Область изобретения
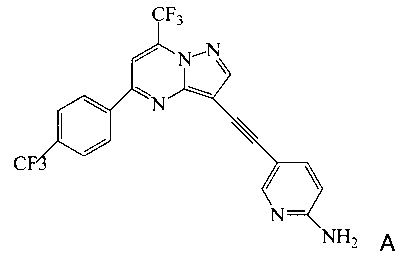
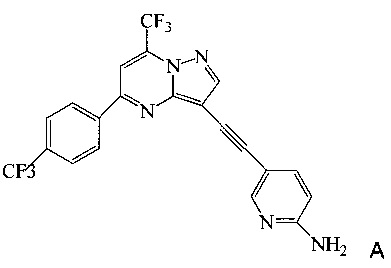
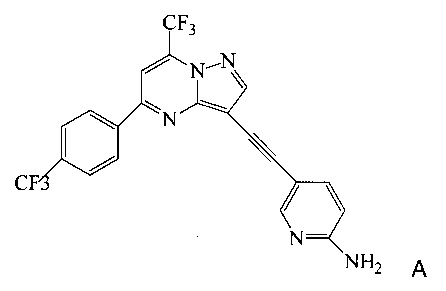
Согласно настоящему изобретению предложены новые способы получения 5-[2-[7-(трифторметил)-5-[4-(трифторметил)фенил]пиразоло[1,5-а]пиримидин-3-ил]этинил]-2-пиридинамина (соединения A). Настоящие способы пригодны для получения соединения A в крупном масштабе на промышленных предприятиях.
Предшествующий уровень техники
5-[2-[7-(Трифторметил)-5-[4-(трифторметил)фенил]пиразоло[1,5-a]пиримидин-3-ил]этинил]-2-пиридинамин (соединение A) представляет собой антагонист mGluR2 (метаботропного рецептора глутамата), который полезен в лечении депрессии и других расстройств ЦНС. WO 2006/099972 описывает синтез соединения A и его возможное применение в лечении расстройств центральной нервной системы (ЦНС). Также в WO 2006/099972 раскрыты аналоги и способы синтеза этих аналогов. Эти способы несовместимы с крупносерийным производством соединения А, необходимом для поддержки клинических программ и извлечения прибыли.
Сущность изобретения
Согласно настоящему изобретению предложены способы получения соединения A, имеющего формулу:
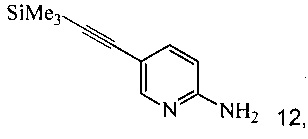
согласно которым:
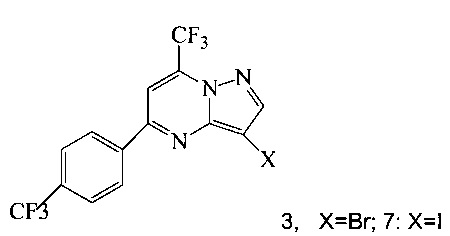
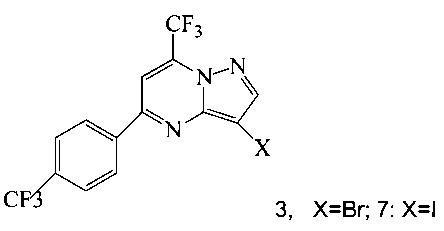
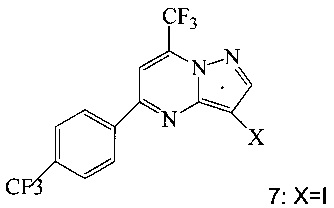
(a) подвергают взаимодействию соединение 3 или 7;
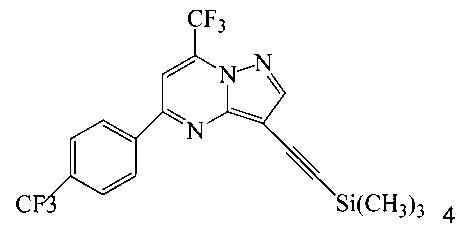
с триметилсилилацетиленом в ходе реакции сочетания по Соногашира в инертном растворителе, что дает соединение 4;
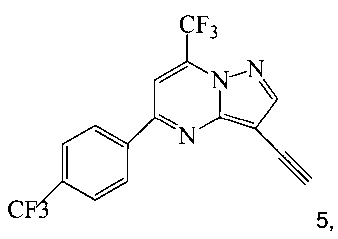
(b) осуществляют десилилирование соединения 4 в инертном растворителе, что дает соединение 5;
 и
и
(c) подвергают взаимодействию соединение 5 с соединением 6 или 8;
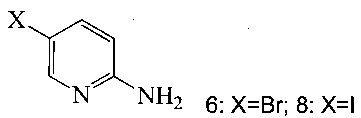
в ходе реакции сочетания по Соногашира в инертном растворителе, что дает соединение A.
Согласно настоящему изобретению предложен способ получения соединения A, имеющего формулу:
согласно которому:
(a) подвергают взаимодействию соединение 3 или 7;
с триметилсилилацетиленом в ходе реакции сочетания по Соногашира в инертном растворителе, что дает соединение 4;
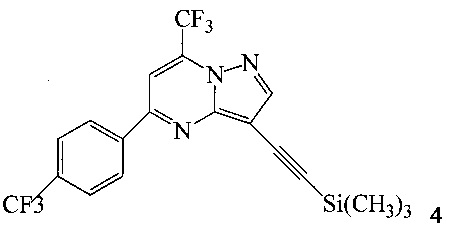
(b) осуществляют десилилирование соединения 4 в инертном растворителе, что дает соединение 5;
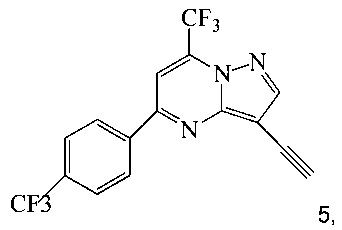 и
и
(c) подвергают взаимодействию соединение 5 с соединением 6 или 8;
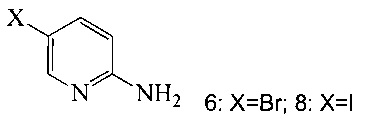
в ходе реакции сочетания по Соногашира в инертном растворителе, что дает соединение A.
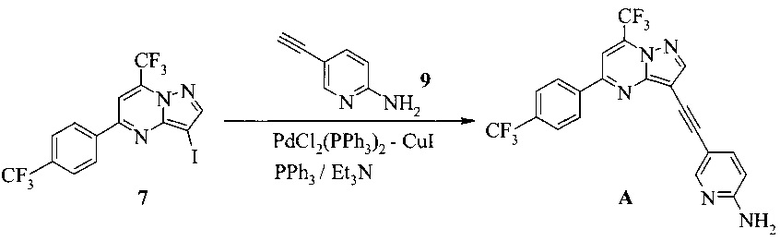
Кроме того, согласно настоящему изобретению предложены способы получения соединения A, имеющего формулу:
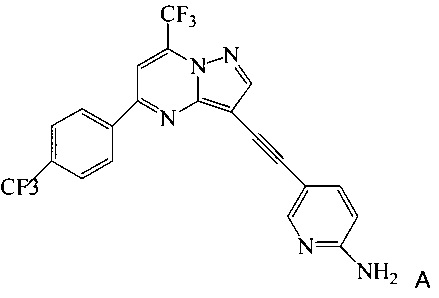
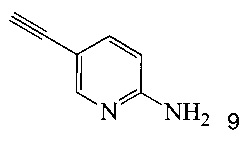
согласно которым подвергают взаимодействию соединение 3 или 7 с соединением 9;
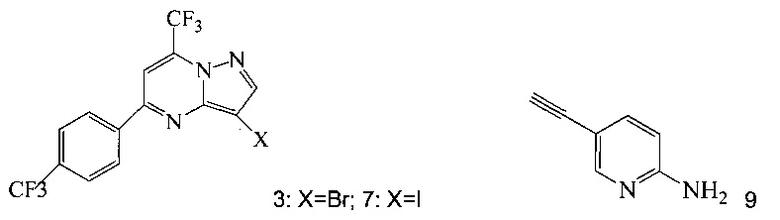
в ходе реакции сочетания по Соногашира в инертном растворителе, что дает соединение A.
Кроме того, согласно настоящему изобретению предложены способы получения соединения А, имеющего формулу:
согласно которым:
(a) подвергают взаимодействию соединение 6 или 8;
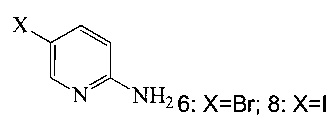
с триметилсилилацетиленом в ходе реакции сочетания по Соногашира в инертном растворителе, что дает промежуточное соединение 12;
 и
и
(b) подвергают взаимодействию соединение 12 с соединением 7;
в инертном растворителе в присутствии фторида калия, что дает соединение A.
Кроме того, согласно настоящему изобретению предложены способы очистки соединения A, имеющего формулу:

согласно которым:
(a) растворяют неочищенное соединение A в тетрагидрофуране с образованием раствора;
(b) обрабатывают раствор со стадии (a) н-трибутилфосфином;
(c) добавляют метансульфоновую кислоту к реакционной смеси со стадии (b), чтобы осадить мезилатную соль соединения A (соединение A-1);
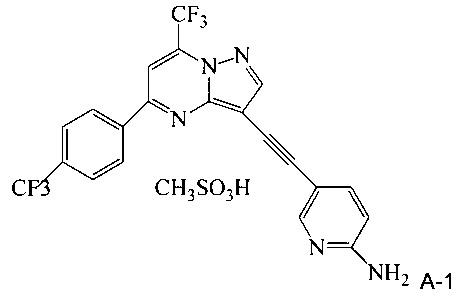
(d) выделяют мезилатную соль со стадии (c);
(e) суспендируют мезилатную соль со стадии (d) в 2-метилтетрагидрофуране с образованием органической смеси;
(f) обрабатывают органическую смесь со стадии (е) водным карбонатом натрия, чтобы превратить мезилатную соль А-1 в соединение А, что дает в результате водную фазу и органическую фазу, содержащую соединение А;
(g) разделяют водную и органическую фазы со стадии (f) и промывают органическую фазу водой; и
(h) осуществляют замену растворителя в органической фазе, чтобы заменить 2-метилтетрагидрофуран на изопропанол, что дает чистое кристаллическое соединение A.
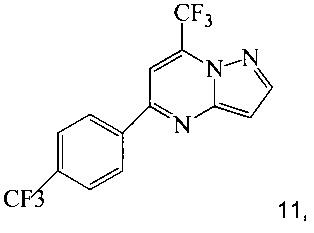
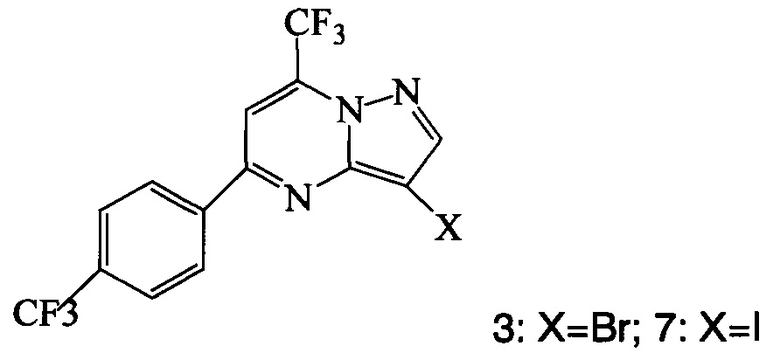
Кроме того, согласно настоящему изобретению предложены способы получения соединения 3 или 7, имеющего формулу:
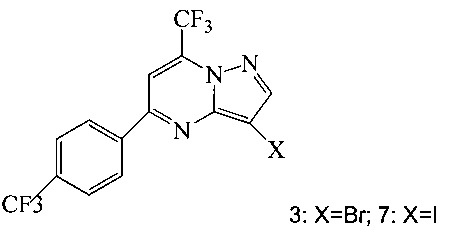
согласно которым:
(a) подвергают взаимодействию соединение 1 с соединением 10; 
в ходе реакции конденсации, что дает соединение 11;
 и
и
(b) подвергают взаимодействию соединение 11 с галогенирующим агентом, что дает соединение 3 или 7.
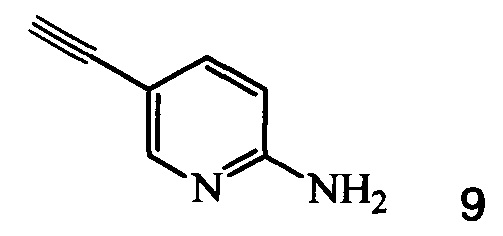
Кроме того, согласно настоящему изобретению предложены способы получения соединения 9, имеющего формулу:
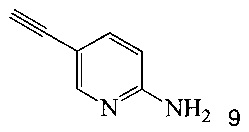
согласно которым:
(a) подвергают взаимодействию соединение 6 или 8;
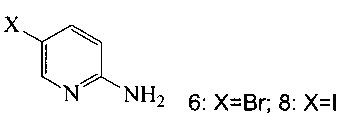
с триметилсилилацетиленом в ходе реакции сочетания по Соногашира в инертном растворителе, что дает соединение 12;
 и
и
(b) осуществляют десилилирование соединения 12 в инертном растворителе, что дает соединение 9.
Кроме того, согласно настоящему изобретению предложены способы получения соединения 9, имеющего формулу:
согласно которым:
(a) подвергают взаимодействию соединение 6 или 8;
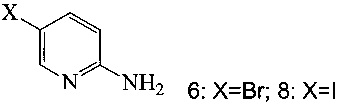
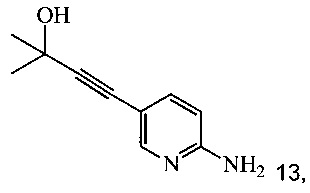
с 2-метил-3-бутин-2-олом в ходе реакции сочетания по Соногашира в инертном растворителе, что дает соединение 13;
 и
и
(b) удаляют защитную группу соединения 13 с помощью основания в инертном растворителе, что дает соединение 9.
Кроме того, согласно настоящему изобретению предложены способы получения соединения 7, имеющего формулу:
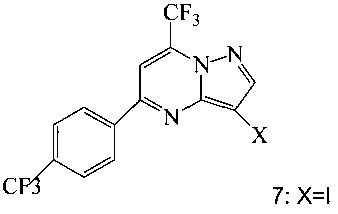
согласно которым:
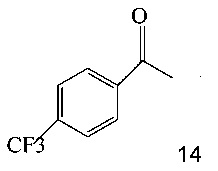
(a) подвергают взаимодействию соединение 14;
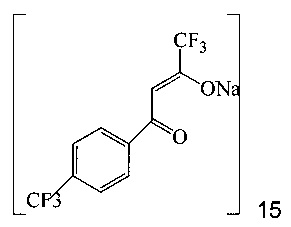
с этил трифторацетатом при основных условиях в инертном растворителе, что дает промежуточное соединение 15;
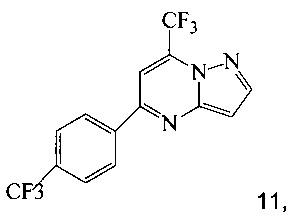
(b) подвергают взаимодействию промежуточное соединение 15 с 3-аминопиразолом в инертном растворителе, что дает промежуточное соединение 11;
 и
и
(c) подвергают взаимодействию промежуточное соединение 11 с йодирующим агентом при кислотных условиях, что дает соединение 7.
Подробное описание изобретения
Согласно настоящему изобретению предложены способы получения 5-[2-[7-(трифторметил)-5-[4-(трифторметил)фенил]пиразоло[1,5-a]пиримидин-3-ил]этинил]-2-пиридинамина (соединения A). Настоящие способы пригодны для получения соединения A в крупном масштабе на промышленных предприятиях.
Как используется здесь, следующие термины имеют значения, приведенные ниже.
Термин «кислотные условия» относится к условиям, касающимся значения pH водного раствора. Чистая вода считается нейтральной, с pH близким к 7,0 при 25°c. Растворы со значением pH меньше 7 считаются кислотными растворами или условиями.
Термин «C1-6-алкил» относится к одновалентной линейной или разветвленной насыщенной углеводородной группе с 1-6 атомами углерода, например, метилу, этилу, пропилу, изопропилу, н-бутилу, изобутилу, втор-бутилу или трет-бутилу.
Термин «арил» относится к одновалентной ароматической карбоциклической моно- или бициклической кольцевой системе, включающей 6-10 кольцевых атомов углерода. Примеры арильных группировок включают фенил и нафтил.
Термин «основные условия» относится к условиям, касающимся значения pH. Чистая вода считается нейтральной, с pH близком к 7,0 при 25°C. Растворы со значением pH больше 7 считаются основными или щелочными растворами или условиями.
Термин «реакция конденсации» относится к химической реакции, в которой две молекулы или группировки (функциональные группы) объединяются с образованием всего одной молекулы вместе с потерей небольшой молекулы. Когда этой небольшой молекулой является вода, тогда реакцию называют реакцией дегидратации.
Термин «десилилирование» относится к удалению силильных защитных групп у молекулы. Фторид-ионы, такие как те, что присутствуют во фториде калия, KF, пригодны для удаления силильных защитных групп в ходе реакций десилилирования.
Термин «галоген» относится к хлору, брому, йоду и фтору, и предпочтительно является йодом и бромом.
Термин «галогенирующий агент» относится к агенту, используемому в реакции галогенирования, который вводит атом галогена в молекулу.
Определенные виды галогенирующих агентов включают фторирующие, хлорирующие, бромирующие и йодирующие агенты. Неограничивающие иллюстративные галогенирующие агенты включают N-бромсукцинимид (NBS), хлорид йода (ICl), N-йодсукцинимид (NIS) и смешанные агенты, такие как I2/NaIO4/HCl.
Термин «гидролизование» относится к реакции гидролиза (гидролиз), в
которой исходная молекула разделяется на две части при добавлении молекулы воды. В ходе реакции гидролиза молекулы воды разделяются на катионы водорода (H+) и гидроксид-анионы (OH-). Одна часть исходной молекулы получает катион водорода от молекулы воды; другая часть получает гидроксид-анион.
Термин «инертный органический растворитель» относится к растворителю, который химически не мешает реакции. Иллюстрирующие неограничивающие примеры инертных органических растворителей в настоящем изобретении включают тетрагидрофуран, 2-метилтетрагидрофуран, диметилформамид, толуол, трет-бутилметиловый эфир и подобные.
Термин «промежуточное соединение» относится к соединению, которое получается в ходе химического синтеза. Само промежуточное соединение не является конечным продуктом, но используется в дальнейших реакциях, которые дают конечный продукт. Оно отличается от исходного вещества и конечного продукта. Промежуточное соединение можно выделять или не выделять. Часто его не выделяют или не очищают, а скорее используют «как есть» в синтезе по экономическим соображениям, особенно в промышленных масштабах.
Термин «мезилат» относится к соли или эфиру метансульфоновой кислоты, CH3SO3H. В солях мезилат находится в виде аниона 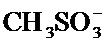
Термины «способ получения» и «процесс получения» можно использовать взаимозаменяемо.
Термин «фармацевтически приемлемый», такой как фармацевтически приемлемый носитель, эксципиент и т.п., означает фармакологически приемлемый и по существу нетоксичный для объекта, которому конкретное соединение вводится.
Термин «фармацевтически приемлемая соль» относится к общепринятым солям присоединения кислоты или солям присоединения основания, которые сохраняют биологическую эффективность и свойства соединений по настоящему изобретению и образуются из подходящих нетоксичных органических или неорганических кислот или органических или неорганических оснований. Примерные соли присоединения кислоты включают те, что получены из неорганических кислот, таких как соляная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, сульфаминовая кислота, фосфорная кислота и азотная кислота, и те, что получены из органических кислот, таких как п-толуолсульфоновая кислота, салициловая кислота, метансульфоновая кислота, щавелевая кислота, янтарная кислота, лимонная кислота, яблочная кислота, молочная кислота, фумаровая кислота и подобные. Примерные соли присоединения основания включают те, что получены из аммония, калия, натрия и гидроксидов четвертичного аммония, таких как, например, гидроксид тетраметиламмония. Химическое изменение фармацевтического соединения (т.е. лекарства) в соль является хорошо известной химикам-фармацевтам методикой получения улучшенной физической и химической устойчивости, гигроскопичности и растворимости соединений. Смотрите, например H. Ansel et. al., Pharmaceutical Dosage Forms and Drug Delivery Systems (6th Ed. 1995) at pp. 196 и 1456-1457.
Термин «защитная группа» относится к группе, вводимой в молекулу в ходе химической модификации, чтобы получить хемоселективность в последующей химической реакции. Во многих получениях органических соединений некоторые функциональные группы в молекуле не могут выдержать реагенты реакции или химическую среду. Следовательно, эти функциональные группы должны быть защищены защитной группой, которая будет защищать функциональную группу в ходе подобной реакции. Как правило, защитную группу легко удалить (снятие защитных групп) после химической реакции. Защитные группы выполняют важную функцию в многостадийном органическом синтезе. Квалифицированный специалист в данной области техники, вероятно, знает, как защитить и удалить защитные группы у конкретной функциональной группы. Во многих учебниках и других ссылочных материалах предожены подобные способы, включая «Protective Groups in Organic Chemistry», J.F.W. McOmie, Plenum Press, 1973; «Greene’s Protective Groups in Organic Synthesis», 4th Edition, Peter G.M. Wuts and Theodora W. Greene, 2006; Wiley, среди прочего.
Термин «реакция сочетания по Соногашира» относится к реакции сочетания концевых алкинов с арил или винилгалогенидами. Обычно для этой реакции необходимо два катализатора: комплекс палладия нулевой валентности и галогенид меди(I). Комплекс палладия активирует органические галогениды в ходе окислительного добавления в связь углерод-галоген. Обычно для этой реакции используют комплексы фосфин-палладия, такие как тетракис(трифенилфосфин)палладий(0) или бис-(трифенилфосфин)-палладий(II) - дихлорид. Галогениды меди(I) взаимодействуют с концевым алкином и дают ацетиленид меди(I), который действует как активированные частицы для реакции сочетания. Реакционная среда должна быть основной, чтобы нейтрализовать галогеноводород, получаемый в качестве побочного продукта этой реакции сочетания. В качестве оснований можно использовать соединения алкиламина, такие кактриэтиламин, диэтиламин или диизопропиламин.
Термин «захватывающий воду агент» или «дегидратирующий агент» относится к агенту, который применяется для удаления воды из реакционной смеси, чтобы ускорить реакцию. В качестве захватывающего воду агента часто используется агент 1,1,1,3,3,3-гексаметилдисилазан (HMDS) для того, чтобы реакции сочетания протекали быстрее и полностью.
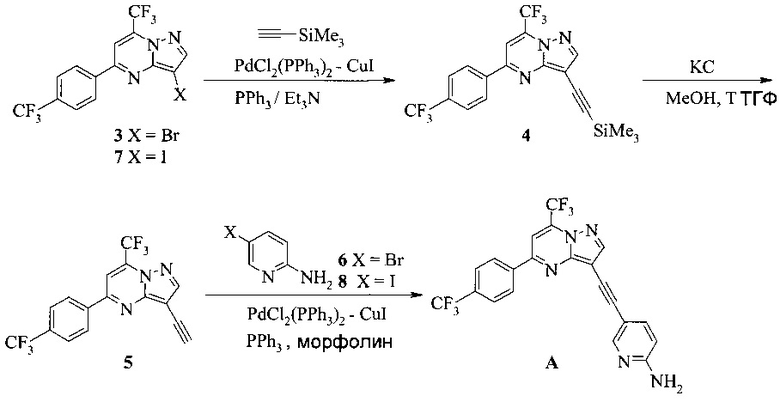
Согласно настоящему изобретению предложены способы получения соединения A. В одном способе связывают соединение 5 с 2-амино-5-галопиридином (соединение 6 или 8), как показано на Схеме 1.
Схема 1
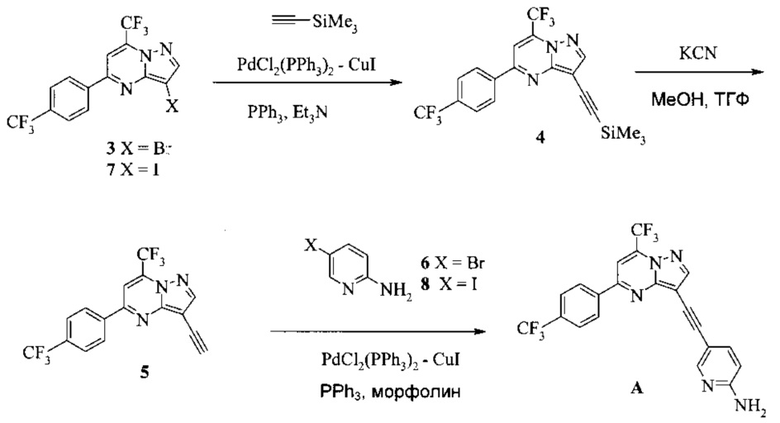
Согласно способам на Схеме 1: (a) связывают соединение 3 или 7 с триметилсилилацетиленом, используя реакцию сочетания по Соногашира, что дает соединение 4; (b) осуществляют десилилирование соединения 4, что дает соединение 5; и (c) связывают соединение 5 с соединением 6 или 8, используя реакцию сочетания по Соногашира, что дает соединение A.
В одном воплощении изобретения предложен способ получения соединения A, согласно которому осуществляют взаимодействие соединения 5 с соединением 6 или 8 в ходе реакции сочетания по Соногашира в инертном растворителе, что дает соединение A. В другом воплощении изобретения предложен способ, где получают соединение 5 в ходе десилилирования соединения 4 в инертном растворителе. В другом воплощении изобретения предложен способ, где получают соединение 4 при взаимодействии соединения 3 или 7 с триметилсилилацетиленом в ходе реакции сочетания по Соногашира в инертном растворителе.
На Схеме 1 представлены предпочтительные реагенты и условия. В реакции по Соногашира можно использовать широкий диапазон условий. Например, несмотря на то, что триметилсилилацетилен является предпочтительным реагентом при получении 5, также можно использовать вместо триметилсилилацетилена другие ацетилены, защищенные силильной группой, с общей структурой, представленной ниже.
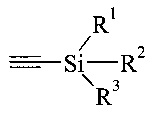
где R1, R2 и R3 могут представлять собой простые алкильные группы, такие как этил, н-пропил и н-бутил; или также могут представлять собой простые арильные группы, такие как фенильная группа.
Несмотря на то, что Pd(Ph3P)2Cl2 является предпочтительным катализатором реакции, также можно использовать другие виды палладиевых катализаторов. Неограничивающие примеры подобных катализаторов включают Pd2(dba)3, Na2PdCl4 и Pd(OAc)2.
Несмотря на то, что Ph3P является предпочтительным лигандом реакции, также можно использовать другие виды лигандов. Неограничивающие примеры подобных лигандов включают P(t-Bu)3, P(o-ToI)3 и другие лиганды без фосфора.
Несмотря на то, что NEt3 является предпочтительным основанием реакции, также можно использовать другие виды оснований в реакции. Неограничивающие примеры подобных оснований включают Et2NH, пирролидин, i-Pr2NH, диизопропилэтиламин, морфолин и Cs2CO3.
Ряд растворителей можно использовать в реакции по Соногашира, такие как ДМФА, ТГФ, 2-Ме-ТГФ, CH3CN, ДМСО, толуол и 1,4-диоксан.
Несмотря на то, что KCN является предпочтительным реагентом превращения 4 в 5, также можно использовать другие реагенты для удаления защитных групп. Неограничивающие примеры подобных реагентов включают гидроксид, алкоголяты, HF, KF, NaF, Bu4NF и другие соли HF-амина. Для реакции можно использовать широкий диапазон растворителей.
Условия (реакции по Соногашира), которые можно использовать для превращения 5 в A, и для всех реакций по Соногашира, описанных здесь, похожи на условия превращения 3/7 в 4.
Согласно настоящему изобретению предложены другие способы получения соединения A, как показано на Схеме 2.
Схема 2
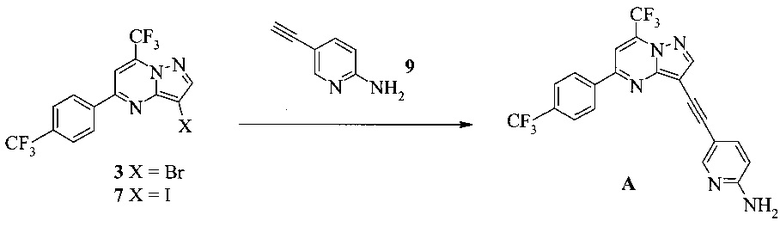
На Схеме 2 соединение 3 или 7 связывают с 2-амино-5-этинил-пиридином (соединение 9), используя реакцию сочетания по Соногашира, что дает соединение A. Диапазон условий, которые можно использовать, схож с условиями Схемы 1.
Также изобретение включает способы получения соединения 3 и 7, как показано на Схеме 3.
Схема 3
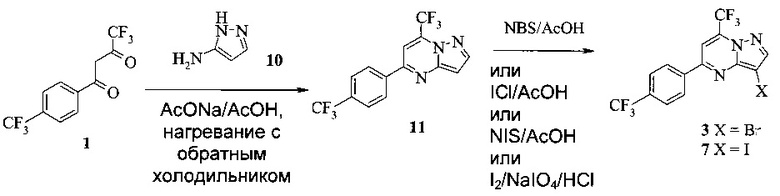
Согласно способам на Схеме 3: (а) конденсируют соединение 1 с 3-аминопиразолом (соединение 10) с образованием соединения 11; (b) подвергают взаимодействию соединение 11 с N-бромсукцинимидом (NBS), что дает соединение 3; или альтернативно, (с) подвергают взаимодействию соединение 11 с хлоридом йода (ICl), что дает соединение 7; или альтернативно, (d) подвергают взаимодействию соединение 11 с 14-йодсукцинимидом (NIS), что дает соединение 7; или альтернативно, (e) подвергают взаимодействию соединение 11 со смешанным реагентом I2/NaIO4/HCl, что дает соединение 7.
В одном воплощении изобретения предложен способ получения соединения, согласно которому подвергают взаимодействию соединение 11 с галогенирующим агентом, что дает соединение 3 или 7. В еще другом воплощении, когда галогенирующий агент представляет собой N-бромсукцинимид, то получают соединение 3, когда представляет собой хлорид йода, то получают соединение 7, когда представляет собой N-йодсукцинимид, то получают соединение 7, или когда представляет собой I2/NaIO4/HCl, то получают соединение 7. В еще другом воплощении соединение 11 получают при взаимодействии соединения 1 с соединением 10 в ходе реакции конденсации, что дает соединение 11.
Также можно использовать другие реагенты для превращения 1 в 11, например NaOEt/HOEt, АсОН / нагревание с обратным холодильником и толуол / перегонка.
Также изобретение включает способы получения соединения 9, как показано на Схеме 4.
Схема 4
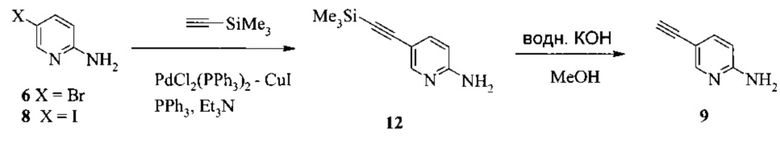
Согласно способам на Схеме 4: (а) связывают соединение 6 или 8 с триметилсилилацетиленом, используя реакцию сочетания по Соногашира, что дает соединение 12; и (b) осуществляют десилилирование соединения 12, что дает соединение 9.
Изобретение также включает способы получения соединения 9, как показано на Схеме 5.
Схема 5
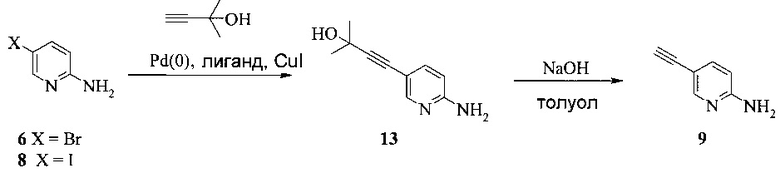
Согласно способам на Схеме 5: (a) связывают соединение 6 или 8 с 2-метил-3-бутин-2-олом, используя реакцию сочетания по Соногашира, что дает соединение 13; и (b) удаляют защитную группу соединения 13, что дает соединение 9.
Для удаления защитной группы также можно использовать сильные основания помимо NaOH. Неограничивающие примеры подобных оснований включают Na2CO3, K2CO3, KOH, K (или Na) OMe, K (или Na) OEt и K (или Na) Ot-Bu или их комбинации. Любой некислотный растворитель, который устойчив при сильных основных условиях, можно использовать для реакции, включая, но не ограничиваясь этим, простые спирты, эфиры и углеводороды.
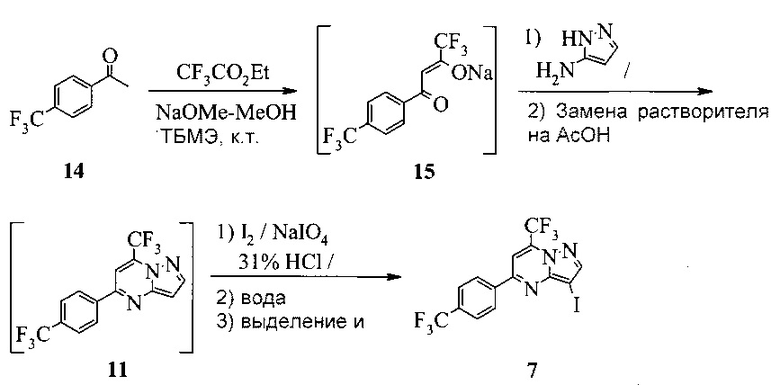
Также изобретение включает «однореакторный» способ (без выделения промежуточных соединений 15 и 11) получения соединения 7, исходя из 4'-(трифторметил)ацетофенона (соединение 14), как показано на Схеме 6.
Схема 6
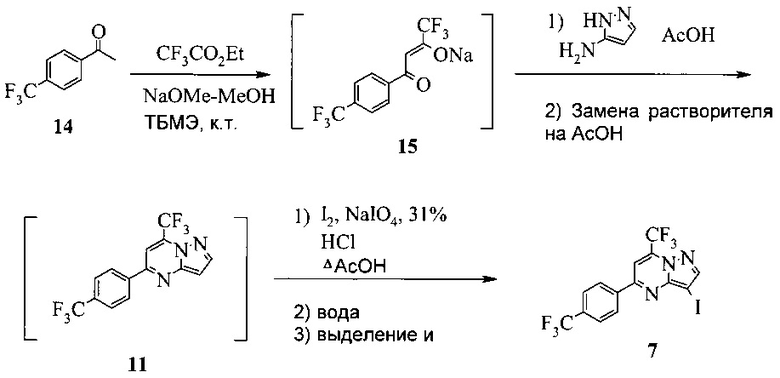
Согласно способу на Схеме 6: (a) подвергают взаимодействию соединение 14 с этил трифторацетатом в NaOMe/МеОН и трет-бутилметиловом эфире (метил-трет-бутиловом эфире, ТБМЭ, МТБЭ), что дает промежуточное соединение 15; (b) смешивают промежуточное соединение 15 с 3-аминопиразолом в уксусной кислоте, что дает промежуточное соединение 11; и (с) смешивают промежуточное соединение 11 со смесью I2/NaIO4/31% HCl (или NIS) в уксусной кислоте, что дает соединение 7. Данный «однореакторный» способ является экономически эффективным и рентабельным, поскольку исключаются обработка и выделение промежуточных соединений, что приводит к снижению расходования растворителя и времени изготовления.
В одном воплощении изобретения предложен способ получения соединения 7, согласно которому подвергают взаимодействию соединение 11 с йодирующим агентом при кислотных условиях, что дает соединение 7. В еще другом воплощении йодирующим агентом является N-йодсукцинимид или I2/NaIO4/HCl. В еще другом воплощении соединение 11 получают, подвергая взаимодействию соединение 15 с 3-аминопиразолом в инертном растворителе, что дает соединение 11. В еще другом воплощении соединение 15 получают, подвергая взаимодействию соединение 14 с этил трифторацетатом при основных условиях в инертном растворителе, что дает соединение 15.
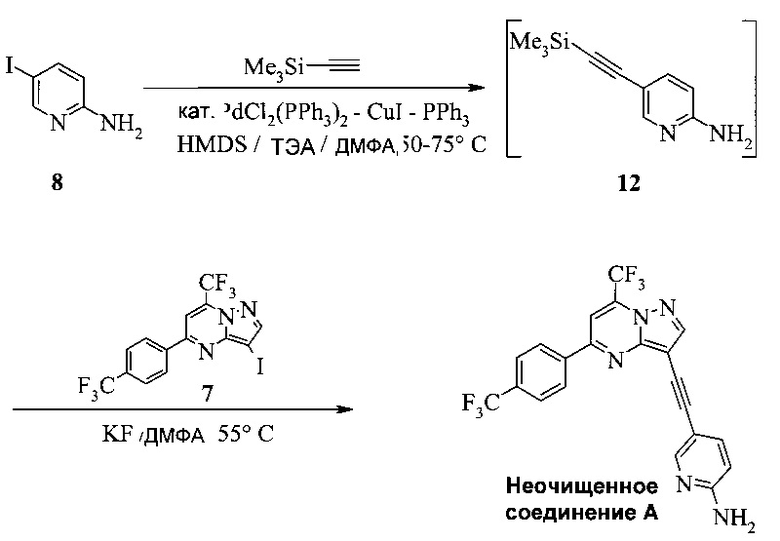
Кроме того, согласно настоящему изобретению предложены способы получения соединения A, как показано на Схеме 7, которые предпочтительно можно осуществлять в виде «однореакторных» способов (без выделения соединений 12 и 9).
Схема 7
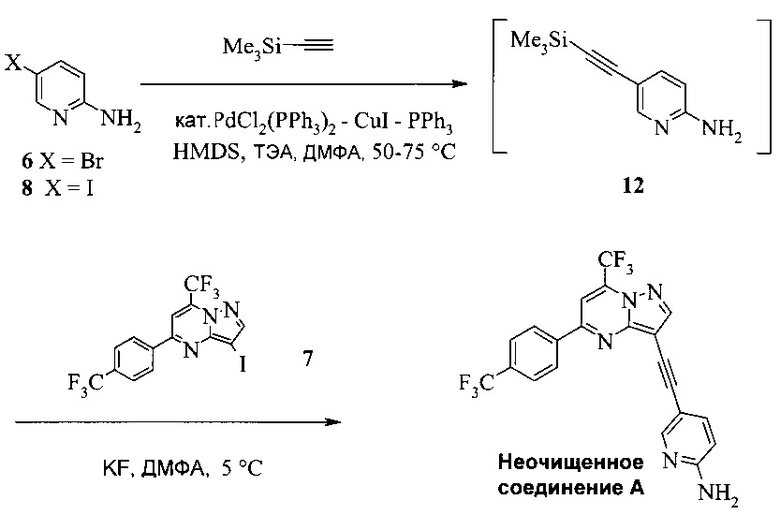
Согласно способам на Схеме 7: (а) подвергают взаимодействию соединение 6 или 8 с триметилсилилацетиленом, используя реакцию сочетания по Соногашира, что дает промежуточное соединение 12; (b) подвергают взаимодействию промежуточное соединение 12 с KF, чтобы десилилировать соединение 12 до соединения 9, в реакционной смеси; и (c) подвергают взаимодействию соединение 9 с соединением 7 в ходе второй реакции сочетания по Соногашира, что дает соединение А. Хотя не требуется, но HMDS (1,1,1,3,3,3-гексаметилдисилазан) предпочтительно использовать в качестве захватывающего воду агента на стадии (a), что делает реакцию сочетания быстрее и без примесей.
В одном воплощении изобретения предложен способ получения соединения А, имеющего формулу, согласно которому подвергают взаимодействию соединение 12 с соединением 7 в инертном растворителе в присутствии фторида калия, что дает соединение А. Реакцию соединения 12 с соединением 7 предпочтительно проводят в присутствии захватывающего воду агента, такого как 1,1,1,3,3,3-гексаметилдисилазан. В еще другом воплощении соединение 12 получают, подвергая взаимодействию соединение 6 или 8 с триметилсилилацетиленом в ходе реакции сочетания по Соногашира в инертном растворителе, что дает соединение 12. В еще другом воплощении соединение 12 получают, подвергая взаимодействию соединение 6 или 8 с триметилсилилацетиленом в ходе реакции сочетания по Соногашира в инертном растворителе, что дает соединение 12.
Этот «однореакторный» способ на Схеме 7 обладает несколькими преимещуствами. Например, выделение промежуточных соединений 12 и 9 исключено. Две реакции сочетания по Соногашира проводят, используя одну группу каталитической системы [PdCl2(PPh3)2-CuI]. Предпочтительно к смеси добавляют воду в конце стадии (с), чтобы неочищенное соединение A выпало в осадок.
Кроме того, согласно настоящему изобретению предложены способы получения соединения A, как показано на Схеме 8.
Схема 8
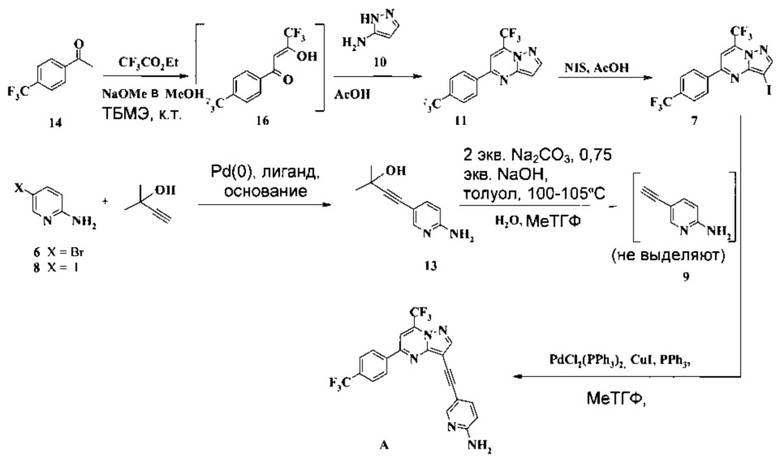
Согласно способам на Схеме 6: (a) связывают соединение 7 с соединением 9, используя реакцию сочетания по Соногашира, что дает неочищенное соединение A, которое затем можно очистить, например, в ходе перекристаллизации в смеси 2-метилтетрагидрофурана и изопропилового спирта.
Согласно способу получения соединения 7: (a) подвергают взаимодействию соединение 14 с этил трифторацетатом в NaOMe/МеОН и трет-бутилметиловом эфире (метил-трет-бутиловом эфире, ТБМЭ, МТБЭ) при комнатной температуре, что дает промежуточное соединение 16; (b) смешивают промежуточное соединение 16 с 3-аминопиразолом в уксусной кислоте, что дает промежуточное соединение 11; (с) смешивают промежуточное соединение 11 с N-йодсукцинимидом (NIS) в уксусной кислоте, что дает промежуточное соединение 7.
Согласно способу получения соединения 9: (а) связывают 2-амино-5-галопиридин (соединение 6 или 8) с 2-метил-3-бутин-2-олом в ходе реакции сочетания по Соногашира, что дает соединение 13 и (b) удаляют защитную группу соединения 13, что дает соединение 9.
Способ получения соединения 13 из Соединения 8 предложен на Схеме 9.
Схема 9
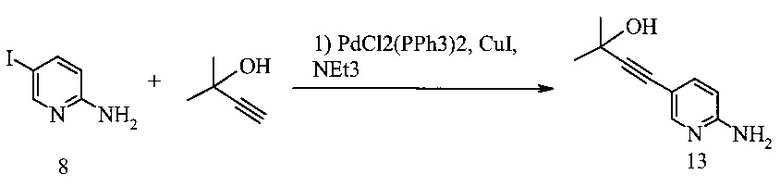
В одном воплощении получение соединения 13 включает (а) взаимодействие 2-амино-5-йодпиридина 8 с 2-метил-3-бутин-2-олом в ходе реакции сочетания по Соногашира при использовании катализатора PdCl2(PPh3)2-CuI с триэтиламином в качестве основания в тетрагидрофуране или 2-метилтетрагидрофуране при температуре около 50°c в течение приблизительно 5 часов.
Способ получения соединения 13 из Соединения 6 предложен на Схеме 10.
Схема 10

В другом воплощении получение соединения 13 включает (а) взаимодействие смеси 2-амино-5-бромпиридина 6; 2-метил-3-бутин-2-ола и катализатора Pd(OAc)2/PPh3-CuI в диизопропиламине при температуре около 80°C в течение 8-22 часов.
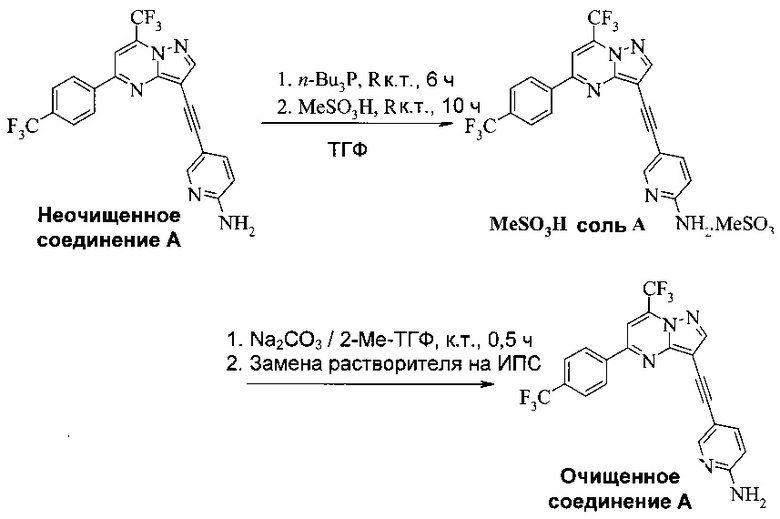
Соединение А может содержать примеси, такие как соли палладия/меди и органические и неорганические примеси. Согласно настоящему изобретению предложены новые способы очистки соединения А, как показано на Схеме 11.
Схема 11
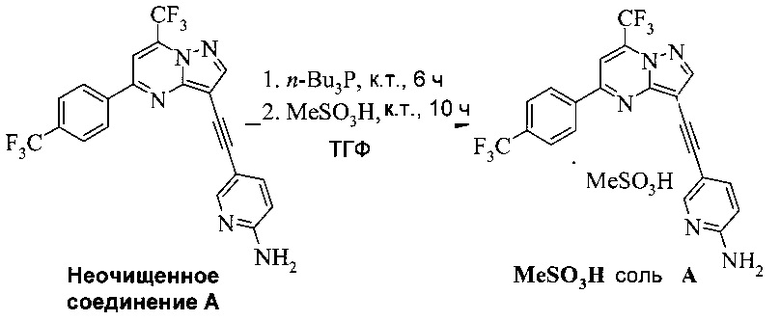
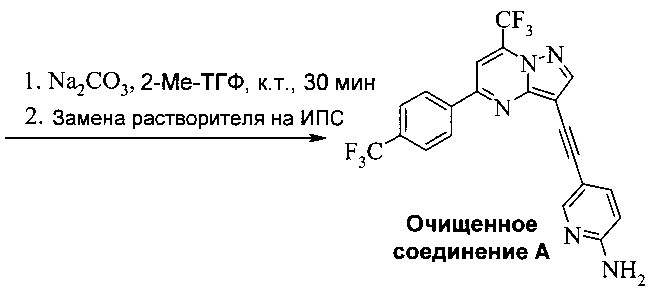
Эти способы очистки включают очистки, опосредованные мезилатной солью, согласно которым: (a) растворяют неочищенное соединение A в тетрагидрофуране (ТГФ) с образованием раствора; (b) обрабатывают раствор со стадии (а) н-трибутилфосфином, чтобы удалить палладий и медь; (c) добавляют метансульфоновую кислоту к реакционной смеси со стадии (b) с образованием мезилатной соли соединения A (соединение A-1), которая выпадает в осадок из раствора; (d) выделяют мезилатную соль со стадии (c); (e) суспендируют мезилатную соль со стадии (d) в 2-метилтетрагидрофуране (2-Me-ТГФ); (f) обрабатывают органическую 2-метилтетрагидрофурановую реакционную смесь со стадии (e) водным карбонатом натрия, чтобы превратить мезилатную соль в свободное основание соединения А, что приводит к водной фазе и органической фазе, содержащей свободное основание соединения A; (g) разделяют водную и органическую фазы и промывают органическую фазу водой; и (h) осуществляют замену растворителя в органической фазе с 2-Me-ТГФ на изопропанол (ИПС), что дает чистое кристаллическое соединение A.
В одном воплощении изобретения предложен способ получения соединения A-1, имеющего формулу
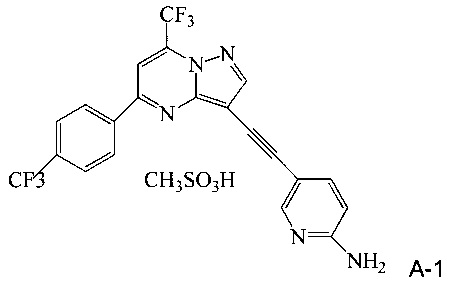
согласно которому (a) растворяют соединение A в тетрагидрофуране с образованием раствора; (b) обрабатывают раствор со стадии (a) н-трибутилфосфином; и (c) добавляют метансульфоновую кислоту к реакционной смеси со стадии (b), чтобы осадить соединение A-1.
В еще другом воплощении изобретения предложен способ очистки соединения A, согласно которому (a) суспендируют соединение A-1 в 2-метилтетрагидрофуране с образованием органической смеси; (b) обрабатывают органическую смесь со стадии (а) водным щелочным раствором, чтобы превратить соединение A-1 в свободное основание соединения A; (c) разделяют водную и органическую фазы со стадии (b) и промывают органическую фазу водой; и (d) отделяют и концентрируют органическую фазу со стадии (c), что дает очищенное соединение А. Предпочтительно водный щелочной раствор представляет собой водный раствор карбоната натрия. Предпочтительно согласно способу, кроме того, осуществляют замену растворителя в органической фазе после стадии (c), чтобы изменить растворитель на изопропанол, что дает кристаллическое соединение A.
Несмотря на то, что н-трибутилфосфин является наиболее эффективным реагентом для удаления палладия и меди, в настоящей реакции можно использовать многие другие фосфины с формулой PR3, где R может представлять собой простые алкильные группы (например, Me, Et, н-пропил и т.п.) или арильные группы (например, фенил, п-толил и т.п.).
Несмотря на то, что метансульфоновая кислота является предпочтительной кислотой для солеобразования, можно использовать многие другие кислоты. Неограничивающие примеры подобных кислот включают HCl, p-TsOH, H2SO4, H3PO4 и HBr.
Несмотря на то, что ТГФ является предпочтительным растворителем для превращения, также можно использовать многие другие инертные растворители. Неограничивающие примеры подобных растворителей включают 2-МеТГФ, этилацетат, метил-трет-бутиловый эфир и ацетонитрил.
Несмотря на то, что карбонат натрия является предпочтительным основанием для превращения MeSO3H соли A в чистое соединение A, также можно использовать другие основания. Неограничивающие примеры подобных оснований включают КОН, NaOH, K2CO3 и Cs2CO3.
Несмотря на то, что 2-Me-ТГФ является предпочтительным растворителем для превращения, также можно использовать многие другие инертные растворители. Неограничивающие примеры подобных растворителей включают ТГФ, этилацетат и метил-трет-бутиловый эфир.
Соединения по настоящему изобретению можно получить согласно примерам, представленным ниже. Примеры представлены в целях демонстрации, а не ограничения, получения соединений по данному изобретению.
Примеры
Согласно настоящему изобретению приведены следующие примеры для иллюстрации предпочтительных способов получения соединений по данному изобретению.
Пример 1
Этот пример иллюстрирует способ получения соединения 4.
Раствор 3 (1,64 г, 4 ммоль), триметилсилилацетилена (1,13 мл, 8 ммоль), комплекса бромида меди(I) - диметилсульфида (34 мг, 0,16 ммоль), ацетата палладия(II) (19 мг, 0,08 ммоль), трифенилфосфина (87 мг, 0,32 ммоль) в дегазированном триэтиламине (16 мл) перемешивали в атмосфере аргона при 50°C в течение 22 часов. Коричневую суспензию разбавляли этилацетатом (EtOAc, 20 мл) и выпаривали при пониженном давлении (200-20 мбар) при 45°C. Остаток распределяли между EtOAc (20 мл) и водой (20 мл) и фильтровали через воронку из спеченного стекла, чтобы удалить нерастворимое вещество. Органический слой отделяли, промывали водой (20 мл), водную фазу снова экстрагировали EtOAc (20 мл) и объединенные органические экстракты сушили над сульфатом натрия (Na2SO4), фильтровали и выпаривали. Коричневый остаток (2,0 г) очищали с помощью растворения в уксусной кислоте (AcOH, 12 мл) при 70°C и осадок выпадал при добавлении через 5 минут воды (5 мл). Густую оранжевую суспензию охлаждали и перемешивали при комнатной температуре (к.т.) в течение 1 часа, затем фильтровали. Продукт 4 промывали дважды водной AcOH (1:1, 3 мл) и один раз водой (5 мл). После сушки при 45°C/20 мбар в течение 2 часов получали 1,68 г (98%, ГЖХ 96%, ВЭЖХ 89% чистоты) оранжевого кристаллического твердого соединения 4.
Пример 2
Этот пример иллюстрирует другой способ получения соединения 4.
Раствор 7 (3,66 г, 8 ммоль), триметилсилилацетилена (2,26 мл, 16 ммоль), комплекса бромида меди(I) - диметилсульфида (67 мг, 0,32 ммоль), ацетата палладия(II) (37 мг, 0,16 ммоль), трифенилфосфина (173 мг, 0,64 ммоль) в дегазированном триэтиламине (30 мл) перемешивали в атмосфере аргона при 50°C в течение 20 часов. Коричневую суспензию разбавляли этилацетатом (EtOAc, 20 мл) и выпаривали при пониженном давлении (200-20 мбар) при 45°C. Остаток распределяли между EtOAc (20 мл) и водой (20 мл) и фильтровали через воронку из спеченного стекла, чтобы удалить нерастворимое вещество. Органический слой отделяли, промывали водой (20 мл), водную фазу повторно экстрагировали EtOAc (20 мл) и объединенные органические экстракты сушили над Na2SO4, фильтровали и выпаривали. Коричневый остаток (3,7 г) очищали в ходе растворения в AcOH (22 мл) при 100°C и осадок выпадал при добавлении через 10 минут воды (8 мл). Густую оранжевую суспензию охлаждали и перемешивали при к.т.в течение 1 часа, затем фильтровали. Продукт 4 промывали дважды водной AcOH (1:1, 10 мл) и дважды водой (10 мл). После сушки при 45°C/20 мбар в течение 16 часов получали 3,0 г (87%, ГЖХ 97% чистоты) оранжевого кристаллического твердого соединения 4.
Пример 3
Этот пример иллюстрирует способ получения соединения 5.
Промежуточное соединение 4 (3,0 г, 7 ммоль) растворяли в смеси ТГФ (8 мл) и MeOH (12 мл). Цианид калия (0,49 г, 7,3 ммоль) добавляли, и темно коричневый раствор перемешивали в атмосфере аргона при к.т.в течение 16 часов, затем выпаривали при пониженном давлении. Остаток распределяли между EtOAc (20 мл) и водой (20 мл). Органическую фазу отделяли и промывали водой (20 мл), сушили над Na2SO4, фильтровали и выпаривали при пониженном давлении. Остаток (2,82 г) очищали при растворении в AcOH (21 мл) при к.т.и осадок выпадал при добавлении через 10 минут воды (7 мл). Бежевую суспензию перемешивали при к.т.в течение 1 часа, затем фильтровали. Продукт 5 промывали дважды водной AcOH (1:1, 10 мл) и дважды водой (10 мл). После сушки при 45°C/20 мбар в течение 16 часов получали 2,0 г (80%, ВЭЖХ 96% чистоты) бежевого кристаллического твердого соединения 5. Фильтрат снова фильтровали, получая дополнительное вещество (0,3 г, 11%, ВЭЖХ 77%) соединения 5.
Пример 4
Этот пример иллюстрирует способ получения соединения A.
Ацетилен 5 (53 мг, 0,15 ммоль) растворяли в дегазированном морфолине (1 мл) и обрабатывали последовательно 2-амино-5-йодпиридином (35 мг, 0,16 ммоль), тетракис(трифенилфосфин)палладием (4 мг, 0,003 ммоль) и комплексом бромида меди(I) - диметилсульфида (1 мг, 0,006 ммоль). Коричневый раствор перемешивали в атмосфере аргона при 75°C в течение 0,5 часа, затем распределяли между EtOAc (10 мл) и водой (10 мл). Органический слой отделяли, водную фазу экстрагировали EtOAc (5 мл) и объединенные органические экстракты промывали водой (5 мл), затем сушили над Na2SO4, фильтровали и выпаривали, получая красно-коричневый остаток (80 мг) соединения A. ВЭЖХ анализ показан 95% чистоты соединения A.
Пример 5
Этот пример иллюстрирует способ получения неочищенного соединения А и его очистку через HCl-соль соединения A.
1,5 литровую 4-горлую круглодонную колбу, оборудованную термометром, механической мешалкой и подводом инертного газа, заполняли 67,3 мл триэтиламина (483,3 ммоль, 2,6 экв.), 1,33 г (1,86 ммоль, 0,01 экв.) бис-(трифенилфосфин)-палладия(II) - дихлорида, 1,03 г (3,72 ммоль, 0,02 экв.) трифенилфосфина, 0,37 г (1,9 ммоль, 0,01 экв.) иодида меди(I) и 170 мл ДМФА. Смесь нагревали до 75°C, получая темно-коричневый раствор. К этому раствору добавляли раствор 85 г (185,9 ммоль) 7 и 24,16 г (204,5 ммоль, 1,1 экв.) 9 в 340 мл ДМФА, содержащих 2,6 мл (18,6 ммоль, 0,1 экв.) триэтиламина, в течение 1 часа при 68-72°C через капельную воронку. Капельную воронку промывали 40 мл ДМФА и продолжали перемешивать в течение 16 часов до завершения реакции. Полученную в результате темно-красную суспензию выпаривали при пониженном давлении (120-50 мбар) при 80°C, удаляя ~170 мл растворителя. Кристаллизацию вызывали, добавляя 850 мл воды при комнатной температуре в течение ~0,5 часа. Красную суспензию перемешивали при комнатной температуре в течение 19 часов, фильтровали, промывали 500 мл воды, затем сушили при 50°C при <10 мбар в течение 48 часов, получая 87,3 г (84%) неочищенного соединения A.
Неочищенное соединение A в количестве 70 г растворяли в 650 мл ТГФ. Раствор разбавляли 650 мл трет-бутилметилового эфира и фильтровали через 490 г нейтрального оксида алюминия III, элюируя смесью, полученной из 1300 мл ТГФ и 1300 мл трет-бутилметилового эфира. Фильтрат концентрировали до объема 400 мл. Затем 325 мл ТГФ добавляли с постоянным объемом перегонки. Добавляли 3,5 г (21,2 ммоль, 0,14 экв.) N-ацетил-L-цистеина и раствор перемешивали при комнатной температуре в течение 1 часа. Добавляли 45 мл (180 ммоль, 1,15 экв.) 4 н гидрохлорида и продолжали перемешивать при комнатной температуре в течение 16 часов. Образовавшуюся кристаллическую суспензию фильтровали и желтый влажный осадок на фильтре промывали 150 мл трет-бутилметилового эфира, что давало HCl-соль соединения A.
100 г (207 ммоль) суспензии неочищенной HCl-соли соединения А в 700 мл ТГФ обрабатывали раствором 34,8 г (414 ммоль, 2 экв.) карбоната натрия в 600 мл воды. Через 0,5 часа добавляли дополнительных 800 мл воды, и кристаллизация завершалась при комнатной температуре в течение 2 часов. Продукт фильтровали и промывали смешанным растворителем 150 мл ТГФ и 300 мл воды, задем 300 мл воды и затем 200 мл 2-пропанола. Продукт влажного осадка на фильтре растворяли в 1480 мл ацетона, и раствор концентрировали до объема 470 мл. После этого добавляли 600 мл 2-пропанола с постоянным объемом перегонки. Кристаллическую суспензию перемешивали при комнатной температуре в течение 16 часов, фильтровали, промывали 600 мл 2-пропанола, затем сушили при 50°C при <10 мбар в течение 24 часов, получая чистое соединение A.
Пример 6
Этот пример иллюстрирует способ получения соединения A.
Раствор 7 (36,0 г), иодида меди(I) (334 мг), бис-(трифенилфосфин)-палладия(II) - дихлорида (557 мг), трифенилфосфина (414 мг) и триэтиламина (25,5 мл) в МеТГФ (145 мл) обрабатывали при 70-75°C в течение 2-3 часов МеТГФ раствором 1,1 эквив. 5-этинилпиридин-2-иламина (9) (полученным согласно Примеру 11) и полученную в результате суспензию потом перемешивали при 70-75°C в течение дополнительных 5-10 часов. Смесь охлаждали до 30°C и обрабатывали водой (150 мл) и 25% водным раствором гидроксида аммония (30 мл). Двухфазную смесь перемешивали в течение 30 минут и слои затем разделяли в течение 20 минут. Водный слой удаляли и МеТГФ слой промывали дважды смесью воды (150 мл) и 25% водного раствора гидроксида аммония (30 мл). МеТГФ слой затем промывали водой (3×150 мл). Органический слой осветляли в ходе фильтрации, и фильтрат обрабатывали н-трибутилфосфином (1,00 мл). МеТГФ затем отгоняли и полностью заменяли на изопропанол (всего 500 мл) при атмосферном давлении. Полученную в результате суспензию (около 250 мл) нагревали с обратным холодильником и перемешивали при нагревании с обратным холодильником в течение 2 часов, затем охлаждали до комнатной температуры в течение ночи. Продукт фильтровали и промывали двумя порциями изопропанола (50 мл). Влажные кристаллы сушили при 50°C и <30 мбар до постоянного веса, получая 29,45 г (выход 84% на основе 7) соединения A в виде красных кристаллов с чистотой 99,7% (ВЭЖХ, площадь-%).
Пример 7
Этот пример иллюстрирует способ получения соединения 7.
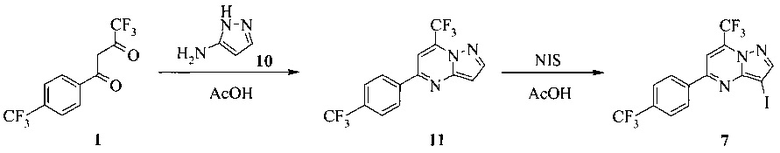
1,5 литровую 4-горлую круглодонную колбу, оборудованную термометром, механической мешалкой и подводом инертного газа, заполняли раствором 1 (85,2 г, 300 ммоль) в 250 мл уксусной кислоты. К смеси добавляли раствор 19,3 г (232 ммоль) 3-аминопиразола (10) и 270 мл уксусной кислоты. Реакционную смесь перемешивали при комнатной температуре в течение 20 часов. Продукт осаждали, добавляя 550 мл воды. Полученную в результате взвесь выдерживали в течение 20 часов. Твердое вещество фильтровали, промывали 300 мл воды, затем сушили при 50°C под <10 мбар в течение 72 часов, получая 74 г соединения 11.
1,5 литровую 4-горлую круглодонную колбу, оборудованную термометром, механической мешалкой и подводом инертного газа, заполняли 71,3 г (215 ммоль) 11 и 715 мл уксусной кислоты. Добавляли 53,52 г (226 ммоль, 1,05 экв.) N-йодсукцинимида (NIS) за один раз и желтую суспензию перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь разбавляли 715 мл воды, чтобы осадить продукт. Взвесь выдерживали в течение 20 часов. Продукт фильтровали, промывали 680 мл воды и сушили при 50°C под <10 мбар в течение 48 часов, что давало 96,7 г соединения 7.
Пример 8
Этот пример иллюстрирует способ получения соединения 3.
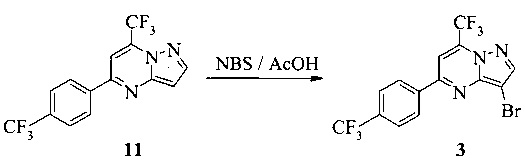
К раствору соединения 11 (1,0 г, 3,0 ммоль) в AcOH (10 мл) добавляли бромсукцинимид (NBS, 0,58 г, 3,2 ммоль). Раствор перемешивали при к.т. в течение 1 часа, затем разбавляли водой (10 мл). Желтую суспензию охлаждали до 5°C, перемешивали в течение 0,5 часа, затем фильтровали. Кристаллический продукт промывали водой (20 мл), затем сушили в течение 16 часов при 40°C/20 мбар, что давало соединение 3 (1,15 г) с чистотой 100% (ВЭЖХ, площадь-%).
Пример 9
Этот пример иллюстрирует способ получения соединения 12.
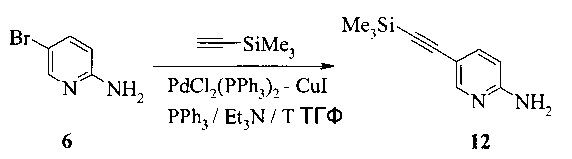
2,5 литровую 4-горлую круглодонную колбу, оборудованную термометром, механической мешалкой и подводом инертного газа, заполняли последовательно 950 мл триэтиламина, 100 г (566,4 ммоль) 2-амино-5-бромпиридина (соединение 6), 8,11 г (11,3 ммоль, 0,02 экв.) бис-(трифенилфосфин)-палладия(II) - дихлорида, 6,26 г (22,7 ммоль, 0,04 экв.) трифенилфосфина и 2,2 г (11 ммоль, 0,02 экв.) иодида меди(I). К перемешиваемой смеси добавляли 96,5 г триметилсилилацетилена (963 ммоль, 1,7 экв.). Полученную в результате темно-коричневую суспензию перемешивали при 75°C в течение 17 часов. При охлаждении до комнатной температуры реакционную смесь фильтровали через 40 г Дикалита с помощью 300 мл триэтиламина. Фильтрат выпаривали при 50°C при пониженном давлении 200 мбар до объема ~600 мл, затем при 200-180 мбар при постоянной замене объема с 850 мл н-гептана. При охлаждении до комнатной температуры добавляли дополнительные 200 мл н-гептана, и кристаллизация завершалась при комнатной температуре в течение 16 часов. Продукт фильтровали, промывали 400 мл н-гептана и сушили при 45°C под <10 мбар в течение 3 часов, что давало 90,4 г неочищенного соединения 12. Неочищенное соединение 12 очищали с помощью колоночной хроматографии (силикагель), элюируя этилацетатом и н-гептаном (2:1, об/об), что давало 83,4 г соединения 12.
Пример 10
Этот пример иллюстрирует способ получения соединения 9.
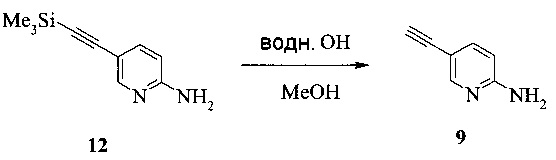
1,5 литровую 4-горлую круглодонную колбу, оборудованную термометром, механической мешалкой и подводом инертного газа, заполняли 80 г (420,3 ммоль) соединения 12, 700 мл этанола и раствором 2,7 г (41,1 ммоль, 0,1 экв.) гидроксида калия в 10 мл воды. Смесь перемешивали при комнатной температуре в течение 1 часа. Смесь концентрировали при 50°C под 260-230 мбар, чтобы удалить около 500 мл растворителя. 750 мл воды добавляли и оставшийся EtOH выпаривали при 60°C под 65 мбар. Смесь экстрагировали один раз 750 мл, дважды 250 мл, всего 1250 мл этилацетата. Объединенные органические фазы, содержащие некоторое нерастворимое вещество, сушили над 80 г сульфата натрия, фильтровали, и влажный осадок на фильтре промывали 50 мл этилацетата. Фильтрат выпаривали при пониженном давлении (240 мбар) при 50°C до объема -350 мл. н-Гептан в количестве 700 мл добавляли с постоянной заменой объема при 50°C под 240-210 мбар. Добавляли дополнительные 100 мл н-гептана, и кристаллизация завершалась при комнатной температуре в течение 1 часа. Продукт фильтровали, промывали 350 мл н-гептана и сушили при комнатной температуре под <10 мбар в течение 3 часов, что давало 49 г соединения 9.
Пример 11
Этот пример иллюстрирует способ получения соединения 9.
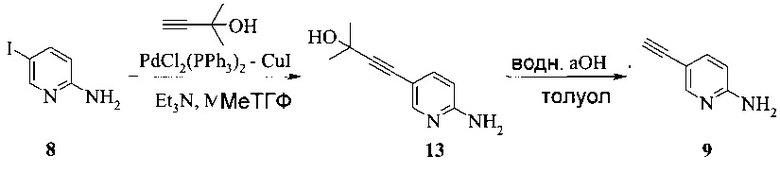
Раствор 2-амино-5-йодпиридина (48,0 г), иодида меди(I) (520 мг), бис-(трифенилфосфин)-палладия(II) - дихлорида (765 мг) и триэтиламина (32,0 г) в МеТГФ (220 мл) обрабатывали при 48-52°C в течение 2-3 часов раствором 2-метил-3-бутин-2-ола (30,0 г) в МеТГФ (40 мл) и полученную в результате суспензию затем перемешивали при 50°C в течение дополнительных 3 часов. Смесь охлаждали до 25-30°C, затем обрабатывали водой (160 мл) и 25% водным раствором гидроксида аммония (40 мл). Двухфазную смесь перемешивали при 25-30°C в течение 20 минут и слои разделяли через 20 минут. Водный слой удаляли и повторно экстрагировали МеТГФ (2×80 мл). Объединенные органические слои разбавляли водой и полученную в результате смесь подкисляли 37% водной соляной кислотой (около 43 г) до pH 1. Нижний водный слой, содержащий продукт, удаляли и МеТГФ-слой экстрагировали водой (1×100 мл). Из объединенных водных слоев МеТГФ отгоняли при пониженном давлении (200-80 мбар). Затем pH полученного в результате водного слоя (около 420 мл) доводили до pH 9-10, добавляя 28% водный раствор гидроксида натрия (около 37 г) при 15-20°C. При pH 5 продукт начинал осаждаться. Полученную в результате суспензию перемешивали в течение 2 часов при 15-20°C и продукт фильтровали и промывали двумя порциями воды (всего 300 мл). Влажную кристаллическую массу (около 46 г) сушили при 40°C и <30 мбар до постоянного веса, получая 33,56 г (выход 87%) соединения 13 в виде желто-бежевых кристаллов с чистотой 99,5% (ВЭЖХ, площадь-%).
Суспензию соединения 13 (17,10 г) и гидроксида натрия (12,2 г) в толуоле (170 мл) нагревали при пониженном давлении (прибл. 750 мбар) до 95-102°C в течение 30-60 минут (прибл. 110°C температура оболочки). Смесь затем перемешивали при этой температуре в течение 4-7 часов. После полного превращения (<2% исходного вещества) смесь охлаждали до 80-86°C и промывали при этой температуре три раза водой (45 мл и 2×30 мл). Объединенные водные слои повторно экстрагировали при комнатной температуре МеТГФ (80 мл). Объединенные толуольные и МеТГФ слои концентрировали почти досуха. Остаток растворяли в 120 мл МеТГФ и затем осветляли в ходе фильтрации. Осадок на фильтре промывали МеТГФ (2×25 мл), получая 157,16 г раствора соединения 9 с концентрацией 6,4% (масс/масс), соответствующей уточненному выходу 88%.
Пример 12
Этот пример иллюстрирует «однореакторный» способ получения соединения 7.
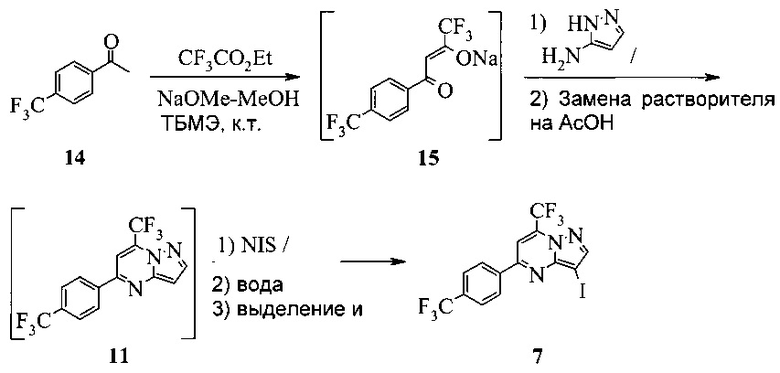
В сухую прозрачную 250 мл колбу Эрленмейера добавляли 45,32 г 4'-трифторметилацетофенона (14) и 94,8 г трет-бутилметилового эфира (МТБЭ). Полученную в результате смесь перемешивали при 20°C в азоте в течение 10-20 минут, что давало прозрачный раствор. Раствор переносили в 250 мл капельную воронку.
В сухую прозрачную 500 мл круглодонную колбу добавляли 70,0 г 25 масс.% метанольного раствора метилата натрия и 39,52 г этил трифторацетата. Полученную в результате смесь перемешивали в азоте и охлаждали до 20°C. Добавляли раствор 4'-трифторметилацетофенона через 15-30 минут. 250 мл колбу Эрленмейера промывали 7,4 г МТБЭ и добавляли раствор для промывки в пробу с помощью той же капельной воронки. Полученную в результате смесь перемешивали в азоте при 20±5°c в течение 3-4 часов, чтобы завершить превращение из 14 в 15. Этот раствор добавляли в 1000 мл 3-горлую круглодонную колбу, содержащую 336 г ледяной уксусной кислоты и 19,42 г 3-аминопиразола. Полученную в результате смесь перемешивали при 40-50°C в азоте в течение 2 часов, чтобы завершить превращение из 15 в 11. Пробу нагревали, чтобы отогнать ~344 г растворителя при атмосферном давлении (конечная температура бани около 115°C). К пробе добавляли 105 г ледяной уксусной кислоты и атмосферную перегонку продолжали до сбора ~105 г растворителя (конечная температура бани около 123°C). Смесь охлаждали до 40±5°C, получая раствор неочищенного соединения 11 в уксусной кислоте.
В 2000 мл 4-горлую круглодонную колбу с рубашкой, оборудованную высокой мешалкой, термопарой и воронкой, добавляли 62,3 г NIS и 336 г ледяной уксусной кислоты. Полученную в результате смесь перемешивали при 40°C, пока добавляли раствор неочищенного соединения 11. Перемешивать продолжали при 40±5°C в течение 3-4 часов. Температуру пробы затем увеличивали до 65±5°C и добавляли 750 г водного раствора тиосульфата натрия (13,7 г в 740 мл воды). Реакционную смесь перемешивали при 65±5°C в течение 1 часа, охлаждали до 20°C и выдерживали в течение 2 часов. Твердое вещество фильтровали через 600 мл воронку грубой фильтрации. Влажный осадок на фильтре промывали 700 г воды, сушили при 65±5°C в вакууме в течение ≥15 часов, получая 104 г соединения 7 в виде светло-желтого твердого вещества.
Пример 13
Этот пример иллюстрирует другой «однореакторный» способ получения соединения 7.
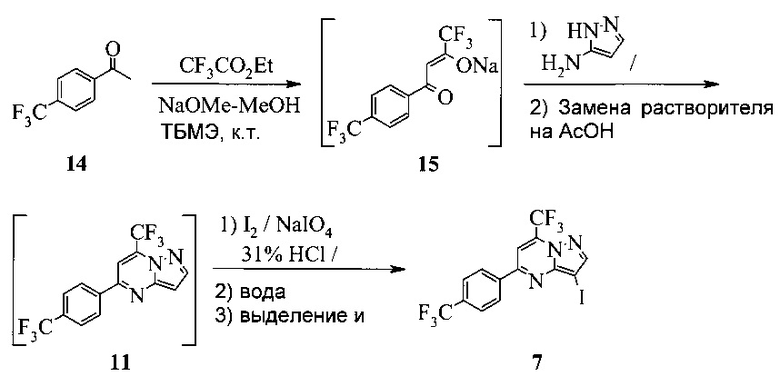
В сухую прозрачную 250 мл колбу Эрленмейера добавляли 45,32 г 4'-трифторметилацетофенона (14) и 94,8 г трет-бутилметилового эфира (МТБЭ). Полученную в результате смесь перемешивали при 20°C в азоте в течение 10-20 минут, что давало прозрачный раствор. Раствор переносили в 250 мл капельную воронку.
В сухую прозрачную 500 мл круглодонную колбу добавляли 70,0 г 25 масс.% метанольного раствора метилата натрия и 39,52 г этил трифторацетата. Полученную в результате смесь перемешивали в азоте и охлаждали до 20°C. Добавляли раствор 4'-трифторметилацетофенона через 15-30 минут. 250 мл колбу Эрленмейера промывали 7,4 г МТБЭ и добавляли раствор для промывки в пробу с помощью той же капельной воронки. Полученную в результате смесь перемешивали в азоте при 20±5°C в течение 3-4 часов, чтобы завершить превращение из 14 в 15. Этот раствор добавляли в 1000 мл 3-горлую круглодонную колбу, содержащую 315 г ледяной уксусной кислоты. К смеси добавляли раствор 19,42 г 3-аминопиразола в 49 г метанола. Полученную в результате смесь перемешивали при 40-50°C в азоте в течение 2 часов, чтобы завершить превращение из 15 в 11. Пробу нагревали, чтобы отогнать ~316 г растворителя при атмосферном давлении (конечная температура бани около 115°C). К пробе добавляли 189 г ледяной уксусной кислоты и атмосферную перегонку продолжали до сбора ~165 г растворителя (конечная температура бани около 126°C). Пробу охлаждали до 40±5°C, получая раствор неочищенного соединения 11 в уксусной кислоте.
В 2000 мл 4-горлую круглодонную колбу с рубашкой, оборудованную высокой мешалкой, термопарой и воронкой, добавляли 33,6 г йода, 8,82 г периодата натрия и 168 г ледяной уксусной кислоты. Полученную в результате смесь перемешивали при 40°C, пока добавляли раствор неочищенного соединения 11. Через 15 минут добавляли 47,5 г 31% водной соляной кислоты с помощью дополнительной воронки, поддерживая температуру пробы при 40±5°C.
Реакционную смесь перемешивали при 40±5°C в течение 1 часа. Температуру пробы затем повышали до 65±5°C и продолжали перемешивать в течение еще часа, чтобы завершить реакцию. Реакционную смесь охлаждали до 40°C и в течение 30-60 минут добавляли 180 г воды при 40±5°C. После дополнительного охлаждения через 1 час до 20°C добавляли 375 г водного раствора тиосульфата натрия (15,1 г в 360 мл воды) при 20°C в течение 30-60 минут. Пробу перемешивали в течение 2 часов при 20°C, затем фильтровали через 600 мл воронку грубой фильтрации. Влажный осадок на фильтре промывали 360 г воды, сушили при 65±5°C в домашнем вакууме в течение ≥15 часов и получали 105 г соединения 7 в виде светло-желтого твердого вещества.
Пример 14
Этот пример иллюстрирует способ получения неочищенного соединения A.
300 мл полимерную колбу, оборудованную охлаждающим устройством, механической мешалкой, обратным холодильником, подводом аргона, термопарой и перегородкой, заполняли 34,06 г 2-амино-5-бромпиридина (1,2 экв.), 2,76 г (PPh3)2PdCl2 (2,4 мол. %), 2,06 г PPh3 (4,8 мол. %), 0,75 г CuI (2,4 мол. %) и 257 г ТЭА (триэтиламина) (15,5 экв.). Смесь перемешивали при 20°C, получая желтую взвесь, затем нагревали до 75°C. К этой смеси добавляли частями 21,3 г ТМС-ацетилена (1,32 экв.). Полный объем разбивали на 4 приблизительно равные части, которые добавляли через t=0, 2, 4 и 6 часов. Пробу выдерживали при 75°C в течение 9-17 часов после завершения добавления ТМС-ацетилена (всего 15-22 часа). Пробу охлаждали до 30°C. Осуществляли вакуумную перегонку до приблизительно минимального перемешиваемого объема (конечная температура перегонки: рубашка <50°C, проба ~30°C, 100 мбар). Рубашку охлаждали до 20°C и добавляли 177 г ДМФА. Пробу фильтровали под давлением через наполненный целитом диск и колбу и осадок на фильтре промывали 71 г ДМФА, получая раствор неочищенного соединения 12.
500 мл реакционный сосуд в рубашке, оборудованный механической мешалкой, термопарой, подводом аргона и заглушкой, заполняли 75,0 г 7 (1,0 экв., ограничивающий реагент), 12,6 г фторида калия (1,32 экв.), 1,66 г ТЭА (10 мол. %) и 107 г ДМФА. Смесь перемешивали, что давало густую желто-зеленую взвесь. К этой смеси добавляли весь раствор 12. Пробу нагревали до 40°C со скоростью 1°C/мин, затем нагревали дополнительно до 55°C со скоростью 0,25°C/мин. Пробу выдерживали при 55°C в течение 2 часов. Пробу охлаждали до 20°C и медленно добавляли к 1240 г воды при 15±5°C, получая ржаво-оранжевую окрашенную взвесь. Полученную в результате смесь перемешивали и выдерживали в течение 2 часов при 15°C, получая темную кроваво-красную окрашенную взвесь. Твердое вещество фильтровали. Реакционный сосуд и влажный осадок на фильтре промывали 450 г воды. Осадок на фильтре сушили при 50°C в вакууме при отборе воздуха в течение ночи, получая неочищенное соединение A в виде темно-красного немного комковатого порошка.
Пример 15
Этот пример иллюстрирует другой способ получения неочищенного соединения А.
300 мл полимерную колбу, оборудованную охлаждающим устройством, механической мешалкой, обратным холодильником, подводом аргона, термопарой и перегородкой, заполняли 11,1 г 2-амино-5-йодпиридина (1,15 экв.), 0,737 г (PPh3)2PdCl2 (2,4 мол. %), 0,551 г PPh3 (4,8 мол. %), 0,20 г CuI (2,4 мол. %) и 94,4 г ДМФА (N,N-диметилформамида). Смесь перемешивали при 20°C, получая светло-янтарную суспензию. К этой смеси добавляли 1,1,1,3,3,3-гексаметилдисилазан (0,24 экв.). К полученному в результате темно-янтарному раствору добавляли 13,3 г ТЭА (триэтиламина) (3,0 экв.). Реакционную смесь нагревали до 50°C и 4,94 г ТМС-ацетилена (1,15 экв.) добавляли частями. Общее количество разделяли на 2 приблизительно равные части, которые добавляли при t=0 и 1 час. Пробу выдерживали при 50°C в течение 3 часов после завершения добавления ТМС-ацетилена (всего 4 часа), затем охлаждали до 20°C, получая раствор неочищенного соединения 12.
К пробе добавляли 20,0 г 7 (1,0 экв.) и 4,32 г фторида калия (1,70 экв.). Твердое вещество, которое оставалось на воронке, промывали ДМФА (~5 мл). Пробу нагревали до 40°C со скоростью 1°C/мин и затем нагревали дополнительно до 55°C со скоростью 0,25°C/мин. Реакционную смесь выдерживали при 55°C в течение 2 часов, охлаждали до 20°C, затем медленно добавляли к 330 г воды в течение ~30 минут, поддерживая температуру пробы при 15±5°C. Образовывалась ржаво-оранжевая окрашенная взвесь, которая темнела до темного кроваво-красного цвета. Пробу перемешивали, выдерживали в течение 2 часов при 15°C, затем фильтровали. Реакционный сосуд и влажный осадок на фильтре промывали 120 г воды. Осадок на фильтре сушили при 50°C в вакууме при отборе воздуха в течение ночи, получая 21,7 г неочищенного соединения A в виде темно-красного сыпучего порошка.
Пример 16
Этот пример иллюстрирует способ получения чистого соединения A.
500 мл четырехгорлый реакционный сосуд с рубашкой (Реакционный сосуд 1), оборудованный высокой мешалкой, датчиком температуры, обратным холодильником и подводом азота, заполняли 35,78 г неочищенного соединения A (80 ммоль, 1,0 экв.) и 160 мл (140,8 г) ТГФ. Начинали перемешивать, получая темно-коричневый раствор, и 20 мл (16,2 г) н-трибутилфосфина (80 ммоль, 1,0 экв.) добавляли с помощью шприца в течение ~5 минут при комнатной температуре. Смесь перемешивали в течение 6 часов при комнатной температуре, затем добавляли 8 мл (11,8 г) метансульфоновой кислоты (120 ммоль, 1,5 экв.) по каплям в течение ~5 минут. Желто-коричневую взвесь перемешивали при комнатной температуре в течение 10 часов, фильтровали через 350 мл фильтр из тонкого стекла и желтый влажный осадок на фильтре промывали 2×30 мл (26,4 г) ТГФ. Метансульфоновую соль сушили при 40°C под полным ваккумом в течение ночи (получали 28,19 г).
Сухую соль добавляли в 500-мл реакционный сосуд (Реакционный сосуд 2) и добавляли 220 мл (189,2 г) 2-Me-ТГФ. Начинали перемешивать, получая желтую взвесь. В колбе Эрленмейера растворяли 12,71 г карбоната натрия (120 ммоль, 1,5 экв.) в 140 мл (140 г) ДИ воды и этот основный раствор добавляли по каплям к желтой взвеси. Твердые вещества растворялись в процессе добавления, давая темно-коричневый раствор (проверка pH: >9). Двухфазный раствор перемешивали при комнатной температуре в течение ~30 минут и пробу фильтровали через заполненный целитом диск. Колбу и подложку из целита промывали 2×30 мл (25,8 г) 2-Me-ТГФ. Фильтрат и жидкость для промывки переносили обратно в реакционный сосуд и слои разделяли. Нижний водный слой сливали, и верхний органической слой промывали 140 мл (140 г) деионизованной (ДИ) воды. Органический слой помещали обратно в прозрачный 500 мл реакционный сосуд (Реакционный сосуд 3) и растворитель отгоняли при пониженном давлении (620 мбар, температура рубашки: 70°C), собирая до ~100 мл (~80 г) дистиллята. Вакуум освобождали и 100 мл (78,5 г) 2-пропанола добавляли по каплям к пробе в течение ~15 минут. Перегонку при пониженном давлении (530 мбар, температура рубашки: 70°C) продолжали до сбора ~190 мл (~160 г) дистиллята. Вакуум освобождали снова и 160 мл (125,6 г) 2-пропанола добавляли по каплям в течение ~15 минут.
После перегонки при пониженном давлении (400 мбар, температура рубашки: 70°C) удаляли дополнительные ~160 мл (~130 г) дистиллята. Вакуум освобождали последний раз и 160 мл (125,6 г) 2-пропанола добавляли по каплям к пробе в течение ~15 минут. Смесь нагревали до ~80°C, перемешивали в течение 2 часов, затем охлаждали до 20°C. Ярко-оранжевую взвесь перемешивали при этой температуре в течение 10 часов и продукт фильтровали через 350 мл фильтр из тонкого стекла. Влажный осадок на фильтре промывали 2×30 мл (23,6 г) 2-пропанола и продукт сушили при 50°C под полным вакуумом в течение ночи, получая 21,8 г очищенного соединения A.
Пример 17
Этот пример иллюстрирует способ получения соединения 13 из соединения 6.
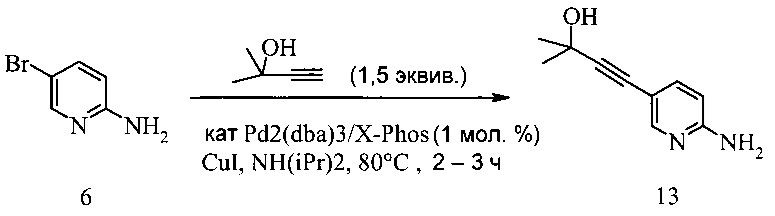
100 мл 4-горлую колбу заполняли 5-бром-2-аминопиридином (5 г, 28,9 ммоль) и диизопропиламином (61,8 мл, 433 ммоль, 15 эквив.). Смесь дегазировали при введении аргона, в то же время последовательно добавляли трис(дибензилиденацетон)дипалладий(0) (132 мг, 144 мкмоль, 1 мол. % Pd), X-Phos (165 мг, 347 мкмоль, 1,2 мол. %), иодид меди (110 мг, 578 мкмоль, 2 мол. %) и 2-метил-3-бутин-2-ол (3,65 г, 43,3 ммоль, 1,5 эквив.). Реакционную смесь нагревали до 80°C и перемешивали при этой температуре в течение 2 часов. Затем нагревание останавливали и черную суспензию охлаждали на водяной ванне до температуры окружающей среды. К этой суспензии добавляли 2-Me-ТГФ (30 мл) и воду (20 мл). Фазы разделяли, водную фазу промывали 2-Me-ТГФ (50 мл) и органическую фазу промывали водой (50 мл). Из объединенных органических фаз растворитель удаляли под вакуумом досуха. Темный остаток суспендировали в воде (30 мл), добавляли HCl (4 мл, 25%) (~pH 1) и смесь перемешивали в течение 30 минут. Темные нерастворимые остатки отфильтровывали и желтый раствор обрабатывали NaOH (4 мл, 32%, pH 10), что вызывало кристаллизацию. Суспензию перемешивали в течение 30 минут, кристаллы отфильтровывали, промывали водой и сушили под вакуумом до постоянного веса, что давало соединение 13 в виде желтых кристаллов (4,8 г, выход 93%, % площади 98,5%, ВЭЖХ).
Пример 18
Этот пример иллюстрирует другой способ получения соединения 13 из соединения 6.
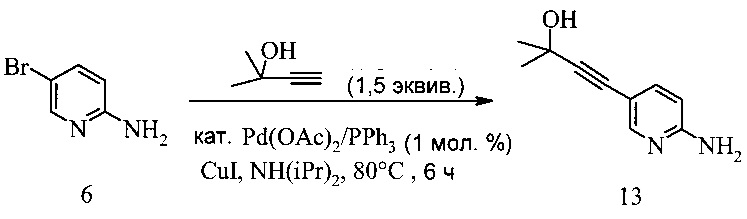
100 мл 4-горлую колбу заполняли 5-бром-2-аминопиридином (5 г, 28,9 ммоль) и диизопропиламином (61,8 мл, 433 ммоль, 15 эквив.). Смесь дегазировали при введении аргона, добавляя в то же время ацетат палладия(II) (64,9 мг, 289 мкмоль, 1 мол. %), трифенилфосфин (303 мг, 1,16 ммоль, 4 мол. %) и иодид меди (110 мг, 578 мкмоль, 2 мол. %) и реакционную смесь нагревали до 80°C. При этой температуре добавляли 2-метил-3-бутин-2-ол (3,65 г, 43,3 ммоль, 1,5 эквив.) в течение 15 минут и реакционную смесь перемешивали при этой температуре в течение 17 часов. Затем нагревание останавливали и черную суспензию охлаждали на водяной ванне до температуры окружающей среды. К этой суспензии добавляли 2-Me-ТГФ (30 мл) и воду (20 мл). Фазы разделяли, водную фазу промывали 2-Me-ТГФ (30 мл) и органическую фазу промывали солевым раствором (30 мл). Из объединенных органических фаз растворитель удаляли под вакуумом досуха. Темный остаток суспендировали в HCl (20 мл, водн. 2 М) для доведения pH до pH 1 и смесь перемешивали в течение 10 минут. Темные нерастворимые остатки отфильтровывали и желтый раствор обрабатывали NH3 (водн., 25%, 4 мл, pH 8,5-9), что вызывало кристаллизацию. Суспензию перемешивали в течение 20 минут, кристаллы отфильтровывали, промывали водой (8 мл) и сушили под вакуумом до постоянного веса, что давало соединение 13 в виде бежевых кристаллов (4,7 г, выход 92%, % площади 99,2%, ВЭЖХ).
Пример 19
Этот пример иллюстрирует другой способ получения соединения 13 из соединения 6.
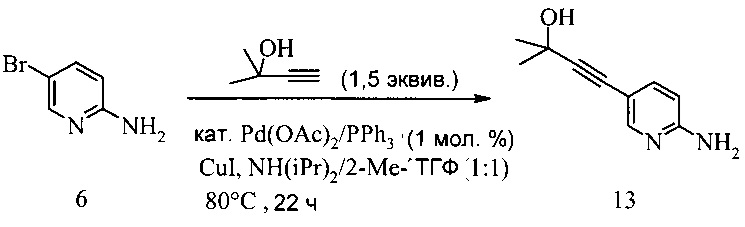
650 мл 4-горлую колбу заполняли 5-бром-2-аминопиридином (30 г, 173,4 ммоль), диизопропиламином (185 мл, 1,3 моль, 7,5 эквив.) и 2-Me-ТГФ (180 мл). Смесь дегазировали при введении аргона, в то же время последовательно добавляя ацетат палладия (II) (389 мг, 1,73 ммоль, 1 мол.% Pd), трифенилфосфин (1,82 г, 6,94 ммоль, 4 мол.%) и иодид меди (660 мг, 3,47 ммоль, 2 мол.%) и реакционную смесь нагревали до 80°C. При этой температуре добавляли в течение 30 минут 2-метил-3-бутин-2-ол (21,9 г, 260 ммоль, 1,5 эквив.) и реакционную смесь перемешивали при 80°C в течение дополнительных 21,5 часа. Затем нагревание останавливали и черную суспензию охлаждали на водяной ванне до температуры окружающей среды. К темной суспензии добавляли воду (100 мл) и 2-Ме-ТГФ (100 мл) и смесь фильтровали через дикалит. После этого фазы разделяли, органическую фазу промывали солевым раствором (200 мл), тогда как водную фазу промывали 2-Me-ТГФ (250 мл). Из объединенных органических фаз растворитель удаляли под вакуумом, что давало коричневое твердое вещество, которое суспендировали в воде (200 мл). Эту суспензию обрабатывали HCl (25%, 24 мл, pH 1) и смесь перемешивали в течение 10 мин. Черный осадок удаляли в ходе фильтрации и маточный раствор обрабатывали гептаном (100 мл). После этого фазы разделяли, водную фазу обрабатывали NH3 (водн., 25%, 30 мл) и pH доводили до pH 10, после чего продукт выпадал в осадок. Суспензию перемешивали в течение 30 минут, кристаллы отфильтровывали, промывали последовательно водой (50 мл) и гептаном (10 мл) и сушили под вакуумом до постоянного веса, что давало соединение 13 в виде желтого твердого вещества (26,5 г, выход 85%, 98 площадь % ВЭЖХ).
Пример 20
Этот пример иллюстрирует способ получения соединения 13 из соединения 8.
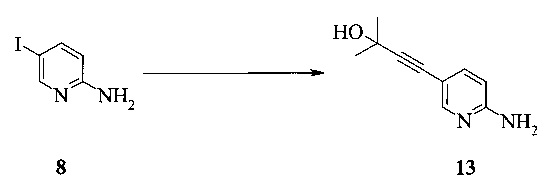
Раствор 2-амино-5-йодпиридина (48,0 г), иодида меди(I) (520 мг), бис-(трифенилфосфин)-палладия(II) - дихлорида (765 мг) и триэтиламина (38,0 г) в МеТГФ (220 мл) обрабатывали при 48-54°C в течение 1-2 часов раствором 2-метил-3-бутин-2-ола (30,0 г) в МеТГФ (40 мл), и полученную в результате суспензию перемешивали при 50°C в течение 3 часов. Смесь охлаждали до 25-30°C и затем обрабатывали водой (80 мл) и 25% водным раствором гидроксида аммония (40 мл). Двухфазную смесь перемешивали при 25-30°C в течение 20 минут, и слои разделяли через 20 минут.Водный слой удаляли, и органический слой промывали смесью воды (40 мл) и 25% водного раствора гидроксида аммония (40 мл). Объединенные водные слои повторно экстрагировали МеТГФ (100 мл). Объединенные органические слои промывали водой (30 мл) и затем концентрировали при пониженном давлении до остаточного объема прибл. 80 мл. Затем добавляли толуол (500 мл) и смесь концентрировали при пониженном давлении до остаточного объема прибл. 400 мл. Смесь обрабатывали изопропанолом (60 мл) и трибутилфосфином (2 мл). Суспензию нагревали до температуры кипячения, чтобы получить прозрачный раствор, и затем охлаждали до 0°C в течение 5 часов, в результате чего продукт кристаллизовался. Полученную в результате суспензию перемешивали при 0°C в течение четырех часов. Продукт фильтровали и промывали 80 мл толуола. Влажные кристаллы сушили при 50°C и <30 мбар до постоянной массы, получая 32,8 г (выход 85%) соединения 13 в виде светло-желтых кристаллов с чистотой 99,9% (ВЭЖХ, площадь-%) и концентрацией 100,0% (ВЭЖХ, масс./масс.-%).
Пример 21
Этот пример иллюстрирует способ получения соединения A из соединения 13.
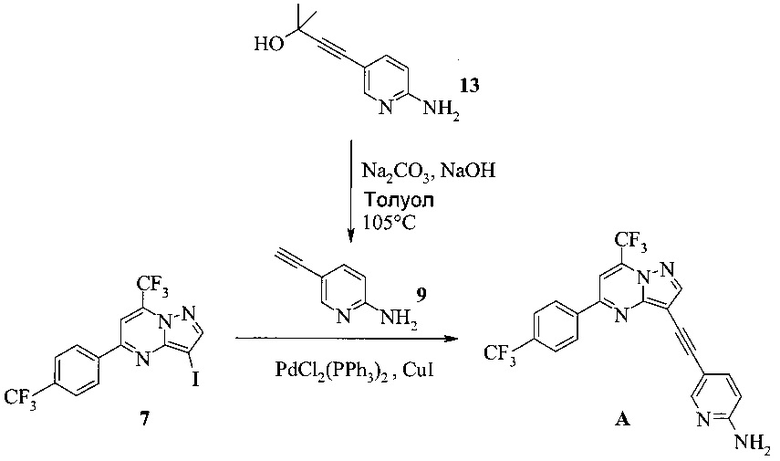
Суспензию соединения 13 (17,6 г), карбоната натрия (23,0 г) и гидроксида натрия (3,0 г) в толуоле (200 мл) нагревали при пониженном давлении (прибл. 800 мбар) до 100-105°C и затем перемешивали при этой температуре в течение 6-10 часов. В ходе реакции отогнанный толуол постоянно заменяли свежим толуолом, поддерживая объем постоянным. После полного превращения (<2% исходных веществ) смесь охлаждали до 50°C и половину толуола отгоняли при пониженном давлении. Добавляли МеТГФ (120 мл) и воду (120 мл) и двухфазную смесь перемешивали в течение 30 минут. Слои разделяли, и нижний водный слой затем удаляли. Органический слой осветляли в ходе фильтрации; фильтрат промывали водой (1×40 мл) и затем концентрировали досуха. Остаток растворяли в 150 мл МеТГФ. Этот раствор затем добавляли в течение 2-3 часов к горячему (70-75°C) раствору соединения 7 (36,0 г), иодида меди(I) (334 мг), бис-(трифенилфосфин)- палладия(II) - дихлорида (557 мг), трифенилфосфина (414 мг) и триэтиламина (25,5 мл) в МеТГФ (145 мл) и полученную в результате суспензию перемешивали при 70-75°C в течение дополнительных 14 часов. Смесь охлаждали до 30°C и обрабатывали водой (150 мл) и 25% водным раствором гидроксида аммония (30 мл). Двухфазную смесь перемешивали в течение 30 минут и слои затем рахделяли в течение 20 минут. Водный слой удаляли и МеТГФ слой промывали дважды смесью воды (150 мл) и 25% водного раствора гидроксида аммония (30 мл). МеТГФ слой затем промывали водой (3×150 мл). Органический слой осветляли в ходе фильтрации, и фильтрат обрабатывали н-трибутилфосфином (1,0 мл). МеТГФ полностью отгоняли и заменяли на этанол (всего 500 мл) при атмосферном давлении. Полученную в результате суспензию (прибл. 300 мл) нагревали с обратным холодильником и перемешивали при нагревании с обратным холодильником в течение 2 часов и затем охлаждали до комнатной температуры в течение ночи. Продукт фильтровали и промывали этанолом (50 мл). Влажные кристаллы сушили при 50°C и<30 мбар до постоянной массы, получая 29,3 г (выход 83%, исходя из 7) соединения A в виде красных кристаллов с чистотой 99,5% (ВЭЖХ, площадь-%) и концентрацией 99,0% (ВЭЖХ, масс./масс.-%).
Кристаллизацию в вышеприведенном примере также можно выполнить с изопропанолом вместо этанола. Продукт может быть дополнительно очищен в ходе повторной обработки (перекристаллизация при растворении продукта в МеТГФ с последующей заменой растворителя на изопропанол или этанол и последующее выделение).
Несмотря на то, что представлен ряд воплощений этого изобретения, очевидно, что базовая конструкция может быть изменена, что дает использовать другие воплощения изобретения, не отклоняясь от сущности и объема изобретения. Предполагается, что все подобные модификации и варианты включены в объем изобретения, как определено в прилагаемой формуле изобретения, а не в конкретных воплощениях, которые представлены с помощью примеров.
| название | год | авторы | номер документа |
|---|---|---|---|
| СИНТЕЗ ТРАНС-8-ХЛОР-5-МЕТИЛ-1-[4-(ПИРИДИН-2-ИЛОКСИ)-ЦИКЛОГЕКСИЛ]-5,6-ДИГИДРО-4Н-2,3,5,10В-ТЕТРААЗАБЕНЗО[E]АЗУЛЕНА И ЕГО КРИСТАЛЛИЧЕСКИЕ ФОРМЫ | 2014 |
|
RU2775690C2 |
| УСОВЕРШЕНСТВОВАННЫЙ СПОСОБ ПОЛУЧЕНИЯ 5-(2,6-ДИ-4-МОРФОЛИНИЛ-4-ПИРИМИДИНИЛ)-4-ТРИФТОРМЕТИЛПИРИДИН-2-АМИНА | 2013 |
|
RU2646760C2 |
| СПОСОБ ПОЛУЧЕНИЯ ХИРАЛЬНЫХ 2-АРИЛМОРФОЛИНОВ | 2014 |
|
RU2677328C1 |
| СОЛЬ (СОЛИ) ДИМЕТИЛАМИДА 7-ЦИКЛОПЕНТИЛ-2-(5-ПИПЕРАЗИН-1-ИЛ-ПИРИДИН-2-ИЛАМИНО)-7Н-ПИРРОЛО[2,3-d]ПИРИМИДИН-6-КАРБОНОВОЙ КИСЛОТЫ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2011 |
|
RU2631243C2 |
| ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ ТРАНС-5-ХЛОР-2-МЕТИЛ-2,3,3f,12b-ТЕТРАГИДРО-1Н-ДИБЕНЗ[2,3:6,7]-ОКСЕПИНО[4,5-c]ПИРРОЛА | 2006 |
|
RU2397164C2 |
| СПОСОБ ПОЛУЧЕНИЯ (5S)-4-[5-(3,5-ДИХЛОРФЕНИЛ)-5-(ТРИФТОРМЕТИЛ)-4H-ИЗОКСАЗОЛ-3-ИЛ]-2-МЕТИЛБЕНЗОЙНОЙ КИСЛОТЫ | 2019 |
|
RU2786390C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1-(4-МЕТАНСУЛЬФОНИЛ-2-ТРИФТОРМЕТИЛБЕНЗИЛ)-2-МЕТИЛ-1Н-ПИРРОЛО[2,3-B]ПИРИДИН-3-ИЛ-УКСУСНОЙ КИСЛОТЫ | 2016 |
|
RU2744976C1 |
| ТВЕРДЫЕ ФОРМЫ (R)-1-(2, 2-ДИФТОРБЕНЗО[d][1, 3]ДИОКСОЛ-5-ИЛ)-N-(1-(2, 3-ДИГИДРОКСИПРОПИЛ)-6-ФТОР-2-(1-ГИДРОКСИ-2-МЕТИЛПРОПАН-2-ИЛ)-1H-ИНДОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДА | 2011 |
|
RU2711481C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ НА ОСНОВЕ 4-ФЕНИЛ-6-(2,2,2-ТРИФТОР-1-ФЕНИЛЭТОКСИ)ПИРИМИДИНА | 2008 |
|
RU2493156C2 |
| ИНДОЛЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ 5-HT | 2007 |
|
RU2449990C2 |
Изобретение относится к усовершенствованному способу получения 5-[2-[7-(трифторметил)-5-[4-(трифторметил)фенил]пиразоло[1,5-a]пиримидин-3-ил]этинил]-2-пиридинамина (соединение A), которое является полезным в лечении депрессии и других расстройств ЦНС. Изобретение также относится к улучшенному способу получения соединения формулы 9, которое используется в синтезе соединения А в качестве промежуточного соединения. Способ получения соединения 9 включает следующие стадии: а) взаимодействие 5-амино-2-йодпиридина 8 с 2-метил-3-бутин-2-олом по реакции Соногашира в присутствии катализатора PdCl2(PPh3)2CuJ с триэтиламином в качестве основания в 2-метилтетрагидрофуране с получением соединения 13 и б) удаление защитной группы в присутствии водного гидроксида натрия в среде толуола по следующей схеме
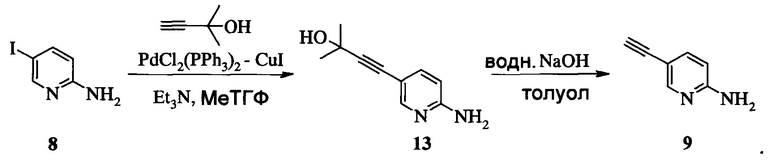
Соединение формулы А получают взаимодействием соединения 3 или 7
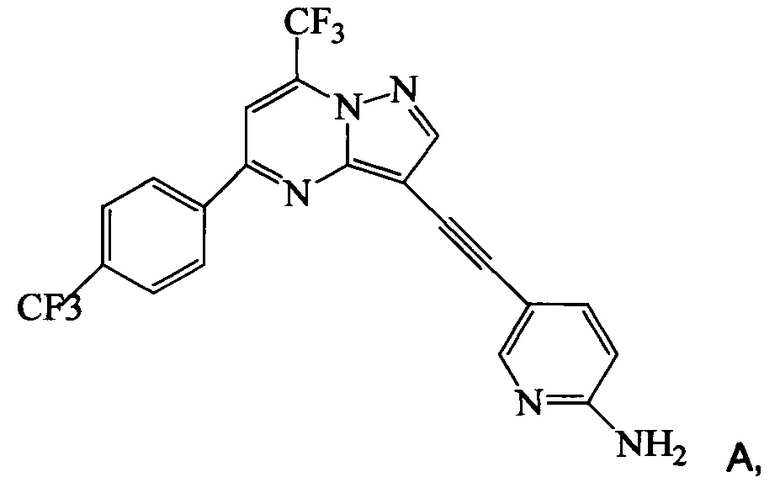
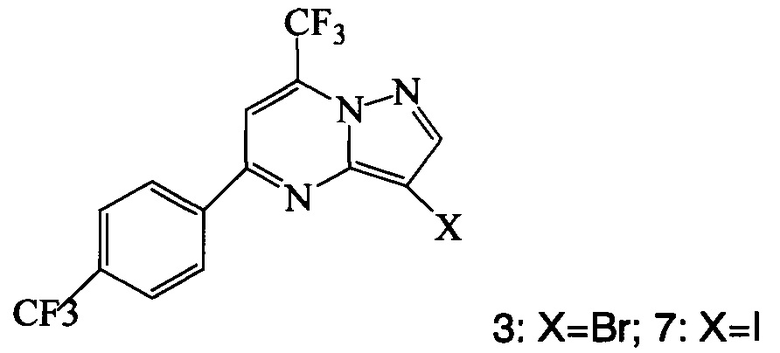
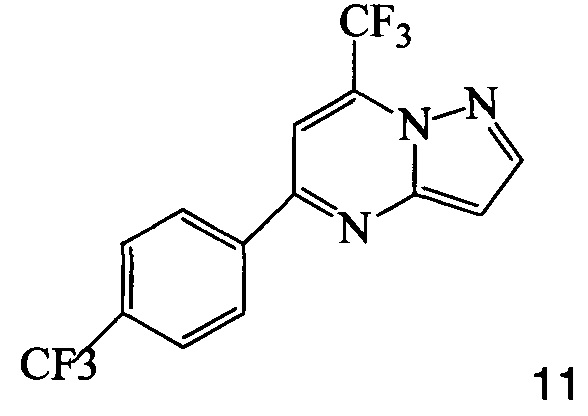
с соединением 9, полученным выше, в условиях реакции сочетания Соногашира в инертном растворителе. Предпочтительное исходное соединение 7 получают взаимодействием соединения 11
с йодирующим агентом, например N-йодсукцинимидом, при кислотных условиях. Способ позволяет получить соединение А с высоким выходом и чистотой и пригоден для использования в крупном масштабе на промышленных предприятиях. 2 н. и 2 з.п ф-лы, 21 пр.
1. Способ получения соединения 9, имеющего формулу
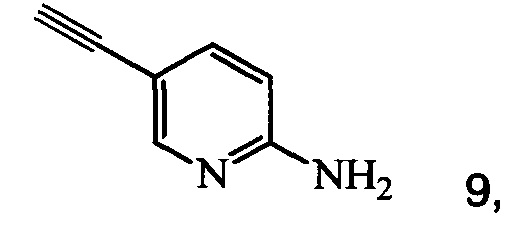
который содержит следующие стадии:
а) взаимодействие 5-амино-2-йодпиридина 8 с 2-метил-3-бутин-2-олом по реакции Соногашира в присутствии катализатора PdCl2(PPh3)2CuJ с триэтиламином в качестве основания в 2-метилтетрагидрофуране с получением соединения 13, и
б) удаление защитной группы в присутствии водного гидроксида натрия в среде толуола по следующей схеме:
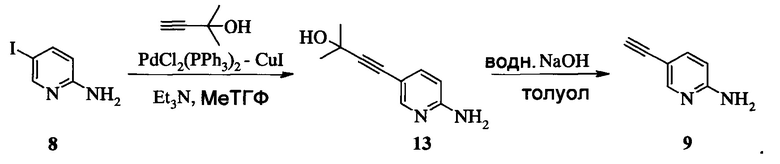
2. Способ получения соединения А, имеющего формулу
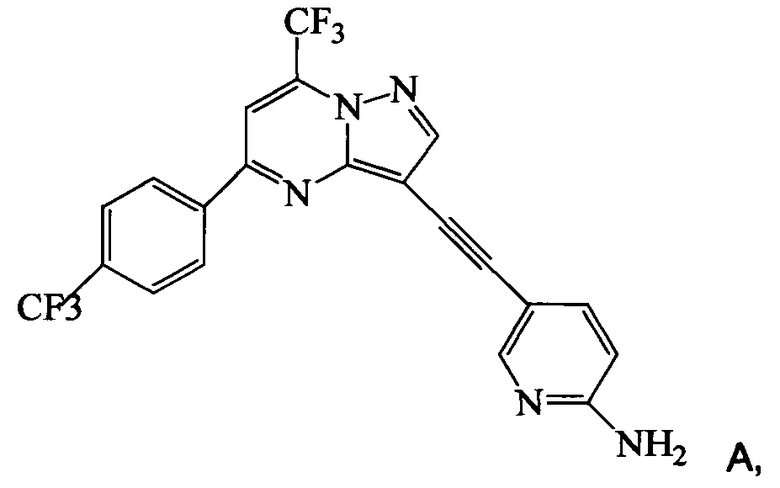
согласно которому подвергают взаимодействию соединение 3 или 7
с соединением 9, полученным по п. 1
по реакции сочетания Соногашира в инертном растворителе, что дает соединение А.
3. Способ по п. 2, где соединение 7 получают, подвергая взаимодействию соединение 11
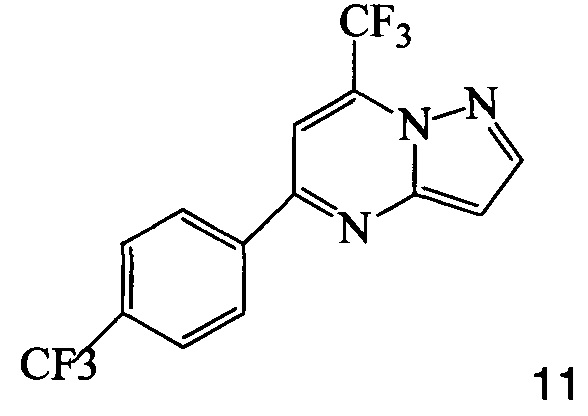
с йодирующим агентом при кислотных условиях, что дает соединение 7.
4. Способ по п. 3, где йодирующий агент представляет собой N-йодсукцинимид.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| ПРОИЗВОДНЫЕ АЦЕТИЛЕНИЛ-ПИРАЗОЛО-ПИРИМИДИНА В КАЧЕСТВЕ АНТАГОНИСТОВ mGluR2 | 2006 |
|
RU2412943C2 |

