Перекрестная ссылка на родственные заявки
В настоящей заявке испрашивается приоритет предварительной заявки на патент США 61/303472, поданной 11 февраля 2010, которая во всей своей полноте вводится в настоящее описание посредством ссылки.
Предшествующий уровень техники
N-метил-D-аспартатный (NMDA) рецептор представляет собой постсинаптический ионотропный рецептор, восприимчивый, inter alia, к возбуждению аминокислотами глутаматом и глицином и к синтезированному соединению NMDA. NMDA-рецептор регулирует поток двухвалентных и моновалентных ионов в постсинаптический нейрон посредством канала, ассоциированного с рецептором (Foster et al, Nature 1987, 329:395-396; Mayer et al, Trends in Pharmacol. Sci. 1990, 11:254-260). NMDA-рецептор участвует в развитии нейронов, в процессе формирования архитектуры нейронов и в синаптической передаче сигнала, и может участвовать в эксперимент-зависимых синаптических модуляциях. Кроме того, очевидно, что NMDA-рецепторы также участвуют в долговременном потенцировании и в расстройствах центральной нервной системы.
NMDA-рецептор играет главную роль в сообщении синаптической системе пластичности, которая лежит в основе многих более сложных когнитивных функций, таких как приобретение информации, способность ее сохранения в памяти и способность к обучению, а также в некоторых когнитивных процессах и в восприятии боли (Collingridge et al., The NMDA Receptor, Oxford University Press, 1994). Кроме того, было высказано предположение, что некоторые свойства NMDA-рецепторов могут играть определенную роль в обработке информации в головном мозге, который сам по себе ответственен за формирование сознания.
NMDA-рецептор представляет особый интерес, поскольку этот рецептор, как очевидно, участвует в индуцировании расстройств ЦНР широкого спектра. Так, например, при ишемии головного мозга, вызываемой инсультом или травмами, избыточное количество возбуждающей аминокислоты, а именно, глутамата, высвобождается из пораженных нейронов или из нейронов, обедненных кислородом. Такой избыток глутамата связывается с рецепторами NMDA, которые открывают дискриминационное окно для лиганда в ионных каналах, в результате чего происходит приток кальция, который продуцирует высокий уровень внутриклеточного кальция, активирующего каскад биохимических реакций, что приводит к разложению белка и к гибели клеток. Также считается, что этот феномен, известный как экситотоксичность, ответственен за различные расстройства, начиная от остановки сердца и до эпилепсии. Кроме того, уже появились сообщения, указывающие на то, что этот феномен ответственен за аналогичные хронические нейродегенеративные расстройства, такие как болезнь Гентингтона, болезнь Паркинсона и болезнь Альцгеймера. Было показано, что NMDA-рецептор ответственен за постишемические судороги, и было также показано, что в некоторых моделях эпилепсии, активация NMDA-рецептора необходима для индуцирования эпилептических припадков. Также известно, что NMDA-рецептор участвует в нейропсихических процессах, поскольку блокада Ca++-канала NMDA-рецептором под действием анестетика PCP (фенилциклидина), взятого у животного, индуцирует у человека психотическое состояние, подобное шизофрении (см. Johnson, K. and Jones, S., 1990). Кроме того, NMDA-рецепторы также участвуют в формировании способности ориентироваться в пространстве.
Очевидно, что NMDA-рецептор состоит из нескольких белковых цепей, находящихся в постсинаптической мембране. Открытые в настоящее время субъединицы первых двух типов образуют крупную внеклеточную область, которая, вероятно, содержит большинство аллостерических сайтов связывания; несколько трансмембранных областей, представляющих собой петлю и имеющих такую укладку, при которой они образуют поры или каналы, являющиеся проницаемыми для Ca++, и карбоксильную концевую область. Открытие и закрытие канала регулируется посредством связывания различных лигандов с доменами (аллостерическими сайтами) белка, локализованного на внеклеточной поверхности. При этом считается, что связывание лигандов влияет на конформационные изменения общей структуры белка, что, в конечном счете, приводит к открытию, частичному открытию, частичному закрытию или к полному закрытию канала.
Соединения, воздействующие на NMDA-рецептор, могут оказывать двойное действие (как агонист/антагонист) на NMDA-рецептор посредством аллостерических сайтов. Такие соединения обычно называются «частичными агонистами». В присутствии главного сайта лиганда, частичный агонист будет вытеснять некоторые лиганды, и тем самым снижать поток Ca++ через рецептор. В отсутствии главного сайта лиганда или при низком уровне этого сайта лиганда, частичный агонист будет увеличивать поток Ca++ через канал рецептора.
В настоящее время существует необходимость в разработке новых и более специфических/активных соединений, которые обладали бы способностью связываться с глицин-связывающим сайтом NMDA-рецепторов или модулировать такой сайт NMDA-рецепторов, например, коровую область, связывающуюся с лигандом NR1 NMDA-рецептора, и обладали бы, например, значительной специфичностью и/или активностью, а в частности, активностью in vivo, способствующей продуцированию фармацевтического эффекта. Кроме того, также необходимо получить фармацевтические формы таких соединений, которые были бы пригодны для пероральной доставки.
Описание сущности изобретения
Настоящее изобретение, по меньшей мере частично, относится к соединениям, которые представляют собой модуляторы NMDA, например, частичные агонисты NMDA. Так, например, настоящее изобретение относится к соединениям, которые могут имитировать бета-складчатую структуру, способную селективно взаимодействовать с глицин-связывающей областью NR1 NMDA-рецептора, например, SEQ ID NO: 1. Описанные пептидомиметики, например, если они связываются с SEQ ID NO: 1, имеют бета-складчатый мотив. В некоторых вариантах изобретения, описанные пептидомиметики, в основном, сохраняют бета-складчатый мотив in vivo или в водном растворе.
В некоторых своих вариантах, настоящее изобретение относится к пептидомиметику, способному связываться или взаимодействовать с коровой областью SEQ ID NO: 1, связывающейся с лигандом NMDA, где указанный пептидомиметик имеет по меньшей мере два альфа-углерода на расстоянии примерно от 6 до 14 Å, например, примерно от 6 до 8 Å. Так, например, описанные миметики могут включать циклическую амидную коровую область, например, спиро-бета-лактам.
В другом варианте изобретения, описанным пептидомиметиком может быть пептид, в котором две или три аминокислоты заменены молекулой, имеющей карбоксильную группу и аминогруппу. В одном из своих вариантов, настоящее изобретение относится к описанному здесь пептидомиметику по любому из пунктов 1-6, где указанный пептидомиметик обладает способностью образовывать водородную связь по меньшей мере в одном, в двух, трех или четырех положениях нижеследующих аминокислот в SEQ ID NO: l: PRO124, THR126, GLU178 и SER180, либо он может обладать способностью образовывать водородную связь со всеми четырьмя аминокислотами.
Так, например, настоящее изобретение относится к пептидомиметику, обладающему способностью связываться с коровой областью SEQ ID NO:1, связывающейся с лигандом NMDA, где указанный пептидомиметик имеет два альфа-углерода на расстоянии примерно от 6 до 14 Å (например, примерно от 6 до 10 Å) и бета-складчатый мотив, содержащий бициклическую амидную коровую область (например, спиро-бета-лактам), расположенную так, что при связывании пептидомиметика с указанной последовательностью SEQ ID NO: 1, такая бициклическая амидная коровая область будет, в основном, сохранять свою конфигурацию. Такой пептидомиметик может включать коровую область, представленную формулой:
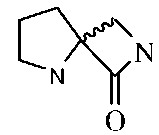 . Репрезентативные пептидомиметики могут, в основном, сохранять бета-складчатый мотив in vivo или в водном растворе, и могут обладать способностью образовывать водородную связь с нижеследующими аминокислотами SEQ ID NO:1: PRO124, THR126, GLU178 и SER180. В некоторых вариантах изобретения, бета-складчатая коровая область может быть конъюгирована с одной или с двумя аминокислотами.
. Репрезентативные пептидомиметики могут, в основном, сохранять бета-складчатый мотив in vivo или в водном растворе, и могут обладать способностью образовывать водородную связь с нижеследующими аминокислотами SEQ ID NO:1: PRO124, THR126, GLU178 и SER180. В некоторых вариантах изобретения, бета-складчатая коровая область может быть конъюгирована с одной или с двумя аминокислотами.
Настоящее изобретение также относится к способам лечения или предупреждения расстройств у пациента, опосредуемых NMDA-рецептором, где указанные способы включают введение пациенту, нуждающемуся в этом, подходящего количества глицин-имитирующего бета-складчатого циклического соединения-пептидомиметика, имеющего циклическую амидную группу, например, бета-лактамовую группу, где указанное соединение является агонистом или антагонистом лиганд-связыващей коровой области NMDA-рецептора.
Настоящее изобретение также относится к способу модуляции активности SEQ ID NO: 1, где такая модуляция обусловлена благоприятной конформацией, сообщаемой данным соединением, и где указанная модуляция происходит в результате взаимодействия водородных связей между указанным соединением и одной, двумя, тремя или четырьмя нижеследующими аминокислотами SEQ ID NO: 1: PRO124, THR126, GLU178 и SER180.
В другом своем варианте, настоящее изобретение относится к способу идентификации соединения, обладающего способностью связываться с SEQ ID NO: 1, где указанный способ включает: a) создание молекулярной модели, содержащей одну или несколько областей-мишеней SEQ ID NO: 1, происходящих по меньшей мере от части SEQ ID NO: 1, и имеющей атомные координаты, полученные путем молекулярного моделирования SEQ ID NO: 1, или атомные координаты модели, депонированной в банке данных о белках под регистрационным номером 1PBQ; b) применение данной молекулярной модели для идентификации соединения, которое может связываться с одной или несколькими областями-мишенями в данной молекулярной модели; и c) продуцирование данного соединения. В некоторых вариантах изобретения, такой способ может также включать дополнительную стадию определения наличия модуляции последовательности SEQ ID NO: 1 с использованием указанного соединения.
Настоящее изобретение также относится к фармацевтически приемлемым композициям, содержащим описанное соединение и фармацевтически приемлемый наполнитель. Так, например, такие композиции могут быть использованы для их перорального введения пациенту.
Способ лечения когнитивного расстройства, такого как расстройство, ассоциированное с потерей памяти или с нарушением способности к обучению, включает введение пациенту, нуждающемуся в этом, эффективного количества описанного соединения. Так, например, настоящее изобретение относится к способам лечения потери памяти или снижения степени потери памяти, или лечения нарушения способности к обучению у пациента, нуждающегося в этом.
В одном из своих вариантов, настоящее изобретение относится к способам устранения боли при невропатии у пациента, нуждающегося в этом, где указанный способ включает введение указанному пациенту эффективного количества описанного соединения.
В настоящей заявке также описаны способы лечения депрессии, синдрома навязчивых состояний или шизофрении у пациента, нуждающегося в этом, где указанные способы включают введение указанному пациенту эффективного количества описанного соединения. В другом своем варианте, настоящее изобретение относится к способам лечения посттравматического стресса, алкогольной зависимости или наркомании у пациента, нуждающегося в этом, где указанные способы включают введение указанному пациенту эффективного количества описанных соединений.
Описание графического материала
На Фиг. 1A-1D показано, что описанное соединение (AK52) осуществляет двухфазную модуляцию возбуждающих постсинаптических токов (e.p.s.c.s), опосредуемых NMDA-рецептором в синапсах коллатералей Шаффера поля CA1 гиппокампа, и селективно усиливает индукцию LTP. 1A: Время продолжительности детектируемого снижения под действием AK52 (1 мкМ; сплошной столбик) NMDA-компонента e.p.s.c.s, индуцированных коллатералями Шаффера в пирамидных нейронах поля CA1. (Каждая точка означает среднее ± ср.ст.ош. амплитуды e.p.s.c. peNRXe для 5 клеток) 1B: Время продолжительности усиления при 10-кратном снижении концентрации AK52 (100 нМ; серый столбик) NMDA-компонента e.p.s.c.s, индуцированных коллатералями Шаффера в пирамидных нейронах поля CA1. (Каждая точка означает среднее ± ср.ст.ош. амплитуды e.p.s.c. peNRXe для 5 клеток). 1C: Время LTD, индуцированного серией низкочастотных стимулов (2 Гц/10 мин; начиная с импульса, указанного стрелкой) в синапсах коллатералей Шаффера поля CA1 в срезах, предварительно обработанных 1 мкМ (заштрихованные кружки; n=10) и 100 нМ (заштрихованные ромбы; n=6) NRX-10,052, по сравнению с контрольными необработанными срезами (незаштрихованные кружки; n=8). (Каждая точка означает среднее ± ср.ст.ош. нормализованного наклона кривой EPSP внеклеточного поля для n срезов). 1D: Время проведения экспериментов, в которых осуществляют сравнение LTP, индуцированного серией высокочастотных импульсов (3×100 Гц/500 мс; стрелка) в синапсах коллатералей Шаффера поля CA1 в срезах, предварительно обработанных 1 мкМ (заштрихованные кружки; n=10) или 100 нМ (заштрихованные ромбы; n=8) NRX-10,052, по сравнению с контрольными необработанными срезами (незаштрихованные кружки; n=15). (Каждая точка означает среднее ± ср.ст.ош. нормализованного наклона кривой e.p.s.р. поля для n срезов).
На Фиг. 2A-2E показано, что низкая концентрация описанного соединения B детектируемо усиливает возбуждающие постсинаптические токи, опосредуемые фармакологически изолированным постсинаптическим NMDA-рецептором (e.p.s.c.s), в синапсах коллатералей Шаффера поля CA1, и потенцирует LTP, тогда как концентрация, в 20 раз превышающая указанную концентрацию, снижает NMDA-индуцированные e.p.s.c.s. 2A: Время продолжительности детектируемого усиления одного импульса e.p.s.c.s., индуцированного фармакологически изолированным NMDA в коллатералях Шаффера и зарегистрированного в пирамидных нейронах поля CA1, под действием соединения В (50 нМ; сплошной столбик). 2B: Время продолжительности усиления e.p.s.c.s. NMDA, индуцированного импульсами (4 импульса/100 Гц) под действием соединения В (50 нМ; сплошной столбик); 2C: Время продолжительности детектируемого снижения одного импульса e.p.s.c.s., индуцированного NMDA в коллатералях Шаффера и зарегистрированного в пирамидных нейронах поля CA1, под действием соединения В (1 мкМ; сплошной столбик). 2D: Время продолжительности снижения e.p.s.c.s. NMDA, индуцированного импульсами (4 импульса/100 Гц) в коллатералях Шаффера и зарегистрированного в пирамидных нейронах поля CA1 под действием соединения В (1 мкМ; сплошной столбик); 2E: Усиление LTP, индуцированное серией высокочастотных импульсов (100 Гц/500×3 мс; сплошная стрелка) в коллатералях Шаффера в синапсах на пирамидных нейронах поля CA1 под действием 50 нМ соединения B (заштрихованные кружки) по сравнению с контрольными необработанными срезами (незаштрихованные кружки). (Каждая точка означает среднее ± ср.ст.ош. амплитуды e.p.s.c. peNRXe для n клеток).
На Фиг. 3A-3C продемонстрировано, что описанное соединение (AK51) в концентрациях 100 нМ и 1 мкΜ усиливает постсинаптические токи, индуцированные фармакологически изолированным NMDA (e.p.s.c.s.) в синапсах коллатералей Шаффера поля CA1, и потенцирует LTP. 3A: Время продолжительности детектируемого усиления одного импульса e.p.s.c.s., индуцированного фармакологически изолированным NMDA в коллатералях Шаффера и зарегистрированного в пирамидных нейронах поля CA1, под действием NRX-10,051 (100 нМ; сплошной столбик) (n=x). 3B: Время продолжительности детектируемого усиления одного импульса e.p.s.c.s., индуцированного фармакологически изолированным NMDA в коллатералях Шаффера и зарегистрированного в пирамидных нейронах поля CA1, под действием AK51 (1 мкΜ; сплошной столбик) (n=y). 3C: Усиление LTP, индуцированное серией высокочастотных импульсов (100 Гц/500×3 мс; сплошная стрелка) в коллатералях Шаффера в синапсах на пирамидных нейронах поля CA1 под действием AK5151 в концентрациях 100 нМ () и 1 мкМ (заштрихованные кружки) по сравнению с контрольными необработанными срезами (незаштрихованные кружки). 3D: Время LTP, индуцированного серией низкочастотных импульсов (2 Гц/10 мин; начиная с импульса, указанного стрелкой) в синапсах коллатералей Шаффера поля CA1 в срезах, предварительно обработанных 1 мкМ (заштрихованные кружки; n=10) и 100 нМ (заштрихованные ромбы; n=6) NRX-10051, по сравнению с контрольными необработанными срезами (незаштрихованные кружки; n=8). (Каждая точка означает среднее ± ср. ст. ош. амплитуды e.p.s.c. peNRXe для n клеток).
На Фиг.4 показано, что описанное соединение усиливает NMDA-индуцированный ток и LTP. A: Время продолжительности воздействия 20-минутного нахождения в бане 100 нМ AK51 (сплошной столбик) на нормализованный ток, индуцированный открытием каналов, опосредуемым фармакологически изолированным NMDA-рецептором, в пирамидных нейронах поля CA1 при регистрации целых клеток (среднее ± ср.ст.ош., n=5). B: Время продолжительности воздействия 20-минутного нахождения в бане 1 мкМ AK51 (сплошной столбик) на нормализованный ток, индуцированный открытием каналов, опосредуемым фармакологически изолированным NMDA-рецептором, в пирамидных нейронах поля CA1 при регистрации целых клеток (среднее ± ср.ст.ош., n=6). C: Время продолжительности воздействия нахождения в бане 100 нМ AK51 (сплошной столбик, заштрихованные кружки, n=8) по сравнению с необработанными контрольными срезами (незаштрихованные кружки, n=6) на величину долговременного потенцирования (LTP), определяемую по наклону кривой внеклеточного возбуждающего постсинаптического потенциала (среднее ± ср.ст.ош., fEPSP), индуцированного стимуляцией коллатералей Шаффера высокочастотными импульсами (стрелка, 2×100 Гц/500 мсек). D: Время продолжительности воздействия нахождения в бане 1 мкМ AK51 (сплошной столбик, заштрихованные кружки, n=8) по сравнению с необработанными контрольными срезами (незаштрихованные кружки, n=6) на величину наклона кривой LTP fEPSP (среднее ± ср.ст.ош.), индуцированного стимуляцией коллатералей Шаффера высокочастотными импульсами (стрелка, 2×100 Гц/500 мсек). E: Время продолжительности воздействия нахождения в бане 1 мкМ AK51 (сплошной столбик, заштрихованные кружки, n=10) по сравнению с необработанными контрольными срезами (незаштрихованные кружки, n=8) на величину наклона кривой долговременной депрессии fEPSP (среднее ± ср.ст.ош.), индуцированной стимуляцией коллатералей Шаффера низкочастотными импульсами (стрелка, 2×100 Гц/500 мсек).
На Фиг.5 представлены результаты теста на обучения крыс в Т-образном лабиринте с использованием описанного соединения.
На Фиг.6 представлены результаты анализа на невропатическую боль у крыс, проводимого с использованием формалина.
На Фиг.7 показано, что один изомер описанного соединения AK-55-A значительно усиливает NMDA-ток и LTP, тогда как само соединение не обладает такой способностью.
На Фиг.8 проиллюстрирован количественный анализ, проводимый с помощью ГХ/МС, и указана площадь под кривой для AK-51 и [2H7]пролина, используемого в качестве внутреннего стандарта, а также ГХ/МС-анализ, проводимый путем селективного мониторинга ионов после дериватизации TBDMS, осуществляемой методами, адаптированными Wood et al. Journal of Chromatography B, 831, 313-9 (2005). Количественный интервал для этого соединения в данном анализе составлял от 0,312 пмоль до 10 пмоль на колонку. Для селективного мониторинга ионов (SIM) использовали ионы 241,2 (данное соединение) и 350,3 (дейтерированный пролин). R2=0,9998 (Квадратическая нелинейная регрессия).
На Фиг.9 представлена последовательность NMDA-рецептора NR1 и различные соединения, связанные водородной связью c конкретными аминокислотами.
На Фиг.10 представлена кристаллическая структура соединения X (GLYX-13) с NMDA-рецептором NR1.
На Фиг.11 представлена модель пептида (GLYX-13), в которой расстояние между атомами альфа-углерода составляет 12,171 Å.
На Фиг.12 представлены 1Н-ЯМР-спектры описанного здесь соединения.
На Фиг.13 представлены 1Н-ЯМР-спектры описанного здесь соединения.
Подробное описание изобретения
В настоящем изобретении, в основном, описаны соединения, способные модулировать NMDA, например, антагонисты или частичные агонисты NMDA, а также их композиции и/или способы применения описанных соединений.
Ниже приводится определение терминов, используемых в описании настоящей заявки:
Используемый здесь термин «алкенил» означает ненасыщенный углеводород с прямой или разветвленной цепью, имеющий по меньшей мере одну углерод-углеродную двойную связь, такую как прямая или разветвленная группа из 2-12, 2-10 или 2-6 атомов углерода, называемая здесь C2-C12алкенилом, C2-C10aлкенилом и C2-C6алкенилом, соответственно. Репрезентативными алкенильными группами являются, но не ограничиваются ими, винил, аллил, бутенил, пентенил, гексенил, бутадиенил, пентадиенил, гексадиенил, 2-этилгексенил, 2-пропил-2-бутенил, 4-(2-метил-3-бутен)пентенил и т.п.
Используемый здесь термин «алкокси» означает алкильную группу, связанную с кислородом (-О-алкил). Репрезентативными алкоксигруппами являются, но не ограничиваются ими, группы, имеющие алкильную группу из 1-12, 1-8 или 1-6 атомов углерода, называемую здесь C1-C12алкокси, C1-C8алкокси и C1-C6алкокси, соответственно. Репрезентативными алкоксигруппами являются, но не ограничиваются ими, метокси, этокси и т.п. Аналогичным образом, репрезентативными «алкенокси»-группами являются, но не ограничиваются ими, винилокси, аллилокси, бутенокси и т.п.
Используемый здесь термин «алкил» означает насыщенный углеводород с прямой или разветвленной цепью. Репрезентативными алкильными группами являются, но не ограничиваются ими, метил, этил, пропил, изопропил, 2-метил-1-пропил, 2-метил-2-пропил, 2-метил-l-бутил, 3-метил-l-бутил, 2-метил-3-бутил, 2,2-диметил-1-пропил, 2-метил-l-пентил, 3-метил-l-пентил, 4-метил-l-пентил, 2-метил-2-пентил, 3-метил-2-пентил, 4-метил-2-пентил, 2,2-диметил-1-бутил, 3,3-диметил-l-бутил, 2-этил-1-бутил, бутил, изобутил, трет-бутил, пентил, изопентил, неопентил, гексил, гептил, октил и т.п.
Алкильная, алкенильная и алкинильная группы могут быть, но необязательно, замещены, если это не оговорено особо, одной или несколькими группами, выбранными из алкокси, алкила, циклоалкила, амино, галогена и -C(О)алкила. В некоторых вариантах изобретения, алкильная, алкенильная и алкинильная группы не замещены, то есть, они являются незамещенными.
Используемый здесь термин «алкинил» означает ненасыщенный углеводород с прямой или разветвленной цепью, имеющий по меньшей мере одну углерод-углеродную тройную связь. Репрезентативными алкинильными группами являются, но не ограничиваются ими, этинил, пропинил и бутинил.
Используемые здесь термины «амид» или «амидо» означают радикал формы -RaC(O)N(Rb)-, -RaC(О)N(Rb)Rc или -C(О)NRbRc-, где каждый из Ra, Rb и Rc независимо выбран из алкокси, алкила, алкенила, алкинила, амида, амино, арила, арилалкила, карбамата, циклоалкила, сложного эфира, простого эфира, формила, галогена, галогеналкила, гетероарила, гетероциклила, водорода, гидроксила, кетона и нитро. Амид может быть связан с другой группой посредством углерода, азота, Rb, Rc или Ra. Амид может быть также циклическим, например, Rb и Rc, Ra и Rb или Ra и Rc, взятые вместе, могут образовывать 3-12-членное кольцо, такие как 3-10-членное кольцо или 5-6-членное кольцо. Термин «карбоксамидо» означает структуру -C(О)NRbRc.
Используемый здесь термин «амин» или «амино» означает радикал формулы -NRdRe, где Rd и Re независимо выбраны из водорода, алкила, алкенила, алкинила, арила, арилалкила, циклоалкила, галогеналкила, гетероарила и гетероциклила. Амино может быть также циклическим, например, Rd и Re, связанные вместе с N, образуют 3-12-членное кольцо, например, морфолино или пиперидинил. Термин «амино» также включает соответствующую соль четвертичного аммония, состоящую из любой аминогруппы, например, -[N(Rd)(Re)(Rf)]+. Репрезентативными аминогруппами являются аминоалкильные группы, где по меньшей мере один из Rd, Re или Rf представляет собой алкильную группу. В некоторых вариантах изобретения, Rd и Re представляют собой водород или алкил.
Используемые здесь термины «галогеногруппа», «галоген» или «Hal» означают F, Cl, Br или I. Используемый термин «галогеналкил» означают алкильную группу, замещенную одним или несколькими атомами галогена.
Термины «гетероциклил» или «гетероциклическая группа» известны специалистам и означают насыщенные или частично ненасыщенные 3-10-членные циклические структуры, и альтернативно, 3-7-членные циклы, которые включают от одного до четырех гетероатомов, таких как азот, кислород и сера. Гетероциклы могут также представлять собой моноциклические, бициклические или другие полициклические системы. Гетероцикл может быть конденсирован с одним или несколькими арильными, частично ненасыщенными или насыщенными циклами. Гетероциклильными группами являются, например, биотинил, хроменил, дигидрофурил, дигидроиндолил, дигидропиранил, дигидротиенил, дитиазолил, гомопиперидинил, имидазолидинил, изохинолил, изотиазолидинил, изоксазолидинил, морфолинил, оксоланил, оксазолидинил, феноксантенил, пиперазинил, пиперидинил, пиранил, пиразолидинил, пиразолинил, пиридил, пиримидинил, пирролидинил, пирролидин-2-онил, пирролинил, тетрагидрофурил, тетрагидроизохинолил, тетрагидропиранил, тетрагидрохинолил, тиазолидинил, тиоланил, тиоморфолинил, тиопиранил, ксантенил, лактоны, лактамы, такие как азетидиноны и пирролидиноны, сультамы, сульфоны и т.п. Гетероциклическое кольцо может быть замещено в одном или нескольких положениях такими заместителями, как алканоил, алкокси, алкил, алкенил, алкинил, амидо, амидино, амино, арил, арилалкил, азидо, карбамат, карбонат, карбокси, циано, циклоалкил, сложный эфир, простой эфир, формил, галоген, галогеналкил, гетероарил, гетероциклил, гидроксил, имино, кетон, нитро, фосфат, фосфонато, фосфинато, сульфат, сульфид, сульфонамидо, сульфонил и тиокарбонил. В некоторых вариантах изобретения, гетероциклическая группа не замещена, то есть, гетероциклическая группа является незамещенной.
Термин «гетероциклоалкил» известен специалистам и означает насыщенную гетероциклильную группу, определенную выше. Используемый здесь термин «гетероциклилалкокси» означает гетероциклил, связанный с алкоксигруппой. Термин «гетероциклилоксиалкил» означает гетероциклил, связанный с кислородом (-О-), который связан с алкильной группой.
Используемые термины «гидрокси» и «гидроксил» означают радикал -OH.
Термины «фармацевтически или фармакологически приемлемый» относятся к молекулам и композициям, которые не вызывают негативных, аллергических или других нежелательных реакций при их введении животному или человеку, нуждающимся в таком введении. Препараты, вводимые человеку, должны удовлетворять требованиям стерильности, апирогенности, общей безопасности и чистоты в соответствии со стандартами, предъявляемыми Комиссией по биологическим стандартам при Управлении по контролю за качеством пищевых продуктов, медикаментов и косметических средств (FDA).
Используемый в настоящем описании термин «частичный агонист NMDA-рецептора» определен как соединение, способное связываться с глицин-связывающим сайтом NMDA-рецептора; причем, при низких концентрациях, агонист NMDA-рецептора действует, в основном, как агонист, а при высоких концентрациях, он действует, в основном, как антагонист. Эти концентрации были экспериментально определены для всех без исключения «частичных» агонистов.
Используемый термин «фармацевтически приемлемый носитель» или «наполнитель» включает все без исключения растворители, дисперсионные среды, материалы для покрытий, антибактериальные и противогрибковые средства, изотонические агенты и агенты, замедляющие всасывание, и т.п., которые являются физиологически совместимыми. В одном из вариантов изобретения, носитель может быть использован для парентерального введения. Альтернативно, носитель может быть использован для внутривенного, внутрибрюшинного, внутримышечного, подъязычного или перорального введения. Фармацевтически приемлемыми носителями являются стерильные водные растворы или дисперсии и стерильные порошки для немедленного приготовления стерильных растворов или дисперсий для инъекции. Применение таких сред и агентов, содержащих фармацевтически активные вещества, хорошо известно специалистам. В настоящем изобретении рассматривается применение всех фармацевтических композиций согласно изобретению, если только они не содержат стандартные среды или стандартные агенты, которые являются несовместимыми с активным соединением. В указанные композиции могут быть также включены дополнительные активные соединения.
Используемый термин «фармацевтически приемлемая(ые) соль(и)» означает соли, содержащие кислотные или основные группы, которые могут присутствовать в соединениях, используемых в композициях согласно изобретению. Соединения, которые входят в состав композиции согласно изобретению, и которые являются основными по своей природе, обладают способностью образовывать соли широкого ряда с различными неорганическими и органическими кислотами. Кислотами, которые могут быть использованы для получения фармацевтически приемлемых кислотно-аддитивных солей таких основных соединений, являются кислоты, образующие нетоксичные кислотно-аддитивные соли, то есть, соли, содержащие фармакологически приемлемые анионы, включая, но не ограничиваясь ими, малат, оксалат, хлорид, бромид, иодид, нитрат, сульфат, бисульфат, фосфат, кислый фосфат, изоникотинат, ацетат, лактат, салицилат, цитрат, тартрат, олеат, таннат, пантотенат, битартрат, аскорбат, сукцинат, малеат, гентизинат, фумарат, глюконат, глюкаронат, сахарат, формиат, бензоат, глютамат, метансульфонат, этансульфонат, бензолсульфонат, п-толуолсульфонат и памоат (то есть, 1,1'-метилен-бис-(2-гидрокси-3-нафтоат)). Соединения, входящие в состав композиций согласно изобретению и включающие аминогруппу, могут образовывать фармацевтически приемлемые соли не только с вышеуказанными кислотами, но и с различными другими аминокислотами. Соединения, которые входят в состав композиций согласно изобретению, и которые являются кислотными по своей природе, могут образовывать соли оснований с различными фармакологически приемлемыми катионами. Примерами таких солей являются соли щелочных или щелочноземельных металлов, а в частности, соли кальция, магния, натрия, лития, цинка, калия и железа.
Соединения согласно изобретению могут содержать один или несколько хиральных центров и/или двойных связей, а поэтому, они могут существовать в природе в виде стереоизомеров, таких как геометрические изомеры, энантиомеры или диастереомеры. Используемый здесь термин «стереоизомеры» включает все геометрические изомеры, энантиомеры или диастереомеры. Эти соединения могут обозначаться символами «R» или «S», в зависимости от конфигурации заместителей, расположенных вокруг стереогенного атома углерода. Настоящее изобретение относится к различным стереоизомерам этих соединений и их смесей. Стереоизомеры включают энантиомеры и диастереомеры. Смеси энантиомеров или диастереомеров могут обозначаться в соответствии с номенклатурой «±», но каждому специалисту известно, что такая структура может обозначаться с указанием хирального центра.
Отдельные стереоизомеры соединений согласно изобретению могут быть получены путем синтеза из коммерчески доступных исходных соединений, имеющих асимметрические или стереогенные центры, или путем приготовления рацемических смесей с последующим применением методов разрешения, хорошо известных среднему специалисту в данной области. Такие методы разрешения включают (1) присоединение смеси энантиомеров к хиральному вспомогательному соединению, разделение полученной смеси диастереомеров путем перекристаллизации или хроматографии, и отделение оптически чистого продукта от вспомогательного соединения, (2) образование соли с использованием оптически активного разделяющего агента, или (3) непосредственное разделение смеси оптических энантиомеров на хиральных хроматографических колонках. Стереоизомерные смеси могут быть также разделены на их стереоизомерные компоненты хорошо известными методами, такими как газовая хроматография с хиральной фазой, высокоэффективная жидкостная хроматография с хиральной фазой, кристаллизация соединения в виде комплекса хиральной соли или кристаллизация соединения в хиральном растворителе. Стереоизомеры могут быть также получены из стереомерно чистых промежуточных соединений с использованием реагентов и катализаторов хорошо известными методами асимметрического синтеза.
Соединения согласно изобретению могут также присутствовать в виде геометрических изомеров. Символ ══ означает связь, которая, как описано здесь, может быть простой, двойной или тройной связью. Настоящее изобретение включает различные геометрические изомеры и их смеси, образующиеся в зависимости от расположения заместителей вокруг углерод-углеродной двойной связи или расположения заместителей вокруг карбоциклического кольца. Заместители вокруг углерод-углеродной двойной связи называются «Z»- или «E»-конфигурациями, где обозначения «Z» и «E» используются в соответствии со стандартами, принятыми ИЮПАК. Если это не оговорено особо, то структуры с двойными связями включают оба «E»- и «Z»-изомера.
Альтернативно, заместители вокруг углерод-углеродной двойной связи могут называться «цис» или «транс», где «цис» означает заместители, расположенные на одной и той же стороне двойной связи, а «транс» означает заместители, расположенные на противоположных сторонах двойной связи. Расположение заместителей вокруг карбоциклического кольца обозначается «цис» или «транс». Термин «цис» относится к заместителям, расположенным на одной и той же стороне плоскости кольца, а термин «транс» относится к заместителям, расположенным на противоположных сторонах плоскости кольца. Смеси соединений, в которых заместители расположены на одной и той же стороне и на противоположных сторонах плоскости кольца, называются «цис/«транс».
Описанные соединения могут присутствовать в сольватированной, а также в несольватированных формах вместе с фармацевтически приемлемыми растворителями, такими как вода, этанол и т.п., при этом подразумевается, что настоящее изобретение охватывает как сольватированную, так и несольватированную формы. В одном из вариантов изобретения, указанное соединение является аморфным. В одном из вариантов изобретения, указанное соединение является полиморфным. В другом варианте изобретения, указанное соединение имеет кристаллическую форму.
Настоящее изобретение также охватывает меченные изотопы соединения согласно изобретению, которые идентичны описанным здесь соединениям, за исключением того, что в этих соединениях, один или несколько атомов заменены атомом, имеющим атомную массу или массовое число, отличающиеся от атомной массы или массового числа атомов, обычно встречающихся в природе. Примерами изотопов, которые могут быть введены в соединения согласно изобретению, являются изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как 2H, 3H, 13C, 14C, 15N, 18О, 17О, 31P, 32P, 35S, 18F и 36Cl, соответственно.
Некоторые описанные меченные изотопами соединения (например, меченные 3H и 14C) могут быть использованы в анализах на распределение соединения и/или субстратов в ткани. Изотопы, меченные тритием (то есть, 3H) и углеродом-14 (то есть, 14C) являются особенно предпочтительными с точки зрения простоты их получения и детектирования. Кроме того, замещение тяжелыми изотопами, такими как дейтерий (то есть, 2H), может давать некоторые терапевтические преимущества, обусловленные более высокой метаболической стабильностью (например, более длительным временем полужизни in vivo или пониженными требованиями к дозам), а поэтому, в некоторых случаях, такое замещение может оказаться предпочтительным. Меченные изотопами соединения согласно изобретению могут быть, в основном, получены с применением нижеописанных методов, аналогичных методам, описанным, например, в имеющемся здесь разделе «Примеры», путем замены реагента, меченного не-изотопами, реагентом, меченным изотопами.
Используемый в настоящем описании термин «NMDA» означает N-метил-D-аспартат.
Используемый в настоящем описании термин «терапевтически эффективное количество» означает количество рассматриваемого соединения, которое продуцирует биологический или терапевтический ответ в ткани, в системе, у животного или человека, где указанный ответ может быть оценен исследователем, ветеринаром, практическим врачом или другим врачом-клиницистом. Соединения согласно изобретению вводят в терапевтически эффективных количествах для лечения заболевания. Альтернативно, терапевтически эффективным количеством соединения является количество, необходимое для достижения желаемого терапевтического и/или профилактического эффекта, например, такое количество, которое определено как количество, необходимое для максимальной коррекции поведения (например, обучения), усиления физиологического ответа (например, индуцирования LTP) или ослабления невропатической боли.
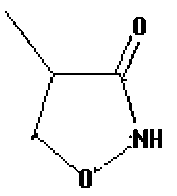
Используемый термин «бета-складчатый мотив» или «бета-складка» означает химическую структуру, имеющую атомы Сα (альфа-углерода) (атомы углерода, находящиеся непосредственно за атомом углерода карбонила), расположенные, в основном, близко друг к другу, например, структуру, имеющую водородную связь между донорным и акцепторным остатком, где донорный и акцепторный остатки находятся на расстоянии друг от друга, которое соответствует длине двух или трех пептидных связей. Описанная химическая структура имеет, например, бета-складчатый мотив, если она включает бициклические кольца (например, бициклический спиролактам), имеющие ограниченное вращение, которое может быть, например, обнаружено по слабым спектрам nOe (ядерный эффект Оверхаузера), например, между атомами H3 и H5.
Соединения
В некоторых вариантах изобретения, соединения, например, описанные пептидомемитики, обладают способностью связываться с коровой областью SEQ ID No. 1, связывающейся с лигандом NDMA. Так, например, описанный пептидомемитик может иметь, например, два альфа-углерода, которые могут быть расположены на расстоянии примерно 6-14 Å, или примерно 9-14 Å, или примерно 10-13 Å друг от друга. В некоторых вариантах изобретения, рассматриваемый пептидомиметик может иметь такие внутренние или конформационные ограничения, которые позволяют этому пептидомиметику имитировать, например, биологически активную конформацию пептида. Так, например, описанный пептидомиметик может включать циклическую амидную коровую область, например, бициклический бета-лактам. Так, например, таким пептидомиметиком может быть описанное соединение, имеющее два модульных звена (например, коровую область бициклического бета-лактама), где каждое звено может быть замещено природной аминокислотой.
Так, например, описанное соединение может иметь бета-складчатый мотив, который сохраняет свою стабильность после введения пациенту, например, он является, в основном, стабильным in vivo или в водном растворе. В некоторых вариантах изобретения, описанное соединение может образовывать водородную связь, либо оно может связываться по меньшей мере с одной, двумя, тремя или четырьмя аминокислотами SEQ ID NO: 1, например, выбранными из группы, состоящей из PRO124, THR126, GLU178 и Ser180.
Описанный здесь пептидомиметик может включать небольшой синтетический (то есть, не-пептидильный) конформационно затрудненный компонент (например, спиролактамовую группу), который может, например, сообщать соединениям частичную агонистическую активность в глициновом сайте. Так, например, описанными здесь соединениями являются соединения, представленные формулой I:
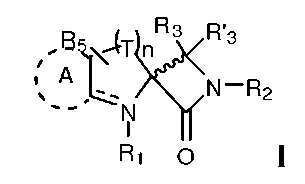
и их фармацевтически приемлемые соли, стереоизомеры и N-оксиды; где T, в каждом случае, независимо представляет CR4R4, а n равно 0, 1, 2 или 3;
A, если он присутствует, выбран из фенила или пиридина, где A необязательно замещен одним или несколькими заместителями, выбранными из Ra;
R1 выбран из группы, состоящей из H, гидроксила, -S(О)2-C1-4алкила; -SO2, C1-4алкила, C2-C4алкенила, фенила, R7, или
 ,
,
где C1-4алкил, C2-4алкенил или фенил необязательно замещены одним или несколькими заместителями, выбранными из Ra;
X представляет собой CH или N;
R3 и R3' независимо выбраны из группы, состоящей из H, галогена, гидроксила, фенила, C1-4алкила, амидо, амина или C2-C4алкенила, где C1-4алкил, C2-4алкенил и фенил необязательно замещены одним или несколькими заместителями, выбранными из Ra;
R4 и R4' независимо выбраны из группы, состоящей из H, галогена, гидроксила, фенила, C1-4алкила, амидо, амина, C1-4алкокси или C2-4алкенила, где C1-4алкил, C2-4алкенил, C1-4алкокси и фенил необязательно замещены одним или несколькими заместителями, выбранными из Ra;
R2 выбран из группы, состоящей из H, R7, -SO2, -S(О)2-C1-4алкила; C1-4алкила, гидроксила или фенила, где C1-4алкил, C2-4алкенил и фенил необязательно замещены одним или несколькими заместителями, выбранными из Ra;
каждый из R5 и R5' независимо выбран из группы, состоящей из H, галогена, C1-4алкила, C1-4алкокси, C2-4алкенила, циано, амино, фенила и гидроксила, где C1-4алкил, C2-4алкенил и фенил необязательно замещены одним или несколькими заместителями, выбранными из Ra;
R7 выбран из группы, состоящей из -C(О)-R9, -C(О)-О-R9 или -C(О)-NRd-R9,
R9 выбран из группы, состоящей из H, C1-4алкила, фенила или гетероциклила, где C1-4алкил, фенил или гетероциклил необязательно замещены 1, 2 или 3 заместителями, выбранными из Rb;
R8 выбран из группы, состоящей из H, -C(O)-C1-4алкила или -C(O)-О-C1-4алкила, где C1-4алкил необязательно замещен 1, 2 или 3 заместителями, выбранными из Ra;
Ra в каждом случае независимо выбран из карбокси, гидроксила, галогена, амино, фенила, C1-4алкила и C1-4алкокси;
Rb в каждом случае независимо выбран из группы, состоящей из карбокси, гидроксила, галогена, амино, фенила, C1-4алкила, C1-4алкокси и -NH-Rc;
Rc в каждом случае независимо выбран из группы, состоящей из -C(O)-О-C1-4алкила и -C(O)-C1-4алкила; и
Rd в каждом случае независимо выбран из группы, состоящей из H и C1-4алкила;
и их фармацевтически приемлемые соли, N-оксиды или стереоизомеры.
Так, например, описанными соединениями могут быть соединения, представленные формулой:
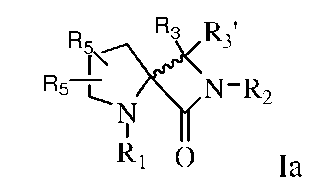 ,
,
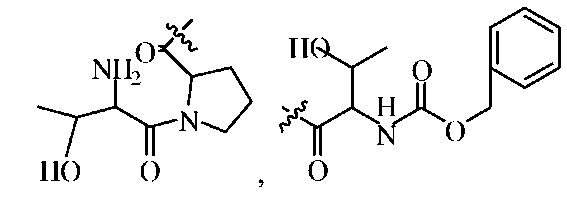
где R1 представляет собой -C(O)-C2-4алкил, где C2-4алкил замещен у одного атома углерода группой NH2 или -N-карбобензилокси, а у другого атома углерода - гидроксилом. Так, например, R1 может представлять собой -C(O)-О-C1-4алкил (например, метил, этил, пропил), где C1-4алкил замещен фенилом, а R3, R3', R5 и R2 определены выше.
В другом варианте изобретения, каждый из R1 и R2 формулы 1а может быть независимо выбран из аминокислоты, например, из L- или D-изомера аминокислоты, например, аланина, аргинина, аспарагина, аспарагиновой кислоты, цистеина, глутаминовой кислоты, глутамина, глицина, гистидина, изолейцина, лейцина, лизина, метионина, фенилаланина, пролина, серина, треонина, триптофана, тирозина и/или валина. Так, например, каждый из R1 и R2 может независимо представлять собой L-Thr или L-Ser, например, соединения, такие как:
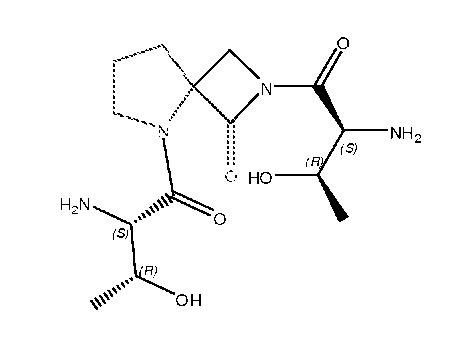
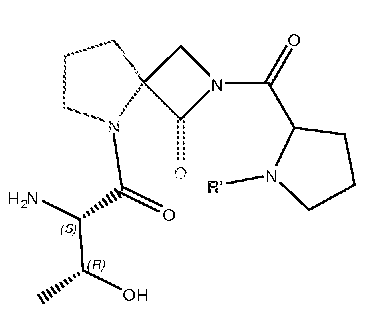
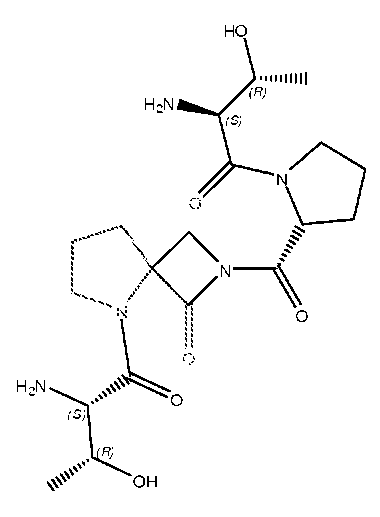
где R' выбран из группы, состоящей из Н или C1-4алкила.
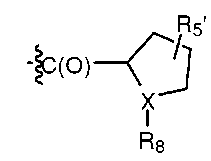
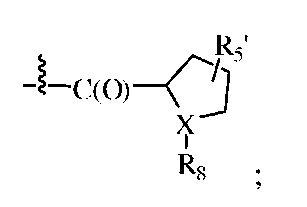
В одном из вариантов изобретения, R1 может представлять собой карбобензилокси, либо он может быть представлен формулой:
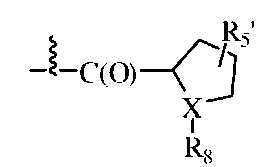 ,
,
где X может представлять собой N; R5' может представлять собой H; а R8 может представлять собой -C(O)-C2-4алкил (например, этил, пропил, н-бутил или трет-бутил), где C2-4 алкил замещен у одного атома углерода группой NH2 или -N-карбобензилокси, а у другого атома углерода - гидроксилом.
В некоторых вариантах изобретения, R3 может представлять собой фенил (необязательно замещенный как описано выше), либо он может представлять собой H. В некоторых вариантах изобретения, R2 может представлять собой -C(O)-C2-4алкил (например, этил, пропил, н-бутил или трет-бутил), необязательно замещенный у одного атома углерода группой NH2, а у другого атома углерода - гидроксилом.
Для любой рассматриваемой группы R, которая включает C1-4алкил (например, R1, R3, R5), алкил может быть выбран из группы, состоящей из метила, этила, пропила, н-бутила или трет-бутила, где указанный C1-4алкил необязательно замещен одним, двумя или тремя заместителями, выбранными из группы, состоящей из F, Cl или Br.
Такие соединения могут иметь различные изомеры, и в некоторых вариантах изобретения, такие изомеры могут быть представлены формулами:
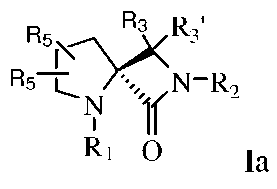 или
или 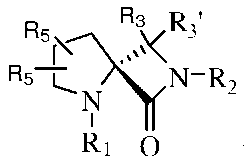 Ib,
Ib,
где R1, R2, R3, R3' и R5 могут быть такими, как они были определены выше.
В другом варианте изобретения рассматриваются соединения, представленные формулой II,
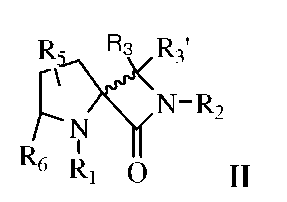
и их фармацевтически приемлемые соли, стереоизомеры и N-оксиды; где:
R1 выбран из группы, состоящей из H, гидроксила, -S(О)2-C1-4алкила; -SO2, C1-4алкила, R7, или
X представляет собой CH или N;
каждый из R3 и R3' независимо выбран из группы, состоящей из H, галогена, гидроксила, фенила, C1-4алкила, амидо, амина или C2-4алкенила, где C1-4алкил, C2-4алкенил и фенил необязательно замещены одним или несколькими заместителями, выбранными из Ra;
R2 выбран из группы, состоящей из H, R7, -SO2, -S(О)2-C1-4 алкила; C1-4алкила, гидроксила или фенила, где C1-4алкил, C2-4 алкенил и фенил необязательно замещены одним или несколькими заместителями, выбранными из Ra;
R5 выбран из группы, состоящей из H, галогена, C1-4алкила, C1-4алкокси, C2-4алкенила, циано, амино, фенила и гидроксила, где C1-4 алкил, C2-4алкенил и фенил необязательно замещены одним или несколькими заместителями, выбранными из Ra;
R6 выбран из группы, состоящей из H, галогена, C1-4алкила, C1-4алкокси, C2-4алкенила, циано, амино, фенила и гидроксила, где C1-4алкил, C2-4алкенил и фенил необязательно замещены 1, 2 или 3 заместителями, выбранными из Ra;
R7 выбран из группы, состоящей из -C(О)-R9, -C(О)-О-R9 или -C(О)-NRd-R9,
R9 выбран из группы, состоящей из H, C1-4алкила, фенила или гетероциклила, где C1-4алкил, фенил или гетероциклил необязательно замещены 1, 2 или 3 заместителями, выбранными из Rb; или
R1 и R6, взятые вместе с соединением формулы II, образуют:
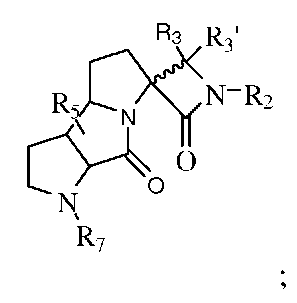
R8 выбран из группы, состоящей из H, -C(O)-C1-4алкила или -C(O)-О-C1-4алкила, где C1-4алкил необязательно замещен 1, 2 или 3 заместителями, выбранными из Ra;
Ra в каждом случае независимо выбран из карбокси, гидроксила, галогена, амино, фенила, C1-4алкила и C1-4алкокси;
Rb в каждом случае независимо выбран из группы, состоящей из карбокси, гидроксила, галогена, амино, фенила, C1-4алкила, C1-4алкокси и -NH-Rc;
Rc в каждом случае независимо выбран из группы, состоящей из -C(O)-О-C1-4алкила и -C(O)-C1-4алкила; и
Rd представляет собой H или C1-4алкил.
В репрезентативном варианте изобретения, группа R1 в соединениях формул I, II, Ia или Ib может быть выбрана из группы, состоящей из:
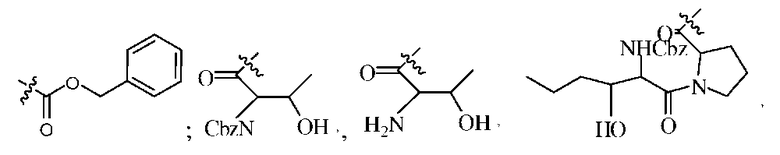
 и
и 
Репрезентативными соединениями являются:
(соединение B)
 и
и 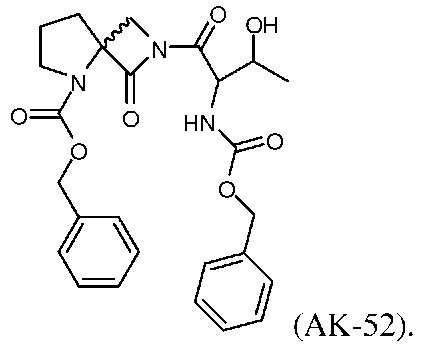
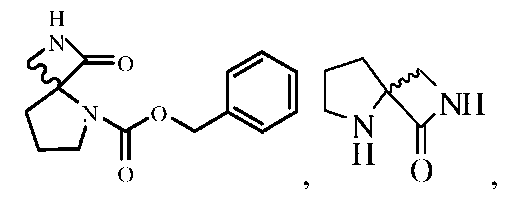
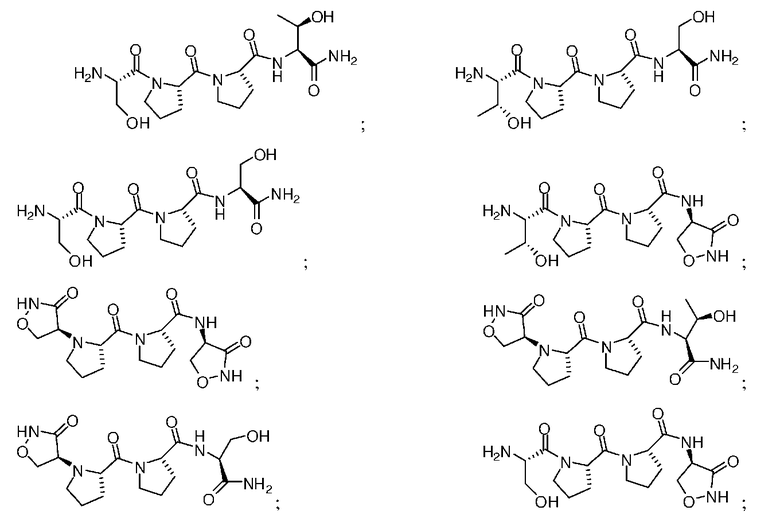
Описанными здесь соединениями являются соединения, выбранные из группы, состоящей из:
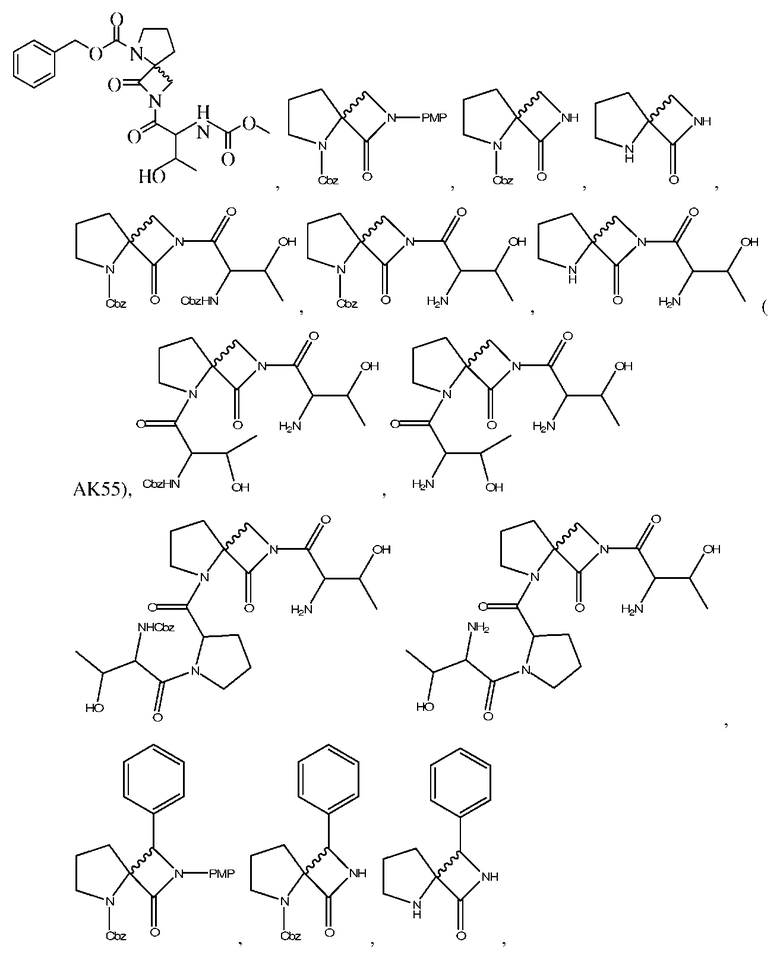
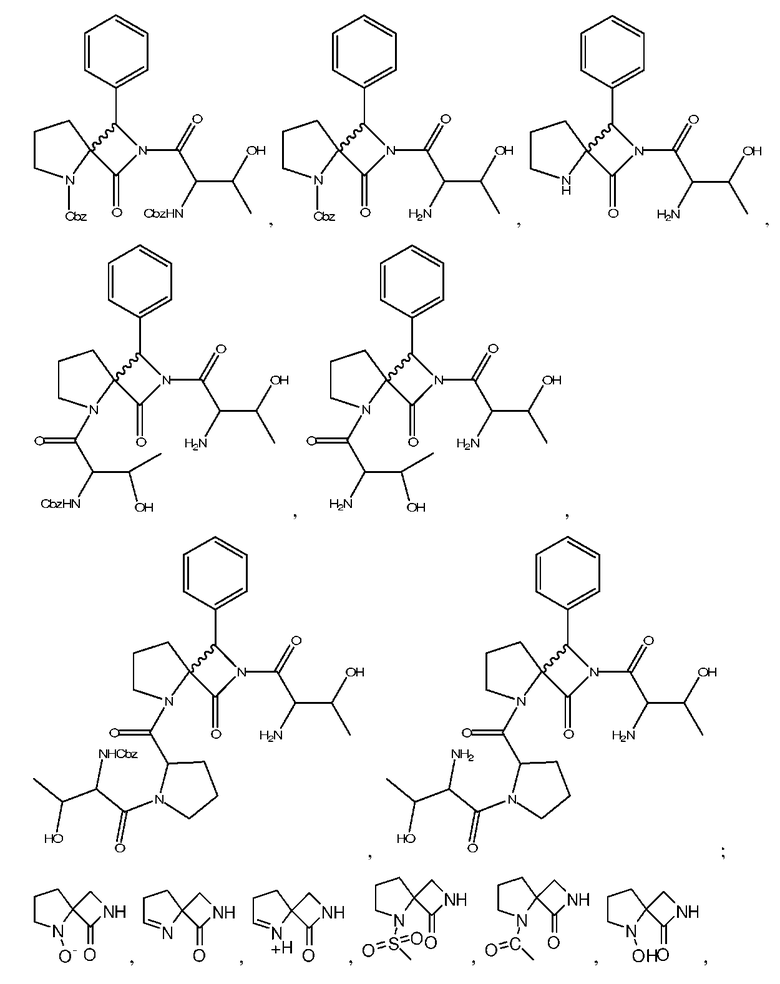
(где n равно 0, 1, 2 или 3)
и  ; или их фармацевтически приемлемые соли
; или их фармацевтически приемлемые соли
и их фармацевтически приемлемые соли, стереоизомеры или N-оксиды.
Соединения согласно изобретению и их композиции включают D-изомерную форму, L-изомерную форму или рацемическую смесь (D- и L-изомерные формы) любого одного или нескольких указанных соединений. Кроме того, композиции указанных соединений включают любую комбинацию или отношение L-изомерных форм к D-изомерным формам одного или нескольких описанных здесь аналогов. Эти и другие композиции описанных соединений, имеющие более высокое отношение форм D- и/или L-изомерных аналогов, могут обладать улучшенными терапевтическими свойствами по сравнению с терапевтическими свойствами рацемических композиций описанных соединений или смесей этих соединений. Так, например, описанными соединениями могут быть их энантиомеры, например:
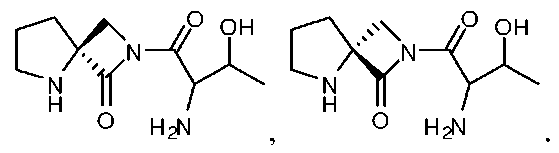 .
.
Описанные соединения могут обеспечивать эффективное открытие катионного канала в сайте NMDA-рецептора, например, они могут связываться или взаимодействовать с глутаматным сайтом NMDA-рецептора, что будет облегчать открытие катионного канала. Описанные соединения могут быть использованы для регуляции функции (включения или выключения) NMDA-рецептора, действующего как агонист.
Другими рассматриваемыми здесь соединениями являются соединения, имеющие циклическую амидную коровую область. В одном из вариантов изобретения, другими репрезентативными соединениями могут быть пептиды, а в другом варианте изобретения - пептидомиметики. Рассматриваемыми соединениями являются соединения, представленные формулой:
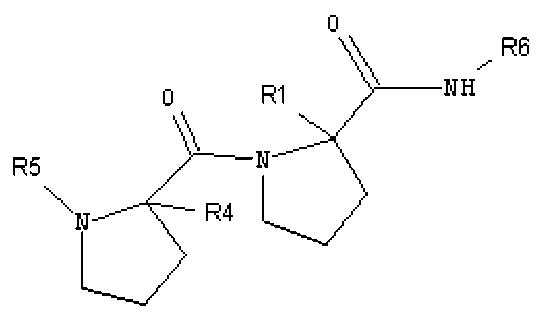 ,
,
где:
R1 представляет собой H или бензильную группу;
R4 представляет собой H или бензильную группу;
R5 представляет собой:
 или
или 
R6 представляет собой:
 или
или 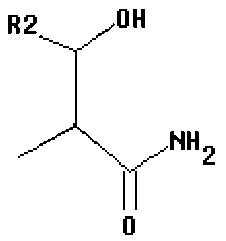
R2 представляет собой H или CH3;
R3 представляет собой H или CH3; и их стереоизомеры или их фармацевтически приемлемые соли или N-оксиды.
Репрезентативными соединениями являются:
 или
или 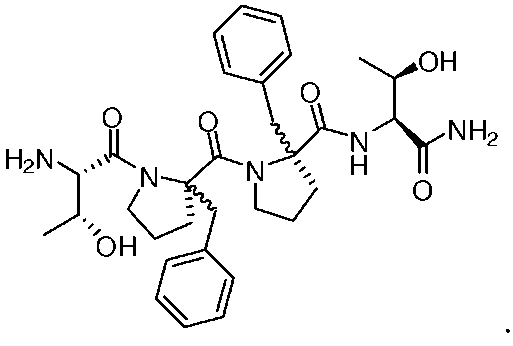
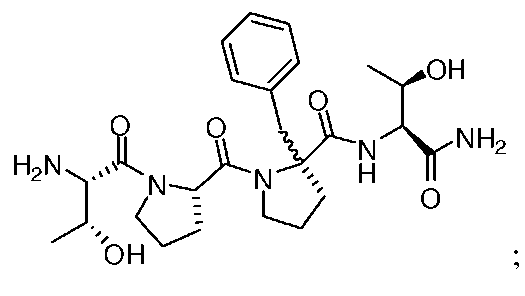
Описанные здесь соединения могут представлять собой частичные агонисты глицинового сайта NMDA-рецептора. Термин «частичный агонист», используемый в контексте настоящего изобретения, означает аналог, который при низкой концентрации действует как агонист, а при высокой концентрации - как антагонист. Связывание с глицином не ингибируется глутаматом или конкурентными ингибиторами глутамата, и кроме того, глицин не связывается в том же сайте, в котором связывается глутамат на NMDA-рецепторе. В NMDA-рецепторе присутствует второй и отдельный сайт связывания с глицином. Таким образом, открываемый под действием лиганда ионный канал NMDA-рецептора находится под контролем по меньшей мере этих двух отличающихся друг от друга аллостерических сайтов. Описанные соединения могут связываться или взаимодействовать с глицин-связывающим сайтом NMDA-рецептора. В некоторых вариантах изобретения, описанные соединения могут обладать активностью, которая в 10 раз или более превышает активность известных частичных агонистов глицинового сайта NMDA-рецептора. Так, например, описанные соединения могут обладать активностью, которая в 10-20 раз превышает активность GLYX-13. GLYX-13 представлен формулой:
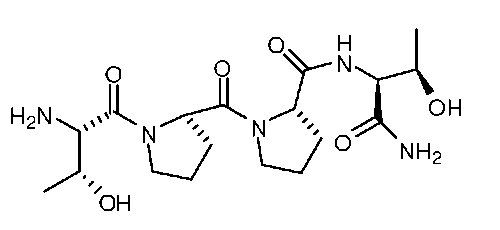
Так, например, настоящее изобретение относится к соединениям, которые могут обладать активностью, по меньшей мере примерно в 20 раз превышающей активность GLYX-13, как было измерено по проводимости одного нейрона (INMDA), индуцированной открытием каналов, опосредуемым NMDA-рецептором, в культуре пирамидных нейронов поля CA1 гиппокампа при концентрации 50 нМ. В другом варианте изобретения, соединение согласно изобретению может обладать способностью генерировать повышенную проводимость одного нейрона (INMDA), индуцированную одним разрядом нейрона с открытием каналов, опосредуемым NMDA-рецептором, в культуре пирамидных нейронов поля CA1 гиппокампа при концентрациях от 100 нМ до 1 мкМ. Описанные соединения могут обладать повышенной активностью по сравнению с GLYX-13, как было измерено по величине долговременного потенцирования (LTP) в синапсах коллатералей Шаффера поля CA-1 в срезах гиппокампа in vitro.
Получение соединений
В некоторых вариантах изобретения описанное соединение, например, пептидомиметик, имеющий бета-складчатый мотив, способный связываться с коровой областью, связывающейся с лигандом NMDA SEQ ID NO: 1, может быть получено путем включения в пептид одной или нескольких дегидро-аминокислот. Так, например, для получения пептидомиметика, имеющего бета-складчатый мотив, могут быть введены пептиды, содержащие дегидрофенилаланин и/или дегидролейцин и/или альфа-аминобис-изомасляную кислоту.
В другом варианте изобретения, описанное соединение может быть получено с использованием стерически затрудненных мотивов бициклического бета-складчатого дипептида (BTD). Этот подход основан на замене дипептидного компонента путем введения карбоксила и аминогруппы в положениях, которые являются геометрически приемлемыми для связывания с пептидом (см. ниже фигуры) и для индуцирования образования складчатых слоев.
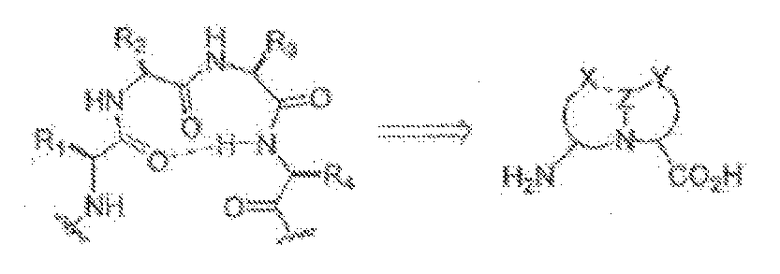
Так, например, может быть включен дипептидный компонент, такой как один или несколько азабициклононанов, с получением структуры, имеющей бета-складчатый мотив. В другом примере, описанный пептидомиметик может включать, например, вместо двух, трех или четырех аминокислот коровую структуру, представленную ниже:
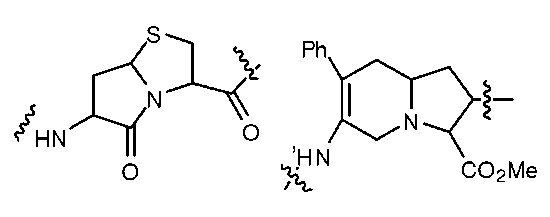
В некоторых вариантах изобретения азациклоалкен может имитировать бета-складчатый миметик, например:
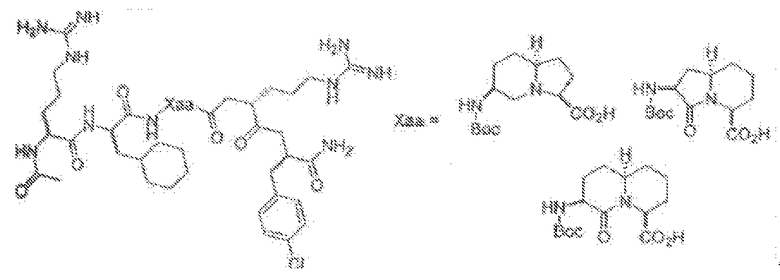
Нижеприведенные схемы представляют собой репрезентативные схемы синтеза, которые могут быть осуществлены для получения описанных соединений и их промежуточных соединений.
Схема 1: Схемы синтеза
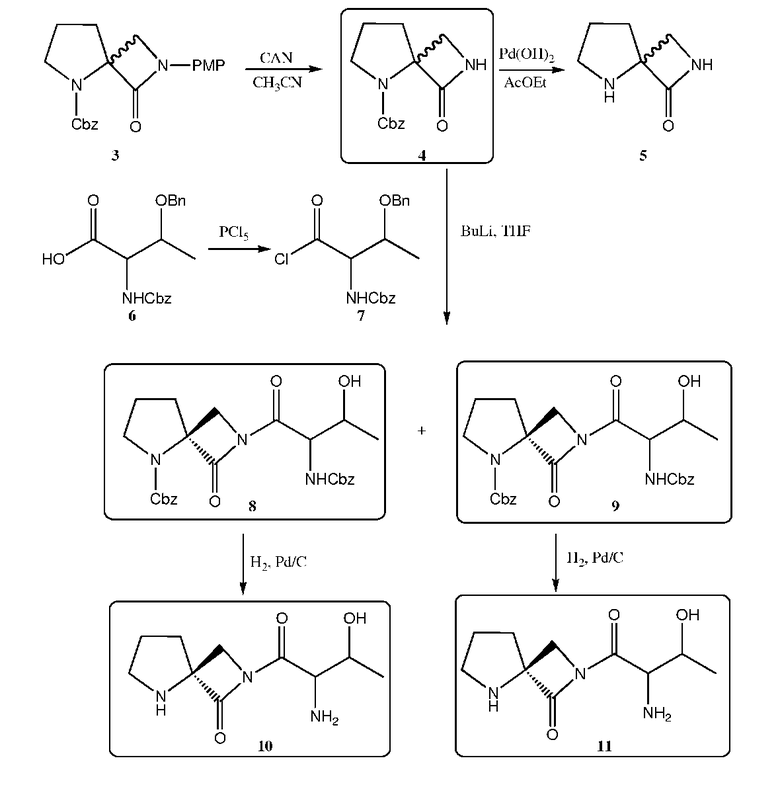
Схема 2
Нитрат церия-аммония или «CAN» представляет собой химическое соединение формулы (NH4)2Ce(NO3)6. Эта водорастворимая соль оранжево-красного цвета широко используется в органическом синтезе в качестве окислителя. Данное соединение используется в количественном анализе в качестве стандартного окислителя.
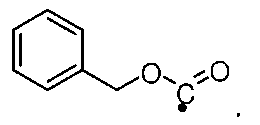
PMP означает п-метоксибензилиден; Cbz означает карбоксибензилокси-радикал, который может быть представлен формулой
Композиции
В других своих аспектах, настоящее изобретение относится к препаратам и композициям, содержащим описанные соединения и, необязательно фармацевтически приемлемый наполнитель. В некоторых вариантах изобретения, рассматриваемый препарат содержит рацемическую смесь одного или нескольких из описанных здесь соединений.
Рассматриваемые препараты могут быть получены в любой из различных обычно используемых форм. Так, например, такими соединениями являются, но не ограничиваются ими, любые соединения, которые могут быть включены в данный препарат, подходящий для перорального введения, для подкожной инъекции или для введения активного агента животному другими способами, известными специалистам-фармацевтам.
Количество описанного здесь соединения, присутствующего в препарате, может варьироваться в зависимости от таких факторов, как патологическое состояние, возраст, пол и масса индивидуума. Схемы введения доз могут быть скорректированы для достижения оптимального терапевтического ответа. Так, например, может быть введена одна ударная доза, либо может быть введено несколько дробных доз в течение определенного периода времени, либо доза может быть пропорционально уменьшена или увеличена в зависимости от срочности принятия терапевтических мер. Для облегчения введения и однородности распределения дозы, особенно предпочтительно, чтобы композиции для парентерального введения были приготовлены в виде унифицированной лекарственной формы. Используемый здесь термин «унифицированная лекарственная форма» означает физически дискретные единицы, подходящие для применения в качестве разовых доз для введения млекопитающим, причем, каждая такая единица содержит предварительно определенное количество активного соединения, вычисленное так, чтобы это соединение в комбинации с необходимым фармацевтическим носителем, продуцировало желаемый терапевтический эффект.
Спецификация унифицированных лекарственных форм согласно изобретению определяется нижеследующими факторами и непосредственно зависит от этих факторов: (a) уникальные свойства выбранного соединения, и конкретный терапевтический эффект, который должен быть достигнут; и (b) ограничения, присущие технологии приготовления лекарственных средств, содержащих активное соединение, которое может вызывать гиперчувствительность у индивидуумов.
Используемый здесь термин «фармацевтически приемлемый носитель» или «наполнитель» включает все без исключения подходящие носители и наполнители.
Обычно, терапевтические композиции должны быть стерильными и стабильными в условиях их производства и хранения. Такая композиция может быть приготовлена в виде раствора, микроэмульсии, липосомы или другой упорядоченной структуры, подходящих для приготовления лекарственного средства в высокой концентрации. Носителем может быть растворитель или дисперсионная среда, содержащая, например, воду, этанол, полиолы (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль и т.п.), и их подходящие смеси. Соответствующая текучесть может поддерживаться, например, путем нанесения покрытия, такого как лецитин, сохранения требуемого размера частиц в случае дисперсии и использования поверхностно-активных веществ. Во многих случаях, в указанную композицию предпочтительно включать изотонические агенты, например, сахара, многоатомные спирты, такие как маннит или сорбит, или хлорид натрия. Пролонгированное поглощение инъецируемых композиций может быть достигнуто путем включения в данную композицию агента, замедляющего поглощение, например, моностеаратных солей и желатина.
Соединения могут быть введены в виде препарата пролонгированного высвобождения, например, композиции, включающей полимер с замедленным высвобождением. Соединения могут быть получены с использованием носителей, защищающих такое соединение от быстрого высвобождения, например, могут быть получены препараты с регулируемым высвобождением, включая имплантаты и микроинкапсулированные системы доставки. При этом, могут быть использованы биологически разлагаемые и биологически совместимые полимеры, такие как этиленвинилацетат, полиангидриды, полигликолевая кислота, коллаген, полиортоэфиры, полимолочная кислота и сополимеры полимолочной и полигликолевой кислоты (PLG). Многие методы приготовления таких препаратов, в основном, известны специалистам.
Стерильные растворы для инъекций могут быть получены путем включения соединения в необходимом количестве в соответствующий растворитель вместе с одним из перечисленных выше ингредиентов или их комбинацией, если это необходимо, с последующим проведением стерилизации на фильтре. Вообще говоря, дисперсии получают путем включения активного соединения в стерильный носитель, содержащий основную дисперсионную среду и другие необходимые ингредиенты из числа ингредиентов, перечисленных выше. В случае стерильных порошков, используемых для приготовления стерильных растворов для инъекций, предпочтительными методами приготовления препаратов являются вакуумная сушка и лиофилизация с получением порошка, содержащего активный ингредиент и любой вспомогательный нужный ингредиент, из предварительного приготовленного раствора, подвергнутого стерильной фильтрации.
В соответствии с альтернативным аспектом изобретения, соединение может быть приготовлено в комбинации с одним или несколькими вспомогательными соединениями, повышающими растворимость данного соединения.
СПОСОБЫ
Настоящее изобретение относится к способам лечения когнитивных расстройств и повышения способности к обучению. Эти способы включают введение фармацевтически приемлемого препарата, содержащего одно или несколько описанных соединений пациенту, нуждающемуся в этом. Рассматриваются также способы лечения пациентов, страдающих возрастной потерей памяти, шизофренией, нарушениями способности к специальному обучению, эпилептическими припадками, постинсультными судорогами, ишемией головного мозга, гипогликемией, асистолией, которая может привести к остановке сердца, эпилепсией, мигренью, а также болезнью Гентингтона, болезнью Паркинсона и болезнью Альцгеймера.
Другие рассматриваемые способы включают лечение ишемии коры головного мозга, инсульта, травмы головного мозга, опухолей головного мозга, острой невропатической боли, хронической невропатической боли, расстройств сна, наркомании, депрессии, некоторых нарушений зрения, синдрома отказа от приема алкоголя, тревожных состояний, а также потери памяти и нарушения способности к обучению. В другом своем аспекте, настоящее изобретение относится к способу улучшения купирования боли и для обезболивания животного.
ПРИМЕРЫ
Нижеследующие примеры приводятся лишь в иллюстративных целях и не рассматриваются как ограничение настоящего изобретения.
Пример 1 - Синтез пирролидиновых производных спиро-β-лактама
Для синтеза спиролактамов была проведена нижеследующая последовательность реакций (Схема A). В качестве исходных соединений использовали гексагидро-1,3,5-триазины, хлорангидрид Cbz-L-пролина и хлорангидрид N-(Cbz)-O-(бензиловый эфир)-L-треонина.
Схема A:
% чистоты
Пример 2 - Синтез соединений и промежуточных соединений
Спиролактам 3. Синтез C4-незамещенного спиролактама 3 осуществляли посредством реакции Штаудингера, то есть, реакции образования метиленимина с использованием триазина 2. Реакцию [2+2]-циклического присоединения между кетеном, полученным из хлорангидрида Cbz-L-пролина, и метиленимином осуществляли следующим образом: сначала получали кетен посредством реакции дегидрохлорирования хлорангидрида с использованием триэтиламина при -40°C в течение 45 минут, а затем добавляли дихлорметановый раствор триазина 2 и этерат трифторида бора (который деполимеризует триазин). Через 12 часов получали соответствующий спиролактам 3 в виде смеси энантиомеров с выходом 30-50%. В результате окислительного отщепления группы PMP от спиролактама 3 в присутствии CAN образовывалось N-незамещенное производное спиролактама 4, из которого, после обработки Pd(OH)2/C, получали соответствующие промежуточные соединения спиролактама 5.
Спиролактам 4 получали после очистки с помощью хроматографии (ВЭЖХ) на силикагеле с чистотой 93%. После проведения хроматографии на силикагеле с элюированием в градиенте 20-70% этилацетата/циклогексана получали спиролактам 5 с чистотой >90% (ЯМР) и с 50% выходом.
Пример 3 - Способы синтеза промежуточных соединений
Триазин 2. К раствору п-анизидина (24,6 г, 200 ммоль) в смеси (500 мл) этилацетата/воды (1:1), охлажденной при 0°C, добавляли водный раствор (17 мл) формальдегида (37%). Реакционную смесь перемешивали в течение 3 часов при 0°C, а затем в течение 1 часа при комнатной температуре, и органический слой отделяли, промывали водой (50 мл) и сушили над Na2SO4. Растворитель удаляли в вакууме, в результате чего образовывалось белое твердое вещество. Это твердое вещество один раз промывали диэтиловым эфиром и получали 26,3 г (твердое вещество сушили при 40°C в течение ночи) чистого триазина 2 с выходом 97%.
Промежуточные соединения спиролактама 3. К перемешанному раствору хлорангидрида N-бензилоксикарбонил-L-пролина (5 г, 18,7 ммоль) в сухом дихлорметане (65 мл), охлажденному до -40°C, по каплям добавляли сухой триэтиламин (10,4 мл, 74,7 ммоль). Раствор становился желтым, что указывало на образование кетена.
После выдерживания в течение 45 минут при -40°C по каплям добавляли пурпурный раствор триазина 2 (2,52 г, 6,16 ммоль) и BF3OEt2 (2,37 мл, 18,7 ммоль), предварительно смешанный в CH2Cl2 (35 мл). Смесь оставляли на ночь для медленного нагревания до комнатной температуры, а затем реакцию гасили насыщенным водным NaHCO3. Водный слой два раза экстрагировали CH2Cl2 (20 мл), а объединенные органические слои промывали раствором соли (20 мл) и сушили над безводным Na2SO4. Затем раствор концентрировали и очищали колоночной хроматографией на силикагеле с элюированием в градиенте 100% циклогексана - 20% этилацетата/циклогексана, в результате чего получали 7,01 г чистого продукта с выходом 37%.
Промежуточные соединения спиролактама 4. К перемешанному раствору спиролактама 3 (2,4 г, 6,55 ммоль) в ацетонитриле (49 мл) при -10°C в течение 1 часа по каплям добавляли CAN (10,8 г, 19,6 ммоль), предварительно растворенный в H2O (30 мл). После завершения добавления, смесь перемешивали в течение 45 минут (ТСХ указывала на отсутствие исходного вещества). Реакционную смесь разводили этилацетатом (100 мл) и насыщенным NaHCO3 (50 мл). К органическому слою добавляли воду (100 мл) и твердый бисульфит натрия (20 экв.) Органический слой промывали раствором соли и сушили над безводным Na2SO4. Затем раствор концентрировали и очищали колоночной хроматографией на силикагеле с элюированием в градиенте 100% циклогексана - 50% этилацетата/циклогексана, в результате чего получали 0,87 г чистого продукта с выходом 50%.
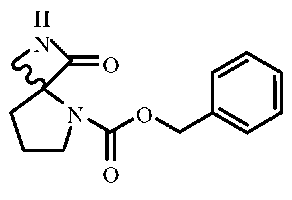
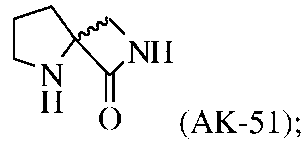
Промежуточные соединения спиролактама 5 (AK-51). 0,5 г соединения 4 растворяли в 20 мл этилацетата и через канюлю переносили в колбу в атмосфере H2 (1 атм.), содержащую 50 мг катализатора 10% Pd(OH)2-C. Смесь перемешивали в течение ночи в атмосфере H2 при 50 фунт/кв. дюйм, а затем катализатор отфильтровывали через целит. Органический слой концентрировали и очищали хроматографией на силикагеле с получением 120 мг продукта с 50% выходом.
Хлорангидрид N-(Cbz)-О-(бензиловый эфир)-L-треонина 7. К перемешанному раствору N-(Cbz)-О-(бензиловый эфир)-L-треонина (0,95 г, 2,7 ммоль) в сухом эфире (27 мл) добавляли PCl5 (0,61 г, 2,9 ммоль), и смесь перемешивали в течение 3 часов при комнатной температуре. Затем растворитель удаляли в высоком вакууме при комнатной температуре. После этого добавляли толуол, а затем его удаляли как описано выше. Неочищенное белое твердое вещество использовали для проведения реакции сочетания без какой-либо очистки.
Промежуточные соединения спиролактамов 8 и 9. К перемешанному раствору спиролактама 4 (200 мг, 0,76 ммоль) в сухом ТГФ (4 мл) при -78°C по каплям добавляли BuLi (0,32 мл, 0,80 ммоль в гексане). После завершения добавления, смесь перемешивали при -78°C в течение 1 часа. Хлорангидрид N-(Cbz)-О-(бензиловый эфир)-L-треонина 7 в ТГФ (4 мл) добавляли при -78°C. Смесь перемешивали в течение ночи при температуре в пределах от -78°C до комнатной.
Реакцию гасили насыщенным NH4Cl (10 мл), и к смеси добавляли этилацетат (10 мл). Водный слой два раза экстрагировали этилацетатом. Объединенные органические слои сушили MgSO4 и концентрировали с получением 0,44 г неочищенного продукта. Неочищенный продукт элюировали через силикагель в градиенте 100% CH2Cl2 - 2% MeOH/CH2Cl2, в результате чего получали фракции с чистотой от 44% до 73%. Эту реакцию повторяли с использованием 0,28 г спиролактама 4, и после хроматографии, получали фракции с чистотой от 50% до 73%.
Пример 4 - Анализ на связывание с NMDA-рецептором
Приготовление тканей:
Неочищенные синаптические мембраны получали из гиппокампа или из переднего мозга крыс (самцов крыс Sprague-Dawley) и тщательно промывали для удаления эндогенных аминокислот, как описано в работе Ransom и Stec (1988). Вкратце, неочищенные синаптические мембраны ресуспендировали в 20 объемах 5 мМ трис-HCl-буфера, pH 7,4 (для использования в экспериментах по связыванию с [3H]TCP), или в 20 объемах 5 мМ трис-ацетатного буфера, pH 7,4 (для использования в экспериментах по связыванию с [3H]глицином) и гомогенизировали на Polytron (Virtis shear; Virtis, NY, U.S.A.). Затем мембраны осаждали путем центрифугирования при 48000 g в течение 20 минут. Эту стадию два раза повторяли, и гомогенат хранили при -70°C в том же буфере. Перед каждым применением, гомогенаты оттаивали при комнатной температуре, осаждали и еще четыре раза промывали. Для проведения эксперимента с использованием [3H]глицина, осадок сначала инкубировали в течение 30 минут при 25°C в 5 мМ трис-ацетатного буфера, содержащего 0,04% тритона X-100, а затем четыре раза промывали путем гомогенизации и центрифугирования. Конечные промытые мембраны ресуспендировали при концентрациях 2-3 мг/мл либо в 5 мМ трис-HCl-буфера, либо в 5 мМ трис-ацетатного буфера.
Анализы на связывание с TCP: Измерение специфического связывания с [3H]TCP осуществляли как описано ранее (Haring et al., 1986, 1987; Kloog et al., 1988a). Конечные реакционные смеси состояли из 50-100 мкг мембранного белка в 200 мкл 5 мМ трис-HCl-буфера и содержали либо [3H]TCP, либо [3H]ТСР, и соответствующую концентрацию лигандов NMDA-рецептора или mAb. Реакции инициировали путем добавления мембран к реакционным смесям. Если это не оговорено особо, анализы на связывание осуществляли в неравновесных условиях при 25°C в течение 1 часа. Неспецифическое связывание одновременно определяли в образцах, содержащих 100 мкΜ немеченного PCP. Реакции связывания оценивали путем фильтрации на стеклянных фильтрах Whatman GF/B, которые были предварительно обработаны 0,1% полиэтиленимином в течение 1 часа.
Диссоциацию [3H]TCP из его сайта связывания с мембраной измеряли после уравновешивания рецепторов 20 нМ [3H]TCP в течение 120 минут. Реакцию диссоциации инициировали путем добавления 100 мкΜ немеченного PCP в присутствии и в отсутствии лигандов NMDA-рецептора или mAb. Реакции сразу завершали (время 0), а затем реакционную смесь инкубировали в течение дополнительного периода времени.
Проводили оценку действия трех соединений 1) на проводимость одного нейрона (INMDA), индуцированную открытием каналов, опосредуемым NMDA-рецептором в пирамидных нейронах поля CA1 гиппокампа, и 2) на величину долговременного потенцирования (LTP) и долговременной депрессии (LTD) в синапсах коллатералей Шаффера поля CA1 или в срезах гиппокампа in vitro. Сообщалось, что GLYX-13 при низких концентрациях (1-10 мкΜ) усиливает активированную импульсами INMDA и LTP, и одновременно снижает LTD и INMDA, индуцированную одним импульсом. GLYX-13 в 100 раз более высокой концентрации, а именно, в концентрации 100 мкМ, он дает снижение LTD и величины INMDA, индуцированной импульсами, но совсем не оказывает воздействия на LTD.
Соединение B обладает в 20 раз большей активностью, чем GLYX-13. 50 нМ этого соединения значительно увеличивало INMDA, индуцированную одним импульсом (1A) и несколькими импульсами (1B), а также в два раза увеличивало величину LTP (1E). В противоположность этому, NRX-10,050 в концентрации 1 мкΜ значительно снижает INMDA, индуцированную одним импульсом (1С) и несколькими импульсами (ID) подобно 100 мкΜ GLYX-13. (см. Фиг.2).
AK-51 обладает более низкой активностью, чем соединение B, но оказывает стимулирующее действие при концентрациях в более широком интервале (Фиг.3). NRX-10,051 в концентрациях 100 нМ (2A) и 1 мкΜ повышает величину INMDA, индуцированного одним импульсом, тогда как NRX-10,051 в концентрации 1 мкМ в два раза увеличивал величину LTP (2D), но не оказывает воздействия на LTD (2E).
AK-52 при низкой концентрации (100 нМ; 3A) продуцирует лишь небольшое увеличение INMDA, индуцированного одним импульсом, а при концентрации 1 мкМ (3B) дает значительное снижение INMDA. AK-52 в концентрации 100 нМ дает увеличение LTP, аналогичное величине, которую давали соединение В и AK-51, но при этом, он дает небольшое, но значимое снижение LTP при концентрации 1 мкΜ, и не оказывает влияния на LTD.
Эти три соединения давали приблизительно в 20 раз большее увеличение активности по сравнению с GLYX-13. Соединение B является самым сильным индуктором INMDA при низких концентрациях (50 нМ). AK-51 дает меньшее увеличение INMDA, и этот эффект сохранялся при 10-кратном увеличении концентрации AK-51 (100 нМ - 1 мкΜ). AK-52 является самым слабым индуктором INMDA, но это его действие быстро меняется на обратное, вызывая заметное снижение величины INMDA.
Эти соединения в одинаковой степени повышали величину LTP, то есть, приблизительно удваивали эту величину. GLYX-13 является единственным соединением, которое обладает способностью одновременно увеличивать LTP и снижать LTD; AK-52 не оказывал влияния на LTD даже при концентрации, которая снижала INMDA. GLYX-13 может селективно увеличивать INMDA, опосредуемый NMDA-рецепторами, содержащими субъединицы NR2A/B, и эти рецепторы были локализованы в экстрасинаптических локусах и более сильно активировались нейронными импульсами, индуцирующими LTP. Хотя все протестированные соединения оказывали сильное воздействие на LTP и INMDA, однако, их меньшее воздействие на LTD позволяет предположить, что они обладают более высокой селективностью по отношению к NR2A/B-содержащим глициновым сайтам NMDA-рецептора, чем GLYX-13.
Пример 5 - Модель обучения с использованием T-образного лабиринта
Эти исследования проводили на 3-месячных самцах крыс, полученных путем скрещивания крыс Fisher 344 и крыс Brown Norway Fl (FBNF1). Т-образный лабиринт с коридорами (длина 45 см × ширина 10 см × высота 10 см) был сконструирован из черного плексигласа, включающего лабиринт. Два пластиковых сосуда с крышками, расположенные вдоль проволочной сетки, были закреплены по концам каждого выхода из коридора, в котором помещался корм в качестве вознаграждения (Cheerios, 100 мг/кусок). Перед началом эксперимента по обучению, животным постепенно уменьшали рацион примерно до 85% от их обычного потребления. За три дня до начала эксперимента, животных содержали в Т-образном лабиринте, в котором повсюду находилась пища. В первый день эксперимента по обучению, животные получали вознаграждение за выбор правого коридора, и критерием полного обучения является 9 правильных выборов подряд из 10 возможных. На второй день обучения, животные получали вознаграждение за выбор левого коридора, и критерием полного обучения является 9 правильных выборов подряд из 10 возможных. На следующий день, животным вводили инъекции AK51 (0,3, 1, 3, 10, 30 мг/кг p.o.) или носитель ДМСО (1 мг/мл; Sigma, Saint Louis MO) слепым методом через желудочный зонд (4”, калибр 16; Braintree Scientific, Braintree MA) за 60 минут до начала проведения эксперимента (n=8-9 на группу). В первом испытании данного эксперимента, корм находился в обоих коридорах, а в последующие 20 испытаний животные получали вознаграждение только за альтернативный выбор (выбор коридора, противоположного коридору, выбранному животным в предыдущем испытании) (интервал между испытаниями составлял ~30 секунд). Для каждого животного, число испытаний подсчитывали по соответствующему критерию (5 правильных выборов подряд). Данные были проанализировали с помощью ANOVA, а затем в соответствии с post hoc критерием Фишера по методу PLSD путем сравнения эффектов отдельных доз лекарственных средств и носителя (α=0,05).
На Фиг.5 представлено среднее число испытаний (± ср.ст.откл.), проводимых на 3-месячных крысах, которые содержались в условиях голодания в Т-образном лабиринте, и которым ставилась задача выбора с чередованием в соответствии с установленным критерием (20 испытаний). Животным вводили p.o. AK051 в дозах 0; 0,3; 1; 3; 10 или 30 мг/кг в носителе ДМСО (n=8-9 на группу) за 60 минут до начала испытания. ***P<0,001, **P<0,01, Сравнение с носителем по post hoc критерию Фишера методом PLSD.
Пример 6 - Тест на невропатическую боль с использованием формалина
Эксперименты проводили как описано в литературе (Abbott et al. Pain, 60, 91-102, 1995; Wood et al., Neuroreport, 19, 1059-1061 2008). В этом исследовании использовали самцов 3-месячных крыс, полученных путем скрещивания крыс Fisher 344 Х и крыс Brown Norway Fl (FBNF1). Перед началом испытаний, животных каждый день в течение последующих 2 дней помещали на 10 минут в камеру для испытаний (из непрозрачного плексигласа размером 30×30×60 см). В день проведения испытаний, животным вводили инъекции AK51 (0,3; 1; 3; 10; 30 мг/кг p.o.) или носителя ДМСО (1 мг/мл; Sigma, Saint Louis MO) слепым методом через желудочный зонд (4", калибром 16; Braintree Scientific, Braintree MA) за 60 минут до инъекций формалина (n=8-9 на группу). За 10 минут до инъекции формалина, животных помещали в камеру для испытаний. Для инъекции формалина, крыс держали руками и в латеральную часть подушечки подошвенной поверхности левой задней лапы подкожно вводили инъекцию 1,5% формалина (50 мкл с иглой калибром 26; Sigma, Saint Louis MO). После инъекций формалина, крыс помещали обратно в камеры для испытаний. После инъекции формалина, поведение животных записывали на видеокамеру в течение 50 минут с помощью углового зеркала. Общее время зализывания лап после инъекции и общее время подергивания лап после инъекции в последней фазе испытаний (через 30-50 минут после инъекции формалина) количественно оценивалось экспериментатором, проводившим испытание слепым методом в режиме «оф-лайн», при этом, результаты, полученные разными наблюдателями при измерении одного и того же параметра (inter-rater reliability), и результаты, полученные одним наблюдателем при повторном измерении одного и того же параметра (intra-rater reliability) имели высокую достоверность (r>0,9) для обоих измерений. Всех животных умерщвляли путем введения СО2 сразу после испытаний. Данные анализировали с помощью ANOVA, а затем по post hoc критерию Фишера методом PLSD путем сравнения отдельных доз лекарственного средства с носителем (α=0,05). На Фиг.6 представлены средние величины (± ср.ст.ош.). % Аналгезии определяли как процент уменьшения подергиваний в поздней фазе испытания (30-50 мин.) после инъекции формалина вовнутрь подошвы (50 мкл 1,5% формалина).
Пример 7 - Препараты для перорального введения, повышающие способность к обучению и улучшающие память
Препарат AK-51 для перорального введения приготавливали в диметилсульфоксиде (ДМСО). Все дозы вводили в объеме 300 мкл. Затем животным p.o. через зонд вводили корм (в ротовое отверстие с помощью иглы для искусственного кормления) в вычисленном объеме, необходимом для доставки животному определенной дозы из расчета массы тела: 0,0 мг/кг 300 мкл ДМСО (носитель); 0,3 мг/кг, 300 мкл в ДМСО; 1,0 мг/кг, 300 мкл в ДМСО; 3,0 мг/кг, 300 мкл в ДМСО; 10,0 мг/кг, 300 мкл в ДМСО; 30,0 мг/кг, 300 мкл в ДМСО.
За 60 минут до начала испытания, животным вводили одну из доз, указанных выше. Затем, для оценки поведения животных, приобретенного в результате обучения, применяли T-образный лабиринт с чередованием задания на выбор (20 испытаний). Этот протокол описан в примере 5. Вкратце, T-лабиринт представляет собой задачу на выбор. Испытуемую крысу помещали в основание лабиринта «T». После короткого перерыва, мышь оставляли для исследования лабиринта и выбора входа в правый или в левый коридор. Выбор оценивали в соответствии с рядом критериев, включая спонтанный выбор, выбор, стимулируемый вознаграждением, или выбор по предпочтительности. Исходя из критериев, используемых в данном исследовании, T-образный лабиринт использовали для оценки способности к обучению и памяти. Пищу, помещаемую на одном конце лабиринта, использовали в качестве подтверждения положительного результата решения животным каждого задания.
У животных, которым перорально вводили дозу 1,0 мг/кг AK-51, наблюдалось статистически значимое повышение способности к обучению в испытании с использованием T-лабиринта (P<0,001). У животных, которым перорально вводили дозу 3,0 мг/кг непептидного аналога NRX-10051, наблюдалось статистически значимое повышение способности к обучению с испытании с использованием T-лабиринта (P<0,01).
Пример 8 - Изомеры
В анализах на связывание с NMDA были использованы два различных изомера AK-55, как в примере 4. Один изомер AK-55 в значительной степени активирует NMDA, а другой изомер не обладает такой способностью. На Фиг.7A указано время продолжительности воздействия 15-минутного нахождения в бане 1 мкМ AK55 (сплошной столбик) на нормализованный ток, индуцированный открытием каналов, опосредуемым фармакологически изолированным NMDA-рецептором, в пирамидных нейронах поля CA1 при регистрации целых клеток (среднее ± ср.ст.ош., n=6). B: Время продолжительности воздействия 15-минутного нахождения в бане 1 мкМ AK55 (сплошной столбик) на нормализованный ток, индуцированный открытием каналов, опосредуемым фармакологически изолированным NMDA-рецептором, в пирамидных нейронах поля CA1 при регистрации целых клеток (среднее ± ср.ст.ош., n=7). C: Время продолжительности воздействия нахождения в бане 1 мкМ AK6 (сплошной столбик, заштрихованные кружки, n=8) по сравнению с необработанными контрольными срезами (незаштрихованные кружки, n=8) на величину долговременного потенцирования (LTP), определяемую по наклону кривой внеклеточного возбуждающего постсинаптического потенциала (среднее ± ср.ст.ош., fEPSP), индуцированного стимуляцией коллатералей Шаффера высокочастотными импульсами (2×100 Гц/500 мсек).
Пример 9 - Биохимические анализы
В таблице B представлены результаты анализов на связывание AK51 с различными мишенями:
Пример 10 - Идентификация β-складчатой структуры в спиросоединениях
Эксперименты, проводимые с помощью одномерного (1-D) протонного резонанса 1H, 13C, DEPT; эксперименты, проводимые с помощью двумерного (2-D) гомонуклеарного резонанса (DQF-COSY, TOCSY, NOESY); и эксперименты, проводимые с помощью гетеронуклеарного резонанса HSQC и HMBC в ДМСО-D6 при 30°C, осуществляли для подтверждения точных химических сдвигов углерода и химических сдвигов протонов в спиросоединении:
Наблюдались следующие химические сдвиги: 1H, ДМСО-d6, 600 МГц, δ в м.д., ТМС при 0,00 м.д.: 8,72 (шир., 1H), 3,47 (дд, 2H), 3,37 (т, 2H), 2,21 (м, 2H), 2,02 (м, 1H), 1,89 (м, 1H) (см. Фиг.12);
13C, ДМСО-d6, 150 МГц, δ в м.д., эталонный ДМСО при 39,5 м.д.: 169,6, 68,7, 45,6, 40,7, 32,9, 22,4.
Химический сдвиг амидного протона наблюдался в области 8,72 м.д. в виде широкого синглета, а между 8,72 и 3,37 м.д. наблюдались перекрещивающиеся пики. Этот факт указывал на химические сдвиги H-3 в области 3,47 м.д. Слабый спектр nOe между 3,37 и 2,21 м.д. указывал на совокупность H-5 в области 2,21 м.д. Общая корреляция наблюдалась в пределах 3,37-2,21 м.д. до 2,02 и 1,89 м.д.; корреляция nOe наблюдалась в области между 2,21, 2,02, 1,89 и 3,37 м.д. Эти наблюдения можно объяснить наличием резонанса H-6 и H-7: H-6 (2,02 и 1,89 м.д.), H-7 (3,37 м.д.). Гетеронуклеарные 2-D-эксперименты (HSQC и HMBC) также подтвердили химические сдвиги протонов и углерода.
Наблюдались следующие химические сдвиги отдельных протонов и углерода: 8,72 (шир., H-2, 1H), 3,47 (дд, H-3, 2H), 3,37 (т, H-7, 2H), 2,21 (м, H-5, 2H), 2,02 (м, H-6,1H), 1,89 (м, Η-6'1Η).
169,6(C-1), 68,7(C-4), 45,6(C-7), 40,7(C-3), 32,9(C-5), 22,4(C-6).
Слабый спектр nOe между H-3 и H-5 позволяет предположить об ограниченном вращении циклов относительно друг друга. Отсутствие водородных связей, в которых имеются донорные и акцепторные остатки i (i±3), а также отсутствие крупной области спектра nOe (атомы Cα<7A°) между двумя циклами указывают на отсутствие какой-либо обнаружимой вторичной укладки.
Пример 11
Эксперименты, проводимые с помощью одномерного (1-D) протонного резонанса 1H, 13C, DEPT; эксперименты, проводимые с помощью двумерного (2-D) гомонуклеарного резонанса (DQF-COSY, TOCSY, NOESY); и эксперименты, проводимые с помощью гетеронуклеарного резонанса HSQC и HMBC в ДМСО-D6 при 30°C, осуществляли для подтверждения точных химических сдвигов углерода и химических сдвигов протонов в спиросоединении:
 .
.
1H-ЯМР в ДМСО представлен на Фиг.13. N15-HSQC-эксперимент, проводимый при 600 МГц, может быть осуществлен для подтверждения химических сдвигов амида.
Эквиваленты
Специалисту в данной области будет очевидно, либо может быть установлено всего лишь рутинным экспериментированием, что в конкретные описанные здесь варианты настоящего изобретения может быть внесено множество эквивалентов. Такие эквиваленты входят в объем прилагаемой формулы изобретения.
Введение посредством ссылки
Полное содержание всех патентов, опубликованных патентных заявок, web-сайтов и других цитируемых здесь работ точно и во всей своей полноте вводится в настоящее описание посредством ссылки, включая нижеследующие работы, вводимые посредством ссылки:
Abbott FV, Franklin KB, Westbrook RF, The formalin test: scoring properties of the first and second phases of the pain response in rats. Pain, 60 (1995) 91-102; Bennett GJ, Xie YK., A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain, 33 (1988) 87-107; патент США № 7273889; патент США № 6821985; патент США № 6667317; патент США № 6635270; патент США № 6521414; патент США № 6197820; патент США № 6147230; патент США № 6007841; патент США № 5952389; патент США № 5902815; патент США № 5741778; патент США № 5605911; патент США № 5523323; патент США № 4959493; Moskal et al. (2005) Neuropharmacology, 49(7): 1077-87; Narahshi, Toshio et al. (2004) Biol. Pharm. Bull., 27(1): 1701-1706; Lynch, Gary et al. (2006), Aging Research Reviews, 5:255-280; Rajashankar et al. J. Am. Chem. Soc. (1992), 114, 9225; Rajashankar et al. J. Am. Chem. Soc. (1995), 117, 10129; A. Karle et al, Biochemistry (1990), 29, 6747; Regan et al, Science (1998), 241, 976; Kaumaya et al, Biochemistry (1990), 29, 13; Hanessian et al, Tetrahedron (1997), 53, 12789; Zhang et al, Org. Lett. (2003), 53115; Xuyuan et al, Org. Lett. (2004), 6, 3285; Halab et al, J. Med. Chem. (2002), 45, 5353.
| название | год | авторы | номер документа |
|---|---|---|---|
| МОДУЛЯТОРЫ НМДА-РЕЦЕПТОРА И ИХ ПРИМЕНЕНИЯ | 2009 |
|
RU2515615C2 |
| КОМБИНАЦИИ СОЕДИНЕНИЙ, МОДУЛИРУЮЩИХ NMDA-РЕЦЕПТОР | 2015 |
|
RU2721948C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ИЛИ ОБЛЕГЧЕНИЯ МИГРЕНИ | 2015 |
|
RU2721401C2 |
| ИСПОЛЬЗОВАНИЕ МОДУЛЯТОРА РЕЦЕПТОРА S1P | 2008 |
|
RU2498796C2 |
| ГУАНИДИНСОДЕРЖАЩИЕ СОЕДИНЕНИЯ, ПРИМЕНИМЫЕ В КАЧЕСТВЕ АНТАГОНИСТОВ МУСКАРИНОВЫХ РЕЦЕПТОРОВ | 2008 |
|
RU2480458C2 |
| СРЕДСТВА ПРОТИВ СЛАБОУМИЯ, СОДЕРЖАЩИЕ ПРОИЗВОДНОЕ 2-АРИЛ-8-ОКСОДИГИДРОПУРИНА В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2001 |
|
RU2277096C2 |
| НЕЙРОАКТИВНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2015 |
|
RU2764702C2 |
| ПТЕРИДИНЫ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ HCV, И СПОСОБЫ ПОЛУЧЕНИЯ ПТЕРИДИНОВ | 2006 |
|
RU2447074C2 |
| НОВЫЕ ЛИГАНДЫ ЭСТРОГЕНОВЫХ РЕЦЕПТОРОВ | 2009 |
|
RU2492164C2 |
| 5-ГЕТЕРОЦИКЛИЛПИРИМИДИНЫ, ИНГИБИРУЮЩИЕ ВИЧ | 2005 |
|
RU2405778C2 |
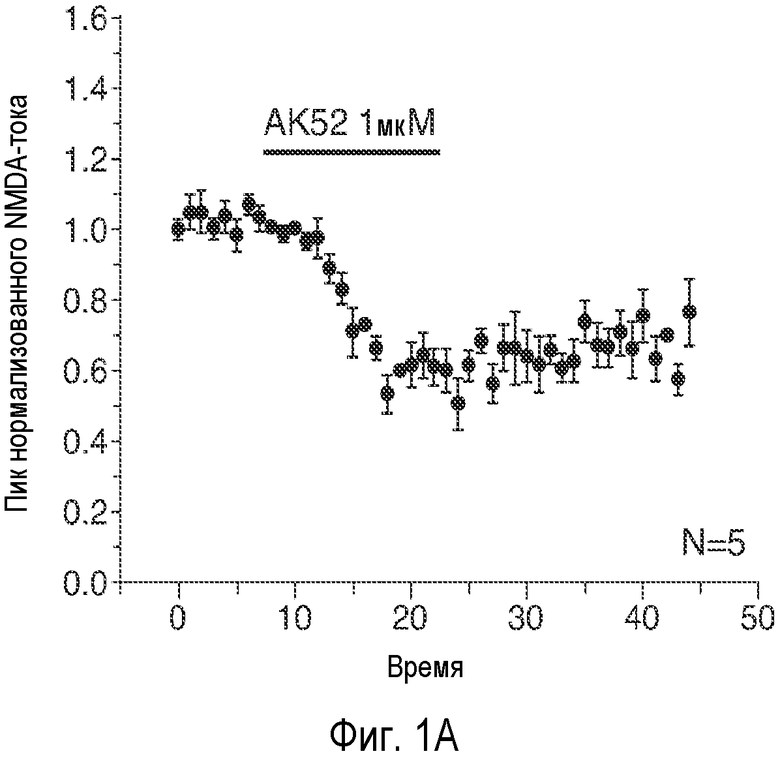
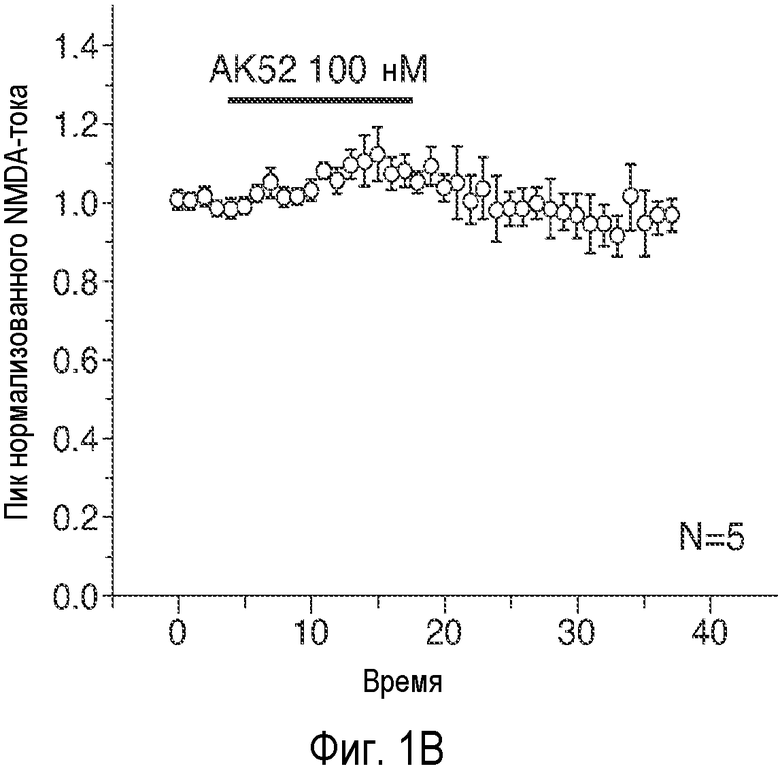
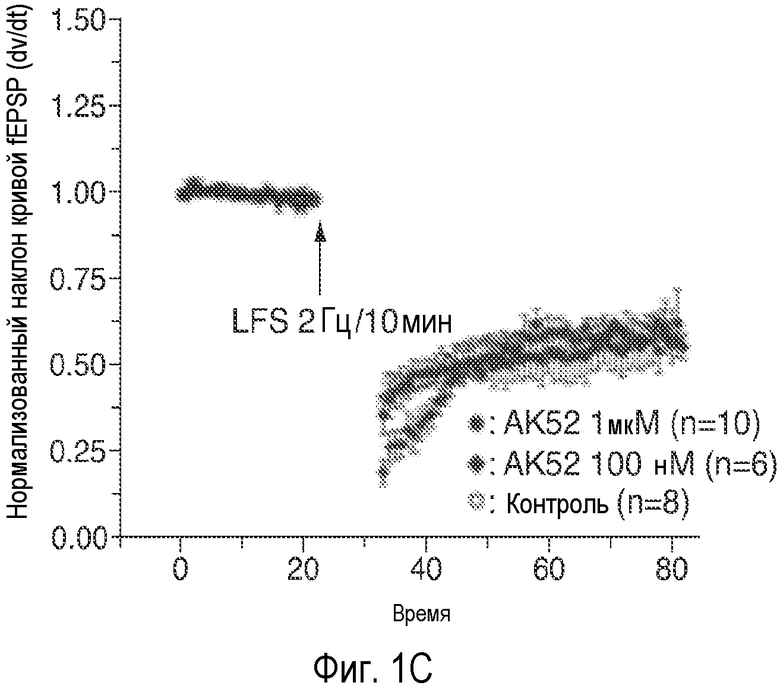
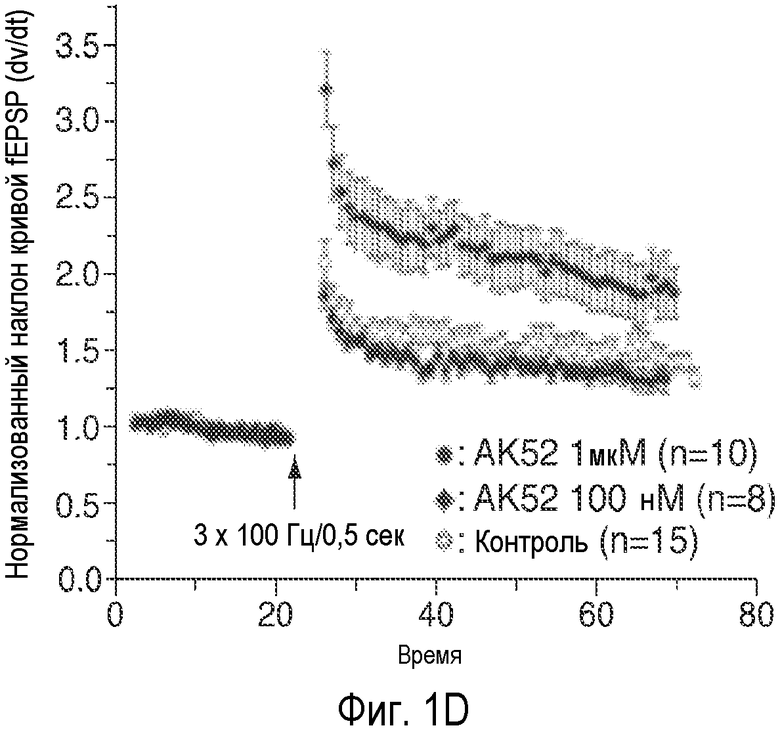
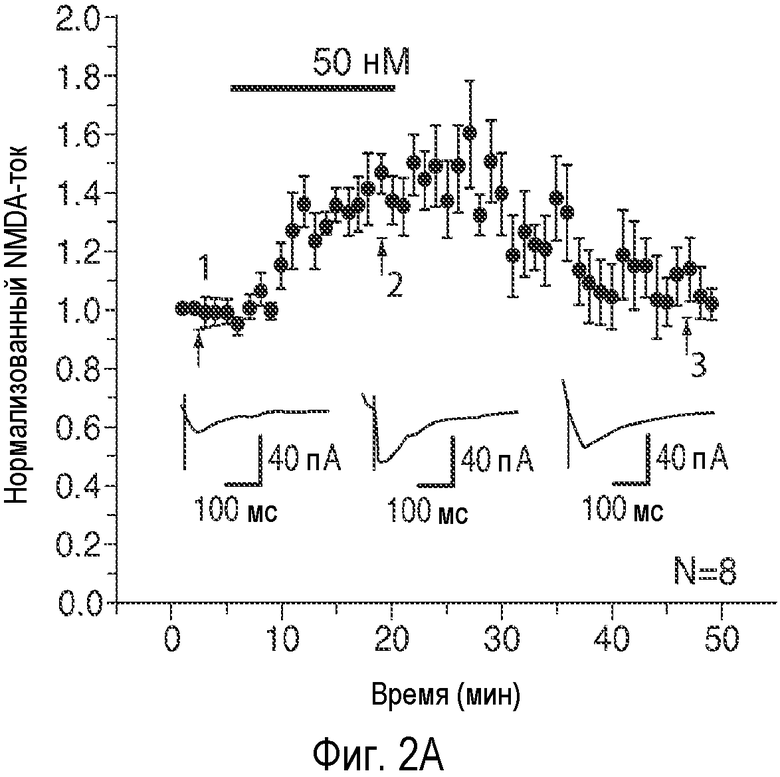
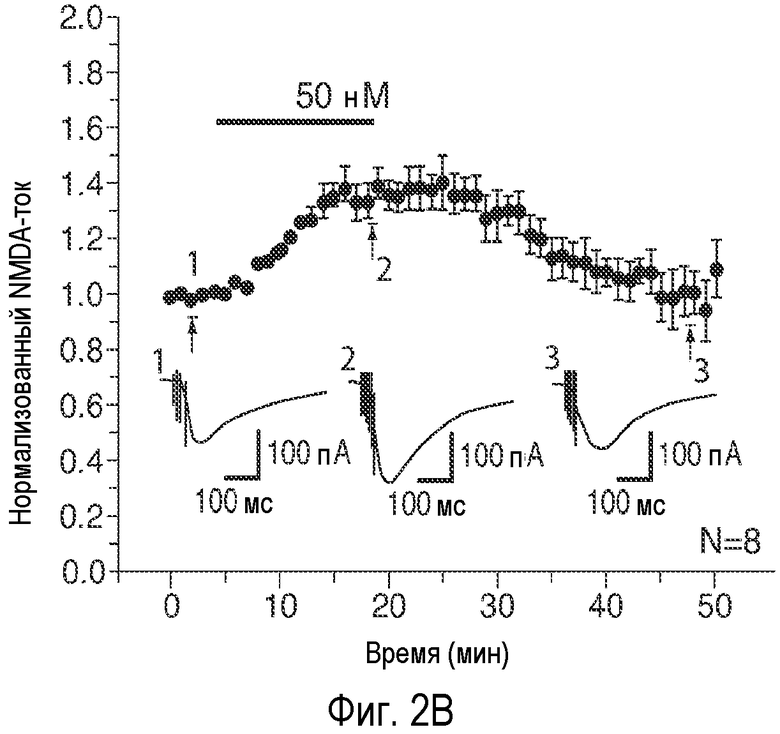
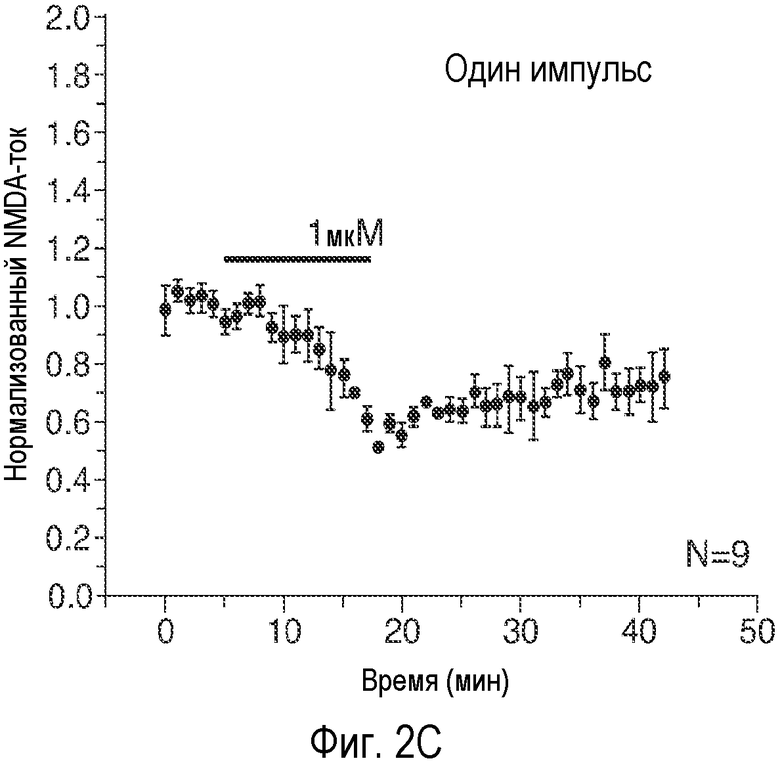
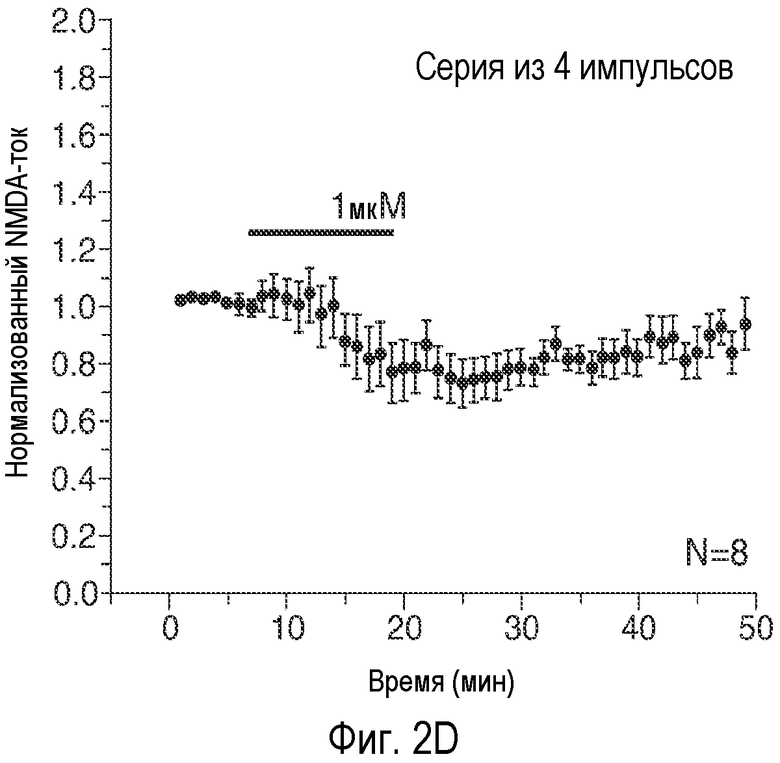
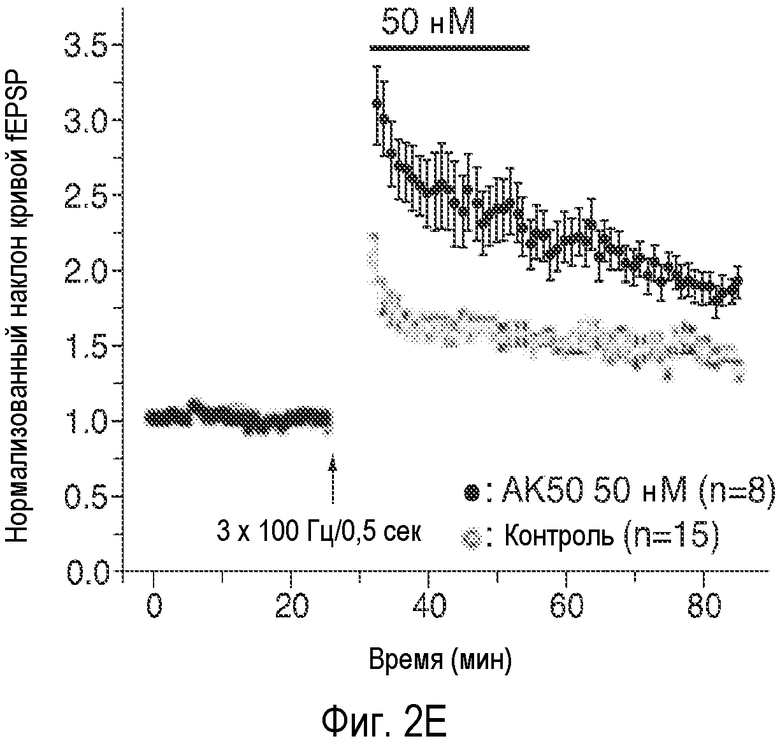
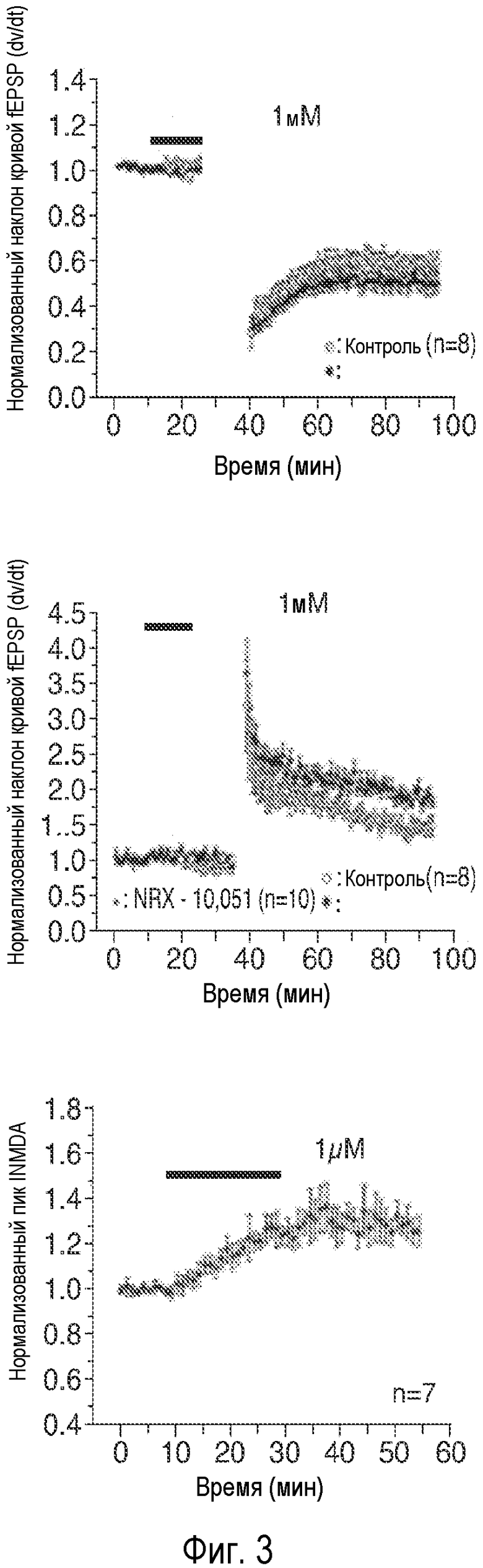
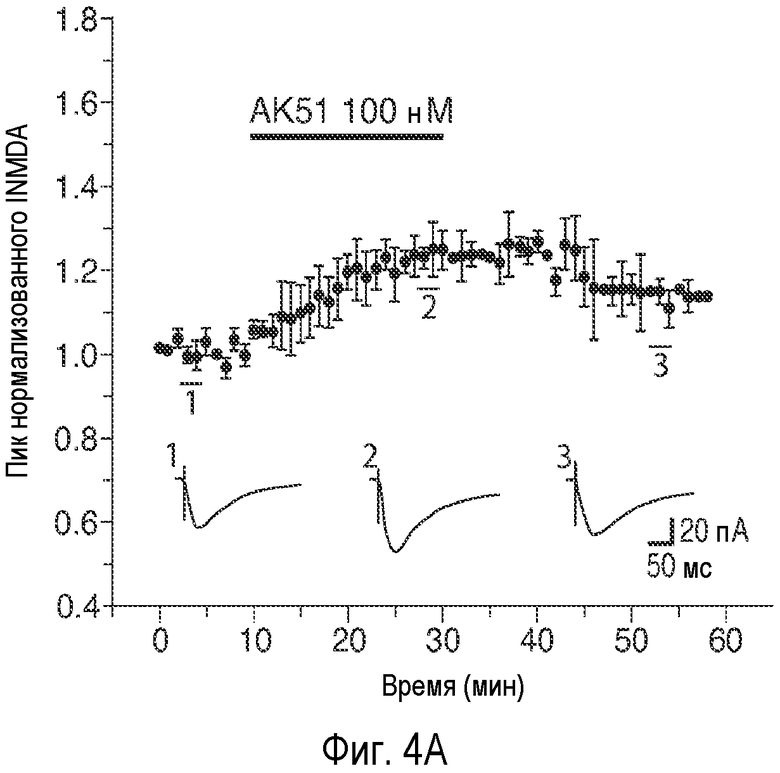
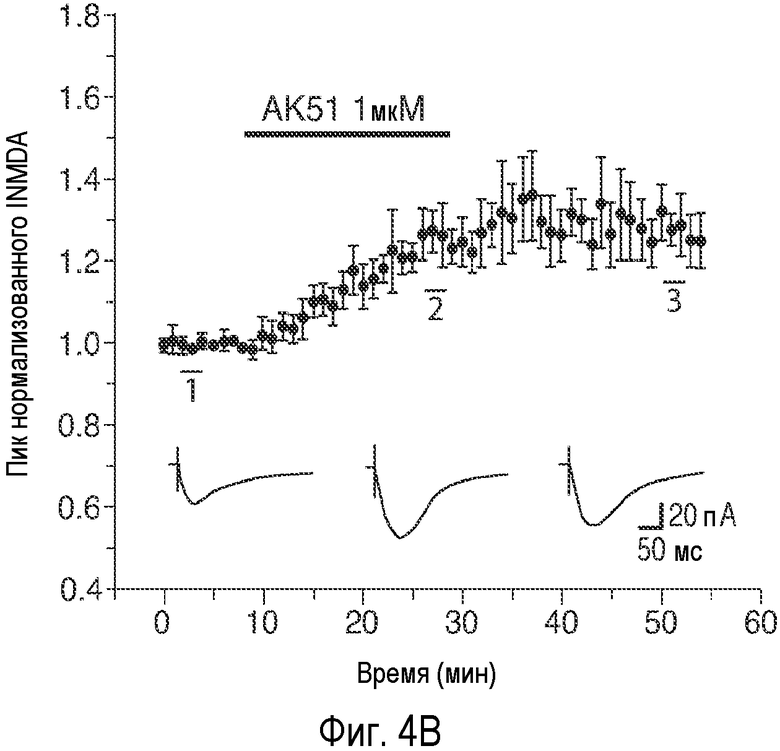
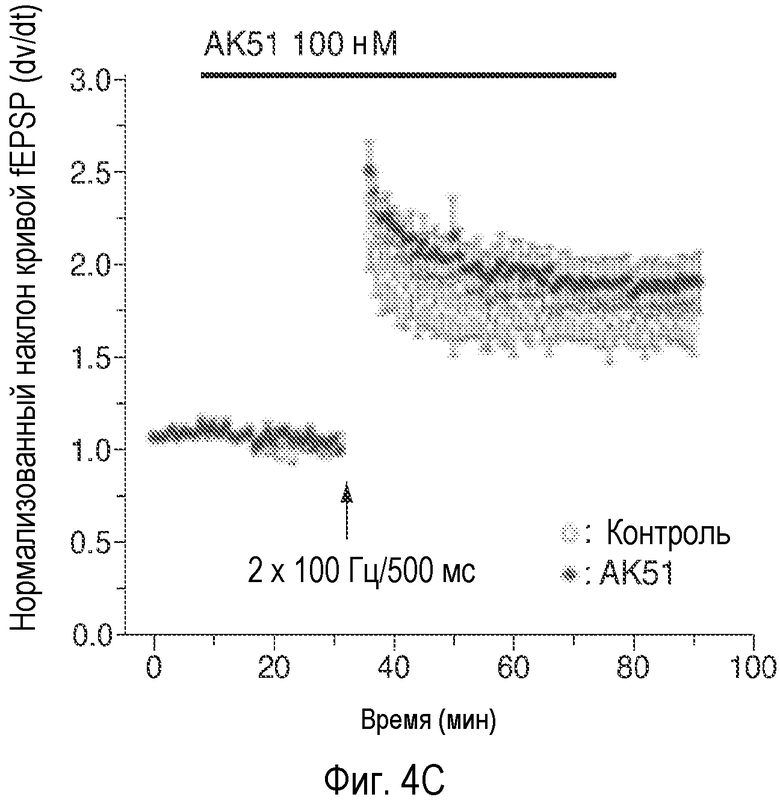
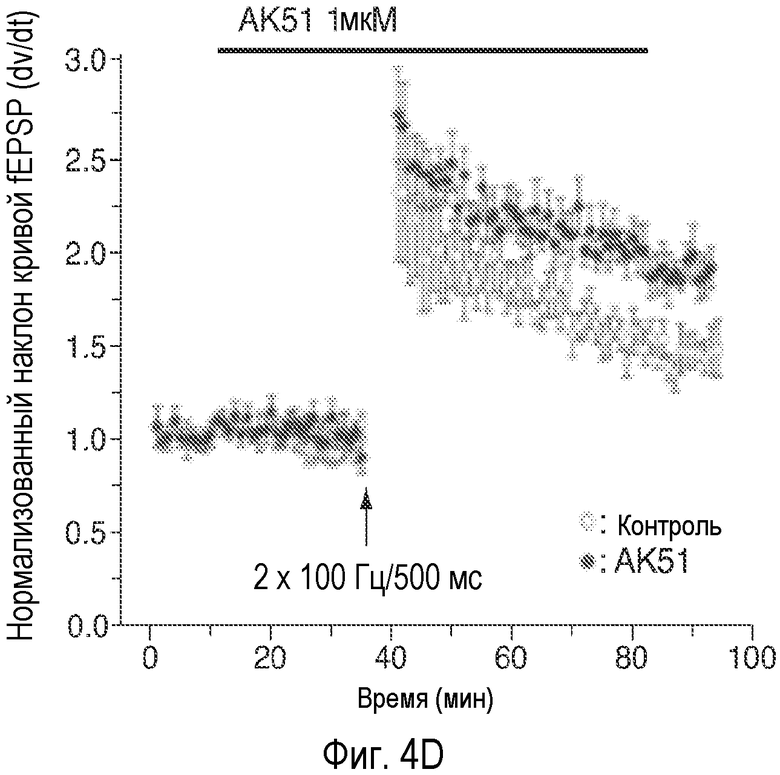
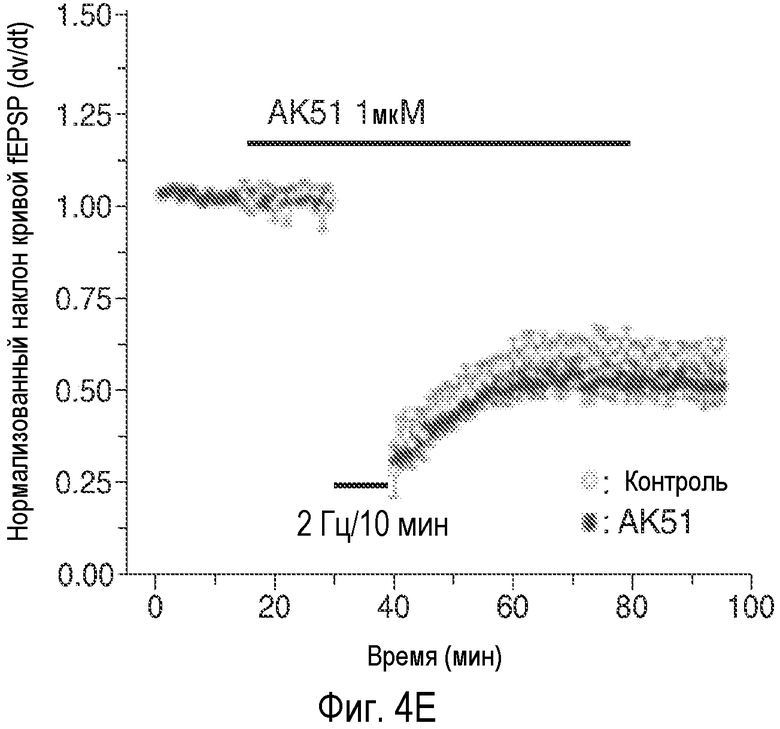
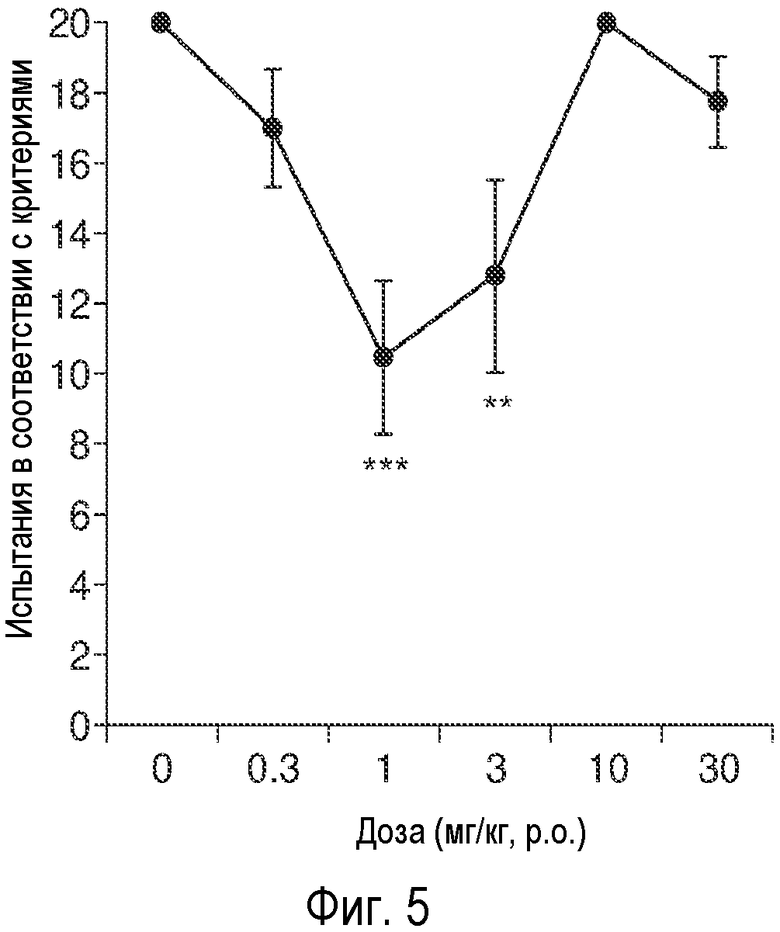
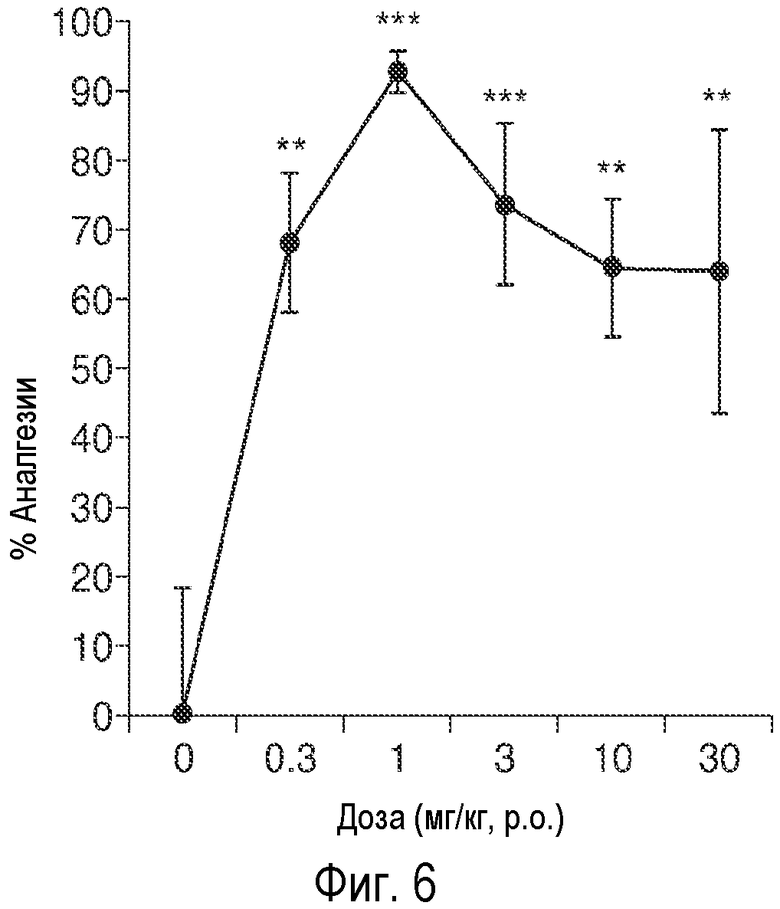
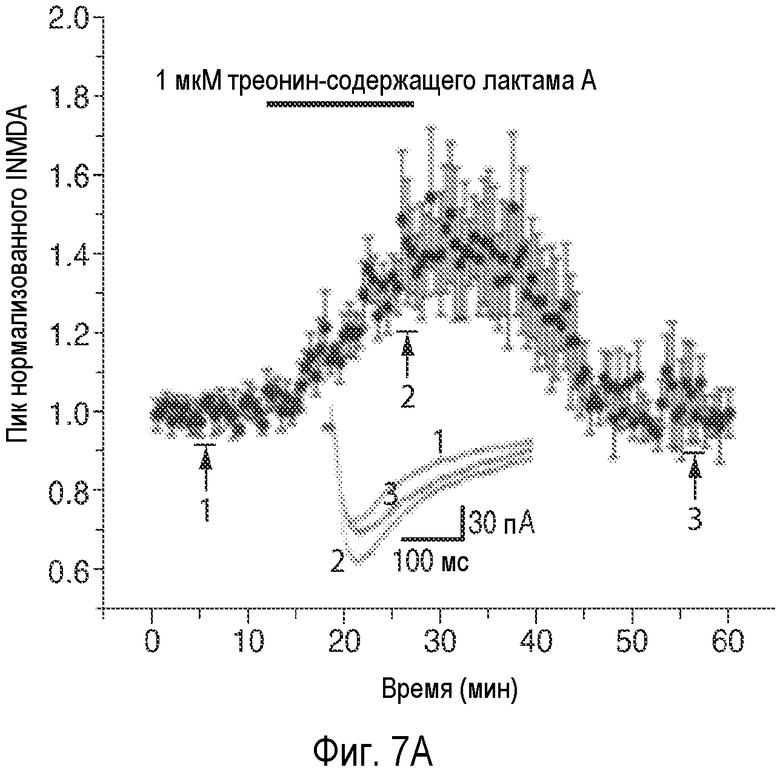
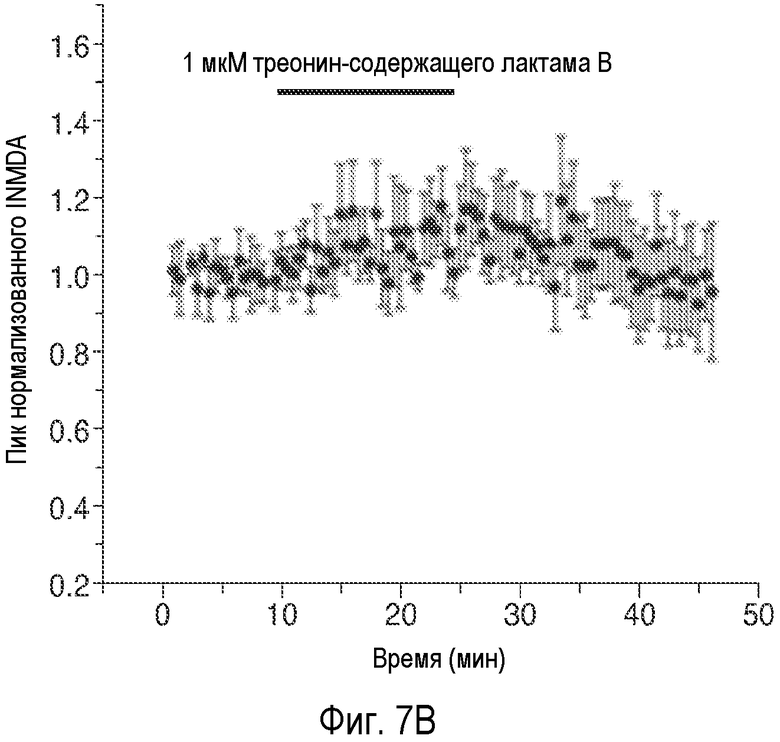
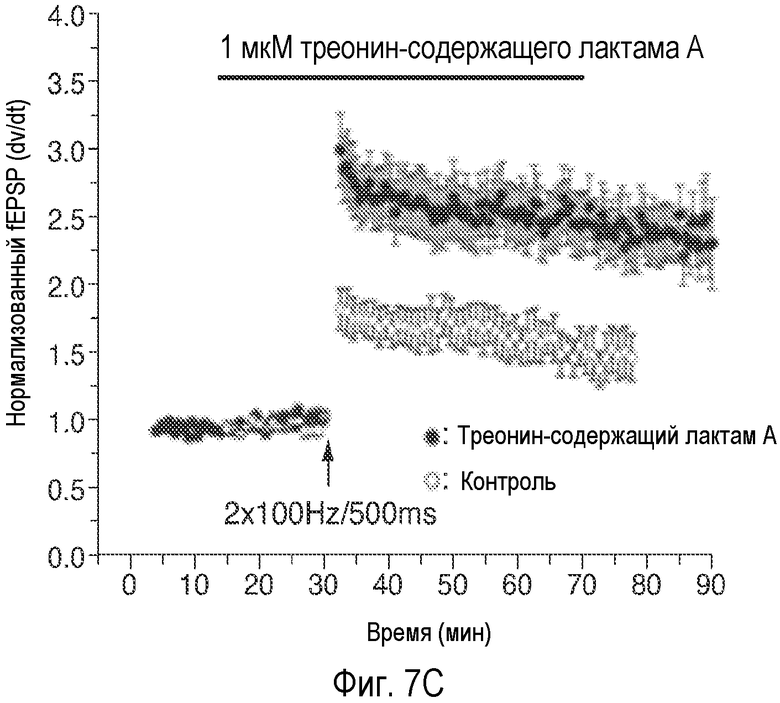
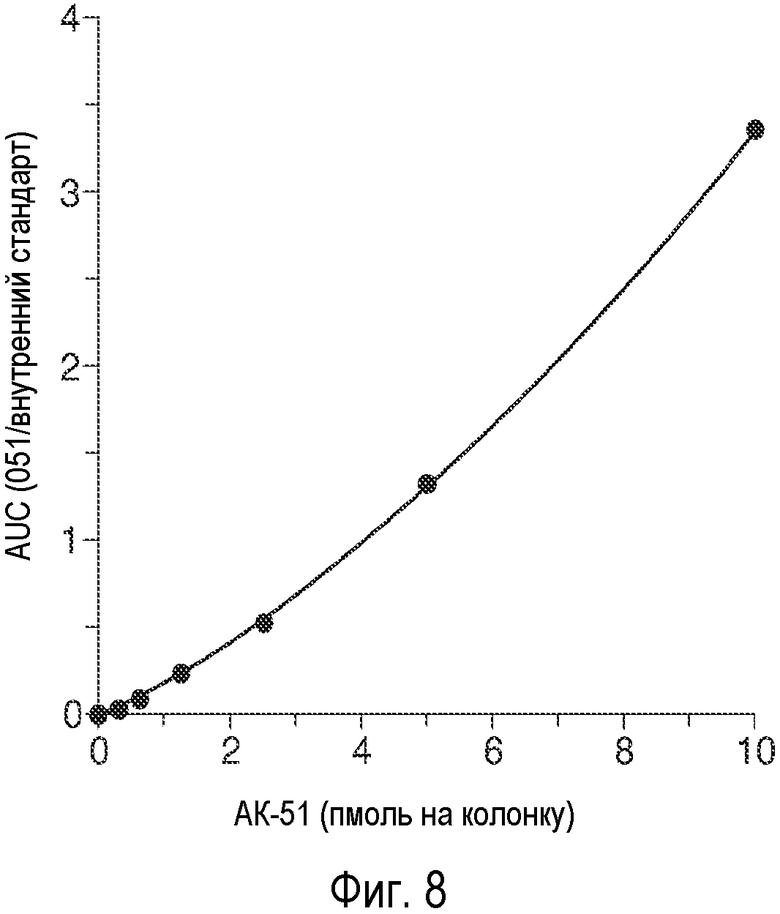

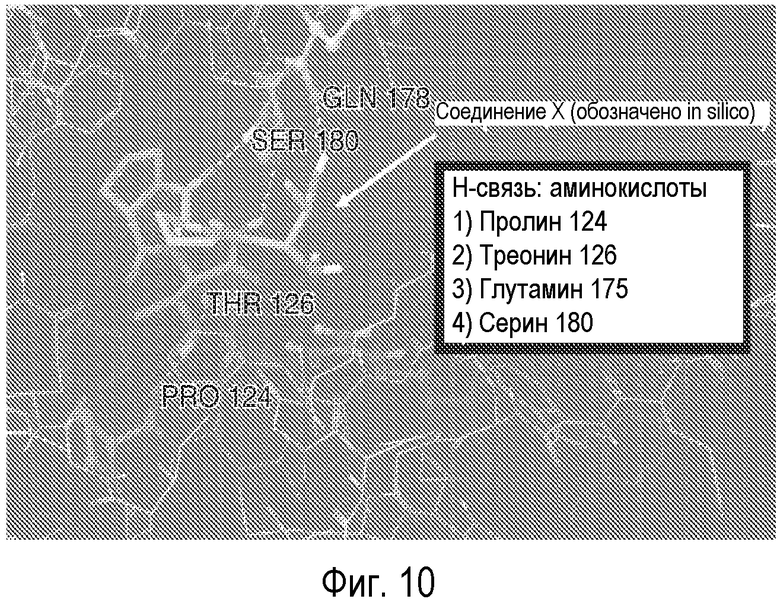
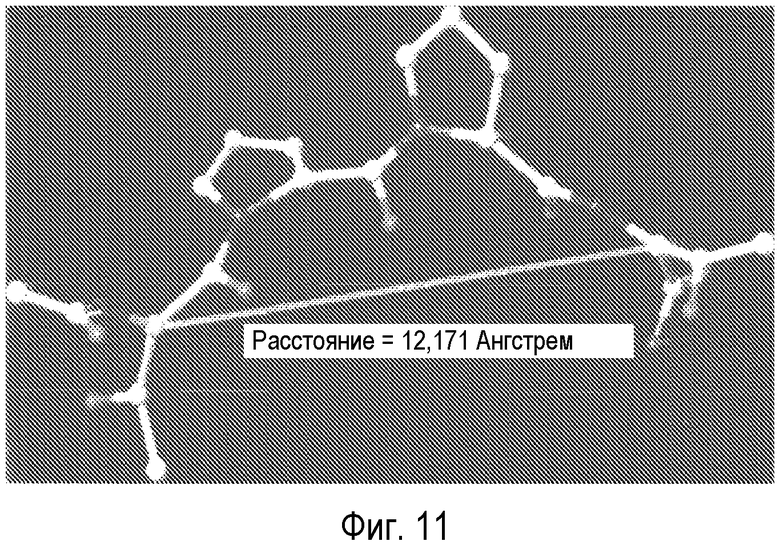
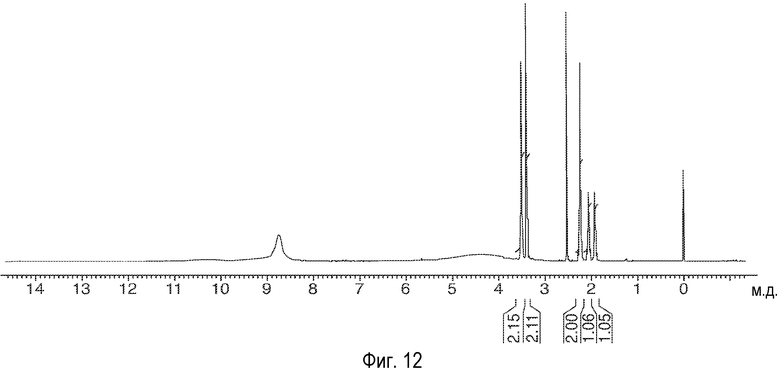
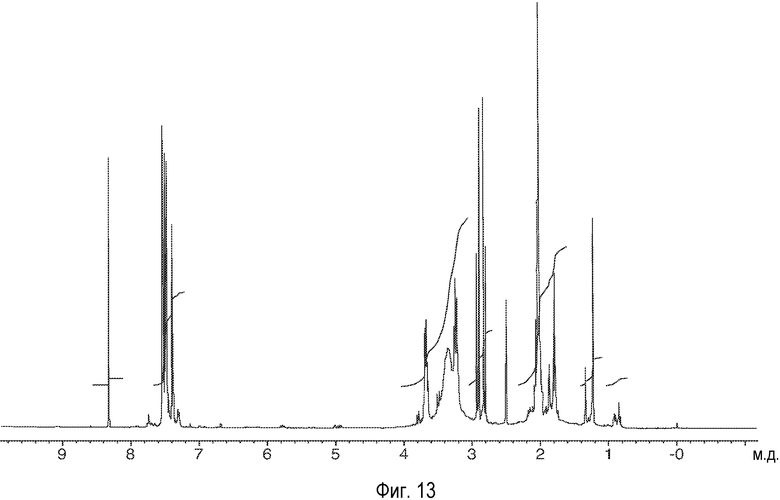
Изобретение относится к соединениям, обладающим повышенной активностью, направленной на модуляцию активности NMDA-рецептора, и их применению для лечения заболеваний и расстройств, таких как нарушение способности к обучению, когнитивные расстройства и аналгезия, а в частности, для ослабления и/или устранения невропатической боли. 7 н. и 2 з.п. ф-лы, 13 ил., 2 табл., 11 пр.
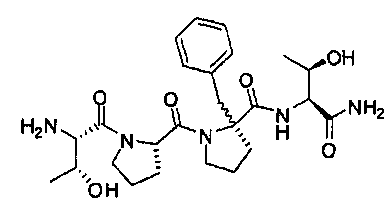
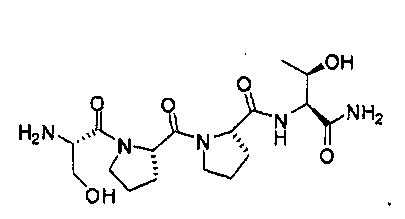
1. Соединение, представленное формулой:
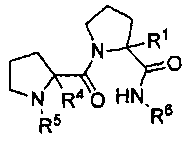
где
R4 представляет собой Н;
R1 представляет собой бензил;
R5 представляет собой
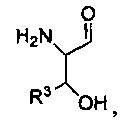
R6 представляет собой:
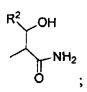
R2 представляет собой Н или СН3;
R3 представляет собой Н или СН3; и их стереоизомеры, N-оксиды или фармацевтически приемлемые соли.
2. Соединение по п. 1, где R2 и R3 представляют собой СН3.
3. Соединение по п. 1, где R2 представляет собой СН3, a R3 представляет собой Н.
4. Соединение, представленное формулой:
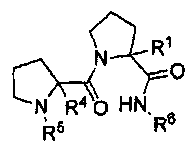
где
R4 представляет собой Н;
R1 представляет собой Н;
R5 представляет собой:
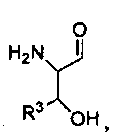
R6 представляет собой:
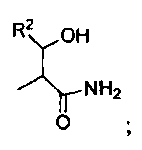
R2 представляет собой СН3, a R3 представляет собой Н или R2 представляет собой Н, a R3 представляет собой СН3; и их стереоизомеры, N-оксиды или фармацевтически приемлемые соли.
5. Соединение, представленное формулами:
или
6. Способ лечения когнитивного расстройства, связанного с потерей памяти или нарушением способности к обучению, где указанный способ включает введение пациенту, нуждающемуся в этом, эффективного количества соединения по одному из пп. 1-5.
7. Фармацевтически приемлемая композиция для лечения невропатической боли, депрессии, расстройства, ассоциированного с навязчивыми состояниями, или шизофрении у пациента, нуждающегося в этом, содержащая соединение по любому из пп. 1-5 и фармацевтически приемлемый наполнитель.
8. Способ лечения невропатической боли у пациента, нуждающегося в этом, где указанный способ включает введение эффективного количества соединения по любому из пп. 1-5.
9. Способ лечения депрессии, расстройства, ассоциированного с навязчивыми состояниями, или шизофрении у пациента, нуждающегося в этом, где указанный способ включает введение эффективного количества соединения по любому из пп. 1-5.
| МОДУЛЯТОРЫ НМДА-РЕЦЕПТОРА И ИХ ПРИМЕНЕНИЯ | 2009 |
|
RU2515615C2 |
| US 2003022253 A1, 30.01.2003 | |||
| Stanton, Patric K | |||
| et al, "Neuroprotection by a novel NMDAR functional glycine site partial agonist, GLYX-13", 2009, NeuroReport, 20(13), 1193-1197 | |||

