ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Платформа деактивированной сердцевины компании King Pharmaceuticals, внедрение связанного налтрексона в сердцевину опиоидной дозированной формы с контролируемым высвобождением, который высвобождается только при разрушении связующей полимерной матрицы, была развита как способ снижения действия избыточно опиоида и лекарственного средства, предпочтительно, когда продукт неправильно используют или злоупотребляют им. Технология деактивированной сердцевины описывается в деталях в патентах США №7,682,633 и 7,682,634, заявках на патент США №US 20080233156, US 20090131466, US 20040131552, US 20100152221, US 20100151014 и US 20100143483 и заявках PCT №PCT/US08/087030, PCT/US08/087043, PCT/US08/87047 и PCT/US08/087055, включенных в данный документ в виде ссылок.
Анальгетическое лекарственное средство Embeda® (также известное как ALO-01) представляет собой пример находящегося на рынке лекарственного препарата, внедряющего технологию деактивированной сердцевины. (Предписывающая информация: Embeda® (морфина сульфат и налтрексона гидрохлорида) капсулы с пролонгированным действием. Alpharma Pharmaceuticals LLC, дочерняя компания, полностью находящаяся в собственности компании King Pharmaceuticals, Inc., Bristol, TN. June 2009.) Коммерциализированное в 2009 Embeda® представляет собой капсульный препарат, содержащий пеллеты с контролируемым высвобождением, которые медленно по прошествии длительного времени высвобождают терапевтические количества морфина сульфата. Налтрексон НС1 изолируют во внутреннем ядре в соотношении 1:20 с морфином и высвобождается он только, когда разрушается связующая полимерная матрица. Когда капсула взята целиком, внутреннее ядро остается неповрежденным, и налтрексон не оказывает воздействия на анальгетический потенциал морфина. Однако, когда Embeda® разжевывают, раздавливают или совершают другие физические манипуляции, налтрексон высвобождается, адсорбируется перорально и конкурентно связывается с мю-опиодным рецептором, таким образом, ослабляя или уменьшая эйфористические эффекты морфина.
Количество налтрексона в основе деактивированной сердцевины варьируется в зависимости от силы опиоидного анальгетического средства. Embeda использует 4% налтрексона (морфин и налтрексон в соотношении 20:1). Исследования продемонстрировали, что 12% налтрексона или больше могут быть оптимальными для оксикодона и гидрокодона. В то время как был исследован ответ дозы по отношению к эйфории и лекарственное средство, находящееся в комбинациях с опиоидами и антагонистами опиоидов, малоизвестно о взаимоотношении доза-ответ налтрексона по отношению к другим фармакологическим действиям опиоидов, включая первичный механизм смертельной передозировки опиоида: угнетение дыхания (White JM and Irvine RJ. Mechanisms of fatal opioid overdose. Addiction. 1999; 94 (7): 961-72; Dahan A, Aarts L, and Smith TW. Incidence, reversal, and prevention of opioid-induced respiratory depression. Anesthesiology. 2010; 112: 226-38).
В настоящее время налоксон представляет собой лекарственное средство из варианта для терапевтического применения как средство экстренной терапии в быстрой отмене активности, вызванной опиоидом, и побочных реакций (Longnecker DE, Grazis PA, and Eggers GWN. Naloxone for antagonism of morphine-induced respiratory depression. Anesthesia and Analgesia Current Researches 1973; 52(3): 447-53). При парентеральном введении были хорошо охарактеризованы фармакодинамические эффекты налоксена по отношению к отмене угнетения дыхания, вызванного опиоидами (Yassen A, Olofsen Е, van Dorp Е, Sarton Е, Teppema L, Danhof M, and Dahan A. Mechanism-based pharmacokinetic-pharmacodynamic modelling of the reversal of buprenorphine-induced respiratory depression by naloxone. Clin Pharmacokinet. 2007; 46 (II): 965-80; Kaufman RD, Gabthuler ML, and Bellville W. Potency, duration of action and pA2 in man of intravenous naloxone measured by reversal of morphine-depressed respiration. J of Pharmacol and Exp.Ther. 1981; 219:156-62). При известной или подозреваемой передозировке опиоида, чтобы аннулировать угнетение дыхания, вызванное опиоидом, обычная IV доза налоксона составляет 0,4-2 мг (Amercian Hospital Formulary Services (AHFS) Information. Naloxone hydrochloride. 2003: 2088-89). Данная первоначальная инфузия может быть дополнена многочисленными инъекциями налоксона при частых интервалах или непрерывной внутривенной инфузией. В послеоперационный период доза болюса налоксона может быть дополнена непрерывной IV инфузией налоксона 3,7 мкг/кг на час, чтобы аннулировать угнетение дыхания.
Патент США №5,834,477 описывает композиции гомогенной смеси, которая содержит, как опиоидный агонист, так и антагонист, которые вызывают минимальное угнетение дыхания. Патент описывает применение суфентанила оксалата и налмефена в молярном соотношении 15:1.
Эффекты от комбинирования гидрокодона битартрата и налтрексона гидрохлорида на угнетение дыхания у крыс были оценены (К. Hew, S. Mason, and Н. Penton, A Respiratory Safety Pharmacology Assessment of Hydrocodone Bitartrate and Naltrexone Hydrochloride). Было проведено сравнение действия оксикодона и морфина по отношению к угнетению дыхания у пациентов (Change et. al., A comparison of the respiratory effects of oxycodone versus morphine: a randomized, double-blind, placebo controlled investigation, Anaesthesia 2010). Данное исследование определило, что степень и скорость возникновения угнетения дыхания, вызванного оксикодоном, была зависимой от дозы большей, чем эквивалентная доза морфина.
Использование налтрексона как средства экстренной терапии у людей является новым применением данного лекарственного средства, так как налтрексон первоначально вводят перорально и постоянно, чтобы лечить опиатную и алкогольную зависимость. Если нет изолирования в препарате с дезактивирующей сердцевиной, например, после дробления или разжевывания препарата и дальнейшего проглатывания, то налтрексон адсорбируется также быстро, как и опиоид (фигура 2) несмотря на то, что опиоид сохраняется дольше, чем налтрексон. Это предполагало бы, что налтрексон имеет столько же потенциала, чтобы предотвратить угнетение дыхания при состоянии острой опиоидной передозировке как и в случае, когда это было или в реверсирующем, или ослабляющем состоянии, зависящем от количества каждого лекарственного средства, которое адсорбировалось. Вследствие этого развитие лучшего понимания взаимосвязи доза-ответ между налтрексоном и угнетением дыхания, вызванным опиоидом, представляет собой вопрос клинической важности.
КРАТКОЕ ОПИСАНИЕ СУТИ ИЗОБРЕТЕНИЯ
Представленное изобретение касается опиоидных композиций, содержащих изолированный опиоидный антагонист, которые, когда приняты внутрь после повреждения (например, раздавливания, разжевывания или растворения), высвобождают опиоидный антагонист и ослабляют угнетение дыхания, когда введены или приняты внутрь после повреждения. Композиции представленного изобретения содержат опиатные анальгетические лекарственные препараты, которые включают твердую, с контролируемым высвобождением, пероральную дозированную форму, содержащую большое количество многослойных пеллет, где каждая пеллета, содержит водорастворимую сердцевину, слой антагониста, содержащий налтрексон или фармацевтически приемлемую соль налтрексона, который покрывает сердцевину, изолирующий полимерный слой, который покрывает слой антагониста, слой агониста, содержащий опиоид или фармацевтически приемлемую соль опиоида, который покрывает изолирующий полимерный слой, и слой контролированного высвобождения, который покрывает слой агониста. Когда композиции вводят человеку нетронутым, что означает, что композиции не были повреждены, что, главным образом, все остатки налтрексона изолированы. Если, однако, композиции повреждены, что означает, что композиции были раздавленными, разжеванными, растворенными или видоизменены любым другим способом так, что налтрексон и опиоид в композиции высвобождаются с исходной дозированной формы, композиции имеют достаточное количество налтрексона, чтобы ослабить угнетение дыхания, опосредованное опиоидом, у индивидуума, который принял поврежденную форму композиций.
Представленное изобретение касается опиатных анальгетических лекарственных препаратов, которые включают твердую, с контролируемым высвобождением, пероральную дозированную форму, содержащую большое количество многослойных пеллет, где каждая пеллета, содержит водорастворимую сердцевину, слой антагониста, содержащий налтрексон или фармацевтически приемлемую соль налтрексона, который покрывает сердцевину, изолирующий полимерный слой, который покрывает слой антагониста, слой агониста, содержащий опиоид или фармацевтически приемлемую соль опиоида, который покрывает изолирующий полимерный слой, и слой контролированного высвобождения, который покрывает слой агониста, в которых налтрексон или фармацевтически приемлемая соль налтрексона в значительной степени не высвобождается при введении неповрежденного препарата человеку, и где минимальное угнетение дыхания вызывается у человека, когда препарат был поврежден до введения человеку.
Представленное изобретение, кроме того, касается способов ослабления угнетения дыхания у человека, опосредованного лекарственным средством, в случае введения человеку лекарственного средства, которое вызывает угнетение дыхания, где способ включает введение человеку опиатного анальгетического лекарственного препарата, который включает твердую, с контролируемым высвобождением, пероральную дозированную форму, содержащую большое количество многослойных пеллет, где каждая пеллета, содержит водорастворимую сердцевину, слой антагониста, содержащий налтрексон или фармацевтически приемлемую соль налтрексона, который покрывает сердцевину, изолирующий полимерный слой, который покрывает слой антагониста, слой агониста, содержащий опиоид или фармацевтически приемлемую соль опиоида, который покрывает изолирующий полимерный слой, и слой контролированного высвобождения, который покрывает слой агониста.
ФИГУРЫ
Фигура 1. График, сравнивающий концентрации налоксона и налтрексона в плазме после IV терапии налоксоном (красная) и при полном высвобождении из 80 мг пероральной дозы ALO-02 или ALO-04, содержащей 12% налтрексона (синяя).
Фигура 2. График, сравнивающий концентрации налтрексона и оксикодона в плазме после теоретического раздавливания дозы ALO-02, содержащей 80 мг оксикодона и 12% (9,6 мг) налтрексона.
Фигура 3. График модифицированной реакции вентиляции возвратного дыхания.
Фигура 4. График средних (±SD) Emax значений парциального давления СO2 в выдыхаемом воздухе в конце выдоха при лечении.
Фигура 5. График средних (+/- SE) насыщенных кислородом (SpO2) уровней в течение времени, определенного из пульсовой оксигемометрии после перорального введения оксикодона 60 мг, оксикодона 60 мг + налтрексона 7,2 мг (12% - текущее соотношение налтрексона в ALO-02) и плацебо.
ДЕТАЛЬНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Предусмотренными данным документом являются композиции и способы введения композиций, содержащие различные активные агенты, млекопитающим в форме и способом, который минимизирует действия одного из активных агентов на другой in vivo. В частности, представленное изобретение касается опиоидных композиций, которые ослабляют угнетение дыхания, когда вводятся человеку. В конкретных вариантах осуществления изобретения, по меньшей мере, два активных агента формулируют как часть фармацевтической композиции. Первый активный опиоидный агент может обеспечивать терапевтическое действие in vivo. Второй активный агент может быть антагонистом первого активного агента и может быть полезным для ослабления угнетения дыхания, если композиция является поврежденной. Композиция остается неповрежденной во время нормального употребления пациентами и антагонист не высвобождается. Однако при повреждении композиции (например, раздавливание, разжевывание или растворение композиции) антагонист может высвобождаться, таким образом, предупреждая, уменьшая или ослабляя действие опиоида при стимулировании значительного угнетения дыхания. В конкретных вариантах осуществления изобретения активные агенты заключены оба в отдельные единицы, такие как пеллеты или гранулы, в форме слоев. Активные агенты могут быть сформулированы, главным образом, с непроницаемым барьером как, например, композиция с контролируемым высвобождением, таким образом, что высвобождение антагониста из композиции минимизируется. В конкретных вариантах осуществления изобретения, антагонист высвобождается при исследовании in vitro, но, главным образом, не высвобождается in vivo. In vitro и in vivo высвобождение активного агента из композиции может быть измерено каким-либо из нескольких хорошо известных методик. Например, высвобождение in vivo может быть определено путем измерения уровней активного агента или его метаболитов в плазме (то есть AUC, Сmax).
В одном варианте осуществления изобретения изобретение предусматривает изолирование субъединицы, содержащей опиоидный антагонист и блокирующий агент, где блокирующий агент в значительной степени предотвращает высвобождение опиоидного антагониста из изолированной субъединицы в желудочно-кишечный тракт на протяжении периода времени, который больше, чем 24 часа. Данную изолированную субъединицу вводят в отдельную фармацевтическую единицу, которая, кроме того, включает опиоидный агонист. Фармацевтическая единица, таким образом, включает сердцевинную часть, на которую нанесен опиоидный антагонист. Запечатанная оболочка затем наносится на антагонист. На запечатанную оболочку затем наносят композицию, содержащую фармацевтически активный агент в способной к высвобождению форме. Дополнительный слой, содержащий такой же или отличный блокирующий агент затем может быть нанесен таким образом, что опиоидный агонист высвобождается в желудочно-кишечный тракт на протяжении времени (то есть контролированное высвобождение). Альтернативно, слой опиоидного агониста может быть в форме непосредственного высвобождения. Таким образом, опиоидный антагонист и опиоидный агонист оба содержатся в отдельной фармацевтической единице, которая, как правило, находится в форме гранулы.
Термин "изолирующая субъединица", как использовано в данном документе, касается какой-либо фармацевтической единицы (например, гранулы или пеллеты), содержащей средства для содержания антагониста и предотвращающей или в значительной степени предотвращающей его высвобождение в желудочно-кишечный тракт, когда она не нарушена, то есть, когда нет повреждений. Термин "блокирующий агент", как использовано в данном документе, касается средств, за счет которых изолирующая единица способна в значительной степени предотвращать высвобождение антагониста. Блокирующий агент может представлять собой связывающий полимер, например, как описано очень детально ниже.
Термины "в значительной степени предотвращает", "предотвращает" или какие-либо слова, которые происходят отсюда, как использовано в данном документе, означают что антагонист в значительной степени не высвобождается из изолированной единицы в желудочно-кишечный тракт. Под термином "в значительной степени не высвобождается" подразумевают, что антагонист может высвобождаться в маленьком количестве, но высвобождаемое количество не оказывает влияние или не оказывает значительного влияния на анальгетическое действие, когда дозированную форму вводят перорально хозяину, например млекопитающему (например, человеку), как предназначено. Термины "в значительной степени предотвращает", "предотвращает" или какие-либо слова, которые происходят отсюда, как использовано в данном документе, не предусматривают обязательно полное или 100% предотвращение. Предпочтительнее, существуют варьирующие степени предотвращения, о которых какой-либо квалифицированный специалист в данной области отдает себе отчет, как имеющих потенциальное преимущество. В этом отношении блокирующий агент в значительной степени предотвращает или предотвращает высвобождение антагониста до предела, при котором предотвращается высвобождение, по меньшей мере, около 80% антагониста из изолирующей субъединицы в желудочно-кишечный тракт на протяжении периода времени, который составляет больше, чем 24 часа. Предпочтительно блокирующий агент предотвращает высвобождение, по меньшей мере, около 90% антагониста из изолирующей субъединицы в желудочно-кишечный тракт на протяжении периода времени, который составляет больше, чем 24 часа. Более предпочтительно блокирующий агент предотвращает высвобождение, по меньшей мере, около 95% антагониста из изолирующей субъединицы. Наиболее предпочтительно блокирующий агент предотвращает высвобождение, по меньшей мере, около 99% антагониста из изолирующей субъединицы в желудочно-кишечный тракт на протяжении периода времени, который составляет больше, чем 24 часа.
По результатам представленного изобретения количество высвобожденного антагониста после перорального введения может быть измерено in vitro путем исследования растворения, как описано в фармакопеи США (United States Pharmacopeia) (USP26) в главе <711> «Растворение». Например, используют 900 мл 0,1 N НС1, аппарат 2 (лопатка), 75 оборотов в минуту, при 37°С, чтобы измерить высвобождение при различных периодах времени из дозированной единицы. Другие способы измерения высвобождения антагониста из изолирующей субъединицы на протяжении заданного периода времени известны в данной области с уровня техники (смотри, например, USP26).
Без привязки к какой-либо определенной теории полагают, что изолирующая субъединица изобретения преодолеет ограничения изолированных форм антагониста, известных с уровня техники, в которых изолирующая субъединица изобретения понижает осмотически регулируемое высвобождение антагониста из изолирующей субъединицы. Кроме того, полагают, что в представленном изобретении изолирующая субъединица уменьшает высвобождение антагониста на протяжении более длительного периода времени (например, больше, чем 24 часа) по сравнению с изолированными формами антагонистов, известных с уровня техники. Факт, что изолированная субъединица изобретения предусматривает более длительное предотвращение высвобождения антагониста, является чрезвычайно релевантным, впоследствии быстрая отмена могла бы происходить после времени, на протяжении которого терапевтический агент высвобождается и действует. Хорошо известно, что время транзита через желудочно-кишечный тракт для индивидуумов значительно варьирует внутри популяции. Отсюда, остаток дозированной формы может удерживаться в тракте на протяжении более длительного времени, чем 24 часа, и в некоторых случаях более длительном, чем 48 часов. Кроме того, хорошо известно, что опиоидные анальгетики становятся причиной снижения моторики кишечника, что в дальнейшем продлевает время транзита через желудочно-кишечный тракт. В настоящее время формы с пролонгированным высвобождением, имеющим действие на протяжении 24 часового периода времени утверждены Food and Drug Administration. В этом отношении изолирующая субъединица представленного изобретения обеспечивает предотвращение высвобождения антагониста на протяжении периода времени, который составляет больше, чем 24 часа, когда изолирующая субъединица не была поврежденной.
Предполагается, что изолирующая субъединица изобретения в значительной степени предотвращает высвобождение антагониста, когда не повреждена. Под "неповрежденной" подразумевают, что дозированная форма не подвергалась повреждению. В качестве такового, антагонист и агонист отделены друг от друга внутри неповрежденной дозированной формы. Подразумевается, что термин "повреждение" включает какую-либо манипуляцию механическим, термическим и/или химическим способами, которые изменяют физические свойства дозированной формы. Повреждением может быть, например, раздавливание (например, с помощью ступки и пестика), разрезание, размалывание, разжевывание, растворение в растворителе, нагревание (например, больше, чем около 45°С) или какое-либо их комбинирование. Когда изолирующая субъединица изобретения повреждается, антагонист непосредственно высвобождается из изолирующей субъединицы. Дозированная форма, которая повредилась, таким образом, что антагонист оттуда высвобождается, рассматривается "в значительной степени разрушенной", где при введении дозированной формы субъекту (например, человеку), антагонист ингибирует или иным способом мешает активности агониста у субъекта, включая вмешательство в способность агониста вызывать угнетение дыхания. В любом случае антагонист ингибирует или иным способом вмешивается в активность агониста, что может быть определено, используя какие-либо фармакодинамические (PD) или фармакокинетические (РК) измерения, имеющиеся в распоряжении квалифицированного специалиста в данной области с уровня техники, включая, но не ограничиваясь, теми, которые описаны в данном документе. Если антагонист вмешивается в действие агониста, то статистически значительная разница в измерениях одного или более измерений PD или РК, как правило, наблюдается между дозированными формами.
Под "субъединицей" подразумевают то, что она включает композицию, смесь, частицу и т.п., что может обеспечить дозированную форму (например, пероральную дозированную форму), когда комбинируют с другой субъединицей. Субъединица может быть в форме драже, пеллеты, гранулы, сфероида или подобного и может быть комбинированным с дополнительными такими же или отличающимися субъединицами, в форме капсул, таблеток или им подобного, чтобы обеспечить дозированную форму, например, пероральную дозированную форму. Субъединица, кроме того, может быть компонентом более большой, отдельной единицы, который образует часть такой единицы, например, слой. Например, субъединица может быть сердцевиной, покрытой антагонистом и защитным слоем; данная субъединица затем может быть покрыта дополнительными композициями, включая фармацевтически активный агент, такой как опиоидный агонист.
Под "антагонистом терапевтического агента" подразумевают какое-либо лекарственное средство или молекулу природного происхождения или синтетическое, которое связывается с такой же молекулой-мишенью (например, рецептором) терапевтического агента, которое не вырабатывает терапевтический, внутриклеточный или in vivo ответ. В этом отношении антагонист терапевтического агента связывается с рецептором терапевтического агента, таким образом, предотвращая терапевтический агент от действия на рецептор. В случае опиоидов антагонист может предотвращать угнетение дыхания.
Стандартные фармакодинамические (PD) и фармакокинетические (РК) измерения могут быть использованы для сравнения действий различных дозированных форм (например, неповрежденных в противоположность "поврежденным" или "в значительной степени разрушенным") на субъект или чтобы определить, если дозированная форма была повреждена или оказалась в значительной степени разрушенной. Стандартные измерения включают, например, известные PD стандарты и шкалы, включая, но не ограничиваясь этим, один или более из визуальной аналоговой шкалы (VAS) сродства препарата (Balster & Bigelow, 2003; Griffiths et al., 2003), VAS общего сродства препарата, ARCI короткой формы (Martin et al., 1971), Cole/ARCI (Cole et al., 1982), Cole/ARCI-стимулированной эйфории, субъективной оценки лекарственного средства (Girffiths, et al., 1993; Griffiths, et al., 1996), Cole/ARCI аддиктивного протенциала, ARCI-морфин-бензедриновой группы (MBG), VAS-хороших эффектов, VAS-испытываемого наслаждения, VAS-нежелательных эффектов, VAS-испытываемой тошноты, VAS-отвращения, ARCI-LSD, Cole/ARCI-вызывания неприятных физических ощущений, Cole/ARCI-вызывания неприятных ощущений дисфории, VAS-каких-либо эффектов, VAS-головокружения, ARCI-амфетамина, ARCI-BG, Cole/ARCI-стимулирования моторики, VAS-сонливости, ARCI-PCAG, Cole/ARCI-успокоения сознания, успокоения моторики, и/или пуппилометрии (Knaggs, et al., 2004), наряду с прочим. Измерения могут включать среднее и/или усредненную площадь под кривой эффекта через 0-2 часа после введения дозы (AUE (0-2 ч)), площадь под кривой эффекта через 0-8 часов после введения дозы (AUE(0-8 ч)), площадь под кривой эффекта через 0-24 часа после введения дозы (AUE(0-24 ч)), возникающий после введения дозы диаметр зрачка (например, PCmin, РАОС(0-2 ч), РАОС(0-8 ч), РАОС(0-24 ч)), исходная оценка через 1,5 часа после введения дозы (HR1,5), максимальный эффект (Еmах), время для достижения максимального эффекта (ТЕmах).
Особенно информативными являются измерения Еmах по VAS-сродства препарата, VAS-общего сродства препарата, Cole/ARCI-стимулированной эйфории, субъективной оценке лекарственного средства, Cole/ARCI-аддитивного потенциала, ARCI-MBG, VAS-хороших эффектов, VAS-испытываемого наслаждения и пуппилометрии.
Для композиций, описанных в данном документе, РК-измерения, относящиеся к высвобождению морфина и налтрексона, могут быть пригодными. Измерения уровней морфина, налтрексона и/или 6-β-налтрексола в крови (например, плазме) или у пациентов, которым были введены различные дозированные формы, являются подходящими. Специфические РК-параметры, которые могут быть измерены, включают, например, среднюю и/или серединную пиковую концентрацию, максимальную концентрацию в плазме (Сmах), время до максимальной концентрации (Тmах), константу скорости элиминации (λz), конечный период полувыведения (Т1/2), площадь под кривой концентрация-время от 0 часов после введения дозы до 8 часов после введения дозы (AUC0-8 ч) (пг*ч/мл), площадь под кривой временной концентрации в плазме от начала отсчета времени до времени последней концентрации, поддающейся количественному определению (AUClast) (пг*ч/мл), и площадь под кривой временной концентрации в плазме от начала отсчета времени, экстраполированного на бесконечность (AUQinf) (пг*ч/мл), скорость элиминации (kе) (1/ч), коэффициент очищения (л/ч), и/или объем распределения (л). Образцы (например, кровь) могут быть забраны с тех, которым вводили дозированную форму в различные точки времени (например, через приблизительно какие-нибудь 0,5, 1, 1,5, 2, 3, 4, 6, 8, 10, 12 часов после введения). Когда образцом является кровь, плазма может быть получена из таких образцов, используя стандартные методики, и измерения могут быть сделаны из этого. Средние и/или серединные измерения в плазме затем могут быть рассчитаны и сравнены для различных дозированных форм.
В конкретных вариантах осуществления изобретения могут быть приняты во внимание одно или более из таких стандартных измерений, которые наблюдались после введения дозированной формы, отличающейся сниженной или повышенной по сравнению с той, которая наблюдалась после введения отличающейся дозированной формы, где различие между действиями дозированных форм отличается на приблизительно какой-либо из следующих диапазонов: 5-10%, 10-15%, 15-20%, 10-20%, 20-25%, 25-30%, 20-30%, 30-35%, 35-40%, 30-40%, 40-45%, 45-50%, 40-50%, 50-55%, 55-60%, 50-60%, 60-65%, 65-70%, 60-70%, 70-75%, 75-80%, 70-80%, 80-85%, 85-90%, 80-90%, 90-95%, 95-100%, и 90-100%. В некоторых вариантах осуществления изобретения могут быть приняты во внимание измерения "подобные" друг другу, где существует меньше, чем приблизительно какой-либо из 0%, 5%, 10%, 15%, 20% или 25% разницы. Разница, кроме того, может быть выражена как дробь или соотношение. Например, измерение, которое наблюдали для неповрежденной дозированной формы или в значительной степени разрушенной дозированной формы может быть выражен как дробь, например, приблизительно какая-либо из 1/2 (одной второй), 1/3 (одной третей), 1/4 (одной четвертой), 1/5 (одной пятой), 1/6 (одной шестой), 1/7 (одной седьмой), 1/8 (одной восьмой), 1/9 (одной девятой), 1/10 (одной десятой), 1/20 (одной двадцатой), 1/30 (одной тридцатой), 1/40 (одной сороковой), 1/50 (одной пятидесятой), 1/100 (одной сотой), 1/250 (одной двести пятидесятой), 1/500 (одной пятисотой) или (1/1000 одной тысячной) из тех, что в значительной степени разрушенной или неповрежденной дозированной формы, соответственно. Разница, кроме того, может быть выражена как соотношение (например, приблизительно какое-либо из 0,001:1, 0,005:1, 0,01:1, 0,1, 0,2:1, 0,3:1, 0,4:1, 0,5:1, 0,6:1, 0,7:1, 0,8:1, 0,9:1, 1:1, 1:2, 1:3, 1:4, 1:5, 1:6, 1:7, 1:8, 1:9 или 1:10).
Следует рассматривать как "значительный", "статистически разный", "значительно уменьшенный" или "значительно выше", например, численные значения или измерения, относящиеся к наблюдаемой(ым) разнице(отличиям), могут быть подвергнуты статистическому анализу. Измерения предельно допустимого содержания вещества в организме могут быть собраны, и может быть определен значительный эффект от предельно допустимого содержания. Эффект от лечения может быть оценен после ковариации предельно допустимого содержания, регулирование было сделано в анализе модели ковариантности (ANCOVA). Модель может включать лечение, период и последовательность как фиксированные эффекты, и объекты вводятся в последовательности как произвольное действие. Для фармакодинамических измерений, которые имеют значения предварительной дозы, модель может включать значение предварительной дозы предельно допустимого содержания, как ковариацию. Модель линейных смешанных эффектов может быть основана из расчета протокола популяции. 5% ошибка типа I рассматривает р-значение меньшее чем 0,05, может считаться "статистически значительной" для всех индивидуальных исследований гипотезы. Все статистические исследования могут быть выполнены, используя двусторонний критерий. Для каждого из основных эффектов нулевой гипотезой может быть "не существовала главная гипотеза" и альтернативной гипотезой может быть "существовала главная гипотеза." Для каждой из противоположностей нулевой гипотезой может быть "не существовала разница от эффектов между исследуемыми парами" и альтернативной гипотезой может быть "существовала разница от эффектов между исследуемой парой." Процедура по Benjamin и Hochberg может быть использована для контролирования ошибки типа I, возникающей при сравнении многократной терапии для всех основных конечных точек.
Статистическое значение, кроме того, может быть измерено, используя анализ вариантности (ANOVA) и критерий Шуирманна двух односторонних проверок по критерию Стьюдента при 5% уровне значения. Например, подвергнутые log-преобразованию РК-параметры Cmax, AUQlast и AUQinf могут быть сопоставлены, чтобы определить статистически значительные различия между дозированными формами. 90% доверительный интервал для соотношения средних геометрических (Тест/Эталон) может быть рассчитан. Как упоминалось, в конкретных вариантах осуществления изобретения дозированные формы могут быть "биоэквивалентными", или "биоэквивалентность" может быть заявлена, если нижний или верхний доверительные интервалы log-преобразованных параметров находятся приблизительно в каком-либо из 70-125%, 80%-125% или 90-125% друг друга. Биоэквивалентный или биоэквивалентность предпочтительно заявляется, когда нижний и верхний доверительные интервалы log-преобразованных параметров находятся около 80%-125%.
Высвобождение морфина, налтрексона и 6-β-налтрексола из разных композиций in vitro может быть определено, используя стандартные методики исследования растворения, такие как описано в Фармакопее США (United States Pharmacopeia (USP26)) в главе <711> Растворение (например, 900 мл 0,1 N НСl, Прибор 2 (лопастный), 75 оборотов в минуту, при 37°С; 37°С и I00 оборотов в минуту) или 72 часа в подходящем буфере, как 500 мл 0,05М pH 7,5 фосфатный буфер), чтобы измерить высвобождение при разных временах из дозированной единицы. Другие способы измерения высвобождения антагониста из изолированной субъединицы на протяжении заданного периода времени известны из уровня техники (смотри, например, USP26) и, кроме того, могут быть использованы. Такие исследования, кроме того, могут быть использованы в модифицированной форме, например, путем применения буферной системы, содержащей поверхностно-активное вещество (например, 72 часа в 0,2% Тритон X-100/0,2% ацетат натрия/0,002 N HCl, pH 5,5). Уровни в крови (включая, например, уровни в плазме) морфина, налтрексона и 6-β-налтрексола могут быть измерены, используя стандартные методики.
Антагонистом может быть какой-либо агент, который сводит на нет действие терапевтического агента или вызывает понижение губительных эффектов опиоидов, которые вызывали угнетение дыхания.
Терапевтический агент может быть опиоидным агонистом. Под "опиоидом" подразумевают, что он включает лекарственное средство, гормон или другое химическое или биологическое вещество, природное или синтетическое, имеющее седативный, наркотический или другой(ие) подобный(е) эффект(ы) к тем, которые содержат опиум или его природные или синтетические производные. Под "опиоидным агонистом" иногда в данном документе взаимозаменяемо использовали с терминами "опиоид" и "опиоидный анальгетик" подразумевают, что он включает один или больше опиоидных агонистов, или самостоятельно, или в комбинации, и, кроме того, подразумевают, что он включает основание опиоида, смешанные или комбинированные агонисты-антагонисты, частичные агонисты, их фармацевтически приемлемые соли, их стереоизомеры, их простые эфиры, их сложные эфиры, и их комбинации.
Опиоидные агонисты включают, например, альфентанил, аллилпродин, альфапродин, анилэридин, бензилморфин, безитрамид, бупренорфин, буторфанол, клонитазен, кодеин, циклазоцин, дезоморфин, декстроморамид, дезоцин, диампромид, дигидрокодеин, дигидроэторфин, дигидроморфин, дименоксадол, димефептанол, диметиламбутен, диоксафетил бутират, дипипанон, эптазоцин, этогептазин, этилметилтиамбутен, этилморфин, этонитазен, эторфин, фентанил, героин, гидрокодон, гидроморфон, гидроксипетидин, изометадон, кетобемидон, леваллорфан, леворфанол, левофенацилморфан, лофентанил, меперидин, мептазинол, метазоцин, метадон, метопон, морфин, мирофин, налбуфин, нарцеин, никоморфин, норлеворфанол, норметадон, налорфин, норморфин, норпипанон, опиум, оксикодон, оксиморфон, папаверетум, пентазоцин, фенадоксон, феназоцин, феноморфан, феноперидин, пиминодин, пиритрамид, профептазин, промедол, проперидин, пропирам, пропоксифен, суфентанил, трамадол, тимидин, их производные или комплексы, их фармацевтически приемлемые соли, и их комбинации. Предпочтительно опиоидный агонист выбирают из группы, состоящей из гидрокодона, гидроморфона, оксикодона, дигидрокодеина, кодеина, дигидроморфина, морфина, бупренорфина, их производных или комплексов, их фармацевтически приемлемых солей и их комбинаций. Наиболее предпочтительно опиоидный агонист представляет собой морфин, гидроморфон, оксикодон или гидрокодон. В предпочтительном варианте осуществления изобретения опиоидный агонист включает оксикодон или гидрокодон и присутствует в дозированной форме в количестве от около 15 до около 45 мг, и опиоидный антагонист включает налтрексон и присутствует в дозированной форме в количестве от около 0,5 до около 5 мг. Эквианальгетические рассчитанные дозы (мг) данных опиоидов по сравнению с 15 мг дозой гидрокодона, являются следующими: оксикодон (13,5 мг); кодеин (90,0 мг), гидрокодон (15,0 мг), гидроморфон (3,375 мг), леворфанол (1,8 мг), меперидин (15,0 мг), метадон (9,0 мг), и морфин (27,0).
Гидрокодон представляет собой полусинтетическое наркотическое анальгетическое и противокашлевое средство с множественными действиями на нервную систему и желудочно-кишечный тракт. Химически гидрокодон представляет собой 4,5-эпокси-3-метокси-17-метилморфинан-6-он, и, кроме того, известен как дигидрокодеинон. Подобно другим опиоидам, гидрокодон может быть характеробразующим и из него может быть получено лекарственное средство в зависимости от морфинового типа. Подобно другим производным опиума избыточные дозы гидрокодона будут угнетать дыхание.
Пероральный гидрокодон, кроме того, является доступным в Европе (например, Бельгии, Германии, Греции, Италии, Люксембурге, Норвегии и Швейцарии) как противокашлевый агент. Парентеральный препарат, кроме того, является доступным в Германии как противокашлевый агент. Для использования как анальгетик, гидрокодона битартрат, как правило, является доступным в Соединенных Штатах только как фиксированная комбинация с неопиатными лекарственными средствами (например, ибупрофеном, ацетаминофеном, аспирином; и т.д.) для облегчения умеренной до умеренно сильной боли.
В вариантах осуществления изобретения, в которых опиоидный агонист включает гидрокодон, пероральные дозированные формы пролонгированного высвобождения могут включать дозы анальгетиков от около 8 мг до около 50 мг гидрокодона на дозированную единицу. В пероральных дозированных формах пролонгированного высвобождения, в которых гидроморфон является терапевтически активным опиоидом, он включен в количестве от около 2 мг до около 64 мг гидроморфона гидрохлорида. В другом варианте осуществления изобретения опиоидный агонист содержит морфин, и пероральные дозированные формы пролонгированного высвобождения изобретения включают от около 2,5 мг до около 800 мг морфина, по массе. В еще другом варианте осуществления изобретения опиоидный агонист содержит оксикодон, и пероральные дозированные формы пролонгированного высвобождения включают от около 2,5 мг до около 800 мг оксикодона.
В предпочтительном варианте осуществления изобретения опиоидный антагонист содержит налтрексон или соль налтрексона. В лечении пациентов, ранее постоянно принимавших опиоиды, налтрексон использовали в больших пероральных дозах (более 100 мг), чтобы предотвратить эффекты, вызывающие эйфорию, опиоидных агонистов. Сообщалось, что налтрексон приводит в действие сильное преимущественно блокирующее действие против мю выше дельта сайтов. Налтрексон известен как синтетический представитель того же рода, что и оксиморфон со свойствами не опиоидного агониста, и отличается по структуре от оксиморфона посредством замещения метильной группы, расположенной при атоме азота, в оксиморфоне на циклопропилметильную группу. Гидрохлоридная соль налтрексона является растворимой в воде вплоть до около 100 мг/см3. Фармакологические и фармакокинетические свойства налтрексона были определены на различных животных и при клинических исследованиях. См., например, Gonzalez et al. Drugs 35:192-213 (1988). При последующем пероральном введении, налтрексон быстро абсорбируется (в течение 1 часа) и имеет пероральную биоэквивалентность в диапазоне 5-40%. Связывание протеина налтрексоном составляет приблизительно 21% и объем распределения следующей однократной дозы введения составляет 16,1 л/кг.
Налтрексон является коммерчески доступным в таблетированной форме (Revia®, DuPont (Wilmington, Del.)) для лечения алкогольной зависимости и для блокирования экзогенно вводимых опиоидов. См., например, Revia (таблетки налтрексона гидрохлорида), Physician's Desk Reference, 51sted., Montvale, N.J.; and Medical Economics 51:957-959 (1997). Дозировка 50 мг Revia® блокирует фармокологические эффекты 25 мг IV введенного героина в течение вплоть до 24 часов. Известно, что при совместном введении с морфином, героином или другими опиоидами на хронической основе налтрексон блокирует развитие физической зависимости к опиоидам. Полагают, что способ, по которому налтрексон блокирует действия героина, представляет собой конкурентное связывание с опиоидными рецепторами. Налтрексон используется для лечения наркотической зависимости посредством полного блокирования действий опиоидов. Обнаружено, что наиболее успешное использование налтрексона при наркотической зависимости происходит у лиц, привычно употребляющих наркотики, имеющих благоприятный прогноз, как часть всесторонней производственной и реабилитационной программы, которая включает поведенческий контроль или другие совместно повышающие способы. Для лечения наркотической зависимости налтрексоном желательным является, чтобы пациент не принимал опиоиды в течение, по меньшей мере, 7-10 дней. Начальная доза налтрексона для таких целей, как правило, составляет около 25 мг, и если не имеет место признаки прекращения приема наркотиков, то доза может быть повышена до 50 мг на день. Дневная доза 50 мг рассматривается для получения адекватного клинического блокирования действий парентерально введенных опиоидов. Налтрексон, кроме того, применяют для лечения алкоголизма как дополнение к социальным и психотерапевтическим способам. Другие предпочтительные опиоидные антагонисты включают, например, циклазоцин и налтрексон, оба из которых имеют циклопропилметильные заместители при азоте, сохраняют многое из их эффективности посредством перорального пути и сохраняют дольше с продолжительностями, приближающимися к 24 часам после перорального введения.
Основываясь на оценках системного клиренса и периода полувыведения налоксона, профили концентрации, следующие после IV инъекции 0,4 мг с и без непрерывной инфузии налоксона в течение 4 часов, могут быть смоделированы, как показано на фигуре 1, где сплошная красная линия показывает профиль концентрации налоксона в плазме после однократной дозы в форме пилюли, и пунктирная линия представляет профиль после дозы в форме пилюли и плюс непрерывной инфузии в течение 4 часов.
Сравниваемая с профилями терапевтическая концентрация налоксона представляет собой профиль концентрации налтрексона, если все из лекарственного средства высвобождалось из 80 мг дозы ALO-02 (оксикодон 80 мг), содержащей 12% налтрексона. Теоретически, пиковые концентрации налтрексона, которые достигаются вплоть до 2500 пг/мл, количество налтрексона, которое достигается при системном циркуляционном действии, как средство экстренной терапии, если препараты оксикодона с изолированным налтрексоном были разжеваны или разрушены при попытке злоупотребления препарата. (Gonzalez JP and Brogden RN. Naltrexone: A review of its pharmacodynamic and pharmacokinetic properties and therapeutic efficacy in the management of opioid dependence. Drugs. 1988; 35: 192-213; Verebey K, Volavka J, Mute SJ, and Resnick RB. Naltrexone: Disposition, metabolism, and effects after acute and chronic dosing. Clin Pharm and Ther. 1976; 20(3): 315-28; Willette RE and Barnett G. Narcotic antagonists: naltrexone pharmacochemistry and sustained-release preparation. Department of Health and Human Services. National Institute on Drug Abuse (NIDA), Division of Research. NIDA Research Monotraph 28, 1981.)
Соотношение опиоидный агонист/налтрексон, которое будет ослаблять вызванное опиоидом угнетение дыхания, будет зависеть частично от опиоидного агониста. Идеально, соотношение является таким, что если препарат подвергают манипуляциям, то количество налтрексона, высвобождаемое при манипуляции, предотвратит индуцирование угнетения дыхания, когда подвергнутый манипуляциям препарат вводят человеку. Препараты представленного изобретения, кроме того, включают соотношения опиоидный агонист/налтрексон, которые уменьшают тяжесть угнетения дыхания, которое вызвано передозировкой опиоидов. В конкретных вариантах осуществления изобретения соотношение оксикодон к налтрексону в композиции составляет от около 2% до около 30%. В другом варианте осуществления изобретения соотношение оксикодон к налтрексону в композиции составляет от около 2% до около 20%. В варианте осуществления изобретения соотношение оксикодон к налтрексону в композиции составляет от около 2:1 (50%) до около 50:1 (2%). В предпочтительном варианте осуществления изобретения соотношение оксикодон к налтрексону в композиции составляет от около 5:1 (20%) до около 25:1 (4%).
В предпочтительном варианте осуществления изобретения соотношение оксикодона к налтрексону в композиции составляет от около 10:1 (10%) до около 20:3 (15%).
В варианте осуществления изобретения соотношение гидрокодон к налтрексону в композиции составляет от около l:l (100%) до около 100:1 (1%). В предпочтительном варианте осуществления изобретения соотношение гидрокодон к налтрексону в композиции составляет от около 5:1 (20%) до около 25:1 (4%). В предпочтительном варианте осуществления изобретения соотношение гидрокодон к налтрексону в композиции составляет от около 10:1 (10%) до около 20:3 (15%).
В варианте осуществления изобретения соотношение морфин к налтрексону в композиции составляет от около 1:1 (100%) до около 100:1 (1%). В предпочтительном варианте осуществления изобретения соотношение морфин к налтрексону в композиции составляет от около 5:1 (20%) до около 25:1 (4%). В предпочтительном варианте осуществления изобретения соотношение морфин к налтрексону в композиции составляет от около 50:1 (2%) до около 20:3 (15%).
Дыхание представляет собой обмен кислорода и углекислого газа. Адекватность дыхания может быть измерена с точки зрения поддержания давления артериального углекислого газа и кислорода в пределах нормы. Вентиляцию, как правило, описывают с точки зрения альвеолярной вентиляции, достаточной для поддержания артериального СO2 и O2. К сожалению, непрерывные, неинвазивные измерения парциального давления газов артериальной крови недоступны. В лучшем случае, является возможным периодический отбор пробы крови, но это требует размещения инвазивной артериальной линии и может считаться клинически неуместным в некоторых популяционных исследованиях. Поэтому искали заменители артериального СO2 и O2, например, парциальное давление СO2 в выдыхаемом воздухе в конце спокойного выдоха (уровень двуоксида углерода в выдыхаемом из организма воздухе, нормальные значения которого составляют от 4% до 6%, что эквивалентно от 35 до 45 мм ртутного столба) и SpO2 (пульсовая оксиметрия дает оценки артериального насыщения оксигемоглобина (SaO2), используя выбранные длины волн света для неинвазивного определения насыщения оксигемоглобином), соответственно.
Вентилирование предусматривает как здоровую дыхательную систему (структурные единицы легких, свободную проходимость дыхательных путей), так и непораженную нейронную активность (дыхательный центр мозга, спинного мозга). Физические компоненты вентилирования могут быть измерены (например, частота дыхания, дыхательный объем) и охарактеризованы по отдельности или в комбинации (минутная вентиляция=частота дыхательных движений х дыхательный объем). Нейронная активация может быть измерена путем измерения вентиляционного ответа к индуцированной гипоксии и/или гиперкапнии. При измерении частоты дыхательных движений могут возникать трудности для наблюдателя, особенно при низких или нерегулярных скоростях. Косвенные измерения частоты дыхательных движений с использованием изменений в электрическом импедансе ЭКГ может дать частоту дыхательных движений, но они предрасположены к ошибке. Измерение следов СO2 в конце спокойного выдоха зависит от свободной проходимости дыхательных путей, так как является измерением объема вдоха на пневмотахографе.
Характерная картина угнетения дыхания, вызванного опиоидами, представляет собой уменьшение частоты дыхательных движений (брадипноэ) с глубокими резкими вдохами при дыхании. Пациенты часто будут находиться в сознании, но не хватает активности дышать. Как только даны вербальные команды, чтобы дышать, пациент будет следовать им и вдыхать, выполняя их. Потеря активности дыхательным центром является характерной для опиоидов, но данное свойство трудно оценить количественно.
Среднее парциальное давление артериального углекислого газа составляет 38 мм ртутного столба и не изменяется со временем. В отличие от него парциальное давление артериального кислорода все же изменяется с возрастом (как правило, 94 мм ртутного столба в возрастном диапазоне 20-29; 81 мм ртутного столба в возрастном диапазоне 60-69). Кроме того, парциальное давление артериального кислорода значительно изменяется в присутствии дополнительного кислорода. Поэтому важно установить содержание кислорода во вдыхаемом воздухе всякий раз, когда сообщается парциальное давление артериального кислорода. Для исследования дыхания предпочтительным является проведение исследования с субъектами, которые дышат воздухом помещения, а не дополнительным кислородом.
Если дыхание представляет собой поддержание адекватного парциального давления артериального СO2 и O2, то далее угнетение дыхания может быть определено как неспособность поддерживать такие парциальные давления артериального СO2 и O2. Несколько работ высветили сложность в определении конкретных порогов угнетения дыхания, так как обычно не существует доступа к газовым данным артериальной крови и, таким образом, выбирают другие параметры дыхания. В настоящее время не существует консенсуса о том, какие конкретные параметры или сочетание параметров адекватно составляют угнетение дыхания.
Таким образом, для целей данного изобретения первичный порог угнетения дыхания может представлять собой развитие гиперкапнии, физического состояния, характеризующимся присутствием аномально высокого уровня диоксида углерода в циркулирующей крови (РаСO2>45 мм ртутного столба). Во время клинически значительного угнетения дыхания гиперкапния обычно происходит в сочетании со снижением дыхательного функционирования, часто проявляющегося как какая-либо комбинация снижения частота дыхательных движений, снижения конечного выдыхаемого объема, снижения минутного объема, снижения pH артериальной крови, снижения насыщенности O2 и увеличения парциальное давление СO2 во выдыхаемом воздухе в конце выдоха (ЕТ СO2) или транскутанных уровней СO2. Ослабление угнетения дыхания, индуцированного опиоидами, налтрексоном может быть свидетельством значительного снижения РЕТСO2, увеличения дыхательного функционирования, повышения pH, увеличения O2 и увеличение наклона взаимоотношения вентиляция-РЕТСO2, основанного на гиперкапническом вентиляционном ответе (HCVR). Ослабление угнетения дыхания, индуцированного опиоидами, может быть определено как, по крайней мере, 5% снижение РЕTСO2 или, по крайней мере, возрастание на 5% вентилирования, или, по крайней мере, увеличение на 5% наклона взаимоотношения вентиляция-РЕТСO2, основанного на гиперкапническом вентиляционном ответе. В предпочтительных вариантах осуществления изобретения ослабление угнетения дыхания, индуцированного опиоидами, обеспечит, по крайней мере, 10%-ное снижение РЕТСO2 или, по крайней мере, 10%-ное увеличение вентилилирования, или, по крайней мере, 10%-ное увеличение наклона взаимоотношения вентиляция-РЕТСO2, основанного на гиперкапническом вентиляционном ответе. В более предпочтительных вариантах осуществления изобретения ослабление угнетения дыхания, индуцированного опиоидами, обеспечит, по крайней мере, 20%-ное снижение РЕТСO2 или, по крайней мере, 20%-ное увеличение вентилилирования, или, по крайней мере, 20%-ное увеличение наклона взаимоотношения вентиляция-РЕТСO2, основанного на гиперкапническом вентиляционном ответе.
Таким образом, представленное изобретение касается опиатных анальгетических лекарственных препаратов и способов введения данных препаратов, при которых у человека ослабляется угнетение дыхания, когда препарат подвергался манипуляции до введения человеку.
Дополнительные варианты осуществления изобретения и характеристики представленного изобретения приведены в следующих неограничивающих примерах.
ПРИМЕРЫ
Пример 1
Эффекты от внутривенно введенного налтрексона на угнетение дыхания, вызванного морфином у здоровых добровольцев
Исследование угнетения дыхания представляет собой двойное, слепое, рандомизированное, четырехлинейное перекрестное исследование здоровых добровольцев, субъектов мужского или женского пола, в возрасте от 21 до 35 лет, включающее, и в основном с хорошим здоровьем, как определено Исследователем.
В части А период дозирования I, с последующим 15-дневным периодом скрининга, группы из 4 субъектов, которые собирают для исследования включение/исключение необходимых условий, вносят в список и рандомизируют в соотношении 3:1, чтобы получать или инъекцию морфина сульфата 10 мг (N=3), или плацебо (N=1).
В течение каждого периода лечения каждого субъекта госпитализируют в клиническое отделение вечером дня - 1. В 1-й день субъект получает исследуемое(ые) лекарственное(ые) средство(а) и подвергают процедурам оценки фармакодинамических, фармакокинетических свойств и безопасности. Субъект остается в клиническом отделении до утра дня 2, в это время его выписывают из клинического отделения в отделение Исследователя.
По завершении части А периода дозирования 1 Исследователь и Спонсор просматривают демаскированные данные по безопасности и конечные данные PD и определяют правильность увеличивающейся дозы морфина сульфата до 20 мг.
Если с медицинской точки зрения считается безопасной и подходящей, то вторую группу из 4 субъектов рандомизируют в соотношении 3:1, чтобы получать или инъекцию морфина сульфата 20 мг (N=3), или плацебо (N=1). По завершении периода дозирования 2, Исследователь и Спонсор просматривают демаскированные данные по безопасности и конечные данные PD и определяют правильность увеличивающейся дозы морфина сульфата до 30 мг.
Если с медицинской точки зрения считается безопасной и подходящей, то третью группу из 4 субъектов рандомизируют в соотношении 3:1, чтобы получать или инъекцию морфина сульфата 30 мг (N=3) или плацебо (N=1). По завершении периода дозирования 3, Исследователь и Спонсор просматривают демаскированные данные по безопасности и конечные данные PD, и выполняют определение приблизительной подходящей дозы инъекции морфина сульфата, чтобы передать на фазу В.
В течение каждого из периодов дозирования в части A (IA-IIIA) субъекты находились изолированными в клиническом отделении на протяжении приблизительно 40 часов (2 ночи и 3 дня) и каждый период дозирования разделяют посредством периода отмены, по меньшей мере, из 7 дней.
Минимальное количество 4 и максимальное количество 12 субъектов принимают участие в части А.
Часть В: Фаза лечения
Часть В представляет собой рандомизированное, двойное слепое, плацебо-контролированное, 4-линейное перекрестное исследование на 12 здоровых добровольцах. В последующей части В 15-дневный период скрининга субъекты, которые собирают для исследования на включение/исключение необходимых условий, вносят в список и рандомизируют в одну из 4 очередных групп лечения (1-4), как показано ниже. Каждый субъект получает все 4 лечения (А, В, С и D), вместе с тем каждое лечение отделено, по меньшей мере, 1-недельным периодом отмены. Доза инъекции морфина сульфата, использованная в части В, представляет собой дозу, определенную в части А, которая с медицинской точки зрения является безопасной и подходящей.
Таблица 1. Схема лечения
Лечение А: Морфина сульфат* внутривенно+Плацебо (солевой раствор) внутривенно
Лечение В: Морфина сульфат* внутривенно+Налтрексон* 4% внутривенно
Лечение С: Морфина сульфат* внутривенно+Налоксон* 4% внутривенно
Лечение D: Плацебо (солевой раствор) внутривенно+Налтрексон* 4% внутривенно
*Доза морфина сульфата (10, 20 или 30 мг) будет определяться из исследования части А. Доза налтрексона HCl и налоксона HCl (антагонист) в части В будет составлять 4% от дозы морфина сульфата, использованной в части В (например 10 мг морфина с 0,4 мг антагониста, 20 мг морфина с 0,8 мг антагониста, и 30 мг морфина с 1,2 мг антагониста).
В течение каждого периода лечения каждого субъекта госпитализируют в клиническое отделение вечером дня - 1. В 1-й день субъект получает исследуемое(ые) лекарственное(ые) средство(а) и подвергается процедурам оценки фармакодинамических, фармакокинетических свойств и безопасности. Субъект остается в клиническом отделении до утра дня 2, в это время его выписывают из клинического отделения в отделение Исследователя. Субъекты остаются в клиническом отделении до утра дня 2, в это время их выписывают из клинического отделения в отделение Исследователя.
В течение каждого из 4-х периодов лечения (I-IV) в части В субъектов удерживали в клиническом отделении в течение приблизительно 40 часов (2 ночи и 3 дня), каждое лечение разделяют посредством периода отмены, по меньшей мере, из 7 дней. Завершающую оценку безопасности выполняли в конце исследования.
Коммерческих поставщиков использовали, чтобы получить внутривенные растворы морфина сульфата, и налоксона НСl и налтрексона. Внутривенные дозированные растворы втягивают в шприцы и разбавляют физиологическим солевым раствором (0,9% хлорида натрия для инъекции) так, чтобы конечный объем дозированного раствора каждого лекарственного средства был одинаковым: морфина сульфат =10 мг в 10 мл солевого раствора; налтрексон=0,4 мг в 10 мл солевого раствора; налоксон =0,4 мг в 10 мл солевого раствора и плацебо=10 мл солевого раствора. Все исследуемые лекарственные средства (то есть морфин + плацебо; морфин+налтрексон; морфин + налоксон; и плацебо+налтрексон) вводят внутривенно, одновременно применяя би-слитое устройство, связанное с ультра-мини объемной трубкой, доставленной инфузионным насосом шприца. Данный способ доставки позволяет для каких-либо двух лекарственных средств быть инъекционно введенными одновременно с минимальным смешиванием, таким образом, понижая риск беспокойств внутривенной совместимости. Каждое лекарственное средство вливают в течение 2-минутного периода времени. Время и план проводимых мероприятий для сопровождения данного исследования представлены в O2.
1. Периоды лечения будут разделены 7-дневным периодом отмены между дозами.
2. Определенный, как приблизительный через 24 часа после дозы части В периода дозирования IV.
3. Медицинский осмотр будет включать рост, вес и BMI.
4. Клинические лабораторные анализы будет выполняться.
5. Скрининг на ВИЧ-1, ВИЧ-2, гепатит В и гепатит С.
6. Основные показатели состояния организма (давление крови, частота сердечных сокращений, частота дыхательных движений) будут измеряться. Во время части А и В периодов дозирования основные показатели состояния организма будут постоянно контролироваться в течение первых 6-часов после введения дозы. Температура в ротовой полости будет измеряться во время скрининга и регистрироваться перед каждым периодом дозирования (части А и В).
7. BIS контролирование будет проводиться постоянно до 6 часов после части В периодов дозирования.
8. Пневмотахография и плетизмография дыхательной индукции (RIP) будет выполняться.
9. Гиперкапнический дыхательный контроль будет выполняться в исходном состоянии (за 1 час до введения дозы) и через 1 и 4 часа после введения дозы. HCVR будут оценивать в исходном состоянии (за 1 час до введения дозы) при максимальном угнетении дыхания и последующем восстановлении дыхания.
10. Будут определяться газы артериальной крови.
11. РК-отбор образцов будет сделан.
12. Пупиллометрия будет сделана.
Как отмечено в плане времени и мероприятий (Таблица 2), в течение периодов дозирования IA на протяжении IIIA (часть А) и I на протяжении IV (часть В) субъекты будут следовать процедурам, указанным ниже, во время каждого 40-часа оставаться в Duke Clinical Research Unit (DCRU). Каждое лечение будет отделено, по меньшей мере, 1-недельным периодом отмены между дозами исследуемого(ых) лекарственного(ых) средства().
Исследование день - 1 (Вечер перед дозированием)
Субъекты, которых собирают по критериям, основанным на оценке скрининга, будут представлены в DCRU, по меньшей мере, за 10 часов перед дозированием. Субъектам может быть предложена еда и/или перекусить по необходимости в зависимости от времени регистрации. Процедуры, указанные ниже, будут выполнены:
Субъектам будет назначена последовательность лечения в соответствии с графиком рандомизации (только часть В).
Анализ мочи на беременность (только женщины).
Анализ мочи на лекарственные средства. Для продолжения у субъекта анализ должен быть отрицательным.
Анализ мочи на алкоголь. Для продолжения у субъекта анализ должен быть отрицательным.
Определяют использование сопутствующего лекарственного средства и записывают на eCRF.
Основные показатели состояния организма, включая температуру ротовой полости.
Все субъекты будут подвергнуты быстрому контролю в течение 6 часов перед лечением. По желанию будет разрешена вода за исключением 2 часового периода до и после введения дозы. Во время периодов нахождения пациентов в стационаре субъекты будут находиться под контролем все время. Штатный врач будет или присутствовать или приходить по вызову на протяжении всего исследования.
День лечения
После наблюдаемого ночного голодания, по меньшей мере, 6-часового будут начаты процедуры исследования. Субъект будет удерживаться в постели под углом примерно 35° в течение, по меньшей мере, 6 часов, во время которых субъект будет спокойно лежать и в полной мере сотрудничать с Исследователем и сотрудником, отвечающим за введение исследуемого(ых) препарата(ов), контролирование безопасности и получение экспериментальных данных. Ондансетрона 0,4 мг будет введен внутривенно за один час до исследования дозирования лекарственных средств в частях А и В. Все исследуемые лекарственные средства будут вводить внутривенно и одновременно в течение 2-минутного периода времени, используя би-слитое мини-насосное устройство, с помощью которого есть способность проведения инфузии двух препаратов одновременно.
Пневмотахограф будут перемещать в течение 15 минут каждые два часа во время шести часового периода после введения дозы (части А и В), на протяжении которого субъект может быть обеспечен полностью жидкой диетой, в зависимости от переносимости.
После 6-часового момента времени по усмотрению Исследователя участник исследования может передвигаться, если разрешено DCRU сотрудником. В тоже время, субъекту будет подан стандартный обед. После этого не будет никаких ограничений на воду или ходьбу, и вечером будет предложен стандартный ужин. Субъект останется в DCRU на 24 часа после введения дозы (день 2), когда субъект будет выписан после выполнения требований исследования.
Каждое лечение будет разделено, по меньшей мере, 1-недельным периодом отмены (выведения препарата из организма) между дозами.
Фармакодинамические измерения
Следующие процедуры выполняют для каждого лечения, описанного в части А и части В. Все периоды времени отбора проб будут определены в зависимости от времени начала инфузии исследуемым(ыми) лекарственным(ыми) средством(ами).
Пневмотахографические измерения выполняют, чтобы определить минутный объем вентиляции легких, частоту дыхательных движений, объем в конце спокойного выдоха и СО2 в период времени: за 30 минут, 10 и 5 минут перед дозированием (исходные значения перед дозированием) и через 5, 15, 30 и 45 минут, и 1, 1,5, 2, 2,5, 3, 3,5, 4 и 6 часов после введения дозы исследуемого(ых) лекарственного(ых) средства().
Периодический отбор образцов артериальной крови проводят в периоды времени: 15 минут (перед введением дозы) и через 5, 15 и 30 минут, и 1, 1,5, 2, 2,5, 3, 3,5, 4, 5, и 6 часов после введения дозы, чтобы измерять уровень диоксида углерода в артериальной крови (РаСО2), pH артериальной крови, и насыщение кислородом (SaO2).
Пульсовую оксиметрию проводят непрерывно, начиная за 30 минут до введения дозы и до 6 часов после введения дозы, чтобы контролировать насыщение кислородом (SpO2). Кроме того, радиоэлектрокардиографию используют, чтобы контролировать частоту сердечных сокращений и артериальное давление, устройство SenTec используют для непрерывного мониторинга чрескожного углекислого газа (PtcCO2), и мониторинг биспектрального индекса (BIS) используют, чтобы контролировать уровень сознания за тот же период времени. Измерения записывают за 15 минут (перед введением дозы) и через 5, 15 и 30 минут, и 1, 1,5, 2, 2,5, 3, 3,5, 4, 5, и 6, 8, 12 и 24 часа после введения дозы.
Устройство SenTec используют для непрерывного мониторинга чрескожного углекислого газа (PtcCO2), и мониторинг биспектрального индекса (BIS) будут использовать, чтобы контролировать уровень сознания за тот же период времени.
Пупиллометрические измерения выполняют за 20 минут перед введением дозы и через 10, 20 и 40 минут и 1, 1,5, 2, 2,5, 3, 3,5, 4, 5, 6, 8, 12 и 24 часа после введения дозы.
Респираторную индуктивную плетизмографию (RIP) применяют как вторичное измерение, чтобы контролировать частоту дыхательных движений и минутный объем начиная с периода времени 30 минут перед введением дозы и вплоть до 6 часов после введения дозы.
Пробу на гиперкапническую реакцию вентиляции (HCVR) выполняют в исходном состоянии (за 1 час перед введением дозы) и через 1 час и 4 часа после введения дозы на усмотрение Исследователя.
Гиперкапническую реакцию вентиляции определяют в исходном состоянии, крайнем состоянии угнетения дыхания и последующем восстановлении после угнетения дыхания.
Радиоэлектрокардиографию будут использовать непрерывно, чтобы контролировать частоту сердечных сокращений, артериальное давление, частоту дыхательных движений от 30 минут до 6 часов после введения дозы. После этого в течение моментов времени 8, 12 и 24 часа после введения дозы основные показатели состояния организма будут взятии у субъекта в положении сидя со ступнями на полу. Субъект должен будет сидеть, не двигаясь, в течение приблизительно 2 минут, перед тем как получить данные измерения давления крови и частоты сердечных сокращений.
Отбор образцов венозной крови выполняют, как описано ниже.
Фармакокинетические измерения
Забор крови и хранение проб
Часть А: Во время части А исследования в общей сложности отбирают вплоть до 195 мл крови (13 образцов на лечение х 5 мл на образец х 3 лечения) с целью количественного определения концентрации морфина, M3G и M6G в плазме. Образцы крови отбирают в пробирки с соответствующей маркировкой K2-EDTA Vacutainer® (забор) в период времени 0 (перед введением дозы) и через 0,25, 0,5, 1, 1,5, 2, 2,5, 3, 4, 6, 8, 12 и 24 часов после введения дозы. Ни налоксон, ни налтрексон не анализировали во время этой части исследования.
Сразу после отбора проб каждую пробирку с собранной кровью осторожно несколько раз перевертывали, чтобы гарантировать, что антикоагулянт тщательно смешивается с кровью, и затем охлаждают в криоблоке (или на ледяной бане). В течение 45 минут после сбора образцы крови центрифугировали при 4°С в течение 10 минут при 3000 оборотов в минуту. Используя соответствующие методики пипетирования плазму из каждого образца переносят в 2 полипропиленовые пробирки для перевозки с завинчивающейся крышкой (одна основная и одна резервная) с указанием информации об исследовании и субъекте (то есть имя поручителя, номер исследования, ID субъекта, дата, номинальное время, анализируемое вещество). Образцы плазмы хранятся в вертикальном положении при 20±10°С или ниже до проведения анализа.
Часть В: Во время части В исследования в общей сложности отбирают вплоть до 520 мл крови (13 образцов на лечение х 10 мл в образце х 4 лечения) с целью количественного определения концентрации морфина и либо налоксона, или налтрексона и соответствующих метаболитов (M3G, M6G, 6-β-налтрексола) в плазме. Образцы крови отбирают в пробирки с соответствующей маркировкой K2-EDTA Vacutainer ® (забор) в момент времени 0 (перед введением дозы) и через 0,25, 0,5, 1, 1,5, 2, 2,5, 3, 4, 6, 8, 12 и 24 часа после введения дозы.
Сразу после отбора проб каждую пробирку с собранной кровью осторожно несколько раз переворачивали, чтобы гарантировать, что антикоагулянт тщательно смешивается с кровью, и затем охлаждают в криоблоке (или на ледяной бане). В течение 45 минут после сбора образцы крови центрифугировали при 4°С в течение 10 минут при 3000 оборотов в минуту. Используя соответствующие методики пипетирования, плазму из каждого образца переносят в 2 полипропиленовые пробирки для перевозки с завинчивающейся крышкой (одна для морфина и одна для налоксона/налтрексона) с указанием информации об исследовании и субъекте (то есть имя поручителя, номер исследования, ID субъекта, дата, номинальное время, анализируемое вещество). Образцы плазмы хранятся в вертикальном положении при -20±10UC или ниже до проведения анализа.
Основные фармакодинамические (PD) параметры, представляющие интерес, будут включать либо максимальный эффект (например, Еmах для РаСO2 и ЕТ СO2), или минимальный эффект (например, Emin для MV, RR, ЕТ СO2, уклона и pH артериальной крови), возникающие в течение 4 часов при дозировании исследуемого лекарственного средства. Дополнительные вспомогательные параметры PaCO2 MV, и будут включать площадь под кривой эффекта в течение времени от исходного (время 0) до 1 часа после введения дозы (AUE0.1 ч), 2 часов после введения дозы (AUE0-2 ч), 3 часов после введения дозы (AUE0-3 ч), 4 часов после введения дозы (AUE0-4 ч) и 6 часов после введения дозы (AUE0-6 ч), и время достижения максимального эффекта (Тmах).
Первичная конечная точка
Максимальный артериальный диоксид углерода (РаСO2)
Вторичная конечная точка
- Минутный объем вентиляции легких (MV)
- Частота дыхательных движений
- СO2 в конце спокойного выдоха (ЕТ СO2)
- Уклон кривой MV против РаСO2 (гиперкапническая реакция вентиляции)
- Артериальное pH
- Артериальное насыщение O2
- Чрескожный уровень диоксида углерода (PtcCO2)
- Диаметр зрачка
- Биспектральный индекс (BIS)
Фармакокинетические конечные точки
Следующие фармакокинетические параметры будут рассчитаны, когда применяли морфин, морфин-3-глюкуронид (M3G), морфин-6-глюкуронид (M6G), налтрексон, 6-β-налтрексол и налоксон:
- Максимальная концентрация (Сmах) и время максимальной концентрации (Тmах)
- Площадь под кривой концентрация плазмы - время (AUC)
- Время полураспределение и полувыведение (t/2α и t/2β) и среднее время удержания (MRT)
- Общий клиренс (CL)
Пример 2
Действие внутривенно введенного налтрексона на угнетение дыхания, вызванное морфином у субъектов мужского пола, преимущественно не имеющих опоидной зависимости.
Однократная доза, трехстороннее перекрестное исследование у 28 опиоидопытных, не имеющих зависимость субъектов мужского пола, показывает, что налтрексон НСl 1,2 мг, введенного внутривенно в комбинации с сульфатом морфина 30 мг (Лечение А), значительно уменьшает угнетение дыхания, вызванное морфином, по сравнению с сульфатом морфина 30 мг, введенным внутривенно самостоятельно (Лечение В) или стандартным солевым раствором (плацебо, Лечение С) (Фигура 4). Все субъекты были рандомизированы на три последовательных дозы лечения с использованием перекрестного дизайна. Субъекты получали одну дозу на каждый день дозирования в двойном слепом, перекрестном способе (с 6 дневной амбулаторной отменой для выведения препарата из организма между дозированиями). Исследовательский анализ EtCO2 обнаружил статистически значительные различия в LS способе во всех группах лечения для Еmах, и частичного AUEs (р<0.0001). Никаких различий не было обнаружено между группой с комбинацией морфин+налтрексон и плацебо группой в уровнях EtCO2 (р=0,3064), что подчеркивает PD-эффект замещение морфина на µ-опиоидном рецепторе налтрексоном.
Пример 3
Исследования диапазона дозы налтрексона для блокирования угнетения дыхания, вызванного оксикодоном
Модель и план исследования:
Исследование является рандомизированным, двойным слепым, 5-сторонним перекрестным исследованием, чтобы оценить действия перорального налтрексона на угнетение дыхания, вызванное оксикодоном у здоровых взрослых добровольцев мужского и женского пола. Пороговую дозу оксикодона, которая вызывает угнетение дыхания, исследуют как две части исследования. В части А (ответ на дозу оксикодона) возрастающие единичные дозы оксикодона в виде таблеток с немедленным высвобождением (IR) будут вводить перорально здоровым добровольцам, чтобы определить соответствующую дозу оксикодона, которая безопасно бы вызывала различимые снижения в дыхательной функции (измеренные как сниженная минутного объема вентиляции легких) у здоровых добровольцев. Дозу оксикодона, выбранную из части А, используют в части В (ответ на дозу налтрексона) у здоровых добровольцев, чтобы оценить соотношение доза-ответ налтрексона по отношению к ослаблению угнетения дыхания, вызванного оксикодоном.
Скрининг
Будет необходимым, чтобы все субъекты подвергались исследованию на критерий включение/исключение и соответствие требованиям скрининга, для того чтобы принимать участие в части А или В исследования. Скрининг будет проведен не более чем за 30 дней до получения исследуемого лекарственного средства.
Часть А: Ответ на дозу оксикодона и "анализируемая" доза налтрексона
Часть А исследования выполняют в дозо-возрастающей манере на 6 здоровых взрослых добровольцах мужского или женского пола. Исследование оценивает безопасность и фармакодинамические (PD) конечные точки, связанные с однократной дозой 40 мг IR оксикодона, введенного перорально при несмешанных условиях дозирования в соответствии с процедурами исследования, описанными ниже. Если однократная доза 40 мг IR оксикодона хорошо переносится, то во втором лечении вводят однократную дозу, состоящую из 80 мг IR оксикодона. Однако, если доза IR оксикодона 40 мг хорошо не переносится, то дозу оксикодона снижают до 20 мг. Все лечения будут разделены, по меньшей мере, 1-недельным периодом перерыва.
Безопасность и PD оценивают перед каждым повышением дозы, однако цель состоит в том, чтобы выбрать максимальную дозу оксикодона для части В, которая могла бы безопасно переноситься и вызывать значительное угнетение дыхания, определяемое как угнетенная минутная вентиляция легких, приводящая к значению РаСO2 большему, чем 45 мм ртутного столба (Фигура 3). Как только соответствующая доза оксикодона будет установлена, вводят 25 мг "тестовой дозы" налтрексон с соответствующей дозой оксикодона, чтобы определить вводящийся налтрексон совместно с оксикодоном, который ослабляет угнетение дыхания, вызванное оксикодоном. Эффективность будут определять путем увеличения минутного объема вентиляции легких с сопутствующим снижением РаСO2 и возвратом к исходным значениям, которые считают "клиническая реверсия" угнетения дыхания.
Часть В: Ответ на дозу налтрексона
Часть В исследование проводят на 12 здоровых взрослых добровольцах мужского и женского пола, используя рандомизированную, пятистороннюю перекрестную схему, в которой стандартную дозу оксикодона (например, 80 мг) вводят совместно с переменной (и слепое) дозой налтрексона, которую определяют как 15 процентов от дозы оксикодона, как описано в таблице 1 и ниже в разделе «исследуемый(ые) препарат(ы) и режим». В конечном счете дозирование налтрексона, использованного для лечения А - Е, зависит от дозы оксикодона (20 мг, 40 мг или 80 мг), выбранного из части А исследования.
Таблица 1. Доза налтрексона для лечения
*количество налтрексона в ALO-02 (12% NTX)
Процедуры исследования
В течение каждого периода дозирования субъектов помещают в отделение клинических исследований (CRU) вечером 1-ого дня. В 1-й день после ночного голодания, по меньшей мере, 10 часов начнут процедуры исследования. Исходные измерения HCVR выполняют при обоих гипероксических и гипоксических условиях задачи. Кроме того, устанавливают исходные значения артериального диоксида углерода (РаСO2), системного pH, чрескожного диоксида углерода (PtcCO2), дыхательного объема и частоты дыхательных движений с использованием респираторной индуктивной плетизмографии (RIP). Субъектов исследуют в положении сидя под углом 35° в течение 6 часов, в течение которых они лежат спокойно и взаимодействуют с Исследователем (и сотрудником), ответственным за контролирование условий исследования, введение исследуемых лекарственных средств, контроль безопасности и получение данных связанных с первичными и вторичными конечными точками.
Исследуемое лекарственное средство, состоящее из фиксированной дозы IR оксикодона±различные количества налтрексона в водном растворе (Лечения А-Е), вводят перорально. Когда применимо, выполняются определенные оценки PD (PtcCO2, частота дыхательных движений, дыхательный объем) и непрерывно регистрируются, в то время как другие (РаСO2, системный pH) определяются в конкретные моменты времени (0, 0,25, 0,5, 1, 1,5, 2, 3, 4, 6, 8, 12 и 24 часа) в соответствии с протоколом. Кроме того, серийный отбор образцов венозной крови проводят перед получением дозы (время 0) через 0,25, 0,5, 1, 1,5, 2, 3, 4, 6, 8, 12 и 24 часа после введения дозы для определения оксикодона, налтрексона и относящиеся к ним концентрации метаболитов в плазме.
Чрескожный диоксид углерода (PtcCO2) измеряют с использованием ушной клипсы в качестве неинвазивного средства оценки артериального РаСO2. Кардиомонитор используют для измерения основных жизненно важных функций организма. Кроме того, субъекты носят рубашку VivoMetrics Life, содержащую эластические ленты, которые измеряют относительное расширение грудной клетки и живота во время дыхания, для измерения дыхательного объема и частоты дыхательных движений на основе респираторной индуктивной плетизмографии (RIP).
HCVR при гипероксических и гипоксических условиях задачи является наиболее трудоемкой процедурой, занимающей до 20 минут, чтобы полностью выполнить каждый анализ. Его делают в момент времени 0 (исходный уровень) и через 1, 2, 4 и 6 часов после введения дозы исследуемого(ых) препарата(ов). Процедура включает обеспечение безопасности, прозрачную пластиковую RespirAct маску одевают на лицо субъекта, а затем контролируют подачу газовой смеси СO2/O2 субъекту. Данная техника "возвратного дыхания", как правило, проводится при двух различных O2 состояниях, гипоксическом (РO2 50 мм ртутного столба) и гипероксическом (РO2 150 мм ртутного столба). Гипоксическое состояние усиливает периферическую активность хеморецепторов таким образом, что дыхательная реакция остается результатом активности как центральных, так и периферических хеморецепторов. В противоположность этому гипероксическое состояние подавляет периферическую активность хеморецепторов, таким образом, отражая (или отделяя) деятельность центральных хеморецепторов, которая является ключевым компонентом, как считается, связанным с неизбежным угнетением дыхания, вызванным опиоидами.
Через 6 часов после введения дозы артериальная линия будет удалена после успешного завершения 6-часового HCVR анализа. Примерно через 8 часов после дозы субъекты принимают стандартизированную пищу по усмотрению Исследователя. После этого субъекты по желанию могут передвигаться. Субъекты остаются в CRU до утра 2-го дня, в это время их выписывают из CRU по усмотрению Исследователя. После перерыва периода выведения препарата из организма, по крайней мере, 7 дней, субъекты возвращаются в CRU и повторяют процедуры исследования, описанные выше в течение периодов лечения II-V. Окончательную оценку безопасности осуществляют по окончании исследования. Во время каждого периода лечения субъектов помещают в CRU на период приблизительно 40 часов (2 ночи и 3 дня).
Продолжительность участия субъекта:
Приблизительно 10 недель, включая скрининг
Население исследования:
Исследование может регистрировать вплоть 24 субъектов в попытке укомплектовать 6 субъектов на часть А и 12 субъектов на часть В.
Исследуемое(ые) лекарственное(ые) средство(а) и режим:
Оксикодон дают в виде таблеток немедленного высвобождения по 5 мг.
Налтрексон дают в виде таблеток по 50 мг, которые используют, чтобы приготовить "стоковый раствор" налтрексона (0,5 мг/мл), из которого готовят дозы налтрексона. Пример лечений налтрексоном, которые сопровождаются 80 мг дозой оксикодона, показаны ниже.
Лечение А 0 мл стокового раствора, добавленного к 150 мл яблочного сока
Лечение В 2,0 мл стокового раствора, добавленного к 148 мл яблочного сока
Лечение С 9,6 мл стокового раствора, добавленного к 140,4 мл яблочного сока
Лечение D 19,2 мл стокового раствора, добавленного к 130,8 мл яблочного сока
Лечение Е 50 мл стокового раствора, добавленного к 100 мл яблочного сока Лечения А - Е следуют с 90 мл воды для общего объема 240 мл жидкой среды, вводимой в каждом лечении.
Статистические методы
Объем выборки
Исследование будет регистрировать вплоть до 24 субъектов в попытке укомплектовать 6 субъектов на фазу А и 12 субъектов на фазу В.
Анализ численности
Безопасная численность состоит из всех пациентов, которые принимали, по меньшей мере, одну дозу оксикодона. PK/PD численность состоит из всех пациентов, которые испытали, по меньшей мере, в течение 6 часов интенсивный отбор образцов на РК и оценку PD.
Эффективность и/или PK/PD анализы
Первичная конечная точка представляет собой минутный объем вентиляции легких, артериальное РаСO2 и наклон кривой дыхательного ответа на СO2. Однако данные для всех PD и РК конечных точек просуммированы графически и распределены по категориям лечения с использованием дискриптивной статистики, включая среднее, стандартное отклонение, срединный, минимальный, максимальный и 95% доверительный интервал (CI) для численности, поддающейся оценке. Дозу ответа налтрексон проверяют графически. Период времени курса для всех PD измерений представлены графически путем обработки.
Все PD конечные точки анализируют, используя модель смешанной эффективности для перекрестного исследования, с лечением, периодом и очередностью в виде фиксированных действий и субъект в рамках очередности, как случайное действие. Статистическое значение всех различий лечения представлено с использованием двустороннего критерия значимости.
Анализы безопасности
Все А-Е закодированы в системно-органный класс и термин предпочтительного употребления, используя медицинский словарь терминологии регулятивной деятельности (MedDRA), и обобщены по возрастным группам и группе лечения. Появившиеся за время лечения АЕ определяют как АЕ, которые начинаются во время или после времени введения оксикодона. Побочные эффекты (АЕ), появившиеся за время лечения можно просуммировать следующим образом:
- Количество пациентов с АЕ, классифицированными по системно-органному классу и термину предпочтительного употребления;
- Количество пациентов с АЕ по максимальной интенсивности, системно-органному классу и термину предпочтительного употребления;
- Количество пациентов с АЕ по отношению с исследуемым лекарственным средством, по системно-органному классу и термину предпочтительного употребления;
- Количество пациентов с SAE, классифицированными по системно-органному классу и термину предпочтительного употребления.
Данные клинических лабораторных анализов (химия, гематология и анализ мочи) просуммированы в скрининговом визите, послеоперативном и лечебном периодах, где последовательно выполняют оценку соответствующей и после лечебной безопасности. Основные показатели состояния организма суммируются в каждый момент времени.
Пример 4
Действие внутривенно введенного налтрексона на угнетение дыхания, вызванное оксикодоном у здоровых волонтеров
Проведено рандомизированное, плацебо контролированное, шестистороннее, перекрестное исследование, чтобы оценить действия налтрексона (12% мас./мас.) на эйфорию, вызванную оксикодоном у испытывающих действие опиоидов взрослых субъектов. Как компонент безопасности в данном исследовании, проводили контроль пульсовой оксиметрией в установленном порядке, чтобы контролировать основные показатели состояния организма и симптомы угнетение дыхания, вызванного оксикодоном. Фигура 5 демонстрирует среднее значение (+/- SE) уровней насыщения кислорода (SpO2) в течение времени, определенного из пульсовой оксиметрии после перорального введения: оксикодона 60 мг; оксикодона 60 мг+налтрексона 7,2 мг (12%); и плацебо.
Результаты демонстрируют, что в дополнение к снижению эффектов эйфории от оксикодона 60 мг, налтрексон снижает эффекты угнетения дыхания, вызванные оксикодоном. Ослабление действия было наиболее ярко выражено приблизительно во время максимальной абсорбции оксикодона и налтрексона, приблизительно через 1 час после введения дозы.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМБИНАЦИИ АГОНИСТ/АНТАГОНИСТ ОПИОИДА | 1998 |
|
RU2241458C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2007 |
|
RU2445077C2 |
| СПОСОБ ПРЕДОТВРАЩЕНИЯ ЗЛОУПОТРЕБЛЕНИЯ СОДЕРЖАЩИМИ ОПИОИДЫ ЛЕКАРСТВЕННЫМИ ФОРМАМИ | 1998 |
|
RU2228180C2 |
| НОВЫЕ И ЭФФЕКТИВНЫЕ ЛЕКАРСТВЕННЫЕ ФОРМЫ ТАПЕНТАДОЛА | 2009 |
|
RU2673882C1 |
| СПОСОБ ЛЕЧЕНИЯ ОПИОИДНОЙ И/ИЛИ АЛКОГОЛЬНОЙ ЗАВИСИМОСТИ | 2006 |
|
RU2310449C1 |
| НОВЫЕ И ЭФФЕКТИВНЫЕ ЛЕКАРСТВЕНЫЕ ФОРМЫ ТАПЕНТАДОЛА | 2009 |
|
RU2599846C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ ПЕРЕДОЗИРОВКИ ОПИОИДАМИ | 2017 |
|
RU2769397C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ОПИОИДНЫЙ АНТАГОНИСТ, ДЛЯ ЛЕЧЕНИЯ ЗАДЕРЖКИ МОЧИ | 2009 |
|
RU2478388C2 |
| ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ, СОДЕРЖАЩИЙ ОКСИКОДОН И НАЛОКСОН | 2003 |
|
RU2295344C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2018 |
|
RU2780002C2 |
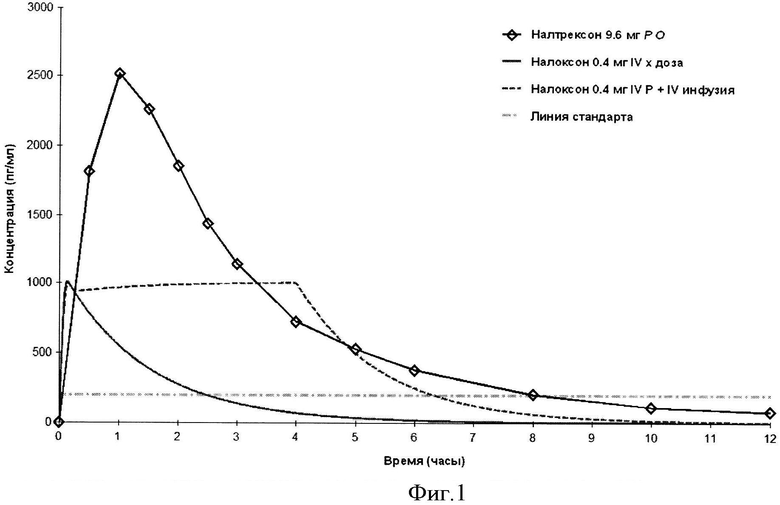
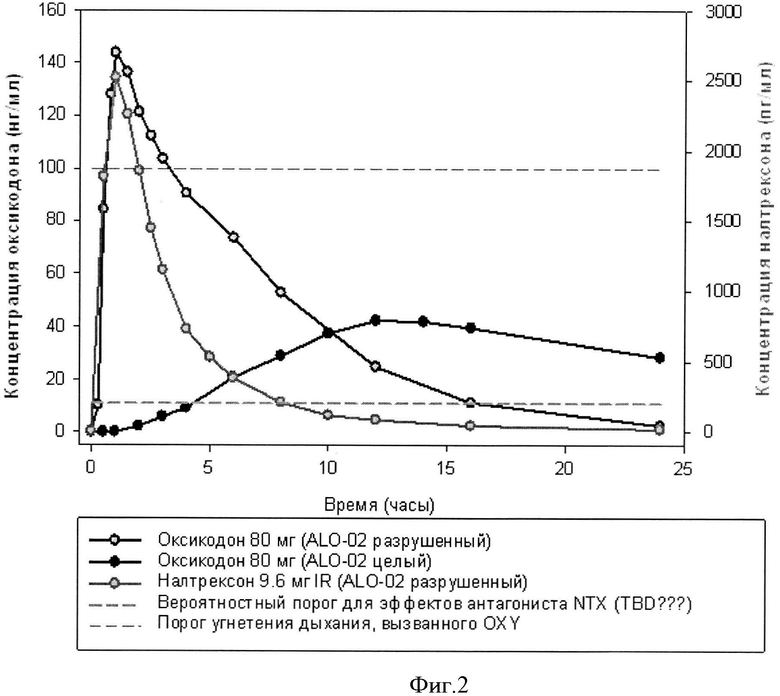
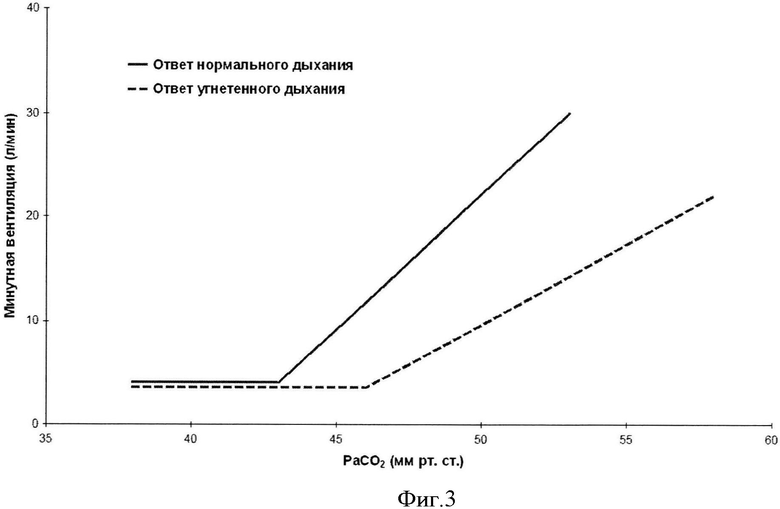
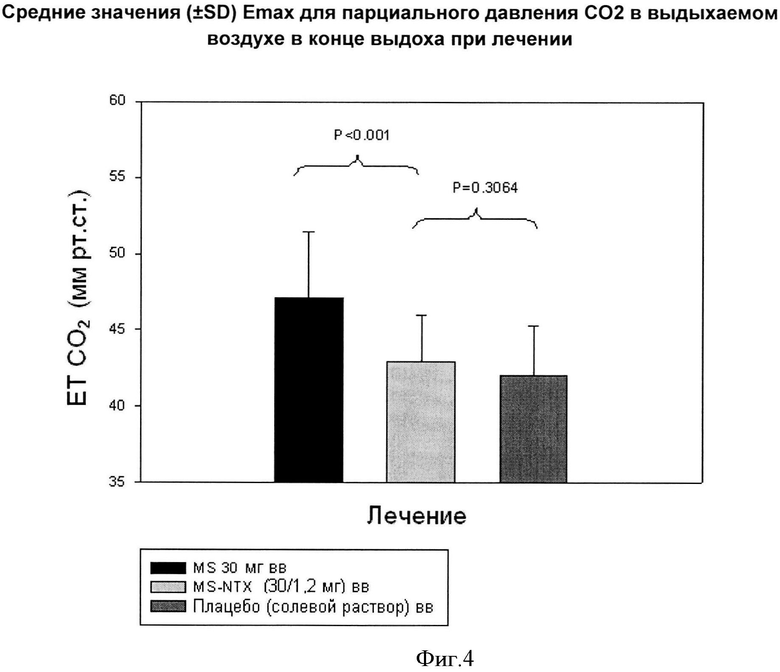
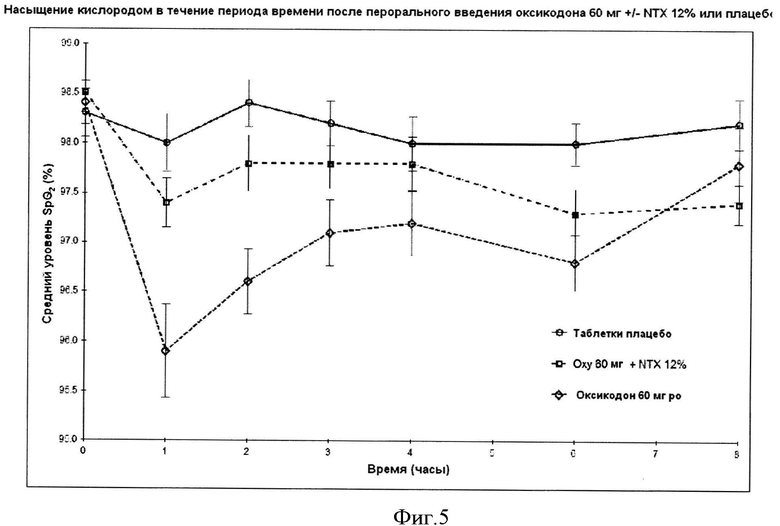
Изобретение относится к опиатному анальгетическому лекарственному препарату, который включает твердую, с контролируемым высвобождением, пероральную лекарственную форму, содержащую большое количество многослойных пеллет. Каждая пеллета содержит водорастворимую сердцевину, слой антагониста, содержащий налтрексон, который покрывает сердцевину, изолирующий полимерный слой, который покрывает слой антагониста, слой агониста, содержащий оксикодон, который покрывает изолирующий полимерный слой, и слой контролируемого высвобождения, который покрывает слой агониста. Лекарственный препарат предназначен для ослабления у человека угнетения дыхания, которое возникает, если препарат был поврежден перед введением человеку, за счет высвобождения налтрексона. 2 н. и 6 з.п. ф-лы, 5 ил., 3 табл., 4 пр.
1. Опиатный анальгетический лекарственный препарат, который включает твердую, с контролируемым высвобождением, пероральную дозированную форму, содержащую большое количество многослойных пеллет, где каждая пеллета содержит:
a) водорастворимую сердцевину;
b) слой антагониста, содержащий налтрексон или фармацевтически приемлемую соль налтрексона, который покрывает сердцевину;
c) изолирующий полимерный слой, который покрывает слой антагониста;
d) слой агониста, содержащий оксикодон или его фармацевтически приемлемую соль, который покрывает изолирующий полимерный слой, и
e) слой контролированного высвобождения, который покрывает слой агониста, из которого в значительной степени не высвобождается налтрексон или фармацевтически приемлемая соль налтрексона, когда человеку введен неповрежденный препарат, и в случае угнетения дыхания, которое возникает у человека, если препарат был поврежден перед введением человеку, оно ослабляется при высвобождении налтрексона или фармацевтически приемлемой соли налтрексона.
2. Препарат по п.1, где ослабление угнетения дыхания измеряют по снижению PETCO2.
3. Препарат по п.2, где снижение PETCO2 составляет, по меньшей мере, 5%.
4. Препарат по п.1, где ослабление угнетения дыхания измеряют по возрастанию уровней насыщения кислородом (SpO2).
5. Применение опиатного анальгетического лекарственного препарата в производстве лекарственного средства для ослабления у человека угнетения дыхания, связанного с лекарственным средством, после введения человеку опиоидного лекарственного средства, которое вызывает угнетение дыхания, где препарат содержит большое количество многослойных пеллет, где каждая пеллета содержит:
a) водорастворимую сердцевину;
b) слой антагониста, содержащий налтрексон или фармацевтически приемлемую соль налтрексона, который покрывает сердцевину;
c) изолирующий полимерный слой, который покрывает слой антагониста;
d) слой агониста, содержащий оксикодон или его фармацевтически приемлемую соль, который покрывает изолирующий полимерный слой, и
e) слой контролированного высвобождения, который покрывает слой агониста, из которого в значительной степени не высвобождается налтрексон или фармацевтически приемлемая соль налтрексона, когда человеку введен неповрежденный препарат, и в случае угнетения дыхания, которое возникает у человека, если препарат был поврежден перед введением человеку, оно ослабляется при высвобождении налтрексона или фармацевтически приемлемой соли налтрексона.
6. Применение препарата по п.5, где ослабление угнетения дыхания измеряют по снижению PETCO2.
7. Применение препарата по п.6, где снижение составляет, по меньшей мере, 5%.
8. Применение препарата по п.5, где ослабление угнетения дыхания измеряют по возрастанию уровней насыщения кислородом (SpO2).
| EMBEDA (morphine sulfate and naltrexone hydrochloride) capsule, extended release, [King Pharmaceuticals, Inc.], Найдено 16.04.2014, Найдено из Интернет:URL: http://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=a7658a2d-b7a9-4fb5-8d65-a20ca1b9ad1f | |||
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |

