимфоцитарную лимфому (PDL), центроцитарную лимфому, диффузную мелкоклеточную лимфому с расщепленными ядрами (DSCCL), фолликулярную лимфому и любой тип лимфом коры головного мозга, которые можно увидеть под микроскопом (нодулярная, диффузная, бластическая лимфома и лимфома из клеток мантии).
Примеры заболеваний и расстройств, связанных с, или характеризующихся неблагоприятным ангиогенезом, включают, но не ограничиваются, воспалительные заболевания, аутоиммунные заболевания, вирусные заболевания, генетические заболевания, аллергенные заболевания, бактериальные заболевания, глазные неоваскулярные заболевания, хориоидальные неоваскулярные заболевания, неоваскулярные заболевания чувствительной оболочки глазного яблока и покраснения (неоваскуляризация уголков). Конкретные примеры заболеваний и расстройств, связанных с, или характеризующихся неблагоприятным ангиогенезом включают, но не ограничиваются, артрит, эндометриоз, болезнь Крона, сердечную недостаточность, запущенную сердечную недостаточность, почечную недостаточность, эндотоксикоз, токсический шок, остеоартрит, деление ретровирусов, атрофию, менингит, фиброз, вызванный диоксидом кремния, фиброз, вызванный асбестом, ветеринарное расстройство, гиперкальцемию, связанную со злокачественным новообразованием, удар, шок кровеносной системы, периодонтит, гингивит, микроцитарную анемию, резистентную анемию и синдром 5q-делеции.
Примеры боли включают, но не ограничиваются, примеры, описанные в патентной публикации США № 2005/0203142, внесенной в реестр 15 сентебря 2005, которая введена в данное изобретение с помощью ссылки. Конкретные виды болей включают, но не ограничиваются, ноцицептивную боль, нейропатическую боль, смешанную боль ноцицептивной и нейропатической боли, висцеральную боль, мигрень, головную боль и постоперационную боль.
Примеры ноцицептивной боли включают, но не ограничиваются, боль, связанную с химическим или термическим ожогами, порезы кожи, закрытую травму кожи, остеоартрит, ревматоидный артрит, тендонит и миофасциальную боль.
Примеры нейропатической боли включают, но не ограничиваются, CRPS I типа, CRPS II типа, симпатическую рефлекторную дистрофию (RSD), рефлекторную нейроваскулярную дистрофию, рефлекторную дистрофию, симпатически поддерживаемый болевой синдром, каузальгию, атрофию Зудека кости, алгонейродистрофию, плечевой синдром, посттравматическую дистрофию, невралгию тройничного нерва, постгерпетическую невролгию, боль, связанную с раком, послеампутационную боль, фибромиалгию, синдром хронической усталости, боль при повреждении спинного мозга, центральную боль после инсульта, радикулопатию, диабетическую нейропатию, боль после инсульта, люэтическую нейропатию и другие болевые нейропатические состояния, такие как состояния, вызванные лекарственным средством, таким как винкристин и велкейд.
Как применяют в настоящем изобретении, термины "комплексный региональный синдром боли", "CRPS" и "CRPS и связанные с ним синдромы" относятся к хроническому болевому расстройству, характеризующемуся одним или более из следующих симптомов: болью, или самопроизвольной или индуцированной, включая аллодинию (болевая ответная реакция на стимул, который обычно не является болевым) и гипералгезию (гипертрофированная ответная реакция на стимул, который обычно являются только слегка болевым); болью, которая не соответствует вызвавшему ее событию (например, годы острой боли после растяжения голеностопного сустава); местной болью, которая не ограничивается распространением только по периферическим нервам; и автономной дисрегуляцией (например, эдемом, изменением в циркуляции крови и гипергидрозом), связанной с трофическими кожными изменениями (аномальный рост волос и ногтей и кожная язва).
Примеры MD и связанных с ним синдромов включают, но не ограничиваются, MD и связанные с ним синдромы, описанные в публикации 2004/0091455, внесенной в реестр 13 мая 2004, которая введена в настоящее изобретение с помощью ссылки. Конкретные примеры включают, но не ограничиваются, атрафическую (сухую форму) MD, эссудативную (влажную форму) MD, возрастную макулопатию (ARM), хориоидальную неоваскуляризацию (CNVM), отслоение ретинального пигментного эпителия (PED) и атрофию ретинального пигментного эпителия (RPE).
Примеры кожных заболеваний включают, но не ограничиваются, кожные заболевания, описанные в публикации США № 2005/0214328A1, внесенной в реестр 29 сентября 2005, которая введена в настоящее изобретение с помощью ссылки. Конкретные примеры включают, но не ограничиваются, кератоз и связанные с ним симптомы, кожные заболевания или расстройства, характеризующиеся чрезмерным ростом эпидермиса, угри и морщины.
Как применяют в настоящем изобретении, термин "кератоз" относится к любому повреждению на эпидермисе, характеризующемуся наличием ограниченного чрезмерного роста рогового слоя, включая, но не ограничиваясь, старческий кератоз, себорейный кератоз, кератоакантому, фолликулярный кератоз (болезнь Дарье), инвертированный фолликулярный кератоз, ладонно-подошвенную кератодермию (PKK, ладонно-подошвенный кератоз), волосяной лишай и штукатурный кератоз. Термин "старческий кератоз" также относится к сенильному кератозу, кератозу старческому, старческой бородавке, юношеской бородавке, солнечному кератозу, кератодерме или кератоме. Термин "себорейный кератоз" также относится к себорейной бородавке, сенильной бородавке или базально-клеточной папилломе. Кератоз характеризуется одним или более из следующих симптомов: появлением шероховатостей, чешуеобразованием, эритематозной папулой, пятнами сыпи, спикулами или утолщениями на обнаженной поверхности (например, лице, руках, ушах, шеи, ногах и грудной клетке), наростом кератина, называемым кожные рога, гиперкератозом, телеангиэктазией, эластозом, пигментированным лентигинозом, акантозом, паракератозом, дискератозом, папилломатозом, избыточной пигментацией базальных клеток, клетотчной атипией, фигурой митоза, патологическим слипанием клеток, плотными воспалительными инфильтратами и небольшим распространением плоскоклеточных карцином.
Примеры кожные заболеваний или расстройств, характеризующихся чрезмерным ростом эпидермиса, включают, но не ограничиваются, любые состояния, заболевания или расстройства, характеризующиеся наличием чрезмерного роста эпидермиса, включая, но не ограничиваясь, инфекции, связанные с вирусом папилломы, арсенокератоз, симптом Лезера-Трела, бородавчатую дискератому (WD), пучкообразные волосы (TS), эритрокератодермию вариабельную (EKV), десквамативную эритродерму новорожденных (ихтиоз Арлекино), врожденную узловатость пальцев кисти, кожную меланоакантому, порокератоз, псориаз, плоскоклеточную карциному, безымянный пигментный папилломатоз (CRP), мягкие бородавки, кожный рог, болезнь Коудена (синдром множественных гамартом), папулезный черный дерматоз (DPN), синдром эпидермального невуса (ENS), вульгарный ихтиоз, заразительный (контагиозный) моллюск, узловатую почесуху, и черный акантоз (AN).
Примеры легочных расстройств включают, но не ограничиваются, легочные расстройства, описанные в публикации США № 2005/0239842 A1, внесенной в реестр 27 октября 2005, которая введена в настоящее изобретение с помощью ссылки. Конкретные примеры включают легочную гипертензию и связанные с ней расстройства. Примеры легочной гипертензии и связанных с ней расстройств включают, но не ограничиваются: первичную легочную гипертензию (PPH); вторичную легочную гипертензию (SPH); семейную PPH; спордическая PPH; прекапиллярную легочную гипертензию; легочную артериальную гипертензию (PAH); гипертензию легочной артерии; идиопатическую легочную гипертензию; тромботическую легочную артериопатию (TPA); плексогенную легочную артериопатию; легочную гипертензию функциональных классов I-IV; и легочную гипертензию, связанную с, относящуюся к, или на фоне, дисфункции левого желудочка, митрального порока сердца, констриктивного перикардита, стеноза устья аорты, кардиомиопатии, средостенного фиброза, аномального дренажа легочных вен, легочной вено-окклюзионной болезни, коллаген-сосудистой болезни, врожденного порока сердца, инфицирования ВИЧ-вирусом, лекарственных средств и токсинов, таких как фенфлурамины, врожденного заболевания сердца, легочной венозной гипертензии, хронического обструктивного легочного заболевания, интерстициального легочного процесса, нарушения дыхания во сне, альвеолярной гиповентиляции, длительного воздействия большой высоты, неонатальной болезни легких, альвеолярно-капиллярной дисплазии, серповидно-клеточной анемии, других расстройство коагулирования, хронической тромбэмболии, болезни соединительных тканей, волчанки, включая системную и кожную волчанку, шистосомоза, саркоидоза или легочного капиллярного гемангиоматоза.
Примеры расстройств, связанных с асбестом включают, но не ограничиваясь, расстройства, описанные в публикации США № 2005/0100529, внесенной в реестр 12 мая 2005, которая введена в настоящее изобретение с помощью ссылки. Конкретные примеры включают, но не ограничиваются, мезотелиому, асбестовый пневмокониоз, злокачественный плевральный выпот, доброкачественный экссудативный выпот, плевральные утолщения, плевральный кальциноз, диффузное плевральное утолщение, дисковидный ателеказ, фиброзные опухолевидные образования и рак легких.
Примеры паразитарных заболеваний включают, но не ограничиваются, паразитарные заболевания, описанные в публикации США № 2006/0154880, внесенной в реестр 13 июля 2006, которая введена в настоящее изобретение с помощью ссылки. Паразитарные заболевания включают заболевания и расстройства, вызванные человеческими внутриклеточными паразитами, такими как, но не ограничиваясь, P. falcifarium, P. ovale, P. vivax, P. malariae, L. donovari, L. infantum, L. aethiopica, L. major, L. tropica, L. mexicana, L. braziliensis, T. Gondii, B. microti, B. divergens, B. coli, C. parvum, C. cayetanensis, E. histolytica, I. belli, S. mansonii, S. haematobium, Trypanosoma ssp., Toxoplasma ssp., и O. volvulus. Также включены в объем настоящего изобретения другие заболевания и расстройства, вызванные нечеловеческими внутриклеточными паразитами, такими как, но не ограничиваясь, Babesia bovis, Babesia canis, Banesia Gibsoni, Besnoitia darlingi, Cytauxzoon felis, Eimeria ssp., Hammondia ssp. и Theileria ssp. Конкретные примеры включают, но не ограничиваются, малярию, бабезиеллез, трипанозомоз, лейшманиоз, токсоплазмоз, менингоэнцефалит, кератит, амебиаз, гиардиоз, криптоспоридиоз, изоспороз, циклоспороз, микроспоридиоз, аскаридозную инвазию, трихиуриаз, анкилостомоз, стронгилоидоз, токсокароз, трихинеллез, филяриатоз лимфоузлов, онхоцеркоз, филяриоз, бильгарциоз и дерматиты, вызванные шистосомами животных.
Примеры иммунодефицитных состояний включают, но не ограничиваются, иммунодефицитные состояния, описанные в заявке США № 11/289723, внесенной в реестр 30 ноября 2005. Конкретные примеры включают, но не ограничиваются, аденозиндезаминазную недостаточность, дефицит антител с нормальным или повышенным количеством иммуноглобулинов, атаксию-телеангиэктазию, синдром лысых лимфоцитов, вариабельный неклассифицируемый иммунодефицит, Ig дефицит с гипер-IgM, делеции тяжелых цепей иммуноглобулинов, IgA дефицит, иммунодефицит тиомом, дефицит стволовых клеток, синдром незелофа, изолированную недостаточность подклассов IgG-типа, транзиторную гипогаммаглобулинемию новорожденных, синдром Уискотта-Олдрича, гаммаглобулинемию, сцепленную с Х-хромосомой, тяжелый комбинированный иммунодефицит, связанный с Х-хромосомой.
Примеры расстройств ЦНС включают, но не ограничиваются, расстройства ЦНС, описанные в публикации США № 2005/0143344, внесенной в реестр 30 июня 2005, которая введена в настоящее изобретение с помощью ссылки. Конкретные примеры включают, но не ограничиваются, амиотрофический латеральный склероз, болезнь Альцгеймера, болезнь Паркинсона, болезнь Хантингтона, множественный склероз, другие нейроиммунологические расстройства, такие как синдром Туретта, делирий, или потерю сознания, которая длится в течение короткого промежутка времени, и амнестическое расстройство, или небольшие нарушения памяти, которые имеют место при отсутствии других нарушений центральной нервной системы.
Примеры повреждений ЦНС и связанных с ними синдромов включают, но не ограничиваются, повреждения ЦНС и связанные с ними синдромы, описанные в публикации США № 2006/0122228, внесенной в реестр 8 июня 2006, которая введена в настоящее изобретение с помощью ссылки. Конкретные примеры включают, но не ограничиваются, повреждение/поражение ЦНС и связанные с ним синдромы, включая, но не ограничиваются, первичную травму мозга, вторичную травму мозга, травматическое повреждение мозга, фокальное повреждение мозга, диффузное аксональное повреждение мозга, травму головы, сотрясение мозга, синдром после сотрясения мозга, ушиб и разрыв мозга, субдуральную гематому, эпидермальную гематому, посттравматическую эпилепсию, хроническое вегетативное состояние, полное SCI, неполное SCI, острое SCI, подострое SCI, хроническое SCI, центромедуллярный синдром, синдром Брауна-Секара, передний спинальный синдром, синдром конуса, синдром «конского хвоста», нейрогенный шок, спинальный шок, измененное состояние сознания, головную боль, тошноту, рвоту, потерю памяти, головокружение, диплопию, расфокусированное зрение, эмоциональную неустойчивость, нарушение сна, раздражительность, неспособность сконцентрироваться, повышенную возбудимость, нарушение поведения, когнитивное расстройство и судороги.
Другие заболевание или расстройств включают, но не ограничиваясь, вирусные, генетические, аллергенные и аутоиммунные заболевания. Конкретные примеры включают, но не ограничиваясь, ВИЧ, гепатит, синдром расстройства дыхания у взрослых, болезни резорбции костной ткани, хронические легочные воспалительные заболевания, дерматит, кистозный фиброз, септический шок, сепсис, эндотоксический шок, гемодинамический шок, сепсис-синдром, постишемическое реперфузионное повреждение, менингит, псориаз, фиброз, кахексию, реакцию трасплантанта против хозяина, отторжение ткани, аутоиммунное заболевание, ревматоидный спондилит, болезнь Крона, язвенный колит, воспалительное заболевание кишечника, множественный склероз, системную красную волчанку, эритему узловатую лепрозную, лучевую болезнь, рак, астму или гипероксийное альвеолярное повреждение.
Примеры атеросклероза и связанных с ним состояний включают, но не ограничиваются, атеросклероз и связанные с ним состояния, описанные в публикации США № 2002/0054899, внесенной в реестр 9, мая 2002, которая введена в настоящее изобретение с помощью ссылки. Конкретные примеры включают, но не ограничиваются, все формы состояний, включая атеросклероз, включая рестеноз после васкулярной интервенции, такой как пластика сосудов, стентирование, атерэктомия и трансплантация. Настоящее изобретение относится ко всем видам васкулярной интервенции, включая заболевания сердечно-сосудистой и почечной системы, такие как, но не ограничиваясь, почечную ангиопластику, чрезкожная коронарная ангиопластика (PCI), чрезкожная транслюминальная коронарная ангиопластика (PTCA), каротидная чрезкожная транслюминальная ангиопластика (PTA), коронарная реваскуляризация, ангиопластика со стентовой имплантацией, периферийная чрезкожная транслюминальная интервенция подвздошной кости, бедра или подколенной артерии, и хирургическое вмешательство, применяя пропитанные искусственные имплантанты. В следующей таблице перечислены главные системные артерии, для которых необходимо лечение, все из которых включены в настоящее изобретение:
Примеры дисфункционального сна и связанных с ним синдромов включают, но не ограничиваются, дисфункциональный сон и связанные с ним синдромы, описанные в публикации США № 2005/0222209A1, внесенной в реестр 6 октября 2005, которая введена в настоящее изобретение с помощью ссылки. Конкретные примеры включают, но не ограничиваются, храп, апноэ во время сна, бессоницу, нарколепсию, синдром беспокойных ног, ночные ужасы, лунатизм, прием пищи во сне и дисфункциональный сон, связанный с хроническими неврологическими или воспалительными состояниями. Хронические неврологические или воспалительные состояния включают, но не ограничиваются, комплексный региональный болевой синдром, хроническую боль в пояснице, костно-мышечную боль, артрит, радикулопатию, боль, связанную с раком, фибромиалгию, синдром хронической усталости, висцеральную боль, боль в мочевом пузыре, хронический панкреатит, невропатию (диабетическую, постгерпетическую, травматическую или воспалительную) и нейродегенеративные расстройства, такие как болезнь Паркинсона, болезнь Альцгеймера, амиотрофический латеральный склероз, множественный склероз, болезнь Хантингтона, брадикинезию; мышечную ригидность; тремор при болезни Паркинсона; нарушение походки при болезни Паркинсона; «застывшее движение»; депрессию; нарушение долговременной памяти, синдром Рабинстайна-Тауби (RTS); деменцию; нарушение ходьбы; гипокинетические расстройства; расстройства, связанные с выработкой синуклеина; множественную системную атрофию; дегенерация черного вещества полосатого тела; оливопонтоцеребеллярную атрофию; синдром Ши-Драгера; заболевание двигательных нейронов с признаками болезни Паркинсона; деменцию с тельцами Леви; тау патологические расстройства; прогрессирующий надъядерный паралич; кортикобазальную дегенерацию; фронтотемпоральную деменцию; амилоидные патологические расстройства; умеренное когнитивное нарушение; болезнь Альцгеймера с паркинсонизмом; болезнь Вильсона; болезнь Галлервордена-Спатца; болезнь Чедиака-Хагаши; SCA-3 спиноцеребеллярную атаксию; X-сцепленный синдром дистонии - паркинсонизма; прионное заболевание; гиперкинетические расстройства; хорею; баллизм; дистонические треморы; амиотрофический латеральный склероз (ALS); травму ЦНС и миоклонус.
Примеры гемоглобинопатии и связанных с ней расстройств включают, но не ограничиваются, гемоглобинопатию и связанные с ней расстройства, описанные в публикации США № 2005/0143420A1, внесенной в реестр 30 июня 2005, которая введена в настоящее изобретение с помощью ссылки. Конкретные примеры включают, но не ограничиваются, гемоглобинопатию, серповидноклеточную анемию и любые другие расстройства, связанные с дифференциацией CD34+ клеток.
Примеры расстройств, связанных с TNFα, включают, но не ограничиваются, расстройства, описанные в WO 98/03502 и WO 98/54170, оба из которых вводятся в настоящее изобретение полностью с помощью ссылки. Конкретные примеры включают, но не ограничиваются: эндотоксемию или синдром токсического шока; кахексию; респираторный дистресс-синдром у взрослых; заболевания, связанные с резорбцией кости, такие как артрит; гиперкальцемию; реакцию «имплантант против хозяина»; церебральную малярию; воспаление; рост опухоли; хронические легочные воспалительные заболевания; реперфузионное повреждение; инфаркт миокарда; инсульт; циркуляторный шок; ревматоидный артрит; болезнь Крона; ВИЧ инфицирование и СПИД; другие расстройства, такие как ревматоидный артрит, ревматоидный спондилит, остеоартрит, псориатический артрит и другие артритные состояния, септический шок, сепсис, эндотоксический шок, реакция «имплантант против хозяина», атрофию, болезнь Крона, язвенные колиты, множественный склероз, системная красная волчанка, эритема узловатая лепрозная, ВИЧ, СПИД и оппортунистические инфекции при СПИДе; расстройства, такие как септический шок, сепсис, эндотоксический шок, гемодинамический шок и сепсис-синдром, постишемическое реперфузионное повреждение, малярия, микобактериальная инфекция, менингит, псориаз, застойная сердечная недостаточность, фибротическое изменение, кахексия, отторжение ткани, онкогенные или раковые состояния, астма, аутоиммунное заболевание, радиационное повреждение и гипероксическое альвеолярное повреждение; вирусные инфекции, такие как инфекции, вызванные вирусами герпеса; вирусный конъюктивит; или атопический дерматит.
В других вариантах осуществления применение соединений, относящихся к настоящему изобретению в различных иммунологических применениях, т.е. применение соединений, относящихся к настоящему изобретению, в комбинации с вакцинацией, например, в виде вспомогательного вещества вакцины. Хотя любые способы и методы применения соединений, относящихся к настоящему изобретению, в комбинации с вакциной, предполагаются в настоящем изобретении, неограничивающим примером таких применений является применение соединений, относящихся к настоящему изобретению, в качестве вспомогательных веществ вакцины, согласно режимам введения, описанным в предварительной заявке США № 60/712823, внесенной в реестр 1 сентября 2005, которая вводится в настоящее изобретение полностью с помощью ссылки. Данные варианты осуществления также относятся к применениям соединений, относящихся к настоящему изобретению, в комбинации с вакцинами для того, чтобы лечить или предотвращать рак или инфекционные заболевания, и к другим различным применениям соединений, относящихся к настоящему изобретению, таким как, но не ограничиваясь, ослабление или снижение чувствительности аллергических реакций.
Дозы соединения, относящегося к настоящему изобретению, или его фармацевтически приемлемой соли, сольвата, клатрата, стереоизомера или пролекарства, изменяется в зависимости от факторов, таких как: конкретное состояние, которое нужно лечить, предотвращать или контролировать; возраст и состояние пациента; и количество применяемого второго активного средства, если его вообще применяют. Обычно, соединение, относящееся к настоящему изобретению, или его фармацевтически приемлемую соль, сольват, клатрат, стереоизомер или пролекарство можно применять в количестве от, приблизительно, 0,1 мг до, приблизительно, 500 мг в день и можно приспосабливать для общепринятого способа введения (например, то же количество, вводимое каждый день периода лечения, предотвращения или менеджемента), циклического способа введения (например, одну неделю вводить, одну нет), или введения в количестве, которое увеличивают или уменьшают в течение курса лечения, предотвращения или менеджемента. В других вариантах осуществления доза может составлять от, приблизительно, 1 мг до, приблизительно, 300 мг, от, приблизительно, 0,1 мг до, приблизительно, 150 мг, от, приблизительно, 1 мг до, приблизительно, 200 мг, от, приблизительно, 10 мг до, приблизительно, 100 мг, от, приблизительно, 0,1 мг до, приблизительно, 50 мг, от, приблизительно, 1 мг до, приблизительно, 50 мг, от, приблизительно, 10 мг до, приблизительно, 50 мг, от, приблизительно, 20 мг до, приблизительно, 30 мг, или от, приблизительно, 1 мг до, приблизительно, 20 мг.
4.3. ВТОРЫЕ АКТИВНЫЕ СРЕДСТВА
Соединение, относящееся к настоящему изобретению, или его фармацевтически приемлемую соль, сольват, пролекарство, клатрат или стереоизомер можно комбинировать с другими фармакологически активными соединениями ("вторыми активными средствами") в способах и композициях, относящихся к настоящему изобретению. Определенные комбинации могут действовать синергически при лечении конкретных типов заболеваний или расстройств и состояний и симптомов, связанных с данными заболеваниями или расстройствами. Соединение, относящееся к настоящему изобретению, или его фармацевтически приемлемая соль, сольват, клатрат, стереоизомер или пролекарство может также действовать, облегчая побочные эффекты, связанные с определенными вторыми активными средствами, и наоборот.
Один или более вторых активных ингредиентов или средств можно применять в способах и композициях, относящихся к настоящему изобретению. Вторые активные средства могут быть большими молекулами (например, белками) или малыми молекулами (например, синтетическими неорганическими, органометаллическими или органическими молекулами).
Примеры активных средств, являющихся большими молекулами, включают, но не ограничиваются, гематопоэтические факторы роста, цитокины и моноклональные и поликлональные антитела. Конкретными примерами активных средств являются анти-CD40 моноклональные антитела (такие как, например, SGN-40); ингибиторы деацелирования гистонов (такие как, например, SAHA и LAQ 824); ингибиторы белка теплового шока-90 (такие как, например, 17-AAG); ингибиторы киназы рецептора инсулиноподобного фактора роста-1; ингибиторы киназы сосудистого эндотелиального фактора роста (такие как, например, PTK787); ингибиторы рецептора инсулинового фактора роста; ингибиторы ацилтрансферазы лизофосфатидиловой кислоты; ингибиторы IkB киназы; p38MAPK ингибиторы; EGFR ингибиторы (такие как, например, гефитиниб и эрлотиниб HCL); HER-2 антитела (такие как, например, трастузумаб (Herceptin®) и пертузумаб (Omnitarg™)); VEGFR антитела (такие как, например, бевацизумаб (Avastin™)); VEGFR ингибиторы (такие как, например, ингибиторы flk-1 специфической киназы, SU5416 и ptk787/zk222584); P13K ингибиторы (такие как, например, вортманнин); C-Met ингибиторы (такие как, например, PHA-665752); моноклональные антитела (такие как, например, ритуксимаб (Rituxan®), тоситумомаб (Bexxar®), эдреколомаб (Panorex®) и G250); и анти-TNF-α антитела. Примеры активных средств, являющихся малыми молекулами, включают, но не ограничиваются, противораковые средства и антибиотики (например, кларитромицин).
Конкретные вторые активные соединения, которые можно комбинировать с соединениями, относящимися к настоящему изобретению, изменяются в зависимости от конкретного состояния, в отношении которого осуществляют лечение, предотвращение или менеджмент.
Например, для лечения, предотвращения или менеджмента рака, вторые активные средства включают, но не ограничиваются: семаксаниб; циклоспорин; этанерцепт; доксициклин; бортезомиб; лапатиниб (Tykerb®); ацивицин; акларубицин; гидрохлорид акодазола; акронин; адозелесин; альдеслеукин; альтретамин; амбомицин; ацетат аметантрона; амсакрин; анастрозол; антамицин; аспарагиназу; асперлин; азацитидин; азетепа; азотомицин; батимастат; бензодепа; бикалутамид; гидрохлорид бисантрена; биснафид димезилат; бизелесин; блеомицинсульфат; бреквинар натрия; бропиримин; бусульфан; кактиномицин; калустерон; карацемид; карбетимер; карбоплатин; кармустин; гидрохлорид карубицина; карзелесин; цедефингол; целекоксиб; хлорамбуцил; циролемицин; цисплатин; кладрибин; криснатол мезилат; циклофосфамид; цитарабин; дакарбазин; диктиномицин; гидрохлорид даунорубицина; децитабин; дексормаплатин; дезагуанин; дезагуанин мезилат; диазиквон; доцетаксел; доксорубицин; гидрохлорид доксорубицина; дролоксифен; дролоксифен цитрат; дромостанолона пропионат; дуазомицин; эдатрексат; гидрохлорид эфлорнитина; элсамитруцин; энлоплатин; энпромат; эпипропидин; гидрохлорид эпирубицина; ербулозол; гидрохлорид эсорубицина; эстрамустин; эстрамустин фосфат натрия; этанидазол; этопозид; этопозид фосфат; этоприн; гидрохлорид фадразола; фазарабин; фенретинид; флоксуридин; флударабин фосфат; фторурацил; фторцитабин; фосквидон; фостриецин натрия; гемцитабин; гидрохлорид гемцитабина; гидроксимочевина; гидрохлорид идарубицина; ифосфамид; илмофосин; ипроплатин; иринотекан; гидрохлорид иринотекана; ланреотид ацетат; летрозол; леупролид ацетат; гидрохлорид лиарозола; лометрексол натрия; ломустин; гидрохлорид лосоксантрона; масопрокол; майтансин; гидрохлорид мехлоретамина; мегестрол ацетат; меленгестрол ацетат; мелфалан; меногарил; меркаптопурин; метотрексат; метотрексат натрия; метоприн; метуредепа; митиндомид; митокарцин; митокромин; митогиллин; митомальцин; митомицин; митоспер; митотан; гидрохлорид митоксантрона; микофеноловая кислота; нокодазол; ногаламицин; ормаплатин; оксисуран; паклитаксел; пегаспаргаза; пелиомицин; пентамустин; пепломицин сульфат; перфосфамид; пипоброман; пипосульфан; гидрохлорид пироксантрона; пликамицин; пломестан; порфимер натрия; порфиромицин; преднимустин; гидрохлорид прокарбазина; пиромицин; гидрохлорид пиромицина; пиразофурин; рибоприн; сафингол; гидрохлорид сафингола; семустин; симтразен; спарфозат натрия; спарсомицин; гидрохлорид спирогермания; спиромустин; спироплатин; стрептонигрин; стрептозоцин; сулофенур; талисомицин; текогалан натрия; таксотер; тегафур; гидрохлорид телоксантрона; темопорфин; тенипозид; тероксирон; тестолактон; тиамиприн; тиогуанин; тиотепа; тиазофурин; тирапазамин; торемифен цитрат; трестолон ацетат; трицирибин фосфат; триметрексат; триметрексат глюкуронат; трипторелин; гидрохлорид тубулозола; урацил мустард; уредепа; вапреотид; вертепофрин; винбластин сульфат; винкристин сульфат; виндезин; виндезин сульфат; винепидин сульфат; винглицинат сульфат; винлеурозин сульфат; винорелбин тартрат; винросидин сульфат; винзолидин сульфат; ворозол; зениплатин; зиностатин; и гидрохлорид зорубицина.
Другие вторые средства включают, но не ограничиваются: 20-эпи-1,25-дигидроксивитамин D3; 5-этинилурацил; абиратерон; акларубицин; ацилфульвен; адеципенол; адозелесин; альдеслеукин; ALL-TK антагонисты; алтретамин; амбамустин; амидокс; амифостин; аминолевулиновая кислота; амрубицин; амсакрин; анагрелид; анастрозол; андрографолид; ингибиторы ангиогенеза; антагонист D; антагонист G; антареликс; анти-дорсализующий морфогенетический белок - 1; антиандроген простатической карциномы; антиэстроген; антинеопластон; олигонуклеотиды, применяемые в антисенс-терапии; афидиколин глицинат; генные модуляторы апоптоза; регуляторы апоптоза; апуриновую кислоту; ара-CDP-DL-PTBA; аргининдеаминазу; асулакрин; атаместан; атримустин; аксинастатин 1; аксинастатин 2; аксинастатин 3; азасетрон; азатоксин; азатирозин; производные баккатина III; баланол; батимастат; BCR/ABL антагонисты; бензохлорины; бензоилстауроспорин; бета-лактамные производные; бета-алетин; бетакламицин B; бетулиновую кислоту; bFGF ингибитор; бикалутамид; бисантрен; бисазиридинилспермин; биснафид; бистратен A; бизелесин; брефлат; бропиримин; будотитан; бутионин сульфоксимин; кальципотриол; калфостин C; камптотециновые производные; капецитабин; карбоксамид-аминотриазол; карбоксамидотриазол; CaRest M3; CARN 700; ингибитор, являющийся производным картилага; карзелесин; ингибиторы казеиновой киназы (ICOS); кастаноспермин; цекропин B; цетрореликс; хлорины; хлорхиноксалин сульфонамид; цикапрост; цис-порфирин; кладрибин; кломифеновые аналоги; клотримазол; коллисмицин A; коллисмицин B; комбретастатин A4; комбретастатиновый аналог; конагенин; крамбесцидин 816; криснатол; криптофицин 8; производные криптофицина A; курацин A; циклопентантрахиноны; циклоплатам; ципемицин; цитарабин окфосфат; цитолитический фактор; цитостатин; дакликсимаб; децитабин; дегидродидемнин B; деслорелин; дексаметазон; дексифосфамид; дексразоксан; дексверапамил; диазиквон; дидемнин B; дидокс; диэтилнорспермин; дигидро-5-азацитидин; дигидротаксол-9; диоксамицин; дифенилспиромустин; доцетаксел; докосанол; доласетрон; доксифлуридин; доксорубицин; дролоксифен; дронабинол; дуокармицин SA; эбселен; экомустин; эделфосин; эдреколомаб; эфлорнитин; элемен; эмитефур; эпирубицин; эпристерид; эстрамустиновый аналог; агонисты эстрогена; антагонисты эстрогена; этанидазол; этопозид фосфат; эксеместан; фадрозол; фазарабин; фенретинид; филграстим; финастерид; флавопиридол; флезеластин; флуастерон; флударабин; гидрохлорид фтордауноруницина; форфенимекс; форместан; фостриецин; фотемустин; тексапирин гадолиния; нитрат галлия; галоцитабин; ганиреликс; ингибиторы желатиназы; гемцитабин; ингибиторы глутатиона; хепсулфам; херегулин; гексаметиленбисацетамид; гиперицин; ибандроновую кислоту; идарубицин; идоксифен; идрамантон; илмофозин; иломастат; иматиниб (Gleevec®), имиквимод; иммуностимулирующие пептиды; ингибитор рецептора инсулиноподобного фактора роста 1; агонисты интерферона; интерфероны; интерлейкины; иобенгуан; йоддоксорубицин; ипомеанол, 4-; ироплакт; ирсогладин; изобенгазол; изогомохаликордин B; итазетрон; джасплакинолид; кахалалид F; ламелларин-N триацетат; ланреотид; леинамицин; ленограстим; лентинан сульфат; лептолстатин; летрозол; фактор ингибирования лейкемии; лейкоцитарный альфа-интерферон; леупролид+эстроген+прогестерон; леупрорелин; левамисол; лиарозол; линейный полиаминовый аналог; липофильный дисахаридный пептид; липофильные платиновые соединения; лиссоклинамид 7; лобаплатин; ломбрицин; лометрексол; лонидамин; лосоксантрон; локсорибин; луртотекан; тексапирин лютеция; лизофиллин; литические пептиды; маитансин; манностатин A; маримастат; масопрокол; маспин; ингибиторы матрилизина; ингибиторы матриксной металлопротеиназы; меногарил; мербарон; метерелин; метиониназа; метоклопрамид; MIF ингибитор; мифепристон; милтефосин; миримостим; митугуазон; митолактол; митомициновые аналоги; митонафид; фактор роста митотоксиновых фибробластов-сапорин; митоксантрон; мофаротен; молграмостим; Эрбитукс (Erbitux), хорионический гонадотропин человека; монофосфорилированный липид A + скелет микобактериальной клеточной стенки; мопидамол; мустардовое противораковое средство; микапероксид B; экстракт микобактериальной клеточной стенки; мириапорон; N-ацетилдиналин; N-замещенные бензамиды; нафарелин; нагрестип; налоксон+пентазоцин; напавин; нафтерпин; нартограстим; недаплатин; неморубицин; неридроновая кислота; нилутамид; нисамицин; модуляторы оксида азота; нитроксидный антиоксидант; нитруллин; облимесен (Genasense®); O6-бензилгуанин; октреотид; окиценон; олигонулкотиды; онапристон; ондансетрон; орацин; пероральный индуктор цитокинов; ормаплатин; осатерон; оксалиплатин; оксауномицин; паклитаксел; аналоги паклитаксела; производные паклитаксела; палауамин; палмитоилризоксин; памидроновая кислота; панакситриол; паномифен; парабактин; пазеллиптин; пегаспаргаза; пелдесин; пентозан полисульфат натрия; пентостатин; пентрозол; перфлуброн; перфосфамид; периллиловый спирт; феназиномицин; фенилацетат; ингибиторы фосфотазы; пикибанил; гидрохлорид пилокарпина; пирарубицин; пиритрексим; плацетин A; плацетин B; ингибитор активатора плазминогена; платиновый комплекс; платиновые соединения; платина-триаминовый комплекс; порфимер натрия; порфиромицин; преднизон; пропил бисакридон; простагландин J2; ингибиторы протеасомы; иммунный модулятор на основе белка A; ингибитор протеинкиназы C; ингибиторы протеинкиназы C, микроалгал; ингибиторы протеин-тирозин-фосфатазы; ингибиторы фосфарилазы пуриновых нуклеозидов; пурпурины; пиразолакридин; конъюгат пиридоксилированного гемоглобина и полиоксиэтилена; raf антагонисты; ралтитрексед; рамосетрон; ингибиторы ras фарнезил протеиновой трансферазы; ras ингибиторы; ras-GAP ингибитор; деметилированный ретеллиптин; этидронат рения Re 186; ризоксин; рибозимы; RII ретинамид; рохитукин; ромуртид; роквинимекс; рубигинон B1; рубоксил; сафингол; саинтопин; SarCNU; саркофитол A; сарграмостим; Sdi 1 миметики; семустин; ингибитор 1, вызывающий старение; олигонуклеотиды, применяемые в сенс-терапии; ингибиторы сигнальной трансдукции; сизофиран; собузоксан; боркаптат натрия; фенилацетат натрия; солверол; соматомедин-связывающий белок; сонермин; спарфосовая кислота; спикамицин D; спиромустин; спленопентин; спронгистатин 1; скваламин; стипиамид; ингибиторы стромелизина; сульфинозин; гиперактивный антагонист вазоактивного интестинальноо пептида; сурадиста; сурамин; сваинсонин; таллимустин; тамоксифен метйодид; тауромустин; тазаротен; текогалан натрия; тегафур; пирилий теллура; ингибиторы теломеразы; темопорфин; тенипозид; тетрахлордекаоксид; тетразомин; талибластин; тиокоралин; тромбопоэтин; тромбопоэтиновый миметик; тималфазин; агонист рецептора тимопоэтина; тимотринан; тиреотропный гормон; этилэтиопурпурин олова; тирапазамин; титаноцен бихлорид; топсентин; торемифен; ингибиторы трансляции; третиноин; триацетилуридин; трицирибин; триметрексат; трипторелин; трописетрон; туростерид; ингибиторы тирозинкиназы; тирфостины; UBC ингибиторы; убенимекс; фактор ингибирования роста, полученый из мочеполового синуса; антагонисты рецептора урокиназы; вапреотид; вариолин B; веларесол; верамин; вердины; вертепорфин; винорелбин; винксалтин; витаксин; ворозол; занотерон; зениплатин; зиласкорб; и зиностатин стималамер.
Конкретные вторые активные средства включают, но не ограничиваются, 2-метоксиэстрадиол, теломестатин, индукторы апаптоза в множественных миеломных клетках (такие как, например, TRAIL), статины, семаксаниб, циклоспорин, этанерцепт, доксициклин, бортезомиб, облимерсен (Genasense®), ремикад, доцетаксел, целекоксиб, мелфалан, дексаметазон (Decadron®), стероиды, гемцитабин, цисплатин, темозоломид, этопозид, циклофосфамид, темодар, карбоплатин, прокарбазин, глиадел, тамоксифен, топотекан, метотрексат, Arisa®, таксол, таксотер, фторурацил, леуковорин, иринотекан, кселоду, CPT-11, интерферон альфа, пелигилированный интерферон альфа (например, PEG INTRON-A), капецитабин, цисплатин, тиотепа, флударабин, карбоплатин, липосомальный даунорубицин, цитарабин, доксетаксол, палицитаксел, винбластин, IL-2, GM-CSF, дакарбазин, винорелбин, золедроновую кислоту, палмитронат, биаксин, бусульфан, преднизон, бифосфонат, триоксид арсения, винкристин, доксорубицин (доксир), паклитаксел, ганцикловир, адриамицин, эстрамустин натрия фосфат (Emcyt®), сулиндак и этопозид.
В другом варианте осуществления примеры конкретных вторых средств, согласно состояниям, в отношении которых проводят лечение, предотвращение или менеджмент, можно найти в следующих документах, все из которых вводятся в настоящее изобретение полностью с помощью ссылки: патент США № 6281230 и 5635517; публикации США № 2004/0220144, 2004/0190609, 2004/0087546, 2005/0203142, 2004/0091455, 2005/0100529, 2005/0214328, 2005/0239842, 2006/0154880, 2006/0122228 и 2005/0143344; и предварительная заявка США 60/631870.
Примеры вторых активных средств, которые можно применять для лечения, предотвращения и/или менеджемента боли, включают, но не ограничиваются, общепринятые терапевтические средства, применяемые для лечения или предотвращения боли, такие как антидепрессанты, противосудорожные средства, гипотензивные средства, анксиолитики, блокаторы кальциевых каналов, миорелоксанты, ненаркотические анальгетики, опиоидные анальгетики, противовоспалительные средства, cox-2 ингибиторы, иммуномодуляторы, агонисты или антагонисты альфа-адренергического рецептора, иммунодепрессанты, кортикостероиды, кислород под повышенным давлением, кетамин, другие анастезирующие средства, NMDA антагонисты и другие терапевтические средства, найденные, например, в Physician 's Desk Reference 2003. Конкретные примеры включают, но не ограничиваются, ацетат салициловой кислоты (Aspirin®), целекоксиб (Celebrex®), Enbrel®, кетамин, габапентин (Neurontin®), фенитоин (Dilantin®),н карбамазепин (Tegretol®), оксакарбазепин (Trileptal®), вальпроевую кислоту (Depakene®), сульфат морфина, гидроморфон, преднизон, грисеофульвин, пентониум, алендронат, дифенгидрамид, гуанетидин, кеторолак (Acular®), тирокальцитоцин, диметилсульфоксид (DMSO), клонидин (Catapress®), бретилиум, кетансерин, резерпин, дроперидол, атропин, фентоламин, бупивакаин, лидокаин, ацетаминофен, нортриптилин (Pamelor®), амитриптилин (Elavil), имипрамин (Tofranil), доксепин (Sinequan®), кломипрамин (Anafranil®), флуоксетин (Prozac®), сертралин (Zoloft®), напроксен, нефазодон (Serzone®), венлафаксин (Effexor®), тразодон (Desyrel®), бупропион (Wellbutrin®), мексилетин, нифедипин, пропранолол, трамадол, ламотригин, виокс, зиконотид, кетамин, декстрометорфан, бензодиазепины, баклофен, тизанидин и феноксибензамин.
Примеры вторых активных средств, которые можно применять для лечения, предотвращения и/или менеджемента дегенерации желтого пятна и связанных с ней синдромов, включают, но не ограничиваются, стероид, сенсибилизатор света, интегрин, антиоксидант, интерферон, ксантиновое производное, гормон роста, нейротрофический фактор, регулятор неоваскуляризации, анти-VEGF антитело, простагландин, антибиотик, фитоэстроген, противовоспалительное соединение или противоангиогенезное соединение, или их комбинация. Конкретные примеры включают, но не ограничиваются, вертепорфин, пурлитин, ангиостатический стероид, rhuFab, интерферон-2α, пентоксифуллин, этиопурпурин олова, мотексафин, луцентис, лутетиум, 9-фтор-11,21-дигидрокси-16,17-1-метилэтилиденбис(окси)прегна-1,4-диен-3,20-дион, латанопрост (смотри патент США № 6225348), тетрациклин и его производные, рифамицин и его производные, микролиды, метронидазол (патент США № 6218369 и 6015803), генистеин, генистин, 6'-О-MaI генистин, 6'-О-Ac генистин, даидзеин, даидзин, 6'-О-MaI даидзин, 6'-О-Ac даидзин, глицитеин, глицитин, 6'-О-MaI глицитин, биоканин A, формононетин (патент США № 6001368), триамцинолон ацетомид, дексаметазон (Патент США № 5770589), талидомид, глутатион (Патент США № 5632984), основной фактор роста фибробластов (bFGF), трансформирующий фактор роста b (TGF-b), мозговой нейротрофический фактор (BDNF), фактор активации плазминогена типа 2 (PAI-2), EYElOl (Eyetech Farmaceutics), LY333531 (Eli Lilly), миравант и ретисерт-имплантант (Bausch & Lomb). Все документы, цитируемые в настоящем изобретении, вводятся полностью с помощью ссылки.
Примеры вторых активных средств, которые можно применять для лечения, предотвращения и/или менеджмента кожных заболеваний, включают, но не ограничиваются, кератолы, ретиноиды, α-гидроксикислоты, антибиотики, коллаген, ботулинический токсин, интерферон, стероиды и иммуномодуляторы. Конкретные примеры включают, но не ограничиваются, 5-фторурацил, масопрокол, трихлоруксусную кислоту, салициловую кислоту, молочную кислоту, лактат аммония, мочевину, третиноин, изотетиноин, антибиотики, коллаген, ботулинический токсин, интерферон, кортикостероид, трансретиноевую кислоту и коллагены, такие как человеческий плацентарный коллаген, животный плацентарный коллаген, Дермалоген, Аллодерм, Фаскию, Циметру, Автологен, Зудерм, Зупласт, Резопласт и Изолаген.
Примеры вторых активных средств, которые можно применять для лечения, предотвращения и/или менеджмента легочной гипертензии и связанных с ней расстройств, включают, но не ограничиваются, антикоагулянты, диуретики, сердечные гликозиды, блокаторы кальциевых каналов, сосудорасширяющие средства, аналоги простациклина, антагонисты эндотелина, ингибиторы фосфодиэстеразы (например, PDE V ингибиторы), ингибиторы эндопептидазы, гиполипидемические средства, ингибиторы тромбоксана и другие терапевтические средства, о которых известно, что они снижают давление в легочной артерии. Конкретные примеры включают, но не ограничиваются, варфарин (Coumadin®), диуретик, сердечный гликозид, диоксин-кислород, дилтиазем, нифедипин, сосудорасширяющее средство, такое как простациклин (например, простагландин 12 (PGI2), эпопростенол (EPO, Floran®), трепростинил (Remodulin®), оксид азота (NO), босентан (Tracleer®), амлодипин, эпопростенол (Floran®), трепростинил (Remodulin®), простациклин, тадалафил (Cialis®), симвастатин (Zocor®), омапатрилат (Vanlev®), ирбесартан (Avapro®), правастатин (Pravachol®), дигоксин, L-аргинин, илопрост, бетапрост и силденафил (Viagra®).
Примеры вторых активных средств, которые можно применять для лечения, предотвращения и/или менеджмента расстройств, связанных с асбестом, включают, но не ограничиваются, антрациклин, платинум, алкилирующее средство, облимерсен (Genasense®), цисплатинум, циклофосфамид, темодар, карбоплатин, прокарбазин, глиадел, тамоксифен, топотекан, метотрексат, таксотере, иринотекан, капецитабин, цисплатин, тиотепу, флударабин, карбоплатин, липосомальный даунорубицин, цитарабин, доксетаксол, пацилитаксел, винбластин, IL-2, GM-CSF, дакарбазин, винорелбин, золедроновую кислоту, палмитронат, биаксин, бусульфан, преднизон, бисфосфат, триоксид арсения, винкристин, доксорубицин (Doxil), паклитаксел, ганцикловир, адриамицин, блеомицин, гиалуронидаза, митомицин C, мепакрин, тиотепа, тетрациклин и гемцитабин.
Примеры вторых активных средств, которые можно применять для лечения, предотвращения и/или менеджмента паразитарных заболеваний, включают, но не ограничиваются, хлороквин, квинин, квинидин, пириметамин, сульфадиазин, доксициклин, клиндамицин, мефлоквин, галофантрин, примаквин, гидроксихлорквин, прогуанил, атоваквон, азитромицин, сурамин, пентамидин, меларсопрол, нифуртимокс, бензнидазол, амфотерецин B, соединения пятивалентной сурьмы (например, стибоглюкуронат натрия), интерферон гамма, итраконазол, комбинацию мертвых промастигот и BCG, леуковорин, кортикостероиды, сульфамид, спирамицин, IgG (serology), триметоприм и сульфометоксазол. Примеры вторых активных средств, которые можно применять для лечения, предотвращения и/или менеджмента иммунодефицитарных состояний, включают, но не ограничиваются: антибиотики (терапевтические или профилактические), такие как, но не ограничиваясь, ампициллин, тетрациклин, пенициллин, цепгалоспорины, стрептомицин, канамицин и эритромицин; антивирусные соединения, такие как, но не ограничиваясь, амантадин, римантадин, ацикловир и рибавирин; иммуноглобулин; плазму; лекарственние средства, повышающие иммунитет, такие как, но не ограничиваясь, левамисол и изопринозин; биопрепараты, такие как, но не ограничиваясь, гамма-глобулин, фактор переноса, интерлейкины и интерфероны; гормоны, такие как, но не ограничиваясь, тимический гормон; и другие иммунологические средства, такие как, но не ограничиваясь, стимуляторы B клеток (например, BAFF/BlyS), цитокины (например, IL-2, IL-4 и IL-5), факторы роста (например, TGF-α), антитела (например, анти-CD40 и IgM), олигонуклеотиды, содержащие неметилированные CpG фрагменты, и вакцины (например, вирусные и опухолевые пептидные вакцины).
Примеры вторых активных средств, которые можно применять для лечения, предотвращения и/или менеджмента расстройств ЦНС, включают, но не ограничиваются: опиоиды; агонист или антагонист допамина, такие как, но не ограничиваясь, Леводопа, L-DOPA, кокаин, α-метилтирозин, резерпин, тетрабеназин, бензотропин, паргилин, фенодолпам мезилат, каберголин, дигидрохлорид прамипексола, ропинорол, гидрохлорид амантадина, гидрохлорид селегилина, карбидопу, перголид мезилат, Синемет CR и Симметрел; ингибитор MAO, такой как, но не ограничиваясь, ипрониазид, клоргилин, фенелзин и изокарбоксамид; COMT ингибитор, такой как, но не ограничиваясь, толкапон и энтакапон; ингибитор холинестеразы, такой как, но не ограничиваясь, физостигмин салицилат, физостигмин сульфат, физостигмин бромид, меостигмин бромид, неостигмин метилсульфат, амбеноним хлорид, эдрофониум хлорид, такрин, пралидоксим хлорид, обидоксим хлорид, тримедоксим бромид, диацетилмоноксим, эндрофоний, пиридостигмин и демекарий; противовоспалительное средство, такое как, но не ограничиваясь, напроксен натрия, диклофенак натрия, диклофенак калия, целекоксиб, сулиндак, оксапрозин, дифлунизал, этодолак, мелоксикам, ибупрофен, кетопрофен, набуметон, рефекоксиб, метотрексат, лефлуномид, сульфасалазин, соли золота, Rho-D иммуноглобулин, микофенилат мофетил, циклоспорин, азатиоприн, такролимус, басиликсимаб, даклизумаб, салициловую кислоту, ацетилсалициловую кислоту, метилсалицилат, дифлунизал, салсалат, олсалазин, сульфасалазин, ацетаминофен, индометацин, сулиндак, мефенамовую кислоту, меклофенамат натрия, толметин, кеторолак, диклофенак, флурбипрофен, оксапрозин, пироксикам, мелоксикам, ампироксикам, дроксикам, пивоксикам, теноксикам, фенилбутазон, оксифенбутазон, антипирин, аминопирин, апазон, зилеутон, ауротиоглюкозу, тиомалат золота натрия, ауранофин, метотрексат, колхицин, аллопуринол, пробенецид, сульфинпиразон и бензбромарон или бетаметазон и другие глюкокортикостероиды; и противорвотное средство, такое как, но не ограничиваясь, метоклопромид, домперидон, прохлорперазин, прометазин, хлорпромазин, триметобензамид, ондансетрон, гранисетрон, гидроксизин, ацетиллейцин моноэтаноламин, ализаприд, азасетрон, бензквинамид, биетанаутин, бромприд, буклизин, клебоприд, циклизин, дименгидринат, дифенидол, доласетрон, меклизин, металлатал, метопимазин, набилон, оксиперндил, пипамазин, скополамин, сулпирид, тетрагидроканнабинол, триэтилперазин, тиопроперазин, тропизетрон и их смеси.
Примеры вторых активных средств, которые можно применять для лечения, предотвращения и/или менеджмента повреждений ЦНС и связанных с ними синдромов, включают, но не ограничиваются, иммуномодуляторы, иммунодепрессанты, гипотензивные средства, противосудорожные средства, фибринолитические средства, антитромбоцитарные средства, нейролептики, антидепрессанты, бензодиазепины, буспирон, амантадин и другие известные или общепринятые средства, применяемые на пациентах с повреждением/поражением ЦНС и связанными с ними синдромами. Конкретные примеры включают, но не ограничиваются: стероиды (например, глюкокортикостероиды, такие как, но не ограничиваясь, метилпреднизолон, дексаметазон и бетаметазон); противовоспалительное средство, включая, но не ограничиваясь, напроксен натрия, дихлофенак натрия, диклофенак калия, целекоксиб, сулиндак, оксапрозин, дифлунизал, этодолак, мелоксикам, ибупрофен, кетопрофен, набуметон, рефекоксиб, метотрексат, лефлуномид, сульфасалазин, соли золота, RHo-D иммуноглобулин, микофенилат мофетил, циклоспорин, азатиоприн, такролимус, басиликсимаб, даклизумаб, салициловую кислоту, ацетилсалициловую кислоту, метилсалицилат, дифлунизал, салсалат, олсалазин, сулфасалазин, ацетаминофен, индометацин, сулиндак, мефенамовую кислоту, меклофенамат натрия, толметин, кеторолак, диклофенак, флурбипрофен, оксапрозин, пироксикам, мелоксикам, ампироксикам, дроксикам, пивоксикам, теноксикам, фенилбутазон, оксифенбутазон, антипирин, аминопирин, апазон, зилеутон, ауротиоглюкозу, тиомалат золота натрия, ауранофин, метотрексат, колхицин, аллопуринол, пробенецид, сульфинпиразон и бензбромарон; cAMP аналог, включая, но не ограничиваясь, db-cAMP; агент, содержащий метилфенидатное лекарственное средство, которое содержит 1-треометилфенидат, d-трео-метилфенидат, dl-трео-метилфенидат, 1-эритро-метилфенидат, d-эритро-метилфенидат, dl-эритро-метилфенидат и их смеси; диуретик, такой как, но не ограничиваясь, маннитол, фуросемид, глицирин и мочевину.
Примеры вторых активных средств, которые можно применять для лечения, предотвращения и/или менеджмента дисфункционального сна и связанных с ним синдромов, включают, но не ограничиваются, трициклический антидепрессант, селективный ингибитор обратного захвата серотонина, противоэпилептическое средство (габапентин, прегабалин, карбамазепин, оксакарбазепин, левитирацетам, топирамат), антиаритмическое средство, средство, блокирующее натриевые каналы, селективный ингибитор медиатора воспаления, опиоидное средство, дополнительное иммуномодулирующее соединение, комбинационное средство и другие известные или общепринятые средства, применяемые в лечении сном. Конкретные примеры включают, но не ограничиваются, Неуронтин, оксиконтин, морфин, топирамат, амитриптилин, нортриптилин, карбамазепин, Леводопа, L-DOPA, кокаин, α-метилтирозин, резерпин, тетрабеназин, бензотропин, паргилин, фенодолпам мезилат, каберголин, дигидрохлорид прамипексола, ропинорол, гидрохлорид амантадина, гидрохлорид селегилина, карбидопа, перголид мезилат, Синемет CR, Симметрел, ипрониазид, клоргилин, фенелзин, изокарбоксазид, толкапон, энтакапон, физостигмин салицилат, физостигмин сульфат, физостигмин бромид, меостигмин бромид, неостигмин метилсульфат, амбеноним хлорид, эдрофоний хлорид, такрин, пралидоксим хлорид, обидоксим хлорид, тримедоксим бромид, диацетил моноксим, эндрофоний, пиридостигмин, демекарий, напроксен натрия, диклофенак натрия, диклофенак калия, целекоксиб, сулиндак, оксапрозин, дифлунизал, этодолак, мелоксикам, ибупрофен, кетопрофен, набуметон, рефекоксиб, метотрексат, лефлуномид, сулфасалазин, соли золота, RHo-D иммуноглобулин, микофенилат мофетил, циклоспорин, азатиоприн, такролимус, басиликсимаб, даклизумаб, салициловую кислоту, ацетилсалициловую кислоту, метилсалицилат, дифлунизал, салсалат, олсалазин, сульфасалазин, ацетаминофен, индометацин, сулиндак, мефенамовую кислоту, меклофенамат натрия, толметин, кеторолак, диклофенак, флурбинпрофен, оксапрозин, пироксикам, мелоксикам, ампироксикам, дроксикам, пивоксикам, теноксикам, фенилбутазон, оксифенбутазон, антипирин, аминопирин, апазон, зилеутон, ауротиоглюкоза, тиомалат золота натрия, ауранофин, метотрексат, колхицин, аллпуринол, пробенецид, сульфинпиразон, бензбромарон, бетаметазон и другие глюкокортикостероиды, метоклопромид, домперидон, прохлорперазин, прометазин, хлорпромазин, триметобензамид, ондансетрон, гранисетрон, гидроксизин, ацетиллейцинмоноэтаноламин, ализаприд, азасетрон, бензквинамид, биетанаутин, бромоприд, буклизин, клебоприд, циклизин, дименгидринат, дифенидол, доласетрон, меклизин, металлатал, метопимазин, набилон, оксиперндил, пипамазин, скополамин, сулпирид, тетрагидроканнабинол, триэтилперазин, тиопроперазин, тропизетрон и их смеси.
Примеры вторых активных средств, которые можно применять для лечения, предотвращения и/или менеджмента гемоглобинопатии и связанных с ней расстройств, включают, но не ограничиваются: интерлейкины, такие как IL-2 (включая рекомбинантный IL-II ("rIL2") и канарипокс IL-2), IL-10, IL-12 и IL-18; интерфероны, такие как интерферон альфа-2a, интерферон альфа-2b, интерферон альфа-n1, интерферон альфа-n3, интерферон бета-Ia и интерферон гамма-Ib; и G-CSF; гидроксимочевину; бутираты или бутиратные производные; оксид азота; гидроксимочевину; HEMOXIN™ (NIPRISAN™; смотри патент США № 5800819); антагонисты каналов Гардоса, такие как клотримазол и триарилметановые производные; Дефероксамин; белок C; и переливание крови или заменителя крови, такого как Hemospan™ или Hemospan™ PS (Sangart).
Введение соединения, относящегося к настоящему изобретению, или его фармацевтически приемлемой соли, сольвата, клатрата, стереоизомера или пролекарства и вторых активных средств пациенту можно осуществлять одновременно или последовательно одинаковыми или различными путями введения. Пригодность конкретного пути введения, применяемого для конкретного активного средства, будет зависеть от самого активного средства (например, можно ли его вводить перорально без разложения перед попаданием его в кровяной поток) и вылечиваемого заболевания. Одним из способов введения соединений, относящихся к настоящему изобретению, является пероральный способ. Пути введения вторых активных средств или ингредиентов являются известными специалистам в данной области техники. Смотри, например, Physicians' Desk Reference (60th ed., 2006).
В одном варианте осуществления дополнительное активное средство вводят внутривенно или подкожно и раз или дважды в день в количестве от, приблизительно, 1 до, приблизительно, 1000 мг, от, приблизительно, 5 до, приблизительно, 500 мг, от, приблизительно, 10 до, приблизительно, 350 мг, или от, приблизительно, 50 до, приблизительно, 200 мг. Конкретное количество дополнительного активного средства будет зависеть от конкретного применяемого средства, типа вылечиваемого или контролируемого заболевания, тяжести и стадии заболевания, и количества (количеств) соединений, относящихся к настоящему изобретению, и любых необязательных вторых активных средств, параллельно вводимых пациенту.
Как обсуждается в другом разделе настоящего описания, настоящее изобретение также включает способ ослабления, лечения и/или предотвращения побочных или неблагоприятных эффектов, связанных с общепринятой терапией, включая, но не ограничиваясь, хирургическую операцию, химиотерапию, лучевую терапию, гормональную терапию, биотерапию и иммунотерапию. Соединения, относящиеся к настоящему изобретению, и другие активные ингредиенты можно вводить пациенту перед, в течение или после появления побочного эффекта, связанного с общепринятой терапией.
4.4 Циклическая терапия
В определенных вариантах осуществления профилактические или терапевтические средства, относящиеся к настоящему изобретению, вводят пациенту циклически. Циклическая терапия включает введение активного средства в течение некоторого периода времени, с последующим перерывом (т.е., прерыванием введения) в течение некоторого периода времени, и повторение циклического введения. Циклическая терапия может замедлять развитие устойчивости к одной или более терапий, избегать или ослаблять побочные эффекты одной из терапий, и/или увеличивать эффективность лечения.
Следовательно, в одном варианте осуществления соединение, относящееся к настоящему изобретению, вводят ежедневно в виде единичной или дробных доз в течение четырех или шести месячного цикла с периодом перерыва в течение, приблизительно, недели или двух недель. Кроме того, периодическая терапия позволяет увеличить частоту, число и длину циклов дозирования. Таким образом, другой вариант осуществления включает введение соединения, относящегося к настоящему изобретению, в течение большого числа циклов, чем обычно, когда его вводят отдельно. В еще другом варианте осуществления соединение, относящееся к настоящему изобретению, вводят в течение большего числа циклов, чем обычно бы обуславливалось ограничивающей дозу токсичностью у пациента, которому не вводят дополнительный активный ингредиент.
В одном варианте осуществления соединение, относящееся к настоящему изобретению, вводят ежедневно и непрерывно в течение трех или четырех недель при дозе от, приблизительно, 0,1 мг до, приблизительно, 500 мг в день, с последующим перерывом в течение одной или двух недель. В других вариантах осуществления доза может составлять от, приблизительно, 1 мг до, приблизительно, 300 мг, от, приблизительно, 0,1 мг до, приблизительно, 150 мг, от, приблизительно, 1 мг до, приблизительно, 200 мг, от, приблизительно, 10 мг до, приблизительно, 100 мг, от, приблизительно, 0,1 мг до, приблизительно, 50 мг, от, приблизительно, 1 мг до, приблизительно, 50 мг, от, приблизительно, 10 мг до, приблизительно, 50 мг, от, приблизительно, 20 мг до, приблизительно, 30 мг, или от, приблизительно, 1 мг до, приблизительно, 20 мг, с последующим перерывом.
В одном варианте осуществления соединение, относящееся к настоящему изобретению, и второе активное средство вводят перорально, причем введение соединения, относящегося к настоящему изобретению, осуществляют за 30-60 минут до введения дополнительного активного ингредиента, в течение цикла от четырех до шести недель. В другом варианте осуществления комбинацию соединения, относящегося к настоящему изобретению, и второго активного средства вводят внутривенным вливанием в течение, приблизительно, 90 минут каждый цикл.
Обычно, число циклов, в течение которых пациенту вводят комбинированную терапию, будет составлять от, приблизительно, одного до, приблизительно, 24 циклов, от, приблизительно, двух до, приблизительно, 16 циклов, или от, приблизительно, четырех до, приблизительно, трех циклов.
4.5 ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ЛЕКАРСТВЕННЫЕ ФОРМЫ Фармацевтические композиции можно применять в получении отдельных, единичных стандартных лекарственных форм. Фармацевтические композиции и лекарственные формы, относящиеся к настоящему изобретению, содержат соединение, относящееся к настоящему изобретению или его фармацевтически приемлемую соль, сольват, стереоизомер, клатрат или пролекарство. Кроме того, фармацевтические композиции и лекарственные формы могут содержать один или более вспомогательных веществ.
Фармацевтические композиции и лекарственные формы, относящиеся к настоящему изобретению, могут также содержать одно или более вторых активных средств. Примеры необязательных вторых или дополнительных активных ингредиентов описывают в параграфе 4.3 выше.
Единичные стандартные лекарственные формы, относящиеся к настоящему изобретению, являются подходящими для перорального, мукозального (например, назального, сублингвального, вагинального, буккального или ректального), парентерального (например, подкожного, внутривенного, болюсной инъекции, внутримышечного или внутриартериального), местного (например, глазные капли или другие офтальмологические препараты), трансдермального или чрезкожного введения пациенту. Примеры лекарственных форм включают, но не ограничиваются: таблетки; каплеты; капсулы, такие как мягкие эластичные желатиновые капсулы; крахмальные капсулы; лепешки; пастилки; дисперсии; суппозитории; порошки; аэрозоли (например, назальные спреи или пульверизаторы); гели; жидкие лекарственные формы, подходящие для перорального или мукозального введения пациенту, включая суспензии (например, водные или неводные жидкие суспензии, эмульсии масла в воде или жидкие эмульсии воды в масле), растворы и эликсиры; жидкие лекарственные формы, подходящие для парентерального введения пациенту; глазные капли или другие офтальмологические препараты, подходящие для местного введения; и стерильные твердые вещества (например, кристаллические или аморфные твердые вещества), которые можно растворять для того, чтобы получить жидкие лекарственные формы, подходящие для парентерального введения пациенту.
Композиция, форма и тип лекарственных форм обычно будет изменяться в зависимости от их применения. Например, лекарственная форма, применяемая при неотложном лечении заболевания, может содержать большие количества одного или более активных ингредиентов, чем содержит лекарственная форма при применении при постоянном лечении того же самого заболевания. Аналогично, парентеральная лекарственная форма может содержать меньшие количества одного или более активных ингредиентов, чем содержит оральная лекарственная форма при применении ее для того, чтобы лечить то же самое заболевание. Данные и другие способы, в которых применяют конкретные лекарственные формы, будут отличаться друг от друга и будут легко понятны специалистам в данной области техники. Смотри, например, Remington's Farmaceutical Sciences, 20th ed., Mack Publishing, Easton PA (2000).
В одном варианте осуществления фармацевтические композиции и лекарственные формы содержат один или более вспомогательных веществ. Подходящие вспомогательные вещества являются известными специалистам в области фармацевтического дела, и неограничивающие примеры подходящих вспомогательных веществ относятся к настоящему изобретению. Является ли конкретное вспомогательное вещество подходящим для введения в фармацевтическую композицию или лекарственную форму зависит от множества факторов, известных в данной области техники, включая, но не ограничиваясь, способ, которым лекарственную форму будут вводить пациенту. Например, оральные лекарственные формы, такие как таблетки, могут содержать вспомогательные вещества, которые не подходят для применения в парентеральных лекарственных формах. Пригодность конкретного вспомогательного вещества может также зависеть от конкретных активных ингредиентов в лекарственной форме. Например, разложение некоторых активных ингредиентов может ускоряться некоторыми вспомогательными веществами, такими как лактоза, или под действием воды. Активные ингредиенты, которые содержат первичные или вторичные амины, особенно подвержены такому ускоренному разложению. Следовательно, настоящее изобретение относится к фармацевтическим композициям и лекарственным формам, которые содержат небольшое количество, если вообще содержат, лактозы и других моно- или дисахаридов. Как применяют в настоящем изобретении, термин "не содержащая лактозу" обозначает то, что количество присутствующей, если вообще присутствующей, лактозы является недостаточным для того, чтобы в значительной степени увеличивать скорость разрушения активного ингредиента.
Композиции, не содержащие лактозу, могут содержать вспомогательные вещества, которые известны в данной области техники, и они перечислены, например, в Фармакопеи США (USP) 25-NF20 (2002). Обычно, композиции, не содержащие лактозу, содержат активные ингредиенты, связующее/наполнитель и смазывающее вещество в фармацевтически совместимых и фармацевтически приемлемых количествах. В одном варианте осуществления лекарственные формы, не содержащие лактозу, содержат активные ингредиенты, микрокристаллическую целлюлозу, предварительно клейстеризованный крахмал и стеарат магния.
Также настоящее изобретение относится к безводным фармацевтическим композициям и лекарственным формам, содержащим активные ингредиенты, т.к. вода может способствовать разрушению некоторых соединений. Например, добавление воды (например, 5%) является широко распространенным в фармацевтических областях техники в качестве средства моделирования продолжительного хранения для того, чтобы определить характеристики, такие как длительность хранения или стабильность составов с течением времени. Смотри, например, Jens T. Carstensen, Drug Stability: Principles & Practice, 2d. Ed., Marcel Dekker, NY, NY, 1995, pp. 379-80. В действительности вода и тепло ускоряют разложение некоторых соединений. Таким образом, действие воды на состав имеет большое значение, т.к. влага обычно присутствует в процессе получения, транспортировки, фасовки, хранения, отправки и применения составов.
Безводные фармацевтические композиции и лекарственные формы можно получить, применяя безводные ингредиенты или ингредиенты, содержащие небольшое количество влаги, и условия с низкой влажностью или количеством влаги. Фармацевтические композиции и лекарственные формы, которые содержат лактозу и, по меньшей мере, один активный ингредиент, который содержит перивичный или вторичный амин, являются безводными, если предполагается существенный контакт с влагой в процессе получения, фасовки и/или хранения.
Безводные фармацевтические композиции следует получать и хранить так, чтобы сохранять их в безводном состоянии. Соответственно, в одном варианте осуществления безводные композиции фасуют, применяя известные материалы для того, чтобы предотвратить воздействие воды так, что их можно включать в подходящие наборы с составом. Примеры подходящих фасовок включают, но не ограничиваются, герметично запечатанную пленку, пластмассу, емкости для единичных доз (например, пробирки), блистерные упаковки и контурные упаковки.
Также настоящее изобретение относится к фармацевтическим композициям и лекарственным формам, которые содержат одно или более соединений, которые снижают скорость, с которой будет разлагаться активный ингредиент. Такие соединения, которые называют в настоящем изобретении "стабилизаторы", включают, но не ограничиваются, антиоксиданты, такие как аскорбиновая кислота, pH буферы или солевые буферы.
Подобно количествам и типам вспомогательных веществ, количества и конкретные типы активных ингредиентов в лекарственной форме могут отличаться в зависимости от факторов, такие как, но не ограничиваясь, путь, которым его вводят пациентам. В одном варианте осуществления лекарственные формы содержат соединение, относящееся к настоящему изобретению, в количестве от, приблизительно, 0,10 до, приблизительно, 500 мг. В других вариантах осуществления лекарственные формы содержат соединение, относящееся к настоящему изобретению в количестве, приблизительно, 0,1, 1, 2, 5, 7,5, 10, 12,5, 15, 17,5, 20, 25, 50, 100, 150, 200, 250, 300, 350, 400, 450 или 500 мг.
В других вариантах осуществления лекарственные формы содержат второе активное средство в количестве от 1 до, приблизительно, 1000 мг, от, приблизительно, 5 до, приблизительно, 500 мг, от, приблизительно, 10 до, приблизительно, 350 мг, или от, приблизительно, 50 до, приблизительно, 200 мг. Конечно, конкретное количество дополнительного активного средства будет зависеть от конкретного применяемого средства, заболеваний или расстройств, которые лечат или контрлируют, и количества (количеств) соединения, относящегося к настоящему изобретению, и любых необязательных вторых активных средств, одновременно вводимых пациенту.
4.5.1 ОРАЛЬНЫЕ ЛЕКАРСТВЕННЫЕ ФОРМЫ
Фармацевтические композиции, которые являются подходящими для перорального введения, можно заготавливать в виде дискретных лекарственных форм, таких как, но не ограничиваясь, таблетки (например, жевательные таблетки), каплеты, капсулы, и жидкости (например, ароматизированные сиропы). Данные лекарственные формы содержат предварительно определенные количества активных ингредиентов, и могут быть получены способами фармацевтики, известными специалистам в данной области техники. Смотри в основном, Remington's Pharmaceutical Sciences, 20th ed., Mack Publishing, Easton PA (2000).
Оральные лекарственные формы, относящиеся к настоящему изобретению, получают смешением активных ингредиентов до однородной смеси, по меньшей мере, с одним вспомогательным веществом, согласно общепринятым фармацевтическим методикам приготовления. Вспомогательные вещества могут иметь большое разнообразие форм в зависимости от формы препарата, требуемой для введения. Например, вспомогательные вещества, подходящие для применения в оральных жидких или аэрозольных лекарственных формах, включают, но не ограничиваются, воду, гликоли, масла, спирты, ароматизирующие средства, консерванты и красящие средства. Примеры вспомогательных веществ, подходящих для применения в твердых оральных лекарственных формах (например, порошках, таблетках, капсулах и каплетах) включают, но не ограничиваются, крахмалы, сахара, микрокристаллическую целлюлозу, разбавители, гранулирующие средства, смазывающие вещества, связующие и разрыхлители.
В одном варианте осуществления оральными лекарственными формами являются таблетки или капсулы, в данном случае применяют твердые вспомогательные вещества. В другом варианте осуществления таблетки можно покрыть стандарным водным или неводным способом. Данные лекарственные формы можно получить любым из способов фармацевтики. Обычно, фармацевтические композиции и лекарственные формы получают однородным и равномерным смешением активных ингредиентов с жидкими носителями, мелкоизмельченными твердыми носителями, или обоими и, затем, формованием продукта до требуемой формы выпуска, в случае необходимости.
Например, таблетку можно получить прессованием или формованием. Прессованные таблетки можно приготовить прессованием в подходящей машине активных ингредиентов в сыпучей форме, такой как порошок или гранулы, не обязательно, смешанных со вспомогательным веществом. Формованные таблетки можно получить формованием в подходящей машине смеси порошкообразного соединения, смоченного инертным жидким разбавителем.
Примеры вспомогательных веществ, которые можно применять в оральных лекарственных формах, относящихся к настоящему изобретению, включают, но не ограничиваются, связующие, наполнители, разрыхлители и смазывающие вещества. Связующие, подходящие для применения в фармацевтических композициях и лекарственных формах, включают, но не ограничиваются, кукурузный крахмал, картофельный крахмал или другие крахмалы, желатин, природные и синтетические смолы, такие как камедь, алгинат натрия, алгиновую кислоту, другие алгинаты, порошкообразный трагакант, гуаровую камедь, целлюлозу и ее производные (например, этилцеллюлозу, ацетат целлюлозы, кальций карбоксиметилцеллюлозу, натрий карбоксиметилцеллюлозу), поливинилпирролидон, метилцеллюлозу, предварительно клейстеризованный крахмал, гидроксипропилметилцеллюлозу, (например, № 2208, 2906, 2910), микрокристаллическую целлюлозу и их смеси.
Подходящие формы микрокристаллической целлюлозы включают, но не ограничиваются, вещества, продаваемые под названием AVICEL-PH-101, AVICEL-PH-103 AVICEL RC-581, AVICEL-PH-105 (полученные от FMC Corporation, American Viscose Division, Avicel Sales, Marcus Hook, PA) и их смеси. Конкретным связующим является смесь микрокристаллической целлюлозы и натрий карбоксиметилцеллюлозы, продаваемая под названием AVICEL RC-581. Подходящие безводные вспомогательные вещества или вспомогательные вещества, содержащие небольшое количество влаги, или добавки включают AVICEL-PH-103™ и Крахмал 1500 LM.
Примеры наполнителей, подходящих для применения в фармацевтических композициях и лекарственных формах, относящихся к настоящему изобретению, включают, но не ограничиваются, тальк, карбонат кальция (например, гранулы или порошок), микрокристаллическую целлюлозу, порошкообразную целлюлозу, декстраты, каолин, маннит, кремниевую кислоту, сорбит, крахмал, предварительно клейстеризованный крахмал и их смеси. В одном варианте осуществления связующее или наполнитель присутствует в фармацевтических композициях в количестве от, приблизительно, 50 до, приблизительно, 99 процентов по весу фармацевтической композиции или лекарственной формы.
Разрыхлители можно применять в композиции для того, чтобы получить таблетки, которые распадаются при воздействии водной среды. Таблетки, которые содержат слишком большое количество разрыхлителя, могут распадаться при хранении, тогда как таблетки, которые содержат слишком маленькое количество разрыхлителя, могут не распадаться с требуемой скоростью или в требуемых условиях. Таким образом, достаточное количество разрыхлителя, которое не является ни слишком большим, ни слишком маленьким для того, чтобы пагубно отразиться на высвобождении активных ингредиентов, можно применять для того получить твердые оральные лекарственные формы. Количество применяемого разрыхлителя изменяется в зависимости от типа состава и является легко определяемым специалистами в данной области техники. В одном варианте осуществления фармацевтические композиции содержат от, приблизительно, 0,5 до, приблизительно, 15 процентов по весу разрыхлителя, или от, приблизительно, 1 до, приблизительно, 5 процентов по весу разрыхлителя.
Разрыхлители, которые можно применять в фармацевтических композициях и лекарственных формах, включают, но не ограничиваются, агар-агар, альгиновую кислоту, карбонат кальция, микрокристаллическую целлюлозу, кроскармеллозу натрия, кросповидон, полакрилин калия, крахмал гликолат натрия, картофельный или маниоковый крахмал, другие крахмалы, предварительно клейстеризованный крахмал, другие крахмалы, глины, другие альгины, другие целлюлозы, смолы и их смеси. Смазывающие вещества, которые можно применять в фармацевтических композициях и лекарственных формах, включают, но не ограничиваются, стеарат кальция, стеарат магния, минеральное масло, светлое минеральное масло, глицерин, сорбит, маннит, полиэтиленгликоль, другие гликоли, стеариновую кислоту, лаурилсульфат натрия, тальк, гидрогенизированное растительное масло (например, кокосовое масло, хлопковое масло, подсолнечное масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло), стеарат цинка, этилолеат, этиллауреат, агар и их смеси. Дополнительные смазывающие вещества включают, например, силоидный силикагель (AEROSIL200, полученный W.R. Grace Co. of Baltimore, MD), коагулированный аэрозоль синтетической двуокиси кремния (продаваемый Degussa Co. of Piano, TX), CAB-O-SIL (пирогенный диоксид кремния, продаваемый Cabot Co. of Boston, MA) и их смеси. Если их вообще применяют, смазывающие вещества можно применять в количестве меньшем, чем, приблизительно, 1 процент по весу фармацевтических композиций или лекарственных форм, в которые их вводят.
В одном варианте осуществления твердая оральная лекарственная форма содержит соединение, относящееся к настоящему изобретению, безводную лактозу, микрокристаллическую целлюлозу, поливинилпирролидон, стеариновую кислоту, колоидный безводный диоксид кремния и желатин.
4.5.2 ЛЕКАРСТВЕННЫЕ ФОРМЫ С КОНТРОЛИРУЕМЫМ ВЫСВОБОЖДЕНИЕМ
Активные ингредиенты, такие как соединения, относящиеся к настоящему изобретению, можно вводить способами с контролируемым высвобождением или средствами доставки, которые хорошо известны специалистам в данной области техники. Примеры включают, но не ограничиваются, способы и средства, описанные в патенте США №: 3845770; 3916899; 3536809; 3598123; и 4008719; 5674533; 5059595; 5591767; 5120548; 5073543; 5639476; 5354556; 5639480; 5733566; 5739108; 5891474; 5922356; 5972891; 5980945; 5993855; 6045830; 6087324; 6113943; 6197350; 6248363; 6264970; 6267981; 6376461; 6419961; 6589548; 6613358; 6699500 каждый из которых вводят в настоящее изобретение с помощью ссылки. Данные лекарственные формы можно применять для того, чтобы добиться медленного или контролируемого высвобождения одного или более активных ингредиентов, применяя, например, гидропропилметилцеллюлозу, другие полимерные матрицы, гели, проницаемые мембраны, осмотические системы, мультислойные покрытия, микрочастицы, липосомы, микросферические частицы или их комбинации для того, чтобы получить требуемый профиль высвобождения при изменяющихся условиях. Подходящие составы с контролируемым высвобождением, известные специалистам в данной области техники, включая составы, описанные в данном изобретении, можно легко подобрать для применения с активными ингредиентами, относящимися к настоящему изобретению. Таким образом, композиции, относящиеся к настоящему изобретению, включают единичные стандартные лекарственные формы, подходящие для перорального введения, такие как, но не ограничиваясь, таблетки, капсулы, гелевые капсулы и каплеты, которые приспособили для контролируемого высвобождения.
Основная цель всех фармацевтических продуктов с контролируемым высвобождением заключается в улучшении лекарственной терапии по сравнению с эффектами, достигнутыми при применении их аналогов без контролируемого высвобождения. Теоретически, применение препарата с оптимально разработанным контролируемым высвобождением при медицинском лечении характеризуется минимальным количеством лекарственного средства, применяемого для лечения или менеджемента состояния за минимальное количество времени. Преимущества составов с контролируемым высвобождением включают длительную активность лекарственного средства, сниженную частоту дозирования и улучшенное соблюдение режима субъектом. Кроме того, составы с контролируемым высвобождением можно применять для того, чтобы влиять на начало действия или другие характеристики, такие как концентрации лекарственного средства в крови, и могут, таким образом, влиять на появление побочных (например, неблагоприятных) эффектов.
Большинство составов с контролируемым высвобождением разработано для первоначального высвобождения количества лекарственного средства (активного ингредиента), которое немедленно производит требуемый терапевтический эффект, и постепенного и непрерывного высвобождения других количеств лекарственного средства для того, чтобы поддерживать данный уровень терапевтического или профилактического эффекта в течение продолжительного периода времени. Для того чтобы поддерживать постоянную концентрацию лекарственного средства в теле, лекарственное средство должно высвобождаться из лекарственной формы со скоростью, при которой высвободившееся лекарственное средство будет замещать количество лекарственного средства, которое подверглось метаболизму и выделилось из тела. Контролируемое высвобождение активного ингредиента можно вызвать различными условиями, включая, но не ограничиваясь, pH, температуру, ферменты, воду или другие физиологические состояния или соединения.
В определенных вариантах осуществления лекарственное средство можно вводить, применяя внутривенное вливание, имплантируемый осмотический насос, трансдермальный пластырь, липосомы или другие режимы введения. В одном варианте осуществления можно применять насос (смотри, Sefton, CRC Crit. Ref. Biomed. Eng. 14:201 (1987); Buchwald et al, Surgery 88:507 (1980); Saudek et al, N. Engl. J. Med. 321:574 (1989)). В другом варианте осуществления можно применять полимерные материалы. В еще другом варианте осуществления систему с контролируемым высвобождением можно помещать в субъекте в подходящем месте, определенном практикующим врачом, т.е., таким образом, требуется только часть общей дозы (смотри, например, Goodson, Medical Applications of Controlled Release, vol. 2, pp. 115-138 (1984)). Другие системы с контролируемым высвобождением рассматриваются в статье Langer (Science 249:1527-1533 (1990)). Активный ингредиент можно диспергировать в твердом внутреннем матриксе, например полиметилметакрилате, полибутилметакрилате, пластифицированном или непластифицированном поливинилхлориде, пластифицированном нилоне, пластифицированном полиэтилентерефталате, природном каучуке, полиизопрене, полиизобутилене, полибутадиене, полиэтилене, этилен-винилацетатных сополимерах, силоксановых каучуках, полидиметилсилоксанах, кремний карбонатных сополимерах, гидрофильных полимерах, такие как гидрогели эфиров акриловой и метакриловой кислоты, коллаген, кросс-сшитом поливиниловом спирте и кросс-сшитом частично гидролизованном поливинилацетате, который окружают внешней полимерной мембраной, например полиэтиленом, полипропиленом, этилен/пропиленовыми сополимерами, этилен/этилакрилатными сополимерами, этилен/винилацетатными сополимерами, силоксановыми каучуками, полидиметилсилоксанами, неопреновой резиной, хлорированным полиэтиленом, поливинилхлоридом, винилхлоридными сополимерами с винилацетатом, винилиденхлоридом, этиленом и пропиленом, иономерным полиэтилентерефталатом, бутилкаучуковыми эпихлоргидриновыми каучуками, этилен/виниловый спирт сополимером, этилен/винилацетат/виниловый спирт терполимером и этилен/винилоксиэтаноловым сополимером, которые являются нерастворимыми в жидкостях организма. Затем активный ингредиент диффундирует через внешнюю полимерную мембрану на стадии с контролируемой скоростью высвобождения. Процент активного ингредиента в данных парентеральных композициях сильно зависит от их особенностей, также как от запросов субъекта.
4.5.3 ПАРЕНТЕРАЛЬНЫЕ ЛЕКАРСТВЕННЫЕ ФОРМЫ
Парентеральные лекарственные формы можно вводить пациентам различными путями, включая, но не ограничиваясь, подкожный, внутривенный (включая болюсную инъекцию), внутримышечный и внутриартериальный пути введения. В некоторых вариантах осуществления введение парентеральной лекарственной формы минует естественную защиту пациентов против заражающих веществ, и таким образом, в данных вариантах осуществления, парентеральные лекарственные формы являются стерильными или их можно стерилизовать перед введением пациенту. Примеры парентеральных лекарственные формы включают, но не ограничиваются, растворы, готовые для инъекции, сухие препараты, готовые для растворения или суспендирования в фармацевтически приемлемой среде для инъекции, готовые для инъекции суспензии и эмульсии.
Подходящие среды, которые можно применять для того, чтобы получить парентеральные лекарственные формы, являются хорошо известными специалистам в данной области техники. Примеры включают, но не ограничиваются: воду для инъекций USP; водные среды, такие как, но не ограничиваясь, раствор хлорида натрия для инъекций, раствор Рингера для инъекций, раствор декстрозы для инъекций, раствор декстрозы и хлорида натрия для инъекций, и лактатный раствор Рингера; среды, смешиваемые с водой, такие как, но не ограничиваясь, этиловый спирт, полиэтиленгликоль и полипропиленгликоль; и неводные смеси, такие как, но не ограничиваясь, кукурузное масло, хлопковое масло, кокосовое масло, кунжутное масло, этилолеат, изопропилмиристат и бензилбензоат.
Соединения, которые увеличивают растворимость одного или более активных ингредиентов, описанных в настоящем изобретении, можно также вводить в парентеральные лекарственные формы. Например, циклодекстрин и его производные можно применять для того, чтобы увеличить растворимость соединения, относящегося к настоящему изобретению. Смотри, например, патент США № 5134127,
который введен в настоящее изобретение с помощью ссылки.
4.5.4 ТОПИЧЕСКИЕ И МУКОЗАЛЬНЫЕ ЛЕКАРСТВЕННЫЕ ФОРМЫ
Топические и мукозальные лекарственные формы, относящиеся к настоящему изобретению, включают, но не ограничиваются, спреи, аэрозоли, растворы, эмульсии, суспензии, глазные капли или другие офтальмологические препараты или другие формы, известные специалистам в данной области техники. Смотри, например, Remington's Pharmaceutical Sciences, 16, 18 и 20th eds., Mack Publishing, Easton PA (1980, 1990 и 2000); и Introduction to Pharmaceutical Dosage Forms, 4th ed., Lea & Febiger, Philadelphia (1985). Лекарственные формы, подходящие для лечения слизистых тканей в полости рта, можно создавать в виде жидкостей для полоскания рта или в виде оральных гелей.
Подходящие вспомогательные вещества (например, носители и разбавители) и другие вещества, которые можно применять для того, чтобы получить топические и мукозальные лекарственные формы, включенные в настоящее изобретение, являются хорошо известными специалистам в области фармацевтики и зависят от конкретной ткани, на которой будут применять данную фармацевтическую композицию или лекарственную форму. В одном варианте осуществления вспомогательные вещества включают, но не ограничиваются, воду, ацетон, этанол, этиленгликоль, пропиленгликоль, бутан-1,3-диол, изопропилмиристат, изопропилпальмитат, минеральное масло и их смеси для того, чтобы образовать растворы, эмульсии или гели, которые являются нетоксичными и фармацевтически приемлемыми. Можно также добавлять к фармацевтическим композициям и лекарственным формам увлажняющие средства или смачивающие вещества. Примеры дополнительных ингредиентов являются известными в данной области техники. Смотри, например, Remington's Pharmaceutical Sciences, 16th, 18th и 20th eds., Mack Publishing, Easton PA (1980, 1990 и 2000).
Можно также регулировать pH фармацевтической композиции или лекарственной формы для того, чтобы улучшить доставку одного или более активных ингредиентов. Также можно регулировать полярность растворителя - носителя, его ионную силу или тоничность для того, чтобы улучшить доставку. Соединения, такие как стеараты, можно также добавлять к фармацевтическим композициям или лекарственным формам для того, чтобы изменить гидрофильность или липофильность одного или более активных ингредиентов так, чтобы улучшить доставку. В других вариантах осуществления стеараты могут служить в качестве липидной среды для состава, в качестве эмульгатора или поверхностно-активного вещества, или в качестве средства, улучшающего доставку или проникновение. В других вариантах осуществления соли, сольваты, пролекарства, клатраты или стереоизомеры активных ингредиентов можно применять для дополнительного регулирования свойств полученной в результате композиции.
4.6 Наборы
В одном варианте осуществления активные ингредиенты, относящиеся к настоящему изобретению, не вводят пациенту в одно и то же время или тем же самым путем введения. В другом варианте осуществления настоящее изобретение относится к наборам, которые могут упрощать введение подходящих количеств активных ингредиентов.
В одном варианте осуществления набор содержит лекарственную форму соединения, относящегося к настоящему изобретению. Кроме того, наборы могут содержать дополнительные активные ингредиенты, такие как облимерсен (Genasense®), мелфалан, G-CSF, GM-CSF, EPO, топотекан, дакарбазин, иринотекан, таксотере, IFN, COX-2 ингибитор, пентоксифуллин, ципрофлоксазин, дексаметазон, IL2, IL8, ILl8, Ara-C, винорелбин, изотретиноин, 13 цис-ретиноевую кислоту или ее фармакологически активный мутант или производное или их комбинацию. Примеры дополнительных активных ингредиентов включают, но не ограничиваются, дополнительные активные ингредиенты, описанные в настоящем изобретении (смотри, например, параграф 4.3).
В других вариантах осуществления наборы могут дополнительно содержать устройства, которые применяют для введения активных ингредиентов. Примеры данных устройств включают, но не ограничиваются, шприцы, пакеты с капельными клизмами, пластыри и пульверизаторы.
Наборы могут дополнительно содержать клетки или кровь для переливания (пересадки), также как фармацевтически приемлемые среды, которые можно применять для введения одного или более активных ингредиентов. Например, если активный ингредиент находится в твердой форме, которую можно растворить для парентерального введения, набор может содержать герметичную емкость с подходящей средой, в которой можно растворить активный ингредиент для того, чтобы получить стерильный раствор, не содержащий частиц, который является подходящим для парентерального введения. Примеры фармацевтически приемлемых сред включают, но не ограничиваются: воду для инъекций USP; водные среды, такие как, но не ограничиваясь, раствор хлорида натрия для инъекций, раствор Рингера для инъекций, раствор декстрозы для инъекций, раствор декстрозы и хлорида натрия для инъекций, и лактатный раствор Рингера; среды, смешиваемые с водой, такие как, но не ограничиваясь, этиловый спирт, полиэтиленгликоль и полипропиленгликоль; и неводные смеси, такие как, но не ограничиваясь, кукурузное масло, хлопковое масло, кокосовое масло, кунжутное масло, этилолеат, изопропилмиристат и бензилбензоат.
5. ПРИМЕРЫ
Определенные варианты осуществления заявленного объекта изобретения проиллюстрированы следующими неограничивающими примерами.
Пример 1
2-(2,6-Диоксопиперидин-3-ил)-4-(2-метоксифенокси)изоиндол-1,3-дион
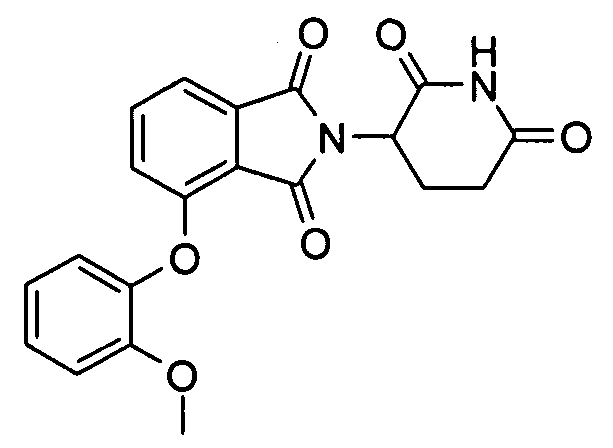
Стадия 1:
Метилйодид (30,2 г, 213 ммоль) добавляли к перемешиваемой смеси 3-нитрофталевой кислоты (15,0 г, 71,0 ммоль) и бикарбоната натрия (23,9 г, 284 ммоль) в DMF (150 мл) при комнатной температуре и затем смесь грели в масляной бане, температура которой была установлена на 60°C, в течение 4 часов. Затем смесь выливали в 700 мл ледяной воды. После того как лед растаял смесь экстрагировали этилацетатом (3×150 мл), органические фазы промывали водой (7×500 мл), сушили (MgSO4) и упаривали, получая 16,2 г диметилового эфира 3-нитрофталевой кислоты в виде бледно-желтого твердого остатка, с выходом 95%; 1H ЯМР (CDCl3) δ 3,95 (с, 3H), 4,02 (с, 3H), 7,69 (т, J=8,1 Гц, 1H), 8,36 (м, 2H).
Стадия 2:
Смесь 3:1 этанол-конц. HCl (200 мл) охлаждали до 0°C и затем добавляли диметиловый эфир 3-нитрофталевой кислоты (15,0 г, 62,8 ммоль). Поддерживая охлаждение, добавляли порциями хлорид олова (II) (70,8 г, 314 ммоль) в течение 15 минут. После завершения добавления охлаждающую баню убирали и продолжали перемешивание при комнатной температуре. Через 2 часа смесь нейтрализовали добавлением твердого бикарбоната натрия и полученную в результате смесь экстрагировали этилацетатом (3×150 мл), объединенные экстракты промывали водой (5×250 мл), сушили (MgSO4) и упаривали, получая 11,3 г диметилового эфира 3-аминофталевой кислоты в виде желтого масла с выходом 86%; 1H ЯМР (CDCl3) δ 3,84 (с, 3H), 3,86 (с, 3H), 5,20 (шир, 2H), 6,78 (дд, J=8,5 Гц, J=1,0 Гц, 1H), 6,90 (дд, 1H, J=7,3 Гц, J=1,0 Гц, 1H), 7,24 (т, J=7,8 Гц, 1H).
Стадия 3:
Раствор диметилового эфира 3-аминофталевой кислоты (9,5 г, 45,4 ммоль) в смеси 1:1 вода-конц. HCl (300 мл) охлаждали до 0°C; при охлаждении образовывался осадок. Затем медленно добавляли раствор NaNO2 (3,5 г, 50,0 ммоль) в 10 мл воды, поддерживая температура в процессе добавления между 0-5°C. После завершения добавления смесь перемешивали при данной температуре в течение 10 минут перед добавлением раствора KI (11,3 г, 68,3 ммоль) в 30 мл смеси 1:1 вода-конц. HCl. Данный раствор добавляли одной порцией и затем реакционную колбу тут же переносили в масляную баню, предварительно нагретую до 65°C. Смесь перемешивали при нагревании в течение 10 минут, затем охлаждали в бане со льдом. Смесь экстрагировали CH2Cl2 (3×150 мл), объединенные органические экстракты промывали водой (3×150 мл), сушили (MgSO4) и упаривали, остаток хроматографировали, применяя градиент этилацетата в гексане. Продукт, который элюировался смесью 17:3 гексан-этилацетат, был светло-фиолетовым твердым остатком, затем его растирали с гексаном, фильтровали и сушили для того, чтобы получить 9,7 г (67%) диметилового эфира 3-йодфталевой кислоты в виде бесцветного твердого остатка; 1H ЯМР (CDCl3) δ 3,90 (с, 3H), 3,99 (с, 3H), 7,19 (т, J=7,9 Гц, 1H), 8,02 (д, J=7,9 Гц, 2H).
Стадия 4:
Смесь гваякола (0,77 г, 6,2 ммоль), бромида меди (I) (0,89 г, 6,2 ммоль) и гидрида натрия (0,3 г 60% дисперсии, 7,5 ммоль) в 100 мл пиридина кипятили с обратным холодильником и перемешивали в атмосфере азота в течение 15 минут. Добавляли диметиловый эфир 3-йодфталевой кислоты (2,0 г, 6,2 ммоль), полученную в результате смесь перемешивали при температуре кипения в течение 20 часов. Смесь охлаждали до комнатной температуры и реакцию прекращали добавлением насыщенного NH4Cl (15 мл). Летучие компоненты удаляли при пониженном давлении. Остаток распределяли между разбавленной водной HCl (100 мл) и этилацетатом (100 мл) и водную фазу экстрагировали этилацетатом (100 мл). Объединенные органические экстракты промывали разбавленной водной HCl (2×100 мл), насыщенным Na2CO3 (2×100 мл), снова разбавленной водной HCl (2×100 мл) и, наконец, водой (100 мл) и упаривали. Хроматография в градиенте этилацетата в гексане давала 0,75 г диметилового эфира 3-(2-метоксифенокси)фталевой кислоты, который элюировался 25-30% этилацетата с выходом 38%; 1H ЯМР (CDCl3) δ 3,79 (с, 3H), 3,91 (с, 3H), 3,95 (с, 3H), 6,86-6,91 (м, 1H), 6,93-7,01 (м, 2H), 7,05 (дд, J=7,9 Гц, J=1,6 Гц, 1H), 7,13-7,20 (м, 1H), 7,30 (т, J=8,0 Гц, 1H), 7,66-7,69 (м, 1H).
Стадия 5:
Смесь диметилового эфира 3-(2-метоксифенокси)фталевой кислоты (0,75 г, 2,4 ммоль) и 3N NaOH (50 мл) в этаноле (100 мл) кипятили с обратным холодильником в течение 2 часов. Смесь охлаждали и растворитель удаляли в вакууме. Остаток растворяли в воде (100 мл), промывали CH2Cl2 (2×100 мл), подкисляли (HCl) и экстрагировали этилацетатом (3×75 мл). Объединенные органические экстракты промывали водой (3×75 мл), сушили (MgSO4) и упаривали, получая 0,53 г 3-(2-метоксифенокси)фталевой кислоты с выходом 78%; 1H ЯМР (DMSO-d 6) δ 3,76 (с, 3H), 6,82 (дд, J=8,4 Гц, J=0,9 Гц, 1H), 6,95-6,98 (м, 2H), 7,16-7,23 (м, 2H), 7,39 (т, J=8,0 Гц, 1H), 7,58 (д, J=8,2 Гц, 1H).
Стадия 6:
Смесь 3-(2-метоксифенокси)фталевой кислоты (0,51 г, 1,8 ммоль) и гидрохлорида рацемического α-аминоглутаримида (0,29 г, 1,8 ммоль) в пиридине (10 мл) кипятили с обратным холодильником в течение 16 часов. Смесь охлаждали и упаривали в вакууме. Остаток растворяли в этилацетате (100 мл), промывали разбавленной водной HCl (2×100 мл) и водой (100 мл) и упаривали. Остаток хроматографировали, применяя градиент CH2Cl2-метанол, заявленное в заголовке соединение элюировалось 95:5 CH2Cl2-метанол, 0,59 г, с выходом 88%; т.пл. 223-225°C; ВЭЖХ, Waters Symmetry C-18, 3,9×150 мм, 5 мкм, 1 мл/мин, 240 нм, 40/60 CH3CNA/0,1% H3PO4, 6,06 (99,46%); 1H ЯМР (DMSO-d 6) δ 2,06-2,11 (м, 1H), 2,53-2,64 (м, 2H), 2,83-2,90 (м, 1H), 3,75 (с, 1H), 5,15 (дд, J=12,4 Гц, J=5,3 Гц, 1H), 6,85 (д, J=8,5 Гц, 1H), 7,06 (т, J=7,3 Гц, 1H), 7,21-7,37 (м, 3H), 7,54 (д, J=7,2 Гц, 1H), 7,72 (т, J=7,9 Гц, 1H), 11,13 (с, 1H); 13C ЯМР (DMSO-d 6) δ 21,9, 31,0, 48,9, 55,7, 113,7, 116,5, 116,7, 120,7, 121,5, 122,3, 127,2, 133,3, 137,0, 141,3, 151,2, 154,7, 165,0, 166,6, 170,0, 172,8; Анал. рассчитанное для C20H16N2O6: C, 63,16; H, 4,24; N, 7,37. Найденное: C, 63,00; H, 4,24; N, 7,29.
Пример 2
3-(4-гидрокси-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион
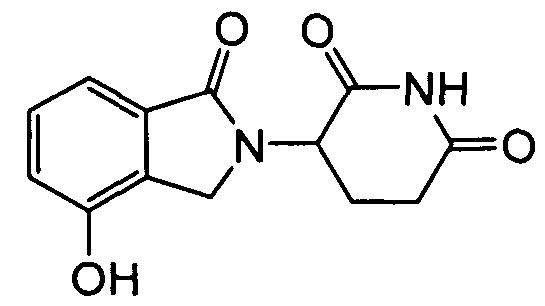
В 250 мл 3N-RBF помещали 3-(4-амино-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион и H2O (10 объемов) и охлаждали до 0-5°C. Добавляли NaNO2 (1,1 экв) и HCl (1,1 экв) и смесь перемешивали в течение 30 минут. Затем смесь грели при 75-80°C в течение 2 часов. Смесь охлаждали до комнатной температуры, фильтровали и сушили в вакууме (18 ч, 35-40°C). Неочищенный продукт очищали препаративной ВЭЖХ (условия: C18 Symmetry Column, 90:10 H2O: MeCN изократический, скорость потока 60 мл/мин, время удержания продукта составляет 30 минут) для того, чтобы получить грязно-белый твердый остаток (240 мг, 2,4%, 99,5 ВЭЖХ AP); т.пл. 296,39°C; ВЭЖХ: Hypersil DBS C8 5m колонка, 250×4,6 мм, 35°C; 99:1-85:15 градиент CH3CN/10 мм водн. KH2PO4, 1,0 мл/мин в течение 20 минут; 7,60 мин, 99,5% AP при 210/240 нм: 1H-ЯМР (DMSO-d 6): 10,97 (1H, шир.с), 10,11 (1H, шир.с), 7,34 (1H, т), 7,17 (1H, д), 7,03 (1H, д), 5,09 (1H, дд), 4,25 (2H, дд), 2,97-2,85 (1H, м), 2,62-2,36 (2H, м), 2,03-1,96 (1H, м) м.д.; 13C-ЯМР (DMSO-d 6): 172,85, 171,04, 168,27, 152,55, 133,41, 129,44, 127,94, 117,97, 113,71, 51,59, 45,09, 31,22, 22,42 м.д.; LC-MS ES+ (M+1) 261; CHN-Анализ, рассчитанное для C13H12N2O4: C, 60,00%; H, 4,65%; N, 10,76%. Найденное: C, 59,54%; H, 4,88%; N, 10,48%.
Пример 3
4-Бензилокси-2-(2,6-диоксопиперидин-3-ил)изоиндол-1,3-дион
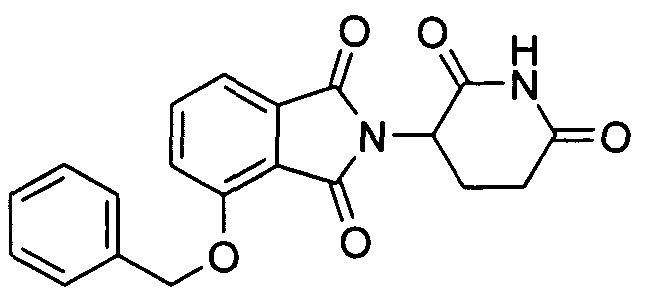
Стадия 1:
Смесь 3-гидроксифталевого ангидрида (4,96 г, 30,2 ммоль) в метаноле (60 мл) кипятили с обратным холодильником в течение 3 часов, охлаждали до комнатной температуры и растворитель упаривали в вакууме. Остаток и бикарбонат натрия (7,11 г, 84,6 ммоль) суспендировали в DMF (40 мл). Добавляли йодметан (4,53 мл, 72,5 ммоль) и реакционную смесь грели при 50°C в течение 2 часов. Растворитель удаляли в вакууме и остаток распределяли между этилацетатом (120 мл) и водой (100 мл). Органическую фазу промывали водой (2×100 мл) и упаривали. Остаток хроматографировали, применяя градиент гексан-этилацетат, продукт элюировался смесью 6:4 гексан-этилацетат, 4,83 г диметилового эфира 3-гидроксифталевой кислоты с выходом 76%; 1H ЯМР (CDCl3) δ 3,89 (с, 3H), 3,92 (с, 3H), 6,97 (дд, J=7,9 Гц, J=0,9 Гц, 1H), 7,09 (дд, J=8,6 Гц, J=1,0 Гц, 1H), 7,46 (т, J=8,3 Гц, 1H), 10,58 (с, 1H).
Стадия 2:
К перемешиваемому раствору диметилового эфира 3-гидроксифталевой кислоты (1,35 г, 6,40 ммоль) в DMF (15 мл) добавляли карбонат калия (1,78 г, 12,9 ммоль) и бензилбромид (0,92 мл, 7,7 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре, затем реакцию прекращали добавлением холодной воды (60 мл). Водный слой экстрагировали этилацетатом (3×40 мл). Объединенные органические слои промывали водой (4×50 мл) и соляным раствором (50 мл), сушили (MgSO4) и растворитель упаривали в вакууме. Остаток хроматографировали, применяя градиент гексан-этилацетат, продукт элюировался со смесью 7:3 гексан-этилацетат, 1,66 г диметилового эфира 3-бензилоксифталевой кислоты с выходом 86%; 1H ЯМР (CDCl3) δ 3,89 (с, 3H), 3,95 (с, 3H), 5,16 (с, 2H), 7,15 (д, J=8,4 Гц, 1H), 7,28-7,41 (м, 6H), 7,62 (д, J=7,9 Гц, 1H).
Стадия 3:
Смесь диметилового эфира 3-бензилоксифталевой кислоты (1,64 г, 5,50 ммоль) и 3N NaOH (50 мл) в этаноле (100 мл) кипятили с обратным холодильником в течение 1 часа и охлаждали до комнатной температуры. Растворитель удаляли в вакууме, остаток растворяли в воде (100 мл), промывали CH2Cl2 (2×100 мл) и подкисляли 6N HCl до pH 1-2. Осадок фильтровали и промывали водой (100 мл) для того, чтобы получить 3-бензилоксифталевую кислоту в виде белого твердого остатка (1,10 г, выход 74%); 1H ЯМР (DMSO-d 6) δ 5,20 (с, 2H), 7,29-7,51 (м, 8H), 13,02 (шир, 2H).
Стадия 4:
Смесь 3-бензилоксифталевой кислоты (1,08 г, 4,00 ммоль) и гидрохлорида рацемического α-аминоглутаримида (0,65 г, 4,0 ммоль) в пиридине (10 мл) кипятили с обратным холодильником в течение 4 часов. Реакционную смесь охлаждали и растворитель упаривали в вакууме. Остаток суспендировали в этилацетате (200 мл) и промывали разбавленной водной HCl (100 мл). Органическую фазу объединяли с нерастворимым осадком и упаривали досуха. Полученный в результате твердый остаток растирали с этилацетатом (100 мл), фильтровали, промывали дополнительным количеством этилацетата (50 мл) и сушили для того, чтобы получить заявленное в заголовке соединение (1,66 г, выход 86%); т.пл. 238-240°C; ВЭЖХ, Waters Symmetry C-18, 3,9×150 мм, 5 мкм, 1 мл/мин, 240 нм, 40/60 CH3CN/0,1% H3PO4, 7,23 (96,71%); 1H ЯМР (DMSO-d 6) δ 1,99-2,08 (м, 1H), 2,45-2,62 (м, 2H), 2,81-2,94 (м, 1H), 5,10 (дд, J=12,6 Гц, J=5,3 Гц, 1H), 5,38 (с, 2H), 7,32-7,53 (м, 6H), 7,60 (д, J=8,5 Гц, 1H), 7,83 (т, J=8,2 Гц, 1H), 11,12 (с, 1H); 13C ЯМР (DMSO-d 6) δ 22,0, 30,9, 48,8, 70,0, 115,6, 116,6, 120,2, 127,3, 128,0, 128,5, 133,3, 136,2, 137,0, 155,5, 165,3, 166,8, 169,9, 172,8; Анализ, рассчитанное для C20H16N2O5: C, 65,93; H, 4,43; N, 7,69. Найденное: C, 65,54; H, 4,35; N, 7,63.
Пример 4
4-(3-Хлорбензилокси)-2-(2,6-диоксопиперидин-3-ил)изоиндол-1,3-дион
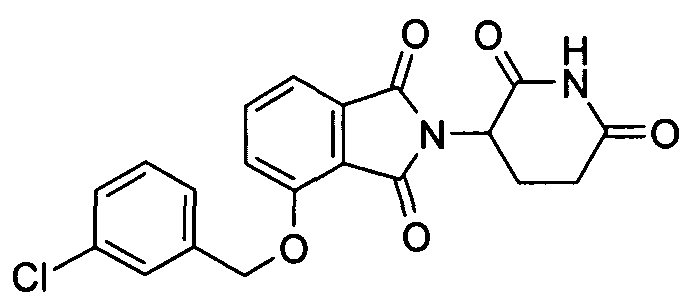
Стадия 1:
К перемешиваемой суспензия диметилового эфира 3-гидроксифталевой кислоты (1,3 г, 6,3 ммоль) в ацетоне (20 мл) и карбоната калия (2,1 г, 15,2 ммоль) добавляли 3-хлорбензилбромид (1,0 мл, 7,6 ммоль) и кипятили с обратным холодильником в течение ночи. Растворитель упаривали, остаток распределяли между водой (100 мл) и этилацетатом (150 мл) и промывали водой (2×100 мл). Объединенные органические фазы сушили, концентрировали и очищали флэш колоночной хроматографией (EtOAc/Гексан) для того, чтобы получить диметиловый эфир 3-(3-хлорбензилокси)фталевой кислоты (1,9 г, выход 92%). Продукт применяли в следующей стадии без дополнительной очистки.
Стадия 2:
Раствор диметилового эфира 3-(3-хлорбензилокси)фталевой кислоты (1,9 г, 5,8 ммоль) в спиртосодержащем реагенте (100 мл) и 3 N гидроксиде натрия (60 мл) кипятили с обратным холодильником в течение 2 часов. Раствор упаривали, остаток растворяли в воде (100 мл) и промывали хлористым метиленом (3×100 мл), затем подкисляли до pH равного, приблизительно, 4. Полученную в результате смесь экстрагировали этилацетатом (2×100 мл), объединенные органические слои промывали водой (2×100 мл), сушили и концентрировали для того, чтобы получить 3-(3-хлорбензилокси)фталевую кислоту в виде грязно-белого твердого остатка (1,6 г, 87% выход). Продукт применяли в следующей стадии без дополнительной очистки.
Стадия 3:
Смесь 3-(3-хлорбензилокси)фталевой кислоты (1,6 г, 5,1 ммоль), гидрохлорида альфа-аминоглутаримида (0,87 г, 5,3 ммоль) в пиридине кипятили с обратным холодильником в течение ночи. Смесь упаривали, остаток очищали флэш колоночной хроматографией (метанол/хлористый метилен) для того, чтобы получить 4-(3-хлорбензилокси)-2-(2,6-диоксопиперидин-3-ил)изоиндол-1,3-дион в виде белого твердого остатка (0,74 г, 37% выход); ВЭЖХ: Waters Symmetry C18, 5 мкм, 3,9×150 мм, 1 мл/мин, 240 нм, 50/50 CH3CN/0,1% H3PO4, 6,44 мин (99,8%); т.пл. 249-251°C; 1H ЯМР (DMSO-d 6) δ 2,02-2,07 (м, 1H, CHH), 2,54-2,62 (м, 2H, CH2), 2,83-2,91 (м, 1Η, CΗH), 5,12 (дд, J=6, 12 Гц, 1H, CH), 5,39 (с, 2Η, CH2), 7,40-7,87 (м, 7Η, Ar), 11,12 (с, 1H, NH); 13C ЯМР (DMSO-d 6) δ 21,96, 30,92, 48,77, 69,05, 115,72, 116,69, 120,10, 125,65, 126,85, 127,84, 130,42, 133,18, 133,26, 137,07, 138,76, 155,19, 165,32, 166,74, 169,89, 172,75. Анализ, рассчитанное для C20H15N2O5Cl + 0,1 H2O: C, 59,96; H, 3,82; N, 6,99; Cl, 8,85. Найденное: C, 59,93; H, 3,54; N, 6,91; Cl, 9,00.
Пример 5
4-(4-Хлорбензилокси)-2-(2,6-диоксопиперидин-3-ил)изоиндол-1,3-дион
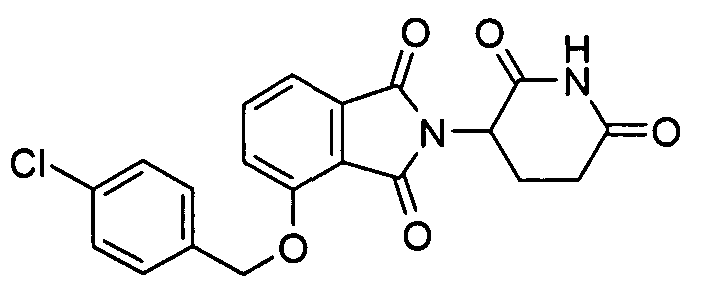
Стадия 1:
К перемешиваемой суспензии диметилового эфира 3-гидроксифталевой кислоты (1,3 г, 6,3 ммоль) в ацетоне (20 мл) и карбоната калия (2,6 г, 19 ммоль) добавляли 4-хлорбензилхлорид (1,1 г, 6,6 ммоль) и кипятили с обратным холодильником в течение ночи. Растворитель упаривали, остаток распределяли между водой (100 мл) и этилацетатом (150 мл) и промывали водой (2×100 мл). Объединенные органические фазы сушили, концентрировали и очищали флэш колоночной хроматографией (EtOAc/Гексан) для того, чтобы получить диметиловый эфир 3-(4-хлорбензилокси)фталевой кислоты (2,3 г, выход неочищенного продукта 110%). Продукт применяли в следующей стадии без дополнительной очистки.
Стадия 2:
Раствор диметилового эфира 3-(4-хлорбензилокси)фталевой кислоты (2,2 г, 6,3 ммоль) в спиртосодержащем реагенте (100 мл) и 3 N гидроксиде натрия (60 мл) кипятили с обратным холодильником в течение 2 часов. Раствор упаривали, остаток растворяли в воде (100 мл) и промывали хлористым метиленом (3×100 мл), затем подкисляли до pH, приблизительно равной 4. Полученную в результате смесь экстрагировали этилацетатом (2×100 мл), объединенные органические слои промывали водой (2×100 мл), сушили и концентрировали для того, чтобы получить 3-(4-хлорбензилокси)фталевую кислоту в виде грязно-белого твердого остатка (1,9 г, 98% выход). Продукт применяли в следующей стадии без дополнительной очистки.
Стадия 3:
Смесь 3-(4-хлорбензилокси)фталевой кислоты (1,9 г, 6,2 ммоль), гидрохлорида альфа-аминоглутаримида (1,1 г, 6,5 ммоль) в пиридине кипятили с обратным холодильником в течение ночи. Смесь упаривали и остаток очищали флэш колоночной хроматографией (метанол/хлористый метилен) для того, чтобы получить 4-(4-хлорбензилокси)-2-(2,6-диоксопиперидин-3-ил)изоиндол-1,3-дион в виде белого твердого остатка (1,2 г, 49% выход); ВЭЖХ: Waters Symmetry C18, 5 мкм, 3,9×150 мм, 1 мл/мин, 240 нм, 50/50 CH3CN/ 0,1% H3PO4, 6,64 мин (99,9%); т.пл. 239-241°C; 1H ЯМР (DMSO-d 6) δ 2,00-2,07 (м, 1H, CHH), 2,54-2,62 (м, 2Η, CH2), 2,83-2,95 (м, 1Η, CΗH), 5,12 (дд, J=6, 12 Гц, 1H, CH), 5,38 (с, 2Η, CH2), 7,48-7,86 (м, 7Η, Ar), 11,12 (с, 1H, NH); 13C ЯМР (DMSO-d 6) δ 21,96, 30,91, 48,75, 69,18, 115,66, 116,65, 120,16, 128,50, 129,02, 132,52, 133,25, 135,22, 137,03, 155,27, 165,30, 166,74, 169,89, 172,75. Анализ, рассчитанное для C20H15N2O5Cl: C, 60,24; H, 3,79; N, 7,02; Cl, 8,89. Найденное: C, 60,41; H, 3,63; N, 7,02; Cl, 8,72.
Пример 6
4-(3,4-Дихлорбензилокси)-2-(2,6-диоксопиперидин-3-ил)изоиндол-1,3-дион
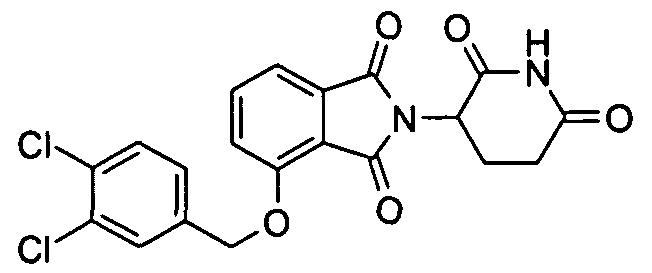
Стадия 1:
Триэтиламин (2,70 мл, 19,4 ммоль) добавляли к смеси 3-гидроксифталевого ангидрида (3,00 г, 18,3 ммоль) и гидрохлорида рацемического α-аминоглутаримида (3,01 г, 18,3 ммоль) в DMF (60 мл). Реакционную смесь грели при 90°C в течение ночи, затем охлаждали до комнатной температуры и растворитель упаривали в вакууме. Остаток перемешивали в CH2Cl2 (100 мл) в течение 30 мин и растворитель удаляли в вакууме. Остаток перемешивали в воде (120 мл) в течение 2 часов, полученный в результате твердый остаток фильтровали, промывали водой (50 мл) и сушили. Добавляли 1,4-диоксан (200 мл) и полученную в результате суспензию перемешивали в течение 16 часов и фильтровали; нерастворимые вещества оставляли. Фильтрат обрабатывали обесцвечивающим углем (2 г) и кипятили с обратным холодильником в течение 1 часа. После охлаждения до 50°C, реакционную смесь фильтровали через целит и фильтр промывали дополнительным количеством 1,4-диоксана (50 мл). Фильтрат объединяли с нерастворимым осадком и упаривали досуха. Полученный в результате твердый остаток растирали с этилацетатом (100 мл), фильтровали и сушили для того, чтобы получить 2-(2,6-диоксопиперидин-3-ил)-4-гидроксиизоиндол-1,3-дион, 4,18 г с выходом 56%; 1H ЯМР (DMSO-d 6) δ 1,99-2,06 (м, 1H), 2,45-2,61 (м, 2H), 2,82-2,96 (м, 1H), 5,08 (дд, J=12,6 Гц, J=5,3 Гц, 1H), 7,23-7,33 (м, 2H), 7,66 (дд, J=8,2 Гц, J=7,2 Гц, 1H), 11,10 (с, 1H), 11,19 (с, 1H).
Стадия 2:
Смесь трифенилфосфина на полимерной подложке (1,46 г, ~ 4,4 ммоль) и 3,4-дихлорбензилового спирта (0,65 г, 3,6 ммоль) перемешивали в THF (10 мл) при 0°C. Поддерживая температуру реакционной смеси равной 0°C, добавляли порциями раствор диизопропилазодикарбоксилата (0,87 мл, 4,4 ммоль) в THF (2,1 мл). Затем добавляли 2-(2,6-диоксопиперидин-3-ил)-4-гидроксиизоиндол-1,3-дион (1,00 г, 3,60 ммоль) в виде твердого остатка, реакционную смесь перемешивали при 0°C в течение 1 часа и затем при комнатной температуре в течение ночи. Растворитель упаривали в вакууме и остаток хроматографировали, применяя градиент метанол-CH2Cl2, продукт элюировался со смесью 95:5 CH2Cl2-метанол. Данное вещество растворяли в этилацетате (150 мл) и добавляли воду (100 мл). Затем органическую фазу промывали 10% разбавленным водным карбонатом натрия (2×50 мл) и водой (3×50 мл). Растворитель удаляли в вакууме, полученный в результате твердый остаток растирали с эфиром, фильтровали и сушили для того, чтобы получить заявленное в заголовке соединение в виде белого твердого остатка (0,41 г, 26% выход); т.пл.245-247°C; ВЭЖХ, Waters Symmetry C-18, 3,9×150 мм, 5 мкм, 1 мл/мин, 240 нм, 60/40 CH3CN/0,1% H3PO4, 3,50 (99,78%); 1H ЯМР (DMSO-d 6) δ 2,02-2,07 (м, 1H), 2,45-2,62 (м, 2H), 2,82-2,96 (м, 1H), 5,12 (дд, J=12,6 Гц, J=5,4 Гц, 1H), 5,38 (с, 2H), 7,49-7,51 (м, 2H), 7,57 (д, J=8,5 Гц, 1H), 7,71 (д, J=8,2 Гц, 1H), 7,81-7,88 (м, 2H), 11,12 (с, 1H); 13C ЯМР (DMSO-d 6) δ 22,0, 30,9, 48,8, 68,5, 115,9, 116,8, 120,1, 127,3, 129,0, 130,5, 130,8, 131,2, 133,3, 137,1, 137,5, 155,1, 165,3, 166,8, 169,9, 172,8; Анал. Рассчитанное для C20H14N2O5Cl2 + 0,3 H2O: C, 54,76; H, 3,35; N, 6,39. Найденное: C, 54,48; H, 3,07; N, 6,29.
Пример 7
4-(3,5-Дихлорбензилокси)-2-(2,6-диоксопиперидин-3-ил)изоиндол-1,3-дион
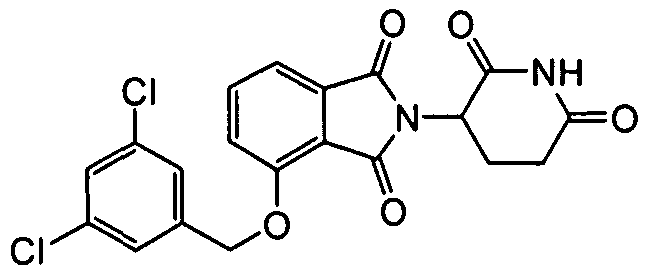
Стадия 1:
К перемешиваемой суспензии диметилового эфира 3-гидроксифталевой кислоты (0,5 г, 2,4 ммоль), (3,5-дихлорфенил)метанола (0,84 г, 4,8 ммоль) и трифенилфосфина на полимерной подложке (1,5 г, 4,8 ммоль) в THF (30 мл) в ледяной бане медленно добавляли диизопропилазодикарбоксилат (1,0 мл, 4,8 ммоль) и перемешивали при комнатной температуре в течение ночи. Смесь фильтровали и твердый остаток промывали этилацетатом (10 мл). Фильтрат упаривали и остаток очищали флэш колоночной хроматографией (EtOAc/Гексан) для того, чтобы получить диметиловый эфир 3-(3,5-дихлорбензилокси)фталевой кислоты (0,42 г, 48% выход). Продукт применяли в следующей стадии без дополнительной очистки.
Стадия 2:
Раствор диметилового эфира 3-(3,5-дихлорбензилокси)фталевой кислоты (0,42 г, 1,2 ммоль) в спиртосодержащем реагенте (10 мл) и 3 N натрия гидроксид (10 мл) кипятили с обратным холодильником в течение 2 часов. Раствор упаривали, остаток растворяли в воде (10 мл) и промывали хлористым метиленом (2×10 мл), затем подкисляли до pH, приблизительно равной 4. Полученную в результате смесь экстрагировали этилацетатом (2×10 мл), объединенные органические слои промывали водой (2×10 мл), сушили и концентрировали для того, чтобы получить 3-(3,5-дихлорбензилокси)фталевую кислоту в виде грязно-белого твердого остатка (0,34 г, 88% выход). Продукт применяли в следующей стадии без дополнительной очистки.
Стадия 3:
Смесь 3-(3,5-дихлорбензилокси)фталевой кислоты (0,3 г, 0,9 ммоль), гидрохлорида альфа-аминоглутаримида (0,15 г, 0,92 ммоль) в пиридине (10 мл) кипятили с обратным холодильником в течение ночи. Смесь упаривали и остаток очищали флэш колоночной хроматографией (метанол/хлористый метилен) для того, чтобы получить 4-(3,5-дихлорбензилокси)-2-(2,6-диоксопиперидин-3-ил)изоиндол-1,3-дион в виде белого твердого остатка (0,30 г, 84% выход); ВЭЖХ: Waters Symmetry C18, 5 мкм, 3,9×150 мм, 1 мл/мин, 240 нм, 70/30 CH3CN/0,1% H3PO4, 3,6 мин (98,0%); т.пл. 278-280°C; 1H ЯМР (DMSO-d 6) δ 2,01-2,08 (м, 1H, CHH), 2,54-2,63 (м, 2H, CH2), 2,84-2,96 (м, 1Η, CHH), 5,13 (дд, J=6, 12 Гц, 1H, CH), 5,40 (с, 2Η, CH2), 7,50-7,89 (м, 6Η, Ar), 11,12 (с, 1H, NH); 13C ЯМР (DMSO-d 6) δ 21,95, 30,92, 48,79, 68,37, 115,90, 116,79, 120,02, 125,58, 127,45, 133,25, 134,18, 137,14, 140,61, 154,93, 165,34, 166,73, 169,88, 172,73. Анал. рассчитанное для C20H14N2O5Cl2: C, 55,45; H, 3,26; N, 6,47; Cl, 16,37. Найденное: C, 55,20; H, 3,13; N, 6,38; Cl, 16,63.
Пример 8
4-(3-Фторбензилокси)-2-(2,6-диоксопиперидин-3-ил)изоиндол-1,3-дион
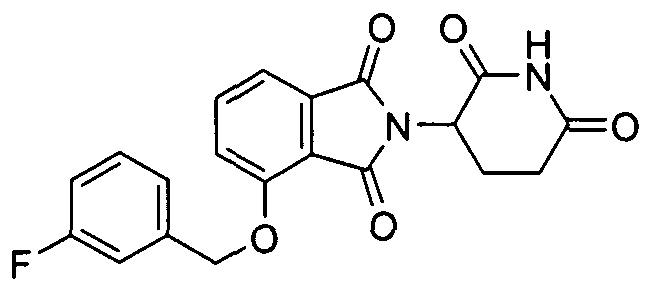
Стадия 1:
К перемешиваемой суспензии диметилового эфира 3-гидроксифталевой кислоты (1,4 г, 6,7 ммоль) в ацетоне (30 мл) и карбоната калия (2,8 г, 20 ммоль) добавляли 3-фторбензилбромид (0,89 мл, 7,0 ммоль) и кипятили с обратным холодильником в течение ночи. Растворитель упаривали, остаток распределяли между водой (100 мл) и этилацетатом (150 мл) и промывали водой (2×100 мл). Объединенные органические фазы сушили, концентрировали и очищали флэш колоночной хроматографией (EtOAc/Гексан) для того, чтобы получить диметиловый эфир 3-(3-фторбензилокси)фталевой кислоты (2,4 г, 113% выход неочищенного продукта). Продукт применяли в следующей стадии без дополнительной очистки.
Стадия 2:
Раствор диметилового эфира 3-(3-фторбензилокси)фталевой кислоты (2,4 г неочищенный, 6,7 ммоль) в спиртосодержащем реагенте (100 мл) и 3 N гидроксиде натрия (60 мл) кипятили с обратным холодильником в течение 2 часов. Раствор упаривали, остаток растворяли в воде (100 мл) и промывали хлористым метиленом (3×100 мл) затем подкисляли до pH, приблизительно равного 4. Полученную в результате смесь экстрагировали этилацетатом (2×100 мл), объединенные органические слои промывали водой (2×100 мл), сушили и концентрировали для того, чтобы получить 3-(3-фторбензилокси)фталевую кислоту в виде грязно-белого твердого остатка (1,9 г, 101% выход неочищенного продукта). Продукт применяли в следующей стадии без дополнительной очистки.
Стадия 3:
Смесь 3-(3-фторбензилокси)фталевой кислоты (1,9 г, 6,7 ммоль), гидрохлорида альфа-аминоглутаримида (1,2 г, 7,0 ммоль) в пиридине кипятили с обратным холодильником в течение ночи. Смесь упаривали и остаток очищали флэш колоночной хроматографией (метанол/хлористый метилен) для того, чтобы получить 4-(3-фторбензилокси)-2-(2,6-диоксопиперидин-3-ил)изоиндол-1,3-дион в виде белого твердого остатка (2,3 г, 89% выход); ВЭЖХ: Waters Symmetry С18, 5 мкм, 3,9×150 мм, 1 мл/мин, 240 нм, 60/40 CH3CN/0,1% H3PO4, 2,22 мин (99,9%); т.пл. 241-243°C; 1H ЯМР (DMSO-d 6) δ 2,01-2,08 (м, 1H, CHH), 2,55-2,62 (м, 2H, CH2), 2,83-2,95 (м, 1Η, CΗH), 5,11 (дд, J=6, 12 Гц, 1H, CH), 5,40 (с, 2Η, CH2), 7,15-7,87 (м, 7Η, Ar), 11,12 (с, 1H, NH); 13C ЯМР (DMSO-d 6) δ 21,96, 30,92, 48,77, 69,11, 113,61, 113,90, 114,52, 114,80, 115,71, 116,70, 120,13, 122,96, 122,99, 130,49, 130,60, 133,25, 137,05, 139,08, 139,18, 155,21, 160,59, 163,81, 165,33, 166,74, 169,89, 172,73. Анал. рассчитанное для C20H15N2O5F: C, 62,83; H, 3,95; N, 7,33; F 4,97. Найденное: C, 62,72; H, 3,75; N, 7,27; F, 5,02.
Пример 9
4-(3-Бромбензилокси)-2-(2,6-диоксопиперидин-3-ил)изоиндол-1,3-дион
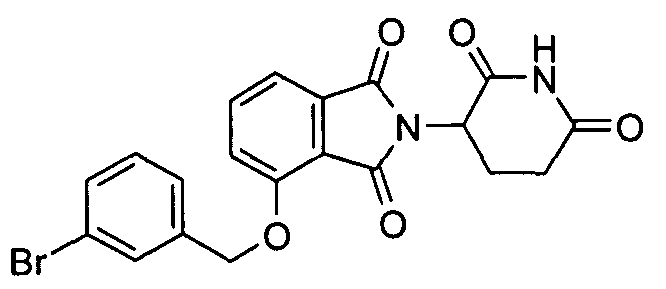
Стадия 1:
К перемешиваемой суспензии диметилового эфира 3-гидроксифталевой кислоты (1,3 г, 6,2 ммоль) в ацетоне (30 мл) и карбоната калия (2,5 г, 18,4 ммоль) добавляли 3-бромбензилбромид (1,6 г, 6,4 ммоль) и кипятили с обратным холодильником в течение ночи. Растворитель упаривали, остаток распределяли между водой (100 мл) и этилацетатом (150 мл) и промывали водой (2×100 мл). Объединенные органические фазы сушили, концентрировали и очищали флэш колоночной хроматографией (EtOAc/Гексан) для того, чтобы получить диметиловый эфир 3-(3-бромбензилокси)фталевой кислоты (2,5 г, выход неочищенного продукта 109%). Продукт применяли в следующей стадии без дополнительной очистки.
Стадия 2:
Раствор диметилового эфира 3-(3-бромбензилокси)фталевой кислоты (1,9 г, 5,8 ммоль) в спиртосодержащем реагенте (100 мл) и 3 N гидроксиде натрия (60 мл) кипятили с обратным холодильником в течение 2 часов. Раствор упаривали, остаток растворяли в воде (100 мл) и промывали хлористым метиленом (3×100 мл), затем подкисляли до pH, приблизительно равного 4. Полученную в результате смесь экстрагировали этилацетатом (2×100 мл), объединенные экстракты промывали водой (2×100 мл), сушили и концентрировали для того, чтобы получить 3-(3-бромбензилокси)фталевую кислоту в виде грязно-белого твердого остатка (2,5 г, 109% выход неочищенного продукта). Продукт применяли в следующей стадии без дополнительной очистки.
Стадия 3:
Смесь 3-(3-бромбензилокси)фталевой кислоты (2,4 г, 6,7 ммоль), гидрохлорида альфа-аминоглутаримида (1,2 г, 7,1 ммоль) в пиридине кипятили с обратным холодильником в течение ночи. Смесь упаривали и остаток очищали флэш колоночной хроматографией (метанол/хлористый метилен) для того, чтобы получить 4-(3-бромбензилокси)-2-(2,6-диоксопиперидин-3-ил)изоиндол-1,3-дион в виде белого твердого остатка (1,8 г, 62% выход); ВЭЖХ: Waters Symmetry C18, 5мкм, 3,9×150 мм, 1 мл/мин, 240 нм, 60/40 CH3CN/0,1% H3PO4, 3,55 мин (99,9%); т.пл. 246-248°C; 1H ЯМР (DMSO-d 6) δ 2,03-2,08 (м, 1H, CHH), 2,54-2,62 (м, 2H, CH2), 2,83-2,95 (м, 1Η, CHH), 5,12 (дд, J=6, 12 Гц, 1H, CH), 5,39 (с, 2Η, CH2), 7,37-7,88 (м, 7Η, Ar), 11,12 (с, 1H, NH); 13C ЯМР (DMSO-d 6) δ 21,96, 30,92, 48,77, 69,00, 115,72, 116,69, 120,10, 121,76, 126,05, 129,75, 130,69, 130,74, 133,26, 137,07, 138,99, 155,19, 165,31, 166,74, 169,89, 172,74. Анал. рассчитанное для C20H15N2O5Br: C, 54,19; H, 3,41; N, 6,32; Br 18,03. Найденное: C, 54,02; H, 3,22; N, 6,27; Br, 17,81.
Пример 10
2-(2,6-Диоксопиперидин-3-ил)-4-(3-метил-бензилокси)изоиндол-1,3-дион
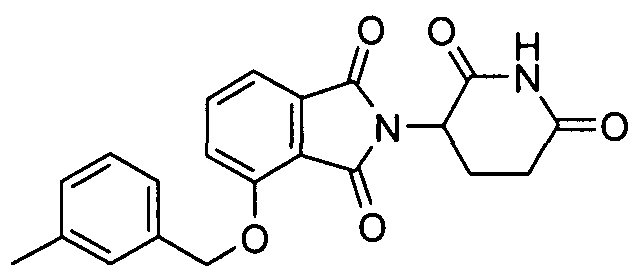
Стадия 1:
К перемешиваемой суспензии диметилового эфира 3-гидроксифталевой кислоты (1,4 г, 6,4 ммоль) в ацетоне (30 мл) и карбоната калия (2,7 г, 19,3 ммоль) добавляли 1-бромметил-3-метилбензол (0,91 мл, 6,7 ммоль) и кипятили с обратным холодильником в течение ночи. Растворитель упаривали, остаток распределяли между водой (100 мл) и этилацетатом (150 мл) и промывали водой (2×100 мл). Объединенные органические фазы сушили, концентрировали и очищали флэш колоночной хроматографией (EtOAc/Гексан) для того, чтобы получить диметиловый эфир 3-(3-метилбензилокси)фталевой кислоты (2,3 г, выход неочищенного продукта 115%). Продукт применяли в следующей стадии без дополнительной очистки.
Стадия 2:
Раствор диметилового эфира 3-(3-метилбензилокси)фталевой кислоты (2,0 г, неочищенный, 6,4 ммоль) в спиртосодержащем реагенте (100 мл) и 3 N гидроксиде натрия (60 мл) кипятили с обратным холодильником в течение 2 часов. Раствор упаривали, остаток растворяли в воде (100 мл) и промывали хлористым метиленом (3×100 мл), затем, подкисляли до pH, приблизительно равного 4. Полученную в результате смесь экстрагировали этилацетатом (2×100 мл), объединенные органические слои промывали водой (2×100 мл), сушили и концентрировали для того, чтобы получить 3-(3-метилбензилокси)фталевую кислоту в виде грязно-белого твердого остатка (3,0 г, выход неочищенного продукта 130%). Продукт применяли в следующей стадии без дополнительной очистки.
Стадия 3:
Смесь 3-(3-метилбензилокси)фталевой кислоты (1,8 г, 6,4 ммоль), гидрохлорида альфа-аминоглутаримида (1,1 г, 6,8 ммоль) в пиридине кипятили с обратным холодильником в течение ночи. Смесь упаривали и остаток очищали флэш колоночной хроматографией (метанол/хлористый метилен) для того, чтобы получить 2-(2,6-диоксопиперидин-3-ил)-4-(3-метилбензилокси)изоиндол-1,3-дион в виде белого твердого остатка (1,2 г, 48% выход); ВЭЖХ: Waters Symmetry C18, 5мкм, 3,9×150 мм, 1 мл/мин, 240 нм, 60/40 CH3CN/0,1% H3PO4, 3,16 мин (99,9%); т.пл. 195-197°C; 1H ЯМР (DMSO-d 6) δ 2,00-2,07 (м, 1H, CHH), 2,33 (с, 3Η, CH3), 2,54-2,62 (м, 2Η, CH2), 2,83-2,95 (м, 1Η, CHH), 5,10 (дд, J=6, 12 Гц, 1H, CH), 5,33 (с, 2Η, CH2), 7,15-7,85 (м, 7Η, Ar), 11,12 (с, 1H, NH); 13C ЯМР (DMSO-d 6) δ 21,00, 21,96, 30,92, 48,74, 70,08, 115,49, 116,58, 120,19, 124,40, 127,85, 128,40, 128,60, 133,26, 136,03, 136,98, 137,64, 155,53, 165,30, 166,77, 169,91, 172,75. Анал. рассчитанное для C21H18N2O5: C, 66,66; H, 4,79; N, 7,40. Найденное: C, 66,50; H, 4,79; N, 7,34.
Пример 11
2-(2,6-Диоксопиперидин-3-ил)-4-(4-метансульфонилбензилокси)изоиндол-1,3-дион
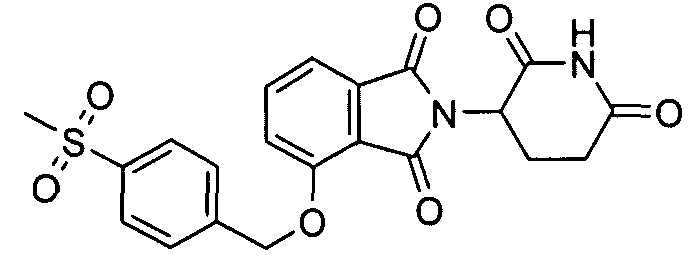
Стадия 1:
К перемешиваемой суспензии диметилового эфира 3-гидроксифталевой кислоты (1,3 г, 6,1 ммоль) в ацетоне (25 мл) и карбоната калия (2,5 г, 18 ммоль) добавляли 1-бромметил-4-метансульфонилбензол (1,6 г, 6,4 ммоль) и кипятили с обратным холодильником в течение ночи. Растворитель упаривали, остаток распределяли между водой (100 мл) и этилацетатом (150 мл) и промывали водой (2×100 мл). Объединенные органические фазы сушили, концентрировали и очищали флэш колоночной хроматографией (EtOAc/Гексан) для того, чтобы получить диметиловый эфир 3-(4-метансульфонилбензилокси)фталевой кислоты (2,4 г, выход неочищенного продукта 104%). Продукт применяли в следующей стадии без дополнительной очистки.
Стадия 2:
Раствор диметилового эфира 3-(4-метансульфонилбензилокси)фталевой кислоты (2,3 г, 6,1 ммоль) в спиртосодержащем реагенте (140 мл) и 3 N гидроксиде натрия (70 мл) кипятили с обратным холодильником в течение 2 часов. Раствор упаривали, остаток растворяли в воде (100 мл) и промывали хлористым метиленом (3×100 мл), затем подкисляли до pH, приблизительно равного 4. Полученную в результате смесь экстрагировали этилацетатом (2×100 мл), объединенные органические слои промывали водой (2×100 мл), сушили и концентрировали для того, чтобы получить 3-(4-метансульфонилбензилокси)фталевую кислоту в виде грязно-белого твердого остатка (2,15 г, выход неочищенного продукта 101%). Продукт применяли в следующей стадии без дополнительной очистки.
Стадия 3:
Смесь 3-(4-метансульфонилбензилокси)фталевой кислоты (2,1 г, 6,1 ммоль), гидрохлорида альфа-аминоглутаримида (1,1 г, 6,4 ммоль) в пиридине кипятили с обратным холодильником в течение ночи. Смесь упаривали и остаток очищали флэш колоночной хроматографией (метанол/хлористый метилен) для того, чтобы получить 2-(2,6-диоксопиперидин-3-ил)-4-(4-метансульфонилбензилокси)изоиндол-1,3-дион в виде белого твердого остатка (1,3 г, 47% выход); ВЭЖХ: Waters Symmetry C18, 5 мкм, 3,9×150 мм, 1 мл/мин, 240 нм, 35/65 CH3CN/0,1% H3PO4, 2,09 мин (99,9%); т.пл. 293-295°C; 1H ЯМР (DMSO-d 6) δ 2,03-2,07 (м, 1H, CHH), 2,54-2,63 (м, 2H, CH2), 2,85-2,90 (м, 1Η, CHH), 3,23 (с, 3Η, CH3SO2), 5,11 (дд, J=6, 12 Гц, 1H, CH), 5,52 (с, 2Η, CH2), 7,49-8,00 (м, 7Η, Ar), 11,13 (с, 1H, NH); 13C ЯМР (DMSO-d 6) δ 21,97, 30,91, 43,46, 48,78, 69,09, 115,82, 116,73, 120,11, 127,21, 127,62, 133,28, 137,10, 140,26, 142,17, 155,12, 165,30, 166,73, 169,89, 172,75. Анал. рассчитанное для C21H18N2O7S + 0,2 H2O: C, 56,55; H, 4,16; N, 6,28; S, 7,19. Найденное: C, 56,32; H, 3,80; N, 6,16; S, 7,20.
Пример 12
2-(2,6-Диоксопиперидин-3-ил)-4-(3-метоксибензилокси)изоиндол-1,3-дион
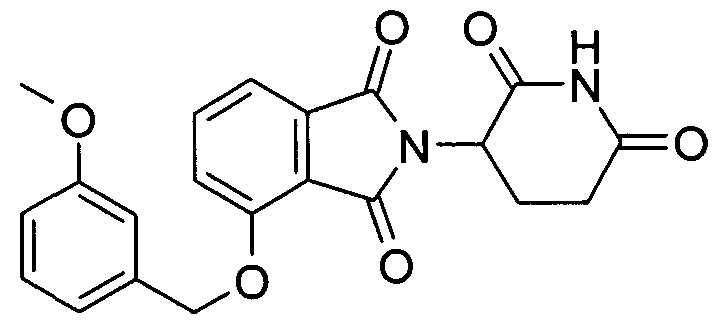
Стадия 1:
К перемешиваемой суспензии диметилового эфира 3-гидроксифталевой кислоты (1,1 г, 5,2 ммоль) в ацетоне (45 мл) и карбоната калия (2,2 г, 15,7 ммоль) добавляли 1-бромметил-3-метоксибензол (0,77 мл, 5,5 ммоль) и кипятили с обратным холодильником в течение 3 часов. Растворитель упаривали, остаток распределяли между водой (50 мл) и этилацетатом (80 мл) и промывали водой (2×50 мл). Объединенные органические фазы сушили, концентрировали и очищали флэш колоночной хроматографией (EtOAc/Гексан) для того, чтобы получить диметиловый эфир 3-(3-метоксибензилокси)фталевой кислоты (2,1 г, выход неочищенного продукта 118%). Продукт применяли в следующей стадии без дополнительной очистки.
Стадия 2:
Раствор диметилового эфира 3-(3-метоксибензилокси)фталевой кислоты (2,0 г, неочищенный, 5,5 ммоль) в спиртосодержащем реагенте (100 мл) и 3 N гидроксиде натрия (35 мл) кипятили с обратным холодильником в течение 2 часов. Раствор упаривали, остаток растворяли в воде (80 мл) и промывали хлористым метиленом (3×70 мл), затем подкисляли до pH, приблизительно равного 4. Полученную в результате смесь экстрагировали этилацетатом (2×60 мл), объединенные органические слои промывали водой (2×70 мл), сушили и концентрировали для того, чтобы получить 3-(3-метоксибензилокси)фталевую кислоту в виде грязно-белого твердого остатка (1,6 г, выход неочищенного продукта 98%). Продукт применяли в следующей стадии без дополнительной очистки.
Стадия 3:
Смесь 3-(3-метоксибензилокси)фталевой кислоты (1,5 г, 5,2 ммоль), гидрохлорида альфа-аминоглутаримида (0,89 г, 5,4 ммоль) в пиридине кипятили с обратным холодильником в течение ночи. Смесь упаривали и остаток очищали флэш колоночной хроматографией (метанол/хлористый метилен) для того, чтобы получить 2-(2,6-диоксопиперидин-3-ил)-4-(3-метоксибензилокси)изоиндол-1,3-дион в виде белого твердого остатка (0,25 г, 12% выход); ВЭЖХ: Waters Symmetry C18, 5 мкм, 3,9×150 мм, 1 мл/мин, 240 нм, 60/40 CH3CN/0,1% H3PO4, 2,41 мин (99,1%); т.пл. 197-201°C; 1H ЯМР (DMSO-d 6) δ 2,02-2,06 (м, 1H, CHH), 2,59-2,62 (м, 2H, CH2), 2,83-2,90 (м, 1Η, CHH), 3,77 (с, 3Η, CH3), 5,10 (дд, J=6, 12 Гц, 1H, CH), 5,35 (с, 2Η, CH2), 6,89-7,85 (м, 7Η, Ar), 11,11 (с, 1H, NH); 13C ЯМР (DMSO-d 6) δ 21,95, 30,92, 48,75, 55,01, 69,80, 112,72, 113,27, 115,55, 116,66, 119,17, 120,21, 129,61, 133,25, 136,98, 137,74, 155,42, 159,35, 165,32, 166,77, 169,90, 172,74. Анал. рассчитанное для C21H18N2O6 + 0,1 H2O: C, 63,67; H, 4,63; N, 7,07. Найденное: C, 63,49; H, 4,40; N, 7,00.
Пример 13
4-(Бензо[1,3]диоксол-5-илметокси)-2-(2,6-диоксопиперидин-3-ил)изоиндол-1,3-дион
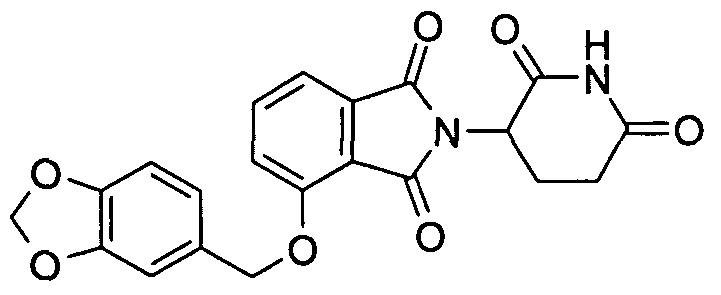
Стадия 1:
К перемешиваемой суспензии диметилового эфира 3-гидроксифталевой кислоты (1,0 г, 4,8 ммоль), бензо[1,3]диоксол-5-илметанола (1,4 г, 9,5 ммоль) и трифенилфосфина на полимерной подложке (3,0 г, 9,5 ммоль) в THF (30 мл) в ледяной бане медленно добавляли диизопропилазодикарбоксилат (1,9 мл, 9,5 ммоль) и перемешивали при комнатной температуре в течение ночи. Смесь фильтровали и фильтр промывали этилацетатом (10 мл). Фильтрат упаривали и остаток очищали флэш колоночной хроматографией (EtOAc/Гексан) для того, чтобы получить диметиловый эфир 3-(бензо[1,3]диоксол-5-илметокси)фталевой кислоты (1,7 г, выход неочищенного продукта 102%). Продукт применяли в следующей стадии без дополнительной очистки.
Стадия 2:
Раствор диметилового эфира 3-(бензо[1,3]диоксол-5-илметокси)фталевой кислоты (1,6 г, неочищенный, 4,8 ммоль) в спиртосодержащем реагенте (100 мл) и 3 N гидроксиде натрия (35 мл) кипятили с обратным холодильником в течение 2 часов. Раствор упаривали, остаток растворяли в воде (80 мл) и промывали хлористым метиленом (3×70 мл), затем подкисляли до pH, приблизительно равного 4. Полученную в результате смесь экстрагировали этилацетатом (2×60 мл), объединенные органические слои промывали водой (2×70 мл), сушили и концентрировали для того, чтобы получить 3-(бензо[1,3]диоксол-5-илметокси)фталевую кислоту в виде грязно-белого твердого остатка (1,2 г, выход неочищенного продукта 80%). Продукт применяли в следующей стадии без дополнительной очистки.
Стадия 3:
Смесь 3-(бензо[1,3]диоксол-5-илметокси)фталевой кислоты (1,2 г, 3,8 ммоль), гидрохлорида альфа-аминоглутаримида (0,66 г, 4,0 ммоль) в пиридине кипятили с обратным холодильником в течение ночи. Смесь упаривали и остаток очищали флэш колоночной хроматографией (метанол/хлористый метилен) для того, чтобы получить 4-(бензо[1,3]диоксол-5-илметокси)-2-(2,6-диоксопиперидин-3-ил)изоиндол-1,3-дион в виде белого твердого остатка (0,45 г, 29% выход); ВЭЖХ: Waters Symmetry C18, 5мкм, 3,9×150 мм, 1 мл/мин, 240 нм, 50/50 CH3CN/0,1% H3PO4, 4,06 мин (98,6%); т.пл. 229-231°C; 1H ЯМР (DMSO-d 6) δ 2,02-2,05 (м, 1H, CHH), 2,55-2,62 (м, 2H, CH2), 2,82-2,94 (м, 1Η, CHH), 5,10 (дд, J=6, 12 Гц, 1H, CH), 5,26 (с, 2Η, CH2), 6,03 (с, 2Η, CH2), 6,93-7,85 (м, 6Η, Ar), 11,11 (с, 1H, NH); 13C ЯМР (DMSO-d 6) δ 21,95, 30,91, 48,74, 70,00, 101,05, 108,10, 108,18, 1 15,49, 116,61, 120,28, 121,25, 129,78, 133,24, 136,95, 147,04, 147,38, 155,44, 165,30, 166,76, 169,89, 172,74. Анал. Рассчитанное для C21Η16N2O7: C, 61,77; H, 3,95; N, 6,86. Найденное: C, 61,44; H, 3,72; N, 6,79.
Пример 14
2-(2,6-Диоксопиперидин-3-ил)-4-(нафталин-2-илметокси)изоиндол-1,3-дион
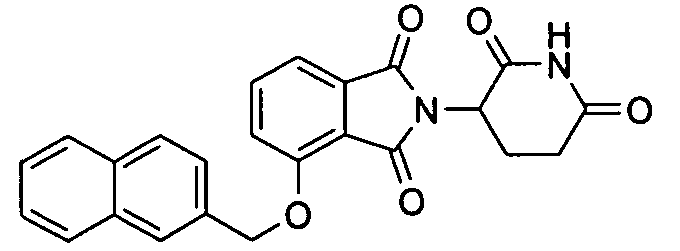
Смесь трифенилфосфина (1,15 г, 4,40 ммоль) и 2-нафталинметанола (0,58 г, 3,6 ммоль) перемешивали в THF (10 мл) при 0°C. Выдерживая реакционную смесь при 0°C, добавляли по каплям раствор диизопропилазодикарбоксилата (0,87 мл, 4,4 ммоль) в THF (2,1 мл). Затем добавляли 2-(2,6-диоксопиперидин-3-ил)-4-гидроксиизоиндол-1,3-дион (1,00 г, 3,60 ммоль) в виде твердого остатка, реакционную смесь перемешивали при 0°C в течение 1 часа и затем при комнатной температуре в течение ночи. Осадок фильтровали, промывали дополнительным количеством THF (10 мл) и сушили. Полученный в результате твердый остаток перемешивали в гексане (50 мл) в течение 2 часов, фильтровали и сушили. Полученный в результате твердый остаток кипятили с обратным холодильником в метаноле (50 мл) в течение 1 часа, фильтровали и сушили для того, чтобы получить продукт в виде белого твердого остатка (0,46 г, 30% выход); т.пл.> 260°C; ВЭЖХ, Waters Symmetry C-18, 3,9×150 мм, 5 мкм, 1 мл/мин, 240 нм, 50/50 CH3CN/0,1% H3PO4, 6,20 (99,48%); 1H ЯМР (DMSO-d 6) δ 2,02-2,07 (м, 1H), 2,44-2,62 (м, 2H), 2,82-2,97 (м, 1H), 5,12 (дд, J=12,5 Гц, J=5,3 Гц, 1H), 5,54 (с, 2H), 7,46-8,05 (м, 10H), 11,13 (с, 1H); 13C ЯМР (DMSO-d 6) δ 22,0, 31,0, 48,8, 70,3, 115,6, 116,7, 120,4, 125,3, 126,1, 126,3, 126,4, 127,7, 127,8, 128,2, 132,6, 132,7, 133,3, 133,8, 137,0, 155,6, 165,4, 166,8, 169,9, 172,8; Анал. рассчитанное для C24H18N2O5: C, 69,39; H, 4,02; N, 6,61. Найденное: C, 69,56; H, 4,38; N, 6,76.
Пример 15
2-(2,6-Диоксопиперидин-3-ил)-4-(хинолин-3-илметокси)изоиндол-1,3-дион
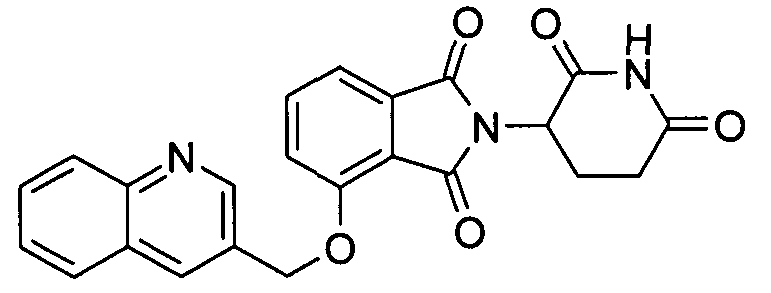
Стадия 1:
3-Хинолинкарбальдегид (2,00 г, 12,7 ммоль) растворяли в 25 мл метанола. К данному раствору добавляли малыми порциями в течение 20 минут боргидрид натрия (0,24 г, 6,4 ммоль). Затем добавляли 2 мл воды и смесь упаривали. Остаток растворяли в этилацетате (75 мл), промывали водой (3×75 мл), сушили (MgSO4) и упаривали, получая 1,8 г хинолин-3-илметанола с выходом 90%; 1H ЯМР (DMSO-d 6) δ 4,89 (с, 2H), 7,53 (т, J=7,1 Гц, 1H), 7,64-7,71 (м, 1H), 7,77 (д, J=8,2 Гц, 1H), 8,04-8,12 (м, 2H), 8,83 (д, J=2,0 Гц, 1H).
Стадия 2:
Смесь PPh3 на полимерной подложке (3,1 г, ~ 9,5 ммоль) и хинолин-3-илметанола (0,76 г, 4,8 ммоль) в 20 мл THF охлаждали до 0°C в атмосфере N2. Добавляли по каплям диизопропилазодикарбоксилат (1,9 г, 9,5 ммоль) и затем добавляли в виде твердого остатка диметиловый эфир 3-гидроксифталевой кислоты (1,0 г, 4,8 ммоль). Смесь перемешивали в течение дополнительного часа при 0°C и затем нагревали до комнатной температуры. После перемешивания в течение 16 часов смесь фильтровали. Фильтр промывали этилацетатом (25 мл) и объединенные фильтраты упаривали.
Стадия 3:
Неочищенный продукт из стадии 2 растворяли в смеси 3N NaOH (50 мл) и этанола (100 мл) и полученный в результате раствор кипятили с обратным холодильником в течение 2 часа. Смесь охлаждали и растворитель удаляли в вакууме. Остаток растворяли в воде (100 мл) и промывали CH2Cl2 (3×100 мл), подкисляли до pH 2-3 (HCl). Полученный в результате осадок фильтровали, промывали дополнительным количеством воды и затем этилацетатом и сушили в вакууме.
Стадия 4:
Неочищенный продукт из стадии 3 и гидрохлорид рацемического α-аминоглутаримида (0,78 г, 4,8 ммоль) в пиридине (10 мл) кипятили с обратным холодильником в течение 16 часов. Смесь охлаждали и упаривали в вакууме. Остаток хроматографировали, применяя градиент CH2Cl2-метанол, заявленное в заголовке соединение элюировалось со смесью 95:5 CH2Cl2-метанол, 0,37 г, с выходом 20% на 3 стадии; т.пл.263-265°C; ВЭЖХ, Waters Symmetry C-18, 3,9×150 мм, 5 мкм, 1 мл/мин, 240 нм, 40/60 CH3CN/вода, 3,75 (97,84%); 1H ЯМР (DMSO-d 6) δ 2,02-2,06 (м, 1H), 2,55-2,62 (м, 2H), 2,81-2,90 (м, 1H), 5,11 (дд, J=12,3 Гц, J=5,3 Гц, 1H), 5,61 (с, 2H), 7,51 (д, J=7,2 Гц, 1H), 7,62-7,71 (м, 2H), 7,77-7,90 (м, 2H), 8,00-8,08 (м, 2H), 8,48 (с, 1H), 9,04 (д, J=1,8 Гц, 1H), 11,12 (с, 1H); 13C ЯМР (DMSO-d 6) δ 22,0, 30,9, 48,8, 68,3, 115,9, 116,8, 120,4, 127,1, 127,2, 128,1, 128,8, 129,3, 129,8, 133,3, 134,5, 137,1, 147,3, 150,3, 155,3, 165,3, 166,8, 169,9, 172,8; Анал. рассчитанное для C23H17N3O5·0,4 H2O: C, 65,37; H, 4,25; N, 9,94. Найденное: C, 65,35; H, 4,06; N, 9,92.
Пример 16
2-(2,6-Диоксопиперидин-3-ил)-4-(хинолин-2-илметокси)изоиндол-1,3-дион
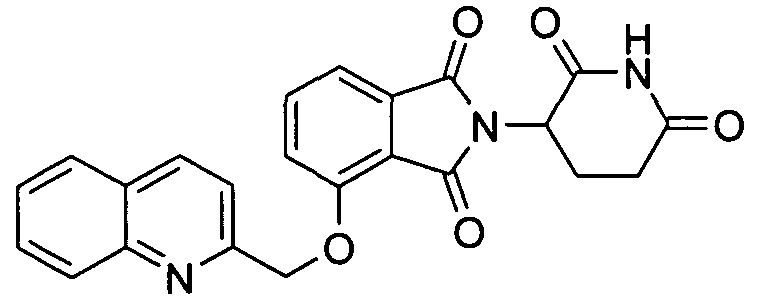
Стадия 1:
2-Хинолинкарбальдегид (2,00 г, 12,7 ммоль) растворяли в 25 мл метанола. К данному раствору добавляли боргидрид натрия (0,24 г, 6,4 ммоль) маленькими порциями в течение 20 минут. Затем добавляли 2 мл воды и смесь упаривали. Остаток растворяли в этилацетате (75 мл), промывали водой (3×75 мл) и упаривали. Остаток хроматографировали в градиенте CH2Cl2-метанол, продукт элюировался со смесью 97:3 CH2Cl2-метанол, получая 1,7 г хинолин-2-илметанола с выходом 85%; 1H ЯМР (CDCl3) δ 4,92 (с, 2H), 7,26 (д, J=2,1 Гц, 1H), 7,30-7,57 (м, 1H), 7,68-7,75 (м, 1H), 7,82 (дд, J=8,0 Гц, J=0,9 Гц, 1H), 8,07 (д, J=8,4 Гц, 1H), 8,13 (д, J=8,5 Гц, 1H).
Стадия 2:
Смесь PPh3 на полимерной подложке (3,1 г, ~ 9,5 ммоль) и хинолин-2-илметанола (0,76 г, 4,8 ммоль) в 20 мл THF охлаждали до 0°C в атмосфере N2. Добавляли по каплям диизопропилазодикарбоксилат (1,9 г, 9,5 ммоль) и затем добавляли в виде твердого остатка диметиловый эфир 3-гидроксифталевой кислоты (1,0 г, 4,8 ммоль). Смесь перемешивали в течение дополнительного часа при 0°C и затем нагревали до комнатной температуры. После перемешивания в течение 16 часов смесь фильтровали. Фильтр промывали этилацетатом (25 мл) и объединенные фильтраты упаривали.
Стадия 3:
Неочищенный продукт из стадии 2 растворяли в смеси 3N NaOH (50 мл) и этанола (100 мл) и полученный в результате раствор кипятили с обратным холодильником в течение 2 часа. Смесь охлаждали и растворитель удаляли в вакууме. Остаток растворяли в воде (100 мл) и промывали CH2Cl2 (3×100 мл), подкисляли до pH 2-3 (HCl). Полученный в результате осадок фильтровали, промывали дополнительным количеством воды и затем этилацетатом и сушили в вакууме.
Стадия 4:
Продукт из стадии 3 и гидрохлорид рацемического α-аминоглутаримида (0,78 г, 4,8 ммоль) в пиридине (10 мл) кипятили с обратным холодильником в течение 16 часов. Смесь охлаждали и упаривали в вакууме. Остаток хроматографировали, применяя градиент CH2Cl2-метанол, продукт элюировался со смесью 95:5 CH2Cl2-метанол. Данное вещество растирали с DMF (5 мл), фильтровали, промывали дополнительными 2 мл DMF и сушили в вакууме. Затем данное вещество очищали препаративной ВЭЖХ, применяя подвижную фазу 35/65 ацетонитрил-вода, получая 75 мг заявленного в заголовке соединения с выходом 4% на 3 стадии; т.пл. 254-256°C; ВЭЖХ, Waters Symmetry C-18, 3,9×150 мм, 5 мкм, 1 мл/мин, 240 нм, 30/70 CH3CN/ 0,1% H3PO4, 7,02 (94,00%); 1H ЯМР (DMSO-d 6) δ 2,03-2,08 (м, 1H), 2,57-2,64 (м, 2H), 2,85-2,96 (м, 1H), 5,13 (дд, J=12,2 Гц, J=4,9 Гц, 1H), 5,62 (с, 2H), 7,50 (д, J=7,1 Гц, 1H), 7,62-7,66 (м, 2H), 7,77-7,87 (м, 3H), 8,00-8,03 (м, 2H), 8,48 (д, J=8,5 Гц, 1H), 11,13 (с, 1H); 13C ЯМР (DMSO-d 6) δ 22,0, 31,0, 48,8, 71,4, 115,8, 116,8, 119,1, 120,3, 126,7, 127,2, 128,0, 128,5, 130,0, 133,3, 137,1, 137,2, 146,9, 155,3, 156,7, 165,4, 166,8, 169,9, 172,8; Анал. рассчитанное для C23H17N3O5·1,6 H2O: C, 62,19; H, 4,58; N, 9,46. Найденное: C, 62,20; H, 3,97; N, 9,15.
Пример 17
4-(Бензофуран-2-илметокси)-2-(2,6-диоксопиперидин-3-ил)изоиндол-1,3-дион
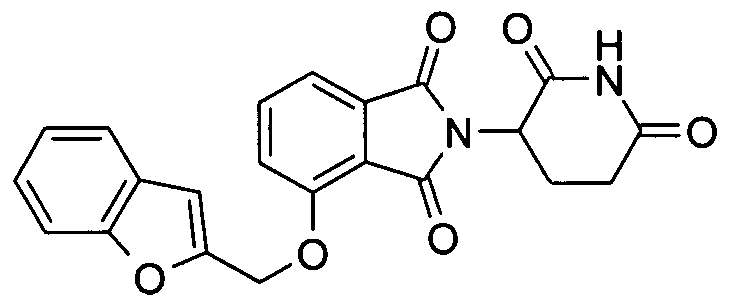
Стадия 1:
2-Бензофуранкарбальдегид (2,2 г, 15 ммоль) растворяли в 25 мл метанола. К данному раствору добавляли маленькими порциями боргидрид натрия (0,28 г, 7,5 ммоль) в течение 20 минут. Затем добавляли 2 мл воды и смесь упаривали. Остаток растворяли в этилацетате (75 мл), промывали водой (3×75 мл), сушили (MgSO4) и упаривали, получая 2,1 г бензофуран-2-илметанола с выходом 95%; 1H ЯМР (CDCl3) δ 2,03 (т, J=6,1 Гц, 1H), 4,77 (д, J=6,1 Гц, 2H), 6,66 (с, 1H), 7,19-7,32 (м, 2H), 7,45-7,50 (м, 1H), 7,53-7,57 (м, 1H).
Стадия 2:
Смесь PPh3 на полимерной подложке (3,1 г, ~ 9,5 ммоль) и бензофуран-2-илметанола (0,70 г, 4,8 ммоль) в 20 мл THF охлаждали до 0°C в атмосфере N2. Добавляли по каплям диизопропилазодикарбоксилат (1,9 г, 9,5 ммоль) и затем добавляли в виде твердый остатка диметиловый эфир 3-гидроксифталевой кислоты (1,0 г, 4,8 ммоль). Смесь перемешивали в течение дополнительного часа при 0°C и затем нагревали до комнатной температуры. После перемешивания в течение 16 часов смесь фильтровали. Фильтр промывали этилацетатом (25 мл) и объединенные фильтраты упаривали. Остаток растворяли в 75 мл этилацетата, промывали Na2CO3 (2×75 мл) и водой (2×75 мл), сушили (MgSO4) и упаривали.
Стадия 3:
Неочищенный продукт из стадии 2 растворяли в смеси 3N NaOH (50 мл) и этанола (100 мл) и полученный в результате раствор кипятили с обратным холодильником в течение 2 часа. Смесь охлаждали и растворитель удаляли в вакууме. Остаток растворяли в воде (100 мл), промывали CH2Cl2 (3×100 мл), подкисляли (HCl) и экстрагировали этилацетатом (3×75 мл). Объединенные органические экстракты промывали водой (3×75 мл), сушили (MgSO4) и упаривали, получая 0,86 г 3-(бензофуран-2-илметокси)фталевой кислоты с выходом 57% на две стадии; 1H ЯМР (DMSO-d 6) δ 5,35 (с, 2H), 7,04 (с, 1H), 7,22-7,37 (м, 2H), 7,45-7,67 (м, 5H).
Стадия 4:
Смесь 3-(бензофуран-2-илметокси)фталевой кислоты (0,55 г, 1,8 ммоль) и гидрохлорида рацемического α-аминоглутаримида (0,30 г, 1,8 ммоль) в пиридине (10 мл) кипятили с обратным холодильником в течение 16 часов. Смесь охлаждали и упаривали в вакууме. Остаток растворяли в CH2Cl2 (100 мл), промывали разбавленной водной HCl (2×100 мл) и водой (2×100 мл) и упаривали. Остаток хроматографировали, применяя градиент CH2Cl2-метанол, заявленное в заголовке соединение элюировалось со смесью 95:5 CH2Cl2-метанол, 0,46 г с выходом 65%; т.пл. 234-236°C; ВЭЖХ, Waters Symmetry C-18, 3,9×150 мм, 5 мкм, 1 мл/мин, 240 нм, 50/50 CH3CN/0,1% H3PO4, 4,16 (98,58%); 1H ЯМР (DMSO-d 6) δ 1,99-2,04 (м, 1H), 2,43-2,61 (м, 2H), 2,81-2,95 (м, 1H), 5,08 (дд, J=12,7 Гц, J=5,3 Гц, 1H), 5,55 (с, 2H), 7,14 (с, 1H), 7,23-7,37 (м, 2H), 7,50 (д, J=7,0 Гц, 1H), 7,60 (д, J=8,1 Гц, 1H), 7,68 (д, J=7,5 Гц, 1H), 7,74 (д, J=8,5 Гц, 1H), 7,86 (т, J=7,8 Гц, 1H), 1 1,11 (с, 1H); 13C ЯМР (DMSO-d 6) δ 23,9, 30,9, 48,8, 63,1, 107,4, 11 1,3, 115,9, 116,7, 120,2, 121,6, 123,1, 125,0, 127,5, 133,3, 137,0, 152,0, 154,6, 155,0, 165,2, 166,7, 169,9, 172,7; Анал. рассчитанное для C22H16N2O6 0,15 H2O: C, 64,91; H, 4,04; N, 6,88. Найденное: C, 64,89; H, 3,99; N, 6,84.
Пример 18
4-(Бензо[b]тиофен-2-илметокси)-2-(2,6-диоксопиперидин-3-ил)изоиндол-1,3-дион
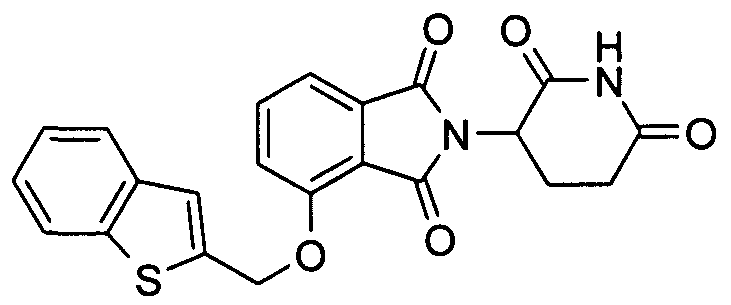
Стадия 1:
Смесь PPh3 на полимерной подложке (3,1 г, ~ 9,5 ммоль) и 1-бензотиофен-2-илметанола (1,0 г, 6,1 ммоль) в 20 мл THF охлаждали до 0°C в атмосфере N2. Добавляли по каплям диизопропилазодикарбоксилат (1,9 г, 9,5 ммоль) и затем добавляли в виде твердого остатка диметиловый эфир 3-гидроксифталевой кислоты (1,0 г, 4,8 ммоль). Смесь перемешивали в течение дополнительного часа при 0°C и затем нагревали до комнатной температуры. После перемешивания в течение 16 часов смесь фильтровали. Фильтр промывали этилацетатом (25 мл) и объединенные фильтраты упаривали.
Стадия 2:
Неочищенный продукт из стадии 1 растворяли в смеси 3 N NaOH (50 мл) и этанола (100 мл) и полученный в результате раствор кипятили с обратным холодильником в течение 2 часа. Смесь охлаждали и растворитель удаляли в вакууме. Остаток растворяли в воде (100 мл), промывали CH2Cl2 (3×100 мл), подкисляли (HCl) и экстрагировали этилацетатом (3×75 мл). Объединенные органические экстракты промывали водой (3×75 мл), сушили (MgSO4) и упаривали.
Стадия 3:
Неочищенный продукт из стадии 2 и гидрохлорид рацемического α-аминоглутаримида (0,40 г, 2,5 ммоль) в пиридине (10 мл) кипятили с обратным холодильником в течение 16 часа. Смесь охлаждали и упаривали в вакууме. Остаток хроматографировали, применяя градиент CH2Cl2-метанол, продукт элюировался со смесью 95:5 CH2Cl2-метанол, давая 0,40 г с выходом 30% на 3 стадии; т.пл. 247-249°C; ВЭЖХ, Waters Symmetry C-18, 3,9×150 мм, 5 мкм, 1 мл/мин, 240 нм, 50/50 CH3CN/0,1% H3PO4, 5,68 (100,00%); 1H ЯМР (DMSO-d6) δ 2,01-2,06 (м, 1H), 2,44-2,61 (м, 2H), 2,82-2,96 (м, 1H), 5,10 (дд, J=12,6 Гц, J=5,3 Гц, 1H), 5,71 (с, 2H), 7,32-7,42 (м, 2H), 7,49 (д, J=7,1 Гц, 1H), 7,58 (с, 1H), 7,68 (д, J=8,5 Гц, 1H), 7,80-7,87 (м, 2H), 7,96 (д, J=8,2 Гц, 1H), 11,11 (с, 1H); 13C ЯМР (DMSO-d 6) δ 22,0, 30,9, 48,8, 66,1, 115,9, 116,9, 120,4, 122,6, 123,8, 123,9, 124,5, 124,7, 133,3, 136,9, 138,9, 139,5, 154,9, 165,2, 166,7, 169,9, 172,8; Анал. рассчитанное для C22H16N2O5S: C, 62,85; H, 3,84; N, 6,66. Найденное: C, 62,88; H, 3,46; N, 6,57.
Пример 19
2-(2,6-Диоксопиперидин-3-ил)-4-(фуран-2-илметокси)изоиндол-1,3-дион
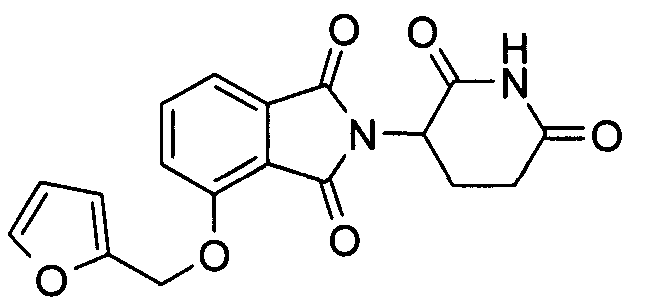
К раствору трифенилфосфина (630 мг, 2,4 ммоль) и фуран-2-илметанола (0,17 мл, 2,0 ммоль) в THF (10 мл) добавляли раствор DEAD (0,38 мл, 2,4 ммоль) в THF (0,6 мл) при 0°C. Через 5 мин добавляли к смеси 4-гидрокси-2-(2,6-диоксо(3-пиперидил))изоиндолин-1,3-дион (550 мг, 2,0 ммоль). Смесь нагревали до комнатной температуры и выдерживали в течение 4 часов. Растворитель удаляли в вакууме и остаток очищали колоночной хроматографией (силикагель) для того, чтобы получить масло. Масло суспендировали в метаноле (10 мл) в течение 3 часов для того, чтобы получить суспензию. Суспензию фильтровали и промывали метанолом (20 мл) для того, чтобы получить 2-(2,6-диоксопиперидин-3-ил)-4-(фуран-2-илметокси)изоиндол-1,3-дион в виде желтого твердого остатка (310 мг, выход 44%): т.пл. 184-186°C; 1H ЯМР (DMSO-d 6) δ 1,99-2,04 (м, 1H, CHH), 2,42-2,61 (м, 2Η, CH2), 2,80-2,95 (м, 1Η, CHH), 5,07 (дд, J=5, 13 Гц, 1H, NCH), 5,35 (с, 2Η, CH2), 6,49 (дд, J=2, 3 Гц, 1H, Ar), 6,68 (д, J=8 Гц, 1H, Ar), 7,47 (д, J=7 Гц, 1H, Ar), 7,68-7,71 (м, 2H, Ar), 7,83 (t J=8 Гц, 1H, Ar), 11,10 (с, 1H, NH); 13C ЯМР (DMSO-d 6) δ 21,96, 30,93, 48,77, 62,55, 110,72, 111,38, 115,72, 116,64, 120,35, 133,35, 136,89, 143,99, 149,15, 155,12, 165,21, 166,73, 169,88, 172,76; Анал. рассчитанное для C18H14N2O6 + 0,1 H2O: C, 60,71; H, 4,02; N, 7,87; H2O, 0,51. Найденное: C, 60,47; H, 3,97; N, 7,73; H2O, 0,38.
Пример 20
4-(3-Хлорбензо[b]тиофен-2-илметокси)-2-(2,6-диоксопиперидин-3-ил)изоиндол-1,3-дион
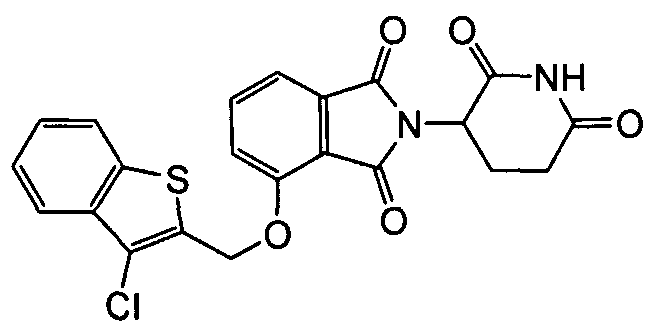
Стадия 1:
К раствору 3-хлорбензо[b]тиофен-2-карбоновой кислоты (3,5 г, 16,6 ммоль) в THF (40 мл) при 0°C добавляли по каплям через капельную воронку 1 M боргидрид в THF (33 мл, 33,2 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Реакцию прекращали добавлением по каплям воды (6 мл). Растворитель упаривали в вакууме и остаток распределяли между насыщ. карбонатом натрия и этилацетатом. Водную фазу экстрагировали этилацетатом (100 мл), объединенные органические фазы промывали водой (3×100 мл), сушили и концентрировали для того, чтобы получить (3-хлорбензо[b]тиофен-2-ил)метанол в виде светло-желтого твердого остатка (3,4 г, выход неочищенного продукта 103%); 1H ЯМР (DMSO-d 6) δ 4,8 (д, J=5,8 Гц, 2H, CH2OH), 5,87 (т, J=5,8 Гц, 1H, CH2OH), 7,42-8,04 (м, 4Η, Ar). Продукт применяли в следующей стадии без дополнительной очистки.
Стадия 2:
К перемешиваемой суспензии диметилового эфира 3-гидроксифталевой кислоты (1,0 г, 4,8 ммоль), (3-хлорбензо[b]тиофен-2-ил)метанола (1,9 г, 9,5 ммоль) и трифенилфосфина на полимерной подложке (3,0 г, 9,5 ммоль) в THF (30 мл) в ледяной бане медленно добавляли диизопропилазодикарбоксилат (1,9 мл, 9,5 ммоль) и перемешивали при комнатной температуре в течение ночи. Смесь фильтровали и твердый остаток промывали этилацетатом (10 мл). Фильтрат упаривали и остаток очищали флэш колоночной хроматографией (EtOAc/Гексан) для того, чтобы получить диметиловый эфир 3-(3-хлорбензо[b]тиофен-2-илметокси)фталевой кислоты (2,1 г, выход неочищенного продукта 109%). Продукт применяли в следующей стадии без дополнительной очистки.
Стадия 3:
Раствор диметилового эфира 3-(3-хлорбензо[b]тиофен-2-илметокси)фталевой кислоты (1,9 г, 4,8 ммоль) в спиртосодержащем реагенте (120 мл) и 3 N гидроксиде натрия (60 мл) кипятили с обратным холодильником в течение 2 часов. Раствор упаривали, остаток растворяли в воде (100 мл) и промывали хлористым метиленом (3×100 мл), затем, подкисляли до pH, приблизительно равного 4. Полученную в результате смесь экстрагировали этилацетатом (2×100 мл), объединенные органические слои промывали водой (2×100 мл), сушили и концентрировали для того, чтобы получить 3-(3-хлорбензо[b]тиофен-2-илметокси)фталевую кислоту в виде грязно-белого твердого остатка (1,6 г, выход 92%). Продукт применяли в следующей стадии без дополнительной очистки.
Стадия 4:
Смесь 3-(3-хлорбензо[b]тиофен-2-илметокси)фталевой кислоты (1,6 г, 4,4 ммоль), гидрохлорида альфа-аминоглутаримида (0,76 г, 4,6 ммоль) в пиридине кипятили с обратным холодильником в течение ночи. Смесь упаривали и остаток очищали флэш колоночной хроматографией (метанол/хлористый метилен) для того, чтобы получить 4-(3-хлорбензо[b]тиофен-2-илметокси)-2-(2,6-диоксопиперидин-3-ил)изоиндол-1,3-дион в виде белого твердого остатка (0,76 г, выход 38%); ВЭЖХ: Waters Symmetry C18, 5 мкм, 3,9×150 мм, 1 мл/мин, 240 нм, 60/40 CH3CN/0,1% H3PO4, 5,26 мин (98,7%); т.пл. 240-242°C; 1H ЯМР (DMSO-d 6) δ 2,02-2,06 (м, 1H, CHH), 2,54-2,62 (м, 2Η, CH2), 2,83-2,95 (м, 1Η, CHH), 5,10 (дд, J=6, 12 Гц, 1H, CH), 5,74 (с, 2Η, CH2), 7,49-8,10 (м, 7Η, Ar), 11,12 (с, 1H, NH); 13C ЯМР (DMSO-d 6) δ 21,94, 30,90, 48,79, 63,99, 116,26, 116,98, 118,36, 120,46, 121,39, 123,36, 125,57, 126,19, 132,93, 133,34, 135,42, 136,91, 137,04, 154,62, 165,05, 166,64, 169,85, 172,73. Анал. рассчитанное для C22H15N2O5SCl + 0,1 H2O: C, 58,09; H, 3,32; N, 6,16; S, 7,05; Cl, 7,79. Найденное: C, 57,77; H, 3,06; N, 6,08; S, 6,87; Cl, 8,05.
Пример 21
2-(2,6-Диоксопиперидин-3-ил)-4-(4-фторбензо[b]тиофен-2-илметокси)изоиндол-1,3-дион
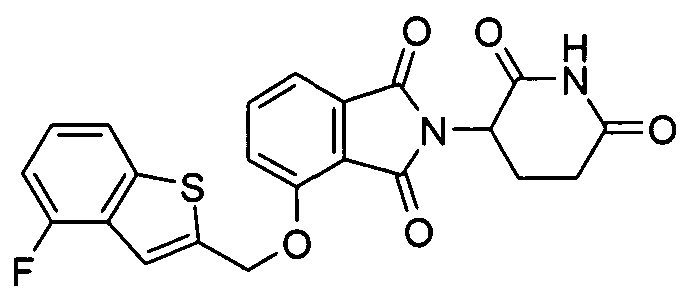
Стадия 1:
К перемешиваемой суспензии диметилового эфира 3-гидроксифталевой кислоты (1,1 г, 5,2 ммоль), (4-фторбензо[b]тиофен-2-ил)метанола (0,96 г, 10,5 ммоль) и трифенилфосфина на полимерной подложке (3,0 г, 10,5 ммоль) в THF (35 мл) в ледяной бане медленно добавляли диизопропилазодикарбоксилат (2,1 мл, 10,5 ммоль) и перемешивали при комнатной температуре в течение ночи. Смесь фильтровали и твердый остаток промывали этилацетатом (10 мл). Фильтрат упаривали и остаток очищали флэш колоночной хроматографией (EtOAc/Гексан) для того, чтобы получить диметиловый эфир 3-(4-фторбензо[b]тиофен-2-илметокси)фталевой кислоты (1,5 г, выход 76%). Продукт применяли в следующей стадии без дополнительной очистки.
Стадия 2:
Раствор диметилового эфира 3-(4-фторбензо[b]тиофен-2-илметокси)фталевой кислоты (1,5 г, 4,0 ммоль) в спиртосодержащем реагенте (120 мл) и 3 N гидроксиде натрия (60 мл) кипятили с обратным холодильником в течение 2 часов. Раствор упаривали, остаток растворяли в воде (100 мл) и промывали хлористым метиленом (3×100 мл), затем подкисляли до pH, приблизительно равного 4. Полученную в результате смесь экстрагировали этилацетатом (2×100 мл), объединенные органические слои промывали водой (2×100 мл), сушили и концентрировали для того, чтобы получить 3-(4-фторбензо[b]тиофен-2-илметокси)фталевую кислоту в виде грязно-белого твердого остатка (1,2 г, выход 84%). Продукт применяли в следующей стадии без дополнительной очистки.
Стадия 3:
Смесь 3-(4-фторбензо[b]тиофен-2-илметокси)фталевой кислоты (1,2 г, 3,4 ммоль), гидрохлорида альфа-аминоглутаримида (0,58 г, 3,6 ммоль) в пиридине кипятили с обратным холодильником в течение ночи. Смесь упаривали и остаток очищали флэш колоночной хроматографией (метанол/хлористый метилен) для того, чтобы получить 2-(2,6-диоксопиперидин-3-ил)-4-(4-фторбензо[b]тиофен-2-илметокси)изоиндол-1,3-дион в виде белого твердого остатка (0,66 г, выход 44%); ВЭЖХ: Waters Symmetry C18, 5 мкм, 3,9×150 мм, 1 мл/мин, 240 нм, 60/40 CH3CN/0,1% H3PO4, 3,08 мин (97,5%); т.пл. 264-266°C; 1H ЯМР (DMSO-d 6) δ 2,01-2,08 (м, 1H, CHH), 2,54-2,95 (м, 2Η, CHHCH2), 5,11 (дд, J=6, 12 Гц, 1H, CH), 5,73 (с, 2Η, CH2), 7,18-7,88 (м, 7H, Ar), 11,12 (с, 1H, NH); 13C ЯМР (DMSO-d 6) δ 21,98, 30,95, 48,82, 65,85, 109,57, 109,81, 116,04, 116,91, 118,02, 119,02, 119,07, 120,37, 125,98, 126,08, 127,39, 127,65, 133,38, 137,02, 140,81, 141,98, 142,06, 154,81, 155,10, 158,41, 165,17, 166,71, 169,91, 172,78. Анал. рассчитанное для C22H15N2O5SF: C, 60,27; H, 3,45; N, 6,39; S, 7,31; F, 4,33. Найденное: C, 60,40; H, 3,26; N, 6,29; S, 7,24; F, 4,32.
Пример 22
4-(5-Хлортиофен-2-илметокси)-2-(2,6-диоксопиперидин-3-ил)изоиндол-1,3-дион
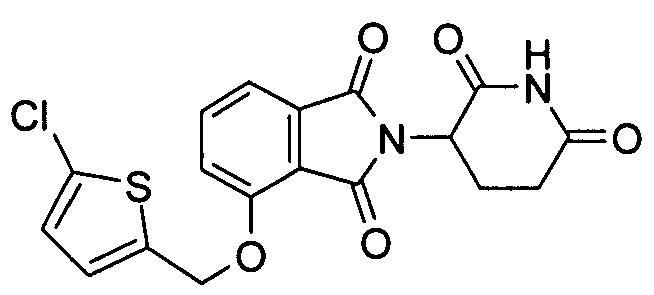
Стадия 1:
К перемешиваемой суспензии диметилового эфира 3-гидроксифталевой кислоты (1,4 г, 6,8 ммоль) в ацетоне (70 мл) и карбоната калия (2,8 г, 20 ммоль) добавляли 2-хлор-5-хлорметилтиофен (0,83 мл, 7,1 ммоль) и кипятили с обратным холодильником в течение 2 часов. Растворитель упаривали, остаток распределяли между водой (100 мл) и этилацетатом (150 мл) и промывали водой (2×100 мл). Объединенные органические фазы сушили, концентрировали и очищали флэш колоночной хроматографией (EtOAc/Гексан) для того, чтобы получить диметиловый эфир 3-(5-хлортиофен-2-илметокси)фталевой кислоты (2,3 г, выход неочищенного продукта 100%). Продукт применяли в следующей стадии без дополнительной очистки.
Стадия 2:
Раствор диметилового эфира 3-(5-хлортиофен-2-илметокси)фталевой кислоты (2,3 г, 6,7 ммоль) в спиртосодержащем реагенте (100 мл) и 3 N гидроксиде натрия (60 мл) кипятили с обратным холодильником в течение 2 часов. Раствор упаривали, остаток растворяли в воде (100 мл) и промывали хлористым метиленом (3×100 мл), затем подкисляли до pH, приблизительно равного 4. Полученную в результате смесь экстрагировали этилацетатом (2×100 мл), объединенные органические слои промывали водой (2×100 мл), сушили и концентрировали для того, чтобы получить 3-(5-хлортиофен-2-илметокси)фталевую кислоту в виде грязно-белого твердого остатка (1,6 г, выход 76%). Продукт применяли в следующей стадии без дополнительной очистки.
Стадия 3:
Смесь 3-(5-хлортиофен-2-илметокси)фталевой кислоты (1,6 г, 5,1 ммоль), гидрохлорида альфа-аминоглутаримида (0,88 г, 5,4 ммоль) в пиридине кипятили с обратным холодильником в течение ночи. Смесь упаривали и остаток очищали флэш колоночной хроматографией (метанол/хлористый метилен) для того, чтобы получить 4-(5-хлортиофен-2-илметокси)-2-(2,6-диоксопиперидин-3-ил)изоиндол-1,3-дион в виде белого твердого остатка (0,76 г, выход 36%); ВЭЖХ: Waters Symmetry C18, 5 мкм, 3,9×150 мм, 1 мл/мин, 240 нм, 60/40 CH3CN/0,1% H3PO4, 2,63 мин (99,3%); т.пл. 217-219°C; 1H ЯМР (DMSO-d 6) δ 2,01-2,07 (м, 1H, CHH), 2,54-2,57 (м, 2H, CH2), 2,62-2,95 (м, 1Η, CHH), 5,10 (дд, J=6, 12 Гц, 1H, CH), 5,50 (с, 2Η, CH2), 7,07-7,87 (м, 5Η, Ar), 11,11 (с, 1H, NH); 13C ЯМР (DMSO-d 6) δ 21,93, 30,90, 48,75, 65,38, 115,94, 116,80, 120,44, 126,55, 127,83, 129,09, 133,31, 136,93, 137,54, 154,76, 165,14, 166,68, 169,77, 169,86, 172,73. Анал. рассчитанное для C18H13N2O5SCl: C, 53,41; H, 3,24; N, 6,92; S, 7,92%; Cl, 8,76. Найденное: C, 53,39; H, 2,95; N, 6,80; S, 7,62%; Cl, 9,01.
Пример 23
2-(2,6-Диоксопиперидин-3-ил)-4-(1-нафталин-2-ил-этокси)изоиндол-1,3-дион
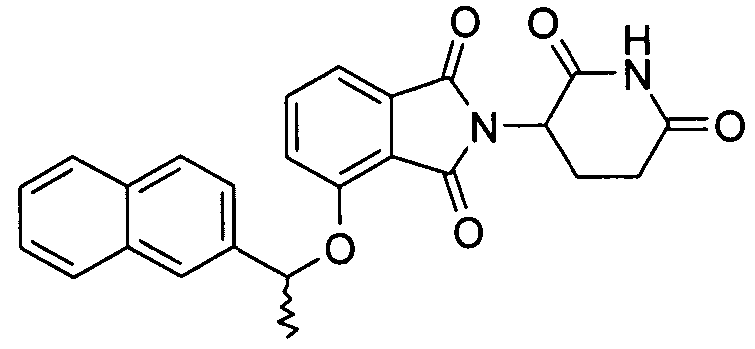
Стадия 1:
Смесь PPh3 на полимерной подложке (3,1 г, ~ 9,5 ммоль) и α-метил-2-нафталинметанола (0,82 г, 4,8 ммоль) в 20 мл THF охлаждали до 0°C в атмосфере N2. Добавляли по каплям диизопропилазодикарбоксилат (1,9 г, 9,5 ммоль) и затем добавляли в виде твердого остатка диметиловый эфир 3-гидроксифталевой кислоты (1,0 г, 4,8 ммоль). Смесь перемешивали в течение дополнительного часа при 0°C и затем нагревали до комнатной температуры. После перемешивания в течение 16 часов смесь фильтровали. Фильтр промывали этилацетатом (20 мл) и объединенные фильтраты упаривали. Остаток хроматографировали в градиенте этилацетата в гексане, 1,2 г диметилового эфира 3-(1-нафталин-2-ил-этокси)фталевой кислоты элюировались 20-30% этилацетата с выходом 66%; 1H ЯМР (DMSO-d 6) δ 1,70 (д, J=6,5 Гц, 3H), 3,88 (с, 3H), 4,03 (с, 3H), 5,49 (кв, J=6,5 Гц, 1H), 6,96 (д, J=8,4 Гц, 1H), 7,18 (т, J=8,0 Гц, 1H), 7,42-7,53 (м, 4H), 7,76-7,84 (м, 4H).
Стадия 2:
Смесь диметилового эфира 3-(1-нафталин-2-илэтокси)фталевой кислоты (0,9 г, 2,5 ммоль) и 3N NaOH (50 мл) в этаноле (100 мл) кипятили с обратным холодильником в течение 2 часа. Смесь охлаждали и растворитель удаляли в вакууме. Остаток растворяли в воде (100 мл), промывали CH2Cl2 (3×100 мл), подкисляли (HCl) и экстрагировали этилацетатом (3×75 мл). Объединенные органические экстракты промывали водой (3×75 мл), сушили (MgSO4) и упаривали, получая 0,50 г 3-(1-нафталин-2-илэтокси)фталевую кислоту с выходом 60%; 1H ЯМР (DMSO-d 6) δ 1,60 (д, J=6,2 Гц, 3H), 5,79 (кв, J=6,2 Гц, 1H), 7,21-7,31 (м, 2H), 7,38 (дд, J=7,1 Гц, J=1,3 Гц, 1H), 7,46-7,57 (м, 3H), 7,83-7,93 (м, 4H).
Стадия 3:
Смесь 3-(1-нафталин-2-илэтокси)фталевой кислоты (0,36 г, 1,0 ммоль) и гидрохлорида рацемического α-аминоглутаримида (0,16 г, 1,0 ммоль) в пиридине (10 мл) кипятили с обратным холодильником в течение 16 часов. Смесь охлаждали и упаривали в вакууме. Остаток растворяли в этилацетате (100 мл) и промывали разбавленной водной HCl (2×100 мл) и водой (100 мл) и упаривали. Остаток хроматографировали, применяя градиент CH2Cl2-метанол, продукт элюировался со смесью 95:5 CH2Cl2-метанол, 0,27 г с выходом 64%; т.пл. 174-176°C; ВЭЖХ, Waters Symmetry C-18, 3,9×150 мм, 5 мкм, 1 мл/мин, 240 нм, 60/40 CH3CN/0,1% H3PO4, 3,69 (99,65%); 1H ЯМР (DMSO-d 6) δ 1,71 (д, J=6,0 Гц, 3H), 1,99-2,09 (м, 1H), 2,51-2,65 (м, 2H), 2,84-2,97 (м, 1H), 5,13 (дд, J=12,5 Гц, J=5,3 Гц, 1H), 6,00 (кв, J=6,0 Гц, 1H), 7,39 (д, J=7,2 Гц, 1H), 7,44-7,53 (м, 3H), 7,62 (д, J=8,5 Гц, 1H), 7,69 (т, J=7,9 Гц, 1H), 7,87-7,96 (м, 3H), 8,00 (с, 1H), 11,15 (с, 1H); 13C ЯМР (DMSO-d 6) δ 22,0, 23,7, 31,0, 48,8, 76,5, 115,5, 117,2, 121,5, 123,6, 124,5, 126,2, 126,4, 127,6, 127,8, 128,5, 132,5, 132,7, 133,4, 136,7, 139,4, 154,8, 165,3, 166,7, 170,0, 172,8; Анал. рассчитанное для C25H20N2O5: C, 70,08; H, 4,71; N, 6,54. Найденное: C, 69,71; H, 4,51; N, 6,28.
Пример 24
2-(2,6-Диоксопиперидин-3-ил)-4-(4-метоксибензилокси)изоиндол-1,3-дион
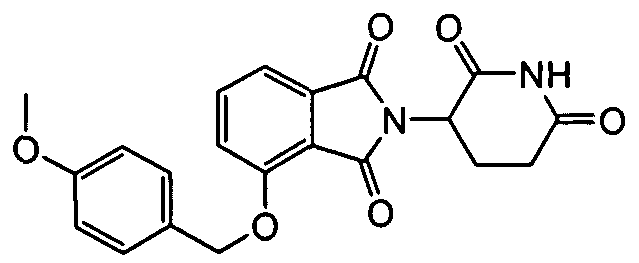
Стадия 1:
Перемешиваемую смесь 3-гидроксифталевого ангидрида (20,5 г, 125 ммоль) в метаноле (100 мл) кипятили с обратным холодильником в течение трех часов. Растворитель упаривали в вакууме и остаток суспендировали в бикарбонате натрия (29,4 г, 350 ммоль) в DMF (250 мл) с последующим добавлением йодметана (19 мл, 300 ммоль) и выдерживанием при 55°C в течение четырех часов. Смесь охлаждали до комнатной температуры, растворитель упаривали в вакууме и остаток распределяли между водой (200 мл) и этилацетатом (200 мл). Органический слой промывали водой (2×200 мл), сушили, концентрировали в вакууме и затем очищали флэш колоночной хроматографией (Silica Gel, EtOAc/Гексан, градиент 0%-100% 30 мин) для того, чтобы получить диметиловый эфир 3-гидроксифталевой кислоты (20,2 г, выход 77%). Продукт применяли в следующей стадии без дополнительной очистки.
Стадия 2:
К перемешиваемой суспензии диметилового эфира 3-гидроксифталевой кислоты (1,1 г, 5,2 ммоль) в ацетоне (45 мл) и карбоната калия (2,2 г, 15,7 ммоль) добавляли 1-бромметил-4-метоксибензол (0,79 мл, 5,5 ммоль). Смесь кипятили с обратным холодильником в течение шести часов. Растворитель упаривали в вакууме и остаток распределяли между водой (50 мл) и этилацетатом (80 мл). Органический слой промывали водой (2×50 мл), сушили, концентрировали в вакууме и очищали флэш колоночной хроматографией (Silica Gel, EtOAc/Гексан, градиент 0%-100% 30 мин) для того, чтобы получить диметиловый эфир 3-(4-метоксибензилокси)фталевой кислоты (2,0 г, выход неочищенного продукта 115%). Продукт применяли в следующей стадии без дополнительной очистки.
Стадия 3:
Перемешиваемый раствор диметилового эфира 3-(4-метоксибензилокси)фталевой кислоты (2,0 г, неочищенный, 5,5 ммоль) в спиртосодержащем реагенте (100 мл) и 3 N гидроксиде натрия (35 мл) кипятили с обратным холодильником в течение 2 часов. Раствор упаривали в вакууме, остаток растворяли в воде (80 мл) и промывали хлористым метиленом (3×70 мл), затем подкисляли HCl до pH, приблизительно равного 4. Полученную в результате смесь экстрагировали этилацетатом (2×60 мл), объединенные органические слои промывали водой (2×70 мл), сушили и концентрировали в вакууме для того, чтобы получить 3-(4-метоксибензилокси)фталевую кислоту в виде грязно-белого твердого остатка (1,6 г, выход неочищенного продукта 100%). Продукт применяли в следующей стадии без дополнительной очистки.
Стадия 4:
Перемешиваемую смесь 3-(4-метоксибензилокси)фталевой кислоты (1,5 г, 5,2 ммоль), гидрохлорида альфа-аминоглутаримида (0,90 г, 5,4 ммоль) в пиридине кипятили с обратным холодильником в течение ночи. Смесь упаривали в вакууме и остаток очищали флэш колоночной хроматографией (Silica Gel, метанол/хлористый метилен, градиент 0%-10%, 30 мин) для того, чтобы получить 2-(2,6-диоксопиперидин-3-ил)-4-(4-метоксибензилокси)изоиндол-1,3-дион в виде белого твердого остатка (0,83 г, выход 40%); ВЭЖХ: Waters Symmetry C18, 5 мкм, 3,9×150 мм, 1 мл/мин, 240 нм, 50/50 CH3CN/0,1% H3PO4, RT = 4,17 мин (98,6%); т.пл. 178-180°C; 1H ЯМР (DMSO-d 6) δ l,99-2,06 (м, 1H, CHH), 2,51-2,82 (м, 2Η, CHHCH2), 3,76 (с, 3Η, CH3), 5,08 (дд, J=6, 12 Гц, 1H, CH), 5,29 (с, 2Η, CH2), 6,95-7,84 (м, 7Η, Ar), 11,11 (с, 1H, NH); 13C ЯМР (DMSO-d6) δ 21,96, 30,91, 48,72, 55,08, 69,92, 113,88, 115,41, 116,55, 120,29, 127,93, 129,21, 133,25, 136,92, 155,57, 159,10, 165,28, 166,77, 169,90, 172,75. Анал. рассчитанное для C21Η18N2O6: C, 63,96; H, 4,60; N, 7,10. Найденное: C, 63,86; H, 4,30; N, 6,92.
5.1 АНАЛИЗЫ
5.1.1 Анализ на ингибирование TNFα в PMBC
Мононуклеарные клетки периферической крови (PBMC) от нормальных доноров получали Ficoll Hypaque (Pharmacia, Piscataway, NJ, USA) центрифугированием в градиенте плотности. Клетки выращивали в RPMI 1640 (Life Technologies, Grand Island, NY, USA), дополненой 10% AB + человеческой плазмой (Gemini Bio-products, Woodland, CA, USA), 2 мМ L-глютамином, 100 U/мл пинициллином, и 100 мкг/мл стрептомицином (Life Technologies).
PBMC (2×105 клеток) помещали в 96-луночные плоскодонные планшеты для тканевых культур фирмы Costar (Corning, NY, USA) в трех экземплярах. Клетки стимулировали липополисахаридами (LPS) (из Salmonella abortus equi, Sigma cat.no. L-1887, St.Louis, MO, USA) при конечной концентрации 1 нг/мл в отсутствие или в присутствии соединений. Соединения, относящиеся к настоящему изобретению, растворяли в DMSO (Sigma) и осуществляли дополнительное разбавление в культуральной среде непосредственно перед применением. Конечная DMSO концентрация во всех анализах может составлять, приблизительно, 0,25%. Соединения добавляли к клеткам за 1 час до LPS стимуляции. Затем клетки выдерживали в течение 18-20 часов при 37°C в 5% CO2, затем собирали кондиционированную среду, разбавляли культуральной средой и анализировали на TNFα концентрации с помощью ELISA (Endogen, Boston, MA, USA). IC50s рассчитывали, применяя нелинейный регрессионный, сигмоидальный дозозависимый эффект, заключенный между верхним значением, составляющим 100%, и нижним 0% значением, допуская переменный наклон (GraphPad Prism v3.02).
5.1.2 Продуцирование IL-2 и MIP-3α T-клетами
PBMC очищали от прилипших моноцитов помещением 1×108 PBMC в 10 мл полной среды (RPMI 1640, дополненной 10% фетальной бычей сывороткой, инактивированной нагреванием, 2 мМ L-глютамином, 100 U/мл пинициллином, и 100 мкг/мл стрептомицином) в 10 см чашке Петри, в 37°C, 5% CO2 инкубаторе в течение 30-60 минут. Чашку промывали средой для того, чтобы удалить все неприлипшие PBMC. T-клетки очищали негативным отбором, применяя следующую смесь антител (Pharmingen) и Dynabead (Dynal) для каждых 1×108 неприлипших PBMC: 0,3 мл овечьи антимышиные IgG гранулы, 15 мкл анти-CD16, 15 мкл анти-CD33, 15 мкл анти-CD56, 0,23 мл анти-CD 19 гранулы, 0,23 мл анти-HLA гранулы класса II и 56 мкл анти-CD 14 гранулы. Клетки и смесь гранул/антител вращали с донышка на крышку в течение 30-60 минут при 4°C. Очищенные T клетки удаляли от гранул, применяя Dynal магнит. Средний выход составлял, приблизительно, 50% T-клеток, 87-95% CD3+, установленный проточной цитометрией.
96-луночные плоскодонные планшеты для тканевых культур покрывали анти-CD3 антителами OKT3 с концентрацией 5 мкг/мл в PBS, 100 мкл на лунку, выдерживали при 37°C в течение 3-6 часов, затем промывали четыре раза полной средой 100 мкл/лунка непосредственно перед добавлением T-клеток. Соединения разбавляли в 20 раз до конечной концентрации в 96-луночном круглодонном планшете для тканевых культур. Конечные концентрации составляли, приблизительно, от 10 мкм до, приблизительно, 0,00064 мкм. 10 мМ стоковый раствор соединений, относящихся к настоящему изобретению, разбавляли 1:50 в полной среде для первого 20× разбавления 200 мкм в 2% DMSO и последовательно разбавляли 1:5 в 2% DMSO. Соединение добавляли в количестве 10 мкл на 200 мкл культуры, для того, чтобы получить конечную DMSO концентрацию, равную 0,1%. Культуры выдерживали при 37°C, 5% CO2 в течение 2-3 дней, и кондиционные среды анализировали на IL-2 и MIP-3α с помощью ELISA (R&D Systems). Концентрации IL-2 и MIP-3α нормализовали по отношению к количеству, продуцированному в присутствии определенного количества соединения, относящегося к настоящему изобретению, и EC50S рассчитывали, применяя нелинейный регрессионный, сигмоидальный дозозависимый эффект, заключенный между верхним значением, составляющим 100%, и нижним 0% значением, допуская переменный наклон (GraphPad Prism v3.02).
5.1.3 Исследование пролиферации клеток
Клеточные линии Namalwa, MUTZ-5 и UT-7 получали у Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (Braunschweig, Germany). Клеточную линию KG-I получали у American Type Culture Collection (Manassas, VA, USA). Клеточную пролиферацию, регистрируемую введением 3H-тимидина, измеряли во всех клеточных линиях следующим образом.
Клетки помещали в 96-луночные планшеты в количестве 6000 клеток на лунку в среде. Клетки предварительно обрабатывали соединениями с концентрацией, приблизительно, 100, 10, 1, 0,1, 0,01, 0,001, 0,0001 и 0 мкм в конечной концентрации, приблизительно, 0,25% DMSO в трех экземплярах при 37°C во влажной камере при 5% CO2 в течение 72 часов. Затем в каждую лунку добавляли один микрокюри 3H-тимидина (Amersham) и клетки выдерживали снова при 37°C во влажной камере при 5% CO2 в течение 6 часов. Клетки собирали на UniFilter GF/C фильтровальные пластины (Perkin Elmer), применяя сборщик клеток (Tomtec), и пластины сушили в течение ночи. Добавляли Microscint 20 (Packard) (25 мкл/лунка), и пластинки анализировали в TopCount NXT (Packard). Радиоактивность каждой пластинки считали в течение одной минуты. Процент ингибирования пролиферации клеток рассчитывали усреднением всех трех экземпляров и нормализацией по отношению к DMSO контролю (0% ингибирование). Каждое соединение исследовали в каждой клеточной линии в трех отдельных экспериментах. Конечную IC50s рассчитывали, применяя нелинейный регрессионный, сигмоидальный дозозависимый эффект, заключенный между верхним значением, составляющим 100%, и нижним 0% значением, допуская переменный наклон (GraphPad Prism v3.02).
5.1.4 Иммунопреципитация и иммуноблот
Namalwa клетки обрабатывали DMSO или определенным количеством соединения, относящегося к настоящему изобретению, в течение 1 часа, затем, стимулировали 10 U/мл Epo (R&D Systems) в течение 30 минут. Получали клеточные лизаты и или иммунопреципитировали с Epo рецептором Ab или незамедлительно отделяли SDS-PAGE. Иммуноблоты исследовали Akt, фосфо-Akt (Ser473 или Thr308), фосфо-Gabl (Y627), Gabl, IRS2, актином и IRF-I Abs и анализировали на Storm 860 Imager, применяя програмное обеспечение ImageQuant (Molecular Dynamics).
5.1.5 Анализ клеточного цикла
Клетки обрабатывали DMSO или определенным количеством соединения, относящегося к настоящему изобретению, в течение ночи. Окрашивание пропидий йодидом для клеточного цикла осуществляли, применяя CycleTEST PLUS (Becton Dickinson) согласно протоколу производителя. После прокрашивания клетки анализировали FACSCalibur проточной цитометрией, применяя програмное обеспечение ModFit LT software (Becton Dickinson).
5.1.6 Исследование апоптоза
Клетки обрабатывали DMSO или определенным количеством соединения, относящегося к настоящему изобретению, в течение различных периодов времени, затем, промывали аннексин-V промывочным буфером (BD Biosciences). Клетки выдерживали с белком, связывающим аннексин-V, и пропидий йодидом (BD Biosciences) в течение 10 минут. Образцы анализировали, применяя проточную цитометрию.
5.1.7 Люциферазный анализ
Namalwa клетки трансфецировали с 4 мкг AP1-люциферазы (Stratagene) на 1×106 клеток и 3 мкл Lipofectamin 2000 (Invitrogen) реагентом согласно инструкции изготовителя. Через шесть часов после трансфекции клетки обрабатывали DMSO или определенным количеством соединения, относящегося к настоящему изобретению. Люциферазную активность анализировали, применяя люциферазный лизирующий буфер и субстрат (Promega), и измеряли, применяя люминометр (Turner Designs).
5.1.8 Ингибирование TNFα и продуцирование IL-2
Применяя методики, в значительной степени аналогичные методикам, представленным в параграфе 5.1.1 выше, определяли точные IC50 величины соединений, относящихся к настоящему изобретению, для TNFα ингибирования. Определенные IC50 величины изменялись от меньше чем 0,2 нМ до, приблизительно, 10-100 мкм. Данные результаты показывают, что соединения, относящиеся к настоящему изобретению, являются пригодными в качестве ингибиторов TNFα.
Применяя методики, в значительной степени аналогичные методикам, представленным в параграфе 5.1.2, выше, также определяли точные EC50 величины соединений, относящихся к настоящему изобретению, для продуцирования IL-2. Определенные EC50 величины изменялись от, больше, чем 1 нМ до, меньше, чем 1 мкм. Данные результаты показывают, что соединения, относящиеся к настоящему изобретению, являются пригодными в качестве стимуляторов продуцирования IL-2.
Предполагается, что варианты осуществления, описанные выше служат просто в качестве примеров, и специалистам в данной области техники будет ясно, или они будут способны определить, применяя, всего-навсего, стандартные эксперименты, многочисленные эквиваленты заданных соединений, материалов и методик. Считают, что все данные эквиваленты включены в объем заявленного изобретения и включены в прилагаемую формулу изобретения.
Все патенты, патентные заявки и публикации, на которые ссылаются в настоящем изобретении, включены польностью в настоящее изобретение. Цитирование или указание любой ссылки в настоящей заявке не является признанием того, что данная ссылка является имеющейся в наличии в качестве известного уровня техники к заявленному изобретению. Полный объем изобретения будет лучше понят на основании прилагаемой формулы изобретения.
Изобретение относится к соединениям общей формулы
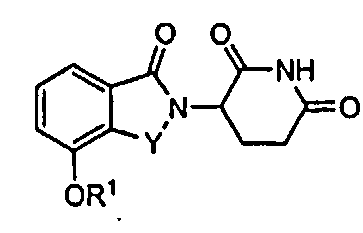
и их фармацевтически приемлемым солям, обладающим свойствами ингибитора TNFα и стимулятора продукции IL-2, а также к фармацевтической композиции, способу лечения и единичной стандартной лекарственной форме и их использованием. В общей формуле Y представляет С=O или СH2; и R1 представляет (С1-С6)алкил, замещенный (С1-С6)алкокси группой, 5-10-членный арил, (5-10-членный арил)-(С1-С6)алкил, 5-10-членный гетероциклил, 5-10-членный гетероциклил-(С1-С6)алкил, 5-10-членный гетероарил, (5-10-членный гетероарил)-(С1-С6)алкил, (C1-С6)алкилкарбонил, ди(С1-С6)алкиламинокарбонил, (С1-С6)алкоксикарбонил, 5-10-членный гетероарилкарбонил или 5-10-членный гетероциклилкарбонил; где гетероциклил содержит 1-3 гетероатома, выбранные из N, S и О, и гетероарил содержит 1-3 гетероатома, выбранные из N, S и О, и где R1 не обязательно замещен одной или более группами, выбранными из (С1-С6)алкокси, гало, (С1-С6)алкила, карбокси, (С1-С6)алкиламинокарбонила и (С1-С6)алкоксикарбонила. 4 н.э., 19 з. п-та ф-лы, 1 табл., 24 пр.
1. Соединение формулы:
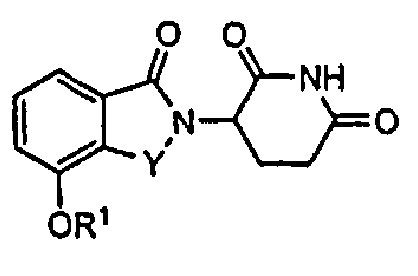
или его фармацевтически приемлемая соль, в котором:
Y представляет С=O или СН2; и
R1 представляет (С1-С6)алкил, замещенный (С1-С6)алкокси группой, 5-10-членный арил, (5-10-членный арил)-(С1-С6)алкил, 5-10-членный гетероциклил, 5-10-членный гетероциклил-(С1-С6)алкил, 5-10-членный гетероарил, (5-10-членный гетероарил)-(С1-С6)алкил, (С1-С6)алкилкарбонил, ди(С1-С6)алкиламинокарбонил, (С1-С6)алкоксикарбонил, 5-10-членный гетероарилкарбонил или 5-10-членный гетероциклилкарбонил; где гетероциклил содержит 1-3 гетероатома, выбранные из N, S и О, и гетероарил содержит 1-3 гетероатома, выбранные из N, S и О, и где R1 необязательно замещен одной или более группами, выбранными из (С1-С6)алкокси, гало, (С1-С6)алкила, карбокси, (С1-С6)алкиламинокарбонила и (С1-С6)алкоксикарбонила.
2. Соединение по п.1, в котором Y представляет С=O.
3. Соединение по п.1, в котором Y представляет СН2.
4. Соединение по п.1, в котором R1 представляет 5-10-членный арил, (5-10-членный арил)-(С1-С6)алкил или (5-10-членный гетероарил)-(С1-С6)алкил.
5. Соединение по любому из пп.1-4, в котором арильная группа в R1 представляет 5- или 6-членную моноциклическую арильную группу.
6. Соединение по п.1, в котором арильная группа в R1 представляет 5- или 6-членную моноциклическую гетероарильную группу, содержащую 1-3 гетероатомов, выбранных из О, N и S.
7. Соединение по п.1, в котором арильная группа в R1 представляет бициклическую арильную или бициклическую гетероарильную группу.
8. Соединение по п.1, в котором R1 замещен одним или более заместителями, выбранными из (С1-С6)алкокси, гало и (С1-С6)алкила.
9. Соединение по п.1, в котором R1 замещен одним или двумя заместителями, выбранными из метокси, хлора, брома, фтора и метила.
10. Соединение по п.1, в котором R1 представляет собой фенил, бензил, нафтилметил, хинолилметил, бензофурилметил, бензотиенилметил, фурилметил или тиенилметил.
11. Соединение по п.1, в котором R1 представляет собой 2-метоксифенил, бензил, 3-хлорбензил, 4-хлорбензил, 3,4-дихлорбензил, 3,5-дихлорбензил, 3-фторбензил, 3-бромбензил, 3-метилбензил, 4-метилсульфонилбензил, 3-метоксибензил, нафтилметил, 3-хинолилметил, 2-хинолилметил, 2-бензофурилметил, 2-бензотиенилметил, 3-хлортиен-2-илметил, 4-фторбензотиен-2-илметил, 2-фурилметил, 5-хлортиен-2-илметил или 1-нафт-2-илэтил.
12. Соединение по п.1, в котором соединение имеет формулу:
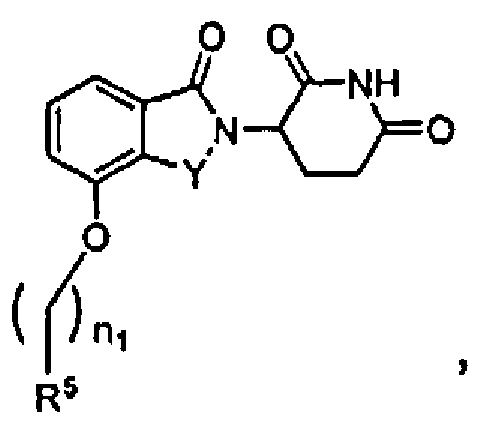
и в котором R5 представляет собой 5-10-членный арил или 5-10-членный гетероарил, необязательно замещенные одной, двумя или тремя группами, выбранными из (С1-С6)алкила, гало, (С1-С6)алкокси, карбокси, (С1-С6)алкиламинокарбонила и (С1-С6)алкоксикарбонила; и n1 равно 0-5.
13. Соединение по п.12, в котором R5 выбирают из фенила, нафтила, фурила, тиенила, бензофурила, бензотиенила и хинолила, необязательно замещенных одной или двумя группами, выбранными из метила, метокси, хлора, фтора и брома.
14. Соединение по п.1, в котором соединение имеет формулу
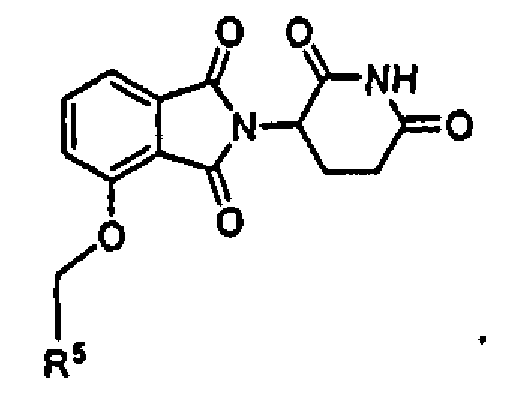
15. Соединение по п.1, выбранное из:
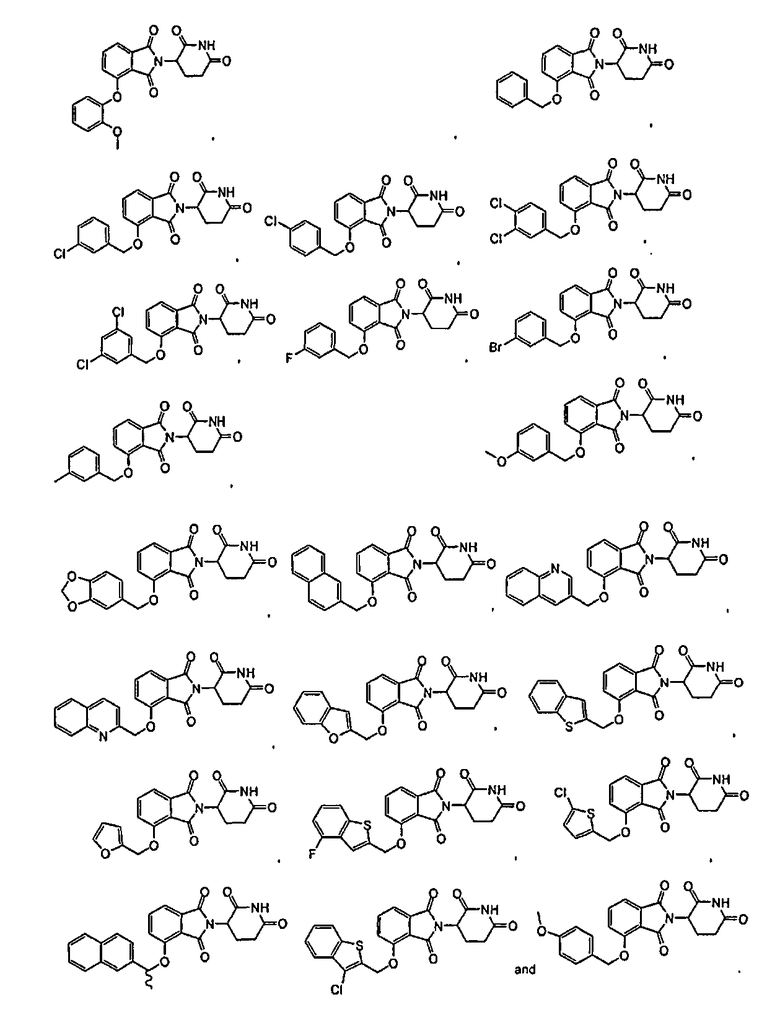
или его фармацевтически приемлемая соль.
16. Соединение по п.1, выбранное из:
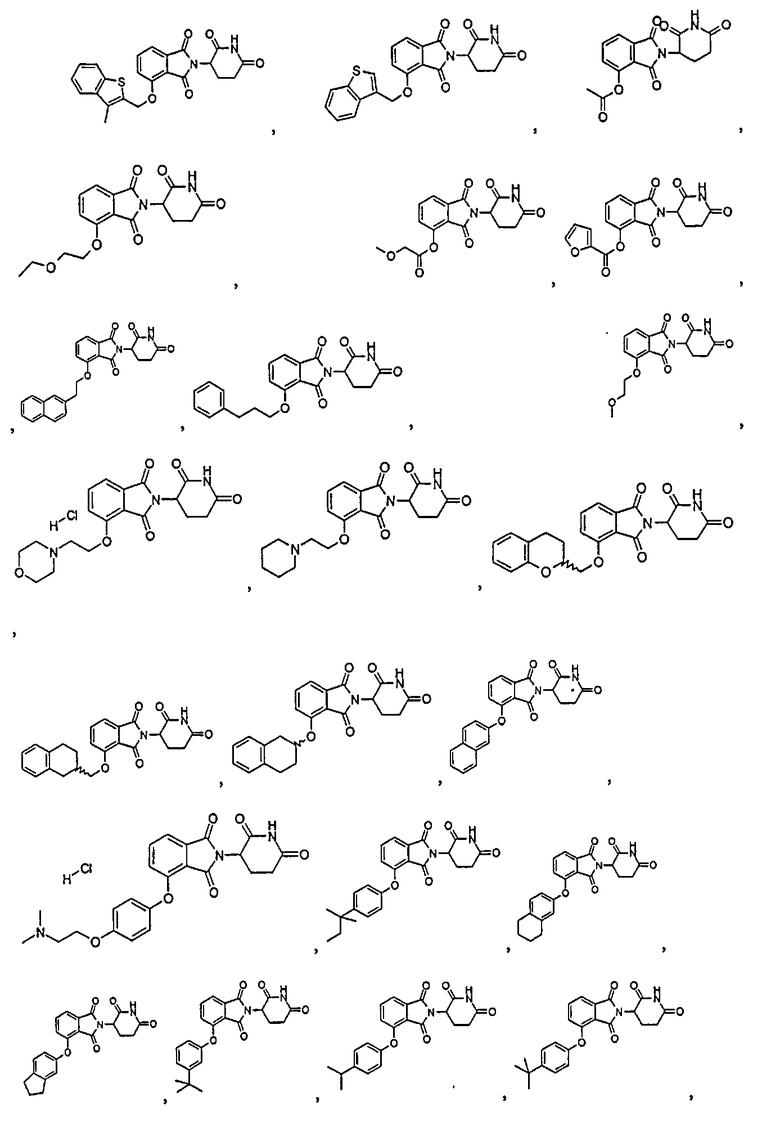
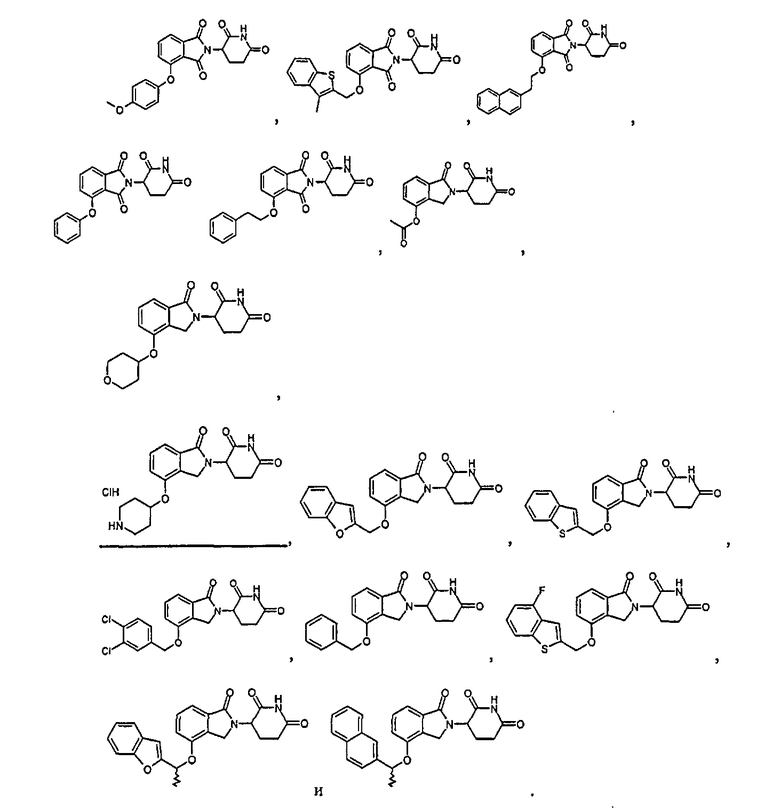
17. Фармацевтическая композиция, ингибирующая TNFα и стимулирующая продукцию IL-2, содержащая соединение по любому одному из пп.1-16 или его фармацевтически приемлемую соль.
18. Способ лечения или предотвращения заболевания или расстройства, включающий введение пациенту соединения по любому одному из пп.1-16 или его фармацевтически приемлемой соли, где заболевание или расстройство является раком, болью, дегенерацией желтого пятна или связанным с ней синдромом, заболеванием кожи, болезнью легких, расстройством, связанным с асбестом, паразитарным заболеванием, расстройством, связанным с иммунодефицитом, расстройством ЦНС, повреждением ЦНС, атеросклерозом или связанным с ним расстройством, дисфункциональным сном или связанным с ним расстройством, инфекционным заболеванием, гемоглобинопатией или связанным с ней расстройством.
19. Способ по п.18, в котором соединение или его фармацевтически приемлемую соль вводят перорально или парентерально.
20. Единичная стандартная лекарственная форма, содержащая соединение по любому одному из пп.1-16, или его фармацевтически приемлемую соль.
21. Единичная стандартная лекарственная форма по п.20, которая является подходящей для перорального или парентерального введения.
22. Единичная стандартная лекарственная форма по п.21, которая является подходящей для перорального введения.
23. Единичная стандартная лекарственная форма по п.22, которая является таблеткой или капсулой.
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| WEINZ С et al, JOURNAL OF CHROMATOGRAPHY В: BIOMEDICAL APPLICATIONS, 674(2), 1995, p.287-292 | |||
| MARKS G.M | |||
| et al, BIOL | |||
| PHARM | |||
| BULL | |||
| Видоизменение пишущей машины для тюркско-арабского шрифта | 1923 |
|
SU25A1 |
| SHOJI A et al, JOURNAL OF AMERICAN CHEMICAL SOCIETY, 129(5), 2007, p.1456-1464. | |||

