Изобретение относится к области органической химии и медицины, а именно к новым пиримидиновым производным бензофенона, обладающим высокой анти-ВИЧ-1 активностью, которые могут быть использованы в качестве ненуклеозидных ингибиторов вирусной репродукции в комплексной терапии СПИД и ВИЧ-1 инфекции.
Пандемия ВИЧ-1 инфекции является самой актуальной и до сих пор не решенной проблемой мирового здравоохранения. По данным Федерального научно-методического центра по профилактике и борьбе со СПИДом, по состоянию на 18 мая 2012 г. в России было зарегистрировано 664976 ВИЧ-инфицированных. Только за первые 3 месяца 2012 года было выявлено 14876 новых случаев ВИЧ-инфекции, а всего за время наблюдения в России от СПИД и оппортунистических инфекций погибло более 85 тысяч человек.
Основу современных методов лечения ВИЧ-1 инфекции составляет высокоактивная антиретровирусная терапия, использующая комбинацию противовирусных препаратов с различным механизмом действия. Неотъемлемой частью этой комбинации являются ненуклеозидные ингибиторы обратной транскриптазы ВИЧ-1 (ННИОТ), которые связываются с аллостерическим сайтом фермента вблизи полимеразного центра и блокируют его [Prajapati D.G., Ramajayam R., Yadav M., Giridhar R. The search for potent, small molecule NNRTIs // Bioorg. Med. Chem. - 2009. - Vol.17. - P.5744-5762]. В настоящее время в клинической практике используется 5 ННИОТ: невирапин, делавирдин, эфавиренц, этравирин и рилпивирин. Несмотря на высокую активность и селективность действия указанных ННИОТ, основным их недостатком, помимо относительно высокой токсичности и ряда побочных эффектов, является быстрое возникновение резистентных штаммов ВИЧ-1, что снижает эффективность дальнейшей терапии этими препаратами. В связи с этим, в мире постоянно ведется поиск новых ННИОТ, активных не только в отношении дикого штамма ВИЧ-1, но и его лекарственно-устойчивых форм [Bollini M., Domaoal R.A., Thakur V.V., Gallardo-Macias R., Spasov K.A., Anderson K.S., Jorgensen W.L. Computationally-guided optimization of a docking hit to yield catechol diethers as potent anti-HIV agents // J. Med. Chem. - 2011. - Vol.54. - P.8582-8591].
Среди большого количества органических веществ гетероциклической природы, синтезированных в качестве потенциальных ННИОТ, наиболее близкими по химическому строению к предлагаемым соединениям являются 1-[2-(2-бензоилфенокси)этил]урацилы [Novikov M.S., Ivanova O.N., Ivanov A.V., Valuev-Elliston V.T., Tembumikar K., Ozerov A.A., Gurskaya G.V., Kochetkov S.N., Pannecouque C., Balzarini J., Seley-Radtke K.S. 1-[2-(2-Benzoyl-and 2-benzylphenoxy)ethyl]uracils as potent anti-HIV-1 agents // Bioorg. Med. Chem. - 2011. - Vol.19. - P.5794-5902; Заявка на изобретение №2011148488 (072724) от 30.11.2011 г. // Ненуклеозидные ингибиторы обратной транскриптазы ВИЧ-1 - производные пиримидинового ряда] общей формулы:
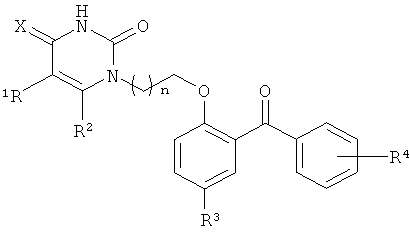
где: n=1-3; X=O, NH; R1=H, алкил C1-С6, бензил, галоген, NH2, морфолино, пиперидино, ариламино; R2=H, алкил С1-С6;
R3=алкил C1-С6, алкокси С1-С6, галоген, CN; R4=H, алкил С1-С6, CN.
Указанные соединения-аналоги обладают выраженной активностью в отношении ВИЧ-1 in vitro в наномолярном диапазоне концентраций и имеют доказанный механизм действия, основанный на прямом ингибировании ОТ ВИЧ-1, в том числе и ее мутантных форм. Однако метод получения этих соединений, основанный на первоначальном O-ацилировании фенолов ароматическими кислотами и последующей перегруппировке по Фрису, не позволяет получать производные, которые бы не содержали в n-положении фенольного фрагмента каких-либо заместителей (во всех соединениях-аналогах R3≠H).
Целью изобретения является получение новых ненуклеозидных ингибиторов обратной транскриптазы ВИЧ-1, обладающих повышенной анти-ВИЧ-1 активностью.
Сущность изобретения заключается в синтезе новых пиримидиновых производных бензофенона, не имеющих дополнительных заместителей в ароматических ядрах, отвечающих общей формуле:
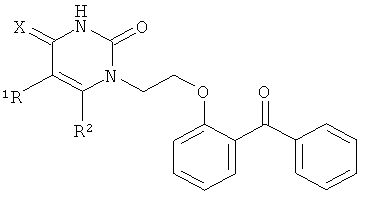
где: R1=H, CH3, F, Br; R2=H, CH3.
Наличие заместителя R3 в n-положении фенольного фрагмента у соединений-аналогов, вероятно, препятствует их связыванию с гидрофобным карманом обратной транскриптазы ВИЧ-1, который расположен на расстоянии 10 А от полимеразного сайта фермента, в связи с чем предлагаемые соединения, не имеющие таких заместителей, обладают более высокой противовирусной активностью. Кроме того, отсутствие дополнительных гидрофобных заместителей во фрагменте бензофенона у новых соединений повышает их растворимость в воде по сравнению с соединениями-аналогами, что в совокупности с меньшими размерами молекул может обеспечить улучшенные фармакокинетические параметры новых веществ.
Следующие примеры иллюстрируют сущность изобретения.
Пример 1. 1-[2-(2-Бензоилфенокси)этил]урацил (I).
(А) Хлорангидрид салициловой кислоты. К 20,0 мл (0,275 моль) тионилхлорида добавляют 1,0 мл (0,013 моль) диметилформамида и при интенсивном перемешивании при температуре 5-10°С порциями в течение 1 ч вносят 25,0 г (0,156 моль) тонкоизмельченного безводного салицилата натрия. Реакционную массу выдерживают в течение 1 сут. при комнатной температуре, избыток тионилхлорида отгоняют в вакууме водоструйного насоса при температуре бани не выше 60°С, к остатку добавляют 200 мл н-гексана, фильтруют, фильтрат упаривают в вакууме при температуре не выше 60°С и получают 19,5 г хлорангидрида салициловой кислоты в виде светло-желтой подвижной жидкости, выход 80%.
(Б) 2-Оксибензофенон. В трехгорлый реактор, снабженный обратным холодильником, термометром, капельной воронкой и эффективной мешалкой, помещают 100 мл (1,125 моль) бензола, 20,0 г (0,150 моль) безводного хлорида алюминия и при интенсивном перемешивании при температуре не выше 25°С добавляют в течение 30 мин раствор 10,0 г (0,064 моль) хлорангидрида салициловой кислоты в 25 мл бензола. Реакционную массу перемешивают при температуре 55-60°С в течение 2 ч, охлаждают до комнатной температуры и выливают при перемешивании в смесь 250 г воды со льдом. Добавляют 100 мл хлороформа, органический слой отделяют, промывают 5% раствором карбоната натрия, водой, сушат сульфатом магния, фильтруют и упаривают на кипящей водяной бане в вакууме водоструйного насоса. Остаток кристаллизуют из смеси н-гексан - диэтиловый эфир (9:1) и получают 9,9 г 2-оксибензофенона, Т.пл. 38-39°С (Т.пл. 38-40°С [Martin R. Hydroxybenzophenones // London: Springer, 2011. - 2913 p.]).
(В) 1-(2-Бензоилфенокси)-2-бромэтан. Смесь 7,5 г (0,038 моль) 2-оксибензофенона, 15,0 мл (0,174 моль) 1,2-дибромэтана, 7,7 г (0,054 моль) безводного карбоната калия, 0,5 г (0,001 моль) дибензо-18-краун-6 и 150 мл безводного ацетона кипятят в течение 1 сут., фильтруют и упаривают на кипящей водяной бане в вакууме водоструйного насоса. Остаток растворяют в 100 мл хлороформа, промывают 5% раствором едкого натра, водой, 5% раствором хлористоводородной кислоты, водой, сушат сульфатом магния, фильтруют и упаривают на кипящей водяной бане в вакууме водоструйного насоса. Получают 9,8 г 1-(2-бензоилфенокси)-2-бромэтана в виде светло-желтой вязкой жидкости, выход 85%. Спектр ПМР, δ, м.д.: 3,83 т (2Н, 4 Гц, CH2-Br); 4,42 т (2Н, 4 Гц, CH2-N); 7,06-7,70 м (9Н, арил).
(Г) 1-[2-(2-Бензоилфенокси)этил]урацил. 2,0 г (0,018 моль) урацила, 25 мл (0,120 моль) гексаметилдисилазана и 0,25 г (0,005 моль) хлорида аммония кипятят с защитой от влаги воздуха до полного растворения осадка (4 ч), избыток гексаметилдисилазана отгоняют на кипящей водяной бане при остаточном давлении не менее 10 мм рт.ст., к остатку добавляют 3,0 г (0,010 моль) 1-(2-бензоилфенокси)-2-бромэтана и нагревают при периодическом перемешивании при температуре 175-180°С в течение 4 ч. Реакционную массу охлаждают, растворяют в 25 мл этилацетата, добавляют 25 мл изопропилового спирта, через 30 мин выделившийся осадок отфильтровывают и фильтрат упаривают в вакууме. Остаток растворяют в 50 мл хлороформа, фильтруют, фильтрат упаривают в вакууме, остаток кристаллизуют из 50 мл изопропилового спирта и получают 1,9 г 1-[2-(2-бензоилфенокси)этил]урацила, Т.пл. 157-159,5°С, выход 57%.
Соединения II-IV получают аналогично. Выход и физико-химические свойства полученных веществ представлены в табл.1, параметры спектров ПМР - в табл.2. Спектры ПМР регистрировали на спектрометре Bruker AMXIII (400 МГц) в растворах диметилсульфоксида-D6, внутренний стандарт - тетраметилсилан. Температуры плавления определяли на приборе Mel-Temp 3.0 (Laboratory Devices Inc). Тонкослойную хроматографию выполняли на пластинах Sorbfil ПТСХ-АФ-А-УФ, подвижная фаза -изопропиловый спирт, проявление - в парах иода.
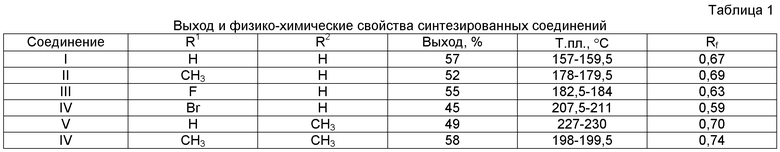
Пример 2. 1-[2-(2-Бензоилфенокси)этил]-6-метилурацил (V). Смесь 1,5 г (0,012 моль) 6-метилурацила, 2,5 г (0,018 моль) безводного карбоната калия и 40 мл безводного диметилформамида перемешивают при температуре 115-120°С в течение 1 ч, добавляют 2,5 г (0,008 моль) 1-(2-бензоилфенокси)-2-бромэтана и перемешивают при той же температуре в течение 6 ч. Реакционную массу охлаждают, фильтруют, фильтрат упаривают на кипящей водяной бане при остаточном давлении 4-5 мм рт.ст., остаток охлаждают, растирают с 25 мл воды и выдерживают при температуре 0-5°С в течение 1 сут. Закристаллизовавшийся остаток отфильтровывают, растворяют в 50 мл хлороформа, раствор фильтруют и упаривают в вакууме. Остаток кристаллизуют из 50 мл изопропилового спирта и получают 1,4 г 1-[2-(2-бензоилфенокси)этил]-6-метилурацила, Т.пл. 227-230°С, выход 49%. Соединение VI получают аналогично.

Пример 3. Исследование анти-ВИЧ-1 активности in vitro. CEM-SS-клетки в количестве 105 клеток/мл суспендировали в питательной среде RPMI-1640, дополнительно содержащей 2 микромоль/мл L-глутамина, 100 ЕД/мл бензилпенициллина и 100 мкг/мл стрептомицина, и инфицировали ВИЧ-1 (штамм HTLV-IIIB) при мультипликации инфекции, равной 0,2. Немедленно после инфицирования вирусом вносили растворы, содержащие различные концентрации исследуемого вещества в ДМСО (6 серийных разведении), и инкубировали в течение 6 сут. в среде 5% CO2 при температуре 37°С. По окончании инкубации количество живых клеток устанавливали спектрофотометрически при длине волны 450 и 650 нм с помощью красителя бромида 3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолия, при этом определяли ингибиторную концентрацию вещества, которая на 50% защищала CEM-SS-клетки от цитопатического эффекта ВИЧ-1 (ИК50). Цитотоксичность соединений изучали параллельно в неинфицированных культурах клеток, при этом определяли цитотоксическую концентрацию вещества, которая на 50% уменьшала количество живых CEM-SS-клеток (ЦК50). Расчетным путем определяли индекс селективности, являющийся отношением цитотоксической концентрации к ингибиторной концентрации: ИС=ЦК50/ИК50. Результаты исследований представлены в табл.3.
Как следует из данных табл.3, все новые соединения I-VI проявляют высокую активность в отношении ВИЧ-1 in vitro, в 2,5-37,5 раза превосходят невирапин, а самые активные соединения (V и VI) не уступают эфавиренцу или в 1,5 раза превосходят его. Кроме того, новые соединения по своей способности подавлять репродукцию ВИЧ-1 значительно превосходят соединения-аналоги, содержащие заместители в фенольном фрагменте (аналоги соединения I при R3=СН3, F и Br имеют величины ИК50 соответственно 0,310, 0,510 и 0,065 µМ). Новые соединения V и VI по уровню противовирусного действия in vitro в среднем в 5-10 раз превосходят самые активные соединения-аналоги, для которых величина ИК50 находится в пределах 0,016-0,020 µМ [Novikov M.S., Ivanova O.N., Ivanov A.V., Valuev-Elliston V.T., Tembumikar K., Ozerov A.A., Gurskaya G.V., Kochetkov S.N., Pannecouque C., Balzarini J., Seley-Radtke K.S. 1-[2-(2-Benzoyl-and 2-benzyl-phenoxy)ethyl]uracils as potent anti-HIV-1 agents // Bioorg. Med. Chem. - 2011. - Vol.19. - P.5794-5902]. При этом цитотоксических свойств у всех новых веществ во всем диапазоне исследованных концентраций (0,001-100 µМ) выявлено не было. Необходимо также отметить, что новые соединения, не содержащие дополнительных заместителей во фрагменте бензофенона, имеют значительно менее сложное химическое строение по сравнению с соединениями-аналогами, а способ их получения основывается на использовании доступных исходных веществ, отличается простотой и технологичностью.
Таким образом, получен ряд новых пиримидиновых производных бензофенона, обладающих повышенной активностью в отношении ВИЧ-1 in vitro по сравнению с известными ННИОТ и соединениями-аналогами.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ПОВЫШЕНИЯ РАСТВОРИМОСТИ ЛЕКАРСТВЕННОГО СРЕДСТВА НА ОСНОВЕ ПИРИМИДИНОВОГО ПРОИЗВОДНОГО БЕНЗОФЕНОНА | 2019 |
|
RU2729792C1 |
| СПОСОБ ПОЛУЧЕНИЯ МОНОЗАМЕЩЕННЫХ ПРОИЗВОДНЫХ УРАЦИЛА | 2017 |
|
RU2648998C1 |
| ПРОИЗВОДНЫЕ ПУРИНА, ОБЛАДАЮЩИЕ ПРОТИВОВИРУСНОЙ АКТИВНОСТЬЮ | 2003 |
|
RU2233842C1 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА, ОБЛАДАЮЩИЕ АНТИДЕПРЕССИВНОЙ, АНКСИОЛИТИЧЕСКОЙ И НООТРОПНОЙ АКТИВНОСТЬЮ | 2012 |
|
RU2507199C1 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА, ОБЛАДАЮЩИЕ НООТРОПНОЙ И АНТИГИПОКСИЧЕСКОЙ АКТИВНОСТЬЮ | 2012 |
|
RU2507198C1 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИН-4(3Н)-ОНА, ОБЛАДАЮЩИЕ НЕЙРО- И КАРДИОПРОТЕКТОРНОЙ АКТИВНОСТЬЮ | 2016 |
|
RU2622638C1 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИН-4(3Н)-ОНА, ИНГИБИРУЮЩИЕ НАТРИЙ-ВОДОРОДНЫЙ ОБМЕН | 2016 |
|
RU2654062C2 |
| СПОСОБ ПОЛУЧЕНИЯ 9-[2-(4-ИЗОПРОПИЛФЕНОКСИ)ЭТИЛ])АДЕНИНА | 2019 |
|
RU2714931C1 |
| Способ получения производных N-алкил- и N,N-диалкилизоцитозина | 2017 |
|
RU2664121C1 |
| 9-[2-(4-ИЗОПРОПИЛФЕНОКСИ)ЭТИЛ]АДЕНИН, ОБЛАДАЮЩИЙ АНТИДЕПРЕССАНТНЫМ И ПРОТИВОСТРЕССОРНЫМ ДЕЙСТВИЕМ | 2013 |
|
RU2529817C1 |
Изобретение относится к новым ненуклеозидным ингибиторам вирусной репродукции, представляющим собой пиримидиновые производные бензофенона: 1-[2-(2-бензоилфенокси)этил]урацилы, соответствующие структурной формуле:
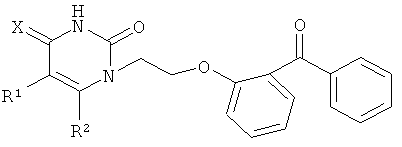
где: R1=H, СН3, F, Br; R2=H, СН3. Новые соединения не содержат дополнительных заместителей в ароматических ядрах бензофенонового фрагмента и проявляют анти-ВИЧ-1 активность in vitro, превосходящую активность невирапина и не уступающую активности эфавиренза. 3 табл., 3 пр.
Пиримидиновые производные бензофенона общей формулы
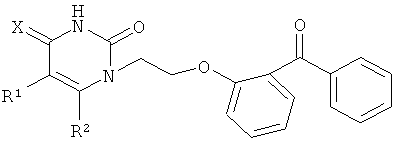
где R1=H, СН3, F, Br, R2=H, СН3,
обладающие анти-ВИЧ-1 активностью.
| BOLLINI MARIELA et al | |||
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| NOVIKOV MIKHAIL S | |||
| et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

