Изобретение относится к методам лазерной десорбции-ионизации и может быть использовано для масс-спектрометрического анализа и идентификации химических соединений в жидких и газообразных пробах. В частности, заявляемый способ может быть использован в аналитической практике для определения состава химических соединений, в медицине и фармакологии для идентификации биологически активных компонентов лекарств, в системах обеспечения безопасности для определения присутствия наркотических и взрывчатых веществ в реальном времени.
Методы лазерной десорбции-ионизации находят широкое применение в масс-спектрометрическом анализе. Технология лазерной десорбции-ионизации основана на нанесении анализируемой пробы на поверхность подложки и воздействии импульсным лазерным излучением на подложку. В результате такого воздействия в газовой фазе образуются ионы химических соединений, которые детектируются в масс-анализаторе.
Известен способ матрично активированной лазерной десорбции-ионизации (MALDI). В известном способе кроме анализируемой пробы на поверхность подложки наносят также специальную матрицу, хорошо поглощающую лазерное излучение. Матрица выполняет две основные функции - перевод молекул аналита в газовую фазу и их ионизацию. Воздействие лазерного излучения приводит к импульсному испарению (абляции) матрицы и сокристаллизованного с ней определяемого соединения. Над поверхностью образца формируется область сравнительно высокого давления, в которой и протекают реакции ионизации [Патент США №5118937, кл. G01N 1/00; G01N 27/447; G01N 27/62; G01N 27/64; G01N 30/72; H01J 49/10; H01J 49/16, опубл. 1992 г.].
Способ применим для определения высокомолекулярных соединений с массой молекул до нескольких сотен тысяч а.е.м. Используется импульсное лазерное излучение с длиной волны 300 нм и выше, в частности лазерное излучение N2-лазера, СО2-лазера, Er-лазера, Er-YAG-лазера. В качестве матриц, поглощающих лазерное излучение, предложено применять бензойную, никотиновую, карбоновые кислоты, их производные, глицерин и ряд других химических соединений.
Известный способ ионизации химических соединений обладает следующими недостатками:
- способ мало применим для определения химических и биохимических соединений с небольшими молекулярными массами вследствие процессов ионизации и сильной фрагментации молекул матрицы, создающих интенсивный химический шум в диапазоне масс до примерно 600 а.е.м.;
- способ позволяет определять лишь качественный состав исследуемых образцов;
- органические матрицы, используемые в MALDI, не универсальны. Каждая из них может быть использована для анализа весьма ограниченного числа классов соединений, и, в частности, для подбора матрицы необходимо знать примерный состав исследуемых образцов;
- способ не применим для анализа веществ, находящихся в газообразном или жидком агрегатном состоянии;
- совместная кристаллизация матрицы и анализируемых биохимических соединений, являющаяся необходимым условием известного способа, может изменять физико-химические свойства соединений (которые в своем естественном состоянии существуют в виде растворов), и в ряде случае известный способ может давать неверную информацию об их составе и структуре.
Наиболее близким техническим решением к предложенному является способ масс-спектрометрического определения химических соединений, включающий нанесение молекул химических соединений на поверхность твердотельного материала путем адсорбции или осаждения, лазерную десорбцию-ионизацию путем воздействия на материал импульсным лазерным излучением и детектирование ионов химических соединений в масс-анализаторе [Патент США №6288390, кл. H01J 49/04; H01J 49/16, опубл. 2001 г.].
В способе анализируемый раствор наносят на поверхность подложки, выполненной из пористого кремния. После испарения растворителя подложку с осаженным на ее поверхность аналитом помещают в ионный источник масс-спектрометра и воздействуют импульсным УФ-лазерным излучением, которое полностью поглощается в слое пористого кремния. В результате лазерной десорбции-ионизации в газовой фазе образуются протонированные молекулы аналита, которых детектируются в масс-спектрометре.
Известный способ ионизации химических соединений характеризуется отсутствием интенсивного химического шума и поэтому позволяет детектировать химические и биохимические соединения с небольшими молекулярными массами. Кроме того, активный слой, выполненный из пористого кремния более универсален, чем матрицы в методе MALDI.
Недостатком прототипа является отсутствие возможности однозначной идентификации определяемых химических соединений (аналитов) в условиях интерференции (наложения) пиков ионов аналита с пиками ионов подложки или сопутствующих компонентов и/или вследствие фрагментации ионов аналитов.
Задачей изобретения является повышение достоверности и надежности идентификации химических соединений, в частности, за счет выделения в масс-спектре пика, характеризующего молекулярную массу соединения, при снижении концентрации анализируемых соединений.
Указанная задача решается тем, что в способе масс-спектрометрического определения химических соединений, включающем нанесение молекул химических соединений на поверхность твердотельного материала путем адсорбции или осаждения, лазерную десорбцию-ионизацию путем воздействия на материал импульсным лазерным излучением и детектирование ионов химических соединений в масс-анализаторе, лазерную десорбцию-ионизацию ведут в присутствии газа-реагента, выбранного из группы соединений общей формулы CnH2nR, где n=1÷4, R=ОН, CN, I.
Предпочтительно использовать поток газа-реагента, направленный на поверхность твердотельного материла.
Целесообразно в качестве газа-реагента использовать одно соединение или смесь соединений, таких как спирты, например, метанол, и нитрилы, например, ацетонитрил, а так же изотопно обогащенные соединения, например, дейтерированный метанол или дейтерированный ацетонитрил.
Целесообразно в качестве полупроводникового материала использовать пористые, нанокристаллические или аморфные полупроводниковые материалы, такие как кремний, германий, оксид цинка, оксид титана, селенид кадмия.
Предпочтительно использовать импульсное лазерное излучение с длиной волны, выбранной из условия поглощения его в твердотельном материале, с интенсивностью, меньшей порога плавления твердотельного материала.
Целесообразно лазерную десорбцию-ионизацию в присутствии газа-реагента проводить в вакууме, при значениях давления, создаваемого газом-реагентом, не превышающих 10-4 мм рт.ст. или при атмосферном давлении, а в качестве масс-анализатора использовать масс-спектрометр или спектрометр ионной подвижности.
Сущность изобретения заключается в следующем. В процессе лазерной десорбции-ионизации, в частности десорбции-ионизации на поверхности пористого кремния, образуются протонированные молекулы химических соединений, предварительно осажденных из раствора или адсорбированных из газовой фазы. Протонирование является основным каналом ионизации в этих способах и обусловлено лазерно-индуцированным процессом переноса протона. При этом ионизуются только химические соединения с высокими значениями энергии сродства к протону. Воздействие излучением на подложку приводит, кроме того, к ее нагреву и десорбции молекул и ионов аналита. В известных способах образующиеся протонированные молекулы химических соединений (аналитов) сразу направляются и детектируются в масс-анализаторе. В заявляемом способе ионы, образующиеся в процессе лазерной десорбции-ионизации, затем вступают в реакции с молекулами газа-реагента. При взаимодействии протонированных молекул (МН)+ с соединениями, имеющими формулу CnH2n+1R (где n=1÷4, R=ОН, CN, SH, I), такого рода реакции можно записать в виде:
(МН)++CnH2n+1R→(MCnH2n+1)++HR.
Например, при воздействии молекулами метанола протекают реакции метилирования:
(МН)++СН3ОН→(МСН3)++H2O.
В результате, в масс-анализатор вводятся ионы, характеризующие аналит, но имеющие другую массу, чем протонированные молекулы, и обладающие другими химическими свойствами. Такой подход имеет ряд преимуществ по сравнению с известными способами, что иллюстрируют приведенные ниже примеры.
Вероятность образования ионов (MCnH2n+1)+ при взаимодействии протонированных молекул аналита с молекулами газа-реагента CnH2n+1R уменьшается с ростом величины n, поэтому в качестве газа-реагента целесообразно использовать соединения с низким значением n, а именно, n≤4.
При необходимости обеспечить высокую воспроизводимость результатов анализа лазерную десорбцию-ионизацию химических соединений обычно проводят в условиях вакуума. При этом твердотельный материал находится в вакууме под воздействием десорбирующего лазерного излучения с интенсивностью, меньшей порога его разрушения, что обеспечивает неизменность физико-химического состава поверхности материала, полную десорбцию всех находящихся на его поверхности химических соединений и, следовательно, высокую воспроизводимость результатов анализа. Добавление газа-реагента приводит к увеличению давления, т.е. к ухудшению вакуума. При этом, с одной стороны, увеличение давления, создаваемого газом-реагентом, увеличивает вероятность образования ионов (MCnH2n+1)+ вследствие увеличения числа столкновений протонированных молекул аналита с молекулами газа-реагента, но, с другой стороны, относительно высокое давление ухудшает качество масс-спектров. Кроме того, возможность увеличения давления газа-реагента ограничена производительностью систем откачки, создающих вакуум. Как следствие этих факторов, в условиях вакуума лазерную десорбцию-ионизацию целесообразно вести в присутствии газа-реагента, создающего давление, не превышающее 10-4 мм рт.ст. При значениях, больших 10-4 мм рт.ст., начинает расти уровень шума, ухудшается разрешение по массам и качество масс-спектров, в то время как величина самого пика ионов (MCnH2n+1)+ уже слабо зависит от давления. Для количественного анализа необходимо, чтобы поток газа-реагента оставался постоянным в процессе анализа и при последовательном анализе различных проб.
Для качественного анализа лазерную десорбцию-ионизацию химических соединений можно проводить при атмосферном давлении. В этом случае отсутствуют ограничения на величину давления, создаваемого газом-регентом, за исключением тех, которые обусловлены конструкцией ввода пробы в масс-анализатор.
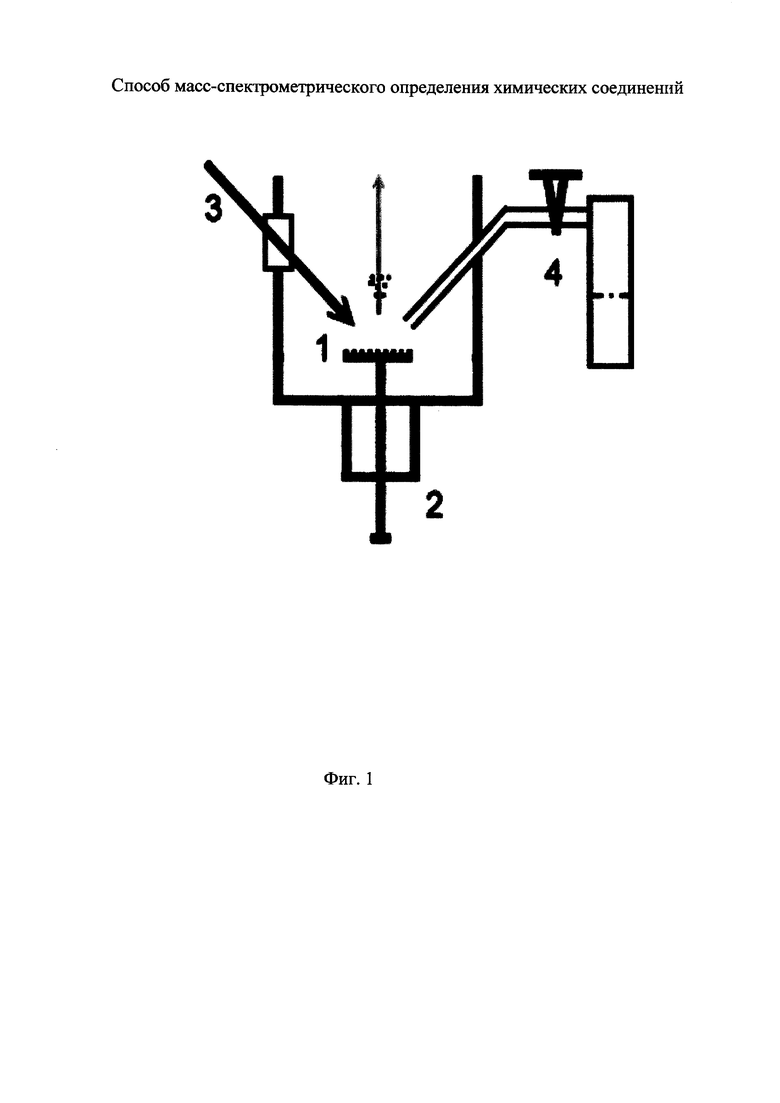
На фиг. 1 представлена принципиальная схема установки для реализации заявляемого способа.
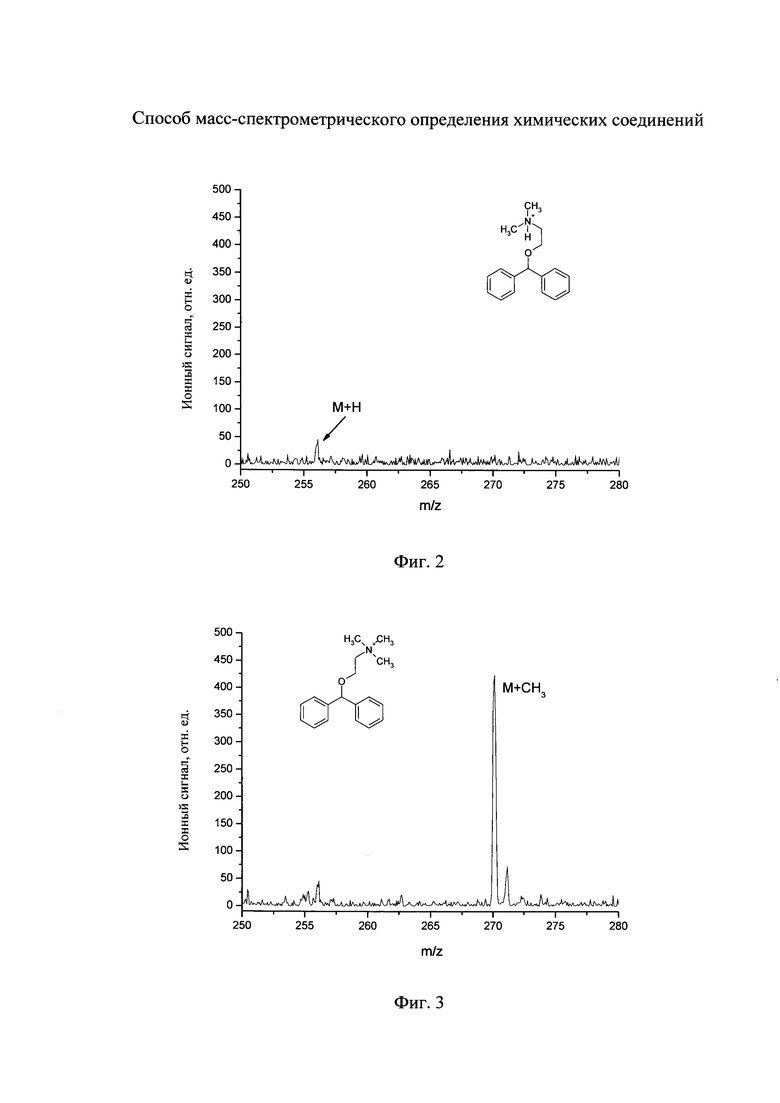
На фиг. 2 - масс-спектр дифенгидрамина, полученный по прототипу.
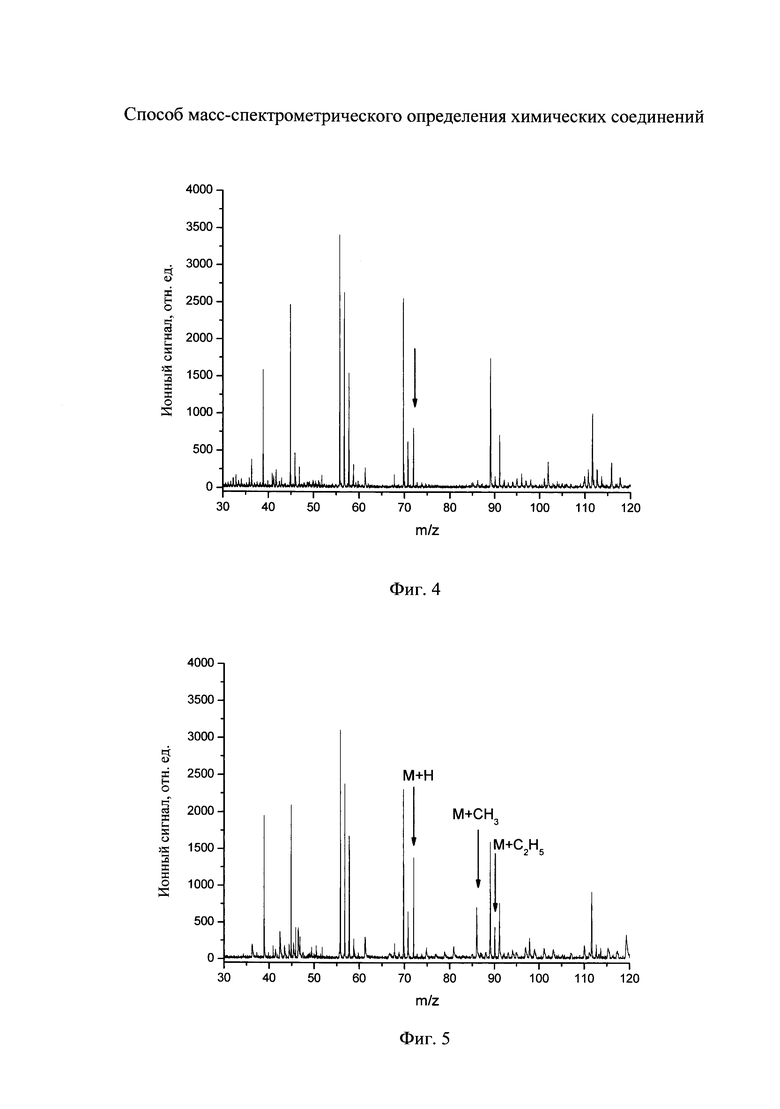
На фиг. 3 - масс-спектр дифенгидрамина, полученный по заявляемому способу.
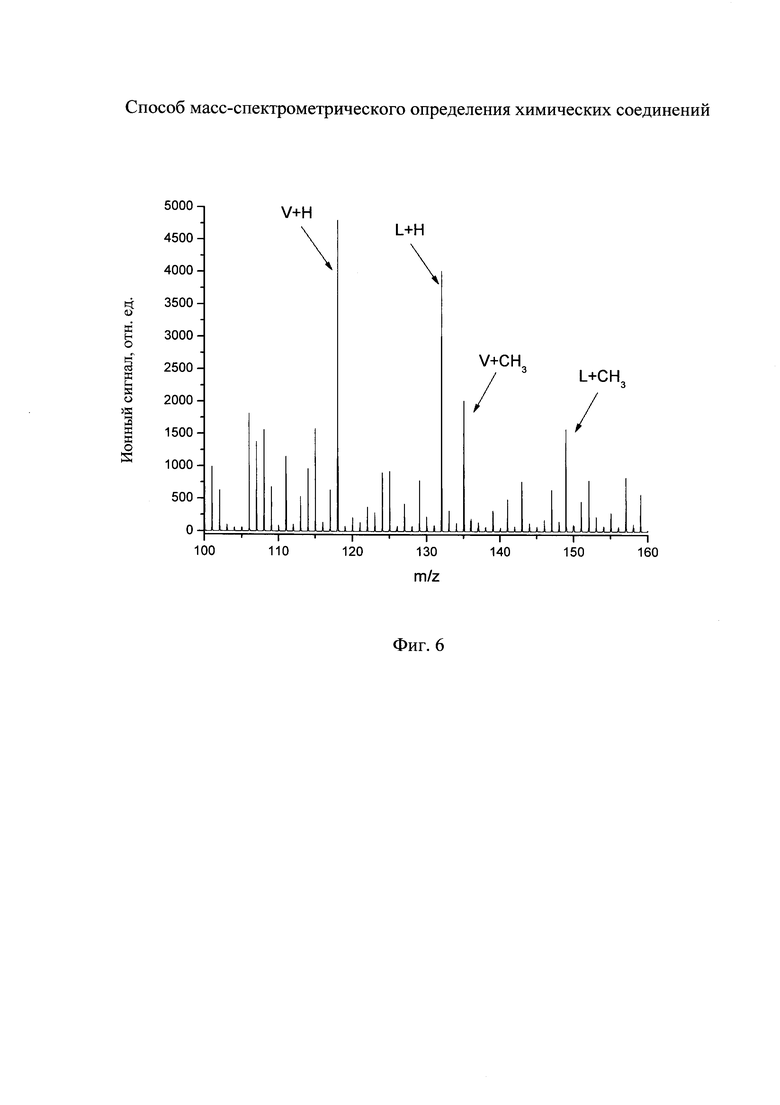
На фиг. 4 - масс-спектр атмосферного воздуха, полученный по заявляемому способу (холостой опыт).
На фиг. 5 - масс-спектр атмосферного воздуха, содержащего пары пирролидина, полученный по заявляемому способу.
На фиг. 6 - масс-спектр валина и лейцина, полученный по заявляемому способу, в условиях лазерной десорбции-ионизации при атмосферном давлении.
Способ реализуется следующим образом.
На фиг. 1 представлена общая схема установки для реализации заявляемого способа. Подложка 1 из твердотельного материала с нанесенными на ее поверхность определяемыми соединениями помещается в ионный источник масс-спектрометра с помощью шлюзовой системы быстрой замены эмиттера ионов 2. При анализе летучих соединений последние могут быть нанесены на поверхность подложки путем адсорбции из газовой фазы как в ионном источнике, так и вне его. На поверхность подложки фокусируется излучение импульсного лазера 3. На ускоряющую сетку ионного источника подается высокое напряжение, которое используется для ускорения ионов. Дозированный ввод газа-реагента осуществляется с помощью системы 4, состоящей из капилляра, расположенного в вакуумной камере масс-спектрометра, регулятора потока газа и источника паров газа реагента, расположенных вне прибора.
Пример 1.
В сравнительном примере представлено определение дифенгидрамина в растворе без использования газа-реагента и с его использованием.
На поверхность подложки из пористого кремния наносят 5 мкл раствора дифенгидрамина (действующего компонента лекарственного препарата «димедрол») в воде с концентрацией 1 мкМ. Нанесенный раствор высушивают в атмосфере воздуха, после чего подложку из пористого кремния помещают в ионный источник линейного времяпролетного масс-анализатора. Остаточное давление газа (воздуха) в ионном источнике составляет 5×10-7 мм рт.ст. На поверхность подложки с нанесенным веществом воздействуют излучением азотного лазера (длина волны лазерного излучения 337 нм) с частотой повторения лазерных импульсов 1 Гц, длительность лазерного импульса 3 нс. Интенсивность лазерного излучения выбирают равную 30 мДж/см2. Регистрацию ионов проводят в положительной ионизационной моде. В результате получают масс-спектр в области протонированных молекул, приведенный на фиг. 2. Видно, что в масс-спектре присутствует пик, соответствующий протонированным молекулам дифенгидрамина с отношением масс к заряду m/z 256 (структура ионов приведена на фиг. 2), однако амплитуда (или площадь) пика мала и примерно соответствует порогу обнаружения. При меньших концентрациях аналита в анализируемой пробе данный пик не регистрируется.
Затем на поверхность подложки направляют поток паров метанола, которые создают давление 2×10-5 мм рт.ст., фокусируют излучение азотного лазера на другой участок поверхности подложки с нанесенным аналитом и регистрируют новый масс-спектр. Полученный масс-спектр приведен на фиг. 3. Видно, что в масс-спектре регистрируется новый пик с m/z 270, который соответствует метилированным молекулам дифенгидрамина (структура ионов приведена на фиг. 3). Амплитуда пика превышает фоновый сигнал более чем в 20 раз. Молекулярную массу соединения определяют из измеренного значения m/z как величину, равную (m/z-15).
Пример 2.
В примере представлено определение пирролидина в воздухе.
Подложку аморфного кремния, полученного стандартным методом радиочастотного распыления, выдерживают в воздухе лаборатории в течение 30 секунд и помещают в ионный источник линейного времяпролетного масс-спектрометра. На поверхность аморфного кремния с помощью линзы фокусируют лазерный пучок Nd:YAG лазера с длиной волны 355 нм (третья гармоника основного излучения). Частота повторения лазерных импульсов 50 Гц, длительность лазерного импульса 0,5 нс. Интенсивность лазерного излучения выбирают равную 25 мДж/см2. Для регистрации ионов используют положительную ионизационную моду. Затем на поверхность подложки направляют поток паров ацетонитрила и этанола. Давление, создаваемое парами ацетонитрила и этанола, составляет 1×10-4 мм рт.ст. В результате получают масс-спектр в холостом опыте, изображенный на фиг. 4.
Затем подложку аморфного кремния помещают в ту же атмосферу, но дополнительно содержащую пары пирролидина с концентрацией 10 ppb. Подложку выдерживают в атмосфере в течение 30 секунд и помещают в ионный источник времяпролетного масс-спектрометра. Затем в ионный источник подают пары ацетонитрила и этанола. Давление, создаваемое парами ацетонитрила и этанола, составляет 1×10-4 мм рт.ст. Для лазерной-десорбции ионизации используют те же параметры лазера, как и в холостом опыте. В результате получают масс-спектр, изображенный на фиг. 5.
Из данных фиг. 4 видно, что в холостом опыте в масс-спектре присутствует ряд пиков, которые могут мешать определению и идентификации аналитов. В частности, в масс-спектре регистрируется пик с m/z 72, который интерферирует с пиком протонированного пирролидина (фиг. 5) и, следовательно, искажает метрологические результаты анализа. Однако при использовании потока паров ацетонитрила и этанола на подложку в масс-спектре регистрируются наряду с пиком протонированного пирролидина также два пика, соответствующих метилированным (М+СН3)+ и этилированным (М+С2Н5)+ ионам пирролидина. Эти пики находятся в области масс-спектра, свободной от интерференции.
Пример 3.
В примере представлено одновременное определение валина и лейцина в растворе.
На поверхность подложки из нанокристаллического оксида цинка ZnO наносят 2 мкл водного раствора, содержащего аминокислоты валин (V) и лейцин (L) с концентрациями 10 мкМ. После испарения растворителя подложку помещают на расстоянии 2 см от заборного устройства масс анализатора. На поверхность подложки с нанесенным веществом воздействуют излучением азотного лазера (длина волны лазерного излучения 337 нм) с частотой повторения лазерных импульсов 5 Гц, длительность лазерного импульса 3 нс. Интенсивность лазерного излучения выбирают равную 40 мДж/см2. Лазерную десорбцию-ионизацию проводят при атмосферном давлении. На поверхность подложки направляют поток паров дейтерированного йодистого метила CD3I. Плотность потока CD3I (количество вещества, попадающего на единицу поверхности в единицу времени) составляет 0.1 мкг/(см2×сек). В качестве масс-анализатора используют комбинацию квадруполь-времяпролетный масс-спектрометр, в которой транспортировка ионов в вакуумную камеру из области ионизации проводится через высокочастотный газонаполненный транспортный квадруполь, а детектирование ионов проводится при помощи времяпролетного рефлектрона. В результате регистрируют масс-спектр, изображенный на фиг. 6.
Валин и лейцин являются гомологами, поэтому пик метилированного валина интерферирует с пиком протонированного лейцина. Для того чтобы избежать интерференции при одновременном определении гомологов, в качестве газа-реагента целесообразно использовать изотопно обогащенные соединения. Из данных фиг. 6 видно, что при использовании дейтерированного йодистого метила, в масс-спектре регистрируются пики аналитов (M+CD3)+, которые не интерферируют с пиками протонированных молекул, что позволяет избежать ошибок при определении и идентификации определяемых соединений.
Присутствие в масс-спектре пика, характеризующего молекулярную массу химического соединения, является важным фактором при определении и идентификации соединения, поскольку позволяет установить его брутто-формулу. Одним из мешающих факторов при определении ряда соединений является сильная фрагментация образующихся ионов аналита. Например, протонированный дифенгидрамин, образующийся в условиях лазерной десорбции-ионизации на поверхности пористого кремния (по способу прототипа), является неустойчивым соединением, которое фрагментирует с образованием низкомолекулярных, и поэтому малоинформативных ионов. Степень фрагментации (отношения числа фрагментных ионов к общему числу ионов соединения) составляет примерно 95%. В свою очередь, ион метилированного дифенгидрамина, образующийся по заявляемому способу (пример 1), является устойчивым соединением и не фрагментирует в условиях лазерной десорбции-ионизации. Столь существенная разница в устойчивости обусловлена следующим: дифенгидрамин, как и целый ряд других лекарственных препаратов, содержит концевую аминогруппу. Соединения таких химических классов, как дифенгидрамин, при присоединении протону стабилизируют свою структуру путем частичного переноса протона к электроотрицательному атому кислорода. Такой перенос заряда приводит к ослаблению связи кислород-углерод и значительному понижению энергии ее диссоциации, что, в свою очередь, приводит к интенсивной фрагментации в условиях лазерной десорбции-ионизации. В свою очередь, при замещении в аминогруппе водорода на метил (т.е. в результате метилирования) процесс переноса протона невозможен. Поэтому метилированный дифенгидрамин, а также аналогичные соединения, не распадается в условиях лазерной десорбции-ионизации. Таким образом, использование заявляемого способа позволяет повысить достоверность идентификации дифенгидрамина при анализе растворов с концентрациями этого соединения менее 1 мкМ.
Другим мешающим фактором является интерференция ионов аналита с пиками ионов подложки и/или сопутствующих компонентов пробы, имеющих такое же значение m/z, как и ионы аналита. Использование заявляемого способа позволяет смещать пик, характеризующий молекулярную массу определяемого соединения, в область, свободную от интерференционных наложений, тем самым обеспечивая достоверность анализа. Следует отметить, что используемые в заявляемом способе газы-реагенты имеют низкое значение энергии сродства к протону и поэтому не ионизуются в условиях лазерной десорбции-ионизации и не мешают анализу. Исследования также показали, что при постоянных условиях анализа величина выхода ионов вида (MCnH2n+1)+ остается постоянной, что позволяет проводить количественные измерения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОПРЕДЕЛЕНИЯ МЕТАЛЛОВ И КОМПЛЕКСНЫХ СОЕДИНЕНИЙ МЕТАЛЛОВ | 2013 |
|
RU2531762C1 |
| СПОСОБ АНАЛИЗА ХИМИЧЕСКОГО СОСТАВА МАТЕРИАЛОВ | 2012 |
|
RU2539740C2 |
| СПОСОБ ДЕСОРБЦИИ-ИОНИЗАЦИИ ХИМИЧЕСКИХ СОЕДИНЕНИЙ | 2005 |
|
RU2285253C1 |
| СПОСОБ ДЕТЕКТИРОВАНИЯ И ИДЕНТИФИКАЦИИ ХИМИЧЕСКИХ СОЕДИНЕНИЙ И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2010 |
|
RU2414697C1 |
| СПОСОБ ДОСТАВКИ АНАЛИЗИРУЕМОГО ВЕЩЕСТВА В СИСТЕМУ РЕГИСТРАЦИИ И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2006 |
|
RU2327244C1 |
| Способ определения химических соединений, принадлежащих к группе тиурамдисульфидов | 2019 |
|
RU2732250C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ИЗОТОПНОГО СОСТАВА МЕТАНА | 2010 |
|
RU2461909C2 |
| СПОСОБ ФОРМИРОВАНИЯ ЭМИТТЕРА ИОНОВ ДЛЯ ЛАЗЕРНОЙ ДЕСОРБЦИИ-ИОНИЗАЦИИ ХИМИЧЕСКИХ СОЕДИНЕНИЙ | 2010 |
|
RU2426191C1 |
| Способ масс-спектрометрического анализа газообразных веществ | 2015 |
|
RU2634926C2 |
| СПОСОБ ПОЛУЧЕНИЯ И АНАЛИЗА ИОНОВ АНАЛИТА | 2010 |
|
RU2434225C1 |
Изобретение относится к способам лазерной десорбции-ионизации, может быть использовано для масс-спектрометрического анализа и идентификации химических соединений в жидких и газообразных пробах. Способ масс-спектрометрического определения химических соединений включает нанесение молекул химических соединений на поверхность твердотельного материала путем адсорбции или осаждения, лазерную десорбцию-ионизацию путем воздействия на материал импульсным лазерным излучением и детектирование ионов химических соединений в масс-анализаторе. Причем лазерную десорбцию-ионизацию ведут в присутствии газа-реагента, выбранного из группы соединений общей формулы CnH2nR, где n=1÷4, R=ОН, CN, I. Техническим результатом является повышение достоверности и надежности идентификации химических соединений, в частности, за счет выделения в масс-спектре пика, характеризующего молекулярную массу соединения при снижении концентрации анализируемых соединений. 11 з.п. ф-лы, 6 ил.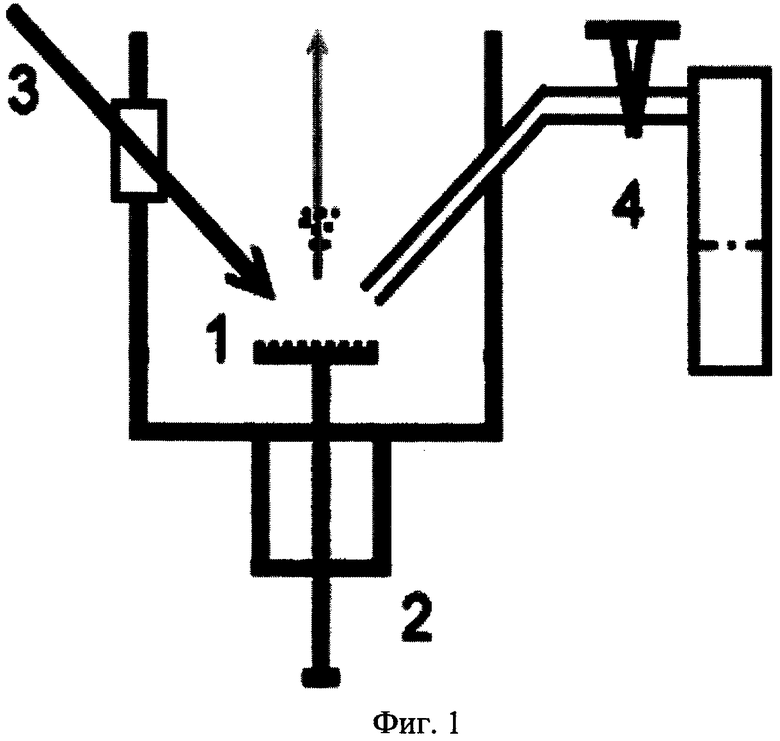
1. Способ масс-спектрометрического определения химических соединений, включающий нанесение молекул химических соединений на поверхность твердотельного материала путем адсорбции или осаждения, лазерную десорбцию-ионизацию путем воздействия на материал импульсным лазерным излучением и детектирование ионов химических соединений в масс-анализаторе, отличающийся тем, что лазерную десорбцию-ионизацию ведут в присутствии газа-реагента, выбранного из группы соединений общей формулы CnH2nR, где n=1÷4, R=ОН, CN, I.
2. Способ по п. 1, отличающийся тем, что используют поток газа-реагента, направленный на поверхность твердотельного материла.
3. Способ по п. 1, отличающийся тем, что в качестве газа-реагента используют одно соединение или смесь соединений.
4. Способ по п. 1, отличающийся тем, что в качестве газа-реагента используют спирты, например метанол, и нитрилы, например ацетонитрил.
5. Способ по пп. 1 и 4, отличающийся тем, что в качестве газа-реагента используют изотопно обогащенные соединения, например дейтерированный метанол или дейтерированный ацетонитрил.
6. Способ по п. 1,отличающийся тем, что в качестве твердотельного материала используют полупроводниковые материалы, например кремний, германий, оксид цинка, оксид титана, селенид кадмия.
7. Способ по пп. 1 и 6, отличающийся тем, что в качестве полупроводникового материала используют пористые, нанокристаллические или аморфные полупроводниковые материалы.
8. Способ по п. 1, отличающийся тем, что используют импульсное лазерное излучение с длиной волны, выбранной из условия поглощения его в твердотельном материале.
9. Способ по п. 1, отличающийся тем, что используют импульсное лазерное излучение с интенсивностью, меньшей порога плавления твердотельного материала.
10. Способ по п. 1, отличающийся тем, что лазерную десорбцию-ионизацию в присутствии газа-реагента ведут в вакууме, при значениях давления, создаваемого газом-реагентом, не превышающих 10-4мм рт.ст.
11. Способ по п. 1, отличающийся тем, что лазерную десорбцию-ионизацию в присутствии газа-реагента ведут при атмосферном давлении.
12. Способ по п. 1, отличающийся тем, что в качестве масс-анализатора используют масс-спектрометр или спектрометр ионной подвижности.
| US 6288390 B1 11.09.2001 | |||
| US 20100301199 A102.12.2010 | |||
| СПОСОБ ДЕСОРБЦИИ-ИОНИЗАЦИИ ХИМИЧЕСКИХ СОЕДИНЕНИЙ | 2005 |
|
RU2285253C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ МЕТАЛЛОВ И КОМПЛЕКСНЫХ СОЕДИНЕНИЙ МЕТАЛЛОВ | 2013 |
|
RU2531762C1 |
| US 20110287550 A1 24.11.2011. | |||

