ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к композициям и способам понижения ЛНП-холестерина и лечения состояний, связанных с повышенными уровнями холестерина. Более конкретно, изобретение относится к композициям и способам ингибирования экспрессии аполипопротеина В в печени.
УРОВЕНЬ ТЕХНИКИ
Коронарная болезнь сердца (CHD) является главной причиной смерти в Соединенных Штатах в течение более ста лет, а осложнения атеросклероза представляют собой наиболее распространенные причины смерти в западном сообществе (Knopp, New Engl. J. Medicine, 1999, 341, 498-511; Davis and Hui, Arterioscler. Thromb. Vasc. Biol., 2001, 21, 887-898; Bonow, Circulation, 2002, 106, 3140-3141). Повышенные уровни холестерина липопротеинов низкой плотности (ЛНП-холестерина) повсеместно признаны в качестве фактора риска CHD. Однако, несмотря на фармакологическое вмешательство, многие субъекты не способны понизить уровни ЛНП-холестерина.
Руководство, касающееся лечения, понижающего уровень липидов, было разработано национальной обучающей программой по холестерину для лечения взрослых людей III (NCEP) в 2001 г. Модификации указанного руководства были рекомендованы координационным комитетом NCEP в 2004 г. и включали в себя более агрессивные цели лечения (Grundy et al., Circulation, 2004, 110, 227-239). Указанное руководство определяет 3 категории риска больших коронарных событий и желательные уровни ЛНП-холестерина. Наибольшему риску подвержены субъекты с CHD или эквивалентом риска CHD, и они должны поддерживать уровень ЛНП-холестерина ниже 100 мг/дл. Последнее руководство NCEP рекомендует субъектам с очень высоким риском CHD применять медикаментозную терапию для достижения уровней ЛНП-холестерина ниже 70 мг/дл. Субъектов с эквивалентом CHD определяют как субъектов с диабетом, заболеванием периферических сосудов, аневризмой брюшного отдела аорты, заболеванием сонных артерий с симптоматикой, а также как субъектов с множественными факторами риска, которые создают 10-летний риск CHD более 20%. Во вторую категорию входят субъекты с умеренным риском CHD, которые имеют множество (2 или более) факторов риска, и для которых 10-летний риск CHD составляет 20%; целью является уровень ЛНП-холестерина ниже 130 мг/дл. Самые последние рекомендации включают в себя терапевтический выбор для понижения уровней ЛНП-холестерина до величин менее 100 мг/дл для категории субъектов с умеренным риском. Третья категория включает в себя субъектов с 0-1 фактором риска, а целью является уровень ЛНП-холестерина ниже 160 мг/дл. Факторы риска включают в себя возраст, курение, гипертензию, низкий уровень ЛВП-холестерина и семейный анамнез CHD. Медикаментозное лечение следует начинать, когда уровень ЛНП-холестерина в сыворотке крови остается на уровне выше 130, 160 и 190 мг/дл в 3 группах риска, соответственно, невзирая на изменения образа жизни в терапевтических целях (Grundy et al., Circulation, 2004, 110, 227-239).
Липопротеины низкой плотности представляют собой один из пяти обширных классов липопротеинов, который включает в себя следующее: хиломикроны, ответственные за транспорт поступающих с пищей жиров из тонкого кишечника в ткани; липопротеины очень низкой плотности (ЛОНП); липопротеины промежуточной плотности (ЛПП); липопротеины низкой плотности (ЛНП); все названные агенты транспортируют триацилглицериды и холестерин из печени в ткани; и липопротеины высокой плотности (ЛВП), которые транспортируют эндогенный холестерин из тканей в печень. Частицы липопротеинов претерпевают непрерывный метаболический процессинг и имеют изменяющиеся свойства и составы. Белковые компоненты липопротеинов известны как аполипопротеины. По меньшей мере девять аполипопротеинов, одним из которых является аполипопротеин В, распределяются в значительных количествах среди различных человеческих липопротеинов.
Аполипопротеин В (известный также как ApoB, аполипопротеин В-100; ApoB-100, аполипопротеин В-48; ApoB-48 и антиген Ag(x)) представляет собой большой гликопротеин, принимающий участие в сборке и секреции жиров и в транспорте и опосредованном рецепторами захвате и доставке различных классов липопротеинов. Аполипопротеин В выполняет ряд функций, включая всасывание и процессинг жиров, поступающих с пищей, а также регуляцию уровней циркулирующих липопротеинов (Davidson and Shelness, Annu. Rev. Nutr., 2000, 20, 169-193). У млекопитающих существуют две формы аполипопротеина В. ApoB-100 представляет белок полной длины, содержащий 4536 аминокислотных остатков, синтезированный, главным образом, в печени человека (Davidson and Shelness, Annu. Rev. Nutr., 2000, 20, 169-193). Усеченная форма, известная как аpoB-48, является коллинеарной концевым 2152 аминокислотным остаткам, и синтезируется в тонком кишечнике всех млекопитающих (Davidson and Shelness, Annu. Rev. Nutr., 2000, 20, 169-193). У людей аpoB-48 циркулирует в ассоциации с хиломикронами и остатками хиломикронов, а указанные частицы устраняются отдельным рецептором, известным как белок, связанный с рецептором ЛНП-холестерина (Davidson and Shelness, Annu. Rev. Nutr., 2000, 20, 169-193). ApoB-48 можно рассматривать как приспособление, посредством которого жир, поступающий с пищей, доставляется из тонкого кишечника в печень, в то время как аpoB-100 участвует в транспорте и доставке холестерина (Davidson and Shelness, Annu. Rev. Nutr., 2000, 20, 169-193). ApoB является главным белковым компонентом ЛНП-холестерина и содержит домен, требующийся для взаимодействия указанного вида липопротеинов с рецептором ЛНП. Помимо этого, ApoB содержит неспаренный остаток цистеина, который опосредует взаимодействие с аполипопротеином (а) и генерирует липопротеин (а) или ЛП(а), другой липопротеин, обладающий атерогенным потенциалом (Davidson and Shelness, Annu. Rev. Nutr., 2000, 20, 169-193). Повышенные уровни липопротеина ЛП(а), содержащего ApoB, связаны с повышенным риском атеросклероза и его проявлений, которые могут включать в себя гиперхолестеринемию (Seed et al., N. Engl. J. Med., 1990, 322, 1494-1499), инфаркт миокарда (Sandkamp et al., Clin. Chem., 1990, 36, 20-23) и тромбоз (Nowak-Gottl et al., Pediatrics, 1997, 99, E11).
Аполипопротеин В участвует в гомеостазе холестерина, а его чрезмерная выработка связана с различными заболеваниями, включая наследственную гиперхолестеринемию, наследственный дефектный АроВ и наследственную комбинированную гиперхолестеринемию (Kane and Havel, The Metabolic and Molecular Bases of Inherited Diseases, 2001, 8-е издание, 2717-2751). Нарушения метаболизма АроВ, которые соответствуют повышенному риску CHD, также наблюдаются при диабете и ожирении (Grundy, Am. J. Cardiol., 1998, 81, 18B-25B; Chan et al., Diabetes, 2002, 51, 2377- 2386; Chan et al., Metabolism, 2002, 51, 1041-1046). Помимо этого, генетические исследования на мышах продемонстрировали корреляцию между повышенными уровнями аполипопротеина В, повышенными уровнями холестерина и атеросклерозом (Kim and Young, J. Lipid Res., 1998, 39, 703-723; Nishina et al., J. Lipid Res., 1990, 31, 859-869).
При изучении субъектов с наследственной гипобеталипопротеинемией (FHBL) было установлено, что у указанных субъектов наблюдается пониженные уровни аполипопротеина В в сыворотке, пониженные уровни ЛНП-холестерина в сыворотке и пониженная частота ишемической болезни сердца (Schonfeld et al., J. Lipid Res., 2003, 44, 878-883). Исследования на мышах показали, что у мышей, имеющих гетерозиготные дефициты аполипопротеина В, наблюдались пониженные уровни ЛНП-холестерина и аполипопротеина В, и, помимо этого, они были защищены от гиперхолестеринемии, вызванной пищей (Farese et al., Proc. Natl. Acad. Sci. USA, 1995, 92, 1774-1778).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В первом аспекте настоящее изобретение относится к способам, включающим в себя введение субъекту фармацевтической композиции, включающей в себя антисмысловой олигонуклеотид, комплементарный нуклеиновой кислоте, кодирующей человеческий аполипопротеин В-100, в которых введение включает в себя фазу индукции, во время которой вводят дозу антисмыслового олигонуклеотида в пределах 100-300 мг один раз в неделю в течение по меньшей мере 13 недель, с последующей поддерживающей фазой, во время которой вводят дозу антисмыслового олигонуклеотида в пределах 80-200 мг один раз в неделю или один раз в две недели в течение такого периода времени, который является необходимым, эффективным и/или переносимым.
В некоторых вариантах осуществления настоящего изобретения доза, которую вводят во время фазы индукции, составляет 100 мг, а доза, которую вводят во время поддерживающей фазы, составляет 200 мг один раз в неделю. В некоторых вариантах осуществления настоящего изобретения доза, которую вводят во время фазы индукции, составляет 200 мг, а доза, которую вводят во время поддерживающей фазы, составляет 300 мг один раз в неделю. В некоторых вариантах осуществления настоящего изобретения доза, которую вводят во время фазы индукции, составляет 100 мг, а доза, которую вводят во время поддерживающей фазы, составляет 200 мг один раз в неделю, и переносимость или эффективность антисмыслового олигонуклеотида оценивают во время или в конце периода индукции или его части один раз в неделю во время поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения доза, которую вводят во время фазы индукции, составляет 200 мг, а доза, которую вводят во время поддерживающей фазы, составляет 300 мг один раз в неделю, и переносимость или эффективность антисмыслового олигонуклеотида оценивают во время или в конце периода индукции или его части.
В некоторых вариантах осуществления настоящего изобретения доза, которую вводят во время фазы индукции, составляет 100 мг, а доза, которую вводят во время поддерживающей фазы, составляет 100 мг один раз в две недели, и переносимость или эффективность антисмыслового олигонуклеотида оценивают во время или в конце периода индукции или его части. В некоторых вариантах осуществления настоящего изобретения доза, которую вводят во время фазы индукции, составляет 200 мг, а доза, которую вводят во время поддерживающей фазы, составляет 200 мг один раз в две недели, и переносимость или эффективность антисмыслового олигонуклеотида оценивают во время или в конце периода индукции или его части. В некоторых вариантах осуществления настоящего изобретения доза, которую вводят во время фазы индукции, составляет от 100 мг до 200 мг, а доза, которую вводят во время поддерживающей фазы, составляет от 200 мг до 300 мг один раз в неделю.
В некоторых вариантах осуществления настоящего изобретения указанное введение включает в себя парентеральное введение. В некоторых вариантах осуществления настоящего изобретения указанное парентеральное введение включает в себя подкожное введение. В некоторых вариантах осуществления настоящего изобретения каждая индукционная доза и каждая поддерживающая доза включают в себя однократную инъекцию. В некоторых вариантах осуществления настоящего изобретения каждая индукционная доза и каждая поддерживающая доза включают в себя две или более инъекции. В некоторых вариантах осуществления настоящего изобретения способы дополнительно включают в себя оценку переносимости или эффективности антисмыслового олигонуклеотида во время или в конце периода индукции или его части. В некоторых вариантах осуществления настоящего изобретения оценивают переносимость и эффективность антисмыслового олигонуклеотида.
В некоторых вариантах осуществления настоящего изобретения переносимость антисмыслового олигонуклеотида оценивают путем мониторинга концентрации АроВ в плазме крови указанного субъекта. В некоторых вариантах осуществления настоящего изобретения переносимость антисмыслового олигонуклеотида оценивают путем мониторинга скорости уменьшения концентрации АроВ и концентрации АроВ в плазме крови указанного субъекта. В некоторых вариантах осуществления настоящего изобретения переносимость антисмыслового олигонуклеотида оценивают путем мониторинга концентраций АЛТ в печени субъекта. В некоторых вариантах осуществления настоящего изобретения переносимость антисмыслового олигонуклеотида оценивают путем мониторинга концентраций АНТ в печени субъекта. В некоторых вариантах осуществления настоящего изобретения переносимость антисмыслового олигонуклеотида оценивают путем мониторинга концентраций билирубина в плазме крови субъекта.
В некоторых вариантах осуществления настоящего изобретения скорость уменьшения концентрации АроВ более приблизительно 30 мг/дл в день указывает, что субъект не переносит введение антисмыслового олигонуклеотида. В некоторых вариантах осуществления настоящего изобретения концентрация АроВ менее приблизительно 60 мг/дл указывает, что субъект не переносит введение антисмыслового олигонуклеотида. В некоторых вариантах осуществления настоящего изобретения скорость уменьшения концентрации АроВ более приблизительно 30 мг/дл в день и концентрация АроВ менее приблизительно 60 мг/дл указывает, что субъект не переносит введение антисмыслового олигонуклеотида. В некоторых вариантах осуществления настоящего изобретения дозу антисмыслового олигонуклеотида уменьшают, после того, как было показано, что введение указанного антисмыслового олигонуклеотида не переносится. В некоторых вариантах осуществления настоящего изобретения частоту введения антисмыслового олигонуклеотида уменьшают, после того, как было показано, что введение указанного антисмыслового олигонуклеотида не переносится. В некоторых вариантах осуществления настоящего изобретения дозу антисмыслового олигонуклеотида увеличивают, после того, как было показано, что введение указанного антисмыслового олигонуклеотида переносится. В некоторых вариантах осуществления настоящего изобретения частоту введения антисмыслового олигонуклеотида увеличивают, после того, как было показано, что введение указанного антисмыслового олигонуклеотида переносится.
В некоторых вариантах осуществления настоящего изобретения эффективность антисмыслового олигонуклеотида оценивают путем мониторинга концентрации АроВ, ЛНП-Х, ЛОНП-Х, ЛПП-Х, не-ЛВП-Х, триглицеридов сыворотки, триглицеридов печени, ЛП(а), окси-ЛНП-Х или малых плотных частиц ЛНП в плазме крови указанного субъекта. В некоторых вариантах осуществления настоящего изобретения уменьшение концентрации АроВ, ЛНП-Х, ЛОНП-Х, ЛПП-Х, не-ЛВП-Х, триглицеридов сыворотки, триглицеридов печени, ЛП(а), окси-ЛНП-Х или малых плотных частиц ЛНП указывает, что антисмысловой олигонуклеотид является эффективным. В некоторых вариантах осуществления настоящего изобретения дозу антисмыслового олигонуклеотида уменьшают, после того, как было показано, что введение указанного антисмыслового олигонуклеотида является эффективным. В некоторых вариантах осуществления настоящего изобретения дозу антисмыслового олигонуклеотида увеличивают, после того, как было показано, что введение указанного антисмыслового олигонуклеотида не является эффективным. В некоторых вариантах осуществления настоящего изобретения частоту введения антисмыслового олигонуклеотида уменьшают, после того, как было показано, что введение указанного антисмыслового олигонуклеотида является эффективным. В некоторых вариантах осуществления настоящего изобретения частоту введения антисмыслового олигонуклеотида увеличивают, после того, как было показано, что введение указанного антисмыслового олигонуклеотида не является эффективным.
В некоторых вариантах осуществления настоящего изобретения указанный субъект имеет повышенный АроВ до указанного введения. В некоторых вариантах осуществления настоящего изобретения указанный субъект имеет повышенный холестерин до указанного введения. В некоторых вариантах осуществления настоящего изобретения указанный повышенный холестерин выбран из повышенного общего холестерина, повышенного ЛНП-холестерина, повышенного ЛОНП-холестерина, повышенного ЛПП-холестерина или повышенного не-ЛВП-холестерина до указанного введения. В некоторых вариантах осуществления настоящего изобретения указанный субъект имеет повышенный ЛП(а) до указанного введения. В некоторых вариантах осуществления настоящего изобретения указанный субъект имеет повышенные триглицериды сыворотки до указанного введения. В некоторых вариантах осуществления настоящего изобретения указанный субъект имеет повышенные триглицериды печени до указанного введения. В некоторых вариантах осуществления настоящего изобретения указанный субъект имеет повышенные уровни малых плотных частиц ЛНП до указанного введения.
В некоторых вариантах осуществления настоящего изобретения указанный субъект имеет гиперхолестеринемию. В некоторых вариантах осуществления настоящего изобретения указанный субъект имеет полигенную гиперхолестеринемию. В некоторых вариантах осуществления настоящего изобретения указанный субъект имеет наследственную гиперхолестеринемию. В некоторых вариантах осуществления настоящего изобретения указанный субъект имеет гомозиготную наследственную гиперхолестеринемию. В некоторых вариантах осуществления настоящего изобретения указанный субъект имеет гетерозиготную наследственную гиперхолестеринемию. В некоторых вариантах осуществления настоящего изобретения указанный субъект имеет смешанную дислипидемию. В некоторых вариантах осуществления настоящего изобретения указанный субъект имеет коронарную болезнь в анамнезе.
В некоторых вариантах осуществления настоящего изобретения указанный субъект имеет один или более факторов риска коронарной болезни. В некоторых вариантах осуществления настоящего изобретения указанный один или более факторов риска выбраны из возраста, курения, гипертензии, низкого уровня ЛВП-холестерина и семейного анамнеза ранней CHD. В некоторых вариантах осуществления настоящего изобретения указанный субъект имеет диабет II типа с дислипидемией. В некоторых вариантах осуществления настоящего изобретения указанного субъекта лечили статином. В некоторых вариантах осуществления настоящего изобретения у указанного субъекта лечение статином не достигло своей мишени - ЛНП-холестерина. В некоторых вариантах осуществления настоящего изобретения указанный субъект не выполнял рекомендованное лечение. В некоторых вариантах осуществления настоящего изобретения у указанного субъекта имели место побочные эффекты лечения статином. В некоторых вариантах осуществления настоящего изобретения указанный субъект имеет низкую активность рецепторов ЛНП. В некоторых вариантах осуществления настоящего изобретения у указанного субъекта лечение, понижающее уровень липидов, не достигло своей мишени - ЛНП-холестерина.
В некоторых вариантах осуществления настоящего изобретения указанная поддерживающая фаза включает в себя введение указанной фармацевтической композиции на протяжении всей жизни субъекта. В некоторых вариантах осуществления настоящего изобретения продолжительность указанной поддерживающей фазы составляет один год, 2 года, 3 года, 4 года, 5 лет, 6 лет, 7 лет, 8 лет, 9 лет, 10 лет, 11 лет, 12 лет, 13 лет, 14 лет, 15 лет, 16 лет, 17 лет, 18 лет, 19 лет или 20 лет. В некоторых вариантах осуществления настоящего изобретения продолжительность указанной поддерживающей фазы составляет от одной недели до двадцати лет.
В некоторых вариантах осуществления настоящего изобретения доза, которую вводят во время фазы индукции, составляет 100 мг. В некоторых вариантах осуществления настоящего изобретения доза, которую вводят во время фазы индукции, составляет 200 мг. В некоторых вариантах осуществления настоящего изобретения доза, которую вводят во время фазы индукции, составляет 300 мг. В некоторых вариантах осуществления настоящего изобретения доза, которую вводят во время поддерживающей фазы, составляет 100 мг. В некоторых вариантах осуществления настоящего изобретения доза, которую вводят во время поддерживающей фазы, составляет 200 мг.
В некоторых вариантах осуществления настоящего изобретения указанное введение указанной фармацевтической композиции обеспечивает минимальные уровни антисмыслового олигонуклеотида в плазме крови от 5 до 100 нг/мл. В некоторых вариантах осуществления настоящего изобретения указанное введение указанной фармацевтической композиции обеспечивает минимальные уровни антисмыслового олигонуклеотида в плазме крови от 5 до 50 нг/мл. В некоторых вариантах осуществления настоящего изобретения указанное введение указанной фармацевтической композиции обеспечивает минимальные уровни антисмыслового олигонуклеотида в плазме крови от 10 до 40 нг/мл. В некоторых вариантах осуществления настоящего изобретения указанное введение указанной фармацевтической композиции обеспечивает минимальные уровни антисмыслового олигонуклеотида в плазме крови от 15 до 35 нг/мл. В некоторых вариантах осуществления настоящего изобретения указанное введение указанной фармацевтической композиции обеспечивает минимальные уровни антисмыслового олигонуклеотида в плазме крови от 20 до 30 нг/мл.
В некоторых вариантах осуществления настоящего изобретения указанное введение указанной фармацевтической композиции обеспечивает понижение АроВ по меньшей мере на 10%. В некоторых вариантах осуществления настоящего изобретения указанное понижение АроВ составляет по меньшей мере 15%, по меньшей мере 20%, по меньшей мере 25%, по меньшей мере 30%, по меньшей мере 35%, по меньшей мере 40%, по меньшей мере 45%, по меньшей мере 50%, по меньшей мере 55%, по меньшей мере 60%, по меньшей мере 65%, по меньшей мере 70%, по меньшей мере 75%, по меньшей мере 80%, по меньшей мере 85%, по меньшей мере 90%, по меньшей мере 95% или по меньшей мере 100%. В некоторых вариантах осуществления настоящего изобретения указанное понижение АроВ составляет от 10% до 80%, от 20% до 70%, от 30% до 60% или от 30% до 70%.
В некоторых вариантах осуществления настоящего изобретения указанное введение указанной фармацевтической композиции обеспечивает понижение ЛНП-холестерина по меньшей мере на 10%. В некоторых вариантах осуществления настоящего изобретения указанное понижение ЛНП-холестерина составляет по меньшей мере 15%, по меньшей мере 20%, по меньшей мере 25%, по меньшей мере 30%, по меньшей мере 35%, по меньшей мере 40%, по меньшей мере 45%, по меньшей мере 50%, по меньшей мере 55%, по меньшей мере 60%, по меньшей мере 65%, по меньшей мере 70%, по меньшей мере 75%, по меньшей мере 80%, по меньшей мере 85%, по меньшей мере 90%, по меньшей мере 95% или по меньшей мере 100%. В некоторых вариантах осуществления настоящего изобретения указанное введение указанной фармацевтической композиции обеспечивает понижение ЛОНП-холестерина по меньшей мере на 10%. В некоторых вариантах осуществления настоящего изобретения указанное понижение ЛОНП-холестерина составляет по меньшей мере 15%, по меньшей мере 20%, по меньшей мере 25%, по меньшей мере 30%, по меньшей мере 35%, по меньшей мере 40%, по меньшей мере 45%, по меньшей мере 50%, по меньшей мере 55%, по меньшей мере 60%, по меньшей мере 65%, по меньшей мере 70%, по меньшей мере 75%, по меньшей мере 80%, по меньшей мере 85%, по меньшей мере 90%, по меньшей мере 95% или по меньшей мере 100%.
В некоторых вариантах осуществления настоящего изобретения указанное введение указанной фармацевтической композиции обеспечивает понижение ЛП(а) по меньшей мере на 10%. В некоторых вариантах осуществления настоящего изобретения указанное понижение ЛП(а) составляет по меньшей мере 15%, по меньшей мере 20%, по меньшей мере 25%, по меньшей мере 30%, по меньшей мере 35%, по меньшей мере 40%, по меньшей мере 45%, по меньшей мере 50%, по меньшей мере 55%, по меньшей мере 60%, по меньшей мере 65%, по меньшей мере 70%, по меньшей мере 75%, по меньшей мере 80%, по меньшей мере 85%, по меньшей мере 90%, по меньшей мере 95% или по меньшей мере 100%. В некоторых вариантах осуществления настоящего изобретения указанное введение указанной фармацевтической композиции обеспечивает понижение малых частиц ЛНП по меньшей мере на 10%. В некоторых вариантах осуществления настоящего изобретения указанное понижение малых частиц ЛНП составляет по меньшей мере 15%, по меньшей мере 20%, по меньшей мере 25%, по меньшей мере 30%, по меньшей мере 35%, по меньшей мере 40%, по меньшей мере 45%, по меньшей мере 50%, по меньшей мере 55%, по меньшей мере 60%, по меньшей мере 65%, по меньшей мере 70%, по меньшей мере 75%, по меньшей мере 80%, по меньшей мере 85%, по меньшей мере 90%, по меньшей мере 95% или по меньшей мере 100%.
В некоторых вариантах осуществления настоящего изобретения указанное введение указанной фармацевтической композиции обеспечивает понижение не-ЛВП-холестерина по меньшей мере на 10%. В некоторых вариантах осуществления настоящего изобретения указанное понижение не-ЛВП-холестерина составляет по меньшей мере 15%, по меньшей мере 20%, по меньшей мере 25%, по меньшей мере 30%, по меньшей мере 35%, по меньшей мере 40%, по меньшей мере 45%, по меньшей мере 50%, по меньшей мере 55%, по меньшей мере 60%, по меньшей мере 65%, по меньшей мере 70%, по меньшей мере 75%, по меньшей мере 80%, по меньшей мере 85%, по меньшей мере 90%, по меньшей мере 95% или по меньшей мере 100%.
В некоторых вариантах осуществления настоящего изобретения указанное введение указанной фармацевтической композиции обеспечивает уменьшение риска коронарной болезни у субъекта. В некоторых вариантах осуществления настоящего изобретения указанное введение указанной фармацевтической композиции замедляет или останавливает прогрессирование атеросклероза у субъекта. В некоторых вариантах осуществления настоящего изобретения указанное введение указанной фармацевтической композиции уменьшает риск развития атеросклероза у субъекта. В некоторых вариантах осуществления настоящего изобретения указанное введение указанной фармацевтической композиции обеспечивает улучшение исхода сердечно-сосудистой патологии у субъекта. В некоторых вариантах осуществления настоящего изобретения указанное улучшение исхода сердечно-сосудистой патологии представляет собой уменьшение риска больших сердечно-сосудистых неблагоприятных событий у субъекта. В некоторых вариантах осуществления настоящего изобретения указанное улучшение исхода сердечно-сосудистой патологии представляет собой улучшение толщины интимальной средней оболочки сонных артерий. В некоторых вариантах осуществления настоящего изобретения указанное улучшение исхода сердечно-сосудистой патологии представляет собой улучшение толщины атеромы. В некоторых вариантах осуществления настоящего изобретения указанное улучшение исхода сердечно-сосудистой патологии представляет собой повышение ЛВП-холестерина.
В некоторых вариантах осуществления настоящего изобретения указанное введение обеспечивает понижение уровня липидов. В некоторых вариантах осуществления настоящего изобретения указанное введение обеспечивает понижение ЛНП-холестерина, триглицеридов или малых частиц ЛНП-холестерина или их комбинаций. В некоторых вариантах осуществления настоящего изобретения указанное введение обеспечивает улучшенное соотношение ЛНП/ЛВП. В некоторых вариантах осуществления настоящего изобретения указанное введение обеспечивает повышение уровня ЛВП-холестерина по меньшей мере на 10%. В некоторых вариантах осуществления настоящего изобретения указанное повышение уровня ЛВП-холестерина составляет по меньшей мере 15%, по меньшей мере 20%, по меньшей мере 25%, по меньшей мере 30%, по меньшей мере 35%, по меньшей мере 40%, по меньшей мере 45%, по меньшей мере 50%, по меньшей мере 55%, по меньшей мере 60%, по меньшей мере 65%, по меньшей мере 70%, по меньшей мере 75%, по меньшей мере 80%, по меньшей мере 85%, по меньшей мере 90%, по меньшей мере 95% или по меньшей мере 100%. В некоторых вариантах осуществления настоящего изобретения указанное введение указанной фармацевтической композиции обеспечивает понижение уровня триглицеридов печени по меньшей мере на 10%.
В некоторых вариантах осуществления настоящего изобретения указанное понижение уровня триглицеридов печени составляет по меньшей мере 15%, по меньшей мере 20%, по меньшей мере 25%, по меньшей мере 30%, по меньшей мере 35%, по меньшей мере 40%, по меньшей мере 45%, по меньшей мере 50%, по меньшей мере 55%, по меньшей мере 60%, по меньшей мере 65%, по меньшей мере 70%, по меньшей мере 75%, по меньшей мере 80%, по меньшей мере 85%, по меньшей мере 90%, по меньшей мере 95% или по меньшей мере 100%. В некоторых вариантах осуществления настоящего изобретения указанное введение указанной фармацевтической композиции обеспечивает понижение уровня сложного эфира холестерина печени по меньшей мере на 10%. В некоторых вариантах осуществления настоящего изобретения указанное понижение уровня сложного эфира холестерина печени составляет по меньшей мере 15%, по меньшей мере 20%, по меньшей мере 25%, по меньшей мере 30%, по меньшей мере 35%, по меньшей мере 40%, по меньшей мере 45%, по меньшей мере 50%, по меньшей мере 55%, по меньшей мере 60%, по меньшей мере 65%, по меньшей мере 70%, по меньшей мере 75%, по меньшей мере 80%, по меньшей мере 85%, по меньшей мере 90%, по меньшей мере 95% или по меньшей мере 100%.
В некоторых вариантах осуществления настоящего изобретения способы дополнительно включают в себя совместное введение указанной фармацевтической композиции и по меньшей мере одного дополнительного лекарственного средства. В некоторых вариантах осуществления настоящего изобретения указанное совместное введение является одновременным. В некоторых вариантах осуществления настоящего изобретения указанную фармацевтическую композицию вводят до введения указанного дополнительного лекарственного средства. Способ по п. 100, в котором указанную фармацевтическую композицию вводят после введения указанного дополнительного лекарственного средства. В некоторых вариантах осуществления настоящего изобретения интервал между введением указанной фармацевтической композиции и указанного дополнительного лекарственного средства составляет приблизительно один час, приблизительно 2 часа, приблизительно 3 часа, приблизительно 4 часа, приблизительно 5 часов, приблизительно 6 часов, приблизительно 7 часов, приблизительно 8 часов, приблизительно 9 часов, приблизительно 10 часов, приблизительно 11 часов или приблизительно 12 часов. В некоторых вариантах осуществления настоящего изобретения интервал между введением указанной фармацевтической композиции и указанного дополнительного лекарственного средства составляет приблизительно 1 день, приблизительно 1 неделю, приблизительно 2 недели, приблизительно 3 недели, приблизительно 4 недели, приблизительно 5 недель, приблизительно 6 недель, приблизительно 7 недель, приблизительно 8 недель, приблизительно 9 недель, приблизительно 10 недель, приблизительно 11 недель или приблизительно 12 недель. В некоторых вариантах осуществления настоящего изобретения интервал между введением указанной фармацевтической композиции и указанного дополнительного лекарственного средства составляет приблизительно 1 месяц, приблизительно 2 месяца, приблизительно 3 месяца, приблизительно 4 месяца, приблизительно 5 месяцев или приблизительно 6 месяцев.
В некоторых вариантах осуществления настоящего изобретения способ дополнительно включает в себя введение одного дополнительного лекарственного средства. В некоторых вариантах осуществления настоящего изобретения способ дополнительно включает в себя введение 2 или более дополнительных лекарственных средств. В некоторых вариантах осуществления настоящего изобретения указанное дополнительное лекарственное средство представляет собой лекарственное средство, понижающее уровень липидов. В некоторых вариантах осуществления настоящего изобретения указанное дополнительное средство лечения, понижающее уровень липидов, представляет собой изменение образа жизни в терапевтических целях. В некоторых вариантах осуществления настоящего изобретения указанное дополнительное лекарственное средство, понижающее уровень липидов, представляет собой ингибитор HMG-CoA-редуктазы. В некоторых вариантах осуществления настоящего изобретения ингибитор HMG-CoA-редуктазы выбран из аторвастатина, розувастатина или симвастатина. В некоторых вариантах осуществления настоящего изобретения указанное дополнительное лекарственное средство, понижающее уровень липидов, представляет собой ингибитор всасывания холестерина. В некоторых вариантах осуществления настоящего изобретения ингибитор всасывания холестерина представляет собой эзетимиб. В некоторых вариантах осуществления настоящего изобретения указанные 2 или более дополнительных лекарственных средств включают в себя ингибитор HMG-CoA-редуктазы и ингибитор всасывания холестерина. В некоторых вариантах осуществления настоящего изобретения ингибитор HMG-CoA-редуктазы представляет собой симвастатин, а указанный ингибитор всасывания холестерина представляет собой эзетимиб. В некоторых вариантах осуществления настоящего изобретения указанное дополнительное средство лечения, понижающее уровень липидов, представляет собой аферез ЛНП. В некоторых вариантах осуществления настоящего изобретения указанное введение указанного дополнительного лекарственного средства включает в себя внутривенное лечение. В некоторых вариантах осуществления настоящего изобретения указанное дополнительное лекарственное средство, понижающее уровень липидов, представляет собой ингибитор МТР.
В некоторых вариантах осуществления настоящего изобретения указанная фармацевтическая композиция включает в себя фармацевтически приемлемый наполнитель. В некоторых вариантах осуществления настоящего изобретения указанный фармацевтически приемлемый наполнитель представляет собой физиологический раствор. В некоторых вариантах осуществления настоящего изобретения дозу антисмыслового олигонуклеотида вводят в концентрации приблизительно 50 мг/мл, приблизительно 75 мг/мл, приблизительно 100 мг/мл, приблизительно 125 мг/мл, приблизительно 150 мг/мл, приблизительно 175 мг/мл, приблизительно 200 мг/мл, приблизительно 225 мг/мл или приблизительно 250 мг/мл.
В некоторых вариантах осуществления настоящего изобретения антисмысловой олигонуклеотид включает в себя по меньшей мере одну модифицированную сахарную часть. В некоторых вариантах осуществления настоящего изобретения модифицированная сахарная часть включает в себя 2'-метоксиэтильную сахарную часть. В некоторых вариантах осуществления настоящего изобретения модифицированная сахарная часть включает в себя сахарную часть бициклической нуклеиновой кислоты.
В некоторых вариантах осуществления настоящего изобретения антисмысловой олигонуклеотид включает в себя 2'-дезоксинуклеотидный гэп-сегмент, расположенный между сегментами-ветвями, в котором каждый нуклеотид сегментов-ветвей включает в себя модифицированную сахарную часть. В некоторых вариантах осуществления настоящего изобретения каждый нуклеотид сегмента-ветви включает в себя 2'-О-метоксиэтильную сахарную часть. В некоторых вариантах осуществления настоящего изобретения каждый нуклеотид сегмента-ветви включает в себя сахарную часть бициклической нуклеиновой кислоты. В некоторых вариантах осуществления настоящего изобретения гэп-сегмент включает в себя десять нуклеотидов, а каждый сегмент-ветвь включает в себя пять нуклеотидов.
В некоторых вариантах осуществления настоящего изобретения по меньшей мере одна межнуклеозидная связь представляет собой фосфортиоатную межнуклеозидную связь. В некоторых вариантах осуществления настоящего изобретения каждая межнуклеозидная связь представляет собой фосфортиоатную межнуклеозидную связь.
В некоторых вариантах осуществления настоящего изобретения по меньшей мере один цитозин представляет собой 5-метилцитозин. В некоторых вариантах осуществления настоящего изобретения каждый цитозин представляет собой 5-метилцитозин.
В некоторых вариантах осуществления настоящего изобретения антисмысловой олигонуклеотид является по меньшей мере на 90% комплементарным нуклеиновой кислоте, кодирующей человеческий АроВ. В некоторых вариантах осуществления настоящего изобретения антисмысловой олигонуклеотид является по меньшей мере на 95% комплементарным нуклеиновой кислоте, кодирующей человеческий АроВ. В некоторых вариантах осуществления настоящего изобретения антисмысловой олигонуклеотид является на 100% комплементарным нуклеиновой кислоте, кодирующей человеческий АроВ.
В некоторых вариантах осуществления настоящего изобретения нуклеиновая кислота, кодирующая человеческий АроВ, включает в себя последовательность, идентифицированную инвентарным номером NM_000384.1.
В некоторых вариантах осуществления настоящего изобретения антисмысловой олигонуклеотид включает в себя от 12 до 30 нуклеотидов. В некоторых вариантах осуществления настоящего изобретения антисмысловой олигонуклеотид включает в себя от 15 до 25 нуклеотидов. В некоторых вариантах осуществления настоящего изобретения антисмысловой олигонуклеотид включает в себя от 17 до 23 нуклеотидов. В некоторых вариантах осуществления настоящего изобретения антисмысловой олигонуклеотид включает в себя от 18 до 22 нуклеотидов. В некоторых вариантах осуществления настоящего изобретения антисмысловой олигонуклеотид включает в себя от 19 до 21 нуклеотида. В некоторых вариантах осуществления настоящего изобретения антисмысловой олигонуклеотид включает в себя 20 нуклеотидов.
В некоторых вариантах осуществления настоящего изобретения антисмысловой олигонуклеотид представляет собой ISIS 301012.
Некоторые варианты осуществления настоящего изобретения относятся к способам, включающим в себя введение субъекту фармацевтической композиции, включающей в себя антисмысловой олигонуклеотид, комплементарный нуклеиновой кислоте, кодирующей человеческий АроВ, в которых введение включает в себя фазу индукции, включающую в себя по меньшей мере одну индукционную дозу, и поддерживающую фазу, включающую в себя по меньшей мере одну поддерживающую дозу, в которых продолжительность фазы индукции превышает пять недель.
Некоторые варианты осуществления настоящего изобретения относятся к способам, включающим в себя введение субъекту фармацевтической композиции, включающей в себя антисмысловой олигонуклеотид, комплементарный нуклеиновой кислоте, кодирующей человеческий АроВ, в которых введение включает в себя фазу индукции, включающую в себя по меньшей мере одну индукционную дозу, и поддерживающую фазу, включающую в себя по меньшей мере одну поддерживающую дозу, в которых индукционная доза меньше поддерживающей дозы.
Некоторые варианты осуществления настоящего изобретения относятся к способам, включающим в себя введение субъекту фармацевтической композиции, включающей в себя антисмысловой олигонуклеотид, комплементарный нуклеиновой кислоте, кодирующей человеческий АроВ, в которых введение включает в себя фазу индукции, включающую в себя по меньшей мере одну индукционную дозу.
Некоторые варианты осуществления настоящего изобретения относятся к способам, включающим в себя введение субъекту, имеющему наследственную гиперхолестеринемию, фармацевтической композиции, включающей в себя антисмысловой олигонуклеотид, комплементарный нуклеиновой кислоте, кодирующей человеческий АроВ, в которых введение включает в себя фазу индукции, включающую в себя по меньшей мере одну индукционную дозу, и поддерживающую фазу, включающую в себя по меньшей мере одну поддерживающую дозу, в которых продолжительность фазы индукции составляет по меньшей мере 8 недель.
Некоторые варианты осуществления настоящего изобретения относятся к способам, включающим в себя введение субъекту фармацевтической композиции, включающей в себя антисмысловой олигонуклеотид, комплементарный нуклеиновой кислоте, кодирующей человеческий аполипопротеин В-100, в которых введение включает в себя поддерживающую фазу, включающую в себя по меньшей мере одну поддерживающую дозу.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Следует понимать, что предыдущее общее описание и последующее подробное описание служат для примера и объяснения и не ограничивают изобретение, как оно заявлено. В настоящем документе использование единственного числа включает в себя множественное число, если особо не указано иное. В настоящем документе используемое «или» означает «и/или», если не указано иное. Помимо этого, термин «включающий в себя», а также другие формы, такие как «включает в себя» и «включавший в себя», не является ограничивающим. Также такие термины как «элемент» или «компонент» охватывают как элементы и компоненты, включающие в себя одну единицу, так и элементы и компоненты, включающие в себя более одной субъединицы, если особо не указано иное.
Заголовки разделов, используемые в настоящем документе, служат лишь организационным целям и не должны истолковываться как ограничивающие описанный предмет изобретения. Все документы, или части документов процитированные в настоящей заявке, включая, без ограничения, патенты, патентные заявки, статьи, книги и монографии, целиком включены в настоящий документ специально в качестве ссылок для любых целей. Патентные заявки США №№ 10/712 795 и 10/200 710 целиком включены в настоящий документ специально в качестве ссылок для любых целей.
А. Определения
Если не представлены особые определения, номенклатура, используемая в связи с процедурами и методиками аналитической химии, синтетической органической химии и лекарственной и фармацевтической химии, описанными в настоящем документе, представляет собой номенклатуру, которая хорошо известна и широко используется специалистами. Стандартные методики могут использоваться для химического синтеза, химического анализа, изготовления и доставки лекарственных средств и лечения субъектов. Указанные методики и процедуры можно найти, например, в «Remington's Pharmaceutical Sciences», Mack Publishing Co., Easton, Pa., 18-е издание, 1990 г. и в других важных ссылках по изготовлению и доставке лекарственных средств, которые включены в настоящий документ в качестве ссылок для любых целей.
Используемый в настоящем документе термин «фармацевтическая композиция» означает смесь веществ, подходящих для введения субъекту. Например, фармацевтическая композиция может включать в себя антисмысловой олигонуклеотид и стерильный водный раствор.
Используемый в настоящем документе термин «антисмысловой олигонуклеотид» означает одноцепочечный олигонуклеотид, имеющий последовательность нуклеотидных оснований, которая позволяет гибридизацию с соответствующей областью нуклеиновой кислоты-мишени. Указанный антисмысловой олигонуклеотид «нацелен» на нуклеиновую кислоту.
Используемый в настоящем документе термин «комплементарность» означает способность образовывать пары между нуклеотидными основаниями первой нуклеиновой кислоты и второй нуклеиновой кислоты.
Используемый в настоящем документе термин «полностью комплементарный» означает, что каждое нуклеотидное основание способно к точному спариванию с соответствующими нуклеотидными основаниями нуклеиновой кислоты-мишени.
Используемый в настоящем документе термин «антисмысловое ингибирование» означает уменьшение уровней нуклеиновой кислоты-мишени в присутствии олигонуклеотида, комплементарного нуклеиновой кислоте-мишени, по сравнению с уровнями нуклеиновой кислоты-мишени в отсутствии олигонуклеотида.
Используемые в настоящем документе термины «нуклеиновая кислота-мишень», «РНК-мишень», «транскрипт РНК-мишени» означают любую нуклеиновую кислоту, способную представлять собой мишень для антисмысловых олигонуклеотидов. Используемые в настоящем документе термины «нуклеиновая кислота-мишень АроВ» и «нуклеиновая кислота, кодирующая АроВ» охватывают нуклеиновую кислоту, включая, например, ДНК (включая, например, кДНК), РНК (включая, например, пре-мРНК и мРНК), транскрибированную с ДНК, кодирующей АроВ, а также кДНК, полученную с использованием указанной РНК. В одном варианте осуществления настоящего изобретения нуклеиновая кислота-мишень АроВ представляет собой последовательность в GENBANK® под номером NM_000384.1, впервые размещенная в GENBANK® 24 марта 1999 г.
Используемый в настоящем документе термин «нуклеиновая кислота, кодирующая человеческий АроВ», означает ДНК, кодирующую АроВ, или РНК, транскрибированную с ДНК, кодирующей АроВ.
Используемый в настоящем документе термин «введение» означает доставку фармацевтического агента в организм субъекта и включает в себя, без ограничения, введение, осуществляемое медицинским работником, или самостоятельное введение.
Используемый в настоящем документе термин «субъект» означает человека или животное, не являющееся человеком, которого выбрали для лечения.
Используемый в настоящем документе термин «фаза индукции» означает фазу дозирования лекарственного средства, во время которой начинают введение и достигают устойчивых концентраций активного фармацевтического агента в ткани-мишени. Например, фаза индукции представляет собой фазу дозирования, во время которой достигают устойчивых концентраций антисмыслового олигонуклеотида в печени.
Используемый в настоящем документе термин «поддерживающая фаза» означает фазу дозирования после того как были достигнуты устойчивые концентрации лекарственного средства.
Используемый в настоящем документе термин «продолжительность» означает период времени, в течение которого активность или событие продолжается. Например, продолжительность фазы индукции представляет собой период времени, в течение которого вводят индукционные дозы. Например, продолжительность поддерживающей фазы представляет собой период времени, в течение которого вводят поддерживающие дозы.
Используемый в настоящем документе термин «парентеральное введение» означает введение посредством инъекции или инфузии. Парентеральное введение включает в себя, без ограничения, подкожное введение, внутривенное введение или внутримышечное введение.
Используемый в настоящем документе термин «подкожное введение» означает введение непосредственно под кожу. Используемый в настоящем документе термин «внутривенное введение» означает введение в вену.
Используемый в настоящем документе термин «поддерживающая доза» означает дозу, которую вводят одним введением во время поддерживающей фазы. Используемый в настоящем документе термин «индукционная доза» означает дозу, которую вводят одним введением во время фазы индукции.
Используемый в настоящем документе термин «доза» означает точно определенное количество фармацевтического агента, которое обеспечивается одним введением. В некоторых вариантах осуществления настоящего изобретения дозу можно вводить двумя или более болюсами, таблетками или инъекциями. Например, в некоторых вариантах осуществления настоящего изобретения, в которых желательно подкожное введение, желательная доза требует такого объема, который плохо подходит для одной инъекции. В указанных вариантах осуществления настоящего изобретения можно использовать две или более инъекций для достижения желательной дозы. В указанных вариантах осуществления настоящего изобретения дозу можно вводить двумя или более инъекциями для минимизации реакции в месте инъекции у субъекта.
Используемый в настоящем документе термин «дозированная единица» означает форму, в которой существует фармацевтический агент. В некоторых вариантах осуществления настоящего изобретения дозированная единица представляет собой флакон, содержащий лиофилизированный ISIS 301012. В некоторых вариантах осуществления настоящего изобретения дозированная единица представляет собой флакон, содержащий повторно растворенный ISIS 301012.
Используемый в настоящем документе термин «схема введения» означает комбинацию доз, разработанную для достижения одного или более желательных эффектов. В некоторых вариантах осуществления настоящего изобретения схема введения разработана для быстрого получения терапевтического эффекта. В некоторых вариантах осуществления настоящего изобретения схема введения разработана для уменьшения нежелательного побочного эффекта, например, гепатотоксичности.
Используемый в настоящем документе термин «фармацевтический агент» означает вещество, которое оказывает благоприятное лечебное действие, когда его вводят субъекту. Например, в некоторых вариантах осуществления настоящего изобретения антисмысловой олигонуклеотид, нацеленный на АроВ, является фармацевтическим агентом.
Используемый в настоящем документе термин «активный фармацевтический ингредиент» означает вещество в фармацевтической композиции, которое оказывает желательное действие. Например, ISIS 301012 представляет собой активный фармацевтический ингредиент в фармацевтической композиции, включающей в себя ISIS 301012 и физиологический раствор.
Используемый в настоящем документе термин «АроВ» означает белок аполипопротеин В-100. Концентрацию АроВ в сыворотке (или плазме) крови обычно выражают в мг/дл или нмоль/л. «АроВ сыворотки» и «АроВ плазмы» означают АроВ в сыворотке и плазме крови, соответственно.
Используемый в настоящем документе термин «холестерин липопротеинов низкой плотности (ЛНП-Х)» означает холестерин, связанный с частицами липопротеинов низкой плотности. Концентрацию ЛНП-Х в сыворотке (или плазме) крови обычно выражают в мг/дл или нмоль/л. «ЛНП-Х сыворотки» и «ЛНП-Х плазмы» означают ЛНП-Х в сыворотке и плазме крови, соответственно.
Используемый в настоящем документе термин «холестерин липопротеинов низкой плотности (ЛОНП-Х)» означает холестерин, связанный с частицами липопротеинов очень низкой плотности. Концентрацию ЛОНП-Х в сыворотке (или плазме) крови обычно выражают в мг/дл или нмоль/л. «ЛОНП-Х сыворотки» и «ЛОНП-Х плазмы» означают ЛОНП-Х в сыворотке и плазме крови, соответственно.
Используемый в настоящем документе термин «холестерин липопротеинов промежуточной плотности (ЛПП-Х)» означает холестерин, связанный с частицами липопротеинов промежуточной плотности. Концентрацию ЛПП-Х в сыворотке (или плазме) крови обычно выражают в мг/дл или нмоль/л. «ЛПП-Х сыворотки» и «ЛПП-Х плазмы» означают ЛПП-Х в сыворотке и плазме крови, соответственно.
Используемый в настоящем документе термин «холестерин не липопротеинов высокой плотности (не-ЛВП-Х)» означает холестерин, связанный с частицами липопротеинов, не являющихся липопротеинами высокой плотности, и включает в себя, без ограничения, ЛНП-Х, ЛОНП-Х и ЛПП-Х.
Используемый в настоящем документе термин «холестерин липопротеинов высокой плотности (ЛВП-Х)» означает холестерин, связанный с частицами липопротеинов высокой плотности. Концентрацию ЛВП-Х в сыворотке (или плазме) крови обычно выражают в мг/дл или нмоль/л. «ЛВП-Х сыворотки» и «ЛВП-Х плазмы» означают ЛПП-Х в сыворотке и плазме крови, соответственно.
Используемый в настоящем документе термин «общий холестерин» означает все типы холестерина, включая, без ограничения, ЛНП-Х, ЛВП-Х, ЛПП-Х и ЛОНП-Х. Концентрацию общего холестерина в сыворотке (или плазме) крови обычно выражают в мг/дл или нмоль/л.
Используемый в настоящем документе термин «липопротеин(а)» или «ЛП(а)» означает частицу липопротеина, которая состоит из ЛНП-Х, частицы аполипопротеина(а) и частицы аполипопротеина В-100.
Используемый в настоящем документе термин «АроА1» означает белок аполипопротеин-А1 в сыворотке. Концентрацию АроА1 в сыворотке крови обычно выражают в мг/дл или нмоль/л.
Используемый в настоящем документе термин «соотношение АроВ:АроА1» означает соотношение концентрации АроВ и концентрации АроА1.
Используемый в настоящем документе термин «АроВ-содержащий липопротеин» означает любой липопротеин, который имеет аполипопротеин В в качестве своего белкового компонента, и включает в себя ЛНП, ЛОНП, ЛПП и липопротеин(а).
Используемый в настоящем документе термин «малые плотные частицы ЛНП» означает подкласс частиц ЛНП, которые характеризуются более маленьким, более плотным размером, по сравнению с другими частицами ЛНП-холестерина.
Используемый в настоящем документе термин «триглицериды» означает липиды, которые представляют собой триэфиры глицерина. «Триглицериды сыворотки» означают триглицериды, присутствующие в сыворотке крови. «Триглицериды печени» означают триглицериды, присутствующие в печени.
Используемые в настоящем документе «липиды сыворотки» включают в себя, без ограничения, холестерин сыворотки и триглицериды сыворотки.
Используемый в настоящем документе термин «содержание эфиров холестерина» означает количество сложного эфира холестерина, присутствующего в ткани печени. В некоторых вариантах осуществления настоящего изобретения концентрацию сложного эфира холестерина в сыворотке крови используют в качестве показателя содержания эфиров холестерина в печени.
Используемый в настоящем документе термин «повышенный общий холестерин» означает общий холестерин у субъекта в концентрации, при которой рекомендуется понижающая уровень липидов терапия, и включает в себя, без ограничения, «повышенный ЛНП-Х», «повышенный ЛОНП-Х», «повышенный ЛПП-Х» и «повышенный не-ЛВП-Х». В некоторых вариантах осуществления настоящего изобретения концентрации общего холестерина менее 200 мг/дл, 200-239 мг/дл и более 240 мг/дл считаются желательными, погранично высокими и высокими, соответственно. В некоторых вариантах осуществления настоящего изобретения концентрации ЛНП-Х 100 мг/дл, 100-129 мг/дл, 130-159 мг/дл, 160-189 мг/дл и более 190 мг/дл считаются оптимальными, приблизительно оптимальными/выше оптимальных, погранично высокими, высокими и очень высокими, соответственно.
Используемый в настоящем документе термин «повышенные триглицериды» означает концентрации триглицеридов в сыворотке крови или печени, при которой рекомендуется понижающая уровень липидов терапия, и включает в себя «повышенные триглицериды сыворотки» «повышенные триглицериды печени». В некоторых вариантах осуществления настоящего изобретения концентрация триглицеридов сыворотки крови 150-199 мг/дл, 200-499 мг/дл и более или равная 500 мг/дл, считается погранично высокой, высокой и очень высокой, соответственно.
Используемый в настоящем документе термин «повышенные уровни малых плотных частиц ЛНП» означает концентрацию малых плотных частиц ЛНП у субъекта, при которой рекомендуется терапия, понижающая уровень липидов.
Используемый в настоящем документе термин «повышенные уровни липопротеина(а)» означает концентрацию липопротеина(а) у субъекта, при которой рекомендуется терапия, понижающая уровень липидов.
Используемый в настоящем документе термин «низкий ЛВП-Х» означает концентрацию ЛВП-Х у субъекта, при которой рекомендуется терапия, понижающая уровень липидов. В некоторых вариантах осуществления настоящего изобретения терапия, понижающая уровень липидов, рекомендуется, когда низкий ЛВП-Х сопровождается повышением не-ЛВП-Х и/или повышениями уровней триглицеридов. В некоторых вариантах осуществления настоящего изобретения концентрации ЛВП-Х менее 40 мг/дл считаются низкими. В некоторых вариантах осуществления настоящего изобретения концентрации ЛВП-Х менее 50 мг/дл считаются низкими.
Используемый в настоящем документе термин «Ctrough» или «минимальная концентрация в плазме» означает минимальную концентрацию в плазме крови, когда концентрации фармацевтического агента в плазме крови находятся в равновесии с концентрациями фармацевтического агента в ткани-мишени. Например, в некоторых вариантах осуществления настоящего изобретения минимальные концентрации ISIS 301012 в плазме достигаются, когда концентрации ISIS 301012 в плазме находятся в равновесии с концентрациями ISIS 301012 в тканях печени.
Используемый в настоящем документе термин «AUCtrough» или «минимальная AUC плазмы» означает площадь под кривой зависимости концентрации от времени на тот момент, когда концентрации фармацевтического агента в плазме крови находятся в равновесии с концентрациями фармацевтического агента в ткани-мишени.
Используемый в настоящем документе термин «соотношение ЛНП/ЛВП» означает отношение ЛНП-Х к ЛВП-Х.
Используемый в настоящем документе термин «окисленный ЛНП» или «окси-ЛНП-Х» означает ЛНП-Х, который является окисленным после воздействия свободных радикалов.
Используемый в настоящем документе термин «гиперхолестеринемия» означает состояние, которое характеризуется повышенными уровнями холестерина в сыворотке крови. В некоторых вариантах осуществления настоящего изобретения гиперхолестеринемия включает в себя, без ограничения, полигенную гиперхолестеринемию, гетерозиготную наследственную гиперхолестеринемию и гомозиготную наследственную гиперхолестеринемию.
Используемый в настоящем документе термин «гиперлипидемия» означает состояние, которое характеризуется повышенными уровнями липидов в сыворотке крови.
Используемый в настоящем документе термин «гипертриглицеридемия» означает состояние, которое характеризуется повышенными уровнями триглицеридов.
Используемый в настоящем документе термин «ненаследственная гиперхолестеринемия» означает состояние, которое характеризуется повышенными уровнями холестерина, которые не являются результатом мутации одного гена.
Используемый в настоящем документе термин «полигенная гиперхолестеринемия» означает состояние, которое характеризуется повышенными уровнями холестерина, которые являются результатом влияния ряда генетических факторов. В некоторых вариантах осуществления настоящего изобретения полигенная гиперхолестеринемия может обостряться приемом липидов с пищей.
Используемый в настоящем документе термин «наследственная гиперхолестеринемия (FH)» означает аутосомное доминантное метаболическое расстройство, которое характеризуется мутацией гена рецептора ЛНП (ЛНП-Р), выраженным повышением ЛНП-Х и преждевременным возникновением атеросклероза. Диагноз наследственной гиперхолестеринемии ставят, если субъект удовлетворяет одному или более следующих критериев: генетическому тестированию, подтверждающему 2 мутировавших гена рецептора ЛНП; генетическому тестированию, подтверждающему один мутировавший ген рецептора ЛНП; зафиксированный в истории болезни нелеченный уровень ЛПН-холестерина в сыворотке крови более 500 мг/дл; сухожильные и/или кожные ксантомы до достижения возраста 10 лет; или у обоих родителей имеется документально зафиксированный повышенный ЛПН-холестерин сыворотки до понижающего уровень липидов лечения, который соответствует гетерозиготной наследственной гиперхолестеринемии.
Используемый в настоящем документе термин «гомозиготная наследственная гиперхолестеринемия» или «HoFH» означает состояние, которое характеризуется мутацией как материнского, так и отцовского гена ЛНП-Р.
Используемый в настоящем документе термин «гетерозиготная наследственная гиперхолестеринемия» или «HeFH» означает состояние, которое характеризуется мутацией материнского или отцовского гена ЛНП-Р.
Используемый в настоящем документе термин «смешанная дислипидемия» означает состояние, которое характеризуется повышенными уровнями холестерина в сыворотке крови и повышенными уровнями триглицеридов в сыворотке крови.
Используемый в настоящем документе термин «диабетическая дислипидемия» или «диабет II типа с дислипидемией» означает состояние, которое характеризуется диабетом II типа, пониженными уровнями ЛВП-Х, повышенными уровнями триглицеридов сыворотки и повышенными уровнями малых плотных частиц ЛНП.
Используемый в настоящем документе термин «эквиваленты риска CHD» означает показатели клинического атеросклеротического заболевания, которое создает высокий риск развития коронарной болезни, и включает в себя клиническую коронарную болезнь, симптоматическую болезнь сонных артерий, болезнь периферических артерий и/или аневризму брюшного отдела аорты.
Используемые в настоящем документе «главные факторы риска», которые вносят свой вклад в высокий риск развития коронарной болезни, включают в себя курение, гипертензию, низкий уровень ЛВП-Х, семейный анамнез коронарной болезни и возраст.
Используемые в настоящем документе «факторы риска CHD» включают в себя эквиваленты риска CHD и главные факторы риска.
Используемый в настоящем документе термин «коронарная болезнь (CHD)» означает сужение малых кровеносных сосудов, которые снабжают сердце кровью и кислородом, которое часто является результатом атеросклероза.
Используемый в настоящем документе термин «пониженный риск коронарной болезни» означает уменьшение вероятности того, что у субъекта разовьется коронарная болезнь.
Используемый в настоящем документе термин «атеросклероз» означает затвердевание артерий, поражающее артерии большого и среднего размера, и характеризуется наличие жировых отложений. Жировые отложения называют «атеромами» или «бляшками», и они состоят, главным образом, из холестерина и других жиров, кальция и рубцовой ткани, и повреждают выстилку артерий.
Используемый в настоящем документе термин «анамнез коронарной болезни» означает эпизод клинически очевидной коронарной болезни в анамнезе субъекта или члена семьи субъекта.
Используемый в настоящем документе термин «раннее начало коронарной болезни» означает диагноз коронарной болезни до достижения возраста 50 лет.
Используемый в настоящем документе термин «субъект с непереносимостью статина» означает субъекта, у которого в результате лечения статином наблюдалось одно или более повышений уровня киназы, отклонения в анализах функции печени, мышечные боли или побочные эффекты со стороны центральной нервной системы.
Используемый в настоящем документе термин «эффективность» означает способность оказывать желаемое действие. Например, эффективность терапии, понижающей уровни липидов, может представлять собой уменьшение концентрации одного или более из ЛНП-Х, ЛОНП-Х, ЛПП-Х, не-ЛВП-Х, АроВ, липопротеина(а) или триглицеридов.
Используемый в настоящем документе термин «приемлемый профиль безопасности» означает паттерн побочных эффектов, который находится в клинически приемлемых пределах.
Используемый в настоящем документе термин «побочные эффекты» означает физиологические реакции, вызываемые лечением, которые отличаются от желаемых эффектов. В настоящем контексте побочные эффекты включают в себя, без ограничения, реакции в месте инъекции, отклонения в анализах функции печени, отклонения в функции почек, гепатотоксичность, токсичность в отношении почек, отклонения со стороны центральной нервной системы и миопатии. Например, повышенные уровни аминотрансферазы в сыворотке крови могут указывать на гепатотоксичность или отклонения в функции печени. Например, повышенные уровни билирубина могут указывать на гепатотоксичность или отклонения в функции печени.
Используемый в настоящем документе термин «реакция в месте инъекции» означает воспаление или ненормальную красноту кожи в месте инъекции у субъекта.
Используемый в настоящем документе термин «соблюдение пациентом режима и схемы лечения субъектом» означает его приверженность рекомендованному или назначенному лечению.
Используемый в настоящем документе термин «понижающая уровень липидов терапия» означает терапевтическую схему, назначаемую субъекту для понижения уровня одного или более липидов. В некоторых вариантах осуществления настоящего изобретения понижающую уровень липидов терапию осуществляют для понижения уровней одного или более из АроВ, общего холестерина, ЛНП-Х, ЛОНП-Х, ЛПП-Х, не-ЛВП-Х, триглицеридов, малых плотных частиц ЛНП и ЛП(а) у субъекта.
Используемый в настоящем документе термин «агент, понижающий уровень липидов» означает фармацевтический агент, назначаемый субъекту для достижения понижения уровней липидов. Например, в некоторых вариантах осуществления настоящего изобретения агент, понижающий уровень липидов, назначают для понижения у субъекта уровней одного или более из АроВ, ЛНП-Х, общего холестерина и триглицеридов.
Используемый в настоящем документе термин «ЛНП-Х-мишень» означает уровень ЛНП-Х, который является желательным после понижающей уровень липидов терапии.
Используемый в настоящем документе термин «соблюдение» означает приверженность субъекта рекомендованному лечению.
Используемый в настоящем документе термин «рекомендованное лечение» означает терапевтическую схему, рекомендованную медицинским работником для лечения, облегчения или профилактики заболевания.
Используемый в настоящем документе термин «низкая активность рецепторов ЛНП» означает активность рецепторов ЛНП, которая является недостаточно высокой, чтобы поддерживать клинически приемлемые уровни ЛНП-Х в кровотоке.
Используемый в настоящем документе термин «исход сердечно-сосудистой патологии» означает возникновение главных неблагоприятных сердечно-сосудистых событий.
Используемый в настоящем документе термин «улучшение исхода сердечно-сосудистой патологии» означает уменьшение распространенности главных неблагоприятных сердечно-сосудистых событий или их риска. Примеры главных неблагоприятных сердечно-сосудистых событий включают в себя, без ограничения, смерть, повторный инфаркт, инсульт, кардиогенный шок, отек легких, остановку сердца и дисритмию предсердий.
Используемый в настоящем документе термин «суррогатные маркеры исхода сердечно-сосудистой патологии» означает косвенные показатели сердечно-сосудистых событий или их риска. Например, суррогатные маркеры исхода сердечно-сосудистой патологии включают в себя толщину интимальной средней оболочки сонных артерий (CIMT). Другой пример суррогатного маркера исхода сердечно-сосудистой патологии включает в себя размер атером. Размер атером можно определить с помощью внутрисосудистого ультразвукового исследования (IVUS).
Используемый в настоящем документе термин «повышенный уровень ЛВП-Х» означает повышение уровня ЛВП-Х в сыворотке крови субъекта с течением времени.
Используемый в настоящем документе термин «понижение уровней липидов» означает уменьшений уровней одного или более липидов в сыворотке крови субъекта с течением времени.
Используемый в настоящем документе термин «совместное введение» означает введение двух или более фармацевтических агентов субъекту. Два или более фармацевтических агентов могут находиться в единой фармацевтической композиции или могут находиться в отдельных фармацевтических композициях. Каждый их двух или более фармацевтических агентов можно вводить одним и тем же или разными путями. Совместное введение включает в себя параллельное или последовательное введение.
Используемый в настоящем документе термин «изменение образа жизни в терапевтических целях» означает изменения питания и образа жизни, предназначенные для понижения уровня холестерина и уменьшения риска развития заболевания сердца, и включает в себя рекомендации по суммарному суточному потреблению с пищей калорий, общего жира, насыщенных жиров, полиненасыщенных жиров, мононенасыщенных жиров, углеводов, белка, холестерина, нерастворимых волокон, а также рекомендации, касающиеся физической активности.
Используемый в настоящем документе термин «статин» означает фармацевтический агент, который ингибирует активность HMG-CoA-редуктазы.
Используемый в настоящем документе термин «ингибитор HMG-CoA-редуктазы» означает фармацевтический агент, который действует посредством ингибирования фермента HMG-CoA-редуктазы.
Используемый в настоящем документе термин «ингибитор всасывания холестерина» означает фармацевтический агент, который ингибирует всасывание экзогенного холестерина, поступающего с пищей.
Используемый в настоящем документе термин «аферез ЛНП» означает форму афереза, посредством которой ЛНП-Х удаляется из крови. Обычно кровь субъекта отбирают из вены и разделяют на эритроциты и плазму. ЛПН-Х отфильтровывают из плазмы, а затем возвращают плазму и эритроциты субъекту.
Используемый в настоящем документе термин «ингибитор МТР» означает фармацевтический агент, который ингибирует фермент белок микросомального переноса триглицеридов.
Используемый в настоящем документе термин «нуклеозид» означает комбинацию основание-сахар.
Используемый в настоящем документе термин «нуклеотидное основание» означает гетероцилическую основную часть.
Используемый в настоящем документе термин «нуклеотид» означает нуклеозид, имеющий фосфатную группу, ковалентно связанную с сахарной частью нуклеозида.
Используемый в настоящем документе термин «межнуклеозидная связь» означает ковалентную связь между соседними нуклеозидами.
Используемый в настоящем документе термин «межнуклеозидная связь, наблюдающаяся в естественных условиях» означает фосфодиэфирную связь между 3' и 5'.
Используемый в настоящем документе термин «олигонуклеотид» означает полимер из связанных между собой нуклеотидов, каждый из которых может быть, независимо друг от друга, модифицированным или немодифицированным.
Используемый в настоящем документе термин «олигонуклеозид» означает олигонуклеотид, в котором межнуклеозидные связи не содержат атома фосфора.
Используемый в настоящем документе термин «немодифицированные» нуклеотидные основания означает пуриновые основания аденин (А) и гуанин (G) и пиримидиновые основания тимин (Т), цитозин (С) и урацил (U).
Используемый в настоящем документе термин «модифицированная сахарная часть» означает сахарную часть, имеющую замещения и/или любое измерение по сравнению с натуральной сахарной частью.
Используемый в настоящем документе термин «натуральная сахарная часть» означает сахарную часть, содержащуюся в ДНК (2'-H) или РНК (2'-OH).
Используемый в настоящем документе термин «2'-O-метоксиэтильная сахарная часть» означает 2'-замещенное фурозильное кольцо, имеющее 2'-O(CH2)2-OCH3 (2'-O-метоксиэтильную или 2'-MOE) замещающую группу.
Используемый в настоящем документе термин «2'-O-метоксиэтильный нуклеотид» означает нуклеотид, включающий в себя 2'-O-метоксиэтильную модифицированную сахарная часть.
Используемый в настоящем документе термин «сахарная часть бициклической нуклеиновой кислоты» означает фурозильное кольцо, модифицированное образованием мостика между двумя негеминальными кольцевыми атомами.
Используемый в настоящем документе термин «сегмент-ветвь» означает множество нуклеозидов, модифицированных таким образом, чтобы придавать олигонуклеотиду такие свойства как повышенная ингибирующая активность, повышенная аффинность связывания в отношении нуклеиновой кислоты-мишени или резистентность к разложению нуклеазами in vivo.
Используемый в настоящем документе термин «гэп-сегмент» означает множество нуклеотидов, которые поддерживают расщепление эндонуклеазой РНКазой Н.
Используемый в настоящем документе термин «ISIS 301012» означает понижающий уровень липидов агент, который представляет собой антисмысловой олигонуклеотид, имеющий последовательность «GCCTCAGTCTGCTTCGCACC», в которой каждая межнуклеозидная связь представляет собой фосфортиоатную межнуклеозидную связь, каждый цитозин представляет собой 5'-метилцитозин, нуклеотиды 6-15 представляют собой 2'-дезоксинуклеотиды, а нуклеотиды 1-5 и 16-20 представляют собой 2'-О-метоксиэтилнуклеотиды. ISIS 301012 является комплементарным нуклеотидам 3249-3268 последовательности, имеющей инвентарный номер NM_00384.1 в GENBANK.
Используемый в настоящем документе термин «метаболический синдром» означает группу липидных и нелипидных факторов сердечно-сосудистого риска метаболического происхождения. В некоторых вариантах осуществления настоящего изобретения метаболический синдром идентифицируется наличием любых трех из следующих факторов: окружностью талии более 102 см у мужчин или более 88 см у женщин; уровнем триглицеридов в сыворотке крови по меньшей мере 150 мг/дл; ЛВП-Х менее 40 мг/дл у мужчин или менее 50 мг/дл у женщин; артериальным давлением по меньшей мере 130/85 мм рт.ст. и уровнем глюкозы натощак по меньшей мере 110 мг/дл.
Используемый в настоящем документе термин «терапевтически эффективное количество» означает количество фармацевтического агента, которое оказывает благоприятное терапевтическое действие у субъекта. Например, терапевтически эффективное количество антисмыслового олигонуклеотида, комплементарного нуклеиновой кислоте, кодирующей человеческий ароВ, представляет собой такое количество, которое обеспечивает понижение уровней ЛНП-Х.
В. Проверенные фармацевтические композиции
В некоторых вариантах осуществления настоящее изобретение относится к фармацевтическим композициям, включающим в себя один или более различных олигонуклеотидов. В некоторых указанных вариантах осуществления настоящего изобретения указанные фармацевтические композиции включают в себя антисмысловой олигонуклеотид, комплементарный нуклеиновой кислоте, кодирующей человеческий ароВ. В некоторых вариантах осуществления настоящего изобретения указанные фармацевтические композиции включают в себя ISIS 301012. ISIS 301012 представляет собой фармацевтический агент, который после введения субъекту обеспечивает дозозависимое уменьшение АроВ, Аро-В-содержащих липопротеинов, включая, без ограничения, ЛНП-Х, триглицериды и ЛП(а). ISIS 301012 обеспечивает эффективность при введении в отдельности, а также обеспечивает эффективность (пропуск в тексте, далее, вероятно: «при введении совместно с другими фармацевтическими агентами» - прим. перев.)
В некоторых вариантах осуществления настоящего изобретения фармацевтические композиции включают в себя олигонуклеотид, обладающий комплементарностью к нуклеиновой кислоте-мишени. В некоторых указанных вариантах осуществления настоящего изобретения достаточное количество нуклеотидных оснований олигонуклеотида могут претерпевать водородное связывание с соответствующими нуклеотидными основаниями нуклеиновой кислоты-мишени, таким образом, что имеет место желаемый эффект. В некоторых указанных вариантах осуществления настоящего изобретения желаемый эффект представляет собой антисмысловое ингибирование нуклеиновой кислоты-мишени. В некоторых указанных вариантах осуществления настоящего изобретения желаемый эффект представляет собой антисмысловое ингибирование ароВ. В некоторых указанных вариантах осуществления настоящего изобретения по меньшей мере 70%, по меньшей мере 70%, по меньшей мере 75%, по меньшей мере 80%, по меньшей мере 85%, по меньшей мере 90%, по меньшей мере 95%, по меньшей мере 96%, по меньшей мере 97%, по меньшей мере 98%, по меньшей мере 99% нуклеотидных оснований олигонуклеотида могут претерпевать водородное связывание с соответствующим нуклеотидным основанием нуклеиновой кислоты-мишени. В некоторых указанных вариантах осуществления настоящего изобретения 100% нуклеотидных оснований олигонуклеотида могут претерпевать водородное связывание с соответствующим нуклеотидным основанием нуклеиновой кислоты-мишени. В указанных вариантах осуществления настоящего изобретения олигонуклеотиды являются полностью комплементарными (т.е., на 100% комплементарными) нуклеиновой кислоте-мишени. В некоторых указанных вариантах осуществления настоящего изобретения олигонуклеотиды являются полностью комплементарными нуклеиновой кислоте, кодирующей АроВ.
В некоторых вариантах осуществления настоящего изобретения нуклеиновая кислота, кодирующая человеческий АроВ, представляет собой мРНК АроВ. В некоторых вариантах осуществления настоящего изобретения указанная мРНК АроВ может включать в себя или может не включать в себя некоторые или все экзоны.
В некоторых вариантах осуществления настоящего изобретения длина олигонуклеотидов составляет от 12 до 30 нуклеотидов, т.е., олигонуклеотиды состоят из 12-30 связанных между собой нуклеотидов. В некоторых указанных вариантах осуществления настоящего изобретения длина олигонуклеотидов составляет от 15 до 25 нуклеотидов. В некоторых указанных вариантах осуществления настоящего изобретения длина олигонуклеотидов составляет от 17 до 23 нуклеотидов. В некоторых указанных вариантах осуществления настоящего изобретения длина олигонуклеотидов составляет от 18 до 22 нуклеотидов. В некоторых указанных вариантах осуществления настоящего изобретения длина олигонуклеотидов составляет от 19 до 21 нуклеотида. В некоторых указанных вариантах осуществления настоящего изобретения длина олигонуклеотидов составляет 20 нуклеотидов.
В некоторых вариантах осуществления настоящего изобретения олигонуклеотиды включают в себя процентную долю идентичности конкретной нуклеотидной последовательности. Олигонуклеотид обладает идентичностью по отношению к другому олигонуклеотиду, если нуклеотидные основания каждого олигонуклеотида обладают способностью образовывать пары в одним и тем же нуклеотидным основанием. В некоторых указанных вариантах осуществления настоящего изобретения олигонуклеотид обладает 90% идентичностью по отношению к другому олигонуклеотиду. В некоторых указанных вариантах осуществления настоящего изобретения олигонуклеотид обладает 95% идентичностью по отношению к другому олигонуклеотиду. В некоторых указанных вариантах осуществления настоящего изобретения олигонуклеотид обладает 100% идентичностью по отношению к другому олигонуклеотиду. В некоторых указанных вариантах осуществления настоящего изобретения идентичность наблюдается на протяжении всей длины олигонуклеотида. В некоторых указанных вариантах осуществления настоящего изобретения идентичность наблюдается на протяжении части олигонуклеотида.
В некоторых вариантах осуществления настоящего изобретения олигонуклеотиды содержат химические модификации. В некоторых указанных вариантах осуществления настоящего изобретения модификации олигонуклеотидов включают в себя замещения или изменения межнуклеотидных связей, сахарных частей или нуклеотидных оснований. Модифицированные олигонуклеотиды часто являются предпочтительными перед нативными формами в силу своих желательных свойств, таких как, например, увеличенный захват клетками, повышенная аффинность в отношении нуклеиновой кислоты-мишени, повышенная стабильность в присутствии нуклеаз или повышенная ингибирующая активность.
В некоторых вариантах осуществления настоящего изобретения химически модифицированные нуклеозиды также можно использовать для увеличения аффинности связывания укороченного или усеченного антисмыслового олигонуклеотида с его нуклеиновой кислотой-мишенью.
В некоторых вариантах осуществления настоящего изобретения олигонуклеотиды включают в себя одну или более модифицированных, т.е., не встречающихся в естественных условиях, межнуклеотидных связей. В некоторых указанных вариантах осуществления настоящего изобретения олигонуклеотиды, имеющие модифицированные межнуклеотидные связи, включают в себя межнуклеотидные связи, которые сохраняют атом фосфора, а также межнуклеотидные связи, которые не имеют атома фосфора. Репрезентативные фосфорсодержащие межнуклеотидные связи включают в себя, без ограничения, фосфодиэфиры, фосфотриэфиры, метилфосфонаты, фосфорамидат и фосфортиоаты. Способы получения фосфорсодержащих и нефосфорсодержащих связей хорошо известны специалистам.
В некоторых вариантах осуществления настоящего изобретения олигонуклеотиды включают в себя один или более нуклеотидов, включающих в себя модифицированные сахарные части. В некоторых указанных вариантах осуществления настоящего изобретения фуранозильное сахарное кольцо нуклеозида модифицировано с использованием ряда способов, включая, без ограничения, добавление замещающей группы, особенно на позиции 2'; образование мостиков между двумя негеминальными кольцевыми атомами с образованием бициклической нуклеиновой кислоты (BNA); и замещение атома или группы, такой как -S-, -N(R)- или -C(R1)(R2) на кислород кольца на позиции 4'. В некоторых указанных вариантах осуществления настоящего изобретения модифицированные сахара включают в себя, без ограничения, замещенные сахара, особенно 2'-замещенные сахара, имеющие 2'-F, 2'-OCH2 (2'-O-Me) или 2'-O(CH2)2-OCH3 (2'-O-метоксиэтильную или 2'-MOE) замещающую группу; или бициклические модифицированные сахара (BNA), имеющие 4'-(CH2)n-O-2' мостик, где n=1 или n=2. Способы получения модифицированных сахаров хорошо известны специалистам.
В некоторых вариантах осуществления настоящего изобретения олигонуклеотиды включают в себя один или более нуклеотидов, включающих в себя модифицированные нуклеотидные основания. В указанных вариантах осуществления настоящего изобретения нуклеотидные основания модифицированы таким образом, чтобы сохранялось водородное связывание. В некоторых указанных вариантах осуществления настоящего изобретения модифицированные нуклеотидные основания включают в себя, без ограничения, 5-метилцитозин (5-meC). Дополнительные модифицированные нуклеотидные основания включают в себя 5-гидроксиметилцитозин, ксантин, гипоксантин, 2-аминоаденин, 6-метильные и другие алкильные производные аденина и гуанина, 6-пропильные и другие алкильные производные аденина и гуанина, 2-тиоурацил, 2-тиотимин и 2-тиоцитозин, 5-галогенурацил и цитозин, 5-пропинил (-C≡C-CH3) урацил и цитозин и другие алкинильные производные пиримидиновых оснований, 6-азоурацил, цитозин и тимин, 5-урацил (псевдоурацил), 4-тиоурацил, 8-галоген-, 8-амино-, 8-тио-, 8-тиоалкил-, 8-гидроксил- и другие 8-замещенные аденины и гуанины, 5-галоген-, особенно, 5-бром-, 5-трифторметил- и другие 5-замещенные урацилы и цитозины, 7-метилгуанин и 7-метиладенин, 2-F-аденин, 2-аминоаденин, 8-азагуанин и 8-азааденин, 7-деазагуанин и 7-деазааденин и 3-деазагуанин и 3-деазааденин.
В некоторых вариантах осуществления настоящего изобретения фармацевтические композиции по настоящему изобретению включают в себя один или более олигонуклеотидов и один или более наполнителей. В некоторых указанных вариантах осуществления настоящего изобретения наполнители выбраны из воды, солевых растворов, спирта, полиэтиленгликолей, желатина, лактозы, амилазы, стеарата магния, талька, кремниевой кислоты, вязкого парафина, гидроксиметилцеллюлозы и поливинилпирролидона.
В некоторых вариантах осуществления настоящего изобретения фармацевтическую композицию по настоящему изобретению изготавливают с использованием известных технологий, включая, без ограничения, процессы смешивания, растворения, гранулирования, изготовления драже, растирания в порошок, эмульгирования, инкапсуляции, улавливания или таблетирования.
В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция по настоящему изобретению представляет собой жидкость (например, суспензию, эликсир и/или раствор). В некоторых указанных вариантах осуществления настоящего изобретения жидкую фармацевтическую композицию изготавливают с использованием известных специалистам ингредиентов, включая, без ограничения, воду, гликоли, масла, спирты, корригенты, консерванты и красители.
В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция по настоящему изобретению представляет собой твердое вещество (например, порошок, таблетку и/или капсулу). В некоторых указанных вариантах осуществления настоящего изобретения твердую фармацевтическую композицию, включающую в себя один или более олигонуклеотидов, изготавливают с использованием известных специалистам ингредиентов, включая, без ограничения, крахмалы, сахара, разбавители, гранулирующие агенты, смазывающие агенты, связующие агенты и разрыхлители.
В некоторых вариантах осуществления настоящего изобретения фармацевтическую композицию по настоящему изобретению изготавливают в виде депо-препарата. Указанные депо-препараты обычно являются более длительно действующими, чем препараты, не являющиеся депо. В некоторых вариантах осуществления настоящего изобретения указанные препараты вводят путем имплантации (например, подкожно или внутримышечно) или путем внутримышечной инъекции. В некоторых вариантах осуществления настоящего изобретения депо-препараты изготавливают с использованием подходящих полимерных или гидрофобных материалов (например, эмульсия в приемлемом масле) или ионообменных смол, или в виде слаборастворимых производных, например, в виде слаборастворимой соли.
В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция по настоящему изобретению включает в себя систему доставки. Примеры систем доставки включают в себя, без ограничения, липосомы и эмульсии. Некоторые системы доставки являются пригодными для изготовления определенных фармацевтических композиций, включая композиции, содержащие гидрофобные соединения. В некоторых вариантах осуществления настоящего изобретения используют проверенные органические растворители, такие как диметилсульфоксид.
В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция по настоящему изобретению включает в себя одну или более специфичных по отношению к ткани молекул, обеспечивающих доставку, подобранных таким образом, чтобы доставлять один или более фармацевтических агентов по настоящему изобретению в конкретные ткани или типы клеток. Например, в некоторых вариантах осуществления настоящего изобретения фармацевтические композиции включают в себя липосомы, покрытые тканевоспецифичным антителом.
В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция по настоящему изобретению включает в себя систему сорастворителей. Некоторые из указанных систем сорастворителей включают в себя, например, бензиловый спирт, неполярное поверхностно-активное вещество, смешивающийся с водой органический полимер и водную фазу. В некоторых вариантах осуществления настоящего изобретения указанные системы сорастворителей используются для гидрофобных соединений. Неограничивающим примером указанной системы сорастворителей является система сорастворителей VPD, которая представляет собой раствор абсолютного этанола, содержащий 3% масс./об. бензилового спирта, 8% масс./об. неполярного поверхностно-активного вещества полисорбата 80.ТМ и 65% масс./об. полиэтиленгликоля 300. Пропорции указанных систем сорастворителей могут значительно варьироваться, без значительного изменения характеристик их растворимости и токсичности. Помимо этого, идентичность компонентов сорастворителей может варьироваться: например, вместо полисорбата 80.ТМ можно использовать другие поверхностно-активные вещества; фракционный размер полиэтиленгликоля может варьироваться; другие биосовместимые полимеры могут заменять полиэтиленгликоль, например, поливинилпирролидон; и другие сахара или полисахариды могут заменять декстрозу.
В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция по настоящему изобретению включает в себя систему с замедленным высвобождением. Неограничивающим примером указанной системы с замедленным высвобождением является полупроницаемый матрикс из твердых гидрофобных полимеров. В некоторых вариантах осуществления настоящего изобретения система с замедленным высвобождением может, в зависимости от ее химической природы, высвобождать фармацевтические агенты в течение часов, дней, недель или месяцев.
В некоторых вариантах осуществления настоящего изобретения фармацевтическую композицию по настоящему изобретению изготавливают для перорального введения. В некоторых указанных вариантах осуществления настоящего изобретения фармацевтическую композицию изготавливают путем объединения одного или более олигонуклеотидов с одним или более фармацевтически приемлемых носителей. Использование некоторых из указанных носителей делает возможным изготовление фармацевтических композиций в виде таблеток, пилюль, драже, капсул, жидкостей, гелей, сиропов, взвесей, суспензий и т.п., для перорального введения субъекту. В некоторых вариантах осуществления настоящего изобретения фармацевтические композиции для перорального применения изготавливают путем смешивания олигонуклеотида и одного или более твердых наполнителей. Подходящие наполнители включают в себя, без ограничения, наполнители, такие как сахара, включая лактозу, сахарозу, маннит или сорбит; препараты целлюлозы, такие как, например, кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, трагакантовую камедь, метилцеллюлозу, гидроксипропилметилцеллюлозу, натрий-карбоксиметилцеллюлозу и/или поливинилпирролидон (PVP). В некоторых вариантах осуществления настоящего изобретения указанную смесь, необязательно, размалывают, и, необязательно, добавляют вспомогательные вещества. В некоторых вариантах осуществления настоящего изобретения изготавливают фармацевтические композиции для получения таблеток или ядер драже. В некоторых вариантах осуществления настоящего изобретения добавляют разрыхлители (например, сшитый поливинилпирролидон, агар или альгиновую кислоту или ее соль, такую как альгина натрия).
В некоторых вариантах осуществления настоящего изобретения на ядра драже наносят покрытия. В некоторых указанных вариантах осуществления настоящего изобретения можно использовать концентрированные растворы сахара, которые могут, необязательно, содержать аравийскую камедь, тальк, поливинилпирролидон, гель карбопол, полиэтиленгликоль и/или диоксид титана, растворы лаков и подходящие органические растворители или смеси растворителей. К покрытиям таблеток или драже можно добавлять красители или пигменты.
В некоторых вариантах осуществления настоящего изобретения фармацевтические композиции для перорального введения представляют собой капсулы плотной насадки, изготовленные из желатина. Некоторые из указанных капсул плотной насадки включают в себя один или более фармацевтических агентов по настоящему изобретению в смеси с одним или более наполнителей, таких как лактоза, связующих агентов, такие как крахмалы, и/или смазывающих агентов, таких как тальк или стеарат магния, и, необязательно, стабилизаторов. В некоторых вариантах осуществления настоящего изобретения фармацевтические композиции для перорального введения представляют собой мягкие, герметично запечатанные капсулы, изготовленные из желатина и пластификатора, такого как глицерин или сорбит. В некоторых мягких капсулах один или более фармацевтических агентов по настоящему изобретению растворены или суспендированы в подходящих жидкостях, таких как жирные масла, жидкий парафин или жидкие полиэтиленгликоли. Помимо этого, можно добавлять стабилизаторы.
В некоторых вариантах осуществления настоящего изобретения фармацевтические композиции изготавливают для буккального введения. Некоторые из указанных фармацевтических композиций представляют собой таблетки или лепешки, изготовленные в обычной манере.
В некоторых вариантах осуществления настоящего изобретения фармацевтические композиции изготавливают для введения путем инъекции (например, внутривенной, подкожной, внутримышечной и т.п.). В некоторых указанных вариантах осуществления настоящего изобретения фармацевтическая композиция включает в себя носитель и изготовлена в водном растворе, таком как вода или физиологически совместимые буферы, такие как раствор Хенкса, раствор Рингера или физиологический раствор. В некоторых вариантах осуществления настоящего изобретения включены другие ингредиенты (например, ингредиенты, которые способствуют растворимости или служат консервантами). В некоторых вариантах осуществления настоящего изобретения изготавливают суспензии для инъекций с использованием подходящих жидких носителей, суспендирующих агентов и т.п. Некоторые фармацевтические композиции для инъекций представлены в виде стандартных лекарственных форм, например, в ампулах или контейнерах, содержащих множество доз. Некоторые фармацевтические композиции для инъекций представляют собой суспензии, растворы или эмульсии в масляных или водных носителях и могут содержать вспомогательные агенты, такие как суспендирующие, стабилизирующие и/или диспергирующие агенты. Некоторые растворители, подходящие для использования в фармацевтических композициях для инъекций, включают в себя, без ограничения, липофильные растворители и жирные масла, такие как кунжутное масло, синтетические сложные эфиры жирных кислот, такие как этилолеат или триглицериды, и липосомы. Водные суспензии для инъекций могут содержать вещества, которые увеличивают вязкость суспензии, такие как натрий-карбоксиметилцеллюлоза, сорбит или декстран. Необязательно, указанные суспензии могут также содержать подходящие стабилизаторы или агенты, которые увеличивают растворимость фармацевтических агентов, чтобы сделать возможным изготовление высококонцентрированных растворов.
В некоторых вариантах осуществления настоящего изобретения фармацевтическую композицию изготавливают для введения через слизистые оболочки. В некоторых из указанных вариантов осуществления настоящего изобретения в композиции используют пенетранты, соответствующие барьеру, через который они должны проникать. Указанные пенетранты хорошо известны специалистам.
В некоторых вариантах осуществления настоящего изобретения фармацевтическую композицию изготавливают для введения путем ингаляции. Некоторые из указанных фармацевтических композиций для ингаляции изготавливают в форме аэрозольного спрея в упаковке под давлением или небулайзере. Некоторые из указанных фармацевтических композиций включают в себя пропеллент, например, дихлордифторметан, трихорфторметан, дихлортетрафторэтан, диоксид углерода или другой подходящий газ. В некоторых вариантах использования аэрозоля под давлением дозированную единицу можно определять с помощью клапана, который доставляет отмеренное количество. В некоторых вариантах осуществления настоящего изобретения можно изготавливать капсулы и картриджи для использования в ингаляторе или инсуффляторе. Некоторые из указанных композиций включают в себя порошкообразную смесь фармацевтического агента по настоящему изобретению и подходящую порошкообразную основу, такую как лактоза или крахмал.
В некоторых вариантах осуществления настоящего изобретения фармацевтическую композицию изготавливают для ректального введения, такую как суппозитории или ретенционные клизмы. Некоторые из указанных фармацевтических композиций включают в себя известные ингредиенты, такие как масло какао и/или другие глицериды.
В некоторых вариантах осуществления настоящего изобретения фармацевтическую композицию изготавливают для местного введения. Некоторые из указанных фармацевтических композиций включают в себя щадящие увлажняющие основы, такие как мази или кремы. Примеры подходящих мазевых основ включают в себя, без ограничения, вазелин, вазелин плюс летучие силиконы, ланолин и эмульсии типа вода в масле, такие как EucerinTM от компании Beiersdorf (Цинцинатти, Огайо). Примеры подходящих кремовых основ включают в себя, без ограничения, крем NiveaTM от компании Beiersdorf (Цинцинатти, Огайо), кольдкрем (USP), Purpose CreamTM от компании Johnson & Johnson (New Brunswick, N.J.), гидрофильную мазь (USP) и LubridermTM от компании Pfizer (Morris Plains, N.J.).
В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция по настоящему изобретению включает в себя олигонуклеотид в терапевтически эффективном количестве. В некоторых вариантах осуществления настоящего изобретения терапевтически эффективное количество является достаточным для профилактики, облегчения или улучшения симптомов заболевания или для продления жизни субъекта, подвергаемого лечению. Определение терапевтически эффективного количества находится в компетенции специалиста.
В некоторых вариантах осуществления настоящего изобретения один или более олигонуклеотидов по настоящему изобретению изготавливают в форме пролекарства. В некоторых вариантах осуществления настоящего изобретения после введения in vivo пролекарство подвергается химическому превращению в биологически, фармацевтически или терапевтически более активную форму олигонуклеотида. В некоторых вариантах осуществления настоящего изобретения пролекарства являются полезными, поскольку их легче вводить, чем соответствующую активную форму. Например, в некоторых случаях пролекарство может быть более биодоступным (например, при пероральном введении), чем соответствующая активная форма. В некоторых случаях пролекарство может обладать улучшенной растворимостью по сравнению с соответствующей активной формой. В некоторых вариантах осуществления настоящего изобретения пролекарства хуже растворяются в воде, чем соответствующая активная форма. В некоторых случаях указанные пролекарства обладают превосходной способностью проникать через клеточные мембраны, когда растворимость в воде ухудшает мобильность. В некоторых вариантах осуществления настоящего изобретения пролекарство представляет собой сложный эфир. В некоторых указанных вариантах осуществления настоящего изобретения сложный эфир после введения подвергается метаболическому гидролизу до карбоновой кислоты. В некоторых случаях соединение, соединение, содержащее карбоновую кислоту, является соответствующей активной формой. В некоторых вариантах осуществления настоящего изобретения пролекарство включает в себя короткий пептид (полиаминокислоту), связанный с кислотной группой. В некоторых указанных вариантах осуществления настоящего изобретения пептид расщепляется после введения с образованием соответствующей активной формы.
В некоторых вариантах осуществления настоящего изобретения пролекарство изготавливают путем модификации фармацевтически активного соединения таким образом, что активное соединение после введения in vivo восстанавливается. Пролекарство может быть разработано таким образом, чтобы изменять метаболическую стабильность или транспортные характеристики лекарственного средства, маскировать побочные эффекты или токсичность, улучшать вкус и запах лекарственного средства или изменять другие характеристики или свойства лекарственного средства. Обладая знаниями фармакодинамических процессов и метаболизма лекарственного средства in vivo, специалисты, поскольку фармацевтически активное соединение известно, могут разработать пролекарства соединения (см., например, Nogrady (1985) Medicinal Chemistry, A Biochemical Approach, Oxford University Press, New Jork, страницы 388-392).
В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция, включающая в себя один или более фармацевтических агентов по настоящему изобретению, является пригодной для лечения состояний или расстройств у млекопитающего и, в частности, у человека. Подходящие пути введения включают в себя, без ограничения, пероральный, ректальный, чресслизистый, интестинальный, энтеральный, местный, с использованием суппозитория, с использованием ингаляции, интратекальный, интравентрикулярный, интраперитонеальный, интраназальный, интраокулярный и парентеральный (например, внутривенный, внутримышечный, интрамедуллярный и подкожный). В некоторых вариантах осуществления настоящего изобретения фармацевтические интратекальные композиции вводят для достижения скорее местного, чем системного воздействия. На фармацевтические композиции можно инъецировать непосредственно в область желаемого эффекта (например, в области почки или сердца).
В некоторых вариантах осуществления настоящего изобретения фармацевтическую композицию по настоящему изобретению вводят в форме дозированной единицы (например, таблетки, капсулы, болюса и т.п.). В некоторых вариантах осуществления настоящего изобретения указанные фармацевтические композиции включают в себя олигонуклеотид в дозе, выбранной из 25 мг, 30 мг, 35 мг, 40 мг, 45 мг, 50 мг, 55 мг, 60 мг, 65 мг, 70 мг, 75 мг, 80 мг, 85 мг, 90 мг, 95 мг, 100 мг, 105 мг, 110 мг, 115 мг, 120 мг, 125 мг, 130 мг, 135 мг, 140 мг, 145 мг, 150 мг, 155 мг, 160 мг, 165 мг, 170 мг, 175 мг, 180 мг, 185 мг, 190 мг, 195 мг, 200 мг, 205 мг, 210 мг, 215 мг, 220 мг, 225 мг, 230 мг, 235 мг, 240 мг, 245 мг, 250 мг, 255 мг, 260 мг, 265 мг, 270 мг, 275 мг, 280 мг, 285 мг, 290 мг, 295 мг, 300 мг, 305 мг, 310 мг, 315 мг, 320 мг, 325 мг, 330 мг, 335 мг, 340 мг, 345 мг, 350 мг, 355 мг, 360 мг, 365 мг, 370 мг, 375 мг, 380 мг, 385 мг, 390 мг, 395 мг, 400 мг, 405 мг, 410 мг, 415 мг, 420 мг, 425 мг, 430 мг, 435 мг, 440 мг, 445 мг, 450 мг, 455 мг, 460 мг, 465 мг, 470 мг, 475 мг, 480 мг, 485 мг, 490 мг, 495 мг, 500 мг, 505 мг, 510 мг, 515 мг, 520 мг, 525 мг, 530 мг, 535 мг, 540 мг, 545 мг, 550 мг, 555 мг, 560 мг, 565 мг, 570 мг, 575 мг, 580 мг, 585 мг, 590 мг, 595 мг, 600 мг, 605 мг, 610 мг, 615 мг, 620 мг, 625 мг, 630 мг, 635 мг, 640 мг, 645 мг, 650 мг, 655 мг, 660 мг, 665 мг, 670 мг, 675 мг, 680 мг, 685 мг, 690 мг, 695 мг, 700 мг, 705 мг, 710 мг, 715 мг, 720 мг, 725 мг, 730 мг, 735 мг, 740 мг, 745 мг, 750 мг, 755 мг, 760 мг, 765 мг, 770 мг, 775 мг, 780 мг, 785 мг, 790 мг, 795 мг и 800 мг. В некоторых указанных вариантах осуществления настоящего изобретения фармацевтическая композиция по настоящему изобретению включает в себя дозу олигонуклеотида, выбранную из 25 мг, 50 мг, 75 мг, 100 мг, 150 мг, 200 мг, 250 мг, 300 мг, 350 мг, 400 мг, 500 мг, 600 мг, 700 мг и 800 мг. В некоторых указанных вариантах осуществления настоящего изобретения фармацевтическая композиция включает в себя дозу олигонуклеотида, выбранную из 50 мг, 100 мг, 150 мг, 200 мг, 250 мг, 300 мг и 400 мг.
В еще одном аспекте фармацевтический агент представляет собой стерильный лиофилизированный олигонуклеотид, который повторно растворяют с использованием подходящего разбавителя, например, стерильной воды для инъекций. Восстановленный продукт вводят в виде подкожной инъекции или в виде внутривенной инфузии после разбавления в физиологическом растворе. Лиофилизированный лекарственный продукт состоит из олигонуклеотида, который был приготовлен в воде для инъекций, с величиной рН, доведенной до 7,0-9,0 с использованием кислоты или основания во время приготовления, а затем лиофилизирован. Лиофилизированный олигонуклеотид может представлять собой 25-800 мг олигонуклеотида. Следует понимать, что в указанное количество входят 25, 50, 75, 100, 125, 150, 175, 200, 225, 250, 275, 300, 325, 350, 375, 425, 450, 475, 500, 525, 550, 575, 600, 625, 650, 675, 700, 725, 750, 775 и 800 мг лиофилизированного олигонуклеотида. Лиофилизированный лекарственный продукт может быть упакован в 2 мл флакон из прозрачного стекла типа I (обработанный сульфатом аммония), закупорен крышкой из бромбутиловой резины и запечатан дополнительным укупорочным средством из алюминиевой FLIP-OFF®. В одном варианте осуществления настоящего изобретения лиофилизированный фармацевтический агент включает в себя ISIS 301012.
Композиции по настоящему изобретению могут дополнительно содержать другие вспомогательные компоненты, которые обычно включают в фармацевтические композиции, в установленных специалистами количествах. Так, например, композиции могут содержать дополнительные, совместимые, фармацевтически активные материалы, такие как, например, противозудные агенты, вяжущие агенты, местные анестетики или противовоспалительные агенты, или могут содержать дополнительные материалы, подходящие для физического составления различных лекарственных форм композиций по настоящему изобретению, такие как красители, корригенты, консерванты, антиоксиданты, агенты, придающие непрозрачность, загустители и стабилизаторы. Однако, указанные материалы, будучи добавленными в композицию, не должны ненадлежащим образом мешать биологической активности компонентов композиций по настоящему изобретению. Композиции можно стерилизовать и, если это желательно, смешивать с вспомогательными агентами, например, смазывающими агентами, консервантами, стабилизаторами, увлажняющими агентами, эмульгаторами, солями для воздействия на осмотическое давление, буферами, красителями, корригентами и/или ароматическими веществами и т.п., которые не взаимодействуют неблагоприятным образом с олигонуклеотидом (олигонуклеотидами) композиции.
С. Некоторые схемы введения
В некоторых вариантах осуществления настоящего изобретения фармацевтические композиции вводят в соответствии со схемой введения. В некоторых указанных вариантах осуществления настоящего изобретения схема введения включает в себя фазу индукции и поддерживающую фазу.
В некоторых вариантах осуществления настоящего изобретения фаза индукции включает в себя одну, две, три, четыре, пять, шесть, семь, восемь, девять, десять, одиннадцать, двенадцать, тринадцать, четырнадцать, шестнадцать, семнадцать, восемнадцать, девятнадцать, двадцать или более двадцати доз.
В некоторых вариантах осуществления настоящего изобретения фаза индукции продолжается от одной недели до пяти месяцев, считая от введения первой дозы фазы индукции до введения первой дозы поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения фаза индукции продолжается от двух недель до пяти месяцев, считая от введения первой дозы фазы индукции до введения первой дозы поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения фаза индукции продолжается от трех недель до четырех месяцев, считая от введения первой дозы фазы индукции до введения первой дозы поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения фаза индукции продолжается от пяти недель до трех месяцев, считая от введения первой дозы фазы индукции до введения первой дозы поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения фаза индукции продолжается пять недель, считая от введения первой дозы фазы индукции до введения первой дозы поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения фаза индукции продолжается шесть недель, считая от введения первой дозы фазы индукции до введения первой дозы поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения фаза индукции продолжается семь недель, считая от введения первой дозы фазы индукции до введения первой дозы поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения фаза индукции продолжается восемь недель, считая от введения первой дозы фазы индукции до введения первой дозы поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения фаза индукции продолжается девять недель, считая от введения первой дозы фазы индукции до введения первой дозы поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения фаза индукции продолжается десять недель, считая от введения первой дозы фазы индукции до введения первой дозы поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения фаза индукции продолжается одиннадцать недель, считая от введения первой дозы фазы индукции до введения первой дозы поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения фаза индукции продолжается двенадцать недель, считая от введения первой дозы фазы индукции до введения первой дозы поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения фаза индукции продолжается тринадцать недель, считая от введения первой дозы фазы индукции до введения первой дозы поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения фаза индукции продолжается четырнадцать недель, считая от введения первой дозы фазы индукции до введения первой дозы поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения фаза индукции продолжается пятнадцать недель, считая от введения первой дозы фазы индукции до введения первой дозы поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения фаза индукции продолжается шестнадцать недель, считая от введения первой дозы фазы индукции до введения первой дозы поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения фаза индукции продолжается семнадцать недель, считая от введения первой дозы фазы индукции до введения первой дозы поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения фаза индукции продолжается восемнадцать недель, считая от введения первой дозы фазы индукции до введения первой дозы поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения фаза индукции продолжается девятнадцать недель, считая от введения первой дозы фазы индукции до введения первой дозы поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения фаза индукции продолжается двадцать недель, считая от введения первой дозы фазы индукции до введения первой дозы поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения фаза индукции продолжается двадцать одну неделю, считая от введения первой дозы фазы индукции до введения первой дозы поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения фаза индукции продолжается двадцать две недели, считая от введения первой дозы фазы индукции до введения первой дозы поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения фаза индукции продолжается двадцать три недели, считая от введения первой дозы фазы индукции до введения первой дозы поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения фаза индукции продолжается двадцать четыре недели, считая от введения первой дозы фазы индукции до введения первой дозы поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения фаза индукции продолжается двадцать пять недель, считая от введения первой дозы фазы индукции до введения первой дозы поддерживающей фазы. В некоторых указанных вариантах осуществления настоящего изобретения дозы, вводимые во время фазы индукции ниже, чем дозы, вводимые во время поддерживающей фазы.
В некоторых вариантах осуществления настоящего изобретения доза, вводимая во время фазы индукции ниже, чем доза, вводимая во время поддерживающей фазы, для того, чтобы избегать нежелательных побочных эффектов. В некоторых вариантах осуществления настоящего изобретения нежелательным побочным эффектом является гепатотоксичность. В некоторых указанных вариантах осуществления настоящего изобретения нежелательным побочным эффектом является повышение уровня АЛТ. В некоторых указанных вариантах осуществления настоящего изобретения более низкая индукционная доза обеспечивает время, необходимое для метаболизма липидов, чтобы компенсировать уменьшенную продукцию АроВ. В некоторых указанных вариантах осуществления настоящего изобретения небольшие повышения уровня АЛТ отражают быструю понижающую уровень липидов активность.
В некоторых вариантах осуществления настоящего изобретения, когда фаза индукции включает в себя более одной дозы, дозы, вводимые во время фазы индукции, являются одинаковыми.
В некоторых вариантах осуществления настоящего изобретения дозы, вводимые во время фазы индукции, не являются одинаковыми. В некоторых вариантах осуществления настоящего изобретения дозы с течением времени увеличиваются. В некоторых вариантах осуществления настоящего изобретения дозы с течением времени уменьшаются.
В некоторых вариантах осуществления настоящего изобретения индукционную дозу вводят парентерально. В некоторых указанных вариантах осуществления настоящего изобретения парентеральное введение представляет собой подкожное введение. В некоторых указанных вариантах осуществления настоящего изобретения парентеральное введение представляет собой внутривенную инфузию.
В некоторых вариантах осуществления настоящего изобретения дозы, вводимые во время фазы индукции, выбраны из 25 мг, 30 мг, 35 мг, 40 мг, 45 мг, 50 мг, 55 мг, 60 мг, 65 мг, 70 мг, 75 мг, 80 мг, 85 мг, 90 мг, 95 мг, 100 мг, 105 мг, 110 мг, 115 мг, 120 мг, 125 мг, 130 мг, 135 мг, 140 мг, 145 мг, 150 мг, 155 мг, 160 мг, 165 мг, 170 мг, 175 мг, 180 мг, 185 мг, 190 мг, 195 мг, 200 мг, 205 мг, 210 мг, 215 мг, 220 мг, 225 мг, 230 мг, 235 мг, 240 мг, 245 мг, 250 мг, 255 мг, 260 мг, 265 мг, 270 мг, 275 мг, 280 мг, 285 мг, 290 мг, 295 мг, 300 мг, 305 мг, 310 мг, 315 мг, 320 мг, 325 мг, 330 мг, 335 мг, 340 мг, 345 мг, 350 мг, 355 мг, 360 мг, 365 мг, 370 мг, 375 мг, 380 мг, 385 мг, 390 мг, 395 мг, 400 мг, 405 мг, 410 мг, 415 мг, 420 мг, 425 мг, 430 мг, 435 мг, 440 мг, 445 мг, 450 мг, 455 мг, 460 мг, 465 мг, 470 мг, 475 мг, 480 мг, 485 мг, 490 мг, 495 мг, 500 мг, 505 мг, 510 мг, 515 мг, 520 мг, 525 мг, 530 мг, 535 мг, 540 мг, 545 мг, 550 мг, 555 мг, 560 мг, 565 мг, 570 мг, 575 мг, 580 мг, 585 мг, 590 мг, 595 мг, 600 мг, 605 мг, 610 мг, 615 мг, 620 мг, 625 мг, 630 мг, 635 мг, 640 мг, 645 мг, 650 мг, 655 мг, 660 мг, 665 мг, 670 мг, 675 мг, 680 мг, 685 мг, 690 мг, 695 мг, 700 мг, 705 мг, 710 мг, 715 мг, 720 мг, 725 мг, 730 мг, 735 мг, 740 мг, 745 мг, 750 мг, 755 мг, 760 мг, 765 мг, 770 мг, 775 мг, 780 мг, 785 мг, 790 мг, 795 мг и 800 мг В некоторых указанных вариантах осуществления настоящего изобретения дозы, вводимые во время фазы индукции, выбраны из 25 мг, 50 мг, 75 мг, 100 мг, 150 мг, 200 мг, 250 мг, 300 мг, 350 мг, 400 мг, 500 мг, 600 мг, 700 мг и 800 мг. В некоторых указанных вариантах осуществления настоящего изобретения дозы, вводимые во время фазы индукции, выбраны из 100 мг, 125 мг, 150 мг, 175 мг, 200 мг, 225 мг, 250 мг, 275 мг, 300 мг, 325 мг, 350 мг, 375 мг и 400 мг. В некоторых указанных вариантах осуществления настоящего изобретения дозы, вводимые во время фазы индукции, выбраны из 100 мг, 125 мг, 150 мг, 175 мг, 200 мг, 225 мг и 250 мг. В некоторых указанных вариантах осуществления настоящего изобретения доза, вводимая во время фазы индукции, составляет 100 мг. В некоторых указанных вариантах осуществления настоящего изобретения доза, вводимая во время фазы индукции, составляет 125 мг. В некоторых указанных вариантах осуществления настоящего изобретения доза, вводимая во время фазы индукции, составляет 150 мг. В некоторых указанных вариантах осуществления настоящего изобретения доза, вводимая во время фазы индукции, составляет 175 мг. В некоторых указанных вариантах осуществления настоящего изобретения доза, вводимая во время фазы индукции, составляет 200 мг. В некоторых указанных вариантах осуществления настоящего изобретения доза, вводимая во время фазы индукции, составляет 225 мг. В некоторых указанных вариантах осуществления настоящего изобретения доза, вводимая во время фазы индукции, составляет 250 мг. В некоторых указанных вариантах осуществления настоящего изобретения доза, вводимая во время фазы индукции, составляет 300 мг. В некоторых указанных вариантах осуществления настоящего изобретения доза, вводимая во время фазы индукции, составляет 325 мг. В некоторых указанных вариантах осуществления настоящего изобретения доза, вводимая во время фазы индукции, составляет 350 мг. В некоторых указанных вариантах осуществления настоящего изобретения доза, вводимая во время фазы индукции, составляет 375 мг. В некоторых указанных вариантах осуществления настоящего изобретения доза, вводимая во время фазы индукции, составляет 400 мг.
В некоторых вариантах осуществления настоящего изобретения, когда желательным является подкожное введение, индукционную дозу можно вводить двумя или более подкожных инъекций. В некоторых указанных вариантах осуществления настоящего изобретения, когда желательная индукционная доза требует объема, не слишком соответствующего однократной инъекции, можно использовать две или более подкожных инъекций для достижения желательной индукционной дозы. В некоторых указанных вариантах осуществления настоящего изобретения две или более подкожных инъекций можно использовать для введения желательной индукционной дозы и минимизировать или устранить реакцию в месте инъекции у субъекта.
В некоторых вариантах осуществления настоящего изобретения дозу, частоту введения и продолжительность фазы индукции можно выбирать для достижения желаемого эффекта. В некоторых вариантах осуществления настоящего изобретения указанные переменные величины подбирают таким образом, чтобы обеспечить желательную концентрацию фармацевтического агента у субъекта. Например, в некоторых вариантах осуществления настоящего изобретения дозу и частоту введения подбирают таким образом, чтобы обеспечить концентрацию фармацевтического агента в плазме крови у субъекта на уровне, достаточном для достижения желаемого эффекта. В некоторых из указанных вариантов осуществления настоящего изобретения концентрацию в плазме крови поддерживают на уровне, превышающем минимальную эффективную концентрацию (МЕС). В некоторых вариантах осуществления настоящего изобретения фармацевтические композиции по настоящему изобретению вводят с использованием схемы введения, разработанной для поддержания концентрации свыше МЕС в течение 10-90% времени, 30-90% времени или 50-90% времени.
В некоторых вариантах осуществления настоящего изобретения дозу, частоту введения и продолжительность фазы индукции можно выбирать для достижения желательной минимальной концентрации фармацевтической композиции в плазме крови. В некоторых указанных вариантах осуществления настоящего изобретения фармацевтическая композиция представляет собой олигонуклеотид. В некоторых указанных вариантах осуществления настоящего изобретения желательная минимальная концентрация в плазме крови составляет 5-100 нг/мл. В некоторых указанных вариантах осуществления настоящего изобретения желательная минимальная концентрация в плазме крови составляет 5-50 нг/мл. В некоторых указанных вариантах осуществления настоящего изобретения желательная минимальная концентрация в плазме крови составляет 10-40 нг/мл. В некоторых указанных вариантах осуществления настоящего изобретения желательная минимальная концентрация в плазме крови составляет 15-35 нг/мл. В некоторых указанных вариантах осуществления настоящего изобретения желательная минимальная концентрация в плазме крови составляет 20-30 нг/мл.
В некоторых вариантах осуществления настоящего изобретения дозу, частоту введения и продолжительность фазы индукции можно выбирать для достижения желаемого эффекта за пять-тринадцать недель. В некоторых указанных вариантах осуществления настоящего изобретения дозы являются одинаковыми, а частота введения изменяется для достижения желаемого эффекта за пять-тринадцать недель. В некоторых указанных вариантах осуществления настоящего изобретения доза возрастает с течением времени, а частота введения остается постоянной. В некоторых указанных вариантах осуществления настоящего изобретения дозу и частоту введения выбирают для достижения желаемого эффекта за шесть недель. В некоторых указанных вариантах осуществления настоящего изобретения дозу и частоту введения выбирают для достижения желаемого эффекта за семь недель. В некоторых указанных вариантах осуществления настоящего изобретения дозу и частоту введения выбирают для достижения желаемого эффекта за восемь недель. В некоторых указанных вариантах осуществления настоящего изобретения дозу и частоту введения выбирают для достижения желаемого эффекта за девять недель. В некоторых указанных вариантах осуществления настоящего изобретения дозу и частоту введения выбирают для достижения желаемого эффекта за десять недель. В некоторых указанных вариантах осуществления настоящего изобретения дозу и частоту введения выбирают для достижения желаемого эффекта за одиннадцать недель. В некоторых указанных вариантах осуществления настоящего изобретения дозу и частоту введения выбирают для достижения желаемого эффекта за двенадцать недель. В некоторых указанных вариантах осуществления настоящего изобретения дозу и частоту введения выбирают для достижения желаемого эффекта за тринадцать недель. В некоторых указанных вариантах осуществления настоящего изобретения одна или более доз фазы индукции превышают одну или более доз поддерживающей фазы. В некоторых указанных вариантах осуществления настоящего изобретения каждая из индукционных доз превышает каждую из поддерживающих доз.
В некоторых вариантах осуществления настоящего изобретения дозу, частоту введения и продолжительность фазы индукции можно выбирать для достижения желаемого эффекта за 13-25 недель. В некоторых указанных вариантах осуществления настоящего изобретения дозы являются одинаковыми, а частота введения изменяется для достижения желаемого эффекта за 13-25 недель. В некоторых указанных вариантах осуществления настоящего изобретения доза возрастает с течением времени, а частота введения остается постоянной. В некоторых указанных вариантах осуществления настоящего изобретения дозу и частоту введения выбирают для достижения желаемого эффекта за тринадцать недель. В некоторых указанных вариантах осуществления настоящего изобретения дозу и частоту введения выбирают для достижения желаемого эффекта за четырнадцать недель. В некоторых указанных вариантах осуществления настоящего изобретения дозу и частоту введения выбирают для достижения желаемого эффекта за пятнадцать недель. В некоторых указанных вариантах осуществления настоящего изобретения дозу и частоту введения выбирают для достижения желаемого эффекта за шестнадцать недель. В некоторых указанных вариантах осуществления настоящего изобретения дозу и частоту введения выбирают для достижения желаемого эффекта за семнадцать недель. В некоторых указанных вариантах осуществления настоящего изобретения дозу и частоту введения выбирают для достижения желаемого эффекта за восемнадцать недель. В некоторых указанных вариантах осуществления настоящего изобретения дозу и частоту введения выбирают для достижения желаемого эффекта за девятнадцать недель. В некоторых указанных вариантах осуществления настоящего изобретения дозу и частоту введения выбирают для достижения желаемого эффекта за двадцать недель. В некоторых указанных вариантах осуществления настоящего изобретения дозу и частоту введения выбирают для достижения желаемого эффекта за двадцать одну неделю. В некоторых указанных вариантах осуществления настоящего изобретения дозу и частоту введения выбирают для достижения желаемого эффекта за двадцать две недели. В некоторых указанных вариантах осуществления настоящего изобретения дозу и частоту введения выбирают для достижения желаемого эффекта за двадцать три недели. В некоторых указанных вариантах осуществления настоящего изобретения дозу и частоту введения выбирают для достижения желаемого эффекта за двадцать четыре недели. В некоторых указанных вариантах осуществления настоящего изобретения дозу и частоту введения выбирают для достижения желаемого эффекта за двадцать пять недель. В некоторых указанных вариантах осуществления настоящего изобретения одна или более доз фазы индукции меньше одной или более доз поддерживающей фазы. В некоторых указанных вариантах осуществления настоящего изобретения каждая из индукционных доз меньше каждой из поддерживающих доз.
В некоторых вариантах осуществления настоящего изобретения желательным является достижение желательного эффекта как можно быстрее. В указанных вариантах осуществления настоящего изобретения может быть желательной фаза индукции с использованием высокой дозы и/или высокой частоты введения. Указанные варианты осуществления настоящего изобретения могут включать в себя введение субъекту, имеющему очень высокие концентрации холестерина.
В некоторых вариантах осуществления настоящего изобретения желательным является смягчение нежелательного побочного эффекта. В некоторых указанных вариантах осуществления настоящего изобретения может быть желательной фаза индукции с использованием низкой дозы и/или низкой частоты введения и/или большой продолжительностью. Например, продолжительная фаза индукции с использованием относительно низких доз может обеспечивать лучшую переносимость фармацевтического агента. Некоторые указанные варианты осуществления настоящего изобретения обеспечивают физиологические изменения, результатом которых является общее уменьшение побочных эффектов. В некоторых вариантах осуществления настоящего изобретения указанная схема введения обеспечивает более низкую гепатотоксичность по сравнению с более высокими первоначальными дозами и/или частотой. Указанные варианты осуществления настоящего изобретения могут включать в себя постепенное увеличение дозы с течением времени.
В некоторых вариантах осуществления настоящего изобретения, в которых фармацевтическую композицию вводят локально, схему введения выбирают для достижения желательной локальной концентрации фармацевтического агента по настоящему изобретению.
В некоторых вариантах осуществления настоящего изобретения дозу, частоту введения и продолжительность фазы индукции можно выбирать для достижения приемлемого профиля безопасности. Например, в некоторых вариантах осуществления настоящего изобретения указанные переменные величины можно выбирать для смягчения токсичности фармацевтической композиции. В некоторых указанных вариантах осуществления настоящего изобретения указанные переменные величины выбирают для смягчения гепатотоксичности. В некоторых указанных вариантах осуществления настоящего изобретения указанные переменные величины выбирают для смягчения токсичности в отношении почек. В некоторых указанных вариантах осуществления настоящего изобретения дозы увеличиваются с течением времени. В некоторых указанных вариантах осуществления настоящего изобретения одна или более доз фазы индукции меньше одной или более доз поддерживающей фазы. В некоторых указанных вариантах осуществления настоящего изобретения профиль безопасности не является приемлемым, если АЛТ в 5-10 раз превышает верхнюю границу нормы. В некоторых указанных вариантах осуществления настоящего изобретения профиль безопасности не является приемлемым, если АЛТ в 5-10 раз превышает верхнюю границу нормы, а билирубин повышен в два или более раз по сравнению с верхней границей нормы. В некоторых указанных вариантах осуществления настоящего изобретения приемлемый профиль безопасности включает в себя повышения АЛТ, которые более чем в три раза превышают верхнюю границу нормы, но не более пятикратного превышения верхней границы нормы. В некоторых указанных вариантах осуществления настоящего изобретения приемлемый профиль безопасности включает в себя повышения уровней АЛТ, которые более чем в три раза превышают верхнюю границу нормы, но не более пятикратного превышения верхней границы нормы, и повышения уровней билирубина, которые составляют не более двукратного превышения верхней границы нормы. В некоторых указанных вариантах осуществления настоящего изобретения, когда введение фармацевтической композиции по настоящему изобретению приводит к повышению уровней АЛТ, которое более чем в три раза превышает верхнюю границу нормы, дозу и/или частоту введения подбирают таким образом, чтобы смягчить повышение уровней АЛТ. В некоторых указанных вариантах осуществления настоящего изобретения, когда введение фармацевтической композиции по настоящему изобретению приводит к повышению уровней АЛТ, которое более чем в три раза превышает верхнюю границу нормы, и к концентрациям билирубина, которые составляют не более двукратного превышения верхней границы нормы, дозу и/или частоту введения подбирают таким образом, чтобы смягчить повышение уровней АЛТ и повышение уровней билирубина. В некоторых указанных вариантах осуществления настоящего изобретения дозу и/или частоту введения подбирают таким образом, чтобы смягчить только повышение уровней билирубина.
В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая фаза включает в себя одну, две, три, четыре, пять, шесть, семь, восемь, девять, десять, одиннадцать, двенадцать, тринадцать, четырнадцать, шестнадцать, семнадцать, восемнадцать, девятнадцать, двадцать или более двадцати доз.
В некоторых вариантах осуществления настоящего изобретения поддерживающая фаза продолжается от одного дня до всей оставшейся жизни субъекта. В некоторых вариантах осуществления настоящего изобретения поддерживающая фаза продолжается от одной недели до двадцати лет, считая от введения последней дозы фазы индукции до введения последней дозы поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения поддерживающая фаза продолжается от двух недель до пятнадцати лет, считая от введения последней дозы фазы индукции до введения последней дозы поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения поддерживающая фаза продолжается от трех недель до десяти лет, считая от введения последней дозы фазы индукции до введения последней дозы поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения поддерживающая фаза продолжается от четырех недель до десяти лет, считая от введения последней дозы фазы индукции до введения последней дозы поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения поддерживающая фаза продолжается столько, сколько доза остается необходимой, эффективной и переносимой.
В некоторых вариантах осуществления настоящего изобретения, когда поддерживающая фаза включает в себя более одной дозы, дозы, вводимые во время поддерживающей фазы, являются одинаковыми. В некоторых вариантах осуществления настоящего изобретения дозы, вводимые во время поддерживающей фазы, не являются одинаковыми. В некоторых вариантах осуществления настоящего изобретения дозы с течением времени уменьшаются.
В некоторых вариантах осуществления настоящего изобретения поддерживающую дозу вводят парентерально. В некоторых указанных вариантах осуществления настоящего изобретения парентеральное введение представляет собой подкожное введение. В некоторых указанных вариантах осуществления настоящего изобретения парентеральное введение представляет собой внутривенную инфузию.
В некоторых вариантах осуществления настоящего изобретения дозы, вводимые во время поддерживающей фазы, выбраны из 25 мг, 30 мг, 35 мг, 40 мг, 45 мг, 50 мг, 55 мг, 60 мг, 65 мг, 70 мг, 75 мг, 80 мг, 85 мг, 90 мг, 95 мг, 100 мг, 105 мг, 110 мг, 115 мг, 120 мг, 125 мг, 130 мг, 135 мг, 140 мг, 145 мг, 150 мг, 155 мг, 160 мг, 165 мг, 170 мг, 175 мг, 180 мг, 185 мг, 190 мг, 195 мг, 200 мг, 205 мг, 210 мг, 215 мг, 220 мг, 225 мг, 230 мг, 235 мг, 240 мг, 245 мг, 250 мг, 255 мг, 260 мг, 265 мг, 270 мг, 275 мг, 280 мг, 285 мг, 290 мг, 295 мг, 300 мг, 305 мг, 310 мг, 315 мг, 320 мг, 325 мг, 330 мг, 335 мг, 340 мг, 345 мг, 350 мг, 355 мг, 360 мг, 365 мг, 370 мг, 375 мг, 380 мг, 385 мг, 390 мг, 395 мг, 400 мг, 405 мг, 410 мг, 415 мг, 420 мг, 425 мг, 430 мг, 435 мг, 440 мг, 445 мг, 450 мг, 455 мг, 460 мг, 465 мг, 470 мг, 475 мг, 480 мг, 485 мг, 490 мг, 495 мг, 500 мг, 505 мг, 510 мг, 515 мг, 520 мг, 525 мг, 530 мг, 535 мг, 540 мг, 545 мг, 550 мг, 555 мг, 560 мг, 565 мг, 570 мг, 575 мг, 580 мг, 585 мг, 590 мг, 595 мг, 600 мг, 605 мг, 610 мг, 615 мг, 620 мг, 625 мг, 630 мг, 635 мг, 640 мг, 645 мг, 650 мг, 655 мг, 660 мг, 665 мг, 670 мг, 675 мг, 680 мг, 685 мг, 690 мг, 695 мг, 700 мг, 705 мг, 710 мг, 715 мг, 720 мг, 725 мг, 730 мг, 735 мг, 740 мг, 745 мг, 750 мг, 755 мг, 760 мг, 765 мг, 770 мг, 775 мг, 780 мг, 785 мг, 790 мг, 795 мг и 800 мг. В некоторых указанных вариантах осуществления настоящего изобретения дозы, вводимые во время поддерживающей фазы, выбраны из 25 мг, 50 мг, 75 мг, 100 мг, 150 мг, 200 мг, 250 мг, 300 мг, 350 мг, 400 мг, 500 мг, 600 мг, 700 мг и 800 мг. В некоторых указанных вариантах осуществления настоящего изобретения дозы, вводимые во время поддерживающей фазы, выбраны из 100 мг, 125 мг, 150 мг, 175 мг, 200 мг, 225 мг, 250 мг, 275 мг, 300 мг, 325 мг, 350 мг, 375 мг и 400 мг. В некоторых указанных вариантах осуществления настоящего изобретения дозы, вводимые во время поддерживающей фазы, выбраны из 100 мг, 125 мг, 150 мг, 175 мг, 200 мг, 225 мг и 250 мг. В некоторых указанных вариантах осуществления настоящего изобретения доза, вводимая во время поддерживающей фазы, составляет 100 мг. В некоторых указанных вариантах осуществления настоящего изобретения доза, вводимая во время поддерживающей фазы, составляет 125 мг. В некоторых указанных вариантах осуществления настоящего изобретения доза, вводимая во время поддерживающей фазы, составляет 150 мг. В некоторых указанных вариантах осуществления настоящего изобретения доза, вводимая во время поддерживающей фазы, составляет 175 мг. В некоторых указанных вариантах осуществления настоящего изобретения доза, вводимая во время поддерживающей фазы, составляет 200 мг. В некоторых указанных вариантах осуществления настоящего изобретения доза, вводимая во время поддерживающей фазы, составляет 225 мг. В некоторых указанных вариантах осуществления настоящего изобретения доза, вводимая во время поддерживающей фазы, составляет 250 мг. В некоторых указанных вариантах осуществления настоящего изобретения доза, вводимая во время поддерживающей фазы, составляет 275 мг. В некоторых указанных вариантах осуществления настоящего изобретения доза, вводимая во время поддерживающей фазы, составляет 300 мг.
В некоторых вариантах осуществления настоящего изобретения, когда желательным является подкожное введение, поддерживающую дозу можно вводить двумя или более подкожных инъекций. В некоторых указанных вариантах осуществления настоящего изобретения, когда желательная поддерживающая доза требует объема, не слишком соответствующего однократной инъекции, можно использовать две или более подкожных инъекций для достижения желательной поддерживающей дозы. В некоторых указанных вариантах осуществления настоящего изобретения две или более подкожных инъекций можно использовать для введения желательной поддерживающей дозы и минимизировать или устранить реакцию в месте инъекции у субъекта.
В некоторых вариантах осуществления настоящего изобретения дозу, частоту введения и продолжительность поддерживающей фазы можно выбирать для достижения желаемого эффекта. В некоторых вариантах осуществления настоящего изобретения указанные переменные величины подбирают таким образом, чтобы обеспечить желательную концентрацию фармацевтического агента у субъекта. Например, в некоторых вариантах осуществления настоящего изобретения дозу и частоту введения подбирают таким образом, чтобы обеспечить концентрацию фармацевтического агента в плазме крови у субъекта на уровне, достаточном для достижения желаемого эффекта. В некоторых из указанных вариантов осуществления настоящего изобретения концентрацию в плазме крови поддерживают на уровне, превышающем минимальную эффективную концентрацию (МЕС). В некоторых вариантах осуществления настоящего изобретения фармацевтические композиции по настоящему изобретению вводят с использованием схемы введения, разработанной для поддержания концентрации свыше МЕС в течение 10-90% времени, 30-90% времени или 50-90% времени.
В некоторых вариантах осуществления настоящего изобретения дозу, частоту введения и продолжительность поддерживающей фазы можно выбирать для достижения желательной минимальной концентрации фармацевтической композиции в плазме крови. В некоторых указанных вариантах осуществления настоящего изобретения фармацевтическая композиция представляет собой олигонуклеотид. В некоторых указанных вариантах осуществления настоящего изобретения желательная минимальная концентрация в плазме крови составляет 5-100 нг/мл. В некоторых указанных вариантах осуществления настоящего изобретения желательная минимальная концентрация в плазме крови составляет 5-50 нг/мл. В некоторых указанных вариантах осуществления настоящего изобретения желательная минимальная концентрация составляет в плазме крови 10-40 нг/мл. В некоторых указанных вариантах осуществления настоящего изобретения желательная минимальная концентрация в плазме крови составляет 15-35 нг/мл. В некоторых указанных вариантах осуществления настоящего изобретения желательная минимальная концентрация составляет в плазме крови 20-30 нг/мл.
В некоторых вариантах осуществления настоящего изобретения дозы, частоту введения и продолжительность поддерживающей фазы можно выбирать для достижения приемлемого профиля безопасности. Например, в некоторых вариантах осуществления настоящего изобретения указанные переменные величины можно выбирать для смягчения токсичности фармацевтической композиции. В некоторых указанных вариантах осуществления настоящего изобретения указанные переменные величины выбирают для смягчения гепатотоксичности. В некоторых указанных вариантах осуществления настоящего изобретения указанные переменные величины выбирают для смягчения токсичности в отношении почек. В некоторых указанных вариантах осуществления настоящего изобретения дозы увеличиваются с течением времени.
В некоторых вариантах осуществления настоящего изобретения дозы, частоту введения и продолжительность поддерживающей фазы время от времени можно корректировать для достижения желаемого эффекта. В некоторых вариантах осуществления настоящего изобретения осуществляют мониторинг субъектов на предмет эффектов (терапевтических и/или токсических эффектов), и дозы, частоту введения и продолжительность поддерживающей фазы можно корректировать на основе результатов указанного мониторинга.
Специалисту следует понимать, что дозы, частоту введения и продолжительность для фазы индукции и для поддерживающей фазы можно изменять независимо друг от друга для достижения желаемого эффекта. Например, в некоторых вариантах осуществления изобретение относится к схемам введения, перечисленных в таблицах 1-6, ниже. Специалисту будет понятно, что переменные величины в таблице можно выбирать и комбинировать независимо друг от друга. Таблица включена только для иллюстрации того, каким образом переменные величины можно комбинировать, и она не ограничивает изобретение. Помимо этого, настоящее изобретение не ограничивается переменными величинами, перечисленными в таблице.
В некоторых вариантах осуществления настоящего изобретения способ введения фармацевтической композиции субъекту включает в себя фазу индукции, во время которой индукционную дозу 200-400 мг вводят один раз в неделю в течение по меньшей мере 8 недель, с последующей поддерживающей фазой, во время которой поддерживающую дозу 100-300 мг вводят с интервалами, варьирующими от одного раза в неделю до одного раза в три месяца, так долго, как это требуется для поддержания желаемого эффекта. В некоторых вариантах осуществления настоящего изобретения индукционную дозу вводят один раз в неделю в течение 8-20 недель. В некоторых вариантах осуществления настоящего изобретения индукционную дозу вводят один раз в неделю в течение 10-15 недель. В некоторых вариантах осуществления настоящего изобретения индукционную дозу вводят один раз в неделю в течение по меньшей мере 12 недель. В некоторых вариантах осуществления настоящего изобретения индукционную дозу вводят один раз в неделю в течение по меньшей мере 14 недель. В некоторых вариантах осуществления настоящего изобретения индукционную дозу вводят один раз в неделю в течение по меньшей мере 16 недель. В некоторых указанных вариантах осуществления настоящего изобретения индукционная доза составляет 200 мг. В некоторых указанных вариантах осуществления настоящего изобретения индукционная доза составляет 300 мг. В некоторых указанных вариантах осуществления настоящего изобретения индукционная доза составляет 400 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 200-300 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 150 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 200 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 250 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 300 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят один раз в неделю. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят один раз в месяц. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят один раз в три месяца. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят в течение по меньшей мере 6 месяцев. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят в течение по меньшей мере одного года. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят в течение периода времени до пяти лет. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят в течение периода времени до десяти лет. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят так долго, как это требуется для поддержания желаемого эффекта. В некоторых указанных вариантах осуществления настоящего изобретения частоту введения поддерживающей дозы подбирают таким образом, чтобы достичь желаемой эффективности и/или желаемого профиля безопасности. В некоторых указанных вариантах осуществления настоящего изобретения частоту введения поддерживающей дозы подбирают таким образом, чтобы достичь желаемой минимальной концентрации олигонуклеотида в плазме крови. В некоторых указанных вариантах осуществления настоящего изобретения минимальная концентрация вводимого антисмыслового олигонуклеотида в плазме крови составляет 15-40 нг/мл. В некоторых указанных вариантах осуществления настоящего изобретения минимальная концентрация вводимого антисмыслового олигонуклеотида в плазме крови составляет 20-30 нг/мл. В некоторых указанных вариантах осуществления настоящего изобретения желаемый эффект выбран из пониженного АроВ, пониженного ЛНП-Х, пониженного ЛОНП-Х, пониженного ЛПП-Х, пониженного не-ЛВП-Х, пониженных триглицеридов сыворотки крови, пониженных триглицеридов печени, пониженного ЛП(а), пониженного окси-ЛНП-Х и пониженных малых плотных частиц ЛНП. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта имеется полигенная гиперхолестеринемия. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта имеется наследственная гиперхолестеринемия. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта имеется гомозиготная наследственная гиперхолестеринемия. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта имеется гетерозиготная наследственная гиперхолестеринемия. В некоторых указанных вариантах осуществления настоящего изобретения фармацевтическую композицию вводят совместно со статином. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта наблюдается непереносимость статина. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта текущая терапия не достигает своей мишени - ЛНП-холестерина. Неограничивающие примеры некоторых схем введения проиллюстрированы в таблице 1.
В некоторых вариантах осуществления настоящего изобретения способ введения фармацевтической композиции субъекту включает в себя фазу индукции, во время которой дозу 200 мг вводят один раз в неделю в течение 13 недель, с последующей поддерживающей фазой, во время которой дозу 80-200 мг вводят с интервалами, варьирующими от одного раза в неделю до одного раза в три месяца, так долго, как это требуется для поддержания желаемого эффекта. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 100-150 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 100 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 125 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 140 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 150 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 175 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 180 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 200 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят один раз в неделю. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят один раз в месяц. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят один раз в три месяца. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят в течение по меньшей мере 6 месяцев. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят в течение по меньшей мере одного года. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят в течение периода времени до пяти лет. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят в течение периода времени до десяти лет. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят так долго, как это требуется для поддержания желаемого эффекта. В некоторых указанных вариантах осуществления настоящего изобретения частоту введения поддерживающей дозы подбирают таким образом, чтобы достичь желаемой эффективности и/или желаемого профиля безопасности. В некоторых указанных вариантах осуществления настоящего изобретения частоту введения поддерживающей дозы подбирают таким образом, чтобы достичь желаемой минимальной концентрации олигонуклеотида в плазме крови. В некоторых указанных вариантах осуществления настоящего изобретения минимальная концентрация вводимого антисмыслового олигонуклеотида в плазме крови составляет 15-40 нг/мл. В некоторых указанных вариантах осуществления настоящего изобретения минимальная концентрация вводимого антисмыслового олигонуклеотида в плазме крови составляет 20-30 нг/мл. В некоторых указанных вариантах осуществления настоящего изобретения желаемый эффект выбран из пониженного АроВ, пониженного ЛНП-Х, пониженного ЛОНП-Х, пониженного ЛПП-Х, пониженного не-ЛВП-Х, пониженных триглицеридов сыворотки крови, пониженных триглицеридов печени, пониженного ЛП(а), пониженного окси-ЛНП-Х и пониженных малых плотных частиц ЛНП. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта имеется полигенная гиперхолестеринемия. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта имеется наследственная гиперхолестеринемия. В некоторых указанных вариантах осуществления настоящего изобретения фармацевтическую композицию вводят совместно со статином. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта наблюдается непереносимость статина. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта текущая терапия не достигает своей мишени - ЛНП-холестерина. Неограничивающие примеры некоторых схем введения проиллюстрированы в таблице 2.
В некоторых вариантах осуществления настоящего изобретения способ введения фармацевтической композиции субъекту включает в себя фазу индукции, во время которой дозу 300 мг вводят один раз в неделю в течение 13 недель, с последующей поддерживающей фазой, во время которой дозу 100-250 мг вводят с интервалами, варьирующими от одного раза в неделю до одного раза в три месяца, так долго, как это требуется для поддержания желаемого эффекта. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 100 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 125 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 150 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 175 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 200 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 250 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят один раз в неделю. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят один раз в месяц. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят один раз в три месяца. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят в течение по меньшей мере 6 месяцев. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят в течение по меньшей мере одного года. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят в течение периода времени до пяти лет. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят в течение периода времени до десяти лет. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят так долго, как это требуется для поддержания желаемого эффекта. В некоторых указанных вариантах осуществления настоящего изобретения частоту введения поддерживающей дозы подбирают таким образом, чтобы достичь желаемой эффективности и/или желаемого профиля безопасности. В некоторых указанных вариантах осуществления настоящего изобретения частоту введения поддерживающей дозы подбирают таким образом, чтобы достичь желаемой минимальной концентрации олигонуклеотида в плазме крови. В некоторых указанных вариантах осуществления настоящего изобретения минимальная концентрация вводимого антисмыслового олигонуклеотида в плазме крови составляет 15-40 нг/мл. В некоторых указанных вариантах осуществления настоящего изобретения минимальная концентрация вводимого антисмыслового олигонуклеотида в плазме крови составляет 20-30 нг/мл. В некоторых указанных вариантах осуществления настоящего изобретения желаемый эффект выбран из пониженного АроВ, пониженного ЛНП-Х, пониженного ЛОНП-Х, пониженного ЛПП-Х, пониженного не-ЛВП-Х, пониженных триглицеридов сыворотки крови, пониженных триглицеридов печени, пониженного ЛП(а), пониженного окси-ЛНП-Х и пониженных малых плотных частиц ЛНП. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта имеется полигенная гиперхолестеринемия. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта имеется наследственная гиперхолестеринемия. В некоторых указанных вариантах осуществления настоящего изобретения фармацевтическую композицию вводят совместно со статином. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта наблюдается непереносимость статина. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта текущая терапия не достигает своей мишени - ЛНП-холестерина. Неограничивающие примеры некоторых схем введения проиллюстрированы в таблице 3.
В некоторых вариантах осуществления настоящего изобретения способ введения фармацевтической композиции субъекту включает в себя фазу индукции, во время которой дозу 100 мг вводят один раз в неделю в течение 13 недель, с последующей поддерживающей фазой, во время которой дозу 100-300 мг вводят с интервалами, варьирующими от одного раза в неделю до одного раза в три месяца, так долго, как это требуется для поддержания желаемого эффекта. В некоторых указанных вариантах осуществления настоящего изобретения доза составляет 150-250 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 100 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 125 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 150 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 175 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 200 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 225 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 250 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 275 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 300 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят один раз в неделю. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят один раз в месяц. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят один раз в три месяца. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят в течение по меньшей мере 6 месяцев. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят в течение по меньшей мере одного года. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят в течение периода времени до пяти лет. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят в течение периода времени до десяти лет. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят так долго, как это требуется для поддержания желаемого эффекта. В некоторых указанных вариантах осуществления настоящего изобретения частоту введения поддерживающей дозы подбирают таким образом, чтобы достичь желаемой эффективности и/или желаемого профиля безопасности. В некоторых указанных вариантах осуществления настоящего изобретения частоту введения поддерживающей дозы подбирают таким образом, чтобы достичь желаемой минимальной концентрации олигонуклеотида в плазме крови. В некоторых указанных вариантах осуществления настоящего изобретения минимальная концентрация вводимого антисмыслового олигонуклеотида в плазме крови составляет 15-40 нг/мл. В некоторых указанных вариантах осуществления настоящего изобретения минимальная концентрация вводимого антисмыслового олигонуклеотида в плазме крови составляет 20-30 нг/мл. В некоторых указанных вариантах осуществления настоящего изобретения желаемый эффект выбран из пониженного АроВ, пониженного ЛНП-Х, пониженного ЛОНП-Х, пониженного ЛПП-Х, пониженного не-ЛВП-Х, пониженных триглицеридов сыворотки крови, пониженных триглицеридов печени, пониженного ЛП(а), пониженного окси-ЛНП-Х и пониженных малых плотных частиц ЛНП. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта имеется полигенная гиперхолестеринемия. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта имеется наследственная гиперхолестеринемия. В некоторых указанных вариантах осуществления настоящего изобретения фармацевтическую композицию вводят совместно со статином. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта наблюдается непереносимость статина. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта текущая терапия не достигает своей мишени - ЛНП-холестерина. Неограничивающие примеры некоторых схем введения проиллюстрированы в таблице 4.
В некоторых вариантах осуществления настоящего изобретения способ введения фармацевтической композиции субъекту включает в себя фазу индукции, во время которой дозу 100-200 мг вводят один раз в неделю в течение 13 недель, с последующей поддерживающей фазой, во время которой дозу 100-300 мг вводят с интервалами, варьирующими от одного раза в неделю до одного раза в три месяца, так долго, как это требуется для поддержания желаемого эффекта. В некоторых указанных вариантах осуществления настоящего изобретения индукционная доза составляет 100 мг. В некоторых указанных вариантах осуществления настоящего изобретения индукционная доза составляет 125 мг. В некоторых указанных вариантах осуществления настоящего изобретения индукционная доза составляет 150 мг. В некоторых указанных вариантах осуществления настоящего изобретения индукционная доза составляет 175 мг. В некоторых указанных вариантах осуществления настоящего изобретения индукционная доза составляет 200 мг. В некоторых указанных вариантах осуществления настоящего изобретения во время фазы индукции вводят четыре дозы по 100 мг, а затем пять доз по 150 мг, с последующим введением четырех доз по 200 мг. В некоторых указанных вариантах осуществления настоящего изобретения во время фазы индукции вводят четыре дозы по 100 мг, а затем четыре дозы по 150 мг, с последующим введением пяти доз по 200 мг. В некоторых указанных вариантах осуществления настоящего изобретения вводят пять доз по 100 мг, а затем четыре дозы по 150 мг, с последующим введением четырех доз по 200 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза превышает индукционную дозу. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 100 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 125 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 150 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 175 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 200 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 225 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 250 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 275 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 300 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят один раз в неделю. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят один раз в месяц. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят один раз в три месяца. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят в течение по меньшей мере 6 месяцев. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят в течение по меньшей мере одного года. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят в течение периода времени до пяти лет. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят в течение периода времени до десяти лет. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят так долго, как это требуется для поддержания желаемого эффекта. В некоторых указанных вариантах осуществления настоящего изобретения количество или частоту введения индукционной дозы подбирают таким образом, чтобы достичь желаемой эффективности и/или желаемого профиля безопасности. В некоторых указанных вариантах осуществления настоящего изобретения количество или частоту введения индукционной дозы подбирают таким образом, чтобы достичь желаемой минимальной концентрации антисмыслового олигонуклеотида в плазме крови. В некоторых указанных вариантах осуществления настоящего изобретения минимальная концентрация вводимого антисмыслового олигонуклеотида в плазме крови составляет 15-40 нг/мл. В некоторых указанных вариантах осуществления настоящего изобретения минимальная концентрация вводимого антисмыслового олигонуклеотида в плазме крови составляет 20-30 нг/мл. В некоторых указанных вариантах осуществления настоящего изобретения количество или частоту введения поддерживающей дозы подбирают таким образом, чтобы достичь желаемой эффективности и/или желаемого профиля безопасности. В некоторых указанных вариантах осуществления настоящего изобретения частоту введения поддерживающей дозы подбирают таким образом, чтобы достичь желаемой минимальной концентрации олигонуклеотида в плазме крови. В некоторых указанных вариантах осуществления настоящего изобретения минимальная концентрация вводимого антисмыслового олигонуклеотида в плазме крови составляет 15-40 нг/мл. В некоторых указанных вариантах осуществления настоящего изобретения минимальная концентрация вводимого антисмыслового олигонуклеотида в плазме крови составляет 20-30 нг/мл. В некоторых указанных вариантах осуществления настоящего изобретения желаемый эффект выбран из пониженного АроВ, пониженного ЛНП-Х, пониженного ЛОНП-Х, пониженного ЛПП-Х, пониженного не-ЛВП-Х, пониженных триглицеридов сыворотки крови, пониженных триглицеридов печени, пониженного ЛП(а), пониженного окси-ЛНП-Х и пониженных малых плотных частиц ЛНП. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта имеется полигенная гиперхолестеринемия. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта имеется наследственная гиперхолестеринемия. В некоторых указанных вариантах осуществления настоящего изобретения фармацевтическую композицию вводят совместно со статином. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта наблюдается непереносимость статина. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта текущая терапия не достигает своей мишени - ЛНП-холестерина. Неограничивающие примеры некоторых схем введения проиллюстрированы в таблице 5.
В некоторых вариантах осуществления настоящего изобретения способ введения фармацевтической композиции субъекту включает в себя фазу индукции, во время которой дозу 100 мг вводят один раз в неделю в течение 14-20 недель, с последующей поддерживающей фазой, во время которой дозу 100-300 мг вводят с интервалами, варьирующими от одного раза в неделю до одного раза в три месяца, так долго, как это требуется для поддержания желаемого эффекта. В некоторых указанных вариантах осуществления настоящего изобретения фаза индукции продолжается 16-20 недель. В некоторых указанных вариантах осуществления настоящего изобретения продолжительность фазы индукции составляет 14 недель. В некоторых указанных вариантах осуществления настоящего изобретения продолжительность фазы индукции составляет 15 недель. В некоторых указанных вариантах осуществления настоящего изобретения продолжительность фазы индукции составляет 16 недель. В некоторых указанных вариантах осуществления настоящего изобретения продолжительность фазы индукции составляет 17 недель. В некоторых указанных вариантах осуществления настоящего изобретения продолжительность фазы индукции составляет 18 недель. В некоторых указанных вариантах осуществления настоящего изобретения продолжительность фазы индукции составляет 19 недель. В некоторых указанных вариантах осуществления настоящего изобретения продолжительность фазы индукции составляет 20 недель. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза превышает индукционную дозу. В некоторых указанных вариантах осуществления настоящего изобретения доза поддерживающая составляет 100-300 мг. В некоторых указанных вариантах осуществления настоящего изобретения доза поддерживающая составляет 100-200 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 100 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 125 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 150 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 175 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 200 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 225 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 250 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 275 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая доза составляет 300 мг. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят один раз в неделю. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят один раз в месяц. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят один раз в три месяца. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят в течение по меньшей мере 6 месяцев. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят в течение по меньшей мере одного года. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят в течение периода времени до пяти лет. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят в течение периода времени до десяти лет. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающую дозу вводят так долго, как это требуется для поддержания желаемого эффекта. В некоторых указанных вариантах осуществления настоящего изобретения частоту введения поддерживающей дозы подбирают таким образом, чтобы достичь желаемой эффективности и/или желаемого профиля безопасности. В некоторых указанных вариантах осуществления настоящего изобретения желаемый эффект выбран из пониженного АроВ, пониженного ЛНП-Х, пониженного ЛОНП-Х, пониженного ЛПП-Х, пониженного не-ЛВП-Х, пониженных триглицеридов сыворотки крови, пониженных триглицеридов печени, пониженного ЛП(а), пониженного окси-ЛНП-Х и пониженных малых плотных частиц ЛНП. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта имеется полигенная гиперхолестеринемия. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта имеется наследственная гиперхолестеринемия. В некоторых указанных вариантах осуществления настоящего изобретения фармацевтическую композицию вводят совместно со статином. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта наблюдается непереносимость статина. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта текущая терапия не достигает своей мишени - ЛНП-холестерина. Неограничивающие примеры некоторых схем введения проиллюстрированы в таблице 6.
В особом варианте осуществления настоящего изобретения способ введения фармацевтической композиции субъекту включает в себя фазу индукции, во время которой дозу 100-300 мг вводят один раз в неделю в течение 13 недель, с последующей поддерживающей фазой, во время которой дозу 80-200 мг вводят один раз в неделю, так долго, как это требуется, эффективно и/или переносится. В некоторых указанных вариантах осуществления настоящего изобретения фармацевтическую композицию вводят подкожно во время фазы индукции и/или поддерживающей фазы. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта имеется наследственная гиперхолестеринемия (гетерозиготная или гомозиготная), ненаследственная гиперхолестеринемия или полигенная гиперхолестеринемия. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая фаза продолжается в течение периода времени от одного дня до конца жизни субъекта или любой ее части, как обсуждалось выше. В некоторых указанных вариантах осуществления настоящего изобретения индукционная доза составляет 100 мг, а поддерживающая доза составляет 80 мг, 100 мг, 140 мг, 180 мг или 200 мг. В некоторых указанных вариантах осуществления настоящего изобретения индукционная доза составляет 200 мг, а поддерживающая доза составляет 80 мг, 100 мг, 140 мг, 180 мг или 200 мг. В некоторых указанных вариантах осуществления настоящего изобретения индукционная доза составляет 300 мг, а поддерживающая доза составляет 80 мг, 100 мг, 140 мг, 180 мг или 200 мг.
В некоторых указанных вариантах осуществления настоящего изобретения введение в конце фазы индукции достигает уменьшения концентрации АроВ в плазме крови приблизительно от -28% до -65%. В некоторых указанных вариантах осуществления настоящего изобретения введение после 13 недель поддерживающей фазы достигает уменьшения концентрации АроВ в плазме крови приблизительно от -32% до -48%, приблизительно от -35% до -52%, приблизительно от -40% до -60%, приблизительно от -43% до -65% или приблизительно от -45% до -67%. В некоторых указанных вариантах осуществления настоящего изобретения введение в конце фазы индукции достигает уменьшения концентрации ЛНП-Х в плазме крови приблизительно от -26% до -60%. В некоторых указанных вариантах осуществления настоящего изобретения введение после 13 недель поддерживающей фазы достигает уменьшения концентрации ЛНП-Х в плазме крови приблизительно от -29% до -44%, приблизительно от -32% до -48%, приблизительно от -37% до -55%, приблизительно от -40% до -61% или приблизительно от -42% до -63%.
В некоторых указанных вариантах осуществления настоящего изобретения введение в конце фазы индукции достигает минимальной концентрации олигонуклеотида в плазме крови, вводимого в качестве части фармацевтической композиции, приблизительно от 11 до 38 нг/мл. В некоторых указанных вариантах осуществления настоящего изобретения введение после 13 недель поддерживающей фазы достигает минимальной концентрации олигонуклеотида в плазме крови, вводимого в качестве части фармацевтической композиции, приблизительно от 7 до 27 нг/мл, приблизительно от 8 до 31 нг/мл, приблизительно от 11 до 38 нг/мл, приблизительно от 13 до 46 нг/мл или приблизительно от 14 до 50 нг/мл. В некоторых указанных вариантах осуществления настоящего изобретения введение в конце фазы индукции достигает концентрации олигонуклеотида в печени, вводимого в качестве части фармацевтической композиции, приблизительно от 55 до 190 мкг/г. В некоторых указанных вариантах осуществления настоящего изобретения введение после 13 недель поддерживающей фазы достигает концентрации олигонуклеотида в печени, вводимого в качестве части фармацевтической композиции, приблизительно от 38 до 133 мкг/г, приблизительно от 44 до 152 мкг/г, приблизительно от 55 до 190 мкг/г, приблизительно от 66 до 228 мкг/г или приблизительно от 7 до 247 мкг/г.
В некоторых указанных вариантах осуществления настоящего изобретения введение в конце фазы индукции достигает уменьшения концентрации АроВ в плазме крови приблизительно от -34% до -77%. В некоторых указанных вариантах осуществления настоящего изобретения введение после 13 недель поддерживающей фазы достигает уменьшения концентрации АроВ в плазме крови приблизительно от -38% до -58%, приблизительно от -43% до -65%, приблизительно от -47% до -70% или приблизительно от -49% до -74%. В некоторых указанных вариантах осуществления настоящего изобретения введение в конце фазы индукции достигает уменьшения концентрации ЛНП-Х в плазме крови приблизительно от -31% до -73%. В некоторых указанных вариантах осуществления настоящего изобретения введение после 13 недель поддерживающей фазы достигает уменьшения концентрации ЛНП-Х в плазме крови приблизительно от -35% до -54%, приблизительно от -40% до -61%, приблизительно от -44% до -66% или приблизительно от -46% до -70%.
В некоторых указанных вариантах осуществления настоящего изобретения введение в конце фазы индукции достигает минимальной концентрации олигонуклеотида в плазме крови, вводимого в качестве части фармацевтической композиции, приблизительно от 16 до 57 нг/мл. В некоторых указанных вариантах осуществления настоящего изобретения введение после 13 недель поддерживающей фазы достигает минимальной концентрации олигонуклеотида в плазме крови, вводимого в качестве части фармацевтической композиции, приблизительно от 10 до 37 нг/мл, приблизительно от 13 до 46 нг/мл, приблизительно от 16 до 55 нг/мл или приблизительно от 18 до 65 нг/мл. В некоторых указанных вариантах осуществления настоящего изобретения введение в конце фазы индукции достигает концентрации олигонуклеотида в печени, вводимого в качестве части фармацевтической композиции, приблизительно от 82 до 285 мкг/г. В некоторых указанных вариантах осуществления настоящего изобретения введение после 13 недель поддерживающей фазы достигает концентрации олигонуклеотида в печени, вводимого в качестве части фармацевтической композиции, приблизительно от 52 до 181 мкг/г, приблизительно от 66 до 228 мкг/г, приблизительно от 80 до 276 мкг/г или приблизительно от 94 до 323 мкг/г.
В другом особом варианте осуществления настоящего изобретения способ введения фармацевтической композиции субъекту включает в себя фазу индукции, во время которой дозу 100-300 мг вводят один раз в неделю в течение 13 недель, с последующей поддерживающей фазой, во время которой дозу 100-200 мг вводят один раз в неделю, так долго, как это требуется, эффективно и/или переносится, когда эффективность и/или переносимость антисмыслового олигонуклеотида мониторируют в течение фазы индукции, поддерживающей фазы или и той, и другой, или любой их части. В некоторых указанных вариантах осуществления настоящего изобретения фармацевтическую композицию вводят подкожно во время фазы индукции и/или поддерживающей фазы. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта имеется наследственная гиперхолестеринемия (гетерозиготная или гомозиготная), ненаследственная гиперхолестеринемия или полигенная гиперхолестеринемия. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая фаза продолжается в течение периода времени от одного дня до конца жизни субъекта или любой ее части, как обсуждалось выше.
В некоторых указанных вариантах осуществления настоящего изобретения скорость уменьшения концентрации АроВ в плазме крови мониторируют по время фазы индукции и/или поддерживающей фазы. В некоторых указанных вариантах осуществления настоящего изобретения концентрацию АроВ в плазме крови мониторируют по время фазы индукции и/или поддерживающей фазы. В некоторых вариантах осуществления настоящего изобретения, если скорость уменьшения концентрации АроВ в плазме крови превышает 30 мг/дл в день, дозу фармацевтической композиции изменяют, например, уменьшают. В некоторых указанных вариантах осуществления настоящего изобретения, если скорость уменьшения концентрации АроВ в плазме крови превышает 30 мг/дл в день, частоту введения фармацевтической композиции изменяют, например, уменьшают. В некоторых указанных вариантах осуществления настоящего изобретения, если концентрация АроВ в плазме крови падает ниже приблизительно 50 мг/дл, дозу фармацевтической композиции изменяют, например, уменьшают. В некоторых вариантах осуществления настоящего изобретения, если концентрация АроВ в плазме крови падает ниже приблизительно 50 мг/дл, частоту введения фармацевтической композиции изменяют, например, уменьшают. В некоторых вариантах осуществления настоящего изобретения, если концентрация АроВ в плазме крови падает ниже приблизительно 60 мг/дл, дозу фармацевтической композиции изменяют, например, уменьшают. В некоторых вариантах осуществления настоящего изобретения, если концентрация АроВ в плазме крови падает ниже приблизительно 60 мг/дл, частоту введения фармацевтической композиции изменяют, например, уменьшают.
В некоторых вариантах осуществления настоящего изобретения, если скорость уменьшения концентрации АроВ в плазме крови превышает 30 мг/дл в день, а концентрация АроВ в плазме крови падает ниже приблизительно 50 мг/дл, дозу фармацевтической композиции изменяют, например, уменьшают. В некоторых вариантах осуществления настоящего изобретения, если скорость уменьшения концентрации АроВ в плазме крови превышает 30 мг/дл в день, а концентрация АроВ в плазме крови падает ниже приблизительно 50 мг/дл, частоту введения фармацевтической композиции изменяют, например, уменьшают. В некоторых вариантах осуществления настоящего изобретения, если скорость уменьшения концентрации АроВ в плазме крови превышает 30 мг/дл в день, а концентрация АроВ в плазме крови падает ниже приблизительно 60 мг/дл, дозу фармацевтической композиции изменяют, например, уменьшают. В некоторых вариантах осуществления настоящего изобретения, если скорость уменьшения концентрации АроВ в плазме крови превышает 30 мг/дл в день, а концентрация АроВ в плазме крови падает ниже приблизительно 60 мг/дл, частоту введения фармацевтической композиции изменяют, например, уменьшают.
В другом особом варианте осуществления настоящего изобретения способ введения фармацевтической композиции субъекту включает в себя фазу индукции, во время которой дозу 100-200 мг вводят один раз в неделю в течение 13 недель, с последующей поддерживающей фазой, во время которой дозу 200-300 мг вводят один раз в неделю, так долго, как это требуется, эффективно и/или переносится, когда переносимость и/или эффективность фармацевтической композиции оценивают во время или в конце фазы индукции или любой ее части. В некоторых указанных вариантах осуществления настоящего изобретения дозу поддерживающей фазы увеличивают относительно дозы фазы индукции, если доза фазы индукции хорошо переносится, а цели лечения не достигнуты. В некоторых указанных вариантах осуществления настоящего изобретения фармацевтическую композицию вводят подкожно во время фазы индукции и/или поддерживающей фазы. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта имеется наследственная гиперхолестеринемия (гетерозиготная или гомозиготная), ненаследственная гиперхолестеринемия или полигенная гиперхолестеринемия. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая фаза продолжается в течение периода времени от одного дня до конца жизни субъекта или любой ее части, как обсуждалось выше. В некоторых указанных вариантах осуществления настоящего изобретения индукционная доза составляет 100 мг, а поддерживающая доза составляет 200 мг. В некоторых указанных вариантах осуществления настоящего изобретения индукционная доза составляет 200 мг, а поддерживающая доза составляет 300 мг. В некоторых вариантах осуществления настоящего изобретения цели лечения оценивают путем мониторинга концентрации в плазме крови АроВ, ЛНП-Х, ЛОНП-Х, не-ЛВП-Х, ЛВП-Х, АроА1, общего холестерина, триглицеридов и ЛП(а). В некоторых вариантах осуществления настоящего изобретения переносимость оценивают путем мониторинга активности АЛТ, активности АСТ и концентраций билирубина в плазме крови.
В некоторых указанных вариантах осуществления настоящего изобретения введение в конце фазы индукции достигает уменьшения концентрации АроВ в плазме крови приблизительно от -17% до -40%. В некоторых указанных вариантах осуществления настоящего изобретения введение после 26 недель поддерживающей фазы достигает уменьшения концентрации АроВ в плазме крови приблизительно от -34% до -63. В некоторых указанных вариантах осуществления настоящего изобретения введение в конце фазы индукции достигает уменьшения концентрации ЛНП-Х в плазме крови приблизительно от -14% до -35%. В некоторых указанных вариантах осуществления настоящего изобретения введение после 13 недель поддерживающей фазы достигает уменьшения концентрации ЛНП-Х в плазме крови приблизительно от -39% до -60%.
В некоторых указанных вариантах осуществления настоящего изобретения введение в конце фазы индукции достигает минимальной концентрации олигонуклеотида в плазме крови, вводимого в качестве части фармацевтической композиции, приблизительно от 5 до 19 нг/мл. В некоторых указанных вариантах осуществления настоящего изобретения введение после 26 недель поддерживающей фазы достигает минимальной концентрации олигонуклеотида в плазме крови, вводимого в качестве части фармацевтической композиции, приблизительно от 12 до 44 нг/мл. В некоторых указанных вариантах осуществления настоящего изобретения введение в конце фазы индукции достигает концентрации олигонуклеотида в печени, вводимого в качестве части фармацевтической композиции, приблизительно от 27 до 95 мкг/г. В некоторых указанных вариантах осуществления настоящего изобретения введение после 26 недель поддерживающей фазы достигает концентрации олигонуклеотида в печени, вводимого в качестве части фармацевтической композиции, приблизительно от 63 до 220 мкг/г.
В другом особом варианте осуществления настоящего изобретения способ введения фармацевтической композиции субъекту включает в себя фазу индукции, во время которой дозу 100-200 мг вводят один раз в неделю в течение 13 недель, с последующей поддерживающей фазой, во время которой дозу 200-300 мг вводят один раз в неделю или два раза в неделю, так долго, как это требуется, эффективно и/или переносится, когда переносимость и/или эффективность фармацевтической композиции оценивают во время или в конце фазы индукции или любой ее части. В некоторых указанных вариантах осуществления настоящего изобретения частоту введения дозы во время поддерживающей фазы уменьшают, если доза фазы индукции не переносится хорошо и/или цели лечения достигнуты. В некоторых указанных вариантах осуществления настоящего изобретения фармацевтическую композицию вводят подкожно во время фазы индукции и/или поддерживающей фазы. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта имеется наследственная гиперхолестеринемия (гетерозиготная или гомозиготная), ненаследственная гиперхолестеринемия или полигенная гиперхолестеринемия. В некоторых указанных вариантах осуществления настоящего изобретения поддерживающая фаза продолжается в течение периода времени от одного дня до конца жизни субъекта или любой ее части, как обсуждалось выше. В некоторых указанных вариантах осуществления настоящего изобретения индукционная доза составляет 100 мг, а поддерживающая доза составляет 200 мг. В некоторых указанных вариантах осуществления настоящего изобретения индукционная доза составляет 200 мг, а поддерживающая доза составляет 300 мг. В некоторых вариантах осуществления настоящего изобретения цели лечения оценивают путем мониторинга концентрации в плазме крови АроВ, ЛНП-Х, ЛОНП-Х, не-ЛВП-Х, ЛВП-Х, АроА1, общего холестерина, триглицеридов и ЛП(а). В некоторых вариантах осуществления настоящего изобретения переносимость оценивают путем мониторинга уровней АЛТ, уровней АСТ, концентраций билирубина в плазме крови или общего билирубина.
D. Проверенные комбинированные лекарственные средства
В некоторых вариантах осуществления настоящего изобретения одну или более фармацевтических композиций по настоящему изобретению вводят совместно с одним или более других фармацевтических агентов. В некоторых вариантах осуществления настоящего изобретения указанный один или более фармацевтических агентов разработаны для лечения того же самого заболевания или состояния, что и одна или более фармацевтических композиций по настоящему изобретению. В некоторых вариантах осуществления настоящего изобретения указанный один или более фармацевтических агентов разработаны для лечения заболевания или состояния, отличающегося от заболевания или состояния, для лечения которых разработаны одна или более фармацевтических композиций по настоящему изобретению. В некоторых вариантах осуществления настоящего изобретения указанный один или более фармацевтических агентов разработаны для лечения нежелательного эффекта одной или более фармацевтических композиций по настоящему изобретению. В некоторых вариантах осуществления настоящего изобретения одну или более фармацевтических композиций по настоящему изобретению вводят совместно с другим фармацевтическим агентом для лечения нежелательного эффекта указанного другого фармацевтического агента. В некоторых вариантах осуществления настоящего изобретения одну или более фармацевтических композиций по настоящему изобретению и один или более других фармацевтических агентов вводят в одно и то же время. В некоторых вариантах осуществления настоящего изобретения одну или более фармацевтических композиций по настоящему изобретению и один или более других фармацевтических агентов вводят в разное время. В некоторых вариантах осуществления настоящего изобретения одну или более фармацевтических композиций по настоящему изобретению и один или более других фармацевтических агентов вводят совместно в виде единой композиции. В некоторых вариантах осуществления настоящего изобретения одну или более фармацевтических композиций по настоящему изобретению и один или более других фармацевтических агентов изготавливают отдельно.
В некоторых вариантах осуществления настоящего изобретения фармацевтические агенты, которые можно совместно вводить с фармацевтической композицией по настоящему изобретению, включают в себя агенты, понижающие уровень липидов. В некоторых указанных вариантах осуществления настоящего изобретения фармацевтические агенты, которые можно совместно вводить с фармацевтической композицией по настоящему изобретению, включают в себя, без ограничения, аторвастатин, симвастатин, розувастатин и эзетимиб. В некоторых указанных вариантах осуществления настоящего изобретения агент, понижающий уровень липидов, вводят до введения фармацевтической композиции по настоящему изобретению. В некоторых указанных вариантах осуществления настоящего изобретения агент, понижающий уровень липидов, вводят после введения фармацевтической композиции по настоящему изобретению. В некоторых указанных вариантах осуществления настоящего изобретения агент, понижающий уровень липидов, вводят в одно и то же время с фармацевтической композицией по настоящему изобретению. В некоторых указанных вариантах осуществления настоящего изобретения доза совместно вводимого агента, понижающего уровень липидов, представляет собой такую же дозу, которая была бы введена, если бы вводили только агент, понижающий уровень липидов, в отдельности. В некоторых указанных вариантах осуществления настоящего изобретения доза совместно вводимого агента, понижающего уровень липидов, меньше дозы, которая была бы введена, если бы вводили только агент, понижающий уровень липидов, в отдельности. В некоторых указанных вариантах осуществления настоящего изобретения доза совместно вводимого агента, понижающего уровень липидов, больше дозы, которая была бы введена, если бы вводили только агент, понижающий уровень липидов, в отдельности.
В некоторых вариантах осуществления настоящего изобретения совместно введенный агент, понижающий уровень липидов, представляет собой ингибитор HMG-CoA редуктазы. В некоторых указанных вариантах осуществления настоящего изобретения ингибитор HMG-CoA редуктазы представляет собой статин. В некоторых указанных вариантах осуществления настоящего изобретения статин выбран из аторвастатина, симвастатина, правастатина, флувастатина и розувастатина.
В некоторых вариантах осуществления настоящего изобретения совместно введенный агент, понижающий уровень липидов, представляет собой ингибитор всасывания холестерина. В некоторых указанных вариантах осуществления настоящего изобретения ингибитор всасывания холестерина представляет собой эзетимиб.
В некоторых вариантах осуществления настоящего изобретения совместно введенный агент, понижающий уровень липидов, представляет собой изготовленные в форме единой композиции ингибитор HMG-CoA редуктазы и ингибитор всасывания холестерина. В некоторых указанных вариантах осуществления настоящего изобретения изготовленный в форме единой композиции агент, понижающий уровень липидов, представляет собой эзетимиб/симвастатин.
В некоторых вариантах осуществления настоящего изобретения совместно введенный агент, понижающий уровень липидов, представляет собой ингибитор белка микросомального переноса триглицеридов.
В некоторых вариантах осуществления настоящего изобретения совместно введенный агент, понижающий уровень липидов, представляет собой олигонуклеотид, выбранный из олигонуклеотида, нацеленного на PCSK9, олигонуклеотида, нацеленного на ACAT-2, олигонуклеотида, нацеленного на эндотелиальную липазу, и олигонуклеотида, нацеленного на СЕТР.
В некоторых вариантах осуществления настоящего изобретения совместно введенный агент, понижающий уровень липидов, представляет собой секвестрант желчных кислот. В некоторых указанных вариантах осуществления настоящего изобретения секвестрант желчных кислот выбран из холестирамина, колестипола и колезевелама.
В некоторых вариантах осуществления настоящего изобретения совместно введенный агент, понижающий уровень липидов, представляет собой никотиновую кислоту. В некоторых указанных вариантах осуществления настоящего изобретения никотиновая кислота выбрана из никотиновой кислоты немедленного высвобождения, никотиновой кислоты продолжительного высвобождения и никотиновой кислоты замедленного высвобождения.
В некоторых вариантах осуществления настоящего изобретения совместно введенный агент, понижающий уровень липидов, представляет собой фибриновую кислоту. В некоторых указанных вариантах осуществления настоящего изобретения фибриновая кислота выбрана из гемфиброзила, фенофибрата, клофибрата, безафибрата и ципрофибрата.
Другие примеры фармацевтических агентов, которые можно вводить совместно с фармацевтической композицией по настоящему изобретению, включают в себя, без ограничения, кортикостероиды, включая, без ограничения, преднизон; иммуноглобулины, включая, без ограничения, внутривенный иммуноглобулин (в/в Ig); анальгетики (например, ацетаминофен); противовоспалительные агенты, включая, без ограничения, нестероидные противовоспалительные лекарственные средства (например, ибупрофен, ингибиторы СОХ-1 и ингибиторы СОХ-2); салицилаты; антибиотики, противовирусные агенты; противогрибковые агенты; противодиабетические агенты (например, бигуаниды, ингибиторы глюкозидазы, инсулины, сульфонилмочевины и тиазолидендионы); адренергические модификаторы; диуретики; гормоны (например, анаболические стероиды, андроген, эстроген, кальцитонин, прогестин, соматостатин и тиреоидные гормоны); иммуномодуляторы; мышечные релаксанты; антигистаминные; агенты для лечения остеопороза (например, бифосфонаты, кальцитонин и эстрогены); простагландины; антинеопластические агенты; психотерапевтические агенты; седативные агенты, продукты сумаха укореняющегося; антитела и вакцины.
В некоторых вариантах осуществления настоящего изобретения фармацевтические композиции по настоящему изобретению можно вводить в сочетании с лечением, понижающим уровень липидов. В некоторых указанных вариантах осуществления настоящего изобретения лечение, понижающее уровень липидов, представляет собой изменение образа жизни в терапевтических целях. В некоторых указанных вариантах осуществления настоящего изобретения лечение, понижающее уровень липидов, представляет собой аферез ЛНП.
Е. Проверенные показания
В некоторых вариантах осуществления изобретение относится к способам лечения субъекта, включающему в себя введение одного или более фармацевтических агентов по настоящему изобретению. В некоторых вариантах осуществления настоящего изобретения указанный субъект имеет гиперхолестеринемию, гиперлипидемию, ненаследственную гиперхолестеринемию, наследственную гиперхолестеринемию, гетерозиготную наследственную гиперхолестеринемию, гомозиготную наследственную гиперхолестеринемию, коронарную болезнь, атеросклероз, смешанную дислипидемию, диабетическую дислипидемию. В некоторых вариантах осуществления настоящего изобретения указанный субъект идентифицирован как имеющий один или более эквивалентов риска CHD. В некоторых вариантах осуществления настоящего изобретения указанный субъект идентифицирован как имеющий главные факторы риска развития коронарной болезни. В некоторых вариантах осуществления настоящего изобретения указанный субъект идентифицирован как имеющий один или более факторов риска CHD. В некоторых вариантах осуществления изобретения указанный субъект идентифицирован как имеющий риск развития атеросклероза. В некоторых вариантах осуществления настоящего изобретения указанный субъект идентифицирован как имеющий коронарную болезнь в анамнезе. В некоторых вариантах осуществления настоящего изобретения указанный субъект идентифицирован как имеющий семейный анамнез раннего возникновения коронарной болезни.
В некоторых вариантах осуществления настоящего изобретения субъект идентифицирован как имеющий повышенный уровень холестерина. В некоторых вариантах осуществления настоящего изобретения субъект идентифицирован как имеющий потребность в лечении, понижающем уровень липидов. В некоторых указанных вариантах осуществления настоящего изобретения субъект идентифицирован как имеющий потребность в лечении, понижающем уровень липидов, согласно руководству, разработанному Национальной программой обучения по холестерину (NCEP), панель III лечения взрослых людей, в 2001 г., и модифицированному Координационным комитетом NCEP в 2004 г. (Grundy et al., Circulation, 2004, 110, 227-239). В некоторых указанных вариантах осуществления настоящего изобретения субъект, нуждающийся в лечении, понижающем уровень липидов, имеет ЛНП-Х выше 190 мг/дл. В некоторых указанных вариантах осуществления настоящего изобретения субъект, нуждающийся в лечении, понижающем уровень липидов, имеет ЛНП-Х выше 160 мг/дл. В некоторых указанных вариантах осуществления настоящего изобретения субъект, нуждающийся в лечении, понижающем уровень липидов, имеет ЛНП-Х выше 130 мг/дл. В некоторых указанных вариантах осуществления настоящего изобретения субъект, нуждающийся в лечении, понижающем уровень липидов, должен поддерживать ЛНП-Х ниже 160 мг/дл. В некоторых указанных вариантах осуществления настоящего изобретения субъект, нуждающийся в лечении, понижающем уровень липидов, должен поддерживать ЛНП-Х ниже 130 мг/дл. В некоторых указанных вариантах осуществления настоящего изобретения субъект, нуждающийся в лечении, понижающем уровень липидов, должен поддерживать ЛНП-Х ниже 100 мг/дл. В некоторых указанных вариантах осуществления настоящего изобретения субъект, нуждающийся в лечении, понижающем уровень липидов, должен поддерживать ЛНП-Х ниже 70 мг/дл.
В некоторых вариантах осуществления настоящее изобретение относится к способам уменьшения концентрации АроВ у субъекта. В некоторых вариантах осуществления настоящее изобретение относится к способу уменьшения концентрации липопротеина, содержащего АроВ, у субъекта. В некоторых вариантах осуществления настоящее изобретение относится к способам уменьшения концентрации ЛНП-Х у субъекта. В некоторых вариантах осуществления настоящее изобретение относится к способам уменьшения концентрации ЛОНП-Х у субъекта. В некоторых вариантах осуществления настоящее изобретение относится к способам уменьшения концентрации ЛПП-Х у субъекта. В некоторых вариантах осуществления настоящее изобретение относится к способам уменьшения концентрации не-ЛВП-Х у субъекта. В некоторых вариантах осуществления настоящее изобретение относится к способам уменьшения концентрации ЛП(а) у субъекта. В некоторых вариантах осуществления настоящее изобретение относится к способам уменьшения концентрации триглицеридов в сыворотке крови у субъекта. В некоторых вариантах осуществления настоящее изобретение относится к способам уменьшения концентрации окси-ЛНП-Х у субъекта. В некоторых указанных вариантах осуществления настоящего изобретения уменьшение АроВ, ЛНП-Х, ЛОНП-Х, ЛПП-Х, общего холестерина, не-ЛВП-Х, ЛП(а), триглицеридов или окси-ЛНП-Х выбрано, независимо, из по меньшей мере 10%, по меньшей мере 15%, по меньшей мере 20%, по меньшей мере 25%, по меньшей мере 30%, по меньшей мере 35%, по меньшей мере 40%, по меньшей мере 45%, по меньшей мере 50%, по меньшей мере 55%, по меньшей мере 60%, по меньшей мере 65%, по меньшей мере 70%, по меньшей мере 75%, по меньшей мере 80%, по меньшей мере 85%, по меньшей мере 90%, по меньшей мере 95% и по меньшей мере 100%. В некоторых указанных вариантах осуществления настоящего изобретения уменьшение АроВ, ЛНП-Х, ЛОНП-Х, ЛПП-Х, общего холестерина, не-ЛВП-Х, ЛП(а), триглицеридов или окси-ЛНП-Х выбрано, независимо, из по меньшей мере 20%, по меньшей мере 30%, по меньшей мере 40%, по меньшей мере 50%, по меньшей мере 60% и по меньшей мере 70%. В некоторых указанных вариантах осуществления настоящего изобретения уменьшение АроВ, ЛНП-Х, ЛОНП-Х, ЛПП-Х, общего холестерина, не-ЛВП-Х, ЛП(а), триглицеридов или окси-ЛНП-Х выбрано, независимо, из по меньшей мере 40%, по меньшей мере 50%, по меньшей мере 60% и по меньшей мере 70%.
В некоторых вариантах осуществления изобретение относится к способу увеличения концентрации ЛВП-Х у субъекта.
В некоторых вариантах осуществления способы по настоящему изобретению не понижают ЛВП-Х. В некоторых вариантах осуществления способы по настоящему изобретению не приводят к накоплению липидов в печени.
В некоторых вариантах осуществления изобретение относится к способам уменьшения концентрации АроВ у субъекта, в то же время уменьшающим побочные эффекты, связанные с лечением. В некоторых указанных вариантах осуществления настоящего изобретения побочный эффект представляет собой гепатотоксичность. В некоторых указанных вариантах осуществления настоящего изобретения побочный эффект представляет собой нарушение функции печени. В некоторых указанных вариантах осуществления настоящего изобретения побочный эффект представляет собой воспаление печени или другое неблагоприятное событие, которое наблюдается в печени. В некоторых указанных вариантах осуществления настоящего изобретения побочный эффект представляет собой повышение уровня аланин-аминотрансферазы (АЛТ). В некоторых указанных вариантах осуществления настоящего изобретения побочный эффект представляет собой повышение уровня аспартат-аминотрансферазы (АСТ). Например, некоторые схемы введения по настоящему изобретению приводят к эффективному уменьшению концентрации АроВ при меньшей гепатотоксичности по сравнению с гепатотоксичностью, которая была установлена в исследованиях с использованием других схем введения. В некоторых вариантах осуществления настоящего изобретения схемы введения по настоящему изобретению приводят к эффективному уменьшению концентрации АроВ при меньшем подъеме АЛТ. В некоторых указанных вариантах осуществления настоящего изобретения количество введенной индукционной дозы меньше количества введенной поддерживающей дозы.
В некоторых вариантах осуществления изобретение относится к способам уменьшения концентрации АроВ у субъекта, у которого не были достигнуты желательные уровни ЛНП-Х в результате лечения, понижающего уровень липидов. В некоторых указанных вариантах осуществления настоящего изобретения ISIS 301012 является единственным агентом, понижающим уровень липидов, введенным субъекту. В некоторых указанных вариантах осуществления настоящего изобретения субъект не выполнял рекомендованное лечение, понижающее уровень липидов. В некоторых указанных вариантах осуществления настоящего изобретения фармацевтическую композицию по настоящему изобретению вводят совместно с дополнительным лечением, понижающим уровень липидов. В некоторых указанных вариантах осуществления настоящего изобретения лечение, понижающее уровень липидов, представляет собой аферез ЛНП. В некоторых указанных вариантах осуществления настоящего изобретения лечение, понижающее уровень липидов, представляет собой статин. В некоторых указанных вариантах осуществления настоящего изобретения лечение, понижающее уровень липидов, представляет собой эзетимиб.
В некоторых вариантах осуществления изобретение относится к способам уменьшения концентрации АроВ у субъекта, который не переносит статин. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта наблюдается увеличение концентрации креатин-киназы в результате введения статина. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта наблюдаются мышечные боли в результате введения статина. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта наблюдаются побочные эффекты со стороны центральной нервной системы в результате введения статина. В некоторых указанных вариантах осуществления настоящего изобретения субъект не выполнял рекомендованное введение статина.
В некоторых вариантах осуществления изобретение относится к способам понижения уровня триглицеридов в печени у субъекта. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта имеется повышенный уровень триглицеридов в печени. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта имеется стеатогепатит. В некоторых указанных вариантах осуществления настоящего изобретения у субъекта имеется стеатоз. В некоторых указанных вариантах осуществления настоящего изобретения уровни триглицеридов в печени измеряют с помощью магнитно-резонансной томографии.
В некоторых вариантах осуществления изобретение относится к способам уменьшения риска развития коронарной болезни у субъекта. В некоторых вариантах осуществления изобретение относится к способам замедления прогрессирования атеросклероза у субъекта. В некоторых указанных вариантах осуществления изобретение относится к способам остановки прогрессирования атеросклероза у субъекта. В некоторых указанных вариантах осуществления изобретение относится к способам уменьшения размера и/или распространенности атеросклеротических бляшек у субъекта. В некоторых вариантах осуществления настоящего изобретения способы обеспечивают уменьшение риска развития атеросклероза у субъекта.
В некоторых вариантах осуществления настоящего изобретения способы обеспечивают улучшение исхода сердечно-сосудистой патологии у субъекта. В некоторых указанных вариантах осуществления настоящего изобретения улучшение исхода сердечно-сосудистой патологии представляет собой уменьшение риска развития коронарной болезни. В некоторых указанных вариантах осуществления настоящего изобретения улучшение исхода сердечно-сосудистой патологии представляет собой уменьшение риска одного или более больших сердечно-сосудистых событий, которые включают в себя, без ограничения, смерть, инфаркт миокарда, повторный инфаркт, инсульт, кардиогенный шок, отек легких, остановку сердца и дисритмию предсердий. В некоторых указанных вариантах осуществления настоящего изобретения улучшение исхода сердечно-сосудистой патологии представляет собой улучшение толщины интимальной средней оболочки сонных артерий. В некоторых указанных вариантах осуществления настоящего изобретения улучшение толщины интимальной средней оболочки сонных артерий представляет собой уменьшение толщины. В некоторых указанных вариантах осуществления настоящего изобретения улучшение толщины интимальной средней оболочки сонных артерий представляет собой профилактику увеличения толщины интимальной средней оболочки.
НЕОГРАНИЧИВАЮЩЕЕ ОПИСАНИЕ И ВКЛЮЧЕНИЕ ССЫЛОК
В то время как описано с использованием определенных вариантов его осуществления, следующие примеры служат лишь для иллюстрации изобретения и не предназначены для его ограничения. Все ссылки, инвентарные номера в GenBank и т.п., упомянутые в настоящей заявке, целиком включены в нее в качестве ссылок.
ПРИМЕРЫ
Пример 1
Субъекты
Трех субъектов идентифицировали как имеющих гомозиготную наследственную гиперхолестеринемию с использованием следующих критериев: (1) задокументированный уровень ЛПН-Х более 500 мг/дл в отсутствии лечения, понижающего уровень липидов в анамнезе; и (2) по меньшей мере одно из: (а) генетическое тестирование, подтверждающее 2 мутировавших гена рецептора ЛНП; (b) сухожильные и/или кожные ксантомы до достижения возраста 10 лет; или (с) у обоих родителей имеется документально зафиксированный повышенный уровень ЛПН-Х до лечения, понижающего уровень липидов, который соответствует гетерозиготной наследственной гиперхолестеринемии (если история болезни родителей была не доступна, то анамнез коронарной болезни первой степени у мужчин моложе 55 лет или первой степени у женщин моложе 60 лет использовали в качестве критерия вместо истории болезни родителей).
Схема введения
Три субъекта получали дозу ISIS 301012 300 мг в дни 1, 4, 8, 11, 15, 22, 29, 36, 43, 50, 57, 64, 71, 78 и 85, как описано в таблице ниже. ISIS 301012 вводили подкожно. Четыре дозы в дни 1, 4, 8 и 11 вводили, чтобы установить уровни ISIS 301012 в ткани печени, которые составляют приблизительно 60-90% устойчивой концентрации. Осуществляли мониторинг концентраций АроВ, ЛНП-Х, ЛОНП-Х, не-ЛВП-Х, ЛВП-Х, АроА1, общего холестерина, триглицеридов и ЛП(а) у субъектов. Также осуществляли мониторинг у субъектов для гарантии приемлемых профилей безопасности. Субъекты также получали один или более видов лечения, понижающего уровень липидов.
Результаты
На день 43, после 9 доз ISIS 301012, АроВ и ЛНП-Х снизились приблизительно на 30% (n=3) по сравнению с исходной концентрацией у субъекта до введения ISIS 301012.
На день 78, после 14 доз ISIS 301012, (n=2; один субъект на момент написания данного документа еще не получил 14-й дозы) АроВ и ЛНП-Х снизились приблизительно на 50%. Концентрация ЛОНП-Х снизилась приблизительно на 30%. Уменьшение концентраций не-ЛВП-Х составило от 32 до 50%. Уменьшение концентраций общего холестерина составило от 30 до 46%. Уменьшение концентраций триглицеридов составило от 29 до 33%. Наблюдалось уменьшение концентраций ЛП(а) 19%, 20% и 54%. Наблюдалось увеличение концентраций ЛВП-Х 5%, 7% и 40%.
Приведенные данные показывают, что ISIS 301012 уменьшает концентрации липидов у субъектов, имеющих наследственную гиперхолестеринемию. Уменьшение ЛНП-Х у субъектов, имеющих наследственную гиперхолестеринемию, наблюдались позже, во время периода введения, по уменьшением ЛНП-Х у субъектов, имеющих ненаследственную гиперхолестеринемию, что наводит на мысль о том, что более продолжительный период индукции у субъектов, имеющих наследственную гиперхолестеринемию, может иметь благоприятный терапевтический эффект у указанных субъектов. Например, период индукции продолжительностью по меньшей мере 8 недель может иметь благоприятный терапевтический эффект у субъектов, имеющих ненаследственную гиперхолестеринемию, в то время как более короткие периоды индукции или периоды индукции с использованием более низких доз могут быть достаточными для субъектов, имеющих ненаследственную гиперхолестеринемию.
Пример 2 - Скорость и величина уменьшения АроВ под действием ISIS 301012
Не будучи связанными с конкретной теорией, полагают, что незначительные повышения уровней АЛТ, наблюдающиеся во время первоначального лечения ISIS 301012 при схемах лечения с использованием нагрузочного периода множеством высоких доз, отражают чрезвычайную понижающую уровень липидов активность. В частности, подъем АЛТ можно отнести на счет скорости и величины понижения липидов. Подъем АЛТ можно уменьшить или предотвратить с использованием схемы введения, которая ограничивает скорость и величину уменьшения АроВ во время индукции или периода первоначального введения, приблизительно во время первых 15-90 дней лечения. Так, в первое время скорость и величина уменьшения АроВ могут коррелировать с уровнями АЛТ.
Субъекты
Субъекты имели гиперхолестеринемию и получали постоянное лечение статином. Для субъектов, участвовавших в испытаниях, определяли уровни АроВ и АЛТ. В таблице 8 представлены уровни АроВ для отдельных субъектов и полученные в результате уровни АЛТ.
Результаты
Представленные данные показывают, что повышения уровней АЛТ могут наблюдаться, когда субъекты имеют показатели ниже двух порогов, минимальной концентрации АроВ приблизительно 60 мг/дл или менее во время первых 15-60 дней и средней скорости уменьшения 1,5 мг/дл в день или более в течение первых 15-60 дней лечения, которые могут вызывать повышение АЛТ приблизительно 100 ЕД/л или более. Это также можно рассматривать в переводе на приблизительно 15% падение в день или более или изменение более чем на 30 мг/дл за первые 30 дней. АЛТ 100 ЕД/л считается в три раза превышающим верхнюю границу нормы. Верхняя граница нормальной концентрации АЛТ составляет приблизительно 30 ЕД/л. Величина, превышающая указанную границу в три раза, должная считаться неблагоприятной.
Оценивали двести сорок четыре субъекта из 6 различных клинических испытаний, включая здоровых добровольцев, субъекта с наследственной гиперхолестеринемией, субъекта с полигенной гиперхолестеринемией и субъекта, получающего лечение статином, и пороговые уровни АроВ были связаны с уровнями АЛТ. Таблица 9 показывает количество субъектов, имеющих положительный наклон кривой (скорость уменьшения АроВ более 1 мг/дл в день), и субъектов, нарушавших порог величины уменьшения (концентрация АроВ менее 60 мг/дл). У 80% указанных субъектов (32 из 41) наблюдалось повышение АЛТ в три раза или более по сравнению с верхней границей нормы.
18(0) - не поддается интерпретации из-за пограничных пороговых уровней
Пример 3 - Уменьшение АроВ при использовании ISIS 301012 у субъектов с полигенной гиперхолестеринемией
Субъекты
Субъектов идентифицировали как имеющих полигенную гиперхолестеринемию. Обычно у субъектов имелись уровни ЛНП-Х выше приблизительно 130 мг/дл в отсутствии лечения, понижающего уровень липидов.
Схема введения
Дозы получали три группы из 8 пациентов. Группы получали дозы 200, 300 или 400 мг ISIS 301012 один раз в неделю в течение 13 недель без первоначальной нагрузки. ISIS 301012 вводили путем подкожной инъекции.
** 4 из 8 пациентов достигли нижней границы обнаружения для АроВ
Результаты
Падение АроВ по сравнению с исходной величиной через 2 недели в группах, получавших дозу 200 и 300 мг/кг, приводило в среднем к падению скорости и/или величины, которое не соответствовало или не превышало пороговые требования, изложенные в примере 2. В трех экспериментальных группах в течение периода введения лекарственного средства не наблюдалось подъемов АЛТ. Падение АроВ по сравнению с исходной величиной через 2 недели в группе, получавшей дозу 400 мг/кг, приводило в среднем к падению скорости и/или величины, которое превышало пороговые требования, изложенные в примере 2, и давало повышение АроВ в 3 ВГН.
Пример 4 - Прогнозируемый эффект длительной индукции с использованием высокой дозы
Субъекты
Субъектов идентифицировали как имеющих наследственную или полигенную гиперхолестеринемию. Обычно у субъектов имелись уровни ЛНП-Х выше приблизительно 120 мг/дл в отсутствии лечения, понижающего уровень липидов.
Схема введения
Сначала субъектам вводили лекарственное средство с использованием длительного периода индукции при более высокой дозе, чем поддерживающая доза. Группа А получала дозу 200 мг ISIS 301012 один раз в неделю в течение 13 недель. Группа В получала дозу 300 мг ISIS 301012 один раз в неделю в течение 13 недель. Через 13 недель субъекты переходили к получению уменьшенной поддерживающей дозы. Группа А получала дозу 100 мг ISIS 301012 один раз в неделю, а группа В получала дозу 200 мг ISIS 301012 один раз в неделю. ISIS 301012 можно вводить подкожно или любым другим способом, описанным в настоящем документе. Индукционные дозы вводили в течение 13 недель, чтобы достичь установившихся уровней ISIS 301012 в ткани печени, которые составляли приблизительно 60-90% устойчивой концентрации. Осуществляли мониторинг концентраций АроВ, ЛНП-Х, ЛОНП-Х, не-ЛВП-Х, ЛВП-Х, АроА1, общего холестерина, триглицеридов и ЛП(а) у субъектов. Также осуществляли мониторинг у субъектов для гарантии приемлемых профилей безопасности, включая АЛТ, АСТ и билирубин.
Несмотря на то, что выше в качестве примера приведены дозы 100 и 200 мг в неделю, поддерживающие дозы могут быть выше или ниже. В таблицах 9, 10, 11 и 12 приведены предсказанные величины, основанные на смоделированных схемах введения. Модели основаны на данных клинических испытаний, полученных к настоящему времени, и, особенно, клинических испытаний, касающихся полигенной гиперхолестеринемии, с использованием монотерапии, примера 3. На основе индукции 200 или 300 мг в неделю, предсказаны уровни АроВ и ЛНП, а также минимальные концентрации в плазме крови и концентрации в печени для фазы индукции и поддерживающей фазы.
Результаты в группе А
Предсказано, что к концу 13-недельной фазы индукции средняя концентрация в печени 301012 в группе А составит приблизительно 123 мкг/г. Указанная концентрация сохраняется при поддерживающей дозе приблизительно 100 мг в неделю. К концу 13-недельной фазы индукции среднее процентное изменение уровней АроВ по сравнению с исходными величинами составляет приблизительно 46,9%. На 13 неделе поддерживающей фазы среднее процентное изменение уровней АроВ по сравнению с исходными величинами сохраняется. К концу 13-недельной фазы индукции среднее процентное изменение уровней ЛНП по сравнению с исходными величинами составляет приблизительно 43,3%. На 13 неделе поддерживающей фазы среднее процентное изменение уровней ЛНП по сравнению с исходными величинами сохраняется. К концу 13-недельной фазы индукции минимальная концентрация в плазме крови составляет приблизительно 24,5 нг/мл. На 13 неделе поддерживающей фазы минимальная концентрация в плазме крови сохраняется.
Результаты в группе В
Предсказано, что к концу 13-недельной фазы индукции средняя концентрация в печени 301012 в группе А составит приблизительно 184 мкг/г. Указанная концентрация сохраняется при поддерживающей дозе приблизительно 200 мг в неделю. К концу 13-недельной фазы индукции среднее процентное изменение уровней АроВ по сравнению с исходными величинами составляет приблизительно 56%. На 13 неделе поддерживающей фазы среднее процентное изменение уровней АроВ по сравнению с исходными величинами сохраняется. К концу 13-недельной фазы индукции среднее процентное изменение уровней ЛНП по сравнению с исходными величинами составляет приблизительно 52,5%. На 13 неделе поддерживающей фазы среднее процентное изменение уровней ЛНП по сравнению с исходными величинами сохраняется. К концу 13-недельной фазы индукции минимальная концентрация в плазме крови составляет приблизительно 36,8 нг/мл. На 13 неделе поддерживающей фазы минимальная концентрация в плазме крови сохраняется.
Пример 5 - Прогнозируемый эффект длительной индукции с использованием низкой дозы
Субъекты
Субъектов идентифицировали как имеющих полигенную или наследственную гиперхолестеринемию. Обычно у субъектов имелись уровни ЛНП-Х выше приблизительно 160 мг/дл в отсутствии лечения, понижающего уровень липидов.
Схема введения
Сначала субъектам вводили лекарственное средство с использованием длительного периода индукции при более низкой дозе, чем поддерживающая доза. Группа А получала дозу 100 мг ISIS 301012 один раз в неделю в течение 13 недель. Группа В получала дозу 200 мг ISIS 301012 один раз в неделю в течение 13 недель. Через 13 недель оценивали состояние субъектов на основе переносимости и эффективности с учетом целей лечения. Если дозы хорошо переносились, а цели лечения не были достигнуты, пациентов переводили на увеличенные поддерживающие дозы. Группа А получала дозу 200 мг ISIS 301012 один раз в неделю, а группа В получала дозу 300 мг ISIS 301012 один раз в неделю. ISIS 301012 вводили подкожно. Индукционные дозы вводили в течение 13 недель, чтобы достичь установившихся уровней ISIS 301012 в ткани печени, которые составляли приблизительно 60-90% устойчивой концентрации. Осуществляли мониторинг концентраций АроВ, ЛНП-Х, ЛОНП-Х, не-ЛВП-Х, ЛВП-Х, АроА1, общего холестерина, триглицеридов и ЛП(а) у субъектов. Также осуществляли мониторинг у субъектов для гарантии приемлемых профилей безопасности (включая АЛТ, АСТ и билирубин).
В таблицах 14 и 15 приведены предсказанные величины, основанные на смоделированных схемах введения. Модели основаны на данных клинических испытаний, полученных к настоящему времени, и, особенно, клинических испытаний, касающихся полигенной гиперхолестеринемии, с использованием монотерапии, примера 3. На основе индукции 100 мг в неделю, предсказаны уровни АроВ и ЛНП, а также минимальные концентрации в плазме крови и концентрации в печени для фазы индукции и поддерживающей фазы.
Результаты в группе А
Предсказано, что к концу 13-недельной фазы индукции средняя концентрация в печени 301012 в группе А составит приблизительно 61,3 мкг/г. Указанная концентрация возросла до 141 после 13 недель поддерживающей дозы 200 мг в неделю. К концу 13-недельной фазы индукции среднее процентное изменение уровней АроВ по сравнению с исходными величинами составляет приблизительно 28,4%. На 26 неделе введения лекарственного средства среднее процентное изменение уровней АроВ по сравнению с исходными величинами составляет 53,1%. К концу 13-недельной фазы индукции среднее процентное изменение уровней ЛНП по сравнению с исходными величинами составляет приблизительно 24,5%. После 13 недель поддерживающей фазы среднее процентное изменение уровней ЛНП по сравнению с исходными величинами возрастает до 49,6. К концу 13-недельной фазы индукции минимальная концентрация в плазме крови составляет приблизительно 12,3 нг/мл. Прогнозируется, что на 13 неделе поддерживающей фазы минимальная концентрация в плазме крови возрастет до 28,2 нг/мл.
Пример 6 - интервал введения раз в две недели
Субъекты
Субъектов идентифицировали как имеющих полигенную гиперхолестеринемию. Обычно у субъектов имелись уровни ЛНП-Х выше приблизительно 120 мг/дл в отсутствии лечения, понижающего уровень липидов.
Схема введения
ISIS 301012 вводили путем п/к инъекции в дозе 200 мг два раза в неделю в течение двух недель, а затем в дозе 200 мг один раз в две недели в течение 11 недель.
** (% изменения по сравнению с исходный уровнем)
Результаты
Результаты показывают эффективность введения ISIS 301012 один раз в две недели, без сопутствующего повышения АЛТ.
Пример 7 - Предсказанный эффект интервала введения один раз в две недели с фазой индукции
Субъекты
Субъектов идентифицировали как имеющих наследственную гиперхолестеринемию, и они представляют собой популяцию с более низкой переносимостью лекарственного средства, такую как страдающие диабетом и имеющие смешанную гиперлипидемию. Обычно у субъектов имелись уровни ЛНП-Х выше приблизительно 130 мг/дл в отсутствии лечения, понижающего уровень липидов.
Схема введения
ISIS 301012 вводили путем п/к инъекции в дозе 200 мг один раз в неделю в течение 13 недель, а затем в дозе 200 мг один раз в две недели в течение 52 недель.
Результаты
Результаты показывают, что введение ISIS 301012 один раз в две недели может представлять собой эффективную схему и, возможно, не приведет к повышению уровней АЛТ.
Пример 8 - Предсказанный эффект интервала введения один раз в месяц с фазой индукции
Субъекты
Субъектов идентифицировали как имеющих наследственную гиперхолестеринемию, и они представляют собой популяцию с более низкой переносимостью лекарственного средства, такую как страдающие диабетом и имеющие смешанную гиперлипидемию. Обычно у субъектов имелись уровни ЛНП-Х выше приблизительно 130 мг/дл в отсутствии лечения, понижающего уровень липидов.
Схема введения
ISIS 301012 вводили путем п/к инъекции или любым другим способом, описанным в настоящем документе. В некоторых вариантах осуществления настоящего изобретения субъектам можно вводить 200, 400 или 800 мг один раз в два месяца в течение 12 месяцев (фиг.1). В некоторых вариантах осуществления настоящего изобретения субъектам вводят 200 мг в неделю в течение 1 месяца, а затем 200, 400 или 600 мг один раз в месяц в течение 11 месяцев (фиг.2). В некоторых вариантах осуществления настоящего изобретения субъектам вводят 200 мг в неделю в течение 3 месяцев, а затем 200, 400 или 600 мг один раз в месяц в течение 9 месяцев (фиг.3).
Результаты
Результаты предсказывают, что введение ISIS 301012 один раз в месяц с периодом индукции или без него может представлять собой эффективную схему и, возможно, не приведет к повышению уровней АЛТ.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОСТАВЫ ДЛЯ ЛЕЧЕНИЯ И ПРЕДОТВРАЩЕНИЯ БОКОВОГО АМИОТРОФИЧЕСКОГО СКЛЕРОЗА | 2019 |
|
RU2824046C2 |
| КОБИТОЛИМОД ДЛЯ ПРИМЕНЕНИЯ ПРИ ЛЕЧЕНИИ ВОСПАЛИТЕЛЬНОГО ЗАБОЛЕВАНИЯ КИШЕЧНИКА | 2018 |
|
RU2766356C2 |
| КОМПОЗИЦИИ И СПОСОБЫ МОДУЛЯЦИИ SMN2 СПЛАЙСИНГА У СУБЪЕКТА | 2010 |
|
RU2683772C2 |
| КОМПОЗИЦИИ И СПОСОБЫ МОДУЛЯЦИИ SMN2 СПЛАЙСИНГА У СУБЪЕКТА | 2010 |
|
RU2566724C9 |
| КОМПОЗИЦИИ И СПОСОБЫ МОДУЛЯЦИИ SMN2 СПЛАЙСИНГА У СУБЪЕКТА | 2019 |
|
RU2793459C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ХОЛЕСТАЗА | 2020 |
|
RU2829466C2 |
| Лечение рака | 2013 |
|
RU2689548C2 |
| ПРИМЕНЕНИЕ ЗАМЕЩЕННЫХ ЦИАНОПИРРОЛИДИНОВ И СОДЕРЖАЩИХ ИХ КОМБИНИРОВАННЫХ ПРЕПАРАТОВ ДЛЯ ЛЕЧЕНИЯ ГИПЕРЛИПИДЕМИИ И АССОЦИИРОВАННЫХ ЗАБОЛЕВАНИЙ | 2003 |
|
RU2362555C2 |
| СПОСОБЫ ПОВЫШЕНИЯ РОСТА ПАЦИЕНТОВ ДЕТСКОГО ВОЗРАСТА С ХОЛЕСТАТИЧЕСКИМ ЗАБОЛЕВАНИЕМ ПЕЧЕНИ | 2020 |
|
RU2822484C2 |
| ПРИМЕНЕНИЕ ТИАОКСОСОЕДИНЕНИЙ ДЛЯ УМЕНЬШЕНИЯ СОДЕРЖАНИЯ АРО С3 | 2015 |
|
RU2705991C2 |
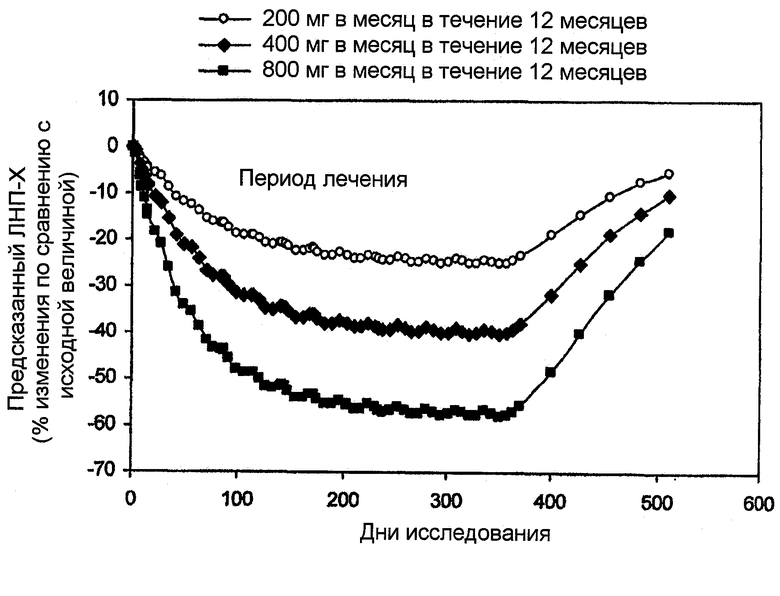
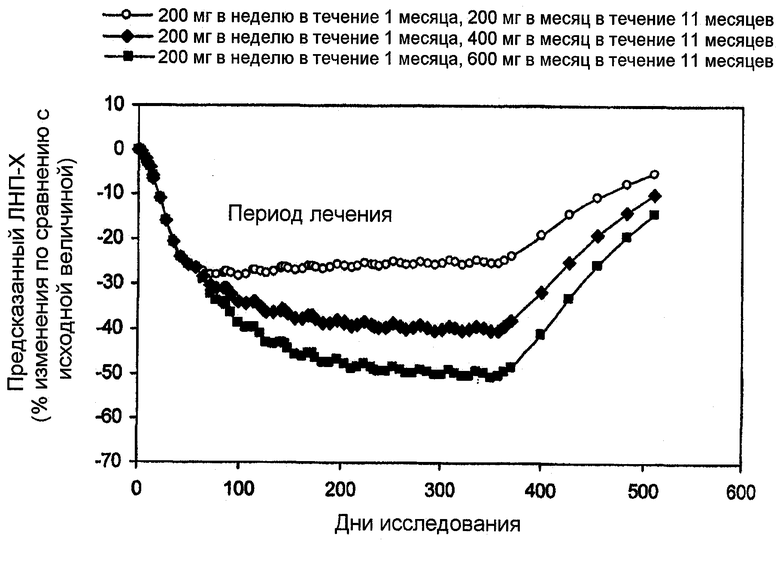
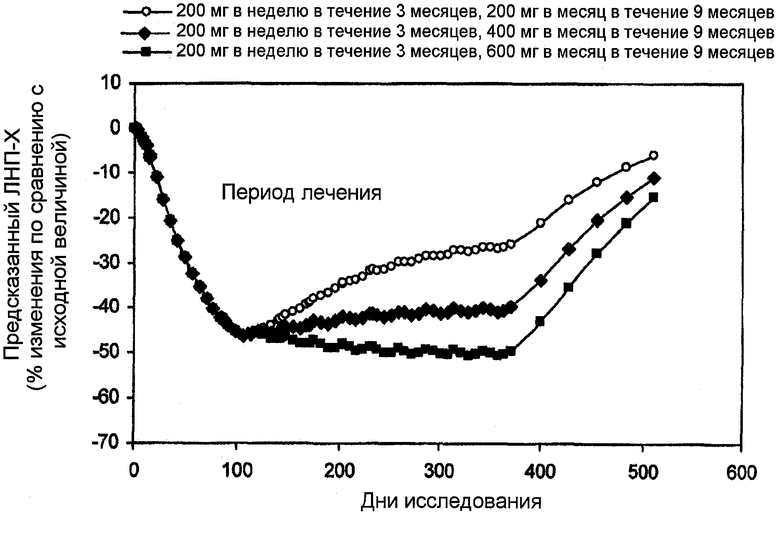
Изобретение относится к области биотехнологии, конкретно к использованию антисмыслового олигонуклеотида ISIS 301012 для долгосрочного понижения уровней АроВ, и может быть использовано в медицине. Способ предусматривает введение субъекту ISIS 301012 в составе фармацевтической композиции, путем введения ISIS 301012 в фазе индукции в дозе 210 мг в неделю в течение по меньшей мере 13 недель, с последующей поддерживающей фазой, во время которой вводят ISIS 301012 в дозе 210 мг в неделю в течение такого периода времени, который является необходимым, эффективным и/или переносимым. При этом каждая индукционная доза и каждая поддерживающая доза независимо включают более двух инъекций. Изобретение позволяет ослабить побочные эффекты терапии, а именно уменьшить эритему в месте введения препарата. 13 з.п. ф-лы, 3 ил., 21 табл., 8 пр.
1. Способ уменьшения концентрации аполипопротеина В-100 (АроВ) и/или АроВ содержащего липопротеина у субъекта, включающий
введение субъекту фармацевтической композиции, включающей антисмысловой олигонуклеотид ISIS 301012, где указанное введение включает фазу индукции, во время которой вводят дозу ISIS 301012 в 210 мг в неделю в течение по меньшей мере 13 недель, с последующей поддерживающей фазой, во время которой вводят дозу ISIS 301012 в 210 мг в неделю в течение такого периода времени, который является необходимым, эффективным и/или переносимым, и где каждая индукционная доза и каждая поддерживающая доза независимо включают более двух инъекций.
2. Способ по п.1, где переносимость или эффективность ISIS 301012 оценивают во время или в конце периода индукции или его части один раз в неделю во время поддерживающей фазы.
3. Способ по п.1, где парентеральное введение включает подкожное введение.
4. Способ по п.1, дополнительно включающий оценку переносимости или эффективности ISIS 301012 во время или в конце периода индукции или его части.
5. Способ по п.4, где эффективность ISIS 301012 оценивают путем мониторинга концентрации АроВ в плазме крови указанного субъекта.
6. Способ по п.1, где указанный субъект имеет гиперхолестеринемию.
7. Способ по п.6, где указанный субъект имеет полигенную
гиперхолестеринемию.
8. Способ по п.6, где указанный субъект имеет наследственную гиперхолестеринемию.
9. Способ по п.8, где указанный субъект имеет гомозиготную наследственную гиперхолестеринемию.
10. Способ по п.8, где указанный субъект имеет гетерозиготную наследственную гиперхолестеринемию.
11. Способ по п.1, где указанная поддерживающая фаза включает введение указанной фармацевтической композиции на протяжении всей жизни субъекта.
12. Способ по п.1, где продолжительность указанной поддерживающей фазы составляет от одной недели до двадцати лет.
13. Способ по п.1, где указанное введение указанной фармацевтической композиции приводит к понижению АроВ по меньшей мере на 10%.
14. Способ по п.13, где указанное понижение АроВ составляет от 10% до 80%, от 20% до 70%, от 30% до 60% или от 30% до 70%.
| US 20060035858 A1, 16.02.2006 | |||
| BURNETT JR., Drug evaluation: ISIS-301012, an antisense oligonucleotide for the treatment of hypercholesterolemia, Curr | |||
| Opin | |||
| Mol | |||
| Ther., 2006, v.8, n.5, p.461-467 | |||
| WO 03097662 A1, 27.11.2003 | |||
| WO2004044181 A2, 27.05.2004 | |||
| KASTELEIN J.J | |||
| et al., Potent reduction of apolipoprotein B and low-density lipoprotein |

