Опиоиды, известные также как опиоидные агонисты, представляют собой группу лекарств, проявляющих опиумные или морфиноподобные свойства. Опиоиды применяются, главным образом, в качестве анальгетиков, но имеют также много других фармакологических воздействий, включающих сонливость, угнетение дыхания, перемены настроения и затемнение сознания без конечной потери сознания. Опиоиды действуют как агонисты, взаимодействуя со стереоспецифичными и насыщаемыми связывающими сайгами в мозгу и других тканях. Эндогенные опиоидноподобные пептиды присутствуют в особенности в областях центральной нервной системы, которые, как предполагается, связаны с ощущением боли, с движением, настроением и поведением, и с регуляцией нейроэндокринологических функций. Опиум содержит более двадцати различных алкалоидов. Морфин, кодеин и папаверин входят в эту группу.
К середине девятнадцатого века в медицинском мире начало распространяться применение чистых алкалоидов, таких как морфин, вместо препаратов опиума-сырца. Парентеральное применение морфина привело к более широкому распространению непреодолимого влечения к использованию наркотиков. Проблема болезненного привыкания к опиоидам стимулировала поиск сильных анальгетиков, которые не были бы потенциально способны вызывать привыкание. К 1967 году исследователи пришли к заключению, что сложное взаимодействие между морфиноподобными лекарствами, антагонистами и тем, что было позднее названо "смешанным агонистом-антагонистом" может быть лучше объяснено при постулировании существования более чем одного типа рецептора для опиоидов и родственных лекарств. С появлением новых полностью синтетических объектов с морфиноподобным действием термин "опиоид" был в целом сохранен как родовое название всех экзогенных веществ, которые связываются стереоспецифично с любым из нескольких подвидов опиоидных рецепторов и производят агонистические действия.
Потенциальное развитие привыкания и физической зависимости при повторном применении опиоидов является характерной отличительной особенностью всех опиоидных лекарств, и возможность развития психологической зависимости (т.е. привыкания к наркотическим средствам) является одним из главных опасений при применении лечения болей опиоидами, даже если ятрогенная наркомания является редкой. Другим главным опасением, связанным с применением опиоидов, является передача этих лекарств от пациента с болями другому (не пациенту) для рекреационных целей, т.е. наркоману.
Все возможные злоупотребления опиоидом не определяются каким-либо одним фактором. Вместо этого имеется сочетание факторов, включающих способность лекарства порождать род физической зависимости, при которой синдром отмены лекарства вызывает сильные страдания, приводящие к поиску лекарства; способность подавлять симптомы синдрома отмены, вызванные отменой других лекарств; степенью, в которой он вызывает эйфорию, подобную той, которая вызывается морфином и другими опиоидами; картинами отравлений, которые наступают, когда дозировка лекарства превышает нормальный терапевтический интервал; физические характеристики лекарства, такие как растворимость в воде. Такие физические характеристики могут определить, возможно ли, что лекарство окажется вредным при парентеральном введении.
В Соединенных Штатах борьба с наркоманами включает попытки контролировать доступность лекарства установлением ограничений на применение опиоидов при лечении болей у пользователей с непреодолимым влечением к препарату. На практике врач часто оказывается перед выбором введения сильных опиоидных аналгезирующих даже людям, которые кажутся предрасположенными к развитию психологической зависимости, т.е. к привыканию к таким лекарствам. В свете этой проблемы было рекомендовано, что такие пациенты не должны получать опиоид, если будет достаточно другого лекарства без потенциального вреда; и, кроме того, таким пациентам не должно разрешаться самолечение парентеральным введением таких лекарств и должна выдаваться за раз только порция лекарства на несколько дней.
Было выделено по меньшей мере три основных схемы использования опиоидов и зависимости от опиоидов. Первая охватывает личности, у которых применение лекарства начинается в рамках медицинского лечения и которые получают свои начальные поставки, например от врача. Другая схема начинается с пробного или "рекреационного" применения препарата и переходит в более интенсивное применение. Третья схема охватывает пользователей, которые начинают тем или иным из предшествующих путей, но позднее переходят к пероральным опиоидам, таким как метадон, получаемых через программы организованного лечения наркомании.
Привыкание означает потребность увеличивать дозу опиоида в течение времени для того, чтобы достигать прежнего уровня аналгезии или эйфории, или наблюдение того, что повторяющееся введение прежней дозы вызывает снижение аналгезии, эйфории или других опиоидных эффектов. Было найдено, что значительная степень привыкания развивается по отношению к угнетающим дыхание, аналгезирующим, седативным, рвотным и эйфоригенным эффектам опиоидов. Однако скорость, с которой такое привыкание может развиваться у наркомана или у пациента, нуждающегося в аналгезии, зависит от схемы применения. Если опиоид используется часто, может быть необходимо увеличить дозу. Привыкание не развивается одинаково или с одной и той же скоростью для всех эффектов опиоидов, и даже у пользователей, которые высоко толерантны к эффектам угнетения дыхания, продолжают проявляться рвота и запор. Привыкание к опиоидам в значительной степени исчезает, когда завершается синдром отмены лекарства.
Физическая зависимость может развиться при повторяющихся приемах или при длительном применении опиоидов. Физическая зависимость постепенно проявляется после прекращения применения опиоида или резко проявляется (например, в течение 20 минут) после введения антагониста наркотика (называемого "резкий выход"). В зависимости от лекарства, к которому установлена зависимость, и от продолжительности применения и дозы симптомы отмены лекарства варьируются по их числу и виду, продолжительности и силе. Наиболее распространенные симптомы синдрома отмены лекарства включают потерю аппетита, потерю веса, расширение зрачков, ознобы, перемежающиеся с избыточным потовыделением, желудочные судороги, тошноту, рвоту, мышечные спазмы, повышенную раздражительность, слезотечение, ринит, гусиную кожу и учащенное сердцебиение. Синдром абстиненции обычно начинает развиваться спустя 24-48 часов после введения последней дозы, достигает своей максимальной интенсивности примерно на третий день и может не начать понижаться до третьей недели.
Психологическая зависимость (т.е. привыкание) от опиоидов характеризуется поведением поиска лекарства, направленным на достижение эйфории и уход от, например, психосоциоэкономического давления. Наркоман будет продолжать применять опиоиды для немедицинских целей и перед лицом саморазрушения.
Фармакологически, опиоидные антагонисты обычно блокируют или меняют на обратное все эффекты опиоидных агонистов. Одним использованием опиоидных антагонистов является применение раз в день налтрексона для того, чтобы блокировать эйфорические эффекты, которые могут быть в противном случае получены при введении опиоидов наркоманам. Малые дозы опиоидных антагонистов использовались для определения того, являются ли люди физически зависимыми от опиоидов. Более часто опиоидные антагонисты использовались для того, чтобы реверсировать эффекты опиоидов у людей, которые были передозированы препаратами опиоидного агониста.
Ранее предпринимались попытки бороться с возможными злоупотреблениями, связанными с опиоидными анальгетиками. Обычно конкретная доза опиоидного анальгетика является более сильной, когда она вводится парентерально, по сравнению с такой же дозой при пероральном введении. Поэтому один распространенный способ злоупотребления пероральными медикаментами включает экстракцию опиоида из лекарственной формы и последующую инъекцию опиоида (с использованием любого подходящего устройства для инъекций) для того, чтобы получить "кайф". Поэтому попытки прекратить злоупотребление обычно были направлены на включение в пероральную лекарственную форму опиоидного антагониста, который не активен при пероральном введении, но который должен существенно блокировать анальгетические эффекты опиоида при попытке растворить опиоид и ввести его парентерально.
Например, сочетание пентазоцина и налоксона было применено в таблетках, доступных в Соединенных Штатах в продаже под названием Talwin®Nx от Sanofi-Winthrop. Talwin®Nx содержит пентазоцин гидрохлорид, эквивалентный 50 мг основания и налоксон гидрохлорид, эквивалентный 0,5 мг основания. Таlwin®Nx показан для облегчения или ослабления острой боли. Количество налоксона, присутствующего в комбинации, не оказывает действия при пероральном приеме и не должно накладываться на фармакологическое действие пентазоцина. Однако это количество налоксона, введенное инъекцией, оказывает полное антагонистическое действие на наркотические анальгетики. Таким образом, введение налоксона предназначено для того, чтобы предотвратить такую форму неправильного использования перорального пентазоцина, которая имеет место при растворении лекарственной формы и инъекции. Поэтому эта дозировка предоставляет меньше возможностей для неправильного парентерального применения, чем предшествующие пероральные формы пентазоцина. Однако она все еще является объектом неправильного применения пациентом и злоупотребления при пероральном введении, например при приеме пациентом нескольких доз сразу.
Sunshine, et al. в "Analgesic Efficacy of Pentazocine versus a Pentazocine-Naloxone Combination Following Oral Administration" Clin. J. Pain, 1988; 4: 35-40 сообщает о влиянии добавки 0,5 мг налоксона на анальгетическую эффективность 50 мг пентазоцина. Было найдено, что комбинация является существенно менее эффективной, чем пентазоцин, по суммарной разнице интенсивности боли (СРИБ) и по облегчению и разнице интенсивности боли (РИБ) на четвертом часе. Для пациентов с умеренным исходным уровнем боли комбинация давала существенно меньшее облегчение боли, чем пентазоцин по СРИБ и по облегчению и РИБ на часах 3 и 4. У пациентов с сильным исходным уровнем боли не было обнаружено существенного различия между пентазоцином и комбинацией пентазоцин плюс налоксон.
Wang, et al. в "Crossover and Parallel Study of Oral Analgesics", J. Clin. Pharmacol., 1981; 21:162-8 изучал комбинацию 0,25 мг налоксона и Percodan® (состоящего из 4,5 мг оксикодон НСl, 0,28 мг оксикодон терефталата, 224 мг аспирина, 160 мг фенацетина и 32 мг кофеина) в сравнении с одним Percodan® и плацебо при раздельном исследовании пациентов с хроническими болями. Комбинация имела в среднем меньшее число баллов, чем один Percodan® по большинству из часовых параметров аналгезии в последние часы опыта. Однако по итоговым переменным комбинация не показала существенного отличия ни от плацебо, ни от Percodan®.
Фиксированная комбинация бупренорфина и налоксона была введена в 1991 в Новой Зеландии (Temgesic®Nx, Reckitt & Colman) для лечения болей.
Терапия фиксированной комбинацией, включающей тилидин (50 мг) и налоксон (4 мг), применялась в Германии для лечения острой боли с 1978 года (Valoron®N, Goedecke). Основной причиной для сочетания этих лекарств является эффективное облегчение боли и предотвращение привязанности к тилидину путем индуцированных налоксоном антагонизмов у рецептора морфина.
Патент США 3773955 (Patcher, et al.) описывает эффективные пероральные анальгетические композиции, которые не вызывают аналгезии, эйфории или физической зависимости при парентеральном введении и тем самым предотвращают злоупотребление аналгезирующими веществами. Такие композиции содержали от примерно 0,1 мг до примерно 10 мг налоксона на пероральную дозу анальгетика. Эта работа не касается перорального злоупотребления опиоидами.
Патент США 3493657 (Lewenstein, et al.) описывает композиции, включающие налоксон и морфин или оксиморфон, где говорится, что композиции обеспечивают сильный аналгезирущий эффект без возникновения нежелательных побочных эффектов, таких как галлюцинации.
Патент США 4457933 (Gordon, et al.) описывает способ снижения и перорального, и парентерального потенциального вреда сильных аналгезирующих агентов, таких как оксикодон, пропоксифен и пентазоцин, путем сочетания аналгезирующей дозы опиоида с налоксоном в специфическом относительно узком соотношении. Предпочтительны композиции оксикодон-налоксон, имеющие соотношение 2,5-5:1 по весу, и композиции пентазоцин-налоксон, имеющие соотношение 16-50:1 по весу. Указано, что доза налоксона, которая должна комбинироваться с опиоидом, существенно устраняет возможность или перорального, или парентерального злоупотребления опиоидом без существенного снижения их пероральной аналгезирующей активности.
Патент США 4582835 (Lewis) описывает способ лечения боли путем введения подъязычно эффективной дозы бупренорфина с налоксоном. Lewis описывает соотношения дозировки налоксона к бупренорфину от 1:3 до 1:1 для парентерального введения и от 1:2 до 2:1 для подъязычного введения.
На практике все более убеждались в том, что пероральными опиоидными композициями злоупотребляют не только при парентеральном введении, но также при пероральном введении, когда пациент или наркоман вводит себе перорально больше предписываемой пероральной дозы в течение одного интервала дозировки. Поэтому существует потребность в разработке рецептуры для лечения боли, которая вводится перорально и которая обеспечивает более низкую возможность перорального злоупотребления.
Насколько известно заявителям, соотношение опиоидного агониста к опиоидному антагонисту, которое должно быть анальгетически эффективным при пероральном введении комбинации, но которое вызывает отвращение у физически зависимого субъекта, на сегодня не определено.
Цели и краткое описание изобретения
Цель настоящего изобретения - предложить пероральную лекарственную форму опиоидного анальгетика, которая предоставляет меньшую возможность злоупотребления при пероральном введении, чем ранее доступные в продаже лекарственные формы.
Другой целью настоящего изобретения является пероральная лекарственная форма опиоидного анальгетика и способ, который обеспечивает терапевтическую аналгезию и который также обеспечивает негативный, "отвращающий" опыт, когда большое количество опиоида, например примерно в 2-3 раза больше обычно прописываемой дозы, принимается или вводится физически зависимому субъекту.
Другой целью изобретения является пероральная лекарственная форма опиоидного анальгетика и способ, который обеспечивает терапевтическую аналгезию таким образом, который не настолько позитивен, чтобы побудить независимого физически субъекта принимать больше обычно предписываемой дозы, например примерно в 2-3 раза больше обычно предписываемой дозы опиоида, по сравнению с таким же количеством опиоида без антагониста.
Другой целью изобретения является разработка способа лечения болей у больных людей пероральной лекарственной формой опиоидного анальгетика, снижая в то же время возможное пероральное злоупотребление лекарственной формой.
Другой целью изобретения является способ получения такой пероральной лекарственной формы опиоидного анальгетика, которая имеет меньшую возможность перорального злоупотребления.
Вышеуказанные и другие цели достигаются в настоящем изобретении, которое основано, в частности, на неожиданном открытии того, что существует соотношение опиоидного антагониста к опиоидному агонисту (анальгетику), которое является анальгетически эффективным при введении комбинации перорально, но которое вызывает отвращение у физически зависимого субъекта. Насколько известно заявителям, такое даже не рассматривалось специалистами в данной области, например наркологами, анестезиологами и клиническими фармакологами. Является неожиданным, что один комбинированный продукт (комбинированный антагонист/агонист) может быть, по существу, терапевтическим для одной популяции (пациентов с болью), являясь в то же время неприемлемым (отвращающим) для другой популяции (например, физически зависимых субъектов) при введении в одинаковой дозе или в более высокой дозе, чем обычно прописываемая дозировка, например примерно в 2-3 раза обычно прописываемой дозы опиоида.
Настоящее изобретение направлено, в частности, на пероральную лекарственную форму, включающую перорально анальгетически эффективное количество опиоидного агониста и опиоидный антагонист в соотношении, которое сохраняет аналгезирующую эффективность опиоидного анальгетика, но которое может до некоторой степени уменьшить аналгезию, что определяется прямыми измерениями у пациентов или путем использования одного или нескольких заменяющих измерений опиоидной эффективности (аналгезии) у людей. Заменяющие измерения опиоидной эффективности (аналгезии) включают успокоение, скорость дыхания и/или размер зрачка (путем скиаметрии) и визуальную аналоговую шкалу ("ВАШ") для "наркотического эффекта". Такие заменяющие измерения двигались в направлении, которое показывает пониженный опиоидный эффект по сравнению с такой же дозой опиоида без сопутствующей дозы опиоидного антагониста.
В некоторых предпочтительных осуществлениях, где опиоидом является гидрокодон и антагонистом является налтрексон, пероральная лекарственная форма включает гидрокодон в виде его двутартратной соли и налтрексон в виде его хлористоводородной соли.
В некоторых предпочтительных осуществлениях, где опиоидом является гидрокодон и антагонистом является налтрексон, соотношение налтрексона к гидрокодону предпочтительно составляет примерно 0,03-0,27:1 по весу, и более предпочтительно примерно 0,05-0,20:1 по весу.
Настоящее изобретение направлено на способ предотвращения перорального злоупотребления субъектом опиоидной композицией для перорального введения, включающий приготовление пероральной лекарственной формы, которая включает анальгетически эффективное количество опиоидного агониста и опиоидный антагонист в соотношении, которое сохраняет аналгезирующую эффективность опиоидного анальгетика, но которое может до некоторой степени уменьшить аналгезию, что определяется прямыми измерениями у пациентов или путем использования одного или нескольких заменяющих измерений опиоидного эффекта у людей. Если пероральная лекарственная форма принимается физически зависимым субъектом в относительно большой дозе, например примерно в 2-3 раза превышающей обычно предписываемую дозу, такое применение вызывает отвращение у физически зависимого субъекта и предпочтительно является не настолько позитивно поощряющим, как опиоид (принятый один) для независимого физически субъекта.
Настоящее изобретение также направлено на способ лечения, включающий пероральное введение перорально аналгетически эффективного количества опиоидного агониста вместе с опиоидным антагонистом в соотношении, которое сохраняет аналгезирующую эффективность опиоидного анальгетика, но которое может до некоторой степени уменьшить аналгезию, что определяется прямыми измерениями у пациентов или путем использования одного или нескольких заменяющих измерений опиоидной эффективности (аналгезии) у людей.
Настоящее изобретение далее направлено частично на пероральные лекарственные формы, включающие комбинацию перорально анальгетически эффективного количества опиоидного агониста и перорально активного опиоидного антагониста, где опиоидный антагонист включен в количестве, которое (i) не вызывает снижение уровня аналгезии, производимого лекарственной формой при пероральном введении до нетерапевтического уровня, и (ii) обеспечивает по меньшей мере средненегативный, "отвращающий" опыт у физически зависимых субъектов (например, резкий синдром абстиненции), когда субъекты пытаются принять по меньшей мере в два раза больше обычно предписываемой дозы за прием (и часто в 2-3 раза больше такой дозы или более), по сравнению с сопоставимой дозой опиоида без присутствия опиоидного антагониста. Предпочтительно количество налтрексона, включенное в пероральную лекарственную форму намного менее позитивно поощрительно (т.е. менее "приятно") для независимого физически опиоидного наркомана, чем сравнимая пероральная лекарственная форма без включенного антагониста. Предпочтительно композиция обеспечивает аналгезию при пероральном введении.
Для целей настоящего изобретения выражение "которое может до некоторой степени уменьшить аналгезию, которая определяется прямыми измерениями у пациентов или путем использования одного или нескольких заменяющих измерений опиоидной аналгезирующей эффективности у людей" означает, что пациент с болью может или не может ощутимо заметить разницу между композицией, введенной по изобретению (т.е. комбинацию опиодного агониста/антагониста) и подобной композицией, которая включает такую же дозу опиоидного агониста без опиоидного антагониста, но будет получать аналгезирующий эффект от комбинации. Фармакодинамический эффект (аналгезия) от композиции, введенной в соответствии с изобретением, может быть описан посредством, например, баллов в анкете по аналгезии, проставленных пациентами во временном ряду после введения лекарственной формы. Суммарные оценки аналгезии включают суммарную разность интенсивности боли (СРИБ) и общее облегчение боли (ООБ).
В некоторых предпочтительных воплощениях изобретения количество опиоидного антагониста, включенного в лекарственную форму, может вызвать клинически значимое понижение уровня аналгезии, произведенного лекарственной формой при пероральном введении, измеренного, например, заменяющими измерениями, такими как "визуальная аналоговая шкала" ("ВАШ") для "наркотического эффекта". В других осуществлениях количество опиоидного антагониста, включенного в пероральную лекарственную форму, может вызвать заметное понижение уровня аналгезии, произведенного лекарственной формой при пероральном введении, но не понижает уровень аналгезии, обеспечивающий субтерапевтический уровень.
Предпочтительно количество антагониста, включенного в пероральную лекарственную форму, является менее позитивно усиливающим (т.е. менее "приятным") для нефизически зависимого субъекта, чем сравнимая пероральная лекарственная форма без включенного антагониста.
Настоящее изобретение направлено также на способ приготовления пероральной лекарственной формы опиоидного анальгетика, предназначенной для лечения болей у пациентов-людей таким образом, чтобы свести к минимуму вероятность перорального злоупотребления лекарственной формой, объединяющий перорально анальгетически эффективное количество опиоидного агониста с опиоидным антагонистом в соотношении, которое поддерживает аналгезирующую эффективность опиоидного анальгетика, но которое может до некоторой степени уменьшить аналгезию, что определяется прямыми измерениями у пациентов или путем использования одного или нескольких заменяющих измерений аналгезии у людей. В некоторых воплощениях изобретения комбинация, когда ее вводят перорально, вызывает клинически значимое понижение уровня аналгезии, вызываемого лекарственной формой при пероральном введении (по сравнению с такой же дозой одного опиоида), и обеспечивает по меньшей мере средненегативный, "отвращающий" опыт у физически зависимых субъектов (например, резкий синдром абстиненции), когда субъект принимает больше обычно предписываемой или обычной дозы опиоида. Субъектом может быть, например, наркоман, который пытается достичь эйфории ("кайфа") путем приема более чем (например, по меньшей мере в 2-3 раза) обычно предписываемой дозы за один раз. Количество опиоидного антагониста, включенного в лекарственную форму, может вызвать или не вызвать заметное понижение уровня аналгезии, вызываемое лекарственной формой при пероральном введении, например, по измерениям фармакодинамических параметров, таких как "визуальная аналоговая шкала" (ВАШ) для "наркотического эффекта", но предпочтительно тем не менее позволяет лекарственной форме обеспечить эффективную аналгезию. В некоторых предпочтительных воплощениях способа доза опиоидного антагониста значительно влияет на замещающее измерение опиоидного аналгезирующего эффекта. В некоторых предпочтительных воплощениях количество антагониста, включенного в пероральную лекарственную форму, является менее позитивно усиливающим (например, менее "приятным") для нефизически зависимого субъекта, чем сравнимая пероральная лекарственная форма без включенного антагониста.
Пероральные фармацевтические композиции, содержащие комбинацию препаратов согласно изобретению, могут быть в форме таблеток, жидкостей, драже, лепешек, водных или масляных суспензий, в виде композиций, содержащих множество частиц, включающих диспергируемые порошки, гранулы, матричные сфероиды, или гранулы с инертным покрытием, эмульсии, твердые или мягкие капсулы или сиропы или эликсиры, микрочастицы (например, микрокапсулы, микросферы и т.п.), защечные таблетки и т.д. Лекарственные формы по настоящему изобретению могут включать любые фармацевтически приемлемые вспомогательные вещества, известные специалистам. Кроме того, лекарственные формы могут обеспечивать немедленное высвобождение опиоидного агониста и опиоидного антагониста. В некоторых предпочтительных воплощениях изобретения лекарственные формы обеспечивают замедленное высвобождение опиоидного агониста и поставляют часть или весь опиоидный антагонист (i) в форме немедленного высвобождения, (ii) в форме замедленного высвобождения, или (iii) в форме и немедленного и замедленного высвобождения. Такие воплощения изобретения могут дополнительно включать часть опиоидного агониста в форме немедленного высвобождения. Замедленное (пролонгированное) высвобождение может быть осуществлено в соответствии с композициями/способами производства, известными специалистам в области фармацевтических рецептур, например, путем включения носителя замедленного высвобождения в матрицу, содержащую опиоидный агонист и опиоидный антагонист, или нанесением покрытия замедленного высвобождения на матрицу, содержащую опиоидный агонист и опиоидный антагонист.
Изобретение может обеспечить получение более безопасного продукта (например, с меньшим угнетением дыхания), а также продукта с более низкой скоростью привыкания и развития физической зависимости.
В некоторых других предпочтительных воплощениях изобретения опиоид, включенный в лекарственную форму, представляет собой перорально активный опиоид, отличающийся от гидрокодона. Соотношение налтрексона, включенного в такие композиции, может быть легко определено простым расчетом, принимая во внимание известные эквианальгетические дозировки различных опиоидных анальгетиков по сравнению с гидрокодоном. Эквианальгетические дозировки различных опиоидных анальгетиков представлены ниже, а остальные известны специалистам, например, из Foley, К., "The Treatment of Cancer Pain", N. Eng1. J. Med., 1985; 313: 84-95, включенной поэтому в качестве ссылки. В других аспектах воплощения данного изобретения налтрексон заменен различными опиоидными антагонистами при использовании их эквиантагонистических доз.
В некоторых воплощениях в композицию включена комбинация двух опиоидных анальгетиков. В других воплощениях включены один или несколько опиоидных анальгетиков и дополнительно включено неопиоидное лекарство в дополнение к опиоидному антагонисту. Такие неопиоидные лекарственные средства должны предпочтительно обеспечивать дополнительную аналгезию и включают, например, аспирин, ацетаминофен, нестероидные противовоспалительные лекарства ("NSAIDS"), антагонисты NMDA и ингибиторы циклооксигеназы-II (ингибиторы СОХ-II). В дополнительных воплощениях может быть включено неопиоидное лекарство, которое обеспечивает отличный от аналгезии желаемый эффект, например противокашлевые, отхаркивающие, противозастойные или антигистаминовые лекарственные средства и т.п.
Термин "парентерально" включает подкожные инъекции, внутривенные, внутримышечные, внутригрудинные инъекции или способы инфузии.
Термин "эффективная аналгезия" определен для целей настоящего изобретения как удовлетворительное снижение или устранение боли при терпимом уровне побочных эффектов по определению пациента.
Термин "замедленное высвобождение" определен для целей настоящего изобретения как высвобождение лекарства (опиоидного анальгетика) из пероральной композиции с такой скоростью, что концентрации (уровни) в крови (например, в плазме) поддерживаются в терапевтическом интервале (выше минимальной эффективной аналгезирующей концентрации или "МЭОК"), но ниже уровней токсичности в течение периода времени, характерного для композиций дважды в день или один раз в день.
Термин "стабильное состояние" относится ко времени, когда скорость разложения лекарства является такой же, как скорость абсорбции этого лекарства в организме.
Для целей настоящего изобретения термин "опиоидный агонист" взаимозаменим с терминами "опиоид" или "опиоидный анальгетик" и должен включать основание опиоида, смеси агонисты/антагонисты, частичные агонисты, их фармацевтически приемлемые соли, их стереоизомеры, их простые и сложные эфиры и их смеси.
Краткое описание чертежей
Следующие чертежи поясняют изобретение и не предназначены для ограничения предмета изобретения в том виде, как он охвачен формулой изобретения.
Фигура 1 показывает антагонизм налтрексона индуцированному гидрокодоном "наркотическому эффекту" ВАШ (визуальной аналоговой шкалы) для примера 1;
Фигура 2 представляет антагонизм налтрексона индуцированному гидрокодоном сокращению зрачка для примера 1;
Фигура 3 представляет средний "наркотический эффект" в баллах ВАШ во времени для каждого из медикаментозных курсов примера 2;
Фигура 4 представляет средний "наркотический эффект" на диаметр зрачка во времени для каждого из медикаментозных курсов примера 2;
Фигуры 5 и 6 представляют средний максимальный "наркотический эффект" в баллах ВАШ (±95% ДИ) и средний минимальный диаметр зрачка (±95% ДИ) как функцию логарифма каждой из доз налтрексона примера 2;
Фигура 7А демонстрирует способность субъекта чувствовать эффект гидрокодона в присутствии изменяющихся количеств налтрексона в примере 3;
Фигуры 7В и 7С показывают субъективное благоприятное или неблагоприятное восприятие субъектом гидрокодона в присутствии изменяющихся количеств налтрексона в примере 3;
Фигура 8А показывает ощущение субъектом синдрома отмены эффекта гидрокодона в присутствии изменяющихся количеств налтрексона в примере 3;
Фигура 8В показывает субъективное ощущение нездоровья в присутствии изменяющихся количеств налтрексона в примере 3;
Фигура 9А показывает влияние гидрокодона на размер зрачка в присутствии меняющихся количеств налтрексона в примере 3;
Фигура 9В показывает видимую степень синдрома отмены от эффекта гидрокодона в присутствии изменяющихся количеств налтрексона в примере 3, с точки зрения наблюдателя;
Фигуры 10А-С представляют площади под кривыми, представленными на фигурах 7А-С, интегрированными за 6 часов периода наблюдения, как функцию дозы налтрексона и 95% доверительные интервалы для реакции на плацебо налтрексон (30 мг гидрокодона, 0 мг налтрексона);
Фигуры 11А-С представляют площади под кривыми, представленными на фигурах 8А-В и фигуре 9А, интегрированными за 6 часов периода наблюдения, как функцию дозы налтрексона и 95% доверительные интервалы для реакции на плацебо налтрексон (30 мг гидрокодона, 0 мг налтрексона).
Подробное описание изобретения
Было предположено, что существуют по меньшей мере три подвида опиоидных рецепторов, обозначенных мю, каппа и дельта. В этих рамках считают, что мю-рецептор участвует в продуцировании повышенной спинномозговой аналгезии, угнетенного дыхания, эйфории и физической зависимости. Считают, что каппа-рецептор участвует в индуцировании спинномозговой аналгезии, рвоты и успокоения. Активация гамма-рецептора вызывает дисфорию и галлюцинации, а также дыхательные и вазомоторные стимуляторные эффекты. Рецептор, отличный от мю-рецептора и обозначенный буквой гамма, был описан у мышей vas deferens, Lord, et al., Nature, 1977, 267, 495-99. Считают, что опиоидные агонисты оказывают свое агонистическое действие прежде всего на мю-рецептор и в меньшей степени на каппа-рецептор. Существует немного лекарств, которые, как считают, действуют как частичные агонисты на тот или иной тип рецептора. Такие лекарства проявляют предельный эффект. Такие лекарства включают налорфин, пропирам и бупренорфин. Другая группа лекарств действует как состязательные антагонисты на мю-рецептор и блокирует эффекты морфиноподобных препаратов путем проявления агонистического действия на каппа- и омега-рецепторы. Термин "агонист-антагонист" был выделен для того, чтобы описать такой механизм воздействия. Понятие антагонизма к воздействию опиоидов рассматривается как сложное.
При введении опиоидных агонистов-антагонистов и частичных агонистов было найдено, что привыкание развивается по отношению к агонистическому эффекту, но не к антагонистическому эффекту препаратов. Даже после продолжительного введения высоких доз прекращение приема налоксона не характеризуется сколько-нибудь значимым синдромом отмены, а синдром отмены налтрексона, другого относительно чистого опиоидного антагониста, вызывает очень слабые признаки и симптомы. Однако после продолжительного введения высоких доз резкое прекращение приема опиоидных агонистов-антагонистов налорфина или циклазоцина вызывает характерный синдром отмены, который одинаков для обоих лекарств.
Налоксон является опиоидным антагонистом, который почти не имеет агонистических эффектов. Подкожные дозы до 12 мг налоксона не дают видимых субъективных эффектов, а 24 мг налоксона вызывают только слабую сонливость. Малые дозы (0,4-0,8 мг) налоксона, введенные внутримышечно или внутривенно человеку предотвращают или резко обращают эффект морфиноподобного опиоидного агониста. Сообщалось, что один мг налоксона внутривенно полностью блокировал эффект 25 мг героина. Эффект налоксона наблюдается почти немедленно после внутривенного введения. Лекарство абсорбируется после перорального введения, но сообщалось, что оно быстро метаболизируется в неактивную форму при своем первом проходе через печень так, что, как сообщалось, оказывается сильным только на 1/15 его воздействия при парентеральном введении. Сообщалось, что пероральная доза в более чем 1 г почти полностью метаболизируется меньше чем за 24 часа.
Другие опиоидные антагонисты, например циклазоцин и налтрексон, оба имеют циклопропилметильные замещения азота, сохраняют больше своей эффективности при пероральном введении, и продолжительность их действия намного больше, достигая 24 часов после пероральных доз. Наиболее предпочтительным опиоидным антагонистом является налтрексон. Однако эквиантагонистические пероральные дозы других опиоидных антагонистов, включающих, но не ограничиваемых этим, налоксон, нальмефен, циклазоцин и леваллорфан, могут быть использованы по настоящему изобретению. Отношение таких других антагонистов к конкретному опиоидному агонисту может быть легко определено специалистом, желающим использовать опиоидный антагонист, отличный от налтрексона, отношение которого к опиоидным агонистам детально проиллюстрировано и обсуждено здесь. Специалисты могут определить такие соотношения других антагонистов к опиоидным агонистам, например, проводя такие же или подобные клинические опыты, как представленные в приложенных здесь примерах. Таким образом, комбинации опиоидный антагонист/опиоидный агонист, которые вводятся перорально в соотношениях, эквивалентных, например, представленному отношению налтрексона к гидрокодону, рассматриваются как входящие в предмет настоящего изобретения и в предмет прилагаемой формулы изобретения. Например, в некоторых воплощениях изобретения в качестве опиоидного антагониста использован налоксон, причем количество налоксона, включенного в лекарственную форму, достаточно велико, чтобы обеспечить эквиантагонистический эффект, как если бы в комбинацию был включен налтрексон.
При лечении пациентов, ранее пристрастившихся к опиоидам, использовали налтрексон в больших пероральных дозах (выше 100 мг), чтобы предотвратить эйфоригенные эффекты опиоидных агонистов. Сообщалось, что налтрексон проявляет сильное блокирующее действие больше преимущественно к мю-сайту, чем к дельта-сайту. Налтрексон известен как синтетический родственник оксиморфона без свойств опиоидного агониста и структурно отличается от оксиморфона заменой метильной группы, размещенной на атоме азота оксиморфона, на циклопропилметильную группу. Хлористоводородная соль налтрексона растворима в воде до примерно 100 мг/см3. Фармакологические и фармакокинетические свойства налтрексона были оценены во многих опытах на животных и в клинике. См., например, Gonzalez J.P., et al., Naltrexone: A review of its Pharmacodynamic and Pharmacokinetic Properties and Therapeuic Efficacy in the Management of Opioid Dependence, Drugs 1988; 35: 192-213, введенную настоящей ссылкой. После перорального введения налтрексон быстро абсорбируется (в пределах 1 часа) и имеет интервал биодоступности от 5 до 40%. Протеиновая связь налтрексона составляет примерно 21% и объем распределения, последующего за введением одиночной дозы, составляет 16,1 л/кг.
Налтрексон имеется в продаже в форме таблеток (Revia®, DuPont) для лечения алкогольной зависимости и блокады экзогенно введенных опиоидов. См., например, Revia (naltrexone hydrochloride tablets) Physician's Desk-Reference 51st ed., Montvale, N.J. "Medical Economics" 1997; 51: 957-959. Дозировка 50 мг Revia® блокирует фармакологический эффект 25 мг героина внутривенно на срок до 24 часов.
Известно, что при совместном введении с морфином, героином или другими опиоидами на постоянной основе налтрексон блокирует развитие физической зависимости от опиоидов. Считается, что способ, которым налтрексон блокирует влияние героина, представляет собой состязательное связывание опиоидных рецепторов. Налтрексон использовали для лечения наркотической зависимости путем полной блокады эффекта опиоидов. Было найдено, что наиболее успешное использование налтрексона для наркотической зависимости имеет благоприятный прогноз для наркоманов как часть широкой программы занятости или реабилитационной программы, включающей контроль за поведением. При лечении наркотической зависимости налтрексоном желательно, чтобы пациент не принимал опиоиды в течение по меньшей мере 7-10 дней. Начальная доза налтрексона для таких целей составляла обычно около 25 мг, если не проявлялись признаки синдрома отмены, доза могла быть увеличена до 50 мг/день. Считают, что ежедневная дозировка 50 мг приводит к адекватной клинической блокаде действия парентерально введенных опиоидов. Налтрексон также использовали для лечения алкоголизма как дополнение к социальным и психотерапевтическим методам.
В лекарственных формах и способах по изобретению количество введенного налтрексона значительно меньше, чем доступные ранее в продаже дозировки. Это так отчасти потому, что согласно настоящему изобретению налтрексон используется другим образом: целью является не блокировка опиоидного эффекта, а обеспечение негативного, "отвращающего" опыта, когда большое количество комбинированного продукта, например в 2-3 раза больше обычно предписываемой дозы, принимается физически зависимым субъектом или вводится ему.
Так, например, в композициях по настоящему изобретению, в которых опиоидом является гидрокодон битартрат, 15 мг, количество налтрексона гидрохлорида, включенного в композицию, составляет от примерно 0,5 мг до примерно 4 мг и предпочтительно от примерно 0,75 мг до примерно 3 мг налтрексона на 15 мг гидрокодона.
Опиоидные анальгетики, которые используются по настоящему изобретению, включают все опиоидные агонисты или смешанные агонисты-антагонисты, частичные агонисты, включающие, но не ограниченные этим, альфентанил, аллилпродин, альфапродин, анилеридин, бензилморфин, бензитрамид, бупренорфин, буторфанол, клонитазен, кодеин, дезоморфин, декстроморамид, дезоцин, диампромид, диаморфон, дигидрокодеин, дигидроморфин, дименоксадол, димефептанол, диметилтиамбутен, диоксафетилбутират, дипипанон, эптазоцин, этогептазин, этилметилтиамбутен, этилморфин, этонитазен, фентанил, героин, гидрокодон, гидроморфон, гидроксипетидин, изометадон, кетобемидон, леворфанол, левофенацилморфан, лофентанил, меперидин, мептазинол, метазоцин, метадон, метопон, морфин, мирофин, нарцеин, никоморфин, норлеворфанол, норметадон, налорфин, налбуфен, норморфин, норпипанон, опиум, оксикодон, оксиморфон, папаверет, пентазоцин, фенадоксон, феноморфан, феназоцин, феноперидин, пиминодин, пиритрамид, профептазин, промедол, проперидин, пропоксифен, суфентанил, тилидин, трамадол, смеси любых предшествующих, соли любых предшествующих и т.п.
В некоторых предпочтительных осуществлениях опиоидный агонист или анальгетик выбирают из группы, состоящей из гидрокодона, морфина, гидроморфона, оксикодона, кодеина, леворфанола, меперидина, метадона, или их солей, или их смесей. В некоторых предпочтительных осуществлениях изобретения опиоидным агонистом является гидрокодон. Эквианальгетические дозы этих опиоидов в сравнении с 15 мг дозой гидрокодона представлены в таблице 1.
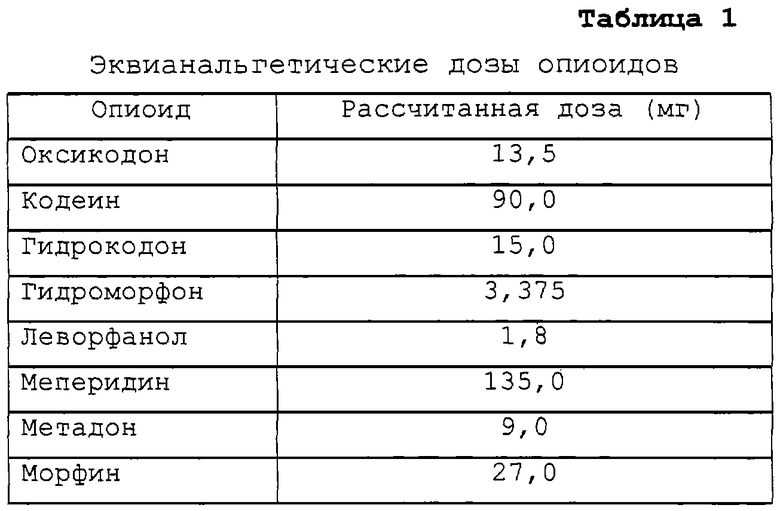
Примерное соотношение налтрексона на 1 мг каждого опиоида представлено в таблице 2, исходя из предпочтительного соотношения налтрексона в количестве от примерно 0,5 до примерно 4 мг на 15 мг гидрокодона.
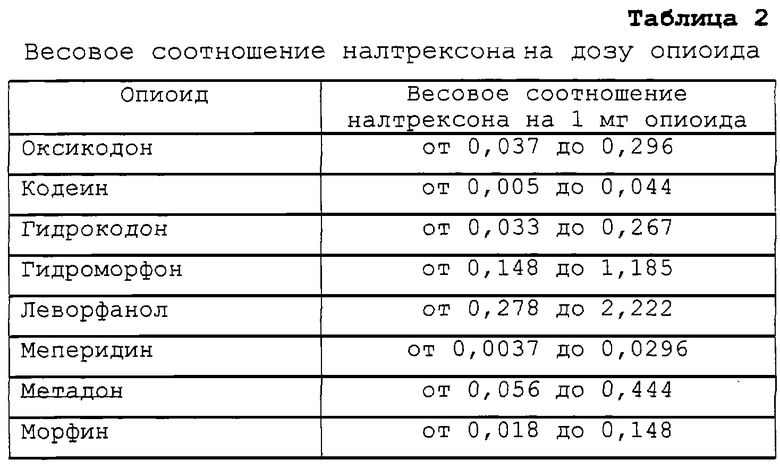
В таблице 3 представлено примерное соотношение налтрексона на 1 мг каждого опиоида исходя из более предпочтительного соотношения от примерно 0,75 мг до примерно 3 мг налтрексона на 15 мг гидрокодона.
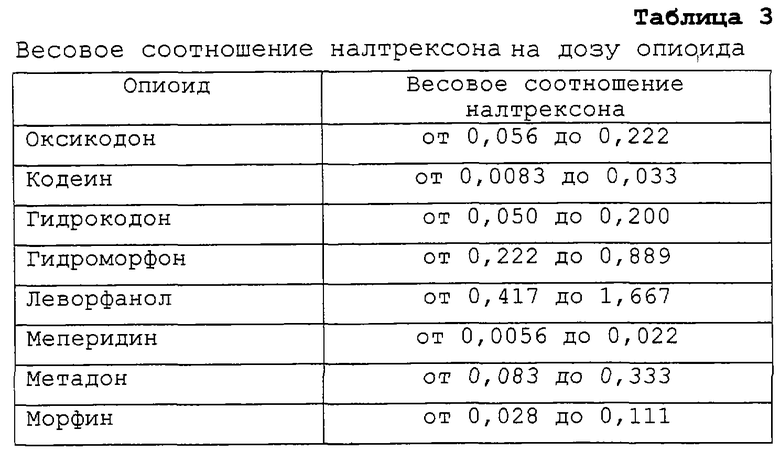
Хотя гидрокодон является эффективным болеутоляющим средством, увеличились злоупотребления им личностями, которые психологически зависимы от опиоидов или которые злоупотребляют им в нетерапевтических целях. Предыдущий опыт с другими опиоидами показал снижение возможности злоупотреблений при введении опиоидов в комбинации с антагонистом наркотика, в особенности у пациентов, являющихся бывшими наркоманами (Weinhold L.L., et al. Buprenorphine Alone and in Combination with Naltrexone in Non-Dependent Humans, Drug and Alcohol Dependence 1992; 30: 263-274; Mendelson J., et al., Buprenorphine and Naloxone Interactions in Opiate-Dependent Volunteers, Clin. Pharm. Ther. 1996; 60: 105-114; обе работы включены в качестве ссылки).
Гидрокодон является полусинтетическим наркотическим анальгетиком и противокашлевым препаратом с множественным действием на центральную нервную систему и желудочно-кишечным действием. Химически гидрокодон представляет собой 4,5-эпокси-3-метокси-17-метилморфинан-6-он, и он также известен как дигидрокодеинон. Подобно другим опиоидам гидрокодон может быть формирующим привычку и может вызвать лекарственную зависимость к препарату типа морфина. При избыточных дозах гидрокодон, подобно другим производным опиума, будет угнетать дыхание.
Пероральный гидрокодон также доступен в Европе (Бельгия, Германия, Греция, Италия, Люксембург, Норвегия и Швейцария) как противокашлевый агент. Парентеральная форма доступна также в Германии в качестве противокашлевого агента. Для использования в качестве анальгетика гидрокодон битартрат доступен в продаже в Соединенных Штатах только в виде фиксированной комбинации с неопиатными лекарствами (например, ибупрофеном, ацетаминофеном, аспирином и т.д.) для облегчения или ослабления острой боли.
Распространенной лекарственной формой гидрокодона является его комбинация с ацетаминофеном, и она имеется в продаже, например, как Lortab® в США от фирмы UCB Pharma, Inc., в виде таблеток гидрокодон/ацетаминофен 2,5/500 мг, 5/500 мг, 7,5/500 мг и 10/500 мг. Таблетки доступны также в соотношении 7,5 мг гидрокодон битартрата и 650 мг ацетаминофена и 7,5 мг гидрокодон битартрата и 750 мг ацетаминофена. Гидрокодон в комбинации с аспирином дают в пероральной лекарственной форме взрослым обычно по 1-2 таблетки каждые 4-6 часов, если требуется облегчить боль. Таблетированная форма составляет 5 мг гидрокодон битартрата и 224 мг аспирина с 32 мг кофеина или 5 мг гидрокодон битартрата и 500 мг аспирина. Относительно новая композиция включает гидрокодон битартрат и ибупрофен. Vicoprofen®, доступный в продаже в США от фирмы Knoll Laboratories, представляет собой таблетку, содержащую 7,5 мг гидрокодон битартрата и 200 мг ибупрофена. Настоящее изобретение предполагает охватить все такие композиции с включением перорально активного опиоидного антагониста в приведенных здесь количествах по изобретению.
Возможность злоупотреблений опиоидными анальгетиками, такими как гидрокодон, неожиданно сокращается найденными комбинациями по настоящему изобретению. Более конкретно, было найдено, что возможно в одной пероральной лекарственной форме объединить опиоидный анальгетик с небольшим количеством опиоидного антагониста для того, чтобы получить продукт, который еще обеспечивает аналгезию, но который практически сводит на нет возможность того, что физически зависимый субъект будет продолжать злоупотреблять препаратом, принимая более одной таблетки за прием, например в 2-3 раза больше обычно предписываемой дозы.
Пероральные лекарственные формы по изобретению включают перорально терапевтически эффективное количество опиоидного агониста вместе с опиоидным антагонистом, таким как налтрексон, в количестве, которое (i) не вызывает снижение уровня аналгезии, производимого лекарственной формой при пероральном введении до нетерапевтического уровня, и (ii) обеспечивает по меньшей мере средне негативное, "отвращающее" действие у физически зависимых субъектов, например у физически зависимых наркоманов (например, резкий синдром абстиненции), когда субъекты пытаются принять больше обычно предписываемой дозы за прием. Предпочтительно, количество антагониста, включенного в пероральную лекарственную форму, является (iii) менее позитивно усиливающим (менее "приятным") для нефизически зависимого субъекта, например опиоидного наркомана, чем сопоставимая пероральная лекарственная форма без включенного антагониста.
Количество антагониста, которое используется для того, чтобы достичь параметры (i)-(iii), представленные в предыдущем абзаце, может быть определено, по меньшей мере частично, например, путем использования "заменяющих" тестов, таких как шкала ВАШ (где субъект оценивает свои ощущения эффекта от лекарственной формы) и/или путем измерений, таких как размер зрачка (измеряется скиаметрией). Такие измерения позволяют специалисту определить дозу антагониста по отношению к дозе агониста, которая вызывает понижение опиатного эффекта агониста. Последовательно специалист может определить уровень опиоидного антагониста, который вызывает отвращающее действие у физически зависимых субъектов, а также уровень опиоидного антагониста, который минимизирует баллы "приятности" или усиливающие свойства опиоида для нефизически зависимых наркоманов. После того как эти уровни опиоидного антагониста определены, можно определить интервал дозировок антагониста на том уровне или ниже того уровня, который должен использоваться для достижения параметров (i)-(iii), представленных в предшествующем абзаце.
Комбинация опиоидного агониста и опиоидного антагониста может быть применена в смесях с обычными вспомогательными веществами, т.е. фармацевтически приемлемыми органическими или неорганическими носителями, пригодными для перорального введения, известными из практики. Пригодные фармацевтически приемлемые носители включают, но не ограничиваются этим, воду, растворы солей, спирты, аравийскую камедь, растительные масла, бензиловые спирты, полиэтиленгликоли, желатин, углеводы, такие как лактоза, амилоза или крахмал, стеарат магния, тальк, кремниевую кислоту, вязкий парафин, душистые масла, моноглицериды и диглицериды жирных кислот, сложные эфиры пентаэритрита и жирных кислот, гидроксиметилцеллюлозу, поливинилпирролидон, и т.д. Фармацевтические препараты могут быть стерилизованы и, если требуется, смешаны с вспомогательными веществами, например смазывающими агентами, консервантами, стабилизаторами, увлажнителями, эмульгаторами, солями для влияния на осмотическое давление, буферами, корригентами и/или ароматическими веществами и т.п. Они могут быть также соединены, если требуется, с другими активными агентами, например, с другими аналгезирующими веществами. Для перорального введения особенно подходящими являются таблетки, драже, жидкости, капли, свечи или капсулы, каплеты и gelcaps. Композиции, предназначенные для перорального применения, могут быть получены любым известным способом, и такие композиции могут содержать один или несколько агентов, выбранных из группы, состоящей из инертных нетоксичных фармацевтически наполнителей, которые пригодны для приготовления таблеток. Такие наполнители включают, например, инертный разбавитель, такой как лактоза, гранулирующие и дезинтегрирующие агенты, такие как кукурузный крахмал, связующие агенты, такие как крахмал, и смазывающие вещества, такие как стеарат магния. Таблетки могут не иметь покрытия или на них может быть нанесено покрытие известными способами для красоты или для того, чтобы замедлить высвобождение активных ингредиентов. Композиции для перорального применения могут также быть представлены в виде жестких желатиновых капсул, в которых активный ингредиент смешан с инертным разбавителем.
Водные суспензии содержат вышеназванную комбинацию лекарств, и такая смесь содержит один или несколько наполнителей, служащих суспендирующими агентам, например фармацевтически приемлемые синтетические смолы, такие как гидроксипропилметилцеллюлоза, или природные смолы. Масляные суспензии могут быть приготовлены путем суспендирования вышеназванной комбинации лекарств в растительном или минеральном масле. Масляные суспензии могут содержать загуститель, такой как пчелиный воск или цетиловый спирт. Сироп, эликсир или подобное могут быть использованы там, где применяется подслащивающий носитель. Могут быть также приготовлены суспензии для инъекций, в каковом случае могут быть применены подходящие жидкие носители, суспендирующие агенты и т.п.
Способ лечения и фармацевтические композиции по настоящему изобретению могут, кроме того, включать одно или несколько лекарств в дополнение к опиоидному анальгетику и опиоидному антагонисту, каковые дополнительные лекарства могут действовать или не действовать синергетически с ними. Так, в некоторых воплощениях в дополнение к опиоидному антагонисту может быть включена комбинация двух опиоидных анальгетиков. Например, лекарственная форма может включать два опиоидных анальгетика, имеющих разные свойства, такие как период полувыведения, растворимость, сила и комбинации любых из перечисленных. В других воплощениях включены один или несколько опиоидных анальгетиков и, кроме того, включено также неопиоидное лекарство в дополнение к опиоидному антагонисту. Такие неопиоидные лекарства предпочтительно должны обеспечивать дополнительную аналгезию и включают, например, аспирин, ацетаминофен, нестероидные противовоспалительные лекарства ("NSAIDS"), например ибупрофен, кетопрофен и т.д.; антагонисты рецептора N-метил-D-аспартата (NMDA), например морфинан, такой как декстроморфан или декстрорфан, или кетамин; ингибиторы циклооксигеназы-II ("ингибиторы COX-II"); и/или антагонисты рецептора глицина.
В некоторых предпочтительных воплощениях настоящего изобретения изобретение дает возможность использовать более низкие дозы опиоидного анальгетика благодаря включению дополнительного неопиоидного агониста, такого как NSAIDS или ингибитор СОХ-2. При использовании более малых количеств одного или обоих лекарств уменьшаются побочные явления, связанные с эффективным ослаблением боли у людей.
Пригодные нестероидные противовоспалительные агенты включают ибупрофен, диклофенак, напроксен, беноксапрофен, флюрбипрофен, фенопрофен, флюбуфен, кетопрофен, индопрофен, пиропрофен, карпрофен, оксапрозин, прамопрофен, муропрофен, триоксапрофен, супрофен, аминопрофен, тиапрофеновую кислоту, флюпрофен, буклоксиновую кислоту, индометацин, сулиндак, толметин, зомепирак, тиопинак, зидометацин, ацеметацин, фентиазак, клиданак, окспинак, мефенаминовую кислоту, меклофенаминовую кислоту, флюфенаминовую кислоту, нифлюминовую кислоту, толфенаминовую кислоту, дифлюризал, флюфенизал, пироксикам, судоксикам или изоксикам, и т.п. Используемые дозировки этих препаратов хорошо известны специалистам.
Антагонисты рецептора N-метил-D-аспартата (NMDA) хорошо известны в практике и охватывают, например, морфинаны, такие как декстроморфан или декстрорфан, кетамин, d-метадон или их фармацевтически приемлемые соли. Для целей настоящего изобретения термин "антагонист NMDA" считается охватывающим также лекарства, которые блокируют главную внутриклеточную последовательность активации рецептора NMDA, например, ганглиозид, такой как GM1 или GM1b, фенотиазин, такой как трифторперазин, или нафталинсульфонамид, такой как N-(6-aминoтексил)-5-хлор-1-нафталинсульфонамид. Указано, что эти лекарства ингибируют развитие привыкания и зависимости от наркотических препаратов, например наркотических анальгетиков, таких как морфин, кодеин и т.д. (патенты США 5321012 и 5556838, оба Mayer, et al.), лечат хронические боли (патент США 5502058, Mayer et al.), что включены в качестве ссылки. Антагонист NMDA, как указано Mayer'ом и др. в патентах, может быть включен один или в сочетании с местными анальгетиками, такими как лидокаин.
Лечение хронических болей путем использования антагонистов рецептора глицина и определение таких лекарств приведены в патенте США 5514680 (Weber, et al.), включенном в качестве ссылки.
Ингибиторы СОХ-2 известны в практике, и известны многие химические структуры, способные ингибировать циклооксигеназу-2. Ингибиторы СОХ-2 описаны, например, в патентах США 5616601, 5604260, 5593994, 5550142, 5536752, 5521213, 5475995, 5639780, 5604253, 5552422, 5510368, 5436265, 5409944 и 5130311, все включены в качестве ссылки. Некоторые предпочтительные ингибиторы СОХ-2 включают целококсиб (SC-58635), DUP-697, флозулид (CGP-28238), мелоксикам, 6-метокси-2-нафтилуксусную кислоту (6-MNA), МК-966, набуметон (пролекарство 6-MNA), нимезулид, NS-398, SC-5766, SC-58215, Т-614 или их сочетания. Уровни дозировки ингибитора СОХ-2 порядка от примерно 0,005 мг до примерно 140 мг на килограмм веса тела являются терапевтически эффективными в сочетании с опиоидным анальгетиком. Альтернативно пациенту вводят от примерно 0,25 мг до примерно 7 г в день в комбинации с опиоидным анальгетиком.
В других воплощениях изобретения может быть включено неопиоидное лекарство, которое обеспечивает иной желаемый эффект, чем аналгезия, например противокашлевые, отхаркивающие, противозастойные, антигистаминовые препараты, местные анальгетики и т.п.
Пероральная лекарственная форма по изобретению может поставляться в виде, например, гранул, сфероидов, бусин, шариков (называемых собирательно "мультичастицами"). Количество мультичастиц, которое является эффективным для поставки желаемой дозы опиоида за нужное время, может быть помещено в капсулу или может быть введено в любую подходящую пероральную твердую форму. Альтернативно пероральная лекарственная форма может быть в виде таблетки.
Лекарственные формы регулируемого высвобождения
Комбинация опиоидного агониста/антагониста может быть приготовлена как пероральная форма контролируемого или замедленного высвобождения в любой подходящей форме, известной специалистам - в таблетках, таблетках с покрытием, в включающей множество частиц форме. Лекарственная форма замедленного высвобождения может, необязательно, включать носитель замедленного высвобождения, который вводят в матрицу вместе с опиоидным агонистом и опиоидным антагонистом или который можно наносить как замедляющее высвобождение покрытие.
В воплощениях, в которых опиоидный анальгетик включает гидрокодон, пероральные лекарственные формы замедленного высвобождения могут включать дозы анальгетиков от примерно 8 мг до примерно 50 мг гидрокодона на единицу дозировки. Пероральные лекарственные формы замедленного высвобождения, в которых терапевтически активным опиоидом является гидроморфон, включают количество от примерно 2 мг до примерно 64 мг гидрохлорида гидроморфона. В других воплощениях опиоидный анальгетик включает морфин, и пероральные лекарственные формы замедленного высвобождения по настоящему изобретению включают от примерно 2,5 мг до примерно 800 мг морфина по весу. В еще одном воплощении опиоидный анальгетик включает оксикодон, и пероральные лекарственные формы замедленного высвобождения по настоящему изобретению включают от примерно 2,5 мг до примерно 800 мг оксикодона. Опиоидный анальгетик может включать трамадол, и пероральные лекарственные формы замедленного высвобождения могут включать от примерно 25 мг до 800 мг трамадола на единицу дозировки. Лекарственная форма может содержать более одного опиоидного анальгетика, обеспечивая практически эквивалентный терапевтический эффект. Альтернативно, лекарственная форма может содержать молярно эквивалентные количества других солей опиоидов, используемых по настоящему изобретению.
В одном предпочтительном воплощении настоящего изобретения пероральная лекарственная форма замедленного высвобождения включает такие частицы, содержащие или включающие активный ингредиент, где частицы имеют диаметр от примерно 0,1 мм до примерно 2,5 мм, предпочтительно от примерно 0,5 мм до примерно 2 мм.
Частицы предпочтительно покрыты пленкой из материала, который дает возможность высвобождения в водной среде комбинации опиоидного агониста/антагониста с замедленной скоростью. Пленочное покрытие выбирают так, чтобы достичь в сочетании с другими указанными свойствами желаемой скорости высвобождения in vitro. Покрытые композиции замедленного высвобождения по настоящему изобретению должны быть способны давать прочную непрерывную пленку, которая является гладкой и красивой, способной быть основой для пигментов и других добавок, нетоксичной, инертной и нелипкой.
В некоторых воплощениях частицы включают матрицы нормального высвобождения, содержащие опиоидный анальгетик с опиоидным антагонистом.
Покрытия
Лекарственные формы по настоящему изобретению могут быть необязательно покрыты одним или несколькими материалами, подходящими для регулирования высвобождения или для защиты композиции. В одном воплощении покрытия предназначены для того, чтобы сделать возможным или рН-зависимое, или рН-независимое высвобождение, например при воздействии желудочно-кишечного сока. рН-зависимые покрытия служат для того, чтобы высвобождать опиоид в желаемых областях желудочно-кишечного (ЖК) тракта, например в желудке или в тонком кишечнике, так, что обеспечивается профиль поглощения, который способен обеспечивать аналгезию пациента в течение по меньшей мере около восьми часов и предпочтительно от примерно двенадцати часов до примерно двадцати четырех часов. Если желательно рН-независимое покрытие, покрытие разрабатывается для того, чтобы достичь оптимального высвобождения независимо от изменений рН в окружающей жидкости, например в ЖК тракте. Возможно также готовить композиции, которые высвобождают часть дозы в одной заданной области ЖК тракта, например в желудке, и высвобождают остаток дозы в другой области ЖК тракта, например, в тонком кишечнике.
Композициям по изобретению, в которых для приготовления композиции используют рН-зависимые покрытия, можно также придать эффект повторного действия, где незащищенное лекарство нанесено поверх кишечного покрытия и высвобождается в желудке, в то время как остаток, будучи защищенным кишечным покрытием, высвобождается далее ниже в желудочно-кишечном тракте. Покрытия, которые являются рН-зависимыми и могут быть использованы по настоящему изобретению, включают шеллак, ацетатфталат целлюлозы (ЦАФ), поливинилацетатфталат (ПВАФ), фталат гидроксипропилметилцеллюлозы, сополимеры сложных эфиров метакриловой кислоты, зейн (zein) и т.п.
В некоторых предпочтительных воплощениях субстрат (например, ядро таблетки, матричная частица), содержащий опиоидный анальгетик (с ингибитором СОХ-2 или без него) покрыта гидрофобным материалом, выбранным из (i) алкилцеллюлозы; (ii) акрилового полимера; или (iii) их смесей. Покрытие может быть нанесено в виде органического или водного раствора или дисперсии. Покрытие может быть нанесено так, чтобы получить массовый привес от примерно 2 до примерно 25% от субстрата для получения желаемого профиля высвобождения. Покрытия, наносимые из водных дисперсий, описаны подробно, например, в патентах США 5273760 и 5286493, принадлежащих патентодержателю настоящего изобретения и включенных в качестве ссылки.
Другие примеры композиций замедленного высвобождения и покрытий, которые могут быть использованы в соответствии с настоящим изобретением, включают патенты США 5324351, 5356467 и 5472712, включенные в качестве ссылки.
Полимеры алкилцеллюлозы
Целлюлозные материалы и полимеры, включая алкилцеллюлозы, представляют собой гидрофобные материалы, хорошо подходящие для покрытия шариков согласно изобретению. Просто в качестве примера, одним предпочтительным алкилцеллюлозным полимером является этилцеллюлоза, хотя специалист должен понимать, что другие целлюлозные и/или алкилцеллюлозные полимеры можно применять отдельно или в любой комбинации в качестве всего или части гидрофобного покрытия по изобретению.
Одной доступной в продаже водной дисперсией этилцеллюлозы является Aquacoat® (FMC Corp., Philadelphia, Pennsylvania, U.S.A). Aquacoat® готовят путем растворения этилцеллюлозы в несмешивающимся с водой органическом растворителе и затем эмульгируют его в воде в присутствии поверхностно-активного вещества и стабилизатора. После гомогенизации для получения субмикронных капелек органический растворитель выпаривают под вакуумом для получения псевдолатекса. На стадии получения в псевдолатекс не вводят пластификатор. Поэтому перед использованием его в качестве покрытия необходимо тщательно смешать Aquacoat® с подходящим пластификатором перед употреблением.
Другая водная дисперсия этилцеллюлозы коммерчески доступна как Surelease® (Colorcon, Inc., West Point, Pennsylvania, U.S.A). Этот продукт готовят путем введения пластификатора в дисперсию во время процесса приготовления. Горячий расплав полимера, пластификатор (дибутилсебацат) и стабилизатор (олеиновая кислота) готовят в виде гомогенной смеси, которую затем разбавляют щелочным раствором для получения водной дисперсии, которую можно непосредственно наносить на субстрат.
Акриловые полимеры
В других предпочтительных воплощениях настоящего изобретения гидрофобный материал, включающий покрытие контролируемого высвобождения, представляет собой фармацевтически приемлемый акриловый полимер, включающий, но не ограниченный этим, сополимеры акриловой и метакриловой кислоты, сополимеры метилметакрилата, этоксиэтилметакрилаты, цианоэтилметакрилат, полиакриловую кислоту, полиметакриловую кислоту, сополимер алкиламида метакриловой кислоты, полиметилметакрилат, полиметакрилат, сополимер полиметилметакрилата, полиакриламид, сополимер аминоалкилметакрилата, поли(метакриловой кислоты ангидрид) и сополимеры глицидилметакрилата.
В некоторых предпочтительных воплощениях акриловый полимер включает один или несколько аммонийно-метакрилатных сополимеров. Аммонийно-метакрилатные сополимеры хорошо известны в технике и описаны в NF XVII как полностью полимеризованные сополимеры сложных эфиров акриловой и метакриловой кислоты с низким содержанием четвертичных аммонийных групп.
Для того чтобы получить желаемый профиль растворения, может быть необходимо ввести два или более аммонийно-метакрилатных сополимера, имеющих разные физические свойства, такие как различные мольные отношения четвертичных аммонийных групп к нейтральным (мет)акриловым сложным эфирам.
Некоторые полимеры типа сложных эфиров метакриловой кислоты используются для получения рН-зависимых покрытий, которые могут быть использованы по настоящему изобретению. Например, существует семейство сополимеров, синтезированных из диэтиламиноэтилметакрилата и других нейтральных метакриловых эфиров, известных также как сополимеры метакриловой кислоты или полимерные метакрилаты, доступных в продаже как Eudragit® от  Tech., Inc. Существует несколько разных типов полимеров Eudragit®. Например, Eudragit® E является примером сополимера метакриловой кислоты, который набухает и растворяется в кислотной среде. Eudragit® L представляет сополимер метакриловой кислоты, который не набухает при примерно рН<5,7 и растворяется при рН>6. Eudragit® S не набухает при рН<6,5 и растворяется при рН>7. Eudragit® RL и Eudragit® RS набухают в воде, и количество воды, поглощаемой этими полимерами, является рН-зависимым, однако лекарственные формы, покрытые Eudragit® RL и RS являются рН-независимыми.
Tech., Inc. Существует несколько разных типов полимеров Eudragit®. Например, Eudragit® E является примером сополимера метакриловой кислоты, который набухает и растворяется в кислотной среде. Eudragit® L представляет сополимер метакриловой кислоты, который не набухает при примерно рН<5,7 и растворяется при рН>6. Eudragit® S не набухает при рН<6,5 и растворяется при рН>7. Eudragit® RL и Eudragit® RS набухают в воде, и количество воды, поглощаемой этими полимерами, является рН-зависимым, однако лекарственные формы, покрытые Eudragit® RL и RS являются рН-независимыми.
В некоторых предпочтительных воплощениях акриловое покрытие включает смесь двух лаков из акриловых смол, выпускаемых фирмой Rhom Pharma под торговыми марками Eudragit® RL30D и Eudragit® RS30D, соответственно. Eudragit® RL30D и Eugradit® RS30D являются сополимерами сложных эфиров акриловой и метакриловой кислоты с низким содержанием четвертичных аммонийных групп, где мольное соотношение аммонийных групп к остаточным нейтральным (мет)акриловым эфирам составляет 1:20 у Eudragit® RL30D и 1:40 у Eudragit® RS30D. Средний молекулярный вес составляет примерно 150000. Условные обозначения RL (высокая проницаемость) и RS (низкая проницаемость) определяют проницаемость этих продуктов. Смеси Eudragit® RL/RS нерастворимы в воде и пищеварительных средах. Однако покрытия, образованные из них, являются набухающими и проницаемыми в водных растворах и пищеварительных жидкостях.
Дисперсии Eudragit® RL/RS по настоящему изобретению могут быть смешаны в любом желаемом соотношении для того, чтобы в конце концов получить композицию замедленного высвобождения, имеющую желаемый профиль растворения. Желаемые композиции замедленного высвобождения могут быть получены, например, из замедляющего покрытия, полученного из 100% Eudragit® RL, 50% Eudragit® RL и 50% Eudragit® RS и 10% Eudragit® RL:90% Eudragit® RS. Конечно, специалист в данной области должен понимать, что могут быть также использованы другие акриловые полимеры, такие как, например, Eudragit® L.
Пластификаторы
В воплощениях настоящего изобретения, в которых покрытие включает водную дисперсию гидрофобного материала, включение в водную дисперсию гидрофобного материала эффективного количества пластификатора должно дополнительно улучшить физические свойства покрытия замедленного высвобождения. Например, поскольку этилцеллюлоза имеет относительно высокую температуру стеклования и не образует гибких пленок при обычных условиях нанесения покрытий, предпочтительно вводить пластификатор в этилцеллюлозное покрытие, содержащее покрытие для замедленного высвобождения перед его использованием в качестве материала для покрытия. Обычно количество пластификатора, включенного в раствор для покрытия, рассчитывают исходя из концентрации пленкообразователя, например наиболее часто от примерно 1 до примерно 50 процентов от массы пленкообразующего вещества. Концентрация пластификатора, однако, может быть определена точно только после тщательных экспериментов с конкретным раствором для нанесения покрытия и способом нанесения.
Примеры подходящих пластификаторов для этилцеллюлозы включают нерастворимые в воде пластификаторы, такие как дибутилсебацат, диэтилфталат, триэтилцитрат, трибутилцитрат и триацетин, хотя возможно, что могут быть использованы другие нерастворимые в воде пластификаторы (такие как ацетилированные моноглицериды, фталатные сложные эфиры, касторовое масло и т.п.). Триэтилцитрат является особо предпочтительным пластификатором для водных дисперсий этилцеллюлозы по настоящему изобретению.
Примеры подходящих пластификаторов для акриловых полимеров по настоящему изобретению включают, но не ограничиваются этим, сложные эфиры лимонной кислоты, такие как триэтилцитрат NF XVI, трибутилцитрат, дибутилфталат и, возможно, 1,2-пропиленгликоль. Другими пластификаторами, для которых было доказано, что они годятся для улучшения эластичности пленок, полученных из акриловых пленок, таких как растворы лаков Eudragit® RL/RS, включают полиэтиленгликоли, пропиленгликоль, диэтилфталат, касторовое масло и триацетин. Триэтилцитрат является особо предпочтительным пластификатором для водных дисперсий этилцеллюлозы по настоящему изобретению.
Кроме того, было найдено, что добавление малого количества талька уменьшает тенденцию водных дисперсий слипаться во время переработки и действует как полирующий агент.
Способы получения шариков с покрытием
Когда гидрофобный материал используют для покрытия фармацевтических шариков, таких как шарики нонпарель 18/20, множество полученных твердых шариков регулируемого высвобождения может быть затем помещено в желатиновую капсулу в количестве, достаточном для обеспечения эффективной дозы регулируемого высвобождения после проглатывания и контакта с окружающей жидкостью, например с желудочным соком или растворяющей средой.
Композиции шариков замедленного высвобождения медленно высвобождают терапевтически активный агент, например, когда они проглочены и подверглись воздействию желудочных жидкостей, а затем кишечных жидкостей. Профиль регулируемого высвобождения композиций по изобретению может быть изменен, например, путем варьирования количества верхнего покрытия гидрофобным материалом, изменения способа, которым к гидрофобному материалу добавляют пластификатор, варьирования количества пластификатора относительно гидрофобного материала путем включения дополнительных ингредиентов или вспомогательных веществ, путем изменения способа получения и т.д. Профиль растворения конечного продукта может также быть модифицирован, например, путем увеличения или уменьшения толщины замедляющего покрытия.
Сфероиды или шарики, покрытые терапевтически активным агентом, готовят, например, растворяя терапевтически активный агент в воде и затем разбрызгивая раствор на субстрат, например, на шарики нонпарель 18/20, используя вставку Вустера (Wuster). Необязательно перед покрытием шариков добавляют также дополнительные ингредиенты для того, чтобы помочь связыванию опиоида с шариками и/или окрасить раствор и т.д. Например, продукт, который включает гидроксипропилметилцеллюлозу и т.д. с красителем (например, Opadry®, выпускаемый в продажу фирмой Colorcon, Inc.) или без него, можно добавить к раствору и перемешать раствор (например, в течение примерно 1 часа) перед нанесением его на шарики. Полученный покрытый субстрат, в данном примере - шарики, может быть затем, необязательно, покрыт сверху барьерным агентом для того, чтобы отделить терапевтически активный агент от гидрофобного покрытия регулируемого высвобождения. Примером подходящего барьерного агента является агент, который включает гидроксипропилметилцеллюлозу. Однако может быть использован любой известный из практики пленкообразователь. Предпочтительно, чтобы барьерный агент не снижал скорость растворения конечного продукта.
После этого шарики могут быть покрыты поверх водной дисперсией гидрофобного материала. Водная дисперсия гидрофобного материала предпочтительно включает дополнительно эффективное количество пластификатора, например триэтилцитрата. Могут быть использованы предварительно образованные водные дисперсии этилцеллюлозы, такие как Aquacoat® или Surelease®. Если используют Surelease®, то не требуется отдельно добавлять пластификатор. Альтернативно могут быть использованы предварительно полученные водные дисперсии акриловых полимеров, такие как Eudragit®.
Растворы для покрытия по настоящему изобретению предпочтительно содержат, в дополнение к пленкообразователю, пластификатору и растворяющей системе (т.е. воде), краситель для того, чтобы придать продукту хороший внешний вид и отличительную окраску. Краситель может быть, вместо этого, добавлен к раствору терапевтически активного агента или дополнительно к водной дисперсии гидрофобного материала. Например, краситель может быть добавлен к Aquacoat® через использование дисперсий красителя на основе спирта или пропиленгликоля, чешуек измельченного алюминия и веществ, делающих материал непрозрачным, таких как двуокись титана, путем добавления красителя со сдвигом к раствору водорастворимого полимера и затем использования слабого сдвига к пластифицированному Aquacoat®. Альтернативно для придания цвета композициям по настоящему изобретению может быть использован любой подходящий способ. Подходящие ингредиенты для придания цвета композициям при использовании водных дисперсий акрилового полимера включают двуокись титана и красящие пигменты, такие как пигменты на основе окислов железа. Введение пигментов, однако, может увеличить замедляющее действие покрытия.
Пластифицированный гидрофобный материал может быть нанесен на субстрат, включающий терапевтически активный агент, путем разбрызгивания при использовании любого подходящего оборудования для разбрызгивания, известного из практики. В предпочтительном способе применяют систему Вустера с псевдоожиженным слоем, в которой воздушное сопло, введенное снизу, флюидизирует материал ядра и вызывает сушку, в то время как происходит набрызгивание покрывающего акрилового полимера. Предпочтительно, наносят достаточное количество гидрофобного материала, чтобы получить заданное регулируемое высвобождение указанного терапевтического агента при экспозиции покрытого субстрата в водных растворах, например в желудочном соке, принимая во внимание физические характеристики терапевтически активного агента, способ введения пластификатора и т.п. После покрытия шариков гидрофобным материалом, необязательно, наносят дополнительный слой пленкообразователя, такого как Opadry®. Это покрытие предусматривается, в случае его нанесения, для того чтобы существенно снизить агломерацию шариков.
На высвобождение терапевтически активного агента из композиции регулируемого высвобождения по настоящему изобретению можно влиять дополнительно, т.е. доводить его до желаемой скорости путем добавления одного или нескольких модифицирующих высвобождение агентов или обеспечения одного или нескольких проходов через покрытие. Отношение гидрофобного материала к водорастворимому материалу определяется, среди прочих факторов, требуемой скоростью высвобождения и характеристиками растворимости выбранных материалов.
Агенты, модифицирующие высвобождение, которые работают как порообразователи, могут быть органическими или неорганическими и включают вещества, которые могут растворяться, экстрагироваться или выщелачиваться из покрытия в используемой окружающей среде. Порообразователи могут включать одно или несколько гидрофильных веществ, таких как гидроксипропилметилцеллюлоза.
Покрытия замедленного высвобождения по настоящему изобретению могут также включать агенты, ускоряющие эрозию, такие как крахмал и смолы.
Покрытия замедленного высвобождения по настоящему изобретению могут также включать вещества, применяемые для создания микропористых пластинок в используемой окружающей среде, такие как поликарбонаты, включающие линейные полиэфиры карбоновых кислот, в которых карбонатные группы повторяются в полимерной цепи.
Агент, модифицирующий высвобождение, может также включать полупроницаемый полимер.
В некоторых предпочтительных воплощениях агент, модифицирующий высвобождение, выбирают из гидроксипропилметилцеллюлозы, лактозы, стеаратов металлов и смесей любого из предшествующих.
Покрытия замедленного высвобождения по настоящему изобретению могут также включать средства для выхода, включающие по меньшей мере один проход, диафрагму или тому подобное. Проход может быть образован такими способами, которые описаны в патентах США 3845770, 3916889, 4063064 и 4088864 (все из которых включены сим в качестве ссылки). Проход может иметь любую форму, такую как круглая, треугольная, квадратная, эллипсоидная, неправильная, и т.д.
Препаративная форма матрицы на основе шариков, включающих матрицу
В других воплощениях настоящего изобретения композицию регулируемого высвобождения получают через матрицу, имеющую покрытие регулируемого высвобождения, как представлено выше. Настоящее изобретение может также использовать матрицу регулируемого высвобождения, которая дает in vitro скорости растворения опиода в предпочтительных пределах и которая высвобождает опиоид рН-зависимым или рН-независимым способом. Материалы, пригодные для включения в матрицу регулируемого высвобождения, должны зависеть от способа, используемого для формирования матрицы.
Например, матрица в дополнение к опиоидному анальгетику и (необязательно) СОХ-2 может включать:
гидрофильные и/или гидрофобные материалы, такие как смолы, простые эфиры целлюлозы, акриловые смолы, вещества, полученные из протеинов; этот список не должен рассматриваться как исключительный, и любой фармацевтически приемлемый гидрофобный материал или гидрофильный материал, который способен обеспечить регулируемое высвобождение активного агента и который плавится (или размягчается до степени, необходимой для экструдирования), может быть использован в соответствии с настоящим изобретением.
Перевариваемые насыщенные или ненасыщенные углеводороды с длинной цепью (С8-С50, предпочтительно C12-C40), такие как жирные кислоты, жирные спирты, сложные глицериновые эфиры жирных кислот, минеральные и растительные масла и воски, стеариловый спирт и полиалкиленгликоли.
Из полимеров предпочтительны акриловые полимеры, в особенности Eudragit® RSPO, простые эфиры гидроксиалкилцеллюлоз и карбоксиалкилцеллюлоз. Пероральная лекарственная форма может содержать между 1% и 80% (по массе) по меньшей мере одного гидрофильного или гидрофобного вещества.
Если гидрофобным веществом является углеводород, углеводород предпочтительно имеет температуру плавления между 25° и 90°С. Из числа углеводородных материалов с длинной цепью предпочтительными являются жирные (алифатические) спирты. Пероральная лекарственная форма может содержать до 60 мас.% по меньшей мере одного перевариваемого углеводорода с длинной цепью.
Предпочтительно пероральная лекарственная форма содержит до 60 мас.% по меньшей мере одного полиалкиленгликоля.
Гидрофобное вещество предпочтительно выбирают из группы, состоящей из алкилцеллюлоз, полимеров и сополимеров акриловой и метакриловой кислоты, шеллака, зейна, гидрированного касторового масла, гидрированного растительного масла или их смесей. В некоторых предпочтительных воплощениях настоящего изобретения гидрофобным веществом является фармацевтически приемлемый акриловый полимер, включающий, но не ограниченный этим, сополимеры акриловой и метакриловой кислоты, метилметакрилат, сополимеры метилметакрилата, этоксиэтилметакрилаты, цианоэтилметакрилат, сополимер аминоалкилметакрилата, полиакриловую кислоту, полиметакриловую кислоту, сополимер алкиламина метакриловой кислоты, полиметилметакрилат, полиангидрид метакриловой кислоты, полиметакрилат, полиакриламид, поли(ангидрид метакриловой кислоты) и сополимеры глицидилметакрилата. В других воплощениях гидрофобное вещество выбирают из таких веществ, как гидроксиалкилцеллюлозы, такие как гидроксипропилметилцеллюлоза, и их смесей.
Предпочтительные гидрофобные вещества являются водорастворимыми с более или менее выраженными гидрофильными и/или гидрофобными свойствами. Предпочтительно гидрофобные вещества, используемые в изобретении, имеют температуру плавления от примерно 30°С до примерно 200°С, предпочтительно от примерно 45°С до примерно 90°С. Предпочтительно гидрофобные материалы могут включать природные или синтетические воски, жирные спирты (такие как лауриловый, миристиловый, стеариловый, цетиловый или, предпочтительно, цетостеариловый спирт), жирные кислоты, включая, но не ограничивая этим, сложные эфиры жирных кислот, глицериды (моно-, ди- и триглицериды) жирных кислот, гидрированные жиры, углеводороды, нормальные воски, стеариновую кислоту, стеариловый спирт и гидрофобные и гидрофильные вещества, имеющие углеводородный скелет. Подходящие воски включают, например, пчелиный воск, гликовоск, касторовый воск и воск карнауба. Для целей настоящего изобретения воскоподобное вещество определено как любой материал, который является обычно твердым при комнатной температуре и имеет температуру плавления от примерно 30°С до примерно 100°С.
Подходящие гидрофобные материалы, которые могут использоваться по настоящему изобретению, включают перевариваемые замещенные или незамещенные углеводороды с длинной цепью (C8-C50, предпочтительно C12-C40), такие как жирные кислоты, жирные спирты, глицериновые сложные эфиры жирных кислот, минеральные и растительные масла и природные или синтетические воски. Предпочтительны углеводороды, имеющие температуру плавления между 25° и 90°С. Из углеводородных материалов с длинной цепью в некоторых воплощениях предпочтительны жирные (алифатические) спирты. Пероральная лекарственная форма может содержать до 60 мас.% по меньшей мере одного перевариваемого углеводорода с длинной цепью.
Предпочтительно в матричные композиции включают сочетание двух или более гидрофобных веществ. Если включен дополнительный гидрофобный материал, его предпочтительно выбирают из природных и синтетических восков, жирных кислот, жирных спиртов и их смесей. Примеры включают пчелиный воск, воск карнауба, стеариновую кислоту и стеариловый спирт. Данный список не является ограничивающим.
Особо предпочтительная матрица включает по меньшей мере одну водорастворимую гидроксиалкилцеллюлозу, по меньшей мере один алифатический спирт С12-С36, предпочтительно C14-C22, и, необязательно, по меньшей мере один полиалкиленгликоль. По меньшей мере одна гидроксиалкилцеллюлоза предпочтительно представляет собой гидрокси(C1-C6)алкилцеллюлозу, такую как гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза и, предпочтительно, гидроксиэтилцеллюлоза. Количество по меньшей мере одной гидроксиалкилцеллюлозы в настоящей пероральной лекарственной форме должно быть определено, среди прочего точным значением желаемой скорости высвобождения опиоида. По меньшей мере один алифатический спирт может быть, например, лауриловым спиртом, миристиловым спиртом или стеариловым спиртом. В особо предпочтительных воплощениях настоящей пероральной лекарственной формы по меньшей мере один алифатический спирт представляет собой цетиловый спирт или цетостеариловый спирт. Количество по меньшей мере одного алифатического спирта в настоящей пероральной лекарственной форме должно быть определено, как и выше, точным значением желаемой скорости высвобождения опиоида. Оно должно также зависеть от того, присутствует или отсутствует в пероральной лекарственной форме по меньшей мере один полиалкиленгликоль. При отсутствии по меньшей мере одного полиалкиленгликоля пероральная лекарственная форма предпочтительно содержит между 20% и 50% (по массе) по меньшей мере одного алифатического спирта. Если по меньшей мере один полиалкиленгликоль присутствует в пероральной лекарственной форме, суммарная масса по меньшей мере одного алифатического спирта и по меньшей мере одного полиалкиленгликоля предпочтительно составляет от 20 до 50 мас.% от всей дозы.
В одном воплощении отношение, например, по меньшей мере одной гидроксиалкилцеллюлозы или акриловой смолы к по меньшей мере одному алифатическому спирту/полиалкиленгликолю в значительной степени определяет скорость высвобождения опиоида из композиции. Предпочтительно отношение по меньшей мере одной гидроксиалкилцеллюлозы к по меньшей мере одному алифатическому спирту/полиалкиленгликолю находится между 1:2 и 1:4, причем отношение между 1:3 и 1:4 является особо предпочтительным.
По меньшей мере один полиалкиленгликоль может быть, например, полипропиленгликолем или, что предпочтительно, полиэтиленгликолем. Величина средней молекулярной массы по меньшей мере одного полиалкиленгликоля предпочтительна между 1000 и 15000, особо предпочтительно между 1500 и 12000.
Другая подходящая матрица регулируемого высвобождения должна включать алкилцеллюлозу (особенно, этилцеллюлозу), алифатический спирт C12-С36 и, необязательно, полиалкиленгликоль.
В другом предпочтительном воплощении матрица включает фармацевтически приемлемую комбинацию по меньшей мере двух гидрофобных материалов.
Дополнительно к вышеназванным ингредиентам матрица регулируемого высвобождения может также содержать нужные количества других веществ, например разбавителей, смазывающих веществ, связующих, веществ, способствующих грануляции, красителей, корригентов и веществ, способствующих скольжению, которые обычны в фармацевтической практике.
Способы приготовления шариков на основе матриц
Для того чтобы облегчить приготовление твердых пероральных лекарственных форм регулируемого высвобождения по изобретению, может быть использован любой известный специалистам способ приготовления матричных композиций. Например, введение в матрицу может быть осуществлено, к примеру, (а) путем формирования гранул, включающих по меньшей мере одну водорастворимую гидроксиалкилцеллюлозу и опиоид или опиоидную соль; (b) путем смешения гранул, содержащих гидроксиалкилцеллюлозу с по меньшей мере одним алифатическим спиртом С12-С36; (с) необязательно, путем прессования и формования гранул. Предпочтительно гранулы формируют путем влажного гранулирования гидроксиалкилцеллюлозы/опиоида с водой. В особо предпочтительном воплощении данного способа количество воды, добавляемой на стадии мокрого гранулирования, предпочтительно между 1,5 и 5 раз и особенно предпочтительно между 1,75 и 3,5 раз больше сухой массы опиоида.
В еще других возможных воплощениях изобретения для образования сфероидов можно использовать сферонизирующий агент вместе с активным ингредиентом. Предпочтительной является микрокристаллическая целлюлоза. Пригодной микрокристаллической целлюлозой является, например, материал, имеющийся в продаже под торговой маркой Avicel РН 101 (FMC Corporation). В таких воплощениях, кроме активного ингредиента и сферонизирующего агента, сфероиды могут также содержать связующее. Пригодные связующие, такие как низковязкостные водорастворимые полимеры, должны быть хорошо известны специалистам в области фармацевтической промышленности. Предпочтительны, однако, водорастворимые низшие гидроксиалкилцеллюлозы, такие как гидроксипропилцеллюлоза. Дополнительно (или альтернативно) сфероиды могут содержать нерастворимый в воде полимер, предпочтительно акриловый полимер, акриловый сополимер, такой как сополимер метакриловой кислоты - этилакрилата, или этилцеллюлозу. В таких воплощениях покрытие замедленного высвобождения должно в общем случае включать гидрофобный материал, такой как (а) воск, отдельно или в смеси с жирным спиртом, или (b) шеллак или зейн.
Матрица, полученная методом экструзии из расплава
Матрицы замедленного высвобождения могут быть также приготовлены способами гранулирования расплава или экструзии из расплава. Обычно способы гранулирования расплава включают плавление обычно твердого гидрофобного материала, например воска, и введение в него порошка лекарства. Для получения лекарственной формы замедленного высвобождения может быть необходимо ввести в расплавленный восковой гидрофобный материал дополнительное гидрофобное вещество, например этилцеллюлозу или нерастворимый в воде акриловый полимер. Примеры композиций замедленного высвобождения, приготовленных способами гранулирования расплава, имеются в патенте США 4861598, включенном в качестве ссылки.
Добавочный гидрофобный материал может включать одно или несколько нерастворимых в воде воскоподобных термопластичных веществ, возможно смешанных с одним или несколькими термопластичными веществами, являющимися менее гидрофобными, чем указанные одно или несколько нерастворимых в воде воскоподобных термопластичных веществ. Для того чтобы добиться постоянного высвобождения, индивидуальные воскоподобные вещества в композиции должны быть практически неразлагаемыми и нерастворимыми в желудочно-кишечных средах во время начальных стадий высвобождения. Полезными водонерастворимыми воскоподобными веществами могут быть вещества с растворимостью в воде, которая ниже чем примерно 1:5000 (вес/вес).
В дополнение к вышеперечисленным ингредиентам матрица замедленного высвобождения может также содержать нужные количества других материалов, например разбавителей, смазывающих веществ, связующих, веществ, способствующих грануляции, красителей, корригентов и веществ, способствующих скольжению, которые обычны в фармацевтической практике. Количества этих дополнительных материалов должны быть достаточными для того, чтобы обеспечить желаемое влияние на желаемую композицию.
В дополнение к указанным материалам матрица замедленного высвобождения, включающая экструдированные из расплава мультичастицы, может также, если требуется, содержать нужные количества других материалов, например разбавителей, смазывающих веществ, связующих, веществ, способствующих грануляции, красителей, корригентов и веществ, способствующих скольжению, которые обычны в фармацевтической практике, в количестве до 50% от массы частиц.
Конкретные примеры фармацевтически приемлемых носителей и вспомогательных веществ, которые могут быть использованы для получения пероральных дозировочных форм, описаны в справочнике Handbook of Pharmaceutical Excipients, American Pharmaceutical Association (1986), включенном в качестве ссылки.
Экструдированные из расплава мультичастицы
Получение подходящей матрицы экструзией из расплава по настоящему изобретению может, например, включать стадии смешения опиоидного анальгетика с по меньшей мере одним гидрофобным материалом и, предпочтительно, с другим гидрофобным материалом для получения гомогенной смеси. Гомогенную смесь затем нагревают до температуры, достаточной для того, чтобы по меньшей мере достаточно размягчить смесь, чтобы ее подвергнуть экструзии. Полученную гомогенную смесь затем экструдируют для образования прядей. Экструдат предпочтительно охлаждают и нарезают на множество частиц известными из практики способами. Пряди охлаждают и режут на множество частиц. Множество частиц затем разделяют на стандартные дозы. Экструдат предпочтительно имеет диаметр от примерно 0,1 до примерно 5 мм и обеспечивает замедленное выделение терапевтически активного агента в течение периода времени от примерно 8 до примерно 24 часов.
Необязательный способ получения экструзий из расплава по настоящему изобретению включает непосредственную дозировку в экструдер гидрофобного материала, терапевтически активного агента и, необязательно, связующего, нагрев гомогенной смеси, экструдирование гомогенной смеси для образования тем самым прядей, охлаждение прядей, содержащих гомогенную смесь, нарезку прядей на частицы, имеющие размер от примерно 0,1 мм до примерно 12 мм и разделение указанных частиц на стандартные дозы. В данном аспекте изобретения реализуется относительно непрерывный способ производства.
Диаметр апертуры экструдера или выходного отверстия может также регулироваться для того, чтобы варьировать толщину экструдированных прядей. Кроме того, выходная часть экструдера не обязана быть круглой, она может быть продолговатой, прямоугольной, и т.д. Выходящие пряди могут быть измельчены до частиц, используя горячий проволочный резак, гильотину и т.п.
Экструдированная из расплава система из мультичастиц может быть, например, в виде гранул, сфероидов или шариков, в зависимости от выходной диафрагмы экструдера. Для целей настоящего изобретения термины "экструдированные из расплава мультичастицы", "экструдированная из расплава система из мультичастиц (системы)" и "экструдированные из расплава частицы" должны означать множественность единиц, предпочтительно в интервале одинакового размера и/или формы и содержащих один или несколько активных агентов и одно или несколько вспомогательных веществ, предпочтительно включающих описанный здесь гидрофобный материал. В этом отношении экструдированные из расплава мультичастицы должны быть в интервале от примерно 0,1 до примерно 12 мм длиной и иметь диаметр от примерно 0,1 до примерно 5 мм. Кроме того, следует понимать, что экструдированные из расплава мультичастицы могут иметь любую геометрическую форму в этом интервале размеров. Альтернативно, экструдат может быть просто нарезан на кусочки желаемой длины и разделен на стандартные дозы терапевтически активного агента без стадии сферонизации.
В одном предпочтительном воплощении получают пероральные лекарственные формы, чтобы включить эффективное количество экструдированных из расплава мультичастиц в капсулы. Например, множество экструдированных из расплава мультичастиц может быть помещено в желатиновую капсулу в количестве, достаточном для того, чтобы обеспечить эффективную дозу замедленного высвобождения после проглатывания и контакта с желудочной жидкостью.
В другом предпочтительном воплощении изобретения подходящее количество экструдата из мультичастиц прессуют в пероральную таблетку, используя обычное таблетировочное оборудование и применяя обычные методы. Способы и композиции для приготовления таблеток (прессованных и оплавленных), капсул (твердых и мягких желатиновых) и пилюль также описано в Remington's Pharmaceutical Sciences (Arthur Osol, editor), 1553-1593 (1980), включенном в качестве ссылки.
В еще одном предпочтительном воплощении экструдат может быть сформован в таблетки так, как представлено в патенте США 4957681 (Klimesch, et al.), описывающим дополнительные подробности вышесказанного и включенном в качестве ссылки.
Необязательно, экструдированные из расплава мультичастичные системы или таблетки могут быть покрыты, или желатиновые капсулы могут быть дополнительно покрыты покрытием замедленного высвобождения, таким как описанные выше покрытия замедленного высвобождения. Такие покрытия предпочтительно включают достаточное количество гидрофобного материала для того, чтобы получить уровень привеса от примерно 2 до примерно 30 процентов, хотя покрытие может быть и больше в зависимости, среди прочего, от физических свойств конкретно использованного опиоидного аналгезирующего соединения и желаемой скорости высвобождения.
Экструдированные из расплава лекарственные формы по настоящему изобретению могут, кроме того, включать комбинации экструдированных из расплава мультичастиц, содержащие один или несколько описанных выше терапевтических агентов, до того как их инкапсулируют. Кроме того, стандартные лекарственные формы могут также включать количество терапевтического агента немедленного высвобождения для немедленного терапевтического эффекта. Терапевтически активный агент немедленного высвобождения может быть введен, например, в виде отдельных шариков в желатиновую капсулу или может быть нанесен на поверхность мультичастиц после приготовления лекарственных форм (например, покрытий регулируемого высвобождения или на основе матриц). Стандартные лекарственные формы по настоящему изобретению могут также содержать комбинацию шариков регулируемого высвобождения и матричных мультичастиц для того, чтобы достичь желаемого эффекта.
Композиции замедленного высвобождения по настоящему изобретению предпочтительно медленно высвобождают терапевтически активный агент, например, когда они проглочены и подвергаются воздействию желудочных жидкостей, а затем кишечных жидкостей. Профиль замедленного высвобождения экструдированных из расплава композиций по изобретению может быть изменен, например, путем варьирования количества замедлителя, т.е. гидрофобного материала, путем варьирования количества пластификатора по отношению к гидрофобному материалу, путем включения дополнительных ингредиентов или вспомогательных веществ, путем изменения способа производства и т.д.
В других воплощениях изобретения экструдированный из расплава материал готовят без включения терапевтически активного агента, который после этого добавляют к экструдату. Такие композиции обычно будут содержать терапевтически активный агент, смешанный с экструдированным матричным материалом, и затем смесь должна таблетироваться для того, чтобы получить композицию медленного высвобождения. Такие композиции могут иметь преимущество, когда, например, включенный в композицию терапевтически активный агент является чувствительным к температурам, необходимым для размягчения гидрофобного материала и/или замедляющего материала.
Подробное описание предпочтительных воплощении
Следующие примеры поясняют различные аспекты настоящего изобретения. Они не должны рассматриваться как ограничивающие каким-либо образом формулу изобретения.
Прямое сравнение состязательных антагонистических свойств налтрексона, сопровождающее его совместное введение с различными опиоидными агонистами, насколько известно заявителям, ранее не предпринималось. Однако были проведены опыты по интервалу дозировки, оценивавшие опиоидно антагонистические свойства у субъектов, получавших разрешенные дозы героина или морфина. В общем, предварительное введение 50 мг налтрексона за 24 часа перед внутривенным введением 25 мг дозы героина полностью блокировало или ослабляло эффекты опиоидного агониста (Смотри Gonzalez J.P., Brogden R.N., "Naltrexone: A Review of its Pharmacodynamic and Pharmacokinetic Properties and Therapeutic Efficacy in the Management of Opioid Dependence", Drugs, 1988; 35: 192-213; Resnick R.R., Valavka J., Freedman A.M., Thomas M., "Studies of EN-1639A (Naltrexone): A New Narcotic Antagonist", Am. J. Psychiatry, 1974; 131: 646-650, настоящим обе работы введены в качестве ссылки).
ПРИМЕР 1.
В примере 1 проводили рандоминизированный односторонне слепой контролируемый по плацебо однодозовый четырехходовой перекрестный опыт для оценки того, может ли пероральное введение раствора 6,4 мг налтрексона блокировать опиоидные агонистические свойства 15 мг гидрокодона у 6 нормальных здоровых женщин-добровольцев. Изучаемая популяция включала только женщин, потому что предыдущие наблюдения показали, что женщины обладают повышенной чувствительностью к эффектам опиоидных агонистов по сравнению с мужчинами. Четырьмя медикаментозными курсами являлись HYIR/APAP (2 таблетки гидрокодона 7,5 и ацетаминофена 750 мг, Vicodin ES®) и пероральный раствор налтрексона 3,2 мг; HYIR/APAP (2×7,5 мг) и пероральный раствор налтрексона 6,4 мг; таблетки компаратора HYIR (2×750 мг таблетки Trilisate®) и пероральный раствор налтрексона (плацебо); и HYIR/APAP (2 таблетки Vicodin ES®) и пероральный раствор налтрексона (плацебо). Все курсы медикаментов вводили в условиях голодания. Между дозами был установлен 48-часовой период вымывания. Субъектов случайным образом распределяли по четырем группам лечения для четырех последовательностей лечения. Субъектов вводили в условия опыта вечером перед первым приемом дозы и продолжали держать в них до полного завершения 24-часовой последозовой оценки последней дозы. Оценки безопасности состояли из регистрации неприятных явлений, дыхания, пульса и температуры, ненормальных лабораторных показателей, ненормальных результатов физического осмотра и результатов ЭКГ. Оценивали также фармакодинамические параметры (размер зрачка и видоизмененный вопросник специфического наркотического эффекта).
Испытуемые медикаментозные курсы
Гидрокодон в таблетках немедленного высвобождения (2×7,5 мг) и пероральный раствор налтрексона 3,2 мг.
Гидрокодон в таблетках немедленного высвобождения (2×7,5 мг) и пероральный раствор налтрексона 6,4 мг.
Компаратор гидрокодона в таблетках немедленного высвобождения и пероральный раствор налтрексона, плацебо.
Плацебо гидрокодона в таблетках немедленного высвобождения (2×7,5 мг) и пероральный раствор налтрексона, плацебо.
Испытуемые продукты
Продукты, испытывавшиеся в этом опыте, включают Vicodin ES® (гидрокодон битартрат 7,5 мг и ацетаминофен 750 мг, Knoll Pharmaceuticals), Trilisate® (холин магнийтрисалицилат, 750 мг, Purdue Frederick), который служил в качестве компаратора, и порошок налтрексона. Vicodin ES® был выбран для активного лечения, поскольку ожидалось, что порция ацетаминофена в этом продукте не должна оказывать влияния на центральную нервную систему или измерения зрачка. Трилизат был выбран для использования в качестве "компаратора", поскольку по внешнему виду он похож на Vicodin ES® и он не влияет на центральную нервную систему или на измерения зрачка. Форма налтрексона в порошке была выбрана вместо принятых в продаже таблеток (Revia® 50 мг таблетки, DuPont) для того, чтобы повысить общую точность при приготовлении перорального раствора. Фармацевт-исследователь готовил по месту пероральный раствор из порошка налтрексона в стерильной среде, используя известные фармацевтические методы. Для приготовления раствора налтрексона использовали порошок налтрексона (Маllinckrodt Chemical). Отдельные основные растворы налтрексона готовили, используя модификацию метода, предложенного Tsang and Holtsman, Tsang В.К., Holtsman R., "Room Temperature Stability of Liquid Naltrexone". Anesthesiology, 1995; 83: A864, включенного настоящим в качестве ссылки. Непосредственно перед (<60 минут) каждым периодом дозирования готовили основной раствор налтрексона путем отвешивания 32 мг и 64 мг порошка налтрексона. Каждую из этих порций растворяли в 50 мл дистиллированной воды и 50 мл простого сиропа, NF до конечного объема 100 мл. Концентрация конечных растворов составляла 0,32 мг/мл (32 мг/100 мл) и 0,64 мг/мл (64 мг/100 мл), соответственно. Эти концентрации давали возможность вводить одинаковый объем (10 мл) перорального раствора налтрексона во время каждого периода дозирования. Пероральный раствор налтрексона плацебо готовили в том же носителе как активный раствор. Добавку порошка агента с горьким вкусом Bitterguard (бензоат денатония, NF) вводили для того, чтобы придать вкус, подобный вкусу активного раствора.
Фармакодинамические измерения
а. Размер зрачка (измерение скиаметрией)
Измерения диаметра зрачков проводили на камере Polaroid CU-5 с линзами 75 мм и встроенной электронной кольцевой вспышкой, используя разовую упаковку 12 пленки Polacolor ER 669. Этот метод получил распространение как безопасный и точный способ изучения зрачков, и его обычно рассматривают как второй метод после методики инфракрасной телевизионной скиаметрии (более универсального и тонкого, но также более дорогого и громоздкого метода). Указывается, что точность метода Polaroid CU-5 находится в пределах 0,1 мм. См. статью Czarnecki J.S., Pilley S.F., Thompson H.S. "The Use of Photography in the Clinical Evaluation of Unequal Pupils". Canad. J. Ophthal., 1979; 14: 297-302, включенную в качестве ссылки.
Диаметр зрачков измеряли следующим образом. Камеру видоизменили путем закрытия двух небольших участков кольцевой вспышки на 3 и 9 часов так, чтобы отражение вспышки в роговице не затемняло горизонтальную границу зрачка. Камеру помещали в центре перед лицом субъекта с 3-дюймовой рамкой (7,5 см) напротив боковых краев орбиты, и глаза занимали только самый верх поля (чтобы минимизировать верхний обзор). Субъекта просили смотреть прямо на корпус камеры и фиксировать взгляд на неопределенной удаленной цели, минимизируя тем самым ближний рефлекс. Когда доброволец фиксировал взгляд на расстоянии, делали снимок. Все фотографии делали при постоянном комнатном освещении. Раздражение зрачка было таким, что вспышка не должна была изменять диаметр зрачков. Наблюдается тоническое сокращение зрачка после вспышки, но оно является коротким, поэтому оно не влияет на измерения, необходимые для данного опыта (см. статью Smith S.A., Dewhist R.R. "A Single Diagnostic Test for Pupillary Abnormality in Diabetic Autonomic Neuropathy". Diabetic Medicine 1988; 3: 38-41, введенную настоящей ссылкой). Проявление отпечатков в течение рекомендованного времени (приблизительно одна (1) минута, изменяясь в зависимости от окружающей температуры) дает фотографию середины лица добровольца в масштабе один к одному с зрачками наверху снимка. Затем измеряют горизонтальный диаметр зрачков, используя простую увеличительную лупу с встроенной шкалой, калиброванной до 0,1 мм. Для измерения зрачкового эффекта в каждый период времени, определенный протоколом, использовали только левый глаз.
b. Видоизмененный вопросник специфического наркотического эффекта
Вопросник является модификацией вопросника из 22 пунктов, использованного Jasinski and Preston, в работах: Jasinski D.R. "Assessment of the Abuse Potential of Mophine-Like Drugs (method used in man)" In: Drug Addiction I (Martin W.R., ed.), 1997: 197-258, Springer-Verlag, New York; Preston K.L., Jasinski D.R., Testa M. "Abuse Potential and Pharmacological Comparison of Tramadol and Morphine", Drug and Alcohol Dependence 1991; 27: 7-17, обе из которых включены в качестве ссылки. Настоящий вопросник состоял из 10 пунктов, заполняемых субъектом за 10 минут перед анализом крови. Вопросы относились к проявлениям опиатных агонистических препаратов и были следующими:
Субъективные вопросы: 1) Вы чувствуете какие-либо эффекты от лекарств? 2) Вы чувствуете мурашки на коже? 3) Вы чувствуете расслабление? 4) Вы чувствуете сонливость? 5) Вы чувствуете опьянение? 6) Вы нервничаете? 7) Вы чувствуете себя полным энергии? 8) Вы чувствуете потребность говорить? 9) Вы чувствуете боль в желудке? 10) Вы чувствуете головокружение? Затем субъект отвечал на вопросы, делая вертикальную отметку на 100 мм визуальной аналоговой шкале (ВАШ), ограниченной на одном конце оценкой "вовсе нет" и на другом конце оценкой "ужасно сильно".
Измеряли размер зрачка левого глаза на исходном уровне (за 30 минут до дозирования) и на 0,5; 1; 2; 4; 6; 9 и 12 часов после введения дозы, и субъект оценивал баллы наркотического эффекта по визуальной аналоговой шкале видоизмененного вопросника специфического наркотического эффекта ("MSDEQ") на исходном уровне и на 0,5; 1; 2; 4; 6; 9 и 12 часов после введения дозы.
Отдельные графики для одиннадцати откликов (вопросы MSDEQ и измерения диаметра зрачка), как функции дозы налтрексона, оценивали визуально и статистически для определения номинально эффективной дозы налтрексона в комбинации с дозой гидрокодона, использованной в опыте.
Зарегистрированные неприятные явления были теми, которые обычно связаны с введением опиоидных анальгетиков и в большинстве классифицировались как "слабые". Не наблюдалось серьезных неприятных явлений или смертей, и ни один пациент не был выведен из опыта из-за неприятных явлений.
Результаты представлены на фигурах 1 и 2.
На фигуре 1 показан антагонизм налтрексона к индуцированному гидрокодоном "наркотическому эффекту" по ВАШ (визуальной аналоговой шкале). Это относится к первому вопросу видоизмененного вопросника специфического наркотического эффекта, который спрашивает субъекта "чувствуете ли Вы какие-либо эффекты от лекарства?". Результаты показывают, что имеется влияние налтрексона на реакцию на дозу, увеличение дозы налтрексона снижает "наркотический эффект" ВАШ гидрокодона. Доза налтрексона 6,4 мг противостоит эффекту 15 мг дозы гидрокодона в большей степени, чем 3,2 мг доза налтрексона. Опиоидный эффект гидрокодона не полностью блокировался 6,4 мг дозой налтрексона. На фигуре 2 показан антагонизм налтрексона индуцированному гидрокодоном сужению зрачка. Эти результаты также подтверждают влияние налтрексона на реакцию на дозу, увеличение дозы налтрексона вызывает меньшее сокращение зрачка у субъектов, которые получили 15 мг гидрокодона. Доза 6,4 мг налтрексона противостоит индуцированному гидрокодоном сокращению зрачка в большей степени, чем доза 3,2 мг налтрексона. Сужение зрачка от гидрокодона не полностью блокировалось дозой 6,4 мг налтрексона. Самое слабое сокращение зрачка наблюдалось в группе, получавшей плацебо. Группа, получавшая гидрокодон плюс плацебо налтрексона, испытывала наибольшее сужение зрачка и поэтому имела самые низкие значения диаметра зрачка.
ПРИМЕР 2.
В примере 2 проводили десятипериодный рандоминизированный перекрестный односторонне слепой опыт на нормальных здоровых женщинах-добровольцах для оценки соотношения перорального налтрексона к пероральному гидрокодону, которое могло бы номинально минимизировать опиоидные агонистические эффекты. В опыт ввели 21 субъекта и 16 субъектов завершили опыт. Десять медикаментозных курсов включали HYIR/APAP (2 таблетки гидрокодона 7,5 и ацетаминофена 750 мг, Vicodin ES®) со следующими дозами перорального раствора налтрексона: 0,4 мг/10 мл, 0,8 мг/10 мл, 1,6 мг/10 мл, 3,2 мг/10 мл, 4,8 мг/10 мл, 6,4 мг/10 мл; 9,6 мг/10 мл, 12,8 мг/10 мл и плацебо пероральным раствором налтрексона, а также таблетки компаратора гидрокодона немедленного высвобождения (2 х 750 мг таблетки Trilisate®) с плацебо пероральным раствором налтрексона. Все медикаменты вводили в условиях голодания. Между дозами был установлен 48-часовой период вымывания. Субъектов случайным образом распределяли по десяти группам лечения для десяти последовательностей лечения. Субъектов вводили в условия опыта вечером перед первым приемом дозы и продолжали держать в них до полного завершения 24-часовой последозовой оценки последней дозы. Оценки безопасности состояли из регистрации неприятных явлений, дыхания, пульса и температуры, ненормальных лабораторных показателей, ненормальных результатов физического осмотра и результатов ЭКГ. Определяли уровень гидрокодона, налтрексона и 6-β-налтрексона в плазме и подсчитывали и анализировали фармакокинетические показатели. Оценивали также фармакодинамические параметры (размер зрачка и видоизмененный вопросник специфического наркотического эффекта).
Режим дозировки
Режим дозировки был следующим:
Таблетки компаратора гидрокодона немедленного высвобождения (плацебо) вводили вместе с 10 мл перорального раствора налтрексона (плацебо) примерно в 8.00 дня введения дозы в периоды от 1 до 10 после 8-часового голодания. Голодание продолжалось в течение дополнительных четырех (4) часов после приема дозы.
Таблетки гидрокодона немедленного высвобождения (2×7,5 мг) вводили вместе с 10 мл перорального раствора налтрексона (плацебо) примерно в 8.00 дня введения дозы в периоды от 1 до 10 после 8-часового голодания. Голодание продолжалось в течение дополнительных четырех (4) часов после приема дозы.
Таблетки гидрокодона немедленного высвобождения (2×7,5 мг) вводили вместе с 10 мл перорального раствора налтрексона (0,4 мг) примерно в 8.00 дня введения дозы в периоды от 1 до 10 после 8-часового голодания. Голодание продолжалось в течение дополнительных четырех (4) часов после приема дозы.
Таблетки гидрокодона немедленного высвобождения (2×7,5 мг) вводили вместе с 10 мл перорального раствора налтрексона (0,8 мг) примерно в 8.00 дня введения дозы в периоды от 1 до 10 после 8-часового голодания. Голодание продолжалось в течение дополнительных четырех (4) часов после приема дозы.
Таблетки гидрокодона немедленного высвобождения (2×7,5 мг) вводили вместе с 10 мл перорального раствора налтрексона (1,6 мг) примерно в 8.00 дня введения дозы в периоды от 1 до 10 после 8-часового голодания. Голодание продолжалось в течение дополнительных четырех (4) часов после приема дозы.
Таблетки гидрокодона немедленного высвобождения (2×7,5 мг) вводили вместе с 10 мл перорального раствора налтрексона (3,2 мг) примерно в 8.00 дня введения дозы в периоды от 1 до 10 после 8-часового голодания. Голодание продолжалось в течение дополнительных четырех (4) часов после приема дозы.
Таблетки гидрокодона немедленного высвобождения (2×7,5 мг) вводили вместе с 10 мл перорального раствора налтрексона (4,8 мг) примерно в 8.00 дня введения дозы в периоды от 1 до 10 после 8-часового голодания. Голодание продолжалось в течение дополнительных четырех (4) часов после приема дозы.
Таблетки гидрокодона немедленного высвобождения (2×7,5 мг) вводили вместе с 10 мл перорального раствора налтрексона (6,4 мг) примерно в 8.00 дня введения дозы в периоды от 1 до 10 после 8-часового голодания. Голодание продолжалось в течение дополнительных четырех (4) часов после приема дозы.
Таблетки гидрокодона немедленного высвобождения (2×7,5 мг) вводили вместе с 10 мл перорального раствора налтрексона (9,6 мг) примерно в 8.00 дня введения дозы в периоды от 1 до 10 после 8-часового голодания. Голодание продолжалось в течение дополнительных четырех (4) часов после приема дозы.
Таблетки гидрокодона немедленного высвобождения (2×7,5 мг) вводили вместе с 10 мл перорального раствора налтрексона (12,8 мг) примерно в 8.00 дня введения дозы в периоды от 1 до 10 после 8-часового голодания. Голодание продолжалось в течение дополнительных четырех (4) часов после приема дозы.
Наблюдаемые субъекты голодали 8 часов до и четыре (4) часа после каждого введения дозы предписанного лекарства каждый день введения дозы. Исходную пробу крови (на содержание гидрокодона, налтрексона и 6-β-налтрексона) отбирали перед (в пределах 30 минут) дозированным введением начальной дозы (0 ч) и на 0,5; 1, 2, 4, 6 и 9 часов после введения дозы. Все пробы отбирали в интервале ±2 мин от назначенного времени. Замеры следующих фармакодинамических параметров делали непосредственно перед отбором пробы крови в точке отсчета (за 30 минут до дозирования) и на 0,5; 1, 2, 4, 6 и 9 часов после введения дозы.
Непосредственно перед каждым периодом дозировки готовили 8 разных основных растворов налтрексона путем отвешивания 4, 8, 16, 32, 48, 64, 96 и 128 мг порошка налтрексона. Каждую из этих порций растворяли в 50 мл дистиллированной воды и в 50 мл простого сиропа. Объем конечного раствора был 100 мл при концентрациях 0,04; 0,08; 0,16, 0,32; 0,48; 0,96 и 1,28 мг/мл. Эти концентрации давали возможность вводить одинаковый объем (10 мл) перорального раствора налтрексона во время каждого периода введения дозы. Пероральный раствор налтрексона плацебо готовили в том же носителе как активный раствор. Добавку порошка агента, придающего горький вкус, Bitterguard (бензоат денатония) вводили для того, чтобы придать вкус, подобный вкусу активного раствора.
Фармакодинамические измерения
Фармакодинамические измерения для примера 2 проводили в соответствии с методиками, представленными выше для примера 1.
Средний "наркотический эффект" в баллах ВАШ и диаметр зрачка во времени для каждого из медикаментозных курсов представлены, соответственно, на фигурах 3 и 4. В целом, введение разовой дозы гидрокодона немедленного высвобождения/ацетаминофена ("HYIR/APAP") с увеличивающимися дозами налтрексона (интервал 0 мг - 12,8 мг) приводит к общему снижению "наркотического эффекта" в баллах ВАШ и уменьшению сокращения зрачка. Фигуры 5 и 6 представляют соответствующий средний максимальный "наркотический эффект" в баллах ВАШ (±95% ДИ) и средний минимальный диаметр зрачка (±95% ДИ) как функцию логарифма каждой из доз налтрексона. Обе фигуры показывают отношения реакции на дозу, причем эффект зрачков показывает более сильную связь реакции с дозой по сравнению с реакцией "наркотического эффекта" по ВАШ.
Результаты позволяют считать, что даже при включении 0,4 мг налтрексона имеет место уменьшение фармакологических эффектов дозы гидрокодона. Приблизительно 0,4 мг налтрексона минимально противостоят дозе 15 мг гидрокодона. Дозы налтрексона выше 0,4 мг начинают показывать увеличенное снижение эффекта дозы гидрокодона.
Зарегистрированные неприятные явления были теми, которые обычно связаны с введением опиоидных анальгетиков, и в большинстве классифицировались как "слабые". Всего пять субъектов (5/21) прервали опыт. Три испытуемых прервали опыт из-за неприятных явлений. Две из них испытывали неприятные ощущения, которые были классифицированы как несерьезные. У одной испытуемой развилась анемия, которая была классифицирована как серьезная и требующая лечения. Две других прервали опыт потому, что их врачи поняли, что в их медицинских картах содержалась информация, которая делала невозможным их участие в экспериментах. При этом исследовании не было смертей.
В целом, однодозовое введение 15 мг таблеток гидрокодона немедленного высвобождения с увеличивающимися дозами перорального раствора налтрексона (интервал 0 мг - 12,8 мг) приводит к общему снижению баллов "наркотического эффекта" ВАШ и к увеличению диаметра зрачка.
ПРИМЕР 3.
Пример 3 представляет результаты опыта по оценке синдрома отмены лекарства у морфино-зависимых добровольцев, получавших таблетки гидрокодона немедленного высвобождения и пероральный раствор налтрексона. Данный опыт был односторонне слепым однодозовым плацебо-контролируемым исследованием увеличения дозы налтрексона у субъектов, физически зависимых от опиоидов. Подопытные субъекты (5) были опиоидно-зависимыми, как было определено по тесту Narcan, баллам Индекса тяжести наркомании, физическому обследованию, наблюдению и результатам анализа мочи на наркотики и в текущий момент не подвергались лечению от наркомании. Для оценки синдрома отмены лекарства, следующей после совместного введения гидрокодона немедленного высвобождения и налтрексона, была выбрана 30 мг доза гидрокодона немедленного высвобождения для симуляции уровня дозы, используемого личностями, которые злоупотребляют гидрокодоном. Эта доза являлась также дозой, которая рассматривается как эквианальгетическая другим, обычно используемым опиоидам у опиоидно-наивных пациентов. Считается, что обезболивающая потенция гидрокодона подобна потенции оксикодона и примерно в два раза выше потенции морфина.
Исследуемые способы лечения
Курсы лечения были следующими:
Таблетки гидрокодон/ацетаминофен немедленного высвобождения (HYIR/APAP) по 30 мг (Lortab® 3×10 мг) и увеличивающиеся дозы перорального раствора налтрексона 0, 0,25 мг, 0,5 мг, 1,0 мг и 2,0 мг.
Таблетки гидрокодон/ацетаминофен немедленного высвобождения (HYIR/APAP) по 30 мг (Lortab® 3×10 мг) и пероральный раствор плацебо налтрексона.
Пероральный раствор налтрексона и раствор плацебо готовили в соответствии с примерами 1 и 2.
Состояние подопытных стабилизировали в течение 5 дней путем введения внутримышечно 15 мг сульфата морфина с регулярными интервалами: в 6 и 10 часов утра и в 4 и 10 часов вечера ежедневно. Доза в 15 мг сульфата морфина внутримышечно эквивалентна 30 мг гидрокодона, введенным перорально. Изучаемые медикаменты вводили после стабилизации в 10 часов утра в дни введения дозы изучаемых медикаментов и вели наблюдение в течение следующих шести часов. Спустя шесть часов, если синдром отмены не наблюдался, введение 15 мг внутримышечно сульфата морфина возобновлялось дозой в 4 часа после полудня. Субъектов стабилизировали 48 часов перед следующим вводом изучаемого препарата. Следуя каждому из медикаментозных курсов (1-4), если синдром отмены не наблюдался, субъект получал изучаемые медикаменты для последующего лечения в следующем возрастающем порядке:
Курс №1: HYIR/APAP таблетки 30 мг (Lortab® 3×10 мг) вводились с пероральным раствором (10 мл) плацебо налтрексона приблизительно в 10.00 дня введения дозы после 8-часового голодания. Голодание продолжалось дополнительно четыре (4) часа после введения дозы.
Курс №2: HYIR/APAP таблетки 30 мг (Lortab® 3×10 мг) вводились с пероральным раствором (10 мл) налтрексона (0,25 мг) приблизительно в 10.00 дня введения дозы после 8-часового голодания. Голодание продолжалось дополнительно четыре (4) часа после введения дозы.
Курс №3: HYIR/APAP таблетки 30 мг (Lortab® 3×10 мг) вводились с пероральным раствором (10 мл) налтрексона (0,5 мг) приблизительно в 10.00 дня введения дозы после 8-часового голодания. Голодание продолжалось дополнительно четыре (4) часа после введения дозы.
Курс №4: HYIR/APAP таблетки 30 мг (Lortab® 3×10 мг) вводились с пероральным раствором (10 мл) налтрексона (1,0 мг) приблизительно в 10.00 дня введения дозы после 8-часового голодания. Голодание продолжалось дополнительно четыре (4) часа после введения дозы.
Пробы крови отбирали за 0,5 часа перед дозой и в 0,5; 1, 2, 4 и 6 часов после дозы. Замеры диаметра зрачка проводили, используя измеритель зрачков Pupilscan и записывали в миллиметрах к ближайшему миллиметру. За каждым опытным периодом следовал 48-часовой период вымывания. Четыре субъекта прошли исследование, один субъект был исключен. Эффектом налтрексона явилась слабая абстиненция (симптомы синдрома отмены) при 1 и 2 мг.
Протокол был изменен, и 12 подопытных субъектов участвовали в опыте, который был идентичен вышеописанному за исключением увеличенного соотношения налтрексона. Дозами налтрексона в измененном протоколе были 0, 1, 2, 4 и 8 мг. Восемь подопытных субъектов завершили исследование, тогда как четверо вышли из него.
Контролировали дыхание, пульс и температуру каждого субъекта, и субъектов контролировали на признаки и симптомы опиоидного синдрома отмены. Признаки синдрома отмены включали заложенный или текущий нос, слезотечение, зевоту, потовыделение, озноб, рвоту, пилоэрекцию, расширение зрачка, раздражительность и беспокойство. Симптомы синдрома отмены включают ощущение изменений температуры, боль в суставах, костях или мышцах, судороги желудка, мурашки по коже, тошноту и сообщения субъекта о субъективном восприятии вышеупомянутых симптомов.
Для того чтобы измерить субъективное восприятие комбинации препаратов, субъекты отвечали на вопросник в период опыта. Ответы на вопросы градуировались по визуальной аналоговой шкале, как описано в примере 1. Субъективные ощущения, которые оценивали, были следующими: нравится/не нравится препарат, возможность ощутить наркотический эффект, потовыделение, беспокойство, озноб, слезящиеся глаза, гусиная кожа, расстройство желудка, заложенный нос, сонливость, холод, жар, мышечная боль, напряжение или расслабление, помешательство, страх, раздражительность, болтливость, ощущения синдрома отмены, ощущение болезни. Субъектов также наблюдали по следующим симптомам: зевота, почесывание, расслабление, заложенный нос, раздражительность, выход. Кроме того, контролировали кровяное давление, пульс, скорость дыхания, размер зрачка и температуру тела.
Данные по пяти субъектам представлены ниже. Фигуры 7А-С показывают средние баллы из вопросников для субъективного восприятия гидрокодона, нанесенные на график как функция времени после введения и как функция дозы налтрексона. Фигура 7А показывает способность субъектов ощущать эффект гидрокодона в присутствии меняющихся количеств налтрексона. Фигуры 7В и 7С показывают, соответственно, благоприятные или неблагоприятные субъективные восприятия гидрокодона в присутствии меняющихся количеств налтрексона.
Фигуры 8А и В показывают средние баллы для субъективного восприятия гидрокодона, нанесенные на график как функция времени после введения и как функция дозы налтрексона. Фигура 8А показывает восприятие субъектами выхода из эффекта гидрокодона в присутствии меняющихся количеств налтрексона. Фигура 8В показывает субъективные ощущения заболевания в присутствии меняющихся количеств налтрексона.
Фигура 9А показывает влияние гидрокодона на размер зрачка в присутствии меняющихся количеств налтрексона. Фигура 9В показывает кажущуюся степень синдрома отмены действия гидрокодона в присутствии меняющихся количеств налтрексона с точки зрения наблюдателя.
Фигуры 10А-С представляют площади под кривыми, представленными на фигурах 7А-С, интегрированными за 6 часов периода наблюдения, как функцию дозы налтрексона и 95% доверительные уровни для ответа на плацебо налтрексон (30 мг гидрокодона, 0 мг налтрексона). Фигура 10А показывает, что доза до 8 мг налтрексона не отменяет способность субъекта воспринимать эффект гидрокодона: экспериментально определенная ППК (площадь под кривой) (от 0 до 6 часов), наблюдавшаяся для каждой дозы налтрексона, полностью лежит в 95% доверительном интервале для реакции на плацебо налтрексон. Фигура 10В показывает ППК (0-6 час) благоприятного восприятия субъектами гидрокодона как функцию дозы налтрексона. Фигура 10В демонстрирует, что благоприятное субъективное восприятие понижается для доз налтрексона >1 мг, то есть экспериментально определенная ППК (от 0 до 6 часов) поднялась выше 95% доверительного интервала для реакции на плацебо налтрексон при примерно 1 мг налтрексона.
Фигуры 11А-С представляют площади под кривыми, представленными на фигурах 8А-В и фигуре 9А, интегрированными за 6 часов периода наблюдения, как функцию дозы налтрексона и 95% доверительные уровни для отклика на плацебо налтрексон (30 мг гидрокодона, 0 мг налтрексона). Фигура 11А демонстрирует ППК (0-6 час) субъективного ощущения выхода в присутствии меняющихся количеств налтрексона. Фигура 11А показывает, что дозы налтрексона больше, чем примерно 0,75 мг приводят к субъективному ощущению заболевания: экспериментально определенная ППК (от 0 до 6 часов), наблюдавшаяся на фигуре 8А для каждой дозы налтрексона, поднялась выше 95% доверительного интервала для реакции на плацебо налтрексон при приблизительно 0,75 мг налтрексона. Фигура 11В демонстрирует ППК (0-6 час) ощущения субъектами заболевания в присутствии меняющихся количеств налтрексона. Фигура 11В показывает, что дозы налтрексона больше, чем приблизительно 0,75 мг, приводят к субъективному ощущению заболевания: экспериментально определенная ППК (от 0 до 6 часов), наблюдавшаяся на фигуре 8В для каждой дозы налтрексона, поднялась выше 95% доверительного интервала для реакции на плацебо налтрексон при приблизительно 0,75 мг налтрексона.
Фигура 11С демонстрирует ППК (0-6 час) экспериментального определенного изменения размера зрачка как функции дозы налтрексона. Фигура 11С показывает, что дозы налтрексона до 8 мг не устраняют эффект сокращения зрачка от гидрокодона: экспериментально определенная ППК (от 0 до 6 часов), наблюдавшаяся на фигуре 9А для каждой дозы налтрексона, полностью лежит в 95% доверительном интервале для реакции на плацебо налтрексон.
Клинические исследования показали, что гидрокодон в комбинации с налтрексоном имеет начало действия <0,5 часа, пик между 0,5 и 1 часом и заметное уменьшение в пределах 3-4 часов. Была получена кривая отклик-доза. Добавление налтрексона понижает субъективное благоприятное восприятие гидрокодона, повышает субъективное неприятное восприятие гидрокодона и повышает субъективное восприятие заболевания и синдрома отмены гидрокодона. Эти ощущения являются явно вызывающими отвращение.
Несмотря на то, что изобретение описано и разъяснено со ссылками на некоторые его предпочтительные воплощения, специалистам должно быть понятно, что в нем могут быть сделаны очевидные видоизменения без нарушения духа и предмета изобретения. Такие вариации рассматриваются как охватываемые предметом изобретения в прилагаемой формуле изобретения.
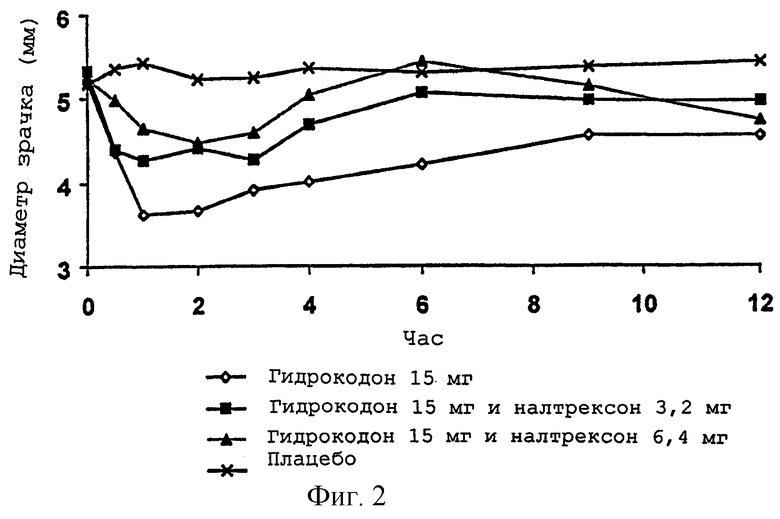
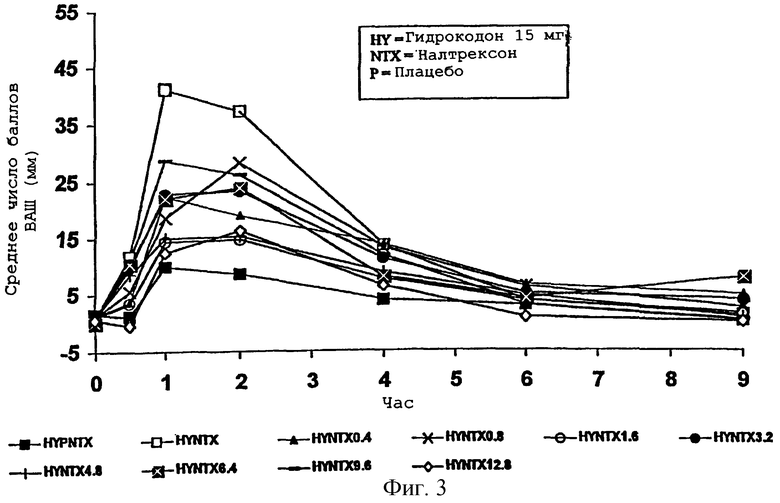
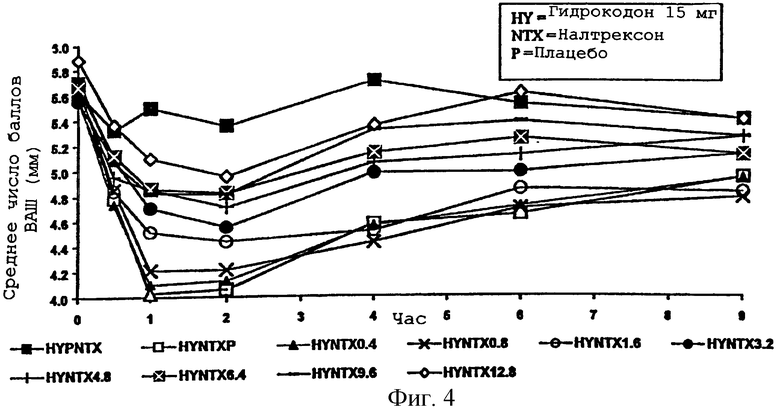
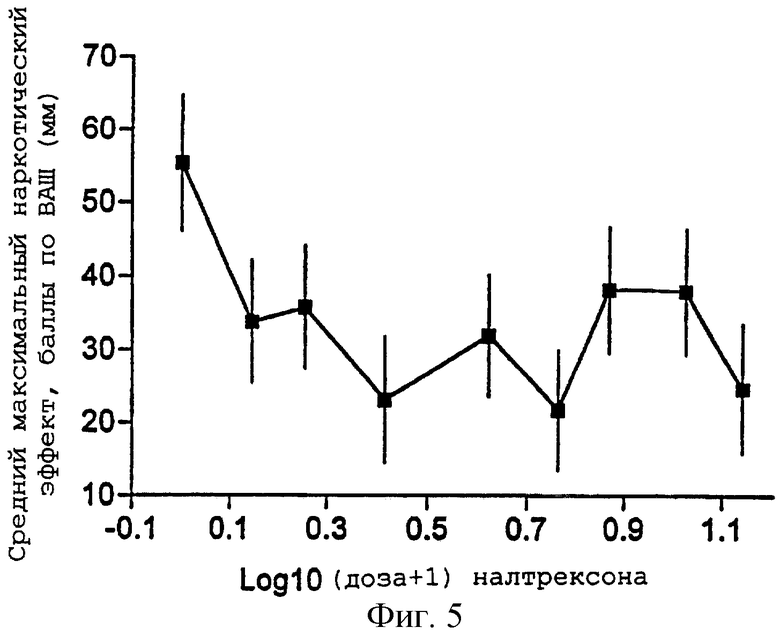
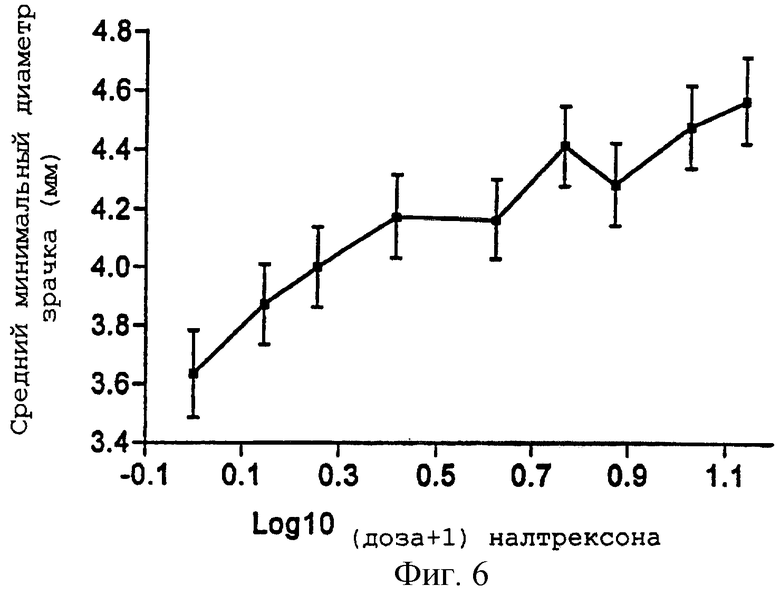
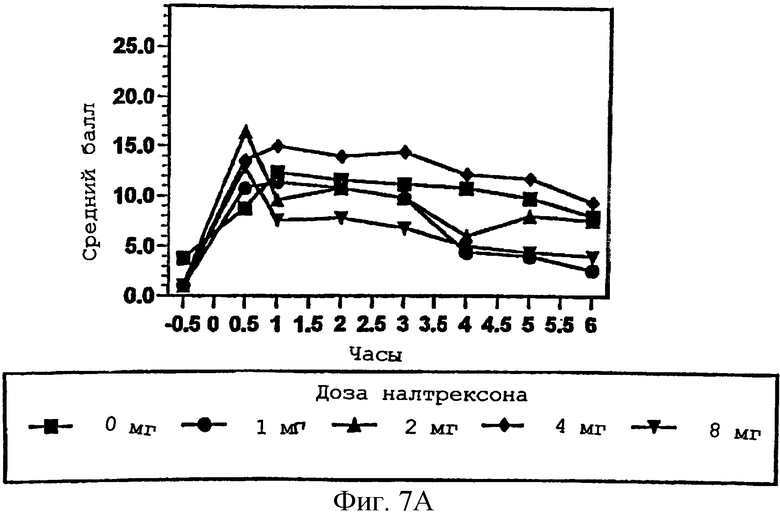
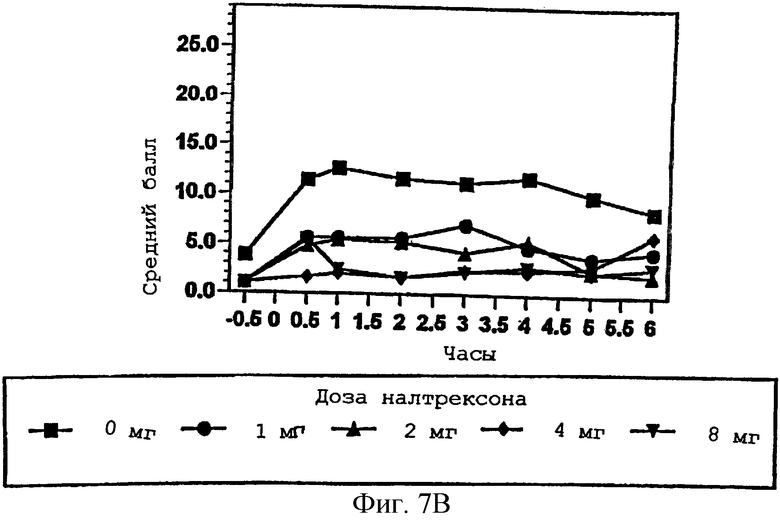
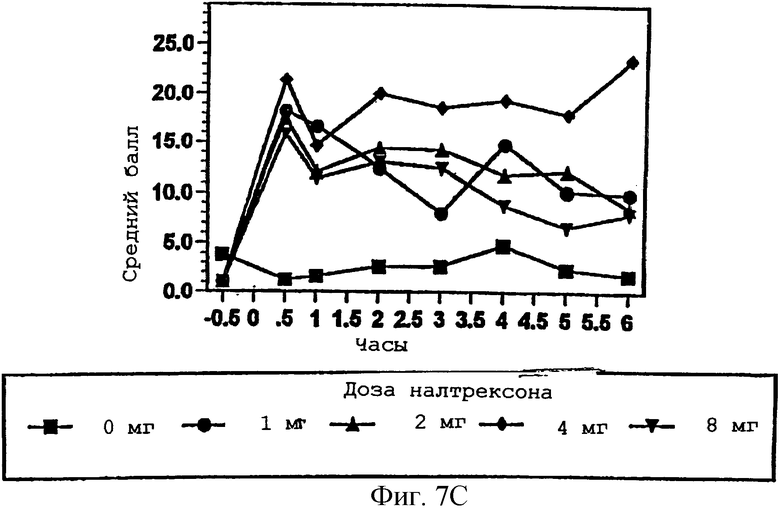
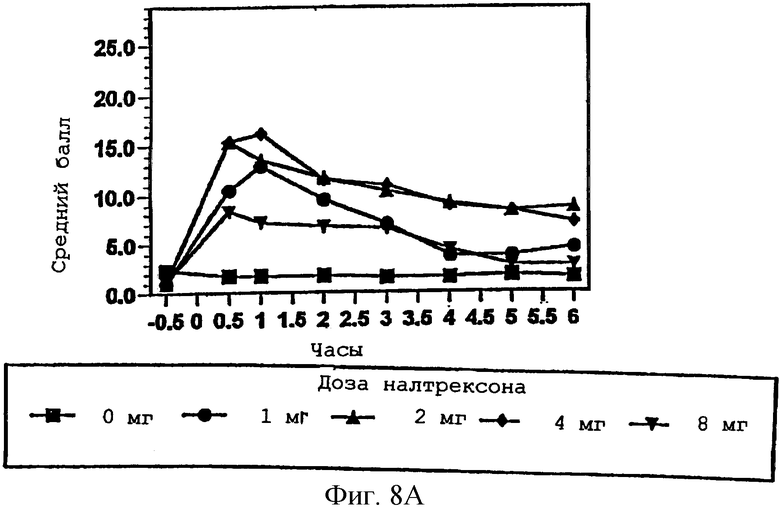
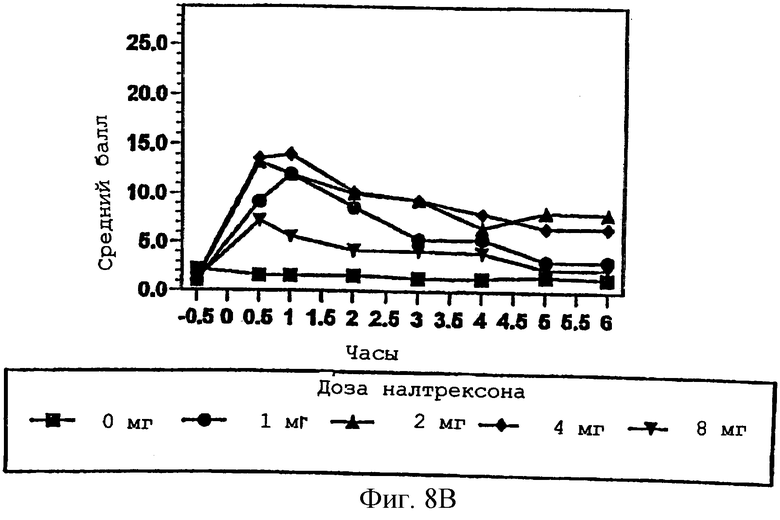
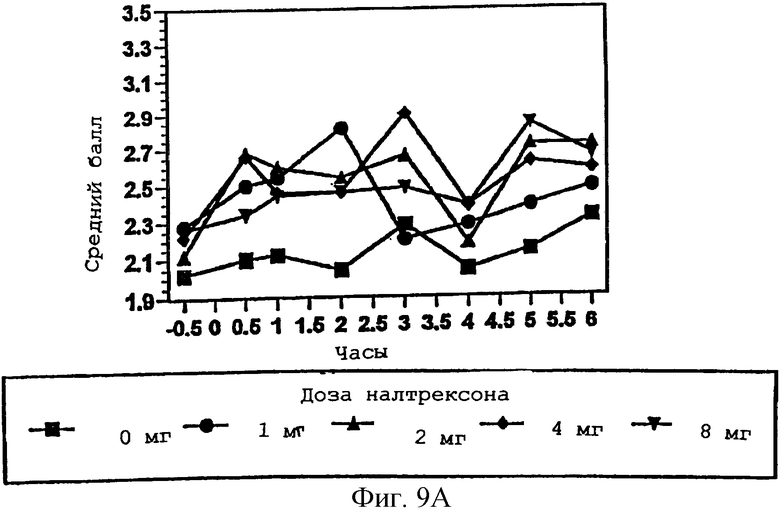
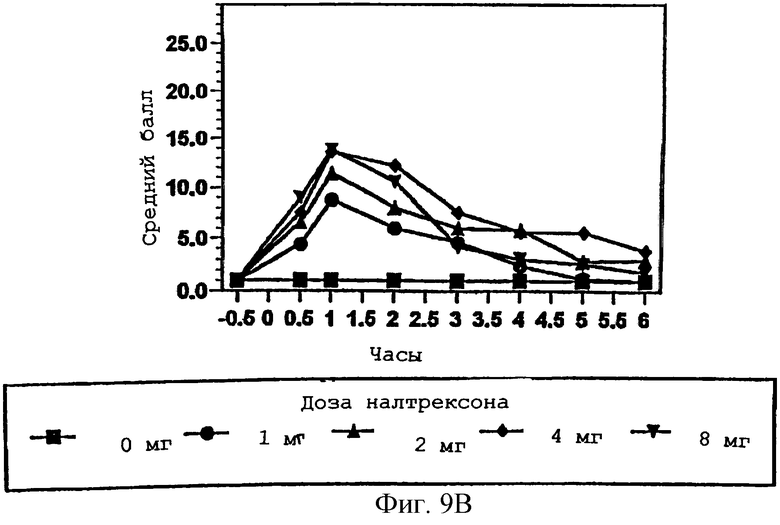
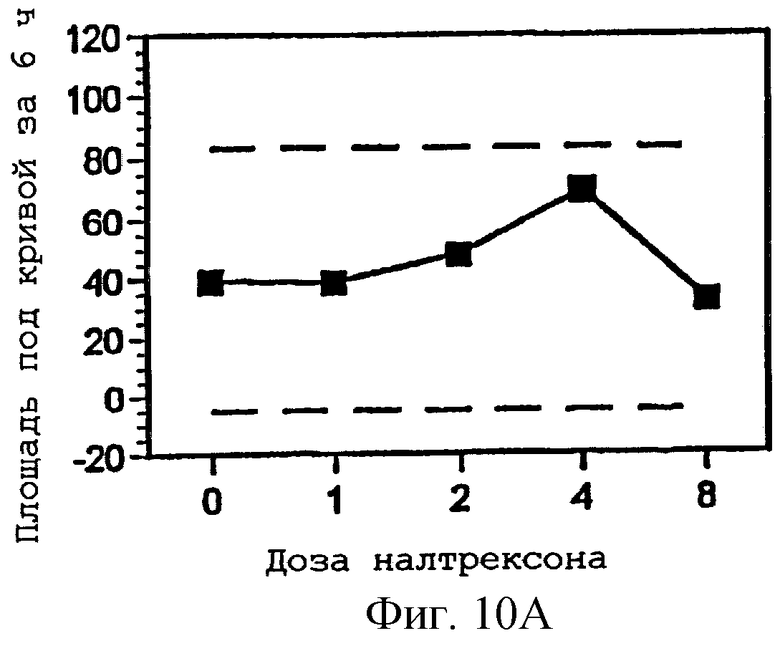
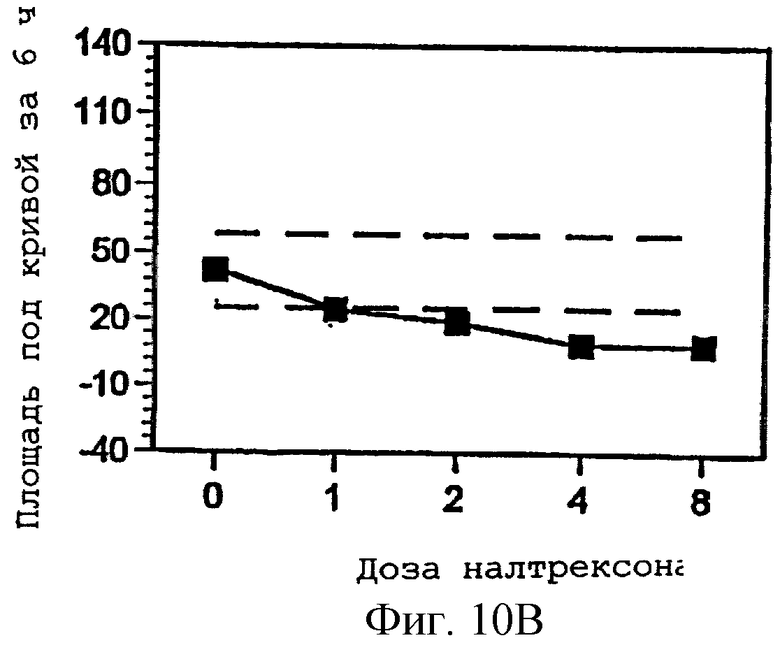
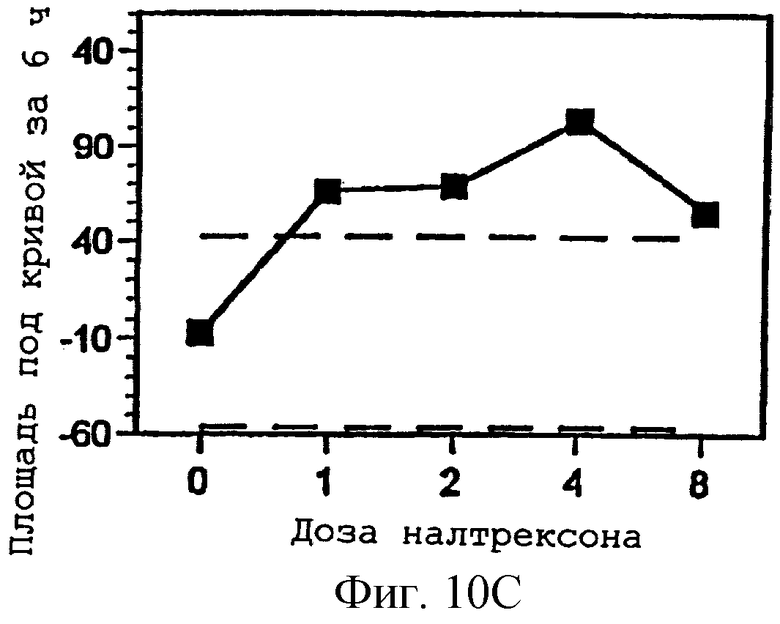
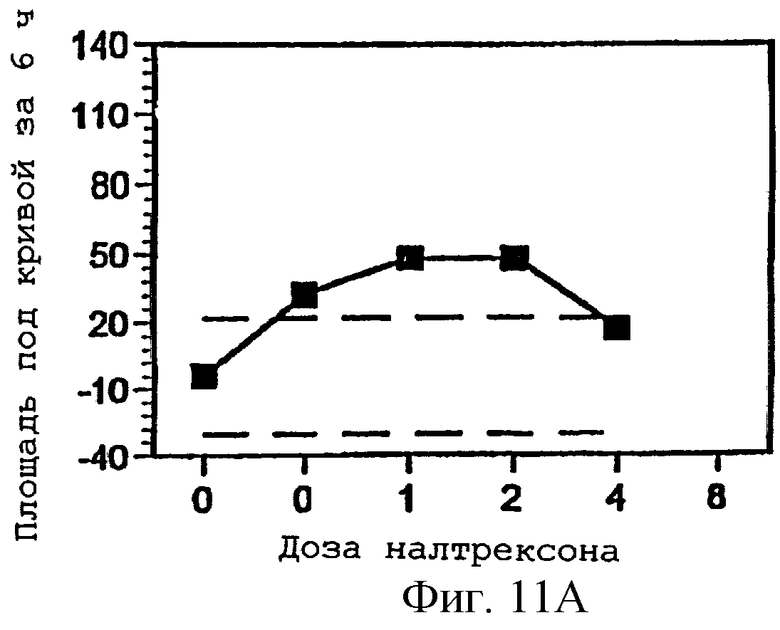
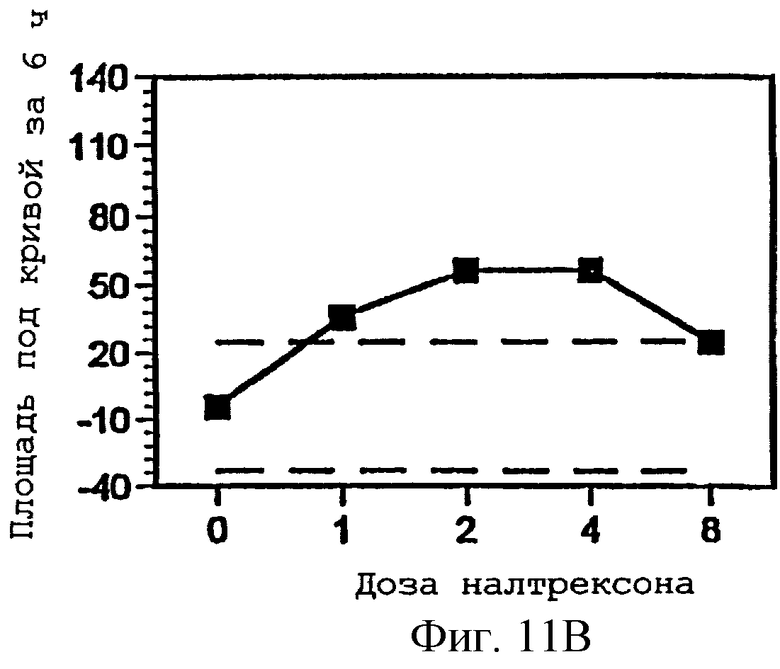
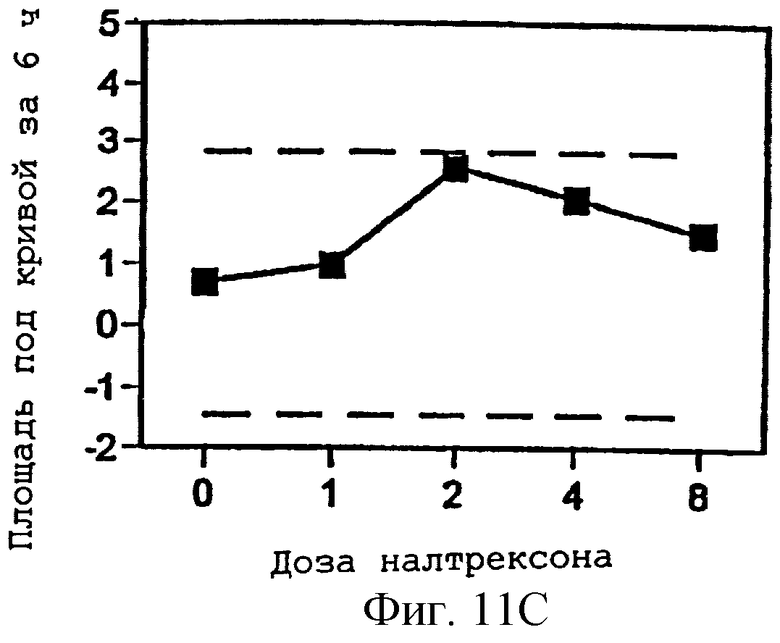
Изобретение относится к пероральным лекарственным формам, включающим комбинацию перорально анальгетически эффективного количества опиоидного агониста и перорально активного опиоидного антагониста, где опиоидный антагонист включен в таком соотношении к опиоидному агонисту, чтобы получить комбинированный продукт, который анальгетически эффективен при пероральном введении, но вызывает отвращение у физически зависимого субъекта. Предпочтительно количество опиоидного антагониста, включенного в комбинированный продукт, вызывает по меньшей мере слабое отвращение у физически зависимых наркоманов (например, синдром отмены). 6 н. и 67 з.п. ф-лы, 11 ил., 3 табл.
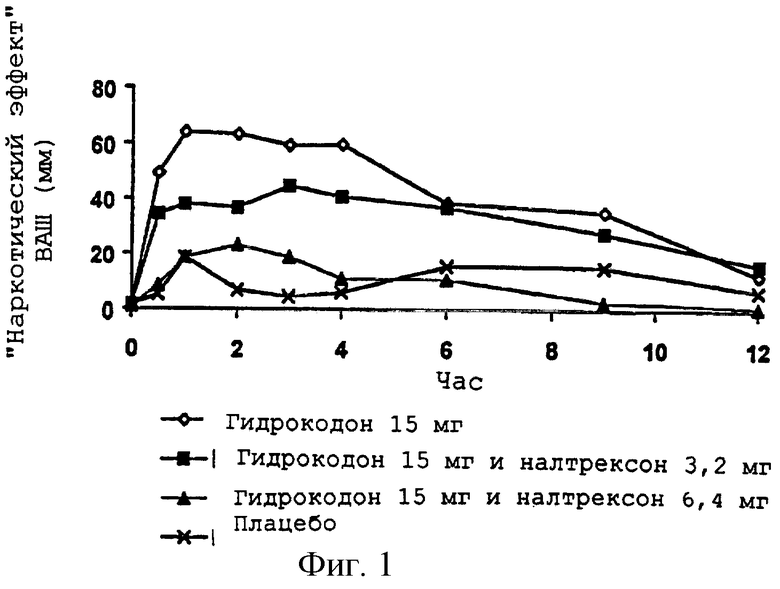
| US 4457933 А, 03.07.1984 | |||
| US 5512578 А, 30.04.1996 | |||
| МАШКОВСКИЙ М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, 1999, ч.1, с.197. |
