Область техники, к которой относится изобретение
Изобретение относится к способу получения материала для катализатора синтеза углеводородов в форме предшественника катализатора (или) катализатора синтеза углеводородов, предпочтительно предшественника катализатора и(или) катализатора синтеза углеводородов Фишера-Тропша. Изобретение также относится к применению предшественника катализатора и(или) катализатора, полученного способом согласно изобретению, в процессе синтеза углеводородов, предпочтительно в процессе синтеза углеводородов Фишера-Тропша.
Предпосылки создания изобретения
Процесс Фишера-Тропша (ФТ), который можно охарактеризовать как катализируемую неоднородной поверхностью реакцию полимеризации, обычно влечет за собой гидрогенизацию окиси углерода (как правило, двуокиси углерода, моноокиси углерода или их смеси) в присутствии катализатора на основе металлов группы VIII, таких как железо, кобальт и рутений. В зависимости от конкретных условий реакции ее продуктами могут являться водные, газообразные, жидкие и парафинистые углеводороды, которые могут быть насыщенными или ненасыщенными. Также могут образовываться оксигенаты углеводородов, такие как спирты, кислоты, кетоны и альдегиды. Распределение углеродного числа у продуктов реакции соответствует хорошо известному распределению Андерсона-Шульца-Флори.
Когда окисью углерода является моноокись углерода, реакция может быть представлена следующим уравнением:
nCO+2nH2→(CH2)n+nH2O
Такие неоднородные процессы Фишера-Тропша обычно именуют высокотемпературным процессом Фишера-Тропша (ВТФТ) или низкотемпературным процессом Фишера-Тропша (НТФТ).
ВТФТ-процесс обычно осуществляют при температурах от 250°С до 400°С, а в качестве катализатора обычно используют катализатор на основе плавленого железа, но также могут применяться катализаторы на основе осажденного железа. При указанных температурах в зоне реакции как реагенты, так и продукты реакции находятся в газообразной фазе, а с учетом того, что катализатор находится в твердой форме, процесс может именоваться двухфазной реакцией ФТ. В промышленном масштабе процесс обычно осуществляется в реакторе с псевдоожиженным слоем, а получаемые продукты имеют относительно высокое содержание олефинов и меньшую длину цепочек (то есть, как у продуктов, относящих к бензиновому и дизельному топливу), чем в случае НТФТ-процессов с использованием железного катализатора.
НТФТ-процесс обычно осуществляют при температурах от 180°С до 310°С, а в качестве катализатора обычно используют катализатор на основе кобальта, хотя также может использоваться катализатор на основе железа. В условиях, в которых осуществляется этот процесс, по меньшей мере один из продуктов в реакторе находится в жидкой фазе. С учетом того, что реагенты находятся в газообразной фазе, по меньшей мере, некоторые из продуктов находятся в жидкой фазе, а катализатор является твердым, этот процесс может именоваться трехфазным процессом. В промышленном масштабе процесс обычно осуществляется в реакторе с неподвижным слоем или реакторе с суспендированным слоем, а получаемыми продуктами являются более тяжелые углеводороды, такие как парафины. Этот процесс не может осуществляться в реакторе с псевдоожиженным слоем, поскольку под действием находящегося в жидкой фазе продукта частицы твердого катализатора слипаются друг с другом, что влияет на псевдоожижающие свойства катализатора.
Поскольку процессы ВТФТ и НТФТ неодинаковы, соответственно, в каждом из процессов также обычно используют неодинаковые катализаторы. Катализатор в целом оптимизируют применительно к конкретному процессу с целью получения конкретного ассортимента продуктов.
В промышленных условиях обычно желательно иметь возможность получать катализаторы на основе железа для реакций как НТФТ, так и ВТФТ из преимущественно чистого источника окиси железа.
Получение таких окисей железа хорошо известно из техники, а в патентах US 1327061 и 1368748 описаны некоторые из соответствующих способов их получения, согласно которым железо, рафинированное от примесей, погружают в раствор растворимой соли двухвалентного или трехвалентного железа, в который введен окислитель, такой как воздух, для осаждения желаемой соли трехвалентного железа.
Одним из недостатков упомянутых известных способов, описанных в этих двух патентах, является то, что растворение, окисление и гидролиз металла происходят в одном и том же реакторе и в одно и то же время. Соответственно, скорости растворения, окисления и гидролиза сложно регулировать по отдельности, в результате чего возможно образование железа в нежелательных формах.
Согласно другому известному из техники способу, описанному в патенте US 6790274, Fe(0) растворяют в кислой среде, в которой соотношение кислоты и железа составляет менее 3:1, окисляют получаемое двухвалентное железо Fe(II) до трехвалентного железа Fe(III) и осуществляют гидролиз и последующее осаждение Fe(III), при этом все стадии протекают в одном и том же реакторе.
Соответственно, в этом способе очень сложно осуществлять быстрое окисление всего железа, а также быстрое осаждение получаемого Fe(III), поскольку определяющей скорость стадией является растворение Fe(0) in situ посредством окислительно-восстановительной пары Fe(0)+2Fe(III)→3Fe(II), в результате чего часть получаемого Fe(III) снова восстанавливается до Fe(II). Одним из недостатков этого является необходимость дополнительного окислителя для повторного преобразования образующегося Fe(II) в Fe(III). В результате часть окислителя косвенно расходуется на окисление Fe(0) до Fe(II).
Еще одним недостатком упомянутого способа по патенту US 6790274 является то, что при его осуществлении в растворе продолжается образование Fe(II), что снижает чистую скорость реакции образования Fe(III) в растворе. Дополнительным недостатком является то, что посредством упомянутой окислительно-восстановительной пары крайне сложно осуществлять гидролиз и(или) осаждение из раствора исключительно Fe(III).
Из уровня техники также известны способы получения катализаторов на основе железа для процесса Фишера-Тропша. Один из таких способов, описанных в патенте US 7199077, включает стадии, на которых получают водный раствор карбоновой кислоты и воды и добавляют в него железо, рафинированное от примесей, после чего принудительно продавливают через кислый раствор окислитель для поглощения железа и получения суспендированной окиси железа. Затем суспензию измельчают и добавляют в нее активаторы. После этого продукт сушат распылением и кальцинируют, чтобы получить катализатор.
Поскольку растворение, окисление и гидролиз металла в этом способе осуществляются в одно и то же время и в одном и том же реакторе, в кислом растворе до окисления может происходить неполное растворение железа, рафинированного от примесей. Это означает, что активаторы, используемые в окислительно-восстановительной паре вместе с Fe(0) (например, медь) можно вводить в процесс только после удаления суспендированного железа из реакционного сосуда, поскольку в противном случае активаторы могут помешать растворению железа.
Заявителем предложен способ получения предшественника катализатора (или) катализатора на основе железа, в котором ослаблены и(или) преодолены, по меньшей мере, некоторые из упомянутых недостатков. Кроме того, как оказалось, при активации такого катализатора его можно успешно применять в процессе ФТ, в частности, НТФТ-процессе синтеза углеводородов из синтез-газа.
Задача изобретения
В основу изобретения положена задача создания нового способа получения материала для катализатора синтеза углеводородов в форме предшественника катализатора (или) катализатора синтеза углеводородов.
Одной из дополнительных задач изобретения является создание способа получения предшественника катализатора (или) катализатора синтеза углеводородов, в котором преодолены или, по меньшей мере, сведены к минимуму упомянутые недостатки.
Еще одной из задач изобретения является применение такого катализатора синтеза углеводородов в процессе Фишера-Тропша.
Краткое изложение сущности изобретения
Предложенный в изобретении способ получения материала для катализатора синтеза углеводородов включает стадии, на которых:
(i) используют раствор карбоксилата Fe(II),
(ii) если молярное отношение карбоксильных и карбоксилатных групп, которые вступили в реакцию или способны вступать в реакцию с железом, и Fe(II) в растворе, используемом на стадии (i), не составляет, по меньшей мере, 3:1, в раствор добавляют источник карбоксильной или карбоксилатной группы, чтобы упомянутое молярное отношение составляло, по меньшей мере, 3:1, до завершения окисления карбоксилата Fe(II) на следующей стадии (iii),
(iii) обрабатывают раствор карбоксилата Fe(II) окислителем, чтобы преобразовать его в раствор карбоксилата Fe(III) в условиях, исключающих такое окисление одновременно с растворением Fe(0),
(iv) осуществляют гидролиз раствора карбоксилата Fe(III), полученного на стадии (iii), и осаждение одного или нескольких продуктов гидролиза Fe(III),
(v) восстанавливают один или несколько продуктов гидролиза, полученных на стадии (iv), и
(vi) добавляют один или несколько активаторов или источников активатора во время или после осуществления любой из предшествующих стадий, чтобы получить материал катализатора в форме предшественника катализатора синтеза углеводородов.
Следует учесть, что такой предшественник катализатора применим для синтеза углеводородов только после активации.
Способ также может включать стадию, на активации предшественника катализатора путем восстановления предшественника катализатора, в результате чего получают материал катализатора в форме катализатора синтеза углеводородов.
Предшественником катализатора может являться предшественник катализатора синтеза Фишера-Тропша. Катализатором может являться катализатор синтеза Фишера-Тропша. Синтезом Фишера-Тропша может являться двухфазный синтез Фишера-Тропша, в качестве альтернативы, трехфазный синтез Фишера-Тропша. Трехфазный синтез Фишера-Тропша может осуществляться в реакторе с неподвижным слоем, но предпочтительно в реакторе с суспендированным слоем.
Раствор карбоксилата Fe(II), используемый на стадии (i), предпочтительно получают на предшествующей стадии растворения железа с состоянием окисления, равным нолю, в кислом растворе, содержащем, по меньшей мере, одну карбоновую кислоту.
Такая предшествующая стадия будет именоваться далее стадией растворения.
Следует учесть, что полученный таким способом раствор карбоксилата Fe(II) часто содержит нерастворенное Fe(0). В таком случае нерастворенное Fe(0) должно быть удалено, как указано в других местах описания.
Раствор карбоксилата Fe(II) может содержать ионы Fe2+, одну или несколько карбоксилатных групп, карбоксилат Fe(II) и необязательно одну или несколько карбоксильных групп.
В одном из вариантов осуществления настоящего изобретения источником карбоксильной группы или карбоксилатной группы является карбоновая кислота.
Продукт, упомянутый на стадии (vi), предпочтительно находится в форме суспензии или пасты, при этом суспензия представляет собой текучую, поддающуюся перекачиванию насосом суспензию тонкодисперсного твердого вещества в жидкости, а паста имеет мягкую и вязкую консистенцию.
В одном из вариантов осуществления изобретения стадия (i) может осуществляться в первом реакционном сосуде, а по меньшей мере, стадия (iii) может осуществляться в отдельном реакционном сосуде.
Стадия (i)
На первой стадии способа используют раствор карбоксилата Fe(II). Этот раствор может быть получен на стадии растворения, на которой источник Fe(0) растворяют в применимом растворе, предпочтительно кислом растворе, предпочтительно водном растворе, содержащем одну или несколько карбоновых кислот, чтобы преобразовать железо с состоянием окисления, равным нолю, в раствор карбоксилата Fe(II), в котором железо имеет состояние окисления, равное двум.
Источником железа может являться железо, рафинированное от примесей. Источник железа может быть выбран из группы, включающей железные опилки, железную стружку, рафинированный от примесей железный лом, распыленное железо, порошковое железо и железную крошку. Источником железа предпочтительно является один из источников железа, не содержащего существенных количеств примесей, таких как Si, Al и Mn.
В одном из вариантов осуществления изобретения, в котором источником железа может по своей природе являться источник мелких частиц железа, эти частицы могут иметь средний диаметр менее 250 микрон, предпочтительно от 30 до 200 микрон. Если источником железа является источник железного лома, железной стружки или железной крошки, частицы железа предпочтительно имеют удельную площадь поверхности более 0,01 м2/кг железа.
В качестве карбоновой кислоты на стадии растворения (или на последующих стадиях, описанных далее) предпочтительно используют карбоновую кислоту с короткой цепью, предпочтительно, содержащей не более трех атомов углерода. Карбоновая кислота может содержать только одну карбоксильную группу, в качестве альтернативы она может содержать несколько карбоксильных групп. Карбоновая кислота может быть выбрана из группы, включающей щавелевую кислоту, муравьиную кислоту, уксусную кислоту, гликолевую кислоту, пировиноградную кислоту, малоновую кислоту и пропионовую кислоту. В одном из предпочтительных вариантов осуществления изобретения карбоновой кислотой может являться муравьиная кислота, уксусная кислота, пропионовая кислота или щавелевая кислота, более предпочтительно уксусная кислота.
В одном из вариантов осуществления изобретения в качестве карбоновой кислоты на упомянутой стадии растворения может использоваться ее водный раствор. Молярное отношение воды и карбоновой кислоты на стадии (i) предпочтительно может составлять от 1:25 до 25:1, предпочтительно от 1:25 до 10:1, более предпочтительно от 1:25 до 1:1.
Раствор карбоксилата Fe(II), используемый на стадии (i), может иметь рН менее 7, предпочтительно от 2 до 5.
Растворение железа может осуществляться при температуре в интервале от 20°С до 200°С, предпочтительно от 40°С до 120°С, более предпочтительно от 50°С до 110°С при давлении окружающей среды. Его предпочтительно осуществляют при давлении окружающей среды ниже давления инертной среды. Инертной средой предпочтительно является среда азота.
Такое растворение может осуществляться до преобразования в раствор карбоксилата Fe(II), по меньшей мере, 90%, предпочтительно, по меньшей мере, 95%, более предпочтительно, по меньшей мере, более 99% железа.
Кроме того, молярное отношение всех карбоксильных и карбоксилатных групп (которые вступили в реакцию или способны вступать в реакцию с железом) карбоновой кислоты и железа на стадии растворения может составлять, по меньшей мере, 2:1, чтобы гарантировать преобразование в раствор карбоксилата Fe(II) преимущественно всего железа, содержащегося в источнике железа.
Стадия (ii)
Вторая стадия способа согласно изобретению является важной, поскольку предполагается, что указанное молярное отношение предотвращает преждевременный гидролиз и последующее осаждение железа из раствора.
Следует учесть, что после образования карбоксилата Fe(II) на стадии растворения, но предпочтительно до окисления Fe(II) на стадии (iii) в кислый раствор может быть добавлен дополнительный источник карбоксильной или карбоксилатной группы с целью достижения упомянутого молярного отношения, по меньшей мере, 3:1.
С целью достижения упомянутого молярного отношения, по меньшей мере, 3:1 карбоновую кислоту предпочтительно добавляют на стадии (ii).
Следует дополнительно учесть, что карбоновая кислота также может добавляться, чтобы гарантировать окисление всего раствора карбоксилата Fe(II) до карбоксилата Fe(III) на стадии (iii).
В качестве альтернативы, кислый раствор может изначально содержать избыток карбоновой кислоты, чтобы гарантировать достижение упомянутого молярного отношения.
Стадия (iii)
На стадии (iii) способа в раствор карбоксилата Fe(II) добавляют окислитель с целью его окисления до раствора карбоксилата Fe(III) таким образом, чтобы исключить такое окисление одновременно с растворением Fe(0).
С этой целью весь способ может осуществляться в виде отдельных стадий, предпочтительно с использованием, по меньшей мере, двух отдельных реакционных сосудов, по меньшей мере, один из которых используется на стадии (iii) и не содержит Fe(0).
В качестве альтернативы такой же результат может быть достигнут путем обеспечения преобразования всего Fe(0) в Fe(II) на стадии (i) или удаления нерастворенного Fe(0) из реакционной среды до осуществления стадии (iii).
Такое нерастворенное Fe(0) может быть удалено одним или несколькими из следующих методов: фильтрацией, декантацией или магнитным разделением. В одном из предпочтительных вариантов осуществления изобретения такое нерастворенное Fe(0) может быть удалено путем фильтрации.
Окислитель на стадии (iii) может быть выбран из группы, включающей одно или несколько из следующего: кислород, перекись водорода, озон, органическую перекись, гидроперекись и газовую смесь, содержащую кислород, такую как, например, воздух.
Окислитель может добавляться в раствор карбоксилата Fe(II) при комнатной температуре, в качестве альтернативы при температуре от 50°С до 100°С, предпочтительно при температуре не выше 70°С.
В одном из вариантов осуществления изобретения молярное отношение окислителя и карбоксилата Fe(II) на стадии (iii) может составлять 2:1, более предпочтительно 1:1 для обеспечения окисления преимущественного всего карбоксилата Fe(II) до карбоксилата Fe(III).
В одном из предпочтительных вариантов осуществления изобретения, по меньшей мере, 90%, предпочтительно 95%, наиболее предпочтительно 99% карбоксилата Fe(II) преобразуют в карбоксилат Fe(III) до осуществления гидролиза на стадии (iv).
Стадия (iv)
На этой стадии осуществляют гидролиз полученного на стадии (iii) раствора карбоксилата Fe(III) водой в известных из техники условиях, обеспечивающих получение осажденной окиси железа или продукта гидролиза гидроксида железа.
Продукты гидролиза могут быть получены на стадии (iv) путем осаждения с использованием основания или путем термического гидролиза (термического разложения).
В одном из вариантов осуществления изобретения продукты гидролиза на стадии (iv) могут быть осаждены из раствора карбоксилата Fe(III) с использованием основания. В качестве основания может использоваться любое основание, выбранное из группы, включающей карбонат натрия, гидроксид натрия, карбонат калия и гидроксид калия, предпочтительно карбонат калия или карбонат натрия.
Описанная реакция осаждения, которая влечет за собой реакцию между карбоксилатом Fe(III) (полученным на стадии (iii)) и основанием в присутствии воды, может быть представлена следующей формулой:
Карбоксилат Fe(III) + основание + H2O→Fe(ОН)3 + соль карбоновой кислоты
Затем полученная соль карбоновой кислоты может быть введена в реакцию с кислотой, такой как HCl, чтобы получить соответствующую карбоновую кислоту. В зависимости от конкретной карбоновой кислоты, используемой в способе, примеры соли карбоновой кислоты могут включать формиат натрия, ацетат натрия, пропионат натрия, калиевую соль щавелевой кислоты, формиат калия, ацетат калия и дикалиевую соль щавелевой кислоты.
В качестве альтернативы, описанная реакция осаждения может осуществляться с использованием аммиака или гидроксида аммония в качестве основания. В этом случае в качестве побочного продукта образуется ацетат аммония, который может быть разложен на аммиак и уксусную кислоту для возможного использования где-либо еще.
В еще одном из вариантов осуществления изобретения путем термического гидролиза могут быть получены продукты гидролиза, образующие выпавшую фазу, которая может находиться в форме окиси железа, оксигидроксидов железа, гидроксидов железа или их сочетания, а также фильтрат карбоновой кислоты. Фильтрат карбоновой кислоты может быть возвращен в повторный цикл или использован где-либо еще.
Следует учесть, что таким способом могут быть получены различные формы окиси железа, оксигидроксида железа и гидроксида железа. Их неограничивающие примеры включают гематит, маггемит, Fe(ОН)3, гетит, фероксигит, лепидокрокит и акаганеит.
В одном из вариантов осуществления изобретения молярное отношение воды и карбоксилата Fe(III) на стадии (iv) гидролиза может составлять от 6:1 до 1:3, предпочтительно от 3:1 до 1:1.
Стадия (v)
На стадии (v) продукт гидролиза, полученный на стадии (iv), восстанавливают путем отделения продуктов гидролиза от раствора, из которого они были осаждены. Отделение может осуществляться путем фильтрации или другими применимыми способами, такими как центрифугирование.
На этой стадии также может осуществляться удаление любых побочных продуктов гидролиза известными из техники методами, такими как промывание и т.п.
Следует дополнительно учесть, что удаление побочных продуктов может происходить одновременно с восстановлением продукта гидролиза.
Если для отделения продуктов гидролиза от раствора используется фильтрация, на фильтре может образовываться осадок.
В одном из дополнительных вариантов осуществления описанный восстановленный продукт гидролиза может быть подвергнут сушке и(или) формованию. В одном из предпочтительных вариантов осуществления изобретения продукт гидролиза подвергают распылительной сушке. В качестве альтернативы, восстановленный продукт гидролиза может быть экструдирован, предпочтительно после сушки.
Восстановленный продукт гидролиза также может быть кальцинирован, предпочтительно с целью окисления продукта гидролиза. Кальцинацию предпочтительно осуществляют после сушки.
Стадия (vi)
На этой стадии в раствор добавляют один или несколько активаторов или источников активатора во время или после любой из стадий с (i) по (v).
Одним или несколькими активаторами или источниками активатора, добавляемыми на стадии (vi), может являться активатор или источник активатора, способный вступать в тесный контакт с железом в материале катализатора. Таким активатором или источником активатора может являться химический активатор, или источник химического и(или) структурного активатора, или источник структурного активатора.
Таким активатором или источником активатора может являться активатор или источник активатора, способствующий восстановлению железа в предшественнике катализатора.
В таком случае источником активатора может являться растворимая соль переходного металла, которая может быть выбрана из группы, включающей свинец, медь, олово, кобальт, никель, хром, ванадий, кадмий, цинк, алюминий, марганец, золото, платину, серебро и смесь двух или более из них. Источником активатора предпочтительно является растворимая соль меди, олова, кобальта, никеля, хрома, кадмия или цинка. Наиболее предпочтительно источником активатора является растворимая соль меди.
Хотя источник активатора может вводиться на любой стадии способа после завершения растворения Fe(0) или после удаления Fe(0), в одном из предпочтительных вариантов осуществления изобретения источник активатора может вводиться до гидролиза на стадии (iv).
Источник активатора может добавляться таким образом, чтобы молярное отношение Fe и активатор составляло от 250:1 до 10:1, предпочтительно от 100:1 до 15:1, более предпочтительно от 40:1 до 20:1.
В одном из предпочтительных вариантов осуществления изобретения источник активатора может вводиться в раствор до, во время или после окисления на стадии (iii) раствора карбоксилата Fe(II) при условии, что в таком раствор отсутствует Fe(0).
Такое добавление предпочтительно осуществляют непосредственно до или во время окисления.
Наиболее предпочтительно добавление источника активатора происходит непосредственно до такого окисления.
Заявитель полагает, что добавление некоторых источников активатора (таких как соли меди) непосредственно до или во время такого окисления является важным, поскольку оно предположительно способствует окислению.
В этом варианте осуществления изобретения окислитель может добавляться в раствор карбоксилата Fe(II) и источник активатора при температуре от 50°C до 100°C, предпочтительно при температуре не выше 70°C.
Заявитель полагает, что возможность добавлять источник активатора на любой стадии способа после полного растворения Fe(0) или после удаления Fe(0), а не только после восстановления продукта гидролиза, как известно из уровня техники, является важной и отличительной особенностью настоящего изобретения.
Изобретение также обеспечивает другие существенные преимущества перед известными способами, которые осуществляются в одном и том же реакторе и не обеспечивают полное растворение Fe(0), которому может помешать активатор.
В отличие от этого, в настоящем изобретении, в котором различные стадии могут осуществляться индивидуально и по отдельности, как это описано выше, может достигаться полное растворение Fe(0) в кислом растворе или осуществляться удаление Fe(0). Соответственно, отсутствует Fe(0), помехой которому могут являться активаторы. Таким образом, источник активатора также может вводиться в карбоксилат до гидролиза карбоксилата Fe(III).
Может добавляться химический активатор. Источником химического активатора может являться растворимая соль щелочного металла или щелочноземельного металла. Эти активаторы обычно влияют на активность и(или) избирательность получаемого катализатора. Источником химического активатора предпочтительно является растворимая соль магнии, калия, бария, стронция, кальция, берилла или лития. Такой источник химического активатора предпочтительно добавляют в один или несколько восстановленных продуктов гидролиза.
Источник химического активатора может добавляться таким образом, чтобы молярное отношение Fe и химического активатора составляло от 170:1 до 5:1. Молярное отношение Fe и химического активатора предпочтительно составляет от 30:1 до 10:1.
Также может добавляться структурный активатор, предпочтительно в форме огнеупорного окисла, такого как SiO2. Источником структурного активатора может являться растворимое стекло, предпочтительно калиевое растворимое стекло. Этот структурный активатор может добавляться в один или несколько восстановленных продуктов гидролиза.
Дополнительно предусмотрено, что структурный активатор добавляют в таком количестве, чтобы молярное отношение Fe и структурного активатора составляло от 10:1 до 0,75:1. Молярное отношение Fe и структурного активатора предпочтительно составляет от 2,5:1 до 1,5:1.
В одном из предпочтительных вариантов осуществления изобретения химический и(или) структурный активатор, добавляемый в один или несколько восстановленных продуктов гидролиза, может добавляться в образующийся на стадии (v) осадок на фильтре.
Активация предшественника катализатора
Предшественник катализатора синтеза углеводородов, полученный на стадии (vi), может активироваться путем восстановления. В одном из вариантов осуществления изобретения предусмотрена стадия восстановления предшественника катализатора с целью восстановления в нем железа до железа с состоянием окисления, равным нолю (например, рафинированного от примесей железа и карбидов железа).
Стадия восстановления может осуществляться в присутствии восстановительного газа. В одном из вариантов осуществления изобретения восстановительный газ может находиться под давлением от 10 до 25 бар. Восстановительным газом может являться водород и(или) моноокиси углерода.
В одном из вариантов осуществления изобретения стадия восстановления может осуществляться при температуре от 200°С до 270°С, предпочтительно 250°С. Стадия восстановления может осуществляться в течение от 3 до 24 часов, предпочтительно 16 часов.
Предпочтительно, по меньшей мере, 40% (в пересчете на соотношение масс железа и суммарного железа), предпочтительно, по меньшей мере, 60%, более предпочтительно, по меньшей мере, 80% содержащегося в катализаторе железа восстанавливают до состояния окисления, равного нолю. Предпочтительно до состояния окисления, равного нолю, восстанавливают преимущественно все железо.
Согласно другой из особенностей изобретения предложен предшественник катализатора и(или) катализатор синтеза углеводородов, полученный описанным в изобретении способом.
Согласно еще одной из особенностей изобретения предложено применение активированного предшественника катализатора синтеза углеводородов, полученного описанным в изобретении способом, в реакции синтеза Фишера-Тропша. Реакцией синтеза может являться двухфазная реакция Фишера-Тропша, но предпочтительно трехфазная реакция синтеза Фишера-Тропша. Трехфазная реакция синтеза Фишера-Тропша может осуществляться в реакторе с неподвижным слоем, но предпочтительно в реакторе с суспендированным слоем.
Согласно другой из особенностей изобретения предложен способ синтеза углеводородов, в котором описанным выше способом получают катализатор синтеза углеводородов и вводят водород в контакт с моноокисью углерода при температуре выше 100°С и давлении, по меньшей мере, 10 бар в присутствии катализатора синтеза углеводородов, в результате чего получают углеводороды и необязательно оксигенаты углеводородов.
Способом синтеза углеводородов может являться процесс синтеза углеводородов Фишера-Тропша. Процессом синтеза углеводородов Фишера-Тропша может являться двухфазный процесс Фишера-Тропша, но предпочтительно трехфазный процесс синтеза углеводородов Фишера-Тропша. Трехфазный процесс синтеза углеводородов Фишера-Тропша может осуществляться в реакторе с неподвижным слоем, но предпочтительно в реакторе с суспендированным слоем.
Температура, при которой осуществляется процесс синтеза углеводородов Фишера-Тропша, может составлять от 180°С до 250°С, предпочтительно от 200°С до 230°С. давление, при котором осуществляется процесс синтеза углеводородов Фишера-Тропша, может составлять от 10 до 40 бар, т.е. от 1000 до 25 4000 Кпа.
При осуществлении способа углеводороды и оксигенаты углеводородов, если они присутствуют, могут подвергаться гидрообработке с целью их преобразования в жидкое топливо и(или) химические продукты.
Рециркуляция фильтрата
Предложенный способ может включать дополнительную стадию восстановления фильтрата и его возврат в повторный цикл в форме карбоновой кислоты, образующегося в результате гидролиза и последующего осаждения с использованием основания или термического разложения раствора карбоксилата Fe(III), полученного на стадии (iv), для возможного использования этого фильтрата на стадиях (i) и(или) (ii) способа.
В одном из вариантов осуществления изобретения таким путем восстанавливают от 60% до 99%, предпочтительно, по меньшей мере, 75% фильтрата карбоновой кислоты в пересчете на исходное количество карбоновой кислоты, использованной на стадиях (i) и (ii).
Фильтрат карбоновой кислоты, восстановленный на этой стадии, может быть возвращен в процесс растворения Fe(0) с целью получения Fe(II) для использования на стадии (i) или на стадии (ii) для достижения упомянутого желаемого молярного отношения.
Краткое описание чертежей
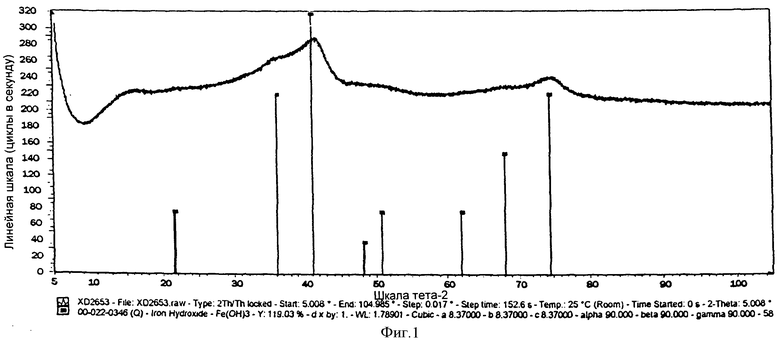
На фиг.1 показана рентгеновская дифракционная (XRD) картина химически активированного предшественника железного катализатора, полученного согласно Примеру 1 изобретения,
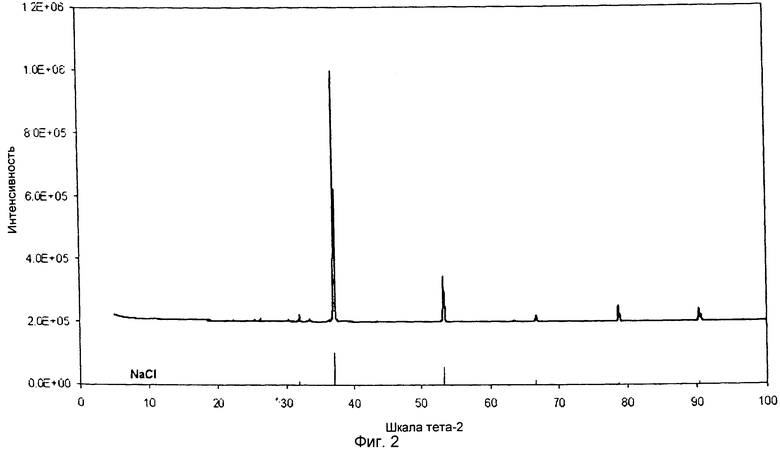
на фиг.2 показана XRD картина хлорида натрия, полученного при рециркуляции уксусной кислоты согласно Примеру 3 изобретения,
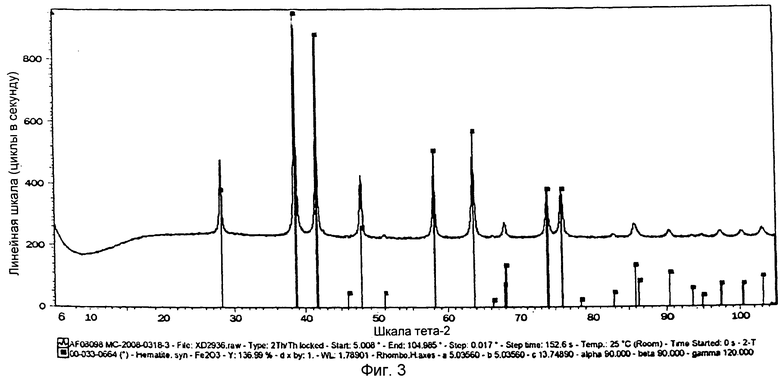
на фиг.3 показана XRD картина химически активированного предшественника железного катализатора, полученного согласно Примеру 4 изобретения,
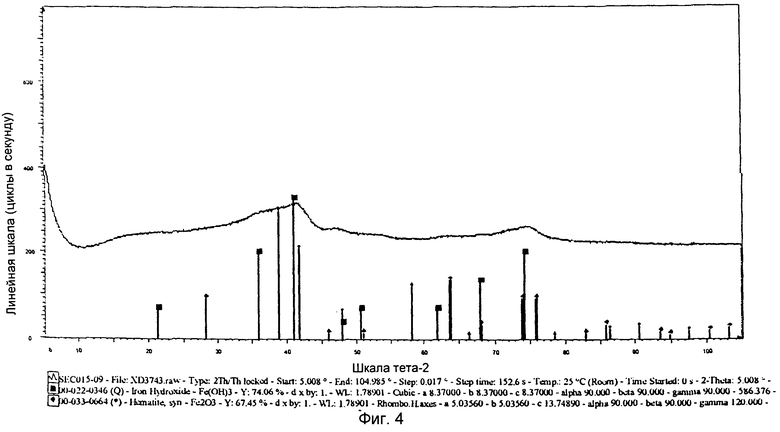
на фиг.4 показана XRD картина химически активированного предшественника железного катализатора, полученного согласно Примеру 7 изобретения,
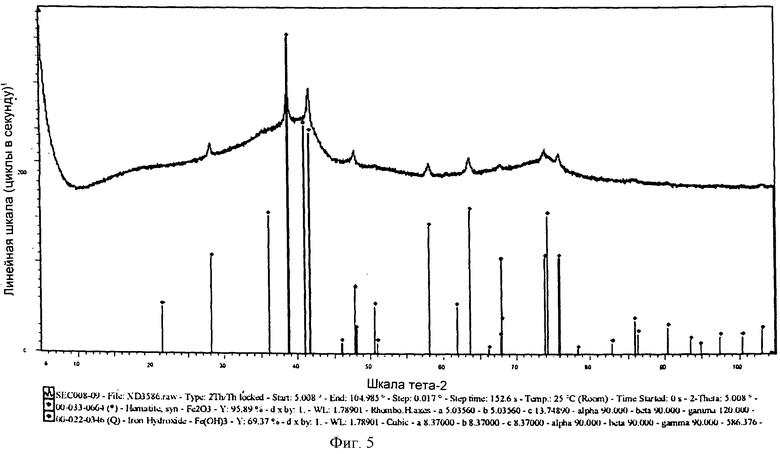
на фиг.5 показана XRD картина химически активированного предшественника железного катализатора, полученного согласно Примеру 9 изобретения,
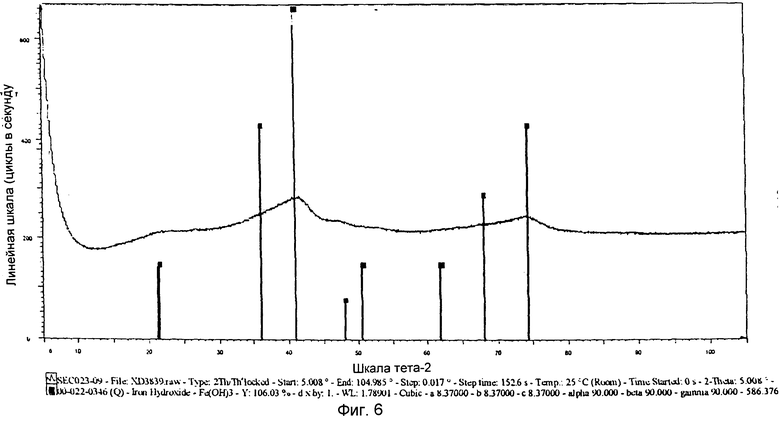
на фиг.6 показана XRD картина химически активированного предшественника железного катализатора, полученного согласно Примеру 11 изобретения, и
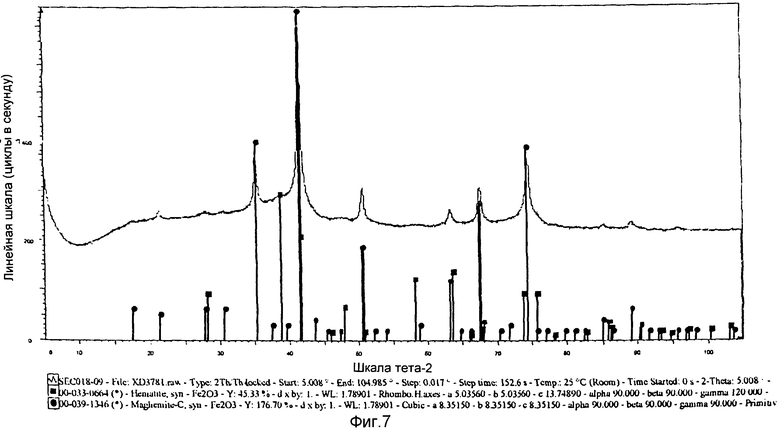
на фиг.7 показана XRD картина химически активированного предшественника железного катализатора, полученного в однореакторной системе с использованием отношения уксусной кислоты и железа менее 3:1 согласно Примеру 13.
Далее настоящее изобретение будет описано на следующих неограничивающих примерах.
Пример 1
Получение предшественника катализатора путем осаждения
Для получения химически активированного предшественника железного катализатора использовали следующую процедуру, в ходе которой получали продукт гидролиза путем осаждения с использованием основания.
В смеси воды (65 мл) и уксусной кислоты (65 мл, 1,048 моля) при температуре 70°С в условиях постоянной подачи азота растворили железо (30 г, 0,5372 моля), чтобы получить раствор ацетата Fe(II) с молярным отношением железа и уксусной кислоты 1:2. После растворения всего железа раствор профильтровали и затем охладили до комнатной температуры, и добавили 7,75 г активатора, содержащего моногидрат ацетата меди(II) (0,03882 моля), и 140 мл раствора уксусной кислоты (1,223 моля) и воды (в объемном соотношении 1:1), в результате чего окончательное молярное отношение железа и уксусной кислоты составляло 1:4. После этого в раствор медленно добавили перекись водорода (30% по весу в воде) (60 мл), чтобы окислить ацетат Fe(II) до ацетата Fe(III). Раствор в течение трех часов перемешивали при комнатной температуре. В процессе окисления молочно-белый цвет раствора поменялся на темно-красный. После завершения окисления раствор, который имел темно-фиолетовый цвет (для подтверждения завершения его окисления использовалась ультрафиолетовая и видимая область спектра), медленно добавили в кипящий раствор карбоната натрия (200 г в 400 мл воды, нагретой то температуры кипения), в результате чего произошло осаждение окиси железа/меди одновременно с образованием раствора ацетата натрия. Тепло отвели, и перемешивали реакционную смесь еще в течение часа. Полученную суспензию профильтровали и промыли, чтобы удалить ацетат натрия. Выпавшую фазу повторно суспендировали в 200 мл дистиллированной воды, и добавили 34 г калиевого растворимого стекла.
Пример 2
Получение катализатора и его применение в синтезе ФТ
Использовали следующую процедуру получения химически активированного железного катализатора.
Выпавшую фазу, полученную в Примере 1, высушили при температуре 150°С и затем кальцинировали при температуре 450°С. На фиг.1 показана XRD картина полученного предшественника катализатора, из которой видно, что предшественник катализатора находится в фазе Fe(ОН)3 и имеет состояние окисления, равное III. Предшественник катализатора просеяли через сито с размером ячеек от 36 до 150 цм, после чего следующим образом оценили в условиях синтеза Фишера-Тропша.
В CSTR (реактор непрерывного действия с механическим перемешиванием) объемом 1 л загрузили 300-350 г воска Фишера-Тропша и расплавили при температуре 160°С. После полного плавления воска добавили 10 г упомянутого предшественника катализатора. Реактор закрыли, установили температуру 255°С и включили мешалку. С помощью аргона медленно снизили давление в системе до 14,5 бар. После стабилизации давления при температуре 255°С ввели Н2 и СО и снижали содержание аргона, пока отношение H2/CO не достигло 1,55, а часовая объемная скорость газа (GHSV, от английского - Gas Hourly Space Velocity) не достигла 10500 млн/грамм катализатора/час. Предшественник катализатора активировали в этих условиях в течение 16 часов. После активации снизили температуру до 245°С и повысили давление до 26,5 бар. Катализатор обеспечил конверсию синтез-газа на уровне 30% (при GHSV 10500 млн/грамм катализатора/час) и избирательность в отношении метана на уровне 2%.
Полученные результаты приведены далее в Таблице.
Пример 3
Рециркуляция побочных продуктов
Для сведения к минимуму себестоимости способа согласно изобретению ацетат натрия, полученный в результате гидролиза в Примере 1, может быть возвращен в повторный цикл путем введения в реакцию с кислотой. Стадия рециркуляции основана на более низкой растворимости NaCl по сравнению с NaOAc (при температуре 25°С растворимость NaCl составляет 26,5 г/100 г Н2О, а растворимость NaOAc составляет 76,5 г/100 г Н2О).
Путем растворения ацетата натрия (18 г) в 50 мл кипящей воды получили его насыщенный раствор. После растворения всего ацетата натрия добавили 50 мл HCl с 32-процентной концентрацией по объему и нагрели раствор до температуры кипения, чтобы вызвать испарение воды и части уксусной кислоты, в результате чего образовался остаток хлорида натрия белого цвета. Этот остаток профильтровали и восстановили остальную уксусную кислоту в качестве фильтрата. Этот фильтрат добавили в восстановленный испаренный раствор воды и уксусной кислоты. Выпавшую фазу подвергли сушке в печи при температуре 150°С в течение 30 минут и подвергли XRD анализу (как показано на фиг.2). Анализ раствора методом ГХ-МС и ВЭЖХ выявил наличие концентрированной уксусной кислоты.
Пример 4
Получение предшественника катализатора путем термического разложения
Была осуществлена такая же процедура, как и в Примере 1, пока не произошло полного окисления ацетата Fe(II) до ацетата Fe(III).
После полного окисления ацетата Fe(II) до ацетата Fe(III) раствор перекачали в автоклав Парра емкостью 1 литр (предварительно нагретый до 250°С) с помощью насоса для ВЭЖХ при одновременном перемешивании с постоянной скоростью 120 об/мин. Раствор ацетата Fe(III) непрерывно нагнетали в реактор со скоростью 10 мл/мин. В результате образования пара в реакторе при этой высокой температуре давление в реакторе повысилось приблизительно до 38 бар. В реакторе поддерживали температуру на уровне 250°С и скорость перемешивания 120 об/мин. Продукт (а именно, суспендированное железо) извлекали из реактора с двухминутными интервалами. Суспензия профильтровали и промыли, чтобы удалить уксусную кислоту. Выпавшую фазу повторно суспендировали в 200 мл дистиллированной воды, и добавили 34 г калиевого растворимого стекла.
Пример 5
Получение катализатора и его применение в синтезе ФТ
Использовали следующую процедуру получения химически активированного железного катализатора.
Выпавшую фазу, полученную в Примере 4, подвергли сушке при температуре 150°С и последующему кальцинированию при температуре 450°С. На фиг.3 показана XRD картина полученного предшественника катализатора, из которой следует, что полученный предшественник железного катализатора находится в фазе гематита. В этой фазе железо имеет состояние окисления, равное III. Предшественник катализатора просеяли через сито с размером ячеек от 36 до 150 µм, после чего следующим образом оценили в условиях синтеза Фишера-Тропша.
В CSTR объемом 1 л загрузили 300-350 г воска Фишера-Тропша и расплавили при температуре 160°С. После полного плавления воска добавили 10 г упомянутого предшественника катализатора. Реактор закрыли, установили температуру 255°С и включили мешалку. С помощью аргона медленно снизили давление в системе до 14,5 бар. После стабилизации давления при температуре 255°С ввели H2 и СО и снижали содержание аргона, пока отношение H2/CO не достигло 1,55, а часовая объемная скорость газа (GHSV) не достигла 10 500 млн/грамм катализатора/час. Предшественник катализатора активировали в этих условиях в течение 16 часов. После активации снизили температуру до 245°С и повысили давление до 26,5 бар. Катализатор обеспечил конверсию синтез-газа на уровне 4,5% (при GHSV 6385 млн/грамм катализатора/час) и избирательность в отношении метана на уровне 7,1%.
Полученные результаты приведены далее в Таблице.
Пример 6
Рециркуляция части продуктов реакции путем термического разложения
Уксусная кислота, полученная в результате гидролиза в Примере 4, может быть возвращена на стадию растворения Fe(0) с целью получения Fe(II) или может быть возращена на стадию (ii) с целью достижения упомянутого желаемого молярного отношения.
Пример 7
Получение предшественника катализатора путем окисления кислородом воздуха
Использовали следующую процедуру получения химически активированного предшественника железного катализатора, в ходе которой окисление ацетата Fe(II) до ацетата Fe(III) осуществляли с использованием воздуха в качестве окислителя.
В смеси воды (65 мл) и уксусной кислоты (65 мл, 1,048 моля) при температуре 70°C в условиях постоянной подачи азота растворили железо (30 г, 0,5372 моля), чтобы получить раствор ацетата Fe(II) с молярным отношением железа и уксусной кислоты 1:2. После растворения всего железа раствор профильтровали, затем охладили до комнатной температуры и добавили 7,75 г активатора, содержащего моногидрат ацетата меди(II) (0,03882 моля), 140 мл раствора уксусной кислоты (1,223 моля) и воды (в объемном соотношении 1:1), в результате чего окончательное молярное отношение железа и уксусной кислоты составляло 1:4. Через раствор в течение 6 часов пропускали пузырьки воздуха при 70°C до завершения окисления раствора, о чем свидетельствовало изменение его цвета на темно-фиолетовый. Для подтверждения завершения окисления раствора использовалась ультрафиолетовая и видимая область спектра. Раствор добавили в кипящий карбонат натрия (200 г в 600 мл воды), раствор профильтровали и промыли полученную выпавшую фазу, пока проводимость промывной воды не стала меньше 500 микросименс. Добавили калиевое растворимое стекло (PWG) (32 г) и в течение 30 минут перемешивали при комнатной температуре.
Пример 8
Получение катализатора и его применение в синтезе ФТ
Использовали следующую процедуру получения химически активированного железного катализатора
Выпавшую фазу, полученную в Примере 7, подвергли сушке при 150°C и последующему кальцинированию при 450°C. На фиг.4 показана XRD картина полученного предшественника катализатора, из которой следует, что полученный предшественник железного катализатора представляет собой смесь фаз, т.е. гематита и Fe(OH)3. Железо в этих фазах имеет состояние окисления, равное III. Предшественник катализатора просеяли через сито с размером ячеек от 36 до 150 µм, после чего следующим образом оценили в условиях синтеза Фишера-Тропша.
В CSTR объемом 1 л загрузили 300-350 г воска Фишера-Тропша и расплавили при температуре 160°C. После полного плавления воска добавили 10 г упомянутого предшественника катализатора. Реактор закрыли, установили температуру 255°C и включили мешалку. С помощью аргона медленно снизили давление в системе до 14,5 бар. После стабилизации давления при температуре 255°C ввели H2 и СО и снижали содержание аргона, пока отношение H2/CO не достигло 1,55, а часовая объемная скорость газа (GHSV) не достигла 10 500 млн/грамм катализатора/час. Предшественник катализатора активировали в этих условиях в течение 16 часов. После активации снизили температуру до 245°С и повысили давление до 26,5 бар. Катализатор обеспечил конверсию синтез-газа на уровне 21% (при GHSV 9018 млн/грамм катализатора/час) и избирательность в отношении метана на уровне 5,6%.
Полученные результаты приведены далее в Таблице.
Пример 9
Получение предшественника катализатора
Использовали следующую процедуру получения химически активированного предшественник железного катализатора, согласно которой добавили медный активатор после пропитки диоксидом кремния и калием.
В смеси воды (65 мл) и уксусной кислоты (65 мл, 1,048 моля) при температуре 70°С в условиях постоянной подачи азота растворили железо (30 г, 0,5372 моля), чтобы получить раствор ацетата Fe(II) с молярным отношением железа и уксусной кислоты 1:2. После растворения всего железа раствор профильтровали и затем охладили до комнатной температуры, и добавили 140 мл раствора уксусной кислоты (1,223 моля) и воды (в объемном соотношении 1:1), в результате чего окончательное молярное отношение железа и уксусной кислоты составляло 1:4. В раствор медленно добавили перекись водорода (30% по весу в воде) (60 мл), чтобы окислить ацетат Fe(II) до ацетата Fe(III). Раствор в течение 2 минут перемешивали в бутыли емкостью 1 л, а затем перемешивали в течение трех часов. Раствор приобрел темно-фиолетовый цвет, что указывало на завершение окисления ацетата Fe(II) до ацетата Fe(III). (Для подтверждения завершения окисления раствора использовалась ультрафиолетовая и видимая область спектра). Раствор медленно добавили в кипящий раствор карбоната натрия (200 г в 400 мл воды, нагретой то температуры кипения), профильтровали и промыли полученную выпавшую фазу, пока проводимость промывной воды не стала меньше 500 микросименс. Добавили калиевое растворимое стекло (PWG) (40 г) и в течение 30 минут перемешивали смесь 30, после чего добавили ацетат Cu(II) (5 г). Смесь дополнительно перемешивали в течение 2 часов при температуре 40°С, после чего профильтровали.
Пример 10
Получение катализатора и его применение в синтезе ФТ
Использовали следующую процедуру получения химически активированного железного катализатора.
Выпавшую фазу, полученную в Примере 9, подвергли сушке при 150°С и последующему кальцинированию при 450°С. На фиг.5 показана XRD картина полученного предшественника катализатора, из которой следует, что полученный предшественник железного катализатора находится в фазе Fe(ОН)3. Железо в этой фазе имеет состояние окисления, равное III. Предшественник катализатора просеяли через сито с размером ячеек от 36 до 150 µм, после чего следующим образом оценили в условиях синтеза Фишера-Тропша.
В CSTR объемом 1 л загрузили 300-350 г воска Фишера-Тропша и расплавили при температуре 160°С. После полного плавления воска добавили 10 г упомянутого предшественника катализатора. Реактор закрыли, установили температуру 255°С и включили мешалку. С помощью аргона медленно снизили давление в системе до 14,5 бар. После стабилизации давления при температуре 255°С ввели Н2 и СО и снижали содержание аргона, пока отношение Н2/СО не достигло 1,55, а часовая объемная скорость газа (GHSV) не достигла 10 500 млн/грамм катализатора/час. Предшественник катализатора активировали в этих условиях в течение 16 часов. После активации снизили температуру до 245°С и повысили давление до 26,5 бар. Катализатор обеспечил конверсию синтез-газа на уровне 16% (при GHSV 8000 млн/грамм катализатора/час) и избирательность в отношении метана на уровне 8,0%.
Полученные результаты приведены далее в Таблице.
Пример 11
Получение предшественника катализатора
Использовали следующую процедуру получения химически активированного предшественника железного катализатора, согласно которой для растворения железа вместе уксусной кислоты использовали пропионовую кислоту.
В смеси воды (65 мл) и пропионовой кислоты (78 мл, 1,048 моля) при температуре 70°С в условиях постоянной подачи азота растворили железо (30 г, 0,5372 моля), чтобы получить раствор ацетата Fe(II) с молярным отношением железа и уксусной кислоты 1:2. После растворения всего железа раствор профильтровали, затем охладили до комнатной температуры и добавили 7,75 г активатора, содержащего моногидрат ацетата меди(II) (0,03882 моля), и 160 мл раствора пропионовой кислоты (1,223 моля) и воды (в объемном соотношении 1:1), в результате чего окончательное молярное отношение железа и уксусной кислоты составляло 1:4. В раствор медленно добавили перекись водорода (30% по весу в воде) (60 мл), чтобы окислить пропионат Fe(II) до пропионата Fe(III). Раствор в течение трех часов перемешивали при комнатной температуре. После полного окисления раствор приобрел темно-фиолетовый цвет (Для подтверждения завершения окисления раствора использовалась ультрафиолетовая и видимая область спектра), добавили раствор в кипящий раствор карбоната натрия (200 г в 600 мл воды). Раствор профильтровали и промывали полученную выпавшую фазу, пока проводимость промывной воды не стала меньше 500 микросименс.
Пример 12
Получение катализатора и его применение в синтезе ФТ
Использовали следующую процедуру получения химически активированного железного катализатора.
Выпавшую фазу, полученную в Примере 11, подвергли сушке при 150°С и последующему кальцинированию при 450°С. На фиг.6 показана XRD картина полученного предшественника катализатора, из которой следует, что полученный предшественник железного катализатора находится в фазе Fe(ОН)3. Железо в этой фазе имеет состояние окисления, равное III. Предшественник катализатора просеяли через сито с размером ячеек от 36 до 150 µм, после чего следующим образом оценили в условиях синтеза Фишера-Тропша.
В CSTR объемом 1 л загрузили 300-350 г воска Фишера-Тропша и расплавили при температуре 160°С. После полного плавления воска добавили 10 г упомянутого предшественника катализатора. Реактор закрыли, установили температуру 255°С и включили мешалку. С помощью аргона медленно снизили давление в системе до 14,5 бар. После стабилизации давления при температуре 255°С ввели Н2 и СО и снижали содержание аргона, пока отношение Н2/СО не достигло 1,55, а часовая объемная скорость газа (GHSV) не достигла 10 500 млн/грамм катализатора/час. Предшественник катализатора активировали в этих условиях в течение 16 часов.
Полученные результаты приведены далее в Таблице.
Пример 13
Сравнительный пример получения предшественника катализатора
Использовали следующую процедуру получения химически активированного предшественника железного катализатора в однореакторной системе с использованием отношения уксусной кислоты и железа менее 3:1.
В бутыль с круглым дном (500 мл) поместили железный порошок (30 г, 0,5372 моля). В железный порошок добавили уксусную кислоту (95 мл) и воду (95 мл), в результате чего отношение железа и уксусной кислоты составляло 1:3. В раствор в течение 5 минут через конденсатор медленно добавляли перекись водорода (30%) (61 мл). Раствор в течение 16 часов дефлегмировали с одновременным перемешиванием при температуре 70°С, а затем добавили в кипящий карбонат натрия (200 г в 600 мл воды). Раствор (рН 7,4, который с помощью уксусной кислоты довели до 6,2) профильтровали, и промывали полученную выпавшую фазу, пока проводимость не стала меньше 500 микросименс. Добавили калиевое растворимое стекло (30,11 г), перемешивали в течение одного часа при температуре 60°С и профильтровали. В выпавшую фазу добавили ацетат меди (4,5 г), профильтровали, сушили в течение двух часов при температуре 145°С, а затем кальцинировали при температуре 400°С. Образец подвергли XRD и ИСП анализу.
Пример 14
Сравнительный пример получения катализатора и его применения в синтезе ФТ
Использовали следующую процедуру получения химически активированного железного катализатора.
Выпавшую фазу, полученную в Примере 13, подвергли сушке при 150°С и последующему кальцинированию при 450°С. На фиг.7 показана XRD картина полученного предшественника катализатора, из которой следует, что предшественник железного катализатора представляет собой смесь фаз, т.е. гематит и маггемит. Железо в этих фазах имеет состояние окисления, равное III. Предшественник катализатора просеяли через сито с размером ячеек от 36 до 150 µм, после чего следующим образом оценили в условиях синтеза Фишера-Тропша.
В CSTR объемом 1 л загрузили 300-350 г воска Фишера-Тропша и расплавили при температуре 160°С. После полного плавления воска добавили 10 г упомянутого предшественника катализатора. Реактор закрыли, установили температуру 255°С и включили мешалку. С помощью аргона медленно снизили давление в системе до 14,5 бар. После стабилизации давления при температуре 255°С ввели Н2 и СО и снижали содержание аргона, пока отношение H2/CO не достигло 1,55, а часовая объемная скорость газа (GHSV) не достигла 10 500 млн/грамм катализатора/час. Предшественник катализатора активировали в этих условиях в течение 16 часов. Катализатор обеспечил конверсию синтез-газа на уровне 5,6% (при GHSV 9018 млн/грамм катализатора/час) и избирательность в отношении метана на уровне 9,5%.
Полученные результаты приведены далее в Таблице.
Обсуждение результатов
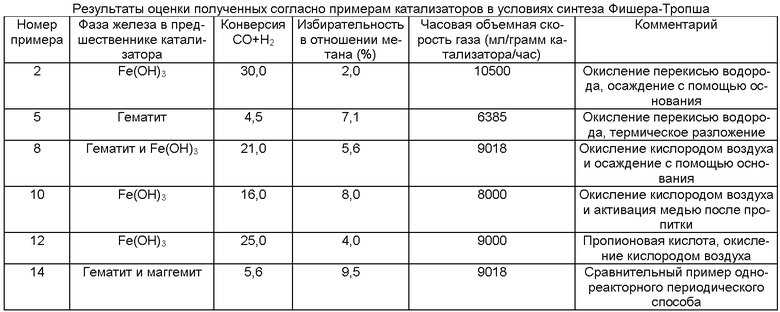
Из Таблицы видно, что катализатор, полученный согласно Примеру 2, обеспечивал очень высокий уровень конверсии синтез-газа (30%) и относительно низкую избирательность в отношении метана (2%) по сравнению с катализаторами согласно другим примерам при достаточно высокой часовой объемной скорости газа (GHSV). В этом примере окисление Fe(II) до Fe(III) на стадии (iii) осуществлялось с помощью перекиси водорода.
Катализатор, полученный согласно Примеру 8, обеспечивал высокий показатель конверсии синтез-газа (21%) и избирательность в отношении метана 5,6%. В этом примере окисление Fe(II) до Fe(III) на стадии (iii) осуществлялось с помощью воздуха.
Таким образом, Примеры 2 и 8 показывают, что предложенный в настоящем изобретении способ может успешно применяться для окисления как перекисью водорода, так и воздухом.
Катализатор, полученный согласно Примеру 5, обеспечивал достаточно низкий показатель конверсии синтез-газа (4,5%) при достаточно высокой избирательности в отношении метана (7,1%). Тем не менее, это пример демонстрирует преимущество способа, состоящее в том, что уксусная кислота может быть возвращена в повторный цикл в существующем виде без необходимости дополнительных стадий подготовки. Следует учесть, что результаты этого примера являлись ожидаемыми с учетом того, что гематит обеспечивает получение катализатора с низким показателем конверсии синтез-газа и относительно высокой избирательностью в отношении метана.
Катализатор согласно Примеру 10 был получен путем добавления медного активатора к предшественнику железного катализатора непосредственно после пропитки диоксидом кремния и калием. Соответственно, этот катализатор отличается меньшим взаимодействием между медным активатором и железом.
Если сравнить результаты использования катализатора, полученного согласно Примеру 10, и результаты использования катализатора, полученного согласно Примеру 2, видно заметное различие в их активности и избирательности, несмотря на то, что оба катализатора содержат железо в одной и той же фазе, а именно Fe(ОН)3.
В частности, у катализатора, полученного согласно Примеру 10, в котором в предшественник железного катализатора непосредственно после пропитки диоксидом кремния и калием был добавлен медный активатор, конверсия синтез-газа составляла 16%, а у катализатор, полученного согласно Примеру 2, в котором осуществлялось совместное осаждение медного активатора, конверсия синтез-газа составляла 30%. Аналогичным образом избирательность в отношении метана значительно выше у катализатора, полученного согласно Примеру 10, чем у катализатора, полученного согласно Примеру 2 (8% против 2%).
В Примере 12 уксусная кислота заменена пропионовой кислотой. Было отмечено, что процесс растворения пропионовой кислотой протекал значительно быстрее, чем уксусной кислотой. Катализатор, полученный согласно Примеру 12, также содержал железо в фазе Fe(ОН)3. Конверсия синтез-газа была вполне сравнима с показателем при использовании уксусной кислоты (25% против 30%), при этом катализатор имел избирательность в отношении метана 4% при GHSV 9000 млн/грамм катализатора/час.
Что касается катализатора, полученного согласно сравнительному примеру (Пример 14), его показатель конверсии синтез-газа составлял лишь 5,6%, при этом он содержал гематит и маггемит, из-за чего катализатор имел высокую избирательность в отношении метана (9,5%).
Эти результаты ясно демонстрируют отчетливые преимущества способа согласно настоящему изобретению. Предложенный в настоящем изобретении способ осуществляется в виде индивидуальных и раздельных стадий, как это описано выше в примерах, за счет чего достигается полное растворение Fe(0) в кислом растворе. Соответственно, отсутствует Fe(0), помехи которому может создавать активатор. За счет этого источник активатора также может вводиться до гидролиза карбоксилата Fe(III). Напротив, эти результаты недостижимы в известном из уровня техники однореакторном способе, примененном в Примере 14, в котором отношение карбоновой кислоты и железа составляет менее 3:1.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ СИНТЕЗА УГЛЕВОДОРОДОВ | 2008 |
|
RU2442815C2 |
| СПОСОБ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА СИНТЕЗА УГЛЕВОДОРОДОВ | 2006 |
|
RU2412001C2 |
| СПОСОБ СИНТЕЗА УГЛЕВОДОРОДОВ | 2008 |
|
RU2450043C2 |
| СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРА СИНТЕЗА ФИШЕРА-ТРОПША | 2009 |
|
RU2481156C2 |
| СПОСОБ ПОЛУЧЕНИЯ НОСИТЕЛЯ ДЛЯ КАТАЛИЗАТОРА С ПОВЫШЕННОЙ ГИДРОТЕРМАЛЬНОЙ СТАБИЛЬНОСТЬЮ (ВАРИАНТЫ), КАТАЛИЗАТОР ДЛЯ СИНТЕЗА УГЛЕВОДОРОДОВ И СПОСОБ СИНТЕЗА УГЛЕВОДОРОДОВ ИЗ СИНТЕЗ-ГАЗА | 2003 |
|
RU2340394C2 |
| КАТАЛИЗАТОР СИНТЕЗА ФИШЕРА-ТРОПША, СОДЕРЖАЩИЙ НИТРИДНЫЙ НОСИТЕЛЬ, И СПОСОБ ЕГО ПОЛУЧЕНИЯ, И ЕГО ПРИМЕНЕНИЕ | 2018 |
|
RU2760904C2 |
| СПОСОБ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА | 2015 |
|
RU2710375C2 |
| КАТАЛИЗАТОР НА ОСНОВЕ Fe ДЛЯ СИНТЕЗА ФИШЕРА-ТРОПША, СПОСОБ ЕГО ПРИГОТОВЛЕНИЯ И ПРИМЕНЕНИЯ | 2010 |
|
RU2468863C1 |
| ПОЛУЧЕНИЕ НИЗКОМОЛЕКУЛЯРНЫХ ОЛЕФИНОВ ИЗ СИНТЕЗ-ГАЗА | 2010 |
|
RU2558954C2 |
| СПОСОБ РЕГЕНЕРАЦИИ КОБАЛЬТОВОГО КАТАЛИЗАТОРА СИНТЕЗА ФИШЕРА-ТРОПША | 2008 |
|
RU2456080C2 |
Изобретение относится к способам получения предшественника катализатора, катализатора синтеза Фишера-Тропша и к самому способу синтеза Фишера-Тропша. Способ получения предшественника катализатора синтеза Фишера-Тропша включает стадии, на которых: (i) используют раствор карбоксилата Fe(II); (ii) если молярное отношение карбоксильных и карбоксилатных групп, которые вступили в реакцию или способны вступать в реакцию с железом, и Fe(II) в растворе, используемом на стадии (i), не составляет, по меньшей мере, 3:1, в раствор добавляют источник карбоксильной или карбоксилатной группы, чтобы упомянутое молярное отношение составляло, по меньшей мере, 3:1, до завершения окисления карбоксилата Fe(II) на следующей стадии (iii); (iii) обрабатывают раствор карбоксилата Fe(II) окислителем, чтобы преобразовать его в раствор карбоксилата Fe(III) в условиях, исключающих такое окисление одновременно с растворением Fe(0); (iv) осуществляют гидролиз раствора карбоксилата Fe(III), полученного на стадии (iii), и осаждение одного или нескольких продуктов гидролиза Fe(III); (v) восстанавливают один или несколько продуктов гидролиза, полученных на стадии (iv); и (vi) добавляют источник активатора в форме растворимой соли переходного металла и химический активатор в форме растворимой соли щелочного металла или щелочноземельного металла во время или после осуществления любой из предшествующих стадий, чтобы получить предшественник катализатора синтеза Фишера-Тропша. Технический результат - достижение полного растворения Fe(0) в кислом растворе; источник активатора может вводиться до гидролиза карбоксилата Fe(III). 3 н. и 12 з.п. ф-лы, 7 ил., 1 табл., 14 пр.
1. Способ получения предшественника катализатора синтеза Фишера-Тропша, включающий стадии, на которых:
(i) используют раствор карбоксилата Fe(II),
(ii) если молярное отношение карбоксильных и карбоксилатных групп, которые вступили в реакцию или способны вступать в реакцию с железом, и Fe(II) в растворе, используемом на стадии (i), не составляет, по меньшей мере, 3:1, в раствор добавляют источник карбоксильной или карбоксилатной группы, чтобы упомянутое молярное отношение составляло, по меньшей мере, 3:1, до завершения окисления карбоксилата Fe(II) на следующей стадии (iii),
(iii) обрабатывают раствор карбоксилата Fe(II) окислителем, чтобы преобразовать его в раствор карбоксилата Fe(III) в условиях, исключающих такое окисление одновременно с растворением Fe(0),
(iv) осуществляют гидролиз раствора карбоксилата Fe(III), полученного на стадии (iii), и осаждение одного или нескольких продуктов гидролиза Fe(III),
(v) восстанавливают один или несколько продуктов гидролиза, полученных на стадии (iv), и
(vi) добавляют источник активатора в форме растворимой соли переходного металла и химический активатор в форме растворимой соли щелочного металла или щелочноземельного металла во время или после осуществления любой из предшествующих стадий, чтобы получить предшественник катализатора синтеза Фишера-Тропша.
2. Способ по п.1, в котором источником карбоксильной группы или карбоксилатной группы является карбоновая кислота.
3. Способ по п.2, в котором карбоновую кислоту выбирают из группы, включающей щавелевую кислоту, муравьиную кислоту, уксусную кислоту, гликолевую кислоту, пировиноградную кислоту, малоновую кислоту и пропионовую кислоту.
4. Способ по п.2, в котором добавляют карбоновую кислоту, чтобы обеспечить окисление всего раствора карбоксилата Fe(II) до карбоксилата Fe(III) на стадии (iii).
5. Способ по п.1, в котором окислитель на стадии (iii) выбирают из группы, включающей одно или несколько из следующего: кислород, перекись водорода, озон, органическую перекись, гидроперекись и газовую смесь, содержащую кислород, такую как, например, воздух.
6. Способ по п.1, в котором продукты гидролиза на стадии (iv) получают путем осаждения с использованием основания или путем термического гидролиза.
7. Способ по п.6, в котором основание выбирают из группы, включающей карбонат натрия, гидроксид натрия, карбонат калия, гидроксид калия, аммиак и гидроксид аммония.
8. Способ по п.6, в котором в результате термического гидролиза образуется выпавшая фаза в форме окиси железа, оксигидроксидов железа, гидроксидов железа или их сочетания вместе с фильтратом карбоновой кислоты.
9. Способ по п.1, в котором растворимую соль переходного металла выбирают из группы, включающей свинец, медь, олово, кобальт, никель, хром, ванадий, кадмий, цинк, алюминий, марганец, золото, платину, серебро и смесь двух или более из них.
10. Способ по п.1, в котором раствор, используемый на стадии (i) карбоксилат Fe(II), получают на предшествующей стадии растворения железа с состоянием окисления, равным нолю, в кислом растворе, содержащем, по меньшей мере, одну карбоновую кислоту.
11. Способ по п.1, необязательно включающий стадию восстановления и рециркуляции фильтрата в форме карбоновой кислоты для использования на стадиях (i) и(или) (ii).
12. Способ получения катализатора синтеза Фишера-Тропша, включающий получение предшественника катализатора синтеза Фишера-Тропша способом по любому из предшествующих пунктов и активацию предшественника катализатора синтеза Фишера-Тропша путем восстановления предшественника катализатора, в результате чего получают катализатор синтеза Фишера-Тропша.
13. Способ синтеза Фишера-Тропша, в котором способом по п.12 получают катализатор синтеза Фишера-Тропша и вводят водород в контакт с моноокисью углерода при температуре выше 100°С и давлении, по меньшей мере, 10 бар в присутствии катализатора синтеза Фишера-Тропша, в результате чего получают углеводороды и необязательно оксигенаты углеводородов.
14. Способ по п.13, в котором способом синтеза Фишера-Тропша является двухфазный способ синтеза углеводородов Фишера-Тропша.
15. Способ по п.13, в котором способом синтеза Фишера-Тропша является трехфазный способ синтеза углеводородов Фишера-Тропша.
| US 20040152791 А1, 05.08.2004 | |||
| КАТАЛИЗАТОР ФИШЕРА-ТРОПША, ПОЛУЧЕННЫЙ ПРИ ИСПОЛЬЗОВАНИИ ВЫСОКОЧИСТОГО ЖЕЛЕЗОСОДЕРЖАЩЕГО ПРЕДШЕСТВЕННИКА (ВАРИАНТЫ) | 2002 |
|
RU2299764C2 |
| Способ получения титаноценов | 1987 |
|
SU1597105A3 |
| US 20040106517 А1, 03.06.2004 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |

