УРОВЕНЬ ТЕХНИКИ
Доставка полинуклеотидов и других по существу не проникающих через клеточные мембраны соединений в живую клетку в значительной степени ограничена сложной системой клеточных мембран. Лекарственные средства, используемые в антисмысловой, РНКи и генной терапии, представляют собой относительно большие гидрофильные полимеры и часто несут сильный отрицательный заряд. Обе указанные физические характеристики значительно ограничивают возможность их непосредственную диффузию через клеточную мембрану. По этой причине основным ограничением для доставки полинуклеотида является доставка указанного полинуклеотида через клеточную мембрану в цитоплазму или ядро клетки.
Один способ, который используется для in vivo доставки малой нуклеиновой кислоты, заключатся в присоединении нуклеиновой кислоты либо к малой нацеливающей молекуле, либо к липиду или стеролу. Несмотря на то, что при использовании указанных конъюгатов наблюдается некоторая доставка и активность, необходимая для этих методов очень высокая доза нуклеиновой кислоты делает их непрактичными.
Также было разработано множество реагентов трансфекции, которые обеспечивали достаточно эффективную доставку полинуклеотидов в клетки в vitro. Однако доставка полинуклеотидов in vivo с использованием указанных реагентов трансфекции является сложной и признана неэффективной из-за токсичности, нежелательных взаимодействий с компонентами сыворотки и слабым направленным действием in vivo. Реагенты трансфекции, которые хорошо работают в vitro, катионные полимеры и липиды, как правило, образуют большие катионные электростатические частицы и дестабилизируют клеточные мембраны. Положительный заряд реагентов трансфекции в vitro облегчает связывание с нуклеиновой кислотой через взаимодействия между зарядами (электростатические), что приводит, к образованию комплекса нуклеиновая кислота/реагент трансфекции. Положительный заряд также является полезным для неспецифического связывания носителя с клеткой и для слияния с мембраной, ее дестабилизации или разрушения. Дестабилизация мембран облегчает доставку по существу не проникающего через клеточные мембраны полинуклеотида через клеточную мембрану. Хотя указанные свойства облегчают перенос нуклеиновой кислоты в vitro, они обуславливают токсичность и обеспечивают неэффективное направленное действие in vivo. Катионный заряд приводит к взаимодействию с компонентами сыворотки, которое вызывает дестабилизацию взаимодействия полинуклеотид-реагент трансфекции, плохую биодоступность и слабое нацеливание. Мембранная активность реагентов трансфекции, которая может являться эффективной в vitro, часто вызывает токсичность in vivo.
Для доставки in vivo носитель (нуклеиновая кислота и связанный агент доставки) должен быть маленьким, менее 100 нм в диаметре, предпочтительно, менее 50 нм. Еще более маленькие комплексы, менее 20 нм или менее 10 нм, были бы более применимыми. Носители для доставки размером более 100 нм имеют очень слабый доступ к клеткам, отличным от клеток кровеносных сосудов, in vivo. Комплексы, образованные за счет электростатических взаимодействий, склонны агрегировать или распадаться под воздействием физиологических концентраций солей или компонентов сыворотки. Кроме того, катионный заряд на носителях для доставки in vivo приводит к нежелательным взаимодействиям с компонентами сыворотки и, таким образом, слабой биодоступности. Интересно, что сильный отрицательный заряд также может ингибировать нацеливание (targeting) in vivo, препятствуя взаимодействию, необходимому для направленного действия, т.е. связыванию нацеливающих лигандов с клеточными рецепторами. Таким образом, для распределения и направленного действия in vivo являются желаемыми почти нейтральные носители. В отсутствие тонкой регуляции разрушающая или дестабилизирующая мембраны активность вызывает токсичность при использовании in vivo. Баланс между токсичностью носителя и доставкой нуклеиновой кислоты легче достигается в vitro, чем in vivo..
Согласно публикации патента США 20080152661, Розема и др. (Rozema et al.) предложили способ обратимой регуляции разрушающей мембраны активности мембраноактивного полиамина с использованием модификации с помощью дизамещенного малеинового ангидрида. Малеаматные связи, образованные путем проведения реакции между малеиновым ангидридом и амином, являются рН-лабильными в диапазоне рН, подходящем для доставки in vivo. Указанный процесс позволял использовать мембраноактивные полимеры для доставки in vivo или нуклеиновых кислот. Согласно настоящему изобретению предложены модифицированные мембраноактивные полимеры, содержащие дипептид-амидобензилкарбаматные связи. Дипептид-амидобензилкарбаматные связи (связывающие фрагменты) являются обратимыми и физиологически чувствительными. В отличие от рН-лабильных малеаматных связей, образованных путем модификации дизамещенным малеиновым ангидридом, присоединение модифицирующих полимер агентов, описанных в настоящей заявке, приводит к образованию расщепляемых ферментами связей, которые являются более стабильными при циркуляции in vivo.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
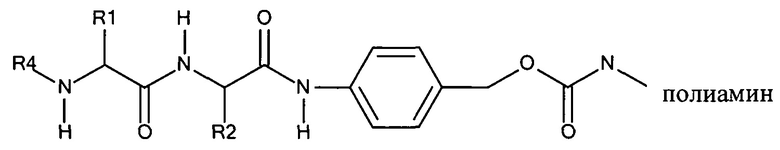
Согласно предпочтительному варианту реализации настоящего изобретения описаны маскирующие агенты для обратимой модификации и ингибирования мембранной активности амфипатического мембраноактивного полиамина, содержащие: стерический стабилизатор или нацеливающий лиганд, присоединенный к дипептид-амидобензилкарбонату, который называются в настоящей заявке дипептидными маскирующими агентами или расщепляемыми протеазы маскирующими агентами. Дипептидные маскирующие агенты имеют общую форму:
R-A1A2-амидобензилкарбонат,
где R представляет собой стерический стабилизатор или нацеливающий лиганд. А1 представляет собой аминокислоту, и А2 представляет собой аминокислоту. Реакция маскирующего агента с полимерным амином приводит к получению карбаматной связи. Маскирующий агент является стабильным до расщепления дипептида in vivo эндогенной протеазой, что приводит, соответственно, к отщеплению стерического стабилизатора или нацеливающего лиганда от полиамина. После ферментативного расщепления за дипептидом (между А и амидобензилом) амидобензилкарбамат претерпевает спонтанную перестройку, которая приводит к регенерации полимерного амина. Предпочтительно R является нейтральным. Более предпочтительно, R является незаряженным. Предпочтительный стерический стабилизатор представляет собой полиэтиленгликоль (PEG). Нацеливающий лиганд может быть выбран из перечня, включающего гаптены, такие как дигоксигенин, витамин, такой как биотин, антитело, моноклональное антитело и лиганд рецептора клеточной поверхности. Нацеливающий лиганд может быть связан с дипептидом через линкер, такой как PEG-линкер. Предпочтительный лиганд рецептора клеточной поверхности представляет собой лиганд рецептора асиалогликопротеина (ASGPr). Предпочтительный лиганд ASGPr представляет собой N-ацетилгалактозамин (NAG). Предпочтительный дипептид состоит из гидрофобной аминокислоты, связанной с гидрофильной незаряженной аминокислотой амидной связью. Предпочтительная амидобензильная группа представляет собой п-амидобензильную группу.
Предпочтительный карбонат представляет собой активированный амин-реактивный карбонат.
Согласно предпочтительному варианту реализации изобретения изобретение относится к композиции для доставки полинуклеотида интерферирующей РНК (РНКи) в клетку in vivo, содержащей: маскированный амфипатический мембраноактивный полиамин (полимер доставки), где указанный полиамин маскирован путем обратимой модификации дипептидными маскирующими агентами, описанными в настоящей заявке, и полинуклеотид РНКи. Полимер доставки может быть ковалентно связан с полинуклеотидом РНКи. Предпочтительная связь для ковалентного присоединения полимера доставки к полинуклеотиду РНКи представляет собой чувствительную к физиологическим условиям ковалентную связь. Согласно одному варианту реализации изобретения указанная связь является ортогональной по отношению к связи дипептидного маскирующего агента. Альтернативно, полимер доставки не является ковалентно связанным с полинуклеотидом РНКи, и полинуклеотид РНКи ковалентно связан с направленной молекулой.
Согласно предпочтительному варианту реализации настоящего изобретения описана композиция, содержащая: амфипатический мембраноактивный полиамин, ковалентно связанный с: a) множеством нацеливающих лигандов или пространственных стабилизаторов дипептид-амидобензилкарбаматными связями; и b) одним или более полинуклеотидами через одну или более обратимых связей. Согласно одному варианту реализации изобретения дипептид-амидобензилкарбаматная связь является ортогональной по отношению к обратимой ковалентной связи полинуклеотида. Конъюгат полинуклеотид-полимер вводят млекопитающему в фармацевтически приемлемом носителе или разбавителе.
Согласно предпочтительному варианту реализации настоящего изобретения описана композиция, содержащая: а) амфипатический мембраноактивный полиамин, ковалентно связанный со множеством нацеливающих лигандов или пространственных стабилизаторов дипептид-амидобензилкарбаматными связями; и b) полинуклеотид РНКи, ковалентно связанный с нацеливающей группой, выбранной из перечня, состоящего из: гидрофобной группы, содержащей 20, или более, атомов углерода и галактозного кластера. Согласно указанному варианту реализации изобретения полинуклеотид РНКи не является ковалентно связанным с модифицированным амфипатическим мембраноактивным полиамином. Конъюгат модифицированного полиамина и нацеленной на полинуклеотид РНКи группы синтезируются раздельно и могут поставляться в отдельных контейнерах или одном контейнере. Конъюгат модифицированного полиамина и нацеленной на полинуклеотид РНКи группы вводят млекопитающему совместно или раздельно в фармацевтически приемлемых носителях или разбавителях.
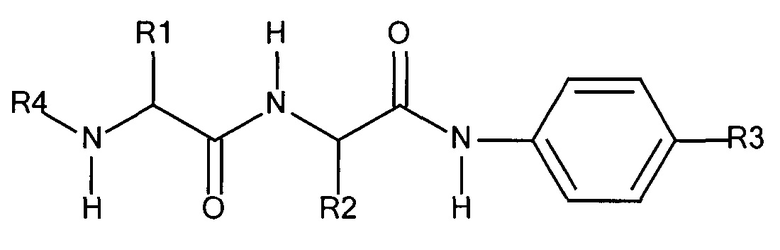
Предпочтительный дипептидный маскирующий агент содержит расщепляемое протеазой (пептидазой) амин-реактивное карбонатное производное дипептид-п-амидобензила. В расщепляемых протеазами маскирующих агентах согласно изобретению используется дипептид, соединенный с активированным амидобензилом карбонатным фрагментом. Нацеливающий лиганд или стерический стабилизатор присоединен к амино-концу дипептида. Активированный амидобензилом карбонатный фрагмент присоединен к карбокси-концу дипептида. Расщепляемые протеазами линкеры, подходящие для применения согласно изобретению, имеют следующую общую структуру:
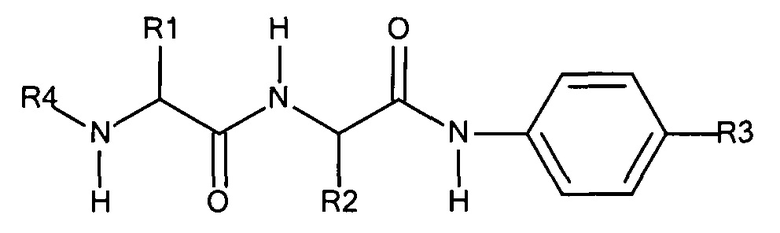
где R4 содержит нацеливающий лиганд или стерический стабилизатор, R3 содержит амин-реактивный карбонатный фрагмент, и R1 и R2 представляют собой боковые цепи аминокислот. Предпочтительный активированный карбонат представляет собой пара-нитрофенол. Однако другие амин-реактивные карбонаты, известные в данной области техники, можно легко использовать вместо пара-нитрофенола. Реакция активированного карбоната с амином приводит к связыванию нацеливающего лиганда или стерического стабилизатора с мембраноактивным полиамином через поддающуюся расщеплению пептидазой дипептид-амидобензилкарбаматную связь. Ферментативное расщепление дипептида между аминокислотой и амидобензильной группой приводит к удалению R4 от полимера и возбуждает реакцию элиминации, которая приводит к регенерации полимерного амина.
Дипептидные маскирующие агенты согласно изобретению применимы для обратимой модификации/ингибирования амфипатических мембраноактивных полиаминов. Ковалентная связь образуется в результате реакции активированного карбоната дипептидного маскирующего агента с полимерным амином, в частности, первичной аминогруппой. Таким образом, согласно настоящему изобретению предложен конъюгат, содержащий дипептид-амидобензилкарбонатный маскирующий агент, описанный в настоящей заявке, и амфипатический мембраноактивный полиамин:
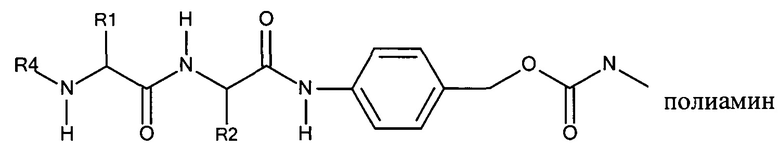
Соединения согласно настоящему изобретению, в целом, могут быть получены с использованием методов, известных специалисту в области органической или медицинской химии. Дополнительные объекты, признаки и преимущества согласно изобретению будут очевидны из следующего подробного описания при его совместном рассмотрении с сопутствующими графическими материалами.
КРАТКОЕ ОПИСАНИЕ ФИГУР.
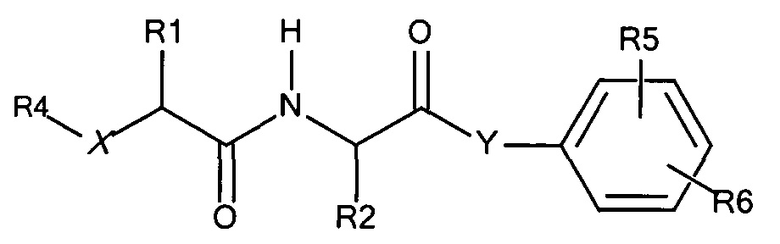
ФИГ.1. Изображение, показывающее структуру дипептидного маскирующего агента, где:
R1 и R2 представляют собой R-группы аминокислот,
R4 представляет собой нацеливающий лиганд стерического стабилизатора,
Х представляет собой -NH-, -О- или -СН2-,
Y представляет собой -NH- или -O-
R5 находится в положении 2, 4 или 6 и представляет собой -CH2-O-C(O)-O-Z, где Z представляет собой карбонат, и
R6 независимо представляет собой водород, алкил или галогенид в каждом из положений 2, 3, 4, 5 или 6, за исключением положения, занимаемого R5.
ФИГ.2. Изображение, показывающее структуру дипептидного маскирующего агента, связанного с полиамином, где: R1 и R2 представляют собой R-группы аминокислот, R4 представляет собой нацеливающий лиганд стерического стабилизатора, Х представляет собой -NH-, -О- или -СН2-, и Y представляет собой -NH- или -О-.
ФИГ.3. Изображение, показывающее структуры различных дипептидных маскирующих агентов.
ФИГ.4. График, показывающий время циркуляции полимеров, модифицированных дипептидными маскирующими агентами, по сравнению с двумя различными маскирующими агентами на основе малеинового ангидрида.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Описаны маскирующие агенты, применимые для обратимой модификации и ингибирования амфипатических мембраноактивных полиаминов, и доставки полимеров, образованных путем модификации полиамина дипептидными маскирующими агентами. Расщепляемые пептидазам связи являются устойчивыми к гидролизу в отсутствие фермента, электрически нейтральными и обеспечивают повышенную стабильность КДП (конъюгатов для доставки полинуклеотидов) при хранении и при циркуляции in vivo. Улучшенное (удлиненное) время полужизни при циркуляции расширяет возможности для лиганд-опосредованного накопления в ткани, такой как опухолевая ткань. Полимеры доставки особенно полезны для доставки полинуклеотидов РНКи in vivo. Доставка полинуклеотидов РНКи in vivo применима для терапевтического ингибирования (нокдауна) экспрессии гена.
Дипептидные маскирующие агенты имеют общую форму:
R-A1A2-амидобензилкарбонат,
где R представляет собой стерический стабилизатор или нацеливающий лиганд, А1 представляет собой аминокислоту, А2 представляет собой аминокислоту, и карбонат представляет собой активированный амин-реактивный карбонат. R предпочтительно является незаряженным. Реакция маскирующего агента карбоната с полимерным амином приводит к образованию карбаматной связи. Маскирующий агент является стабильным до расщепления дипептида эндогенной протеазой in vivo, что приводит, соответственно, к отщеплению стерического стабилизатора или нацеливающего лиганда от полиамина. После ферментативного расщепления за дипептидом (между А2 и амидобензилом), амидобензилкарбамат подвергается спонтанной перестройке, которая приводит к регенерации полимерного амина. Предпочтительный стерический стабилизатор представляет собой полиэтиленгликоль (PEG). Предпочтительный нацеливающий лиганд для доставки в печень представляет собой лиганд ASGPr. Предпочтительный лиганд ASGPr представляет собой N-ацетилгалактозамин (NAG). Предпочтительная амидобензильная группа представляет собой п-амидобензильную группу.
Дипептиды дипептидных маскирующих агентов, обозначенные как А1А2 (или АА), представляют собой димеры аминокислот, соединенных амидными связями. Аминокислоты, включая α- и β-аминокислоты, хорошо известны в области биологии и химии и представляют собой молекулы, содержащие аминогруппу, карбоксильную группу и боковую цепь, которая различается у разных аминокислот. Предпочтительная аминокислота представляет собой α-аминокислоту, имеющую общую формулу H2NCHRCOOH, где R представляет собой органический заместитель. Предпочтительная α-аминокислота представляет собой незаряженную природную аминокислоту. В предпочтительном дипептиде А1 представляет собой гидрофобную аминокислоту, и А2 представляет собой незаряженную гидрофильную аминокислоту. Предпочтительная гидрофобная аминокислота представляет собой фенилаланин, валин, изолейцин, лейцин, аланин или триптофан. Предпочтительная незаряженная гидрофильная аминокислота представляет собой аспарагин, глутамин или цитруллин. Более предпочтительная гидрофобная аминокислота представляет собой фенилаланин или валин. Более предпочтительная незаряженная гидрофильная аминокислота представляет собой цитруллин. Несмотря на то, что дипептиды являются предпочтительными, возможно включение дополнительной аминокислоты между А1 и R. Также возможно использовать одну аминокислоту вместо дипептида путем исключения аминокислоты А1. Любые природные аминокислоты, применимые согласно изобретению, в настоящей заявке обозначены обычными аббревиатурами. Несмотря на то, что могут использоваться заряженные аминокислоты, предпочтительно, чтобы маскирующий агент был незаряженным.
Согласно предпочтительному варианту реализации изобретения амфипатический мембраноактивный полиамин обратимо модифицирован путем проведения реакции с дипептид-амидобензилкарбонатным маскирующим агентом согласно изобретению с получением не обладающего мембранной активностью полимера доставки. Дипептидные маскирующие агенты могут защищать полимер от неспецифических взаимодействий, повышать время циркуляции в кровотоке, усиливать специфические взаимодействия, подавлять токсичность или изменять заряд полимера.
Обратимо маскированные полимеры согласно изобретению включают следующую структуру:
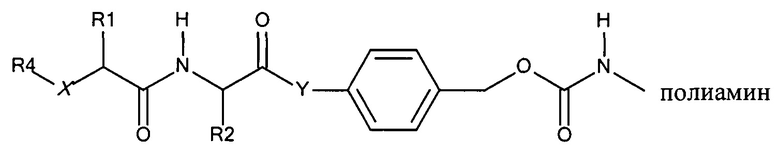
где:
Х представляет собой -NH-, -О- или -СН2-
Y представляет собой -NH- или -O-
R1 предпочтительно представляет собой
-(СН2)k-фенил (k представляет собой 1, 2, 3, 4, 5, 6; k=1, фенилаланин),
-СН-(СН3)2 (валин),
-СН2-СН-(СН3)2 (лейцин),
-СН(СН3)-СН2-СН3 (изолейцин),
-СН3 (аланин),
-(СН2)2-СООН (глутаминовая кислота),
или
R2 предпочтительно представляет собой
водород (глицин)
-(СН2)3-NH-С(O)-NH2 (цитруллин),
-(CH2)4-N-(CH3)2 (лизин(СН3)2),
-(CH2)k -C(O)-NH2; (k представляет собой 1, 2, 3, 4, 5, 6),
-CH2-C(O)-NH2 (аспарагин),
-(CH2)2-C(O)-NH2 (глутамин),
-CH2-C(O)-NR1R2 (амид аспарагиновой кислоты),
-(CH2)2-C(O)-NR1R2 (амид глутаминовой кислоты),
-CH2-C(O)-OR1 (эфир аспарагиновой кислоты) или
-(CH2)2-C(O)-OR1 (эфир глутаминовой кислоты),
R1 и R2 представляют собой алкильные группы
R4 включает полиэтиленгликоль или нацеливающий лиганд; и
полиамин представляет собой амфипатический мембраноактивный полиамин.
Несмотря на то, что на структуре, приведенной выше, показан один дипептидный маскирующий агент, связанный с полимером, при осуществлении настоящего изобретения от 50% до 90%, или более, полимерных аминов модифицированы дипептидными маскирующими агентами.
Согласно предпочтительному варианту реализации изобретения обратимо маскированный полимер согласно изобретению включают структуру:
где R1, R2, R4 и полиамин являются такими, как описано выше.
Обратимо маскированные полимеры согласно изобретению образованы путем проведения реакции между дипептидными маскирующими агентами согласно изобретению и аминами полимера. Дипептидные маскирующие агенты согласно изобретению имеют следующую структуру:
где:
X, Y, R1, R2, и R4 являются такими, как описано выше
R5 находится в положении 2, 4 или 6 и представляет собой -CH2-O-C(O)-O-Z, где Z представляет собой
- Галогенид,
 ,
,
 ,
,
 или
или
 ; и
; и
R6 независимо представляет собой водород, алкил, -(СН2)n-СН3 (где n=0-4), -(СН2)-(СН3)2 или галогенид в каждом из положений 2, 3, 4, 5 или 6, за исключением положения, занимаемого R5.
Согласно предпочтительному варианту реализации изобретения Х представляет собой -NH-, Y представляет собой -NH-, R4 является незаряженным, R5 находится в положении 4, и R6 представляет собой водород, как показано на следующей схеме:
 .
.
Согласно другому варианту реализации изобретения R4 представляет собой:
R-(O-CH2-CH2)s-O-Y1-, где
R представляет собой водород, метил или этил; и s представляет собой целое число от 1 до 150,
и Y1 представляет собой линкер, выбранный из перечня, включающего:
-O-Y2-NH-C(O)-(CH2)2-C(O)-, где Y2 представляет собой
-(СН2)3-
-C(O)-N-(CH2-CH2-O)p-CH2-CH2- (p представляет собой целое число от 1 до 20), и
-О-.
Нацеливающий лиганд может быть выбран из перечня, включающего гаптен, витамин, антитело, моноклональное антитело и лиганд рецептора клеточной поверхности. Нацеливающий лиганд может быть связан с дипептидом через линкер, такой как PEG-линкер.
Неограничивающие примеры мембраноактивных полимеров, подходящих для применения согласно изобретению, были ранее описаны в публикациях патентов США 20080152661, 20090023890, 20080287630, и 20110207799. Подходящий амфипатический мембраноактивный полиамин также может представлять собой маленький пептид, такой как пептид мелитина.
Полимерные амины обратимо модифицировали с использованием расщепляемых ферментами линкеров, описанных в настоящей заявке. Амин является обратимо модифицированным, если расщепление модифицирующей группы приводит к восстановлению амина. Реакция маскирующего агента на основе активированного карбоната с полимерным амином приводит к связыванию нацеливающего лиганда или стерического стабилизатора с полимером через поддающуюся расщеплению пептидазой дипептид-амидобензильную карбаматную связь, как показано далее:
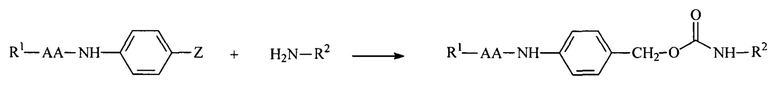
R1 включает нацеливающий лиганд (содержащий или не содержащий защитные группы) или PEG,
R2 представляет собой амфипатический мембраноактивный полиамин,
АА представляет собой дипептид (содержащий или не содержащий защитные группы), и
Z представляет собой амин-реактивный карбонат.
Защитные группы можно использовать в ходе синтеза дипептидных маскирующих агентов. В случае присутствия защитных групп, их можно удалять до или после модификации амфипатического мембраноактивного полиамина.
Обратимая модификация достаточного процента полимерных аминов дипептидными маскирующими агентами ингибирует мембранную активность мембраноактивного полиамина. Модификация полимерных аминов дипептидными маскирующими агентами также предпочтительно приводит к нейтрализации заряда амина. Дипептид-амидобензилкарбаматная связь чувствительна к расщеплению протеазой (или пептидазой). В присутствии протеазы анилидная связь расщепляется, что приводит к образованию промежуточного соединения, которое сразу подвергается реакции 1,6-элиминации с высвобождением свободного полимера:
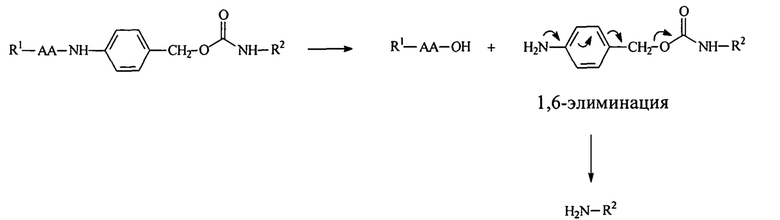
На приведенной выше схеме реакции АА представляет собой дипептид, R1 содержит нацеливающий лиганд или стерический стабилизатор, и R2 представляет собой амфипатический мембраноактивный полиамин. Важно, что свободный полимер является немодифицированным и, таким образом, мембранная активность восстанавливается.
В маскированном состоянии обратимо маскированный мембраноактивный полиамин не обладает разрушающей мембраны активностью. Может требоваться обратимая модификация дипептидными маскирующими агентами более чем 50%, более чем 55%, более чем 60%, более чем 65%, более чем 70%, более чем 75%, более чем 80% или более чем 90% аминов на полиамине для ингибирования мембранной активности и обеспечения функции нацеливания на клетки, т.е. образования обратимо маскированного мембраноактивного полимера (полимера доставки).
Настоящее изобретение также обеспечивает способ доставки биологически активного вещества в клетки. Более конкретно, настоящее изобретение относится к соединениям, композициям и способам, применимым для доставки полинуклеотидов РНКи в клетки млекопитающих in vivo.
Согласно одному варианту реализации изобретения полинуклеотид РНКи связан с полимером доставки согласно изобретению чувствительной к физиологическим условиям ковалентной связью. Благодаря использованию чувствительной к физиологическим условиям связи полинуклеотид можно отщеплять от полимера, высвобождая указанный полинуклеотид для его участия в функциональных взаимодействиях с клеточными компонентами.
Изобретение включает систему доставки конъюгата, имеющую следующую общую структуру:
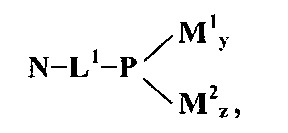
где N представляет собой полинуклеотид РНКи, L1 представляет собой чувствительную к физиологическим условиям ковалентную связь, P представляет собой амфипатический мембраноактивный полиамин, М1 представляет собой нацеливающий лиганд, связанный с Р дипептид-амидобензилкарбаматной связью, и М2 представляет собой стерический стабилизатор, связанный с P дипептид-амидобензилкарбаматной связью, каждый из y и z представляет собой целое число,, большее или равное нулю, при условии, что y+z имеет значение больше 50%, больше чем 60%, больше 70%, больше 80% или больше 90% первичных аминов на полиамине P, как определено на основе количества аминов на P в отсутствие любых маскирующих агентов. В немодифицированном состоянии P представляет собой мембраноактивный полиамин. Полимер доставки 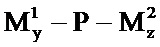
Согласно другому варианту реализации изобретения полинуклеотид РНКи вводят совместно с полимером доставки согласно изобретению in vivo. Таким образом, изобретение включает композиции, которые имеют следующую общую структуру:
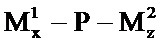
где N представляет собой полинуклеотид РНКи, Т представляет собой нацеливающую группу, P представляет собой амфипатический мембраноактивный полиамин, М1 представляет собой нацеливающий лиганд, связанный с P дипептид-амидобензилкарбаматной связью, и М2 представляет собой стерический стабилизатор, связанный с P дипептид-амидобензилкарбаматной связью. Каждый из y и z представляет собой целое число, большее или равное нулю, при условии, что y+z имеет значение больше 50%, больше 60%, больше 70%, больше 80% или больше 90% первичных аминов на полиамине P, как определено на основе количества аминов на Р в отсутствие любых маскирующих агентов. В немодифицированном состоянии Р представляет собой мембраноактивный полиамин. Полимер доставки M1y-P-M2z не является мембраноактивным. Обратимая модификация первичных аминов Р путем присоединения М1 и/или М2 приводит к обратимому ингибированию или инактивации мембранной активности Р. Показано, что некоторые маленькие амфипатические мембраноактивные полиамины, такие как пептид мелитина, содержат только 3-5 первичных аминов. Таким образом, предполагается, что изменение процента аминов отражает изменение процента аминов в популяции полимеров. При расщеплении М1 и М2 амины полиамина регенерируют, что приводит, соответственно, к возвращению Р в его немодифицированное мембраноактивное состояние. N связывается с Т через ковалентную связь с образованием конъюгата полинуклеотид РНКи-нацеливающая группа с использованием стандартных способов, известных в данной области техники. Предпочтительная ковалентная связь представляет собой чувствительную к физиологическим условиям ковалентную связь. N-T. Полимер доставки и N-T синтезируются или производятся раздельно. Т и N не являются напрямую или не напрямую ковалентно связанными с Р, М1 или М2. Электростатическое или гидрофобное соединение полинуклеотида или конъюгата полинуклеотида с маскированным или немаскированным полимером не является необходимым для доставки полинуклеотида в печень in vivo. Маскированный полимер и конъюгат полинуклеотида могут поставляться в одном контейнере или в отдельных контейнерах. Их можно объединять до введения, вводить совместно или вводить раздельно.
Для доставки в гепатоциты, независимо от того, является ли полинуклеотид РНКи связанным с полимером доставки через ковалентную связь или его вводят совместно с полимером доставки, у имеет значение больше 50% и до 100% первичных аминов на полимере Р. z, таким образом, имеет значение, больше или равное нулю процентов (0%), но меньше чем 50% первичных аминов на полимере Р.
В случае доставки к опухолевым клеткам печени z может иметь значение вплоть до 100% первичных аминов на полимере Р. Согласно предпочтительному варианту реализации изобретения для доставки к опухолевым клеткам z имеет значение более 50%, более 60%, более 70%, более 80% или более 90% первичных аминов на полиамине Р, и y равен нулю.
Мембраноактивные полиамины способны разрушать плазматические мембраны или лизосомальные/эндоцитозные мембраны. Указанная мембранная активность является существенным свойством для доставки в клетки полинуклеотида. Однако мембранная активность обуславливает токсичность при введении полимера in vivo. Полиамины также легко взаимодействуют со многими анионными компонентами in vivo, что обуславливает нежелательное биораспределение. Таким образом, обратимое маскирование мембранной активности полиамина является существенным для применения in vivo.
Маскирование осуществляют путем обратимого присоединения описанных дипептидных маскирующих агентов к мембраноактивному полиамину с образованием обратимо маскированного мембраноактивного полимера, т.е. полимера доставки. Помимо ингибирования мембранной активности, маскирующие агенты защищают полимер от неспецифических взаимодействий, снижают взаимодействия с компонентами сыворотки, нейтрализуют полиамин, снижая положительный заряд и приводя к образованию почти нейтрально заряженного полимера, повышают время циркуляции в кровотоке и/или обеспечивают клеточно-специфичные взаимодействия, т.е. направленность.
Существенным признаком маскирующих агентов является то, что при агрегации они ингибируют мембранную активность полимера. Маскирующие агенты могут защищать полимер от неспецифических взаимодействий (снижать взаимодействия с компонентами сыворотки, повышать время циркуляции в кровотоке). Мембраноактивный полиамин является мембраноактивным в немодифицированном (немаскированном) состоянии и не обладает мембранной активностью (является инактивированным) в модифицированном (маскированном) состоянии. Для достижения желаемой степени инактивации достаточное количество маскирующих агентов должно быть связано с полимером. Желаемая степень модификации полимера путем присоединения маскирующего агента (агентов) легко определяется с использованием подходящих тестов активности полимера. Например, если полимер в конкретном тесте обладает мембранной активностью, с указанным полимером связывают количество маскирующих агентов, достаточное для достижения желаемой степени ингибирования мембранной активности в указанном анализе. Маскирование требует модификации ≥50%, ≥60%, ≥70%, ≥80% или ≥90% первичных аминогрупп на популяции полимера, что определяют на основе количества первичных аминов на полимере в отсутствие любых маскирующих агентов. Также предпочтительной характеристикой маскирующих агентов является то, что их присоединение к полимеру снижает положительный заряд полимера, приводя, соответственно, к образованию более нейтрального полимера доставки. Желательно, чтобы маскированный полимер оставался растворимым в воде.
Мембраноактивный полиамин может быть конъюгирован с маскирующими агентами в присутствии избытка маскирующих агентов. Указанный избыток маскирующего агента может быть удален от конъюгированного полимера доставки до введения указанного полимер доставки.
В настоящей заявке «стерический стабилизатор» представляет собой неионный гидрофильный полимер (природный, синтетический или неприродный), который предотвращает или ингибирует внутримолекулярные или межмолекулярные взаимодействия полимера, к которому он присоединен, в отличие от полимера, не содержащего стерический стабилизатор. Стерический стабилизатор препятствует вовлечению полимера, к которому он присоединен, в электростатические взаимодействия. Электростатическое взаимодействие представляет собой нековалентное связывание двух или более веществ благодаря силам притяжения между положительными и отрицательными зарядами. Стерические стабилизаторы могут ингибировать взаимодействие с компонентами крови и, таким образом, ингибировать опсонизацию, фагоцитоз и захват ретикулоэндотелиальной системой. Стерические стабилизаторы, таким образом, могут повышать время циркуляции в кровотоке молекул, к которым они присоединены. Стерические стабилизаторы также могут предотвращать агрегацию полимера. Предпочтительный стерический стабилизатор представляет собой полиэтиленгликоль (PEG) или производное PEG. В настоящей заявке предпочтительный PEG может содержать примерно 1-500 или 2-25 этиленгликолиевых мономеров. В настоящей заявке средняя молекулярная масса предпочтительного PEG может также составлять примерно 85-20,000 Дальтон (Да), примерно 85-1000 Да. В настоящей заявке стерические стабилизаторы предотвращают или ингибируют внутримолекулярные или межмолекулярные взаимодействия полимера, к которому они присоединены, в водном растворе, по сравнению с полимером, не содержащим стерический стабилизатор.
«Нацеливающие лиганды» улучшают фармакокинетические свойства или биораспределение конъюгата, к которому они присоединены, для улучшения клеточно- или тканеспецифического распределения и клеточно-специфического захвата конъюгата. Для ясности, В настоящей заявке термин «нацеливающий лиганд» применяется для обозначения нацеливающего лиганда, который присоединен к дипептидному маскирующему агенту, и «нацеливающая группа» представляет собой нацеливающий лиганд, который связан с полинуклеотидом РНКи в конъюгате полинуклеотид РНКи-нацеливающая группа. Нацеливающие лиганды усиливают связывание молекулы с клеткой-мишенью. Таким образом, нацеливающие лиганды могут улучшать фармакокинетические свойства или биораспределение конъюгата, к которому они присоединены, обеспечивая улучшение клеточного распределения и клеточного захвата указанного конъюгата. Связывание нацеливающего лиганда с клеткой или клеточным рецептором может вызывать эндоцитоз. Нацеливающие лиганды могут являться моновалентными, бивалентными, тривалентными, тетравалентными или иметь более высокую валентность. Нацеливающие лиганды могут быть выбраны из группы, включающей: соединения, обладающие аффинностью к молекуле клеточной поверхности, лиганды клеточных рецепторов, антитела, моноклональное антитела, фрагменты антител и миметики антител, обладающие аффинностью к молекулам клеточной поверхности. Предпочтительный нацеливающий лиганд включает лиганд клеточного рецептора. Различные лиганды используют для обеспечения нацеливания лекарственных средств и генов на клетки и на специфические клеточные рецепторы. Лиганды клеточных рецепторов могут быть выбраны из группы, включающей: углеводы, гликаны, сахариды (включая, но не ограничиваясь указанными: галактозу, производные галактозы, маннозу и производные маннозы), витамины, фолат, биотин, аптамеры и пептиды (включая, но не ограничиваясь указанными: RGD-содержащие пептиды, инсулин, ЭФР и трансферрин).
Для нацеливания на гепатоциты предпочтительный нацеливающий лиганд представляет собой сахарид, обладающий аффинностью к рецептору асиалогликопротеина (ASGPr). Галактоза и производные галактозы использовали для обеспечения нацеливания молекул на гепатоциты in vivo путем их связывания с ASGPr, экспрессируемым на поверхности гепатоцитов. В настоящей заявке «ASGPr-нацеливающий лиганд» содержит галактозу и производное галактозы, обладающее аффинностью к ASGPr, равной или большей, чем аффинность галактозы. Связывание содержащих галактозу нацеливающих лигандов с рецептором (рецепторами) ASGPr облегчает клеточно-специфическую нацеливание полимера доставки на гепатоциты и эндоцитоз указанного полимера доставки гепатоцитами.
ASGPr-нацеливающие лиганды могут быть выбраны из группы, включающей: лактозу, галактозу, N-ацетилгалактозамин (NAG), галактозамин, N-формилгалактозамин, N-ацетилгалактозамин, N-пропионилгалактозамин, N-н-бутаноилгалактозамин и N-изо-бутаноилгалактозамин (Iobst, S.T. and Drickamer, K. J.B.C. 1996, 277, 6686). ASGPr-направленные фрагменты могут являться мономерными (например, содержать один галактозамин) или мультимерными (например, содержать много галактозаминов).
Согласно одному варианту реализации изобретения мембраноактивный полиамин обратимо маскирован путем присоединения маскирующих агентов, содержащих ASGPr-нацеливающий лиганд, к ≥50%, ≥60%, ≥70%, ≥80% или ≥90% первичных аминов на полиамине. Согласно другому варианту реализации изобретения мембраноактивный полиамин обратимо маскирован путем присоединения маскирующих агентов, содержащих ASGPr-нацеливающий лиганд, и маскирующих агентов, содержащих PEG, к ≥50%, ≥60%, ≥70%, ≥80% или ≥90% первичных аминов на полимере. В случае, когда присутствуют и маскирующие агенты, содержащие ASGPr-нацеливающий лиганд, и маскирующие агенты, содержащие PEG, отношение PEG к ASGPr-нацеливающему лиганду составляет примерно 0-4:1, более предпочтительно примерно 0,5-2:1.
«Амфипатические», или амфифильные, полимеры хорошо известны и распространены в данной области техники и имеют как гидрофильную (полярную, растворимую в воде), так и гидрофобную (неполярную, липофильную, не растворимую в воде) группу или часть.
«Гидрофильные группы» означают, в качественном отношении, что указанный химический фрагмент предпочитает воду. Как правило, такие химические группы являются растворимыми в воде и являются донорами или акцепторами водородных связей с водой. Гидрофильная группа может быть заряженной или незаряженной. Заряженные группы могут быть положительно заряженными (анионными) или отрицательно заряженными (катионными) или заряженными как положительно, так и отрицательно (цвиттерионными). Примеры гидрофильных групп включают следующие химические фрагменты: углеводы, полиоксиэтилен, конкретные пептиды, олигонуклеотиды, амины, амиды, алкоксиамиды, карбоновые кислоты, сульфаты и гидроксилы.
«Гидрофобные группы» означают, в качественном отношении, что химический фрагмент избегает воду. Как правило, такие химические группы не растворимы в воде и не склонны образовывать водородные связи. Липофильные группы растворяются в жирах, маслах, липидах и неполярных растворителях и имеют слабую способность или не имеют способности образовывать водородные связи. Углеводороды, содержащие два (2) или более атомов углерода, некоторые замещенные углеводороды, холестерин и производные холестерина, являются примерами гидрофобных групп и соединений.
Гидрофобные группы предпочтительно представляют собой углеводороды, содержащие только атомы углерода и водорода. Однако допускается присутствие неполярных заместителей или неполярных гетероатомов, которые поддерживают гидрофобность и включают, например, атом фтора. Термин включает алифатические группы, ароматические группы, ацильные группы, алкильные группы, алкенильные группы, алкинильные группы, арильные группы, аралкильные группы, аралкенильные группы и аралкинильные группы, каждая из которых может быть прямой, разветвленной или циклической. Термин «гидрофобная группа» также включает: стерины, стероиды, холестерин и производные стероидов и холестерина.
При использовании в настоящей заявке в отношении амфипатических полимеров, часть определяется как молекула, образованная при разрушении одной ковалентной связи и замещении на водород. Например, в молекуле бутиламина разрыв связи между атомом углерода и атомом азота и замещение атомами водорода приводит к образованию аммиака (гидрофильного) и бутана (гидрофобного). Если в молекуле 1,4-диаминобутана расщепляются связи между атомами азота и атомами углерода с замещением атомами водорода, то полученные молекулы также представляют собой аммиак (2×) и бутан. Однако 1,4,-диаминобутан не считается амфипатическим, так как для образования гидрофобной части требуется разрушение двух связей.
В настоящей заявке поверхностно-активный полимер снижает поверхностное натяжение воды и/или натяжение на границе раздела с другими фазами, и, соответственно, хорошо адсорбируется на поверхности раздела жидкости и пара. Свойство поверхностной активности обычно связано с тем, что молекулы вещества являются амфипатическими или амфифильными.
В настоящей заявке «мембраноактивные» полимеры представляют собой поверхностно-активные, амфипатические полимеры, которые способны оказывать один или более из следующих эффектов на биологическую мембрану: изменение или разрушение мембраны, которое позволяет не способным проникать через мембраны молекулам проходить в клетку или преодолевать мембрану, образование пор в мембране, разделение мембран или разрушение или растворение мембраны. В настоящей заявке мембрана или клеточная мембрана содержит липидный бислой. Изменение или разрушение мембраны можно функционально определить по активности пептида, по меньшей мере, в одном из следующих анализов: лизис эритроцитов (гемолиз), подтекание липосом, слияние липосом, клеточное слияние, лизис клеток и высвобождение из эндосом. Мембраноактивные полимеры, которые вызывают лизис клеточных мембран, также называются мембранолитическими полимерами. Полимеры, которые предпочтительно вызывают разрушение эндосом или лизосом по сравнению с плазматическими мембранами, считаются эндосомолитическими. Влияние мембраноактивных полимеров на клеточную мембрану может быть временным. Мембраноактивные полимеры обладают аффинностью в отношении мембраны и вызывают денатурацию или деформацию бислойных структур. Мембраноактивный полимеры могут представлять собой синтетические или неприродные амфипатические полимеры.
В настоящей заявке мембраноактивные полимеры отличаются от класса полимеров, которые называют проникающими в клетку пептидами, или полимеров, представленных соединениями, такими как обогащенный агринином пептид, произошедший из белка ТАТ ВИЧ, пептид Antennapedia, пептид VP22, транспортан, аргинин-багатые синтетические пептиды, малые гуанидиний-богатые синтетические полимеры и т.д. В то время как проникающие через клетку соединения могут переносить некоторые молекулы через мембрану от одной стороны липидного бислоя на другую сторону липидного бислоя, очевидно не нуждаясь в эндоцитозном процессе и не нарушая целостности мембраны, механизм их действия остается неизвестным.
Доставка полинуклеотида в клетку опосредуется мембраноактивным полимером, разрушающим или дестабилизирующим плазматическую мембрану или мембрану внутриклеточной везикулы (например, эндосомы или лизосомы), включая образование поры в мембране или разрушение эндосомальных или лизосомальных везикул, приводящее, таким образом, к высвобождению содержимого везикул в цитоплазму клетки.
Амфипатические мембраноактивные полиаминные сополимеры согласно изобретению представляют собой продукты сополимеризации двух или более типов мономеров. Согласно одному варианту реализации изобретения амфипатические мембраноактивные гетерополимеры согласно изобретению имеют следующую общую структуру:
-(А)а-(В)b-
где А содержит боковые функциональные группы первичного или вторичного амина, и В содержит боковую гидрофобную группу, а и b представляют собой целые числа >0.
Полимеры могут представлять собой полимеры с нерегулярных чередованием мономеров, блок-полимеры или полимеры с регулярным чередованием мономеров. Возможно включение дополнительных мономеров.
«Эндосомолитические полимеры» представляют собой полимеры, которые в ответ на специфические для эндосом факторы окружения, такие как присутствие литических ферментов, способны приводить к разрушению или лизису эндосомы или обеспечивать высвобождение в норме не проникающего через клеточные мембраны соединения, такого как полинуклеотид, из внутриклеточных заключенных в мембрану везикул, таких как эндосома или лизосома. В эндосоме происходит изменение физико-химических свойств эндосомолитических полимеров. Указанное изменение может представлять собой изменение растворимости полимера или способности взаимодействовать с другими соединениями или мембранами в результате изменения заряда, гидрофобности или гидрофильности. Обратимо маскированный мембраноактивный полиамин согласно изобретению считается эндосомолитическим полимером.
«Мелитин» представляет собой маленький амфипатический мембраноактивный пептид, который в природе встречается в пчелином яде. Мелитин может быть выделен из биологического источника или может являться синтетическим. Синтетический полимер получен или произведен человеком с помощью химического процесса, а не получается в результате природного биологического процесса. В настоящей заявке мелитин включает природные пептиды пчелиного яда семейства мелитина, которые могут быть обнаружены, например, в яде следующих видов: Apis mellifera, Apis cerana, Vespula maculifrons, Vespa magnifica, Vespa velutina nigrithorax, Polistes sp. HQL-2001, Apis florae, Apis dorsata, Apis cerana cerana, Polistes hebraeus. В настоящей заявке мелитин также включает синтетические пептиды, содержащие аминокислотную последовательность, идентичную или подобную природным пептидам мелитина. В частности, аминокислотная последовательность мелитина включает последовательности, показанные в Таблице 1. Синтетические пептиды мелитина могут содержать природные аминокислоты в L-форме или энантиомерные аминокислоты в D-форме (инвертированные). Однако пептид мелитинаа должен содержать по существу все аминокислоты в L-форме или все аминокислоты в D-форме, но при этом может содержать аминокислоты с противоположным стереоцентром, присоединенные к любому из амино- или карбокси-концов. Аминокислотная последовательность мелитина также может быть обращенной (ретро). Обращенный мелитин может содержать аминокислоты в L-форме или аминокислоты в D-форме (ретроинвертированный). Два пептида мелитина также могут быть ковалентно связаны с образованием димера мелитина. Мелитин может содержать модифицирующие группы, присоединенные к амино- или карбокси-концу, отличные от маскирующих агентов, которые усиливают тканевую нацеливание или облегчают циркуляцию in vivo.
Соединение или «линкер» представляет собой соединение между двумя атомами, которое связывает одну химическую группу или интересующий сегмент с другой химической группой или интересующим сегментом с помощью одной или более ковалентных связей. Например, связь может соединять маскирующий агент или полинуклеотид с полимером. Лабильная связь включает лабильную химическую связь. Связь может необязательно включать спейсер, который увеличивает расстояние между двумя соединяемыми атомами. Спейсер может также обеспечивать дополнительную подвижность и/или длину связи. Спейсеры могут включать, но не ограничиваются указанными, алкильные группы, алкенильные группы, алкинильные группы, арильные группы, аралкильные группы, аралкенильные группы, аралкинильные группы; каждая из которых может содержать один или более гетероатомов, гетероциклов, аминокислот, нуклеотидов и сахаридов. Спейсерные группы хорошо известны в данной области техники, и предшествующий перечень не ограничивает объем настоящего изобретения.
«Лабильная химическая связь» представляет собой ковалентную связь, отличную от ковалентной связи с атомом водорода, которая способна селективно разрушаться или расщепляться в условиях, которые не приводят к разрушению или расщеплению других ковалентных связей в той же молекуле. Более конкретно, лабильная химическая связь представляет собой ковалентную связь, которая является менее стабильной (термодинамически) или более быстро разрушается (кинетически) при соответствующих условиях, чем другие не лабильные ковалентные связи в той же молекуле. Расщепление лабильной связи внутри молекулы может приводить к образованию двух молекул. Специалисты в данной области техники обычно описывают расщепление или лабильность связи с помощью времени полужизни (t½) для расщепления связи (времени, требуемого для расщепления половины связей). Таким образом, лабильные связи включают связи, которые могут селективно расщепляться быстрее, чем другие связи в молекуле.
В настоящей заявке «чувствительная к физиологическим условиям» представляет собой лабильную связь, которая поддается расщеплению в условиях, которые в норме встречаются или аналогичны встречающимся в теле млекопитающего. Группы чувствительных к физиологическим условиям связей выбираются таким образом, что они претерпевают химическое превращение (например, расщепление) при определенных физиологических условиях.
В настоящей заявке «чувствительная к физиологическим условиям в клетке» представляет собой лабильную связь, которая поддается расщеплению в условиях, существующих в клетках млекопитающих. Условия внутри клеток млекопитающих включают химические условия, такие как рН, температура, окислительные или восстановительные условия или агенты и концентрация солей, встречающиеся или аналогичные встречающимся в клетках млекопитающих. Условия внутри клеток млекопитающих также включают наличие ферментативной активности, в норме присутствующей в клетках млекопитающих, например, активности протеолитических или гидролитических ферментов. Клеточно-чувствительная к физиологическим условиям может также расщепляться в ответ на введение фармацевтически приемлемого экзогенного агента.
Конъюгат интерферирующая РНК-нацеливающая группа: Нацеливающая группа может быть связана с 3’- или 5’-концом полинуклеотида РНКи. В случае полинуклеотидов миРНК нацеливающая группа может быть связана со смысловой или антисмысловой нитью, хотя смысловая нить является предпочтительной.
Согласно одному варианту реализации изобретения нацеливающая группа состоит из гидрофобной группы. Более конкретно, нацеливающая группа состоит из гидрофобной группы, содержащей, по меньшей мере, 20 атомов углерода. Гидрофобные группы, используемые в качестве направленных на полинуклеотид фрагментов в настоящей заявке называются гидрофобными направленными фрагментами. Примеры подходящих гидрофобных групп могут быть выбраны из группы, включающей: холестерин, дихолестерин, токоферол, дитокоферол, дидецил, дидодецил, диоктадецил, дидодецил, диоктадецил, изопреноид и холеамид. Гидрофобные группы, содержащие 6 или меньше, атомов углерода, не являются эффективными в качестве направленных на полинуклеотид фрагментов, тогда как гидрофобные группы, содержащие от 8 до 18 атомов углерода, обеспечивают повышение эффективности доставки полинуклеотида с повышением размера гидрофобной группы (т.е. повышением числа атомов углерода). Присоединение гидрофобной нацеливающей группы к полинуклеотиду РНКи не обеспечивает эффективную функциональную in vivo доставку полинуклеотида РНКи в отсутствие совместного введения полимера доставки. Несмотря на то, что другими авторами было описано, что конъюгаты миРНК-холестерин доставляют миРНК (миРНК-холестерин) в клетки печени in vivo в отсутствие каких-либо дополнительных носителей для доставки, требуются высокие концентрации миРНК, и наблюдается низкая эффективность доставки. При комбинации с полимерами доставки, описанными в настоящей заявке, наблюдается значительное улучшение доставки полинуклеотида. При обеспечении конъюгата миРНК-холестерин вместе с полимером доставки согласно изобретению эффективность указанного конъюгата миРНК-холестерин повышается примерно в 100 раз.
Гидрофобные группы, применимые в качестве направленных на полинуклеотид фрагментов, могут быть выбраны из группы, состоящей из: алкильной группы, алкенильной группы, алкинильной группы, арильной группы, аралкильной группы, аралкенильной группы и аралкинильной группы, каждая из которых может быть прямой, разветвленной или циклической, холестерина, производного холестерина, стерина, стероида и стероидного производного. Гидрофобные направленные группы предпочтительно представляют собой углеводороды, содержащие только атомы углерода и водорода. Однако возможно присутствие заместителей или гетероатомов, которые поддерживают гидрофобность, например, фтора. Гидрофобная нацеливающая группа может быть присоединена к 3’- или 5’- концу полинуклеотида РНКи с использованием методов, известных в данной области техники. Для полинуклеотидов РНКи, содержащих 2 нити, таких как миРНК, гидрофобная группа может быть присоединена к любой из указанных нитей.
Согласно другому варианту реализации изобретения нацеливающая группа содержит галактозный кластер (направленный фрагмент на основе галактозного кластера). В настоящей заявке «галактозный кластер» включает молекулу, содержащую от двух до четырех концевых производных галактозы. В настоящей заявке термин производное галактозы включает как галактозу, так и производные галактозы, обладающие аффинностью к ASGPr, равной или большей, чем аффинностью галактозы. Концевое производное галактозы присоединено к молекуле через ее С-1 атом углерода. Предпочтительный галактозный кластер содержит три концевых галактозамина или производных галактозамина, каждый из которых обладает аффинностью к рецептору асиалогликопротеина. Более предпочтительный галактозный кластер содержит три концевых N-ацетилгалактозамина. Другие термины, общепринятые в данной области техники, включают галактозу, содержащую три ветви, трехвалентную галактозу и тример галактозы. Известно, что кластеры производных галактозы, имеющие три ветви, связываются с ASGPr с более высокой аффинностью, чем структуры производных галактозы, содержащие две ветви или одну ветвь (Baenziger и Fiete, 1980, Cell, 22, 611-620; Connolly et al., 1982, J. Biol. Chem., 257, 939-945). Мультивалентность необходима для достижения наномолярной аффинности. Присоединение одного производного галактозы, имеющего аффинность к рецептору асиалогликопротеина, не обеспечивает функциональную доставку полинуклеотида РНКи в гепатоциты in vivo при совместном введении с полимером доставки.
Галактозный кластер содержит два-четыре, предпочтительно три производных галактозы, каждое из которых связано с центральной точкой ветвления. Производные галактозы присоединены к центральной точке ветвления через атомы углерода сахаридов С-1. Производное галактозы предпочтительно связано с точкой ветвления через линкеры или спейсеры. Предпочтительный спейсер представляет собой подвижный гидрофильный спейсер (U.S. Patent 5885968; Biessen et al. J. Med. Chem. 1995 Vol.39 p.1538-1546). Предпочтительный подвижный гидрофильный спейсер представляет собой PEG-спейсер. Предпочтительный PEG-спейсер представляет собой PEG3-спейсер. Точка ветвления может представлять собой любую малую молекулу, которая допускает присоединение трех производных галактозы и также допускает присоединение указанной точки ветвления к полинуклеотиду РНКи. Пример группы точки ветвления представляет собой ди-лизин. Молекула ди-лизина содержит три аминогруппы, через которые могут быть присоединены три производных галактозы, и карбоксильную реактивную группу, через которую ди-лизин может присоединяться к полинуклеотиду РНКи. Присоединение точкой ветвления к полинуклеотиду РНКи может происходить через линкер или спейсер. Предпочтительный спейсер представляет собой подвижный гидрофильный спейсер. Предпочтительный подвижный гидрофильный спейсер представляет собой PEG-спейсер. Предпочтительный PEG-спейсер представляет собой PEG3-спейсер (три этиленовые единицы). Галактозный кластер может быть присоединен к 3’- или 5’-концу полинуклеотида РНКи с использованием методов, известных в данной области техники. В случае полинуклеотидов РНКи, содержащих 2 нити, таких как миРНК, галактозный кластер может быть присоединен к любой из указанных нитей. Подходящие галактозные кластеры описаны в публикации патента США 20110207799.
Термин «полинуклеотид» или нуклеиновая кислота или полинуклеиновая кислота представляет собой специальный термин, который относится к полимеру, содержащему по меньшей мере два нуклеотида. Нуклеотиды представляют собой мономерные единицы полинуклеотидных полимеров. Полинуклеотиды, содержащие менее 120 мономерных единиц, часто называют олигонуклеотидами. Природные нуклеиновые кислоты имеют дезоксирибозо- или рибозофосфатный скелет.Неприродные или синтетические полинуклеотиды представляют собой полинуклеотиды, которые были полимеризованы в vitro или в бесклеточной системе и содержат такие же или подобные основания, но могут иметь скелет, отличный от природного рибозо- или дезоксирибозофосфатного скелета. Полинуклеотиды можно синтезировать с использованием любого метода, известного в данной области техники. Полинуклеотидные скелеты, известные в данной области техники включают: ПНК (пептидонуклеиновые кислоты), фосфоротиоаты, фосфородиамидаты, морфолины и другие варианты фосфатного скелета нативных нуклеиновых кислот. Основания включают пурины и пиримидины, которые также включают природные соединения аденин, тимин, гуанин, цитозин, урацил, инозин и природные аналоги. Синтетические производные пуринов и пиримидинов включают, но не ограничиваются указанными, модификации, в результате которых к нуклеотиду добавляются новые реактивые группы, включая, но не ограничиваясь указанными, амины, спирты, тиолы, карбоксилаты и алкилгалогениды. Термин «основание» включает любые из известных аналогов оснований ДНК и РНК. Полинуклеотид может включать рибонуклеотиды, дезоксирибонуклеотиды, синтетические нуклеотиды или любую подходящую комбинацию. Полинуклеотиды могут быть полимеризованы в vitro, могут являться рекомбинантными, содержать гибридные последовательности или производные указанных групп. Полинуклеотид может содержать концевой кэпирующий фрагмент на 5’-конце, 3’-конце или на обоих, 5’- и 3’-, концах. Кэпирующий фрагмент может представлять собой, но не ограничивается указанными, инвертированный дезоксифрагмент, лишенный азотистого основания, инвертированный дезокситимидиновый фрагмент, тимидиновый фрагмент или 3’-глицерильную модификацию.
«Полинуклеотид интерферирующей РНК (РНКи)» представляет собой молекулу, способную индуцировать РНК-интерференцию путем взаимодействия с системой пути РНК-интерференции клеток млекопитающих для разрушения или ингибирования трансляции транскриптов информационной РНК (иРНК) трансгена последовательность-специфичным образом. Два первичных полинуклеотида РНКи представляют собой малые (или короткие) интерферирующие РНК (миРНК) и микро РНК (микроРНК). Полинуклеотиды РНКи могут быть выбраны из группы, включающей: миРНК, микроРНК, двунитевую РНК (днРНК), короткую РНК, образующую «шпильку», (РНК-шпилька) и кассеты экспрессии, кодирующие РНК, способные вызывать РНК-интерференцию. миРНК имеет двунитевую структуру, как правило, включающую 15-50 пар оснований и предпочтительно 21-25 пар оснований и содержащую последовательность нуклеотидов, идентичную (абсолютно комплементарную) или почти идентичную (частично комплементарную) кодирующей последовательности в экспрессируемом гене-мишени или РНК в клетке. миРНК может содержать динуклеотидные 3’-«липкие концы». миРНК может состоять из двух гибридизованных полинуклеотидов или одного полинуклеотида, который образует структуру «шпильки». Молекула миРНК согласно изобретению содержит смысловой участок и антисмысловой участок. Согласно одному варианту реализации изобретения миРНК-конъюгат состоит из двух олигонуклеотидных фрагментов, где один фрагмент содержит нуклеотидную последовательность антисмысловой нити молекулы миРНК, и второй фрагмент содержит нуклеотидную последовательность смыслового участка молекулы миРНК. Согласно другому варианту реализации изобретения смысловая нить связана с антисмысловой нитью через линкерную молекулу, такую как полинуклеотидный линкер или не нуклеотидный линкер. Микро РНК (микроРНК) представляют собой генные продукты - малые некодирующие РНК, содержащие примерно 22 нуклеотида в длину, которые направлены на разрушение или подавление трансляции их мРНК-мишеней. Если комплементарность между микроРНК и мРНК-мишенью является частичной, то трансляция мРНК-мишени подавляется. Если комплементарность высокая, то мРНК-мишень расщепляется. В случае микроРНК комплекс связывается с сайтами-мишенями, обычно расположенными в 3’ UTR мРНК, которые, как правило, имеют только частичную гомологию с микроРНК. «Seed»-область - участок, состоящий примерно из (7) следующих друг за другом нуклеотидов на 5’-конце микроРНК, в котором наблюдается абсолютное спаривание оснований с его мишенью - играет ключевую роль в специфичности микроРНК. Связывание комплекса RISC/микроРНК с мРНК может приводить либо к подавлению трансляции белка, либо к расщеплению и деградации мРНК. Недавно полученные данные указывают на то, что расщепление мРНК происходит преимущественно в том случае, если существует абсолютная гомология по всей длине микроРНК с ее мишенью, а не точное спаривание оснований только в «seed»-области (Pillai et al. 2007).
Кассеты экспрессии полинуклеотида РНКи могут транскрибироваться в клетке с образованием малых РНК-«шпилек», которые могут функционировать как миРНК, отдельно смысловая и антисмысловая нить линейной миРНК или микроРНК. Транскрибируемая РНК-полимеразой III ДНК содержит промоторы, выбранные из перечня, включающего: промоторы U6, H1-промоторы и тРНК-промоторы. Промоторы РНК-полимеразы II включают промоторы U1, U2, U4 и U5, промоторы РНК-«шпильки», промоторы микроРНК и промоторы мРНК.
Перечни известных последовательностей микроРНК можно найти в базах данных, которые поддерживаются исследовательскими организациями, такими как Институт Сенгера (Wellcome Trust Sanger Institute), Центр биоинформатики Пенна (Penn Center for Bioinformatics), Мемориальный онкологический центр им. Слоуна-Кеттеринга (Memorial Sloan Kettering Cancer Center), и Европейская молекулярно-биологическая лаборатория (European Молекулу Biology Laboratory) и другие. Известные эффективные последовательности миРНК и распознаваемые сайты связывания также хорошо представлены в соответствующей литературе. Молекулы РНКи легко конструируют и получают с помощью методов, известных в данной области техники. Кроме того, существуют автоматические инструменты, помогающие найти эффективные и специфические мотивы последовательностей (Pei et al. 2006, Reynolds et al. 2004, Khvorova et al. 2003, Schwarz et al. 2003, Ui-Tei et al. 2004, Heale et al. 2005, Chalk et al. 2004, Amarzguioui et al. 2004).
Полинуклеотиды согласно изобретению могут быть химически модифицированы. Неограничивающие примеры указанных химических модификаций включают: включение фосфоротиоатных межнуклеотидных связей, 2’-O-метилрибонуклеотидов, 2’-дезокси-2’-фторрибонуклеотидов, 2’-дезоксирибонуклеотидов, нуклеотидов с «универсальными основаниями», 5-С-метилнуклеотидов и инвертированного лишенного азотистого основания дезокси-остатка. Показано, что указанные химические модификации при использовании в различных конструкциях полинуклеотидов ингибируют активность полинуклеотида в клетках, в то же время повышая сывороточную стабильность указанных соединений. Химически модифицированные миРНК также могут минимизировать вероятность активации интерферона у людей.
Согласно одному варианту реализации изобретения химически модифицированный полинуклеотид РНКи согласно изобретению содержит дуплекс, включающий две нити, одна или обе из которых могут быть химически модифицированы, причем каждая нить содержит от примерно 19 до примерно 29 нуклеотидов. Согласно одному варианту реализации изобретения полинуклеотид РНКи согласно изобретению содержит один или более модифицированных нуклеотидов способности сохраняет способность опосредовать РНК интерференцию внутри клетки или в воспроизведенной системе в vitro. Полинуклеотид РНКи может быть модифицирован, причем химически модифицированным может быть один или более (например, примерно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более) нуклеотидов. Полинуклеотид РНКи согласно изобретению может содержать модифицированные нуклеотиды как процент от общего числа нуклеотидов, присутствующих в полинуклеотиде РНКи. В таком случае полинуклеотид РНКи согласно изобретению может в общем содержать модифицированные нуклеотиды в от примерно 5 до примерно 100% положений нуклеотида (например, 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95% или 100% положений нуклеотида). Действительный процент модифицированных нуклеотидов, присутствующих в конкретном полинуклеотиде РНКи зависит от общего числа нуклеотидов, присутствующих в полинуклеотиде РНКи. Если полинуклеотид РНКи является однонитевым, то процент модификации может быть основан на общем числе нуклеотидов, присутствующих в указанном однонитевом полинуклеотиде РНКи. Подобным образом, если полинуклеотид РНКи является двунитевым, то процентная модификация может быть основана на общем числе нуклеотидов, присутствующих в смысловой нити, антисмысловой нити или в смысловой и антисмысловой нитях. Кроме того, действительный процент модифицированных нуклеотидов, присутствующих в конкретном полинуклеотиде РНКи, также может зависеть от общего числа пуриновых и пиримидиновых нуклеотидов, присутствующих в полинуклеотиде РНКи. Например, когда все пиримидиновые нуклеотиды и/или все пуриновые нуклеотиды, присутствующие в полинуклеотиде РНКи, являются модифицированными.
Полинуклеотид РНКи модулирует экспрессию РНК, кодируемой геном. Так как множество генов могут иметь некоторую степень гомологии по последовательности друг с другом, может быть сконструирован полинуклеотид РНКи, направленный на класс генов с достаточной гомологией последовательности. Таким образом, полинуклеотид РНКи может содержать последовательность, которая комплементарна последовательностям, которые содержатся в различных генах-мишенях, или которые являются уникальными для специфического гена-мишени. Таким образом, может быть сконструирован полинуклеотид РНКи, направленный на консервативные участки последовательности РНК, по которым различные гены имеют гомологию, таким образом, направленный на несколько генов в семействе генов (например, различные изоформы генов, сплайс-варианты, мутантаные гены и т.д.). Согласно другому варианту реализации изобретения может быть сконструирован полинуклеотид РНКи, направленный на последовательность, которая является уникальной для специфической последовательности РНК одного гена.
Термин «комплементарность» относится к способности полинуклеотида образовывать водородную связь (связи) с другой полинуклеотидной последовательностью либо по классическому уотсон-криковскому типу, либо по не классическим типам. В случае молекул полинуклеотида согласно настоящему изобретению свободная энергия связывания молекулы полинуклеотида с ее мишенью (эффекторный сайт связывания) или комплементарной последовательностью является достаточной для возможности поддержания соответствующей функции полинуклеотида, например, ферментативного расщепления мРНК или ингибирования трансляции. Способы определение свободных энергий связывания для молекул нуклеиновой кислоты хорошо известно в данной области техники (Frier et al. 1986, Turner et al. 1987). Процент комплементарности указывает на процент оснований в непрерывной цепи первой молекулы полинуклеотида, которые могут образовывать водородные связи (например, путем спаривания оснований по типу Уотсона-Крика) со второй последовательностью полинуклеотида (например, 5, 6, 7, 8, 9, 10 из 10 являются на 50%, 60%, 70%, 80%, 90% и 100% комплементарными). Абсолютная комплементарность означает, что все основания непрерывной цепи последовательности полинуклеотида образуют водородные связи с таким же числом следующих друг за другом оснований во второй последовательности полинуклеотида.
Под терминами ингибировать, подавлять или снижать экспрессию гена подразумевается, что экспрессия гена, измеренная по уровню РНК, транскрибируемой с указанного гена, или по уровню полипептида, белка или субъединицы белка, транслируемой с указанной РНК, снижается ниже уровня, наблюдаемого в отсутствие ингибирующего конъюгата полинуклеотида согласно изобретению. В результате ингибирования, подавления или снижения экспрессии гена с использованием полинуклеотида, который доставляется с помощью композиций согласно изобретению, уровень экспрессии предпочтительно ниже уровня, наблюдаемого в присутствии контрольной неактивной нуклеиновой кислоты, нуклеиновой кислоты с зашифрованной последовательностью или приводящими к инактивации ошибками или в отсутствие конъюгации полинуклеотида с маскированным полимером.
Было обнаружено, что устойчивость миРНК к деградации расположенными в эндосомах/лизосомах нуклеазами, таким как DNAаза II, обеспечивает значительное усиление снижения экспрессии гена-мишени. Такая устойчивость может непосредственно влиять на количество миРНК, высвобождаемой в цитоплазму, где располагается клеточный аппарат РНК-интерференции. Только доступная в цитоплазме порция миРНК вызывает эффект РНКи.
Помимо слабых фармакокинетических характеристик, миРНК чувствительна к действию нуклеаз в биологическом окружении при введении в кровоток без обеспечивающего защиту носителя доставки. Соответственно, многие миРНК быстро деградируют либо во внеклеточном пространстве в ткани и кровеносном русле, либо после внутриклеточного захвата (в эндосоме). Расщепление нуклеазами может подавляться нуклеотидами, не содержащими 2’-ОН-группы, такими как 2’-дезокси, 2’-O-метил (2’-ОМе) или 2’-дезокси-2’-фтор (2’-F)-нуклеотиды, и 5’-концевыми ненуклеотидными фрагментами полинуклеотида, такими как холестерин, аминоалкильный линкер или фосфотиоат в первой межнуклеотидной связи. Предпочтительно, полинуклеотид РНКи не содержит ни одного 2’-ОН-нуклеотида в цепи, начиная с 2’-ОМе-нуклеотида на 5’-конце, связанного с помощью фосфоротиоатной (РТО) связи со вторым нуклеотидом.
миРНК можно стабилизировать в значительной степени при использовании следующей схемы модификации, где антисмысловая нить олигонуклеотида имеет следующий тип модификации: 5’-(w)-(Z1)-(Z2)-(Z3)na-3’, и смысловая нить имеет модификацию 5’-(Z3)ns-3’, где
w независимо представляет собой 5’-фосфат или 5’-фосфотиоат или Н,
Z1 независимо представляет собой 2’-модифицированный нуклеозид.
Z2 независимо представляет собой 2’-дезоксинуклеозид или 2’-фтор-модифицированный нуклеозид,
Z3 независимо представляет собой 2’-модифицированный нуклеозид,
na равен 8-23, и ns равен 8-25.
Согласно одному предпочтительному варианту реализации изобретения олигонуклеотид имеет антисмысловую нить со схемой модификации: 5’-(w)-(Z1)-(Z2)-(Z3)na-3’, и смысловую нить со схемой модификации 5’-(Z3)ns-3’, где Z1 представляет собой 2’-фтор-модифицированный нуклеозид или 2-дезоксинуклеозид, и все остальные заместители, а также переменные na и ns имеют значения, приведенные выше.
Согласно одному предпочтительному варианту реализации изоберетения, олигонуклеотид имеет антисмысловую нить со схемой модификации: 5’-(w)-(Z1)-(Z2)-(Z3)na-3’, и смысловуюя нить со схемой модификации 5’-(Z3)ns-3’, где Z3 представляет собой 2’-O-метил-модифицированный нуклеозид, 2’-фтор-модифицированный нуклеозид или 2-дезоксинуклеозид, и все остальные заместители, а также переменные na и ns имеют значения, приведенные выше.
Согласно одному предпочтительному варианту реализации изобретения олигонуклеотид имеет антисмысловую нить со схемой модификации: 5’-(w)-(Z1)-(Z2)-(Z3)na-3’ и смысловую нить со схемой модификации 5’-(Z3)ns-3’, где Z1 представляет собой 2’-фтор-модифицированный нуклеозид или 2-дезоксинуклеозид и Z3 представляет собой 2’-O-метил-модифицированный нуклеозид, 2’-фтор-модифицированный нуклеозид или 2-дезоксинуклеозид, и все остальные заместители, а также переменные na и ns имеют значения, приведенные выше.
Нуклеозиды в последовательности нуклеиновых кислот олигонуклеотида с новой схемой модификации могут быть связаны с помощью 5’-3’ фосфодифирных или 5’-3’ фосфоротиоатных связей.
В настоящей заявке «антисмысловая» нить представляет собой нить миРНК, которая комплементарна мРНК-мишени и которая связывается с мРНК, если указанная миРНК расплетена. Смысловая нить указанной миРНК, имеющая новую схему модификации, комплементарна антисмысловой нити.
По существу, сайт расщепления нуклеазой между полинуклеотидом РНКи и направленным фрагментом или полимером доставки, к которому он ковалентно присоединен, может вводиться с помощью 3’- или 5’-«липких» концов, содержащих, по меньшей мере, один 2’-ОН-нуклеотид, на смысловой или антисмысловой нити. Конечная активная миРНК получается в результате внутриклеточного процессинга под действием нуклеаз. Также возможно использование определенных сайтов расщепления, которые обеспечиваются 2’-ОН-нуклеотидами в участке со спаренными основаниями, что может осуществляться с использованием, по меньшей мере, одного 2’-ОН-нуклеотида, комплементарного противоположной нити, или путем введения либо, по меньшей мере, одного несоответствующего 2’-ОН-нуклеотида, либо «шпильки»/петли, содержащей по меньшей мере, один 2’-ОН-нуклеотид.
Связь полинуклеотида с полимером доставки
Согласно одному варианту реализации изобретения полинуклеотид РНКи связан с полимером доставки через чувствительную к физиологическим условиям связь или линкер. Физиологически лабильный линкер выбран таким образом, что он подвергается химическому превращению (например, расщеплению) при определенных физиологических условиях, (например, дисульфидная связь расщепляется в восстановительных условиях цитоплазмы клетки). Высвобождение полинуклеотида из полимера путем расщепления чувствительной к физиологическим условиям связи облегчает взаимодействие указанного полинуклеотида с соответствующими клеточными компонентами, обеспечивающее его активность.
Конъюгат полинуклеотид-полимер образован путем ковалентного связывания полинуклеотида с полимером. Полимер полимеризован или модифицирован таким образом, что он содержит реактивную группу А. Полинуклеотид также полимеризован или модифицирован таким образом, что он содержит реактивную группу В. Реактивные группы А и В выбраны таким образом, что они могут быть связаны через обратимую ковалентную связь с использованием методов, известных в данной области техники.
Конъюгация полинуклеотида с полимером может осуществляться в присутствии избытка полимера. Так как полинуклеотид и полимер могут иметь противоположный заряд во время конъюгации, присутствие избытка полимера может приводить к снижению или устранению агрегации конъюгата. Альтернативно, может использоваться избыток полимерного носителя, такого как поликатион. Избыток полимера может быть удален от конъюгированного полимера до введения конъюгата животному или в клеточную культуру. Альтернативно, избыток полимера можно вводить совместно с конъюгатом животному или в клеточную культуру.
In vivo введение
В фармакологии и токсикологии способ введения представляет собой путь, по которому лекарственное средство, жидкость, яд или другое вещество приводят во взаимодействие с организмом. В целом, способы введения лекарственных средств и нуклеиновых кислот для лечения млекопитающих хорошо известны в данной области техники и могут применяться для введения композиций согласно изобретению. Соединения согласно настоящему изобретению могут вводиться с помощью любого подходящего способа, наиболее предпочтительно, парентерально, в композиции, соответствующим образом адаптированной для указанного способа. Таким образом, соединения согласно настоящему изобретению можно вводить путем инъекции, например, внутривенной, внутримышечной, внутрикожной, подкожной или внутрибрюшинной. Соответственно, согласно настоящему изобретению также предложены фармацевтические композиции, содержащие фармацевтически приемлемый носитель или наполнитель.
Парентеральные пути введения включают внутрисосудистые (внутривенные, внутриартериальные), внутримышечные, интрапаренхиматозные, внутридермальные, субдермальные, подкожные, внутриопухолевые, внутрибрюшинные, интратекальные, субдуральные, эпидуральные и внутрилимфатические инъекции, для осуществления которых используют шприц и иглу или катетер. «Внутрисосудистый», В настоящей заявке относится к трубчатой структуре, называемой сосудом, которая связана с тканью или органом внутри организма. В полости указанной трубчатой структуры биологическая жидкость течет в направлении к части тела или от нее. Примеры биологических жидкостей включают кровь, спинномозговую жидкость (СМЖ), лимфу или желчь. Примеры сосудов включают артерии, артериолы, капилляры, венулы, синусоидные капилляры, вены, лимфатические сосуды, желчные протоки и протоки слюнных или других экзокринных желез. Внутрисосудистый способ включает доставку через кровеносные сосуды, такие как артерия или вена. Кровеносная система обеспечивает системное распространение фармацевтического средства.
Описанные композиции вводят в фармацевтически приемлемых растворах-носителях. Термин «фармацевтически приемлемый» относится к таким свойствам и/или веществам, которые приемлемы для млекопитающего с фармакологической/токсикологической точки зрения. Фраза «фармацевтически приемлемый» относится к молекулярным субстанциям, композициям и свойствам, которые являются физиологически переносимыми и, как правило, не вызывают аллергической реакции или других нежелательных или токсических реакций при введении млекопитающему. Предпочтительно, в настоящей заявке термин «фармацевтически приемлемый» означает одобренный регулирующим федеральным органом или государственным правительством или перечисленный в Фармакопее США или другой общепризнанной фармакопее для применения у животных и, более конкретно, у человека.
Указанные носители также могут содержать вспомогательные соединения, такие как консерванты, смачивающие агенты, эмульгаторы и диспергирующие агенты. Для предупреждения появления микроорганизмов можно использовать способы стерилизации, описанные выше, а также включение различных антибактериальных и противогрибковых агентов, например, парабена, хлорбутанола, фенола, сорбиновой кислоты и т.д. Также может являться желательным включение в композиции изотонических агентов, таких как сахара, хлорид натрия и т.д. Кроме того, замедленная абсорбция фармацевтической формы для инъекции может обеспечиваться путем включения агентов, задерживающих абсорбцию, таких как моностеарат алюминия и желатин.
Согласно одному варианту реализации изобретения конъюгат полинуклеотид РНКи-нацеливающая группа вводят совместно с полимером доставки согласно изобретению. Совместное введение означает, что конъюгат полинуклеотида РНКи и полимер доставки вводят млекопитающему таким образом, что они присутствуют в теле млекопитающего в одно и то же время. Конъюгат олигонуклеотид РНКи-нацеливающая группа и полимер доставки можно вводить одновременно, или они могут доставляться последовательно. Для одновременного введения их можно смешивать до введения. Для последовательного введения первым можно вводить конъюгат полинуклеотид РНКи-направленный фрагмент или полимер доставки.
Терапевтический эффект
Полинуклеотиды РНКи можно доставлять в исследовательских целях или для достижения изменения в клетке, которое является терапевтическим. In vivo доставка полинуклеотидов РНКи применима для реагентов исследований и для различных терапевтических, диагностических целей, для подтверждения мишени, исследования генома, генной инженерии и фармакогеномного применения. Изобретатели настоящего изобретения описали доставку полинуклеотида РНКи, приводящую к подавлению эндогенной экспрессии гена в гепатоцитах. Уровень экспрессии репортеного (маркерного) гена, измеренной после доставки полинуклеотида, дает достаточные основания ожидать близкий уровень экспрессии гена после доставки других полинуклеотидов. Уровень после проведения лечения, который считается благоприятным специалистами в данной области техники, различается в зависимости от заболевания. Например, гемофилия А и гемофилия В вызваны дефицитом Х-связанных факторов свертывания VIII и IX, соответственно. На течение указанных болезней сильно влияет процент фактора VIII или IX в сыворотке от нормального уровня: <2%, тяжелая; 2-5%, средней тяжести; и 5-30%, легкая форма. Таким образом, повышение уровня на 1%-2% от нормального уровня циркулирующего фактора у пациентов с тяжелой формой гемофилией может считаться благоприятным. Уровень более 6% приводит к предотвращению спонтанных кровотечений, но не вторичных по отношению к хирургическому вмешательству или повреждению кровотечений. Подобным образом, подавление гена не должно составлять 100% для обеспечения терапевтического благоприятного эффекта. Специалист в области генной терапии сможет обоснованно прогнозировать благоприятный уровень экспрессии гена, специфичного для конкретного заболевания, на основе результатов достаточного уровня маркерного гена. В случае гемофилии, если уровень экспрессии маркерных генов с получением белка сравним по объему с 2% от нормального уровня фактора VIII, то имеются достаточные основания ожидать, что ген, кодирующий фактор VIII, также будет экспрессироваться на подобном уровне. Таким образом, репортерные или маркерные гены служат для получения полезного представления об экспрессии внутриклеточных белков в общем.
Печень представляет собой одну из наиболее важных тканей-мишеней для генной терапии, учитывая ее центральную роль в метаболизме (например, метаболизме липопротеинов при различных гиперхолестеринемиях) и секреции циркулирующих белков (например, факторов свертывания крови при гемофилии). Кроме того, распространены приобретенные нарушения, такие как хронический гепатит и цирроз, которые также потенциально можно лечить с помощью терапевтического лечения печени на основе полинуклеотидов. Ряд заболеваний или состояний, оказывающие влияние на печень или связанные с печенью, потенциально можно лечить путем снижения (подавления) экспрессии гена в печени. Такие заболевания и состояния печени могут быть выбраны из перечня, включающего: рак печени (включая гепатоклеточную карциному, НСС), вирусные инфекции (включая гепатит), метаболические нарушения, (включая гиперлипидемию и диабет), фиброз и острые повреждения печени.
Реальный уровень дозы активных ингредиентов в фармацевтических композициях согласно настоящему изобретению может меняться для получения такого количества активного ингредиента, которое является эффективным для достижения желаемого терапевтического эффекта для конкретного пациента, композиции и способа введения, и не вызывает токсичности у указанного пациента. Выбранный уровень дозы будет зависеть от различных фармакокинетических факторов, включая активность конкретной используемой композиции согласно настоящему изобретению, способ введения, время введения, скорость выведения конкретного используемого соединения, продолжительность лечения, другие лекарственные средства, соединения и/или материалы, используемых в комбинации с конкретной используемой композицией, возраст, пол, масса тела, состояние, общее состояние здоровья и предшествующая истории болезни пациента, подвергаемого лечению, и других факторов, хорошо известных в области медицины.
Количество (доза) полимера доставки, конъюгата полинуклеотид РНКи-нацеливающая группа или конъюгата полимер доставки-полинуклеотид РНКи, которое предполагается вводить, можно определить эмпирическим путем. Согласно изобретению, показано эффективное снижение экспрессии гена при использовании миРНК в концентрации 0,1-10 мг/кг массы тела животного и полимера доставки в концентрации 1,5-60 мг/кг массы тела животного. Предпочтительная концентрация для мышей миРНК-конъюгата составляет 0,25-2,5 мг/кг, и полимера доставки - 10-40 мг/кг. Более предпочтительно, вводят примерно 1,5-20 мг/кг полимера доставки. Количество конъюгата полинуклеотида РНКи можно с легкостью увеличивать, так как указанный конъюгат, как правило, не проявляет токсичности в более высоких дозах.
При использовании в настоящей заявке, in vivo означает происходящий внутри организма, и более конкретно, относится к процессу, который осуществляется в живой ткани или на живой ткани целого живого многоклеточного организма (животного), такого как млекопитающее, в отличие от части организма или мертвого организма.
В настоящей заявке «фармацевтическая композиция» включает конъюгаты согласно изобретению, фармацевтический носитель или разбавитель и любую другую среду или агент, необходимый для получения композиции.
В настоящей заявке «фармацевтический носитель» включает все без исключения растворители, дисперсионные среды, покрытия, антибактериальные и противогрибковые агенты, изотонические и замедляющие абсорбцию агенты и т.д., которые являются физиологически совместимыми. Предпочтительно, носитель подходит для внутривенного, внутримышечного, подкожного, парентарального, спинального или эпидермального введения (например, путем инъекции или инфузии).
ПРИМЕРЫ
Пример 7. Синтез расщепляемых протеазами (пептидазами) маскирующих агентов.
Все реакции, за исключением сочетания аминокислот в водном растворе НаНСОз и удаления силильной защитной группы, проводили в безводных условиях с использованием свежеприготовленных безводных растворителей. Колоночную очистку осуществляли на силикагеле с использованием специальных элюентов. Масс-спектры (МС) были получены с помощью ионизации электрораспылением.
Для получения активных п-нитрофенил-п-ациламидобензилкарбонатных производных NAG и PEG (NAG-L-AA-PABC-PNP и PEG-AA-PABC-PNP) использовали NHS-эфиры соответствующих PEG или NAG-содержащих производных для ацилирования амино-конца предшественника дипептид-п-ациламинобензилового спирта. На следующих этапах бензильные гидроксильные группы превращали в п-нитрофенилкарбонат с последующим удалением защитных групп с аминокислоты и фрагмента NAG. В некоторых случаях, когда использовали паранитрофенол (PNP)-карбонаты для модификации определенных полимеров, защитные группы удаляли до модификации полимера.
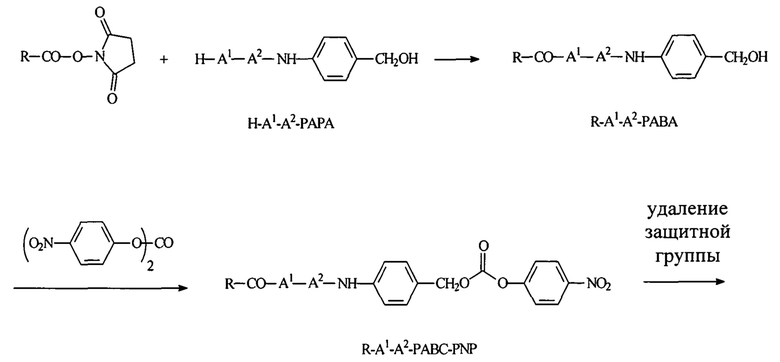
R содержит лиганд ASGPr (защищенный или незащищенный) или PEG, и А1 и А2 представляют собой аминокислоты (защищенные или незащищенные)
Синтез начинали с получения производных H-A1A2-PABA (Таблица 1). Указанные продукты присоединения получали с использованием схемы синтеза, описанной Дубовчиком (Dubowchik at al., 2002) с такими же модификациями. Fmoc-защищенные аминокислоты, Fmoc-A1-OH, активировали путем превращения в эфиры N-гидроксисукцинимида, Fmoc-A1-NHS, в реакции с дициклогексилкарбодиимидом (DCC) и N-гидроксисукцинимидом (NHS). Указанные реактивные NHS-эфиры подвергали реакции сочетания с защищенными аминокислотами А2 в присутствии водного растовра NaHCO3, который добавляли для поддержания реактивности аминогруппы. Для получения 1е и 1f (Таблица 1) вместо NHS-эфиров для сочетания использовали коммерчески доступные пентафторфениловые эфиры (OPfp).
Синтез Fmoc-дипептидов 1a-h.
а) NHS-эфиры АА получали из соответствующих аминокислот с использованием NHS и DCC и использовали без дополнительной очистки.
Условия: (i) N-гидроксисукцинимид (NHS), N-N’-дициклогексилкарбодиимид (DCC), 0-20°С.
Для получения Fmoc-Ala-NHS к ледяному раствору Fmoc-Ala-OH (412 мг, 1,32 ммоль) и NHS (160 мг, 1,38 ммоль) в ДХМ (13 мл) добавляли DCC (286 мг, 1,38 ммоль), перемешивали в течение 30 мин. и затем при 20°С в течение 16 часов. Твердую дициклогексилмочевину (DCU) отфильтровывали и растворитель удаляли в вакууме.
Для получения Fmoc-Asn(DMCP)-NHS к ледяному раствору Fmoc-Asn(DMCP)-OH (298 мг, 0,68 ммоль) и NHS (83 мг, 0,72 ммоль) в ДХМ (13 мл) добавляли DCC (148 мг, 0,72 ммоль), перемешивали в течение 30 мин., и затем при 20°С в течение 16 часов. Твердую DCU отфильтровывали и растворитель удаляли в вакууме.
Для получения Fmoc-Gly-NHS Fmoc-Gly-OH (891 мг, 3 ммоль) и NHS (380 мг, 3,3 ммоль) перемешивали в ТГФ (10 мл) при 0°С в течение 5 мин. и обрабатывали раствором DCC (650 мг, 3,15 ммоль) в ТГФ (5 мл). Охлаждающую баню удаляли в течение 30 мин. и реакционную смесь перемешивали при 20°С в течение 10 часов. Твердый DCU отфильтровывали, промывали ТГФ и растворитель удаляли на роторном вакуумном испарителе. Продукт взвешивали и растворяли в DME с получением 0,2 мМ раствора.
Для получения Fmoc-Glu(O-2PhiPr)-NHS к ледяному раствору Fmoc-Glu(O-2PhiPr)-OH (487 мг, 1 ммоль) и NHS (127 мг, 1,1 ммоль) в ТГФ (5 мл) добавляли DCC (217 мг, 1,05 ммоль), перемешивали в течение 15 мин. и затем при 20°С в течение 10 часов. Обработку проводили, как описано для Fmoc-Gly-NHS.
Для получения Fmoc-Phe-NHS к ледяному раствору Fmoc-Phe-OH (2,11 г, 5,45 ммоль) и NHS (664 мг, 5,77 ммоль) в ДХМ (50 мл) добавляли DCC (1,181 г, 5,72 ммоль), перемешивали в течение 30 мин. и затем при 20°С в течение 10 часов. Твердый DCU отфильтровывали и растворитель удаляли в вакууме.
Для получения Fmoc-Val-NHS, к ледяному раствору Fmoc-Val-OH (339 мг, 1 ммоль) и NHS (127 мг, 1,1 ммоль) в ДХМ (13 мл) добавляли DCC (227 мг, 1,1 ммоль), перемешивали в течение 30 мин. и затем при 20°С в течение 16 часов. Твердый DCU отфильтровывали и растворитель удаляли в вакууме.
b) Аминокислоты H-Asn(DMCP)-OH и H-Lys(MMT)-OH получали из доступных Fmoc-защищенных производных
Условия: (i) Триэтиламин (Et3N) в диметилформамиде (ДМФА).
H-Asn(DMCP)-OH
Fmoc-Asn(DMCP)-OH (576 мг, 1,32 ммоль) перемешивали в ДМФА (9 мл) с Et3N (3,7 мл, 26,4 ммоль) в течение 15 часов. Все летучие вещества удаляли на роторном вакуумном испарителе при 40°С/с помощью масляного вакуумного насоса. Осадок три раза растирали с эфиром (30 мл) и сушили в вакууме. Выход: 271 мг (96%). МС: 643,6 [3М+1]+; 451,3 [2M+Na]+; 429,5 [2M+1]+; 236,7 [M+Na]+; 215,3 [M+1]+; 132,8 [M-DMCP+1]+.
H-Lys(MMT)-OH. Fmoc-Lys(MMT)-OH (4,902 г, 7,65 ммоль) перемешивали в ДМФА (100 мл) с Et3N (32 мл, 30 экв. 229,4 ммоль) в течение 10 часов. Все летучие вещества удаляли на роторном вакуумном испарителе при 40°С/с помощью масляного вакуумного насоса. Осадок дважды растирали с эфиром и сушили в вакууме. Выход: 3,1 г (97%). МС (в режиме отрицательных ионов): 455, 453,3 [М+С1]-; 417,8 [M-1]-.
с) Синтез Fmoc-A1A2-OH.
Условия: (i) Н-А2-ОН, NaHCO3, смесь диметоксиэтана (DME), тетрагидрофурана (ТГФ) и H2O. (ii) H-A2-OH, NaHCO3, DME/ТГФ/Н2О. (iii) H-Cit-OH, NaHCO3, ТГФ в H2O.
Для получения Fmoc-GlyGly-OH 1a глицин (75 мг, 1 ммоль) и NaHCO3 (100 мг, 1,2 ммоль) растворяли в H2O (10 мл) и диметоксиэтане (DME) (5 мл). Добавляли раствор Fmoc-Gly-NHS в DME (5 мл, 1 ммоль). Добавляли ТГФ (2,5 мл), смесь обрабатывали ультразвуком для ее гомогенизации и перемешивали в течение 20 часов. Все летучие вещества удаляли на роторном вакуумном испарителе, осадок обрабатывали EtOAc и 5% раствором KHCO3 в H2O. Продукт экстрагировали четыре раза EtOAc, промывали солевым раствором при рН=3, сушили (Na2SO4), концентрировали и сушили в вакууме. Выход: 321 мг (90%). МС: 775.0 [2М+2Na]+; 377,4 [M+Na]+; 355,1 [M+1]+.
Для получения Fmoc-Glu(O-2PhiPr)Gly-OH 1b глицин (75 мг, 1 ммоль) и NaHCO3 (84 мг, 1 ммоль) растворяли в смеси Н2О (2 мл), ТГФ (4 мл) и DME (5 мл). Добавляли раствор Fmoc-Glu(O-2PhiPr)-NHS в DME (5 мл, 1 ммоль) и перемешивали в течение 10 часов. Все летучие вещества удаляли на роторном вакуумном испарителе, добавляли 20 мл 0,1 М буфера MES (рН=5) с последующим добавлением EtOAc (25 мл). Реакционную смесь перемешивали на льду и окисляли до рН=5 с помощью 5% раствора KHSO4. Продукт экстрагировали четыре раза EtOAc, промывали солевым раствором при рН=5, сушили (Na2SO4), концентрировали и сушили в вакууме. Выход: 528 мг (96%). МС: 567 [M+Na]+; 562 [M+NH4]+; 545,0 [M+1]+; 427,1 [М-2PhiPr]+.
Для получения Fmoc-Asn(DMCP)Gly-OH соединение 1c получали из Fmoc-Asn(DMCP)-NHS и H-Gly-OH, как описано выше для соединения 1b. Выход: 96%. МС: 987,4 [2М+1]+; 516,3 [M+Na]+; 494,4 [M+1]+; 412,2 [M-DMCP+1]+.
Для получения Fmoc-PheLys(MMT)-OH соединение 1d получали из Fmoc-Phe-NHS и Н-Lys(MMT)-OH, как описано выше для соединения 1b. Выход: 94%. МС: 788,5 [М+1]+, 273,1 [М-ММТ+1]+.
Для получения Fmoc-PheCit-OH 1e:
i) Fmoc-Phe-NHS (4,96 г, 10,26 ммоль) в DME (40 мл) добавляли к раствору, содержащему L-цитруллин (1,80 г, 10,26 ммоль) и NaHCO3 (0,86 г, 10,26 ммоль) в смеси Н2О (40 мл) и ТГФ (20 мл). Реакционную смесь перемешивали в течение 15 часов. Остаточный DCC после активации фильтровали и органический растворитель удаляли на роторном вакуумном испарителе. К осадку добавляли H2O (100 мл) и iPrOH (10 мл). Суспензию окисляли до рН=3 с помощью 5% KHSO4, продукт экстрагировали раствором EtOAc:iPrOH=9:1 (3×, 500 мл), промывали смесью солевой раствор:iPrOH=9:1 (2×, 50 мл), сушили (Na2SO4), фильтровали и концентрировали и сушили с помощью масляного насоса. В результате растирания с эфиром получали чистый продукт 1е. Выход: 3,84 г (68%). МС: 545,6 [M+Na]+; 528,5 [М-H2O]+; 306,3 [M-Fmoc+H2O]+.
ii) Раствор Fmoc-Phe-OPfp (553 мг, 1 ммоль) в ТГФ (5 мл) добавляли к раствору H-Cit-ОН (184 мг, 1,05 ммоль) и NaHCO3 (88,2 мг, 1,05 ммоль) в H2O (2,6 мл). Добавляли ТГФ (2 мл) для обеспечения гомогенности раствора и перемешивали в течение 10 часов. ТГФ удаляли на роторном вакуумном испарителе, осадок разбавляли Н2О (10 мл) и iPrOH (1 мл) и окисляли до рН=1 с помощью 3% HCl. Продукт экстрагировали пять раз раствором EtOAc:iPrOH=9:1, промывали смесью солевой раствор:iPrOH=9:1, сушили (Na2SO4) и концентрировали в вакууме. В результате растирания с эфиром получали 313 мг чистого продукта 1е (57%).
Fmoc-AlaCit-OH 1f получали из Fmoc-Ala-NHS и H-Cit-OH, как описано выше для соединения 1е-(а). Выход: 77%. МС: 959,8 [2M+Na]+; 938,1 [2М+1]+; 491,4 [M+Na]+; 469,9 [М+1]+.
Неочищенный Fmoc-ValCit-OH 1g получали из Fmoc-Val-NHS и H-Cit-OH, как описано выше для соединения 1b. Конечную очистку проводили путем растирания с эфиром. Общий выход: 76%. МС: 1060,3 [2М+3Na]+; 1015,7 [2M+Na]+; 519,7 [M+Na]+; 497,9 [M+1]+.
Fmoc-Ala-Asn(DMCP)-OH 1h получали из Fmoc-Ala-NHS и H-Asn(DMCP)-OH, как описано выше для 1b. Выход: 95%. МС: 530,2 [M+Na]+; 508,2 [M+1]+; 426,0 [М-DMCP+1]+.
Сочетание с п-аминобензиловым спиртом, получение Fmoc-AA-PABA и Fmoc-A-PABA 2а-m.
Продукты 1a-h подвергали реакции сочетания с п-аминобензиловым спиртом (РАВА) в присутствии 2-этокси-1-этоксикарбонил-1,2-дигидрохинолина (EEDQ) с образованием 2а-h. Также получали четыре типичных соединения 3 j-1, содержащих только одну присоединенную к РАВА фрагменту аминокислоту.
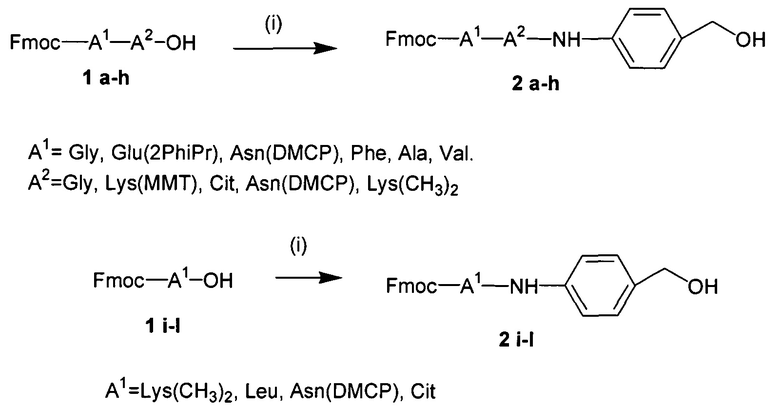
Условия: (i) PABA, EEDQ, ТГФ
Для получения Fmoc-GlyGly-PABA 2а раствор соединения 1а (318 мг, 0,9 ммоль) и PABA (220 мг, 1,8 ммоль) в ДХМ (17 мл) и МеОН (6 мл) перемешивали с EEDQ (444 мг, 1,8 ммоль) в течение 10 часов. Все летучие вещества удаляли на роторном вакуумном испарителе, осадок растирали с Et2O и продукт отфильтровывали и сушили в вакууме. Выход: 348 мг (84%).
Для получения Fmoc-Glu(O-2PhiPr)Gly-PABA 2b раствор соединения 1b (524 мг, 0,96 ммоль) и PABA (142 мг, 1,55 ммоль) в ДХМ (10 мл) перемешивали с EEDQ (357 мг, 1,44 ммоль) в течение 10 часов. Обработку проводили, как описано выше для соединения 2а. Выход: 462 мг (74%).
Fmoc-Asn(DMCP)Gly-PABA 2 с получали, как описано выше для соединения 2а. Выход: 64%. МС: 621,5 [M+22]+; 599,3 [M+1]+.
Fmoc-PheLys(MMT)-PABA 2d получали, как описано выше для соединения 2b. Выход: 70%.
Для получения Fmoc-PheCit-PABA 2e раствор соединения 1е (5,98 г, 10,97 ммоль) и PABA (2,70 г, 21,95 ммоль) в ДХМ (150 мл) и МеОН (50 мл) обрабатывали EEDQ (5,43 г, 21,95 ммоль) и перемешивали в течение 15 часов. Обработку проводили, как описано выше для соединения 2а. Выход: 6,14 г (86%). МС: 650,7 [М+1]+; 527,3 [М-РАВА+1]+.
Для получения Fmoc-AlaCit-PABA 2f раствор соединения 1f (2,89 г, 6,17 ммоль) и PABA (1,52 г, 12,34 ммоль) в ДХМ (45 мл) и МеОН (15 мл) обрабатывали EEDQ (3,05 г, 12,34 ммоль) и перемешивали в течение 15 часов. Обработку проводили, как описано выше для 2а. Выход: 4,56 г (74%). МС (ИЭР, в режиме отрицательных ионов): 307,4 [М-263,6-1]-; 349,9 [M-Fmoc-1]-; 610, 608,4 [M+HCl-1]-.
Fmoc-ValCit-PABA 2g получали, как описано выше для соединения 2b. (98%).
Fmoc-AlaAsn(DMCP)-PABA 2h получали, как описано выше для соединения 2а. Выход: 59%. МС: 613,2 [М+1]+; 531,4 [M-DMCP+1]]+; 408,2 [M-205+1]+.
Для получения Fmoc-Lys(СН3)2-РАВА 2i соль Fmoc-Lys(СН3)2-ОН⋅HCl (433 мг, 1 ммоль) и PABA (246 мг, 2 ммоль) растворяли в ДХМ (10 мл) и МеОН (1,5 мл), охлаждали до 5°С и добавляли EEDQ (495 мг, 2 ммоль). Охлаждающую баню удаляли и смесь перемешивали в течение 10 часов при комнатной температуре. Все летучие вещества удаляли на роторном вакуумном испарителе, осадок растирали с Et2O и неочищенный продукт отфильтровывали. Указанный продукт заново растворяли в смеси ДХМ (2 мл) и МеОН (1 мл) и снова осаждали путем добавления по каплям в Et2O (40 мл). Продукт фильтровали и сушили в вакууме. Выход: 448 мг (83%).
Для получения Fmoc-Leu-PABA 2j раствор Fmoc-Leu-OH (353 мг, 1 ммоль), EEDQ (495 мг, 2 ммоль) и РАВА (222 мг, 1,8 ммоль) в ДХМ (10 мл) перемешивали в течение 10 часов. Все летучие вещества удаляли на роторном вакуумном испарителе, осадок растворяли в Et2O (40 мл), охлаждали на сухом льду в течение 2 часов и твердое вещество отделяли посредством центрифугирования. Полученный неочищенный материал очищали на колонке, элюент: градиент МеОН (1-2%) в CHCl3. Выход: 444 мг (97%). МС: 459,4 [М+1]+.
Fmoc-Asn(DMCP)-PABA 2k получали, как описано для соединения 2j. Во время обработки вместо колоночной очистки после удаления ДХМ осадок растирали с Et2O, охлаждали до 0°С и неочищенный продукт отфильтровывали. Указанную обработку повторяли еще раз с последующим высушиванием в вакууме. Выход: 77%. МС: 542,5 [M+1]+.
Для получения Fmoc-Cit-PABA 21, раствор Fmoc-Cit-OH (345,7 мг, 0,87 ммоль) и РАВА (214 мг, 1,74 ммоль) в ДХМ (10 мл) и МеОН (4 мл) обрабатывали EEDQ (430 мг, 1,74 ммоль) и перемешивали в течение 15 часов. Твердый продукт растирали три раза с эфиром и полученный продукт фильтровали и сушили. Выход: 288 мг (67%). МС: 502,3 [М+1]+; 485,5 [M-H2O+1]+; 263 [M-Fmoc-H2O+1]+; 179,0 [M-306+1]+; 120,2 [М-365,3+1]+.
Продукт 2m получали с использованием другой схемы: сочетание производного Н-Lys(CH3)2-PABA 3 с Fmoc-Phe-NHS.
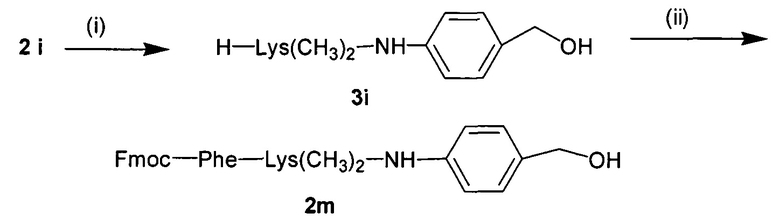
Условия: (i) Триэтиламин (Et3N) в ДМФА, 10 часов, (ii) Fmoc-Phe-NHS, диизопропилэтиламин (DIEA), ДМФА.
Для получения Fmoc-PheLys(CH3)-PABA 2m защитную группу Fmoc удаляли от Fmoc-Lys(СН3)2-РАВА (2i) (448 мг, 0,83 ммоль) путем перемешивания с Et3N (3,5 мл) в ДМФА (11 мл) в течение 10 часов. Все летучие вещества удаляли на роторном вакуумном испарителе при 40°С/с помощью масляного вакуумного насоса с получением неочищенного продукта 3i. Указанный продукт растворяли в ДМФА (7 мл), добавляли Fmoc-Phe-NHS (482 мг, 0,996 ммоль) с последующим добавлением DIEA (0,42 мл, 2,2 ммоль) и смесь перемешивали в течение 10 часов. Растворитель с DIEA удаляли на роторном вакуумном испарителе при 40°С/с помощью масляного вакуумного насоса с получением неочищенного соединения 2m, которое использовали без дополнительной очистки. МС: 549,4 [M+1]+.
Получение Н-АА-РАВА 3а-h, m и Н-А-РАВА 3j-l.
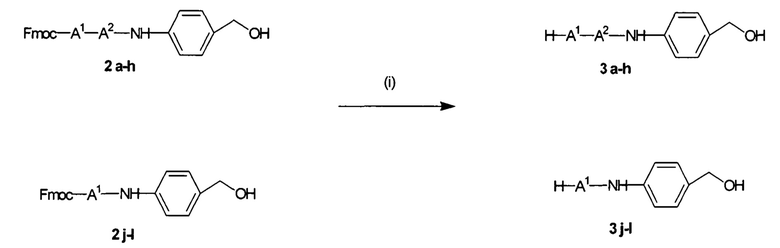
Условия: (i) Et3N в ДМФА, 10 часов.
Fmoc-производные 2a-h, j-l обрабатывали Et3N в ДМФА, как описано выше для соединения 3i, с последующим концентрированием и сушкой в вакууме. Неочищенные продукты растворяли в ДМФА с получением 0,1 М раствора и использовали без дополнительной очистки.
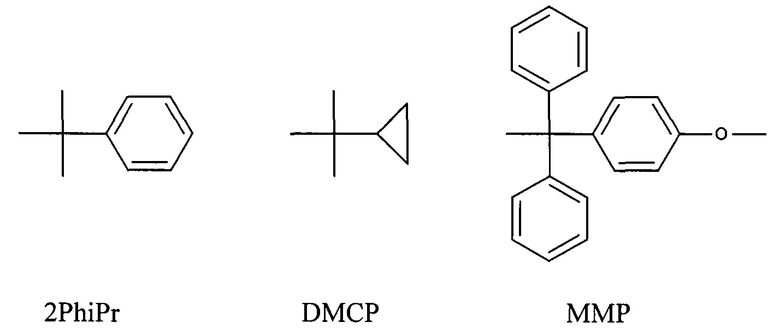
Получение расщепляемых протеазами NAG-маскирующих реагентов.
Получение NAG(Rl,R2,R3)-L-AA-PABC-PNP (Таблицы 2, 3)
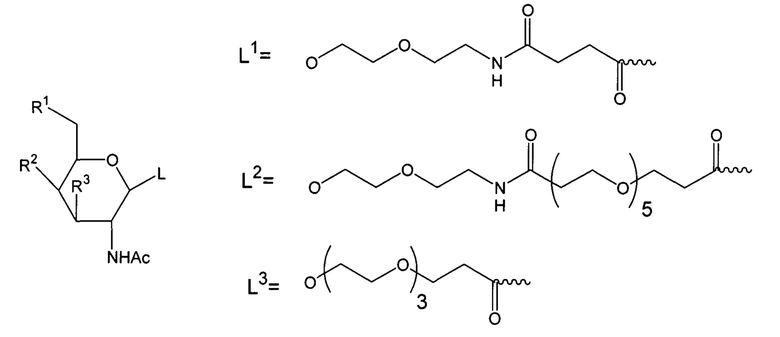
Получение NAG(R1,R2,R3)-L-AA-PABC-PNP, где R1, R2 и R3 представляют собой защитные группы, и L представляет собой связь между фрагментом галактозамина (NAG) и дипептидом (АА), начинали с получения NAG-L-CO2H-кислот 6, 10а,b, 13 и 17, которые после превращения в NHS-эфир использовали для ацилирования Н-АА-РАВА 2. В карбонатах 21a-f созданные для чувствительных к основным условиям полимеров защитные группы должны были быть удалены до модификации полимера. Для указанной цели при получении соединений 10а,b, 13 и 17 Ас-защитные группы в GAL-фрагменте замещали лабильными триэтилсилильными (TES) и трет-бутилдиметилсилильными (TBDMS) группами. Указанные группы можно удалять с использованием 70% раствора трифторуксусной кислоты (ТФУ) в Н2О при 0°С, не затрагивая чувствительный к основным условиям PNP-карбонатный фрагмент.
а) В случае получения NAG-L1-CO2H 6, где R1=R2=R3=OAc, от Z-защищенного NAG-тетраацетата 4 [3-5] удаляли защитную группу Z (H2, Pd/C(10%), MeOH, CHCl3 (20%) с получением NAG-амина 5, который затем ацилировали ангидридом янтарной кислоты. (ангидрид янтарной кислоты, Et3N, ДХМ, 1 час).
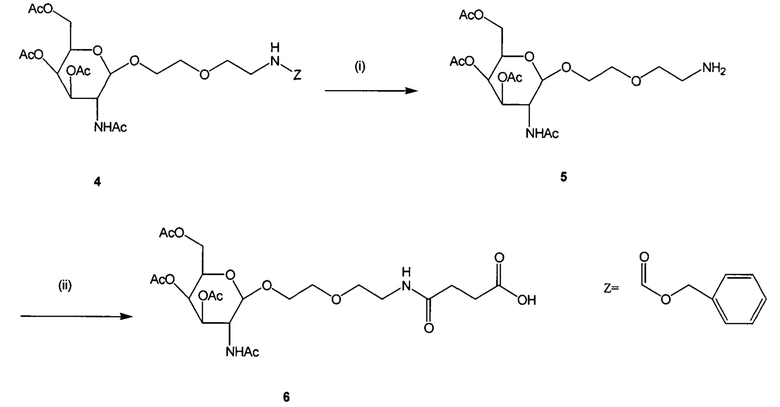
Условия: (i) H2, Pd/C (10%), MeOH, CHCl3, (20%), (ii), ангидрид янтарной кислоты, Et3N, CDM, 1 час.
NAG-амин 5: Для получения соединения 5 раствор NAG 4 (6,74 г, 11,85 ммоль) в MeOH (144 мл) и CHCl3 (36 мл) гидрогенировали в присутствии 10% Pd/C (674 мг) при 1 атм. в течение 10 часов. Катализатор отфильтровывали через целит, продукт концентрировали и сушили в вакууме. Выход: 5,04 г (98%).
NAG-L1-ОН 6: Для получения соединения 6 раствор янтарного ангидрида (966 мг, 9,65 ммоль) в ДХМ (30 мл) добавляли к NAG-амину 5 (4 г, 9,15 ммоль) в ДХМ (50 мл) с последующим добавлением Et3N (1,964 мл, 14 ммоль). Через 1 час реакционную смесь концентрировали и сушили в вакууме. Продукт очищали на колонке, элюент: градиент МеОН (5-7%) в CHCl3. Выход: 3,1 г (63%). МС: 535,3 [M+1]+; 330,3 [продукт дегликозилирования]+.
b) Производные NAG с легко удаляемыми защитными группами силилового эфира получали путем O-деацетилирования соединения 4 в смеси триэтиламина в водном растворе метанола с последующей обработкой триалкилсилилхлоридами.
i) NAG-L1-OH 10а,b. 10а: R1=OTES и OTBDMS, R2=OH, R3=OTES; 10b: R1=R3=OTBDMS, R2=OH.
Получение NAG 8a,b.
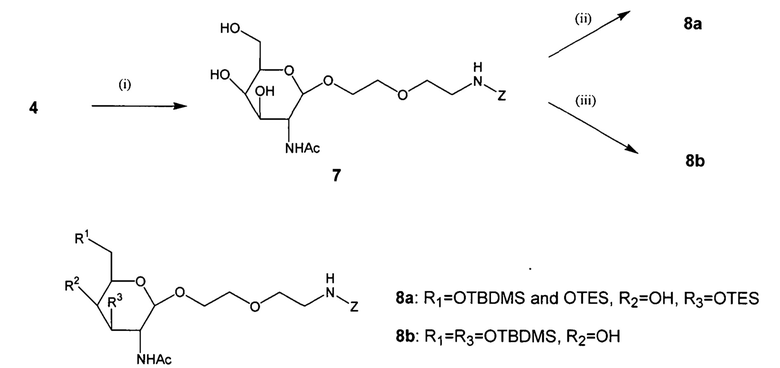
Условия: (i) Et3N, МеОН, H2O (5:7:6) 10 часов, (ii) TBDMSC1 (1 экв.), имидазол, 1 час, затем TESC1 (3 экв.), 10 часов в ДМФА. (iii) TBDMSC1 (3 экв.), имидазол, 10 часов в ДМФА.
Производное NAG 7.
Получение 10a,b.
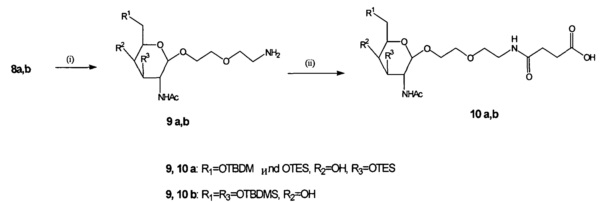
Условия: (i) H2, Pd/C (10%), ТГФ. (ii) ангидрид янтарной кислоты, Ее3Т ДХМ, 1 час.
Для получения соединения 7 NAG 4 (2 г, 3,52 ммоль) O-деацетилировали путем перемешивания в растворе МеОН (10 мл), H2O (32 мл) и Et3N (25 мл) в течение 10 часов. Все летучие вещества удаляли на роторном вакуумном испарителе при 40°С и осадок сушили посредством двукратного выпаривания толуола из реакционной смеси. Продукт 7 непосредственно использовали в следующем этапе. МС: 544,3 [M+Et3N+1]+; 443,7 [M+1]+; 204 [продукт дегликозилирования]+.
Для получения соединения 8а продукт 7 (1,76 ммоль) в ДМФА (15 мл) обрабатывали имидазолом (718 мг, 10,54 ммоль) и TBDMSC1 (265 мг, 1,76 ммоль), перемешивали в течение 2 часов и реакционную смесь охлаждали до 0°С. Добавляли TESC1 (531 мг, 3,52 ммоль), перемешивали в течение 10 часов, концентрировали и сушили в вакууме. Осадок отбирали в смеси EtOAc (110 мл) и H2O (30 мл). Органический слой отделяли, охлаждали до 5°С, промывали лимонной кислотой (5%), Н2О, NaHCO3 и сушили (Na2SO4). Неочищенный продукт пропускали через колонку, элюент: МеОН 2% в CHCl3 с получением смеси TBDMS и TES ди-Si-защищенных производных NAG 8а. Выход: 575 мг (49%). МС: 672,0 [М+1]+; 432,5 [продукт дегликозилирования]+.
Для получения 8b раствор 7 (1,76 ммоль) в ДМФА (15 мл) перемешивали с имидазолом (718 мг, 10,56 ммоль) и TBDMSC1 (1,061 г, 7 ммоль) в течение 10 часов. Реакционную смесь обрабатывали, как описано выше для получения соединения 8а. Выход: после колоночной очистки 767 мг (65%). М: 672,0 [М+1]+; 432,7 [продукт дегликозилирования]+.
Продукт 10а получали в виде смеси TBDMS и TES ди-Si-защищенных производных NAG в соответствии со способом, описанным для соединения 10b, ниже.
Для получения соединения 10b соединение 8b (920 мг, 1,37 ммоль) гидрогенировали в ТГФ (20 мл) в присутствии 10% Pc/С (150 мг) при 1 атм. в течение 10 часов. Катализатор отфильтровывали через целит, продукт 9b концентрировали и сушили в вакууме.
NAG-амин 9b без дополнительной очистки растворяли в ДХМ (12 мл), добавляли раствор янтарного ангидрида (140 мг, 1,40 ммоль) в ДХМ (7 мл) с последующим добавлением Et3N (0,236 мл, 1,676 ммоль) и перемешивали в течение 2 часов. Растворитель удаляли на роторном вакуумном испарителе и продукт очищали на колонке, элюент: 1% АсОН, 10% МеОН в CHCl3. Выход: 614 мг (72%).
ii) NAG-L2-OH 13. R1=R3=OTBDMS, R2=OH. Производные NAG с более длинным PEG-спейсером.
Для получения аналогов с более длинным PEG-спейсерами сначала удаляли защитную группу ацетила предшественника 5 с получением соединения 11 (Et3N, MeOH, H2O (5:7:6) 10 часов). Соединение 11 затем ацилировали бис-dPEG5полу-бензильным-полу-NHS эфиром (Quanta Product cat. #10237) с получением бензилового эфира 12 (NHS-PEG5-CO2Bn, Et3N, ДХМ). Соединение 12 последовательно бис-силилировали TBDMSC1 (TBDMSC1 (3 экв.), имидазол, 10 часов в ДМФА) и дебензилировали путем гидрогенизации (Н2, Pd/C (10%), ТГФ) с получением кислоты 13.
Получение NAG-производного 12.
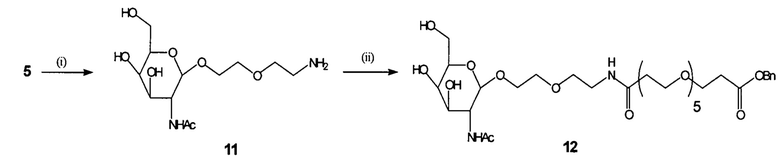
Условия: (i) Et3N, МеОН, Н2О (5:7:6) 10 часов, (ii) NHS-PEG5-CO2Bn, Et3N, ДХМ.
Получение NAG-L2-OH 13.
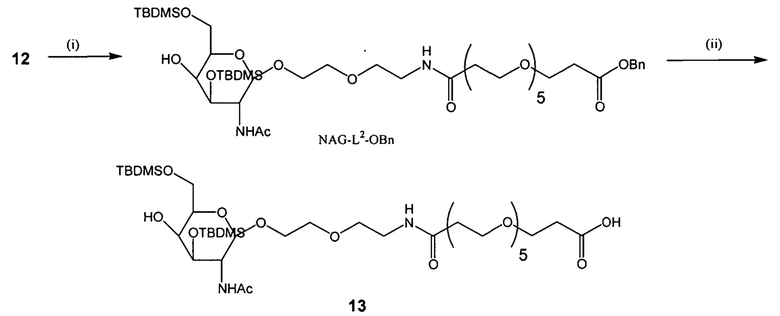
Условия: TBDMSC1 (3 экв.), имидазол, 10 часов в ДМФА. (vi) H2, Pd/C (10%), ТГФ.
Бензиловый эфир NAG-PEG8-SA 12. Для получения NAG-амина 11 NAG-амин 5 (0,381 ммоль) O-деацетилировали, как описано для предшественника 7 (способ для 8а,b). Продукт 11 сушили посредством двукратного выпаривания толуола на роторном вакуумном испарителе и растворяли в ДМФА (25 мл). бис-dPEG5полу-бензиловый-полу-NHS-эфир (200 мг, 0,381 ммоль) добавляли к реакционной смеси с последующим добавлением DIEA (0,079 мл, 0,457 ммоль), перемешивали в течение 8 часов и концентрировали на роторном вакуумном испарителе при 40°С/с помощью масляного вакуумного насоса. Неочищенный продукт 12 использовали в следующем этапе без дополнительной очистки. МС: 719,4 [М+1]+; 516,4 [продукт дегликозилирования]+.
Для получения NAG-L2-OBn сухой продукт 12 растворяли в ДМФА (5 мл), обрабатывали TBDMSC1 (230 мг, 1,524 ммоль), затем имидазолом (156 мг, 2,29 ммоль). Реакционную смесь перемешивали в течение 10 часов, все летучие вещества удаляли на роторном вакуумном испарителе при 40°С/с помощью масляного вакуумного насоса и осадок отбирали в EtOAc (85 мл) и промывали раствором HCl (1%), H2O. Водные фазы объединяли и заново экстрагировали EtOAc. Объединенные органические растворы сушили (Na2SO4), концентрировали и очищали на колонке, элюент: градиент МеОН (3-6%) в CHCl3. Выход бензилового эфира: 291 мг (80%). МС: 965,3 [М+NH4]+; 948,0 [М+1]+; 516,4 [продукт дегликозилирования]+.
Для получения NAG-L2-ОН 13 сложный эфир NAG-L2-OBn гидрогенировали, как описано для 9b (способ для 10b). Выход: 98%. МС: 858,0 [М+1]+; 426,1 [продукт дегликозилирования]+. Продукт использовали без дополнительной очистки.
iii) NAG-L3-OH 17. R1=R3=OTBDMS, R2=OH. Производные NAG с более длинным PEG спейсером.
Соединение 17 получали путем гликозилирования пентаацетата 14 [3-5] моно-tBu-эфиром PEG4 (TMCOTf, DCE/HO-PEG4-CO2tBu, SnCl4, ДХМ) с получением соединения 15. Соединение 15 подвергали гидролизу (HCO2H, 10 часов) с получением кислоты 16, которую затем O-деацетилировали (Et3N, MeOH, H2O (5:7:6) 10 часов) и обрабатывали TBDMSC1 (TBDMSC1 (3 экв.), имидазол, 10 часов в ДМФА) с получением бис-силилированной NAG-кислоты 17.
Получение сложного эфира NAG(OAc)3-L3-O-tBu 15.
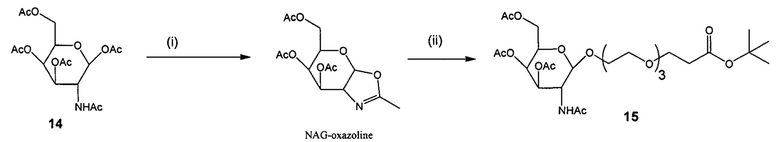
Условия: (i) триметилсилилтрифторметансульфонат (TMCOTf), дихлорэтан (DCE). (ii) т-бутил-12-гидрокси-4,7,10-триоксадодеканоат (HO-PEG4-CO2tBu), SnCl4, дихлорметан (ДХМ).
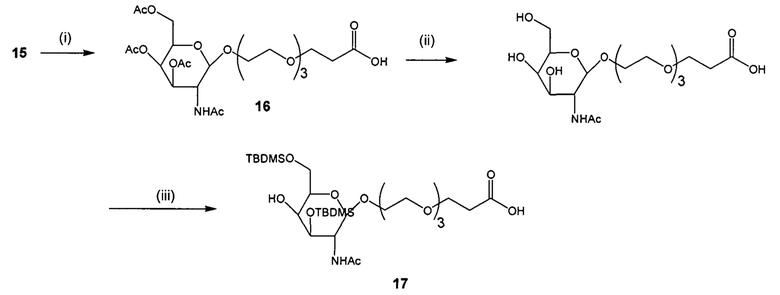
Условия: (i) HCO2H, 10 часов, (ii) Et3H MeOH, H2O (5:7:6) 10 часов. (iii) TBDMSC1 (3 экв.), имидазол, 10 часов в ДМФА.
Для получения сложного эфира NAG(OAc)3-L3-O-tBu 15 пентаацетильное производное галактозамина 14 (10 г, 25,64 ммоль) сушили посредством двукратного выпаривания из толуола. Полученное белое стекло обрабатывали TMSTf(5,18 мл, 28,6 ммоль) в DCE (223 мл) и перемешивали при 60°С в течение 16 часов. Реакционную смесь охлаждали до 0°С, гасили TEA (2,6 мл), разбавляли CHCl3 (300 мл) и дважды промывали раствором NaHCO3 и солевым раствором. Отделенный органический раствор обрабатывали MgSO4, концентрировали и сушили в вакууме. Неочищенное производное оксазолина использовали без дополнительной очистки. Выход: 8,14 г (96%). МС: 368,1 [М+K]+; 352,2 [M+Na]+; 330,2 [M+1]+.
К перемешиваемой смеси производного оксазолина (5,28 г, 16 ммоль), т-бутил-12-гидрокси-4,7,10-триоксадодеканоата (5,12 г, 18,4 ммоль) и CaSO4 (20 г) в ДХМ (270 мл) добавляли по каплям SnCl4 (0,84 мл, 0,84 ммоль). Раствор перемешивали в течение 16 часов, фильтровали, разбавляли CHCl3 (250 мл), дважды промывали раствором NaHCO3 и солевым раствором. Продукт сушили с помощью MgSO4 и концентрировали. Неочищенный продукт очищали на колонке, элюент: градиент МеОН (0-7%) в этилацетате. Выход: 4,83 г (50%). МС: 630,8 [M+Na]+; 625,5 [N+NH4]+; 608,4 [M+1]+; 552,6 [M-t-Bu+1]+; 330,2 [продукт дегликозилирования]+.
Для получения NAG(OAc)3-L3-OH 16 трет-бутиловый эфир 15 (1,99 г, 3,27 ммоль) перемешивали в чистой муравьиной кислоте (54 мл) в течение 16 часов и все летучие вещества удаляли в вакууме с последующими тремя выпариваниями толуола. Продукт сушили с помощью масляного вакуумного насоса в течение 2 часов и использовали без дополнительной очистки. Выход: 1,77 г (98%). МС: 330,2 [продукт дегликозилирования]+; 590,4 [М+K]+; 574,6 [M+Na]+; 569,6 [M+NH4]+; 552,6 [M+1]+.
Для получения NAG(R1,R2,R3)-L3-OH 17 (R1=R3=OTBDMS, R2=OH) продукт 16 O-деацетилировали, обрабатывали TBDMSC1, как в получении соединения 8b, и очищали на колонке, элюент: 3% МеОН, 0,5% АсОН в CHCl3. Выход: 18%. МС: 1228,7 [М+1]+, 796,7 [продукт дегликозилирования]+.
Все пять полученных кислот 6, 10a,b, 13, 17 превращали в NHS-эфиры 18а-е в ходе реакции с NHS и DCC (NHS, DCC, ДХМ, 10 часов).
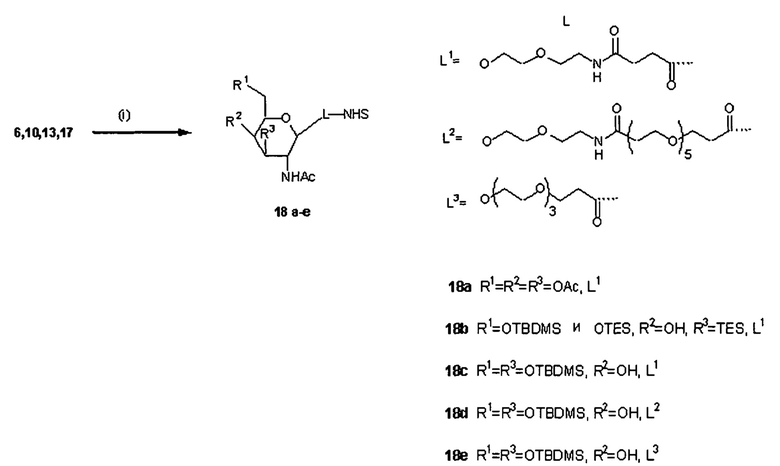
Условия: (i) NHS, DCC, DXCM, 10 часов.
Для получения NAG-L-NHS 18а-е использовали способ, описанный для соединения 18c, ниже. Для получения продукта 18c ледяной раствор 10b (614 мг, 0,964 ммоль) и NHS (122 мг, 1,061 ммоль) в ДХМ (15 мл) обрабатывали DCC (219 мг, 1,061 ммоль), перемешивали в течение 30 мин. на льду и 8 часов при 20°С. Реакционную смесь охлаждали до 0°С, DCU отфильтровывали, осадок концентрировали и сушили в вакууме. Неочищенный продукт растворяли в ДМФА с получением 0,05 М раствора и использовали без дополнительной очистки.
Продукты 18b-е получали, как описано для соединения 18а.
с) Получение соединения 20а-l, соединение 3а-h ацилировали NHS-эфиром гидроксил-защищенных NAG-производных 18а-е (DIEA, ДМФА, 5-10 часов) с получением 19а-l. Продукты 19а-l затем обрабатывали 5 эквивалентами бис(п-нитрофенил)карбоната ((PNP)2CO) ((PNP)2CO, диоксана или ДХМ, 40-50°С, 15-24 часов) с получением содержащих в качестве защитной группы O-ацетил-PNP-карбонатных производных 20а-l. Продукты 20а-е непосредственно использовали для модификации пептидов. Ацетильные группы и защитные группы 2PhiPr, DMCP, ММТ удаляли от аминокислоты после модификации во время последовательной обработки КДП ТФУ и Et3N.
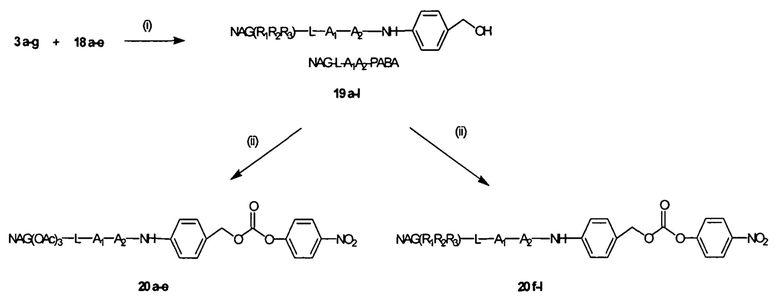
Условия: (i) DIEA, ДМФА, 5-10 часов, (ii) (PNP)2CO, диоксан или ДХМ, 25-60°С, 16-48 часов.
NAG(R1R2R3)-L-AA-PABA 19a-l.
Для получения продукта 19а (R1=R2=R3=OAc, L=L1, AA=GlyGly) раствор NAG-NHS-эфира 18а в ДМФА (0,05М) (0,282 ммоль) обрабатывали 0,1 М раствором соединения 3а (0,282 ммоль) в ДМФА и DIEA (59 мкл, 0,338 ммоль). В течение 3 часов все летучие вещества удаляли на роторном вакуумном испарителе при 40°С/с помощью масляного вакуумного насоса, растирали с Et2O и очищали на колонке, элюент: градиент EtOAc:CHCl3:МеОН=8:7:5-8:7:6. Выход: 114 мг (53%). МС: 754,4 [M+1]+.
Продукт 19b (R1=R2=R3=ОАс, L=L1, AA=Glu(2PhiPr)Gly) получали, как описано для соединения 19а, и очищали на колонке, элюент: EtOAc:CHCl3:МеОН=8:7:3. Выход: 64%. МС: 944,5 [М+1]+.
Для получения продукта 19c (R1=R2=R3=OAc, L=L1 AA=Asn(DMCP)Gly) к раствору 3c (0,43 ммоль) и DIEA (83 мкл, 0,476 ммоль) в ДМФА (2,15 мл) добавляли раствор 18а (0,43 ммоль) в ДМФА (2,15 мл). Смесь перемешивали в течение 16 часов, фильтровали и все летучие вещества удаляли на роторном вакуумном испарителе при 40°С/с помощью масляного вакуумного насоса. Неочищенный продукт растирали с Et2O и очищали на колонке, элюент: CHCl3:ацетон:МеОН (5:5:1). Выход: 242 мг (62%). МС: 915,3 [М+Na]+; 910,6 [M+NH4]+; 893,6 [M+1]+.
Продукт 19d (R1=R2=R3=OAc, L=L1 AA=PheLys(MMT)) получали, как описано для 19а, и очищали на колонке, элюент: градиент МеОН (5-6%) в CHCl3. Выход: 56%. МС: 1187,9 [М+1]+.
Для получения продукта 19е (R1=R2=R3=OAc, L=L1, AA=PheCit) к раствору соединения 3e (0,57 ммоль) и DIEA (119 мкл, 0,684 ммоль) в ДМФА (3 мл) добавляли раствор соединения 18а (0,57 ммоль) в ДМФА (3 мл). Смесь перемешивали в течение 16 часов, фильтровали и все летучие вещества удаляли на роторном вакуумном испарителе при 40°С/с помощью масляного вакуумного насоса. Неочищенный продукт осаждали в Et2O (45 мл) из смеси CHCl3:МеОН (5 мл) и использовали без дополнительной очистки. Выход: 392 мг (73%). МС: 966,8 [M+Na]+; 944,7 [M+1]+; 926,8 [M-H2O]+; 821,5 [M-PABA+1]+; 615,6 [M-NAcGal+1]+; 492,3 [M-PABA-NAcGal+1]+.
Продукт 19f (R1=R3=OTBDMS, R2=OH, L=L1, AA=AlaCit) получали, как описано для соединения 19е, и использовали без дополнительной очистки. Выход: 50%. МС: 993,2 [M+Na]+; 971,0 [M+1]+; 539,6 [продукт дегликозилирования]+.
Продукт 19g (R1=R3=OTBDMS, R2=OH L=L1, AA=ValCit) получали, как описано для соединения 19f, и использовали в следующем этапе без дополнительной очистки. Выход: 67%: 998,9 [M+1]+.
Продукт 19h (R1=R3=OTBDMS, R2=OH L=L1, AA=Glu(2PhiPr)Gly) получали, как описано для соединения 19а, и очищали на колонке, элюент: раствор NH4OH 3% и МеОН 7,5% в ДХМ. Выход: 15%. МС: 1047,2 [М+1]+, 615,7, 432,6 [продукт дегликозилирования]+.
Продукт 19i (R1=R3=OTBDMS, R2=OH, L=L1, AA=PheCit) получали, как описано для получения соединения 19е, и использовали без дополнительной очистки. Выход: 50%. МС: 1068,7 [M+Na]+; 1047,3 [M+1]+; 615,4 [продукт дегликозилирования]+; 432,5 [продукт дегликозилирования]+.
Продукт 19j (R1=OTBDMS и OTES, R2=OH, R3=OTES, L=L1, AA=PheCit) получали из 3е и 18b в виде смеси С-3 и С-6 O-TBDMS и содержащих в качестве защитной группы O-TES производных NAG, как описано для получения соединения 19е, и использовали в следующем этапе без дополнительной очистки. Выход: 76%. МС: 1047,4 [М+1]+, 615,8 [продукт дегликозилирования]+.
Продукт 19k R1=R3=OTBDMS, R2=OH, L=L2, AA=PheCit получали, как описано для соединения 19е, и использовали в следующем этапе без дополнительной очистки. Выход: 67%: 1268,2 [M+1]+; 835,9 [продукт дегликозилирования]+.
Продукт 19l R1=R3=OTBDMS, R2=OH, L=L3, AA=PheCit получали, как описано для соединения 19е, и очищали на колонке, элюент: 5% МеОН раствора в CHCl3 Выход: 60%. МС: 1064,0 [М+1]+; 632,7 [продукт дегликозилирования]+.
NAG-AA-PABC-PNP 20а-l
Для получения продукта 20а (R1=R2=R3=OAc, L=L1, AA=GlyGly) суспензию соединения 19а (100 мг, 0,132 ммоль), (PNP)2CO (202 мг, 0,663 ммоль) и DIEA (0,07 мл, 0,396 ммоль) в диоксане (5 мл) перемешивали в течение 8 часов в темноте при 40°С. Добавляли дополнительную порцию (PNP)2CO (121 мг, 0,397 ммоль) и DIEA (0,04 мл, 0,226 ммоль) и продолжали перемешивать в течение дополнительных 8 часов при 40°С. Все летучие вещества удаляли на роторном вакуумном испарителе и продукт очищали на колонке, элюент: CHCl3:EtOAc:МеОН=7:8:3. Выход: 84 мг (69%).
Для получения продукта 20b (R1=R2=R3=OAc, L=L1, AA=Glu(2PhiPr)Gly) раствор соединения 19b (160 мг, 0,169 ммоль), (PNP)2CO (258 мг, 0,847 ммоль) и DIEA (0,09 мл, 0,507 ммоль) в ДХМ (10 мл) перемешивали в темноте в течение 10 часов, концентрировали на роторном вакуумном испарителе и продукт очищали на колонке, элюент: 5-6% МеОН раствора в CHCl3. Выход: 174 мг (92%).
Для получения продукта 20c (R1=R2=R3=OAc, L=L1 AA=Asn(DMCP)Gly) раствор соединения 19c (127 мг, 0,142 ммоль), (PNP)2СО (216 мг, 0,710 ммоль) и DIEA (74 мкл, 0,426 ммоль) в ДХМ (5 мл) перемешивали в темноте в течение 16 часов, концентрировали на роторном вакуумном испарителе и продукт очищали на колонке, элюент: CHCl3:EtOAc:МеОН (7:2,2:0,8). Выход: 110,6 мг (74%). МС: 1080,9 [M+Na]+; 1058,7 [M+1]+.
Продукт 20d (R1=R2=R3=OAc, L=L1 AA=PheLys(MMT)) получали, как описано для соединения 20b, и очищали на колонке, элюент: CHCl3:EtOAc:МеОН=9:7:1 Выход: 76 мг (47%).
Для получения продукта 20е (R1=R2=R3=OAc, L=L1, AA=PheCit) раствор соединения 19e (164 мг, 0,173 ммоль), (PNP)2CO (528 мг, 1,73 ммоль) и DIEA (182 мкл, 1,04 ммоль) в диоксане (17 мл) перемешивали в темноте при 60°С в течение 16 часов и все летучие вещества удаляли на роторном вакуумном испарителе. Остаточный DIEA удаляли посредством двух последовательных выпариваний ДМФА на роторном вакуумном испарителе при 40°С/с помощью масляного вакуумного насоса и продукт очищали на колонке, элюент: CHCl3:EtOAc:МеОН (8:1,5:0,5), затем CHCl3:МеОН (7:1). Выход: 85 мг (44%). МС: 1132,0 [M+Na]+; 1110,1 [M+1]+; 780,8 [продукт дегликозилирования]+.
Продукт 20f (R1=R3=OTBDMS, R2=OH, L=L1, AA=AlaCit) получали, как описано для соединения 20е, и очищали на колонке, элюент: CHCl3:EtOAc:МеОН=9:10:1. Выход: 153 мг (36%).
Продукт 20g (R1=R3=OTBDMS, R2=OH, L=L1, AA=ValCit) получали, как описано для соединения 20е, и очищали на колонке, элюент: CHCl3:EtOAc:МеОН=16:3:1. Выход: 44%. МС: 1164,5 [М+1]+.
Продукт 20h (R1=R3=OTBDMS, R2=OH, L=L1, AA=Glu(2PhiPr)Gly) получали, как описано для соединения 20b, и очищали на колонке, элюент: CHCl3:EtOAc:МеОН=8:7:1. Выход: 77%.
Для получения продукта 20i (R1=R3=OTBDMS, R2=OH, L=L1, AA=PheCit), раствор соединения 19i (316 мг, 0,297 ммоль), (PNP)2CO (913 мг, 2,97 ммоль) и DIEA (310 мкл, 1,78 ммоль) в диоксане (7 мл) перемешивали в темноте при 65°С в течение 40 часов и все летучие вещества удаляли на роторном вакуумном испарителе. Остаточный DIEA удаляли посредством двух последовательных выпариваний ДМФА на роторном вакуумном испарителе при 40°С/с помощью масляного вакуумного насоса и продукт очищали на колонке, элюент: CHCl3:EtOAc:МеОН (8:1,5:0,5) с последующим CHCl3:МеОН (92:08). Выход: 297 мг (81%). МС: 1246,7 [M+NH4]+; 1228,7 [M+1]+; 797,6 [продукт дегликозилирования]+; 432,7 [продукт дегликозилирования]+.
Продукт 20j (R1=OTBDMS и OTES, R2=OH, R3=OTES, L=L1, AA=PheCit), продукт 20j в виде смеси С-3 и С-6 O-TBDMS и содержащие в качестве защитной группы O-TES производные NAG получали, как описано для соединения 20е, и очищали на колонке, элюент: CHCl3:EtOAc:МеОН=16:3:1 с последующим 10% МеОН в CHCl3. Выход: 50%. МС: 1212,0 [М+1]+, 480,0 [продукт дегликозилирования]+.
Продукт 20k (R1=R3=OTBDMS, R2=OH, L=L2, AA=PheCit) получали, как описано для соединения 20е, и очищали на колонке, элюент: градиент МеОН (8-10%) в CHCl3. Выход: 85%. Продукт непосредственно использовали в следующем этапе.
Для получения продукта 20l (R1=R3=OTBDMS, R2=OH, L=L3, AA-PheCit), раствор соединения 19l (316 мг, 297 ммоль), (PNP)2CO (912 мг, 3 ммоль) и DIEA (0,31 мл, 1,78 ммоль) в диоксане (8 мл) перемешивали в атмосфере аргона в темноте при 60°С в течение 48 часов. Все летучие вещества удаляли на роторном вакуумном испарителе и продукт очищали на колонке, элюент: CHCl3:EtOAc:МеОН=16:3:1. Выход: 297 мг (81%).
NAG-L-AA-PABC-PNP 21a-f (R1=R2=R3=OH), удаление защитных групп от соединения 20f-l
Образование соединения 21a-f. Для модификации чувствительных к основным условиям полиакрилатов защитные группы на NAG и дипептиде АА удаляли до момента присоединения к полимеру путем обработки соединения 20f-l смесью ТФУ:H2O=3:1(ТФУ/H2O=3:1, 5°С, 2-3 часов) с получением NAG-дипептидных маскирующих реагентов 21a-f.
Условия: ТФУ/H2O=3:1, 5°С, 2-3 часа
Для получения продукта 21а (AA=AlaCit, L=L1) соединение 20f (150 мг, 0,132 ммоль) перемешивали в ледяном растворе ТФУ:H2O=3:1 (2 мл) в течение 4 часов и добавляли по каплям к перемешиваемому Et2O (20 мл). Осадок отделяли и сушили посредством выпаривания толуола на роторном вакуумном испарителе/30°С и затем в вакууме. Выход: 112 мг (94%). МС: 907,2 [M+1]+; 704,4 [продукт дегликозилирования]+.
Для получения продукта 21b (AA=ValCit, L=L1) соединение 20g (305 мг, 0,26 ммоль) перемешивали в ледяном растворе ТФУ:H2O=3:1 (5 мл) в течение 1 часа и добавляли по каплям к перемешиваемому Et2O (45 мл). Твердый продукт отделяли и сушили посредством выпаривания толуола на роторном вакуумном испарителе/30°С и затем в вакууме. Выход: 193 мг (79%). МС: 935,8[М+1]+, 732,7 [продукт дегликозилирования]+.
Продукт 21c (AA=GluGly, L=L1) получали, как описано для соединения 21b. Выход: 55 мг (98%). МС: 865,5 [М+1]+, 662,3 [продукт дегликозилирования]+.
Продукт 21d (AA=PheCit, L=L1) получали, как описано для соединения 21b. МС: 983,7 [М+1]+, 780,9 [продукт дегликозилирования]+.
Для получения продукта 21е (AA=PheCit, L=L2) соединение 20k перемешивали ледяном растворе ТФУ:H2O=3:1 (5 мл) в течение 1,5 часов в условиях, описанных для соединения 21b. Выход: 25% (исходя из 19k). МС: 1203,9 [М+1]+, 1001,0 [продукт дегликозилирования]+.
Продукт 21f (AA=PheCit L=L3) получали из соединения 201 путем удаления защитных групп в течение 3 часов в условиях, описанных для 21b. Выход: 75%. МС: 1203,9 [М+1]+, 1001,0 [продукт дегликозилирования]+.
Получение расщепляемых протеазами PEG-маскирующих реагентов.
Аминогруппы любого из соединений Н-АА-РАВА 3b,e,g,h,j,k-m ацилировали NHS-эфиром PEG-кислоты (DIEA, ДМФА, 5-10 часов) с получением 22a-k. Гидроксильные группы в продукте 22a-k затем превращали в п-нитрофенилкарбонат ((PNP)2CO, диоксан или ТГФ, 40-60°С, 10 часов) с получением 23a-k. Для 23a,d,g защитные группы на Asn и Glu удаляли путем обработки водным раствором ТФУ (ТФУ/H2O=3:1, 5°С, 2-3 часа) с получением желаемых продуктов 24а-с. (Можно ли сохранить соответствие путем превращения 23а, d, g в 24а, d, g).
Получение PEGn-AA-PABA 22a-k.
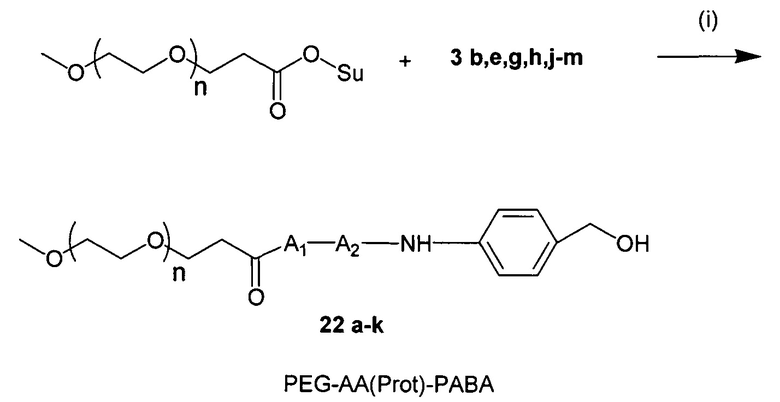
Условия: (i) DIEA, ДМФА, 5-10 часов.
Продукт 22а (n=11, AA=GluGly). А 0,1М раствора соединения 3b в ДМФА (3,5 мл, 0,35 ммоль) перемешивали в течение 10 часов с PEG11-NHS-эфиром (240 мг, 0,35 ммоль) и DIEA (0,061 мл, 0,35 ммоль). Все летучие вещества удаляли на роторном вакуумном испарителе при 40°С/с помощью масляного насоса и продукт очищали на колонке, элюент: CHCl3:МеОН:АсОН=38:2:1. Выход: 274 мг (78%) МС: 1015,6 [M+NH4]+, 998,7 [M+1]+.
Продукт 22b (n=11, AA=PheCit). К раствору соединения 3е (0,88 ммоль) и DIEA (167 мкл, 0,96 ммоль) в ДМФА (3 мл) добавляли раствор PEG11-NHS-эфира (0,80 ммоль) в ДМФА (3 мл). Смесь перемешивали в течение 16 часов, фильтровали и все летучие вещества удаляли на роторном вакуумном испарителе при 40°С/с помощью масляного вакуумного насоса. Неочищенный продукт осаждали в Et2O (45 мл) из CHCl3:МеОН (5 мл) и очищали на колонке, элюент: градиент МеОН (10-16%) в CHCl3. Выход: 420 мг (53%). МС: 1015,9 [M+H2O]+; 998,8 [М+1]+; 981,1 [M-H2O]+.
Продукт 22c (n=11, AA=ValCit). Продукт 22f получали из неочищенного соединения 3g (полученного из 300 мг, 0,5 ммоль 2g), PEG11-NHS-эфира (298 мг, 0,435 ммоль) и DIEA (0,09 мл, 0,522 ммоль), как описано для соединения 22а. После концентрирования на роторном вакуумном испарителе при 40°С/с помощью масляного насоса продукт суспендировали в смеси МеОН:ДХМ=1:1 (6 мл), обрабатывали ультразвуком, фильтровали и осаждали в Et2O (50 мл). Твердое вещество отделяли и процедуру повторяли. Остаточные растворители удаляли в вакууме. Выход: 283 мг (60%). МС: 951,5 [М+1]+.
Продукт 22d (n=11, AA=AlaAsn(DMCP)). К раствору 3h (0,56 ммоль) и DIEA (116 мкл, 0,67 ммоль) в ДМФА (3 мл) добавляли раствор PEG11-NHS-эфира (0,56 ммоль) в ДМФА (3 мл). Смесь перемешивали в течение 16 часов, фильтровали и все летучие вещества удаляли на роторном вакуумном испарителе при 40°С/с помощью масляного вакуумного насоса. Осадок растворяли в смеси CHCl3:МеОН=1:1 (5 мл) и осаждали в охлажденном (0°С) Et2O (45 мл). Твердое вещество очищали на колонке, элюент: градиент МеОН (3-14%) в ДХМ. Выход: 261 мг (49%). МС: 983,7 [M+Na]+; 979,1 [M+NH4]+; 961,8 [M+1]+; 943,9 [M-H2O+1]+.
Продукт 22е (n=11, AA=PheLys(Me2)). Продукт 22е получали, как описано для 22а. Очищали с использованием колонки для ВЭЖХ Nucleodur C-18, 250×4,6, элюент: ACN-Н2О (0,1% ТФУ), градиент 15-30%. МС: 998,1 [М+1]+. Выделенный продукт обессоливали на смоле Dowex 1×8, элюент: H2O. Выход: 40%.
Продукт 22f (n=11, AA=Leu). Продукт 22f получали, как описано для соединения 22а, и очищали на колонке, элюент: CHCl3:EtOAc:МеОН:АсОН=9:7:2:0,04. Выход: 48%. МС: 824,9 [M+NH4]+.
Продукт 22g (n=11, AA=Asn(DMCP). Неочищенный 3k (полученный из 419 мг, 0,77 ммоль 2k), Peg11NHS-эфира (200 мг, 0,292 ммоль) и DIEA (0,06 мл, 0,35 ммоль) перемешивали в ДХМ (5 мл) в течение 10 часов. Растворитель удаляли на роторном вакуумном испарителе и продукт очищали на колонке, элюент: CHCl3:EtOAc:МеОН АсОН=4,5:3,5:1:0,02. Выход: 254 мг (37%). МС: 891,1 [М+1]+.
Продукт 22h (n=11 AA=Cit). К раствору соединения 3l (0,50 ммоль) и DIEA (104 мкл, 0,60 ммоль) в ДМФА (2,5 мл) добавляли раствор PEG11-NHS-эфира (0,50 ммоль) в ДМФА (2,5 мл). Смесь перемешивали в течение 16 часов, фильтровали и все летучие вещества удаляли на роторном вакуумном испарителе при 40°С/ с помощью масляного вакуумного насоса. Осадок растворяли в смеси CHCl3:МеОН=1:1 (5 мл) и осаждали в Et2O (45 мл). Осаждали еще два раза и продукт использовали без дополнительной очистки. Выход: 340 мг (80%). МС: 869,4 [М+NH4]+; 851,9 [M+1]+.
Продукт 22i (n=23, AA=PheCit). К раствору соединения 3е (0,72 ммоль) и DIEA (130 мкл, 0,74 ммоль) в ДМФА (3 мл) добавляли раствор PEG23-NHS-эфира (0,60 ммоль) в ДМФА (3 мл). Смесь перемешивали в течение 16 часов, фильтровали и все летучие вещества удаляли на роторном вакуумном испарителе при 40°С/ с помощью масляного вакуумного насоса. Осадок растворяли в смеси CHCl3:МеОН=1:1 (5 мл) и осаждали в Et2O (45 мл). Твердый продукт очищали на колонке, элюент: градиент МеОН (7-12%) в CHCl3. Выход: 487 мг (53%). МС: 1555,2 [M+Na]+; 1544,7 [M+NH4]+; 1527,7 [M+1]+.
Продукт 22j (PEG со средней молекулярной массой MW 1000. AA=PheCit). Смесь mPEG-1000-спирта (Fluka) (0,173 г, 0,173 ммоль), N,N-дисукцинимидилкарбоната (62 мг, 0,242 ммоль) и TEA (0,101 мл, 0,726 ммоль) перемешивали в MeCN (1 мл) в течение 16 часов. Все летучие вещества удаляли на роторном вакуумном испарителе и неочищенный осадок растворяли в CHCl3 (10 мл). Органический слой промывали H2O (1 мл, рН=5), затем солевым раствором, сушили над Na2SO4 и концентрировали с получением PEG-1000-NHS-карбоната. Указанный продукт перемешивали в течение 16 часов с 3е (0,121 ммоль) и DIEA (30 мкл, 0,173 ммоль) в ДМФА (1 мл), фильтровали и все летучие вещества удаляли на роторном вакуумном испарителе при 40°С/с помощью масляного вакуумного насоса. Осадок растворяли в смеси CHCl3:МеОН=1:1 (5 мл) и осаждали в Et2O (45 мл). Осаждали еще два раза и продукт использовали без дополнительной очистки. Выход: 134 мг (79%).
Продукт 22k (n=23, AA=ValCit). К раствору соединения 3g (1,0 ммоль) и DIEA (183 мкл, 1,04 ммоль) в ДМФА (4 мл) добавляли раствор PEG23-NHS-эфира (0,87 ммоль) в ДМФА (4 мл). Смесь перемешивали в течение 16 часов, фильтровали и все летучие вещества удаляли на роторном вакуумном испарителе при 40°С/с помощью масляного вакуумного насоса. Осадок растворяли в смеси CHCl3:МеОН=1:1 (5 мл) и осаждали в Et2O (45 мл). Осаждали еще два раза и продукт использовали без дополнительной очистки. Выход: 1,0 г (77%). МС: 1496,1 [M+NH4]+; 1479,3 [M+1]+.
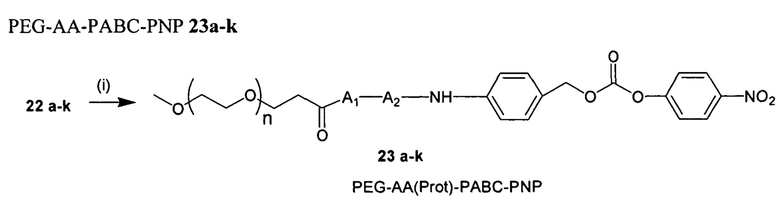
Условия: (i) (PNP)2CO, диоксан или ТГФ, 40-60°С, 10 часов.
Для получения продукта 23а (n=11, AA=Glu(2PhiPr)Gly) продукт 22а (274 мг, 0,274 ммоль) в ДХМ (15 мл) перемешивали в темноте с (PNP)2CO (418 мг, 1,372 ммоль) и DIEA (0,143 мл, 0,823 ммоль) в течение 15 часов. Растворитель удаляли на роторном вакуумном испарителе и продукт очищали на колонке, элюент: 4% МеОН, 0,2% АсОН в CHCl3. Выход: 260 мг (81%). МС: 1180,7 [М+NH4]+.
Для получения продукта 23b (n=11, AA=PheCit) раствор соединения 22b (419 мг, 0,42 ммоль), (PNP)2CO (766 мг, 2,52 ммоль) и DIEA (263 мкл, 1,52 ммоль) в диоксане (4 мл) перемешивали в темноте при 50°С в течение 15 часов и все летучие вещества удаляли на роторном вакуумном испарителе. Остаточный DIEA удаляли посредством двух последовательных выпариваний ДМФА на роторном вакуумном испарителе при 40°С/с помощью масляного вакуумного насоса и продукт очищали на колонке, элюент: CHCl3:EtOAc:МеОН (4,5:5:0,5) с последующим CHCl3:МеОН (9:1). Выход: 390 мг (80%). МС: 1181,2 [M+NH4]+, 1164,2 [M+1]+.
Для получения продукта 23c (n=11, AA=ValCit) раствор соединения 22c (273 мг, 0,287 ммоль), (PNP)2CO (874 мг, 2,88 ммоль) и DIEA (0,3 мл, 1,72 ммоль) в 1,4-диоксане (22 мл) перемешивали в темноте в течение 24 часов при 50°С. Растворитель удаляли на роторном вакуумном испарителе при 40°С/масляном насосе и продукт очищали на колонке, элюент: CHCl3:EtOAc:МеОН=16:3:1 с последующим 12-15% МеОН в CHCl3 Выход: 163 мг (51%). МС: 1116,0 [M+1]+.
Продукт 23d (n=11, AA=AlaAsn(DMCP)) получали, как описано в получение соединения 23b. Продукт очищали на колонке, элюент: CHCl3:EtOAc:МеОН (9:2:1). Выход: 77%. МС: 1144,0 [M+NH4]+; 1127,3 [M+1]+.
Продукт 23е (n=11, AA=PheLys(Me)2) получали, как описано для соединения 23a, и очищали на колонке, элюент: 10% МеОН, 0,2% АсОН в CHCl3. Выход: 63%. МС: 1163,1 [М+1]+.
Продукт 23f (n=11, AA=Leu) получали, как описано для соединения 23c, с использованием только 5 эквивалентов (PNP)2CO и 3 эквивалентов DIEA, с нагревом в течение 24 часов. Продукт очищали на колонке, элюент: градиент МеОН (7-12%) в CHCl3. Выход: 75%. МС: 972 [М+1]+.
Продукт 23g (n=11, AA=Asn(DMCP)) получали, как описано для соединения 23f, и неочищенный продукт использовали в следующем этапе без дополнительной очистки. МС: 1073,4 [М+18]+.
Для получения продукта 23h (n=11, AA=Cit), раствор 22h (340 мг, 0,40 ммоль), (PNP)2CO (608 мг, 2,00 ммоль) и DIEA (208 мкл, 1,20 ммоль) в ДХМ (4 мл) перемешивали в темноте при 30°С в течение 15 часов и все летучие вещества удаляли на роторном вакуумном испарителе. Остаточный DIEA удаляли посредством двух последовательных выпариваний ДМФА на роторном вакуумном испарителе при 40°С/с помощью масляного вакуумного насоса и продукт очищали на колонке, элюент: CHCl3:EtOAc:МеОН (7:2,5:0,5) с последующим использованием градиента МеОН (8-14%) в CHCl3. Выход: 390 мг (80%). МС: 1034,3 [M+NH4]+; 1016,9 [M+1]+.
Продукт 23i (n=23, AA=PheCit) получали, как описано в получении соединения 23b и очищали на колонке, элюент: CHCl3:EtOAc:МеОН (4,5:5:0,5) с последующим градиентом МеОН (6-12%) в CHCl3. Выход: 86%. МС: 1711,4 [M+NH4]+; 1694,4 [M+1]+.
Продукт 23j (PEG 1000K AA=PheCit) получали, как описано в получении соединения 23b и очищали на колонке, элюент: CHCl3:EtOAc:МеОН (4,5:5:0,5) с последующим использованием градиента МеОН (6-12%) в CHCl3. Выход: 72%.
Продукт 23k (n=23, AA=ValCit) получали, как описано в получении соединения 23b, и продукт очищали с помощью ВЭЖХ. Колонка: Luna (Phenomenex) 5 мкм, С-8, 100 А. Подвижная фаза: ACN-H2O (F3CO2H 0,01%), градиент ACN 30-37%, 31 мин. Выход: 530 мг(48%). МС: 1666,4 [M+Na]+; 1644,2 [M+1]+.
PEG-AA-PABC-PNP 24а-с, удаление защитных групп АА.
Условия: (i) ТФУ/H2O=3:1, 5°С, 2-3 ч.
Продукт 24а (n=11, AA=GluGly). Продукт 23а (250 мг, 0,215 ммоль) перемешивали в 3% растворе ТФУ CHCl3 (16 мл) в течение 35 мин., концентрировали на роторном вакуумном испарителе и сушили в вакууме. Выход: 224 мг (100%) (МС: 1062,6 [M+NH4]+; 1045,9 [M+1]+.
Продукт 24b (n=11, AA=AlaAsn-PABC-PNP). Соединение 23d перемешивали в течение 1,5 часов в смеси ТФУ:ДХМ (3:1) и все летучие вещества удаляли на роторном вакуумном испарителе при 20°С. Продукт очищали на колонке, элюент: градиент МеОН (6-12%) в CHCl3. Выход: 30%. МС: 1066,7 [M+Na]+, 1062,0 [M+NH4]+; 1045,2 [M+1]+.
Продукт 24c (n=11, AA=Asn). Реакционную колбу, содержащую соединение 23g (160 мг, 0,143 ммоль), охлаждали до 0°С и добавляли холодную смесь ТФУ:H2O (9:1) (12,5 мл). Смесь перемешивали в течение 1,5 часов и разбавляли холодной Н2О (50 мл). Продолжали перемешивать в течение 20 мин. при 20°С. Осадок отфильтровывали и промывали Н2О. Все летучие вещества удаляли на роторном вакуумном испарителе при 40°С и продукт очищали на колонке, элюент: CHCl3:EtOAc:МеОН:АсОН=4,5:3,5:1,2:0,02. Выход: 43 мг (30%). МС: 974,0 [М+1]+.
Пример 2. Соединение расщепляемых протеазами маскирующих агентов с аминсодержащими полимерами - формирование п-ациламидобензилкарбаматных связей.
А. Модификация мелитина поддающимися расщеплению протеазами маскирующими агентами. 1х мг пептида мелитина и 10х мг основания HEPES при концентрации 1-10 мг/мл пептида маскировали путем добавления 2-6х мг амин-реактивного п-нитрофенилкарбонатного или N-гидроксисукцинимидкарбонатного производного NAG-содержащего поддающегося расщеплению протеазами субстрата. Раствор затем инкубировали в течение по меньшей мере 1 часа при комнатной температуре (RT) до введения животным.
В. Модификация полиаминов поддающимися расщеплению протеазами маскирующими агентами. Активированные (амин-реактивные) карбонатные производные п-ациламидобензилового спирта подвергали реакции с аминогруппами амфипатических мембраноактивных полиаминов в Н2О при рН>8 с получением п-ациламидобензильного карбамата.
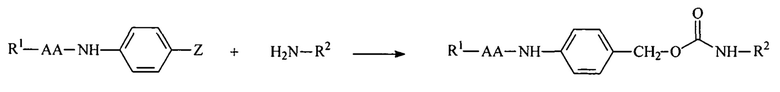
R1 содержит лиганд ASGPr (либо защищенный, либо незащищенный) или PEG,
R2 представляет собой амфипатический мембраноактивный полиамин,
АА представляет собой дипептид (либо защищенный, либо незащищенный), и
Z представляет собой амин-реактивный карбонат.
К х мг полимерф добавляли 12х мг свободного основания HEPES в изотонической глюкозе. К буферному раствору полимера добавляли от 24 до 16х мг дипептидного маскирующего агента в концентрации 200 мг/мл в ДМФА. В некоторых случаях, полимер модифицировали 2х мг дипептидным маскирующим агентом с последующим присоединением миРНК. Конъюгат полимер-миРНК затем дополнительно модифицировали 6ч до 8х мг дипептидного маскирующего агента.
Пример 3. миРНК. миРНК имели следующие последовательности:
МиРНК фактора VII
смысловая: (Chol)-5’ GfcAfaAfgGfcGfuGfcCfaAfcUfcAf(invdT) 3’ (Seq ID 1)
антисмысловая: 5’ pdTsGfaGfuUfgGfcAfcGfcCfuUfuGfcdTsdT 3’ (Seq ID 2) или
смысловая: 5’ GGAUfCfAUfCfUfCfAAGUfCfUfUfACfdTsdT 3’ (Seq ID 3) антисмысловая: 5’ GUfAAGACfUfUfGAGAUfGAUfCfCfdTsdT 3’ (Seq ID 4) или
смысловая: (NH2C6)GfuUfgGfuGfaAfuGfgAigCfuCfaGf(mvdT) 3’ (Seq ID 5)
антисмысловая: pCfsUfgAfgCfuCfcAfuUfcAfcCfaAfcdTsdT 3’ (Seq ID 6) или
смысловая: 5’ (NH2C6)uGuGfcAfaAfgGfcGfuGfcCfaAfcUfcAf(invdT) 3’ (Seq ID 23)
антисмысловая: 5’ pdTsGfaGfuUfgGfcAfcGfcCfuUfuGfcdTsdT 3’ (Seq ID 24)
миРНК фактора VII(примат)
смысловая: (chol)-5’ uuAGGfuUigGniGfaAfuGfgAigCfuCfaGf(invdT) 3’ (Seq ID 7)
антисмысловая: 5’ pCfsUfgAfgCfuCfcAfuUfcAfcCfaAfcdTsdT 3’ (Seq ID 8)
миРНК АроВ:
смысловая: (cholC6SSC6)-5’ GGAAUCuuAuAuuuGAUCcAsA 3’ (Seq ID 9)
антисмысловая: 5’ uuGGAUcAAAuAuAAGAuUCcscsU 3’ (Seq ID 10)
миРНК AhaI:
смысловая: (NH2C6)GfgAfuGfaAfgUfgGfaGfaUfuAfgUf(invdT) 3’ (Seq ID 11)
антисмысловая: pdAsCfuAfaUfcUfcCfaCfuUfcAfuCfcdTsdT 3’ (Seq ID 12)
миРНК Luc
смысловая: (chol) 5’-uAuCfuUfaCfgCfuGfaGfuAfcUfuCfgAf(invdT)-3’ (Seq ID 13)
антисмысловая: 5’-UfcGfaAfgUfaCfuCfaGfcGfuAfaGfdTsdT-3’ (Seq ID 14) или
смысловая: (NH2C6)cuuAcGcuGAGuAcuucGAdTsdT 3’ (Seq ID 15)
антисмысловая: UCGAAGuACUcAGCGuAAGdTsdT 3’ (Seq ID 16)
Eg5-KSP
смысловая: (NH2C6)UfcGfaGfaAfuCfuAfaAfcUfaAfcUf(invdT) 3’ (Seq ID 17)
антисмысловая: pAfGfuUfaGfuUfuAfgAfuUfcUfcGfadTsdT 3’ (Seq ID 18) или
смысловая: AGUuAGUUuAGAUUCUCGAdTsdT 3’ (Seq ID 19)
антисмысловая: (NH2C6)ucGAGAAucuAAAcuAAcudTsdT 3’ (Seq ID 20)
EFRP
смысловая: 5’ (NH2C6)AuAucAuGGccGAcAAGcAdTsdT 3’ (Seq ID 21)
антисмысловая: 5’ UGCUUGUCGGCcAUGAuAUdTsdT 3’ (Seq ID 22)
строчные буквы = 2’-O-СН3 замещение
s = фосфоротиоатная связь
f после нуклеотида = 2’-F-замещение
d перед нуклеотидом = 2’-дезокси
Синтез РНК осуществляли на твердофазном носителе путем стандартной фосфороамидитной реакции на приборе ДКТА Oligopilot 100 (GE Healthcare, Фрайбург, Германия) с использованием стекла с контролируемым размером пор (CPG) в качестве твердофазного носителя.
Пример 4. Введение полинуклеотидов РНКи in vivo и доставка в гепатоциты. КДП получали, как описано выше. Шести-восьми недельных мышей (линия C57BL/6 или ICR, каждое животное массой ~18-20 г) получали из компании Harlan Sprague Dawley (Индианаполис, Индиана). Мышей содержали в течение по меньшей мере 2 дней до инъекции. Животные имели неограниченный доступ к пище, представляющей собой корм для грызунов Harlan Teklad (Harlan, Мэдисон, Висконсин). КДП синтезировали, как описано в настоящей заявке. Растворы конъюгата (0,4 мл) вводили путем инфузии в хвостовую вену. Композиции являлись растворимыми и не образовывали агрегатов при физиологических условиях. Предполагается, что инъекция в другие сосуды, например, ретро-орбитальная инъекция, являются равно эффективными. Крыс линии Wistar Han массой 175-200 г получали из компании Charles River (Уилмингтон, Массачусетс). Крыс содержали в течение по меньшей мере 1 недели до инъекции. Объем инъекции для крыс, как правило, составлял 1 мл. Если иное не указано, образцы сыворотки и/или образцы печени собирали через 48 часов после введения инъекции.
Определения уровня аполипопротеина В (АроВ) в сыворотке. Мышей не кормили в течение 4 часов (16 ч для крыс) до забора сыворотки путем кровопускания из подчелюстной области. У крыс кровь собирали из яремной вены. Уровень белка АроВ в сыворотке определяли с помощью стандартных методов «сэндвич»-ИФА. Вкратце, поликлональные антитела козы к АроВ мыши и антитела к АроВ мыши (Biodesign International) использовали в качестве иммобилизированных и идентифицирующих антител, соответственно. После этого наносили конъюгированные с пероксидазой хрена (HRP) козьи анти-кроличьи антитела IgG (Sigma) для связывания комплекса АроВ/антитело. Затем измеряли поглощение в колориметрической реакции с тетраметилбензидином (ТМВ, Sigma) с помощью ридера для микропланшетов Tecan Safire2 (Австрия, Европа) при 450 нм.
Измерение активности плазматического Фактора VII (F7). Образцы плазмы животных готовили путем забора крови (9 объемов) (путем кровопускания из подчелюстной области для мышей или из яремной вены для крыс) в центрифужные микропробирки, содержащие 0,109 моль/л антикоагулянта цитрата натрия (1 объем) в соответствии со стандартными методами. Активность F7 в плазме измеряли хромогенным методом с использованием набора BIOPHEN VII (Hyphen BioMed/Aniara, Мэйсон, Огайо) в соответствии с рекомендациями производителя. Абсорбцию в ходе колориметрического анализа измеряли с помощью ридера для микропланшетов Tecan Safire2 при 405 нм.
Пример 5. Доставка миРНК в клетки печени in vivo с использованием амфипатического мембраноактивного полиакрилат-полиамина, обратимо модифицированного дмпептидными поддающимися расщеплению маскирующими агентами. Полиакрилат Ant-41658-111 в 100 мМ буфере HEPES, рН 7,5, модифицировали 0,5 масс.% активированного дисульфидного реагента сукцинимидилоксикарбонил-альфа-метил-альфа(2-пиридилдитио)толуола (SMPT) (Pierce) с получением тиол-реактивных групп для последующего присоединения миРНК. Тиол-реактивный полимер затем разбавляли до концентрации 5 мг/мл в 60 мг/мл основания HEPES. К указанному раствору добавляли 10 мг/мл различных описанных расщепляемых ферментами маскирующих реагентов. Указанное количество соответствовало молярному отношению, составляющему 1 полимерный амин к 2 маскирующим реагентам. Для реакции модификации полимера предпочтительное молярное отношение полимерных аминов к маскирующим реагентам составляет от 1:1 до 1:5. Более предпочтительное отношение составляет от 1:2 до 1:4. Более предпочтительное отношение составляет 1:2. Через 1 час миРНК эндогенного фактора VII грызунов (от 0,1 до 0,2 эквивалентов по массе по отношению к полимеру), содержащую ацетат-защищенный тиол, добавляли к раствору полимера. После инкубации в течение ночи конъюгаты дополнительно модифицировали путем добавления N-ацетилгалактозаминового производного малеинового ангидрида (NAG-CDM; Таблица 5). NAG-CDM добавляли до достижения концентрации 25 мг/мл и инкубировали в течение от 30 минут до 4 часов.
Для модификации NAG-CDM-полимера NAG-CDM лиофилизировали из 0,1% водного раствора ледяной уксусной кислоты. К высушенному NAG-CDM добавляли раствор полимера. После полного растворения ангидрида полученный раствор инкубировали в течение, по меньшей мере, 30 мин. при комнатной температуре до введения животному. Реакция NAG-CDM с полимером приводила к получению:
где R представляет собой полимер, и R1 содержит лиганд ASGPr (например, N-ацетилгалактозамин).
Как показано в Таблице 5, экспрессия фактора VII снижалась на 49-85% у животных, которых лечили маскированными дипептидными агентами КДП.
Пример 6. Доставка миРНК in vivo с использованием NAG/PEG-AA-п-нитрофенилкарбаматных поли(акрилат)КДП.
A) Модификации PEG плюс NAG. Полиакрилат Ant-41658-111 в 100 мМ буфере HEPES, рН 7,5, модифицировали 0,5 масс.% активированным дисульфидным реагентом сукцинимидилоксикарбонил-альфа-метил-альфа(2-пиридилдитио)толуолом (SMPT) от компании Pierce. Тиол-реактивный полимер разводили до концентрации 5 мг/мл в 60 мг/мл основания HEPES. К указанному раствору добавляли 10 мг/мл различных PEG-AA-п-нитрофенилкарбонатных маскирующих реагентов. Через 1 час миРНК фактора VII, содержащую ацетат-защищенный тиол, добавляли к раствору полимера с отношением полимера к миРНК в диапазоне от 5-10 до 1. После инкубации в течение ночи добавляли NAG-AA-п-нитрофенилкарбонатные маскирующие реагенты до концентрации 40 мг/мл. После инкубации в течение, по меньшей мере, 30 минут, но не более 4 часов, КДП инъецировали в хвостовую вену мышей линии ICR массой 20 г. Через 48 часов после инъекции собирали образец сыворотки и измеряли уровень фактора VII.
B) Модификации только NAG. Полиакрилат ANT-41658 111 в 100 мМ буфере HEPES, рН 7,5, модифицировали 0,5 масс.% активированного дисульфидного реагента сукцинимидилоксикарбонил-альфа-метил-альфа(2-пиридилдитио)толуола (SMPT) от компании Pierce. Тиол-реактивный полимер разводили до 5 мг/мл в 60 мг/мл основания HEPES. миРНК фактора VII, содержащую ацетат-защищенный тиол, добавляли к раствору полимера в отношении полимера к миРНК в диапазоне от 5-10 до 1. После инкубации в течение ночи добавляли NAG-AA-п-нитрофенил-карбонатные маскирующие реагенты до концентрации 50 мг/мл. После инкубации в течение, по меньшей мере, 30 минут, но не более 4 часов, конъюгированную с полимером миРНК инъецировали в хвостовую вену мышей линии ICR массой 20 г. Через 48 часов после инъекции, собирали образцы сыворотки и измеряли уровень фактора VII.
Пример 7. Доставка миРНК in vivo с использованием поли(виниловый эфир)NAG/PEG-AA-п-нитрофенилкарбаматных КДП. Амфипатический мембраноактивный полиамин поли(виниловый эфир) DW1360 модифицировали 10× масс. эквивалентами дипептидных поддающихся расщеплению маскирующих агентов, как описано выше для полиакрилата Ant-41658-111, за исключением того, что маскирующие агенты сохраняли защитные группы во время модификации полимера. После модификации полимера защитные группы аминокислот удаляли с помощью ТФУ и NAG-ацетатные защитные группы удаляли путем инкубации в присутствии раствора, содержащего 30 объемных % триэтиламина, 50% метанола и 20% воды. Раствор для удаления ацетатных защитных групп удаляли посредством ротационного выпаривания. Маскированный полимер вводили мышам совместно с конъюгатом холестерин-миРНК АроВ.
Пример 8. in vivo снижение уровня эндогенного АроВ после доставки миРНК АроВ с помощью пептида доставки мелитина у мышей; поддающиеся ферментативному расщеплению маскирующие агенты. Мелитин обратимо модифицировали указанным количеством расщепляемых ферментами маскирующих агентов, как описано выше. Затем вводили 200-300 мкг маскированного мелитина совместно с 50-100 мкг конъюгата миРНК АроВ - холестерин. Эффект на уровень АроВ определяли, как описано выше. Модифицированный поддающимся расщеплению пептидазой дипептид-амидобензилкарбаматом мелитин являлся эффективным пептидом для доставки миРНК. Предпочтительным является применение пептида мелитина в D-форме в комбинации с поддающимися ферментативному расщеплению маскирующими агентами. Несмотря на то, что требовалось большее количество пептида для соответствующего уровня при подавлении экспрессии гена-мишени, терапевтический индекс был либо не изменен, либо улучшен (по сравнению с тем же пептидом, маскированным CDM-NAG) из-за более стабильного маскирования пептида.
Пример 9. Обеспечение опухолевой нацеливания с помощью расщепляемых протеазами КДП.
А) Измерение снижения экспрессии гена-мишени. Для всех исследований, представленных ниже, миРНК специфична к транскрипту гена Aha1, гена-мишени. миРНК усовершенствованного мутанта зеленого флуоресцентного белка (EGFP) использовали в качестве нецелевого контроля. миРНК Aha1 являлась комплементарной мотиву последовательности Aha1, для которого наблюдается 100% гомология между геном человека и мыши. Таким образом, доставка миРНК Aha1 как в клетки хозяина, так и в опухолевые клетки человеческого ксенотрансплантата, приводила к расщеплению и деградации мРНК. С использованием мотивов последовательности, различающихся в генах Aha1 мыши и человека, конструировали ПЦР-праймеры, которые обеспечивали количественное измерение уровня мРНК как человеческого, так и мышиного Aha1 в образцах ткани, которые содержали смешанную популяцию типов клеток. Через 24, 48 или 72 часа после доставки миРНК опухоли собирали вместе с небольшой частью прилежащей здоровой ткани печени мыши и обрабатывали на приборе Tri-Reagent (Invitrogen) для выделения общей РНК. Затем измеряли уровень мРНК человеческого и мышиного Aha1 с помощью количественных ПЦР-анализов с использованием гена человеческого Сус-А и мышиного β-актина в качестве внутреннего контроля. Уровень мРНК Aha1 у ложно-инъецированных животных или у мышей, которые получали нецелевую контрольную миРНК GFP, принимали за 100%. Результаты выражены в виде процента уровня мРНК Aha1 по отношению к контролю и показаны в Таблицах, ниже.
B) Модель ортотопической гепатоклеточной карциномы (НСС) у мышей. Клетки гепатоклеточной карциномы HegG2, Нер3В или HuH7 ко-трансфецировали 2 векторами экспрессии, pMIR85, вектором человеческой плацентарной щелочной фосфатаза (SEAP) и pMIR3, вектором гена устойчивости к неомицину/канамицину, для получения клеточных линий со стабильной экспрессией SEAP. Клетки растили на среде DMEM с добавлением 10% ЭБС и 300 мкг/мл G418), собирали, подсчитывали и смешивали с матригелем (BD Biosciences) (50% по объему). Бестимусных («nude») мышей или мышей линии Scid анестезировали ~3% изофлураном и помещали в горизонтальное положение вверх грудиной. Делали небольшой (1-2 см) разрез брюшной стенки по средней линии непосредственно под мечевидным отростком. С использованием влажного ватного тампона левую долю печени аккуратно выводили на поверхность тела. Указанную левую долю печени осторожно оттягивали и в ее середину вводили иглу для шприца. Иглу для шприца с концевым срезом примерно 0,5 см вводили непосредственно под капсулу печени. 10 мкл смеси клетки/матригель, содержащей 100,000 клеток, инъецировали в печень с использованием шприцевого насоса. Иглу оставляли в печени на некоторое время (15-20 секунд) для уверенности в завершении инъекции. Клетки SEAP-HepG2 вводили бестимусным «голым» мышам. Клетки SEAP-Нер3В и SEAP-HuH7 вводили мышам линии Scid. Затем шприц удаляли из печени и ватный тампон помещали на место инъекции для предотвращения вымывания клеток или кровотечения. Смесь матригель/клетки образовывала массу, которая являлась видимой и не исчезала после удаления иглы. Печеночную долю аккуратно помещали обратно в брюшную полость и брюшную стенку закрывали. Образцы сыворотки собирали через неделю после имплантации опухоли и подвергали анализу на SEAP для отслеживания опухолевого роста. Для большинства исследований опухоль мышей использовали через 4-5 недель после имплантации, когда размер указанной опухоли предположительно составлял примерно 4-8 мм на основании значений уровня SEAP.
C) Модель колоректального метастатического рака. Клетки НТ29 растили на среде МсСоу 5a с добавлением 10% ЭБС, собирали, подсчитывали и смешивали с матригелем (BD Biosciences) (50% по объему). Бестимусных («nude») мышей анестезировали ~3% изолураном и помещали в горизонтальное положение вверх грудиной. Делали небольшой (1-2 см) разрез брюшной стенки по средней линии непосредственно под мечевидным отростком. С использованием влажного ватного тампона левую долю печени аккуратно выводили на поверхность тела. Указанную левую долю печени осторожно оттягивали и в ее середину вводили иглу для шприца. Иглу для шприца с концевым срезом примерно 0,5 см вводили непосредственно под капсулу печени. 5 мкл смеси клетки/матригель, содержащей ~40,000 клеток, инъецировали в печень с использованием шприцевого насоса. Иглу оставляли в печени на некоторое время (15-20 секунд) для уверенности в завершении инъекции. Затем шприц удаляли из печени и ватный тампон помещали на место инъекции для предотвращения выхода клеток или кровотечения. Смесь матригель/клетки образовывала массу, которая являлась видимой и не исчезала после удаления иглы. Печеночная доля аккуратно помещали обратно в брюшную полость и брюшную стенку закрывали. Опухоль у мышей использовали через 4-5 недель после имплантации.
Пример 10. Снижение экспрессии гена-мишени in vivo в модели ортотопической гепатоклеточной карциномы (НСС) HepG2-SEAP после введения PEG24-Val-Cit КДП. (2011062805) Ant-129-1 полимерные КДП модифицировали (маскировали) либо с использованием 18× избытка (по массе) PEG24-Phe-Cit-маскирующего агента (или PEG24-Val-Cit-маскирующего агента), либо с использованием 7× PEG550-CDM, как описано выше. Aha1-миРНК (RD-09070) или GFP-миРНК (RD-05814; нецелевой контроль) присоединяли к полимеру, как описано выше (массовое отношение 4:1). КДП не очищали с помощью гель-фильтрации до осуществления доставки и не добавляли нацеливающий лиганд. 320 мкг (масса полимера) конъюгата КДП в 200 мкл изотонической глюкозы на животное вводили путем инъекции в хвостовую вену (n=3 на группу). Через 24 часа животным вводили вторую инъекцию, содержащую 320 мкг (масса полимера) конъюгата КДП в 200 мкл изотонической глюкозы. Через 48 часов после второй инъекции собирали образцы сыворотки для оценки токсичности путем измерения уровня ферментов печени (ALT и AST) и азота мочевины крови (BUN), затем собирали образцы ткани и проводили и количественный ПЦР-анализ.
Использование PEG24-Val-Cit-Ant-129-1-миРНК-КДП для доставки миРНК Aha1 приводило к снижению экспрессии гена Aha1 в опухолевых клетках человека на 46% (Таблица 9). В отличие от снижения экспрессии Aha1 у человека, у мышей экспрессия Aha1 снижалась на 70% в ответ на ведение PEG24-Val-Cit-Ant-129-1-миРНК-КДП (Таблица 1). По сравнению с подобными КДП, полученными с использованием маскирующих агентов на основе дизамещенного малеинового ангидрида (PEG550-CDM), эндогенный уровень Aha1 в гепатоцитах снижался. По результатам измерения уровня ALT, AST и BUN PEG24-Val-Cit-КДП хорошо переносились и не вызывали токсичности (Таблица).
Пример 11. Подавлении экспрессии гена-мишени с использованием нацеленных на биспецифические антитела (bsAb) КДП (2011090701). Полимер Ant-129-1 модифицировали 5× Dig-PheCit (Dig-FCit) маскирующим агентом, как описано выше. миРНК затем присоединяли к конъюгату. Наконец, конъюгат Dig-FCit-Ant-129-1-миРНК также модифицировали 8× (по массе) PEG12-FCit. Aha1-миРНК (RD-09070) или миРНК GFP (RD-05814) присоединяли к полимеру с массовым отношением полимер:миРНК, составляющим 4:1. PEG12-FCit КДП очищали на спин-колонке Sephadex G50 для удаления несвязанных реагентов.
Создавали нацеленные на клетки биспецифические антитела (bsAb), специфичные к протеогликану гепарансульфату Glypican-3 (GPC3), протеогликану гепарансульфату клеточной поверхности, который, как известно, экспрессируется на высоком уровне в клетках HepG2-SEAP, и дигоксигенину (Dig). В качестве контроля создавали биспецифические антитела, специфичные к белку CD33 (маркеру гематопоэтическихз стволовых клеток костно-мозгового происхождения) и Dig. CD33 не экспрессируется в клетках HepG2-SEAP. BsAbs объединяли с модифицированными КДП с массовым отношением, составляющим 1,25:1, с получением рассчитанного молярного отношения 1:1. Комплексы образовывали в ФБР, по меньшей мере, за 30 минут до их доставки.
КДП вводили мышам, несущим опухоль HepG2-SEAP, вместе с bsAb-нацеленным агентом или без него. Каждое животное (n=3 на группу) получало одну дозу 250 мкг (по массе полимера) КДП. КДП вводили мышам в хвостовую вену в 200 мкл стерильного ФБР. Образцы сыворотки и ткани собирали через 48 часов и анализировали, как описано выше. Как показано в Таблице 11, введение одной дозы bsAb-направленного КДП (250 мкг полимера, 62,5 мкг миРНК) приводило к снижению экспрессии гена-мишени на 21-32%.
Пример 12. подавление экспрессии гена-мишени с использованием направленных с помощью биспецифического антитела (bsAb) КДП. КДП получали, как описано выше, за исключением того, что a) PEG24-FCit использовали вместо PEG12-FCit и b) Dig-PEG12-NHS использовали для присоединения Dig к полимеру. PEG24-FCit-КДП агрегировали менее интенсивно по сравнению с PEG12-FCit-КДП и являлись меньшими по размеру и более гомогенными. Помимо того, что Dig-PEG12-NHS представлял собой нелабильную связь, он также содержал более длинный PEG. КДП представляли собой комплексы с bsAb, их вводили животным, как описано выше. Образцы сыворотки и ткани собирали либо через 24, либо через 48 часов после инъекции. Как показано в Таблице 12, введение однократной дозы КДП (250 мкг полимер, по массе) приводило к подавлению экспрессии Aha1 у человека на 46-56% через 24 часа после инъекции.
Пример 13. Обеспечение нацеливания КДП на опухолевую ткань метастазов колоректальной аденокарциномы в печени с помощью bsAb-направленных КДП. Ant-129-1-полимер модифицировали 5× молярным избытком Dig-PEG12-NHS и 8× избытком по массе PEG24-FCit. миРНК Aha1 или миРНК GFP присоединяли к модифицированному полимеру с массовьм отношением полимер:миРНК, составляющим 4:1. КДП очищали на спин-колонке Sephadex G50 для удаления несвязанных реагентов. Dig-КДП объединяли с эквимолярным количеством IGF1R-Dig-bsAb или CD33-Dig-bsAb или без bsAb в стерильном ФБР, по меньшей мере, за 30 минут до введения инъекций. Животным, содержащим опухолевые клетки НТ29 (колоректальная аденокарцинома человека; номер в АТСС НТВ-38), вводили КДП. В клетках НТ29 наблюдается повышенная экспрессия рецепторного белка инсулин-подобного фактора роста-1 (IGF1R), и они могут связывать и интернализировать биспецифические антитела к IGF1R-Dig. Животные получали (n=3) КДП (320 мкг полимера). Инъекции повторяли через 24 часа. Образцы сыворотки и ткани собирали через 48 часов после введения второй дозы. Подавление экспрессии Aha1 человека в опухолевых клетках составляло 26-38% (Таблица 13). По сравнению с CDM-КДП, FCit-КДП показали меньшее нецелевое снижение уровня Aha1 в печени (78-83% по сравнению с 24-36%). FCit-КДП также показали сниженное накопление в печени по сравнению с CDM-КДП.
Пример 14. Снижение in vivo эндогенного уровня Aha1 в опухоли печени. 400 мкг Lau41648-106 модифицировали 8× (по массе) PEG12-ValCit или 16× PEG24-PheCit. 100 мкг миРНК Aha1 или 100 мкг контрольной миРНК Eg5 присоединяли к модифицированному полимеру, как описано выше. Животным, содержащим опухолевые клетки Hep3B-SEAP, вводили КДП. Образцы сыворотки и ткани собирали через 48 часов после инъекции. Снижение человеческого Aha1 в клетках опухоли составляло 26-38%.
Пример 15. Циркуляция in vivo и тканевая нацеливание маскированного полимера. Полиакрилат Lau24AB (100 мкг) обрабатывали реагентом 125I-Bolton-Hunter (BH) (50 мкКи) в 50 мМ буфере HEPES, рН 8,0, в течение 1 часа при комнатной температуре. Меченный полимер очищали на спин-колонке, содержащей 2 мл Sephadex QEA в воде. Раствор меченного полимера хранили при 4°С. К немеченому полимеру добавляли 125I-меченный полимер для инъекции примерно 1 мг полимера, содержащей 0,2 мкКи на крысу массой 200 г. Смесь меченного и немеченого полимера (рассчитано для ~3,5 животных) модифицировали, как описано выше, PEG24-FCit или PEG-CDM (2 мг/мл полиакрилата, 14 мг/мл PEG-CDM-реагента, 14 мг/мл NAG-PEG-CDM-реагента, 16 мг/мл PEG24-FCit-реагента). Время инкубации составляло 1 час. Реакционную смесь затем разбавляли изотонической глюкозой с получением дозы инъекции на животное в объеме 1 мл. Инъекцию вводили 3 животным на группу. В определенное время у животных отбирали образцы крови (0,1-0,2 мл). Количество полимера, присутствующего в образцах, определяли путем подсчета на счетчике гамма-излучения. Как показано на ФИГ.4, полимер, модифицированный поддающимся расщеплению протеазами маскирующим агентом, медленнее вымывался из сыворотки по сравнению с полимером, маскированным рН-лабильным маскирующим агентом на основе малеинового ангидрида. Повышение времени циркуляции в кровотоке является благоприятным для нацеливания на ткань, отличную от ткани печени.
Пример 16. Синтез амфипатического мембраноактивного полимера.
А) Поли(винилакрилат)
i) RAFT-сополимеризация N-Boc-этилэтоксиакрилата и пропилметакрилата:
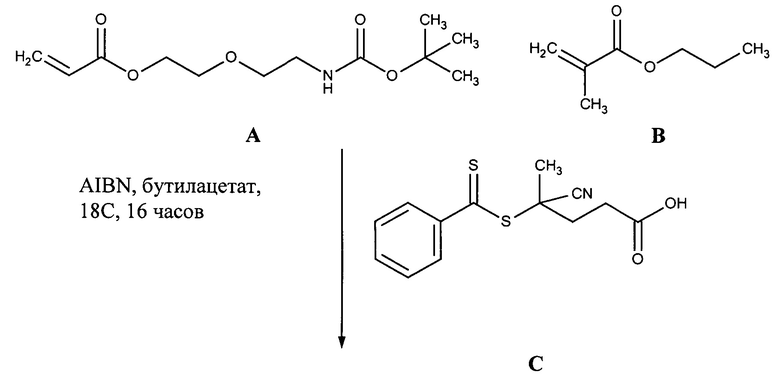
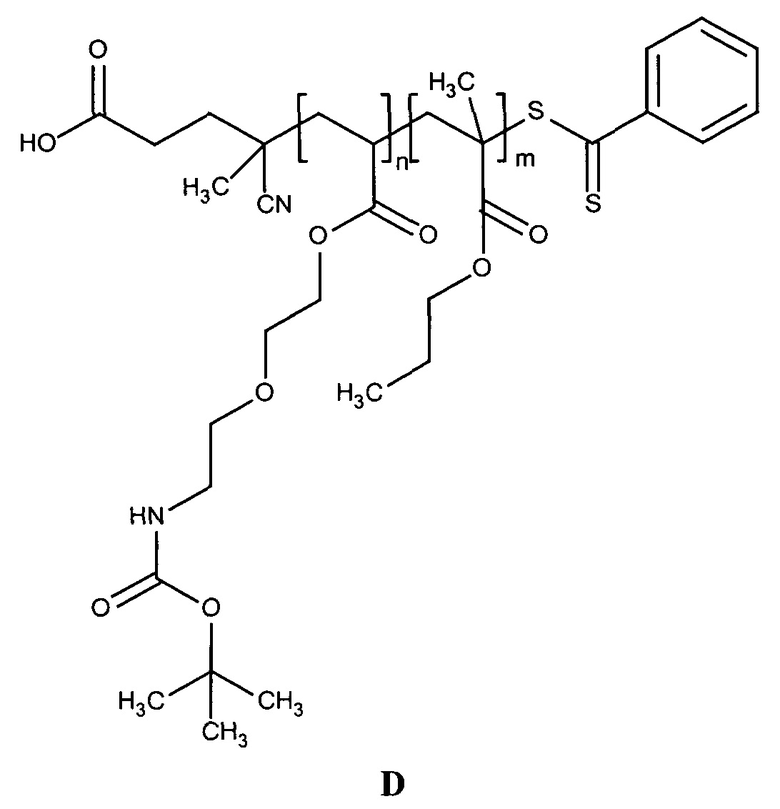
где: А представляет собой boc-защищенный этилэтоксиаминоакрилат
В представляет собой пропилметакрилат
С представляет собой RAFT-агент СРСРА (4-циано-4-(фенилкарбонотиоилтио)пентановую кислоту),
n и m представляют собой целые числа.
Удаление boc-защитной группы после синтеза приводит к получению мономеров амина.
Для других мембраноактивных полимеров А также может представлять собой защищенный этил-, пропил- или бутиламинакрилат. В может представлять собой более гидрофобный акрилат (содержащий 10-24 атома углерода, показан С18-акрилат), менее гидрофобный акрилат (содержащий 1-6 атомов углерода, показан С4-акрилат) или комбинацию мене и более гиброфобных акрилатов.
Сополимеры, состоящие из аминоакрилата/С3-метакрилата, синтезировали, как следует далее. Мономеры и RAFT-агент взвешивали и переносили в бутилацетат в указанном отношении. Добавляли AIBN (азобисизобутиронитрил) и через реакционную смесь барботировали азот при комнатной температуре в течение 1 часа. Реакционную смесь затем помещали на масляную баню при 80°С на 15 часов. Полимер затем осаждали с помощью гексана и далее частично осаждали с использованием системы растворителей ДХМ/гексан (см. ниже). Полимер затем сушили при пониженном давлении. Защитную группу полимера удаляли с помощью 7 мл 2М HCl в уксусной кислоте в течение 30 мин. при комнатной температуре. Через 30 мин. к реакционной смеси добавляли 15 мл воды и смесь переносили в диализную трубку MWCO с размером пор 3,5 кДа. Полимер подвергали диализу против NaCl в течение ночи и затем в течение следующего дня против dH2O. Затем воду удаляли посредством лиофилизирования и полимер растворяли в dH2O.
Синтез мономера для (Ant 41658-111). 2,2’-Азобис(2-метилпропионитрил) (AIBN, радикальный инициатор), 4-циано-4-(фенилкарбонотиоилтио)пентановая кислота (СРСРА, RAFT-агент) и бутилацетат были получены от компании Sigma Aldrich. Мономер пропилметакрилата (Alfa Aesar) фильтровали для удаления ингибиторов.
В круглодонной колбе на 2 л, снабженной магнитной мешалкой, растворяли 2-(2-аминоэтокси)этанол (21,1 г, 202,9 ммоль, Sigma Aldrich) в 350 мл дихлорметана. В отдельной колбе на 1 л ВОС-ангидрид (36,6 г, 169,1 ммоль) растворяли в 660 мл дихлорметана. Круглодонную колбу на 2 л снабжали капельной воронкой и в указанную колбу добавляли раствор ВОС-ангидрида в течение 6 часов. Реакционной смеси позволяли перемешиваться в течение ночи. В разделительной воронке на 2 л продукт промывали 10% лимонной кислотой, 10% K2CO3, насыщенным раствором NaHCO3 и насыщенным раствором NaCl (по 300 мл). Продукт, ВОС защищенный 2-(2-аминоэтокси)этанол, сушили над Na2SO4, фильтровали посредством гравитационной фильтрации, и ДХМ выпаривали с использованием ротационного выпаривания и высокого вакуума.
В Круглодонную колбу на 500 мл, снабженную магнитной мешалкой и наполненную аргоном, добавляли ВОС-защищенный 2-(2-аминоэтокси)этанол (27,836 г, 135,8 ммоль) с последующим добавлением 240 мл безводного дихлорметана. Добавляли диизопропилэтиламин (35,5 мл, 203,7 ммоль) и систему помещали на баню сухой лед/ацетон. Акрилоилхлорид (12,1 мл, 149,4 ммоль) разбавляли с помощью 10 мл дихлорметана и добавляли по каплям к системе, наполненной аргоном. Систему хранили в атмосфере аргона, позволяли ей достигать комнатной температуры и перемешивали в течение ночи. Продукт промывали dH2O, 10% лимонной кислотой, 10% K2CO3, насыщенным раствором NaHCO3 и насыщенным раствором NaCl (по 100 мл). Продукт, ВОС-аминоэтилэтоксиакрилат (ВАЕЕА), сушили над Na2SO4, фильтровали посредством гравитационной фильтрации и ДХМ выпаривали посредством ротационного выпаривания. Продукт очищали с помощью колоночной хроматографии на силикагеле (29 см) с использованием колонки диаметром 7,5 см. Используемая система растворителей представляла собой 30% этилацетат в гексане. Rf: 0,30. Фракции собирали и растворитель удаляли с использованием ротационного выпаривания и высокого вакуума. ВАЕЕА получали с выходом 74%. ВАЕЕА хранили в морозильной камере.
Полимер Ant-41658-111: Получали растворы AIBN (1,00 мг/мл) и RAFT-агента (4-циано-4(фенилкарбонотиоилтио)пентановой кислоты (СРСРА), 10,0 мг/мл) в бутилацетате. Соотношение мономеров в исходной смеси составляло 75 ВАЕЕА: 25 пропилметакрилат (CAS:2210-28-8) и 0,108 RAFT-агента СРСРА и 0,016 катализатора AIBN (общее количество молей 0,00562).
В стеклянную пробирку на 20 мл с магнитной мешалкой добавляли ВАЕЕА (1,09 г, 4,21 ммоль) (А), пропилметакрилат (,180 г, 1,41 ммоль) (В), раствор СРСРА (,170 мл,, 00609 ммоль) (С), раствор AIBN (,150 мл,, 000915 ммоль) и бутилацетат (5,68 мл). Пробирку герметизировали с помощью резиновой пробки и через раствор барботировали азот в течение 1 часа с помощью длинной иглы для шприца и использованием второй короткой иглы для шприца в качестве выходного отверстия. Иглы для шприца удаляли и систему нагревали до 80°С в течение 15 часов с использованием масляной бани. Раствору позволяли охлаждаться до комнатной температуры и переносили его в центрифужную пробирку н 50 мл, затем к указанному раствору добавляли гексан (35 мл). Раствора центрифугировали в течение 2 мин. При скорости 4,400 об./мин. Супернатант осторожно декантировали и нижний слой (твердый или геле-подобный) промывали гексаном. Указанный нижний слой затем заново растворяли в ДХМ (7 мл), осаждали в гексане (35 мл) и центрифугировали еще раз. Супернатант декантировали и нижний слой промывали гексаном, затем полимер сушили при пониженном давлении в течение нескольких часов. Молекулярную массу определяли с помощью MALS: 73,000 (PDI 1,7); состав полимера, определенный с помощью Н1 ЯМР: 69:31 амин:алкил.
Преципитация фракций. Высушенный, осажденный продукт растворяли в ДХМ (100 мг/мл). Гексан добавляли до достижения температуры помутнения (~20 мл). Полученный мутный раствор центрифугировали. Нижний слой (вязкая жидкость, содержащая ~60% полимера) экстрагировали и полностью осаждали в гексане. Оставшийся верхний слой раствора также полностью осеждали путем добавления дополнительного гексана. Обе фракции центрифугировали, затем полимер выделяли и сушили в вакууме. Фракция 1: Mw 87,000 (PDI 1,5); Фракция 2: Mw 52,000 (PDI 1,5-1,6).
MALS-анализ. Приблизительно 10 мг полимера растворяли в 0,5 мл 89,8% дихлорметана, 10% тетрагидрофуране, 0,2% триэтиламине. Молекулярную массу и полидисперсность (PDI) измеряли с использованием многоугольного детектора светорассеяния Wyatt Helos II, присоединенного к установке для ВЭЖХ Shimadzu Prominence, использующего колонку Jordi (5 мкм, 7,8×300 Mixed Bed LS DVB). Неочищенный полимер: MW: 73,000 (PDI 1,7), Фракция 1: MW 87,000 (PDI: 1,5), Фракция 2: MW 52,000 (PDI 1,5-1,6)
Очищенный ВОС-защищенный полимер подвергали реакции с 2М HCl в уксусной кислоте (7 мл) в течение 0,5 часов для удаления защитных групп ВОС и получения аминов. К реакционной смеси добавляли 15 мл dH2O, раствор переносили на целлюлозную трубку, пропускающую соединения с молекулярной массой 3500, подвергали диализу против раствора высокой соли в течение 24 часов, затем против dH2O в течение 18 часов. Содержимое лиофилизировали, затем растворяли в DI H2O в концентрации 20 мг/мл. Раствор полимера хранили при 2-8°С.
ii) Полимер Lau24B получали, как описано выше, за исключением того, что отношение исходной полимерной смеси составляло 72,5 ВАЕЕА: 27,5 пропил метакрилат.
iii) Ant-129-1 получали по существу, как описано выше, за исключением того, что использовали следующие мономеры:
 ,
,
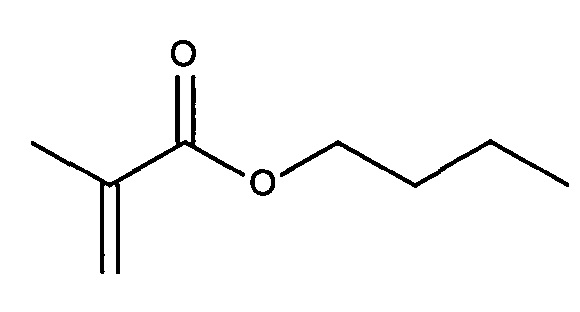 ,
,
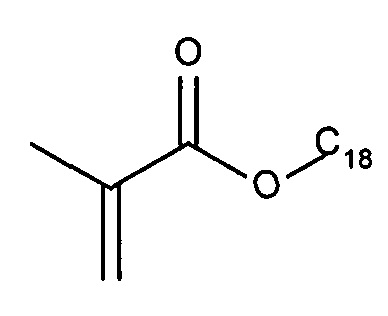 , и
, и
 .
.
Для получения N-Boc-аминопропилакрилата (ВАРА) в круглодонную колбу на 500 мл, снабженную магнитной мешалкой и наполенную аргоном, добавляли 3-(ВОС-амино)1-пропанол (TCI) (135,8 ммоль) с последующим добавлением 240 мл безводного дихлорметана. Добавляли диизопропилэтиламин (203,7 ммоль) и систему помещали на баню сухой лед/ацетон. Акрилоилхлорид (149,4 ммоль) разбавляли с помощью 10 мл дихлорметана и по каплям добавляли в систему, наполненую аргоном. Указанную систему хранили в атмосфере аргона, позволяли ей достигать комнатной температуры и перемешивали в течение ночи. Продукт промывали dH2O, 10% лимонной кислотой, 10% K2CO3, насыщенным NaHCO3 и насыщенным NaCl (по 100 мл). Продукт, ВОС-аминопропилакрилат (ВАРА), сушили над Na2SO4, фильтровали путем гравитационной фильтрации и ДХМ выпаривали посредством ротационного выпаривания. Продукт очищали путем колоночной хроматографии на силикагеле (29 см) с использованием колонки диаметром 7,5 см. Используемая система растворителя представляла собой 30% этилацетат в гексане. Rf: 0,30. Фракции собирали и растворитель удаляли посредством ротационного выпаривания и с использованием высокого вакуума. ВАРА получали с выходом 74%. ВАРА хранили в морозильной камере.
iv) Сополимеризация с нерегулярным чередованием мономеров N-Boc-этилэтоксиакрилата и пропилметакрилата. Сополимеры, состоящие из аминакрилата/Cn-метакрилата синтезировали, как описано ниже. Мономеры взвешивали и переносили в диоксан в указанных отношениях. Добавляли AIBN (азобисизобутиронитрил) и через реакционную смесь барботировали азот при комнатной температуре в течение 1 часа. Реакционную смесь затем помещали на масляную баню при 60°С на 3 часа. Полимер затем сушили при пониженном давлении. Полимер очищали с помощью GPC, после чего удаляли защитные группы от фракций полимера с помощью 7 мл 2М HCl в уксусной кислоте в течение 30 мин. при комнатной температуре. Через 30 мин. к указанной реакционной смеси добавляли 15 мл воды и смесь переносили в диализную трубку MWCO с размером пор 3,5 кДа. Полимер подвергали диализу в течение ночи против NaCl и затем в течение следующего дня против dH2O. Затем удаляли воду посредством лиофилизирования и полимер растворяли в dH2O.
Полимер Lau41648-106. Молярное соотношение мономеров исходной смеси составляло 80 ВАЕЕА: 20 пропилметакрилат (CAS:2210-28-8) и 3% катализатора AIBN на основе общего количества молей мономеров. В стеклянную пробирку на 50 мл с магнитной мешалкой добавляли ВАЕЕА (6,53 г, 25,2 ммоль) (А), пропилметакрилат (0,808 г, 6,3 ммоль) (В), AIBN (0,155 г, 0,945 ммоль) и диоксан (34,5 мл). Соединения А и В получали, как описано выше в Примере 16Ai. Реакцию проводили три раза. Через каждый раствор барботировали азот в течение 1 часа с помощью длинной пипетки. Пипетку убирали и каждую пробирку осторожно герметизировали. Затем каждый раствор нагревали при 60°С в течение 3 ч на масляной бане. Каждому раствору позволяли охлаждаться до комнатной температуры и указанные растворы объединяли в круглодонной колбе. Неочищенный полимер сушили при пониженном давлении. Молекулярную массу определяли с помощью MALS: 55,000 (PDI 2,1); состав полимера, определенный с помощью Н1 ЯМР: 74:26 амин:алкил.
Фракционирование GPC. Высушенный неочищенный полимер переносили в концентрации 50 мг/мл в смесь 75% дихлорметана, 25% тетрагидрофурана и 0,2% триэтиламина. Затем полимер фракционировали на колонке Jordi Gel DVB, 104 Å - 500 мм/22 мм, со скоростью потока 5 мл/мин, и вводом 10 мл. Раннюю фракцию собирали на 15-17 минуте, и позднюю фракцию собирали на 17-19 минуте. Фракция 15-17: Mw 138,000 (PDI 1,1); Фракция 17-19: Mw 64,000 (PDI 1,2).
MALS-анализ. Приблизительно 10 мг полимера растворяли в смеси 0,5 мл 89,8% дихлорметана, 10% тетрагидрофурана, 0,2% триэтиламина. Молекулярную массу и полидисперсность (PDI) измеряли с использованием многоугольного детектора светорассеяния Wyatt Helos II, присоединенного к установке для ВЭЖХ Shimadzu Prominence, использующей колонку Jordi (5 мкм, 7,8×300 Mixed Bed LS DVB).
Неочищенный полимер: MW: 55,000 (PDI 2,1), Фракция 15-17: MW 138.000 (PDI:1,1), Фракция 17-19: MW 64,000 (PDI 1,2)
Очищенный ВОС-защищенный полимер подвергали реакции с 2М HCl в уксусной кислоте (7 мл) в течение 0,5 часов для удаления защитных групп ВОС и получения аминов. К реакционной смеси добавляли 15 мл dH2O, раствор переносили на целлюлозную трубку, пропускающую соединения с молекулярной массой 3,500, подвергали диализу против высокой соли в течение 24 часов, затем снова против dH2O в течение 18 часов. Содержимое лиофилизировали, затем растворяли в DI H2O в концентрации 20 мг/мл. Раствор полимера хранили при 2-8°С.
v) Синтез растворимых в воде, амфипатических, мембраноактивных поли(виниловый эфир)полиаминных терполимеров. Х мол.% амин-защищенного винилэфира (например, 2-винилоксиэтилфталимида) добавляли в высушенную в печи круглодонную колбу под слоем азота в безводном дихлорметане. К указанному раствору добавляли Y мол.% винилового эфира низшей гидрофобной группы (например, пропила, бутила) и необязательно Z мол.% винилэфира высшей гидрофобной группы (например, додецила, октадецила) (ФИГ.1). Раствор помещали на баню при -50 - -78°С и позволяли 2-винилоксиэтилфталимиду осаждаться. К указанному раствору добавляли 10 мол. % BF3⋅(ОСН2СН3)2 и реакции позволяли протекать в течении 2-3 часов при -50 - -78°С.Полимеризацию останавливали путем добавления гидроксида аммония в растворе метанола. Полимер высушивали при пониженном давлении и затем переносили в смесь 1,4-диоксан/метанол (2/1). Добавляли 20 мол. эквивалентов гидразина на фталимид для удаления защитной группы от амина. Раствор нагревали до обратной конденсации в течение 3 часов и затем высушивали при пониженном давлении. Полученное твердое вещество растворяли в 0,5 моль/л HCl и нагревали в течение 15 мин. с образованием гидрохлорида полимера, разбавляли дистиллированной водой и нагревали до обратной конденсации в течение дополнительного часа. Затем раствор нейтрализовали NaOH, охлаждали до комнатной температуры (RT), переносили на молекулярную целлюлозную трубку, подвергали диализу против дистиллированной воды, и лиофилизировали. Полимер далее можно очищать с использованием эксклюзионной хроматографии или другого типа хроматографии. Молекулярную массу полимеров оценивали с использованием колонок в соответствии со стандартными способами, включая аналитическую эксклюзионную хроматографию и эксклюзионную хроматографию с многоугольным светорассеиванием (SEC-MALS).
Полимер DW1360. Терполимер амин/бутил/октадецилполи(виниловый эфир) синтезировали из мономеров 2-винилоксиэтилфталимида (5 г, 23,02 ммоль), бутилвинилэфира (0,665 г, 6,58 ммоль) и октадецилвинилэфира (0,488 г, 1,64 ммоль). 2-Винилоксиэтилфталимид в 36 мл безводного дихлорметана добавляли в круглодонную высушенную в печи колбу на 200 мл с магнитной мешалкой под слоем аргона. К указанному раствору добавляли бутилвиниловый эфир и н-октадецилвиниловый эфир. Мономеры полностью растворяли при комнатной температуре (RT) с получением прозрачного гомогенного раствора. Реакционный сосуд, содержащий чистый раствор, затем помещали на баню при -50°С, полученную путем добавления сухого льда к раствору денатурированного спирта степени чистоты стандарта ACS и этиленгликоля (1:1) и позволяли образовываться видимому осадку мономера фталимида. После охлаждения в течение примерно 1,5 мин. добавляли BF3⋅(ОСН2СН3)2 (0,058 г, 0,411 ммоль) для инициации реакции полимеризации. После инициации полимеризации мономер фталимида растворяли. Реакции позволяли протекать в течение 3 часов при -50°С. Полимеризацию останавливали путем добавления 5 мл 1% гидроксида аммония в метаноле. Растворители затем удаляли посредством ротационного выпаривания.
Полимер затем растворяли в 30 мл смеси 1,4-диоксан/метанол (2/1). К указанному раствору добавляли гидразин (0,147 г, 46 ммоль) и смесь нагревали до температуры обратной конденсации в течение 3 часов. Растворители затем удаляли посредством ротационного выпаривания и полученное твердое вещество отбирали в 20 мл 0,5 моль/л HCl и нагревали до обратной конденсации в течение 15 минут, разбавляли 20 мл дистиллированной воды и нагревали до обратной конденсации в течение дополнительного часа. Указанный раствор затем нейтрализовали NaOH, охлаждали до комнатной температуры, переносили на целлюлозную трубку, пропускающую соединения с молекулярной массой 3,500, подвергали диализу в течение 24 часов (2×20 л) против дистиллированной воды и лиофилизировали.
В) Мелитин. Все пептиды мелитина получали с использованием стандартных методов синтеза пептидов, известных в данной области техники. Подходящие пептиды мелитина могут состоять из аминокислот только в L-форме, из аминокислот только в D-форме (инвертированные). Независимо от L-или D-формы, последовательность пептида мелитина может быть обратной (ретро).
| название | год | авторы | номер документа |
|---|---|---|---|
| Композиция системы доставки на основе конъюгата для доставки полинуклеотида РНК-интерференции в клетку печени и способ ее получения | 2011 |
|
RU2623160C9 |
| НИЗКОМОЛЕКУЛЯРНЫЕ КОНЪЮГАТЫ ДЛЯ ВНУТРИКЛЕТОЧНОЙ ДОСТАВКИ БИОЛОГИЧЕСКИ АКТИВНЫХ СОЕДИНЕНИЙ | 2011 |
|
RU2629957C2 |
| НИЗКОМОЛЕКУЛЯРНЫЕ КОНЪЮГАТЫ ДЛЯ ВНУТРИКЛЕТОЧНОЙ ДОСТАВКИ НУКЛЕИНОВЫХ КИСЛОТ | 2011 |
|
RU2582235C2 |
| НОВЫЕ КОНЪЮГАТЫ ОЛИГОНУКЛЕОТИДОВ И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2599449C1 |
| ОДНОРАЗОВАЯ СИСТЕМА ДЛЯ СТЕРИЛЬНОГО ПОЛУЧЕНИЯ ЧАСТИЦ ИЗ ЛИПИДОВ И НУКЛЕИНОВЫХ КИСЛОТ | 2012 |
|
RU2642640C2 |
| ПОЛИКОНЪЮГАТЫ ДЛЯ ВВЕДЕНИЯ IN VIVO ПОЛИНУКЛЕОТИДОВ | 2007 |
|
RU2430740C2 |
| НАЦЕЛЕННЫЕ КОНЪЮГАТЫ И ЧАСТИЦЫ И ИХ СОСТАВЫ | 2015 |
|
RU2695220C2 |
| НАЦЕЛИВАЮЩИЕ АМИНОКИСЛОТНЫЕ ЛИПИДЫ | 2013 |
|
RU2654210C2 |
| КОНЪЮГАТЫ АНТАГОНИСТОВ ИНТЕГРИНА ДЛЯ НАЦЕЛЕННОЙ ДОСТАВКИ К КЛЕТКАМ, ЭКСПРЕССИРУЮЩИМ АЛЬФА-V-БЕТА-3 | 2013 |
|
RU2623441C2 |
| КОМПОЗИЦИИ, СОДЕРЖАЩИЕ КОМПЛЕКСЫ ВКЛЮЧЕНИЯ | 2001 |
|
RU2288921C2 |




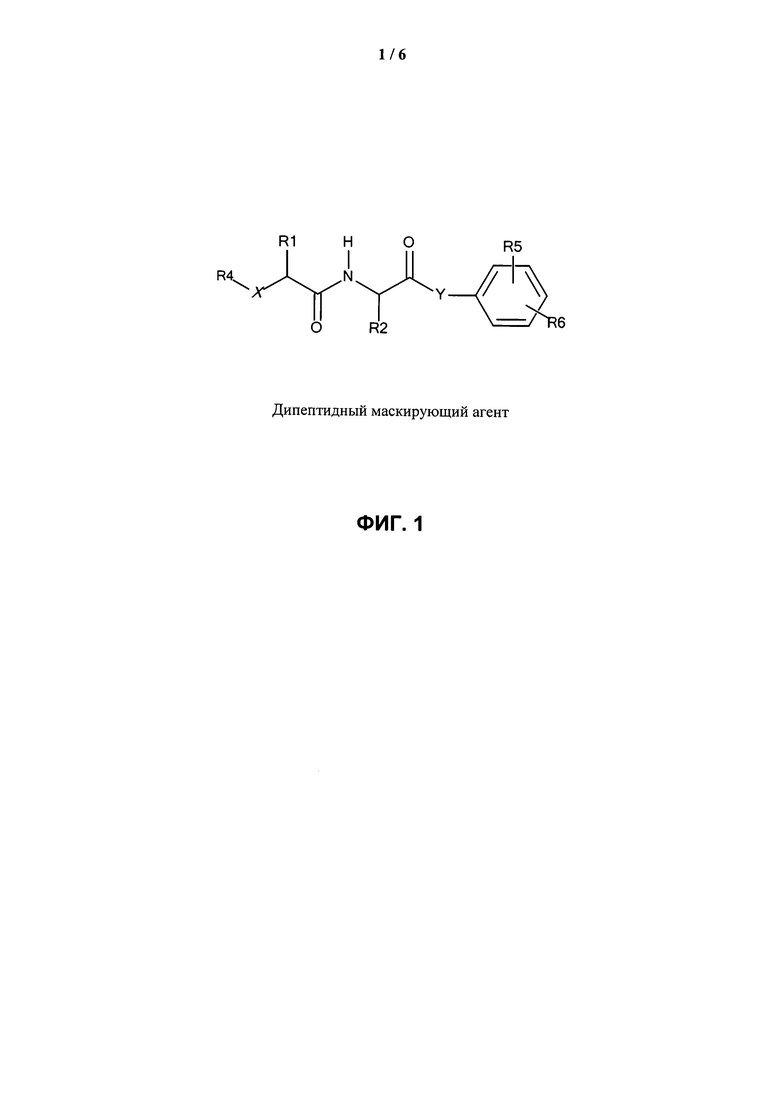
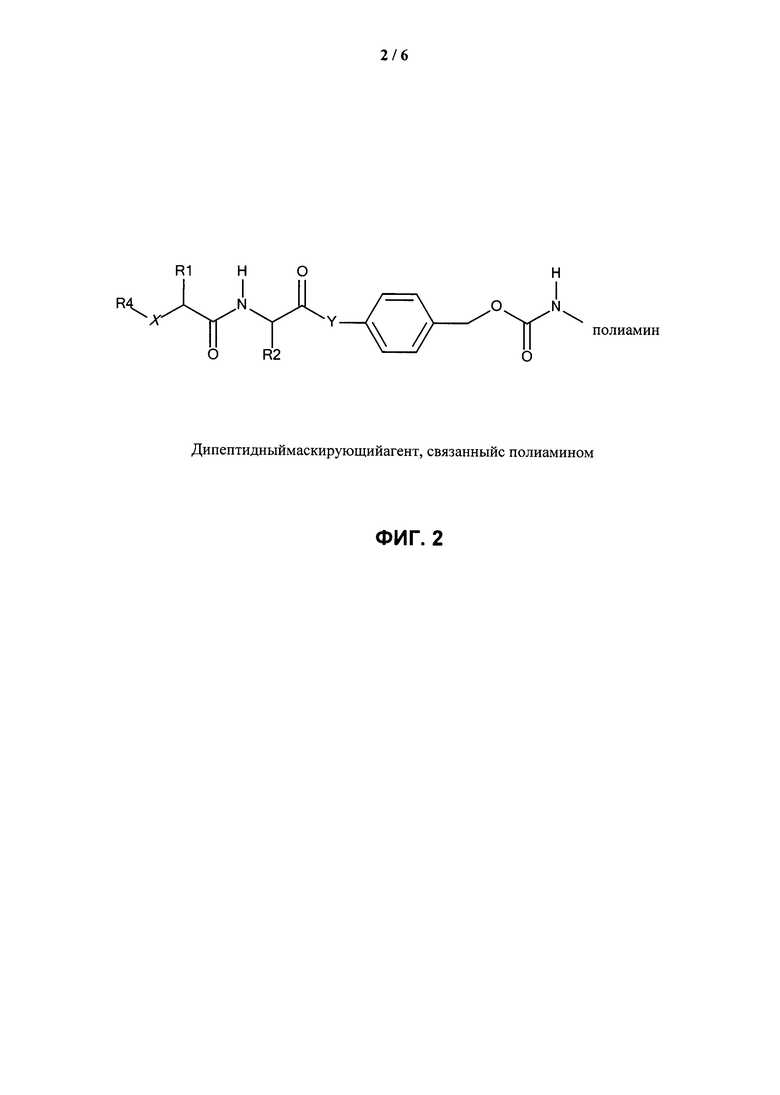
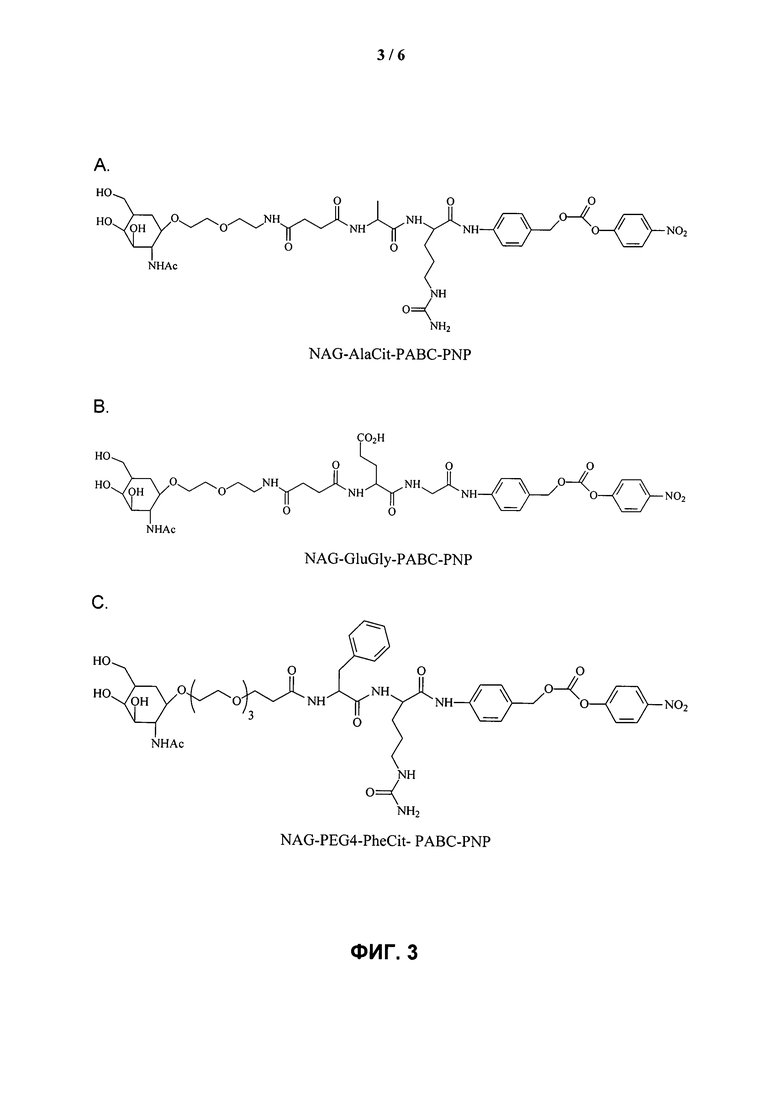
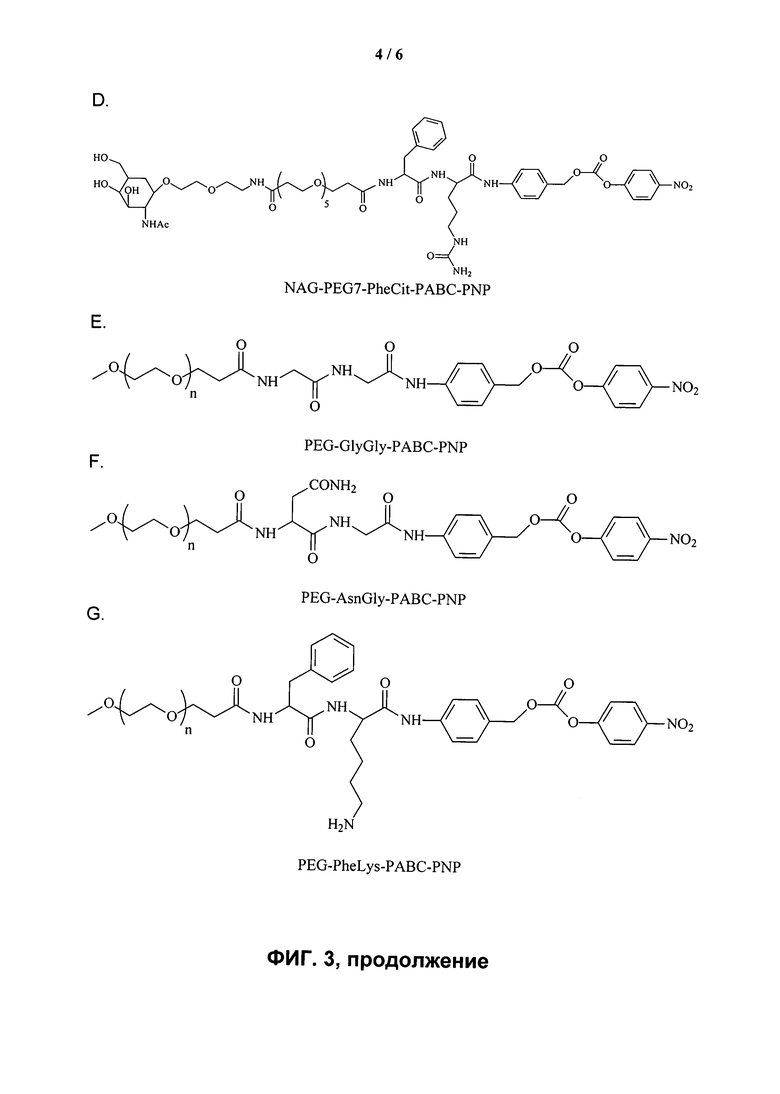
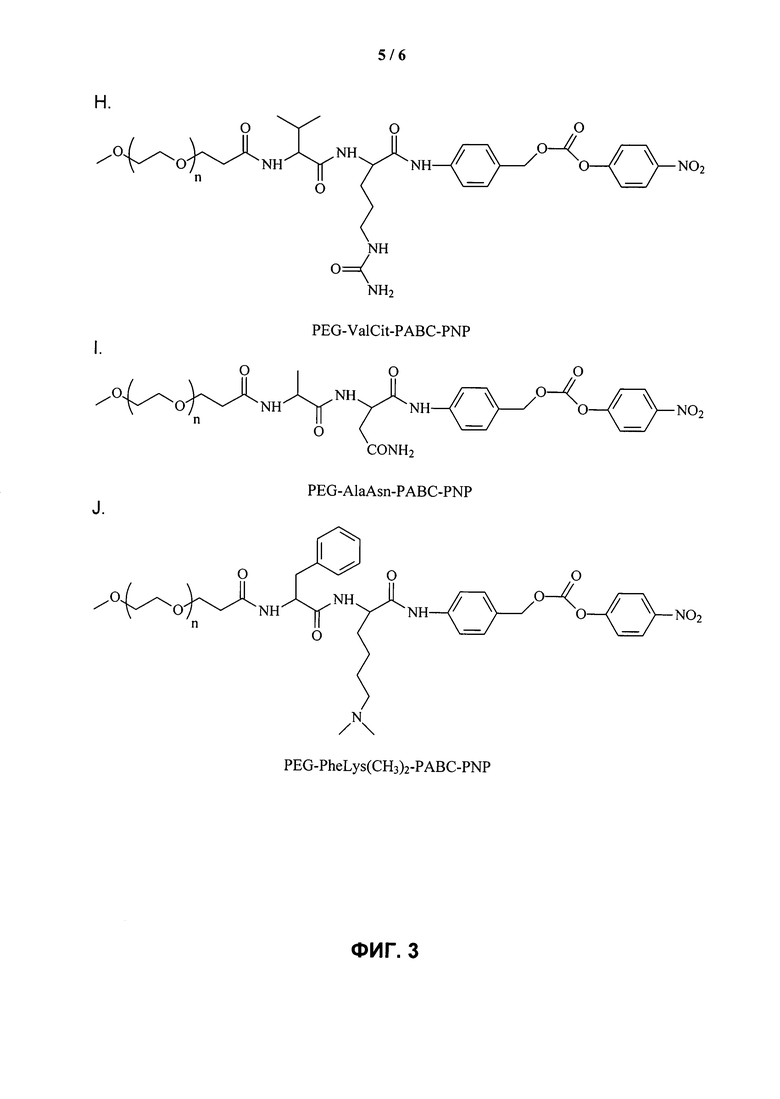
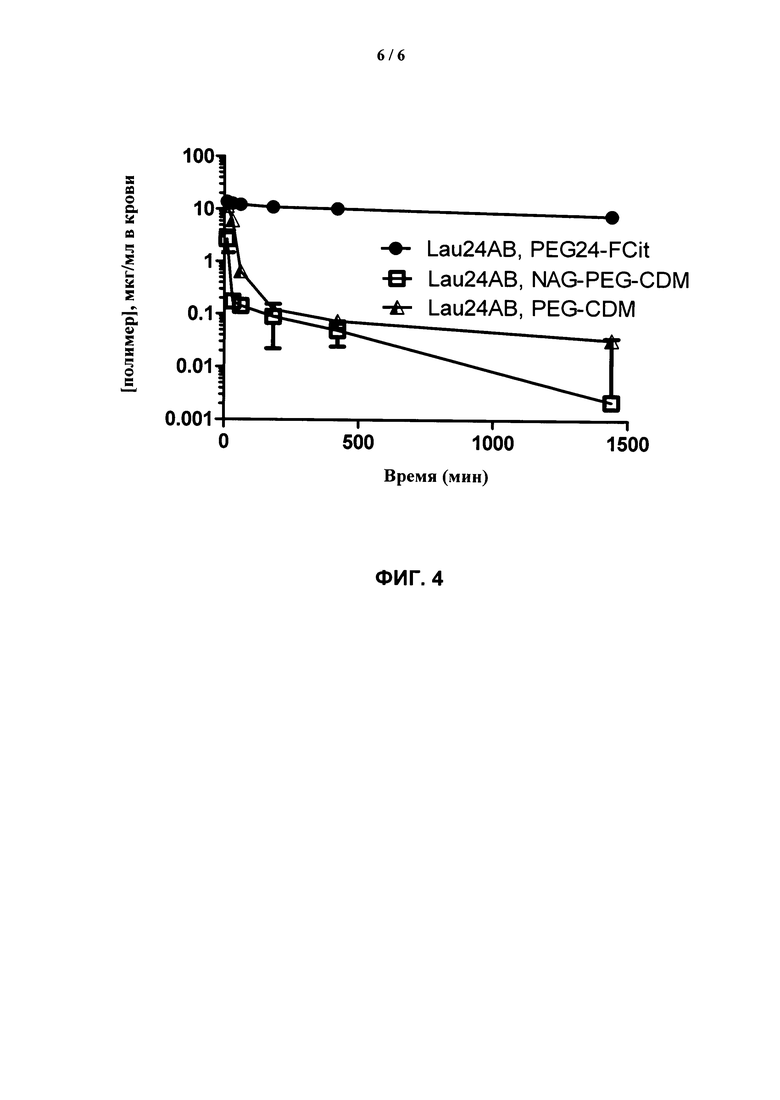
Изобретение относится к соединениям для обратимой модификации амфипатического мембраноактивного полиамина и полимеру для доставки полинуклеотида в клетку in vivo. Амфипатические мембраноактивные полиамины обратимо модифицированы поддающимися ферментативному отщеплению дипептидными амидобензилкарбонатными маскирующими агентами. Указанная модификация приводит к маскированию мембранной активности полимера, а обратимость обеспечивает чувствительность к физиологическим условиям. Обратимо модифицированные полиамины (динамичные поликонъюгаты, или КДП) также ковалентно связаны с полинуклеотидом РНКи или вводятся совместно с направленным конъюгатом полинуклеотид РНКи-нацеливающая молекула. 4 н. и 13 з.п. ф-лы, 15 табл., 4 ил., 16 пр.
1. Соединение для обратимой модификации амфипатического мембраноактивного полиамина, содержащее нацеливающий лиганд, ковалентно связанный с дипептидамидобензилкарбонатом, имеющее следующую структуру:
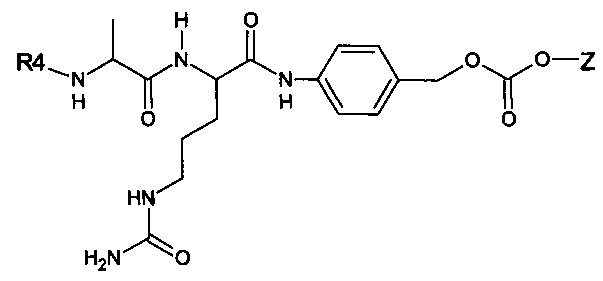
где R4 является незаряженным и содержит лиганд рецептора клетки и
Z представляет собой
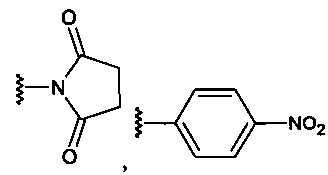 или
или 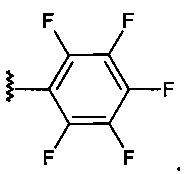
2. Соединение по п. 1, отличающееся тем, что лиганд содержит лиганд рецептора асиалогликопротеина (ASGPr).
3. Соединение по п. 2, отличающееся тем, что лиганд ASGPr выбран из перечня, состоящего из лактозы, галактозы, N-ацетилгалактозамина, галактозамина, N-формилгалактозамина, N-пропионилгалактозамина, N-н-бутаноилгалактозамина и N-изо-бутаноилгалактозамина.
4. Соединение по любому из пп. 1-3, отличающееся тем, Z представляет собой
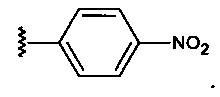
5. Соединение для обратимой модификации амфипатического мембраноактивного полиамина, содержащее
R-А1А2-п-амидобензилкарбонат,
где R содержит полиэтиленгликоль,
А1 представляет собой гидрофобную аминокислоту, выбранную из группы, состоящей из аланина, фенилаланина, валина, изолейцина или триптофана,
А2 представляет собой гидрофильную незаряженную аминокислоту, связанную с А1 амидной связью, причем указанная гидрофильная незаряженная кислота выбрана из группы, состоящей из цитрулина, глицина, треонина, диметиллизина, аспарагина или глютамина, и
карбонат представляет собой активированный амин-реактивный карбонат.
6. Соединение по п. 5, отличающееся тем, что R является нейтральным.
7. Соединение для обратимой модификации амфипатического мембраноактивного полиамина, имеющее следующую структуру:
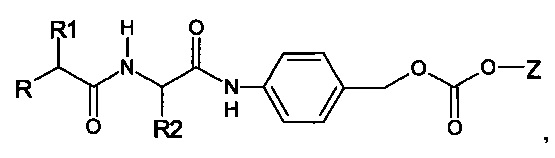
где R содержит полиэтиленгликоль,
R1 представляет собой боковую цепь гидрофобной аминокислоты, выбранной из группы, состоящей из аланина, фенилаланина, валина, изолейцина или триптофана,
R2 представляет собой боковую цепь гидрофильной незаряженной аминокислоты, выбранной из группы, состоящей из цитрулина, треонина, диметиллизина, аспарагина или глютамина,
Z представляет собой
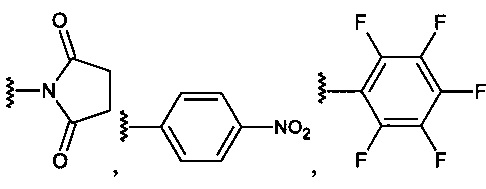 или
или 
8. Полимер доставки для доставки полинуклеотида в клетку in vivo, содержащий
М1у-Р-М2z,
где Р представляет собой амфипатический мембраноактивный полиамин,
М1 содержит структуру, представленную следующей формулой:
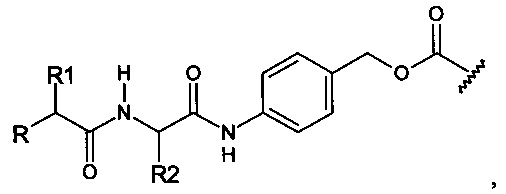
где R содержит лиганд рецептора клетки, R1 представляет собой боковую цепь гидрофобной аминокислоты, выбранной из группы, состоящей из аланина, фенилаланина, валина, изолейцина или триптофана, и R2 представляет собой боковую цепь гидрофильной незаряженной аминокислоты, выбранной из группы, состоящей из цитрулина, глицина, треонина, диметиллизина, аспарагина или глутамина,
М2 содержит структуру, представленную следующей формулой:
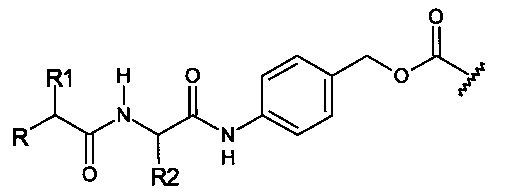
где R содержит полиэтиленгликоль, R1 представляет собой боковую цепь гидрофобной аминокислоты, выбранной из группы, состоящей из аланина, фенилаланина, валина, изолейцина или триптофана, и R2 представляет собой боковую цепь гидрофильной незаряженной аминокислоты, выбранной из группы, состоящей из цитрулина, глицина, треонина, диметиллизина, аспарагина или глутамина,
каждый из у и z представляет собой целое число, большее или равное нулю,
у+z имеет значение, составляющее более 50% первичных аминов на полиамине Р, определенное по количеству аминов на Р в отсутствие любых маскирующих агентов, и
Р представляет собой амфипатический мембраноактивный полиамин.
9. Полимер доставки по п. 8, отличающийся тем, что М1 и М2 независимо являются незаряженными или заряженными нейтральными.
10. Полимер доставки по п. 8, отличающийся тем, что лиганд содержит лиганд ASGPr.
11. Полимер доставки по п. 10, отличающийся тем, что лиганд ASGPr выбран из перечня, состоящего из лактозы, галактозы, N-ацетилгалактозамина, галактозамина, N-формилгалактозамина, N-пропионилгалактозамина, N-н-бутаноилгалактозамина и N-изо-бутаноилгалактозамина.
12. Полимер доставки по п. 10, отличающийся тем, что лиганд ASGPr представляет собой N-ацетилгалактозамин.
13. Полимер доставки по любому из пп. 8-12, отличающийся тем, что амфипатический мембраноактивный полиамин выбран из перечня, состоящего из полимера с нерегулярным чередованием мономеров, блок-полимера или синтетического полимера с регулярным чередованием мономеров.
14. Полимер доставки по любому из пп. 8-12, отличающийся тем, что амфипатический мембраноактивный полиамин представляет собой пептид мелитина.
15. Полимер доставки по любому из пп. 8-12, отличающийся тем, что амфипатический мембраноактивный полиамин дополнительно ковалентно связан с полинуклеотидом.
16. Полимер доставки по п. 15, отличающийся тем, что полинуклеотид включает полинуклеотид интерферирующей РНК (РНКи).
17. Полимер доставки по п. 16, отличающийся тем, что полинуклеотид интерферирующей РНК выбран из группы, состоящей из ДНК, РНК, дцРНК, миРНК и микроРНК.
| US 6180095 B2, 30.01.2001 | |||
| ПОЛИКОНЪЮГАТЫ ДЛЯ ВВЕДЕНИЯ IN VIVO ПОЛИНУКЛЕОТИДОВ | 2007 |
|
RU2430740C2 |
| US 20080293800 A1, 27.11.2008 | |||
| WO 2002083180 A1, 24.10.2002 | |||
| WO 2009141240 A1, 26.11.2009 | |||
| US 7091186 B2, 15.08.2006. | |||

