Изобретение относится к способу получения оксида хрома(III), исходя из хроматов щелочных металлов и газообразного аммиака, а также к использованию полученного оксида хрома(III) для различных применений.
Оксид хрома(III) представляет собой многосторонний продукт с широкой палитрой применения. Так его можно использовать в качестве пигмента для окрашивания в различных областях применения, таких как, например, строительные материалы, пластмассы, краски и лаки, стекла или керамика. Для этих областей применения предъявляется требование, чтобы присутствовало по возможности низкое количество растворимых в воде примесей.
Кроме того, оксид хрома(III) также применяется в шлифовальных средствах и в устойчивых при высоких температурах технических материалах. В случае использования оксида хрома(III) в устойчивых при высоких температурах технических материалах желательно по возможности низкое содержание щелочного металла, чтобы по возможности подавить благоприятное при высоких температурах и в присутствии ионов щелочных металлов окисление Cr(III) в хромат щелочного металла.
Другая важная промышленная область применения оксида хрома(III) состоит в использовании в качестве исходного материала для получения металлического хрома и/или хромсодержащих высокоэффективных сплавов. В этом случае используются, как правило, только оксиды хрома(III), которые отличаются низким содержанием серы и низким содержанием углерода. Понятие "оксид хрома(III) с низким содержанием серы" в связи с этим часто используют в качестве синонима для "оксида хрома(III) для металлургических целей".
Оксид хрома(III) можно получить согласно уровню техники различными способами. В большинстве случае его получают при более высоких температурах из шестивалентных соединений хрома, причем могут достигаться различные степени чистоты. В качестве исходных соединений шестивалентного хрома используют хромовую кислоту, хроматы аммония или хроматы щелочных металлов. Реакцию можно проводить с добавлением или без добавления восстанавливающего средства. В качестве восстанавливающих средств используются органические или не органические восстанавливающие средства, такие как опилки, меласса, целлюлозные отработанные растворы, ацетилен, метан, сера и ее соединения, фосфор, углерод, водород и тому подобные. Такого рода способы описаны в многочисленных патентах. В качестве примера можно назвать US 1893761 и DE-A-2030510. В US 1893761 сообщается о получении оксида хрома(III) путем восстановления хроматов щелочных металлов органическими веществами. В случае применения углерода или органических соединений в качестве восстанавливающих средств можно процесс осуществить таким образом, что в результате получают карбонат натрия в качестве побочного продукта, как упоминается в US 1893761. Его можно при необходимости возвращать в процесс производства бихромата натрия в том случае, когда бихромат натрия получают оксидативным щелочным разложением, исходя из хромовой руды. Однако, полученный таким образом оксид хрома(III) имеет высокое содержание углерода, который делает его не пригодным для металлургического применения. В DE-A-2030510 описан способ непрерывного получения очень чистого с низким содержанием серы оксида хрома(III), путем восстановления хроматов щелочных металлов водородом при высокой температуре, а также описано подходящее для этого устройство. Температура реакции лежит между 1000-1800°С, предпочтительно между 1100-1400°С, и полученный продукт отделяют с помощью щелочно установленной дисперсии от отходящих газов. В описанных в DE-A-2416203 и US 4052225 способах, также используется водород для восстановления хроматов щелочных металлов. В обоих способах хромат щелочного металла восстанавливают в мелкодисперсной форме в обогреваемой, содержащей водород реакционной зоне при температуре от 900 до 1600°С, причем восстановление можно также проводить в присутствии газа, который связывает ионы щелочного металла, образующиеся при восстановлении хромата щелочного металла, с образованием соли, и причем образующийся оксид хрома(III) осаждается в виде щелочно заданной дисперсии. В качестве солеобразующих газов предпочтительно применяют хлор или хлористый водород, в результате чего образуется хлористый натрий. Однако, в связи с тем, что температура плавления хлористого натрия лежит около 800°С, может происходить его расплавление в реакторе, в результате чего при более длительном процессе могут образоваться комки и происходить пригорания.
Недостаток всех этих способов, которые проводятся с восстановительным средством, состоит в том, что в результате применения восстановительного средства обязательно образуется побочный продукт, который подлежит переработке.
Термическое разложение чистого бихромата аммония, напротив, само не приводит ни к какому заметному образованию побочного продукта, так как оно протекает в идеальном случае по уравнению реакции

и при температуре около 200°С. Во всяком случае, практикуемые в настоящее время технические способы получения бихромата аммония исходят из бихроматов щелочных металлов - чаще всего из бихромата натрия. В этом случае бихромат натрия с помощью хлорида аммония или сульфата аммония превращают в бихромат аммония и хлористый натрий, соответственно, в бихромат аммония и сульфат натрия. Оксид хрома(III) для металлургических целей получали раньше в промышленных масштабах путем кальцинирования в печи смеси бихромата аммония и хлористого натрия, которая получается в результате in-situ реакции бихромата натрия и хлорида аммония в практически стехиометрически эквивалентных количествах. Температура кальцинирования должна быть выше 700°С, для того чтобы гарантировать, что в реакционной смеси содержится высокая доля оксида хрома(III); при слишком высокой температуре, как правило, повышается риск образования шлака в печи, и в связи с этим температуру поддерживают, как правило, ниже 850°С.
Использование сульфата аммония вместо хлорида аммония часто является более предпочтительным, так как хлорид аммония в связи с его низкой температурой сублимации при кальцинировании сублимируется в виде NH3 и НСl и таким образом может переходить в отходящие газы. По этой причине применение хлорида аммония в настоящее время более не имеет хозяйственного значения. Недостаток использования сульфата аммония состоит, во всяком случае, в том, что таким путем сера вовлекается в процесс получения, хотя желательным является оксид хрома(III) с по возможности низким содержанием серы.
В DE-A-2635086 (US-A-4235862) описан способ получения оксида хрома(III) с низким содержанием серы, который отличается отжигом смеси бихромата щелочного металла и сульфата аммония при температуре кальцинирования от 800 до 1100°С и отделением образовавшегося оксида хрома(III) от образовавшейся соли щелочного металла, причем на 1 моль хромата щелочного металла используют от 0,7 до 0,89, предпочтительно от 0,7 до 0,84 моля сульфата аммония. Переработку оксида хрома(III) после отжига проводят непрерывным способом путем вымывания водорастворимых солей и сушки. Таким путем могут быть достигнуты содержания серы в оксиде хрома(III) от 50 до 100 млн долей. Недостаток при осуществлении этого способа состоит в том, что для достижения низких содержаний серы исходные вещества приходится смешивать не в стехиометрическом отношении и сульфат аммония берется в отчетливо меньшем количестве. Это приводит к низким превращениям в области около 90%, и требует поддержания более высоких температур отжига. Находящийся в избытке бихромат щелочного металла разлагается термически в хромат щелочного металла, оксид хрома(III) и кислород. Таким образом, при реакции, наряду с большим количеством сульфата щелочного металла (например, сульфата натрия), образуется также хромат щелочного металла (например, хромат натрия), который при позднейшем вымывании переходит в маточный раствор или в промывающую жидкость и должен быть отделен, для того чтобы при необходимости возвратить обратно в процесс. Маточный раствор в этом случае также содержит избыточный осадившийся сульфат щелочного металла, очистка которого является затратной в связи с тем, что он загрязнен хроматом щелочного металла. Кроме того, на практике оказалось, что предложенные условия получения оксида хрома(III) с низким содержанием серы оказались трудновыполнимыми в связи с тем, что содержащийся в реакционной смеси сульфат натрия при требуемой высокой температуре испытывает спекание (температура плавления сульфата натрия около 885°С), а это приводит к нарушению протекания процесса получения.
Что касается получения оксида хрома(III) с низким содержанием серы, то в US 4296076 описан способ, при котором среди прочего используют бихромат натрия и хлорид аммония, соответственно, бихромат натрия и сульфат аммония. В отличие от DE-A-2635086 при этом берется в существенной мере стехиометрическое отношение или предпочтительно избыток аммониевого соединения. На первой стадии реакции исходные соединения превращают в бихромат аммония и хлористый натрий, соответственно, в бихромат аммония и сульфат натрия. В опубликованных примерах эта стадия реакции происходит при температуре от 400 до 800°С, затем следует обработка водой и после этого второй процесс отжига при температуре выше 1100°С. При этом способе добиваются содержания серы в оксиде хрома(III) ниже 40 млн долей. При этом способе образуются большие количества хлористого натрия, соответственно, сульфата натрия, очистка которых является затратной. Кроме того, применение указанных аммониевых соединений, в особенности хлорида аммония, не беспроблемно, в связи с тем, что они легко сублимируются и тем самым могут попасть в отходящие газы.
Другой описанный в уровне техники способ получения качественно высокоценного оксида хрома (III) описан в RU 2258039. И здесь используют бихромат аммония, получаемый при взаимодействии бихромата натрия с сульфатом аммония в водной фазе, для получения оксида хрома(III), при этом образующийся при взаимодействии сульфат натрия отделяют от реакционной смеси, так что относительно чистый, то есть бедный серой, бихромат аммония термически разлагается в оксид хрома(III). В качестве побочного продукта образуется сульфат натрия, очистка которого является затратной, так как он загрязнен Cr(VI).
Термическое разложение чистой хромовой кислоты (2) описано среди прочего в литературе в виде реакции (например, в Ullmann's Encyclopedia of Industrial Chemistry, Vol.A7, p.87, VCH Verlag, 1986)

Также и в случае использования хромовой кислоты в качестве исходного материала для получения оксида хрома(III), как правило, на первой стадии хроматы щелочного металла подвергают взаимодействию с серной кислотой и/или с гидросульфатсодержащими соединениями с образованием бихроматов щелочного металла (3) и затем с помощью дополнительной серной кислоты переводят в хромовую кислоту (4)
 ,
,
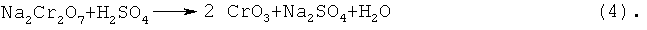
И при этих способах получения оксида хрома(III) образуется существенное количество сульфатов щелочных металлов, например, сульфата натрия, в качестве побочного продукта. В упомянутом способе термического разложения чистой хромовой кислоты, исходя из хромата натрия, образуются на один килограмм оксида хрома(III) около 1,9 кг сульфата натрия (комбинация из реакций (3), (4) и (2)). Сульфат натрия, однако, загрязнен хроматом натрия, так что у него худшее качество и его перед продажей надо еще подвергнуть затратной очистке. Кроме того, хромовая кислота является очень сильным окисляющим средством и является сильно коррозионно действующим соединением. В связи с этим обращение с этим веществом в технических процессах при более высоких температурах соответственно затруднено.
Описаны и другие способы получения оксида хрома(III) с небольшим содержанием серы, при которых используют исходные вещества, в значительной мере свободные от углерода и серы.
В DE-A-2852198 (US-A-4230677) получение монохромата аммония осуществляют превращением бихромата натрия или монохромата натрия и экстракцией из раствора органическим растворителем. Примыкающее кальцинирование в оксид хрома(III) происходит при температуре 500°С. Недостаток этого способа состоит в том, что работают с очень разбавленными водными растворами. Так концентрация хрома - в пересчете на Cr2O3 - в подлежащем экстрагированию водном растворе лежит в интервале от 1 г/л до 25 г/л, причем в качестве более предпочтительной указана концентрация 8,2 г/л. Но и в органической фазе после двух стадий экстракции может быть достигнута только концентрация Cr2O3, равная 10 г/л. В результате приходится иметь дело с большими количествами жидкости, их переработкой и новым введением в кругооборот. В качестве органических растворителей используют бензол, ксилол или толуол сами по себе или в виде смеси с изопарафиновым углеводородом. В случае всех этих веществ имеются в виду опасные вещества, которые легко воспламеняются, так что осуществление этого способа следует проводить с соблюдением многих мер по защите сотрудников и окружающей среды. Кроме того, экстракция проводится при рН-значении между 1 и 2, для чего используют соляную кислоту. В результате образуется незначительное количество хлористого натрия, который загрязняет сточные воды. Используемые органические растворители все обнаруживают заметную растворимость в воде (растворимость при температуре 20°С в воде: бензола 1,77 г/л, толуола 0,47 г/л, ксилола 0,2 г/л), так что сточные воды дополнительно содержат высокую долю органических соединений, и очистка сточных вод является затратной. В связи с многочисленными недостатками этот способ до сих пор не приобрел хозяйственного значения.
Термическая обработка бихромата натрия при более высоких температурах приводит и в отсутствии восстанавливающих средств к оксиду хрома(III). Так Na2Cr2O7*2H2O согласно исследованиям S. Sampath и др. (Thermochimica Acta, 159 (1990), p.327-335), начиная с температуры 500°С медленно разлагается на Na2CrO4 и Cr2O3

Согласно уравнению химической реакции в идеальном случае максимум 50 мол. процентов использованного Cr(VI) превращаются в оксид хрома(III). Во время процесса нагревания вначале при температуре 83°С происходит переход содержащего кристаллизационную воду бихромата натрия в безводное соединение. Не содержащий кристаллизационной воды бихромат натрия плавится при температуре 357°С, так что происходит разложение расплава. В результате этого степень превращения при реакции еще раз отчетливо понижается. Скорость реакции при температуре около 500°С еще очень низка, так что приходится применять более высокие температуры для того, чтобы достигнуть приемлемых скоростей реакции. Так, например, при разложении безводного бихромата натрия при температуре 750°С только около 25 мол. процентов Cr(VI) превращаются в Cr2О3. В связи с низким выходом этот способ получения оксида хрома(III) не представляет интереса в промышленных масштабах.
В CN-A-1310132 получение хромата аммония проводят путем превращения хромата натрия в присутствии двуокиси углерода и аммиака. Получаемый согласно этому способу хромат аммония затем используют для получения оксида хрома(III). Однако опубликованный способ получения хромата аммония обнаруживает много недостатков. С одной стороны, используемый раствор хромата натрия перед началом необходимо перекристаллизовать и профильтровать. То есть необходима - только не полностью описанная - стадия очистки, при которой образуется хлористый натрий в качестве побочного продукта. С другой стороны, взаимодействие с двуокисью углерода и аммиаком проходит в две стадии реакции, при которых в каждом случае добавляют двуокись углерода и аммиак. Отделение образовавшегося при первом взаимодействии гидрокарбоната натрия происходит при охлаждаемой кристаллизации, причем скорость охлаждения составляет от около 1°С/час до 4°С/час. В связи с этим кристаллизация представляет собой очень медленный и затратный по времени процесс, так как во всех опубликованных примерах перед фильтрованием имеет место двухчасовая стадия старения. Условия, при которых из полученного хромата аммония должен быть получен оксид хрома(III), однако не описаны в CN-A-1310132.
Использование чистого хромата аммония или бихромата аммония для термического разложения с целью получения чистого оксида хрома(III), как правило, не является не критическим, так как разложение в сухом состоянии может происходить взрывообразно. Бихромат аммония в связи с этим является опасным веществом с символом опасности "Е" (взрывоопасен). В связи с этим реакция разложения является трудно управляемой. Полученный при этой реакции продукт разложения Cr2O3 отличается экстремально низкой насыпной плотностью, которая может находиться в интервале от 0,1 до 0,2 г/см3. В связи с этим полученный продукт разложения Cr2O3 проявляет очень сильную склонность к распылению. В промышленном процессе отходящие газы должны очищаться от большого количества пыли. Пыль содержит, кроме того, еще не превращенные доли Cr(VI).
В CN-A-1418822 сообщается об одновременном получении бихроматов щелочных металлов и оксида хрома(III), которое отличается тем, что смешивают хромат щелочного металла с хроматом аммония или бихроматом аммония в молярном отношении хромат щелочного металла: хромат аммония или бихромат аммония=(0,3-3):1 и смесь отжигают в температурном интервале от 650°С до 1200°С в течение 0,5-3 часов. Отожженный продукт растворяют в воде. После разделения твердое вещество/жидкость твердый остаток содержит оксид хрома(III) с низким содержанием серы. Из сконцентрированного маточного раствора путем охлаждения выкристаллизовывают бихромат щелочного металла. Твердый бихромат щелочного металла отделяют при разделении твердое вещество/жидкость от еще не прореагировавшего хромата щелочного металла. В опубликованных примерах применяют смеси хромата натрия (Na2CrO4*4H2O) и хромата аммония, хромата натрия (Na2CrO4) и бихромата аммония, хромата калия (K2CrO4) и хромата аммония, а также хромата калия (K2CrO4) и бихромата аммония. Выход оксида хрома(III) в пересчете на содержащийся в исходных смесях Cr(VI) колеблется в примерах 1-3 между 36 и 40%. Кроме того, продукт реакции, который получают, например, в примерах 1 и 2, является очень клейким. Это сильно затрудняет техническую реализацию, например, во вращающейся печи.
Из CN 1418821 известно, что не содержащий серы оксид хрома получают кальцинированием при температуре 650-1200°С двойной соли 1:1 хромата аммония-щелочного металла. Недостаток описанного там способа, однако, состоит в том, что выход оксида хрома составляет только около 23% в пересчете на исходное вещество, содержащее Cr(VI), и способ тем самым не является экономичным способом получения оксида хрома. Другой недостаток состоит в том, что доля Na - в пересчете на металлический Na - в полученном оксиде хрома, составляющая 1900 млн долей, очень высока. Кроме того, реакционная смесь, начиная с температуры около 700°С, при которой происходит кальцинирование, становится сильно клейкой, что, в частности, сильно затрудняет техническую реализацию, например, во вращающейся трубчатой печи.
В GB 748610 описано в общем виде восстановление водородом хроматов щелочных металлов и последующее превращение в Cr2O3. Выходы при такого рода восстановлении являются, во всяком случае, не очень низкими. Так выход Cr2O3, исходя из свободного от щелочи монохромата натрия, составляет менее 67%, что делает этот способ превращения с такими реактивными исходными веществами не представляющим интерес в техническом масштабе.
В CN 1907865А опубликован способ получения оксида хрома, причем применяют хроматную соль в качестве исходного вещества и восстанавливающий газ, такой как водород, природный газ, каменноугольный газ или их смеси в качестве восстановительного средства при температуре 300-850°С и проводят реакцию в течение 0,5-3 часов. После охлаждения реакционную смесь промывают водой и после просушивания кальцинируют при температуре 400-1100°С в течение 1-3 часов. Способ, описанный в CN 1907865A, в частности пример 1, исходящий из хромата и водорода, соответствует способу, приведенному в GB 748,610, причем повторение этого примера при указанных условиях не приводило ни к экзотермии, ни к измеряемому превращению.
В CN-101475217 получают пигментный оксид хрома путем взаимодействия бихромата натрия с аммиаком при 350°С с последующим гидролизом, выделением промежуточного продукта и последующим кальцинированием в присутствии оксидных добавок при температуре 1100°С. Выход оксида хрома, полученного этим способом, с соответствующими оксидными побочными продуктами, составляющий менее 45%, однако, очень низок и тем самым не представляет технического интереса (смотри пример для сравнения 2).
Поляк и Девятовская описали еще в 1957 году лабораторные опыты по взаимодействию монохромата натрия и бихромата натрия с газообразным аммиаком (Труды Урал. научно-исследовательского хим. инст.4, 1957, стр.30-32). Согласно их исследованиям хромат натрия и бихромат натрия могут только при температуре 700°С быть восстановлены газообразным аммиаком в хромит натрия NaCrO2. Гидролиз хромита натрия описывается, как труднопротекающий, а по поводу дальнейшей переработки в оксид хрома авторы не упоминают.
Задача данного изобретения состояла в том, чтобы создать способ получения оксида хрома, который является экономически выгодным и, наряду с этим, получить оксид хрома, который можно применять для металлургических целей, то есть содержит очень малые количества серы и щелочных металлов, в частности, имеет малое содержание натрия, а также по возможности малое содержание побочных продуктов.
Неожиданно было открыто, что газообразный аммиак может быть применен в качестве реагента для хроматов щелочных металлов и таким путем создается возможность получения оксида хрома(III) через хромит щелочного металла. Изобретение относится в связи с этим к способу получения оксида хрома(III), включающему стадии:
a) взаимодействия хромата щелочного металла с газообразным аммиаком, в частности, при температуре от 200 до 800°С,
b) гидролиза продукта реакции, полученного на стадии а), причем рН-значение воды для гидролиза перед гидролизом и/или предпочтительно щелочного маточного раствора во время или после гидролиза понижают с помощью кислоты до значения 4-11, предпочтительно до значения 5-10,
c) выделения продукта гидролиза, выпавшего в осадок на стадии Ь), предпочтительно при рН-значении от 4 до 11, более предпочтительно при рН-значении от 5 до 10, и при необходимости промывания и при необходимости сушки и
d) кальцинирование продукта гидролиза, полученного на стадии с) при температуре от 700 до 1400°С, более предпочтительно при температуре от 800 до 1300°С.
Стадия а)
В качестве исходного материала для получения оксида хрома(III) могут применяться хроматы щелочных металлов. При этом не существенно, применяются ли хроматы щелочных металлов в виде монохроматов, бихроматов, трихроматов и так далее или в виде полихроматов. Далее не существенно, применяются ли хроматы щелочных металлов в виде безводных соединений или в виде их гидратов. Предпочтительным хроматом щелочного металла является хромат натрия, дигидрат бихромата натрия или бихромат калия, более предпочтителен бихромат натрия и дигидрат бихромата натрия.
Понятие "хроматы щелочных металлов" по смыслу данного изобретения также охватывает группу так называемых хромихроматов щелочных металлов, у которых хром присутствует не только в окислительной стадии +VI (в виде хромата), но и одновременно в окислительной стадии +III. В качестве примера такого рода хромихромата щелочного металла здесь можно привести, например, NaCr3O, который также может быть описан суммарной формулой NaCr(CrO4)2. Хромат щелочного металла может применяться или в виде раствора, в частности, в виде водного раствора, или в виде суспензии, или в виде твердого вещества. В способе согласно данному изобретению предпочтительно применяют твердые вещества, причем эти вещества предпочтительно имеют остаточную влажность менее 4,0 вес. процентов, предпочтительно менее 2,0 вес. процентов. Также предпочтительно, когда применяемый хромат щелочного металла имеет содержание гидроксида щелочного металла менее 2 вес. процентов, более предпочтительно менее 1 вес. процента, еще более предпочтительно менее 0,5 вес. процента.
Нет необходимости применения в чистом виде хромата щелочного металла, используемого на стадии а). Хромат может предпочтительно применяться и в виде смеси. Так, например, более предпочтительны смеси, состоящие из монохроматов щелочного металла и оксида хрома(III). Такие смеси можно получить синтетически путем смешивания обоих компонентов, однако, их можно также получить путем твердотельной реакции, исходя из хромита щелочного металла и бихромата щелочного металла. Предпочтительные смеси содержат монохроматы щелочного металла и оксид хрома(III), например, в соотношении от 99:1 до 1:99, предпочтительно от 90:10 до 10:90, еще более предпочтительно от 40:60 до 60:40 в пересчете на содержащийся в соединениях хром(III), соответственно, хром(VI) получают при твердотельных реакциях в указанных выше количественных соотношениях. Твердотельные реакции между хромитом щелочного металла и бихроматом щелочного металла происходят предпочтительно при температуре выше 300°С, более предпочтительно при температуре от 300°С до 500°С.
Реакция хроматов щелочных металлов с газообразным аммиаком происходит предпочтительно при температуре от 200 до 800°С, более предпочтительно при температуре от 200 до 600°С, еще более предпочтительно при температуре от 200 до 500°С.При этом не обязательно, чтобы реакция проходила при одной и той же температуре. Оказалось предпочтительным, когда температура в ходе реакции повышается. Предпочтительно реакцию начинают при температуре от 200 до 300°С и эту температуру поддерживают до тех пор, пока не будет замечено повышение температуры. Для дальнейшего превращения после этого можно повысить температуру, причем это повышение может происходить непрерывно или ступенчато.
Реакция хроматов щелочных металлов с газообразным аммиаком происходит предпочтительно в не непосредственно нагреваемом реакторе, в частности, во вращающейся трубчатой печи или в вихревом слое.
Время реакции составляет, как правило, от 0,5 до 10 часов и зависит среди прочего от температуры реакции, от использованного хромата щелочного металла и от размеров кристалла использованного хромата щелочного металла. По этой причине может оказаться предпочтительным, когда хромат щелочного металла измельчают перед тем, как его подают на стадию а). Предпочтительны частицы менее чем 1000 мкм, более предпочтительно менее чем 500 мкм, еще более предпочтительно менее 300 мкм.
Реакция хроматов щелочных металлов с газообразным аммиаком приводит к образованию промежуточного соединения, которое при приведенных температурах в конце концов превращается в хромитовую соль щелочного металла в качестве продукта реакции. Как будет показано на примере реакции бихромата натрия с газообразным аммиаком, можно для общей реакции исходить из следующего брутто-превращения:


Доказательство существования промежуточного соединения можно получить косвенно, опираясь на изменение температуры, которое наблюдается во время реакции: примерно через 50 минут после введения аммиака при внутренней температуре реактора 220°С температура скачкообразно возрастает без повышения подачи тепла извне до примерно 370°С. Образовавшийся хромит натрия NaCrO2 можно обнаружить в реакционном продукте с помощью рентгенограммы порошка, как удалось обнаружить в одном из описанных ниже примеров.
Для того чтобы гарантировать по возможности полное превращение при реакции, газообразный аммиак предпочтительно применяют не в точно стехиометрическом соотношении, как показано в обоих уравнениях (6) и (7), а в избытке. Предпочтительно избыток аммиака составляет как минимум 5%, более предпочтительно как минимум 10%, еще более предпочтительно от 10 до 30%, в пересчете на стехиометрическое количество.
Предпочтительно вначале завершают превращение согласно стадии а), а после этого реакционный продукт гидролизуют четырехкратным количеством воды с образованием суспензии с рН-значением как минимум 11, более предпочтительно как минимум 12, еще более предпочтительно как минимум 13. Для установления предпочтительного завершения превращения предпочтительно делают забор проб реакции, подвергают их гидролизу водой, как описано, и определяют рН-значение полученной суспензии.
Продукт реакции, полученный на стадии а), можно перед проведением стадии b) также подвергнуть измельчению, чтобы обеспечить по возможности быстрый и полный гидролиз.
Стадия b)
Продукт реакции, полученный на стадии а), гидролизуют водой, причем образуется осадок и маточный щелок.
Гидролиз можно проводить при комнатной температуре или также при повышенной температуре. При гидролизе в качестве осадка образуется гидроксид хрома(III) и/или оксидгидроксид хрома(III) и щелочь, так что образующийся маточный щелок при применении чистой воды показывает очень высокое рН-значение. Гидролиз на примере хромита натрия NaCrO2 можно формально описать двумя следующими уравнениями реакций:
 ,
,
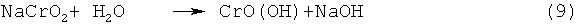 .
.
Выпадающий при этом в осадок продукт гидролиза является рентгеноаморфным, так что установление его точной структуры пока не было возможно. Предпочтительно применяют для гидролиза продукта реакции, полученного на стадии а), как минимум равное по весу количество воды. В связи с тем, что вязкость полученной суспензии может оказаться слишком высокой, предпочтительно применяют для гидролиза как минимум двойное по весу количество воды, в пересчете на вес продукта реакции, полученного на стадии а). Независимо от точной структуры и состава продукт гидролиза при применении чистой воды суспендируют в сильно щелочном маточном щелоке. Кроме того, продукт гидролиза, как правило, является мелкочастичным, так что разделение твердое вещество/жидкость является затруднительным и сильно времязатратным. Фильтрационные свойства и качество продукта (чистота, в частности, содержание Na) конечного полученного оксида хрома(III), а также его выход могут быть улучшены в способе согласно данному изобретению. Предпочтительно понижение рН-значения происходит при температуре от 20 до 100°С, более предпочтительно от 40 до 100°С. Еще более предпочтительно понижение рН-значения происходит во время или после гидролиза. В альтернативном варианте изобретения можно самой воде, которую используют для гидролиза, задать пониженное рН-значение, которое предпочтительно выбирают таким, чтобы после гидролиза получалось рН-значение от 4 до 11, предпочтительно от 5 до 10. Более предпочтительно понижение рН-значения проводят перед происходящим на стадии с) отделением выпавшего в осадок продукта гидролиза. Для понижения рН-значения предпочтительно применяют не органические кислоты или органические кислоты. К не органическим кислотам по смыслу данного изобретения относятся и кислореагирующие в воде газы, такие как, например, двуокись углерода. Эти кислореагирующие газы могут вводиться при нормальном или повышенном давлении в маточный щелок. Более предпочтительно применяют органические кислоты, в частности, низкомолекулярные органические кислоты, такие как, например, муравьиная кислота и уксусная кислота. Эти органические кислоты имеют то преимущество, что они - в случае если остатки остаются в продукте гидролиза и эти остатки не удается удалить при промывке на стадии с) - при позднейшем кальцинировании на стадии о.) путем окисления разлагаются без остатков и не остаются в виде примесей в кальцинированном продукте.
Наиболее предпочтительно применяют двуокись углерода для задания рН-значения, которую можно вводить при нормальном давлении или при повышенном давлении в маточный щелок. Предпочтительно рН-значение после его установления лежит в области от 4 до 11, более предпочтительно в области от 5 до 10. Установление рН-значения может происходить одноступенчато или многоступенчато.
Преимущество применения СO2 для установления рН-значения состоит в том, что таким путем может быть получен карбонат натрия или гидрокарбонат натрия из щелочного маточного щелока. При этом карбонат натрия или гидрокарбонат натрия может быть отделен различными непрерывными или прерывистыми способами твердо/жидкого разделения. Из непрерывно работающих агрегатов более предпочтительны, например, вакуумные барабанные фильтры или вакуумные ленточные фильтры, или центрифуги. Карбонат натрия можно напрямую опять применять для получения хроматов и бихроматов щелочных металлов путем оксидативного разложения хромжелезной руды. В случае гидрокарбоната натрия, то его можно отжигом перевести в карбонат натрия, а после этого применять для получения хроматов и бихроматов щелочных металлов путем оксидативного разложения хромжелезного штейна. Двуокись углерода, высвобождающаяся при отжиге гидрокарбоната натрия с образованием карбоната натрия, можно возвратить в процесс с целью понижения рН-значения.
Стадия с)
Полученный на стадии b), выпавший в осадок продукт гидролиза, содержащий хром, отделяют от маточного щелока. Специалистам известно много подходящих агрегатов и способов твердо/жидкого разделения. Является несущественным, происходит ли твердо/жидкое разделение и при необходимости примыкающее промывание непрерывно или прерывисто. Также несущественно, проводятся ли они при повышенном или при пониженном давлении.
В случае непрерывно работающих фильтровальных и промывающих агрегатов более предпочтительны, например, вакуумные барабанные фильтры или вакуумные ленточные фильтры. Из непрерывно работающих фильтровальных и промывочных агрегатов более предпочтительны фильтр-прессы.
Выделенный, предпочтительно отфильтрованный продукт гидролиза можно промыть или - при необходимости после сушки - подать на стадию d). Специалистам известно большое число подходящих агрегатов для стадии сушки. Здесь следует упомянуть только канальные, ленточные, этажные, валковые, барабанные, трубчатые, лопастные, распылительные сушилки (распылительные сушилки с шайбами или форсунками), сушилки с вихревым слоем или прерывисто работающие камерные сушилки Гордена. Предпочтительно влажный фильтровальный осадок, в частности, без промывки напрямую подают на кальцинирование на стадию d).
Выделенный продукт гидролиза можно также промыть одноступенчато или многоступенчато. Промывание можно проводить непосредственно в воде. Для того чтобы улучшить промывные и фильтровальные свойства полученного твердого вещества, может оказаться предпочтительным уменьшение рН-значения воды для промывания. К промывной воде предпочтительно перед промыванием или во время промывания добавляют кислоту для уменьшения рН-значения. Для этого предпочтительно добавляют неорганические кислоты или органические кислоты, или двуокись углерода, как описано выше. Более предпочтительно применяют органические кислоты, в частности, низкомолекулярные органические кислоты, такие как, например, муравьиная кислота и уксусная кислота. Еще более предпочтительно применяют двуокись углерода для задания рН-значения, двуокись углерода подают в промывную воду при нормальном или повышенном давлении. Предпочтительно рН-значение промывной воды после промывки лежит в интервале от 4 до 11, более предпочтительно в интервале от 5 до 10. Задание рН-значения можно проводить одноступенчато или многоступенчато.
Также может оказаться предпочтительным, когда перед фильтрованием или перед промыванием применяют хлопьеобразующие средства или вспомогательные хлопьеобразующие средства. Введение органических хлопьеобразующих средств или вспомогательных хлопьеобразующих средств особенно предпочтительно, так как они при последующем кальцинировании на стадии а) разлагаются при оксидации без образования остатков и не остаются в кальцинированном продукте в качестве примесей. Предпочтительными хлопьеобразующими средствами являются анионные электролиты, например, на основе полиакрилата, полиакриламида, полиэтиленимина и полиэтиленоксида с различной длиной цепи. Наряду с этим, могут применяться также неионные синтетические и природные соединения (например, крахмал или глина) в качестве вспомогательных хлопьеобразующих средств.
Влажный фильтровальный остаток, полученный после проведенного выделения и при необходимости проведенного промывания продукта гидролиза может или напрямую подаваться на кальцинирование согласно стадии d) или перед этим еще раз подвергнуться сушке. Для стадии сушки применяются многочисленные подходящие агрегаты, которые известны специалистам и описаны выше.
В том случае, когда при промывании также используют двуокись углерода для уменьшения рН-значения, то полученные на стадии с) промывные воды можно также использовать для получения карбоната натрия или гидрокарбоната натрия, как описано в стадии b). Конечно, можно также объединить маточный щелок со стадии b) и промывную воду со стадии с) и совместно - при необходимости после концентрирования - применять для получения карбоната натрия или гидрокарбоната натрия. В принципе также возможно оба отдельных потока перед объединением раздельно один от другого концентрировать и применять для получения карбоната натрия или гидрокарбоната натрия. Такой порядок действий, однако, включает риск того, что при слишком сильном концентрировании из маточного щелока будет выкристаллизовываться гидрокарбонат натрия в связи с тем, что маточный щелок будет показывать слишком высокую концентрацию натрия.
Стадия d)
Термическая обработка при повышенной температуре, то есть кальцинирование, согласно стадии d) происходит при температуре от 700 до 1400°С, более предпочтительно при температуре от 800 до 1300°С, предпочтительно в течение более чем 20 минут, более предпочтительно более чем 30 минут, еще более предпочтительно в течение от 30 минут до 4 часов. Специалистам известны многочисленные подходящие аппараты для кальцинирования при таких высоких температурах. Здесь следует упомянуть кольцевые нагревательные печи, вращающиеся трубчатые печи, реакторы с вихревым слоем или прерывисто работающие камерные печи. Кальцинирование проводят предпочтительно в напрямую нагреваемой вращающейся трубчатой печи. Время нахождения в печи кальцинируемого материала в зависимости от устройства и длины печи предпочтительно составляет от 30 минут до 4 часов. Кальцинирование предпочтительно проводят на воздухе или в атмосфере чистого кислорода или в воздушной атмосфере, обогащенной кислородом.
Полученный на стадии с) и при необходимости промытый продукт гидролиза, который кальцинируют на стадии d), не склонен к прилипанию во время кальцинирования, так что кальцинирование может происходить без проблем.
В более предпочтительном варианте способа согласно данному изобретению перед кальцинированием добавляют один или несколько галоидов щелочных металлов или галоидов аммония, или галоидов щелочноземельных металлов, предпочтительны фториды, хлориды, бромиды или йодиды натрия или калия, или аммония, или гидроксиды щелочных металлов, предпочтительно гидроксид натрия или гидроксид калия, или хромовую кислоту в количестве от 0,01 вес. процентов до 3,0 вес. процентов, более предпочтительно от 0,02 вес. процентов до 1,0 вес. процентов, в пересчете на количество продукта гидролиза, вводимого для кальцинирования. В результате такого рода добавок можно оказать влияние на технические свойства при применении, в частности, на повышение насыпной плотности полученного оксида хрома(III). Однако, предпочитают не добавлять такие добавки во время кальцинирования.
Полученный после кальцинирования согласно стадии d) оксид хрома(III) предпочтительно охлаждают и при необходимости размалывают. В более предпочтительном варианте способа согласно данному изобретению кальцинированный продукт после стадии d) суспендируют в воде, причем образуется маточный щелок, и при необходимости промывают водой одноступенчато или многоступенчато, и в заключение опять сушат. В результате этого можно удалить еще присутствующие в оксиде хрома(III) растворимые в воде примеси (растворимые в воде соли) - в существенной мере хроматы щелочных металлов, например, хромат натрия, который образуется в результате оксидации оксида хрома(III) при высоких температурах - известными способами вымывания одноступенчато или многоступенчато с помощью воды или водных сред и отделяют твердое вещество от жидкости. Действуют предпочтительные варианты для промывания, которые приведены выше для стадии с).
Оксид хрома(III) обладает, как правило, хорошими фильтровальными и промывными свойствами, так что отпадает необходимость в регулировании рН-значения или в добавлении хлопьеобразующего вещества или вспомогательного хлопьеобразующего вещества. Твердый оксид хрома(III), полученный после твердо/жидкого разделения, после этого сушат. Высушенный оксид хрома(III) после этого предпочтительно напрямую расфасовывают или при необходимости перед расфасовкой размалывают.
Для сушки могут применяться упомянутые выше агрегаты. В зависимости от выбранного агрегата для сушки может оказаться необходимым, чтобы примыкала стадия размалывания. Но даже в том случае, когда не проводят выщелачивания, промывания и сушки кальцинированного продукта, может оказаться предпочтительным размалывание. Предпочтительно кальцинированный и при необходимости суспендированный в воде и промытый, и высушенный продукт подвергают еще и размалыванию. Для этого пригодны агрегаты для размалывания различных конструкций, такие как, например, валковые мельницы, бегуны, маятниковые мельницы, молотковые дробилки, штифтовые мельницы, турбомельницы, шаровые мельницы или струйные мельницы. В том случае, когда кальцинированный продукт был промыт, то более предпочтительно можно применять мельницу с сушкой, в которой сушка и размалывание происходят на одной рабочей стадии. Выбор подходящего мельничного агрегата зависит среди прочего также от соответствующей области применения полученного оксида хрома(III).
Когда промывают кальцинированный оксид хрома(III), то соответствующие маточный щелок и промывная вода содержат в обоих случаях в существенной мере хромат щелочного металла и/или бихромат щелочного металла. Оба эти ценные вещества можно снова возвратить в процесс производства, при котором они, например, опять могут применяться для получения бихромата щелочного металла или, например, для получения двойной соли хромата щелочного металла - аммония. Более предпочтительно маточные щелоки и промывную воду, которая образуется при промывке кальцинированных продуктов, снова применяют для получения бихромата щелочного металла или, например, для получения двойной соли хромата щелочного металла - аммония.
Оксид хрома(III), получаемый способом согласно данному изобретению, является высокочистым. Он в высокой степени пригоден для металлургических целей, таких как получение металлического хрома или хромсодержащих высокоэффективных сплавов, в частности, путем восстановления в присутствии металлического алюминия в ходе алюминотермического способа, и для получения технических материалов, устойчивых при высокой температуре, однако, его можно также применять в качестве красящего пигмента для пигментарных применений, так как он отличается низким содержанием растворимых в воде солей.
Изобретение также охватывает применение оксида хрома(III), полученного согласно данному изобретению, в качестве красящего пигмента, шлифовального средства, а также в качестве исходного материала для получения устойчивых при высоких температурах технических материалов, металлического хрома или хромсодержащих высокоэффективных сплавов, в частности, путем восстановления в присутствие металлического алюминия с применением алюминотермического способа.
Способ согласно данному изобретению получения высокочистого, с малым содержанием серы оксида хрома(III) обладает некоторыми существенными преимуществами по сравнению со способами, описанными в уровне техники. Преимущество способа согласно данному изобретению состоит в том, что в качестве побочных продуктов образуются хромат щелочного металла и/или бихромат щелочного металла, которые без проблем можно снова возвратить в процесс получения. Образующийся при гидролизе сильно щелочной маточный щелок можно подкислить двуокисью углерода и таким образом или перевести напрямую в карбонат натрия, или вначале перевести в гидрокарбонат натрия, который затем кальцинируют в карбонат натрия. Карбонат натрия можно снова применить для оксидативного разложения хромжелезного штейна с образованием хромата натрия. Большое преимущество по сравнению со способом, описанным в патенте CN 1907865A, состоит в том, что выход улучшается и в особенности в случае уменьшения рН-значения могут быть далее повышены выход и чистота полученного оксида хрома. Одновременно удается снизить содержание Na в продукте гидролиза, что оказывается предпочтительным при примыкающем кальцинировании, так как при низком содержании Na меньше Cr(III) оксидируется в хромат и переводится в хромат натрия. В связи с этим способ согласно данному изобретение дает отчетливо более высокие выходы оксида хрома(III). Образующийся при способе согласно данному изобретению оксид хрома(III) является высокочистым. Он имеет бедное содержание серы, так как в процесс получения не вводят никаких серусодержащих соединений. Далее он беден содержанием щелочных металлов. "Бедными содержанием серы" по смыслу данного изобретения являются оксиды хрома(III), которые имеют содержание серы менее чем 200 млн долей, предпочтительно менее 50 млн долей, еще более предпочтительно менее 40 млн долей. "Бедными содержанием щелочных металлов" по смыслу данного изобретения являются оксиды хрома(III), которые имеют содержание щелочного металла - в пересчете на щелочной металл - менее чем 1500 млн долей, предпочтительно менее 500 млн долей.
Изобретение более подробно поясняется приведенными ниже примерами, которые ни в коем случае не ограничивают данное изобретение.
Примеры
Пример 1
60 г безводного измельченного бихромата натрия Na2Cr2O7 помещают в стеклянный сосуд, к которому сделаны подвод и отвод для газа со стеклянной фриттой. Дно покрыто полностью и датчик температуры погружают в насыпанную массу. Стеклянный сосуд помещают в регулируемую печь. Стеклянный сосуд нагревают в атмосфере азота до 300°С температуры рубашки. Азот при внутренней температуре 220°С мощным потоком заменяют на аммиак, который устремляется потоком снизу через продукт. Через 50 минут внутренняя температура возрастает в результате экзотермической реакции в течение нескольких минут от 220°С до 371°С. Через 60 минут в течение 10 минут повышают температуру рубашки до 400°С для завершения реакции. Через следующие 6,5 часов аммиак снова вытесняют азотом и проводят охлаждение до комнатной температуры. Получают 50,3 г продукта реакции.
Грубо измельченный продукт реакции помещают в 200 мл воды, образуют шлам и гидролизуют. Сильно щелочную суспензию с помощью ледяного уксуса доводят до рН-значения 6, фильтруют через фильтр Нутча и фильтровальный остаток без дальнейшего промывания сушат при температуре 120°С. После этого его отжигают в течение 2 часов при температуре 1250°С. Подвергнутый отжигу оксид хрома(III) суспендируют в воде, промывают водой и в заключение сушат при температуре 120°С.
Полученный таким путем оксид хрома(III) имеет содержание Na - в пересчете на Na-металл - равное 210 млн долей. Выход оксида хрома(III) составляет 92% в пересчете на хром, содержавшийся в исходном материале Na2Cr2O7.
Пример 2
Взаимодействие 60 г измельченного бихромата натрия Na2Cr2O7 с газообразным аммиаком происходит так же, как описано в примере 1.
Измельченный реакционный продукт помещают в 200 мл воды, образуют шлам и гидролизуют, в результате чего образуется суспензия с рН-значением, равным 14. После этого в суспензию подают двуокись углерода при нормальном давлении до тех пор, пока не будет достигнуто рН-значение, равное 9,4, которое уже более не удалось понизить. После этого суспензию кратковременно нагревают до 95°С и фильтруют через фильтр Нутча. Фильтровальный остаток снова суспендируют в 200 мл воды и рН-значение суспензии устанавливают равным 7,7 путем введения двуокиси углерода при нормальном давлении. Дальнейшее понижение рН-значения было невозможно. После этого суспензию опять кратковременно нагревают до 95°С и фильтруют через фильтр Нутче. Фильтровальный остаток снова суспендируют в 200 мл воды и рН-значение суспензии устанавливают равным 6,8 путем введения двуокиси углерода при нормальном давлении. Дальнейшее понижение рН-значения было невозможно. После этого суспензию опять кратковременно нагревают на 95°С и фильтруют через фильтр Нутче. Фильтровальный остаток сушат при температуре 120°С. Затем проводят отжиг в течение 2 часов при температуре 1250°С. Подвергнутый отжигу оксид хрома(III) суспендируют в воде, промывают водой и в заключение сушат при 120°С.
Полученный таким путем оксид хрома(III) имеет содержание Na - в пересчете на Na-металл - 130 млн долей. Исходя из содержания Cr, степень чистоты составляет 98,9%. Выход оксида хрома(III) составляет 91% в пересчете на содержание хрома в исходном материале Na2Cr2O7.
Пример 3
Взаимодействие 60 г измельченного бихромата натрия Na2Cr2O7 с газообразным аммиаком происходит так же, как описано в примере 1.
Реакционный продукт, измельченный до размера частиц менее чем 600 мкм, помещают в 200 мл воды, образуют шлам и гидролизуют. Затем добавляют 2,0 г 0,1-процентного водного раствора вспомогательного хлопьеобразующего средства (сополимер из акриламида и акрилата натрия). После этого вводят при перемешивании при комнатной температуре двуокись углерода при нормальном давлении до тех пор, пока не установится рН-значение, равное 9,9. Дальнейшее понижение рН-значения оказалось невозможно. Затем суспензию кратковременно кипятят и фильтруют через фильтр Нутче. Фильтровальный остаток снова суспендируют в 200 мл воды, добавляют 2,0 г упомянутого выше 0,1-процентного водного раствора вспомогательного хлопьеобразующего средства и устанавливают рН-значение суспензии путем пропускания двуокиси углерода при нормальном давлении равным 7,4. Затем суспензию снова кратковременно кипятят и фильтруют через фильтр Нутче. Фильтровальный остаток снова суспендируют в 200 мл воды, добавляют 2,0 г упомянутого выше 0,1-процентного водного раствора вспомогательного хлопьеобразующего средства и устанавливают рН-значение суспензии путем пропускания двуокиси углерода при нормальном давлении равным 6,5. После этого суспензию опять кратковременно кипятят и фильтруют через фильтр Нутче. Фильтровальный остаток сушат при температуре 120°С. Затем проводят отжиг в течение 2 часов при температуре 1250°С. Подвергнутый отжигу оксид хрома(III) суспендируют в воде, промывают водой и в заключение сушат при 120°С.
Полученный таким путем оксид хрома(III) имеет содержание Na - в пересчете на Na-металл - 310 млн долей. Исходя из содержания Cr, степень чистоты составляет 99,7%. Выход оксида хрома(III) составляет 96% в пересчете на содержание хрома в исходном материале Na2Cr2O7.
Пример 4
Измельченный хромит натрия NaCrO2 и измельченный бихромат натрия Na2Cr2O7 смешивают в молярном соотношении Cr(III): Cr(VI) I: 1 и нагревают в атмосфере инертного газа до температуры 350°С. Через один час температуру ступенчато повышают со скоростью 3°С/мин до 450°С и выдерживают еще 30 минут при температуре 450°С. Полученный реакционный продукт имеет темно-зеленый цвет. Согласно рентгенограммы порошка он состоит из оксида хрома(III) и монохромата натрия Na2CrO4:
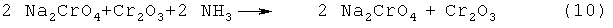 .
.
Реакционный продукт подвергают взаимодействию с газообразным аммиаком, который вводится в виде смеси 13,6 объемных процентов аммиака и инертного газа, при температуре 500°С, причем восстановление начинается уже при температуре около 350°С. При восстановлении происходит потеря веса в 10,65%, что находится в хорошем согласии с ожидаемой реакцией образования хромита натрия:
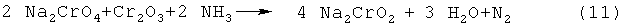 .
.
Полученный после взаимодействия с аммиаком согласно уравнению (11) хромит натрия NaCrO2 может быть переработан таким путем, как описано в примерах 1-3.
Пример для сравнения 1 (без уменьшения рН-значения при гидролизе)
Взаимодействие 60 г измельченного бихромата натрия Na2Cr2O7 с газообразным аммиаком происходит, как описано в примере 1.
Измельченный реакционный продукт помещают в 100 мл воды, образуют шлам и гидролизуют, в результате чего образуется щелочная суспензия. Затем фильтруют через фильтр Нутче и фильтровальный остаток снова суспендируют на 1 час в 100 мл воды и фильтруют через фильтр Нутче. Фильтровальный остаток сушат при температуре 120°С и затем отжигают в течение 2 часов при температуре 1250°С. Подвергнутый отжигу оксид хрома(III) суспендируют в воде, промывают водой и в заключение сушат при температуре 120°С.
Полученный таким путем оксид хрома(III) показывает содержание Na - в пересчете на Na-металл - равное 540 млн долей. Выход оксида хрома(III) составляет 65%, в пересчете на хром, содержащийся в исходном материале Na2Cr2O7.
Пример для сравнения 2 (согласно CN-101475217 без уменьшения рН-значения при гидролизе)
Бихромат натрия Na2Cr2O7, как описано в CN-101475217, в течение одного часа подвергают взаимодействию с газообразным аммиаком при температуре 350°С. Полученный реакционный продукт охлаждают и три раза промывают при температуре 80°С приведенными количествами воды. После фильтрования твердый продукт обнаруживает содержание влаги 29% и его смешивают с 2 вес. процентами эквимолярной смеси, состоящей из P2O3, Аl2O3, ZnO, Sb2O3, ВаО и TiO2 и подвергают отжигу в течение 1 часа при температуре 1100°С. В заключение опять многократно промывают, сушат и измельчают. Выход оксида хрома(III) составляет 44,6%, в пересчете на хром, содержавшийся в исходном материале Na2Cr2O7.
Пример для сравнения 3 (опираясь на CN-101475217 без уменьшения рН-значения при гидролизе) Бихромат натрия Na2Cr2O7, как описано в примере 1, подвергают взаимодействию с газообразным аммиаком. Полученный реакционный продукт охлаждают и после этого далее перерабатывают и подвергают превращению согласно CN-101475217, как описано в примере для сравнения 2. Выход оксида хрома(III) составляет 65%, в пересчете на хром, содержавшийся в исходном материале Na2Cr2O7.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ОКСИДА ХРОМА (III) | 2011 |
|
RU2591245C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОКИСИ ХРОМА | 2004 |
|
RU2258039C1 |
| СПОСОБ ПОЛУЧЕНИЯ РАСТВОРА БИХРОМАТА НАТРИЯ | 1991 |
|
RU2008262C1 |
| СПОСОБ ПОЛУЧЕНИЯ α -ОКСИДА АЛЮМИНИЯ | 1992 |
|
RU2015105C1 |
| КАТАЛИЗАТОР ДЕГИДРИРОВАНИЯ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ ОЛЕФИНОВЫХ УГЛЕВОДОРОДОВ C-C С ИСПОЛЬЗОВАНИЕМ ЭТОГО КАТАЛИЗАТОРА | 2011 |
|
RU2463109C1 |
| СПОСОБ ВЫДЕЛЕНИЯ ВАНАДИЯ И ХРОМА ИЗ ВАНАДИЕВО-ХРОМОВОГО ШЛАКА | 2019 |
|
RU2726540C1 |
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНЫХ ОКСИДНЫХ МАТЕРИАЛОВ НА ОСНОВЕ ХРОМА | 1995 |
|
RU2081837C1 |
| СПОСОБ ПОЛУЧЕНИЯ СМЕШАННЫХ ОКИСЛОВ НА ОСНОВЕ ХРОМА | 1971 |
|
SU319553A1 |
| СПОСОБ ПОЛУЧЕНИЯ ВАНАДИЕВО-ХРОМОВОГО СПЛАВА ПУТЕМ ИЗВЛЕЧЕНИЯ ВАНАДИЯ ИЗ ВАНАДИЕВО-ХРОМОВОГО ШЛАКА ПОСРЕДСТВОМ ОБЖИГА И КИСЛОТНОГО ВЫЩЕЛАЧИВАНИЯ | 2021 |
|
RU2792060C1 |
| СПОСОБ ПОЛУЧЕНИЯ ХРОМАТА КАЛЬЦИЯ | 1971 |
|
SU305136A1 |
Изобретение относится к способу получения оксида хрома (III), включающему стадии: a) взаимодействия хромата щелочного металла или бихромата щелочного металла с газообразным аммиаком, в частности, при температуре от 200 до 800°C, b) гидролиза реакционного продукта, полученного на стадии а), причем pH-значение воды для гидролиза перед гидролизом или щелочного маточного щелока во время или после гидролиза устанавливают равным от 4 до 11, понижая с помощью кислоты, c) выделения продукта гидролиза, выпавшего в осадок на стадии b), d) сушки продукта, полученного на стадии с), и e) кальцинирования продукта гидролиза, полученного на стадии d) при температуре от 700 до 1400°C. Получаемый оксид хрома (III), который можно применять для металлургических целей, содержит очень малые количества серы и щелочных металлов. 10 з.п. ф-лы, 7 пр.
1. Способ получения оксида хрома (III), включающий стадии:
a) взаимодействия хромата щелочного металла или бихромата щелочного металла с газообразным аммиаком, в частности, при температуре от 200 до 800°C,
b) гидролиза реакционного продукта, полученного на стадии а), причем pH-значение
- воды для гидролиза перед гидролизом или
- щелочного маточного щелока во время или после гидролиза устанавливают равным от 4 до 11, понижая с помощью кислоты,
c) выделения продукта гидролиза, выпавшего в осадок на стадии b),
d) сушки продукта, полученного на стадии с), и
e) кальцинирования продукта гидролиза, полученного на стадии d) при температуре от 700 до 1400°C.
2. Способ по п. 1, отличающийся тем, что взаимодействие хромата щелочного металла или бихромата щелочного металла с газообразным аммиаком проводят в косвенно обогреваемом реакторе, в частности во вращающейся трубчатой печи или в вихревом слое.
3. Способ по п. 1, отличающийся тем, что в качестве бихромата щелочного металла используют бихромат натрия, дигидрат бихромата натрия или бихромат калия.
4. Способ по п. 1, отличающийся тем, что pH-значение на стадии b) устанавливают равным от 5 до 10.
5. Способ по п. 1, отличающийся тем, что понижение pH-значения осуществляют добавлением неорганических или органических кислот, в частности введением двуокиси углерода.
6. Способ по п. 1, отличающийся тем, что выделение продукта гидролиза на стадии с) проводят при pH-значении от 4 до 11, в частности при pH-значении от 5 до 10.
7. Способ по п. 1, отличающийся тем, что на стадии с) дополнительно проводят промывание.
8. Способ по п. 1, отличающийся тем, что кальцинирование на стадии е) проводят при температуре от 800 до 1300°C.
9. Способ по п. 1, отличающийся тем, что дополнительно кальцинированный продукт согласно стадии е) суспендируют в воде и при необходимости промывают водой одноступенчато или многоступенчато и затем высушивают.
10. Способ по п. 1, отличающийся тем, что дополнительно кальцинированный продукт согласно стадии е) подвергают размалыванию при необходимости после предварительной промывки водой и сушки.
11. Способ по одному или нескольким пп. 1-10, отличающийся тем, что дополнительно перед кальцинированием согласно стадии е) добавляют один или несколько галоидов щелочных металлов, или галоидов аммония, или галоидов щелочноземельных металлов, предпочтительно добавляют фториды, хлориды, бромиды или йодиды натрия, или калия, или аммония, или гидроксиды щелочных металлов, предпочтительно гидроксид натрия или гидроксид калия, или хромовую кислоту в количестве от 0,01 вес.% до 3,0 вес.%, в особенности от 0,02 вес.% до 1,0 вес.%, в пересчете на количество продукта, подаваемого на кальцинирование.
| CN 101475217 A, 08.07.2009 | |||
| US 4804528 A, 14.02.1989 | |||
| СПОСОБ ПОЛУЧЕНИЯ ОКИСИ ХРОМА | 2004 |
|
RU2258039C1 |
| US 20090136402 A, 28.05.2009. | |||

