Область техники
Настоящее изобретение предлагает способы получения замещенных 1-[4-(пиридин-2-илокси)-циклогексил]-5,6-дигидро-4H-2,3,5,10b-тетрааза-бензо[e]азуленов. Также раскрыты соединения, применимые в качестве промежуточных соединений в способах по изобретению.
Предшествующий уровень техники
Расстройства аутистического спектра (ASD, от англ. - autistic spectrum disorders) являются клинически гетерогенными заболеваниями, характеризующимися нарушениями социализации и речи. ASD включает широкий спектр нарушений, в том числе истинную неспособность устанавливать эмоциональные связи, нарушения поведения при социальном взаимодействии, вербальных и невербальных коммуникациях, ограниченную заинтересованность в окружающей среде, ассоциированную со стереотипными движениями и повторяющимися ролями (Bourreau et al., 2009)1. Исследования показывают, что в это может быть вовлечена генетическая предрасположенность, но также необходимо учитывать факторы окружающей среды (Bourgeron, 2009)2. В настоящее время отсутствует эффективное биологическое/фармацевтическое лечение ASD.
1-[4-(пиридин-2-илокси)-циклогексил]-5,6-дигидро-4H-2,3,5,10b-тетрааза-бензо[e]азулены ранее описаны в области техники3.
Кроме того, в WO2004074291 и WO20050684664 раскрыты соединения триазола и способ их получения.
Неожиданно было обнаружено, что при применении способов по настоящему изобретению 8-хлор-5-метил-1-[4-(2-пиридилокси)циклогексил]-4,6-дигидро-[1,2,4]триазоло[4,3-a][1,4]бензодиазепин и его фармацевтически приемлемые соли могут быть получены более экономично с меньшим числом этапов способа при умеренных условиях реакции и с хорошим выходом. Кроме того, неочищенные промежуточные продукты обычно могут быть применены на последующих этапах реакции без необходимости дополнительных этапов очистки.
Кроме того, некоторые формы были идентифицированы и неожиданно было обнаружено, что форма F является наиболее предпочтительной.
Определения
Следующие определения общих терминов настоящего описания применяют вне зависимости от того, употреблены ли термины поодиночке или в сочетании с другими группами.
Термин «комнатная температура» (КТ) относится к температуре от 18 до 30°C, предпочтительно от 20 до 25°C, более предпочтительно к 20°C.
«Раствор», как употреблено в данном документе, охватывает жидкости, в которых реагент или реактант представляет собой растворитель в растворенной форме (в качестве растворенного вещества) или присутствует частично в нерастворенной форме или обеих. Таким образом, «раствор» предполагает, что растворенное вещество может быть растворено в нем не полностью и твердое растворенное вещество может присутствовать в виде дисперсии или суспензии. Соответственно, «раствор» отдельного реагента или реактанта подразумевает суспензии и дисперсии, а также растворы таких реагентов или реактантов. «Раствор» или «суспензия» могут быть употреблены взаимозаменяемо.
«Растворитель», как употреблено в данном документе, охватывает жидкости, которые полностью растворяют реагент или реактант, при взаимодействии с растворителем, а также жидкости, только частично растворяющие реагент или реактант, или действующие в качестве дисперсантов в отношении реагента или реактанта. Таким образом, когда отдельную реакцию проводят в «растворителе», предполагается, что не все реагенты или реактанты присутствуют в растворенной форме.
Термин «приблизительно» в сочетании со значениями градусов 2-тета относится к ±0,2 градусов 2-тета.
Термины «кристаллическая форма» или «форма» относятся к полиморфным формам и сольватам соединения.
Термин «фармацевтически приемлемые соли» относится к солям, которые пригодны для применения в контакте с тканями человека и животных. Примерами подходящих для образования солей неорганических и органических кислот являются, но не ограничиваются этим, уксусная кислота, лимонная кислота, муравьиная кислота, фумаровая кислота, соляная кислота, молочная кислота, малеиновая кислота, яблочная кислота, метан-сульфоновая кислота, азотная кислота, ортофосфорная кислота, п-толуолсульфоновая кислота, янтарная кислота, серная кислота, винная кислота, трифторуксусная кислота и т.п. Предпочтительными являются муравьиная кислота, трифторуксусная кислота и соляная кислота. Наиболее предпочтительной является соляная кислота.
Термины “аутистический спектр” и “расстройства аутистического спектра” объединяют состояния, классифицируемые как первазивные расстройства развития, которые включают, но не ограничены, аутизм, синдром Аспергера, неуказанное конкретно первазивное расстройство развития (PDD-NOS), дезинтегративное расстройство детского возраста, синдром Ретта и синдром ломкой Х-хромосомы, в частности аутизм. Данные заболевания обычно характеризуются социальными нарушениями, сложностями коммуникации, стереотипным или повторяющимся поведением и интересами и задержкой развития когнитивных функций.
Номенклатура, примененная в данной заявке, основана на систематической номенклатуре IUPAC, если не указано иное.
Описание изобретения
В деталях, настоящее изобретение относится к способу синтеза кристаллической формы соединения формулы I
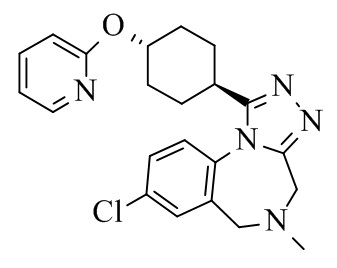 I.
I.
Конкретное воплощение изобретения относится к кристаллической форме A соединения формулы I, как описано в данном документе, порошковая рентгеновская дифрактограмма которого имеет характеристические пики, выраженные в значениях градусов 2-тета, приблизительно при
Конкретное воплощение изобретения относится к кристаллической форме A соединения формулы I, как описано в данном документе, характеризующейся порошковой рентгеновской дифрактограммой, показанной на фиг. 1.
Конкретное воплощение изобретения относится к кристаллической форме А соединения формулы I, как описано в данном документе, характеризующейся спектром инфракрасного излучения (ИК-спектром), показанным на фиг. 2.
Конкретное воплощение изобретения относится к кристаллической форме А соединения формулы I, как описано выше, характеризующейся спектром Рамана, показанным на фиг. 3.
Конкретное воплощение изобретения относится к кристаллической форме В соединения формулы I, как описано в данном документе, характеризующейся порошковой рентгеновской дифрактограммой, имеющей характеристические пики, выраженные в значениях градусов 2-тета, приблизительно при
Конкретное воплощение изобретения относится к кристаллической форме В соединения формулы I, как описано в данном документе, характеризующейся порошковой рентгеновской дифрактограммой, показанной на фиг. 4.
Конкретное воплощение изобретения относится к кристаллической форме В соединения формулы I, как описано в данном документе, характеризующейся ИК-спектром, показанным на фиг. 5.
Конкретное воплощение изобретения относится к кристаллической форме В соединения формулы I, как описано в данном документе, характеризующейся спектром Рамана, показанным на фиг. 6.
Конкретное воплощение изобретения относится к кристаллической форме B соединения формулы I, как описано в данном документе, характеризующейся следующими параметрами элементарной ячейки
Конкретное воплощение изобретения относится к кристаллической форме С соединения формулы I, как описано в данном документе, характеризующейся порошковой рентгеновской дифрактограммой, имеющей характеристически пики, выраженные в значениях градусов 2-тета, приблизительно при
Конкретное воплощение изобретения относится к кристаллической форме С соединения формулы I, как описано в данном документе, характеризующейся порошковой рентгеновской дифрактограммой, показанной на фиг. 7.
Конкретное воплощение изобретения относится к кристаллической форме С соединения формулы I, как описано в данном документе, характеризующейся ИК-спектром, показанным на фиг. 8.
Конкретное воплощение изобретения относится к кристаллической форме С соединения формулы I, как описано в данном документе, характеризующейся спектром Рамана, показанным на фиг. 9.
Конкретное воплощение изобретения относится к кристаллической форме С соединения формулы I, как описано в данном документе, характеризующейся следующими параметрами элементарной ячейки
Конкретное воплощение изобретения относится к кристаллической форме D соединения формулы I, как описано в данном документе, характеризующейся порошковой рентгеновской дифрактограммой, имеющей характеристически пики, выраженные в значениях градусов 2-тета, приблизительно при
Конкретное воплощение изобретения относится к кристаллической форме D соединения формулы I, как описано в данном документе, характеризующейся порошковой рентгеновской дифрактограммой, показанной на фиг. 10.
Конкретное воплощение изобретения относится к кристаллической форме D соединения формулы I, как описано в данном документе, характеризующейся ИК-спектром, показанным на фиг. 11.
Конкретное воплощение изобретения относится к кристаллической форме D соединения формулы I, как описано в данном документе, характеризующейся спектром Рамана, показанном на фиг. 12.
Конкретное воплощение изобретения относится к кристаллической форме D, характеризующейся следующими параметрами
Конкретное воплощение изобретения относится к кристаллической форме Е соединения формулы I, как описано в данном документе, характеризующейся порошковой рентгеновской дифрактограммой, имеющей характеристические пики, выраженные в значениях градусов 2-тета, приблизительно при
Конкретное воплощение изобретения относится к кристаллической форме Е соединения формулы I, как описано в данном документе, характеризующейся порошковой рентгеновской дифрактограммой, показанной на фиг. 13.
Конкретное воплощение изобретения относится к кристаллической форме Е соединения формулы I, как описано в данном документе, характеризующейся ИК-спектром, показанным на фиг. 14.
Конкретное воплощение изобретения относится к кристаллической форме Е соединения формулы I, как описано в данном документе, характеризующейся спектром Рамана, показанным на фиг. 15.
Конкретное воплощение изобретения относится к кристаллической форме F соединения формулы I, как описано в данном документе, характеризующейся порошковой рентгеновской дифрактограммой, имеющей характеристические пики, выраженные в значениях градусов 2-тета, приблизительно при
Конкретное воплощение изобретения относится к кристаллической форме F соединения формулы I, как описано в данном документе, характеризующейся порошковой рентгеновской дифрактограммой, показанной на фиг. 16.
Конкретное воплощение изобретения относится к кристаллической форме F соединения формулы I, как описано в документе, характеризующейся ИК-спектром, показанном на фиг. 17.
Конкретное воплощение изобретения относится к кристаллической форме F соединения формулы I, как описано в данном документе, характеризующейся спектром Рамана, показанном на фиг. 18.
Конкретное воплощение изобретения относится к кристаллической форме F соединения I, как описано в данном документе, характеризующейся следующими параметрами элементарной ячейки
Конкретное воплощение изобретения относится к кристаллической форме G соединения формулы I, как описано в данном документе, характеризующейся порошковой рентгеновской дифрактограммой, имеющей характеристические пики, выраженные в значениях градусов 2-тета, приблизительно при
Конкретное воплощение изобретения относится к кристаллической форме G соединения формулы I, как описано в данном документе, характеризующейся порошковой рентгеновской дифрактограммой, показанной на фиг. 19.
Конкретное воплощение изобретения относится к кристаллической форме G соединения формулы I, как описано в данном документе, характеризующейся ИК-спектром, показанным на фиг. 20.
Конкретное воплощение изобретения относится к кристаллической форме G соединения формулы I, как описано в данном документе, характеризующейся спектром Рамана, показанным на фиг. 21.
Конкретное воплощение изобретения относится к кристаллической форме G соединения формулы I, как описано в данном документе, характеризующейся следующими параметрами элементарной ячейки
Конкретное воплощение изобретения относится к кристаллической форме Н соединения формулы I, как описано в данном документе, характеризующейся порошковой рентгеновской дифрактограммой, имеющей характеристические пики, выраженные в значениях градусов 2-тета, приблизительно при
Конкретное воплощение изобретения относится к кристаллической форме H соединения формулы I, как описано в данном документе, характеризующейся порошковой рентгеновской дифрактограммой, показанной на фиг. 22.
Конкретное воплощение по изобретению относится к кристаллической форме H соединения формулы I, как описано в данном документе, характеризующейся спектром Рамана, показанным на фиг. 23.
Конкретное воплощение изобретения относится к кристаллической форме H соединения формулы I, как описано в данном документе, характеризующейся следующими параметрами элементарной ячейки:
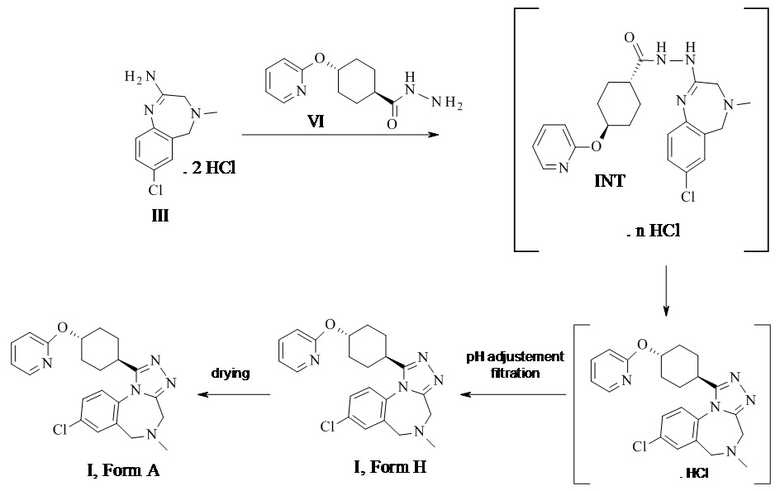
Конкретное воплощение изобретения относится к способу трансформации формы А в форму F.
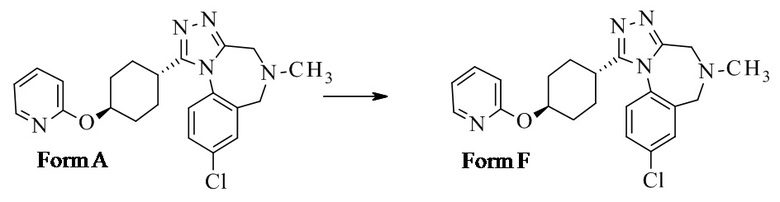 .
.
Конкретное воплощение изобретения относится к тригидрату соединения формулы I.
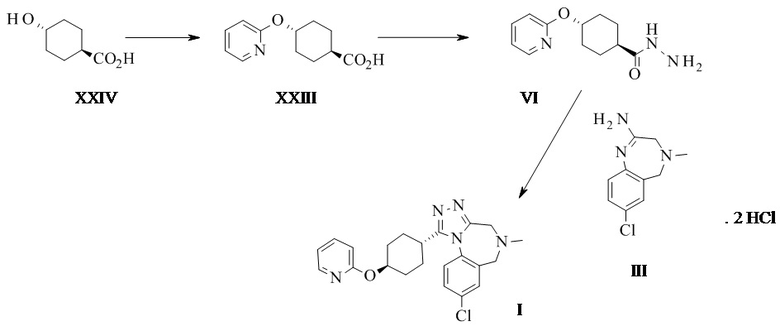
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, как описано в данном документе, включающему реакцию соединения формулы II с соединением формулы VI
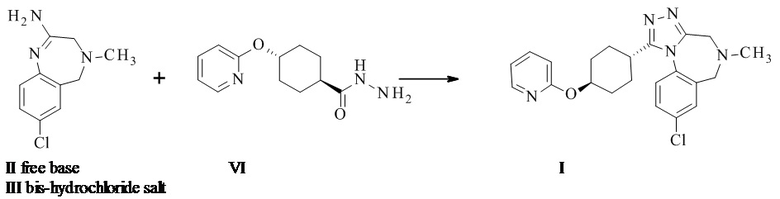 .
.
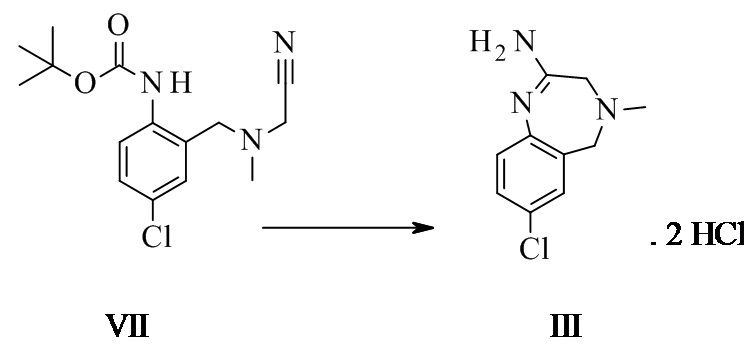
Амидиновое свободное основание II может реагировать термически с соединением формулы VI с образованием соединения формулы I. Присутствие кислоты повышает реакционную способность и чистоту неочищенного активного фармацевтического ингредиента. Этого обычно достигают посредством применения амидин-бис-гидрохлорида III в качестве субстрата. III может быть выделен в качестве кристаллического промежуточного соединения, что таким образом обеспечивает точку тонкой очистки в данном синтезе.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, как описано в данном документе, включающему реакцию соединения формулы III с соединением формулы VI.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, как описано в данном документе, включающему реакцию соединения формулы III с соединением формулы VI, при этом они реагируют термически, в частности при температуре 95°C плюс/минус 35°C, в частности 85°C плюс/минус 15°C, в частности 80°C плюс/минус 5°C. Конкретными температурами являются 75°C, 76°C, 77°C, 78°C, 79°C, 80°C, 81°C, 82°C, 83°C, 84°C и 85°C.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, как описано в данном документе, включающему реакцию соединения формулы III с соединением формулы VI в органическом растворителе, таком как ТГФ, диоксан, ДМФ, НМП, ацетонитрил и спирты, в частности в спиртовом растворителе, таком как этанол, н-пропанол, изопропанол, н-бутанол, особенно изопропанол и н-пропанол, наиболее предпочтителен изопропанол. Соединение I может быть непосредственно выделено в виде гидрохлорида посредством фильтрования, когда реакцию проводят в подходящем растворителе, таком как изопропанол. Альтернативно, I в виде свободного основания может быть выделено посредством добавления водного основания, такого как водный гидроксид натрия, водный гидроксид калия, водный бикарбонат натрия, водный бикарбонат калия, водный карбонат натрия, водный карбонат калия, в частности водный гидроксид натрия, водный гидроксид калия, особенно, водный гидроксид натрия. Соединение I затем выделяют в виде тригидрата (форма H), который переходит в безводную форму А после высушивания.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, как описано в данном документе, включающему реакцию соединения формулы II с соединением формулы VI, в котором свободное основание продукта I выделяют при pH более 8, в частности при pH более 10, в частности при pH более 12.
Конкретное воплощение изобретения относится к выделению свободного основания продукта I при pH более 8, в частности при pH более 10, в частности при pH более 12 при помощи соответствующей смеси растворителей, такой как смесь спирт/вода, в частности этанол/вода, изопропанол/вода, н-пропанол/вода, в частности изопропанол/вода, свободная от нежелательного побочного продукта 4-(2-пиридилокси)-N'-[4-(2-пиридилокси)циклогексанкарбонил]циклогексан-карбонгидразид (VI').
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, как описано в данном документе, включающему реакцию соединения формулы II с соединением формулы VI, в котором 4-(2-пиридилокси)-N'-[4-(2-пиридилокси)циклогексанкарбонил]циклогексан-карбогидразид VI' является побочным продуктом.
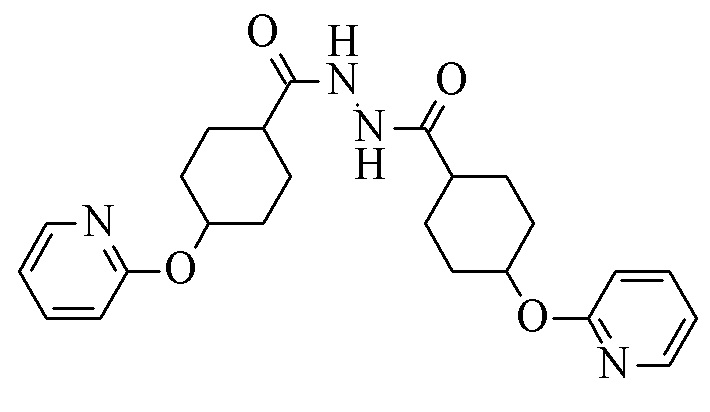 VI'.
VI'.
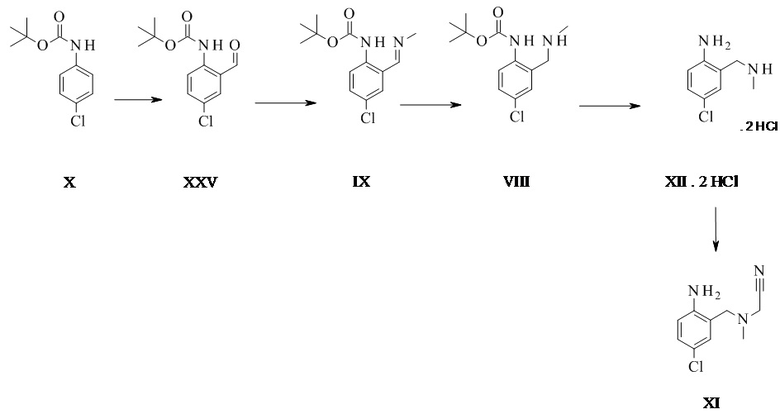
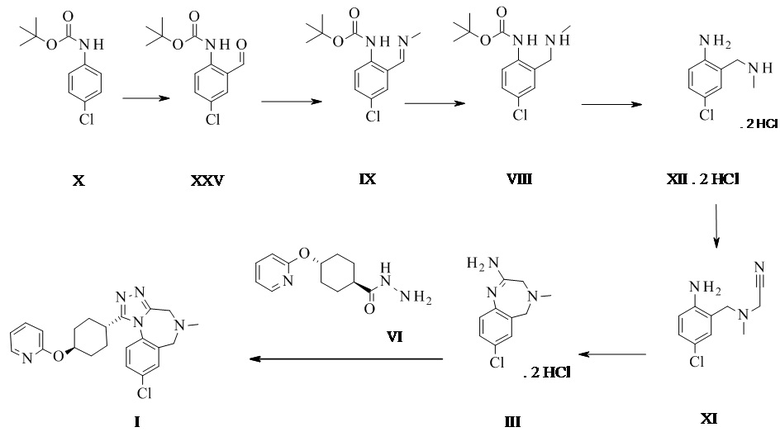
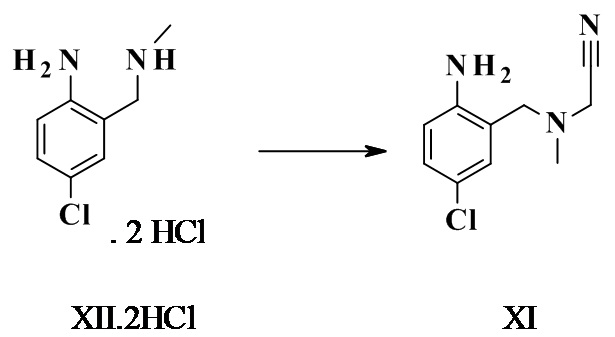
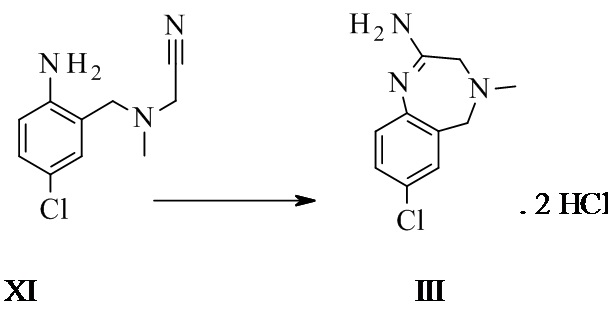
Конкретное воплощение изобретения относится к способу, как описано выше, дополнительно включающему превращение соединения формулы XI или его хлористоводородной соли в соединение формулы III:
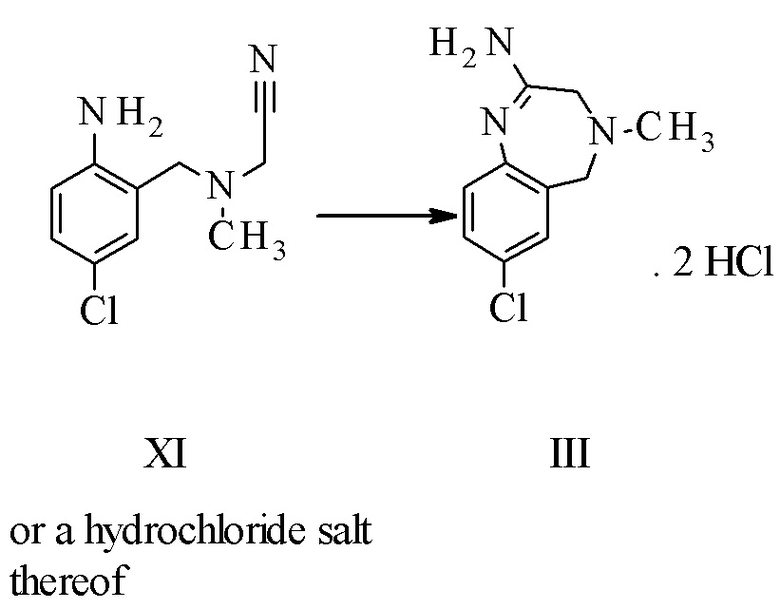 .
.
Конкретное воплощение изобретения относится к способу, как описано выше, дополнительно включающему превращение соединения формулы X в соединение формулы XI через следующие этапы:
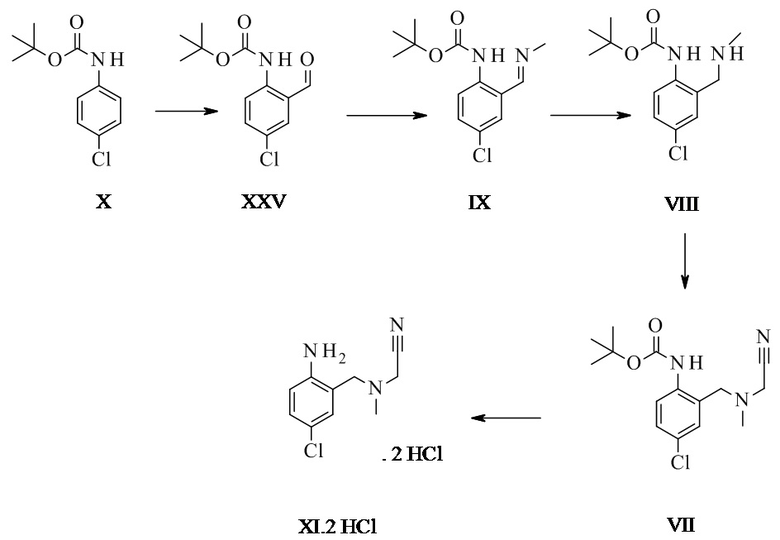
Соединение формулы XI может быть выделено в виде бис-гидрохлорида. Альтернативно, оно может быть получено in-situ и непосредственно в дальнейшем превращено в соединение формулы III.
Конкретное воплощение изобретения относится к способу, как описано выше, дополнительно включающему превращение соединения формулы X в соединение формулы III через следующие этапы:
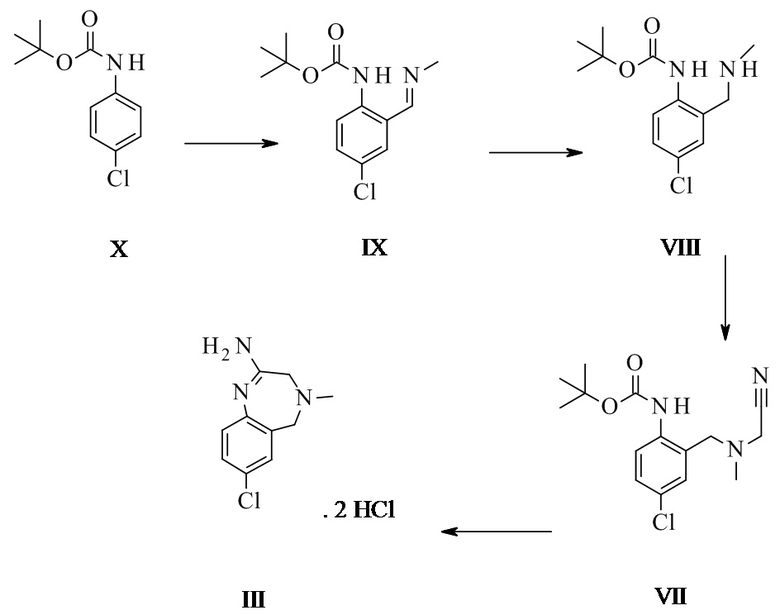
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, включающему следующие этапы:
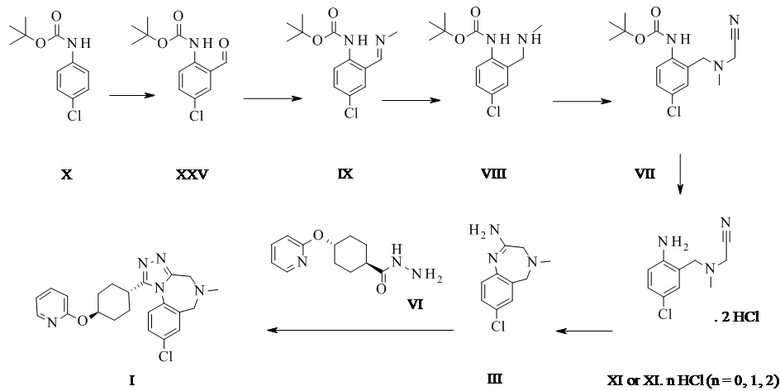
Альдегид формулы XXV описан в уровне техники (Aubé et al.)5, причем его получали посредством орто-литирования с втор-бутиллитием (s-BuLi) при -78°C, затем повышения температуры до -20°C перед гашением посредством ДМФ. Продукт был получен с 54% выходом после хроматографии. Настоящую реакцию проводят при более высокой температуре (до -30°C) и с н-бутиллитием (н-BuLi) с более высоким выходом, превышающим 80%, без хроматографии и после кристаллизации. Способ, описанный в данном документе, является намного более эффективным и масштабируемым.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы XXV из соединения формулы X, при этом литирование проводят в тетрагидрофуране (ТГФ), 2-метил-тетрагидрофуране (2-Me-ТГФ) или метил-трет-бутиловом эфире (МТБЭ), в частности ТГФ и МТБЭ, в частности МТБЭ.
Конкретное воплощение по изобретению относится к способу синтеза соединения формулы XXV из соединения формулы X, при этом литирование проводят при температуре от -60°C до -10°C, в частности от -40°C до -20°C, в частности при -30 плюс/минус 2°C.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы XXV из соединения формулы X, при этом литирование проводят в присутствии добавки, подобной (но не ограниченной) тетраметилэтилендиамину (ТМЭДА) или пентаметилдиэтилентриамину (ПМДТА), в частности ТМЭДА.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы XXV из соединения формулы X, при этом литирование проводят при помощи н-бутиллития, н-гексиллития или втор-бутиллития, в частности н-бутиллития.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы XXV из соединения формулы X, при этом литирование проводят при помощи н-BuLi, в присутствии тетраметилэтилендиамина (ТМЭДА) в МТБЭ и при -30 плюс/минус 2°C.
Соединение формулы XXV может быть выделено в виде кристаллического промежуточного соединения и затем преобразовано на втором этапе в имин формулы IX. Кристаллизация может быть выполнена, например, в этаноле или изопропаноле.
Альтернативно, неочищенный экстракт соединения XXV может быть введен в этап образования имина посредством замены растворителя на целевой растворитель с последующим образованием имина и выделением соединения формулы IX.
Конкретное воплощение изобретения относится к способу, как описано выше, включающему превращение соединения формулы XXV в соединение формулы IX, при этом образование имина проводят в спирте, таком как метанол, этанол, изопропанол или н-пропанол, в частности, в этаноле или метаноле или их смеси.
Имин формулы IX выделяют в виде кристаллического промежуточного соединения посредством прямой кристаллизации из реакционной смеси. Обнаружено, что кристаллизация имина обеспечивает очень эффективную точку очистки при синтезе.
Имин формулы IX может быть восстановлен посредством каталитической гидрогенизации для обеспечения промежуточного соединения формулы VIII.
Конкретное воплощение изобретения относится к способу, как описано выше, включающему превращение соединения формулы IX в соединение формулы VIII, при этом восстановление проводят при помощи водорода в присутствии катализатора, такого как платина на угле (Pt/C), в частности водорода и Pt/C в метаноле.
Конкретное воплощение изобретения относится к способу, как описано выше, включающему правкращение соединения формулы IX в соединение формулы VIII, при этом восстановление проводят при помощи водорода над Pt/C при температуре между 15°C и 50°C, в частности между 20 и 30°C, в частности между 20 и 25°C.
Конкретное воплощение изобретения относится к способу, как описано выше, включающему превращение соединения формулы IX в соединение формулы VIII, при котором проводят восстановление при помощи водорода над Pt/C при давлении между 1 и 10 бар (между 100000 и 1000000 Па), в частности при 5 бар (500000 Па).
Конкретное воплощение изобретения относится к способу, как описано выше, включающему превращение соединения формулы IX в соединение формулы VIII, при котором восстановление проводят при помощи водорода над Pt/C в метаноле при давлении 5 бар (500000 Па) и комнатной температуре.
Альтернативно, имин формулы IX может быть восстановлен до промежуточного соединения формулы VIII за счет применения боргидрида натрия.
Несмотря на то, что восстановление проводят в апротонном растворителе, таком как ТГФ, в присутствии карбоновой кислоты (уксусной кислоты, капроновой кислоты, 2-этилгексановой кислоты и пивалевой кислоты, в частности уксусной кислоты и пивалевой кислоты), лучшие результаты могут быть получены в протонных органических растворителях, таких как метанол или этанол, в частности в метаноле.
Проведение реакции в гомогенной реакционной системе, подобной смесям ТГФ/метанол, и в присутствии метиламина в качестве добавки минимизирует образование следующих 2 основных побочных продуктов димера 1 и димера 2.
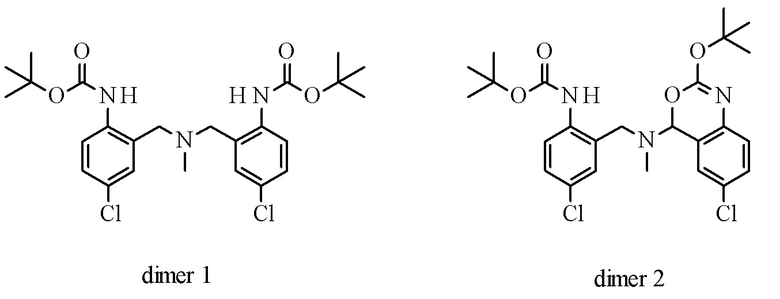
Гомогенная система максимизирует концентрацию имина формулы IX в растворе, таким образом повышая скорость продуктивного восстановления vs образования димеров. Из-за того, что иминовый субстрат имеет растворимость в метаноле от средней до низкой, применяют добавку, такую как, например, ТГФ, для обеспечения прозрачного раствора перед дозированием восстановителя.
Присутствие метиламина конкурирует с продуктом формулы VIII при реакции с иминовым субстратом формулы IX, таким образом, снижая количество побочных продуктов димера 1 и/или димера 2.
Конкретное воплощение изобретения относится к способу, как описано выше, включающему превращение соединения формулы IX в соединение формулы VIII, при котором восстановление выполняют при помощи боргидрида натрия в смеси ТГФ и метанола, в частности с достаточным количеством метанола, чтобы обеспечить реакционную способность, и достаточным количеством ТГФ, чтобы обеспечить растворимость иминового субстрата.
Конкретное воплощение изобретения относится к способу, как описано выше, включающему превращение соединения формулы IX в соединение формулы VIII, при котором восстановление выполняют при помощи боргидрида натрия в метаноле или смеси ТГФ и метанола, в частности смеси ТГФ и метанола, в частности в смеси метанола и ТГФ 2:1.
Конкретное воплощение изобретения относится к способу, как описано выше, включающему превращение соединения формулы IX в соединение формулы VIII, при котором восстановление боргидридом натрия проводят в присутствии карбоновой кислоты, такой как (но не ограничиваясь этим) уксусная кислота или пивалевая кислота, в частности в присутствии уксусной кислоты.
Конкретное воплощение изобретения относится к способу, как описано выше, включающему превращение соединения формулы IX в соединение формулы VIII, при котором восстановление боргидридом натрия проводят в присутствии метиламина. Конкретное воплощение изобретения относится к способу, как описано выше, включающему превращение соединения формулы IX в соединение формулы VIII, при котором восстановление боргидридом натрия проводят в смеси метанол/ТГФ 2:1, в присутствии уксусной кислоты и метиламина.
Конкретное воплощение изобретения относится к способу, включающему восстановление имина формулы IX до промежуточного соединения формулы VIII, при котором димер 1 и димер 2 образуются в качестве побочных продуктов в количествах менее 1%. Промежуточное соединение VIII может быть выделено посредством кристаллизации, например, из смеси iPrOH и воды или в виде соли, например, его уксуснокислой соли.
Экстракция неочищенного продукта формулы VIII (с этапа восстановления боргидридом натрия) в водной фазе при кислом рН (например, но не ограничиваясь этим, рН от 4 до 6), с последующим вымыванием примесей при помощи органического растворителя, с последующей экстракцией продукта в органическом растворителе при рН от нейтрального до основного дает продукт очень высокой чистоты. Затем экстракт может быть введен в следующий этап (алкилирование) без необходимости в проведении этапов кристаллизации и сушки.
Конкретное воплощение изобретения относится к способу, как описано выше, включающему превращение соединения формулы IX в соединение формулы VIII, в котором очистку неочищенного промежуточного соединения формулы проводят при помощи экстрагирующей обработки, в частности экстрагирования кислотой продукта в водной фазе, с последующей промывкой органическим растворителем, с последующей экстракцией продукта органическим растворителем при рН от нейтрального до основного.
Алкилирование соединения формулы VIII для получения соединения формулы VII может быть проведено при помощи хлор-, бром-, или йод-ацетонитрила. Реакционная способность хлорацетонитрила может быть повышена при помощи источника бромида или йодида, такого как, например, йодид или бромид калия.
Конкретное воплощение изобретения относится к способу, как описано выше, включающему превращение соединения формулы VIII в соединение формулы VII, при котором алкилирование проводят при помощи хлорацетонитрила, в частности хлорацетонитрила в присутствии йодида калия или бромида калия, в частности при помощи хлорацетонитрила в присутствии йодида калия.
Несмотря на то, что алкилирование может быть выполнено в полярных апротонных растворителях, подобных ДМФ, НМП, ДМА или ДМСО, для лучшей обработки все видов отходов предпочтительны альтернативные растворители. К подходящим растворителям относятся ТГФ, 2-Ме-ТГФ, ацетон, толуол, ацетонитрил или этилацетат. По кинетическим причинам, в частности, применяют ацетонитрил, ацетон и этилацетат, в частности этилацетат.
Конкретное воплощение изобретения относится к способу, как описано выше, включающему превращение соединения формулы VIII в соединение формулы VII, при котором алкилирование проводят при помощи хлорацетонитрила и йодида калия, в ацетоне, ацетонитриле или этилацетате, в частности в этилацетате. Этилацетат дает дополнительное преимущество, позволяя проводить прямое выделение экстракцией без замены растворителя перед экстракцией или применения дополнительного растворителя, разделяющего фазы.
Конкретное воплощение изобретения относится к способу, как описано выше, включающему превращение соединения формулы VIII в соединение формулы VII, при котором алкилирование проводят при помощи хлорацетонитрила в присутствии подходящего основания, такого как гидрокарбонат натрия, карбонат натрия, гидрокарбонат калия, гидрокарбонат натрия, гидрокарбонат цезия или карбонат цезия, в частности при помощи гидрокарбоната натрия или гидрокарбоната калия, в частности при помощи гидрокарбоната натрия.
Конкретное воплощение изобретения относится к способу, как описано выше, включающему превращение соединения формулы VIII в соединение формулы VII, при котором алкилирование проводят при помощи хлорацетонитрила, в этилацетате с обратным холодильником, в присутствии йодида и гидрокарбоната натрия в качестве основания.
Продукт формулы VII может быть выделен посредством кристаллизации, например, в изопропаноле или смесях этанол/вода.
Конкретное воплощение изобретения относится к способу, как описано выше, включающему превращение соединения формулы VII в соединение формулы III, при котором реакцию проводят в присутствии избытка HCl, в спирте, таком как метанол, этанол, трифторэтанол, изопропанол, в частности изопропанол или трифторэтанол, в частности изопропанол, или в смеси спирт/дихлорметан, в частности трифторэтанол/дихлорметан (для применения трифторэтанола в качестве растворителя для получения амидинов из нитрила см. Caron et al.6).
Конкретное воплощение изобретения относится к способу, как описано выше, включающему превращение соединения формулы VII в соединение формулы III, при котором соединение формулы VII преобразуют в соединение формулы XI. 2 HCl, которое не выделяют, а in situ превращают впоследствии в соединение формулы III.
Конкретное воплощение изобретения относится к способу, как описано выше, включающему превращение соединения формулы VII в соединение формулы III, при котором образуются алкил-2-[(2-амино-5-хлор-фенил)метил-метил-амино]ацетат, соответствующие побочные продукты на основе имидата или ортоэфира, при этом используется RO фрагмент, приходящий из спирта.
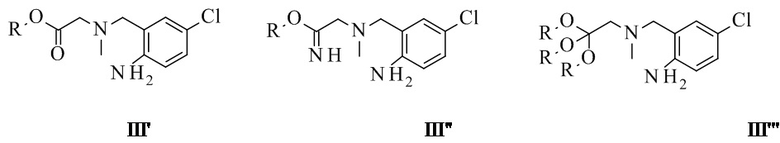
По сравнению с применением линейных спиртов, подобных этанолу, количество этих побочных продуктов (III', III'', III''') снижено за счет применения менее нуклеофильных спиртов, подобных изопропанолу или трифторэтанолу. Изопропанол является новой и более дешевой альтернативой трифторэтанолу.
Конкретное воплощение изобретения относится к способу, как описано выше, включающему превращение соединения формулы VII в соединение формулы III, при котором реакцию проводят в присутствии избытка HCl, в изопропаноле.
Конкретное воплощение изобретения относится к способу, как описано выше, включающему превращение соединения формулы VII в соединение формулы III, при котором исходный материал дозируют в раствор, после чего происходит удаление защитных групп Boc в соединении формулы XI (в виде хлористоводородной соли) способом, позволяющим контролировать отходящий CO2.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, дополнительно включающему превращение соединения формулы X в соединение формулы XI через следующие этапы:
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, включающему следующие этапы:
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, дополнительно включающему превращение соединения формулы XV в соединение формулы XI через следующие этапы:
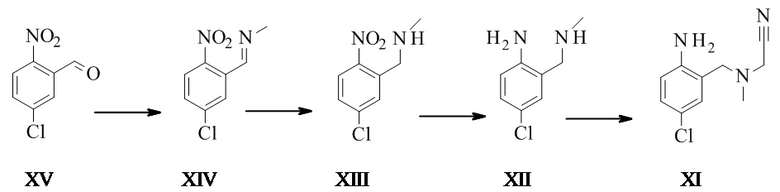 .
.
Соединение формулы XII также может быть выделено в виде хлористоводородной соли.
Превращение соединения формулы XV в соединение формулы XII производилось на основе WO2005/684667.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, включающему следующие этапы:
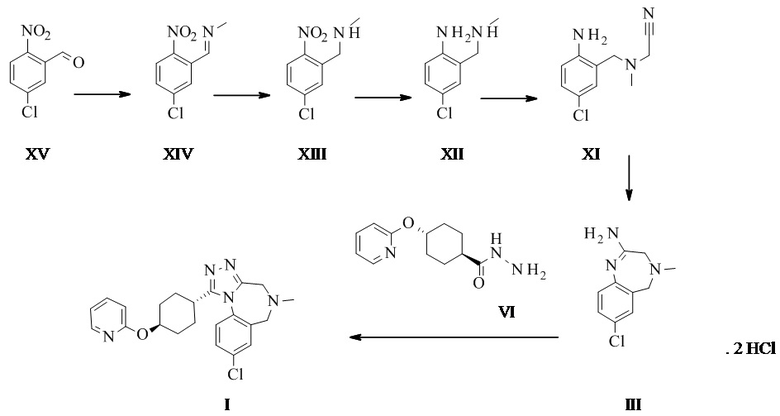
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, дополнительно включающему превращение соединения формулы XVI в соединение формулы XI через следующие этапы:
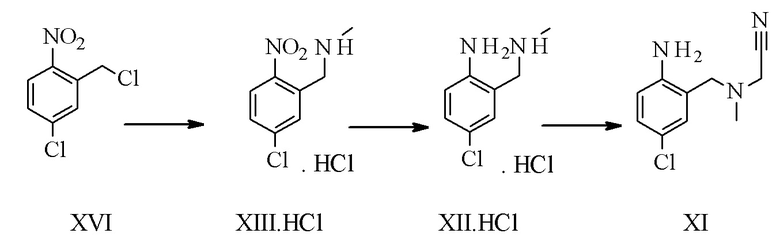
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, включающему следующие этапы:
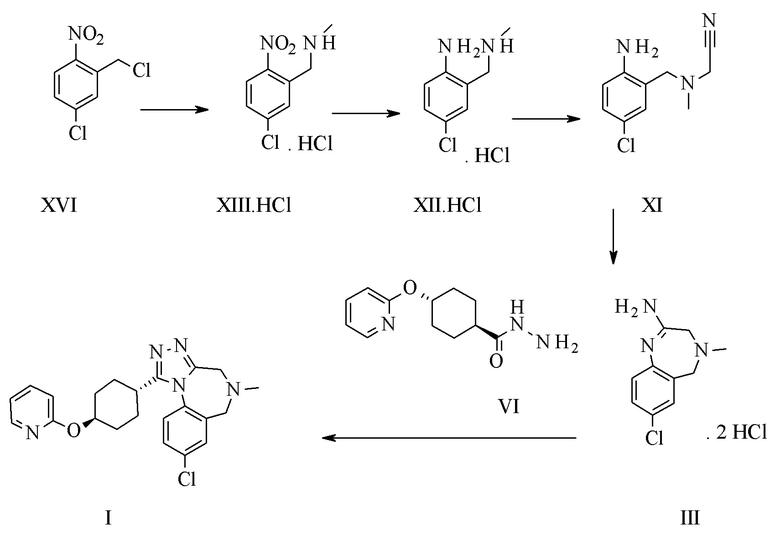
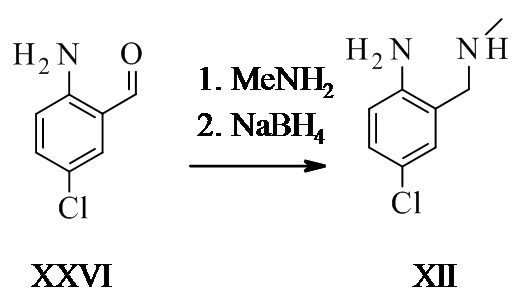
Конкретное воплощение изобретения относится к синтезу соединения формулы I, включающему превращение соединения формулы XXVI в соединение формулы XI через следующие этапы:
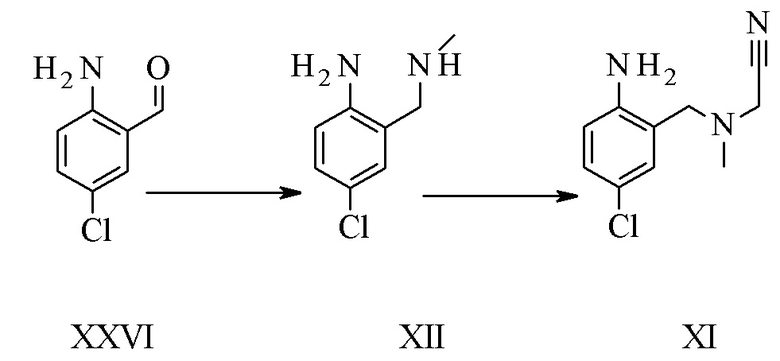 .
.
Соединение формулы XII описано в уровне техники Venkov et al.8 в качестве промежуточного соединения, которое не выделяли и использовали непосредственно в последующей реакции.
Конкретное воплощение изобретения относится к способу, как описано выше, включающему превращение соединения формулы XXVI в соединение формулы XII, в котором соединение формулы XII выделяют из реакционной смеси.
Конкретное воплощение изобретения относится к способу, как описано выше, включающему превращение соединения формулы XXVI в соединение формулы XI, в котором восстановительное аминирование и этап алкилирования выполняют в одном сосуде.
Конкретное воплощение изобретения относится к промежуточному соединению XI.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, включающему следующие этапы:
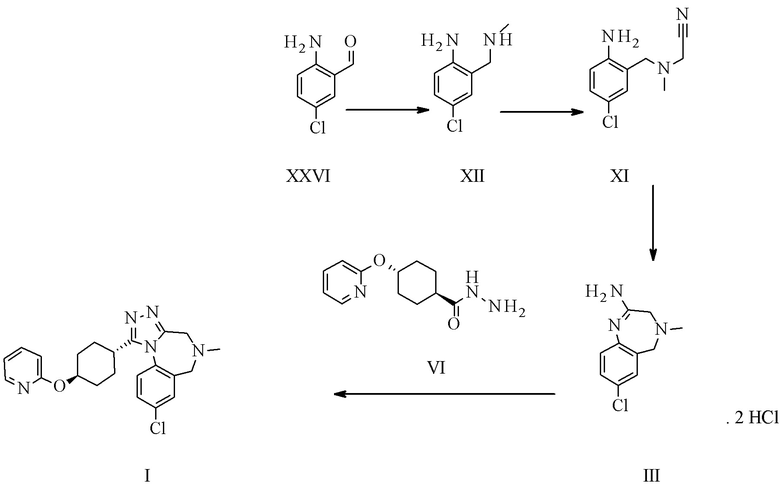 .
.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, как описано в данном документе, включающему следующие этапы:
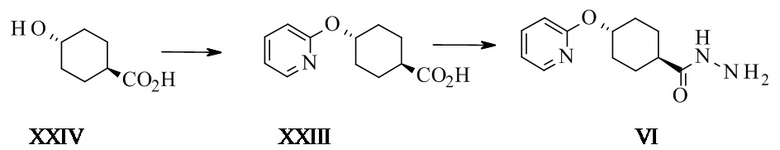
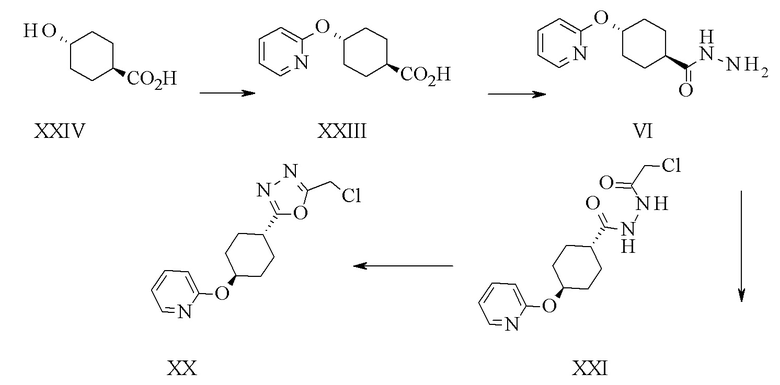
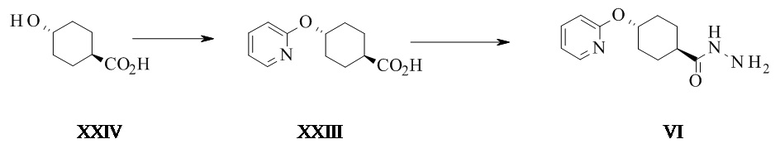
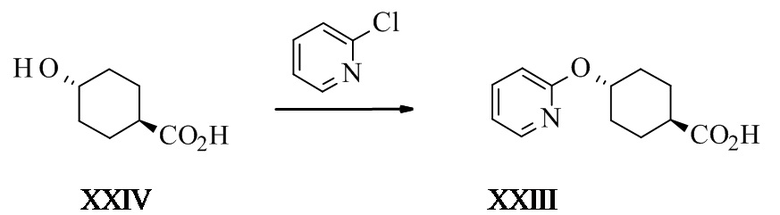
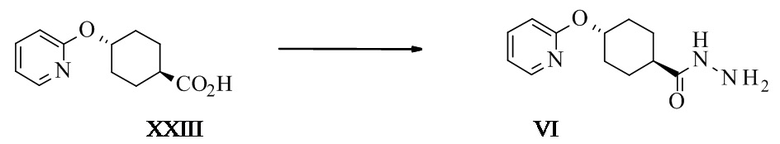
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, как описано в данном документе, включающему превращение соединения формулы XXIII в соединение формулы VI путем ароматического нуклеофильного замещения 2-галопиридина 4-гидроксициклогексанкарбоновой кислотой.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, как описано в данном документе, включающему превращение соединения формулы XXIV в соединение формулы XXIII, в котором могут использоваться основания, такие как трет-амилалкоголят натрия (tAmONa), трет-амилалкоголят калия (tAmOK), трет-бутоксид натрия (tBuONa), трет-бутоксид калия (tBuOK), в частности tAmONa.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, как описано в данном документе, включающему превращение соединения формулы XXIV в соединение формулы XXIII, в котором растворитель представляет собой N-метил-2-пирролидон (НМП) или диметилацетамид (ДМА), в частности НМП.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, как описано в данном документе, включающему превращение соединения формулы XXIV в соединение формулы XXIII, в котором реакцию проводят при температуре от 80 до 120°C, в частности от 88 до 92°C.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, как описано в данном документе, включающему превращение соединения формулы XXIV в соединение формулы XXIII, в котором 2-галопиридины выбраны из 2-фторпиридина и 2-хлорпиридина, в частности 2-хлорпиридина.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, как описано в данном документе, включающему превращение соединения формулы XXIV в соединение формулы XXIII, в котором соединение формулы XXIV реагирует с 2-хлорпиридином в НМП в присутствии трет-амилоксида при температуре от 85 до 95°C.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, как описано в данном документе, включающему превращение соединения формулы XXIII в соединение формулы VI, в котором XXIII активируют реакцией с подходящим алкилхлорформиатом, таким как изобутил-, этил- или метилхлорформиат, в частности изобутилхлорформиат.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, как описано в данном документе, включающему превращение соединения формулы XXIII в соединение формулы VI, в котором XXIII активируют при помощи подходящего алкилхлорформиата в присутствии подходящего основания, такого как триэтиламин, основание Хенига, пиридин, коллидин или N-метилморфолин, в частности N-метилморфолин.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, как описано в данном документе, включающему превращение соединения формулы XXIII в соединение формулы VI, в котором XXIII активируют карбонилдимимидазолом (КДИ) для получения соответствующего промежуточного соединения ацилимидазола, которое затем реагирует с гидразином.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, как описано в данном документе, включающему превращение соединения формулы XXIII в соединение формулы VI, в котором проводят реакцию в подходящем растворителе, таком как ДМФ, НМП, ТГФ, 2-МеТГФ, в частности ТГФ.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, как описано в данном документе, включающему превращение соединения формулы XXIII в соединение формулы VI, в котором активацию КДИ проводят при температуре от 10°C до 50°C, предпочтительно от 20°C до 30°C, более предпочтительно при 25°C.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, как описано в данном документе, включающему превращение соединения формулы XXIII в соединение формулы VI, в котором промежуточное соединение ацилимидазола затем реагирует с гидразином, в частности применяют избыток гидразина, более предпочтителен по меньшей мере 2-кратный избыток КДИ на этапе активации.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, как описано в данном документе, включающему превращение соединения формулы XXIII в соединение формулы VI, в котором порядок прибавления включает прибавление активированной кислоты к гидразину.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, как описано в данном документе, включающему превращение соединения формулы XXIII в соединение формулы VI, в котором реакционная смесь ацилимидазола может быть дегазирована после активации и до реакции с гидразином для удаления растворенного CO2.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, как описано в данном документе, включающему превращение соединения формулы XXIII в соединение формулы VI, в котором в качестве побочного продукта образуется 4-(2-пиридилокси)-N'-[4-(2-пиридилокси)циклогексанкарбонил]циклогексан-карбогидразид (VI').
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, включающему следующие этапы:
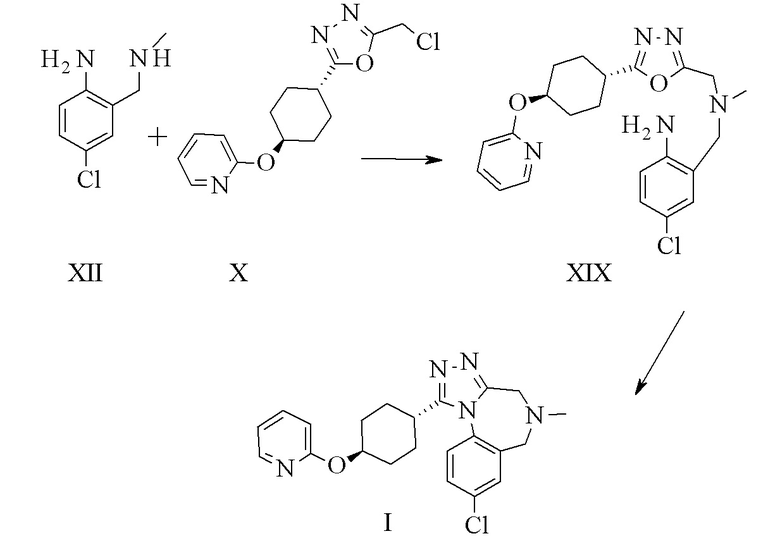
Конкретные предшественники оксадиазола описаны в уровне техники9.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, как описано в данном документе, включающему следующие этапы:
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, включающему следующие этапы:
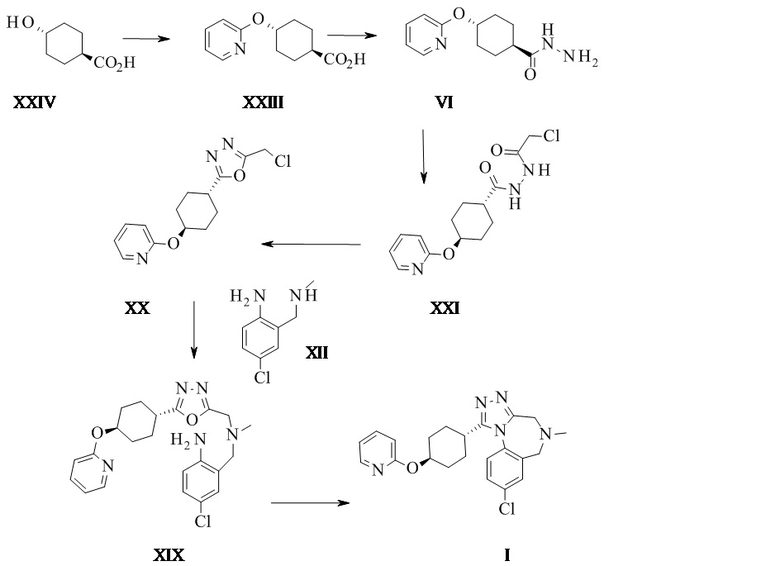 .
.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, в котором в качестве промежуточного соединения образуется соединение формулы INT, его таутомер или соль:
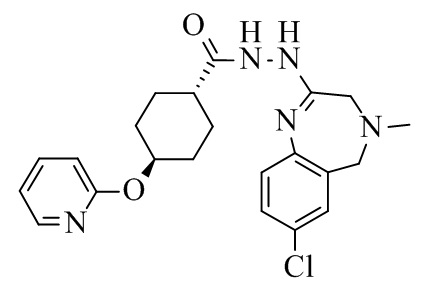 INT.
INT.
Конкретное воплощение изобретения относится к промежуточному соединению INT, его таутомеру или соли. Конкретное воплощение изобретения относится к промежуточному соединению INT.
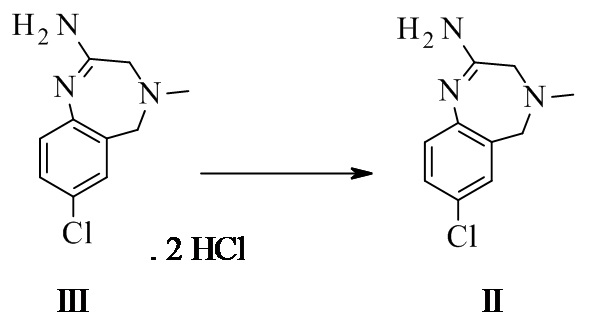
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, в котором в качестве промежуточного соединения образуется соединение формулы III.
Конкретное воплощение изобретения относится к промежуточному соединению II, его таутомеру или соли:
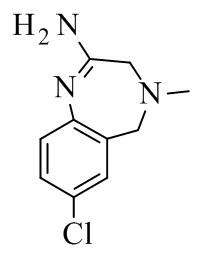 II: свободное основание, III: 2HCl.
II: свободное основание, III: 2HCl.
Конкретное воплощение изобретения относится к промежуточному соединению II или его соли.
Конкретное воплощение изобретения относится к промежуточному соединению III.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы VI.
 .
.
Конкретное воплощение изобретения относится к способу синтеза соединения формулы I, включающему следующие этапы:
 .
.
Конкретное воплощение изобретения относится к соединению формулы I или его фармацевтически приемлемой соли, во всех случаях полученные способом, описанным в данном документе.
Конкретное воплощение изобретения относится к соединению формулы I, как описано в данном документе, для применения в качестве лекарственного препарата.
Конкретное воплощение изобретения относится к соединению формулы I, как описано в данном документе, для применения при терапевтическом и/или профилактическом лечении неадекватной секреции вазопрессина, тревоги, депрессивных расстройств, обсессивно-компульсивного расстройства, аутистических расстройств, шизофрении, агрессивного поведения и расстройств, связанных со сдвигом фазы сна, в частности десинхроноза.
Краткое описание графических материалов
Данные инфракрасной спектроскопии с преобразованием Фурье (FTIR) собраны в суспензии в вазелиновом масле, так что в области ИК-спектра видны дополнительные пики за счет агента, диспергирующего минеральное масло.
Фиг. 1: порошковая рентгеновская дифрактограмма формы А.
Фиг. 2: ИК-спектр формы А.
Фиг. 3: спектр Рамана формы А.
Фиг. 4: порошковая рентгеновская дифрактограмма формы B.
Фиг. 5: ИК-спектр формы B.
Фиг. 6: спектр Рамана формы B.
Фиг. 7: порошковая рентгеновская дифрактограмма формы C.
Фиг. 8: ИК-спектр формы C.
Фиг. 9: спектр Рамана формы C.
Фиг. 10: порошковая рентгеновская дифрактограмма формы D.
Фиг. 11: ИК-спектр формы D.
Фиг. 12: спектр Рамана формы D.
Фиг. 13: порошковая рентгеновская дифрактограмма формы E.
Фиг. 14: ИК-спектр формы E.
Фиг. 15: спектр Рамана формы E.
Фиг. 16: порошковая рентгеновская дифрактограмма формы F.
Фиг. 17: ИК-спектр формы F.
Фигура 18: спектр Рамана формы F.
Фиг. 19: порошковая рентгеновская дифрактограмма формы G.
Фиг. 20: ИК-спектр формы G.
Фиг. 21: спектр Рамана формы G.
Фиг. 22: порошковая рентгеновская дифрактограмма формы H.
Фиг. 23: спектр Рамана формы H.
Экспериментальная часть
Следующие эксперименты приведены в качестве иллюстраций изобретения. Они не должны рассматриваться как ограничивающие объем изобретения, а лишь в качестве его иллюстрации.
Форма А соединения I
100 мг I растворяли в закрытом сосуде при 22°C в 5,0 мл смеси этанол/вода 1:1 (о/о). После растворения раствор фильтровали через 0,45 мкм фильтр. Затем прозрачный раствор выпаривали при 22°C в течение 10 дней. После полного выпаривания продукт высушивали (50°C/менее 20 мбар (2000 Па) в течение более 24 ч) и анализировали.
Форма B соединения I
100 мг соединения I растворяли в закрытом сосуде при 22°C в 3,0 мл этилацетата. После растворения раствор фильтровали через 0,45 мкм фильтр. Затем прозрачный раствор выпаривали при 22°C в течение 10 дней. Эксперимент давал единичные кристаллы формы В, подходящие для анализа монокристаллической структуры. После полного выпаривания продукт высушивали (50°C/менее 20 мбар (2000 Па) в течение более 24 ч) и анализировали.
Форма C соединения I
100 мг I растворяли в закрытом сосуде при 22°C в 1,4 мл смеси воды, насыщенной бутанолом (приблизительно 20% о/о). После растворения раствор фильтровали через 0,45 мкм фильтр. Затем прозрачный раствор выпаривали при 22°C в течение 1 месяца. Эксперимент давал монокристаллы формы C, подходящие для анализа монокристаллической структуры. После полного выпаривания продукт высушивали (50°C/менее 20 мбар (2000 Па) в течение более 24 ч) и анализировали.
Форма D (гемисольват п-ксилола) соединения I
100 мг соединения I суспендировали в закрытом сосуде при 22°C в 0,35 мл п-ксилола и оставляли перемешиваться при 60°C. После 14 дней уравновешивания при 60°C, суспензию фильтровали и продукт высушивали (50°C/менее 20 мбар (2000 Па) в течение более 24 ч) и анализировали. Выпаривание фильтрата (3 дня при 22°C) давало монокристаллы формы D, подходящие для анализа монокристаллической структуры.
Форма E (гемисольват уксусной кислоты) соединения I
100 мг соединения I растворяли в закрытом сосуде при 22°C в 0,4 мл уксусной кислоты. После растворения раствор фильтровали через 0,45 мкм фильтр. Затем прозрачный раствор выпаривали при 22°C в течение 14 дней. Эксперимент давал маслянистый остаток, который превращался в порошок после соскабливания шпателем. Продукт высушивали (50°C/менее 20 мбар (2000 Па) в течение более 24 ч) и анализировали.
Форма F соединения I
100 мг формы В суспендировали в закрытом сосуде при 22°C в 0,3 мл изопропанола и перемешивали при 22°C. После однодневного перемешивания добавляли 10 мг API /формы C и суспензию перемешивали при 22°C. После 14 дней уравновешивания при 22°C суспензию фильтровали, продукт высушивали (50°C/менее 20 мбар (2000 Па) в течение более 24 ч) и анализировали.
Форма G (сольват бутиронитрила) соединения I
100 мг соединения I растворяли в закрытом сосуде при 22°C в 1,5 мл бутиронитрила. Сразу после растворения раствор начинал при перемешивании выпадать в осадок. Суспензию частично выпаривали, продолжая перемешивать, при 22°C в течение 10 дней. После частичного выпаривания (приблизительно 50%) суспензию фильтровали, продукт высушивали (50°C/менее 20 мбар (2000 Па) в течение более 24 ч) и анализировали. Выпаривание фильтрата (2 недели при 22°C) давало монокристаллы формы G, подходящие для анализа монокристаллической структуры.
Форма H (тригидрат) соединения I
100 мг соединения I растворяли в 1,9 мл смеси этанол/вода 1:1 (о/о) при 65°C в закрытом сосуде. Прозрачный раствор линейно охлаждали с 65°C до -20°C в течение 8 ч без перемешивания. Эксперимент давал монокристаллы формы H, подходящие для анализа монокристаллической структуры. Продукт выделяли посредством удаления маточной жидкости при помощи пипетки и анализировали во влажном состоянии.
Трет-бутил-N-(4-хлор-2-формилфенил)карбамат XXV
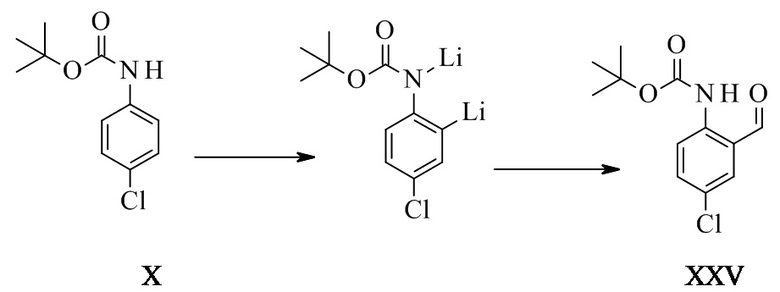
Трет-бутил-4-хлорфенилкарбамат (40 г, 175 ммоль, Eq: 1,00) растворяли в ТГФ (248 г, 280 мл). Раствор охлаждали до -30°C. N,N,N',N'-тетраметилэтилендиамин (44,5 г, 57,8 мл, 379 ммоль, Eq: 2,17) добавляли по капле. Спустя 5 мин н-бутиллитий 2,5 M в гексане (210 мл, 524 ммоль, Eq: 3,00) добавляли по капле в течение 60 мин при -30°C до -20°C. Спустя 5 ч при -30° ДМФ (38,4 г, 40,5 мл, 524 ммоль, Eq: 3,00) добавляли в течение 35 мин. Спустя 1 ч при -30°C добавляли холодный (от 0 до 5°C) метил-трет-бутиловый эфир (МТБЭ) (207 г, 280 мл) (0°C). 25% водный хлористый водород (HCl) (178 г, 149 мл, 1,22 моль, Eq: 7,0) добавляли в течение 30 мин при от -30° до 0°C. Водную фазу отделяли и экстрагировали при помощи МТБЭ (74,0 г, 100 мл). Органические фазы промывали последовательно при помощи 10% водного хлорида натрия (NaCl) (100 мл), 5% водного гидрокарбоната натрия (NaHCO3) (100 мл) и полунасыщенного водного NaCl (100 мл). Органические фазы комбинировали, высушивали над сульфатом магния (MgSO4) и концентрировали при пониженном давлении (40°C/ снижали до 10 мбар (1000 Па)) до получения 45,2 г неочищенного продукта. Неочищенный продукт растворяли в 2-пропаноле (157 г, 200 мл) при 80°C. Прозрачный раствор медленно охлаждали до 0°C, во время чего продукт начинал кристаллизоваться. Суспензию перемешивали в течение 1 ч при 0°C и фильтровали. Осадок на фильтре промывали холодным (от 0 до 5°C) 2-пропанолом (15,7 г, 20 мл), высушивали при 50°C/10 мбар (1000 Па) до получения 38,8 г титульного соединения.
Трет-бутил-N-[4-хлор-2-[(E)-метилиминометил]фенил]карбамат IX
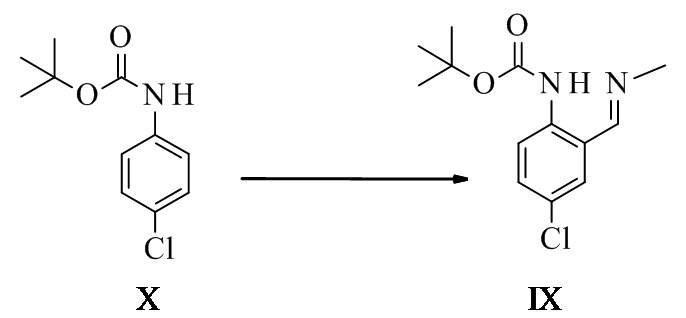
Способ МТБЭ
N-Boc-4-хлоранилин (121 г, 531 ммоль, Eq: 1,00) растворяли в МТБЭ (648 г, 875 мл). Раствор охлаждали до -25°C. Добавляли TMEDA (тетраметилэтилендиамин) (72 г, 92,9 мл, 620 ммоль, Eq: 1,17). 2,5 M н-бутиллитий (BuLi) в гексане (398 г, 572 мл, 1,43 моль, Eq: 2,69) добавляли через 70 мин, поддерживая температуру ниже -20°C. Спустя 2,5 ч добавляли диметилформамид (ДМФ) (113 г, 120 мл, 1,55 моль, Eq: 2,91) в течение 30-45 мин, поддерживая температуру в диапазоне от -30°C до -20°C. Спустя 1 ч 25% водный HCl (526 г, 470 мл, 3,61 моль, Eq: 6,79) добавляли с такой скоростью, что внутренняя температура поддерживалась между -30°C и 0°C. Реакционную смесь нагревали до комнатной температуры (КТ) в течение 30 мин. Водную фазу отделяли и экстрагировали при помощи МТБЭ (333 г, 450 мл). Органические фазы объединяли и промывали последовательно насыщенным водным NaCl (600 мл), 10% водным NaHCO3 (600 мл) и водным NaCl (600 мл). Органическую фазу концентрировали до приблизительно 550 мл и растворитель МТБЭ заменяли на этанол (EtOH) при постоянном объеме (Tj max 55°C). Суспензию неочищенного альдегида разбавляли EtOH (250 мл). Добавляли 33% метиламин в EtOH (150 г, 1,59 моль, Eq: 3), и реакционную смесь перемешивали в течение более 2 ч при 25°C (до тех пор, пока не оставалось менее 2% альдегида, IPC). При необходимости в реакционную смесь вводили затравку при 20°C. Полученную суспензию охлаждали в течение 1 ч до -10°C. Спустя 3 ч при -10°C суспензию фильтровали. Осадок на фильтре промывали холодным (приблизительно -10°C) EtOH и высушивали при 60°C/5 мбар (500 Па) для получения 109 г титульного соединения в виде светло-желтых кристаллов.
Способ ТГФ
Альтернативно, трет-бутил-4-хлорфенилкарбамат (120 г, 511 ммоль, Eq: 1,00) растворяли в тетрагидрофуране (ТГФ) (745 г, 840 мл). Раствор охлаждали до -30°C. Добавляли N,N,N',N'-тетраметилэтилендиамин (129 г, 168 мл, 1,1 моль, Eq: 2,15). N-бутиллитий 2,5 M в гексане (613 мл, 1,53 моль, Eq: 3.00) добавляли в течение 60 мин при температуре между -30°C и -20°C. Спустя 5 ч при -30°C добавляли ДМФ (112 г, 118 мл, 1,53 моль, Eq: 3,00) в течение 45 мин при температуре между -30° и -20°C. 25% HCl (522 г, 435 мл, 3,58 моль, Eq: 7,0) добавляли в течение 30 мин при температуре от -30°C до 0°C (pH от 4 до 5). Водную фазу отделяли и экстрагировали при помощи смеси ТГФ (106 г, 120 мл) и гексана (79,1 г, 120 мл). Органические фазы последовательно промывали полунасыщенным водным NaCl (240 мл), 5% водным NaHCO3 (240 мл) и полунасыщенным водным NaCl (240 мл). Органические фазы объединяли и концентрировали до приблизительно 300 мл и разделяли на две части.
Часть 1 разводили ТГФ (887 г, 1 л) и азеотропировали при 45°C / 400 мбар (40000 Па). Раствор представлял собой растворитель, заменяемый на метанол для получения 285 г желтой суспензии (остаточная вода: 0,14 %). Добавляли 9,8 M метиламина в метаноле (36,5 мл, 358 ммоль, Eq: 1,4 относительно теоретического содержания альдегида). Получали прозрачный желтый раствор. После 15 мин имин начинал кристаллизоваться (в случае если спонтанная кристаллизация не наблюдалась, вводили затравку). Спустя 2 ч при температуре от 20 до 25°C суспензию перемешивали в течение 1 ч при 40°C, охлаждали до -10°C в течение 1 ч и фильтровали. Осадок на фильтре промывали холодным (-10°C) метанолом (47,5 г, 60 мл) и высушивали при 40°C при пониженном давлении с получением 57 г титульного соединения в виде светло-желтого порошка.
Часть 2 азеотропировали и растворитель заменяли на этанол при 45°C / 200 мбар (20000 Па) для получения 281 г желтой суспензии (вода: менее 0,1 %). Добавляли 9,8 M метиламина в метаноле (36,5 мл, 358 ммоль, Eq: 1,4 к теоретическому содержанию альдегида) при КТ. Спустя 4 ч при КТ и 1 ч при -10°C суспензию фильтровали. Осадок на фильтре промывали холодным (-10°C) этанолом (47,4 г, 60 мл) и высушивали при 40°C при пониженном давлении с получением 51,5 г титульного соединения в виде желтого порошка.
Трет-бутил-N-[4-хлор-2-[(E)-метилиминометил]фенил]карбамат IX
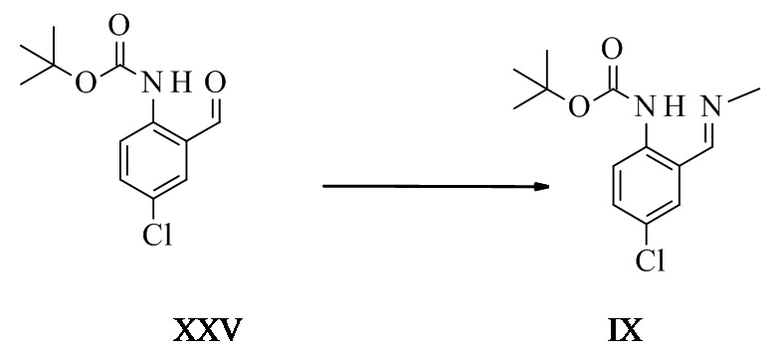
Трет-бутил-4-хлор-2-формилфенилкарбамат (38 г, 149 ммоль, Eq: 1,00) суспендировали в метаноле (195 г, 247 мл). Добавляли 9,8 M раствор метиламина в метаноле (21,2 мл, 208 ммоль, Eq: 1,40) в течение 30 мин при КТ. Реакционную смесь перемешивали 1 ч и полученный раствор охлаждали до -10°C (при приблизительно 0°C продукт начинал спонтанно кристаллизоваться). Спустя 2 ч при -10°C суспензию фильтровали. Осадок на фильтре промывали холодным (-10°C) метанолом (15,0 г, 19,0 мл) и высушивали при пониженном давлении (10 мбар (1000 Па) /50°C) с получением 36,4 г титульного соединения в виде белого кристаллического порошка.
Трет-бутил-N-[4-хлор-2-(метиламинометил)фенил]карбамат VIII
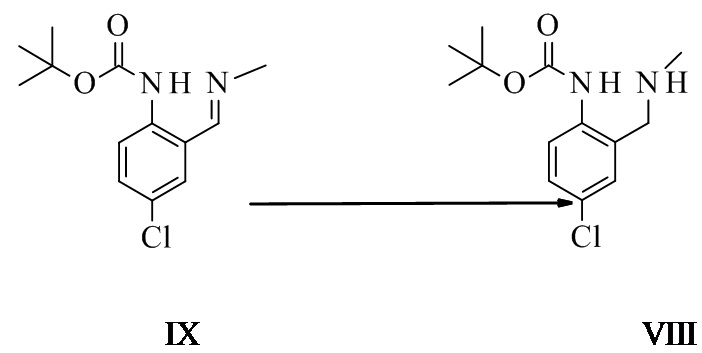
Трет-бутил-N-[4-хлор-2-[(E)-метилиминометил]фенил]карбамат (50 г, 184 ммоль, Eq: 1,00) растворяли в смеси метанола (253 г, 320 мл) и ТГФ (142 г, 160 мл). Раствор охлаждали до КТ. Добавляли 40% раствор метиламина в метаноле (MeOH) (14,4 г, 185 ммоль, Eq: 1,01), а затем уксусную кислоту (AcOH) (22,0 г, 21,0 мл, 365 ммоль, Eq: 1,98). Venpure 20-20 (боргидрид натрия (NaBH4) 20% / гидроксид натрия (NaOH) 20% в воде, 35 г, 28,8 мл, 185 ммоль, Eq: 1,00) добавляли при 0°C в течение 45-60 мин. Спустя 30 мин добавляли ацетон (21,4 г, 27,0 мл, 366 ммоль, Eq: 1,99) в течение 30 мин при 0°C. Спустя более 0,5 ч при 0°C реакционную смесь добавляли к смеси, состоящей из 5% водного Na2CO3 (500 мл), полунасыщенного водного NaCl (125 мл) и МТБЭ (370 г, 500 мл). Органическую фазу отделяли и промывали 10% водным NaCl (210 г, 200 мл). Органическую фазу экстрагировали дважды смесью, состоящей из 9 мл муравьиной кислоты в 0,5 л воды. Водные фазы объединяли и промывали дважды МТБЭ (370 г, 500 мл). Органические фазы удаляли. Добавляли МТБЭ (0,5 л) и pH доводили до 12-13, добавляя 32% водный NaOH (41,9 г, 31 мл, 335 ммоль, Eq: 1,82). Водную фазу отделяли и экстрагировали МТБЭ (250 мл). Органические фазы объединяли и промывали насыщенным водным NaHCO3 (209 г, 200 мл) и 10% водным NaCl (210 г, 200 мл) (pH 7-8). Раствор неочищенного продукта концентрировали до приблизительно половины объема (KFT менее 0,5% воды). Смесь неочищенного продукта фильтровали для удаления солей. Раствор концентрировали при пониженном давлении с получением 51 г неочищенного продукта (более 99,5 абс.% согласно высокоэффективной жидкостной хроматографии (ВЭЖХ), содержит приблизительно 8% остаточного МТБЭ). В растворе неочищенного продукта заменяли растворитель на этилацетат (AcOEt) и вводили в следующий этап без дополнительной очистки.
Продукт может быть перекристаллизован из смеси изопропанол (iPrOH)/вода:
1,0 г трет-бутил-N-[4-хлор-2-(метиламинометил)фенил]карбамата растворяли при 40°C в 2-пропаноле (3,92 г, 5 мл). Прозрачный раствор охлаждали до КТ и добавляли воду (3,00 г, 3 мл). Раствор затравливали (неочищенный высушенный продукт медленно кристаллизовался при выстаивании, давая первые затравочные кристаллы), и кристаллизация медленно начиналась. Спустя 30 мин добавляли воду (7,00 г, 7 мл) по каплям в течение 10 мин. Белую суспензию перемешивали 1 ч при КТ и фильтровали. Осадок на фильтре промывали водой и высушивали при 40°C/5 мбар (500 Па) с получением 1 г продукта в виде белых кристаллов. Альтернативно, трет-бутил-N-[4-хлор-2-[(E)-метилиминометил]фенил]карбамат (2 г, 7,29 ммоль, Eq.: 1) суспендировали в метаноле (20 мл). Добавляли Pt/C 5% (185 мг), смесь продували водородом (5 бар (500000 Па)) и перемешивали при КТ. После завершения реакции катализатор отфильтровывали и раствор концентрировали при пониженном давлении с получением 1,85 г неочищенного трет-бутил-N-[4-хлор2-(метиламинометил)-фенил]карбамата. Титульное соединение кристаллизовали, как описано выше.
Трет-бутил-N-[4-хлор-2-[[цианометил(метил)амино]метил]фенил]-карбамат VII
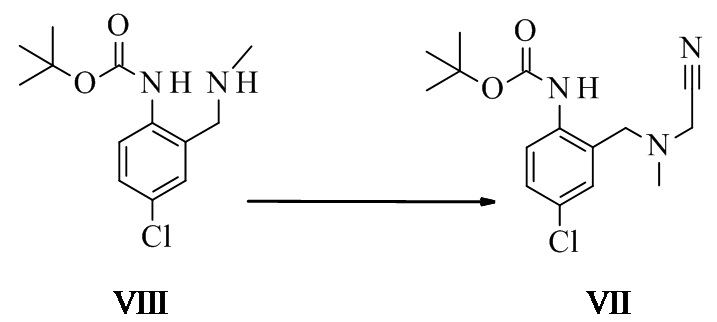
Трет-бутил-N-[4-хлор-2-(метиламинометил)фенил]карбамат (49,9 г, 184 ммоль, Eq: 1,00) растворяли в AcOEt (226 г, 250 мл). Гидрокарбонат натрия (16,6 г, 198 ммоль, Eq: 1,07) и йодид калия (KI) (6 г, 36,0 ммоль, Eq: 0.196) добавляли одной навеской. Добавляли одной навеской 2-хлорацетонитрил (15,4 г, 13,0 мл, 200 ммоль, Eq: 1,09) и реакционную смесь нагревали с обратным холодильником в течение 15 ч (менее 2% исходного материала). Реакционную смесь охлаждали до КТ. Добавляли 10% водный NaCl (262 г, 250 мл). Органическую фазу отделяли и промывали полунасыщенным водным NaHCO3 (261 г, 250 мл). Органическую фазу перемешивали в течение ночи с 10% водным тиосульфатом натрия (291 г, 250 мл, 184 ммоль, Eq: 1,00) и хлоридом тетрабутиламмония (1 г, 3,6 ммоль, Eq: 0,02). Органическую фазу отделяли и промывали 10% водным NaCl (262 г, 250 мл). Органическую фазу концентрировали приблизительно до половины объема и фильтровали. Объем доводили приблизительно до 200 мл EtOH и растворитель в растворе заменяли на EtOH при постоянном объеме. Раствор охлаждали приблизительно до температуры 28-30°C и вводили затравку. Спустя 30 мин суспензию охлаждали до КТ и добавляли воду (40 мл) по каплям. Суспензию перемешивали в течение ночи при КТ и 2 ч при температуре от 0 до 5°C. Суспензию фильтровали. Осадок на фильтре промывали EtOH/вода 1:1 (100 мл) и высушивали при 60°C/5 мбар (500 Па) с получением 46,8 г титульного соединения в виде белых кристаллов.
трет-бутил-N-[4-хлор-2-[[цианометил(метил)амино]метил]фенил]-карбамат VI
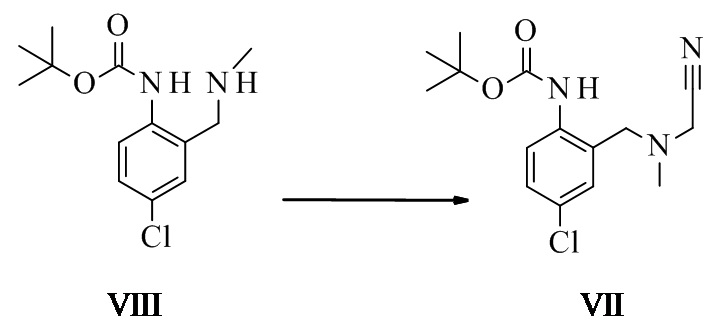
Трет-бутил-4-хлор-2-((метиламино)метил)фенилкарбамат (9,0 г, 31,6 ммоль, Eq: 1,00) растворяли в этилацетате (40,6 г, 45,0 мл). Добавляли бикарбонат натрия (3,18 г, 37,9 ммоль, Eq: 1,2), а затем йодид калия (1,06 г, 6,34 ммоль, Eq: 0,201). Добавляли 2-хлорацетонитрил (2,92 г, 2,46 мл, 37,9 ммоль, Eq: 1,2), суспензию нагревали до 78°C (масляная баня 80°C) и перемешивали в течение ночи. Реакционную смесь охлаждали до КТ и добавляли воду (22,5 г, 22,5 мл). Органическую фазу отделяли и промывали полунасыщенным водным NaHCO3 (22,5 мл), 10% водным раствором тиосульфатата натрия (22,5 мл) и водой (22,5 г, 22,5 мл). Органическую фазу концентрировали при пониженном давлении (45°C/ 180 мбар (18000 Па), приблизительно 50 мл) до приблизительно половины объема. Растворитель в растворе неочищенного продукта заменяли на 2-пропанол (конечный объем приблизительно 30 мл). Раствор 2-пропанола затравливали и перемешивали в течение 1 ч при КТ, затем белую суспензию охлаждали до температуры 0°-2°C, перемешивали в течение еще одного часа и фильтровали через стеклокерамическую воронку. Кристаллы промывали холодным 2-пропанолом (7,84 г, 10 мл) и высушивали до постоянной массы (5 мбар (500 Па)/ 50°C) с получением 8,8 г титульного соединения в виде белого кристаллического порошка.
7-хлор-4-метил-3,5-дигидро-1,4-бензодиазепин-2-амин дигидрохлорид III
2-пропанол (312 г, 400 мл) загружали в реактор при температуре от 20 до 25°C. Добавляли ацетилхлорид (AcCl) (255 г, 231 мл, 3,22 моль, Eq: 9.97) по каплям в течение 45 мин. Спустя 15 мин добавляли теплый (45-55°C) раствор трет-бутил-N-[4-хлор-2-[[цианометил(метил)амино]метил]фенил]карбамата в 2-пропаноле (468 г, 600 мл) в течение 45-60 мин, поддерживая температуру в диапазоне от 20 до 40°C, в ходе чего большая часть Boc-защитных групп удаляется и начинается этап циклизации. Спустя 2 ч при 40°C добавляли AcCl (127 г, 115 мл, 1,6 моль, Eq: 4,97) по каплям при температуре от 35 до 40°C. Спустя 4 ч при 40°C добавляли AcCl (127 г, 115 мл, 1,6 моль, Eq: 4,97) при температуре от 35 до 40°C. Суспензию перемешивали в течение ночи при 40°C. Реакционную смесь концентрировали при Tj=60°C, при пониженном давлении до объема приблизительно 400 мл. Растворитель в суспензии заменяли при постоянном объеме дополнительным 2-пропанолом (936 г, 1,2 л) и перемешивали более 1 ч при КТ. Суспензию фильтровали, и осадок на фильтре промывали 2-пропанолом (195 г, 250 мл). Кристаллы высушивали при 60°C/10 мбар (1000 Па) с получением 85,8 г продукта в виде белых кристаллов (99,2 абс.% чистоты согласно ВЭЖХ).
8-хлор-5-метил-1-[4-(2-пиридилокси)циклогексил]-4,6-дигидро-[1,2,4]триазоло[4,3-a][1,4]бензодиазепин I, форма A
В реактор загружали 7-хлор-4-метил-3,5-дигидро-1,4-бензодиазепин-2-амина дигидрохлорид (92,3 г, 326 ммоль, Eq: 1,00) и 4-(2-пиридилокси)сциклогексанкарбогидразид (76,8 г, 326 ммоль, Eq: 1,00), а затем 2-пропанол (504 г, 646 мл). Суспензию нагревали с обратным холодильником в течение 18 ч при температуре от 80 до 83°C (до полного превращения амидина и промежуточного соединения). Реакционную смесь охлаждали до КТ при добавлении воды (775 г, 775 мл). Почти прозрачный раствор фильтровали. Фильтр промывали водой (24,9 г, 24,9 мл) с получением 1,5 л раствора неочищенного продукта (pH 4).
Фильтрат (1,5 л) делили на две части: 1 л в реактор B (217 ммоль теоретически) и 0,5 л в реактор A (109 ммоль теоретически).
Соединение I наилучшим образом выделяется в виде свободного основания. Однако его гидрохлорид также может быть выделен: после полного превращения амидина и промежуточного соединения реакционную смесь охлаждали до температуры от 0 до 5°C. Полученную суспензию перемешивали в течение 1 ч при температуре от 0 до 5°C и фильтровали. Осадок на фильтре промывали холодным изопропанолом и высушивали при пониженном давлении 50°C/10 мбар (1000 Па) с получением I.HCl.
Реактор A, кристаллизация при pH 9-10.- 1,7 экв. NaOH
8% водный NaOH (приблизительно 95 г, соответствует приблизительно 1,7 экв.) добавляли в течение 15 мин, поддерживая температуру от 20 до 25°C (спонтанная кристаллизация при добавлении 79 г, pH 10 в конце добавления). Добавляли затравочные кристаллы I, формы A (75 мг) (в случае отсутствия спонтанной кристаллизации). Светло-желтую суспензию перемешивали в течение 1,5 ч при КТ и охлаждали до температуры 0-5°C в течение 30 мин. После 5 ч перемешивания при температуре 0-5°C суспензию фильтровали. Осадок на фильтре (форма H) промывали холодной (0-5°C) смесью 2-пропанол/вода 1:2 (123 мл) и водой (42,0 г, 42 мл) и высушивали при 60°C при пониженном давлении с получением 38,4 г титульного соединения в виде белого кристаллического порошка (кристаллическая форма A по данным порошкового рентгеноструктурного анализа, 99,3 абс.% чистота согласно ВЭЖХ, 0,4 абс.% соединения формулы VI').
Реактор B, кристаллизация при pH, равном или больше 12:
рН доводили до значения, равного или больше 12, добавляя 222 г приблизительно 8% водного NaOH (приблизительно 2 экв.) в течение 30 мин, поддерживая температуру от 20 до 25°C (pH 10-11 после добавления 201 г, спонтанная кристаллизация после добавления 130 г). Добавляли затравочные кристаллы I, формы A (75 мг) (в случае отсутствия спонтанной кристаллизации). Желтую суспензию перемешивали в течение 2 ч при КТ, затем охлаждали до температуры 0-5°C в течение 30 мин. После 5 ч перемешивания при температуре 0-5°C суспензию фильтровали. Осадок на фильтре (форма H) промывали холодной (0-5°C) смесью 2-пропанол/вода 1:2 (246 мл) и водой (83,0 г, 83 мл) и высушивали при 60°C при пониженном давлении с получением 74,6 г титульного соединения в виде белого кристаллического порошка (99,7 абс.% чистоты согласно ВЭЖХ, соединение формулы VI' не было обнаружено, форма A по данным порошкового рентгеноструктурного анализа).
8-хлор-5-метил-1-[4-(2-пиридилокси)циклогексил]-4,6-дигидро-[1,2,4]триазоло[4,3-a][1,4]бензодиазепин I, форма F
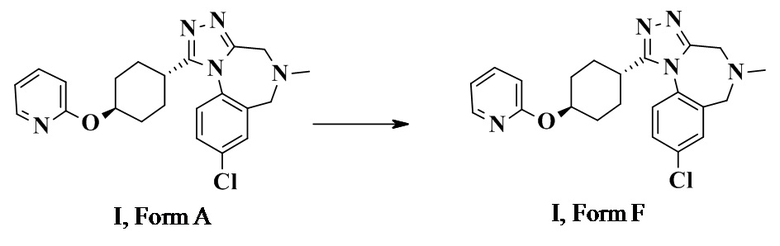
8-хлор-5-метил-1-[4-(2-пиридилокси)циклогексил]-4,6-дигидро-[1,2,4]триазоло[4,3-a][1,4]бензодиазепин (38,1 г, 92,8 ммоль, Eq: 1,00) суспендировали в метилацетате (698 г, 750 мл), суспензию нагревали до 55°C. Полученный мутный раствор фильтровали и охлаждали до температуры 43-45°C в течение 30 мин. Раствор затравливали при помощи 0,75 г соединения формулы I, формы F и охлаждали в течение 2 ч до КТ. Суспензию перемешивали в течение ночи и заменяли приблизительно 550 мл метилацетата (MeOAc) при постоянном объеме (Tj max 45°C / от 400 до 450 мбар (45000 Па)) н-гептаном (374 г, 550 мл), с достижением приблизительно 45-55% мас./мас. содержания MeOAc.
Суспензию охлаждали до 0°C и перемешивали при 0°C в течение более 4 ч. Суспензию фильтровали. Осадок на фильтре промывали н-гептаном (102 г, 150 мл) и высушивали при 60°C при пониженном давлении с получением 36 г титульного соединения в виде кристаллической формы F (по данным порошкового рентгеноструктурного анализа).
8-хлор-5-метил-1-[4-(2-пиридилокси)циклогексил]-4,6-дигидро-[1,2,4]триазоло[4,3-a][1,4]бензодиазепин I, форма F
8-хлор-5-метил-1-[4-(2-пиридилокси)циклогексил]-4,6-дигидро-[1,2,4]триазоло[4,3-a][1,4]бензодиазепин (13,6 г) растворяли в 2-пропаноле (213 г, 272 мл) при 55°C. Горячий раствор фильтровали. Раствор концентрировали до приблизительно 130-140 мл. Добавляли н-гептан (93,0 г, 136 мл) при 55°C в течение 15 мин. Прозрачный раствор охлаждали до приблизительно 45°C и затравливали 300 мг кристаллического соединения I, формы F. Смесь охлаждали в течение 20 ч до 0°C. Полученную суспензию фильтровали. Осадок на фильтре промывали холодной (0°C) смесью 2-пропанол/н-гептан 1:1 (54,4 мл) и высушивали с получением 11,7 г титульного соединения в виде кристаллической формы F (по данным порошкового рентгеноструктурного анализа).
Транс-4-(2-пиридилокси)циклогексанкарбоновая кислота XXIII
В реактор загружали трет-амилоксид натрия (tAmONa) (444 г, 3,83 моль, Eq: 2,26), N-метил-2-пирролидон (НМП) (2,06 кг, 2 л) и нагревали при Tj=90°C. Добавляли раствор транс-4-гидроксициклогексанкарбоновой кислоты (244 г, 1,69 моль, Eq: 1,00) в НМП (515 г, 500 мл) в течение 15 мин при температуре от 80 до 85°C. Добавляли 2-хлорпиридин (239 г, 2,11 моль, Eq: 1,24) в течение 5 мин при температуре от 80 до 85°C. Спустя более 60 ч реакционную смесь охлаждали до 50°C и добавляли воду (8,00 кг, 8 л) при 50°C. Реакционную смесь охлаждали до КТ. pH доводили до приблизительно 5, добавляя 25% водную HCl (280 г, 250 мл). Суспензию охлаждали до температуры 0-5°C, перемешивали в течение более 2 ч и фильтровали. Осадок на фильтре промывали водой (8,00 кг, 8 л) и высушивали при 50°C при пониженном давлении с получением 245 г титульного соединения (более 99 абс.% чистоты по данным газовой хроматографии (GC)).
Транс-4-(2-пиридилокси)циклогексанкарбогидразид VI
1,1'-карбонилдиимидазол (КДИ) (215 г, 1,32 моль, Eq: 1,21) суспендировали в ТГФ (1,07 кг, 1,2 л) при 20°C. Добавляли раствор транс-4-(2-пиридилокси)циклогексанкарбоновой кислоты (243 г, 1,1 моль, Eq: 1,00) в ТГФ (1,07 кг, 1,2 л, Eq: -) в течение 70 мин. Спустя 16 ч реакционную смесь дегазировали (циклы вакуум/N2). Приблизительно 100 мл растворителя отгоняли при пониженном давлении при Tr менее 30°C. Полученный раствор активированной кислоты добавляли при температуре от 15 до 25°C к раствору гидразинмоногидрата (75,2 г, 73 мл, 1,5 моль, Eq: 1,4) в смеси ТГФ (1,07 кг, 1,3 л) /вода (1,2 кг, 1,3 л). После более 2 ч перемешивания при температуре 20-25°C отгоняли 3,2 л растворителя при Tj 50-55°C / 300-200 мбар (30000-20000 Па), непрерывно добавляя 3,5 л воды. Полученную суспензию перемешивали в течение ночи при КТ и фильтровали. Осадок на фильтре промывали водой (750 г, 0,75 л) и высушивали при 50°C при пониженном давлении с получением 223 г титульного соединения (98,9 абс.% по данным ВЭЖХ, 0,4% соединения формулы VI').
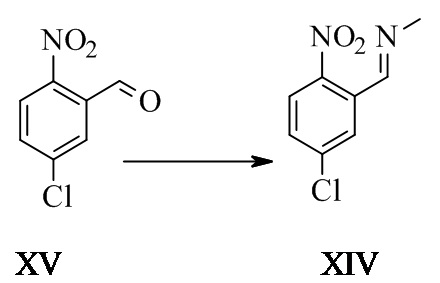
1-(5-хлор-2-нитрофенил)-N-метил-метанимин XIV
5-хлор-2-нитробензальдегид (45 г, 243 ммоль, Eq: 1,00) обрабатывали 2 M раствором метиламина в MeOH (141 г, 180 мл, 360 ммоль, Eq: 1,48). Реакционную смесь перемешивали при КТ в течение 5 ч и концентрировали при пониженном давлении с получением 48,06 г титульного соединения. Неочищенный продукт вводили непосредственно в следующий этап без дополнительной очистки.
1-(5-хлор-2-нитрофенил)-N-метил-метанамин XIII
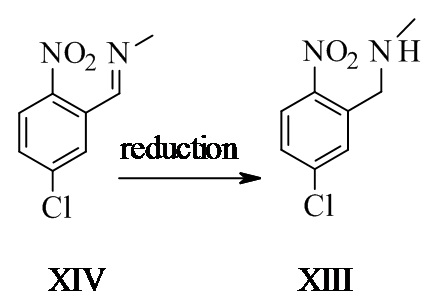
(E)-N-(5-хлор-2-нитробензилиден)метанимин (47,5 г, 239 ммоль, Eq: 1,00) растворяли в метаноле (447 г, 565 мл). Раствор охлаждали до 0°C и добавляли боргидрид натрия (7,64 г, 194 ммоль, Eq: 0,811) по частям в течение 25 мин. Реакционную смесь перемешивали в течение ночи при КТ (приблизительно 98% превращение). Добавляли дополнительный боргидрид натрия (1,77 г, 44,9 ммоль, Eq: 0,19) и реакционную смесь перемешивали в течение 3 ч. Растворитель заменяли на дихлорметан (ДХМ) (конечный объем приблизительно 400 мл) и промывали насыщенным водным NaHCO3 (200 мл). Водную фазу отделяли и экстрагировали дважды ДХМ (318 г, 240 мл). Органические фазы дважды последовательно промывали полунасыщенным водным NaHCO3 (200 мл). Органические фазы объединяли, высушивали над сульфатом магния (MgSO4) и концентрировали при пониженном давлении с получением 47,1 г титульного соединения.
4-хлор-2-(метиламинометил)анилин XII
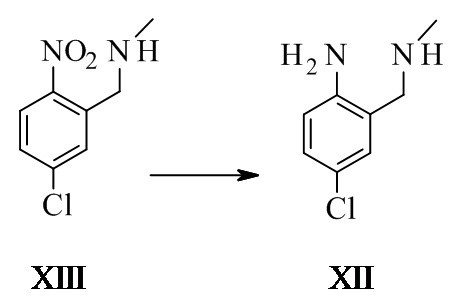
1-(5-хлор-2-нитрофенил)-N-метил-метанамин (23 г, 109 ммоль, Eq.: 1) растворяли в метаноле (690 мл), добавляли 46% никелевый катализатор Ренея (6,91 г, 55 ммоль, 0,5 экв.) и смесь перемешивали в атмосфере водорода (1 бар (100000 Па)) при КТ. После завершения реакции суспензию фильтровали и фильтрат концентрировали при пониженном давлении с получением 19 г неочищенного титульного соединения.
1-(5-хлор-2-нитрофенил)-N-метил-метанамина гидрохлорид XIII.HCl
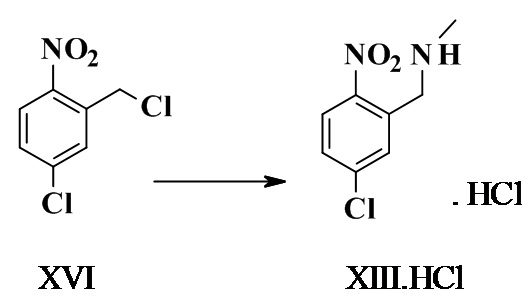
40% метиламин в метаноле (90,0 мл, 882 ммоль, Eq: 12,1) загружали в реактор и добавляли по каплям раствор 4-хлор-2-(хлорметил)-1-нитробензола (15 г, 72,8 ммоль, Eq: 1,00) в MeOH (94,8 г, 120 мл) в течение 50 мин при КТ. Светло-желтый раствор перемешивали при КТ в течение 5,5 ч (до завершения реакции). Реакционную смесь концентрировали при пониженном давлении с получением 21,5 г желтого твердого вещества, которое поглощалось в AcOEt (108 г, 120 мл). Полученную суспензию фильтровали. Осадок на фильтре (метиламина гидрохлорид) промывали три раза AcOEt (135 г, 150 мл). Фильтрат выпаривали до получения 14,6 г желтого масла. Неочищенный 1-(5-хлор-2-нитро-фенил)-N-метил-метанамин растворяли в AcOEt (108 г, 120 мл). Медленно добавляли 4,4 M соляной кислоты (HCl) в AcOEt (33,6 мл, 147 ммоль, Eq: 2,02). Полученную бледно-желтую суспензию перемешивали в течение ночи при КТ. Суспензию фильтровали. Осадок на фильтре промывали дважды AcOEt и высушивали при 10 мбар (1000 Па) 50°C с получением 15,6 г титульного соединения в виде светло-желтого порошка.
4-хлор-2-(метиламинометил)анилина гидрохлорид XII.HCl
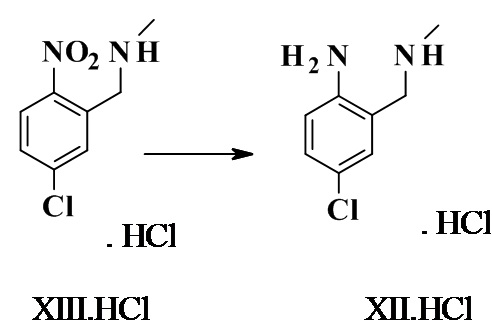
1-(5-хлор-2-нитрофенил)-N-метил-метанамина гидрохлорид (50 г, 208 ммоль, Eq.: 1) растворяли в метаноле (790 мл), добавляли 46% никелевый катализатор Ренея (13 г,104 ммоль, 0,5 экв.) и смесь перемешивали в атмосфере водорода (1 бар (100000 Па)) при КТ. После завершения реакции суспензию фильтровали и фильтрат концентрировали при пониженном давлении с получением 43 г неочищенного титульного соединения.
Неочищенный продукт может быть перекристаллизован:
Неочищенный продукт (22,5 г) растворяли в метаноле (400 мл). Добавляли воду (3,7 мл) и активированный уголь (2,5 г). Суспензию нагревали до 50°C, затем охлаждали до КТ и фильтровали. Фильтрат концентрировали при пониженном давлении до приблизительно половины объема. Добавляли изопропанол (200 мл) и раствор концентрировали при пониженном давлении до приблизительно 220 г, в течение чего начиналась кристаллизация, приводящая к образованию густой суспензии. Добавляли изопропанол (50 мл). Суспензию перемешивали в течение 2 ч при КТ и фильтровали. Осадок на фильтре промывали изопропанолом (30 мл) и высушивали при 50°C/10 мбар (1000 Па) с получением 15 г титульного соединения в виде грязно-белого порошка.
2-[(2-амино-5-хлор-фенил)метил-метил-амино]ацетонитрил XI
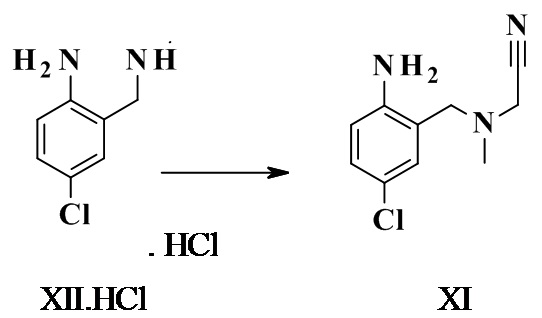
4-хлор-2-((метиламино)метил)анилина гидрохлорид (10 г, 48,3 ммоль, Eq: 1,00) суспендировали в ацетонитриле (78,0 г, 100 мл). Добавляли гидрокарбонат натрия (8,92 г, 106 ммоль, Eq: 2,2) и суспензию нагревали до 85°C. Добавляли 2-хлорацетонитрил (3,91 г, 3,28 мл, 50,7 ммоль, Eq: 1,05) и реакционную смесь перемешивали в течение 24 ч. Реакционную смесь охлаждали до КТ и добавляли воду (150 г, 150 мл). Добавляли толуол (173 г, 200 мл) и большую часть ацетонитрила удаляли при помощи ротационного испарителя. Водную фазу отделяли и экстрагировали толуолом (86,7 г, 100 мл). Органические фазы промывали полунасыщенным водным NaHCO3 (100 мл) и полунасыщенным водным NaCl. Органические фазы объединяли, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 9,95 г титульного соединения в виде светло-желтого твердого вещества. Альтернативно, алкилирование также может быть выполнено с использованием свободного основания XII в качестве исходного материала.
Транс-N'-(2-хлорацетил)-4-(2-пиридилокси)циклогексанкарбо-гидразид XXI
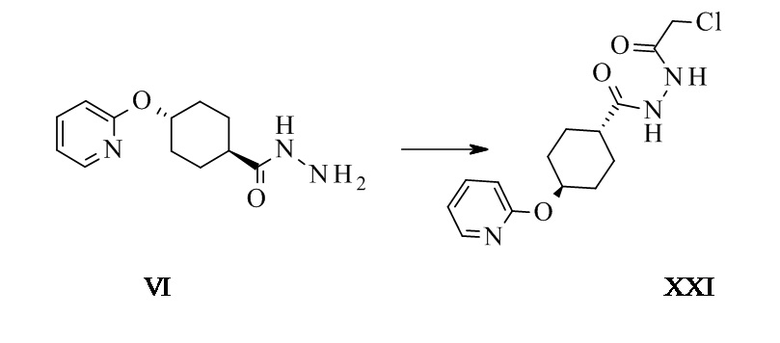
Транс-4-(2-пиридилокси)циклогексанкарбогидразид (4 г, 17,0 ммоль, Eq: 1,00) суспендировали в ДХМ (66,2 г, 50,0 мл). Добавляли 2,4,6-триметилпиридин (сим-коллидин) (2,29 г, 2,5 мл, 18,7 ммоль, Eq: 1,1). Суспензию охлаждали до 0°C и добавляли по каплям 2-хлорацетилхлорид (2,04 г, 1,43 мл, 17,9 ммоль, Eq: 1,05) в течение 30 мин при температуре от 0 до 5°C. Спустя 1 ч при температуре 0-5°C суспензию фильтровали. Осадок на фильтре промывали холодным дихлорметаном (40 мл) и высушивали при пониженном давлении при 40°C с получением 5,1 г титульного соединения.
Транс-2-(хлорметил)-5-[4-(2-пиридилокси)циклогексил]-1,3,4-оксадиазол XX
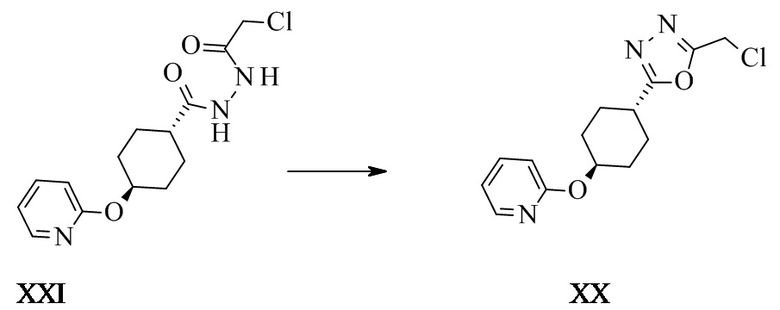
N'-(2-хлорацетил)-4-(2-пиридилокси)циклогексанкарбогидразид (44 г, 141 ммоль, Eq: 1,00) суспендировали в ацетонитриле (257 г, 330 мл, Eq: -). Суспензию охлаждали до 0°C и добавляли трифторметансульфоновый альдегид (48,8 г, 28,7 мл, 169 ммоль, Eq: 1,2) в течение 30 мин. Реакционную смесь перемешивали при КТ до тех пор, пока степень превращения не превысит 95% (более 15 ч). Полученный раствор охлаждали до 0°C и добавляли раствор гидрокарбоната натрия (27,0 г, 322 ммоль, Eq: 2,28) в воде (440 г, 440 мл), а затем дихлорметан (437 г, 330 мл). Водную фазу экстрагировали дважды дихлорметаном (662 г, 500 мл). Органические фазы промывали последовательно полунасыщенным водным NaCl (500 мл). Органические фазы объединяли, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 44,0 г неочищенного титульного соединения.
Кристаллизация: неочищенный продукт (39,0 г) кристаллизовали из изопропанола с получением 19,08 г титульного соединения.
Транс-4-хлор-2-[[метил-[[5-[4-(2-пиридилокси)циклогексил]-1,3,4-оксадиазол-2-ил]метил]амино]метил]анилин XIX
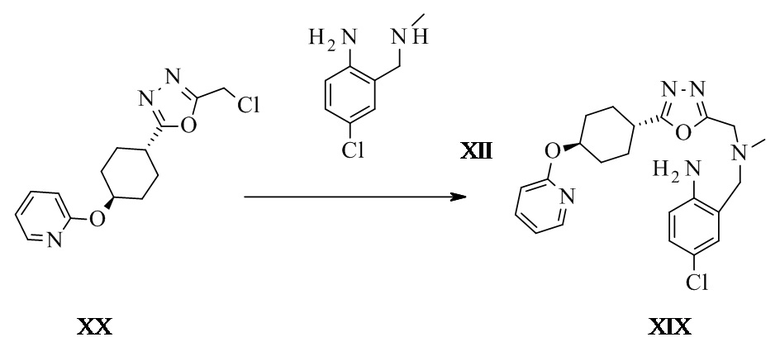
Транс-2-(хлорметил)-5-[4-(2-пиридилокси)циклогексил]-1,3,4-оксадиазол (6,7 г, 21,9 ммоль, Eq: 1,00), 4-хлор-2-((метиламино)метил)анилин (4,33 г, 24,1 ммоль, Eq: 1,1), гидрокарбонат натрия (2,21 г, 26,3 ммоль, Eq: 1,2) и ацетонитрил (54,8 г, 70,3 мл) загружали в реактор и нагревали с обратным холодильником в течение 4 ч. Добавляли дополнительный 4-хлор-2-((метиламино)метил)анилин (393 мг, 2,19 ммоль, Eq: 0,1) и реакционную смесь перемешивали в течение 20 ч с обратным холодильником. Реакционную смесь охлаждали до КТ. Добавляли воду (20,0 г, 2,0 мл) и дихлорметан (79,5 г, 60,0 мл). Водную фазу отделяли и экстрагировали дихлорметаном (26,5 г, 20,0 мл). Органические фазы промывали последовательно полунасыщенным водным хлоридом аммония (NH4Cl) (25,0 мл), 10% водным NaCl (25,0 мл) и насыщенным водным NaCl (25,0 мл). Органические фазы объединяли, высушивали над MgSO4 и фильтровали. Фильтрат фильтровали через 25 г силикагеля (SiO2) и концентрировали при пониженном давлении с получением 5,3 г титульного соединения.
Транс-8-хлор-5-метил-1-[4-(2-пиридилокси)циклогексил]-4,6-дигидро-[1,2,4]триазоло[4,3-a][1,4]бензодиазепин I
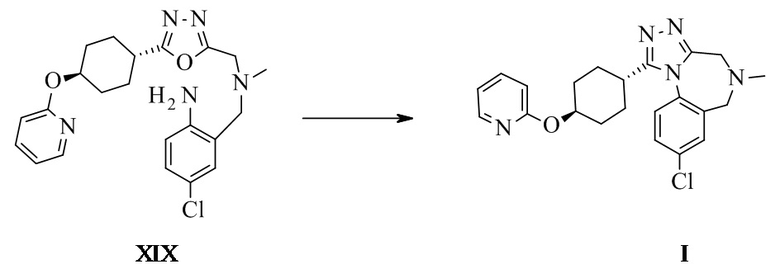
Транс-4-хлор-2-[[метил-[[5-[4-(2-пиридилокси)циклогексил]-1,3,4-оксадиазол-2-ил]метил]амино]метил]анилин (5 г, 10,1 ммоль, Eq: 1,00) растворяли в тетрагидрофуране (44,4 г, 50 мл). Добавляли трифторуксусную кислоту (2,02 г, 1,36 мл, 17,4 ммоль, Eq: 1,72) и реакционную смесь нагревали до 60°C в течение 2,5 ч. Реакционную смесь охлаждали до КТ, добавляли насыщенный водный NaHCO3 (25 мл) (pH = 8) и смесь перемешивали в течение 15 мин (образование желтой суспензии). Добавляли воду (25,0 г, 25 мл) и AcOEt (36,1 г, 40 мл). После 30 мин перемешивания водную фазу отделяли и экстрагировали AcOEt (18,0 г, 20 мл). Органические фазы промывали дважды насыщенным водным NaCl (17 мл) (pH примерно 7). Органические фазы объединяли, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 5,03 г неочищенного титульного соединения. Неочищенный продукт поглощали в изопропаноле (20 мл) и выпаривали, вновь растворяли в изопропаноле (20 мл) и выпаривали. Осадок растворяли в изопропаноле (11,8 г, 15 мл) и затравливали формой F соединения I. Начиналась кристаллизация, и суспензию перемешивали в течение 18 ч при КТ. Суспензию фильтровали. Осадок на фильтре промывали дважды изопропанолом (7,84 г, 10 мл) и высушивали при пониженном давлении с получением 3,11 г титульного соединения (форма F по данным порошкового рентгеноструктурного анализа).
4-хлор-2-(метиламинометил)анилина дигидрохлорид XII.2HCl
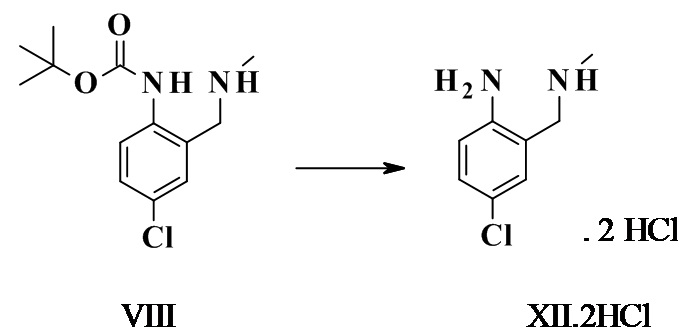
Трет-бутил-4-хлор-2-((метиламино)метил)фенилкарбамат (1,0 г, 3,69 ммоль, Eq: 1,00) растворяли в AcOEt (4,5 г, 5,00 мл). Добавляли 4 M HCl в AcOEt (4,62 мл, 18,5 ммоль, Eq: 5,00). Полученную суспензию нагревали в течение ночи при 40°C. Суспензию охлаждали до КТ, перемешивали в течение 1 ч и фильтровали. Осадок на фильтре промывали AcOEt (20 мл) и высушивали при пониженном давлении при 50°C с получение 0,9 г титульного соединения.
2-[(2-амино-5-хлорфенил)метил-метил-амино]ацетонитрил
4-хлор-2-((метиламино)метил)анилина дигидрохлорид с предыдущего этапа (0,8 г, 3,28 ммоль, Eq: 1,00) суспендировали в ацетонитриле (6,24 г, 8,00 мл). Добавляли гидрокарбонат натрия (883 мг, 10,5 ммоль, Eq: 3,2). Белую суспензию нагревали до 85°C. Добавляли 2-хлорацетонитрил (266 мг, 223 мкл, 3,45 ммоль, Eq: 1,05) и перемешивали в течение ночи при 85°C. Реакционную смесь охлаждали до КТ, добавляли воду (12,0 г, 12,0 мл) и смесь перемешивали в течение 10 мин. Добавляли толуол (13,9 г, 16,0 мл) и большую часть ацетонитрила удаляли при пониженном давлении. Водную фазу отделяли и экстрагировали толуолом (6,94 г, 8,00 мл). Органические фазы промывали полунасыщенным водным NaHCO3 (8,00 мл) и полунасыщенным NaCl (8,00 мл), высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 840 мг титульного соединения. Неочищенный продукт растворяли в МТБЭ (5 мл) с обратным холодильником. Бесцветный раствор медленно охлаждали до КТ. Полученную белую суспензию фильтровали. Осадок на фильтре промывали н-гептаном (20 мл) и высушивали при пониженном давлении с получением 410 мг титульного соединения в виде белого порошка.
4-хлор-2-(метиламинометил)анилин XII
2-амино-5-хлорбензальдегид (500 мг, 3,12 ммоль, Eq: 1,00) растворяли при КТ в этаноле (5,93 г, 7,50 мл). Добавляли 41% водный раствор метиламина (472 мг, 527 мкл, 6,23 ммоль, Eq: 2,00) и желтый раствор перемешивали в течение 1 ч при КТ. Добавляли NaBH4 (118 мг, 3,12 ммоль, Eq: 1,00) и суспензию перемешивали в течение 18 ч при КТ. Добавляли этилацетат (18,0 г, 20 мл) и полунасыщенный водный NaCl (20 мл). Органическую фазу отделяли, высушивали над MgSO4, фильтровали и выпаривали до сухости с получением 550 мг титульного соединения.
7-хлор-4-метил-3,5-дигидро-1,4-бензодиазепин-2-амин II
7-хлор-4-метил-4,5-дигидро-1H-бензо[e][1,4]диазепин-2(3H)-имина дигидрохлорид (1,75 г, 6,19 ммоль, Eq: 1,00) суспендировали в AcOEt (50 мл). Добавляли насыщенный водный гидрокарбонат натрия (30 мл) и смесь перемешивали в течение 30 мин при КТ. Водную фазу отделяли и экстрагировали дважды AcOEt (20 мл). Органические фазы объединяли, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 930 мг титульного соединения.
7-хлор-4-метил-3,5-дигидро-1,4-бензодиазепин-2-амина дигидрохлорид III
2-((2-амино-5-хлорбензил)(метил)амино)ацетонитрил (11,1 г, 51,4 ммоль, Eq: 1,00) растворяли трифторэтанолом (138 г, 100 мл). Добавляли 4M HCl в диоксане (38,5 мл, 154 ммоль, Eq: 3,0). Реакционную смесь перемешивали 6 ч при 40°C до завершения реакции, затем концентрировали при пониженном давлении с получением 17,95 г титульного соединения (содержит приблизительно 9% диоксана и 11% остаточного трифторэтанола).
Альтернативно, соединение формулы XI может быть превращено в соединение формулы III в условиях, похожих на условия, что использовались для прямого преобразования соединения формулы VII в соединение формулы III, как описано в предыдущем примере.
Уксуснокислая соль трет-бутил-N-[4-хлор-2-(метиламинометил)фенил]карбамата VIII.AcOH
Трет-бутил-4-хлор-2-((метиламино)метил)фенилкарбамат (1,0 г, 3,1 ммоль, Eq: 1,00) растворяли в МТБЭ (8,21 г, 12 мл) при КТ. Уксусную кислоту (206 мг, 196 мкл, 3,41 ммоль, Eq: 1,1) добавляли по каплям, в течение чего продукт начинал кристаллизоваться. Спустя 2 ч при КТ суспензию фильтровали. Осадок на фильтре промывали МТБЭ и высушивали при пониженном давлении (10 мбар (1000 Па)/50°C) с получением 0,58 г титульного соединения.
Ссылки
1 Genes, Brain and Behavior (2011) 10: 228-235
2 Curr. Opin. Neurobiol. 19, 231-234 (2009)
3 WO 2010060836
4 WO 2004074291 и WO 2005068466
5 Aubé et al, J. Org. Chem., Vol. 65, No. 3, 2000
6 Caron, L. Wei, J. Douville, A. Ghosh, J. Org. Chem. 2010, 75, 945-947
7 WO2005/68466
8 Venkov et al., Synthesis, 1990, 253
9 WO 2004074291, WO 2005068466 и WO 2006021882.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ И ПРОЦЕСС ПРИГОТОВЛЕНИЯ И ПРОИЗВОДСТВА ДЕЙТЕРИРОВАННОЙ ОМЕГА-ДИФЕНИЛМОЧЕВИНЫ | 2011 |
|
RU2527037C2 |
| СПОСОБ ПОЛУЧЕНИЯ ХИРАЛЬНЫХ 2-АРИЛМОРФОЛИНОВ | 2014 |
|
RU2677328C1 |
| БЕНЗОИЛАМИНОГЕТЕРОЦИКЛИЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АКТИВАТОРОВ ГЛЮКОКИНАЗЫ (GLK) | 2007 |
|
RU2440992C2 |
| ТВЕРДЫЕ ФОРМЫ (1,1-ДИОКСО-4-ТИОМОРФОЛИНИЛ)-[6-[[3-(4-ФТОРФЕНИЛ)-5-МЕТИЛ-4-ИЗОКСАЗОЛИЛ]МЕТОКСИ]-3-ПИРИДИНИЛ]-МЕТАНОНА | 2012 |
|
RU2618524C2 |
| НОВЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ МОНОАЦИЛГЛИЦЕРИНЛИПАЗЫ | 2019 |
|
RU2801190C2 |
| НОВЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ МОНОАЦИЛГЛИЦЕРИНЛИПАЗЫ | 2019 |
|
RU2769507C1 |
| ТАРТРАТ 3-((1R,3R)-1-(2,6-ДИФТОР-4-((1-(3-ФТОРПРОПИЛ)АЗЕТИДИН-3-ИЛ)АМИНО)ФЕНИЛ)-3-МЕТИЛ-1,3,4,9-ТЕТРАГИДРО-2H-ПИРИДО[3,4-b]ИНДОЛ-2-ИЛ)-2,2-ДИФТОРПРОПАН-1-ОЛА, ЕГО ТВЕРДЫЕ ФОРМЫ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2809220C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2-ТРИФТОРМЕТИЛИЗОНИКОТИНОВОЙ КИСЛОТЫ И ЕЕ ЭФИРОВ | 2013 |
|
RU2654486C2 |
| МОДУЛЯТОРЫ ФОСФОДИЭСТЕРАЗЫ 10 | 2012 |
|
RU2567396C1 |
| ТВЕРДЫЕ ФОРМЫ ВНУТРЕННЕЙ СОЛИ ПИРИДОПИРИМИДИНИЯ | 2013 |
|
RU2638137C2 |
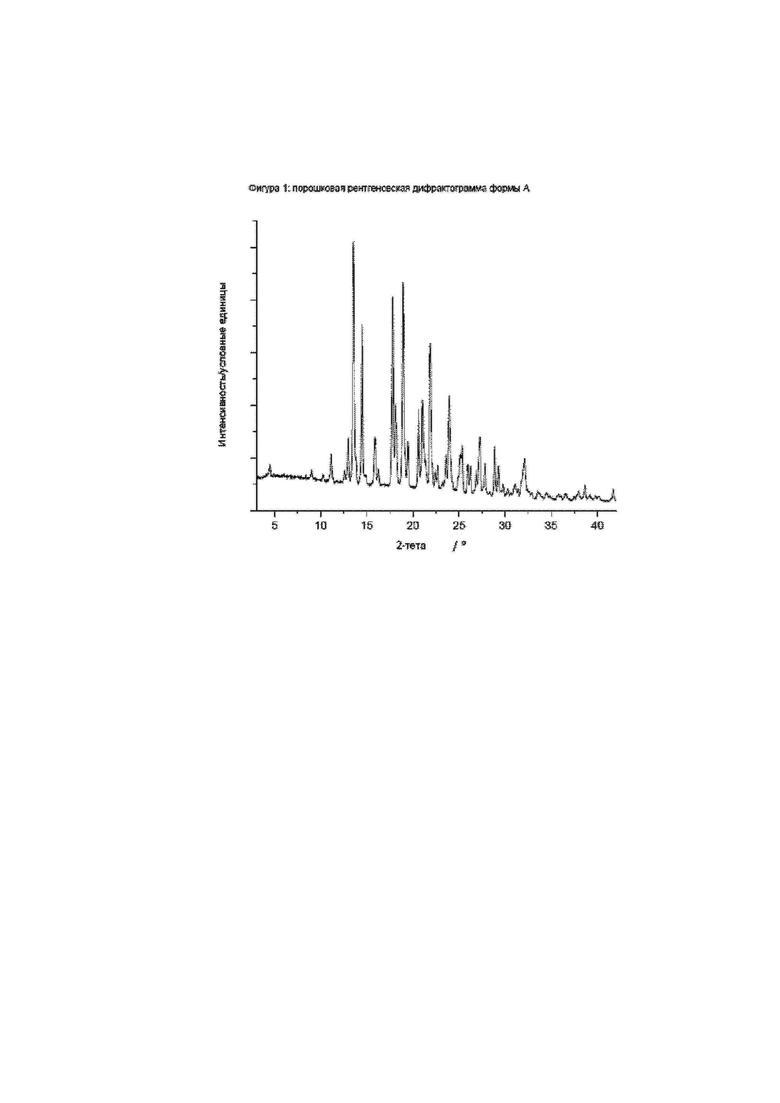
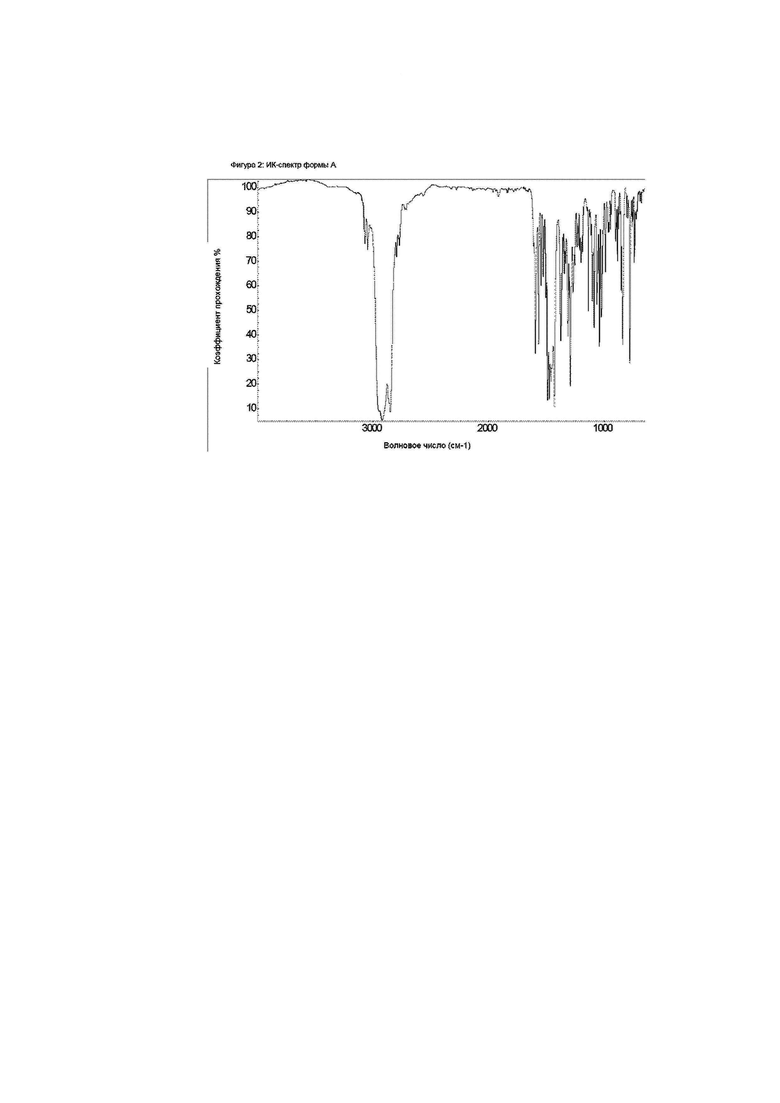
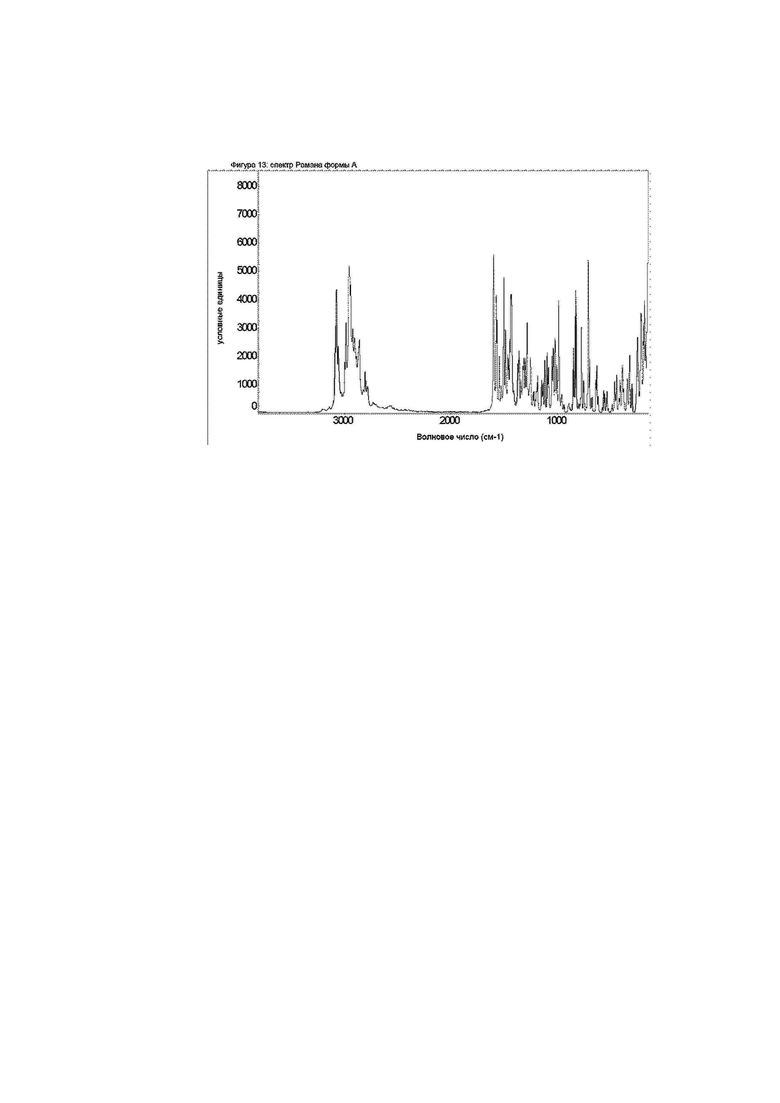
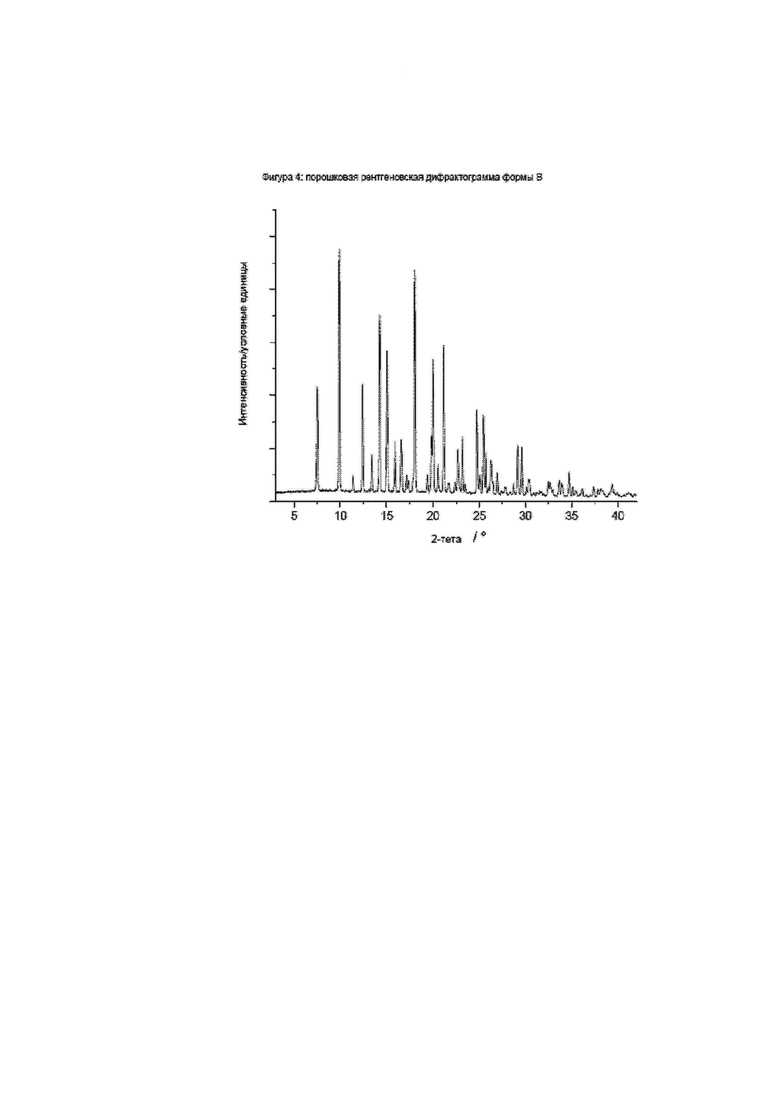
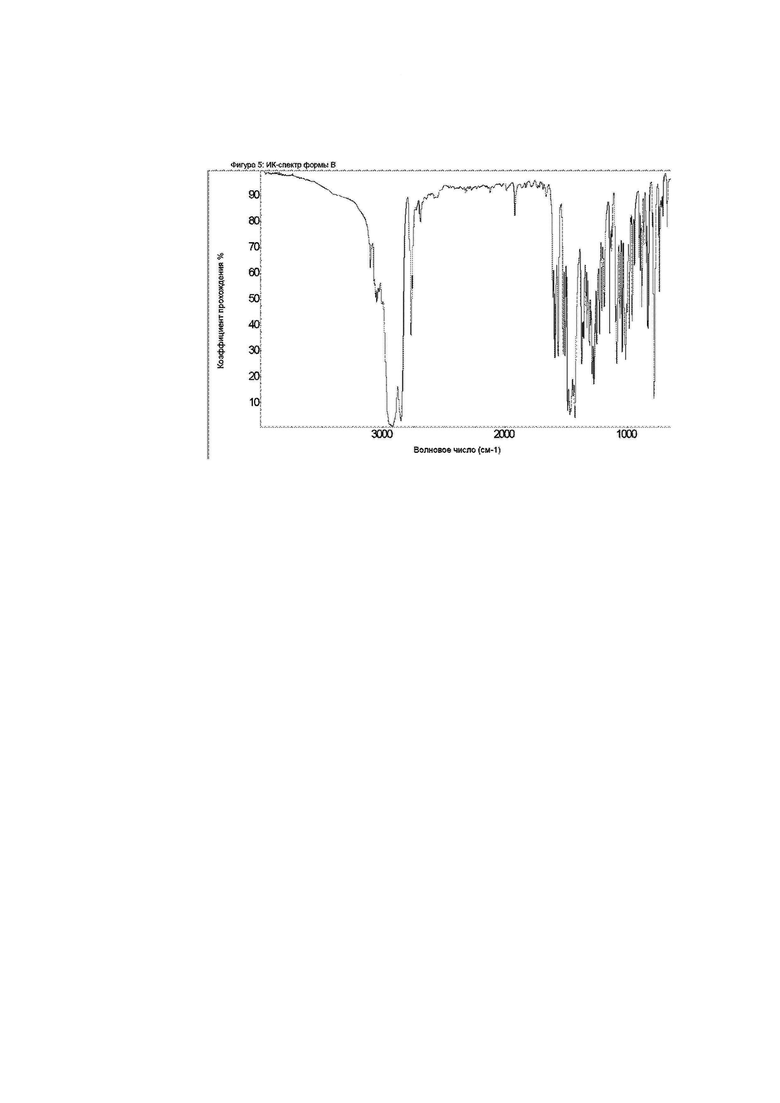
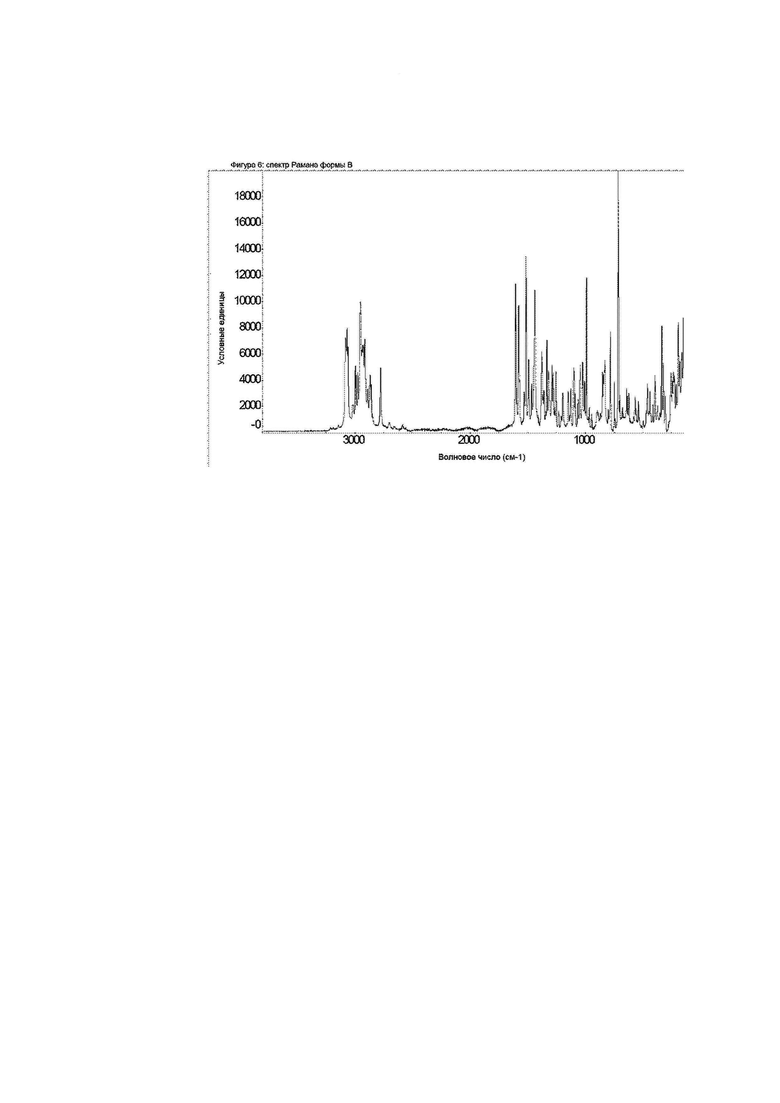
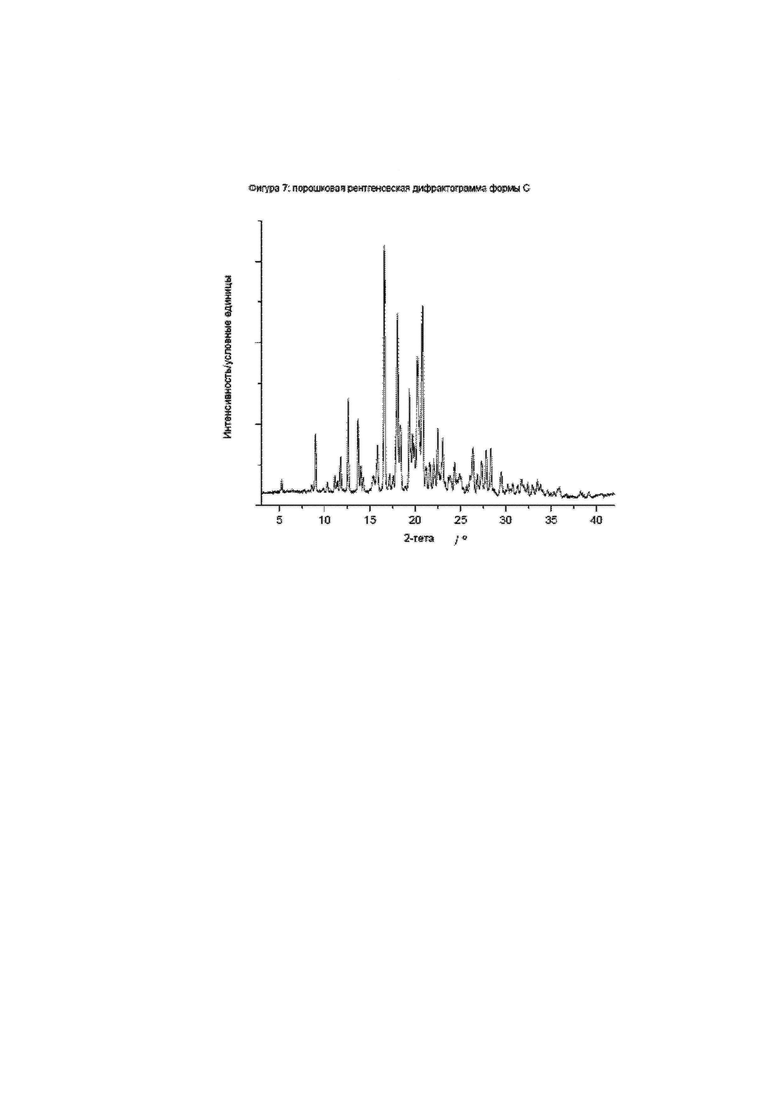
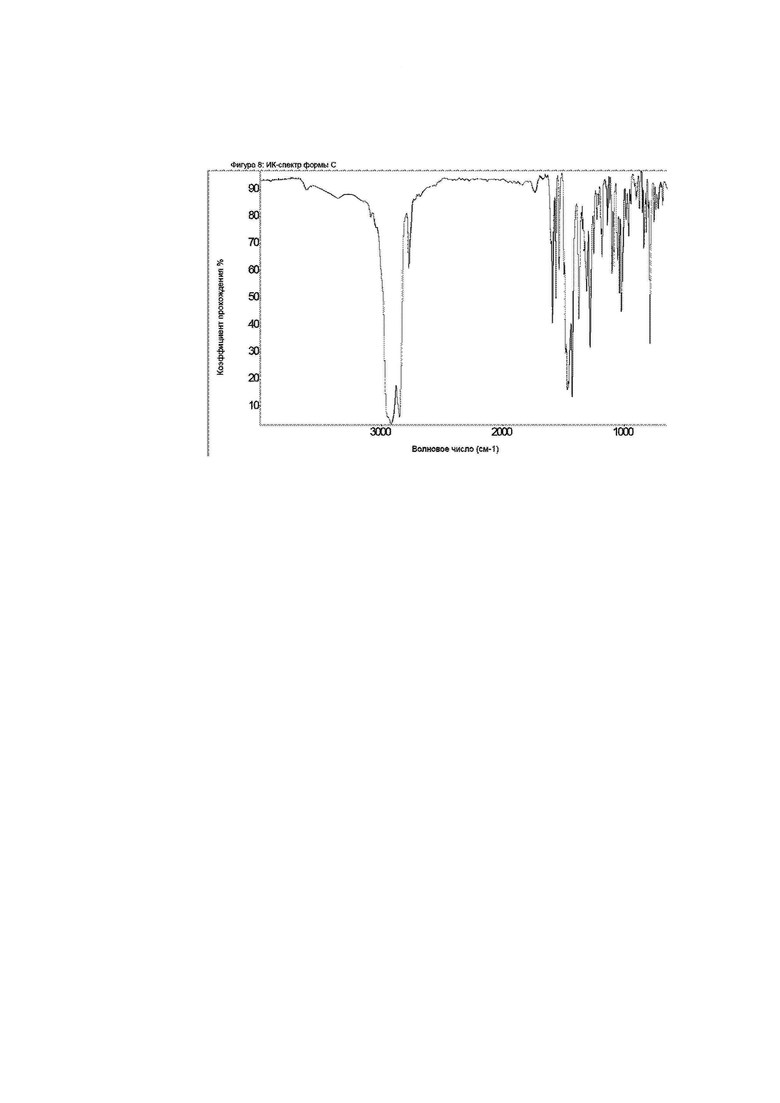
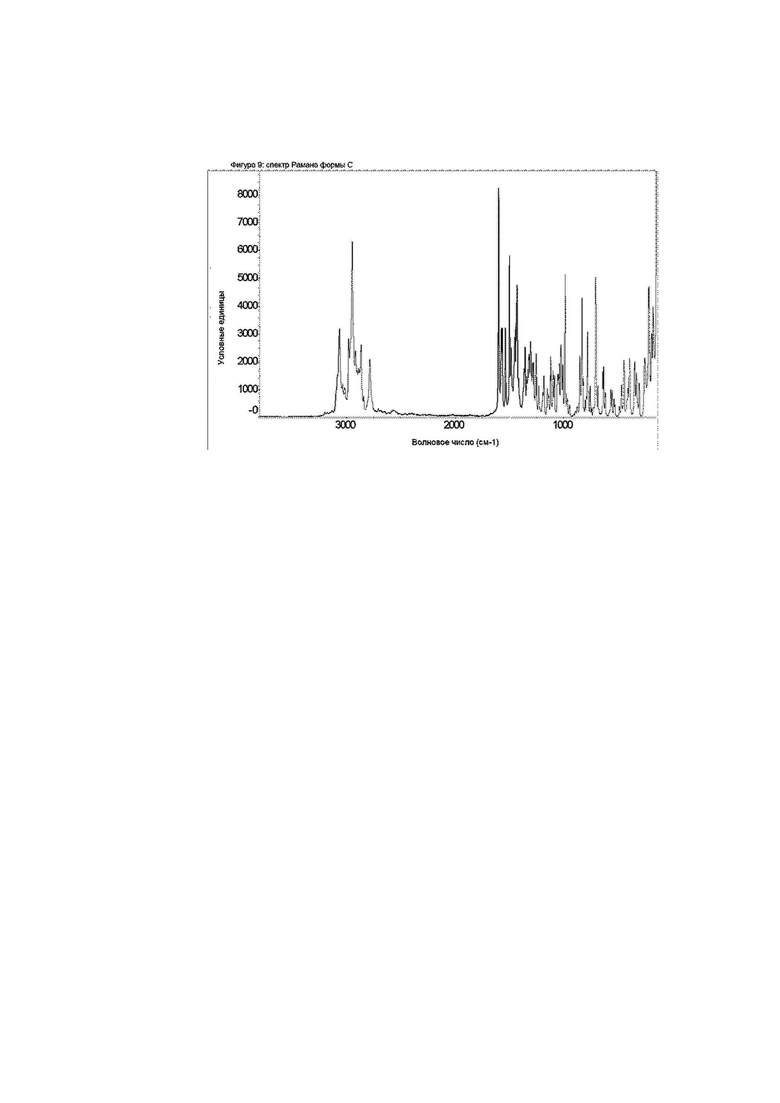
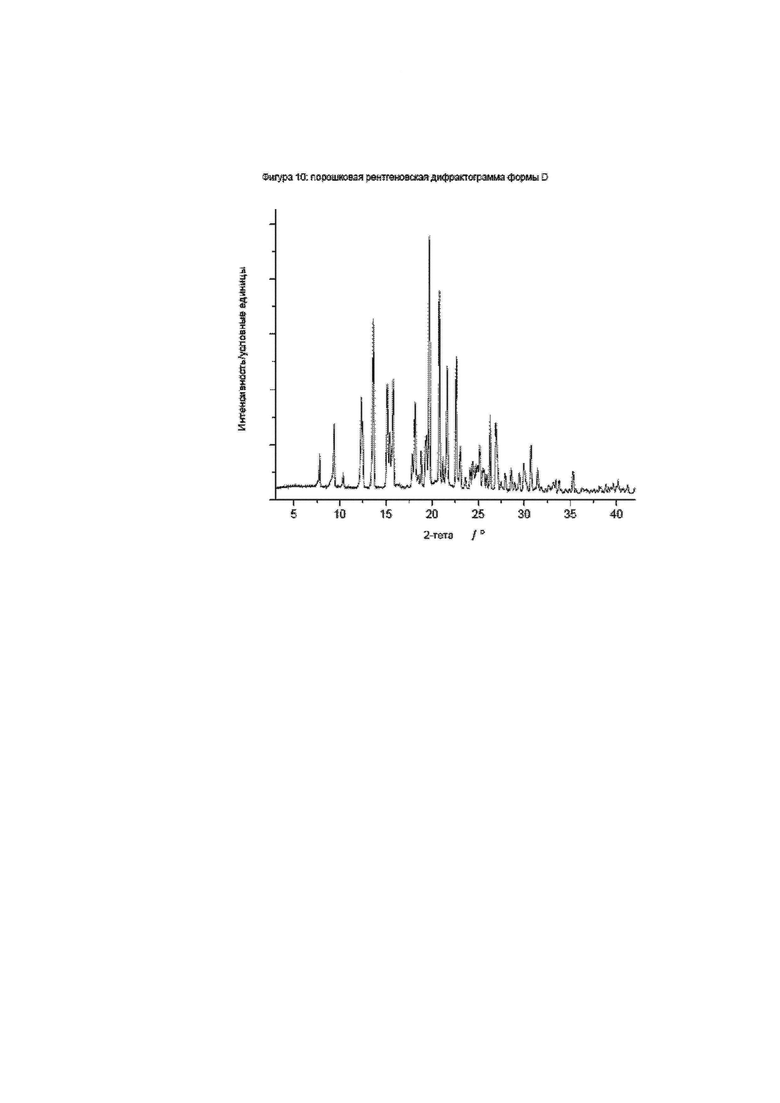
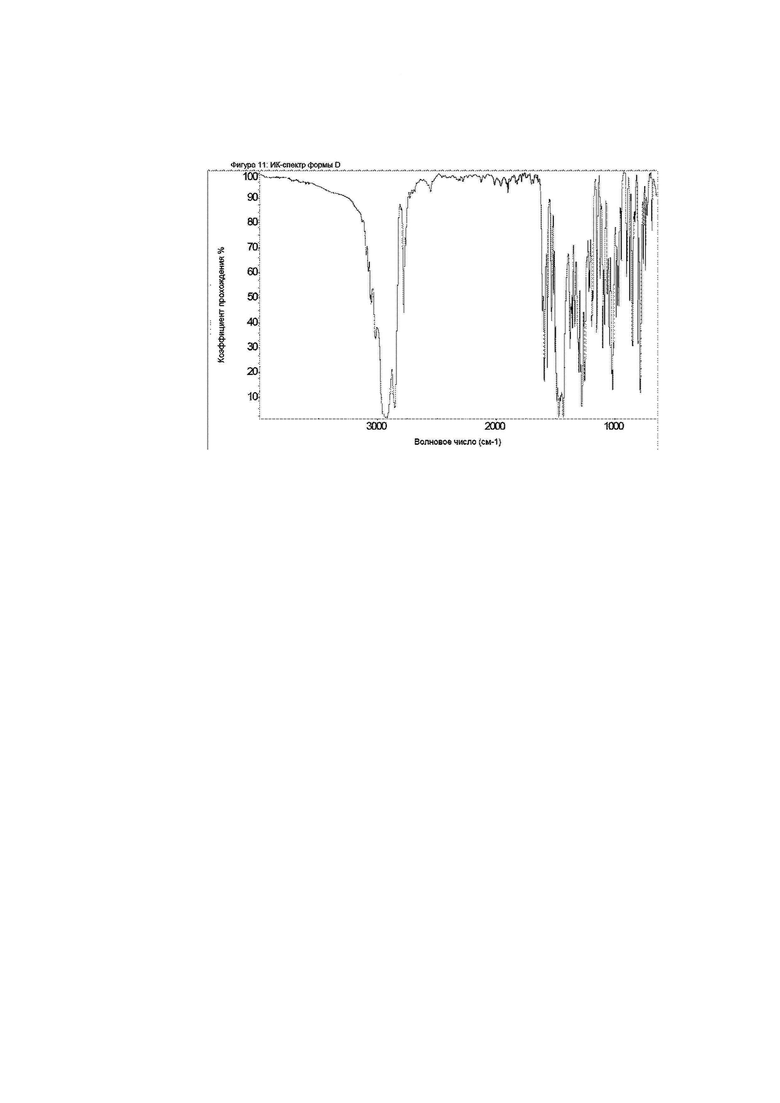
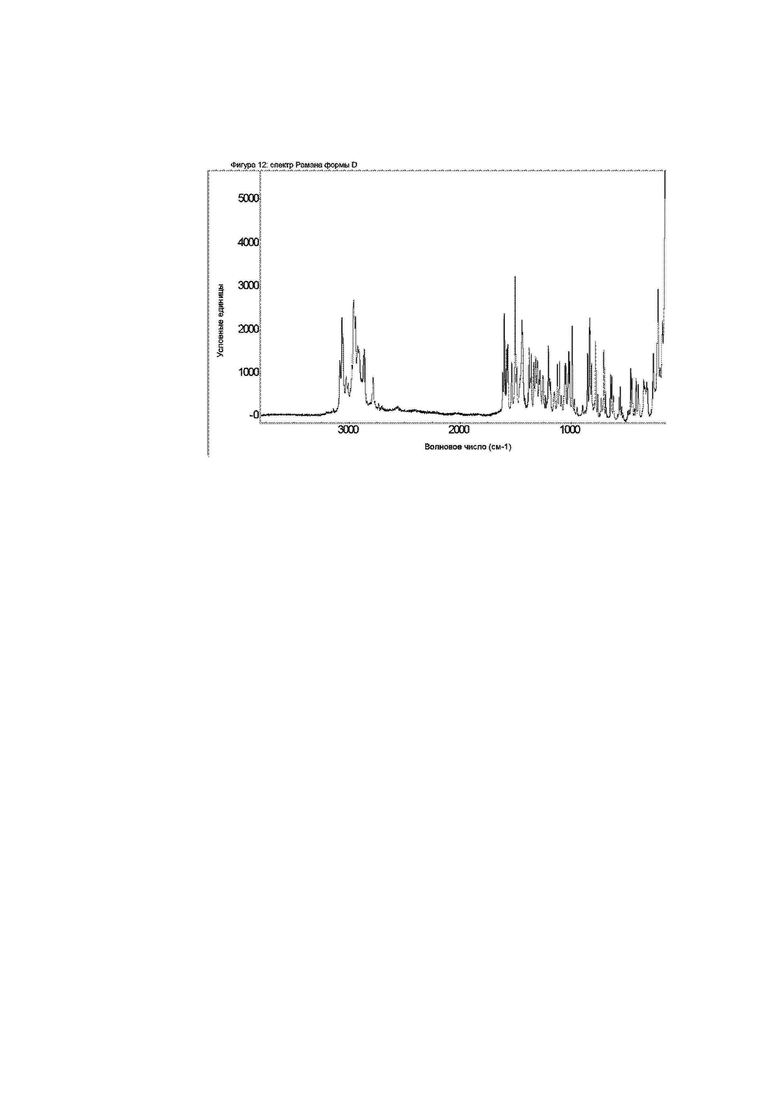
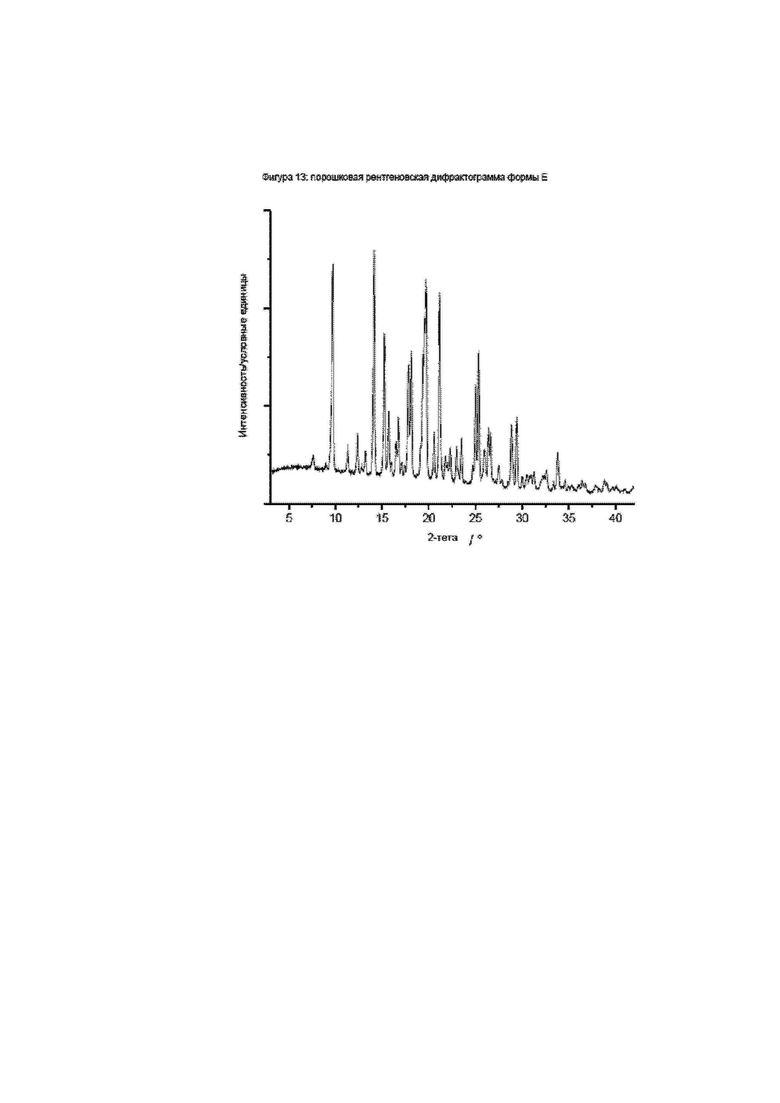
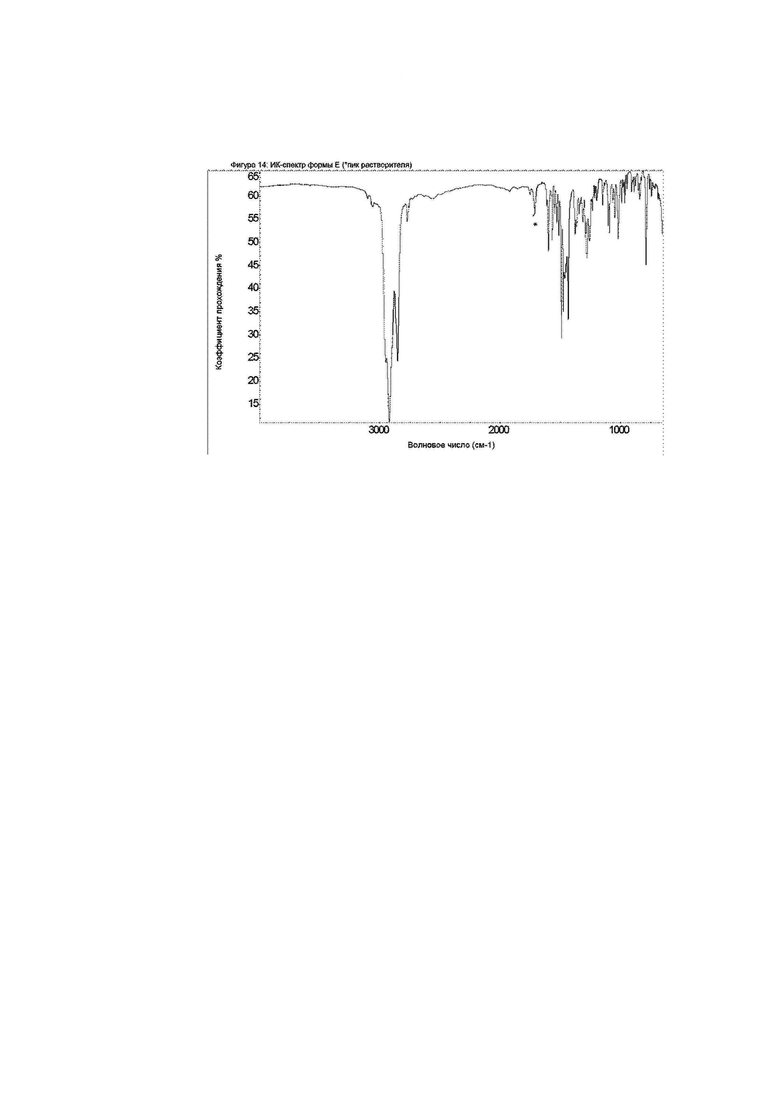
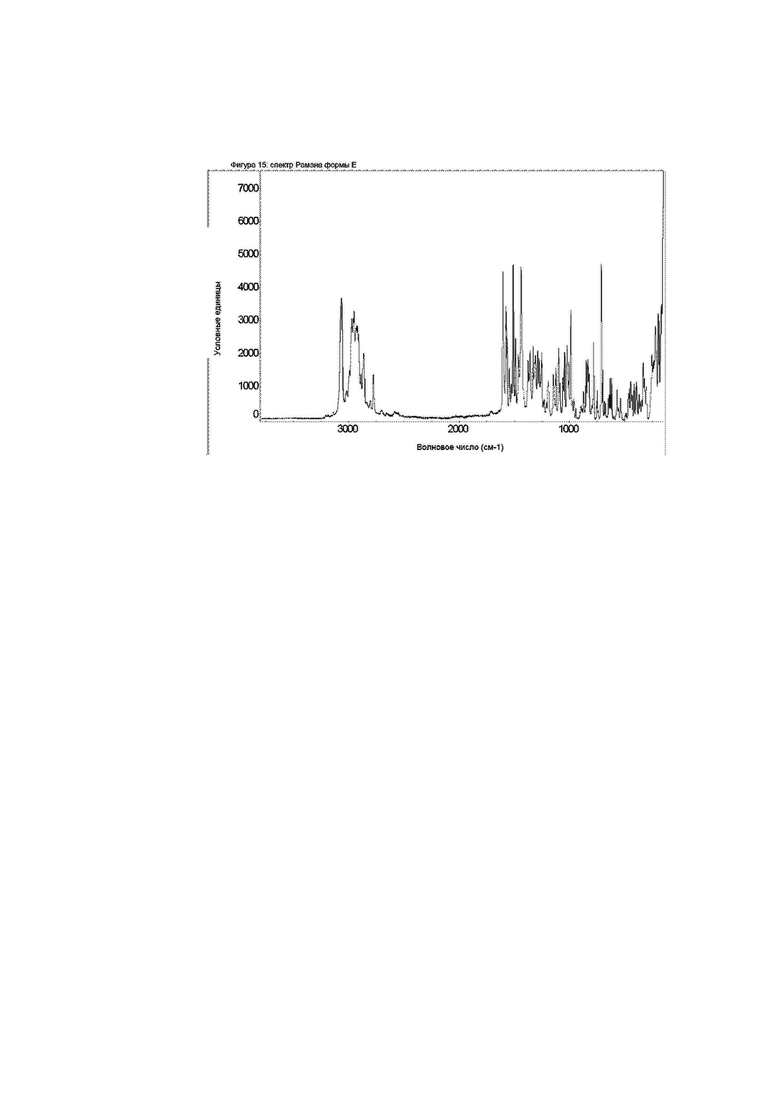
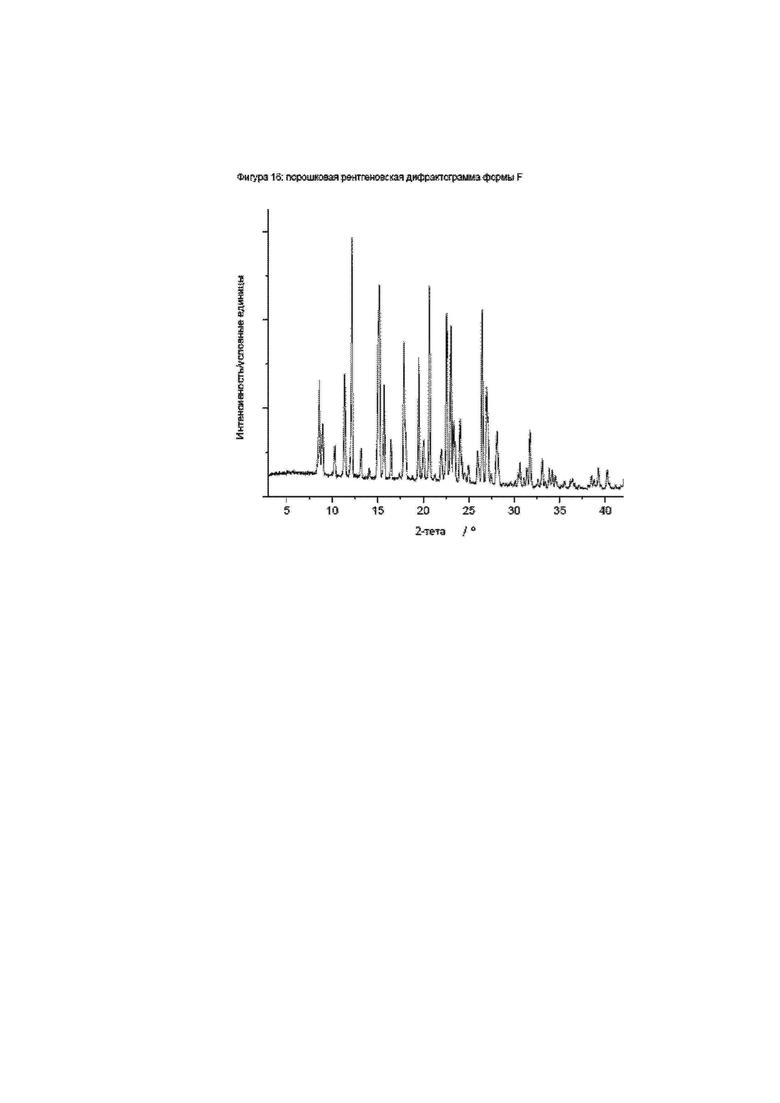
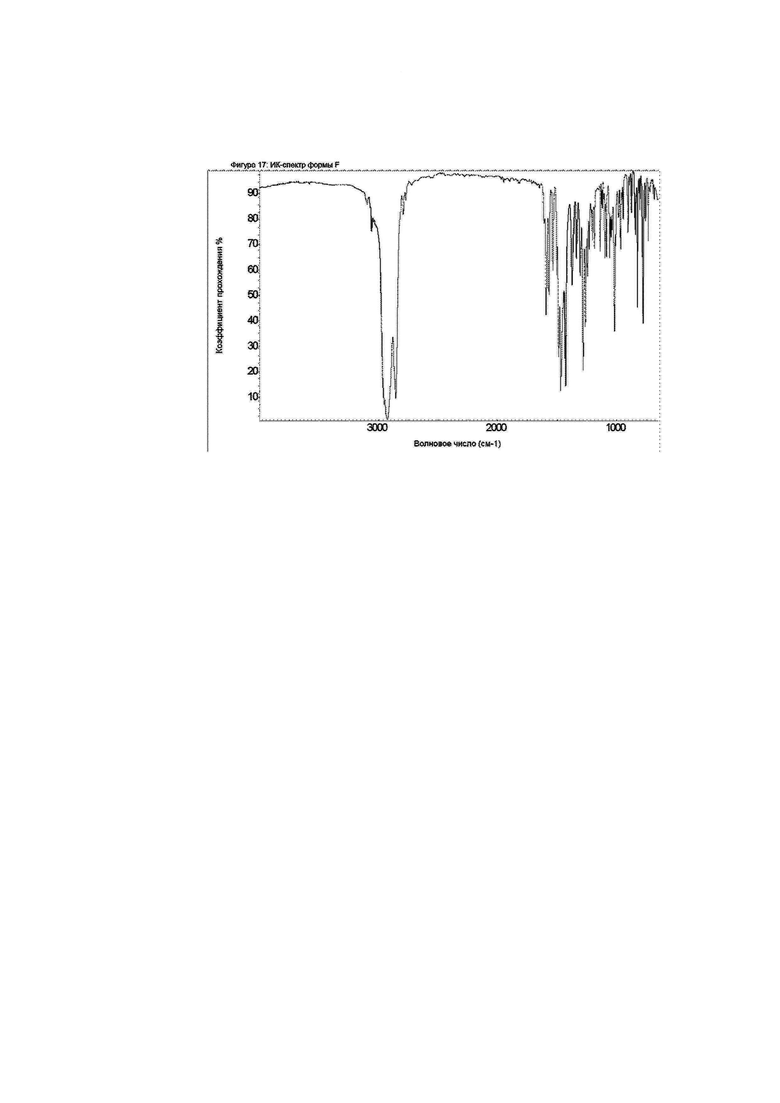
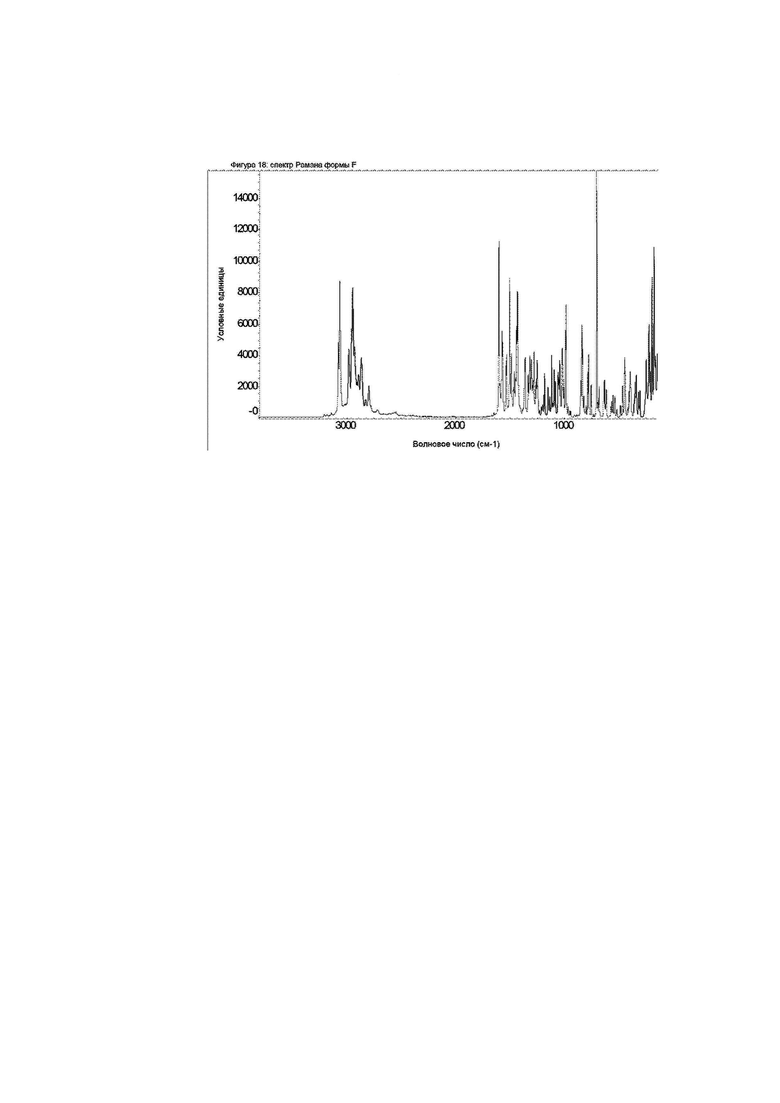
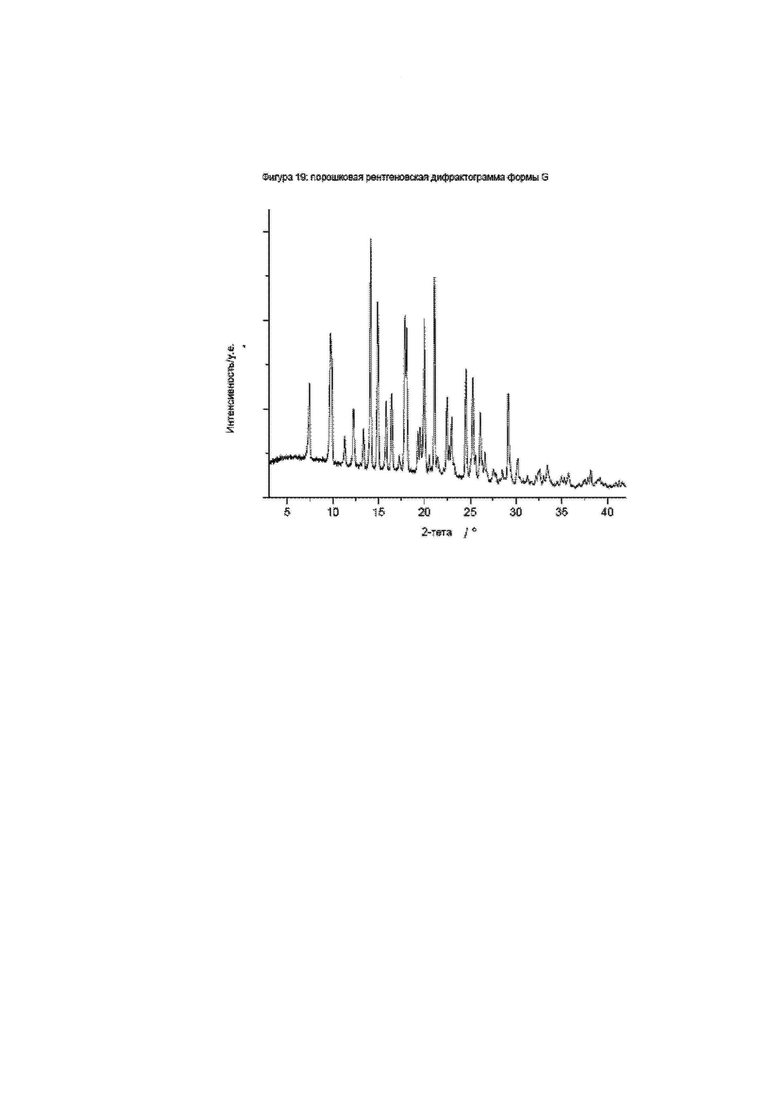
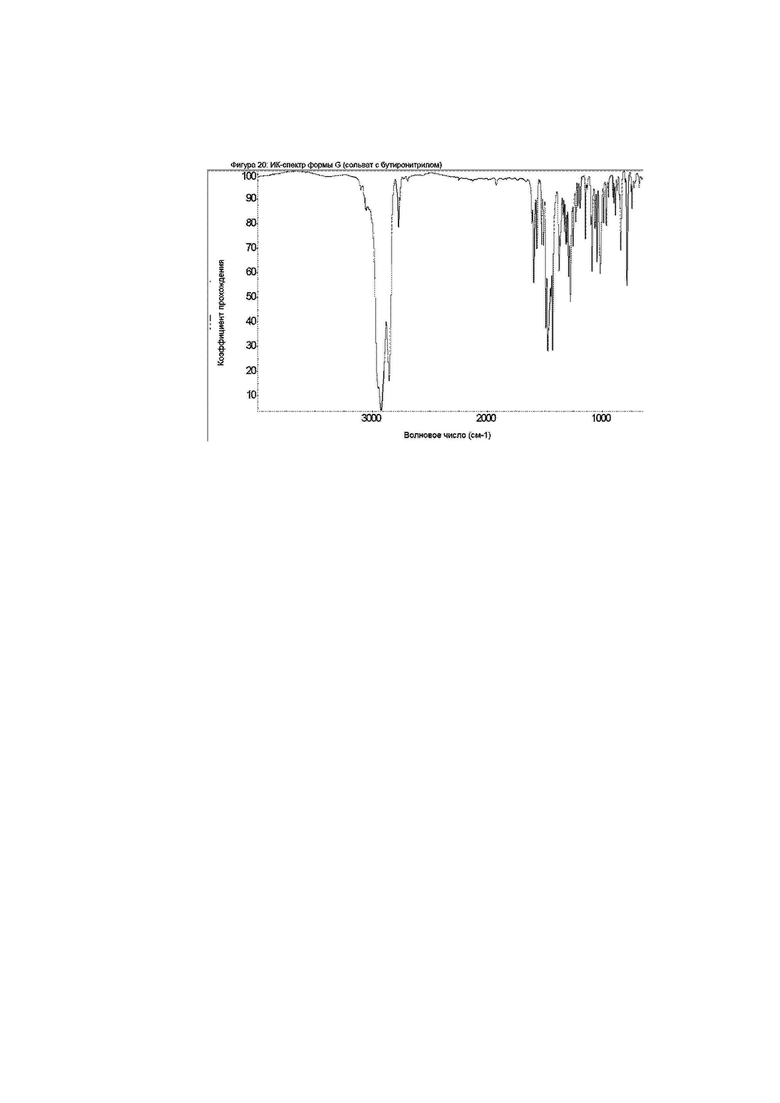
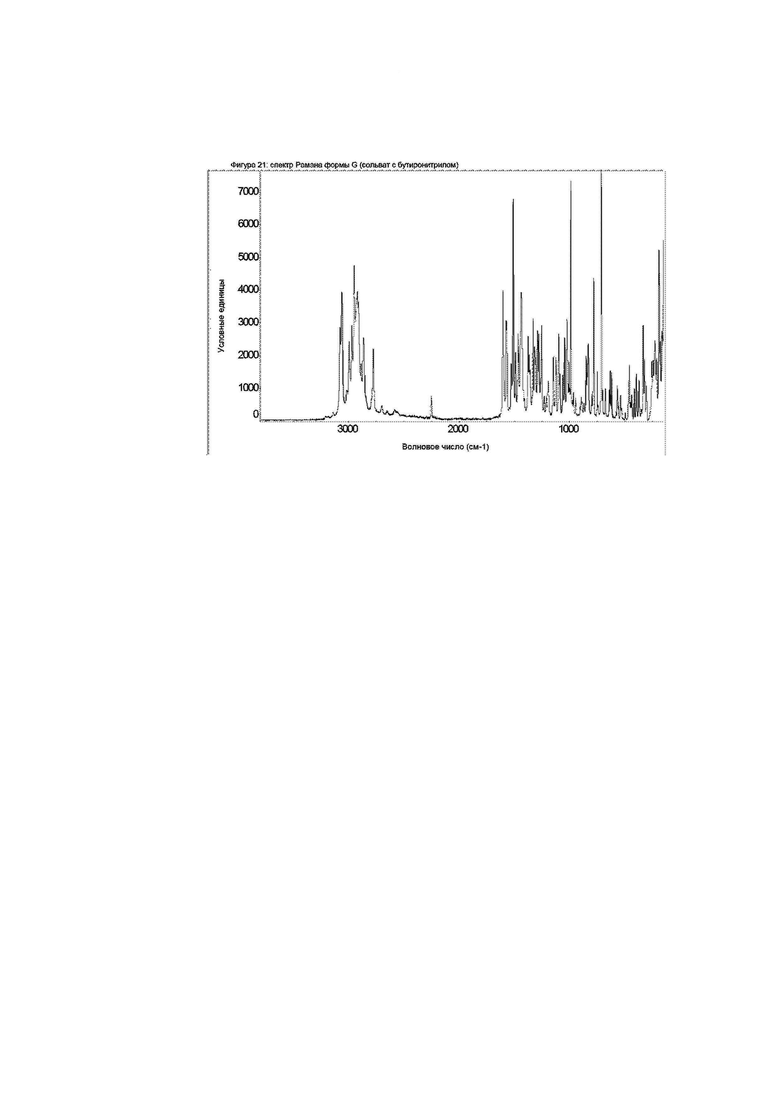
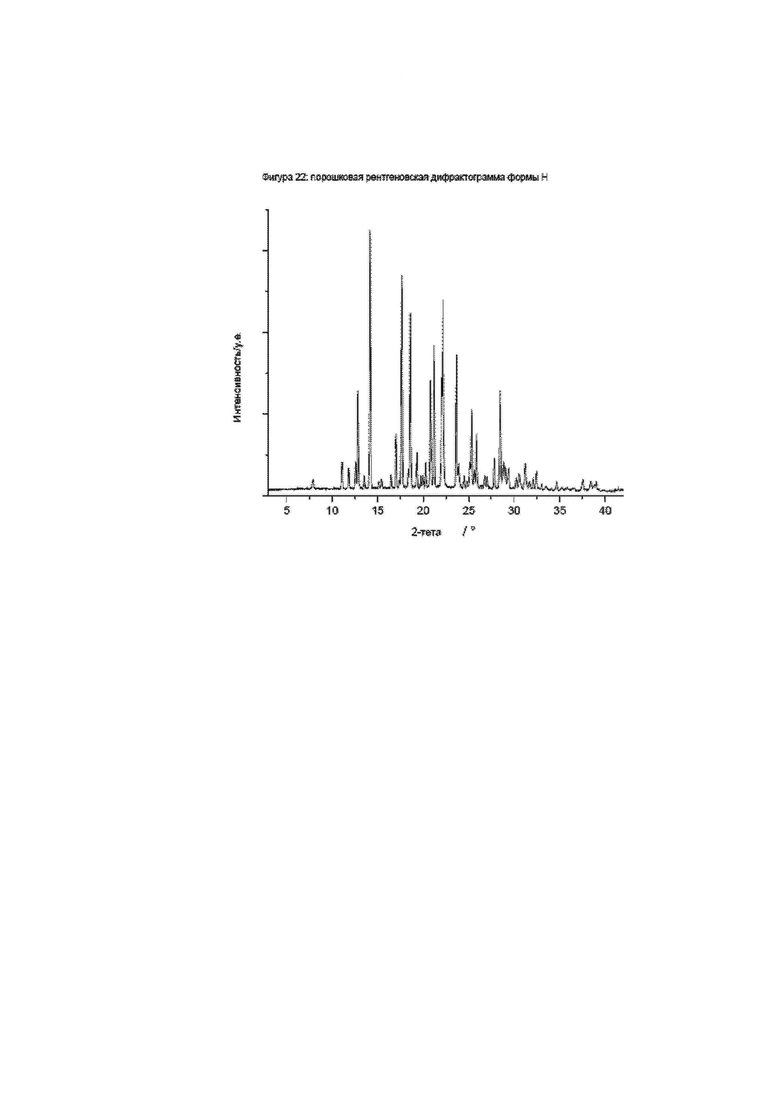
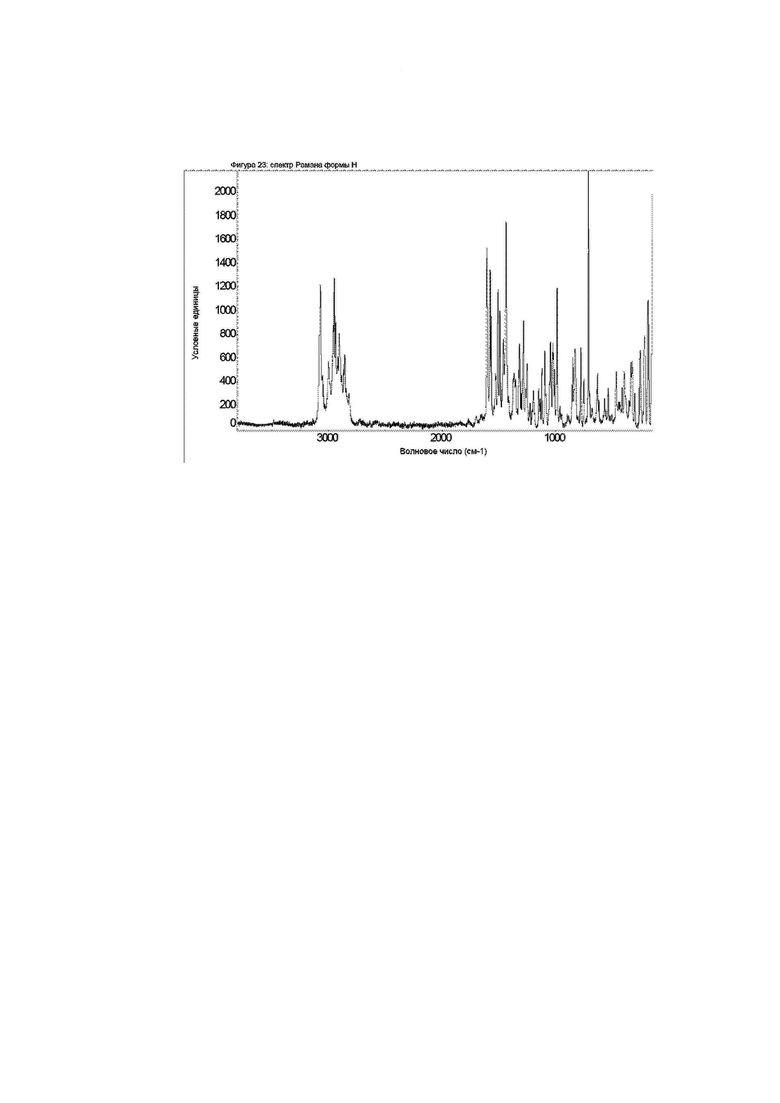
Настоящее изобретение предлагает способы получения замещенных 1-[4-(пиридин-2-илокси)-циклогексил]-5,6-дигидро-4H-2,3,5,10b-тетрааза-бензо[e]азуленов. Способ синтеза соединения формулы I, включает реакцию соединения формулы II с соединением формулы VI. Технический результат - получение 8-хлор-5-метил-1-[4-(2-пиридилокси)циклогексил]-4,6-дигидро-[1,2,4]триазоло[4,3-a][1,4]бензодиазепин с более экономично с меньшим числом этапов способа при умеренных условиях реакции и с хорошим выходом, также неочищенные промежуточные продукты обычно могут быть применены на последующих этапах реакции без необходимости дополнительных этапов очистки. 2 н. и 5 з.п. ф-лы, 23 ил.
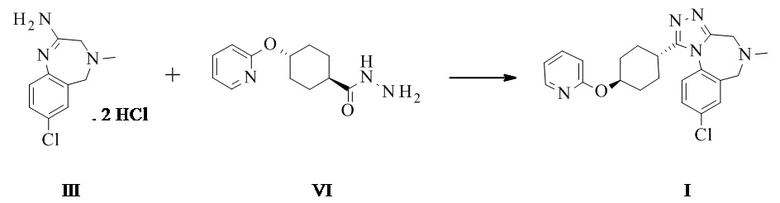
1. Способ синтеза соединения формулы I, включающий реакцию соединения формулы II с соединением формулы VI
 .
.
2. Способ по п. 1, дополнительно включающий превращение соединения формулы XI в соединение формулы III:
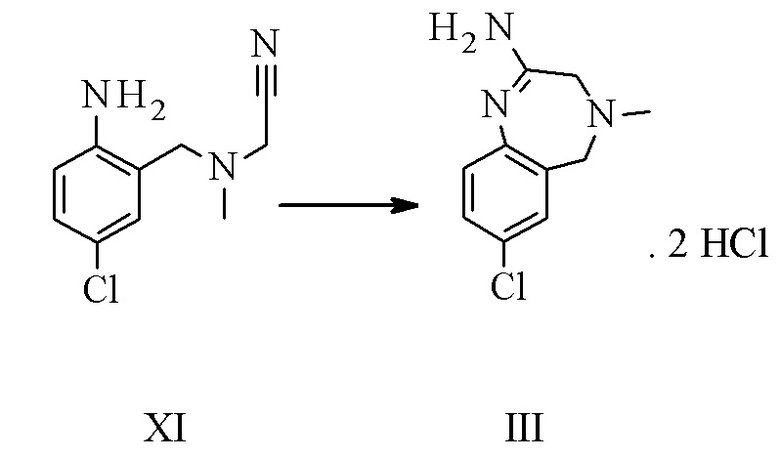 .
.
3. Способ по п. 1 или 2, дополнительно включающий превращение соединения формулы X в соединение формулы XI или его фармацевтически приемлемую соль через следующие этапы:
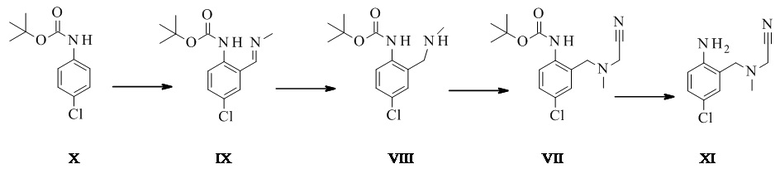 .
.
4. Способ по п. 1 или 2, дополнительно включающий превращение соединения формулы XV в соединение формулы XI или его фармацевтически приемлемую соль через следующие этапы:
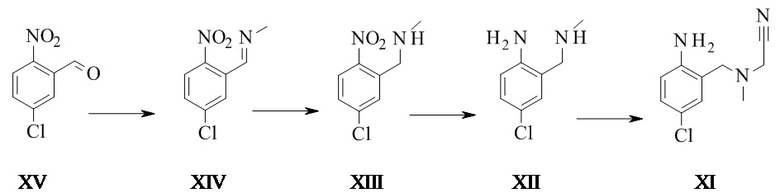 .
.
5. Способ по п. 1 или 2, дополнительно включающий превращение соединения формулы XXVI в соединение формулы XI или его фармацевтически приемлемую соль через следующие этапы:
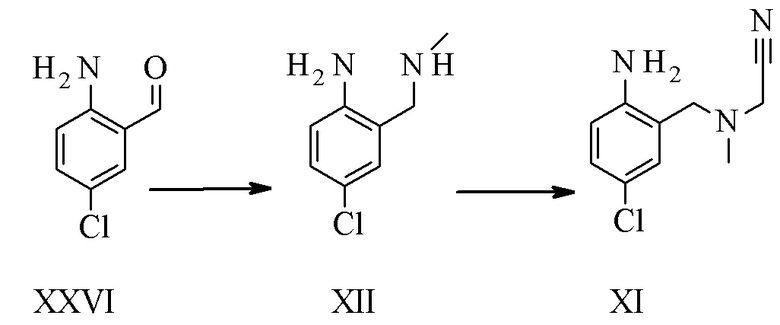 .
.
6. Способ по п. 1 или 2, в котором в качестве промежуточного соединения образуется соединение формулы INT:
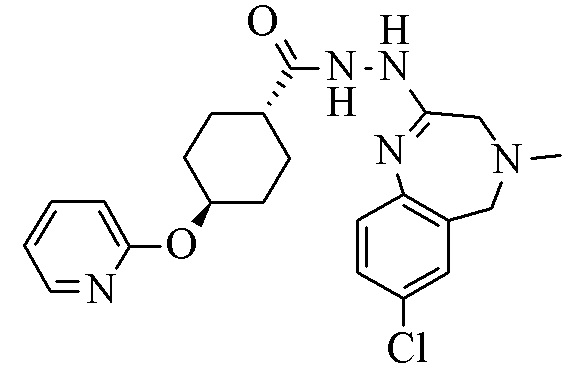 INT.
INT.
7. Промежуточное соединение II или его соль III:
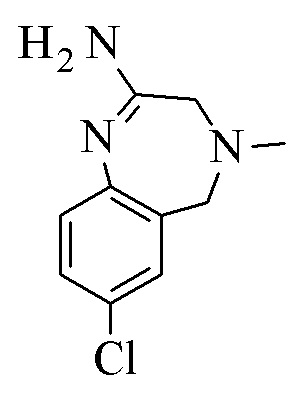 II: свободное основание, III: .2HCl.
II: свободное основание, III: .2HCl.
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| ПРОИЗВОДНЫЕ БЕНЗОДИАЗЕПИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2278865C2 |

