Область техники, к которой относится изобретение
Настоящее изобретение относится к диарилгидантоиновым соединениям, включающим диарилтиогидантоины, способам их синтеза и их применению при лечении гормонорезистентного рака простаты. Эта заявка включает путем отсылки заявку PCT/US 2006/011417 того же правообладателя.
Уровень техники
Рак простаты - наиболее часто встречающийся тип рака и вторая причина смерти от рака у мужчин в западных странах. Если рак локально ограничен, заболевание может быть вылечено хирургическим путем или облучением. Однако в 30% случаев такой рак возникает вновь в виде удаленных метастазов, а в других случаях при диагностировании выявляют запущенное заболевание. Запущенное заболевание лечат путем кастрации и/или введения антиандрогенов, с помощью так называемой андрогенной депривационной терапии. Кастрация снижает уровень циркулирующих андрогенов и уменьшает активность рецептора андрогенов (AR). Введение антиандрогенов блокирует функцию AR путем конкуренции и прекращения связывания андрогенов, следовательно, путем снижения активности AR. Хотя и эффективные вначале, эти способы лечения быстро прекращают действие, и рак становится гормонорезистентным.
Недавно было обнаружено и подтверждено, что суперэкспрессия AR является причиной гормонорезистентного рака простаты. Смотри работу Chen, C.D., Welsbie, D.S., Tran, С., Baek, S.H., Chen, R., Vessella, R., Rosenfeld, M.G., and Sawyers, C.L., Molecular determinants of resistance to antiandrogen therapy, Nat. Med., 10: 33-39, 2004, которая включена сюда путем отсылки. Суперэкспрессии AR достаточно для того, чтобы вызвать прогрессирование рака простаты от гормоночувствительной до гормонорезистентной стадии, что позволяет предположить, что более совершенные ингибиторы AR по сравнению с использующимися в настоящее время лекарственными средствами могут замедлить прогрессирование рака простаты. Было показано, что AR и связывание его лигандов необходимы для роста гормонорезистентного рака простаты, что указывает на то, что AR по-прежнему является мишенью этого заболевания. Также было продемонстрировано, что суперэкспрессия AR превращает антиандрогены из антагонистов в агонисты в случае гормонорезистентного рака простаты (антагонист AR ингибирует активность AR, а агонист стимулирует активность AR). Результаты этой работы объясняют, почему эмаскуляция и антиандрогены не в состоянии предотвратить прогрессирование рака простаты и раскрывают неизвестные свойства гормонорезистентного рака простаты.
Бикалутамид (торговая марка: касодекс) - наиболее часто применяемый антиандроген. Несмотря на то, что он обладает ингибирующим действием на AR в случае гормоночувствительного рака простаты, он прекращает подавлять AR, когда рак становится гормонорезистентным. Две слабые стороны современных антиандрогенов являются причиной неспособности предупреждать прогрессирование рака простаты из гормоночувствительной стадии в гормонорезистентное заболевание и эффективно лечить гормонорезистентный рак простаты. Одна сторона связана с их слабыми антагонистическими активностями, а другая - с их сильными агонистическими активностями, когда AR суперэкспрессируется в случае гормонорезистентного рака простаты. Поэтому, более совершенные ингибиторы AR с более сильными антагонистическими активностями и минимальными агонистическими активностями необходимы для того, чтобы отсрочить прогрессирование заболевания, и для того, чтобы лечить смертельно опасный гормонорезистентный рак простаты.
Нестероидные антиандрогены, такие как бикалутамид, имели преимущества по сравнению со стероидными соединениями, применяемыми при раке простаты, так как они более селективны и имели меньше побочных эффектов. Этот класс соединений был описан во многих патентах, таких как Патент США №4,097,578, Патент США №5,411,981, Патент США №5,705,654, Международных заявок по процедуре РСТ WO 97/00071 и WO 00/17163 и опубликованной патентной заявке США №2004/0009969, все они включены сюда путем отсылки.
Патент США №5,434,176 включает широкую формулу изобретения, которая включает большое количество соединений, но пути синтеза представлены только лишь для небольшой части этих соединений, а фармакологические данные представлены только для двух из них, и специалист в этой области техники не смог бы легко додумать их для других специфических соединений.
Так как механизм гормонорезистентного рака простаты неизвестен, не существует биологической системы для тестирования этих соединений, описанных в этих патентах, в отношении их действия в случае гормонорезистентного рака простаты. В частности, способность суперэкспрессии AR в случае гормонорезистентного рака простаты переключать ингибиторы из антагонистов в агонисты не установлена. Некоторые новые свойства гормонорезистентного рака простаты сообщены в заявках РСТ US 04/42221 и US 05/05529, которые включены сюда путем отсылки. В международной заявке РСТ US 05/05529 представлена методология идентификации антагонистических и агонистических свойств соединений в отношении рецепторов андрогенов. Однако для каждого полученного соединения необходимо выбирать требующий большого времени способ определения антагонистических и агонистических свойств соединения. То есть не существует способа для точного предсказания характеристик, имеющих значение для лечения рака простаты, только лишь на основании химической структуры соединения.
Было сообщено, что некоторые соединения являются ингибиторами лиганд-связывающего домена (LBD) рецептора андрогенов (AR). Некоторые из них применяли в качестве лекарственных средств при лечении рака простаты, например, бикалутамид (касодекс). Были обнаружены некоторые соединения, связывающиеся с AR LBD, например, тиогидантоины, RU59063 и BTID (Teutsch, G.; Goubet, F.; Battmann, Т.; Bonfils, A.; Bouchoux, F.; Cerede, E.; Gofflo, D.; Gaillard-Kelly, M.; Philibert. D.J. Steroid Boichem. Molec. Biol. 1994, 48, 111-119; Van Dort, M.E.; Robins, D.M.; Waybum, B.J. Med. Chem. 2000, 43, 3344-3347).
Существует необходимость в новых тиогидантоиновых соединениях, обладающих желаемыми фармакологическими свойствами, и синтетических путях их получения. Так как активности чувствительны к небольшим структурным изменениям, одно из соединений может быть эффективно при лечении рака простаты, тогда как второе соединение может быть неэффективным, даже если оно отличается от первого соединения лишь немного, скажем, заменой единственного заместителя.
Обнаружение соединений, которые обладают высокой эффективностью в качестве антагонистов андрогенной активности и которые обладают минимальной агонистической активностью, должно победить гормонорезистентный рак простаты (HRPC) и избавиться или замедлить прогрессирование гормоночувствительного рака простаты (HSPC). Таким образом, в этой области техники существует необходимость обнаружения селективных модуляторов рецептора андрогенов, таких как модуляторы, которые являются нестероидными, нетоксичными и тканеспецифичными.
Раскрытие изобретения
Изобретение предоставляет серию соединений, обладающих сильными антагонистическими активностями с минимальными агонистическими активностями в отношении AR. Эти соединения ингибируют рост гормонорезистентного рака простаты.
Специфические соединения изобретения включают:
 .
.
 .
.
 .
.
Изобретение также обеспечивает фармацевтическую композицию, включающую терапевтически эффективное количество соединения, соответствующего любому из предшествующих соединений, или их фармацевтически приемлемой соли и фармацевтически приемлемый носитель или разбавитель.
Изобретение включает способ лечения гиперпролиферативного нарушения, включающий введение такой фармацевтической композиции субъекту, нуждающемуся в таком лечении, осуществляющий, таким образом, лечение гиперпролиферативного нарушения. Гиперпролиферативное нарушение может быть гормонорезистентным раком простаты. Дозировка может быть в диапазоне примерно от 0,001 мг на кг массы тела в день до примерно 100 мг на кг массы тела в день, примерно от 0,01 мг на кг массы тела в день до примерно 100 мг на кг массы тела в день, примерно от 0,1 мг на кг массы тела в день до примерно 10 мг на кг массы тела в день, или около 1 мг на кг массы тела в день.
Соединение можно вводить с помощью внутривенной инъекции, с помощью инъекции в ткань, внутрибрюшинно, перорально или назально. Композиция может быть в форме, выбираемой из группы, состоящей из раствора, дисперсии, суспензии, порошка, капсулы, таблетки, пилюли, капсулы с высвобождением во времени, таблетки с высвобождением во времени и пилюли с высвобождением во времени.
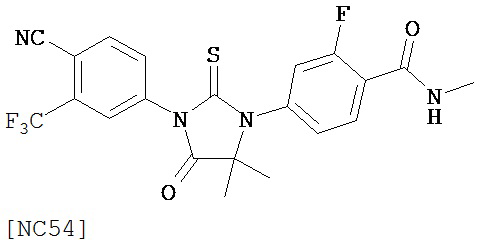
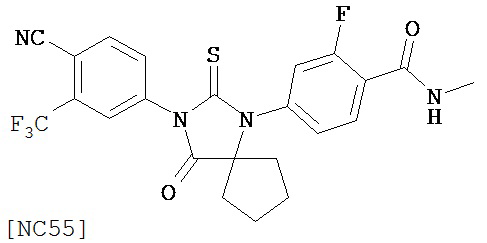
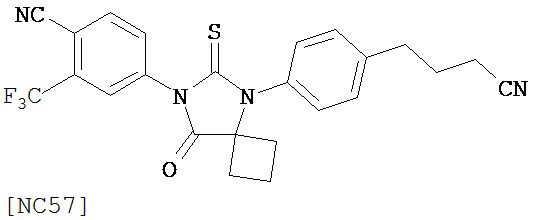
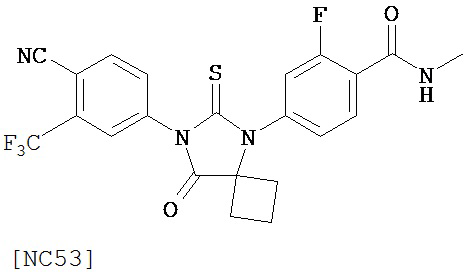
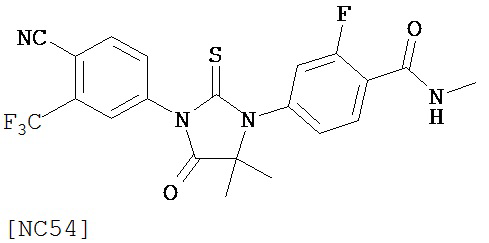
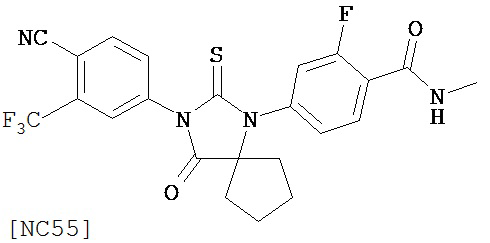
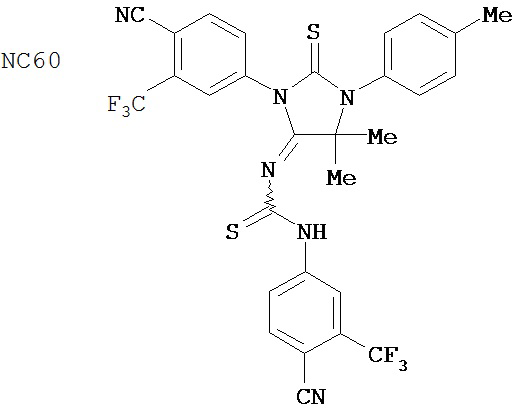
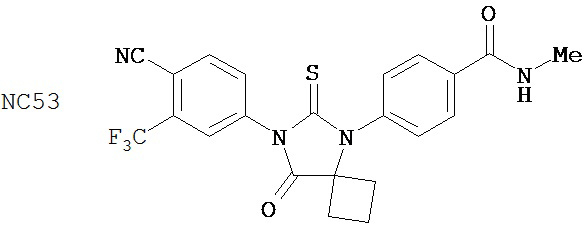
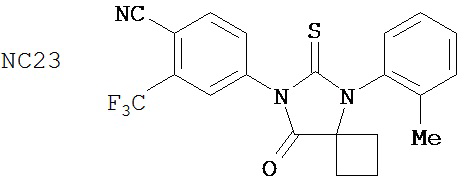
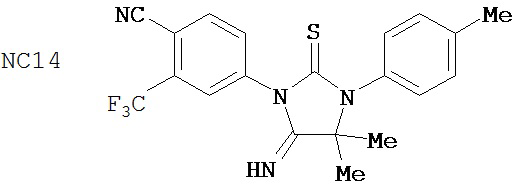
Вводимое соединение может быть выбрано из группы, состоящей из NC54, NC55, NC56 или NC57 или их фармацевтически приемлемой соли. Вводимое соединение может быть NC53 или его фармацевтически приемлемой солью.
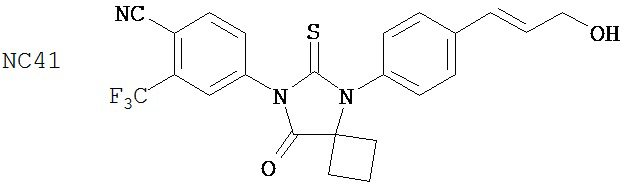
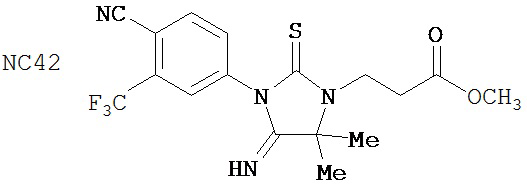
Изобретение обеспечивает способ синтеза NC54, включающий смешивание N-метил-2-фтор-4-(1,1-диметил-цианометил)-аминобензамида и 4-изотиоцианат-2-трифторметилбензонитрила в DMF и нагревание для образования первой смеси и обработку, указанную выше.
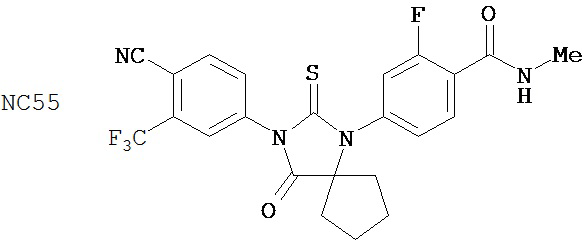
Изобретение также обеспечивает способ синтеза NC55, включающий смешивание N-метил-2-фтор-4-(1-цианоциклопентил)аминобензамида, 4-изотиоцианат-2-трифторметил бензонитрила и DMF и нагревание с обратным холодильником для образования первой смеси и обработку, указанную выше.
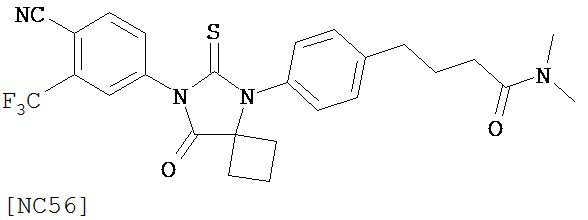
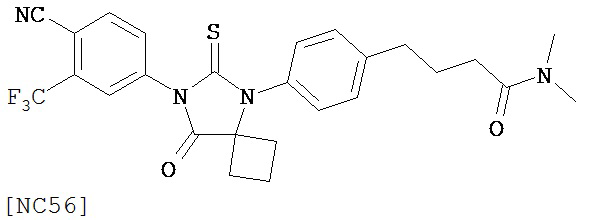
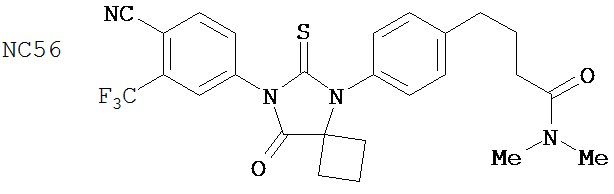
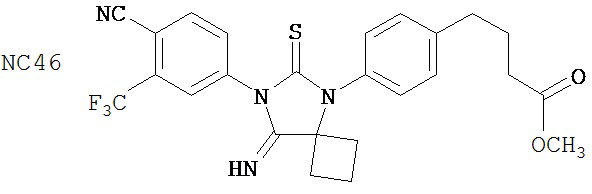
Изобретение, кроме того, обеспечивает способ синтеза NC56, включающий смешивание N,N-диметил 4-[4-(1-цианоциклобутиламино)фенил]бутанамида, 4-изотиоцианат-2-трифторметилбензонитрила и DMF и нагревание с обратным холодильником для образования первой смеси и обработку, указанную выше.
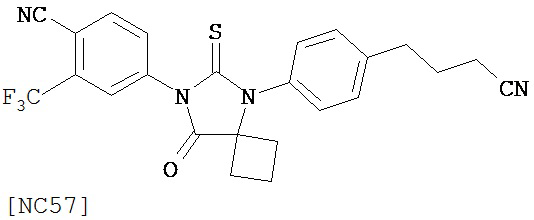
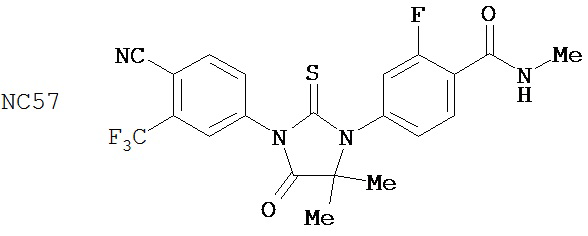
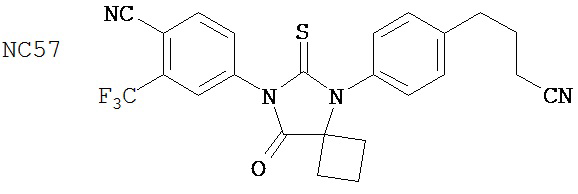
Изобретение обеспечивает способ синтеза NC57, включающий смешивание DMSO, дихлорметана и оксалилхлорида для образования первой смеси, добавление 4-(4-(7-(4-циано-3-(трифторметил)фенил)-8-оксо-6-тиоксо-5,7-диазаспиро[3.4]октан-5-ил)фенил)бутанамида к первой смеси для образования второй смеси; добавление триэтиламина ко второй смеси для образования третьей смеси; нагревание третьей смеси и остановку реакции с помощью водного раствора NH4Cl для образования четвертой смеси; экстракцию органического слоя из четвертой смеси и выделение соединения из органического слоя.
В одном воплощении соединение имеет формулу:
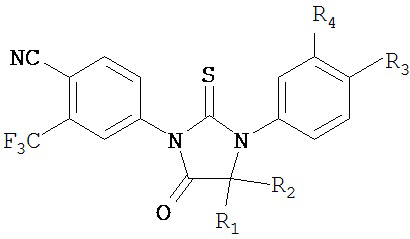
R1 и R2 независимо являются метилом или вместе с атомом углерода, к которому они присоединены, циклоалкильной группой, состоящей из 4-5 атомов углерода, R3 выбирают из группы, состоящей из карбамоила, алкилкарбамоила, карбамоилалкила, алкилкарбамоилалкила, циано и цианоалкила, и R4 представляет собой водород или фтор.
В одном воплощении фармацевтическая композиция включает терапевтически эффективное количество соединения по п.1 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель или разбавитель.
Соединение, например, может иметь формулу
 или
или
Фармацевтическая композиция может включать терапевтически эффективное количество соединения NC54 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель или разбавитель. Фармацевтическая композиция может включать терапевтически эффективное количество соединения формулы NC55 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель или разбавитель.
В одном воплощении способ лечения гиперпролиферативного нарушения включает введение фармацевтической композиции субъекту, нуждающемуся в таком лечении, осуществляющий, таким образом, лечение гиперпролиферативного нарушения.
Композиция, например, может быть в форме, выбираемой из группы, состоящей из раствора, дисперсии, суспензии, порошка, капсулы, таблетки, пилюли, капсулы с высвобождением во времени, таблетки с высвобождением во времени и пилюли с высвобождением во времени. Соединение можно вводить с помощью внутривенной инъекции, с помощью инъекции в ткань, внутрибрюшинно, перорально или назально. Композицию можно вводить при дозировке соединения, составляющей примерно от 0,001 мг на кг массы тела в день до примерно 100 мг на кг массы тела в день. Композицию можно вводить при дозировке соединения, составляющей примерно от 0,01 мг на кг массы тела в день до примерно 100 мг на кг массы тела в день. Композицию можно вводить при дозировке соединения, составляющей примерно от 0,1 мг на кг массы тела в день до примерно 10 мг на кг массы тела в день. Композицию можно вводить при дозировке соединения, составляющей около 1 мг на кг массы тела в день.
Существует способ лечения рака простаты, включающий введение фармацевтической композиции субъекту, нуждающемуся в таком лечении, осуществляющий, таким образом, лечение рака простаты. Фармацевтическая композиция может предотвращать транскрипцию мРНК простат-специфического антигена. Фармацевтическая композиция может предотвращать транслокацию в ядро белка рецептора андрогенов. Фармацевтическая композиция может дестабилизировать белок рецептора андрогенов. Композицию можно вводить перорально. Композиция может быть в форме, выбираемой из группы, состоящей из капсулы, таблетки и пилюли.
В одном воплощении соединение может быть представлено NC54, NC55, NC56, NC57, фармацевтически приемлемой солью любого из них или их смесью.
Способ синтеза диарильного соединения, имеющего формулу
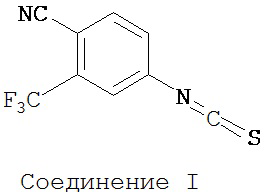
включает смешивание соединения I
с соединением II
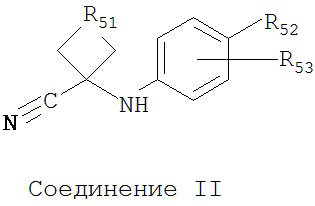
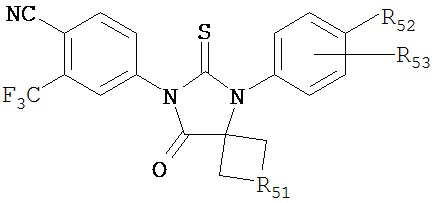
в первом полярном растворителе для образования смеси, нагревание смеси, добавление к смеси второго полярного растворителя, того же самого или отличающегося от первого полярного растворителя, и водного раствора кислоты, нагревание смеси с обратным холодильником, охлаждение смеси и смешивание с водой, и отделение диарильного соединения из смеси. R51 может включать алкильную цепь, состоящую из 1-4 атомов углерода. R52 может быть циано, гидрокси, метилкарбамоилом, метилкарбамоил-замещенным алкилом, метилсульфонкарбамоил-замещенным алкилом, метиламинометилом, диметиламинометилом, метилсульфонилоксиметилом, метоксикарбонилом, 3-циано-4-трифторметилфенилкарбамоилом, карбамоил-замещенным алкилом, карбоксиметилом, метоксикарбонилметилом, метансульфонилом, 4-циано-3-трифторметилфенилкарбамоил-замещенным алкилом, карбокси-замещенным алкилом, 4-метансульфонил-1-пиперазинилом, пиперазинилом, гидроксиэтилкарбамоил-замещенным алкилом или гидроксиэтоксикарбонил-замещенным алкилом. R53 может быть выбран из группы, состоящей из F и Н.
В одном воплощении R51 включает алкильную цепь, состоящую из 1-2 атомов углерода, R52 выбирают из группы, состоящей из карбамоила и метилкарбамоила, a R53 представляет собой F.
Способ синтеза соединения, имеющего формулу:
может включать смешивание 4-изотиоцианат-2-трифторметилбензонитрила и N-метил-4-(1-цианоциклобутиламино)-2-фторбензамида в диметилформамиде для образования первой смеси, нагревание первой смеси для образования второй смеси, добавление спирта и кислоты ко второй смеси для образования третьей смеси, нагревание с обратным холодильником третьей смеси для образования четвертой смеси, охлаждение четвертой смеси, смешивание четвертой смеси с водой и экстракцию органического слоя и выделение соединения из органического слоя.
Способ синтеза соединения [NC54] может включать смешивание N-метил-2-фтор-4-(1,1-диметил-цианометил)-аминобензамида и 4-изотиоцианат-2-трифторметилбензонитрила в DMF и нагревание для образования первой смеси, добавление спирта и кислоты к первой смеси для образования второй смеси, нагревание с обратным холодильником второй смеси, охлаждение второй смеси, смешивание второй смеси с водой и экстракцию органического слоя и выделение соединения из органического слоя.
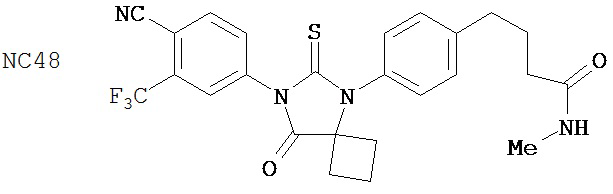
Способ синтеза соединения [NC55] может включать смешивание N-метил-2-фтор-4-(1-цианоциклопентил)аминобензамида, 4-изотиоцианат-2-трифторметилбензонитрила и DMF и нагревание с обратным холодильником для образования первой смеси, добавление спирта и кислоты к первой смеси для образования второй смеси, нагревание с обратным холодильником второй смеси, охлаждение второй смеси, смешивание второй смеси с водой и экстракцию органического слоя и выделение соединения из органического слоя.
Способ синтеза соединения [NC56] может включать смешивание N,N-диметил 4-[4-(1-цианоциклобутиламино)фенил]бутанамида, 4-изотиоцианат-2-трифторметилбензонитрила и DMF и нагревание с обратным холодильником для образования первой смеси, добавление спирта и воды к первой смеси для образования второй смеси, нагревание с обратным холодильником второй смеси, охлаждение второй смеси, смешивание второй смеси с водой и экстракцию органического слоя и выделение соединения из органического слоя.
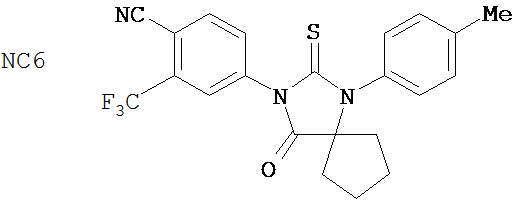
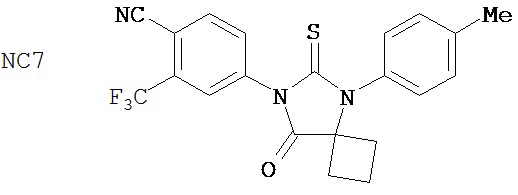
Способ синтеза соединения [NC57] может включать смешивание DMSO, дихлорметана и оксалилхлорида для образования первой смеси, добавление 4-(4-(7-(4-циано-3-(трифторметил)фенил)-8-оксо-6-тиоксо-5,7-диазаспиро[3.4]октан-5-ил)фенил)бутанамида к первой смеси для образования второй смеси, добавление триэтиламина ко второй смеси для образования третьей смеси, нагревание третьей смеси и остановку реакции с помощью водного раствора NH4Cl для образования четвертой смеси, экстракцию органического слоя из четвертой смеси, выделение соединения из органического слоя.
Способ может включать обеспечение по меньшей мере одного диарилтиогидантоинового соединения; количественную оценку ингибирования активности рецептора андрогенов для соединения и определение того, превышает ли ингибирование первый заданный уровень; количественную оценку стимуляции активности рецептора андрогенов в гормонорезистентных раковых клетках для соединений и определение того, находится ли стимуляция ниже второго заданного уровня; отбор соединения, если ингибирование превышает первый заданный уровень, а стимуляция находится ниже второго заданного уровня. Заданные уровни могут быть такими, как у бикалутамида. Количественная оценка ингибирования может включать измерение ингибирующей концентрации (1C 50) в AR-чувствительной репортерной системе или в системе, секретирующей простат-специфический антиген. Количественная оценка стимуляции может включать измерение кратности индукции увеличивающимися концентрациями в AR-чувствительной репортерной системе или в системе, секретирующей простат-специфический антиген. Количественная оценка ингибирования и/или стимуляции может включать оценку влияния соединения на рост опухоли у животного. Стадия количественной оценки ингибирования и/или стимуляции активности рецептора андрогенов может включать измерение аффинности связывания рецептора андрогенов в отношении соединения. Стадия количественной оценки ингибирования и/или стимуляции активности рецептора андрогенов может включать количественную оценку предотвращения вовлечения рецептора андрогенов во взаимодействие по меньшей мере с одним из энхансера простат-специфического антигена и промотора простат-специфического антигена. Стадия количественной оценки ингибирования и/или стимуляции активности рецептора андрогенов может включать количественную оценку предотвращения транслокации рецептора андрогенов в ядро. Стадия количественной оценки ингибирования и/или стимуляции активности рецептора андрогенов может включать количественную оценку дестабилизации белка рецептора андрогенов.
Способ может включать контакт клетки млекопитающих, способной экспрессировать простат-специфический антиген с достаточным количеством диарилтиогидантоинового соединения для того, чтобы предотвратить транскрипцию мРНК простат-специфического антигена. Диарилтиогидантоиновое соединение может быть выбрано из группы, состоящей из NC53, NC54, NC55, NC56 и NC57. Соединение может предотвратить образование транскрипционного комплекса гена простат-специфического антигена. Соединение может предотвратить образование комплекса андроген-рецепторного белка с геном простат-специфического антигена. Соединение может предотвратить образование комплекса РНК-полимеразы II с геном простат-специфического антигена.
Способ включает контакт клетки млекопитающих с достаточным количеством диарилтиогидантоинового соединения для того, чтобы предотвратить транслокацию андроген-рецепторного белка в ядро и/или дестабилизировать белок рецептора андрогенов.
Краткое описание чертежей
Чертежи, приведенные ниже, представляют результаты фармакологического исследования определенных соединений.
Фиг. 1 представляет собой график, показывающий, что бикалутамид демонстрирует агонистическое действие на LNCaP-AR. Агонистические активности бикалутамида при AR-суперэкспрессирующем гормонорезистентном раке простаты. AR-суперэкспрессирующие клетки LNCaP обрабатывали увеличивающимися концентрациями DMSO в качестве среды-растворителя или бикалутамида в отсутствие R1881. Измеряли активности AR-чувствительного репортера.
Фиг. 2 представляет собой график, показывающий анализ антагонистического действия бикалутамида на LNCaP-AR. Агонистические активности бикалутамида при гормоночувствительном раке простаты. Клетки LNCaP обрабатывали увеличивающимися концентрациями DMSO в качестве среды-растворителя или бикалутамида в отсутствие R1881. Измеряли активности AR-чувствительного репортера.
Фиг. 3 представляет собой график, показывающий действие соединений на LNCaP-AR.
Фиг. 4 представляет собой график, показывающий ингибирующий эффект на LNCaP-AR.
Фиг. 5 - ингибирующий эффект на экспрессию ПСА в AR-суперэкспрессирующей модели ксенотрансплантата LNCaP. Мышам перорально вводили среду-растворитель, 0,1, 1 или 10 мг образца 7-3b (NC7) на кг массы тела в течение 44 дней один раз в день. Через 44 дня воздействия изолировали опухоли, экстрагировали опухолевые лизаты и с помощью ELISA в опухолевом лизате определяли уровень ПСА.
Фиг. 6 представляет собой график зависимости объема опухоли как функции от времени воздействия средой-растворителем, касодексом и NC53.
Фиг. 7 представляет собой график размера опухоли. AR-суперэкспрессирующие клетки LNCaP вводили подкожно в боковую поверхность живота кастрированных мышей SCID. Когда опухоли достигали примерно 100 кубических мм, животных случайным образом разделяли на пять групп. В каждой группе было по девять животных. После того, как опухоль у них достигала этого объема, им каждый день перорально давали или среду-растворитель, или бикалутамид, или NC53 в количестве 10 или 50 мг/кг. Опухоли измеряли в трех направлениях, по ширине, длине и глубине, используя штангенциркуль.
Фиг. 8 (A-D) демонстрирует экспериментальные результаты измерения размеров опухоли. На 18 день животных фотографировали с помощью оптической CCD-камеры спустя 3 часа после приема последней дозы. ROI перемещали над опухолью для измерения активности люциферазы в фотонах/секунду. Правые панели представляют собой измерения ROIs.
Фиг. 9 представляет собой график, показывающий фармакокинетические кривые для NC53 при внутривенном (верхняя кривая) и пероральном введении (нижняя кривая).
Фиг. 10 представляет собой график поглощения флуоресценции как функции от логарифма концентрации, который отражает сродство связывания некоторых соединений с рецепторами андрогенов крысы.
Фиг. 11 представляет изображения, отражающие состояние комплексообразования рецептора андрогенов и PHK-полимеразы II с энхансером ПСА и промотором ПСА, если добавляли касодекс или NC53.
Фиг. 12 представляет изображения, отражающие то, что рецептор андрогенов переносится в ядро в присутствии касодекса, но не в присутствии NC53.
Фиг. 13 представляет изображения, отражающие то, что рецептор андрогенов переносится в ядро в присутствии касодекса, но не в присутствии NC53.
Фиг. 14 представляет изображения, отражающие то, что рецептор андрогенов разрушается в присутствии NC53.
Фиг. 15 представляет собой гистограмму, показывающую массу простаты после обработки различными соединениями. 10, 25, или 50 мг соединения на килограмм массы тела в день вводили, как указано в обозначениях гистограммы. Соединения вводили здоровым мышам FVB. После обработки соединением в течение 14 дней определяли массу урогенитального тракта путем удаления и взвешивания семенных пузырьков, простаты и мочевого пузыря. Каждое соединение вводили трем мышам, чтобы получить результаты, представленные с помощью «усов» на гистограмме. Часть мышей не обрабатывали соединением: данные приведены в обозначениях как «необработанные». Другой набор мышей обрабатывали только средой-растворителем: данные представлены в обозначениях как «среда-растворитель».
Фиг. 16 представляет собой график, показывающий анализ ПСА, проведенный в соответствии с экспериментальным протоколом, представленным на фиг. 6.
Фиг. 17 представляет собой график, показывающий эффект различных дозовых режимов NC53 на объем опухоли.
Фиг. 18 (A-C) представляет собой график, показывающий скорость эмиссии фотонов, ассоциированную с активностью люциферазы на 17 день в сравнении со скоростью в день 0 после обработки соединением NC53 в дозах 0,1; 1и 10 мг на килограмм массы тела в день и без обработки соединением NC53.
Фиг. 19 (A-C) представляет результаты эксперимента, в котором мышам SCID путем инъекции вводили клеточную линию LN-AR (HR) для индукции роста опухоли. Одну группу мышей обрабатывали соединением NC53 в дозе 10 мг на килограмм массы тела в день; другую группу мышей обрабатывали только средой-растворителем. (А) Относительный объем опухоли как функция времени показан для каждой группы мышей. (В) Изображения для каждой группы мышей, выполненные с использованием фотонной эмиссии, связанной с активностью люциферазы на 31 день, показаны в виде цветных контуров. (С) Скорость фотонной эмиссии, связанной с активностью люциферазы, показана для нескольких промежутков времени для каждой группы мышей.
Фиг. 20 (A, B) представляет собой график, показывающий поглощение ПСА, ассоциированное с клетками LN-AR, обработанными различными концентрациями соединений NC53, NC54, NC55 и NC57 и средой-растворителем.
Фиг. 21 представляет собой график, показывающий поглощение ПСА, ассоциированное с клетками LN-CaP, обработанными различными концентрациями соединений NC7, NC48, NC53, бикалутамида и DMSO.
Фиг. 22 (A, B) представляет результаты эксперимента, проведенного с нетрансгенными мышами дикого типа (WT), кастрированными мышами с трансгеном люциферазы (Cast) и некастрированными мышами с трансгеном люциферазы (Intact). Показаны результаты для кастрированных мышей с трансгеном люциферазы, обработанных имплантированной тестостероновой гранулой, отдающей 12,5 мг на килограмм массы тела в день с 90-дневным периодом высвобождения (T/Cast), и показаны результаты для некастрированных мышей с трансгеном люциферазы, обработанных имплантированной тестостероновой гранулой, отдающей 12,5 мг на килограмм массы тела в день с 90-дневным периодом высвобождения (Intact+T). Показаны результаты для кастрированных мышей с трансгеном люциферазы, обработанных имплантированной тестостероновой гранулой и бикалутамидом (BIC+T/Cast) или соединением NC53 (NC53+T/Cast) в количестве 10 мг на килограмм массы тела в день. (А) Урогенитальный тракт, взвешенный на 14 день. (В) Эмиссия фотонов на 14 день. Во всех случаях не была индуцирована гормонорезистентная стадия заболевания.
Фиг. 23 представляет собой график, показывающий поглощение ПСА, измеренное в клетках LN-AR после обработки различными дозами нескольких соединений.
Фиг. 24 (A, B) - таблица, в которой представлены некоторые характеристики соединений. Фиг. 15 также представляет собой график фармакокинетических характеристик нескольких соединений в виде функции концентраций соединений в сыворотке крови от времени.
Фиг. 25 представляет собой график люциферазной активности клеточной линии LIAR, к которой добавляли различные соединения, вводимые в диапазоне концентраций от 125 нМ до 1000 нМ.
Осуществление изобретения
Ниже детально обсуждаются воплощения изобретения. В описанных воплощениях для ясности применяют специальную терминологию. Однако изобретение не ограничено выбранной таким образом специальной терминологией. Специалисту в этой области техники будет ясно, что без отступления от сущности и объема изобретения можно использовать другие эквивавлентные термины. Все источники, процитированные в тексте заявки, включены сюда путем отсылки, как если бы каждая такая публикация в индивидуальном порядке была включена в описание путем отсылки.
Синтез диарилгидантоиновых соединений
Пример 56 [NC54]
Реакции, чувствительные к воздуху или влаге, приведенные ниже, проводили в атмосфере аргона, с применением высушенной в сушильном шкафу стеклянной посуды и стандартных методик, использующих шприцы и мембраны. За реакциями следили с помощью кремнеземных пластинок для тонкослойной хроматографии (SiO2, TLC) в ультрафиолетовом свете (254 нм) с последующей визуализацией с помощью окрашивающего раствора пара-анизальдегида или нингидрина. Колоночную хроматографию проводили на силикагеле 60. Спектр 1H-ЯМР, если специально не оговорено, измеряли при 400 МГц в CDCl3 и результаты, как указано ниже, приведены в м.д. (δ) относительно внутреннего стандарта (TMS, 0,0 м.д.): химический сдвиг (мультиплетность, интегрирование, константа взаимодействия в Гц).
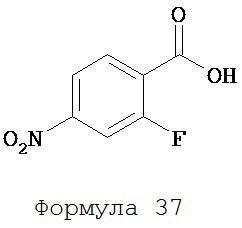
Периодную кислоту (1,69 г, 7,41 ммоль) растворяли в ацетонитриле (25 мл) при энергичном перемешивании, затем в полученном растворе растворяли триоксид хрома (0,16 г, 1,60 ммоль). К указанному выше раствору при перемешивании добавляли 2-фтор-4-нитротолуол (0,33 г, 2,13 ммоль). В экзотермической реакции сразу же образовывался белый осадок. После перемешивания в течение 1 часа жидкий супернатант реакционной среды декантировали в колбу, и растворитель удаляли с помощью выпаривания. Остатки экстрагировали дихлорметаном (2×30 мл) и водой (2×30 мл). Органический слой высушивали над MgSO4 и концентрировали для получения 2-фтор-4-нитробензойной кислоты (формула 37) (0,32 мг, 81%) в виде твердого вещества белого цвета. 1Н-ЯМР (8,06 (ddd, 1H, J=9,9, 2,2 и 0,3), 8,13 (ddd, 1H, J=8,6, 2,2 и 0,9), 8,25 (ddd, 1H, J=8,6, 7,0 и 0,3).
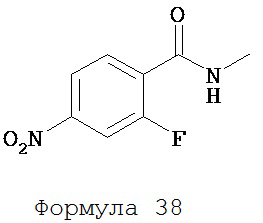
К раствору 2-фтор-4-нитробензойной кислоты (формула 37) (0,20 г, 1,10 ммоль) в DMF (5 мл), охлажденному до -5°C, медленно добавляли тионилхлорид (0,15 г, 1,30 ммоль). Смесь дополнительно перемешивали в течение еще 1 часа при -5°C. В реакционную среду добавляли избыток метиламина (свежеперегнанного из 40%-ного водного раствора). Вторую смесь перемешивали дополнительно еще в течение 1 часа. К смеси, которую промывали солевым раствором (2×50 мл), добавляли этилацетат (50 мл).
Органический слой высушивали над MgSO4 и концентрировали для получения N-метил-2-фтор-4-нитробензамидина (формула 38) (0,18 г, 85%) в виде твердого вещества желтого цвета. 1H-ЯМР (ацетон-d6) (3,05 (d, 3H, J=4,3), 6,31 (dd, 1H, J=13,5 и 2,1), 6,40 (dd, 1H, J=8,6 и 2,1), 7,64 (dd, 1H, J=8,6 и 8,6).
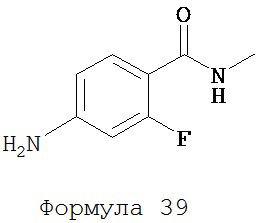
Смесь N-метил-2-фтор-4-нитробензамида (формула 38) (0,18 г, 0,91 ммоль) и железа (0,31 г, 5,60 ммоль) в этилацетате (5 мл) и уксусной кислоте (5 мл) нагревали с обратным холодильником в течение 1 часа. Частицы твердого вещества отфильтровывали. Фильтрат промывали водой и экстрагировали этилацетатом. Органический слой высушивали над MgSO4, концентрировали и остаток очищали с помощью колоночной хроматографии на SiO2 (дихлорметан:ацетон, 95:5) для получения N-метил-2-фтор-4-аминобензамида (формула 39) (0,14 г, 92%) в виде твердого вещества не совсем белого цвета. 1H-ЯМР (ацетон-d6) (2,86 (d, 3H, J=4,3), 5,50 (br s, 2H), 6,37 (dd, 1H, J=14,7 и 2,1), 6,50 (dd, 1H, J=8,6 и 2,1), 7,06 (br s, 1H), 7,68 (dd, 1H, J=8,8 и 8,8).
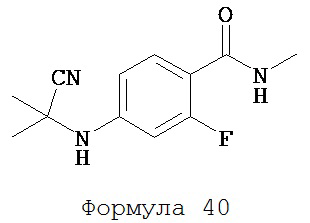
Смесь N-метил-2-фтор-4-аминобензамида (формула 39) (96 мг, 0,57 ммоль), ацетонцианогидрина (0,3 мл, 3,14 ммоль) и сульфата магния (50 мг) нагревали до 80°C и перемешивали в течение 12 часов. К среде добавляли этилацетат (25 мл) и затем промывали водой (2×25 мл). Органический слой высушивали над MgSO4 и концентрировали, и остаток очищали с помощью колоночной хроматографии на SiO2 (дихлорметан:ацетон, 95:5) для получения N-метил-2-фтор-4-(1,1-диметил-цианометил)-аминобензамида (формула 40) (101 мг, 75%) в виде твердого вещества белого цвета. 1H-ЯМР (1,74 (s, 6H), 2,98 (dd, 3H, J=4,8 и 1,1), 6,58 (dd, 1H, J=14,6 и 2,3), 6,63 (dd, 1H, J=8,7 и 2,3), 6,66 (br s, 1H), 7,94 (dd, 1H, J=8,7 и 8,7).
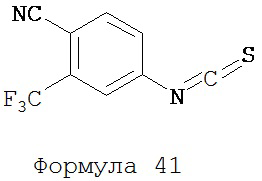
4-Амино-2-трифторметилбензонитрил (2,23 г, 12 ммоль) добавляли порциями в течение 15-17 минут в хорошо перемешиваемую гетерогенную смесь тиофосгена (1 мл, 13 ммоль) в воде (22 мл) при комнатной температуре. Перемешивание продолжали дополнительно в течение еще 1 часа. Реакционную среду экстрагировали хлороформом (3×15 мл). Объединенную органическую фазу высушивали над MgSO4 и выпаривали досуха при пониженном давлении для получения желаемого продукта 4-изотиоцианат-2-трифторметилбензонитрила (формула 41) в виде твердого вещества коричневатого цвета и применяли в таком виде на следующей стадии (2,72 г, 11,9 ммоль, 99%). 1H-ЯМР δ 7,49 (dd, 1H, J=8,3 и 2,1), 7,59 (d, 1H, J=2,1), 7,84 (d, 1H, J=8,3).
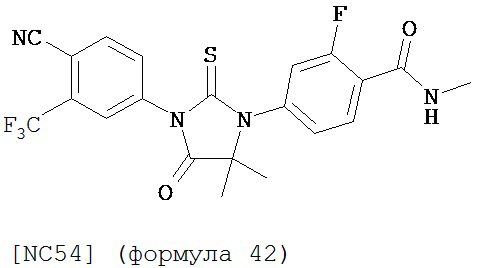
Смесь N-Метил-2-фтор-4-(1,1-диметил-цианометил)-аминобензамида (формула 40) (30 мг, 0,13 ммоль) и 4-изотиоцианат-2-трифторметилбензонитрила (формула 41) (58 мг, 0,26 ммоль) в DMF (1 мл) нагревали при микроволновом излучении при 100°C в течение 11 часов. К смеси добавляли метанол (20 мл) и водный раствор 1 N HCl (5 мл). Вторую смесь нагревали с обратным холодильником в течение 1,5 часов. После охлаждения до комнатной температуры реакционную смесь выливали в холодную воду (50 мл) и экстрагировали этилацетатом (50 мл). Органический слой высушивали над MgSO4, концентрировали и остаток очищали с помощью колоночной хроматографии на SiO2 (дихлорметан:ацетон, 95:5) для получения NC54 (формула 42) (15 мг, 25%) в виде бесцветного кристаллического вещества. 1Н-ЯМР (1,61 (s, 6H), 3,07 (d, 3H, J=4,1), 6,71 (m, 1H), 7,15 (dd, 1H, J=11,7 и 2,0), 7,24 (dd, 1H, J=8,4 и 2,0), 7,83 (dd, 1H, J=8,2 и 2,1), 7,95 (d, 1H, J=2,1), 7,99 (d, 1H, J=8,2), 8,28 (dd, 1H, J=8,4 и 8,4).
Пример 57
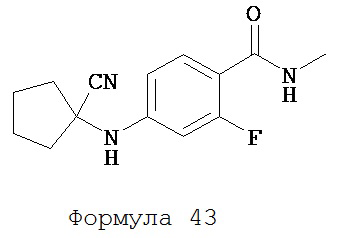
Смесь N-метил-2-фтор-4-аминобензамида (формула 39) (62 мг, 0,37 ммоль), циклопентанона (0,07 мл, 0,74 ммоль) и TMSCN (0,1 мл, 0,74 ммоль) нагревали до 80°C и перемешивали в течение 13 часов. К среде добавляли этилацетат (2×20 мл) и затем промывали водой (2×20 мл). Органический слой высушивали над MgSO4 и концентрировали и остаток очищали с помощью колоночной хроматографии на силикагеле (дихлорметан: ацетон, 95:5) для получения N-метил-2-фтор-4-(1-цианоциклопентил)аминобензамида (формула 43) (61 мг, 63%) в виде твердого вещества белого цвета. 1H-ЯМР (7,95 (dd, 1H, J=8,8, 8,8 Гц), 6,65 (br s, 1H), 6,59 (dd, 1H, J=8,8, 2,3 Гц), 6,50 (dd, 1H, J=14,6, 2,3 Гц), 4,60 (br s, 1H), 2,99 (dd, 3H, J=4,8, 1,1 Гц), 2,36-2,45 (m, 2H), 2,10-2,18 (m, 2H), 1,82-1,95 (m, 4H).
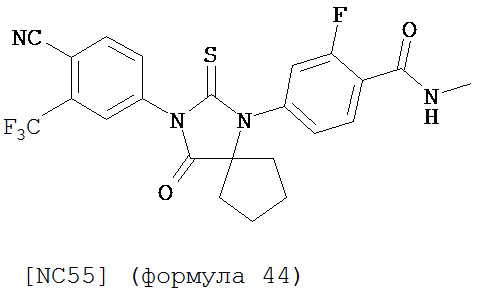
Смесь N-Метил 2-фтор-4-(1-цианоциклопентил)аминобензамида (формула 43) (57 мг, 0,22 ммоль) и 4-изотиоцианат-2-трифторметил бензонитрила (0,15 г, 0,65 ммоль) в DMF (3 мл) нагревали при микроволновом излучении (открытый сосуд) при 130°C в течение 12 часов. К этой смеси добавляли метанол (20 мл) и водный раствор 1 N HCl (5 мл). Вторую смесь нагревали с обратным холодильником в течение 1,5 часов. После охлаждения до комнатной температуры реакционную смесь выливали в холодную воду (50 мл) и экстрагировали этилацетатом (50 мл). Органический слой высушивали над MgSO4, концентрировали и остаток очищали с помощью колоночной хроматографии на силикагеле (дихлорметан:ацетон, 95:5) для получения 4-(3-(4-циано-3-(трифторметил)фенил)-4-оксо-2-тиоксо-1,3-диазаспиро[4.4]нонан-1-ил)-2-фтор-N-метилбензамида, NC55 (формула 44) (8 мг, 7%) в виде твердого вещества бледно-желтоватого цвета. 1H-ЯМР (8,28 (dd, 1H, J=8,4, 8,4 Гц), 7,98 (d, 1H, J=8,3 Гц), 7,96 (d, 1H, J=1,8 Гц), 7,84 (dd, 1H, J=8,3, 1,8 Гц), 7,27 (dd, 1H, J=8,4, 1,8 Гц), 7,17 (dd, 1H, J=11,7, 1.8 Гц), 6,67-6,77 (m, 1H), 3,07 (d, 3H, J=4,3 Гц), 2,32-2,41 (m, 2H), 2,13-2,21 (m, 2H), 1,85-1,96 (m, 2H), 1,49-1,59 (m, 2H).
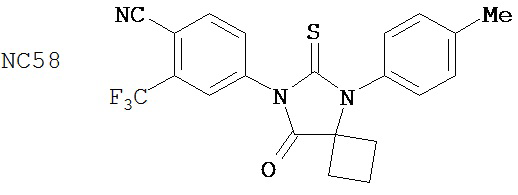
Пример 58
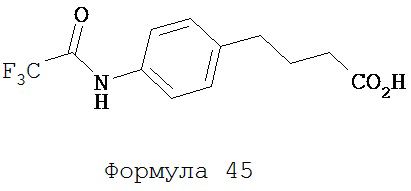
Трифторуксусный ангидрид (0,85 мл, 6,14 ммоль) добавляли к раствору 4-(4-аминофенил)масляной кислоты (0,5 г, 2,79 ммоль) в хлороформе (10 мл) при 0°C. Смесь нагревали до комнатной температуры и перемешивали в течение 3 часов. Смесь разделяли хлороформом (20 мл) и водой (20 мл). Органический слой высушивали над MgSO4, концентрировали и остаток очищали с помощью колоночной хроматографии на силикагеле (дихлорметан:ацетон, 9:1) для получения 4-[4-(2,2,2-трифторацетиламино)фенил]масляной кислоты (формула 45) (0,53 г, 69%). 1Н-ЯМР (7,81 (br s, 1H), 7,48 (d, 2H, J=8,5 Гц), 7,22 (d, 2H, J=8,5 Гц), 2,68 (t, 2H, J=7,5 Гц), 2,38 (t, 2H, J=7,5 Гц), 1,96 (p, 2H, J=7,5 Гц).
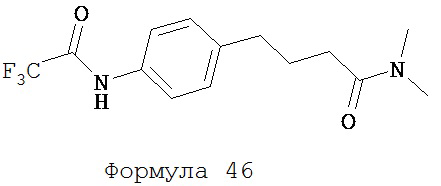
Тионилхлорид (71 мг, 0,60 ммоль) медленно добавляли к раствору 4-[4-(2,2,2-трифторацетиламино)фенил]масляной кислоты (формула 45) (0,15 г, 0,55 ммоль) в DMF (5 мл), охлажденного до -5°C. Смесь перемешивали дополнительно еще в течение 1 часа при -5°C. К реакционной среде добавляли избыток диметиламина (свежеперегнанного из его 40%-ного водного раствора). Вторую смесь перемешивали дополнительно в течение еще 1 часа. Этилацетат (50 мл) добавляли к смеси, которую промывали солевым раствором (2×50 мл). Органический слой высушивали над MgSO4 и концентрировали для получения N,N-диметил-4-[4-(2,2,2-трифторацетиламино)фенил]бутанамида (формула 46) (0,17 г, количественно) в виде твердого вещества желтого цвета. 1H-ЯМР (9,70 (br s, 1H), 7,55 (d, 2H, J=8,6 Гц), 7,11 (d, 2H, J=8,6 Гц), 2,91 (s, 2H), 2,89 (s, 3H), 2,60 (t, 2H, J=7,7 Гц), 2,27 (t, 2H, J=7,7 Гц), 1,89 (p, 2H, J=7,7 Гц).
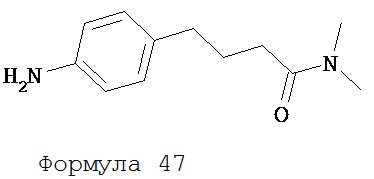
1 N раствор NaOH (3 мл) добавляли к раствору N,N-диметил-4-[4-(2,2,2-трифторацетиламино)фенил]бутанамида (формула 46) (0,17 г, 0,55 ммоль) в метаноле (2 мл) при комнатной температуре. Смесь перемешивали в течение 14 часов. Смесь разделяли хлороформом (25 мл) и водой (25 мл). Органический слой высушивали над MgSO4 и концентрировали и остаток очищали с помощью колоночной хроматографии на силикагеле (дихлорметан: ацетон, 9:1) для получения N-диметил-4-(4-аминофенил)бутанамида (формула 47) (74 мг, 66%) в виде твердого вещества белого цвета. 1H-ЯМР (6,97 (d, 2Н, J=8,3 Гц), 6,61 (d, 2H, J=8,3 Гц), 3,56 (br s, 2H), 2,92 (s, 6H), 2,56 (t, 2H, J=7,7 Гц). 2,28 (t, 2H, J=7,7 Гц), 1,91 (p, 2H, J=7,7 Гц).
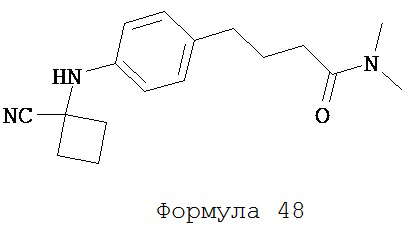
Формула 48
Смесь N,N-диметил-4-(4-аминофенил)бутанамида (формула 47) (74 мг, 0,36 ммоль), циклобутанона (54 мг, 0,78 ммоль) и TMSCN (77 мг, 0,78 ммоль) нагревали до 80°C и перемешивали в течение 15 часов. К среде добавляли этилацетат (2×20 мл) и затем промывали водой (2×20 мл.). Органический слой высушивали над MgSO4 и концентрировали и остаток очищали с помощью колоночной хроматографии на силикагеле (дихлорметан:ацетон, 9:1) для получения N,N-диметил-4-[4-(1-цианоциклобутиламино)фенил]бутанамида (формула 48) (58 мг, 57%) в виде твердого вещества белого цвета. 1H-ЯМР (7,07 (d, 2H, J=8,5 Гц), 6,59 (d, 2H, J=8,5 Гц), 3,94 (br s, 1H), 2,94 (s, 3H), 2,93 (s, 3H), 2,75-2,83 (m, 2H), 2,60 (t, 2H, J=7,6 Гц), 2,33-2,42 (m, 2H), 2,30 (t, 2H, J=7,6 Гц). 2,11-2,28 (m, 2H), 1,93 (p, 2H, J=7,6 Гц).
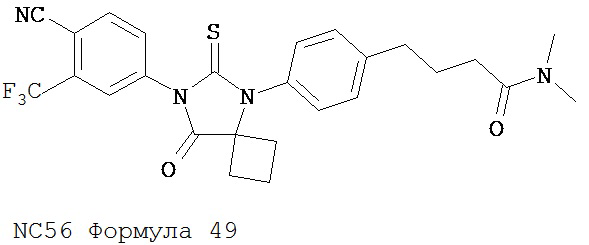
Смесь N-диметил-4-[4-(1-цианоциклобутиламино)фенил]бутанамида (формула 48) (58 мг, 0,20 ммоль) и 4-изотиоцианат-2-трифторметилбензонитрила (74 мг, 0,32 ммоль) в DMF (3 мл) нагревали с обратным холодильником в течение 2 часов. К этой смеси добавляли метанол (20 мл) и водный раствор 1 N HCl (5 мл). Вторую смесь нагревали с обратным холодильником в течение 1,5 часов. После охлаждения до комнатной температуры реакционную смесь выливали в холодную воду (50 мл) и экстрагировали этилацетатом (50 мл). Органический слой высушивали над MgSO4, концентрировали и остаток очищали с помощью колоночной хроматографии на силикагеле (дихлорметан: ацетон, 95:5) для получения 4-(4-(7-(4-циано-3-(трифторметил)фенил)-8-оксо-6-тиоксо-5,7-диазаспиро[3.4]октан-5-ил)фенил)-N-диметилбутанамида, NC56 (формула 49) (44 мг, 42%) в виде твердого вещества бледно- желтоватого цвета. 1H-ЯМР (7,98 (s, 1Н), 7,97 (d, 1H, J=8,2 Гц), 7,86 (d, 1H, J=8,2 Гц), 7,42 (d, 2H,.7=8,3 Гц), 7,22 (d, 2H, J=8,3 Гц), 2,99 (s, 3H), 2,96 (s, 3H), 2,78 (t, 2H, J=7,5 Гц), 2,62-2-70 (m, 2H), 2,52-2,63 (m, 2H), 2,40 (t, 2H, J=7,5 Гц), 2,15-2,30 (m, 1H), 2,04 (p, 2H, J=7,5 Гц), 1,62-1,73 (m, 1H).
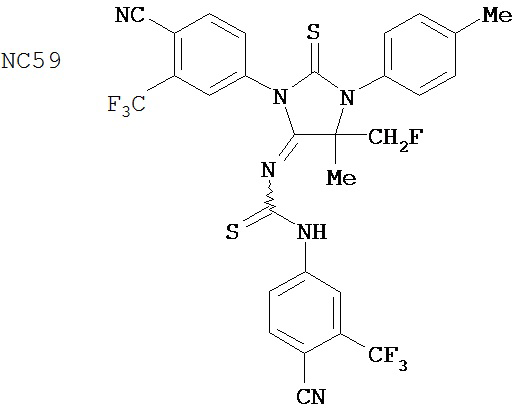
Пример 59
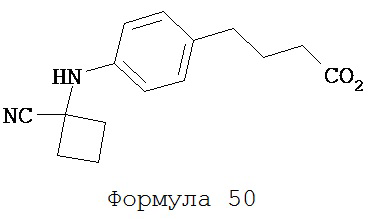
Смесь 4-(4-аминофенил)масляной кислоты (0,20 г, 1,12 ммоль), циклобутанона (0,17 мл, 2,23 ммоль) и TMSCN (0,30 мл, 2,23 ммоль) нагревали до 80°C и перемешивали в течение 13 часов. К среде добавляли этилацетат (2×30 мл) и затем промывали водой (2×30 мл). Органический слой высушивали над MgSO4 и концентрировали и остаток очищали с помощью колоночной хроматографии на силикагеле (дихлорметан: ацетон, 9:1) для получения 4-[4-(1-цианоциклобутиламино)фенил]масляной кислоты (формула 50) (0,21 г, 74%) в виде твердого вещества желтого цвета. 1H-ЯМР (7,06 (d, 2H, J=8,6 Гц), 6,59 (d, 2H, J=8,6 МГц), 2,75-2,83 (m, 2H), 2,59 (t, 2H, J=7,5 Гц). 2,37 (t, 2H, J=7,5 Гц), 2,33-2,42 (m, 2H), 2,11-2,28 (m, 2H), 1,92 (p, 2H, J=7,5 Гц).
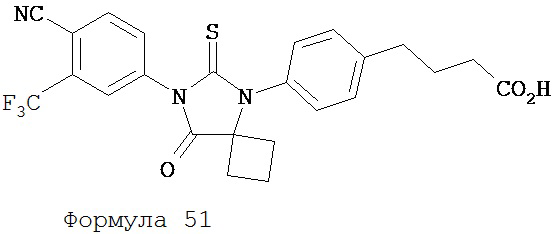
Смесь 4-[4-(1-цианопиклобутиламино)фенил]масляной кислоты (формула 50) (0,21 г, 0,83 ммоль) и 4-изотиоцианат-2-трифторбензонитрила (0,25 г, 1,08 ммоль) в толуоле (10 мл) нагревали с обратным холодильником в течение 1 часа. К этой смеси добавляли водный 1 N HCl (5 мл). Вторую смесь нагревали с обратным холодильником в течение 1,5 часов. После охлаждения до комнатной температуры реакционную смесь выливали в холодную воду (50 мл) и экстрагировали этилацетатом (50 мл). Органический слой высушивали над MgSO4, концентрировали и остаток очищали с помощью колоночной хроматографии на силикагеле (дихлорметан: ацетон, 95:5) для получения 4-(4-(7-(4-циано-3-(трифторметил)фенил)-8-оксо-6-тиоксо-5,7-диазаспиро[3.4]октан-5-ил)фенил)масляной кислоты, NC122 (формула 51) (60 мг, 15%). 1H-ЯМР (7,98 (d, 1H, J=1,8 Гц), 7,97 (d, 1H, J=8,3 Гц), 7,86 (dd, 1H, J=8,3, 1,8 Гц), 7,42 (d, 2H, J=8,5 Гц), 7,24 (d, 2H, J=8,5 Гц), 2,79 (t, 2H, J=7,5 Гц), 2,62-2,68 (m, 2H), 2,51-2,59 (m, 2H), 2,47 (t, 2H, J=7,5 Гц), 2,14-2,26 (m, 1H), 2,06 (p, 2H, J=7,5 Гц), 1,60-1,70 (m, 1H).
Пример 61
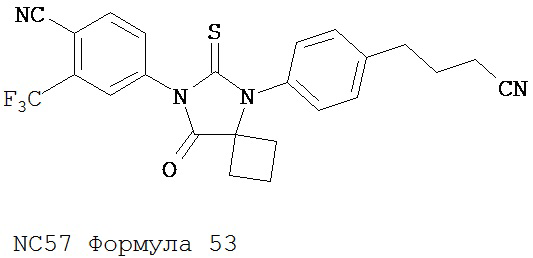
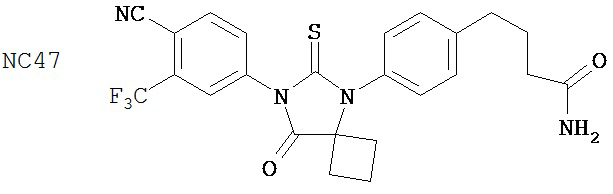
Раствор DMSO (0,01 мл, 0,12 ммоль) в безводном дихлорметане (1 мл) добавляли при перемешивании к раствору оксалилхлорида (0,01 мл, 0,09 ммоль) в безводном дихлорметане (2 мл) при -78°C. Через 15 минут в реакционную смесь добавляли дихлорметановый раствор 4-(4-(7-(4-циано-3-(трифторметил)фенил)-8-оксо-6-тиоксо-5,7-диазаспиро[3.4)октан-5-ил)фенил)бутанамида, NC47 (формула 52) (35 мг, 0,07 ммоль). Перемешивание продолжали в течение 20 минут при -78°C и затем добавляли триэтиламин (0,03 мл, 0,22 ммоль). Через 30 минут при -78°C реакционную смесь нагревали до комнатной температуры и затем реакцию останавливали насыщенным водным раствором NH4Cl. Реакционную смесь разводили дихлорметаном и экстрагировали дихлорметаном. Органический слой высушивали над MgSO4, концентрировали и хроматографировали (дихлорметан: ацетон, 95:5) для получения 4-(5-(4-(3-цианопропил)фенил)-8-оксо-6-тиоксо-5,7-диазаспиро[3.4]октан-7-ил)-2-(трифторметил)бензонитрила, NC57 (формула 53) (29 мг, 87%) в виде вязкого масла. 1Н-ЯМР (7,98 (d, 1H, J=1,8 Гц), 7,98 (d, 1H, J=8,3 Гц), 7,86 (dd, 1H, J=8,3, 1,8 Гц), 7,43 (d, 2H, J=8,4 Гц), 7,27 (d, 2H, J=8,4 Гц), 2,90 (t, 2H, J=7,3 Гц), 2,63-2,73 (m, 2H), 2,52-2,62 (m, 2H), 2,42 (t, 2H, J=7,3 Гц), 2,18-2,30 (m, 1H), 2,07 (p, 2H, J=7,3 Гц), 1,63-1,73 (m, 1H).
Специалист в этой области техники сможет модифицировать и/или комбинировать синтезы, описанные в тексте заявки, чтобы получить другие диарилгидантоиновые соединения.
Фармакологическое исследование соединений
Соединения, для которых выше были описаны пути синтеза, исследовали путем скрининга гормонорезистентных раковых клеток простаты на антагонистические и агонистические активности по отношению к AR с применением методик отбора, сходных с теми, которые описаны в заявках РСТ US 04/42221 и US 05/05529, которые включены сюда путем отсылки. Многие соединения демонстрировали мощные активности при минимальных агонистических активностях при суперэкспрессии AR в случае гормонорезистентного рака простаты.
Биологический анализ in vitro
Эффект соединений на AR в репортерном анализе
Соединения тестировали с помощью искусственной системы, реагирующей на ответ AR, в линии клеток гормонорезистентного рака простаты. В такой системе клетки рака простаты LNCaP конструировали таким образом, чтобы они стабильно экспрессировали AR на уровне, примерно в пять раз превышающем эндогенный уровень. Экзогенный AR обладает сходными свойствами с эндогенным AR, заключающимися в том, что оба рецептора стабилизируются синтетическим андрогеном R1881. Клетки, суперэкспрессирующие AR, также были сконструированы таким образом, чтобы стабильно включать AR-чувствительный репортер, и репортерная активность этих клеток демонстрирует свойства гормонорезистентного рака простаты. Она реагирует на низкие концентрации синтетического андрогена R1881, ингибируется только высокими концентрациями бикалутамида (смотри таблицу 1) и демонстрирует агонистическую активность по отношению к бикалутамиду (фиг. 1 и таблица 2). В соответствии с опубликованными данными бикалутамид ингибировал AR-чувствительный репортер и не обладал агонистической активностью в клетках гормоночувствительного рака простаты (фиг. 2).
Таблица 1
Мы исследовали антагонистическую активность соединений, синтез которых был описан выше, в присутствии 100 пМ R1881. Сконструированные клетки LNCaP (LNCaP-AR, также сокращенные как LN-AR) поддерживали в среде Искова, содержащей 10%-ную фетальную сыворотку теленка (FBS). Два дня до обработки лекарственным средством клетки растили на среде Искова, содержащей 10%-ную очищенную на активированном угле FBS (CS-FBS), чтобы удалить андрогены. Клетки разделяли и выращивали в среде Искова, содержащей 10%-ную CS-FBS с 100 пМ R1881 и увеличивающимися концентрациями тестируемых соединений. После двух дней инкубации анализировали репортерные активности.
В таблице 1 приведены значения IC50 тех соединений, которые ингибируют AR в случае гормонорезистентного рака простаты. Контрольное соединение бикалутамид обладает IC50, равной 889 нМ. Большинство исследованных соединений (диарилтиогидантоинов) обладали величинами IC50, находящимися в диапазоне от 100 до 200 нМ при ингибировании AR в случае гормонорезистентного рака простаты. В противоположность этому, антиандрогенные соединения, перечисленные в качестве примеров в патенте US patent No. 5,705,654, например, такие как 30-2, 30-3, 31-2, 31-3 и 24-3 (NC24-NC28), не обладают активностями, ингибирующими AR, в этой системе.
Антагонистические активности по отношению к AR в случае гормонорезистентного рака простаты измеряли с помощью AR-чувствительного репортера и с помощью экспрессии эндогенного ПСА.
Таблица 2
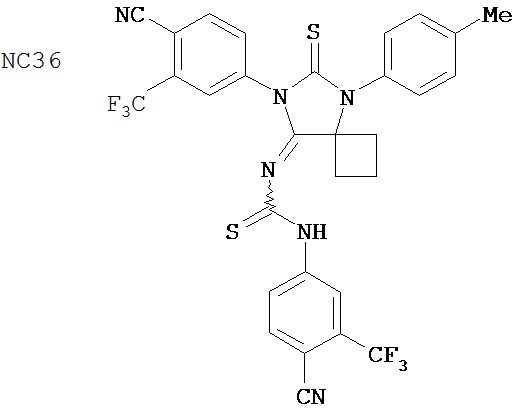
Одним ранее неизвестным свойством суперэкспрессии AR в случае гормонорезистентного рака простаты является ее способность переключать антагонисты в агонисты. Поэтому в качестве антиандрогенов для этого заболевания признавали только те соединения, которые обладали минимальными агонистическими активностями или у которых эти активности полностью отсутствовали. Чтобы определить агонистические активности различных соединений, мы исследовали их способности стимулировать AR с помощью AR-чувствительного репортера в качестве критерия в системе LN-AR в отсутствие R1881. В таблице 2 представлены агонистические активности различных соединений. В соответствии с предыдущими результатами бикалутамид активировал AR в случае гормонорезистентного рака простаты. Производные диарилтиогидантоина, такие как примеры 7-3b (NC7), 33 (NC34), 34 (NC35)и 35 (NC36), не обладают агонистической активностью. В противоположность этому, RU59063 и другие антиандрогенные соединения, перечисленные в качестве примеров в патенте US Patent Number 5,705,654, например, 30-2, 30-3, 31-2, 31-3 и 24-3 (NC24-NC28), сильно активировали AR в случае гормонорезистентного рака простаты.
Чтобы исследовать специфичность ингибиторов AR, отобранные соединения тестировали в клетках LNCaP с суперэкспрессией глюкокортикоидного рецептора (GR), ближайшего представителя AR в семействе ядерных рецепторов. Эти клетки также несут в себе GR-чувствительный репортер, активность репортера индуцировали дексаметазоном, агонистом GR, а его индукцию блокировали RU486, ингибитором GR.
Пример 7-3b (NC7) (4-(8-оксо-6-тиоксо-5-(4-метилфенил)-5,7-диазаспиро[3.4]окт-7-ил)-2-трифторметилбензонитрил) не оказывал влияния на GR в этой системе.
Влияние соединений на AR, определенное с помощью измерения уровня секреции простат-специфического антигена (ПСА)
Хорошо установлено, что уровень ПСА является индикатором активностей AR при раке простаты. Чтобы исследовать, влияют ли соединения на функцию AR в физиологическом окружении, мы определили уровень секреции эндогенного ПСА, индуцируемый R1881, в клетках LNCaP, суперэкспрессирующих AR (LNCaP-AR, также сокращаемых как LN-AR). Клетки LNCaP-AR представляют собой линию клеток метастатически пораженных лимфатических узлов при карциноме простаты, которые были трансдуцированы плазмидой, обеспечивающей экспрессию рецепторов андрогенов. Клетки LNCaP-AR поддерживали в среде Искова, содержащей 10%-ную FBS. Два дня до обработки лекарственными средствами клетки выращивали в среде Искова, содержащей 10%-ную CS-FBS, чтобы удалить андрогены. Клетки разделяли и выращивали в среде Искова, содержащей 10%-ную CS-FBS с подходящими концентрациями R1881 и тестируемыми соединениями. После четырех дней инкубации измеряли уровень секреции ПСА с помощью наборов PSA ELISA (American Qualex, San Clemente, CA).
Уровень ПСА, секретируемого клетками LNCaP-AR, сильно индуцировался 25 пМ R1381. В противоположность этому, ПСА не индуцировался в исходных клетках LNCaP, пока концентрация R1881 не достигала 100 пМ. Это согласуется с нашим предыдущим сообщением о том, что AR в случае гормонорезистентного рака простаты гиперчувствителен к андрогенам. Чтобы определить величины IC50 для различных соединений при ингибировании экспрессии ПСА, проводили дозозависимое ингибирование активности AR, эти результаты приведены в таблице 1. Величины IC50 при действии отобранных соединений на экспрессию ПСА строго соответствуют значениям, измеренным с помощью репортерного анализа, это подтверждает, что производные диарилгидантоина являются мощными ингибиторами AR в случае гормонорезистентного рака простаты.
Мы также исследовали агонистические активности отобранных соединений в отношении AR в случае гормонорезистентного рака простаты, используя в качестве маркера-имитатора секретируемый ПСА. Чтобы сделать это, истощенные по андрогенам клетки LNCaP, суперэкспрессирующие AR, инкубировали с увеличивающими концентрациями соединений, синтез которых был описан выше, в отсутствие R1881, и спустя 4 дня измеряли ПСА, секретируемый в культуральную среду.
В таблице 3 приведены агонистические активности отобранных соединений. В соответствии с результатами, полученными в репортерном анализе, производные диарилтиогидантоина, такие как примеры 7-3b (NC7), 33 (NC34), 34 (NC35)и 35 (NC36), не обладают агонистическими активностями. В противоположность этому, RU59063 и другие антиандрогенные соединения, перечисленные в качестве примеров в патенте US patent no. 5,705,654, такие как 30-2 (NC24), 30-3 (NC25) и 31-2 (NC26), стимулировали экспрессию ПСА в случае гормонорезистентного рака простаты.
Эффект соединений, нацеленных на AR, на митохондриальную активность в анализе MTS
Клетки LNCaP-AR поддерживали в среде Искова, содержащей 10%-ную FBS. Исследовали влияние соединений на рост клеток гормонорезистентного рака простаты. Использовали суперэкспрессирующие клетки LNCaP, так как они ведут себя как клетки гормонорезистентного рака простаты in vitro и in vivo (1). Мы исследовали митохондриальную активность в анализе MTS, являющуюся суррогатным индикатором роста. Клетки LNCaP с суперэкспрессированным AR (LN-AR) поддерживали в среде Искова, содержащей 10%-ную FBS. Два дня до обработки лекарственными средствами клетки выращивали в среде Искова, содержащей 10%-ную CS-FBS, чтобы удалить андрогены. Клетки разделяли и выращивали в среде Искова, содержащей 10%-ную CS-FBS с подходящими концентрациями R1881 и увеличивающимися концентрациями тестируемых соединений. После четырех дней инкубации с помощью анализа MTS (Promega, Madison, WI) следили за ростом клеток.
В соответствии с результатами репортерного анализа и анализа ПСА, рост AR-суперэкспрессирующих клеток LNCaP стимулировался 25 мкМ R1881, но исходные клетки не стимулировались до тех пор, пока концентрация R1881 не достигала 100 мкМ. Фиг. 2 показывает ингибирующий эффект отобранных соединений на рост гормонорезистентного рака простаты в присутствии 100 пМ R1881. Применяемое в настоящее время клиническое средство бикалутамид не подавлял гормонорезистентный рак простаты. В противоположность этому, пример 5-3b (NC2) (4-[3-(4-метилфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил]-2-трифторметил-бензонитрил) и пример 7-3b (NC7) (4-(8-оксо-6-тиоксо-5-(4-метилфенил)-5,7-диазаспиро[3.4]окт-7-ил)-2-трифторметилбензонитрил) подавлял гормонорезистентный рак простаты с высокой эффективностью.
Мы исследовали, происходит ли подавление роста в анализе MTS, если мишенью действия является AR; пример 5-3b (NC2) (4-[3-(4-метилфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил]-2-трифторметилбензонитрил) и пример 7-3b (NC7) (4-(8-оксо-6-тиоксо-5-(4-метилфенил)-5,7-диазаспиро[3.4]окт-7-ил)-2-трифторметилбензонитрил) были протестированы в клетках DU-145, клеточной линии рака, в которых отсутствует экспрессия AR. Эти соединения не обладали подавляющим действием на клетках DU-145. Соединения не подавляли клетки, которые не были AR-экспрессирующими клетками рака простаты, так как они не оказывали влияния на рост клеток MCF7 и SkBr3, двух широко применяемых клеток рака молочной железы, или ЗТЗ, клеточной линии нормальных мышиных фибробластов.
Примеры биологической активности производных диарилтиогидантоина in vitro показаны на фиг. 3 и 4. Например, если исходить из относительной люциферазной активности, фиг. 3 показывает, что в концентрации 500 нМ соединения классифицируются в порядке от наиболее активных соединений к наименее активным следующим образом: NC67>NC68>NC66>NC69>NC77=NC53>бикалутамид. Например, если исходить из относительного уровня ПСА, было обнаружено, что в концентрации 500 нМ соединения классифицируются в порядке от наиболее активных к наименее активным следующим образом: NC50>NC48>NC7>NC43>NC44>NC49>NC50>NC45>бикалутамид. Например, если исходить из относительных единиц MTS, фиг. 4 показывает, что в концентрации 500 нМ соединения классифицируются в порядке от наиболее активных к наименее активным следующим образом: NC70>NC7>NC122>NC53>бикалутамид.
Подавляющий эффект на ксенотрансплантатные опухоли гормонорезистентного и гормоночувствительного рака простаты
Соединения изобретения применяли, чтобы исследовать, обладают ли производные диарилгидантоина эффектами на гормонорезистентный рак простаты in vivo. Вначале мы исследовали такое соединение на ксенотрансплантатных опухолях, сформированных из AR-суперэкспрессирующих клеток LNCaP. Клетки, заключенные в матригель (Collaborative Biomedical), подкожно инъецировали в боковые поверхности живота кастрированных мышей-самцов SCID. Размер опухоли в трех измерениях определяли каждую неделю с помощью штангенциркуля. После того, как ксенотрансплантатные опухоли были сформированы (размер опухоли, по меньшей мере, 40 мм3), мышей с опухолями распределяли случайным образом и один раз в день перорально вводили различные дозы соединений. Подавляющий эффект на рост AR-суперэкспрессирующей ксенотрансплантатной модели LNCaP изучали следующим образом. Мышей со сформированными ксенотрансплантатными опухолями LN-AR распределяли случайным образом и один раз в день перорально вводили указанные соединения. С помощью штангенциркуля измеряли размер опухолей.
В соответствии с клиническими наблюдениями применяемое в настоящее время клиническое средство бикалутамид не подавлял рост гормонорезистентного рака простаты (как и в случае среды-растворителя). В противоположность этому, соединения в соответствии с изобретением подавляют рост этих опухолей и подавление было дозозависимым. Кроме того, соединения подавляют экспрессию ПСА, клинического маркера гормонорезистентного рака простаты.
Соединения изобретения также тестировали в другой ксенотрансплантатной модели гормонорезистентного рака простаты, в гормонорезистентной линии LAPC4. Эту модель формируют путем перевивания гормоночувствительного рака простаты кастрированным мышам, которое имитирует клиническое прогрессирование рака простаты (2). Также как и в результатах с использованием AR-суперэкспрессирующей ксенотрансплантатной модели LNCaP, применяемое в настоящее время клиническое средство бикалутамид не подавлял рост и экспрессию ПСА в гормонорезистентной ксенотрансплантатной модели LAPC4 (как и в случае среды-растворителя). В противоположность этому, соединения изобретения подавляли рост и экспрессию ПСА в этих опухолях.
Фиг. 6 демонстрирует результаты эксперимента, в котором клетки из гормоночувствительной модели LNCaP были трансплантированы в мышей (мышам инъецировали 106 клеток LNCaP). Первую группу мышей обрабатывали соединением NC53, вторую группу мышей обрабатывали касодексом и третью группу мышей обрабатывали средой-растворителем. Каждая группа включала 6 мышей. Мышам вводили по 10 мг вещества/кгмассы тела в день. Фиг. 6 представляет результаты в виде графика объема опухолей как функции времени. Контрольные мыши, обработанные средой-растворителем, демонстрировали наиболее быстрое увеличение объема опухолей. Мыши, обработанные касодексом, и мыши, обработанные соединением NC53, демонстрировали сходные скорости роста опухолей, более медленные по сравнению с мышами, обработанными средой-растворителем.
Эффект подавления роста клеток гормоночувствительного рака простаты
Чтобы определить, подавляют ли также производные диарилтиогидантоина клетки гормоночувствительного рака простаты, мы тестировали действие некоторых отобранных соединений на рост клеток LNCaP, измеряя с помощью MTS митохондриальные активности. Истощенные по андрогенам клетки LNCaP обрабатывали увеличивающимися концентрациями DMSO в качестве среды-растворителя или тестируемых соединений в присутствии 1 пМ R1881. Через 4 дня инкубации рост клеток измеряли с помощью анализа MTS. Соединения изобретения подавляли гормоночувствительный рак простаты с большей эффективностью, чем бикалутамид.
Биологический анализ in vivo
Все эксперименты на животных проводили в соответствии с рекомендациями Комитета по исследованиям на животных в Университете Калифорнии в Лос-Анжелесе. Животных покупали в Taconic и содержали в клетке с ламинарным потоком воздуха с определенным составом флоры. Векторные клетки LNCaP-AR и LNCaP поддерживали в среде RPMI, дополненной 10%-ной FBS. Интактным или кастрированным мьшам SCID подкожно в бок инъецировали 106 клеток в 100 мкл смеси матригель: среда RPMI (1:1). Размер опухолей измеряли в трех направлениях (длина, ширина, глубина) каждую неделю с помощью штангенциркуля. Мышей разделяли случайным образом на группы для обработки, когда размер опухоли достигал примерно 100 мм3. Лекарственные средства давали перорально каждый день в количестве 10 мг/кг и 50 мг/кг. Чтобы получить фармакодинамическое считывание показаний, животных снимали с помощью оптической CCD-камеры, спустя 3 часа после приема последней дозы. ROI перемещали над опухолью для измерения люциферазной активности в фотонах/секунду. Правые панели были изображением измерений ROIs. Результаты приведены на фиг. 7 и 8. В пределах 18 дней соединение NC53 было эффективным для предупреждения роста опухоли и даже для сокращения размера опухоли и было отчетливо более эффективным, чем бикалутамид.
Фармакокинетику бикалутамида, 4-[7-(4-циано-3-трифторметилфенил)-8-оксо-6-тиоксо-5,7-диаза-спиро[3.4]окт-5-ил]-толуола [NC7], N-метил-4-{4-[7-(4-циано-3-трифторметилфенил)-8-оксо-6-тиоксо-5,7-диаза-спиро[3.4]окт-5-ил]фенил}бутанамида [NC48] и N-метил-4-[7-(4-циано-3-трифторметилфенил)-8-оксо-6-тиоксо-5,7-диаза-спиро[3.4]окт-5-ил]-2-фторбензамида (52d) [NC53] оценивали in vivo, используя 8-недельных мышей FVB, которых получали из Charles River Laboratories. Мышей делили на группы по три животных на каждую временную точку. Двух мышей не обрабатывали лекарственным средством и двух других мышей обрабатывали средой-растворителем. Каждую группу обрабатывали соединением в количестве 10 мг на килограмм массы тела.
Лекарственное средство растворяли в смеси DMSO:PEG400:H2O (среда-растворитель) (1:5:14) и вводили мышам через хвостовую вену. Животных согревали под горячей лампой приблизительно в течение 20 минут перед обработкой, чтобы расширить их хвостовую вену. Каждую мышь помещали в приспособление для фиксирования мыши (Fisher Sci. Cat# 01-288-32A) и инъецировали 200 мкл лекарственного средства в среде-растворителе в расширенную хвостовую вену. После введения лекарственного средства животных подвергали эвтаназии путем ингаляции CO2 в различные промежутки времени: 5 минут, 30 минут, 2 часа, 6 часов, 16 часов. Сразу же после обработки CO2 у животных спускали кровь с помощью прокола сердца (BD-шприц на 1 мл + игла 27G 5/8). Для пероральной дозировки лекарственное средство растворяли в смеси ОМ80:карбоксиметилцеллюлоза:твин-80:Н20 (50:10:1:989) перед пероральным введением с помощью шприца-дозатора.
Анализировали образцы сыворотки крови, чтобы определить концентрацию лекарственных средств с помощью ВЭЖХ (насос Waters 600. контроллер Waters 600 и детектор Waters 2487), которая была оснащена колонкой Alltima С 18 (3μ, 150 мм (4,6 мм). Соединения NC7, NC48 и NC53 детектировали при длине волны 254 нм, а бикалутамид детектировали при длине волны 270 нм.
Образцы для ВЭЖХ-анализа получали в соответствии с процедурой, приведенной ниже:
- Клетки крови отделяли от сыворотки с помощью центрифугирования.
- К 400 мкл сыворотки добавляли 80 мкл 10 мкМ раствора внутреннего стандарта и 520 мкл ацетонитрила. Происходило образование осадка.
- Смесь перемешивали на Вортексе в течение 3 минут и затем помещали в ультразвуковой дезинтегратор на 30 минут.
- Частицы твердого вещества отфильтровывали или отделяли с помощью центрифугирования.
- Фильтрат высушивали в токе аргона досуха. Перед анализом с помощью ВЭЖХ образец растворяли в 80 мкл ацетонитрила, чтобы определить концентрацию лекарственного средства.
- Чтобы повысить точность, использовали стандартную кривую для лекарственного средства.
Концентрация соединения NC53 в плазме крови как функция от времени при внутривенном и пероральном введении показана на фиг. 9. Стационарная концентрация (Css) бикалутамида, NC48 и NC53 показана в таблице 4. Концентрация в стационарном состоянии соединения NC53 является в основном такой же подходящей, как и в случае бикалутамида, и значительно лучше, чем для соединения NC48.
Активность рецептора андрогенов может включать в себя несколько аспектов стимуляции и ингибирования свойств рецептора андрогенов, включающих без ограничения следующие свойства: ингибирующую концентрацию (IC50) в AR-чувствительной репортерной системе или в системе секреции простат-специфического антигена; кратность индукции, связанной с увеличивающимися концентрациями в AR-чувствительной репортерной системе или в системе секреции простат-специфического антигена; ассоциированный рост опухоли у животного; аффинность связывания рецептора андрогенов с соединением; вовлечение рецептора андрогенов во взаимодействие с энхансером простат-специфического антигена и промотором простат-специфического антигена; перенос рецептора андрогенов в ядро и дестабилизацию андроген-рецепторного белка.
Анализы in vitro
Фиг. 10 представляет величины относительного сродства связывания соединений с лиганд-связывающими доменами рецептора андрогенов крысы (крысиного AR), определяемые с помощью наборов для анализа конкурентного связывания (Invitrogen). Использовали показания поляризации флуоресценции. Каждую дозу гормона давали в трех повторах и относительную ошибку измерения определяли, рассчитывая из средней величины стандартную ошибку трех измерений. В исследовании контролировали минимальную конкуренцию (только среда-растворитель), отсутствие рецептора, отсутствие флуоресцентного лиганда и максимальную конкуренцию (10-5 М R1881, прогестерон, Е2 или дексаметазон). Подгонку кривых осуществляли с помощью модели с единственным местом конкурентного связывания (использовали пакет программ для статистического анализа Prism). R1881 обладал самой низкой равновесной константой диссоциации. Ki=4 нМ (и, таким образом, рецептор андрогенов крысы имел самое высокое сродство к R1881 из четырех протестированных соединений). RU59063 обладал равновесной константой диссоциации Ki=20 нМ и NC53 обладал равновесной константой диссоциации Ki=0,8 мкМ. Касодекс обладал равновесной константой диссоциации Ki=0,4 мкМ (и, таким образом, рецептор андрогенов крысы имел самое низкое сродство к касодексу из четырех протестированных соединений). NC53 и касодекс обладали сходными равновесными константами диссоциации и, таким образом, рецептор андрогенов крысы имел сходное сродство к этим соединениям.
NC53 предотвращал взаимодействие рецептора андрогенов (AR) и взаимодействие РНК-полимеразы II (Pol II) с энхансером ПСА и промотором ПСА. Фиг. 11 представляет результаты исследования. В качестве материалов использовали хроматин IP (Chromatin IP) с AR (Upstate, cat# 06-680) и Pol II (Covance. cat# MMS-126R). Клетки LNCaP (ATCC) высевали в планшете в полной сыворотке. В день эксперимента планшет однократно промывали 1 × PBS и в течение 3 дней добавляли 5%-ную CSS. В первой серии экспериментов добавляли 10 MKMNC53, во второй серии экспериментов добавляли 10 мкМ бикалутамид (С) и в третьей серии экспериментов добавляли 1 нМ R1881 (+). Каждое из этих соединений добавляли в течение 6 часов. В четвертой серии экспериментов (контроль) дополнительное соединение не добавляли (-). Временную точку 6 часов проводили в 28 циклов. Использовали наборы СЫР фирмы Upstate (cat# 17-295). Праймеры для энхансера и промотора получали от Louie (PNAS 2003 Vol.100, pp.2226-2230) и Shang (Molecular Cell 2002 vol. 9, pp.601-610), соответственно. Темное изображение в экспериментах, в которых добавляли NC53, указывает на то, что NC53 мешает рецептору андрогенов и мешает РНК-полимеразе II образовывать транскрипционный комплекс с геном простат-специфического антигена (ПСА). В противоположность этому, светлое изображение в экспериментах, в которых добавляли бикалутамид (касодекс, С), указывает на то, что в присутствии бикалутамида рецептор андрогенов и РНК-полимераза II все еще взаимодействовали с элементами ПСА для транскрипции мРНК ПСА.
NC53 ингибировал перенос рецептора андрогенов в ядро в клетках LNCaP. Фиг. 12 и 13 представляют результаты исследования. Клетки LNCaP высевали в планшет в 5%-ной CSS. В первой серии экспериментов клетки обрабатывали 10 мкМ NC53, во второй серии экспериментов клетки обрабатывали 10 мкМ бикалутамидом (С) и в третьей серии экспериментов клетки обрабатывали 1 нМ R1881 (+). Четвертая серия экспериментов служила контролем (-). ТОРО I (Santa Cruz, cat# sc-32736) использовали как контроль для ядерной фракции и GAPDH (Santa Cruz, cat# sc-20357) использовали как контроль для цитоплазматической фракции. Клетки LNCaP получали для субклеточного фракционирования и окрашивали с помощью FITC-меченых (Santa Cruz) антител к рецептору андрогенов (AR) (Santa Cruz, cat# sc-815). Для субклеточных фракций получали изображения, как показано на фиг. 12. Темное изображение в ядерной фракции образца, обработанного бикалутамидом (касодекс, С), указывает на то, что бикалутамид индуцировал перенос рецептора андрогенов в ядро. Светлое изображение образца, обработанного NC53, указывает на то, что NC53 предотвращал перенос в ядро. В анализе AR-FITC на предметные стекла монтировали покровные стекла, используя среду, содержащую DAPI, и спустя 24 часа получали изображение клеток с помощью флуоресцентного микроскопа Nikon при Х60 с фильтрами для DAPI и FITC. В анализе AR-FITC ядра клеток, обработанных R1881 и бикалутамидом, были отчетливо зелеными, как показано на фиг. 13, указывающей на то, что произошел перенос рецептора андрогенов в ядро. В противоположность этому, ядра в клетках, обработанных DMSO и NC53, были менее зелеными.
NC53 дестабилизировал белки рецептора андрогенов в клетках LNCaP. Фиг. 14 показывает результаты исследования. Исследование проводили путем высевания 105 клеток LNCaP (fgc) в планшет в 5%-ной CSS в течение 3 дней. 100 пМ R1881 добавляли к первой группе клеток (+), 10 мкМ бикалутамид добавляли ко второй группе клеток (В), 10 мкМ NC53 добавляли к третьей группе клеток (NC53), 100 пМ R1881 и 10 мкМ бикалутамид добавляли к четвертой группе клеток (В+) и 100 пМ R1881 и 10 мкМ NC53 добавляли к пятой группе клеток (R1881+NC53). К шестой группе клеток не добавляли ни R1881, ни бикалутамид, ни NC53 (-). Клетки оставляли с добавленным бикалутамидом, NC53 и/или R1881 на 24 часа (или в случае группы (-) без добавления этих веществ на 24 часа). На фиг. 14 темное изображение для группы, к которой добавляли бикалутамид (В), и для группы, к которой добавляли бикалутамид и R1881 (В+), указывает на то, что если добавляли эти комбинации соединений, то белок рецептора андрогенов поддерживался на определенном уровне. В противоположность этому, светлое изображение для группы, к которой добавляли NC53 (NC53), и для группы, к которой добавляли NC53 и R1881 (R1881+NC53), указывает на то, что добавление NC53 приводит к деградации белков рецептора андрогенов, независимо от того, присутствовал или нет R1881.
Классифицирование соединений по группам
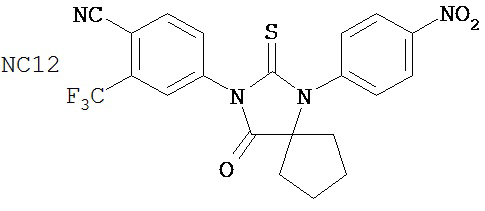
Таблицы 5-10 представляют диарилгидантоиновые соединения, распределенные по группам 1-6. Таблица 11 представляет диарилгидантоиновые соединения, которые не были помещены в группу. Расположение соединений по группам было основано на имеющихся данных в сочетании с аналитическими рассуждениями. Принятые во внимание данные включали анализы in vitro (AR-чувствительная репортерная система в клеточной линии LNCaP, измерение уровня ПСА, митохондриальный анализ MTS) и эксперименты in vivo (размер опухоли, измеряемый непосредственно, или с помощью эмиссии, индуцированной репортерным геном люциферазы, фармакокинетические анализы, основанные на определении уровня веществ в плазме крови). Не каждое соединение тестировали в каждом анализе. Приведены не все полученные данные. При ранжировании соединений относительно друг друга решение принимали на основе их применимости при лечении рака простаты, в частности, так поступали при ранжировании двух соединений, для которых не проводили одни и те же эксперименты. Характеристики, принимаемые во внимание при ранжировании, включали антагонистическую активность AR, потерю антагонистической активности AR в гормонорезистентных клетках, предупреждение роста опухоли, уменьшение размера опухоли и фармакокинетическое поведение с увеличенным временем пребывания в крови, которое является предпочтительным.
Группа 1
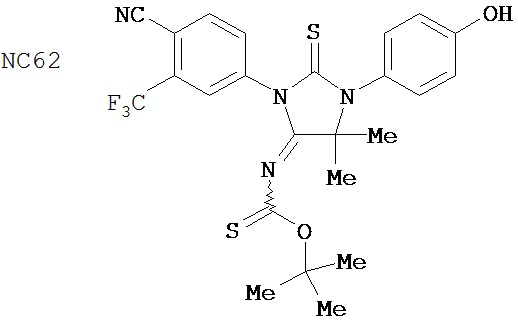
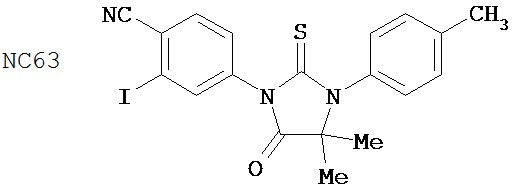
В основном соединения группы 1 представляют собой диарилтиогидантоины с двухзамещенным левым арильным кольцом, которые имеют два заместителя у правого атома углерода в гидантоиновом кольце и содержат или атом кислорода, или N-замещенную группу у левого атома углерода в гидантоиновом кольце. Ожидается, что амидный заместитель гидролизуется в водных растворах до кислорода, как происходит в биологических системах, in vitro и in vivo. NC63 обладает хорошей активностью, если слева арильное кольцо замещено на иод вместо CF3.
Был сделан вывод, что соединения группы 1 (смотри таблицу 5) намного лучше, чем бикалутамид при лечении рака простаты. Однако было обнаружено, что NC7 и NC48 метаболизируются быстрее, то есть обладают более коротким временем пребывания в крови. NC53 обладает желаемой фармакокинетикой.
Фиг. 16 показывает, что при обработке бикалутамидом, уровень ПСА в клетках LNCaP оставался тем же самым или повышенным по сравнению с уровнем при обработке средой-растворителем, тогда как при обработке соединением NC53, уровень ПСА снижался. Фиг. 17 демонстрирует, что при обработке средой-растворителем опухоли продолжали увеличиваться в размерах. В противоположность этому, при обработке соединением NC53 в дозировке 1 мг на кг массы тела в день, скорость увеличения опухоли снижалась, и оказалось, что размер опухоли стабилизировался после примерно 17 дней. При обработке соединением NC53 в дозировке 10 мг на кг массы тела в день, со временем размер опухоли уменьшался. Фиг. 18 демонстрирует, что при обработке соединением NC53 в дозировке 10 мг на кг массы тела в день, фотонная эмиссия, связанная с люциферазной активностью, уменьшалась. Фиг. 19 демонстрирует, что обработка соединением NC53 в этой дозировке приводила к уменьшению или стабилизации размера опухоли и уменьшению фотонной эмиссии, связанной с люциферазной активностью.
Фиг. 20 показывает, что при обработке соединениями NC53, NC54, NC55, NC56 и NC57 в дозировках 100, 200, 500 и 1000 нМ, уровень ПСА в клетках LN-AR снижался. Кроме того, более высокая дозировка снижала уровень ПСА сильнее. Фиг. 22 представляет результаты взвешивания урогенитального тракта и скорость фотонной эмиссии, связанную с люциферазной активностью, вначале и через 14 дней после обработки бикалутамидом или соединением NC53 интактных или кастрированных мышей. Масса и скорость фотонной эмиссии увеличивались как у интактных, так и у кастрированных мышей. Обработка кастрированных мышей соединением NC53 приводила к снижению массы и фотонной эмиссии по сравнению с необработанными кастрированными мышами, также как и в случае обработки бикалутамидом.
Таким образом, соединения группы 1 являются особенно полезными для применения их в качестве антагонистов AR и в качестве терапевтических средств при лечении гормонорезистентного рака простаты. Они могут быть полезны для лечения других ассоциированных с AR заболеваний или состояний, таких как доброкачественная гиперплазия простаты, потеря волос и акне. Эти и родственные соединения также могут быть полезными в качестве модуляторов других ядерных рецепторов, таких как глюкокортикоидный рецептор, рецептор эстрогенов и рецептор активатора пролиферации пероксисом, и в качестве терапевтических средств при заболеваниях, в которых играют роль ядерные рецепторы, таких как рак молочной железы, рак яичников, диабет, заболевания сердца и заболевания, связанные с обменом веществ. Они могут быть полезными в анализах, например, в качестве стандартов, или в качестве промежуточных продуктов или пролекарственных средств.
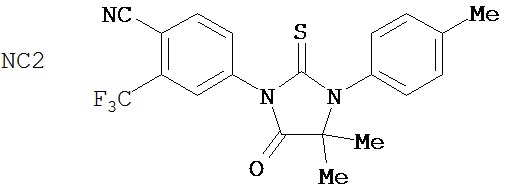
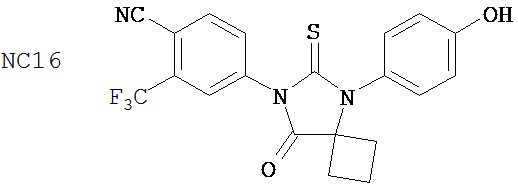
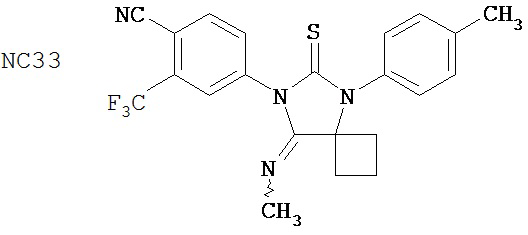
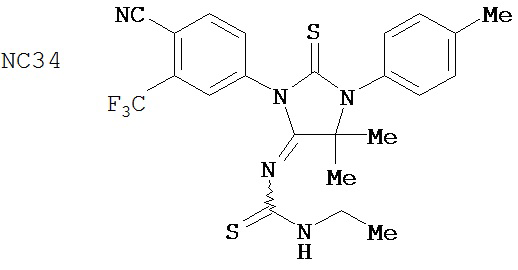
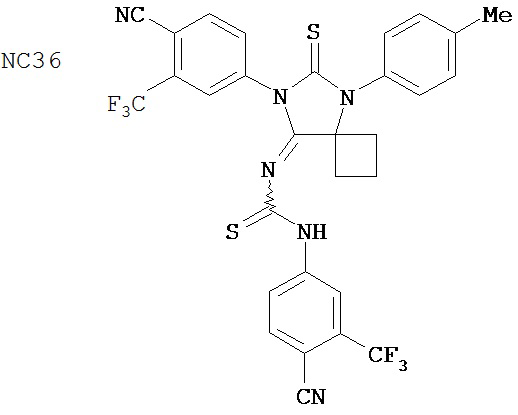
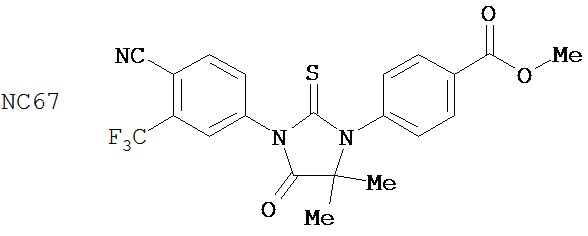
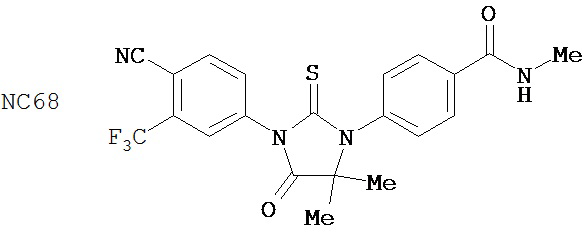
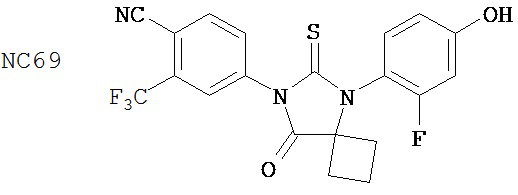
Группа 2
Соединения группы 2 (смотри таблицу 6) были значительно лучше бикалутамида при лечении рака простаты, хотя были указания на то, что соединение NC12 могло действовать в качестве агониста. Фиг. 3 иллюстрирует то, что соединения NC66, NC67, NC68, NC53 и NC69 из группы 1 и соединение NC77 из группы 2, дозированные в диапазоне концентраций от 125 нМ до 1000 нМ, действуют, снижая люциферазную активность в клетках LNCaP-AR, тогда как контрольные растворы DMSO и бикалутамида оказывают небольшой эффект или не влияют совсем. Было обнаружено, что в концентрации 1000 нМ соединения NC7 и NC48 из группы 1 вызывали более значительное снижение уровня ПСА в клетках LNCaP-AR, чем соединения NC43, NC44 и NC50 из группы 2. Фиг. 7 представляет изменение объема опухоли во времени и демонстрирует, что при обработке бикалутамидом или средой-растворителем опухоли продолжали расти, тогда как при обработке соединением NC53 из группы 1 опухоли уменьшались в размере. Фиг. 8 демонстрирует, что фотонная эмиссия, связанная с люциферазной активностью, оставалась примерно такой же или увеличивалась при обработке бикалутамидом по сравнению с обработкой средой-растворителем, тогда как фотонная эмиссия снижалась при обработке соединением NC53. Фиг. 23 демонстрирует, что при обработке бикалутамидом было небольшое снижение уровня ПСА или снижение уровня отсутствовало, тогда как при обработке соединениями NC48 и NC53 уровень ПСА снижался. Фиг. 24 демонстрирует, что величины IC50 для NC7, NC48 и NC53 из группы 1 были намного ниже, чем величина IC50 для бикалутамида.
В основном соединения группы 2 структурно похожи на соединения группы 1, но отличаются другими заместителями правого арильного кольца. Соединения группы 2 полезны для применения в качестве антагонистов AR и в качестве терапевтических средств в случае гормонорезистентного рака простаты. Они могут быть полезны при лечении других ассоциированных с AR заболеваний или состояний, таких как доброкачественная гиперплазия простаты, потеря волос и акне. Эти и родственные соединения также могут быть полезными в качестве модуляторов других ядерных рецепторов, таких как рецептор эстрогенов и рецептор активатора пролиферации пероксисом, и в качестве терапевтических средств при заболеваниях, в которых играют роль ядерные рецепторы, таких как рак молочной железы, рак яичников, диабет, заболевания сердца и заболевания, связанные с обменом веществ. Они могут быть полезными в анализах, например, в качестве стандартов, или в качестве промежуточных продуктов или пролекарственных средств.
 (для сравнения)
(для сравнения)
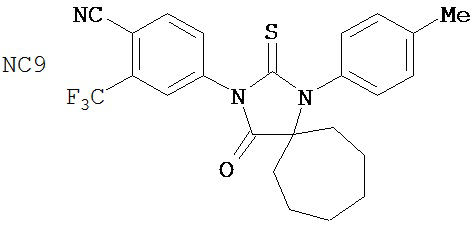
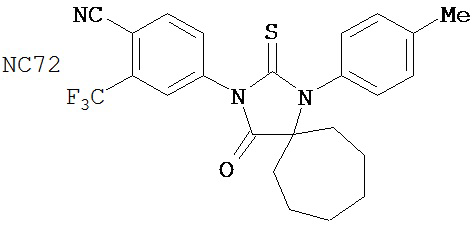
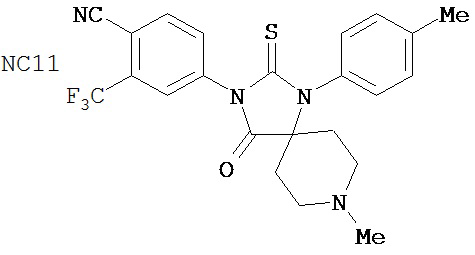
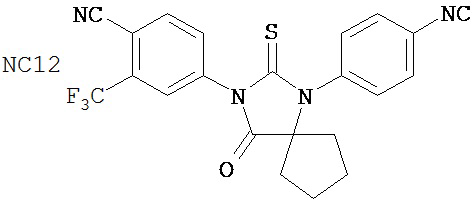
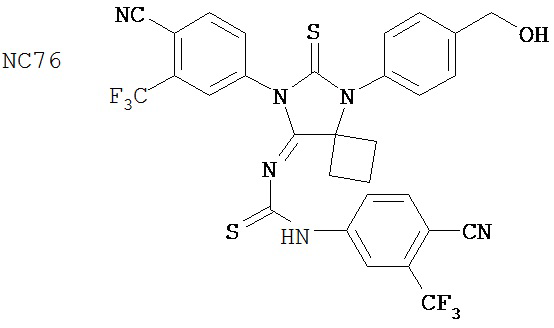
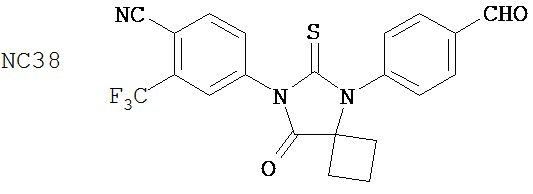
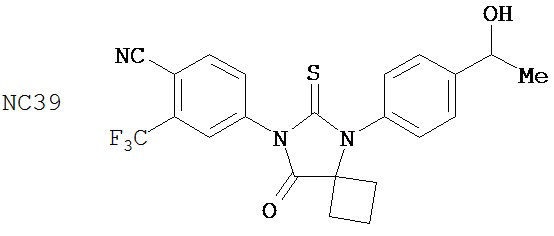
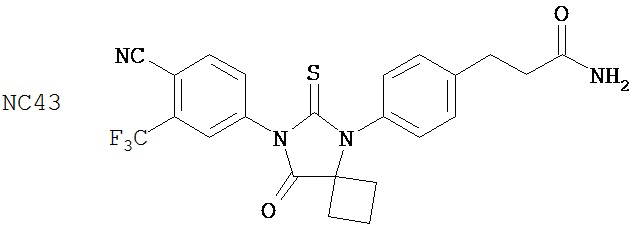
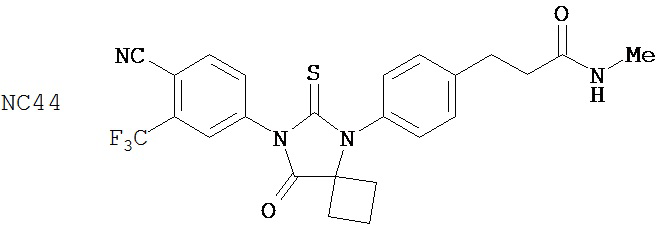
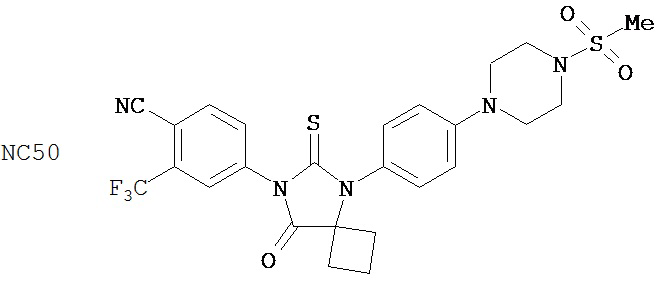
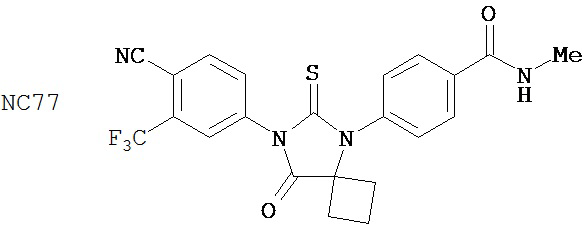
Группа 3
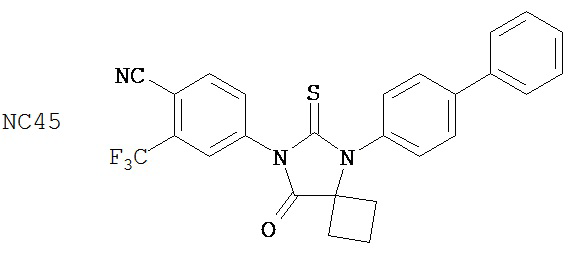
Был сделан вывод, что соединения группы 3 (смотри таблицу 7) были слегка лучше, чем бикалутамид при лечении рака простаты. NC43, NC44 и NC50 (в группе 2) вызывали более значительное снижение уровня ПСА в клетках LNCaP-AR, чем NC45 и NC49 из группы 3. Все эти соединения вызывали более значительное снижение уровня ПСА, чем бикалутамид.
Другие соединения группы 3 (не показаны) не были диарилтиогидантоинами и были сравнимы по активности с известными в этой области техники моноарилгидантоиновыми соединениями NC83, NC79 и NC80.
Таким образом, соединения группы 3 полезны в качестве антагонистов AR и в качестве терапевтических средств в случае гормонорезистентного рака простаты. Они могут быть полезны при лечении других ассоциированных с AR заболеваний или состояний, таких как доброкачественная гиперплазия простаты, потеря волос и акне. Эти и родственные соединения также могут быть полезными в качестве модуляторов других ядерных рецепторов, таких как рецептор эстрогенов и рецептор активатора пролиферации пероксисом, и в качестве терапевтических средств при заболеваниях, в которых играют роль ядерные рецепторы, таких как рак молочной железы, рак яичников, диабет, заболевания сердца и заболевания, связанные с обменом веществ. Они могут быть полезными в анализах, например, в качестве стандартов, или в качестве промежуточных продуктов или пролекарственных средств.
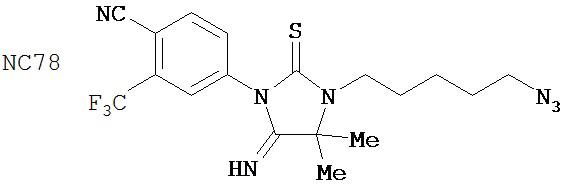 (для сравнения)
(для сравнения)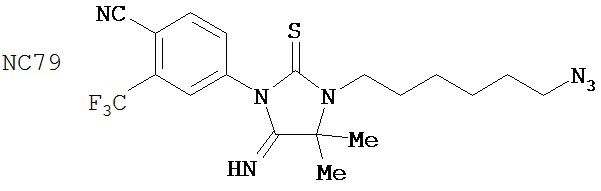 (для сравнения)
(для сравнения)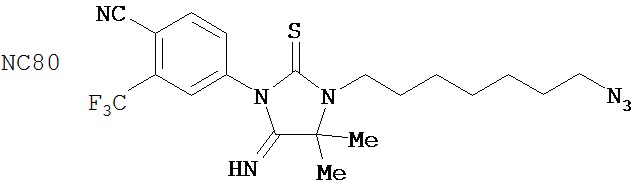 (для сравнения)
(для сравнения)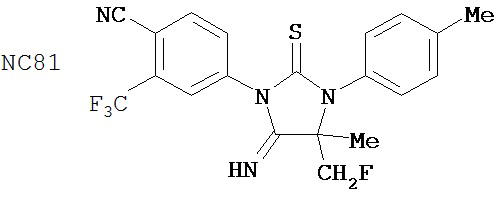
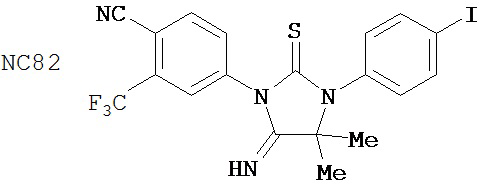
Группа 4
Был сделан вывод, что соединения группы 4 (смотри таблицу 8) не были лучше, чем бикалутамид при лечении рака простаты. NC93 и NC94 из группы 4 и NC7 из группы 1, например, отличались лишь заместителем у нижнего правого атома углерода в гидантоиновом кольце. Заместители в правом арильном кольце могут также влиять на активность.
Некоторые соединения группы 4 (включая те, которые показаны, и другие, которые не показаны) не были диарильными соединениями (отсутствие правого арильного кольца), не были тиогидантоинами, не имели двух заместителей у нижнего правого атома углерода в гидантоиновом кольце и/или имели заместители, отличные от кислорода или амидо, у нижнего левого атома углерода в гидантоиновом кольце. Это предоставляет доказательство неожиданных преимуществ диарилтиогидантоинов, которые имеют два заместителя у нижнего правого атома углерода в гидантоиновом кольце и имеют кислород или амидо у нижнего левого атома углерода в гидантоиновом кольце.
Таким образом, соединения группы 4 могут быть полезны в качестве антагонистов AR и в качестве терапевтических средств в случае гормонорезистентного рака простаты, по меньшей мере, в той степени, в которой они сравнимы с бикалутамидом. Они могут быть полезны при лечении других ассоциированных с AR заболеваний или состояний, таких как доброкачественная гиперплазия простаты, потеря волос и акне. Эти и родственные соединения также могут быть полезными в качестве модуляторов других ядерных рецепторов, таких как рецептор эстрогенов и рецептор активатора пролиферации пероксисом, и в качестве терапевтических средств при заболеваниях, в которых играют роль ядерные рецепторы, таких как рак молочной железы, рак яичников, диабет, заболевания сердца и заболевания, связанные с обменом веществ. Они могут быть полезными в анализах, например, в качестве стандартов, или в качестве промежуточных продуктов или пролекарственных средств.
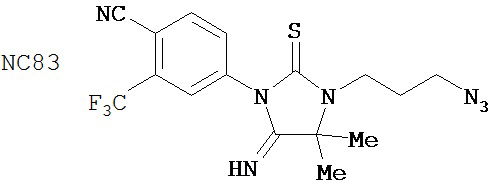 (для сравнения)
(для сравнения)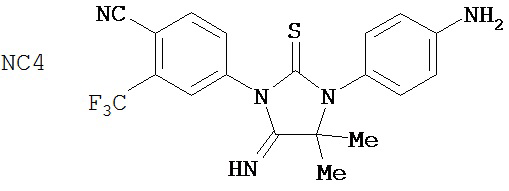
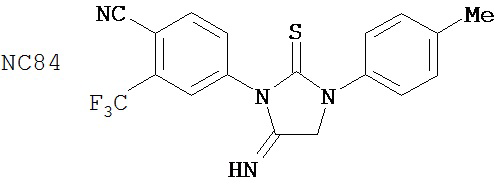
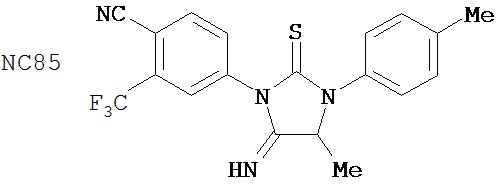
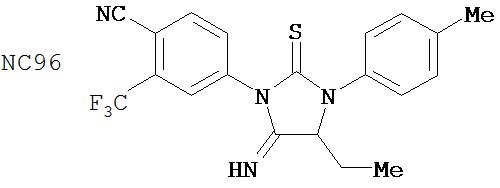
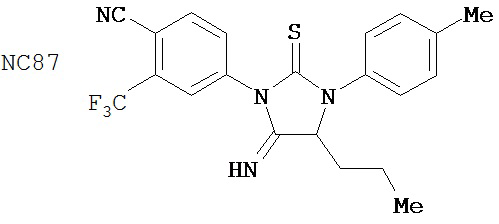
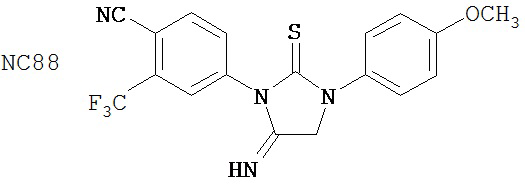
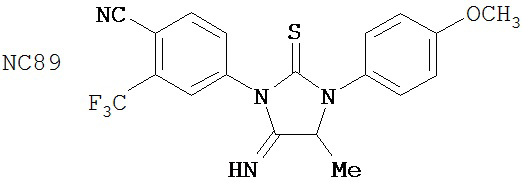
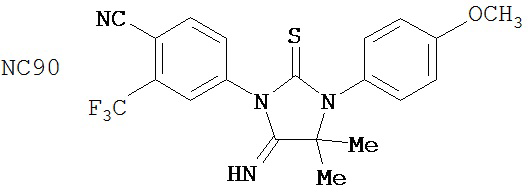
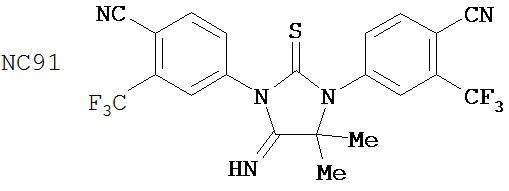
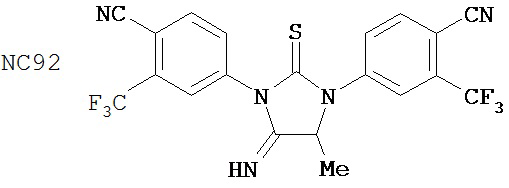
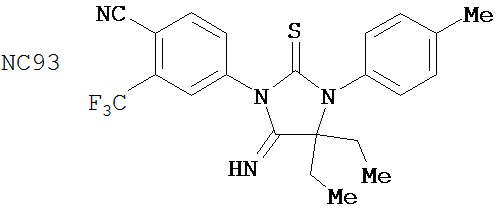
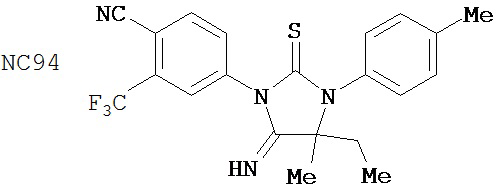
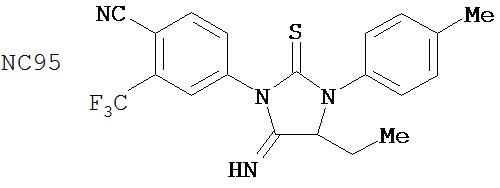
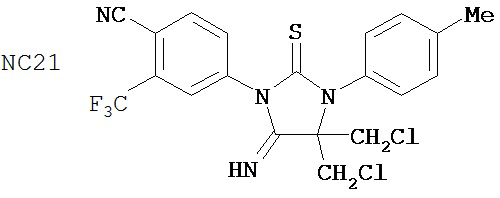
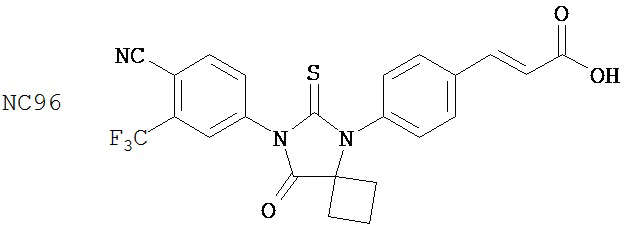
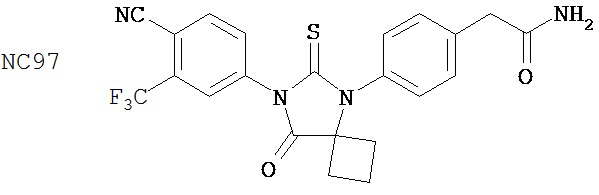
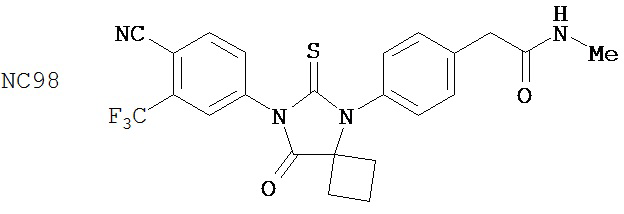
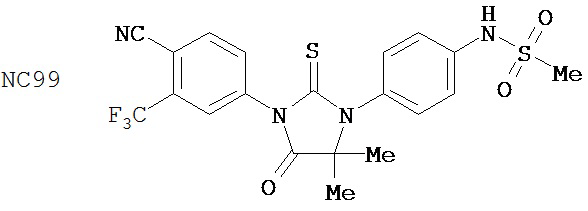
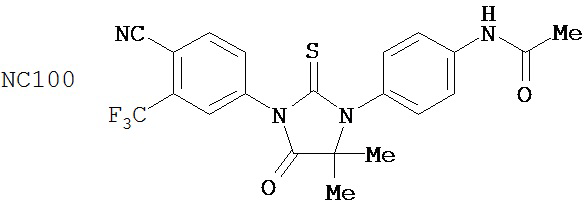
Группа 5
Соединения группы 5 (смотри таблицу 9) были неактивными или почти неактивными и, таким образом, они были хуже, чем бикалутамид при лечении рака простаты. Заместители правого арильного кольца важны для детерминирования активности.
Некоторые соединения группы 5 (некоторые из них показаны, а другие не показаны) не были диарильными соединениями (отсутствие правого арильного кольца), не были тиогидантоинами, не имели двух заместителей у нижнего правого атома углерода в гидантоиновом кольце и/или имели заместители, отличные от кислорода или амидо, у нижнего левого атома углерода в гидантоиновом кольце. Это предоставляет доказательство неожиданных преимуществ диарилтиогидантоинов, которые имеют два заместителя у нижнего правого атома углерода в гидантоиновом кольце и имеют кислород или амидо у нижнего левого атома углерода в гидантоиновом кольце. В частности, концевой заместитель в NC103, NC104 и NC106 (CH2NRxRy, где Rx,y=H или метил) не дает вклад в активность этих соединений.
Соединения группы 5 не желательны при лечении рака простаты или в качестве антагонистов AR, хотя эти и родственные соединения могут быть полезными в качестве модуляторов других ядерных рецепторов, таких как рецептор эстрогенов и рецептор активатора пролиферации пероксисом, и в качестве терапевтических средств при заболеваниях, в которых играют роль ядерные рецепторы, таких как рак молочной железы, рак яичников, диабет, заболевания сердца и заболевания, связанные с обменом веществ. Они могут быть полезными в анализах, например, в качестве стандартов, или в качестве промежуточных продуктов или пролекарственных средств.
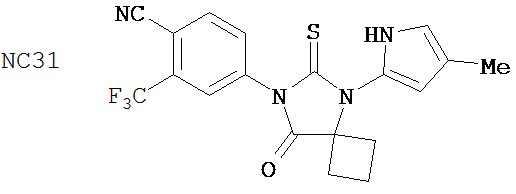
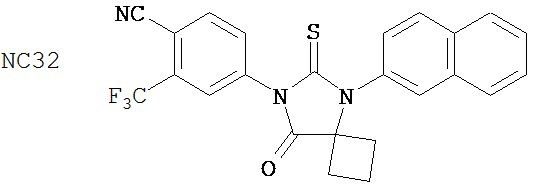
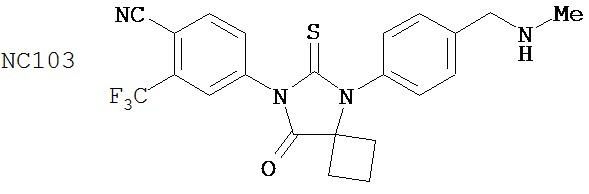
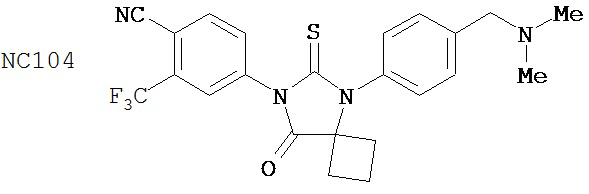
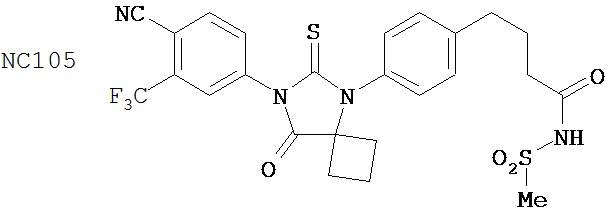
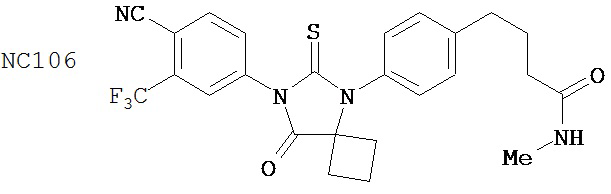
Группа 6
Соединения группы 6 (смотри таблицу 10) были неактивными или почти неактивными и, кроме того, были сильными агонистами и, таким образом, были намного хуже, чем бикалутамид при лечении рака простаты. Классифицированные сравнительные соединения очень плохо соотносятся с соединениями изобретения. В частности, NC107 с хлорным заместителем в левом арильном кольце обладает очень низкой активностью, тогда как NC2 с трифторметаном и NC63 с атомом йода был отнесен к группе 1. Результаты, полученные для соединений группы 6, обеспечивают доказательство неожиданных преимуществ диарилтиогидантоинов, которые имеют два заместителя у нижнего правого атома углерода в гидантоиновом кольце и имеют кислород или амидо у нижнего левого атома углерода в гидантоиновом кольце и имеют определенные заместители в левом арильном кольце.
Соединения группы 6 нежелательны при лечении рака простаты или в качестве антагонистов AR.
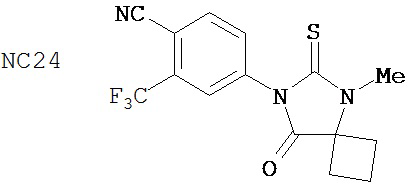 (для сравнения)
(для сравнения) 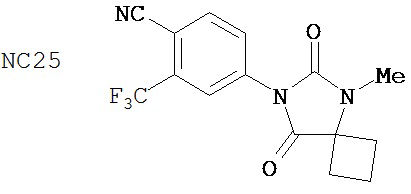 (для сравнения)
(для сравнения)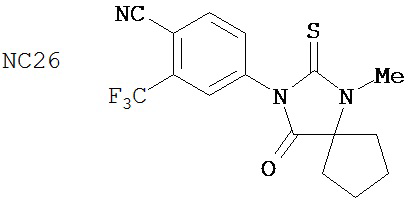
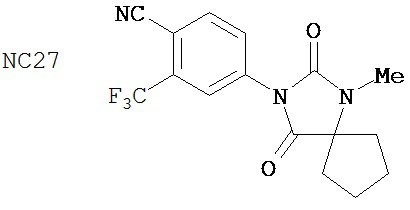 (для сравнения)
(для сравнения)
Неклассифицированные соединения
Для некоторых соединений не было достаточных экспериментальных данных, чтобы их классифицировать. Эти неклассифицированные соединения представлены в таблице 11.
На основании полученных результатов и способов изобретения и вывода о применимости, сделанного на основании обзора многих соединений, включающих те, которые не показаны в этой заявке, можно сделать некоторые наблюдения относительно неклассифицированных соединений. Можно ожидать, что сравнительный пример NC108 относится к группе 3 вместе со сравнительными примерами NC78-NC80. Можно ожидать, что NC113 гидролизуется до NC7 (группа 1) и должен, таким образом, обладать сравнимой активностью. Можно ожидать, что NC114 гидролизуется до NC16 (группа 1) и должен, таким образом, обладать сравнимой активностью. Можно ожидать, что NC115 гидролизуется до NC3 (группа 1), и можно ожидать, что NC120 и NC121 гидролизуются до NC50 (группа 2), и они, таким образом, должны обладать сравнимой активностью.
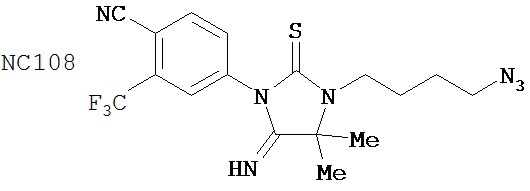 (для сравнения)
(для сравнения)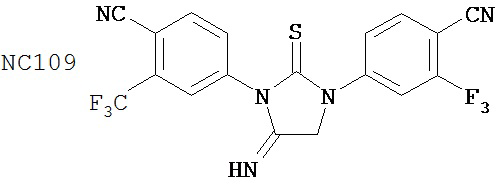
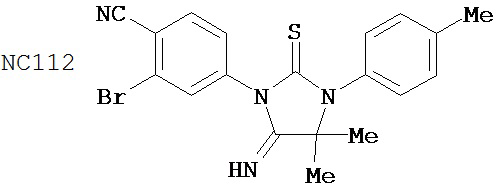
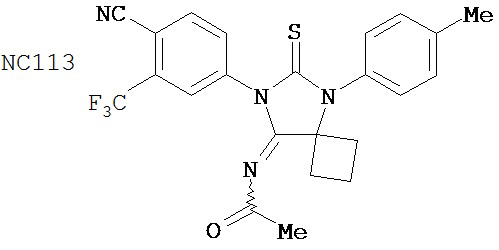
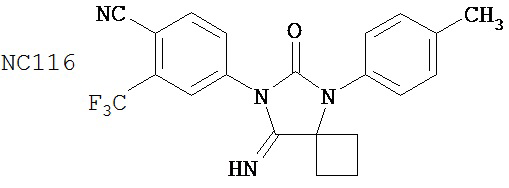
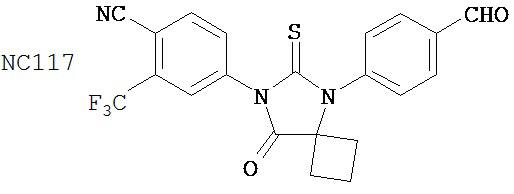
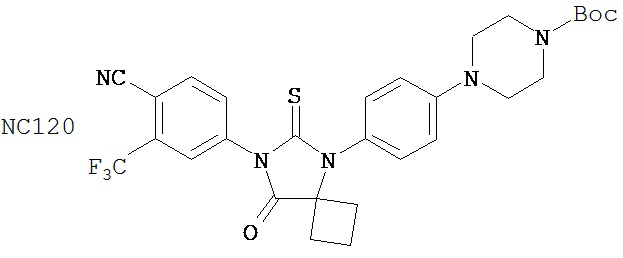
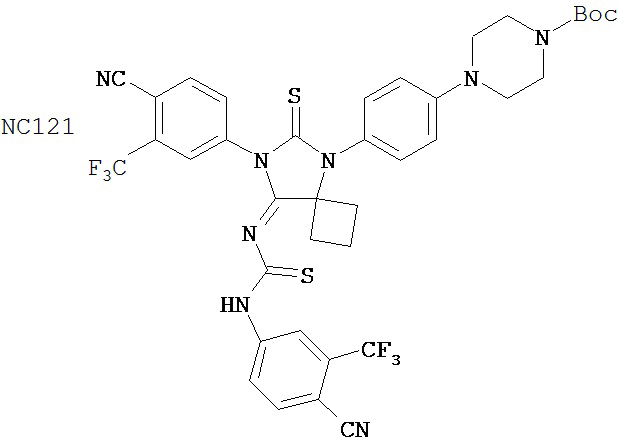
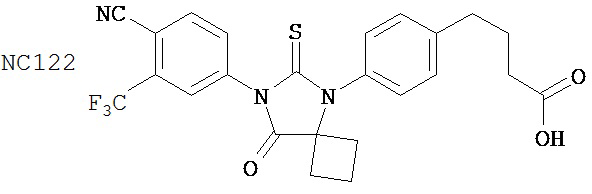
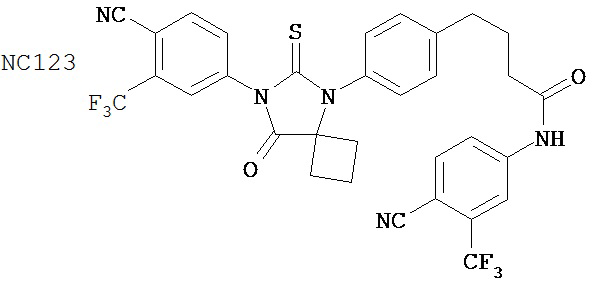
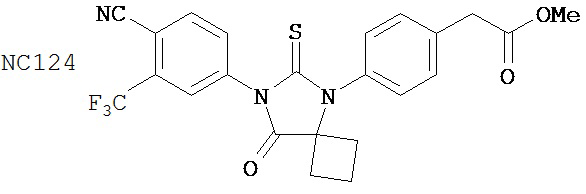
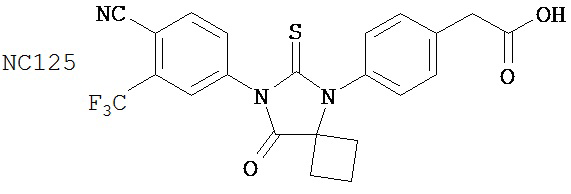
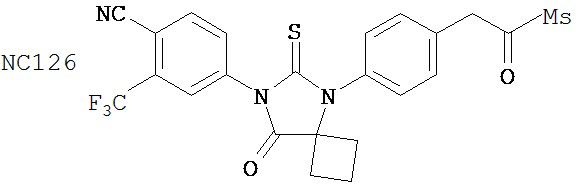
Короче говоря, были обнаружены и изготовлены новые соединения, которые демонстрируют доказательства того, что они намного превосходят бикалутамид при лечении рака простаты.
Чувствительность противораковой активности соединений к структурным различиям
Авторы изобретения обнаружили, что небольшие изменения в структуре гидантоиновых соединений могут привести к большим изменениям эффективности этих соединений при лечении рака простаты. Например, NC77 и NC53 отличаются только по единственному фтор-содержащему заместителю в арильном кольце, и NC53 относится к группе 1, тогда как NC77 относится к группе 2, оба лучше, чем бикалутамид при лечении рака простаты, но соединение NC53 превосходит NC77. Однако соединение NC98, которое отличается от NC77 только тем, что имеет дополнительный атом углерода между метилкарбамоильной группой и арильным кольцом, не лучше, чем бикалутамид при лечении рака простаты, и его относят к группе 4. Влияние соединений NC77, NC53 и NC98 на люциферазную активность можно видеть на фиг. 25. При заданных концентрациях соединения люциферазная активность после воздействия соединениями NC77 и NC53 меньше, чем люциферазная активность после воздействия соединением NC98.
Соединение NC4 отличается от NC3 только тем, что аминогруппа заменена на гидроксильную группу. Однако, несмотря на то, что соединение NC3 принадлежит группе 1 и оно намного лучше бикалутамида при лечении рака простаты, соединение NC4, принадлежащее группе 4, не лучше, чем бикалутамид. Влияние соединений NC3 и NC4 на люциферазную активность в клеточной линии 1AR изучали путем введения различных соединений в диапазоне концентраций от 1,25 до 10 мкМ и измерения уровня ПСА. При заданной дозировке люциферазная активность после воздействия соединением NC3 была меньше, чем люциферазная активность после воздействия соединением NC4. Влияние соединений NC3 и NC4 на люциферазную активность в клеточной линии 4AR изучали путем введения различных соединений в диапазоне концентраций от 1,25 до 10 мкМ и измерения люциферазной активности. При заданной дозировке люциферазная активность после воздействия соединением NC3 была меньше, чем люциферазная активность после воздействия соединением NC4. Влияние соединений NC3 и NC4 на уровень ПСА в клеточной линии LN/AR изучали путем введения различных соединений в диапазоне концентраций от 1,25 до 10 мкМ и измерения люциферазной активности. При заданной дозировке уровень ПСА после воздействия соединением NC3 был меньше, чем уровень ПСА после воздействия соединением NC4.
Соединения NC47 и NC48 отличаются друг от друга только метальным заместителем на конце карбамоильной группы, и оба соединения классифицированы в группу 1, хотя было обнаружено, что соединение NC48 является особенно предпочтительным. Соединение NC46 практически идентично с соединением NC47, за исключением того, что метоксигруппа заменена на аминогруппу. Однако соединение NC46 отнесено к группе 3. Соединение NC42 практически идентично с соединением NC46, но имеет на один углеродный атом меньше в цепи, связывающей сложноэфирную группу с арильным кольцом; NC42 отнесено к группе 3. Влияние соединений NC47, NC48, NC42 и NC46 на уровень ПСА в клеточной линии LN/AR изучали путем введения различных соединений в диапазоне концентраций от 125 нМ до 1000 нМ и измерения уровня ПСА. При заданной концентрации уровень ПСА после воздействия соединением NC47 и NC48 был меньше, чем уровень ПСА после воздействия соединениями NC42 и NC46.
Соединения NC68 и NC103 отличаются друг от друга тем, что первое соединение имеет метилкарбамоильную группу, присоединенную к арильному кольцу, а диметильный заместитель присоединен к тиогидантоиновой группе, тогда как последнее соединение имеет метиламиногруппу, присоединенную к правому арильному кольцу, а циклобутильный заместитель присоединен к тиогидантоиновой группе. Хотя соединение NC68, отнесенное к группе 1, намного лучше, чем бикалутамид при лечении рака простаты, соединение NC103, отнесенное к группе 5, неактивно или практически неактивно при лечении рака простаты. Влияние соединений NC68 и NC103 на люциферазную активность в клеточной линии LN/AR изучали путем введения различных соединений в диапазоне концентраций от 125 нМ до 1000 нМ и измерения люциферазной активности. При заданной концентрации люциферазная активность после воздействия соединением NC68 была меньше, чем люциферазная активность после воздействия соединением NC 103.
Соединения NC16 и NC18 отличаются друг от друга заменой тио на оксогруппу и заменой диметила на циклобутильный заместитель. Несмотря на то, что соединение NC16 отнесено к группе 1, соединение NC18 отнесено к группе 4.
Фармацевтические композиции и их введение
Соединения изобретения полезны в виде фармацевтических композиций, получаемых с помощью терапевтически эффективного количества соединения изобретения, как описано в тексте заявки, и приемлемого носителя или разбавителя.
Диарилгидантоиновые соединения изобретения могут быть составлены в виде фармацевтических композиций и введены субъекту, нуждающемуся в лечении, например, млекопитающему, такому как пациент-человек, в различных формах, адаптированных к выбранному пути введения, например, пероральному, назальному, внутрибрюшинному или парентеральному, с помощью внутривенного, внутримышечного, местного или подкожного способа введения или с помощью инъекции в ткань.
Таким образом, диарилгидантоиновые соединения изобретения можно вводить систематически, например, перорально в комбинации с фармацевтически приемлемой средой-растворителем, такой как инертный разбавитель или ассимилируемый пищевой носитель, или с помощью ингаляции или инсуффляции. Они могут быть помещены в твердые или мягкие покрытые желатиновые капсулы, могут быть спрессованы в таблетки или могут быть непосредственно включены в пищу пациента. Для перорального терапевтического введения диарилгидантоиновые соединения можно смешивать с одним или несколькими эксципиентами и применять в форме проглатываемых таблеток, буккальных таблеток, пастилок, капсул, эликсиров, суспензий, сиропов, облаток и т.п. Диарилгидантоиновые соединения можно смешивать с мелким инертным порошковым носителем и делать субъекту ингаляцию или инсуффляцию. Такие композиции и препараты должны содержать по меньшей мере 0,1% диарилгидантоинового соединения. Процентный состав композиций и препаратов, конечно, может различаться и обычно может быть в диапазоне от приблизительно 2% до приблизительно 60% по массе в заданной стандартной лекарственной форме. Количество диарилгидантоиновых соединений в таких терапевтически полезных композициях таково, чтобы был достигнут эффективный дозировочный уровень.
Таблетки, пастилки, пилюли, капсулы и т.п. могут также содержать следующие компоненты: связующие средства, такие как трагакант, гуммиарабик, кукурузный крахмал или желатин; эксципиенты, такие как дикальцийфосфат; дезинтегрирующее средство, такое как кукурузный крахмал, картофельный крахмал, альгиновая кислота и т.п.; смазывающее средство, такое как магния стеарат; и подслащивающее вещество, такое как сахароза, фруктоза или аспартам, или ароматизирующее средство, такое как перечная мята, винтергреневое масло, или можно добавлять вишневый ароматизатор. Если стандартной лекарственной формой является капсула, она кроме веществ, указанных выше типов, может содержать жидкий носитель, такой как растительное масло или полиэтиленгликоль. Могут присутствовать различные другие материалы в качестве покрывающих оболочек или для того, чтобы иным способом изменить физическую форму твердой стандартной лекарственной формы. Например, таблетки, пилюли или капсулы могут быть покрыты желатином, парафином, шеллаком или сахаром и т.п. Сироп или эликсир могут содержать активное соединение, сахарозу или фруктозу в качестве подсластителя, метил и пропилпарабены в качестве консервирующих средств, краситель и отдушку, такую как вишневый или апельсиновый ароматизатор. Безусловно, любой материал, применяемый при получении любой стандартной лекарственной формы, должен быть фармацевтически приемлемым и в значительной степени нетоксичным в используемых количествах. Кроме того, диарилгидантоиновые соединения могут быть включены в препараты и приспособления для замедленного высвобождения. Например, диарилгидантоиновые соединения могут быть включены в капсулы с высвобождением во времени, таблетки с высвобождением во времени и пилюли с высвобождением во времени.
Диарилгидантоиновые соединения можно также вводить внутривенно и внутрибрюшинно с помощью инфузии или инъекции. Растворы диарилгидантоиновых соединений могут быть получены в воде, необязательно смешанной с нетоксичными поверхностно-активными веществами. Дисперсии также могут быть получены в глицерине, жидких полиэтиленгликолях, триацетине и их смесях и в маслах. В обычных условиях хранения и применения препараты могут содержать консервирующее средство для предотвращения роста микроорганизмов.
Фармацевтические лекарственные формы, пригодные для инъекции или инфузии, могут включать стерильные водные растворы или дисперсии, или стерильные порошки, включающие диарилгидантоиновые соединения, которые адаптированы для немедленного получения стерильных растворов для инъекции или инфузии, необязательно включенных в липосомы. Во всех случаях окончательная лекарственная форма должна быть стерильной, жидкой и стабильной в условиях изготовления и хранения. Жидкий носитель или среда-растворитель могут быть растворяющей или жидкой дисперсионной средой, включающей, например, воду, этанол, полиол (например, глицерин, пропиленгликоль, жидкие полиэтиленгликоли и т.п.), растительные масла, нетоксичные сложные эфиры глицерина и их пригодные смеси. Можно поддерживать подходящую текучесть, например, с помощью образования липосом, с помощью поддержания необходимого размера частиц в случае дисперсий или с помощью использования поверхностно-активных веществ. Профилактику действия микроорганизмов можно осуществить с помощью различных антибактериальных и противогрибковых средств, например, с помощью парабенов, хлорбутанола, фенола, сорбиновой кислоты, тимеросала и т.п. Во многих случаях предпочтительно включать изотонические средства, например, сахара, буферы или натрия хлорид. Длительное всасывание проглатываемых композиций может быть обусловлено применением в композициях агентов, замедляющих всасывание, например, алюминия моностеарата и желатина.
Стерильные инъецируемые растворы получают путем включения диарилгидантоиновых соединений в необходимом количестве в подходящем растворителе с различными ингредиентами, перечисленными выше, если это необходимо, с последующей стерилизацией путем фильтрования. В случае стерильных порошков для получения стерильных инъецируемых растворов предпочтительными методами получения являются методы вакуумной сушки и лиофилизации, которые дают порошок активного ингредиента плюс любой дополнительный желаемый ингредиент, присутствовавший в растворах, предварительно простерилизованных с помощью фильтрования.
Для местного нанесения диарилгидантоиновые соединения можно применять в очищенном виде. Однако, как правило, желательно наносить их на кожу в виде композиций и составов вместе с дерматологически приемлемым носителем, который может быть твердым или жидким.
Полезные твердые носители включают тонко диспергированные твердые вещества, такие как тальк, глина, микрокристаллическая целлюлоза, кремнезем, окись алюминия и т.п. Другие твердые носители включают нетоксичные полимерные наночастицы или микрочастицы. Полезные жидкие носители включают воду, спирты или гликоли, или смеси вода/спирт/гликоль, в которых диарилгидантоиновые соединения могут быть растворены или диспергированы на эффективном уровне, необязательно с помощью нетоксичных поверхностно-активных соединений. Для заданного применения, чтобы оптимизировать свойства, можно добавлять вспомогательные вещества, такие как ароматизаторы и дополнительные антимикробные средства. Полученные в результате жидкие композиции можно наносить из абсорбирующего тампона, используемого для того, чтобы пропитывать бинты и другие перевязочные материалы, или распылять на обрабатываемую поверхность с помощью распылителей насосного типа или аэрозолей.
Для создания растекаемых паст, гелей, мазей, мыл и т.п. для применения прямо на коже пользователя вместе с жидкими носителями также можно применять загустители, такие как синтетические полимеры, жирные кислоты, соли и сложные эфиры жирных кислот, жирные спирты, модифицированные целлюлозы или модифицированные минеральные вещества.
Примеры полезных дерматологических композиций, которые можно применять для доставки диарилгидантоиновых соединений на кожу, известны в этой области техники; например, смотри: Jacquet et al. (U.S. Pat. No. 4,608,392), Geria (U.S. Pat. No. 4,992,478), Smith et al. (U.S. Pat. No. 4,559,157) и Wortzman (U.S. Pat. No. 4,820,508), каждый из которых включен сюда путем отсылки.
Полезные дозировки соединений, имеющих формулу I, могут быть определены путем сравнения их активности in vitro и активности in vivo в моделях на животных. Методы экстраполяции эффективных дозировок, определенных для мышей и других животных, к дозировкам для человека известны в этой области техники; например, смотри патент U.S. Pat No. 4,938,949, который включен сюда путем отсылки.
Например, концентрация диарилгидантоиновых соединений в жидкой композиции, такой как лосьон, может составлять примерно от 0,1-25% по массе или примерно от 0,5-10% по массе. Концентрация в полутвердой или твердой композиции, такой как гель или порошок, может составлять примерно 0,1-5% по массе или примерно 0,5-2,5% по массе.
Количество диарилгидантоиновых соединений, необходимых для применения при лечении, будет различаться не только от выбранной соли, но также от способа введения, природы состояния, которое подвергают лечению, и возраста и состояния пациента, и, в конечном счете, будет зависеть от выбора лечащего терапевта или клинициста.
Эффективные дозировки и способы введения средств изобретения общеприняты. Точное количество (эффективная доза) средства будет различаться от субъекта к субъекту, будучи зависимой, например, от вида, возраста, массы тела и основного или клинического состояния субъекта, тяжести или механизма любого расстройства, которое подвергают лечению, применяемого специфического средства или среды-растворителя, способа и режима введения и т.п. Терапевтически эффективную дозу можно определить эмпирически, с помощью общепринятых процедур, известных специалистам в этой области техники. Смотри, например, The Pharmacological Basis of Therapeutics, Goodman, and Oilman, eds., Macmillan Publishing Co., New York. Например, эффективную дозу можно оценить в самом начале или в анализах клеточных культур, или в подходящих моделях на животных. Чтобы определить приблизительные диапазоны дозировок и способы введения, можно также использовать модель на животных. Такую информацию затем можно использовать, чтобы определить полезные дозы и способы введения для людей. Терапевтическую дозу также можно выбрать путем аналогии с дозировками сопоставимых терапевтических средств.
Специфический способ введения и режим дозировок выбирает лечащий клиницист, принимающий во внимание определенные обстоятельства (например, личность субъекта, заболевание, осложненное состояние заболевания, и является ли лечение профилактическим). Лечение может включать ежедневный прием однократных или многократных дозировок соединения(соединений) в течение периода, составляющего от нескольких дней до месяцев и даже лет.
Обычно, однако, подходящая доза будет находиться в диапазоне от примерно 0,001 до примерно 100 мг/кг, например, от примерно 0,01 до примерно 100 мг/кг массы тела в день, так как было описано выше, примерно 0,1 мг на килограмм, или в диапазоне от примерно 1 до примерно 10 мг на килограмм массы тела реципиента в день. Например, подходящая доза может быть примерно 1 мг/кг, 10 мг/кг или 50 мг/кг массы тела в день.
Диарилгидантоиновые соединения обычно вводят в виде стандартной лекарственной формы; например, содержащей от 0,05 до 10000 мг, от 0,5 до 10000 мг, от 5 до 1000 мг или примерно 100 мг активного ингредиента на стандартную лекарственную форму.
Диарилгидантоиновые соединения можно вводить для того, чтобы достигнуть максимальных концентраций в плазме крови, например, от примерно 0,5 до примерно 75 мкМ, от примерно 1 до 50 мкМ, от примерно 2 до примерно 30 мкМ или от примерно 5 до примерно 25 мкМ. Примеры желаемых концентраций в плазме крови включают по меньшей мере или не более чем 0,25; 0,5; 1; 5; 10; 25; 50; 75, 100 или 200 мкМ. Например, уровень в плазме может быть в диапазоне от примерно 1 до 100 мкМ или от примерно 10 до примерно 25 мкМ. Этого можно достигнуть, например, с помощью внутривенной инъекции от 0,05 до 5%-ного раствора диарилгидантоиновых соединений, необязательно в физиологическом растворе, или путем перорального введения в виде болюса, содержащего около 1-100 мг диарилгидантоиновых соединений. Желаемый уровень в крови может быть поддержан с помощью непрерывной инфузии для обеспечения примерно 0,00005-5 мг на кг массы тела в час, например, по меньшей мере или не более чем 0,00005; 0,0005; 0,005; 0,05; 0,5 или 5 мг/кг/час. Альтернативно такой уровень можно достичь с помощью перемежающихся инфузий, содержащих около 0,0002-20 мг на кг массы тела, например по меньшей мере или не более чем 0,0002; 0,002; 0,02; 0,2; 2, 20 или 50 мг диарилгидантоиновых соединений на кг массы тела.
Диарилгидантоиновые соединения обычно могут быть в виде стандартной дозы или могут быть разделены на дозы, которые вводят через определенные промежутки времени, например, в виде двух, трех, четырех или более субдоз в день. Субдозу, в том виде как она есть, можно дополнительно разделить, например, на некоторое свободно выбираемое количество отдельных введений, таких как многократные ингаляции из инсуффлятора.
Некоторое количество указанных выше соединений демонстрируют небольшие агонистические активности или лишены их в отношении клеток гормонорезистентного рака простаты. Так как эти соединения являются мощными ингибиторами AR, их можно применять не только при лечении рака простаты, но также при лечении других ассоциированных с AR заболеваний или состояний, таких как доброкачественная гиперплазия простаты, потеря волос и акне. Так как AR принадлежит к семейству ядерных рецепторов, эти соединения также могут служить в качестве основы для синтеза лекарственных средств, нацеленных на другие ядерные рецепторы, такие как рецептор эстрогенов и рецептор активатора пролиферации пероксисом. Следовательно, их можно в дальнейшем усовершенствовать для других заболеваний, таких как рак молочной железы, рак яичников, диабет, заболевания сердца и заболевания, связанные с обменом веществ, в которых играют роль ядерные рецепторы.
Последовательность химического синтеза для нескольких соединений в соответствии с изобретением приведена ниже. Цианогидрины 10abc превращают в четыре различные цианоамина 12abcd с помощью реакции с тремя различными анилинами 11abc (10а и 11а дают 12а, 10b и 11а дают 12b, 10с и 11b дают 12с, и 10с и 11с дают 12d). В отдельном способе анилин 13 превращают в одну стадию в изотиоцианат 14. Добавление 12abcd к 14с с последующей обработкой мягкой кислотой приводит к образованию желаемых тиогидантоинов 4 (NC54), 5 (NC55), 6 (NC56) и 7 с хорошим выходом.
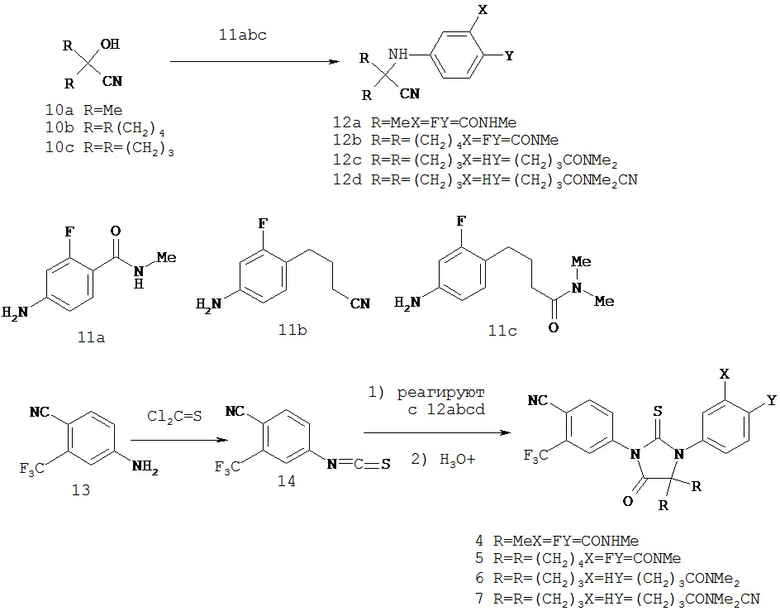
Воплощения, проиллюстрированные и обсужденные в этом описании, предназначены только для обучения специалистов в этой области техники лучшему, по мнению авторов заявки, способу изготовления и применения изобретения. Ничто в этом описании не должно быть рассмотрено как ограничение объема изобретения. Все представленные примеры являются типичными и не ограничительными. Как должно быть понятно специалистам в этой области техники, описанные выше воплощения изобретения могут быть модифицированы или изменены без отступления от изобретения в свете указанных выше наставлений. Поэтому ясно, что в рамках пунктов формулы изобретения и их эквивалентов изобретение может быть осуществлено другим способом, отличным от того, который был специфически описан.
| название | год | авторы | номер документа |
|---|---|---|---|
| ДИАРИЛТИОГИДАНТОИНОВЫЕ СОЕДИНЕНИЯ | 2007 |
|
RU2449993C2 |
| ДИАРИЛГИДАНТОИНЫ | 2011 |
|
RU2638833C2 |
| ДИАРИЛГИДАНТОИНЫ | 2021 |
|
RU2828689C2 |
| НИЗКОМОЛЕКУЛЯРНЫЕ ИНГИБИТОРЫ N-КОНЦЕВОЙ АКТИВАЦИИ РЕЦЕПТОРА АНДРОГЕНОВ | 2009 |
|
RU2519948C2 |
| ТЕРАПЕВТИЧЕСКИЕ СРЕДСТВА НА ОСНОВЕ ПРОИЗВОДНЫХ ДИГЛИЦИЛИЛОВЫХ ПРОСТЫХ ЭФИРОВ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2009 |
|
RU2572596C2 |
| ПРОИЗВОДНЫЕ ТИОГИДАНТОИНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРА АНДРОГЕНА | 2012 |
|
RU2598854C2 |
| СЕЛЕКТИВНЫЕ ЛИГАНДЫ-РАЗРУШИТЕЛИ АНДРОГЕННЫХ РЕЦЕПТОВ (SARD) И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2724103C2 |
| НЕСТЕРОИДНЫЕ МОДУЛЯТОРЫ АНДРОГЕННОГО РЕЦЕПТОРА, СПОСОБ ПОЛУЧЕНИЯ, ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2006 |
|
RU2378250C2 |
| СЕЛЕКТИВНЫЕ ЛИГАНДЫ-РАЗРУШИТЕЛИ АНДРОГЕННЫХ РЕЦЕПТОРОВ (SARD) И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2689988C2 |
| ЛИГАНДЫ, СЕЛЕКТИВНО РАЗРУШАЮЩИЕ АНДРОГЕННЫЕ РЕЦЕПТОРЫ (SARD), И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2017 |
|
RU2795431C2 |

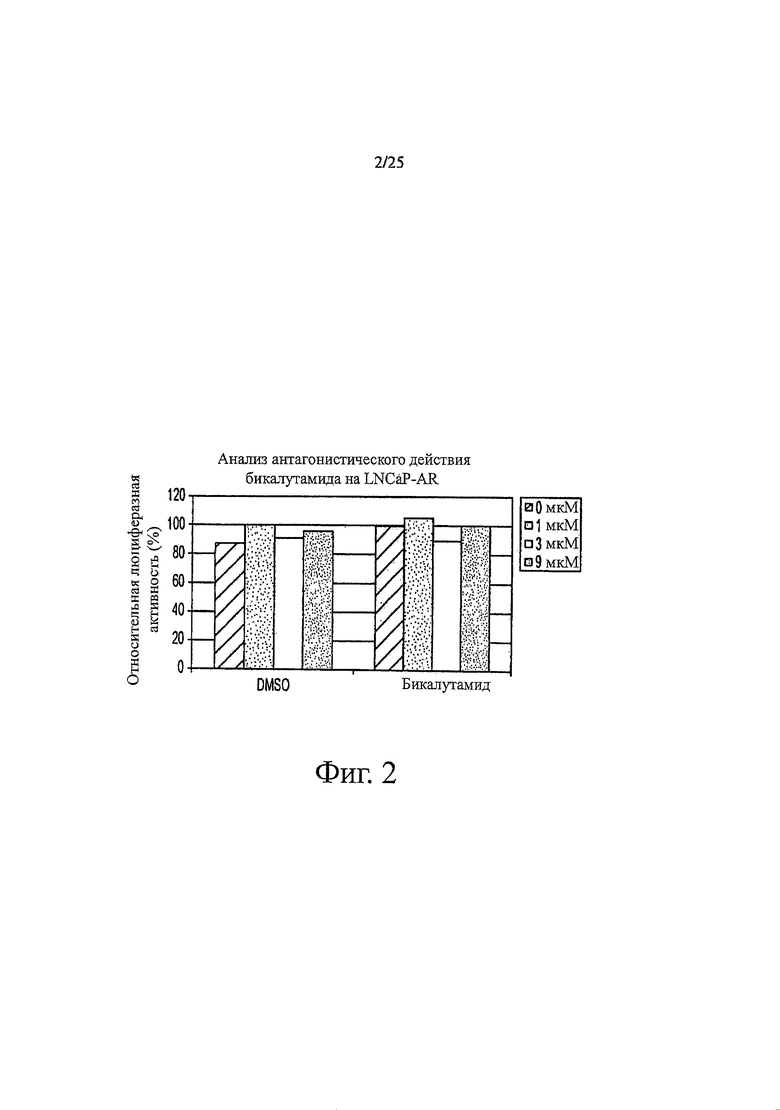
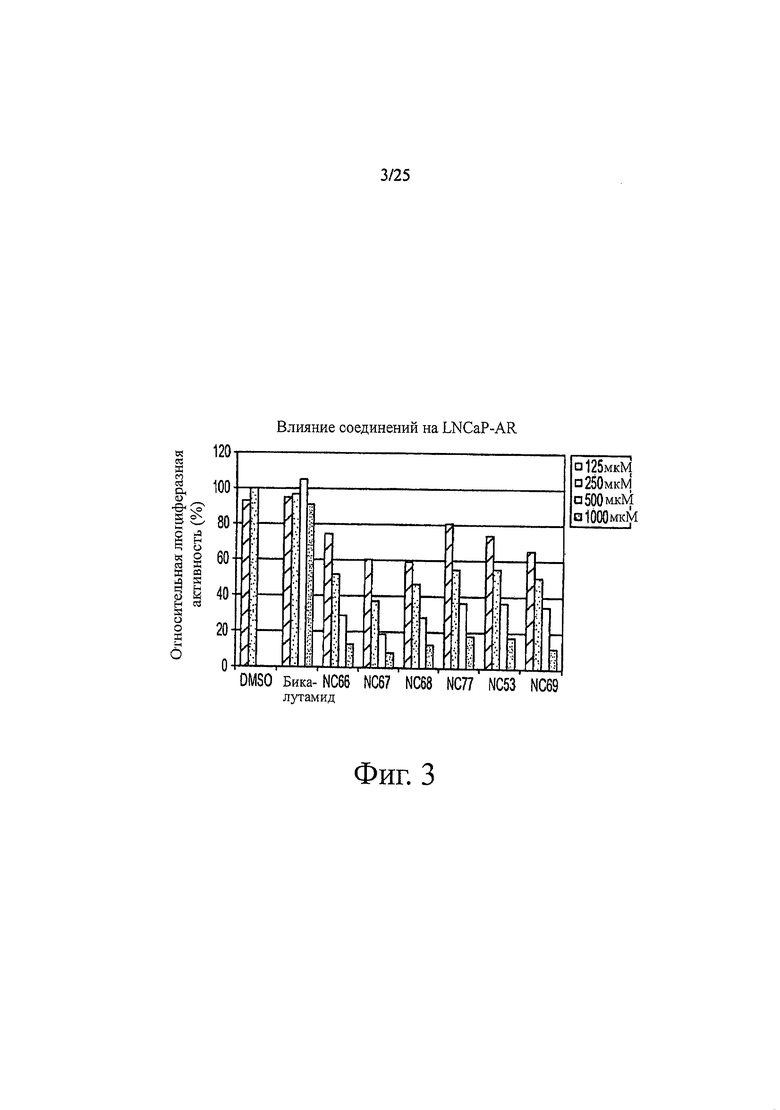
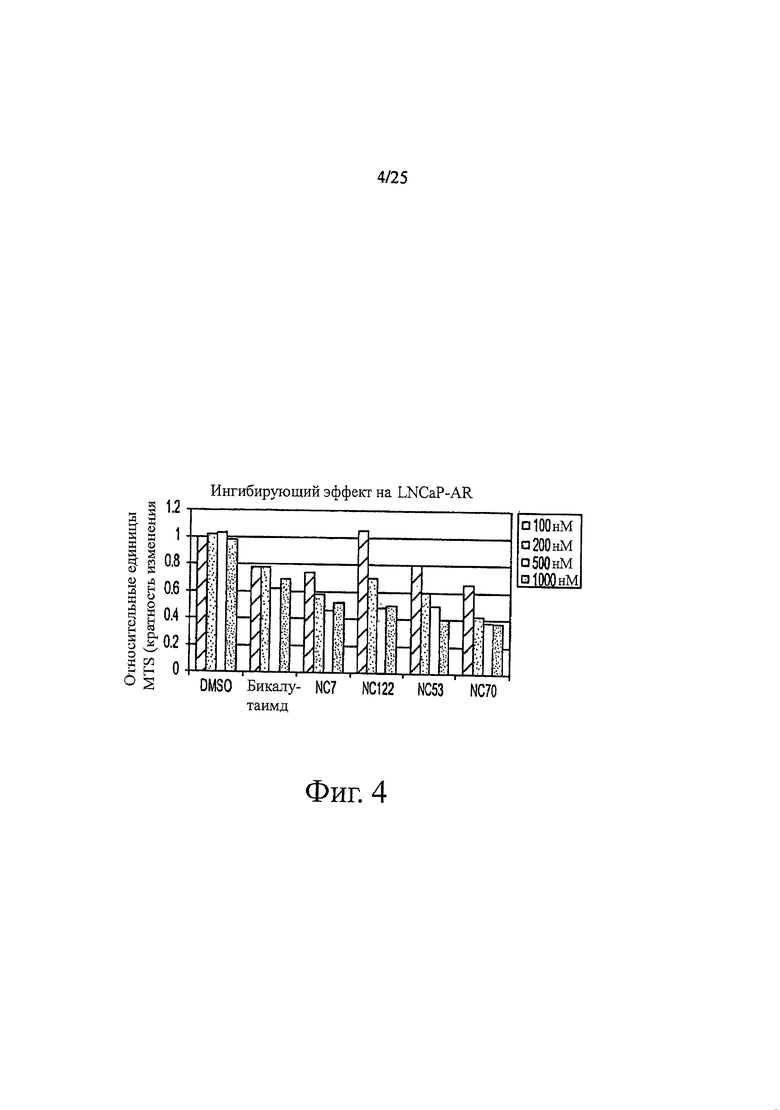
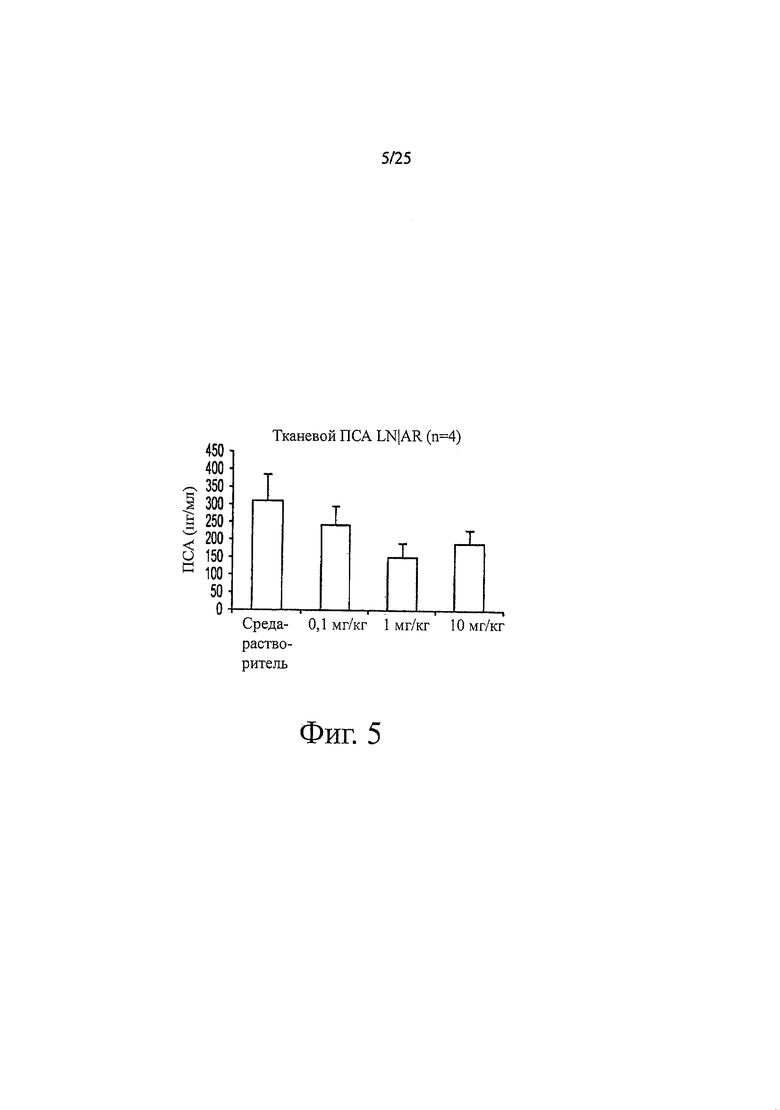

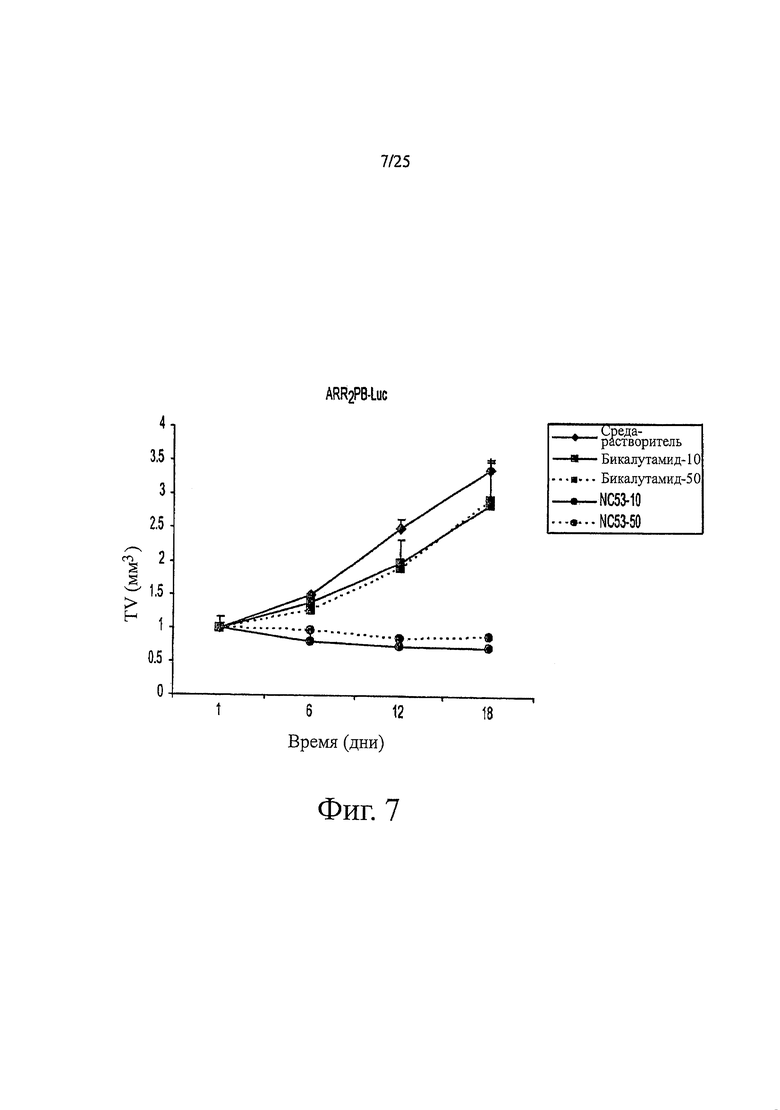
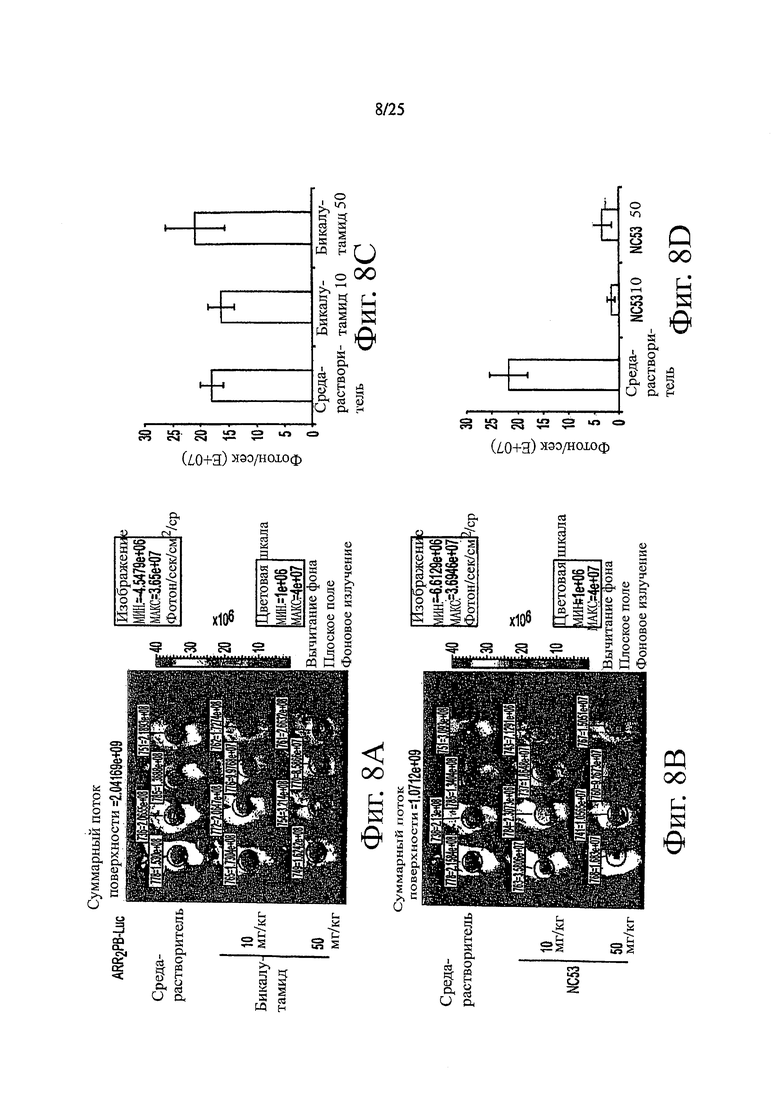
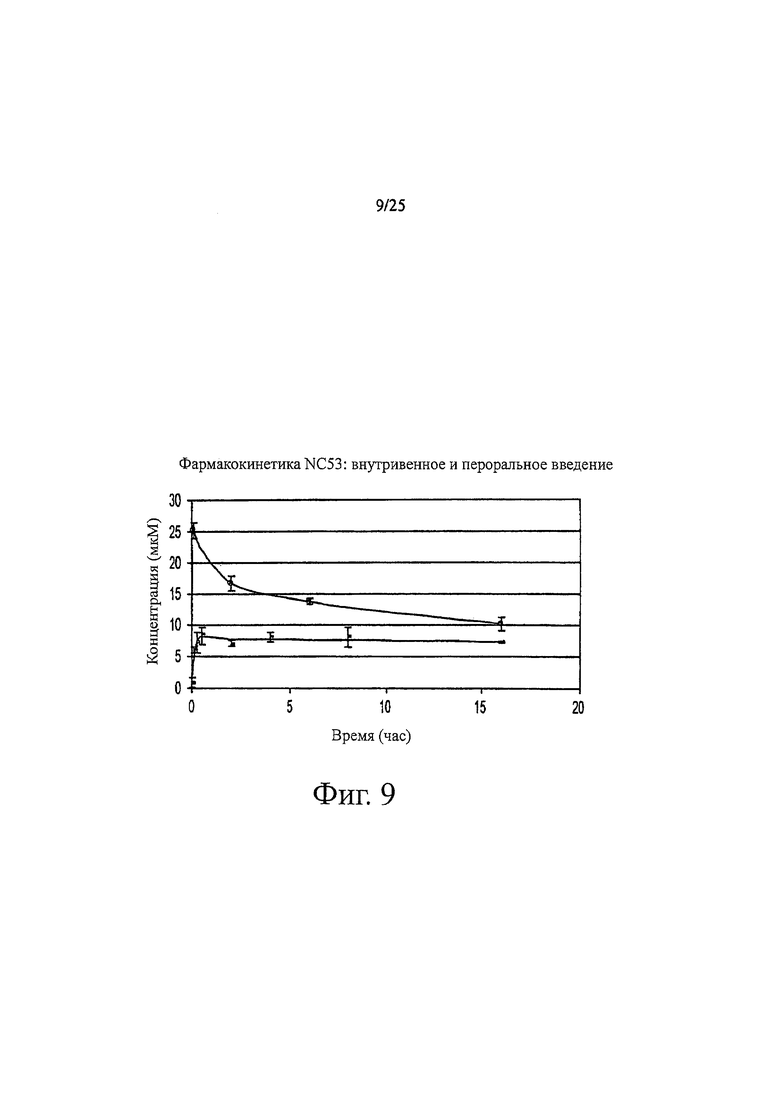
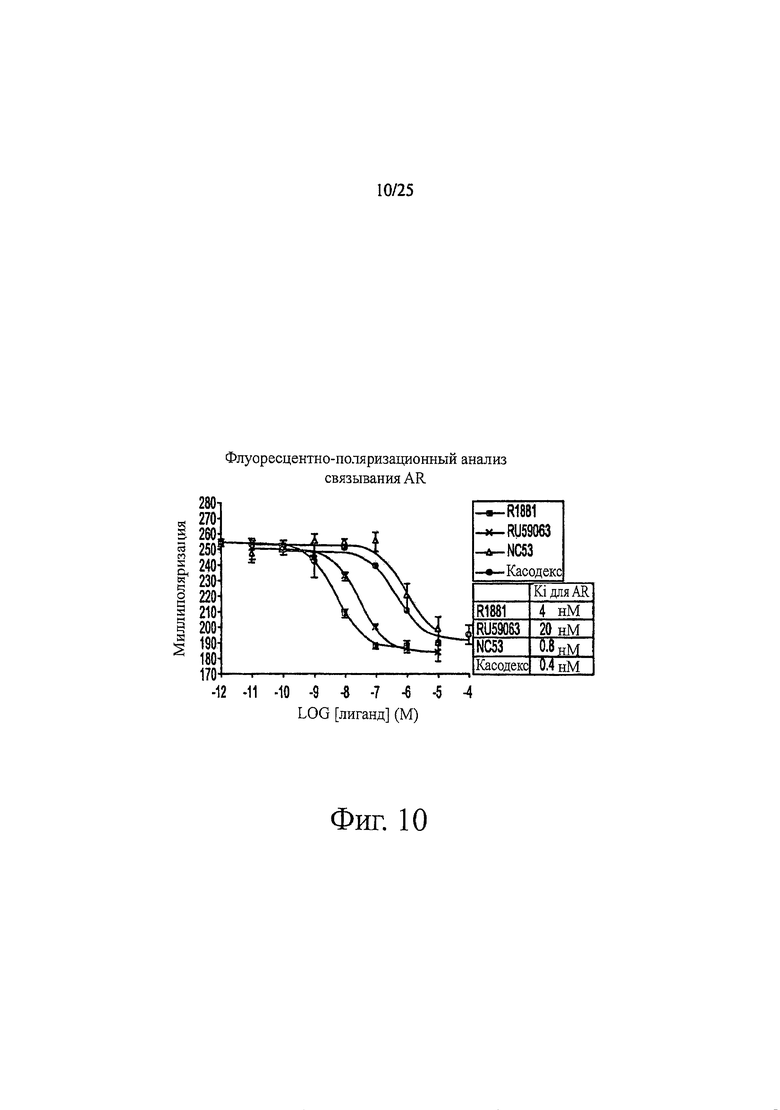

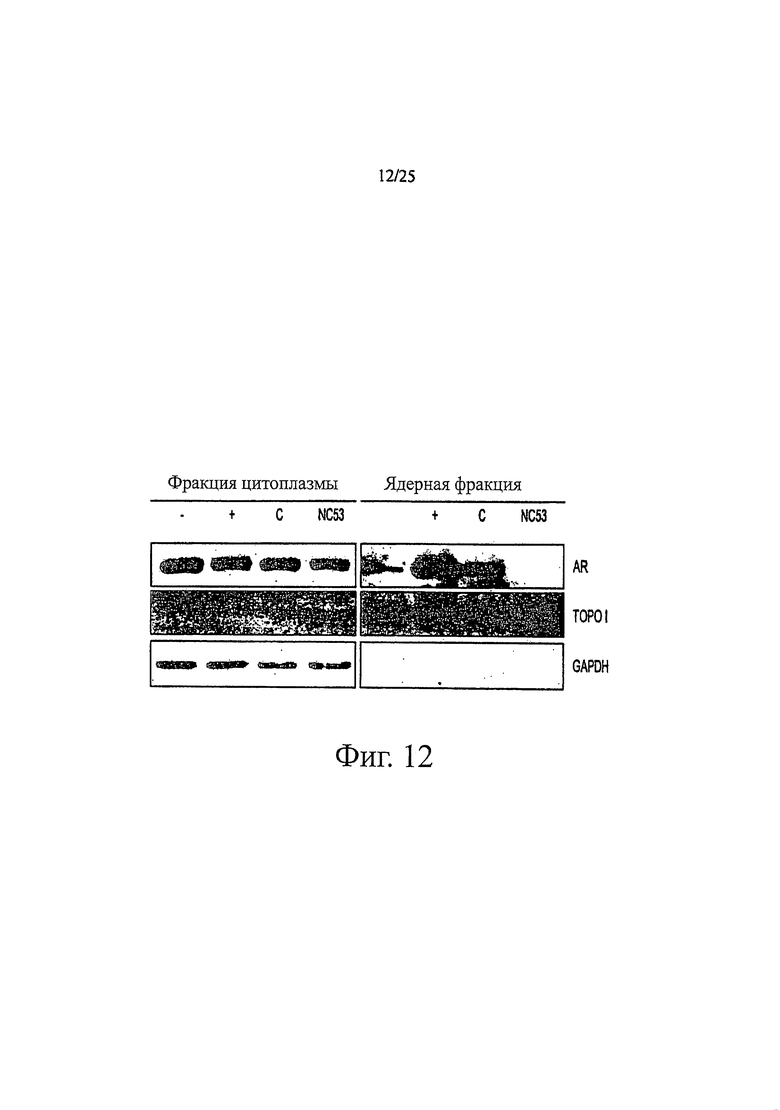
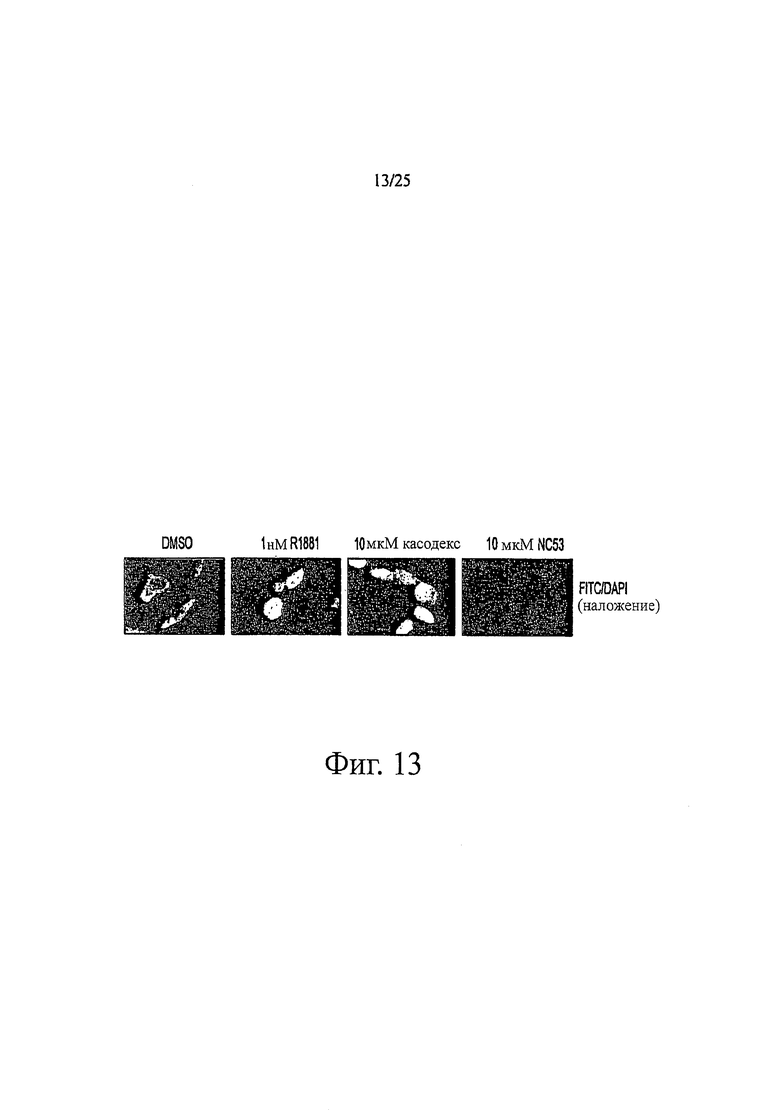
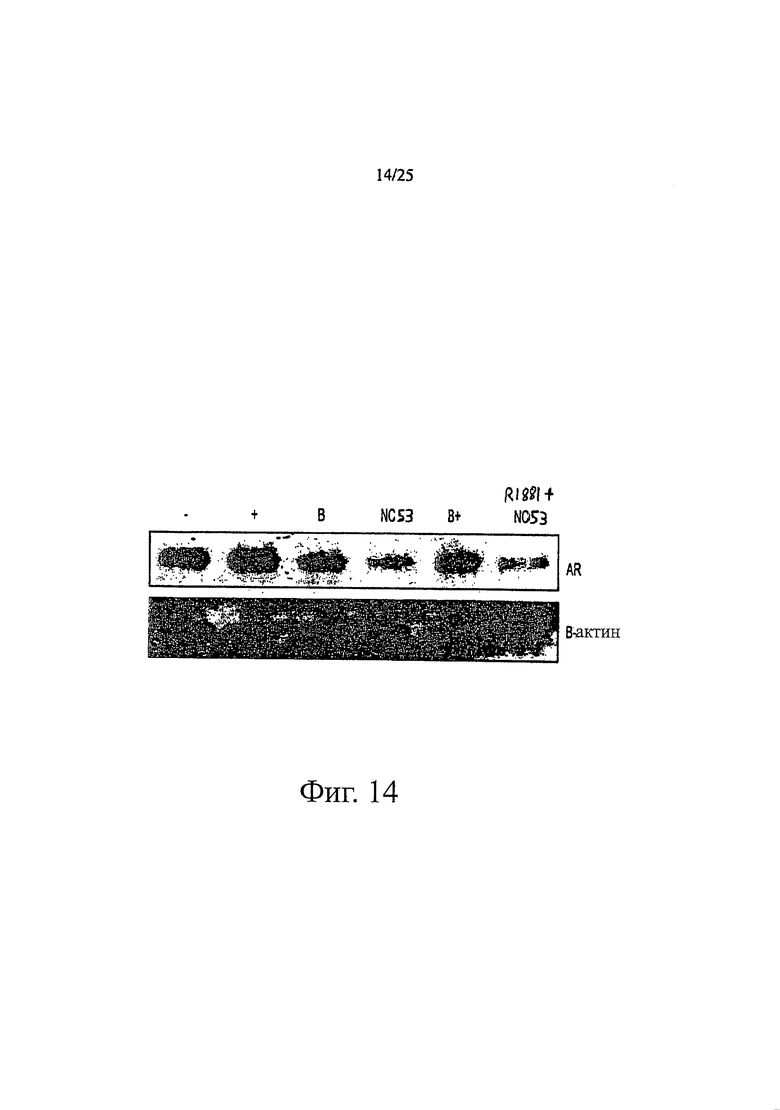
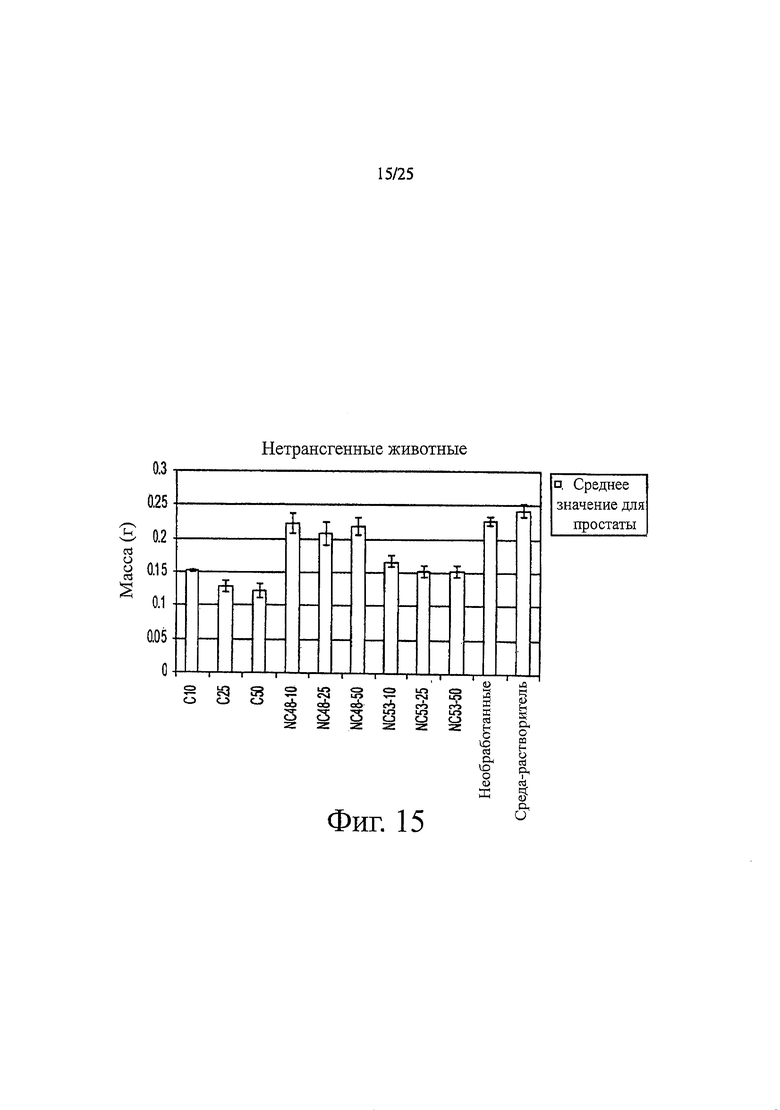
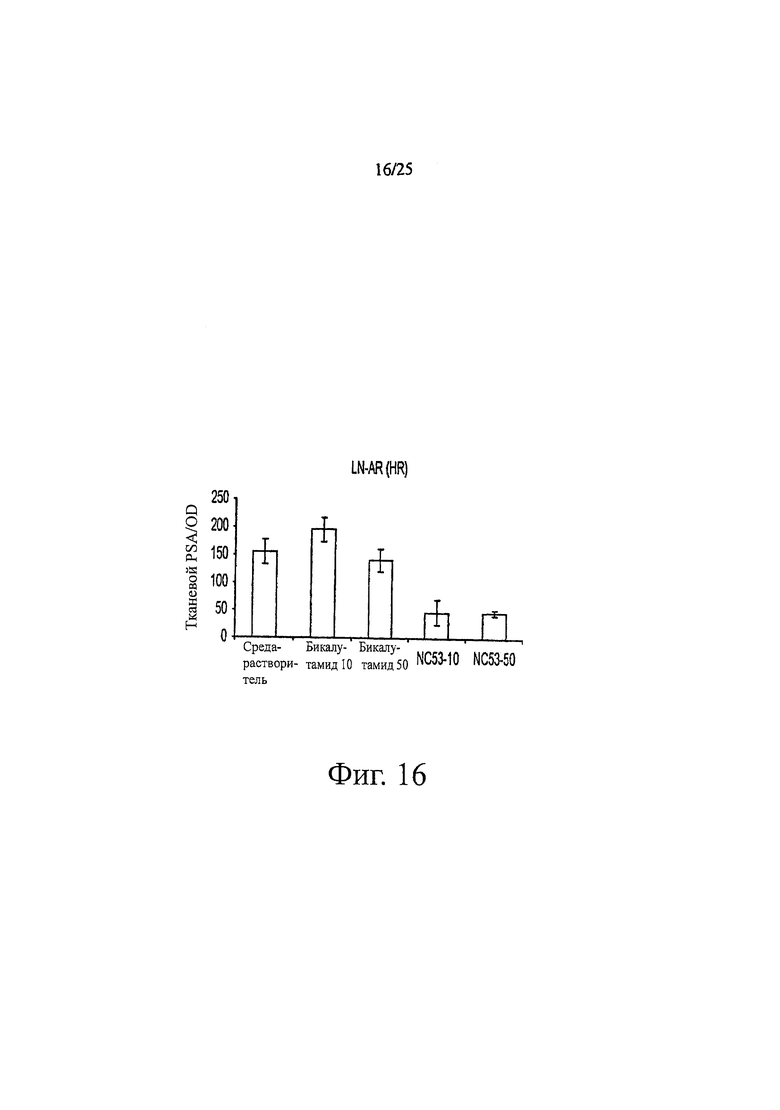
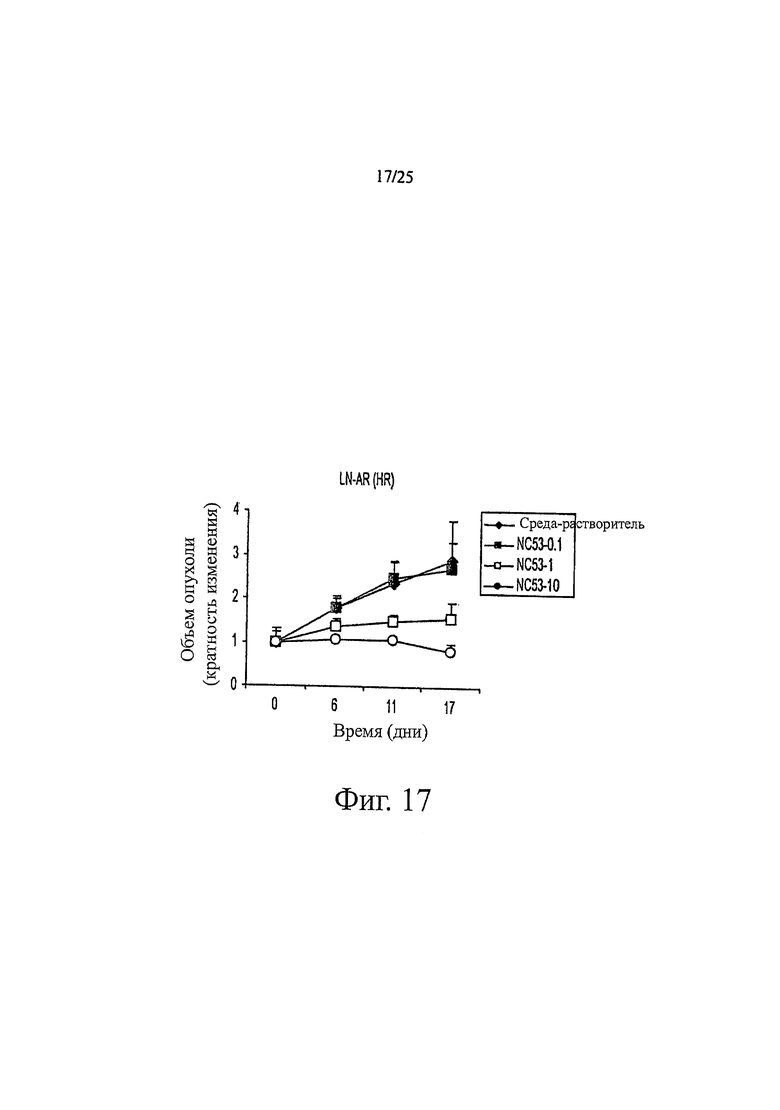
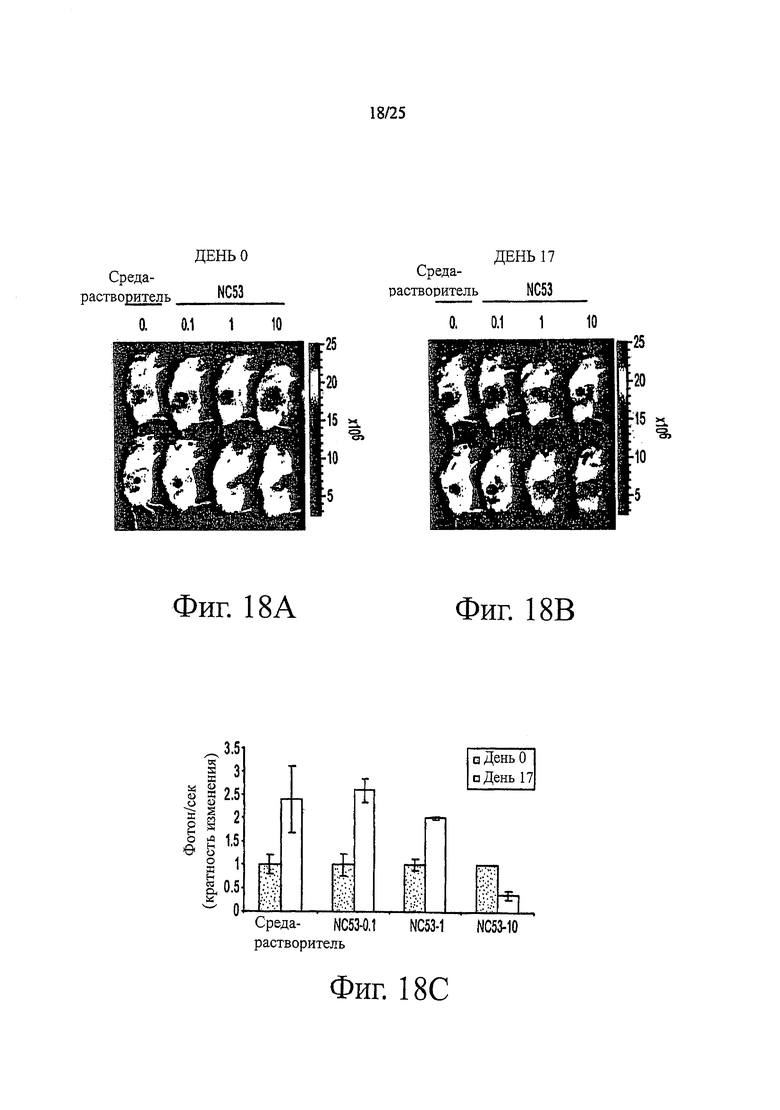
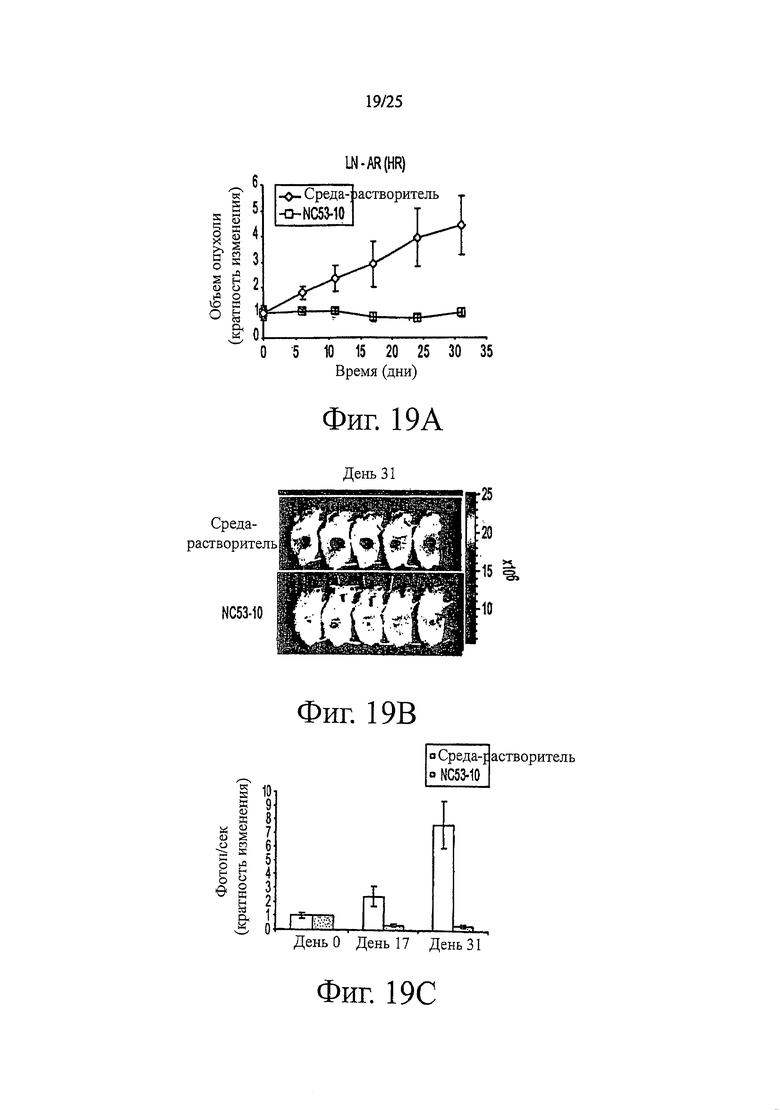
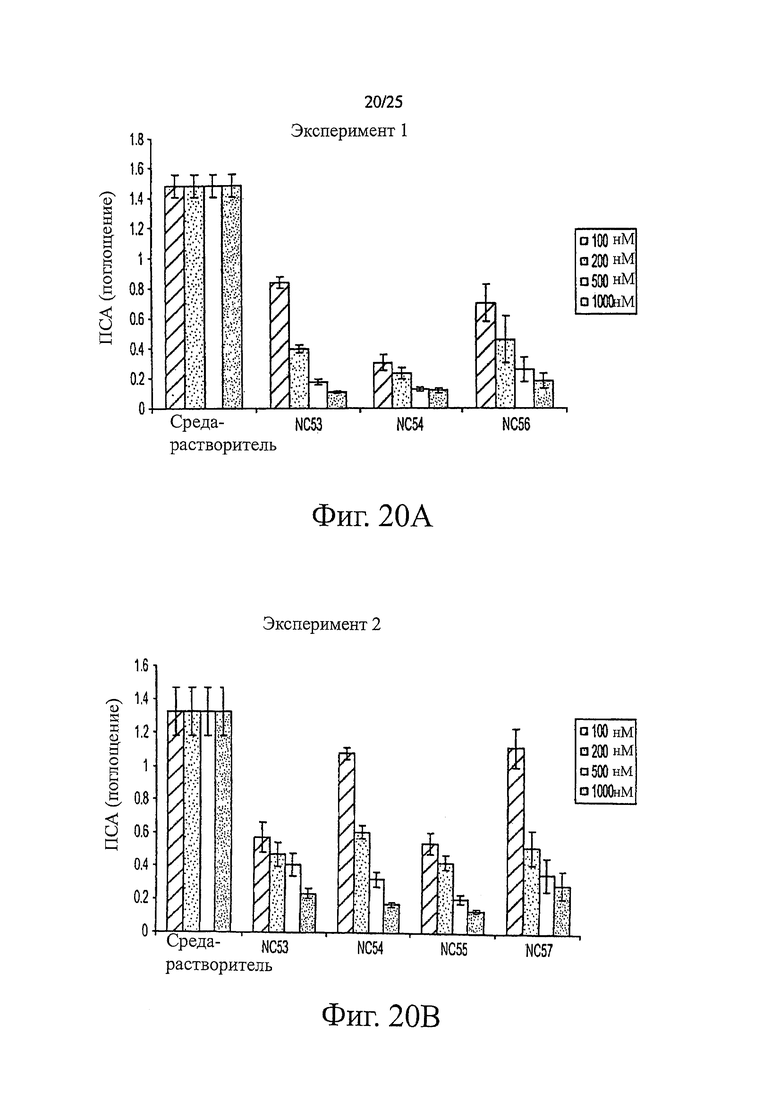
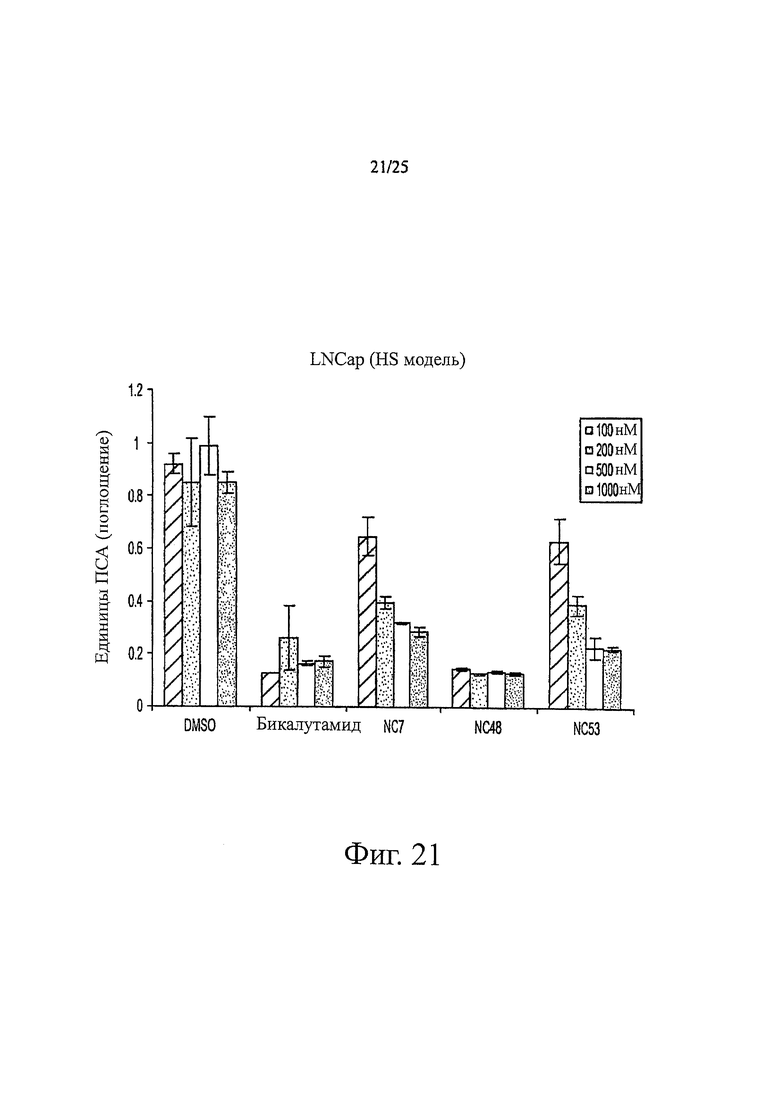
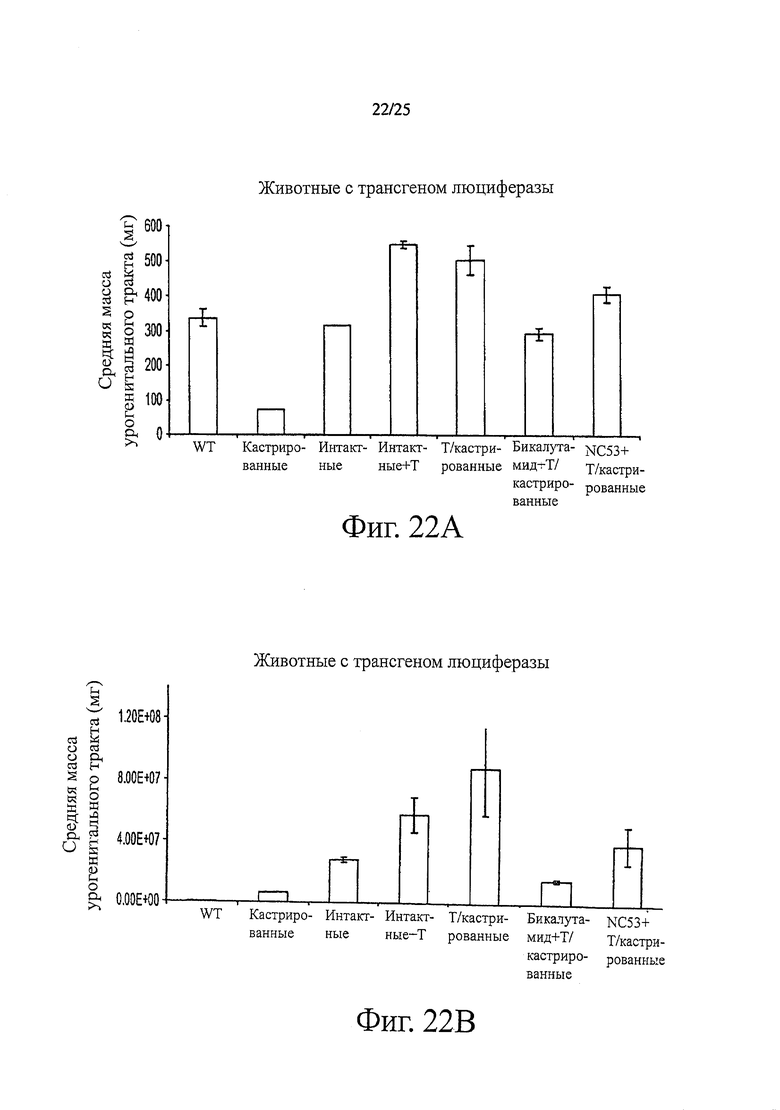
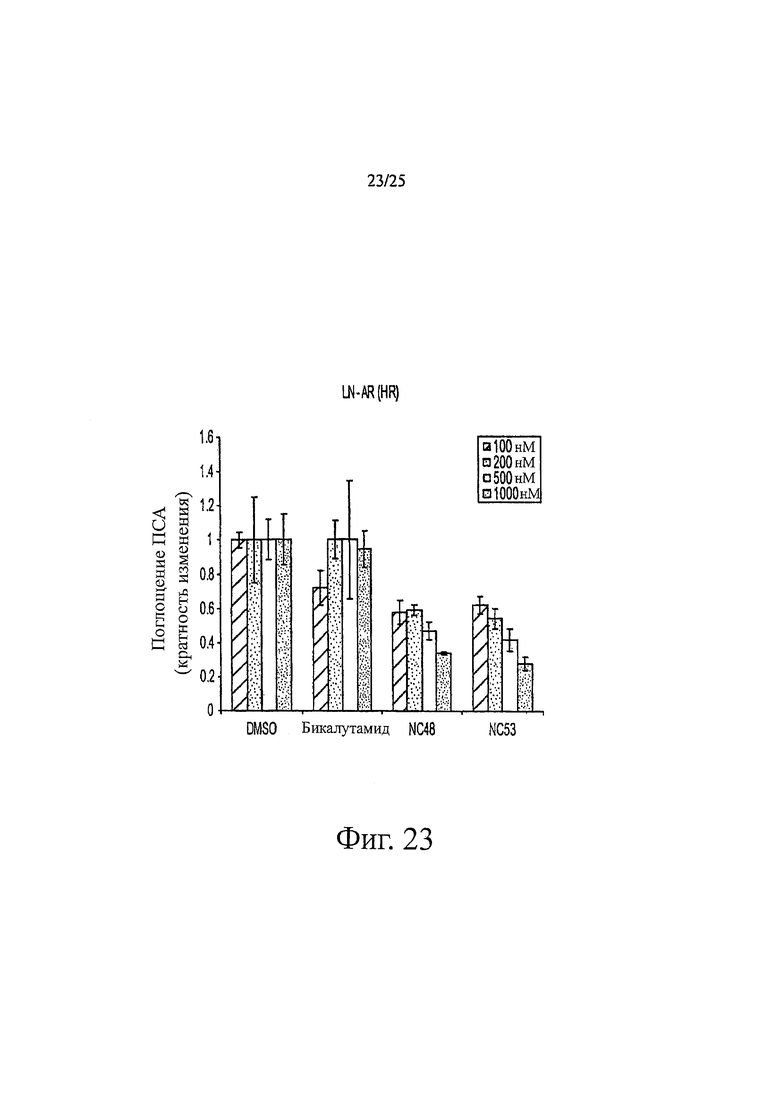
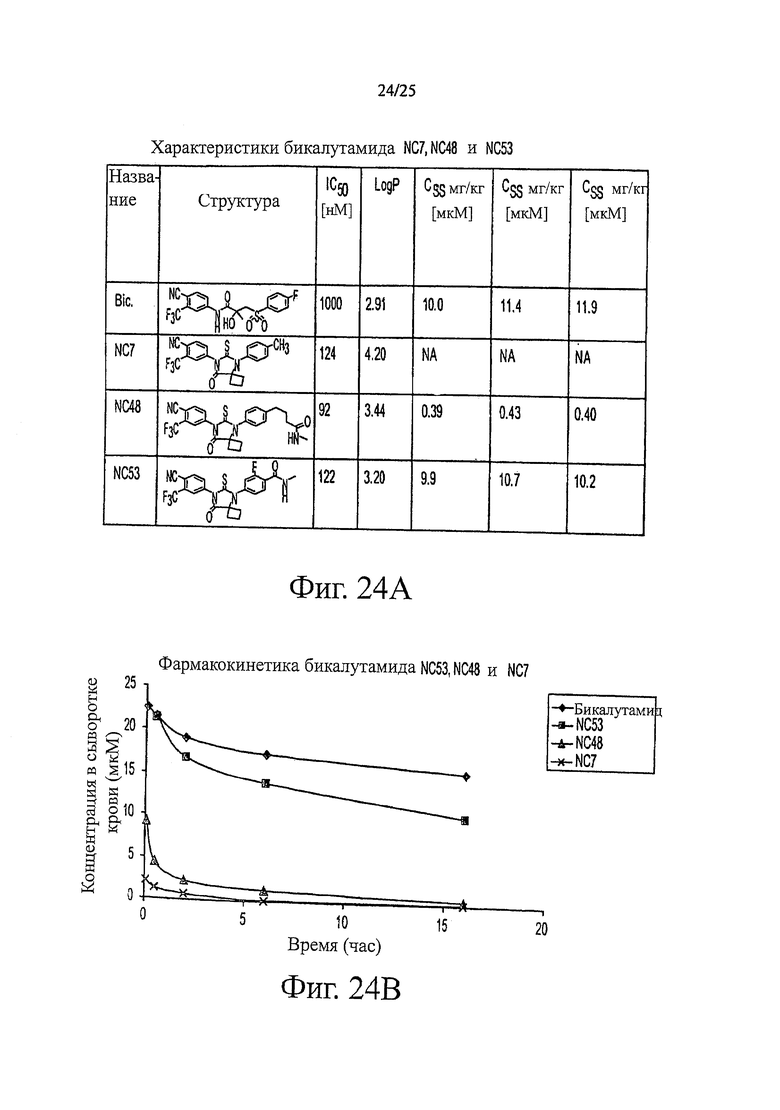
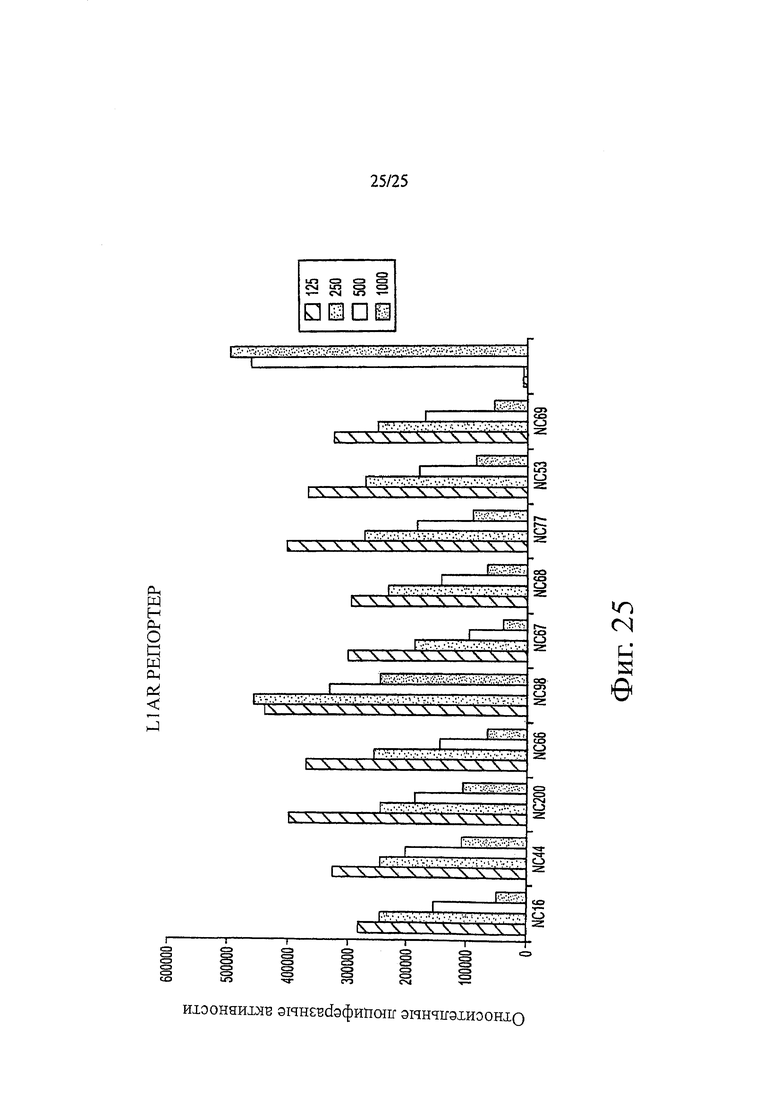
Изобретение относится к диарилтиогидантоиновому производному формулы [NC96/RD111], полезному при лечении некоторых видов рака. Технический результат: получено новое соединение, полезное при лечении некоторых видов рака, в том числе и гормонорезистентного рака простаты. 5 з.п. ф-лы, 25 ил., 11 табл., 5 пр.

1. Соединение, имеющее формулу
2. Соединение по п.1 для применения при лечении организма человека или животного путем терапии.
3. Соединение по п.1 для применения при лечении гормонорезистентного рака простаты.
4. Соединение по п.1 для применения при лечении доброкачественной гиперплазии простаты.
5. Соединение по п.1 для применения при лечении рака молочной железы.
6. Соединение по п.1 для применения при лечении рака яичников.
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| ЗАМЕЩЕННЫЕ ФЕНИЛИМИДАЗОЛИДИНЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТИАНДРОГЕННОЙ АКТИВНОСТЬЮ | 1993 |
|
RU2116298C1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| ЗАМЕЩЕННЫЕ ФЕНИЛИМИДАЗОЛИДИНЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1995 |
|
RU2152934C1 |

