Уровень техники
Акромегалия является гормональным заболеванием, которое возникает тогда, когда гипофиз вырабатывает слишком много гормона роста (GH). Чаще всего она поражает людей среднего возраста и может привести к серьезному заболеванию и преждевременной смерти. Если поставлен диагноз акромегалии, то она поддается лечению у большинства пациентов, но из-за медленного и зачастую скрытого развития ее часто не удается правильно диагностировать. Наиболее серьезными последствиями акромегалии для здоровья являются сахарный диабет, гипертензия и повышенный риск возникновения сердечно-сосудистых заболеваний. Больные акромегалией также подвергаются повышенному риску возникновения полипов толстой кишки, которые могут перерасти в рак. Если вырабатывающая GH опухоль появляется в детском возрасте, то возникающее заболевание называется гигантизм, а не акромегалия. После достижения половой зрелости происходит слияние пластинок роста длинных трубчатых костей, поэтому у взрослых при чрезмерной выработке GH не происходит повышения роста. А длительное воздействие чрезмерного уровня GH до слияния пластинок роста вызывает усиление роста трубчатых костей и повышение роста.
Акромегалия возникает при длительной чрезмерной выработке гормона роста (GH) гипофизом. Гипофиз представляет собой небольшую железу у основания мозга, которая вырабатывает несколько важных гормонов, контролирующих такие функции организма, как рост и развитие, размножение и метаболизм. GH является частью каскада гормонов, которые, как это видно из названия, регулируют физический рост организма. Этот каскад начинается в той части мозга, что называется гипоталамус, который вырабатывает гормоны, регулирующие гипофиз. Один из них - гормон высвобождения гормона роста (GHRH) - стимулирует вырабатывание GH гипофизом. Другой гормон гипоталамуса - соматостатин - ингибирует вырабатывание и выделение GH. Секреция GH из гипофиза в кровоток вызывает выработку другого гормона - инсулиноподобного фактора роста 1 (IGF-1) в печени. IGF-1 является фактором, вызывающим рост костей и других тканей организма. В свою очередь IGF-1 подает сигнал гипофизу об уменьшении выработки GH. Уровни GHRH, соматостатина, GH и IGF-1 в организме жестко регулируются друг другом, и на них влияют такие внешние раздражители, как сон, упражнения, стресс, прием пищи и уровень сахара в крови. Если гипофиз вырабатывает GH независимо от нормальных регуляторных механизмов, то уровень IGF-1 будет возрастать, что приведет к росту костей и увеличению органов. Избыток GH также вызывает изменения метаболизма сахаров и липидов и может вызвать диабет.
У свыше 90% больных акромегалией чрезмерная выработка GH вызывается доброкачественной опухолью гипофиза, которая именуется аденомой. Эти опухоли вырабатывают избыток GH и по мере их увеличения сдавливают окружающие ткани мозга, как-то зрительные нервы. Такое увеличение вызывает головные боли и зрительные нарушения, которые часто являются симптомами акромегалии. Кроме того, сдавливание окружающей нормальной ткани гипофиза может изменить вырабатывание других гормонов, что приводит к изменениям месячных и выделениям из молочной железы у женщин и импотенции у мужчин.
У некоторых больных акромегалия вызывается не опухолью гипофиза, а опухолью поджелудочной железы, легких или надпочечников. Эти опухоли вызывают избыток GH либо потому, что они сами вырабатывают GH, либо, что встречается чаще, потому, что они вырабатывают GHRH, то есть гормон, стимулирующий выработку GH гипофизом. У этих больных отмечается избыток GHRH в крови, который свидетельствует, что причиной акромегалии не является дефект гипофиза. При удалении этих негипофизарных опухолей уровень GH падает и симптомы акромегалии улучшаются.
Схемы лечения акромегалии включают уменьшение продукции GH до нормального уровня, чтобы уменьшить давление, которое растущая опухоль гипофиза оказывает на окружающие зоны мозга, сохранить нормальное функционирование гипофиза и обратить или ослабить симптомы акромегалии. Возможные способы лечения включают хирургическое удаление опухоли, лекарственную терапию и лучевую терапию гипофиза.
Было показано, что октреотид эффективен при лечении акромегалии. Уровень GH обычно снижается уже через 2 часа после подкожного введения октреотида. Октреотид приводит к снижению уровней GH и IGF-1 у большинства пациентов с нормализацией уровня IGF-1 у вплоть до 60% пациентов, что означает биохимическую ремиссию. У большинства пациентов отмечается заметное улучшение симптомов акромегалии, включая головные боли, боли в суставах и потоотделение, сразу же после начала лечения октреотидом. Сейчас октреотид доступен в виде Sandostatin LAR® Depot, который после восстановления представляет собой суспензию микросфер, содержащих октреотида ацетат. Sandostatin LAR® Depot является единственным медикаментом, показанным для продолжительной поддерживающей терапии у больных акромегалией. Он также показан для продолжительного лечения тяжелой диареи и гиперемии при метастатических карциноидных опухолях и профузной водянистой диареи при VIP-секретирующих опухолях. Sandostatin LAR® Depot вводится путем внутримышечной инъекции раз в 4 недели после периода титрования. Октреотида ацетат также доступен в форме для немедленного высвобождения - раствора Sandostatin® Injection, который нужно вводить уколом 3 раза в день.
У больных, не проявляющих существенного снижения уровня GH в ответ на периодическое введение октреотида, более частое введение октреотида может привести к большему клиническому ответу. Больным рефрактерной акромегалией можно вводить октреотид непрерывно с помощью подкожного насоса, чтобы предотвратить утечку GH между инъекциями.
В свете эффективности октреотида при лечении акромегалии и отсутствия способа лечения и лекарственной формы октреотида с контролируемым высвобождением существует явная потребность в такой лекарственной форме и способе введения, которые способны вводить октреотид в течение определенного времени с контролируемой скоростью, чтобы избежать осложнений у больных, которым приходится страдать, к примеру, от множественных периодических уколов.
Сущность изобретения
Настоящее изобретение в общем касается фармацевтической композиции октреотида, которая может применяться для лечения лиц, страдающих гормональными заболеваниями. Настоящее изобретение предпочтительно выполняется в виде формы с контролируемым высвобождением. В частности, настоящее изобретение основывается на неожиданном открытии того, что октреотид может высвобождаться с контролируемой скоростью из имплантируемого устройства, например имплантируемого устройства, не требующего заправки перед имплантированием.
Одно воплощение касается способа доставки октреотида субъекту с профилем высвобождения практически нулевого порядка в течение длительного времени, но не менее 6 месяцев, при этом субъектом является млекопитающее, кроме собак, а способ включает подкожное имплантирование субъекту по меньшей мере одного имплантируемого устройства, причем данное по меньшей мере одно имплантируемое устройство включает композицию, содержащую октреотид, а композиция заключена в гидрофильный полимер, и при этом имплантируемое устройство имплантируется в сухом состоянии так, чтобы субъект на протяжении по меньшей мере 6 месяцев получал в пересчете на суточную дозу такое количество октреотида, которое эффективно для лечения субъекта. В предпочтительном воплощении гидрофильный полимер включает один или несколько полимеров на основе полиуретана либо один или несколько полимеров на основе метакрилата. В предпочтительном воплощении октреотид находится в виде свободного вещества, соли или в виде комплекса, например, в котором октреотид представляет собой ацетат октреотида. В предпочтительном воплощении субъект страдает расстройством гормона GH или IGF-1 либо его симптомами, например акромегалии. В предпочтительном воплощении субъект получает октреотид со средней скоростью от 75 мкг в сутки до 300 мкг в сутки на протяжении по меньшей мере 6 месяцев. В предпочтительном воплощении дозировка октреотида, получаемого субъектом, дает уровень октреотида в сыворотке от 0,5 нг/мл до 2 нг/мл, от 0,5 нг/мл до 1,4 нг/мл, от 0,6 нг/мл до 1,2 нг/мл, от 0,8 нг/мл до 1,2 нг/мл или от 0,9 нг/мл до 1,0 нг/мл. В предпочтительном воплощении субъект получает эффективное количество октреотида на протяжении по меньшей мере 12 месяцев. В целях определения уровня в сыворотке интервал может быть указан в виде среднего за какой-то период времени, например от 3 дней до 150 дней, от 3 дней до 120 дней, от 5 дней до 100 дней, от 10 дней до 75 дней и т.д. В предпочтительном воплощении дозировка октреотида, получаемого субъектом, дает величину Cmax для уровня октреотида в сыворотке менее 1,2 нг/мл. В предпочтительном воплощении дозировка октреотида, получаемого субъектом, дает величину Cmax для уровня октреотида в сыворотке менее 1,0 нг/мл. В предпочтительном воплощении высвобождение октреотида происходит по меньшей мере от 3 до 10 дней после имплантирования. В предпочтительном воплощении субъект страдает заболеванием, выбранным из группы, состоящей из карциноидного синдрома, VIP-омы, нейроэндокринных опухолей, пролиферативной диабетической ретинопатии, красных угрей, панкреатита, желудочно-кишечных кровотечений, свищей поджелудочной железы и кишечных свищей, базедовой болезни (офтальмопатии Грейвса), глаукомы и симптомов, связанных с химиотерапией или СПИД.
Одно воплощение касается имплантируемого устройства, включающего композицию с контролируемым высвобождением, содержащую октреотид, которая прочно заключена в полимер на основе полиуретана или метакрилата, при этом имплантируемое устройство выделяет октреотид для субъекта с профилем высвобождения практически нулевого порядка в течение длительного времени, но не менее 6 месяцев, а субъектом является млекопитающее, кроме собак, причем имплантируемое устройство имплантируется субъекту в сухом состоянии. В предпочтительном воплощении гидрофильный полимер включает один или несколько полимеров на основе полиуретана. В предпочтительном воплощении октреотид находится в виде свободного вещества, соли или в виде комплекса, например, ацетата октреотида. В предпочтительном воплощении при имплантировании субъекту имплантируемого устройства субъект получает октреотид со средней скоростью от 75 мкг в сутки до 300 мкг в сутки на протяжении по меньшей мере 6 месяцев. В предпочтительном воплощении при имплантировании имплантируемого устройства субъекту дозировка октреотида, получаемого субъектом, дает уровень октреотида в сыворотке от 0,5 нг/мл до 2 нг/мл. В предпочтительном воплощении при имплантировании имплантируемого устройства субъекту дозировка октреотида, получаемого субъектом, дает уровень октреотида в сыворотке от 0,8 нг/мл до 1,8 нг/мл. В предпочтительном воплощении при имплантировании имплантируемого устройства субъекту дозировка октреотида, получаемого субъектом, дает величину Cmax для уровня октреотида в сыворотке менее 1,2 нг/мл. В предпочтительном воплощении при имплантировании имплантируемого устройства субъекту дозировка октреотида, получаемого субъектом, дает величину Cmax для уровня октреотида в сыворотке менее 1,0 нг/мл. В предпочтительном воплощении при имплантировании имплантируемого устройства субъекту высвобождение октреотида происходит по меньшей мере от 3 до 10 дней после имплантирования.
Одно воплощение касается применения октреотида для изготовления медикамента для лечения субъекта из млекопитающих, кроме собак, при этом медикамент представляет собой композицию с контролируемым высвобождением, которая прочно заключена в полимер на основе полиуретана или метакрилата, тем самым образуя имплантируемое устройство, причем при имплантировании субъекту имплантируемое устройство выделяет октреотид для субъекта с профилем высвобождения практически нулевого порядка в течение длительного времени, но не менее 6 месяцев, а субъектом является млекопитающее, кроме собак, причем имплантируемое устройство имплантируется субъекту в сухом состоянии.
Настоящее изобретение обеспечивает терапевтически эффективное количество октреотида в течение длительного времени, предпочтительно по меньшей мере 2 месяцев, более предпочтительно 6 месяцев и вплоть до 2 лет. Настоящее изобретение также предусматривает композиции, обеспечивающие контролируемое высвобождение октреотида на протяжении по меньшей мере 2 месяцев, предпочтительно 6 месяцев и вплоть до 2 лет.
Воплощения настоящего изобретения касаются фармацевтической композиции, содержащей октреотид либо его соли, пролекарственные формы или производные, которые могут применяться при эффективном лечении различных заболеваний.
Краткое описание чертежей
На фиг.1 приведен график, представляющий линейную зависимость между равновесным водосодержанием и процентным содержанием звеньев гидроксипропилметакрилата (НРМА) в сшитых полимерах НЕМА/НРМА в состоянии максимальной гидратации.
На фиг.2 приведен график, представляющий высвобождение октреотида из композиции импланта настоящего изобретения.
На фиг.3 приведен график, представляющий высвобождение октреотида из композиции импланта настоящего изобретения.
На фиг.4 приведен график, представляющий высвобождение октреотида из композиции импланта настоящего изобретения.
На фиг.5 приведен график, представляющий высвобождение октреотида из композиции импланта настоящего изобретения.
На фиг.6 приведен график, представляющий уровни октреотида и IGF-1 в сыворотке у здоровой собаки, которой имплантировали композицию октреотида по настоящему изобретению.
На фиг.7 приведен график, представляющий уровни октреотида и IGF-1 в сыворотке на протяжении 6 месяцев в группе из 3 здоровых собак, которым имплантировали по одной композиции октреотида настоящего изобретения.
На фиг.8 приведен график, представляющий уровни октреотида и IGF-1 в сыворотке на протяжении 6 месяцев в группе из 3 здоровых собак, которым имплантировали по две композиции октреотида настоящего изобретения.
На фиг.9A и 9B приведены графики, представляющие уровень IGF-1 в сыворотке и его изменения на протяжении 6 месяцев у 11 субъектов с акромегалией, которым имплантировали композицию октреотида по настоящему изобретению.
На фиг.10 приведен график, представляющий уровень октреотида в сыворотке на протяжении 6 месяцев у 11 субъектов с акромегалией, которым имплантировали композицию октреотида по настоящему изобретению.
На фиг.11 приведен график, представляющий уровень октреотида в сыворотке на протяжении 6 месяцев у 2 собак, которым имплантировали композицию октреотида по настоящему изобретению.
На фиг.12 приведен график, представляющий уровень IGF-1 в сыворотке на протяжении 6 месяцев у 2 собак, которым имплантировали композицию октреотида по настоящему изобретению.
На фиг.13 приведен график, представляющий уровни октреотида в сыворотке при выделении его гидратированным имплантом и сухим имплантом (также см. табл.6).
На фиг.14 приведен график, представляющий уровни октреотида в сыворотке при выделении его гидратированным имплантом и сухим имплантом (также см. табл.6).
На фиг.15 приведены графики, представляющие уровень гормона роста при выделении октреотида гидратированными и сухими имплантами (в верхней части - концентрация GH, в нижней части - уменьшение GH в %).
На фиг.16 приведены графики, представляющие уровень инсулиноподобного фактора роста 1 (IGF-1) при выделении октреотида гидратированными и сухими имплантами (в верхней части - концентрация IGF-1, в нижней части - стандартное отклонение).
На фиг.17 приведены графики, представляющие уровень инсулиноподобного фактора роста 1 (IGF-1) при выделении октреотида гидратированными и сухими имплантами (в обеих частях представлены данные опытов, а значения выражены в виде % от нормального уровня).
Раскрытие сущности изобретения
Перед описанием композиций и способов настоящего изобретения следует уяснить, что данное изобретение не ограничивается описываемыми конкретными молекулами, композициями, методами или методиками, поскольку они могут варьироваться. Следует также уяснить, что используемая при описании терминология предназначается только для описания конкретных вариантов или воплощений и не предназначается для ограничения сферы действия настоящего изобретения, которая должна ограничиваться только прилагаемой формулой изобретения. Используемые термины имеют значения, признанные и известные специалистам, однако, для удобства и полноты определенные термины и их значения излагаются ниже.
Формы единственного числа включают и значения множественного, если из контекста четко не следует иное. Если не указано иначе, все технические и научные термины в настоящем изобретении имеют те же значения, что обычно подразумеваются рядовыми специалистами в этой области. Хотя при выполнении или тестировании воплощений настоящего изобретения можно применять способы и материалы, аналогичные или эквивалентные описанным в нем, но предпочтительные способы, устройства и материалы описаны ниже. Все публикации, приведенные в изобретении, включены путем ссылки в пределах того, что они подтверждают настоящее изобретение. Ничто в настоящем изобретении не должно восприниматься как допущение того, что оно не может предвосхищать такое описание по факту предшествующего изобретения.
В настоящем изобретении термин "примерно" означает плюс-минус 10% от численного значения того числа, к которому оно относится. Например, примерно 50% означает в пределах 45-55%.
"Композиция с контролируемым высвобождением" означает композицию, предназначенную для последовательного высвобождения заданного, терапевтически эффективного количества препарата или иного активного агента, как-то полипептида или синтетического соединения, в течение длительного времени, в результате чего уменьшается количество обработок, необходимых для достижения требуемого терапевтического эффекта. Композиция с контролируемым высвобождением уменьшает количество обработок, необходимых для достижения требуемого терапевтического эффекта, в смысле снижения уровня гормона роста или снижения уровня IGF-1 либо улучшения симптомов, связанных, к примеру, с акромегалией, в том числе аномального роста, карциноидного синдрома, VIP-омы (аденомы, секретирующей вазоактивный интестинальный пептид), нейроэндокринных опухолей (в частности лечения симптомов гиперемии и диареи), пролиферативной диабетической ретинопатии, красных угрей, панкреатита, желудочно-кишечных кровотечений, свищей поджелудочной железы и кишечных свищей, базедовой болезни (офтальмопатии Грейвса), глаукомы или лечения симптомов при химиотерапии и СПИД. Композиции с контролируемым высвобождением по настоящему изобретению достигают желательного фармакокинетического профиля у субъекта, предпочтительно начинают высвобождать активное средство практически немедленно после помещения в рабочую среду с последующим постоянным, непрерывным высвобождением активного средства, предпочтительно нулевого или почти нулевого порядка.
В настоящем изобретении термин "контролируемое высвобождение" обозначает заранее установленное постоянное высвобождение активного средства из дозовой формы с такой скоростью, чтобы поддерживался терапевтически полезный уровень активного средства в крови ниже токсического уровня на протяжении, к примеру, по меньшей мере 2 месяцев, 6 месяцев или больше (например, вплоть до 2 лет).
Термины "пациент" или "субъект" охватывают всех животных, включая человека. Примеры пациентов или субъектов включают людей, коров, собак, кошек, коз, овец и свиней.
Термин "фармацевтически приемлемые соли, эфиры, амиды и пролекарственные формы" в настоящем изобретении относится к тем солям карбоновых кислот, солям с аминокислотами, эфирам, амидам и пролекарственным формам соединений настоящего изобретения, которые, в пределах здравого медицинского суждения, подходят для применения в контакте с тканями пациентов без излишней токсичности, раздражения, аллергических реакций и т.п. Их применение соразмерно с удовлетворительным соотношением между выгодой и риском и эффективно для их предназначения. Цвиттерионные формы, по возможности, тоже являются полезными соединениями изобретения. Кроме того, соединения настоящего изобретения могут существовать, к примеру, в несольватированных, равно как и сольватированных формах с фармацевтически приемлемыми растворителями, такими, к примеру, как вода, этанол и др. В общем, сольватированные формы считаются эквивалентными несольватированным формам в целях настоящего изобретения.
Термин "пролекарственная форма" относится к таким соединениям, которые подвергаются быстрому превращению in vivo с образованием исходных соединений вышеприведенной формулы, например, при гидролизе в крови. Всестороннее обсуждение приведено в работах Т. Higuchi and V. Stella, "Pro-drags as Novel Delivery Systems," Vol.14 of the A.C.S. Symposium Series, и Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987, которые обе включены путем ссылки во всей полноте.
Термин "соли" относится к сравнительно нетоксичным солям соединений настоящего изобретения с неорганическими и органическими кислотами. Такие соли могут быть получены in situ при окончательном выделении и очистке соединений или отдельно проведением реакции очищенного соединения в виде свободного основания с подходящей органической или неорганической кислотой и выделением образовавшейся при этом соли. Примеры солей включают соли ацетата, гидробромида, гидрохлорида, сульфата, бисульфата, нитрата, ацетата, оксалата, валерата, олеата, пальмитата, стеарата, лаурата, бората, бензоата, лактата, фосфата, тозилата, цитрата, малеата, фумарата, сукцината, тартрата, нафтилата, мезилата, глюкогептоната, лактобионата, лаурилсульфоната и др. Они могут включать катионы щелочных и щелочноземельных металлов, как-то натрия, лития, калия, кальция, магния и др., а также нетоксические катионы аммония, тетраметиламмония, тетраэтиламмония, метиламина, диметиламина, триметиламина, триэтиламина, этиламина и др. (например, см. S.M.Barge et al., "Pharmaceutical Salts," J. Pharm. Sci., 1977, 66:1-19, которая включена путем ссылки во всей полноте).
"Лечение" означает введение лекарства или проведение медицинских процедур в отношении пациента, либо для профилактики, либо для лечения заболевания или болезни в том случае, когда пациент этим страдает.
"Терапевтически эффективное количество" есть такое количество, которое достаточно для уменьшения, предотвращения или ослабления симптомов, связанных с медицинским заболеванием. В плане гормональной терапии оно может означать и такое количество, которое достаточно для нормализации функций организма или уровня гормонов при заболевании. Например, терапевтически эффективное количество композиции октреотида с контролируемым высвобождением есть предварительно установленное количество, рассчитанное на достижение требуемого эффекта, например, на эффективное снижение уровня гормона роста или IGF-1 у пациента.
Настоящее изобретение может применяться для лечения целого ряда гормональных расстройств, в том числе акромегалии и других заболеваний, нарушений или симптомов, которые эффективно лечатся, к примеру, октреотидом, например, карциноидного синдрома, VIP-омы (аденомы, секретирующей вазоактивный интестинальный пептид), нейроэндокринных опухолей (в частности, лечения симптомов гиперемии и диареи), пролиферативной диабетической ретинопатии, красных угрей, панкреатита, желудочно-кишечных кровотечений, свищей поджелудочной железы и кишечных свищей, базедовой болезни (офтальмопатии Грейвса), глаукомы или лечения симптомов при химиотерапии и СПИД.
Акромегалия характеризуется рядом клинических симптомов, включающих увеличение рук и ног, изменения лица, включая выступание лобных бугров, увеличение нижней челюсти и расстояния между зубами, артралгию, обильное потоотделение, апноэ во сне, гипертензию, сахарный диабет и гипертрофическую кардиомиопатию. Опухоли, вызывающие акромегалию, зачастую вызывают местное анатомическое сдавливание, которое приводит, к примеру, к ограничению поля зрения, головным болям, гипопитуитаризму и парезу черепно-мозговых нервов. При этом у больных акромегалией в 2-5 раз повышается смертность, в основном из-за сердечно-сосудистых и цереброваскулярных заболеваний. Также повышается степень злокачественности в связи с акромегалией, причем лучше всего изучен рак толстой кишки.
Карциноидные опухоли обычно встречаются в аппендиксе, бронхиолах, толстом или тонком кишечнике и секретируют химические вещества, вызывающие расширение кровеносных сосудов типа серотонина. Расширение сосудов может отвечать за симптомы, которые обычно наблюдаются при карциноидных опухолях, таких, к примеру, как диарея, гиперемия и астма. В зависимости от гормонов и биохимических веществ, секретируемых карциноидными опухолями, может отмечаться целый ряд симптомов. Они собирательно известны как "карциноидный синдром". В биохимическом отношении люди с карциноидными опухолями склонны вырабатывать больше серотонина исходя из аминокислоты триптофана, а серотонин далее разрушается в организме с образованием 5-гидрокси-индолуксусной кислоты (5-HIAA), которая обнаруживается в моче у большинства таких больных. Диагностические анализы крови и мочи показывают, что больные с карциноидными опухолями проявляют повышение уровня 5-HIAA в моче, понижение триптофана в крови, повышение хромогранина A и серотонина в крови. Также применяются анализы крови на содержание гистамина, брадикинина, нейроноспецифичной енолазы, кальцитонина, субстанции P, нейрокинина A и панкреатического полипептида.
"ОктреоСкан" означает сканирующий тест для идентификации карциноидных опухолей и нейроэндокринных опухолей. В нем применяется радиоактивное производное октреотида под названием пентетреотид. После инъекции он концентрируется в тканях, экспрессирующих соматостатиновый рецептор. Нейроэндокринные опухоли суперэкспрессируют этот рецептор и выявляются при таком тестировании.
В настоящем изобретении термин "октреотид" относится вообще ко всем соединениям, содержащим приведенную структуру, включая разнообразные соли. Октреотид включает октапептид со следующей последовательностью аминокислот: L-цистеинамид-D-фенилаланил-L-цистеинил-L-фенилаланил-D-триптофанил-L-лизил-L-треонил-N-[2-гидрокси-1-(гидроксиметил)пропил]-цикло(2→7)дисульфид; [R-(R*,R*)]. Ниже представлена структура октреотида:
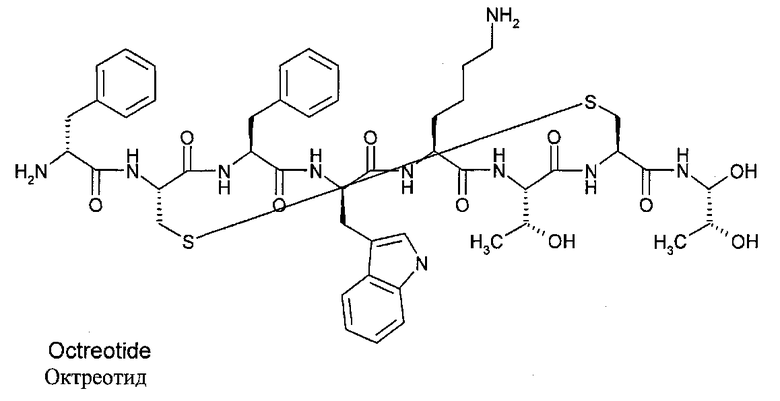
Его химическая формула - C49H66N10O10S2, а молекулярный вес - 1019,3 Да. Терапевтическая категория - желудочное антисекреторное средство. Октреотид по настоящему изобретению может существовать, к примеру, в виде свободного вещества, соли или в виде комплекса. Соли с кислотами могут образовываться, к примеру, с органическими кислотами, полимерными кислотами и неорганическими кислотами. К ним относятся, к примеру, гидрохлориды и ацетаты. Комплексы образуются, к примеру, из октреотида при добавлении неорганических веществ, например, неорганических солей или гидроокисей типа солей Ca и Zn, и/или при добавлении полимерных органических веществ. Предпочтительной солью для композиций настоящего изобретения является ацетатная соль.
Воплощениями настоящего изобретения предусмотрено устройство для доставки лекарств, которое может выполнять следующие цели: контролируемая скорость высвобождения (кинетика высвобождения нулевого или почти нулевого порядка), чтобы максимально увеличить терапевтические эффекты и свести к минимуму нежелательные побочные эффекты; удобный способ извлечения устройства, если нужно прекратить лечение; и повышение биодоступности с меньшими отклонениями всасывания и отсутствием пресистемного метаболизма.
Фармацевтическая композиция с контролируемым высвобождением, содержащая ацетат октреотида, может быть частью гидрогелевого устройства для контролируемого высвобождения. Композиция настоящего изобретения способна обеспечить, при введении пациенту, такой профиль высвобождения октреотида, который растянется по меньшей мере на 2 месяца, предпочтительно на 6 месяцев или больше, например, вплоть до 2 лет. Октреотид может находиться, к примеру, внутри гидрогеля, а композиция будет выделять терапевтически эффективное количество октреотида в течение длительного времени. Гидрогель может включать полимер, выбранный из полимеров на основе метакрилата, полимеров на основе полиуретана и их комбинаций. Терапевтически эффективное количество есть такое количество октреотида, предпочтительно ацетата октреотида, которое при введении пациенту или субъекту ослабляет симптомы акромегалии. Композиция может дополнительно включать фармацевтически приемлемые наполнители.
При введении композиций настоящего изобретения пациентам концентрация октреотида в плазме пациента по времени (профиль высвобождения) может сохраняться на протяжении по меньшей мере 2 месяцев, предпочтительно 6 месяцев и вплоть до 2 лет. Композиции могут обеспечить среднюю концентрацию октреотида в плазме человека в стационарном состоянии от 0,1 до 9 нг/мл, от 5 до 1 нг/мл, от 1 до 2 нг/мл или от 1,2 до 1,6 нг/мл. Стационарное состояние - это точка, в которой количество введенного за данный интервал препарата равна количеству препарата, подвергнувшегося элиминации за тот же самый период.
Гидрофильный имплант, содержащий композицию октреотида, может быть сформирован из ксерогеля с тем, чтобы он хорошо впитывал воду. В гидратированном состоянии ксерогель именуется гидрогелем. В любой форме, гидратированной или не гидратированной, он биосовместим и не токсичен для организма и не подвержен биодеградации. Он набухает, но не растворяется в воде. Когда гидрогель достигает максимальной степени гидратации, его водосодержание именуется "равновесным водосодержанием" (EWC). Относительное водосодержание гидрогеля (в любой степени гидратации) определяется следующим образом:
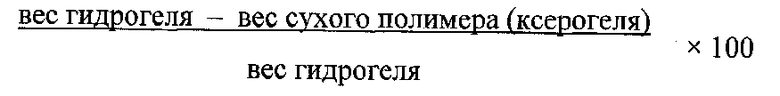
Гидрогель может представлять собой гомогенный гомополимер или сополимер с заданной величиной равновесного водосодержания (EWC), образовавшийся при полимеризации смеси из этиленового ненасыщенного мономера A и этиленового ненасыщенного мономера B, например 2-гидроксиэтилметакрилата (HEMA) и гидроксипропилметакрилата (HPMA). Заданную величину EWC можно рассчитать путем определения значений EWC гомополимера гидрогеля из гидрофильного мономера A (гомополимера A) и гомополимера гидрогеля из гидрофильного мономера В (гомополимера B); определения зависимости значений EWC гомогенных сополимеров AB от химического состава данных сополимеров АВ; выбора намеченного значения EWC и определения химического состава сополимера AB с намеченным значением EWC; составления полимеризуемой смеси из мономера A и мономера B в количествах, достаточных для получения сополимера АВ с намеченным значением EWC; и выполнения реакции полимеризации, дающей сополимер AB, характеризующийся намеченным значением EWC.
В настоящем изобретении "сополимер AB" или "сополимер AB, состоящий в основном из звеньев мономера A и звеньев мономера В", означает то, что полиприсоединение мономера A и мономера B осуществлялось через полимеризуемую этиленовую связь мономеров. Для примера, если мономером A является 2-гидроксиэтилметакрилат, а мономером B является N-метилакриламид, то сополимер AB содержит повторяющиеся звенья мономера A и повторяющиеся звенья мономера B.
Если из контекста не следует иное, термин "сополимер" охватывает полимеры, полученные при полимеризации смеси по меньшей мере двух ненасыщенных этиленовых мономеров.
В настоящем изобретении "звено HEMA" означает повторяющуюся структуру в полимере, полученном при полимеризации гидрофильного материала, содержащего 2-гидроксиэтилметакрилат ("HEMA"). Термином "звено HEMA" обозначается структура:
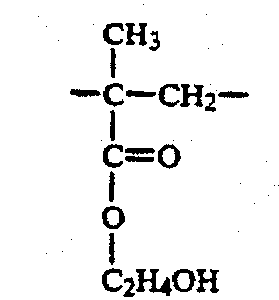
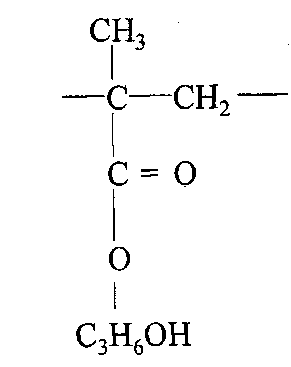
В настоящем изобретении "звено HPMA" означает повторяющуюся структуру в полимере, полученном при полимеризации гидрофильного материала, содержащего гидроксипропилметакрилат ("HPMA"). Термином "звено НРМА" обозначается структура:
Жидкие полимеризуемые материалы, используемые в гидрофильных продуктах, включают широкий круг полимеризуемых гидрофильных, ненасыщенных этиленовых соединений, в частности гидрофильных мономеров, таких, к примеру, как моноэфиры акриловой кислоты или метакриловой кислоты с полигидроксисоединениями, содержащими эфирообразующую гидроксильную группу и по меньшей мере еще одну гидроксильную группу, как-то, к примеру, моноалкиленовые и полиалкиленовые полиоли метакриловой кислоты и акриловой кислоты, например, 2-гидроксиэтилметакрилат и -акрилат, диэтиленгликольметакрилат и -акрилат, пропиленгликольметакрилат и -акрилат, дипропиленгликольметакрилат и -акрилат, глицидилметакрилат и -акрилат, глицерилметакрилат и -акрилат и др.; 2-алкенамиды, например акриламид, метакриламид и др.; N-алкил- и N,N-диалкилзамещенные акриламиды и метакриламиды типа N-метилметакриламида, N,N-диметилметакриламида и др.; N-винилпирролидон; алкилзамещенные N-винилпирролидоны, например метилзамещенный N-винилпирролидон; N-винилкапролактам; алкилзамещенные N-винилкапролактамы, например N-винил-2-метилкапролактам, N-винил-3,5-диметилкапролактам и др. В этих композициях также могут использоваться акриловая и метакриловая кислота.
В реакции полимеризации используются смеси гидрофильных мономеров. Тип и пропорции мономеров выбираются так, чтобы получить гомогенный полимер, предпочтительно сшитый гомогенный полимер, который при гидратации будет иметь требуемую величину EWC для предусмотренного применения. Такую величину EWC можно определить заранее путем приготовления ряда сополимеров с использованием различных соотношений мономеров, например смесей НЕМА и НРМА в различных пропорциях; установления величины EWC этих сополимеров; и составления графика зависимости относительной величины EWC этих сополимеров от % звеньев НРМА (или НЕМА) в сополимерах НРМА/НЕМА (фиг.1).
В некоторых случаях полимеризация определенных смесей гидрофильных мономеров приводит к образованию таких гомогенных гидрофильных сополимеров, которые в некоторой степени растворяются в водной среде. В таких случаях в смесь мономеров можно включить небольшое количество, например до 3% сополимеризуемого ненасыщенного многоэтиленового сшивающего реагента, получая гомогенные сшитые сополимеры, которые нерастворимы в воде, но набухают в воде. Малосшитые гомополимеры НЕМА могут иметь значение EWC, к примеру, около 38%. Сшитые сополимеры НЕМА и НРМА имеют значения EWC менее 38%. С другой стороны, сшитые сополимеры НЕМА и акриламида проявляют значения EWC более 38%, например, вплоть до 75% и выше. Таким образом, в зависимости от полезной или эффективной скорости выделения активного соединения, например лекарственного препарата, которая требуется от гидрогелевой системы доставки для конкретного применения, специалист в этой области, следуя изложенным в настоящем изобретении предписаниям, может приспособить гидрогелевые мембраны из сополимера для выделения препарата с нужной скоростью. Сополимеры могут содержать, к примеру, от 15% до 70% масс, звеньев НЕМА и от 85 до 30% масс. звеньев второго этиленового мономера будут иметь заданные значения EWC в пределах от 20% до 75%, предпочтительно около 25%. Гомогенные сополимеры могут включать сополимеры, сделанные из смеси гидрофильных мономеров, содержащей около 80% масс. НРМА и около 20% масс. НЕМА. В дополнительных воплощениях смесь может дополнительно содержать небольшое количество многоэтиленового ненасыщенного сшивающего реагента, например триметилолпропантриметакрилата ("ТМРТМА").
Различные аспекты изобретения включают гомогенные гидрофильные сополимеры, у которых структура гомогенного полимера образуется при полимеризации смеси гидрофильных мономеров, описанных ранее; и устройство для доставки препаратов, в котором используются картриджи из гомогенного полимера в системе доставки. Полимеризация смеси из гидрофильных мономеров и гидрофобных мономеров дает гетерогенные полимеры. Если в полимере есть гидрофобные сегменты, то межфазная свободная энергия возрастает, что повышает всасывание белка и минерализацию после имплантирования животному. При измерении межфазной свободной энергии у гидрогелей из поли-НЕМА, к примеру, она оказалась близкой к нулю. В соответствии с интерпретацией межфазной свободной энергии гидрогели из чисто гидрофильных компонентов должны оказаться биосовместимыми с тканями организма. Малосшитый НЕМА является гомогенным гидрофильным гомополимером (не учитывая сравнительное небольшое количество в нем полимеризованного сшивающего реагента) с относительно фиксированными характеристиками или параметрами. Методы изменения "гомополимерного" поли-НЕМА для придания ему дополнительных характеристик или свойств являются трудными, отнимают много времени и зачастую приводят к неустойчивому их поведению. С другой стороны, при полимеризации смесей НЕМА с различным количеством других полимеризуемых гидрофильных сомономеров можно получить предсказуемые гомогенные гидрофильные сополимеры с (заданными) нестандартными свойствами.
Полезные сшивающие реагенты, которые можно включать в среду для реакции полимеризации, включают, к примеру, многоэтиленовые ненасыщенные соединения, содержащие по меньшей мере два полимеризуемых этиленовых участка, как-то ди-, три- и тетраэтиленовые ненасыщенные соединения, в частности трижды ненасыщенные сшивающие реагенты с дважды ненасыщенными сшивающими соединениями или без них, например дивинилбензол, этиленгликольдиметакрилат и -диакрилат, пропиленгликоль-диметакрилат и -диакрилат, и ди-, три- и тетраакриловые или метакриловые эфиры следующих полиолей: триэтаноламина, глицерина, пентаэритритола, 1,1,1-триметилолпропана и др.
Реакция полимеризации может проводиться в массе или с инертным растворителем. Подходящие растворители включают, к примеру, воду, органические растворители (например, водорастворимые низшие алифатические одноатомные спирты, а также многоатомные спирты, например гликоль, глицерин, диоксан и др.; а также их смеси).
Соединения, полезные при катализе полимеризуемых этиленовых ненасыщенных соединений, включают свободнорадикальные соединения и/или инициаторы типа тех, что обычно применяются при винильной полимеризации, как-то органические пероксиды, перкарбонаты, гидропероксиды и сульфаты щелочных металлов. Показательные примеры включают гидропероксид кумена, трет-бутилгидропероксид, бензоилпероксид, бис(4-трет-бутилциклогексил)пероксидикарбонат, пероксид водорода, 2,4-дихлорбензоил-пероксид, ацетилпероксид, ди-н-пропилпероксидикарбонат, ди-трет-бутилпероксид, ди-втор-бутилпероксидикарбонат, сульфат аммония, сульфат калия и сульфат натрия. Предпочтительным катализатором является такой, который эффективен при умеренно низкой температуре, такой, к примеру, как 20-80°C (например, трет-бутилпероктоат, бензоилпероксид и ди-втор-бутилпероксидикарбонат).
Также можно использовать стандартный окислительно-восстановительный катализатор полимеризации. Полимеризация этиленовых соединений может осуществляться, к примеру, с помощью облучения, например, ультрафиолетом, рентгеновского облучения, гамма-облучения, микроволнового или иного хорошо известного облучения. Предпочтительным катализатором при облучении ультрафиолетом является метиловый эфир бензоина. Для оптимизации реакции полимеризации катализаторы и/или инициаторы и/или облучение применяются в каталитически эффективном количестве.
Настоящее изобретение обращено на использование полимеров, термопластов или термореактивных пластмасс на основе полиуретана, на создание имплантируемых устройств с препаратами для выделения биологически активных соединений с контролируемой скоростью в течение длительного времени. Из полиуретановых полимеров предпочтительно делают цилиндрические полые трубочки с одним или двумя открытыми концами посредством экструзии, (реактивного) литьевого прессования, прямого формования либо отливки с центрифугированием (например, см. US Pat. Nos. 5266325 и 5292515, которые включены путем ссылки во всей полноте), в зависимости от типа полиуретана.
Термопластичный полиуретан можно обрабатывать посредством экструзии, литьевого прессования или прямого формования. Термореактивный полиуретан можно обрабатывать посредством реактивного литьевого прессования, прямого формования или отливки с центрифугированием. Размеры цилиндрической полой трубочки можно установить и точно подогнать.
Полимеры на основе полиуретана синтезируют из многофункциональных полиолей, изоцианатов и удлинителей цепи. Характеристики каждого полиуретана определяются его структурой.
Термопластичный полиуретан получают из макродиолов, диизоцианатов и двухфункциональных удлинителей цепи (например, см. US Pat. Nos. 4523005 и 5254662, которые включены путем ссылки во всей полноте). Макродиолы составляют мягкие домены. Диизоцианаты и удлинители цепи составляют твердые домены. Твердые домены служат физическими центрами сшивания для полимеров. Варьирование пропорций этих двух доменов может изменить физические характеристики полиуретана.
Термореактивный полиуретан можно получить из многофункциональных (более чем двухфункциональных) полиолов и/или диизоцианатов и/или удлинителей цепи (например, US Pat. Nos. 4386039 и 4131604, которые включены путем ссылки во всей полноте). Термореактивный полиуретан также можно получить путем введения ненасыщенных связей в цепи полимера и химической сшивки с помощью соответствующих сшивающих реагентов и/или инициаторов (например, US Pat. No. 4751133, который включен путем ссылки во всей полноте). Контролируя количество центров сшивания и то, как они распределены, можно контролировать скорость высвобождения активных веществ.
В зависимости от требуемых свойств, можно ввести различные функциональные группы в полимерные цепи полиуретана посредством модификации остова полиолей. Если устройство используется для доставки водорастворимых препаратов, то в полиоли можно встроить гидрофильные боковые группы типа ионных, карбоксильных, эфирных или гидроксильных групп, чтобы повысить гидрофильность полимера (например, US Pat. Nos. 4743673 и 5354835, которые включены путем ссылки во всей полноте). Если же устройство используется для доставки гидрофобных препаратов, то в полиоли можно встроить гидрофобные боковые группы типа алкильных и силоксановых, чтобы повысить гидрофобность полимера (например, US Pat. No. 6313254, который включен путем ссылки во всей полноте). Скорость высвобождения активных веществ также можно контролировать путем изменения гидрофильности/гидрофобности полиуретановых полимеров.
Небольшие импланты цилиндрической формы по изобретению могут содержать внутри октреотид, предпочтительно ацетат октреотида, и необязательно фармацевтически приемлемый носитель. Толщина мембраны (между внутренней и внешней поверхностью) у импланта практически одинакова, и она служит лимитирующим скорость барьером для высвобождения содержащегося внутри средства. Такие импланты можно пластифицировать или гидратировать и превращать в изделия другой геометрической формы для разнообразного медицинского применения.
При изготовлении имплантируемой формы нужно учитывать несколько факторов. Определяется профиль высвобождения (время задержки, скорость и продолжительность высвобождения); устанавливается гидрофильный полимерный материал; и измеряется диффузивность активного средства через нее (в качестве лимитирующей скорость мембраны). Профиль гидратации лимитирующей скорость мембраны для данного активного средства можно легко определить путем изготовления пленки из выбранного полимера и исследования ее на диффузию с помощью вертикальной стеклянной кюветы из двух отсеков, как это хорошо известно в данной области.
Определяется коэффициент диффузии и водосодержание, при котором начинается диффузия (ниже которого диффузия практически не происходит - в дальнейшем "% Hd"). Готовится серия мембран из различных полимеров. Затем мембраны подвергают гидратации до отказа и измеряют значения EWC. Полностью гидратированные мембраны помещают в вертикальные стеклянные кюветы из двух отсеков для измерения и строят график диффузии макромолекулярной композиции через материал мембраны при различных значениях EWC. Равновесное водосодержание наиболее гидратированной мембраны, через которую не отмечается никакой диффузии (например, никакой диффузии активного агента в принимающую ячейку), и составляет % Hd для тестируемой системы. Это осуществляется построением графика зависимости проницаемости от EWC.
Данные о проницаемости (коэффициенты диффузии) получают согласно первому закону диффузии Фика при помощи уравнения
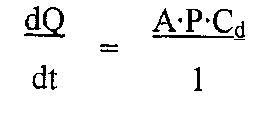
где dQ/dt означает поток через материал мембраны (мкг/ч), который измеряется по наклону линейной части графика зависимости кумулятивного переноса от времени; A - площадь мембраны (см2); P - коэффициент проницаемости мембраны (см2/ч), то есть DKd, где D означает диффузивность мембраны (см2/ч), a Kd - коэффициент распределения между мембраной и донорным раствором; l - толщина мембраны, измеренная по окончании эксперимента (см); a Cd - концентрация донорного раствора (мкг/см3).
Затем можно определить профиль задержки высвобождения. Готовится другая серия полимерных мембран, опять же с различным количеством сшивающего реагента и мономеров. Затем эти мембраны подвергают гидратации, но только частично, например, до водосодержания, меньшего или равного величине %Hd. Частично гидратированные мембраны помещают в вертикальные стеклянные кюветы из двух отсеков для измерения и строят графики диффузии активного соединения через мембраны в зависимости от времени. Буферные растворы для донорных и принимающих ячеек выбираются так, чтобы они контактировали с частично гидратированными мембранами и дополнительно их гидратировали примерно с такой же скоростью, с которой они будут гидратироваться в среде доставки. Время между началом опыта по диффузии, т.е. добавлением активного агента в донорную ячейку, и выявлением фармацевтически эффективной концентрации активного агента в принимающей ячейке и составляет время задержки высвобождения для этого сочетания полимера и исходного процента гидратации.
Для определения физических размеров устройства цилиндрической формы определяется общее количество подлежащего доставке активного агента. Это есть произведение требуемой суточной дозы на продолжительность выделения. В предпочтительных воплощениях продолжительность выделения составляет по меньшей мере 2 месяца, более предпочтительно 6 месяцев и вплоть до 2 лет. Требуемая суточная доза составляет, к примеру, от 10 до 1000 мкг октреотида в сутки, предпочтительно от 20 до 800 мкг октреотида в сутки, более предпочтительно от 30 до 300 мкг октреотида в сутки.
Объем цилиндрического резервуара (сердцевины) устройства цилиндрической формы равняется πri 2·h, где ri означает радиус резервуара, a h - его высоту. Формула для стационарного высвобождения из цилиндра такова:
[dQ/dt]=[2π·h·D·Kd·Cd]/[ln(ro/ri)]
где ro означает внешний радиус цилиндрического устройства, a Cd - концентрация препарата в донорном растворе, т.е. носителе. Стационарное высвобождение происходит тогда, когда Cd поддерживается на уровне насыщения. Следовательно, толщина мембраны, необходимая для нужного непрерывного высвобождения, составляет ro-ri.
Количество используемого активного агента будет зависеть не только от требуемой суточной дозы, но и от того, сколько дней нужно поддерживать этот уровень дозы. Хотя это количество можно рассчитать эмпирически, однако фактическая выделяемая доза зависит от любых взаимодействий с материалами и носителем, если он применяется в устройстве.
Как только будет выбран надлежащий полиуретановый полимер, специалист в этой области сможет установить наилучший метод изготовления имплантов цилиндрической формы для достижения подходящих характеристик доставки, как описано в настоящем изобретении.
Для термопластичного полиуретана можно использовать прецизионную экструзию и литьевое прессование для получения полых трубочек с двумя открытыми концами и соответственными физическими размерами. Резервуар можно свободно наполнить соответствующими составами, содержащими активные вещества и носители, либо заполнить изготовленными заранее гранулами, чтобы увеличить до максимума содержание активных веществ. Для заделывания двух открытых концов можно использовать две изготовленные заранее торцевые заглушки. Операция заделывания может выполняться с применением нагревания или растворителя или иного средства для заделывания концов, предпочтительно навсегда.
Для термореактивного полиуретана предпочтительным методом является прецизионное реактивное литьевое прессование или отливка с центрифугированием, в зависимости от механизма отверждения. Реактивное литьевое прессование применяется, если отверждение проводится посредством термообработки, а отливка с центрифугированием применяется, если отверждение проводится посредством световой обработки и/или термообработки. Предпочтительно полые трубочки с одним открытым концом делают методом отливки с центрифугированием. Предпочтительно полые трубочки с двумя открытыми концами делают методом реактивного литьевого прессования. Резервуар можно заполнять таким же образом, как и для термопластичного полиуретана.
Предпочтительно для заделывания открытого конца применяется заполнение открытого конца соответствующей композицией термореактивного полиуретана, подлежащей световой и/или термообработке, которая подвергается отверждению с помощью света и/или нагревания. Более предпочтительно для заделывания открытого конца можно использовать изготовленную заранее торцевую заглушку путем наложения соответствующей композиции термореактивного полиуретана, подлежащей световой и/или термообработке, на стыке между готовой торцевой заглушкой и открытым концом и отверждения ее с помощью света и/или нагревания либо иного средства для заделывания концов, предпочтительно навсегда. Твердые активные средства и необязательные наполнители можно прессовать в виде гранул, чтобы увеличить до максимума загрузку активных ингредиентов.
Перед имплантированием имплантируемые композиции можно необязательно гидратировать или "примировать" в течение заданного времени. Примирование может способствовать тому, что активный ингредиент начнет проникать в стенки из гидрогеля и насыщать их и может начать просачиваться из гидрогеля еще до имплантации в зависимости от продолжительности примирования импланта. Примированный имплант начнет выделять активный ингредиент практически сразу после имплантирования, поэтому максимальное высвобождение препарата может установиться вскоре после имплантирования. Напротив, слабое примирование или его отсутствие может привести к тому, что активный ингредиент практически не будет высвобождаться после имплантации какое-то время до того, как произойдет гидратация импланта и активный ингредиент начнет выделяться, однако эти характеристики примирования зависят от конкретного состава композиции. Так, изобретение предусматривает способ введения композиции октреотида с контролируемым высвобождением, включающий имплантирование субъекту дегидратированной композиции октреотида.
В зависимости от типа активного ингредиента - гидрофильного или гидрофобного - нужно выбрать соответствующие среды кондиционирования и примирования. Водные среды предпочтительны для гидрофильных активных ингредиентов, а масляные среды предпочтительны для гидрофобных активных ингредиентов. С другой стороны, некоторые импланты изобретения не нуждаются в примировании перед имплантацией. В некоторых случаях примирование улучшает доставку активного агента контролируемым образом, но в других случаях примирование перед имплантацией субъекту не влияет на доставку так, чтобы это оправдывало возрастание времени и трудов, необходимых для примирования импланта.
Гидратирующая жидкость, используемая при практическом применении изобретения, как правило, это жидкость, напоминающая ту среду, в которую будет выделяться активное соединение, например жидкость организма, стерильная вода, слезная жидкость, физиологический солевой раствор, раствор фосфатного буфера и др. Хотя в качестве гидратирующей жидкости применяются и другие жидкости, чем вода, однако степень гидратации гидрофильной мембраны обозначается как ее "водосодержание".
Примирование и кондиционирование устройств доставки лекарств включает введение активного ингредиента (препарата) в полимер, окружающий резервуар, что предотвращает потери активного ингредиента до фактического применения импланта. Условия, используемые для стадии кондиционирования и примирования, зависят от активного агента, температуры и среды, в которой они проводятся. В некоторых случаях условия кондиционирования и примирования могут быть одинаковыми.
Стадия кондиционирования и примирования в процессе подготовки устройств доставки лекарств выполняется для того, чтобы добиться заданной скорости высвобождения определенного препарата. Стадия кондиционирования и примирования импланта, содержащего гидрофильный препарат, предпочтительно выполняется в водной среде, более предпочтительно в физрастворе. Для гидрофобных препаратов среда может представлять собой среду, похожую на плазму, в том числе цикл о декстрин. Операции кондиционирования и примирования выполняются с контролированием 3 специфических факторов, а именно температуры, среды и продолжительности.
Специалист в этой области должен понимать, что на стадию кондиционирования и примирования устройств доставки лекарств влияет среда, в которую будет помещено устройство. Например, гистрелиновые и налтрексоновые импланты можно кондиционировать и примировать в физрастворе, более предпочтительно кондиционировать в физрастворе с 0,9% NaCl и примировать в физрастворе с 1,8% NaCl.
Температура при кондиционировании и примировании устройств доставки лекарств может колебаться в широких пределах, но в некоторых случаях используют 37°C.
Продолжительность кондиционирования и примирования устройств доставки лекарств может колебаться от одного дня до нескольких недель в зависимости от того, какая скорость высвобождения требуется для конкретного импланта или препарата.
Специалист в этой области должен понимать, что стадии кондиционирования и примирования имплантов, когда это нужно, служат для оптимизации скорости высвобождения препарата, содержащегося внутри импланта. При этом сокращение продолжительности кондиционирования и примирования устройства доставки лекарств может привести, к примеру, к снижению скорости высвобождения препарата по сравнению с аналогичным устройством доставки лекарств, подвергавшимся более длительной стадии кондиционирования и примирования. Однако в отсутствие примирования неожиданно оказалось, что эффективное высвобождение происходит в течение более длительного времени (например, 28 недель и больше), а меньшие концентрации активного ингредиента в сыворотке оказывают улучшающее действие.
Температура на стадии кондиционирования и примирования также может повлиять на скорость высвобождения с тем, что снижение температуры ведет к снижению скорости высвобождения препарата, содержащегося в устройстве доставки лекарств, по сравнению с аналогичным устройством доставки лекарств, подвергавшимся обработке при более высокой температуре.
Аналогичным образом в случае водных растворов, которые в некоторых случаях предпочтительно являются солевыми растворами, содержание NaCl в растворе также определяет то, какая скорость высвобождения будет у устройства доставки лекарств. В частности, снижение содержания NaCl может привести к повышению скорости высвобождения препарата по сравнению с устройством доставки лекарств, подвергавшимся операции кондиционирования и примирования при более высоком содержании NaCl.
Для того чтобы установить локализацию импланта, в устройство доставки может быть включен рентгеноконтрастный материал путем введения его в резервуар либо при изготовлении торцевой заглушки, используемой для заделывания картриджа.
Композиция настоящего изобретения может содержать фармацевтически приемлемый носитель, который может включать, к примеру, суспендирующие среды, растворители, водные системы и твердые субстраты или матриксы.
Суспендирующие среды и растворители, применимые в качестве носителя, включают, к примеру, масла, как-то силиконовое масло (особенно медицинское), кукурузное масло, касторовое масло, арахисовое масло и кунжутное масло; продукты конденсации касторового масла с этиленоксидом; жидкие эфиры триглицерила с низкомолекулярными жирными кислотами; низшие алканолы; гликоли; и полиалкиленгликоли.
Водные системы включают, к примеру, стерильную воду, физраствор, декстрозу, декстрозу в воде или физрастворе и др. Присутствие электролитов в водных системах может уменьшить растворимость в них макромолекулярных препаратов.
Твердые субстраты или матриксы включают, к примеру, крахмал, желатин, сахара (например, глюкозу), природные камеди (например, гуммиарабик, альгинат натрия), карбоксиметилцеллюлозу и др. В предпочтительном воплощении фармацевтическая форма дополнительно содержит от 2% до 20%, более предпочтительно 10% гидроксипропилцеллюлозы. Кроме того, фармацевтические формы могут содержать и гидроксипропилцеллюлозу, гидроксиэтилцеллюлозу, метилцеллюлозу, натриевую карбоксиметилцеллюлозу, модифицированный крахмал или сшитый поливинилпирролидон.
Носитель также может содержать вспомогательные вещества, как-то консерванты, стабилизирующие, увлажняющие и эмульгирующие вещества и др.
В одном воплощении фармацевтическая форма настоящего изобретения содержит композицию октреотида ацетата внутри сополимера из смеси НЕМА и НРМА, предпочтительно 20% НЕМА и 80% НРМА. Фармацевтическая форма может содержать, к примеру, от 20 до 150 мг октреотида, предпочтительно от 40 до 90 мг. Форма может дополнительно содержать от 2 до 20% наполнителей. Форма также может содержать около 10% гидроксипропилцеллюлозы и/или 2% стеарата магния.
Фармацевтическая форма настоящего изобретения может содержать композицию из 50 мг октреотида внутри сополимера из смеси НЕМА и НРМА, предпочтительно 20% НЕМА и 80% НРМА. В следующем воплощении форма дополнительно содержит около 10% гидроксипропилцеллюлозы и 2% стеарата магния вместе с ацетатом октреотида.
Фармацевтическая форма настоящего изобретения может содержать и композицию из 83 мг октреотида внутри сополимера из смеси НЕМА и НРМА, предпочтительно 40% НЕМА и 60% НРМА. В следующем воплощении форма дополнительно содержит около 10% гидроксипропилцеллюлозы и 2% стеарата магния вместе с ацетатом октреотида. Фармацевтическая форма также может содержать стеариновую кислоту, растительный стеарин, тальк и диоксид кремния.
Фармацевтическая форма настоящего изобретения также может содержать композицию от 20 мг до 150 мг, более предпочтительно от 40 мг до 90 мг октреотида в ксерогеле, предпочтительно гидрогеле или полимере на основе полиуретана.
Предусмотрен способ лечения заболеваний, связанных с гормональными нарушениями. Способ может включать введение октреотида и поддержание концентрации октреотида в плазме в стационарном состоянии от 0,1 нг/мл до 9 нг/мл в течение длительного времени, предпочтительно по меньшей мере 2 месяцев, более предпочтительно около 6 месяцев и вплоть до 2 лет. В предпочтительном воплощении концентрация октреотида в плазме в стационарном состоянии составляет от 1 нг/мл до 2 нг/мл, более предпочтительно от 1,2 нг/мл до 1,6 нг/мл, в течение длительного времени. Гормональные нарушения включают, к примеру, акромегалию.
Изобретение также касается способов снижения уровня GH путем введения октреотида и поддержания стационарной концентрации октреотида в плазме от 0,1 нг/мл до 9 нг/мл, от 0,5 нг/мл до 1 нг/мл, от 1 нг/мл до 2 нг/мл либо от 1,2 нг/мл до 1,6 нг/мл в течение длительного времени, предпочтительно по меньшей мере 2 месяцев, более предпочтительно около 6 месяцев и вплоть до 2 лет.
Изобретение также касается способов снижения уровня IGF-1 путем введения октреотида и поддержания стационарной концентрации октреотида в плазме от 0,1 нг/мл до 9 нг/мл, от 0,5 нг/мл до 1 нг/мл, от 1 нг/мл до 2 нг/мл либо от 1,2 нг/мл до 1,6 нг/мл в течение длительного времени, предпочтительно по меньшей мере 2 месяцев, более предпочтительно около 6 месяцев и вплоть до 2 лет.
Изобретение также касается способов лечения акромегалии, включающих введение по меньшей мере одного импланта настоящего изобретения, предпочтительно двух имплантов настоящего изобретения. В этом способе каждый вводимый имплант может содержать от 20 до 150 мг октреотида, предпочтительно от 40 до 90 мг октреотида, более предпочтительно около 50 мг октреотида, и выделять терапевтически эффективное количество октреотида на протяжении по меньшей мере 2 месяцев, более предпочтительно около 6 месяцев и вплоть до 2 лет.
Изобретение также касается способов лечения симптомов, связанных с карциноидными опухолями и VIP-омами. Настоящее изобретение также охватывает способы лечения тяжелой диареи и эпизодов гиперемии в связи с карциноидными опухолями путем введения имплантируемой композиции октреотида, выделяющей терапевтически эффективное количество октреотида на протяжении по меньшей мере 2 месяцев, более предпочтительно около 6 месяцев и вплоть до 2 лет. Настоящее изобретение также охватывает способы лечения водянистого поноса в связи с VIP-омами путем введения имплантируемой композиции октреотида, выделяющей терапевтически эффективное количество октреотида на протяжении по меньшей мере 2 месяцев, более предпочтительно около 6 месяцев и вплоть до 2 лет.
Лекарственные формы настоящего изобретения проявляют специфический, желательный профиль высвобождения, который увеличивает до максимума терапевтический эффект и при этом уменьшает до минимума отрицательные побочные эффекты импланта. Желательный профиль высвобождения может быть описан в терминах максимальной концентрации препарата или активного агента в плазме (Cmax) и концентрации препарата или активного агента в плазме в стационарном состоянии (Css).
Настоящее изобретение также касается терапевтических композиций гидрогеля и октреотида, из которой после имплантирования октреотид выделяется со скоростью, обеспечивающей и/или поддерживающей величину Css от 0,1 нг/мл до 9 нг/мл, от 0,5 нг/мл до 1 нг/мл, от 1 нг/мл до 2 нг/мл или от 1,2 нг/мл до 1,6 нг/мл. Следующее воплощение составляет терапевтическая композиция гидрогеля и октреотида, из которой после имплантирования октреотид выделяется со скоростью от 10 мкг до 1000 мкг в сутки в течение длительного времени, предпочтительно от 20 мкг до 800 мкг в сутки, более предпочтительно от 30 мкг до 300 мкг в сутки. Октреотид может выделяться на протяжении по меньшей мере 2 месяцев, около 6 месяцев или вплоть до 2 лет. Гидрогель может включать полимеры на основе метакрилата или полимеры на основе полиуретана.
Следующее воплощение составляет лекарственная форма с контролируемым высвобождением, включающая октреотид и гидрофильный полимер, которая позволяет высвобождение октреотида со скоростью от 30 мкг до 250 мкг в сутки на протяжении по меньшей мере 2 месяцев, около 6 месяцев или вплоть до 2 лет in vitro. В некоторых воплощениях доставка составляет от 100 мкг до 130 мкг в сутки. В следующем воплощении гидрофильный полимер лекарственной формы позволяет высвобождение октреотида со средней скоростью около 100 мкг в сутки in vitro. Гидрофильный полимер может быть выбран из полимеров на основе полиуретана и полимеров на основе метакрилата.
Следующее воплощение настоящего изобретения составляет содержащая октреотид лекарственная форма для имплантирования, в которой октреотид содержится в гидрофильном полимере, эффективно позволяющем высвобождение из нее in vitro не более 20% октреотида за 6 недель и около 60% октреотида за 6 месяцев.
Количество фармацевтически приемлемого октреотида (например, различных солей, сольватационных состояний или его пролекарственных форм), включенного в фармацевтическую композицию настоящего изобретения, будет зависеть от целого ряда факторов, в том числе, к примеру, от конкретного октреотида, нужного уровня дозы, типа и количества гидрогеля и присутствия, типа и количества других материалов, включенных в композицию. Количество октреотида или его производных в композиции зависит от желательной дозы для эффективной доставки препарата, молекулярного веса и активности соединения. Фактическое количество используемого препарата может зависеть от возраста пациента, его веса, пола, медицинского состояния, заболевания или иных медицинских критериев. Фактическое количество препарата определяется согласно его медицинскому предназначению известными методами. Фармацевтическая доза в составе формы по изобретению может вводиться, к примеру, раз в 6 месяцев, что устанавливается лечащим врачом.
Как правило, октреотид входит в состав импланта или иной фармацевтической композиции в количестве от 20 мг до 150 мг, предпочтительно от 40 до 90 мг октреотида, более предпочтительно от 50 до 85 мг. Для взрослых суточная доза при лечении акромегалии обычно составляет от 300 мкг до 600 мг октреотида с немедленным высвобождением в сутки (100 мкг или 200 мкг Sandostatin®). Количество октреотида в композиции можно выбрать, к примеру, так, чтобы высвобождалось от 10 мкг до 1000 мкг в сутки в течение длительного времени, от 20 мкг до 800 мкг в сутки или от 30 мкг до 300 мкг в сутки. Такая скорость высвобождения поддерживает требуемый терапевтический уровень в крови пациента от 0,1 до 9 нг/мл в течение длительного времени.
Гидрогелевое устройство, в котором содержится октреотид, обеспечивает контролируемое высвобождение октреотида в плазму пациента. Гидрогели, пригодные для контролирования скорости высвобождения октреотида из фармацевтических композиций настоящего изобретения, включают полимеры из гидрофильных мономеров, в том числе НРМА, НЕМА и др. Такие гидрогели также способны предотвратить деградацию и потери октреотида из композиции.
Фармацевтическая форма настоящего изобретения может содержать ацетат октреотида внутри гидрофильного сополимера 2-гидроксиэтилметакрилата и гидроксипропилметакрилата. Сополимер фармацевтической формы может содержать, к примеру, 20% НЕМА и 80% НРМА. С другой стороны, сополимер фармацевтической формы может содержать, к примеру, 40% НЕМА и 60% НРМА.
Размер, форму и площадь поверхности импланта можно модифицировать так, чтобы увеличить или уменьшить скорость высвобождения октреотида из импланта.
Фармацевтическая композиция настоящего изобретения также может содержать вспомогательные вещества или наполнители, например скользящие вещества, растворяющие вещества, поверхностно-активные вещества (ПАВ), разбавители, связующие вещества, в том числе низкотемпературные связующие, дезинтегрирующие вещества и/или смазывающие вещества. Растворяющие вещества повышают скорость растворения октреотида из дозовой формы и действуют путем повышения растворимости октреотида. Подходящие растворяющие вещества включают, к примеру, такие органические кислоты, как лимонная, фумаровая, винная, янтарная, аскорбиновая, уксусная, яблочная, глутаровая и адипиновая кислота, которые можно использовать поодиночке или в сочетании. Эти вещества можно также комбинировать с солями кислот, например цитрат натрия с лимонной кислотой, получая буферную систему.
Другие вещества, которые могут изменять pH микроокружения при растворении и установление терапевтически эффективного профиля концентрации октреотида в плазме включают соли неорганических кислот и гидроксид магния. Другие вещества, которые можно использовать, - это поверхностно-активные вещества (ПАВ) и другие солюбилизирующие вещества. Пригодные для применения в фармацевтических композициях настоящего изобретения ПАВ включают, к примеру, лаурилсульфат натрия, полиэтиленстеараты, жирнокислотные эфиры полиэтиленсорбитана, производные полиоксиэтилена с касторовым маслом, алкиловые эфиры полиоксиэтилена, бензилбензоат, цетримид, цетиловый спирт, докозат натрия, глицерилмоноолеат, глицерилмоностеарат, глицерилпальмитостеарат, лецитин, среднецепочечные триглицериды, моноэтаноламин, олеиновую кислоту, полоксамер, поливиниловый спирт и жирнокислотные эфиры сорбитана.
Разбавители, пригодные для применения в фармацевтических композициях настоящего изобретения, включают, к примеру, такие фармацевтически приемлемые инертные заполнители, как микрокристаллическая целлюлоза, лактоза, сахароза, фруктоза, глюкоза, декстроза и другие сахара, двухосновный фосфат кальция, сульфат кальция, целлюлоза, этилцеллюлоза, производные целлюлозы, каолин, маннитол, лактитол, мальтитол, ксилитол, сорбитол и другие сахароспирты, сухой крахмал, сахариды, декстрин, мальтодекстрин и другие полисахариды, инозитол или их смеси. Разбавитель может представлять собой водорастворимый разбавитель. Примеры разбавителей включают, к примеру, микрокристаллическую целлюлозу типа Avicel PH112, Avicel РН101 и Avicel PH102 фирмы FMC Corporation; лактозу типа моногидрата лактозы, безводной лактозы и Pharmatose DCL 21; двухосновный фосфат кальция типа Emcompress фирмы Penwest Pharmaceuticals; маннитол; крахмал; сорбитол; сахарозу; и глюкозу. Разбавители тщательно выбирают так, чтобы они соответствовали конкретной композиции, обращая внимание на свойства сжатия. Разбавитель предпочтительно применяется в количестве от 2% до 80% по весу, предпочтительно от 20% до 50% массы композиции с контролируемым высвобождением.
Скользящие вещества используются для улучшения текучести и прессуемости ингредиентов во время обработки. Подходящие скользящие вещества включают, к примеру, коллоидный диоксид кремния - сверхтонкий плавленый кремнезем, который получают, к примеру, парофазным гидролизом кремниевых соединений, таких, к примеру, как тетрахлорид кремния. Коллоидный диоксид кремния представляет собой сверхтонкий аморфный порошок, который коммерчески доступен из ряда источников, включая Cabot Corporation (под фирменным названием Cab-O-Sil®); Degussa, Inc. (под фирменным названием Aerosil®); и E.I. DuPont & Со. Коллоидный диоксид кремния так же известен как коллоидный кремнезем, плавленый кремнезем, легкая безводная кремниевая кислота, кремниевый ангидрид и плавленый диоксид кремния, среди прочего. В одном воплощении скользящее вещество составляет Aerosil® 200.
Дезинтегрирующие вещества, подходящие для применения в фармацевтических композициях настоящего изобретения, включают, к примеру, крахмал, крахмальный гликолат натрия, кросповидон, кроскармелозу, микрокристаллическую целлюлозу, слабозамещенную гидроксипропилцеллюлозу, пектин, сополимер метакрилата калия и дивинилбензола, поливиниловый спирт, тиламид, бикарбонат натрия, карбонат натрия, производные крахмала, декстрин, бета-циклодекстрин, производные декстрина, оксид магния, глины, бентонит и их смеси.
Активный ингредиент настоящего изобретения можно смешивать с теми наполнителями, которые фармацевтически приемлемы и совместимы с активным ингредиентом, в количестве, подходящем для применения в терапевтических способах, описанных в настоящем изобретении. Различные наполнители можно однородно смешивать с октреотидом настоящего изобретения, как это должно быть известно специалистам. Например, октреотид можно смешивать или комбинировать с такими наполнителями, как микрокристаллическая целлюлоза, коллоидный диоксид кремния, лактоза, крахмал, сорбитол, циклодекстрин и их сочетания.
Смазывающие вещества, подходящие для применения в фармацевтических композициях настоящего изобретения, включают реагенты, воздействующие на сыпучесть прессуемого порошка, в том числе диоксид кремния, такой, к примеру, как Aerosil® 200, тальк; стеариновая кислота, стеарат магния, стеарат кальция, гидрогенизированное растительное масло, бензоат натрия, хлорид натрия, белый вазелин, лаурилсульфат магния и глицерилмоностеарат.
Изобретение также касается имплантируемых дозовых форм с контролируемым высвобождением, содержащих эффективное количество октреотида в гидрогеле, которые при введении пациенту или в качестве составной части режима лечения обеспечивают профиль высвобождения (терапевтически эффективного уровня октреотида в плазме крови) с продолжительностью по меньшей мере 2 месяцев, около 6 месяцев или вплоть до 2 лет.
Дозовые формы настоящего изобретения могут включать один или несколько фармацевтически приемлемых наполнителей. В предпочтительных воплощениях дозовая форма должна содержать разбавители и смазывающее вещество наряду с единицей дозы октреотида и контролирующим скорость полимером. Для этой цели подходящим наполнителем является стеарат магния. При использовании этих материалов компонент стеарата магния может составлять от 0,5 до 5% массы дозовой формы (например, около 2%), а гидрогель и октреотид составляют остальную часть формы.
Другим подходящим наполнителем является гидроксипропилцеллюлоза. При ее использовании компонент гидроксипропилцеллюлозы может составлять от 0,5 до 20% массы дозовой формы (например, около 10%), а гидрогель и октреотид составляют остальную часть формы.
В одном воплощении форма включает и стеарат магния, и гидроксипропилцеллюлозу, предпочтительно 2% стеарата магния и 10% гидроксипропилцеллюлозы, а гидрогель и октреотид составляют остальную часть формы.
Композиции настоящего изобретения могут применяться для лечения гормональных заболеваний, например акромегалии или ее симптомов, характеризующихся повышением уровня GH и IGF-1, путем введения пациенту имплантируемой формы настоящего изобретения. Имплант может вводиться, к примеру, раз в 6 месяцев и будет высвобождать терапевтически эффективное количество октреотида. Имплантируемая композиция выделяет такую концентрацию октреотида у пациента, которая на минимальном терапевтически эффективном уровне ослабляет гормональное нарушение, но сравнительно меньшую, чем максимальная концентрация, чтобы увеличить период покоя для пациента в течение дня. Композиции можно вводить субъекту в такой дозе и на такой срок, которые достаточны для того, чтобы субъект переносил дозу, не проявляя отрицательных эффектов, а затем, повышая дозу активного агента, если нужно, через отдельные промежутки времени, пока не будет достигнута терапевтическая доза у субъекта. Активное средство можно вводить, к примеру, в дозе от 10 мкг до 1000 мкг, от 20 мкг до 800 мкг или от 30 мкг до 300 мкг октреотида в сутки на протяжении по меньшей мере 2 месяцев, около 6 месяцев или вплоть до 2 лет.
Композиции настоящего изобретения, в которых октреотид представляет собой ацетат октреотида, подходят для применения при лечении гормональных заболеваний, характеризующихся повышением уровня GH и IGF-1, напримеракромегалии. Октреотида ацетат в соответствии с изобретением также подходит для лечения симптомов, связанных с карциноидным синдромом и VIP-омами.
Другие особенности и воплощения настоящего изобретения раскрываются на следующих неограничивающих примерах.
Примеры
Пример 1. Скорость высвобождения октреотида in vitro
Данный пример иллюстрирует подготовку имплантируемых форм октреотида по настоящему изобретению и высвобождение из них октреотида in vitro. В данном исследовании тестировали серию имплантов для определения устойчивости и характеристик высвобождения октреотида in vitro из гидрогелевых форм на протяжении 22 недель (№146), 28 недель (№136) и 33 недель (все остальные формы). Каждый имплант содержал 50 мг октреотида ацетата и 2% стеариновой кислоты, но полимерные картриджи содержали различное количество НЕМА и НРМА, поэтому они проявляли различные значения %EWC, приведенные в табл.1.
На фиг.2, 3 и 4 представлено высвобождение октреотида из имплантов за сутки для каждой из форм, приведенных выше. Как видно из фиг.2, у формы №136 начальная скорость была высокой, но сравнительно быстро падала. Как видно из фиг.3, у формы №146 начальная скорость высвобождения была сравнительно низкой. На фиг.4 представлен профиль высвобождения у форм №№145, 147, 133, 144, 143 и 142. Как видно из фиг.4, начальная скорость высвобождения проявляла хорошую зависимость от %EWC, составляя от 20 до 450 мкг в сутки для значений % EWC от 22,9 до 27,6%. Однако возникали проблемы в связи с перепадом осмотического давления между имплантом и средой вымывания. Чтобы стабилизировать лекарственные формы октреотида, были разработаны эксперименты с использованием наполнителей, обеспечивающих большую устойчивость по принципу "предпочтительной гидратации".
Пример 2. Исследование лекарственных форм в телячьей сыворотке
Чтобы определить влияние осмотического давления на проблему с набуханием, два импланта настоящего изобретения, соответствующие форме №136 и форме №143, вымывали в телячьей сыворотке. В частности, тестировали форму №136, состоящую из 40% НЕМА и 60% НРМА и содержащую октреотида ацетат с 2% стеариновой кислоты, и форму №143, состоящую из 30% НЕМА и 70% НРМА и содержащую смесь из 20% PEG 3300 и 80% октреотида ацетата. Через 3 месяца импланты имели нормальный вид - они были сравнительно ровными и только слегка набухшими.
Пример 3. Исследование лекарственных форм
Вследствие перепада осмотического давления импланты, описанные в Примере 1, существенно набухали, что в конечном счете приводило к разрыву имплантов. В данном примере приведены формы, разработанные для скрининга агентов, применимых для стабилизации имплантов октреотида. Отслеживали ряд имплантов, чтобы определить влияние наполнителя на форму и прочность импланта. Каждый из полимерных картриджей состоял из 28% НЕМА, 66,5% НРМА и 5% глицерина. Внутри содержался ацетат октреотида с различными наполнителями, приведенными в табл.2.
Гидрофобные вещества, такие как кунжутное масло и МСС, отделялись от лекарственной формы и не обеспечивали "предпочтительную гидратацию", поэтому они были менее предпочтительны по настоящему изобретению. Гидрофильные вещества типа PEG 3300 увеличивали перепад осмотического давления и усиливали набухание. Низкомолекулярные добавки типа маннитола и гликолевой кислоты не обеспечивали стабилизирующего эффекта и приводили к уменьшению целостности. Ни одно из этих веществ не обеспечивало удовлетворительной стабилизации лекарственных форм октреотида.
Пример 4. Исследование лекарственных форм и скорости высвобождения октреотида in vitro
Данное исследование проводилось для оценки устойчивости октреотида в гидро-гелевых имплантах при использовании различных наполнителей, приведенных в табл.3. Наполнители выбирались так, чтобы они имели большой молекулярный вес и некоторую гидрофильность. Каждый имплант состоял из полимерного картриджа, состоящего из 20% НЕМА и 80%) НРМА. Отслеживали внешний вид имплантов в физрастворе и оценивали на протяжении 9 недель. Результаты представлены в табл.3.
Как видно из фиг.5, содержащая декстран форма имела самую высокую скорость вымывания. Формы, содержащие пектин, AcDiSol и Carbopol, проявляли далеко не удовлетворительное высвобождение после 2-недельной гидратации и 9-недельного вымывания. Соответственно, предпочтительное воплощение с хорошим стабилизирующим эффектом, сочетанием хорошего вымывания и внешнего вида, было достигнуто с гидроксипропилцеллюлозой.
Пример 5. Исследование по 1-месячной имплантации на здоровой собаке
Данный пример иллюстрирует приготовление лекарственных форм настоящего изобретения и высвобождение из них октреотида или его фармацевтически приемлемых солей. Здоровой собаке имплантировали один подкожный имплант октреотида по настоящему изобретению. Композиция подкожного импланта октреотида имела водосодержание 26,6% и содержала 44 мг ацетата октреотида. Скорость высвобождения in vitro составляла около 500 мкг в сутки в 1-ю неделю и снижалась до 300 мкг в сутки в 4-ю неделю при общем высвобождении около 10 мг октреотида на протяжении всего исследования. Имплант удаляли через 28 дней после имплантации. Использовали имплант длиной 3,5 см. Для определения концентрации октреотида ацетата, IGF-1 и GH в сыворотке брали пробы крови (1,5 мл) в дни 0, 1-7, 11, 14, 18, 21, 25 и 28 из яремной вены без анестезии и не натощак.
Клинические наблюдения показали, что композиция импланта октреотида хорошо переносится, потребление пищи было в норме и не отмечалось аномального поведения.
Анализы сыворотки показали пик октреотида ацетата на 4-й день и различимые концентрации октреотида ацетата во все измеренные промежутки времени. Концентрация IGF-1 снижалась после имплантации вплоть до 4-го дня, а затем возвращалась на исходный уровень на 25-й день. Уровень IGF-1 снижался на 40-90% от исходного уровня, как это видно из фиг.6.
Пример 6. Исследование по 6-месячной имплантации на 6 здоровых собаках
Данный пример иллюстрирует приготовление лекарственных форм настоящего изобретения и высвобождение из них октреотида или его фармацевтически приемлемых солей. Шесть здоровых собак разделяли на 2 группы и имплантировали им один или два подкожных импланта октреотида по настоящему изобретению, соответственно. Подкожные импланты октреотида имели водосодержание 25,2% и содержали 60 мг ацетата октреотида. Импланты удаляли через 6 месяцев после имплантации. Для определения концентрации октреотида ацетата, IGF-1 и GH в сыворотке брали пробы крови (10 мл) раз в сутки в первые 7 дней после имплантации, затем 2 раза в неделю на протяжении 3 недель, а затем раз в неделю до истечения 6-месячного срока. За 4 дня до имплантации брали исходные образцы сыворотки в качестве контроля.
Результаты показали, что уровень октреотида в сыворотке составлял от 200 до 700 мкг/мл у собак, получивших один имплант, и от 400 до 1000 мкг/мл у собак, получивших два импланта. Уровень IGF-1 снижался на целых 90% в обеих опытных группах, как это видно из фиг.7 и 8. Измерение уровня GH в сыворотке прекратили после первого месяца исследования, так как уровень GH оказался слишком низким для обнаружения еще большего снижения. Клинические наблюдения показали, что композиция импланта октреотида хорошо переносится, потребление пищи было в норме и не отмечалось аномального поведения.
Пример 7. Исследование по 6-месячной имплантации на людях
Данный пример иллюстрирует приготовление лекарственных форм настоящего изобретения и высвобождение из них октреотида или его фармацевтически приемлемых солей. Проводилось 6-месячное исследование на 11 больных акромегалией. Имплантировали подкожно один или два импланта настоящего изобретения 11 больным с диагнозом акромегалии, которых ранее лечили коммерчески доступным препаратом октреотида LAR. Измеряли уровни GH и IGF-1 в исходном состоянии и каждый месяц после этого на протяжении 6 месяцев. Каждый имплант содержал примерно 60 мг октреотида ацетата в сополимере из 20% НЕМА и 79,5% НРМА со значением EWC около 25,2%. Использовались импланты длиной 44 мм в сухом состоянии и 50 мм в гидратированном состоянии. Диаметр имплантов составлял 2,8 мм в сухом состоянии и 3,5-3,6 мм в гидратированном состоянии. Импланты подвергали гидратации в течение 1 недели перед имплантированием.
Контрольная область значений для GH составляет до 2,5 мг/л, независимо от возраста. В табл.4 приведены уровни GH в мг/л на протяжении 6 месяцев после имплантации имплантов октреотида настоящего изобретения. Пациент №11 не участвовал в исследовании, так как он не удовлетворял критериям отбора. Видно, что на 6-й месяц у 89%о субъектов проявлялась нормализация уровня гормона роста.
Контрольная область значений для IGF-1 составляет: (i) в возрасте 17-24 лет - 180-780 нг/мл; (ii)) в возрасте 25-39 лет - 114-400 нг/мл; (iii)) в возрасте 40-54 лет - 90-360 нг/мл; и (iv)) более 54 лет - 70-290 нг/мл. В табл.5 приведены уровни IGF-1 в нг/л на протяжении 6 месяцев после имплантации имплантов октреотида настоящего изобретения. Видно, что на 6-й месяц у 22% субъектов проявлялась нормализация уровня IGF-1.
На фиг.9A и 9B представлено сравнение импланта октреотида настоящего изобретения с коммерчески доступной формой октреотида ацетата. Эффективность импланта оказалась по крайней мере не хуже, чем у коммерчески доступной формы октреотида LAR. Терапевтический эффект этих имплантов успешно сохранялся на протяжении всех 6 месяцев исследования.
Уровень IGF-1 снижался у всех пациентов при нормализации у 2 пациентов. Снижение наблюдалось уже через 1 месяц терапии, а средний уровень IGF-1 сохранялся на протяжении последующих 5 месяцев. У 8 из 9 пациентов можно было провести сравнение со снижением, наблюдавшимся ранее при лечении коммерчески доступной формой октреотида LAR. У 6 из 8 пациентов процент снижения IGF-1 при импланте оказался больше, чем при лечении коммерчески доступной формой октреотида LAR, тогда как у 2 он был меньше. После 6 месяцев лечения с помощью импланта уровень GH у 3 пациентов составил <1 нг/мл, а у 5 других <2,5 нг/мл. Это хорошо согласуется с результатами по коммерчески доступной форме октреотида LAR, при которой уровень GH только у 2 пациентов составил <1 нг/мл, а у 2 других меньше 2,5 нг/мл.
Также измеряли уровень октреотида в сыворотке пациентов, который представлен в табл.6. Сравнение уровней октреотида, достигаемых при одном или двух имплантах, представлено на графике на фиг.10. В целом результаты показывают, что имплант октреотида настоящего изобретения снижает уровни GH и IGF-1 у больных акромегалией по крайней мере столь же эффективно, как и коммерчески доступная форма октреотида ацетата LAR.
Пример 8. Доставка октреотида с помощью сухих имплантов in vitro
Данный пример иллюстрирует приготовление лекарственных форм настоящего изобретения и высвобождение из них октреотида или его фармацевтически приемлемых солей. Двум здоровым собакам имплантировали по одному подкожному импланту октреотида по настоящему изобретению. Импланты не подвергали гидратации перед имплантированием. Подкожные импланты октреотида состояли из 59,5% НРМА и 40% НЕМА и имели равновесное водосодержание 27,6%. Импланты содержали 84 мг ацетата октреотида, гидроксипропилцеллюлозу и стеарат магния. Импланты удаляли через 6 месяцев после имплантации. Для определения концентрации октреотида ацетата и IGF-1 в сыворотке брали пробы крови (10 мл) раз в сутки через день в первые 4 недели после имплантации, затем 2 раза в неделю на протяжении 4 недель, а затем раз в неделю до истечения 6-месячного срока. За 2 дня до имплантации брали исходные образцы сыворотки в качестве контроля.
На фиг.11 представлен уровень октреотида в сыворотке собак, а на фиг.12 представлен уровень IGF-1 у собак.
Пример 9. Состав имплантов
Возможные составы имплантов по изобретению включают, к примеру, те, что приведены ниже в табл.7. В процессе формирования картриджей имплантов размером более 3,2-3,4 мм (сухих) применялись разделительные вещества, например TPGS витамина Е.
Пример 10. Открытое исследование для оценки фармакокинетики и фармакодинамики гидратированных и негидратированных имплантов с 84 мг октреотида на больных акромегалией
Было включено 30 больных акромегалией после получения письменного информированного согласия. Пациентов разделяли на 2 группы по рандомизированной схеме: 15 пациентов получали по одному гидратированному импланту с 84 мг октреотида, а 15 пациентов получали по одному негидратированному импланту с 84 мг октреотида. Данные пациенты получали имплант в пределах 7 дней от отборочного посещения. Имплант октреотида вставляли подкожно на внутренней стороне неосновной руки под местной анестезией. Для определения концентрации IGF-1, GH и октреотида в сыворотке брали пробы крови через заданные промежутки времени в первые 6 недель после имплантации. Затем пациенты снова приходили через 8, 12, 16, 20 и 24 недели для отбора проб крови для определения концентрации IGF-1, GH и октреотида в сыворотке, а также для оценки безопасности. Импланты удаляли по окончании 6-месячной (24-недельной) фазы лечения. После удаления импланта пациентам велели прийти через 4 недели для заключительного обследования (28 недель). На всем протяжении исследования тщательно отслеживали безопасность и эффективность.
Исследуемые препараты:
гидратированный имплант октреотида (84 мг октреотида ацетата) для подкожной имплантации;
негидратированный имплант октреотида (84 мг октреотида ацетата) для подкожной имплантации.
Продолжительность лечения:
участвующие пациенты получали по одному импланту, гидратированному либо не гидратированному. По окончании 6-месячной (24-недельной) фазы лечения импланты удаляли. После удаления импланта пациентам велели прийти через 4 недели для заключительного обследования.
Критерии для включения:
1) мужчины и женщины, больные акромегалией;
2) должно исполниться >18 лет;
3) подтвержденный диагноз опухоли, секретирующей гормон роста (повышение уровня IGF-1 на ≥20% выше верхнего предела стандартизованной по возрасту и полу нормальной величины, и либо уровень GH после глюкозы ≥1,0 нг/мл, либо опухоль гипофиза, выявленная методом MRI). Если пациент подвергался операции на гипофизе и у него осталась остаточная опухоль, то она должна находиться на расстоянии не менее 3 мм от перекреста зрительных нервов (если только пациент не является кандидатом на операцию), а уровень IGF-1 должен быть повышен, как описано выше. Если же нет остаточной опухоли или пациент не подлежит операции, то он должен соответствовать обоим критериям на IGF-1 и GH, как описано выше.
4) должны полностью или частично реагировать на октреотид по данным лабораторных анализов в истории болезни, как изложено ниже:
a) полное реагирование: подавление IGF-1 в сыворотке до нормального стандартизованного по возрасту и полу уровня и подавление GH в сыворотке до<1,0 нг/мл после OGTT;
b) частичное реагирование: снижение уровня IGF-1 и GH на >30% по сравнению с уровнем до лечения, но еще не соответствует критериям полного реагирования;
c) либо
d) должны реагировать на октреотид по данным лабораторных анализов, полученных методом острого водного тестирования во время отборочного посещения у пациентов, не получавших октреотид, или пациентов, у которых реакция на октреотид не известна, как изложено ниже:
e) реагирование при остром водном тестировании: снижение уровня GH на ≥30% в любой точке времени 4-часового срока тестирования в ответ на подкожную инъекцию 100 мкг водного препарата октреотида;
5) должны быть в состоянии общаться, дать письменное информированное согласие, высказывать желание участвовать и выполнять требования исследования;
6) если пациент подходит для участия по мнению исследователя.
Критерии для исключения:
1) женщины, которые беременны, кормят грудью или способные родить ребенка и не применяющие приемлемый медицинский способ регулирования рождаемости;
2) пациенты после операции на гипофизе менее чем за 12 недель до отбора;
3) пациенты с заболеваниями печени (например, циррозом, хроническим активным или стойким гепатитом либо стойкими отклонениями ALT, AST (уровень в >2 раза больше нормы), щелочной фосфатазы (уровень в >2 раза больше нормы) или несвязанного билирубина (уровень в >1,5 раза больше нормы);
4) другие лабораторные анализы, которые исследователь или спонсор посчитают клинически значимыми;
5) пациенты с нестабильной стенокардией, стойкой желудочковой аритмией, сердечной недостаточностью (NYHA III и IV) или историей острого инфаркта миокарда за 3 месяца до отбора;
6) пациенты с симптомами желчнокаменной болезни;
7) пациенты с историей злоупотребления лекарственными средствами или алкоголем за 6 месяцев до отбора;
8) пациенты, получавшие исследуемый препарат за 1 месяц до отбора;
9) пациенты, получавшие радиотерапию по поводу опухоли гипофиза в любое время перед отбором;
10) пациенты, прекратившие лечение октреотидом из-за проблем переносимости или эффективности.
Определяли уровень октреотида в сыворотке (см. графики на фиг.13 и 14).
Эффективность модулирования концентрации цитокина после введения импланта октреотида, в сухом виде либо после гидратации, представлена на фиг.15, 16 и 17.
Пример 11. Лечение опухолей с помощью октреотида
Sandostatin LAR® Depot, как указано на этикетке FDA (все содержание которой включено путем ссылки), представляет собой дозовую форму продолжительного действия, состоящую из микросфер биоразложимого полимера крахмала и глюкозы, сополимера D,L-молочной и гликолевой кислоты, которые содержат октреотид. Она сохраняет все клинические и фармакологические характеристики дозовой формы с немедленным высвобождением Sandostatin® (октреотида ацетат) для инъекций с добавлением замедленного высвобождения октреотида по месту инъекции, что уменьшает необходимость частого введения. Такое замедленное высвобождение происходит по мере биоразложения полимера, главным образом посредством гидролиза. Sandostatin LAR® Depot предназначается для внутримышечных инъекций (в ягодицы) раз в 4 недели.
Октреотид оказывает фармакологическое действие, аналогичное природному гормону - соматостатину. Он является еще более сильным ингибитором гормона роста, глюкагона и инсулина, чем соматостатин. Подобно соматостатину, он также подавляет реакцию LH на GnRH, уменьшает спланхнический кровоток и ингибирует высвобождение серотонина, гастрина, вазоактивного интестинального пептида, секретина, мотилина и панкреатического полипептида. На основании такого фармакологического действия октреотид применялся для лечения симптомов, связанных, к примеру, с метастатическими карциноидными опухолями (гиперемии и диареи) и секретирующими вазоактивный интестинальный пептид (VIP) аденомами (водянистой диареи). Октреотид существенно снижает и во многих случаях нормализирует уровень гормона роста и/или IGF-1 (сомато-медина C) у больных акромегалией. Однократные дозы Sandostatin® для инъекций при подкожном введении, как оказалось, подавляют сократимость желчного пузыря и уменьшают секрецию желчи у нормальных добровольцев.
Фармакокинетика у больных акромегалией слегка отличается от таковой у здоровых добровольцев. Средняя максимальная концентрация в 2,8 нг/мл (при дозе в 100 мкг) достигается через 0,7 ч после введения. Объем распределения (Vdss) составляет 21,6±8,5 л, а общий клиренс в организме возрастал до 18 л/ч. Среднее содержание связанного препарата составляло 41,2%. Полупериоды ликвидации и элиминации были близки к норме.
Лечение этих опухолей, как правило, включает хирургию в качестве первоочередной терапии. При неудачной операции пациентам обычно вводят октреотид для инъекций (типа S-LAR). Химиотерапия также может оказаться полезной, как это бывает у 30% пациентов. Пациентов с карциноидными опухолями лечили 6 дозами S-LAR в 10, 20 или 30 мг при внутримышечном введении раз в 4 недели. Концентрации в сыворотке составили 1,2, 2,5 и 4,2 нг/мл. Стационарное состояние достигалось после двух доз в 20 или 30 мг и после 3 доз в 10 мг.
Лечение с помощью S-LAR приводило к снижению частоты стула до 2-2,5 раз в день, уменьшению среднего числа эпизодов гиперемии до 0,5-1 раза в день и снижению медианы 24-часового уровня 5-HIAA в моче на 38-50%. Следует отметить, что за все 6 месяцев испытания у 50-70% пациентов, прошедших испытание, потребовались дополнительные инъекции Sandostatin, чтобы приостановить ухудшение симптомов.
Хотя настоящее изобретение и было описано достаточно подробно в отношении некоторых предпочтительных его воплощений, однако возможны и другие варианты. Поэтому суть и объем прилагаемой формулы изобретения не следует ограничивать описанием и предпочтительными вариантами, содержащимися в данном описании.
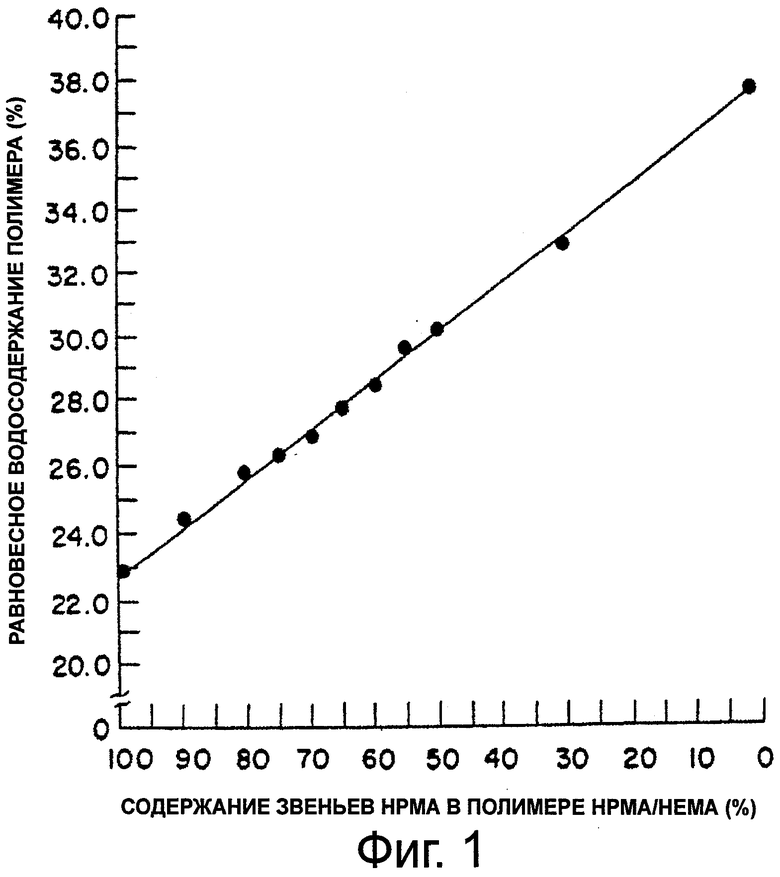
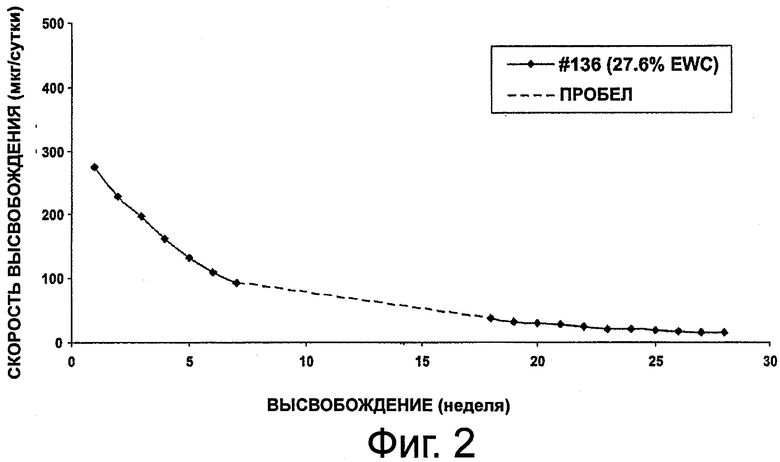
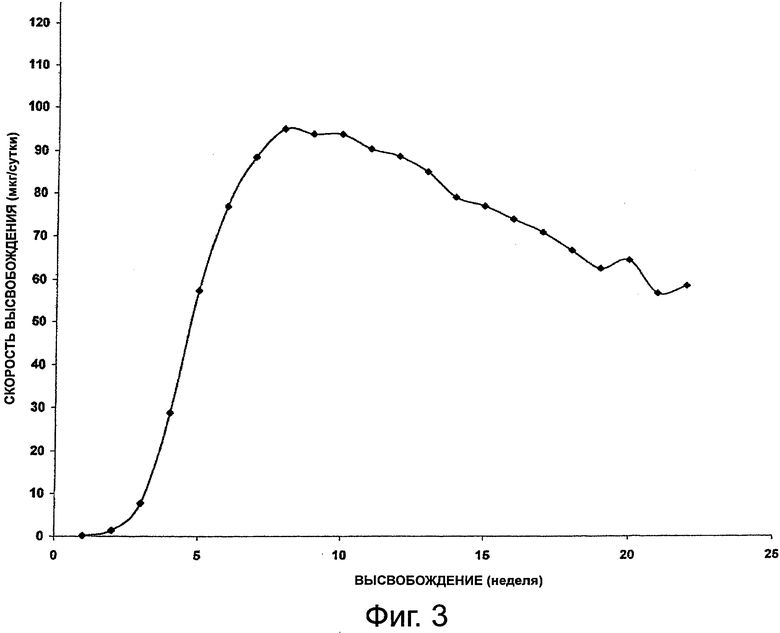
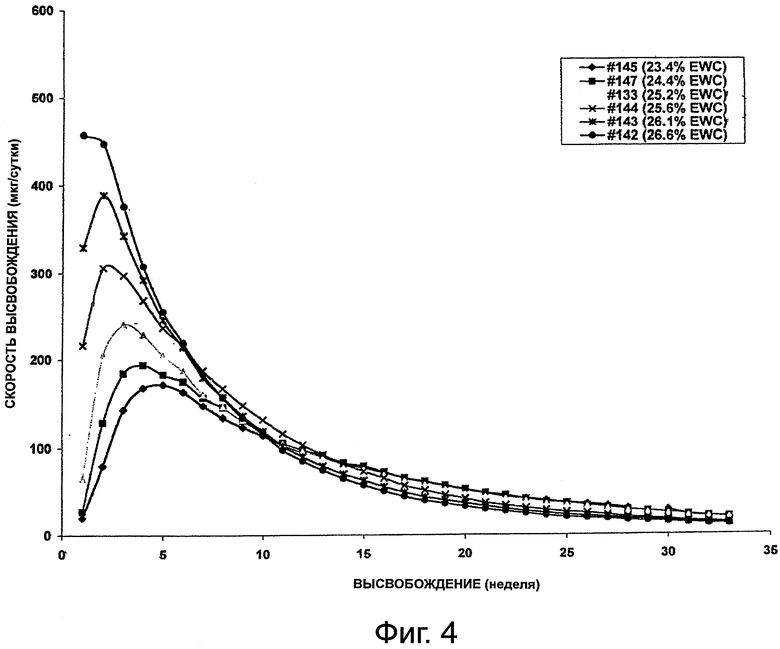
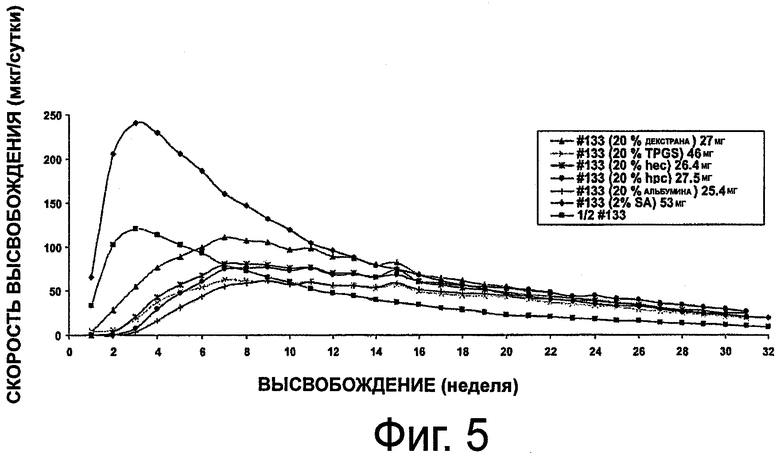
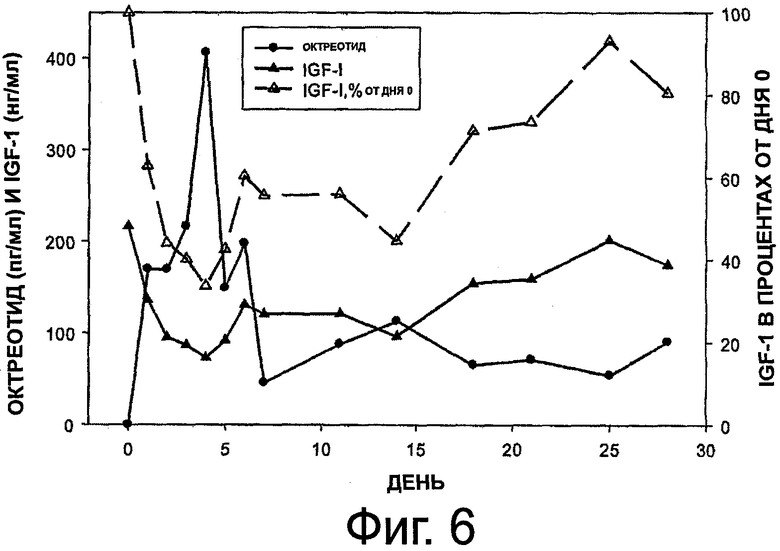
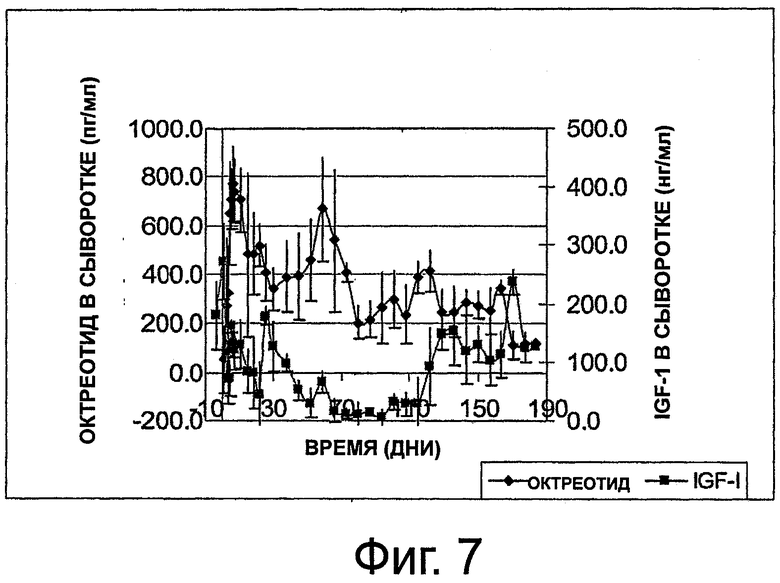
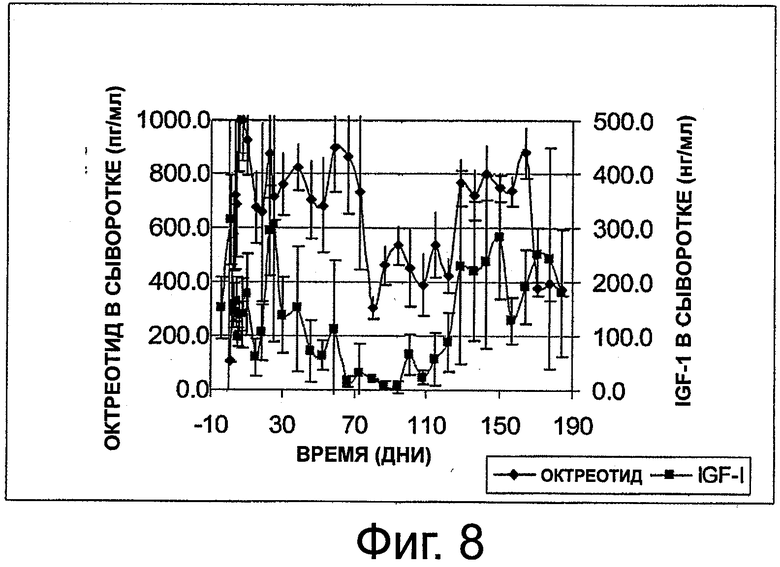
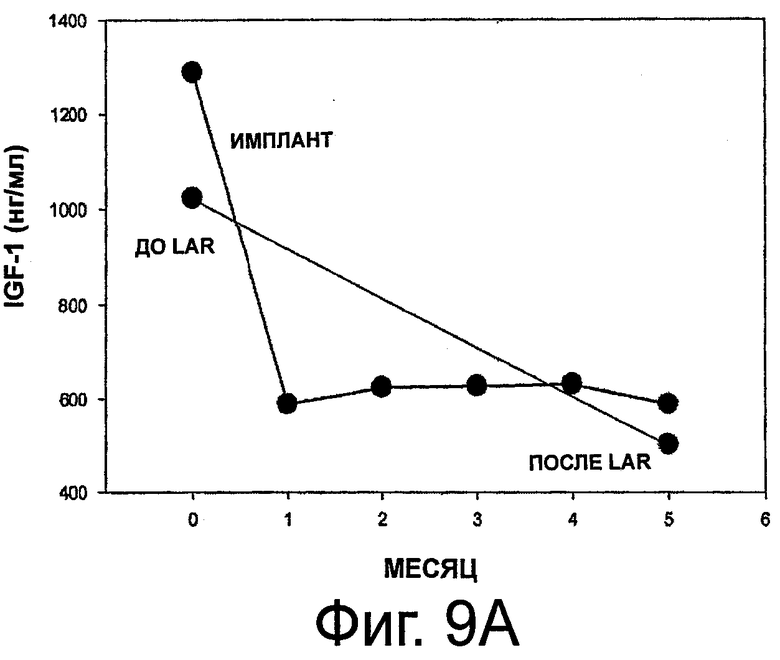
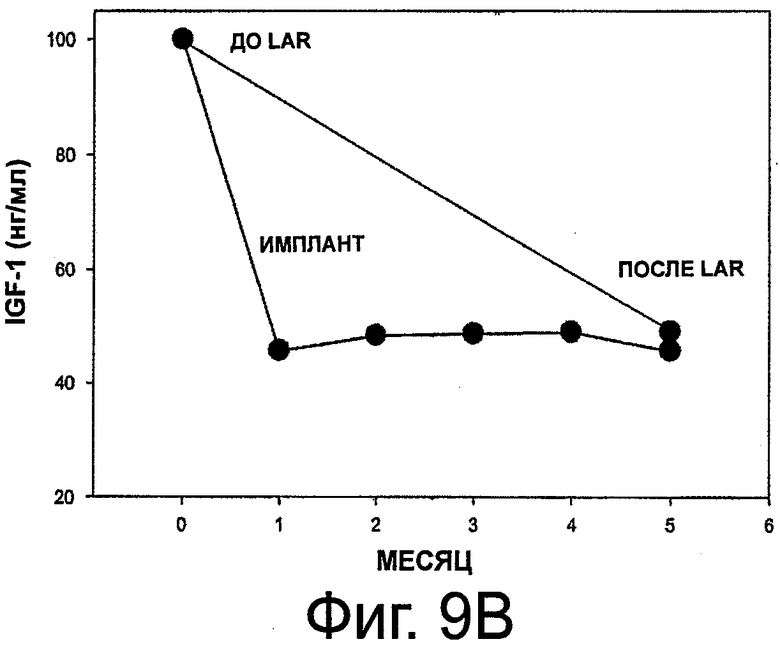
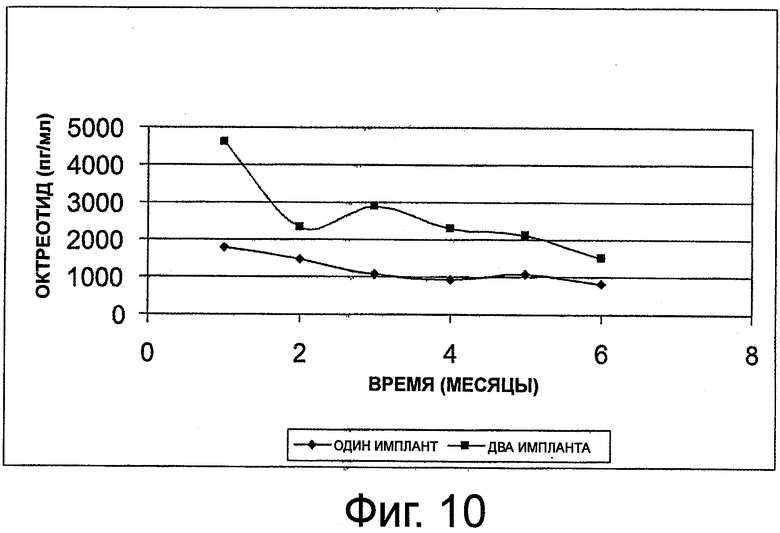
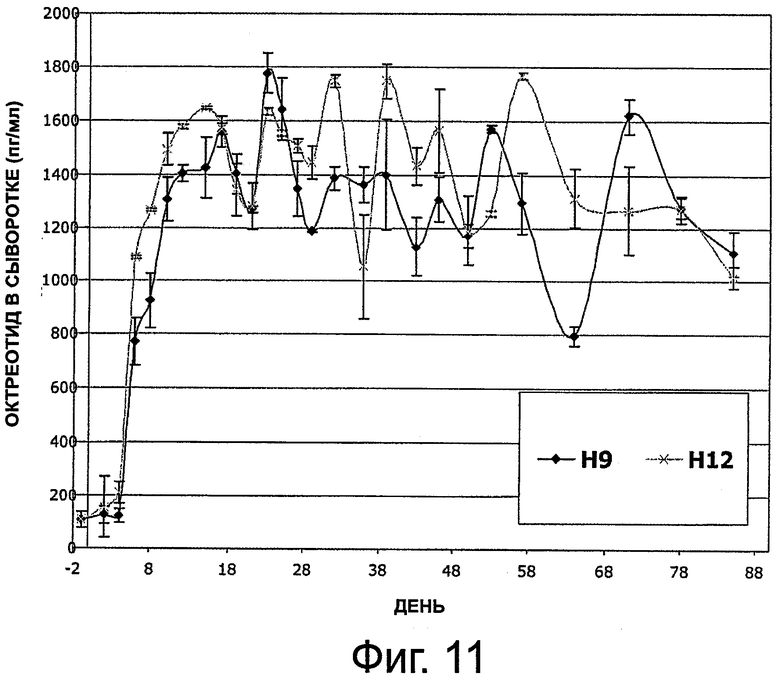
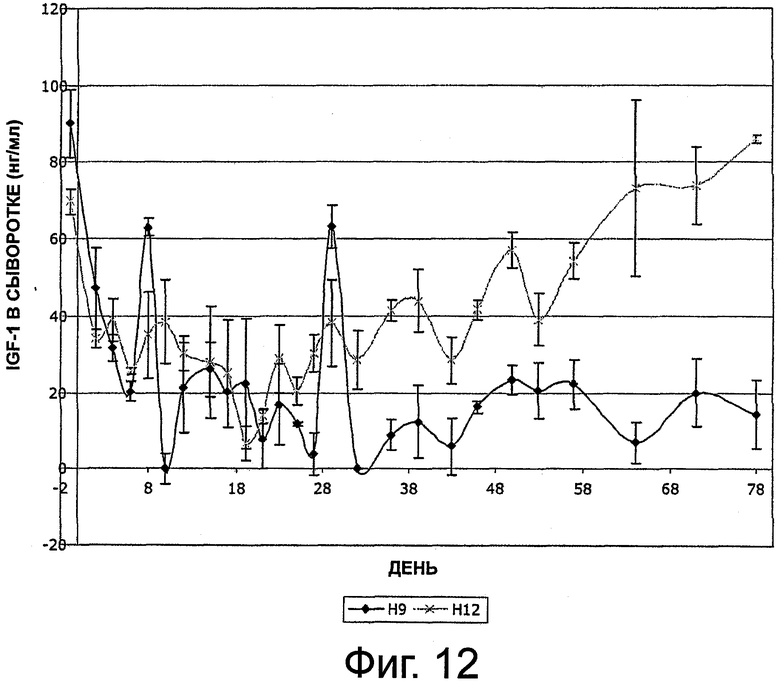
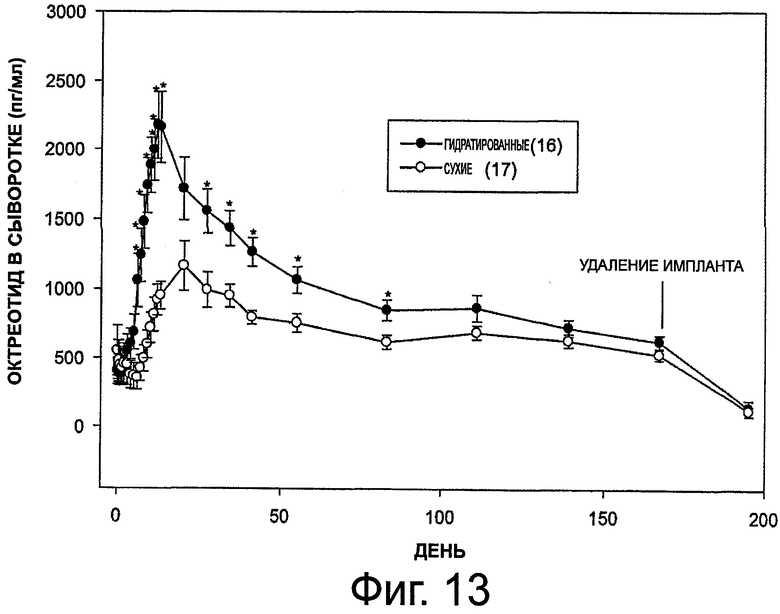
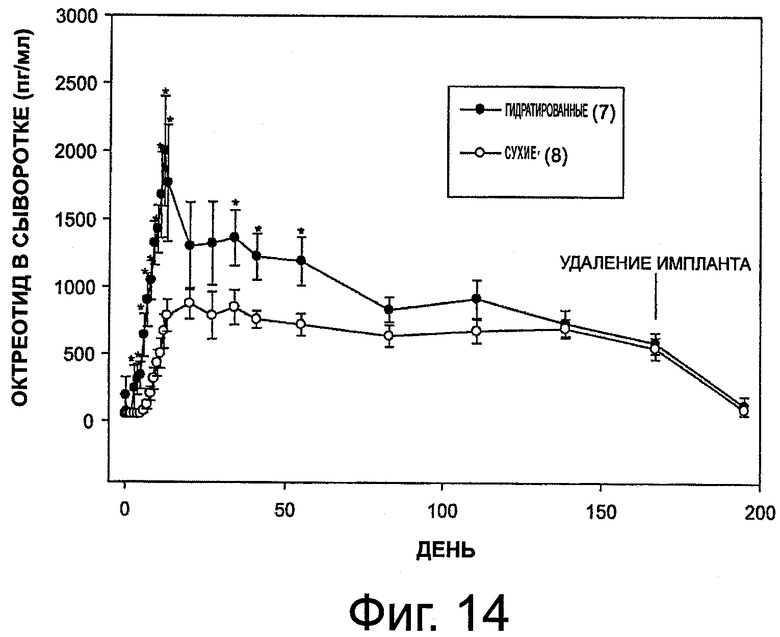
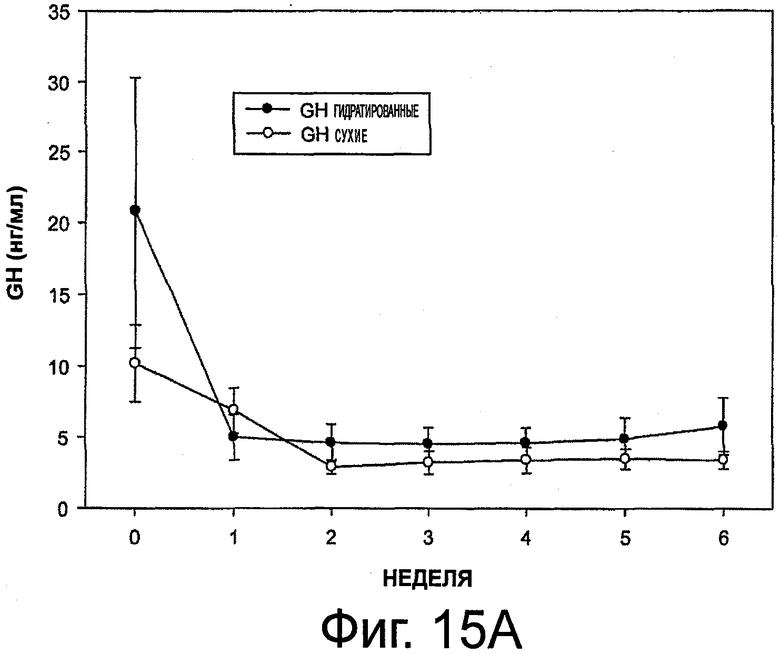
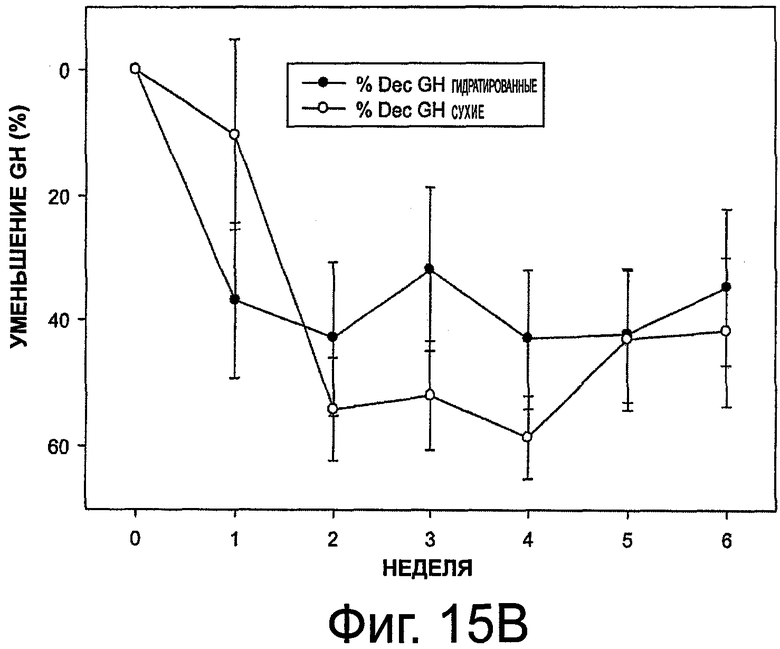
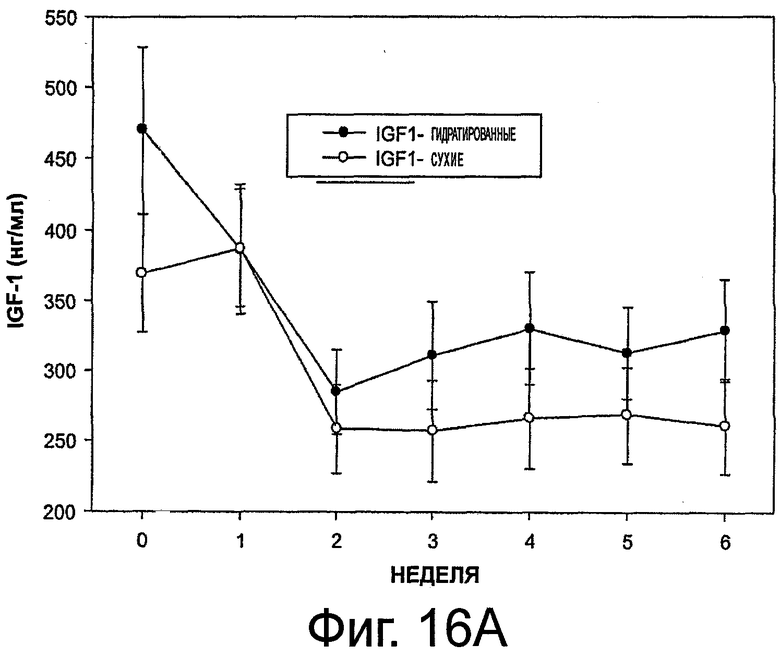
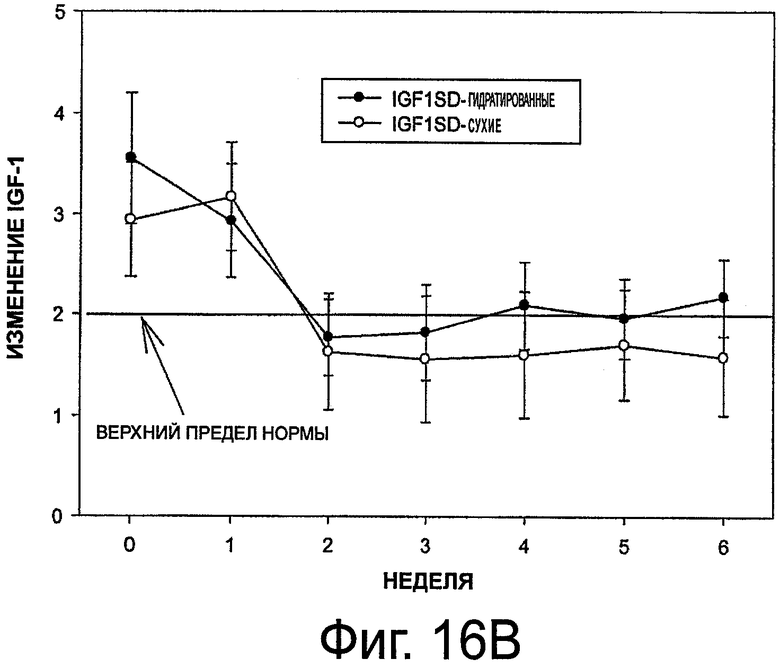
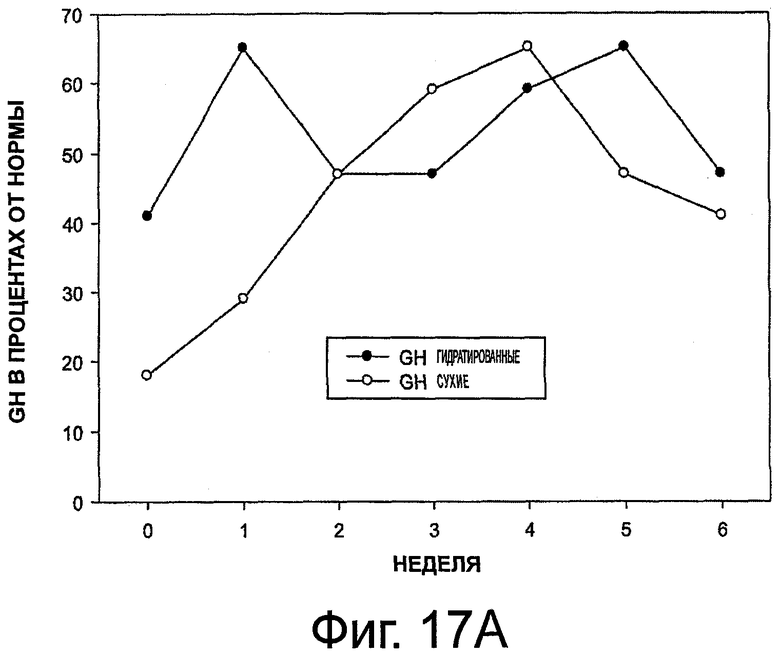
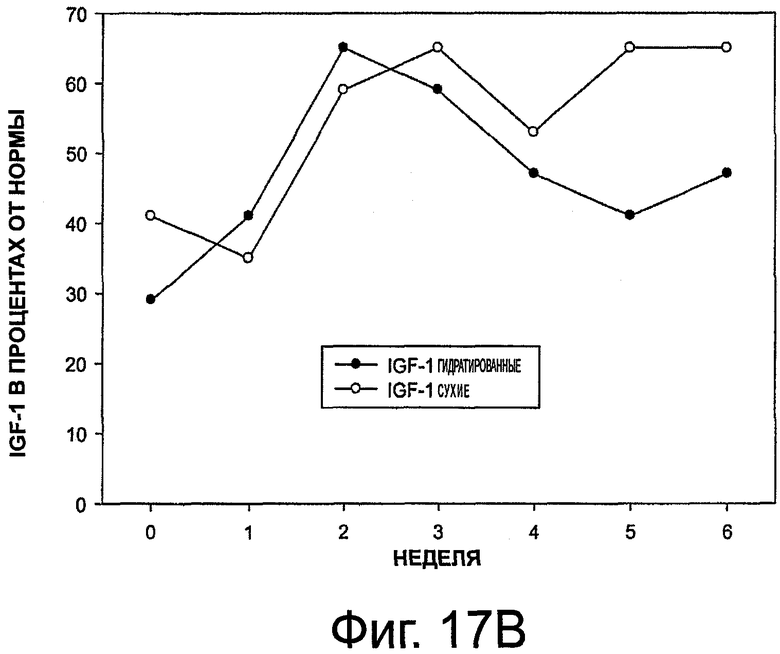
Изобретение относится к медицине и применяется для лечения лиц, страдающих гормональными заболеваниями. Описаны способ и устройство для доставки октреотида пациентам, включающие имплантирование композиции с контролируемым высвобождением для доставки октреотида, причем композиция не требует гидратирования перед имплантацией и при этом композиция необязательно содержит разделительное вещество. 2 н. и 8 з.п. ф-лы, 11 пр., 7 табл., 17 ил.
1. Способ доставки октреотида субъекту с профилем высвобождения практически нулевого порядка в течение длительного времени, но не менее 6 месяцев, при этом субъектом является млекопитающее, кроме собак, а способ включает подкожное имплантирование субъекту, по меньшей мере, одного имплантируемого устройства, причем данное, по меньшей мере, одно имплантируемое устройство включает композицию, содержащую 84 мг октреотид ацетата, 9,5 мг гидроксипропилцеллюлозы и 2 мг стеарата магния, где композиция заключена в гидрофильный полимер, изготовленный полимеризацией смеси, содержащей 40% HEMA, 59,5% HPMA, 0,5% TMPTMA и 1% витамина E TPGS, и при этом имплантируемое устройство имплантируется в сухом состоянии так, чтобы субъект на протяжении, по меньшей мере, 6 месяцев получал в пересчете на суточную дозу такое количество октреотида, которое эффективно для лечения субъекта, но которое дает более низкие сывороточные концентрации октреотида по сравнению с имплантацией имплантируемого устройства в гидратируемом состоянии.
2. Способ по п.1, в котором субъект страдает чувствительным к октреотиду заболеванием или расстройством.
3. Способ по п.1, в котором субъект страдает расстройством гормона GH или IGF-1 либо его симптомами.
4. Способ по п.6, в котором расстройство гормона GH или IGF-1 представляет собой акромегалию.
5. Способ по п.1, в котором субъект получает октреотид со средней скоростью около 250 мкг в сутки 6 месяцев.
6. Имплантируемое устройство по п.1, в котором размеры имплантируемого устройства составляют 3,4 мм×43 мм.
7. Способ по п.1, в котором субъект страдает заболеванием, выбранным из группы, состоящей из карциноидного синдрома, VIP-омы, нейроэндокринных опухолей, пролиферативной диабетической ретинопатии, красных угрей, панкреатита, желудочно-кишечных кровотечений, свищей поджелудочной железы и кишечных свищей, базедовой болезни (офтальмопатии Грейвса), глаукомы и симптомов, связанных с химиотерапией или СПИД.
8. Имплантируемое устройство, включающее композицию с контролируемым высвобождением, содержащую 84 мг октреотид ацетата, 9,5 мг гидроксипропилцеллюлозы и 2 мг стеарата магния, которая прочно заключена в полимер на основе метакрилата, изготовленный полимеризацией смеси, содержащей 40% HEMA, 59,5% HPMA, 0,5% TMPTMA и 1% витамина E TPGS, при этом имплантируемое устройство выделяет октреотид для субъекта с профилем высвобождения практически нулевого порядка в течение длительного времени, но не менее 6 месяцев, а субъектом является млекопитающее, кроме собак, причем имплантируемое устройство имплантируется субъекту в сухом состоянии.
9. Имплантируемое устройство по п.8, причем при имплантировании субъекту имплантируемого устройства субъект получает октреотид со средней скоростью около 250 мкг в сутки 6 месяцев.
10. Имплантируемое устройство по п.8, в котором размеры имплантируемого устройства составляют 3,4 мм×43 мм.
| US 2006204540 A1, 14.09.2006 | |||
| RU 2011102602 A, 27.07.2012 | |||
| WO 2005021023 A1, 10.03.2005 |

