Родственная заявка
Для данной заявке испрашивается приоритет на основании раздела 35 Кодекса законов США, §119(е), по предварительной заявке на патент США № 61/772095, поданной 4 марта 2013 г., полное содержание которой включено в настоящее описание изобретения посредством ссылки.
Область техники, к которой относится изобретение
Настоящее изобретение относится к сухим фармацевтическим композициям, предназначенным для введения через слизистую оболочку, в частности, для интраназального и интравагинального введения, а также для других путей введения, содержащим сухие частицы, включающие наночастицы активного агента, связующее вещество и фармацевтически приемлемый носитель. Настоящее изобретение также относится к способам приготовления и применения таких композиций.
Уровень техники
Многие активные агенты, предназначенные для системного воздействия, вводят внутривенно. Однако внутривенное введение не всегда является удобным, требует специального медицинского оборудования и квалифицированного медицинского персонала. Кроме того, многие составы для внутривенного введения являются неустойчивыми и должны быть приготовлены непосредственно перед применением. Введение через слизистую оболочку, такое как интраназальное или интравагинальное введение, является альтернативой внутривенному введению. Однако, для введения через слизистую оболочку необходимы специальные составы. Например, композиции для интраназального введения должны обеспечивать всасывание активного агента через слизистую оболочку носа, а не через легкие.
Известны многие различные терапевтические применения прогестерона. Например, прогестерон может быть использован для регулирования менструального цикла, для усиления фазы лютеинизации при оплодотворении in vitro и в гормонозаместительной терапии. Композиции, предназначенные для интравагинального введения, особенно пригодны для вышеуказанных и подобных применений, так как преимуществом интравагинального введения является быстрая доставка лекарственного средства непосредственно к предполагаемому месту действия, что позволяет избежать эффекта первичного прохождения, связанного, например, с пероральным введением.
Прогестерон может быть также использован для лечения поражений центральной нервной системы, включающих травматические поражения центральной нервной системы (такие как травматическое поражение головного мозга (TBI)) и ишемический инсульт. Например, недавно выполненные исследования показали, что лечение прогестероном ограничивает поражение ткани и улучшает функцию после тупой травмы головного мозга, инсульта, повреждения спинного мозга, диабетической невропатии и других типов острых нейропоражений. Sayeed & Stein, in PROGRESS IN BRAIN RES. Vol. 175:21-37 (J. Verhaagen et al., eds.) (Elsevier B.V. 2009). При выполнении клинических исследований прогестерон вводили внутривенно, используя составы, приготовленные непосредственно перед использованием. Такое введение ограничивает возможность применения прогестерона в экстренных ситуациях, например, в случае TBI или инсульта.
Поэтому существует потребность в альтернативных составах, содержащих активные агенты, такие как прогестерон, в частности, в сухих составах, предназначенных для введения через слизистую оболочку, например, для интраназального или интравагинального введения.
Также существует потребность в альтернативных составах, содержащих другие активные агенты, такие как мометазон, в том числе фуроат мометазона, в частности, в сухих составах, предназначенных для введения через слизистую оболочку, например, для назального введения.
Сущность изобретения
Настоящее изобретение относится к сухим фармацевтическим композициям, содержащим сухие частицы, включающие наночастицы фармацевтически активного агента, связующее вещество и фармацевтически приемлемый носитель, в которых наночастицы активного агента связаны с частицами носителя. В конкретных вариантах осуществления изобретения сухие фармацевтические композиции предназначены для введения через слизистую оболочку и содержат высушенные распылением частицы, включающие наночастицы фармацевтически активного агента, связующее вещество и фармацевтически приемлемый носитель, при этом наночастицы активного агента до сушки распылением имеют средний диаметр частицы менее примерно 1 мкм, и где до 10% высушенных распылением частиц имеют размер частицы, равный по меньшей мере примерно 15 мкм, и по меньшей мере 90% высушенных распылением частиц имеют размер до около 55 мкм. В некоторых вариантах осуществления изобретения активный агент является прогестероном, его метаболитом, производным или пролекарством. В некоторых вариантах осуществления изобретения активный агент является мометазоном, его метаболитом, производным или пролекарством, таким как фуроат мометазона.
В некоторых вариантах осуществления изобретения носитель выбран из группы, состоящей из микрокристаллической целлюлозы, кристаллической целлюлозы, целлюлозы, α-целлюлозы, перекрестносшитой натрий-карбоксиметилцеллюлозы, перекрестносшитого винилполивинилпирролидона, перекрестносшитого карбоксивинильного полимера или его солей, перекрестносшитого поливинилового спирта, перекрестносшитого полигидроксиэтилметакрилата, гидроксипропилового крахмала, карбоксиметилового крахмала, перекрестносшитого крахмала, амилазы, амилопектина, пектина, желатина, казеина, аравийской камеди, траганта и глюкоманнана. В некоторых вариантах осуществления изобретения носитель включает микрокристаллическую целлюлозу (МСС).
В некоторых вариантах осуществления изобретения связующее вещество выбрано из группы, состоящей из гидроксипропилметилцеллюлозы (НРМС), натрий-карбоксиметилцеллюлозы (NaCMC), полиакрилатов и их солей, низших алкиловых эфиров целлюлозы, таких как кальций-карбоксиметилцеллюлоза, гидроксипропилцеллюлозы (НРС, НРС-SL и HPC-L), гидроксипропилметилцеллюлозы (НРМС), гипромеллозы 2910, натрий-карбоксиметилцеллюлозы (NaCMC), метилцеллюлозы, гидроксиэтилцеллюлозы, гидроксипропилцеллюлозы, фталата гидроксипропилметилцеллюлозы, некристаллической целлюлозы, олеиновой кислоты, глицерина, белого вазелина, сложного эфира глицерина гидрированной канифоли, полиэтиленгликолей, пропиленгликоля и полиэтиленгликоля 400. В некоторых вариантах осуществления изобретения связующее вещество выбирают из группы, состоящей из гидроксипропилметилцеллюлозы (НРМС) и натрий-карбоксиметилцеллюлозы (NaCMC).
В некоторых вариантах осуществления изобретения фармацевтическая композиция далее включает интенсификатор размола, необязательно выбираемый из группы, состоящей из полисорбата 20, полисорбата 80 и хлорида бензалкония.
В некоторых вариантах осуществления изобретения активный агент составляет от около 10 до около 75 мас. %, от около 40 до около 75 мас. %, от около 25 до около 50 мас. %, от около 1,0 до около 25,0 мас. % или от около 5% до около 10 мас. % частиц. В некоторых вариантах осуществления изобретения связующее вещество независимо составляет от около 1 до около 15 мас. % или около 10,0-50,0 мас. % высушенных распылением частиц. В некоторых вариантах осуществления изобретения интенсификатор размола независимо составляет от около 0,1 до около 5 мас. % высушенных распылением частиц.
В некоторых вариантах осуществления изобретения высушенные распылением частицы включают от около 40 до около 75 мас. % прогестерона, от около 25 до около 50 мас. % МСС, от около 1 до около 15 мас. % НРМС и от около 0,1 до около 5,0 мас. % полисорбата 20. В других конкретных вариантах осуществления изобретения высушенные распылением частицы включают от около 5 до около 10 мас. % фуроата мометазона, от около 60 до около 80 мас. % МСС, от около 20 до около 50 мас. % НРМС и от около 0,05 до около 0,1 мас. % хлорида бензалкония.
В некоторых вариантах осуществления изобретения композиции далее включают один или несколько фармацевтически приемлемых наполнителей, таких как микрокристаллическая целлюлоза со средним диаметром частиц от около 30 до около 150 мкм, крахмал или их смесь и/или трехосновный фосфат кальция. В некоторых вариантах осуществления изобретения композиции далее включают примерно 5,0-30,0% мас./мас. микрокристаллической целлюлозы со средним диаметром частиц от около 30 до около 150 мкм, крахмал или их смесь и примерно 0,1-10% мас./мас. трехосновного фосфата кальция.
В некоторых вариантах осуществления изобретения более 90% активного агента высвобождается из композиции в течение 60 минут по результатам испытания на растворение.
В некоторых вариантах осуществления изобретения указанные композиции предназначены для интраназального введения. В некоторых вариантах осуществления изобретения указанные композиции предназначены для интравагинального введения.
Настоящее изобретение также относится к способам получения сухой фармацевтической композиции по настоящему изобретению, содержащей сухие частицы, включающие наночастицы фармацевтически активного агента, связующее вещество и фармацевтически приемлемый носитель, которые включают образование частиц, включающих наночастицы фармацевтически активного агента, связанные с частицами носителя. В некоторых вариантах осуществления изобретения сухие частицы являются высушенными распылением частицами, полученными способом, включающим сушку распылением водной композиции, содержащей наночастицы фармацевтически активного агента, связующее вещество и фармацевтически приемлемый носитель, с образованием высушенных распылением частиц, при этом средний диаметр наночастиц активного агента до сушки распылением составляет менее примерно 1 мкм, до 10% высушенных распылением частиц имеют размер менее 10 мкм, по меньшей мере 50% высушенных распылением частиц имеют размер, равный по меньшей мере примерно 15 мкм, и по меньшей мере 90% высушенных распылением частиц имеют размер до около 55 мкм. В некоторых вариантах осуществления изобретения указанный способ включает одно или несколько размалываний (необязательно с интенсификатором размола), грохочение, просеивание и центрифугирование.
В некоторых вариантах осуществления изобретения массовое соотношение активного агента и носителя в водной композиции, подвергаемой сушке распылением, составляет от около 1:1 до около 2:1 и массовое соотношение активного агента и связующего вещества в водной композиции, подвергаемой сушке распылением, составляет от около 5:1 до 20:1. В некоторых вариантах осуществления изобретения водная композиция, подвергаемая сушке распылением, содержит в расчете на массу примерно 5-20% активного агента, примерно 2-10% носителя, примерно 0,3-3,0% связующего вещества, примерно 0,2-1,0% интенсификатора размола и воду. В других вариантах осуществления изобретения водная композиция, подвергаемая сушке распылением, содержит в расчете на соотношение массы примерно 0,6 или 0,7% активного агента, примерно 6-7% носителя, примерно 1,8-3% связующего вещества, примерно 0,06-0,07% интенсификатора размола и воду.
Настоящее изобретение также относится к сухим фармацевтическим композициям, полученным способами по настоящему изобретению.
Настоящее изобретение также относится к способам введения активного агента, которые включают введение нуждающемуся субъекту сухой фармацевтической композиции по настоящему изобретению через слизистую оболочку.
Таким образом, сухие фармацевтические композиции по настоящему изобретению предназначены для применения в способе введения активного агента нуждающемуся субъекту через слизистую оболочку. Настоящее изобретение также относится к использованию сухой фармацевтической композиции по настоящему изобретению для приготовления лекарственного средства, предназначенного для применения в способе введения активного агента нуждающемуся субъекту через слизистую оболочку.
Краткое описание чертежей
На фиг. 1 показано распределение по размерам наночастиц прогестерона по настоящему изобретению. Средний диаметр частиц равен примерно 0,2 мкм, диаметр частиц D10 равен примерно 0,1 мкм и диаметр частиц D90 равен примерно 0,8 мкм.
На фиг. 2 показаны профили высвобождения прогестерона из высушенных распылением частиц по настоящему изобретению (“образец”) по сравнению с высвобождением из сухой смеси тонкоизмельченного прогестерона (средний диаметр частиц >1 мкм) и микрокристаллической целлюлозы (МСС) с массовым соотношением 1:1. Высушенные распылением частицы полностью высвобождают прогестерон в течение примерно 15-20 минут, при этом полное высвобождение из смеси 1:1 происходит в течение примерно 60 минут.
На фиг. 3 показаны профили высвобождения прогестерона из четырех образцов (5, 6, 7 и 8) высушенных распылением частиц по настоящему изобретению по сравнению с высвобождением из сухой смеси тонкоизмельченного прогестерона (средний диаметр частиц >1 мкм) и МСС с массовым соотношением 1:1. Образцы 5 и 6 характеризуются более быстрым высвобождением и достижением более высоких концентраций прогестерона в принимающей жидкости (например, достижение большего высвобождения лекарственного средства), в то время как образцы 7 и 8 демонстрируют более медленное высвобождение и более низкие концентрации прогестерона в принимающей жидкости, но все же высвобождение является более быстрым и концентрации прогестерона более высокими по сравнению со смесью 1:1.
На фиг. 4 представлены данные профилей высвобождения, изображенных на фиг. 3, в виде процентного значения высвобожденного прогестерона, которые показывают, что образцы 5, 6 и 8 высвободили больше прогестерона в процентном выражении (примерно 100%) в течение более короткого времени по сравнению со смесью 1:1.
На фиг. 5 показаны уровни прогестерона в сыворотке обезьян после внутривенного введения (i.v.) или интраназального введения композиции, включающей высушенные распылением частицы по настоящему изобретению, (FID-3207) или композиции, включающей тонкоизмельченный прогестерон (средний диаметр частиц >1 мкм) и микрокристаллическую целлюлозу (МСС) с массовым соотношением 1:1 (смесь 1:1). Показанные данные свидетельствуют о быстром всасывании и элиминации после назального введения при доставке большего количества лекарственного средства из композиции, содержащей высушенные распылением частицы, по настоящему изобретению.
На фиг. 6 представлены значения площади под кривой, полученные при выполнении исследований с использованием обезьян, показанных на фиг. 5. Представленные данные указывают на более высокое высвобождение прогестерона из композиции, включающей высушенные распылением частицы, по настоящему изобретению по сравнению со смесью 1:1. (Данные, полученные при внутривенном введении, не подлежат прямому сравнению из-за разной дозы, как описано в приведенном ниже примере 5.
На фиг. 7 представлены значения максимальной концентрации (Cmax), полученные при выполнении исследований с использованием обезьян, показанных на фиг. 5. Обнаруженные максимальные концентрации в плазме отличались в разных группах (F(2,8) = 22,86, р<0,01). Интраназальное введение сухой смеси 1:1 характеризовалось гораздо более низкими максимальными концентрациями по сравнению с введением композиции, включающей высушенные распылением частицы, по настоящему изобретению (FID-3207) (p<0,01) или внутривенным введением (р<0,01), при этом показатели введения композиции, включающей высушенные распылением частицы, существенно не отличались от показателей внутривенного введения (р>0,05).
На фиг. 8 представлены значения времени до достижения максимальной концентрации (Tmax), полученные при выполнении исследований с использованием обезьян, показанных на фиг. 5. Статистический анализ выполнен не был; однако полученные данные свидетельствуют о коротком периоде Tmax для внутривенного введения и более длительном периоде Tmax для обоих интраназальных введений.
Подробное описание изобретения
Настоящее изобретение относится к сухим фармацевтическим композициям, предназначенным для введения через слизистую оболочку, в частности, для интраназального или интравагинального введения активного агента. Сухие композиции содержат сухие частицы, такие как высушенные распылением частицы, состоящие из наночастиц активного агента, связующего вещества и носителя, при этом наночастицы активного агента связаны с частицами носителя. Как более подробно описано ниже, размер сухих частиц, таких как высушенные распылением частицы, и наночастиц активного агента может быть выбран и отрегулирован для достижения интраназального введения, интравагинального введения и других способов введения.
Технические и научные термины, использованные в настоящем описании изобретения, имеют значения, известные специалистам в данной области, за исключением особо оговоренных случаев. В настоящем описании изобретения сделаны ссылки на разные методики, известные специалистам в данной области. Публикации и другие материалы, содержащие описание известных методик, представленных в настоящем описании изобретения, полностью включены в настоящее описание изобретения в качестве ссылки. При осуществлении настоящего изобретения могут быть использованы любые приемлемые материалы и/или методы, известные специалистам в данной области. Однако в настоящем описании изобретения рассмотрены конкретные материалы и методы. Материалы, реагенты и тому подобные вещества, указанные в нижеследующем описании и примерах, могут быть приобретены в коммерческих источниках за исключением особо оговоренных случаев.
В использованном здесь значении формы единственного числа включают как формы единственного числа, так и формы множественного числа за исключением специального указания об использовании только формы единственного числа.
Термин “примерно” и использование диапазонов, определяемых термином “примерно”, означает, что данное число не ограничено точно указанным числом, а включает значения в диапазоне, не выходящем за пределы объема изобретения. Термин “примерно” в использованном здесь значении известен специалистам в данной области и может несколько изменяться в зависимости от контекста, в котором данный термин использован. В тех случаях, когда значение термина “примерно” не ясно из контекста специалистам в данной области, значение данного термина предполагает плюс или минус 10% от указанного значения.
Термин “сухая фармацевтическая композиция” в использованном здесь значении означает фармацевтическую композицию, по существу не содержащую воду. Фраза “по существу не содержащая” в использованном здесь значении означает, что описанная композиция (например, фармацевтическая композиция) содержит менее примерно 5%, менее примерно 3% или менее примерно 1 мас. % исключаемого компонента (например, воды) в расчете на общую массу рассматриваемой композиции. В некоторых вариантах осуществления изобретения сухая фармацевтическая композиция содержит не более примерно 0,1 мас. % воды или не более примерно 0,01 мас. % воды.
Термин “сухая частица” в использованном здесь значении означает сухую частицу, включающую наночастицы фармацевтически активного агента, связующее вещество и носитель, при этом наночастицы активного агента связаны с частицами носителя.
Термин “высушенная распылением частица” в использованном здесь значении имеет общепринятое значение, используемое в области фармацевтических составов, и означает частицу, представляющую собой высушенный распылением агрегат из фармацевтически активного агента, связующего вещества и носителя.
Термины “фармацевтически активный агент”, “активный агент”, “активный фармацевтический ингредиент” и “API” имеют взаимозаменяемые значения.
Термин “наночастицы” в использованном здесь значении означает частицы со средним диаметром, равным примерно 1 мкм или меньше. Наночастицы могут быть получены из более крупных частиц методами, известными в данной области, которые включают размалывание, просеивание и т.д.
Размер частиц может быть измерен при помощи лазерной дифракции, известной в данной области. Метод лазерной дифракции основан на явлении, при котором угол дифракции света частицей обратно пропорционален размеру данной частицы. В указанном приборе использован источник лазерного излучения, который облучает суспензию частиц и измеряет интенсивность света, отражаемого под разными углами относительно падающего света. Основываясь на данных распределения интенсивности, прибор использует алгоритм, созданный на основании решения Миэ уравнений Максвелла, относящихся к рассеянию электромагнитного излучения сферой, для преобразования картины интенсивности рассеянного света в распределение частиц по размерам.
В использованном здесь значении фраза “терапевтически эффективное количество” означает дозу лекарственного средства, вызывающую специфическую фармакологическую реакцию, для достижения которой указанное лекарственное средство вводят субъекту, нуждающемуся в таком лечении. “Терапевтически эффективное количество” лекарственного средства не всегда обеспечивает эффективное лечение состояния/ заболевания у данного субъекта, даже если, по мнению специалистов в данной области, такая доза представляет собой терапевтически эффективное количество. Для удобства и иллюстрации типичные дозы и терапевтически эффективные количества указаны применительно к взрослым людям. Специалисты в данной области могут привести указанные количества в соответствие со стандартной практикой, применяемой для лечения конкретного субъекта и/или состояния/заболевания.
Фармацевтические композиции
Настоящее изобретение относится к сухим фармацевтическим композициям, предназначенным для введения активного агента через слизистую оболочку, в частности, для интраназального или интравагинального введения, а также для других способов введения. Сухие композиции включают сухие частицы, такие как высушенные распылением частицы, состоящие из наночастиц активного агента, связующего вещества и фармацевтически приемлемого носителя, при этом наночастицы активного вещества связаны с частицами носителя. В некоторых вариантах осуществления изобретения по меньшей мере примерно 50% наночастиц активного вещества связано с частицами носителя, в том числе по меньшей мере 75% и по меньшей мере 80%.
Как более подробно описано ниже, размеры сухих частиц, таких как высушенные распылением частицы, и наночастиц активного агента могут быть выбраны и отрегулированы для достижения интраназальной доставки. Таким образом, сухие (например, высушенные распылением) частицы композиций по настоящему изобретению могут характеризоваться одним или несколькими нижеследующими распределениями по размерам:
до 10% высушенных распылением частиц имеют размер менее 10 мкм;
по меньшей мере 50% высушенных распылением частиц имеют размер, равный по меньшей мере примерно 15 мкм; и
по меньшей мере 90% высушенных распылением частиц имеют размер примерно до 55 мкм.
В конкретных вариантах осуществления изобретения композиции по настоящему изобретению обладают одной или несколькими благоприятными характеристиками, такими как быстрая доставка активного агента, биоадгезия, лучшая устойчивость при хранении и возможность упаковки в готовой к использованию форме.
Наночастицы активного агента
Фармацевтические композиции по настоящему изобретению содержат сухие частицы, такие как высушенные распылением частицы, включающие наночастицы активного агента. Активный агент может быть любым активным агентом, пригодным для интраназального введения, местного или системного воздействия, или любым активным агентом, который желательно использовать в сухой фармацевтической композиции, содержащей частицы, включающие активный агент и связующее вещество. В некоторых вариантах осуществления изобретения активный агент предназначен для интраназального введения с целью достижения системного воздействия. В других вариантах осуществления изобретения активный агент предназначен для интравагинального введения с целью достижения системного воздействия.
В соответствии с некоторыми вариантами осуществления изобретения активный агент является прогестероном, его производным, метаболитом или пролекарством. Типичные неограничивающие примеры метаболитов прогестерона включают аллопрегнанолон, прегнандиол, прегнанолон, прегнандион, 20-α-дигидропрогестерон и 17-ОН-прогестерон. Типичные неограничивающие примеры производных прогестерона включают 5-альфа-дигидропрогестерон, 6-дигидроретропрогестерон (дидрогестерон), капроат гидроксипрогестерона, левоноргестрел, норэтиндрон, ацетат норэтиндрона, норэтинодрел, норгестрел, медроксипрогестерон, хлормадинон и мегестрол, их фармацевтически приемлемые соли или сложные эфиры. Другие типичные производные прогестерона включают производные прогестерона, содержащие такие заместители как 6-α-метил, 6-метил, 6-ен или 6-хлор, введенные в структуру прогестерона, и 19-норпрогестероны. Типичные неограничивающие примеры пролекарств прогестерона включают сложные эфиры прогестерона, такие как 17-альфа-ОН эфиры прогестерона.
В соответствии с другими вариантами осуществления изобретения активный агент является мометазоном, его производным, метаболитом или пролекарством. В некоторых вариантах осуществления изобретения активный агент является фуроатом мометазона, который представляет собой пролекарство мометазона с химическим названием (11β,16α)-9,21-дихлор-11-гидрокси-16-метил-3,20-диоксопрегна-1,4-диен-17-ил-2-фуроат. Фуроат мометазона является глюкокортикостероидом, используемым для уменьшения воспаления кожи или дыхательных путей.
В соответствии с другими вариантами осуществления изобретения активный агент является любым активным агентом, который желательно использовать в сухой фармацевтической композиции, содержащей частицы, включающие активный агент и связующее вещество, таким как противовоспалительное средство, антибактериальное средство, антивирусное средство, средство, используемое при лечении рака, противорвотное средство, средство, используемое при лечении сердечно-сосудистых заболеваний и состояний (например, сердечно-сосудистое средство), сосудосуживающее средство, сосудорасширяющее средство, антипсихотическое средство, антидепрессант, анестетик, стероид, стимулятор, седативное средство, бронхорасширитель или любое другое средство, используемое при лечении заболевания или состояния, в том числе аллергических и других состояний.
В некоторых вариантах осуществления изобретения наночастицы активного агента имеют средний диаметр до сушки распылением (или другой обработки) менее примерно 1 мкм. Наночастицы активного агента могут быть получены из более крупных частиц активного агента методами, известными в данной области. Например, более крупные частицы могут быть размолоты (путем размалывания в дробящей среде или размалывания в струйной мельнице), измельчены, просеяны и/или центрифугированы для получения наночастиц со средним диаметром менее примерно 1 мкм.
В некоторых вариантах осуществления изобретения наночастицы активного агента получают методом, включающим размалывание, такое как размалывание в дробящей среде, размалывание под высоким давлением или размалывание в струйной мельнице.
При размалывании в дробящей среде или размалывании под высоким давлением активный агент получают в суспензии, которая может включать интенсификатор размола (то есть диспергатор), такой как поверхностно-активное вещество. В использованном здесь значении термины “интенсификатор размола” и “диспергатор” имеют взаимозаменяемые значения. Суспензия может быть водной или неводной, например, включающей органическую жидкость (например, масло или спирт). В конкретных вариантах осуществления изобретения суспензия является водной суспензией. Суспензию объединяют с дробящей средой, которая обычно представляет собой сферические гранулы твердого инертного материала. Частицы активного агента разбивают с образованием более мелких частиц путем механического истирания вследствие перемешивания гранул. Размалывание в лабораторных условиях может быть произведено в маломощном устройстве, например, путем прокатывания суспензии в баке роликовой мельницы, и в высокомощном устройстве, например, путем смешивания при помощи роторного смесителя. Приемлемые размалывающие устройства включают стандартные мельницы с мокрыми гранулами, например, производимые компаниями Nylacast (поставляются компанией Nylacast Components, Leicester, UK), Netzsch (поставляются компанией NETZSCH GmbH & Co. Selb, Germany), Drais (поставляются компанией Draiswerke, Inc., 40 Whitney Road, Mahwah, N.J., 07430, USA) и другими.
В качестве примера можно привести способ получения наночастиц активного агента, который включает:
(а) образование суспензии, включающей активный агент, интенсификатор размола и воду;
(b) размалывание суспензии с использованием первой дробящей среды;
(c) размалывание суспензии с использованием второй дробящей среды, при этом диаметр гранул первой дробящей среды больше диаметра гранул второй дробящей среды; и
(d) удаление размолотых наночастиц активного агента из суспензии, при этом наночастицы активного агента имеют средний диаметр менее примерно 1 мкм.
Например, гранулы первой дробящей среды могут иметь диаметр от около 1 мм до около 2 мм, в то время как гранулы второй дробящей среды могут иметь диаметр от около 0,1 до около 0,5 мм.
Как указано выше, размалывание может быть произведено с использованием интенсификатора размола, такого как поверхностно-активное вещество. Использование одного или нескольких интенсификаторов размола может предотвращать или уменьшать агрегацию частиц активного агента во время размалывания, а также может способствовать образованию наночастиц активного вещества требуемого размера. И наоборот было установлено, что использование связующего вещества при размалывании в дробящей среде может увеличивать вязкость размалывающей суспензии и может способствовать или усиливать образование агрегатов частиц активного агента, что затрудняет возможность получения наночастиц активного агента требуемого размера. Используя руководство, приведенное в настоящем описании изобретения, специалисты в данной области могут выбрать компоненты и отрегулировать условия размола с целью получения наночастиц активного агента требуемого размера.
В соответствии с некоторыми вариантами осуществления изобретения интенсификатор размола является поверхностно-активным веществом, таким как сложный эфир жирной кислоты полиоксиэтиленсорбита, например, одним или несколькими коммерчески доступными твинами®, такими как, например, твин 20® и твин 80® (ICI Specialty Chemicals). В некоторых вариантах осуществления изобретения интенсификатор размола выбирают из группы, состоящей из полисорбата 20, полисорбата 80 и хлорида бензалкония.
Как правило, поверхностно-активное вещество может быть ионогенным поверхностно-активным веществом или неионогенным поверхностно-активным веществом. Ионогенное поверхностно-активное вещество может быть анионогенным поверхностно-активным веществом или катионогенным поверхностно-активным веществом. Примеры анионогенных поверхностно-активных веществ включают алкилсульфаты, например, лаурилсульфат натрия, додецилсульфат натрия и сульфосукцинат диоктилнатрия, и алкилкарбоксилаты, например, стеарат кальция. Примеры катионогенных поверхностно-активных веществ включают хлорид цетилпиридиния, бромид цетилтриметиламмония, бромид додецилтриметиламмония и хлорид бензалкония. В конкретных вариантах осуществления изобретения поверхностно-активное вещество является лаурилсульфатом натрия, таким как дюпонол Р® (DuPont); тритоном Х-200®, который является алкиларилполиэфирсульфонатом (Rohm and Haas); Crodestas F-110®, который является смесью стеарата сахарозы и дистеарата сахарозы (Croda Inc.). Примеры неионогенных поверхностно-активных веществ включают полиоксиэтиленовые (РОЕ) алкилфенолы, РОЕ спирты с прямой цепью, РОЕ полиоксипропиленгликоли, РОЕ меркаптаны, сложные эфиры карбоновых кислот с длинной цепью, такие как глицериловый и полиглицериловый эфиры природных жирных кислот, например, моностеарат глицерина, пропиленгликоль, сорбит и сложные эфиры РОЕ сорбита, сложные эфиры полиоксиэтиленгликоля, полоксамеры, например, Pluronic F680® и F108®, которые являются блоксополимерами этиленоксида и пропиленоксида, полоксамины, например, Tetronic 908®, также известный как Poloxamine 908®, который является тетрафункциональным блоксополимером, полученным в результате последовательного добавления пропиленоксида и этиленоксида к этилендиамину (BASF Wyandotte Corporation, Parsippany, N.J.).
Дополнительно или альтернативно поверхностно-активное вещество может быть фосфолипидом. Типичные фосфолипиды включают, не ограничиваясь ими, фосфатидилхолин, фосфатидилэтаноламин, фосфатидилглицерин, фосфатидную кислоту и их смеси. Указанные фосфолипиды обычно содержат от около 4 до около 22 атомов углерода, в частности, от около 10 до около 18 атомов углерода, и характеризуются разными степенями насыщения.
В некоторых вариантах осуществления изобретения размалываемая суспензия включает примерно 0,01-5,0% мас./мас. интенсификатора размола, такого как поверхностно-активное вещество, в том числе примерно 0,05-2,0% мас./мас., примерно 0,5-1,0% мас./мас., например, примерно 0,5% мас./мас. или примерно 1,0% мас./мас. В конкретных вариантах осуществления изобретения соотношение массы активного агента и интенсификатора размола в размалываемой суспензии составляет от около 10:1 до около 20:1. Конкретные варианты осуществления изобретения рассмотрены в приведенных ниже примерах.
Выбор типа и количества интенсификатора размола, такого как поверхностно-активное вещество, производят в соответствии со стандартной практикой получения наночастиц фармацевтически активного агента. Конкретные варианты осуществления изобретения рассмотрены в приведенных ниже примерах.
Дополнительно или альтернативно размалывание может включать размалывание в струйной мельнице. Размалывание в струйной мельнице обычно включает создание циклонного потока подаваемого под давлением газа (например, азота) внутри стальной цилиндрической камеры размалывания. Подлежащий размалыванию материал вводят в указанную камеру в условиях вакуума, создаваемого с помощью отверстия для отвода воздуха на входе. Центробежная сила, создаваемая циклонным потоком газа, направляет частицы на внутренние стенки камеры, сталкивая их друг с другом и разбивая на более мелкие частицы. Когда частицы достигают размера, при котором воздействующие на частицы аэродинамические силы, создаваемые выходящим потоком газа, начинают превышать аэродинамические силы, создаваемые центробежной силой, частицы выносятся из мельницы и улавливаются фильтром. Типичные струйные мельницы включают спиральные струйные мельницы, контурные струйные мельницы и струйные мельницы с псевдоожиженным слоем.
Дополнительно или альтернативно наночастицы активного агента могут быть получены любыми другими методами уменьшения размера частиц, такими как гомогенизация под высоким давлением, перекристаллизация, дробление, просеивание и/или центрифугирование, позволяющими получить наночастицы со средним диаметром менее примерно 1 мкм.
В некоторых вариантах осуществления изобретения наночастицы активного агента состоят из фармацевтически активных частиц, которые получают, осуществляя размалывание без использования интенсификатора размола или других наполнителей. В некоторых вариантах осуществления изобретения наночастицы активного агента состоят из фармацевтически активного агента и интенсификатора размола, которые получают, осуществляя размалывание с использованием интенсификатора размола. В конкретных вариантах осуществления изобретения соотношение массы активного агента и интенсификатора размола в наночастицах активного агента составляет от около 10:1 до около 20:1. В конкретных вариантах осуществления изобретения наночастицы активного агента не содержат носителя. В конкретных вариантах осуществления изобретения наночастицы активного агента не содержат связующего вещества. Таким образом, наночастицы активного агента могут быть получены без какого-либо носителя или связующего вещества со средним размером менее примерно 1 мкм.
В некоторых вариантах осуществления изобретения наночастицы активного агента имеют средний диаметр, равный примерно 0,01 мкм, 0,05 мкм, 0,1 мкм, 0,2 мкм, 0,3 мкм, 0,4 мкм, 0,5 мкм, 0,6 мкм, 0,7 мкм, 0,8 мкм, 0,9 мкм, менее примерно 1 мкм, или средний диаметр в диапазоне между любыми двумя указанными значениями. В конкретных вариантах осуществления изобретения наночастицы активного агента имеют средний диаметр больше примерно 0,1 мкм и меньше примерно 1,0 мкм. В конкретных вариантах осуществления изобретения наночастицы активного агента имеют срединный диаметр, равный примерно 0,2 мкм. В конкретных вариантах осуществления изобретения примерно до 10% наночастиц активного агента имеют диаметр около 0,1 мкм или меньше. В конкретных вариантах осуществления изобретения по меньшей мере примерно 90% наночастиц активного агента имеют диаметр до около 0,8 мкм. В конкретных вариантах осуществления изобретения по меньшей мере примерно 80% наночастиц активного агента имеют диаметр от около 0,1 мкм до около 0,8 мкм. В наиболее конкретных вариантах осуществления изобретения наночастицы активного агента имеют срединный диаметр около 0,2 мкм, при этом примерно до 10% наночастиц активного агента имеют диаметр около 0,1 мкм или меньше и 90% наночастиц активного агента имеют диаметр до около 0,8 мкм или по меньшей мере 80% наночастиц активного агента имеют диаметр от около 0,1 мкм до около 0,8 мкм.
Носитель
В соответствии с любыми вариантами осуществления изобретения носитель может быть любым носителем, пригодным для использования в качестве носителя для сухой частицы, такой как высушенная распылением частица, для сухого фармацевтического порошкообразного состава, такого как состав, предназначенный для введения через слизистую оболочку, в частности, для назального или интравагинального введения. Носитель может быть выбран с учетом распределения частиц по размерам, совместимого с целевым распределением по размерам сухих (например, высушенных распылением) частиц по настоящему изобретению. Например, может быть выбран носитель с распределением частиц по размерам меньше целевого распределения по размерам сухих (например, высушенных распылением) частиц, как более подробно описано ниже.
В некоторых вариантах осуществления изобретения носитель поглощает воду примерно при рН 7,4 и температуре около 36°С, но предпочтительно не является водорастворимым в указанных условиях. В других вариантах осуществления изобретения носитель поглощает воду и/или предпочтительно является водорастворимым в указанных условиях.
Типичные носители включают, например, водопоглощающие и не растворимые в воде целлюлозы, такие как кристаллическая целлюлоза (в том числе микрокристаллическая целлюлоза), целлюлоза, α-целлюлоза и перекрестносшитая натрий-карбоксиметилцеллюлоза; перекрестносшитые винильные полимеры, такие как перекрестносшитый винилполивинилпирролидон, перекрестносшитый карбоксивинильный полимер или его соли, перекрестносшитый поливиниловый спирт и полигидроксиэтилметакрилат. Дополнительные примеры носителей включают, например, водопоглощающие и не растворимые в воде крахмалы, такие как гидроксипропиловый крахмал, карбоксиметиловый крахмал, перекрестносшитый крахмал, амилазу, амилопектин и пектин; водопоглощающие и не растворимые в воде белки, такие как желатин, казеин; водопоглощающие и не растворимые в воде камеди, такие как аравийская камедь, трагант и глюкоманнан.
В соответствии с любыми вариантами осуществления изобретения носитель может быть микрокристаллической целлюлозой (МСС). Микрокристаллическая целлюлоза может быть приобретена под торговым названием Ceolus® в компании Asahi Kasein Corporation, включая продукт, продаваемый как РН-F20JP. Кроме того, в композициях по настоящему изобретению может быть использована МСС корпорации FMC, продаваемая под торговым названием Avicel®, включая Avicel® PH-105.
Связующее вещество
В соответствии с любыми вариантами осуществления изобретения связующее вещество может быть любым связующим веществом, пригодным для использования в качестве связующего вещества для сухой частицы, такой как высушенная распылением частица, для сухого фармацевтического порошкообразного состава, такого как состав, предназначенный для назального введения. В некоторых вариантах осуществления изобретения связующее вещество объединяют с наночастицами активного агента до их обработки (например, размалывания и т.д.) с целью получения частиц со средним размером менее примерно 1 мкм. Таким образом, в таких вариантах осуществления изобретения наночастицы активного агента обрабатывают в присутствии связующего вещества для получения частиц со средним размером менее примерно 1 мкм.
Как указано выше, в некоторых вариантах осуществления изобретения связующее вещество поглощает воду примерно при рН 7,4 и температуре около 36°С и обычно является водорастворимым в указанных условиях.
Типичные связующие вещества включают полиакрилаты и их соли (например, соли натрия, калия, аммония), низшие алкиловые эфиры целлюлозы, такой как кальций-карбоксиметилцеллюлоза, гидроксипропилцеллюлоза (НРС, НРС-SL и HPC-L), гидроксипропилметилцеллюлоза (НРМС), гипромеллоза 2910, натрий-карбоксиметилцеллюлоза (NaCMC), метилцеллюлоза, гидроксиэтилцеллюлоза, гидроксипропилцеллюлоза, фталат гидроксипропилметилцеллюлозы, некристаллическая целлюлоза, олеиновую кислоту, глицерин, белый вазелин, сложный эфир глицерина гидрированной канифоли, пропиленгликоль, полиэтиленгликоль 400, полиэтиленгликоли (например, CarboWax 3350® и 1450® (The Dow Chemical co.) и Carbopol 934® (Lubrizol Corp.)), поливинилпирролидон (не являющийся перекрестносшитым), амилазу, алюмосиликаты магния, спирт (дегидратированный), дихлордифторметан, дихлортетрафторэтан, норфлуран, триолеат сорбита, трихлормонофторметан, хлорид бензалкония, цитрат натрия, уксусную кислоту, гидроксианизол, хлорбутанол, лимонную кислоту, моногидрат лимонной кислоты, динатрийэдетат, хлористоводородную кислоту, метилпарабен, полиэтиленгликоль 3350, одноосновный фосфат калия, пропилпарабен, ацетат натрия, хлорид натрия, гидроксид натрия, фосфат натрия, двухосновный безводный фосфат натрия, гептагидрат двухосновного фосфата натрия, сорбит, сахаралозу, безводную декстрозу, декстрозу, фенилэтиловый спирт, полисорбат 80, серную кислоту, аллил-альфа-ионон, безводный тринатрийцитрат, хлорид бензэтония, бензиловый спирт, бутилированный гидроксианизол, бутилированный гидрокситолуол, кофеин, диоксид углерода, декстрозу, азот, полиоксилстеарат 400, полисорбат 20, сорбат калия, додекагидрат двухосновного фосфата натрия, гептагидрат двухосновного фосфата натрия, одноосновный безводный фосфат натрия, дегидрат одноосновного фосфата натрия, мукоадгезивные полимеры, такие как описанные в публикации Chaturvedi et al., J. Adv. Pharm. Technol. Res. 2(4): 215-222 2011; желатинизаторы F-127, такие как описанные в публикации Khairnar et al., Int. J. Pharm. Sci. 3: 250 (2011); производные хитозаны, такие как описанные в заявке на патент США 2010/0256091; и связующие вещества, описанные в патенте США № 5958458.
Типичные связующие вещества включают полиакрилаты и их соли (например, соли натрия, калия, аммония), низшие алкиловые эфиры целлюлозы, такой как кальций-карбоксиметилцеллюлоза, гидроксипропилцеллюлоза (НРС, НРС-SL и HPC-L), гидроксипропилметилцеллюлоза (НРМС), гипромеллоза 2910, натрий-карбоксиметилцеллюлоза (NaCMC), метилцеллюлоза, гидроксиэтилцеллюлоза, гидроксипропилцеллюлоза, фталат гидроксипропилметилцеллюлозы, некристаллическая целлюлоза, олеиновую кислоту, глицерин, белый вазелин, сложный эфир глицерина гидрированной канифоли, полиэтиленгликоли, пропиленгликоль и полиэтиленгликоль 400.
Высушенные распылением частицы
Как указано выше, сухие композиции по настоящему изобретению могут включать сухие частицы, такие как высушенные распылением частицы, состоящие из наночастиц активного агента, связующего вещества и фармацевтически приемлемого носителя.
Высушенные распылением частицы могут быть получены известными в данной области методами получения высушенных распылением частиц для фармацевтических составов. См., например, публикацию Masters et al., The Spray-Drying Handbook, 5th Ed., Longman Scientific & Technical (1991). Как правило, сушка распылением позволяет получить частицы с относительно однородным распределением по размерам благодаря аэрозолизации раствора или суспензии материала в нагретой камере, при этом растворитель испаряется, оставляя сухие частицы растворенного вещества.
Например, во время сушки распылением распылительная сушилка (например, Buchi B290) может разделять жидкий поток на твердое вещество (растворенное вещество или компоненты суспензии) и пар (компоненты растворителя). Твердое вещество обычно собирают в барабане или циклоне. Входящий поток жидкости может быть распылен через сопло в поток горячего пара (например, воздуха или азота) и выпарен. Твердые вещества быстро выделяются из капель во влажном состоянии. Сопло обычно служит для получения очень мелких капель, обеспечения максимальной теплопередачи и быстрого испарения воды. Например, в типичном процессе сушки распылением размер капель в зависимости от сопла находится в пределах от около 20 мкм до 180 мкм, от около 20 мкм до 100 мкм или от около 20 мкм до 50 мкм. В некоторых вариантах осуществления изобретения использовано одно распылительное сопло высокого давления (50-300 бар). В других вариантах осуществления изобретения использованы два распылительных сопла, из которых одно распылительное сопло предназначено для сушки жидкости и второе распылительное сопло предназначено для подачи сжатого газа (обычно воздуха под давлением 1-7 бар).
В типичном способе наночастицы активного агента, связующее вещество и фармацевтически приемлемый носитель объединяют с образованием водной композиции (то есть суспензии) (то есть исходного материала) и сушат распылением, получая при этом высушенные распылением частицы. В некоторых вариантах осуществления изобретения водная композиция может быть разбавлена водой до сушки распылением. Например, в одном типичном варианте осуществления изобретения, представленном в нижеследующей таблице 8, получают водную композицию, включающую прогестерон (20% мас./мас.), PS20 (1% мас./мас.), МСС (10% мас./мас.), НРМС (3% мас./мас.) и воду (66% мас./мас.), которую разбавляют водой (например, 50-75% мас./мас.) до сушки распылением.
В некоторых вариантах осуществления изобретения в процессе получения наночастиц активного агента используют интенсификатор размола, такой же как в процессе размалывания, который может служить в качестве диспергатора в процессе сушки распылением. К водной суспензии может быть необязательно добавлен диспергатор, аналогичный известным в данной области, включая вышеописанные поверхностно-активные вещества, такие как полисорбат 20.
В качестве примера наночастицы активного агента могут составлять примерно 0,1-25,0% мас./мас. водной композиции, подвергаемой сушке распылением, включая примерно 0,5-5,0% мас./мас. наночастиц активного агента, в том числе примерно 1,0% мас./мас. наночастиц активного агента. В конкретных вариантах осуществления изобретения водная композиция, подвергаемая сушке распылением, включает примерно 5,0-20,0% мас./мас. наночастиц активного агента, в том числе примерно 5,0, 6,0, 7,0, 8,0, 9,0, 10,0, 11,0, 12,0, 13,0, 14,0 и 15,0% мас./мас. наночастиц активного агента, например, 9,0, 10,0, 15,0 или 20,0% мас./мас. наночастиц активного агента. В других конкретных вариантах осуществления изобретения водная композиция, подвергаемая сушке распылением, включает примерно 0,3-1,0% мас./мас. наночастиц активного агента, в том числе примерно 0,3, 0,4, 0,5, 0,6, 0,7, 0,8, 0,9 и 1,0% мас./мас. наночастиц активного агента.
В качестве примера носитель (такой как МСС) может составлять примерно 0,1-25,0% мас./мас. водной композиции, подвергаемой сушке распылением. В конкретных вариантах осуществления изобретения водная композиция, подвергаемая сушке распылением, включает примерно 2,0-15,0% мас./мас. носителя, в том числе примерно 2,5-5,0% мас./мас., примерно 5,0-7,0% мас./мас., примерно 7,0-12,0% мас./мас. носителя, включая 8,0, 9,0, 10,0 и 11,0% мас./мас., например, 9,0 или 10,0% мас./мас. носителя. В других конкретных вариантах осуществления изобретения водная композиция, подвергаемая сушке распылением, включает примерно 5,0, 6,0, 7,0% мас./мас. носителя.
В качестве примера связующее вещество (такое как НРМС или NaCMC) может составлять примерно 0,01-5,0% мас./мас. водной композиции, подвергаемой сушке распылением. В конкретных вариантах осуществления изобретения водная композиция, подвергаемая сушке распылением, включает примерно 0,3-4,0% мас./мас. связующего вещества, в том числе 0,375, 0,75, 1,0, 1, 2,0, 2,5 или 3,0% мас./мас. связующего вещества, например, примерно 1,0% мас./мас. связующего вещества. В других конкретных вариантах осуществления изобретения водная композиция, подвергаемая сушке распылением, включает примерно 1,8-3,0% мас./мас. связующего вещества.
Относительные количества активного агента, связующего вещества и носителя могут изменяться в зависимости от конкретно используемого активного агента, связующего вещества и носителя и требуемого метода получения сухих частиц и сухой фармацевтической композиции. В типичных вариантах осуществления изобретения соотношение массы активного агента и носителя в водной композиции, подвергаемой сушке распылением, составляет от около 1:1 до около 2:1, в том числе примерно 1,5:2, при этом масса активного агента больше массы носителя. В типичных вариантах осуществления изобретения соотношение массы активного агента и связующего вещества в водной композиции, подвергаемой сушке распылением, составляет от около 5:1 до около 20:1, в том числе от около 10:1,5 до 20:1,5, включая примерно 10:1. В других типичных вариантах осуществления изобретения соотношение массы активного агента и связующего вещества в водной композиции, подвергаемой сушке распылением, составляет от около 1:2 до около 1:20, например, от около 1:5 до около 1:15, в том числе примерно 1:10, при этом масса активного агента меньше массы носителя.
В конкретных вариантах осуществления изобретения активный агент, интенсификатор размола, носитель и связующее вещество представляют собой соответственно прогестерон, полисорбат 20, МСС и НРМС.
В других конкретных вариантах осуществления изобретения активный агент, интенсификатор размола, носитель и связующее вещество представляют собой соответственно фуроат мометазона, хлорид бензалкония, МСС и НРМС.
В типичных вариантах осуществления изобретения высушенные распылением частицы включают примерно 10,0-75,0% мас./мас. активного агента (такого как прогестерон), в том числе примерно 40-70,0% мас./мас. активного агента, в частности, примерно 45,0 (в том числе 45,5), примерно 50,0 (в том числе примерно 52), примерно 55, примерно 60 (в том числе примерно 59, 61 и 62) и примерно 65% мас./мас. активного агента. В других вариантах осуществления изобретения высушенные распылением частицы включают по меньшей мере примерно 10%, 25,0%, 40,0%, 45,0%, 50,0%, 75,0% или более активного агента (мас./мас.).
В других вариантах осуществления изобретения высушенные распылением частицы включают примерно 1,0-25,0% мас./мас. активного агента, в частности, от около 5% до около 10% мас./мас., в том числе от около 6,0% до около 7% мас./мас. активного агента. Указанные последними варианты осуществления изобретения представлены ниже с использованием фуроата мометазола в качестве активного агента.
В типичных вариантах осуществления изобретения высушенные распылением частицы включают примерно 10,0-60,0% мас./мас. носителя (такого как МСС), например, примерно 25,0-50,0% мас./мас. носителя или примерно 30,0-46,0% мас./мас. носителя, в частности, примерно 31%, 36% или 45,5% мас./мас. носителя. В других типичных вариантах осуществления изобретения высушенные распылением частицы включают примерно 30, 31, 34, 35 или 36% мас./мас. носителя. Указанные варианты осуществления изобретения рассмотрены ниже с использованием прогестерона в качестве активного агента.
В других типичных вариантах осуществления изобретения высушенные распылением частицы включают примерно 60,0-80,0% мас./мас. носителя (такого как МСС), например, примерно, 61,0-70,0% мас./мас. носителя. Указанные варианты осуществления изобретения представлены ниже с использованием фуроата мометазона в качестве активного агента.
В типичных вариантах осуществления изобретения высушенные распылением частицы включают примерно 1,0-15,0% мас./мас. связующего вещества (такого как НРМС или NaCMC), в том числе примерно 1,0-8,0% мас./мас. или примерно 2,5-5,0% мас./мас., в частности, примерно 3,1% мас./мас. или примерно 3,7% мас./мас. связующего вещества. В других типичных вариантах осуществления изобретения высушенные распылением частицы включают примерно 4,0-6,0% мас./мас., в частности, примерно 4,0% мас./мас. или примерно 4,5% мас./мас. связующего вещества. В других типичных вариантах осуществления изобретения высушенные распылением частицы включают примерно 5,5, примерно 9 или примерно 10,5% мас./мас. связующего вещества. Указанные варианты осуществления изобретения рассмотрены ниже с использованием прогестерона в качестве активного агента.
В других типичных вариантах осуществления изобретения высушенные распылением частицы включают большее количество связующего вещества, в частности, примерно 10,0-50,0% мас./мас. связующего вещества (такого как НРМС или NaCMC), в том числе примерно 20,0 или примерно 30,0% мас./мас. связующего вещества. Указанные варианты осуществления изобретения рассмотрены ниже с использованием фуроата мометазона в качестве активного агента.
В некоторых вариантах осуществления изобретения количество связующего вещества (например, НРМС) выбирают и регулируют в зависимости от диаметра высушенных распылением частиц. Например, как показано в приведенном ниже примере 4, увеличение содержания НРМС с около 1,5% до около 3,0% мас./мас. вызывает образование высушенных распылением частиц с большим диаметром. Указанные частицы также характеризуются более медленной скоростью высвобождения лекарственного средства.
Как указано выше и проиллюстрировано в нижеследующих примерах, при получении наночастиц активного агента может быть использован интенсификатор размола или поверхностно-активное вещество, тогда высушенные распылением частицы могут включать интенсификатор размола или поверхностно-активное вещество (такое как хлорид бензалкония, твин 20® или твин 80®). В указанных вариантах осуществления изобретения такие компоненты могут составлять примерно до 2,0% мас./мас. водной суспензии и/или примерно до 5,0% мас./мас. высушенных распылением частиц. В некоторых вариантах осуществления изобретения такие компоненты могут составлять примерно до 0,1% мас./мас. водной суспензии, например, от около 0,05 до около 0,07% мас./мас., и/или примерно до 1,0% мас./мас. высушенных распылением частиц, например, от около 0,5 до около 0,7% мас./мас. Указанные последними варианты осуществления изобретения рассмотрены ниже с использованием фуроата мометазона в качестве активного агента.
В некоторых вариантах осуществления изобретения водная композиция, подвергаемая сушке распылением, включает в расчете на массу примерно 9% активного агента, примерно 9% носителя, примерно 0,9% связующего вещества, примерно 0,9% интенсификатора размола и воду. В других вариантах осуществления изобретения водная композиция, подвергаемая сушке распылением, включает в расчете на массу примерно 10, 15 или 20% активного агента, примерно 10% носителя, примерно 1, 1,5 или 3% связующего вещества, примерно 1% интенсификатора размола и воду. Указанные варианты осуществления изобретения рассмотрены ниже с использованием прогестерона в качестве активного агента.
В других вариантах осуществления изобретения водная композиция, подвергаемая сушке распылением, включает в расчете на массу примерно 0,6 или 0,7% активного агента, примерно 6-7% носителя, примерно 1,8-3% связующего вещества, примерно 0,06-0,07% интенсификатора размола и воду. Указанные варианты осуществления изобретения рассмотрены ниже с использованием фуроата мометазона в качестве активного агента.
В соответствии с любыми вариантами осуществления изобретения, рассмотренными в настоящем описании изобретения, активный агент может составлять от около 40 до около 75% мас./мас., в том числе примерно 60%, примерно 65% и примерно 70% мас./мас. высушенных распылением частиц. В конкретных вариантах осуществления изобретения носитель может составлять от около 25 до около 50% мас./мас. (в том числе примерно 30% мас./мас.) высушенных распылением частиц. В других конкретных вариантах осуществления изобретения связующее вещество может составлять от около 1 до около 15% мас./мас. (в том числе примерно 5% или примерно 10% мас./мас.) высушенных распылением частиц. В конкретных вариантах осуществления изобретения интенсификатор размола может составлять от около 0,1 до около 5,0% мас./мас. (в том числе примерно 2% и примерно 4% мас./мас.) высушенных распылением частиц. Указанные варианты осуществления изобретения рассмотрены ниже с использованием прогестерона в качестве активного агента.
Альтернативно, в соответствии с любыми вариантами осуществления изобретения, рассмотренными в настоящем описании изобретения, активный агент может составлять примерно 6-7% мас./мас. высушенных распылением частиц. В конкретных вариантах осуществления изобретения носитель может составлять от около 60 до около 70% (в том числе от около 61 до около 70% мас./мас.) высушенных распылением частиц. В других конкретных вариантах осуществления изобретения связующее вещество может составлять от около 20 до около 30% (в том числе от около 21 до около 31% мас./мас.) высушенных распылением частиц. В других конкретных вариантах осуществления изобретения интенсификатор размола может составлять примерно 0,6-0,7% мас./мас. высушенных распылением частиц. Указанные варианты осуществления изобретения рассмотрены ниже с использованием фуроата мометазона в качестве активного агента.
В конкретных вариантах осуществления изобретения размер высушенных распылением частиц выбирают и регулируют с возможностью достижения назальной доставки активного агента при отсутствии или минимальной доставке в легкие. Например, частицы со средним диаметром, равным примерно 100 мкм, являются достаточно крупными для успешного воздействия на слизистую оболочку носа, но не могут в значительной степени всасываться через слизистую оболочку носа. С другой стороны, более мелкие частицы (например, частицы со средним диаметром около 2 мкм) могут слишком легко вдыхаться в легкие (доставка в легкие). Как правило, частицы со средним диаметром около 10 мкм или больше могут всасываться интраназально, в то время как частицы со средним диаметром около 5 мкм или меньше могут всасываться в легких. Таким образом, высушенные распылением частицы композиций по настоящему изобретению могут характеризоваться следующим распределением частиц по размерам:
до 10% высушенных распылением частиц имеют размер менее 10 мкм;
по меньшей мере 50% высушенных распылением частиц имеют размер, равный по меньшей мере примерно 15 мкм и
по меньшей мере 90% высушенных распылением частиц имеют размер примерно до 55 мкм.
В конкретных вариантах осуществления изобретения размер высушенных распылением частиц выбирают и регулируют в зависимости от параметров сушки распыления, известных в данной области, в соответствии с представленным выше руководством и приведенными примерами. Типичные неограничивающие примеры параметров, которые могут изменяться в процессе сушки распылением, включают растворители, концентрации исходных веществ, скорости потоков подаваемых веществ и/или воздуха и температуры процесса. Дополнительно или альтернативно размер высушенных распылением частиц может быть выбран и отрегулирован с помощью методов, включающих просеивание и/или центрифугирование.
В некоторых вариантах осуществления изобретения высушенные распылением частицы характеризуются хорошим высвобождением активного агента. Например, испытание на растворение может быть выполнено в соответствии с USP II (лопастное устройство 2) при скорости 75 оборотов/мин., 37°С, в растворяющей среде, содержащей 900 мл 1,0% твина® 80 в физиологическом растворе с фосфатным буфером (PBS) при рН 7,4. В некоторых вариантах осуществления изобретения высушенные распылением частицы высвобождают более 90% активного агента в течение 60 минут, в течение 30 минут, в течение 20 минут, в течение 15 минут или более короткого периода времени при испытании указанным методом, как показано в приведенных ниже примерах.
Другие сухие частицы
Хотя объектом приведенного выше описания являются высушенные распылением частицы, сухие композиции по настоящему изобретению могут включать сухие частицы, полученные методами, отличными от сушки распылением. Например, сухие частицы, включающие активный агент, связанный с носителем, могут быть получены любым методом, известным в данной области и предназначенным для физического связывания частиц друг с другом, таким как мокрая грануляция, сухая грануляция и обработка в псевдоожиженном слое.
Типы и относительные количества активного агента, связующего вещества и носителя, используемые для получения сухих частиц методами, отличными от сушки распылением, могут соответствовать параметрам, приведенным выше для высушенных распылением частиц. Таким образом, типы и относительные количества активного агента, связующего вещества и носителя, указанные выше для высушенных распылением частиц, применимы в вариантах осуществления изобретения, относящихся к получению сухих частиц методами, отличными от сушки распылением, хотя такие компоненты и относительные количества могут быть изменены в соответствии со стандартной практикой, применяемой в данной области.
При мокрой грануляции гранулы (то есть частицы) образуются в результате добавления гранулирующей жидкости (например, включающей связующее вещество и растворитель) в слой порошка, подвергаемый воздействию ротора (такого как шнеки в грануляторе с высоким сдвигающим усилием или воздух в грануляторе с псевдоожиженным слоем). В некоторых вариантах осуществления изобретения наночастицы вводят в частицы носителя и диспергируют в гранулирующей жидкости (например, со связующим веществом). Смачивание и перемешивание в процессе мокрой грануляции позволяют получить наночастицы активного агента, связанные с частицами носителя (например, МСС). Растворитель гранулирующей жидкости (который обычно является водой или летучим растворителем, таким как этанол или изопропанол) удаляют, получая при этом сухие частицы, включающие наночастицы активного агента, связанные с носителем.
При сухой грануляции гранулирующая жидкость не используется. Гранулы образуются путем прессования и уплотнения сухих компонентов, например, под высоким давлением. Гранулятор Свеинга или гранулятор-смеситель с высоким сдвигающим усилием являются примерами оборудования, которое может быть использовано для сухой грануляции. В результате осуществления указанного процесса образуются сухие частицы, включающие наночастицы активного агента, связанные с носителем.
При обработке в псевдоожиженном слое псевдоожиженный слой сухой порошкообразной композиции помещают в условия, заставляющие сухую порошкообразную композицию вести себя как жидкость. Такое состояние обычно достигается путем нагнетания сжатого воздуха, газа или других текучих сред через слой сухой порошкообразной композиции. Вследствие этого сухая порошкообразная композиция приобретает свойства и признаки, подобные свойствам обычных жидкостей, и находится в состоянии, известном как псевдоожижение. Таким образом, обработка в псевдоожиженном слое может быть использована для образования сухих частиц, включающих наночастицы активного агента, связанные с носителем.
Необязательные добавки
Композиции по настоящему изобретению могут необязательно включать один или несколько фармацевтически приемлемых наполнителей. Приемлемые наполнители описаны в справочнике Handbook of Pharmaceutical Excipients, Pharmaceutical Press, 2012, опубликованном Американской фармацевтической ассоциацией и Королевским фармацевтическим обществом Великобритании.
Типичные фармацевтически приемлемые наполнители включают подкисляющие, алкализирующие, связывающие, хелатообразующие, комплексообразующие и/или солюбилизирующие агенты, антисептики, консерванты, в том числе антимикробные средства, например, метил- и пропилгидроксибензоаты, антиоксиданты, например, α-токоферол или аскорбилпальмитат, стабилизирующие агенты, агенты, изменяющие вязкость, растворители, разбавители, смазывающие вещества, такие как тальк, стеарат магния и минеральное масло, смачивающие вещества, эмульгирующие и суспендирующие агенты, ароматизаторы, терапевтические средства, полиолы, буферы и инертные наполнители.
В некоторых вариантах осуществления изобретения сухие частицы, такие как высушенные распылением частицы, получают с наполнителями, выбираемыми для улучшения текучести сухой композиции, которые описаны, например, в европейском патенте ЕР 2116264, который полностью включен в настоящее описание изобретения в качестве ссылки. Например, композиция может включать микрокристаллическую целлюлозу с меньшим диаметром частиц, микрокристаллическую целлюлозу с большим диаметром частиц и трехосновный фосфат кальция. В частности, состав может включать первую кристаллическую целлюлозу, которая имеет объемную плотность в интактном состоянии 0,13-0,29 г/см3, удельную площадь поверхности 1,3 м2/г или больше, средний диаметр частиц 30 мкм или меньше и угол отражения 55° или больше; вторую кристаллическую целлюлозу, которая имеет объемную плотность в интактном состоянии 0,26-0,48 г/см3, удельную площадь поверхности 1,3 м2/г или меньше, угол отражения 50° или меньше и средний диаметр частиц 150 мкм или меньше, например, от около 30 до около 150 мкм, от около 30 до около 100 мкм, в том числе от около 40 до около 75 мкм, и трехосновный фосфат кальция или любой один или несколько таких компонентов. В частности, считается, что микрокристаллическая целлюлоза с частицами большего диаметра улучшает текучесть композиции. В конкретных вариантах осуществления изобретения композиция включает примерно 5,0-30,0% мас./мас. микрокристаллической целлюлозы с частицами большего диаметра или крахмала (либо их смеси), например, примерно 10% мас./мас. микрокристаллической целлюлозы с частицами большего диаметра или крахмала (либо их смеси) и примерно 0,1-10% мас./мас. трехосновного фосфата кальция, в том числе примерно 1% мас./мас. трехосновного фосфата кальция в расчете на общую массу фармацевтической композиции. Альтернативно сухие частицы, такие как высушенные распылением частицы по настоящему изобретению, вводят в композицию, улучшающую текучесть, которая включает первую микрокристаллическую целлюлозу, вторую микрокристаллическую целлюлозу и трехосновный фосфат кальция, при этом трехосновный фосфат кальция составляет примерно 0,1-10% мас./мас. улучшающей текучесть композиции и вторая микрокристаллическая целлюлоза составляет от около 5 до около 30% мас./мас. улучшающей текучесть композиции. В некоторых вариантах осуществления изобретения первая микрокристаллическая целлюлоза является компонентом сухих (например, высушенных распылением) частиц или необязательно отсутствует, поэтому композиция включает сухие (например, высушенные распылением) частицы, микрокристаллическую целлюлозу со средним диаметром частиц от около 30 до около 150 мкм и трехосновный фосфат кальция.
Таким образом, сухие частицы, такие как высушенные распылением частицы по настоящему изобретению, могут быть необязательно объединены с одним или несколькими необязательными фармацевтически приемлемыми наполнителями с образованием конечной сухой фармацевтической композиции.
В конкретных вариантах осуществления изобретения композиции являются стерильными. В некоторых вариантах осуществления изобретения композиции удовлетворяют требованиям, указанным в одной или нескольких главах фармакопеи США, в частности, включающих главу USP <71> (стерильность), главу USP <85> (“тест на бактериальный эндотоксин”) и главу USP <151> (“тест на пироген”).
Композиции по настоящему изобретению могут быть получены в готовой к применению форме. Термин “готовый к применению” в использованном здесь значении означает, что композиция не требует дальнейшей обработки, такой как разведение или смешивание нескольких компонентов.
Композиции могут быть получены в герметичной упаковке, пригодной для сухих составов. В конкретных вариантах осуществления изобретения композиция упакована в герметичную емкость. Указанная емкость может быть завернута для защиты от воздействия физической среды.
В некоторых вариантах осуществления изобретения композиции получают в стерильной, готовой к применению форме и могут храниться по меньшей мере один год, три года или семь лет при комнатной температуре.
Интравагинальное введение
Сухие фармацевтические композиции по настоящему изобретению могут быть введены интравагинально при помощи методов, инструментов для местного применения лекарственного средства и/или устройств, известных в данной области.
Интраназальное введение
Сухие фармацевтические композиции по настоящему изобретению могут быть введены интраназально при помощи методов, инструментов для местного применения лекарственного средства и/или устройств, известных в данной области.
В некоторых вариантах осуществления изобретения сухая композиция предназначена для применения в назальных распылителях, оснащенных системами распыления однократной дозы, компании Aptar Inc., (Crystal Lake, IL); устройствах для назальной доставки лекарственного средства, приводимых в действие дыханием, компании OptiNose Inc. (Yardley, Pennsylvania); устройствах для доставки лекарственного средства через “назальную соломинку” TriVair™ компании Trimel Inc. (Mississauga, Ontario); ингаляторах сухого порошка (DPI) MicroDose™, распылителях сухого порошка (DPN) MicroDose™, “электрических” назальных распылителях компании MicroDoseTherapeutx Inc. (Monmouth Junction, NJ) и инсуффляторах однократной дозы компании MIAT S.p.A. (Milan, Italy).
В некоторых вариантах осуществления изобретения сухая композиция предназначена для применения в устройстве DPI, таком как вышеописанные устройства DPI, или диспергирована в пропелленте, используемом в находящихся под давлением дозирующих ингаляторах (PMDI). Действие DPI основано на силе вдыхаемого воздуха, который, проходя через устройство, доставляет дозу лекарственного средства. Такие устройства описаны, например, в патенте США № 4807814, в котором представлен пневматический эжектор порошка, характеризующийся наличием стадии всасывания и стадии впрыскивания, и в патенте США № 5785049, в котором представлены устройствам доставки сухого порошка для лекарственных средств. Композиции, предназначенные для применения в PDMI, могут быть получены в виде суспензии в приемлемом пропелленте, таком как галогенированный углеводород. PMDI описаны, например, в публикации Newman, S.P., Aerosols and the Lung, Clarke et al., eds., pp. 197-224 (Butterworth’s, London, England, 1984). Устройства PDMI высвобождают определенную дозу при каждом приведении в действие.
В некоторых вариантах осуществления изобретения композицию вводят при помощи интраназального инсуффлятора. Инсуффлятор может включать механизм впрыскивания фиксированной дозы для доставки по существу постоянной дозы композиции.
Другие способы введения
Композиции по настоящему изобретению могут быть также введены другими способами. В некоторых вариантах осуществления изобретения сухие фармацевтические композиции по настоящему изобретению могут быть использованы, например, для перорального, трансбуккального или внутриглазного введения. Дополнительно или альтернативно могут быть получены композиции, предназначенные для инъекций или вливания, в частности, для внутривенного, подкожного или внутримышечного введения.
Методы лечения
Сухие фармацевтические композиции по настоящему изобретению пригодны для введения через слизистую оболочку при лечении любого состояния, подлежащего лечению путем введения активного агента через слизистую оболочку с достижением местного, регионарного или системного действия. Как указано выше, сухие фармацевтические композиции по настоящему изобретению также пригодны для введения другими способами при лечении любого состояния, подлежащего лечению данным активным агентом с достижением местного, регионарного или системного действия.
В конкретных вариантах осуществления изобретения при использовании прогестерона в качестве активного агента сухие фармацевтические композиции могут быть использованы в любом терапевтическом методе применения прогестерона для достижения местного, регионарного или системного действия, например, при лечении низких уровней прогестерона у нуждающихся субъектов, в том числе у женщин, подвергаемых искусственному оплодотворению, при возникновении или опасности возникновения преждевременных родов, при необходимости регулирования менструального цикла и т.д.
Сухие фармацевтические композиции по настоящему изобретению, включающие прогестерон, также пригодны для лечения у субъекта, в том числе человека, травматического или ишемического поражения центральной нервной системы, такого как травматическое поражение головного мозга или инсульт, путем интраназальной доставки композиции субъекту, имеющему травматическое или ишемическое поражение центральной нервной системы, в частности, травматическое поражение головного мозга или инсульт.
В конкретных вариантах осуществления изобретения, в которых активный агент является мометазоном или фуроатом мометазона, сухие фармацевтические композиции могут быть использованы в любом терапевтическом методе применения мометазона или фуроата мометазона для достижения местного, регионарного или системного действия, например, при лечении астмы или воспалительных нарушений, таких как воспаление пазух носа и т.д.
В некоторых вариантах осуществления изобретения сухие фармацевтические композиции по настоящему изобретению могут быть использованы для введения субъектам при стационарном лечении, например, в отделении интенсивной терапии. В других вариантах осуществления изобретения сухие фармацевтические композиции по настоящему изобретению могут быть использованы для введения субъектам при неотложной терапии, например, на месте получения травмы, в пункте первой помощи, поликлинике, при отсутствии медицинского работника (например, дома). Например, сухие фармацевтические композиции по настоящему изобретению могут быть введены субъекту интраназально или интравагинально до поступления субъекта в больницу, после выписки субъекта из больницы либо до, во время или после амбулаторного лечения, в кабинете врача, в любой клинике или при отсутствии медицинского работника. В некоторых вариантах осуществления изобретения сухие фармацевтические композиции по настоящему изобретению могут быть введены интраназально или интравагинально в качестве послебольничного лечения субъектов, прошедших в больнице курс внутривенного, внутримышечного или другого введения прогестерона. В некоторых вариантах осуществления изобретения субъект является амбулаторным больным с поражением центральной нервной системы (CNS), не требующим пребывания в больнице, либо был выписан из больницы или прошел курс амбулаторного лечения.
Термин “лечение” предполагает любое улучшение здоровья субъекта, в том числе морфологическое восстановление (то есть повышение жизнеспособности тканей), поведенческую активацию и/или уменьшение интенсивности симптомов заболевания, регресс заболевания или выздоровление после заболевания или состояния. Улучшение может характеризоваться повышением интенсивности и/или степени поведенческого, анатомического и/или физиологического восстановления после поражения, заболевания или состояния. Результатом лечения может быть “положительная терапевтическая реакция”, которая включает полную и частичную реакцию.
Методы лечения могут включать введение терапевтически эффективного количества активного агента, такого как прогестерон. Фраза “терапевтически эффективное количество прогестерона” означает количество прогестерона, достаточное для оказания терапевтического воздействия, например, количество, которое эффективно повышает уровни прогестерона в сыворотке и/или оказывает нейрозащитное действие. Фраза “терапевтически эффективное количество мометазона или фуроата мометазона” означает количество мометазона или фуроата мометазона, достаточное для оказания терапевтического воздействия, например, количество, которое повышает уровни иометазона в сыворотке и/или оказывает противовоспалительное действие.
Как правило, в каждую ноздрю можно ввести примерно до 20-50 мг порошка. Таким образом, доза может включать примерно 35 мг активного агента в каждую ноздрю, при этом будет введено 50 мг высушенных распылением частиц, включающих примерно 70% активного агента. В некоторых вариантах осуществления изобретения доза включает примерно 1 мг, примерно 5 мг, примерно 10 мг, примерно 15 мг, примерно 20 мг, примерно 25 мг, примерно 30 мг, примерно 35 мг или примерно 40 мг активного агента в каждую ноздрю или любое количество в диапазоне указанных значений. Указанные дозы особенно приемлемы, когда активным агентом является прогестерон. Когда активным агентом является мометазон, могут потребоваться более низкие дозы активного агента, например, примерно 100 мкг (0,1 мг) в каждую ноздрю в сутки для взрослого субъекта и примерно 50 мкг (0,05 мг) в каждую ноздрю в сутки для ребенка.
Настоящее изобретение, общее описание которого было дано выше, будет лучше понято со ссылкой на определенные конкретные примеры, приведенные в настоящем описании изобретения только с целью иллюстрации и не ограничивающие объем изобретения.
Примеры
Пример 1. Размалывание прогестерона в дробящей среде с образованием наночастиц
Было создано несколько экспериментальных систем для оценки способности образования наночастиц прогестерона (со средним диаметром частиц <1 мкм) путем размалывания в дробящей среде, как показано в приведенной ниже таблице 1. Размер частиц измеряли методом лазерной дифракции (Horiba LA-950 V2).
Размалывание прогестерона в дробящей среде производили, используя гранулы из диоксида циркония, стабилизированные оксидом иттрия (YTZ). Суспензии 1-9 размалывали, используя дробящую среду с размером гранул 0,5 мм. Суспензии 10 и 11 размалывали последовательно, при этом сначала использовали среду с размером гранул 2 мм и после экстракции использовали среду с размером гранул 0,5 мм. Такое последовательное размалывание позволило получить требуемые наночастицы прогестерона.
Наночастицы были получены из 10% суспензий прогестерона в присутствии 0,5% хлорида бензилалкония (ВАС) или 1% поверхностно-активных веществ, представляющих собой полисорбаты, такие как полисорбат 20 (PS20) или полисорбат 80 (PS80). Оценивали суспензии со связующими веществами и без связующих веществ, таких как гидроксипропилметилцеллюлоза (НРМС) и натрий-карбоксиметилцеллюлоза (NaCMC). В присутствии 1% NaCMC или НРМС были получены более крупные частицы, не соответствующие требованиям. См. суспензии 1, 2, 4, 5, 7 и 8. Из суспензий без связующих веществ были получены наночастицы или частицы почти требуемого наноразмера. См. суспензии 3, 6, 9, 10 и 11.
Из суспензий 10 и 11 были получены наночастицы прогестерона со средним диаметром менее примерно 1 мкм, что соответствовало требованиям. Суспензия наночастиц 10 была использована на следующей стадии сушки распылением.
Пример 2. Образование высушенных распылением частиц
Как указано выше, сушке распылением были подвергнуты наночастицы, полученные из суспензии 10 в соответствии с приведенным выше описанием. (Распределение по размерам указанных наночастиц показано на фигуре 1). Наночастицы экстрагировали и объединяли с микрокристаллической целлюлозой (например, Ceolus PH-F20), НРМС или водой для сушки распылением в соотношениях, указанных в нижеследующей таблице 2, в которой приведены номинальные значения компонентов до и после сушки распылением. В соответствии с условиями, представленными в таблице 3, были получены две партии, А и В, высушенных распылением частиц.
В таблице 3 представлены параметры процесса сушки распылением, использованные при получении двух партий, А и В, высушенных распылением частиц.
В нижеследующей таблице 4 показаны результаты сравнения распределения по размерам высушенных распылением частиц из партий А и В и контрольной партии, содержащей только микрокристаллическую целлюлозу в качестве носителя. Относительные распределения частиц по размерам измеряли в полидиметилсилоксане во избежание растворения связующего вещества НРМС. Часть высушенных распылением частиц из испытуемой партии А испытывали на устойчивость. После хранения в течение одного месяца при -20°С, 40°С/75% относительной влажности или 60°С содержание высушенных распылением частиц было таким же, как и до хранения в пределах обнаружения. В использованной здесь значении D10, D50 и D90 означают диаметр частиц в микронах в соответствии с 10-м, 50-м (срединный) и 90-м процентилями.
Таким образом, в обеих партиях А и В не более 10% высушенных распылением частиц имеют размер менее 10 мкм, по меньшей мере 10% высушенных распылением частиц имеют размер, равный по меньшей мере 10 мкм, по меньшей мере 50% частиц имеют размер, равный по меньшей мере 15 мкм, и по меньшей мере 90% частиц имеют размер примерно до 55 мкм.
Пример 3. Размалывание прогестерона в струйной мельнице
Размалывание в струйной мельнице также было исследовано в качестве метода измельчения прогестерона.
Материал из партии С прогестерона обрабатывали при относительно высоких давлениях (7 кг/см2/6,7 кг/см2) (100 ф/кв.дюйм/95 ф/кв.дюйм). Размеры частиц, полученные в результате первоначального размалывания в струйной мельнице, суммированы в таблице 5. Следующую партию (партия материала D) размалывали при самом низком давлении, создаваемом в струйной мельнице, и значения распределения частиц по размерам, полученные в данном случае, представлены в таблице 5 в виде данных размера частиц после каждой последующей стадии обработки (например, просеивание, грохочение и центрифугирование) партии материала D. Две дополнительные партии (Е и F) были получены в результате размалывания прогестерона в струйной мельнице при низком давлении с последующим просеиванием частиц, размолотых в струйной мельнице, через 45-микронное сито; в таблице 5 также представлены значения распределения частиц по размерам для партий Е и F. В указанных условиях размалывание в струйной мельнице не было таким же эффективным в отношении выхода наночастиц прогестерона как размалывание в дробящей среде.
(100 ф/кв.дюйм/95 ф/кв.дюйм)
(5 ф/кв.дюйм/5 ф/кв.дюйм)
(5 ф/кв.дюйм/5 ф/кв.дюйм) и 45 мкм сито
(5 ф/кв.дюйм/5 ф/кв.дюйм) и 45 мкм сито
Пример 4. Растворение/высвобождение прогестерона
Анализ растворения in vitro (USP II, устройство 2 (лопастное)) был выполнен для определения скорости растворения/высвобождения прогестерона из испытуемых композиций в имитированных физиологических условиях. Параметры анализа растворения представлены в таблице 6.
Анализ высвобождения был выполнен при помощи ВЭЖХ при длине волны обнаружения 243 нм, что соответствует максимальному значению ультрафиолетового спектра прогестерона. Образцы и эталоны разводили смесью воды/ацетонитрила с соотношением 50/50 (об./об.) до рабочей концентрации ввода, равной примерно 50 мг/мл. Прогестерон элюирует примерно через 4,4 мин. Стадию вымывания всего органического содержимого использовали во избежание накопления поверхностно-активного вещества и засорения колонки. Параметры анализа представлены в таблице 7.
Вымывание 100% ацетонитрила (5-7 минут) с последующим повторным уравновешиванием до соотношения 25/75
Высвобождение прогестерона из высушенных распылением частиц (средний диаметр частиц >1 мкм) по настоящему изобретению, включающих наночастицы прогестерона (средний диаметр частиц <1 мкм), и МСС (образец FID-3207), сравнивали с высвобождением из композиции, включающей сухую смесь тонкоизмельченного прогестерона (средний диаметр частиц >1 мкм) и МСС РН-F20 в массовом соотношении 1:1 (FID-3237 ниже). В качестве среды для испытания на растворение был использован физиологический раствор с фосфатным буфером, рН 7,4, с 1,0% PS80. Результаты показаны на фигуре 2.
Как показано на фигуре 2, высушенные распылением частицы (FID-3207) полностью высвобождают прогестерон в течение примерно 15-20 минут, при этом в смеси 1:1 (F-3237) полное высвобождение было достигнуто примерно через 60 минут.
Дополнительные композиции, включающие высушенные распылением частицы, были получены в соответствии с таблицей 8, в которой показано получение водных композиций до сушки распылением, диаметры частиц после сушки распылением, теоретические и действительные композиции после сушки распылением.
(PH-F20)
Высвобождение прогестерона из образцов 5, 6, 7 и 8 сравнивали с высвобождением из композиции, включающей сухую смесь тонкоизмельченного прогестерона (средний диаметр частиц >1 мкм) и МСС РН-F20 с массовым соотношением 1:1 (FID-3237). Полученные результаты показаны на фигуре 3. Как показано на фигуре 3, образцы 5 и 6 характеризуются более быстрым высвобождением прогестерона с достижением больших концентраций прогестерона в принимающей жидкости (например, большее высвобождение лекарственного средства), при этом образцы 7 и 8 характеризовались более медленным высвобождением и более низкими концентрациями прогестерона в принимающей жидкости, но все же высвобождение было более быстрым и концентрации были более высокими по сравнению с аналогичными показателями для смеси 1:1. На фигуре 4 приведены данные профиля высвобождения, показанного на фигуре 3, в виде процентного значения высвобожденного прогестерона, а также показано, что образцы 5, 6 и 8 высвободили больше прогестерона в процентном выражении (примерно 100%) и быстрее по сравнению со смесью 1:1.
Сравнение образцов 5/6 и 7/8 показывает, что связующее вещество в массовом отношении может быть использовано для регулирования скорости высвобождения прогестерона из высушенных распылением частиц по настоящему изобретению. Кроме того, образцы 5, 6, 7 и 8 обеспечивали достижение более высоких концентраций прогестерона (например, высвобождали больше прогестерона), чем смесь 1:1.
Пример 5. Сравнение интраназального и внутривенного введения прогестерона in vivo
Обезьян Cynomolgus исследовали с целью оценки и сравнения концентрации прогестерона в плазме после внутривенного и интраназального введения прогестерона при использовании высушенной распылением композиции по настоящему изобретению и вышеописанной смеси 1:1.
В использованном липидном составе для внутривенного введения концентрация прогестерона была равна 0,25 мг/мл. Указанный состав получали за 24 часа до введения, хранили при комнатной температуре и защищали от воздействия света. Состав для внутривенного введения вводили путем вливания в дозе 1 мг/кг в течение 2 часов в объеме 4 мл/кг со скоростью 2 мл/кг/час, что соответствует дозе, равной 0,5 мг/кг/час.
Составы в виде сухого порошка, предназначенные для интраназального введения, вводили в количестве 25 мг/ноздрю, используя ингалятор унифицированной дозы.
Животным вводили состав для внутривенного введения в 1-й день исследования, смесь 1:1 вводили в 8-й день исследования и высушенный распылением состав вводили в 15-й день исследования.
Периодически брали пробы крови, которые анализировали в отношении концентрации прогестерона.
На фигуре 5 показаны уровни прогестерона в сыворотке обезьян после внутривенного введения (i.v.) или интраназального введения композиции, включающей высушенные распылением частицы, по настоящему изобретению (FID-3207) или композиции, включающей тонкоизмельченный прогестерон (средний диаметр частиц >1 мкм) и микрокристаллическую целлюлозу (МСС РН-F20) с массовым соотношением 1:1 (смесь 1:1). Полученные данные свидетельствуют о быстром всасывании и элиминации после назального введения при большей доставке лекарственного средства из композиции, включающей высушенные распылением частицы, по настоящему изобретению.
На фигуре 6 приведены значения площади под кривой (AUC), полученные при исследовании обезьян, показанном на фигуре 5. Полученные данные свидетельствуют о большем высвобождении прогестерона из композиции, включающей высушенные распылением частицы, по настоящему изобретению, по сравнению со смесью 1:1. (Значения внутривенного введения не подлежат прямому сравнению из-за разной дозы, как указано в приведенном ниже примере 5).
На фигуре 7 приведены значения максимальной концентрации (Cmax), полученные при исследовании обезьян, показанном на фигуре 5. Максимальная концентрация в плазме отличалась в разных группах (F(2, 8) = 22,86, р<0,01). Интраназальное введение сухой смеси 1:1 позволило получить гораздо более низкие концентрации, чем введение композиции, включающей высушенные распылением частицы, по настоящему изобретению (FID-3207) (p<0,01) или внутривенное введение (р<0,01), в то время как показатели, полученные при введении композиции, включающей высушенные распылением частицы, существенно не отличались от внутривенного введения (р>0,05).
На фигуре 8 приведены значения времени до достижения максимальной концентрации (Tmax), полученные при исследовании обезьян, показанном на фигуре 5. Статистический анализ не выполняли; однако, на основании полученных данных прослеживается тенденция сокращения значения Tmax для внутривенного введения и увеличения значения Tmax для обоих интраназальных введений.
Пример 6. Определение in vivo концентрации прогестерона в плазме обезьян после однократного назального введения нескольких составов
Было выполнено подобное исследование обезьян Cynomolgus in vivo для сравнения концентраций прогестерона в плазме после интраназального введения прогестерона с использованием образцов 5 и 7, показанных в таблице 8. Полученные результаты позволили произвести оценку биодоступности.
Образцы 5 и 7 высушенных распылением составов, приведенные в таблице 8, вводили с помощью ингалятора унифицированной дозы в правую и левую ноздри пяти обезьян в дозе 0,25 мг/ноздрю. В частности, образец 5 высушенного распылением состава для интраназального введения вводили животным в 8-й день исследования и образец 7 высушенного распылением состава вводили в 15-й день исследования. У каждой обезьяны брали пробы крови из бедренной вены через 5, 10, 15, 30 и 45 минут и через 1, 1,5, 2, 3, 4, 6, 8 и 12 часов после введения дозы и исследовали в отношении концентрации прогестерона. Анализы проб крови суммированы в приведенной ниже таблице 9.
животное
Стандартное отклонение
Пример 7. Сухие частицы фуроата мометазона
Сухие частицы, включающие фуроат мометазона, связанный с носителем, получали путем сушки распылением наноизмельченного фуроата мометазона на микрокристаллической целлюлозе (МСС) с использованием НРМС в качестве связующего вещества. Состав водных композиций для сушки распылением (например, исходный материал для сушки распылением) и условия сушки распылением изменяли для оценки эффективности прикрепления наночастиц к носителю МСС. Составы наноизмельченных суспензий и исходных материалов для сушки распылением представлены в нижеследующих таблицах.
Условия сушки распылением. Температура на входе = 110°С, температура на выходе = 54°С, вытяжной вентилятор = 100%, скорость насоса = 20%, поток Q = 50%, колпачок сопла = 1,5 мм.
Условия размалывания в роликовой мельнице. Фуроат мометазона = первоначальный срединный размер = 19,7 мкм. Образцы размалывали в 250 мл цилиндрах в течение 3 часов в среде 0,5 мм YTZ. Диаметр частиц (PSD) после 3-часового размалывания измеряли в воде. После размалывания суспензию экстрагировали (игла 22G) из цилиндров и смешивали в соответствии с приведенными в таблице данными и применяемым методом.
Метод получения суспензий для сушки распылением. Количества используемых НРМС, МСС и Н2О определяли в расчете на экстрагированное количество суспензии (таблица II). НРМС добавляли в виде раствора в Н2О. Раствор НРМС добавляли к экстрагированной суспензии и смешивали в течение 15 минут. Добавляли МСС (сорт РН-F20JP, 20 мкм) и смешивали в течение 15 минут. Диаметр частиц (PSD) измеряли после сушки распылением путем диспергирования высушенного распылением порошка в жидкость, такую как изопропилпальмитат или силиконовое масло, которые не являются растворителями компонентов, не изменяют размер частиц и не вызывают дезинтеграции композиционной частицы.
Материалы. фуроат мометазона (Hovione, East Windsor, NJ), безводная лимонная кислота (Fisher, Hampton, New Hampshire), хлорид бензалкония (Fluka, St. Louis, Missouri), микрокристаллическая целлюлоза (AsahKasei, Glenview, Illinois), гидроксипропилметилцеллюлоза (Colorcon, Harleysville, Pennsylvania).
Оборудование. Роликовая мельница (US Stoneware, East Palestine, Ohio), весы (Sartorius, Bohemia, NY), лазерный анализатор PSD (Horiba, Kyoto, Japan), распылительная сушилка Buchi В-290 (Buchi, Flawil, Switzerland).
Оптические микрофотографии составов, включающих сухой порошок (после сушки распылением), показывают, что фуроат мометазона ассоциирован с частицами МСС. Также были обнаружены некоторые коалесцированные частицы фуроата мометазона, не ассоциированные с частицами МСС. На относительную долю фуроата мометазона, ассоциированного с частицами МСС, могут влиять условия сушки распылением.
(Компоненты показанной суспензии мометазона)
| название | год | авторы | номер документа |
|---|---|---|---|
| ДЕАМОРФИЗАЦИЯ ВЫСУШЕННЫХ РАСПЫЛЕНИЕМ СОСТАВОВ ПОСРЕДСТВОМ СМЕШИВАНИЯ РАСПЫЛЕНИЕМ | 2014 |
|
RU2698331C2 |
| СУХИЕ ПОРОШКОВЫЕ КОМПОЗИЦИИ В ВИДЕ ЧАСТИЦ, КОТОРЫЕ СОДЕРЖАТ ДВА ИЛИ БОЛЕЕ АКТИВНЫХ ИНГРЕДИЕНТА, ДЛЯ ЛЕЧЕНИЯ ОБСТРУКТИВНЫХ ИЛИ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ ДЫХАТЕЛЬНЫХ ПУТЕЙ | 2012 |
|
RU2629333C2 |
| Агрегированные частицы | 2013 |
|
RU2666963C2 |
| КОМБИНАЦИЯ АЗЕЛАСТИНА И СТЕРОИДОВ | 2003 |
|
RU2361593C2 |
| НАПРАВЛЕННАЯ ДОСТАВКА ВЫСУШЕННЫХ РАСПЫЛЕНИЕМ КОМПОЗИЦИЙ В ЛЕГКИЕ | 2016 |
|
RU2731212C2 |
| КОМПОЗИЦИИ, СПОСОБЫ И СИСТЕМЫ ДЛЯ РЕСПИРАТОРНОЙ ДОСТАВКИ ДВУХ ИЛИ БОЛЕЕ АКТИВНЫХ АГЕНТОВ | 2010 |
|
RU2586297C2 |
| СПОСОБ ОБРАБОТКИ ЧАСТИЦ АКТИВНЫХ ФАРМАЦЕВТИЧЕСКИХ ИНГРЕДИЕНТОВ | 2011 |
|
RU2597790C2 |
| ЛЕЧЕНИЕ АЛЛЕРГИЧЕСКОГО РИНИТА У СУБЪЕКТОВ ДЕТСКОЙ ВОЗРАСТНОЙ КАТЕГОРИИ С ПРИМЕНЕНИЕМ КОМБИНАЦИИ МОМЕТАЗОНА И ОЛОПАТАДИНА | 2019 |
|
RU2798030C2 |
| ИНГАЛЯЦИОННАЯ ПРЕПАРАТИВНАЯ ФОРМА, СОДЕРЖАЩАЯ ПРОСТОЙ СУЛЬФОАЛКИЛОВЫЙ ЭФИР ГАММА-ЦИКЛОДЕКСТРИНА И КОРТИКОСТЕРОИД | 2004 |
|
RU2390330C2 |
| ИНГАЛЯЦИОННАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ СУЛЬФОАЛКИЛОВЫЙ ЭФИР ЦИКЛОДЕКСТРИНА И КОРТИКОСТЕРОИД | 2004 |
|
RU2388462C2 |
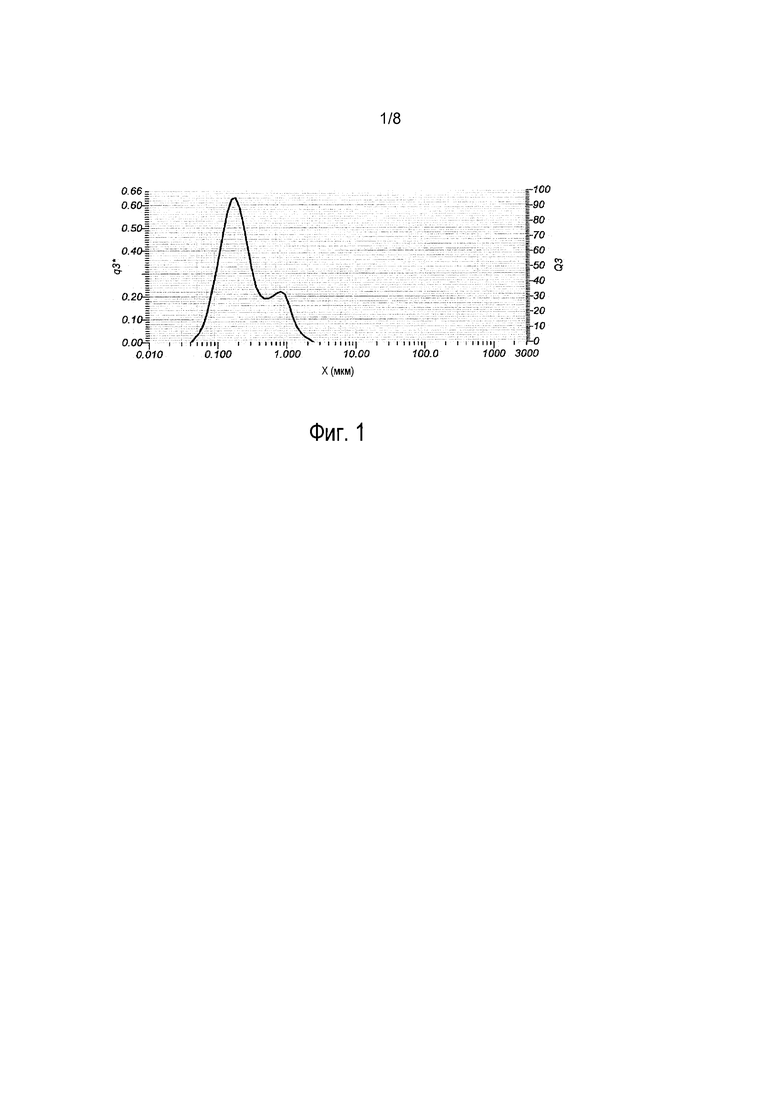
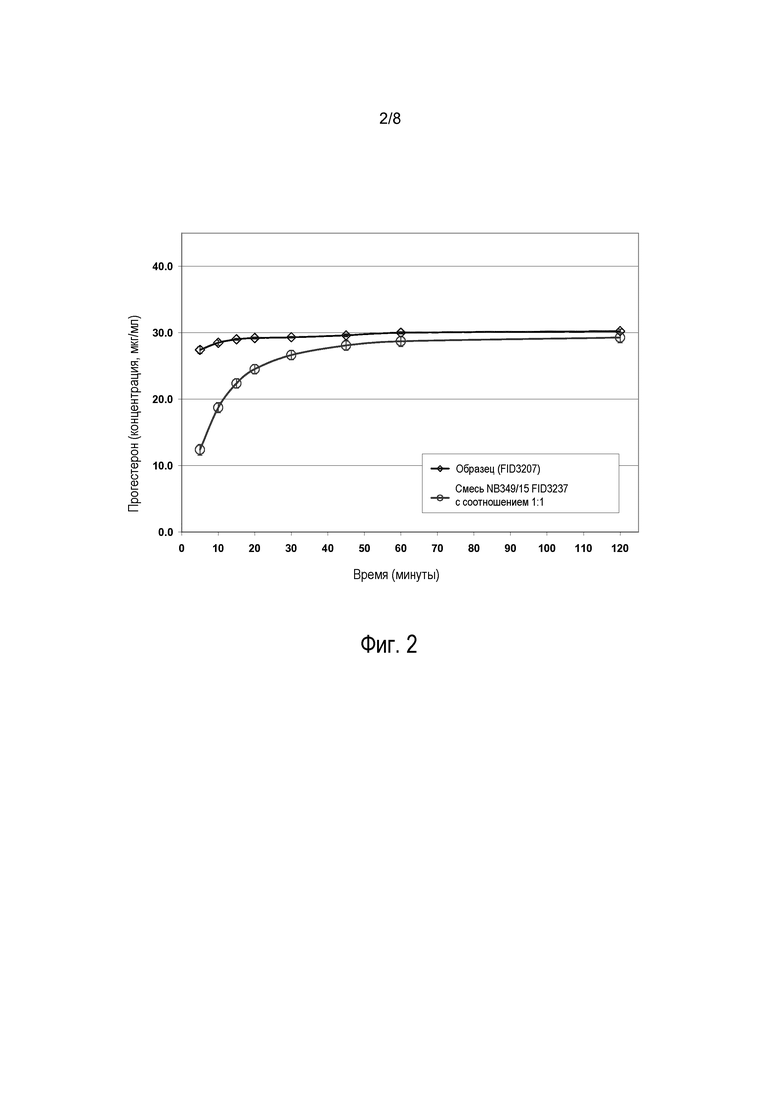
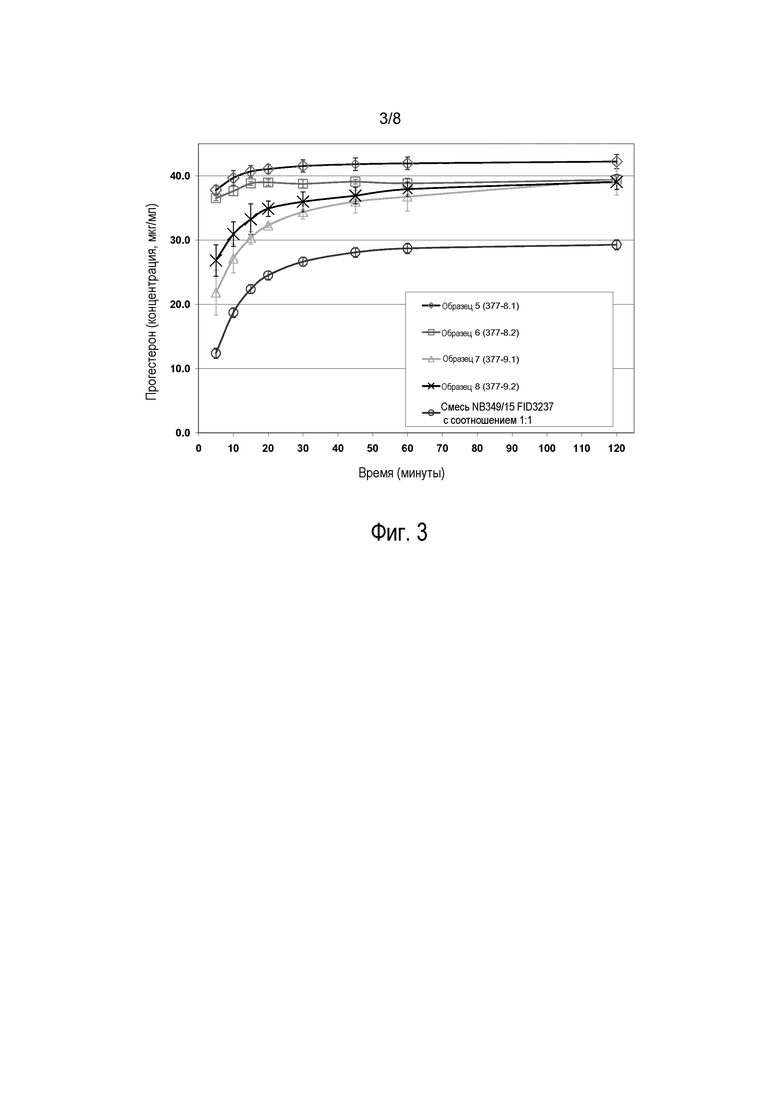
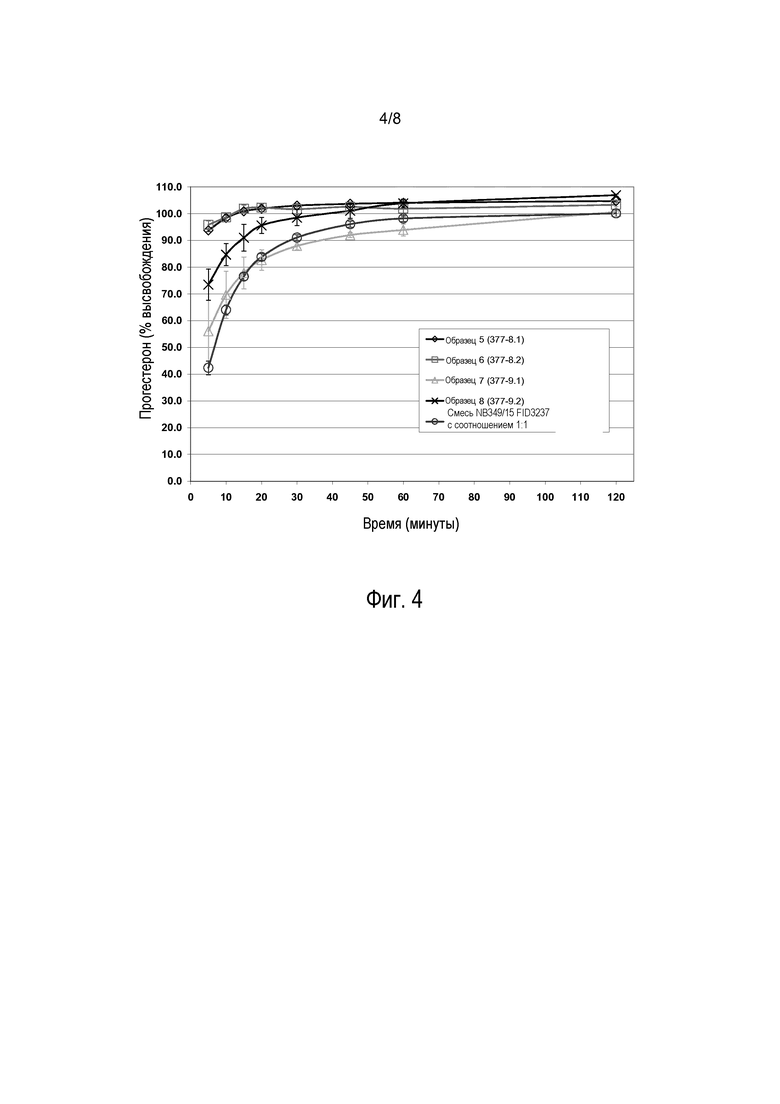
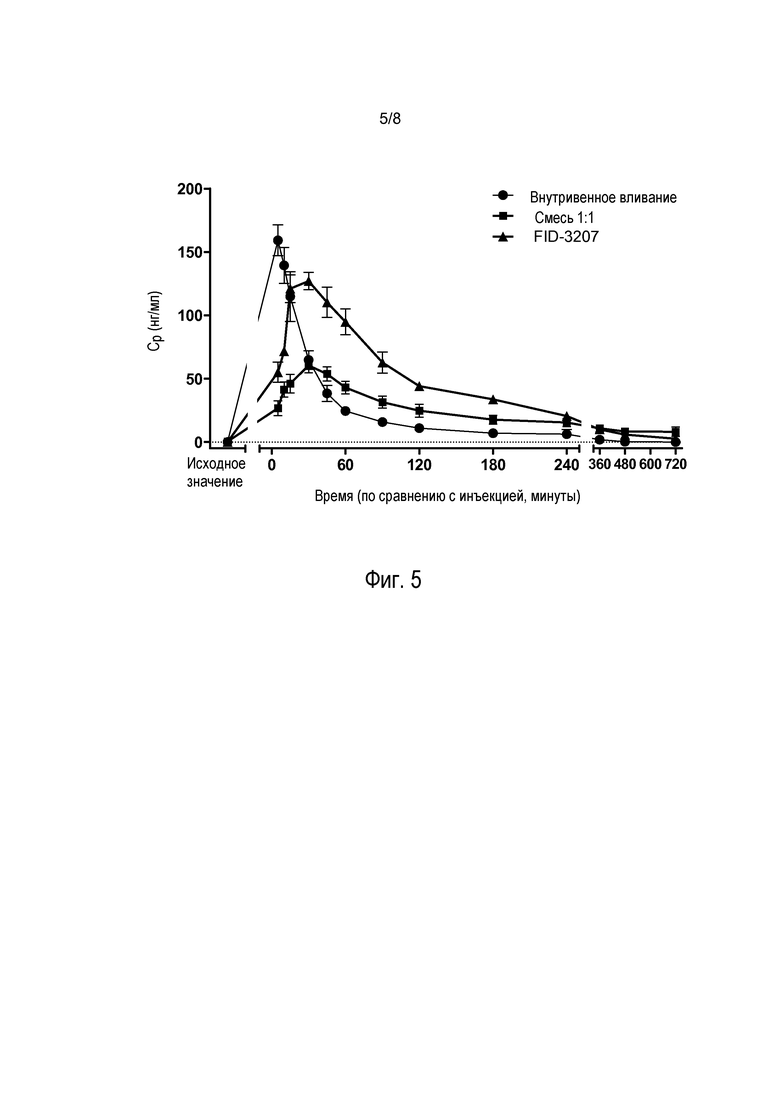
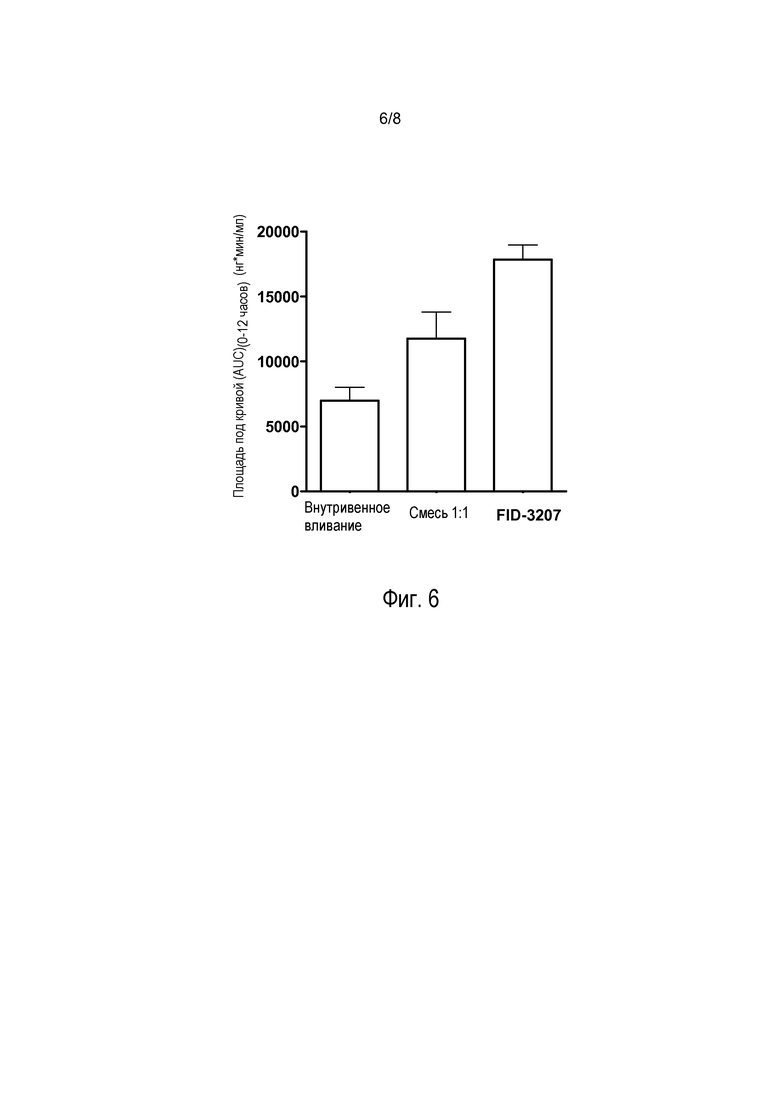
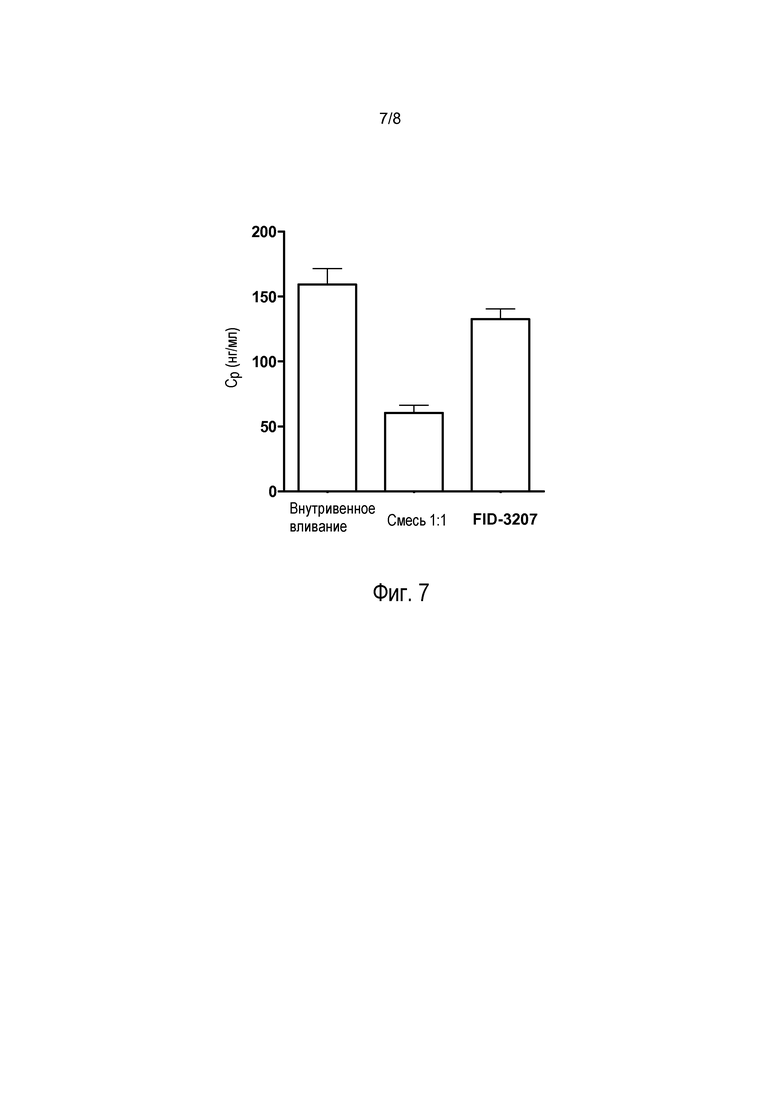
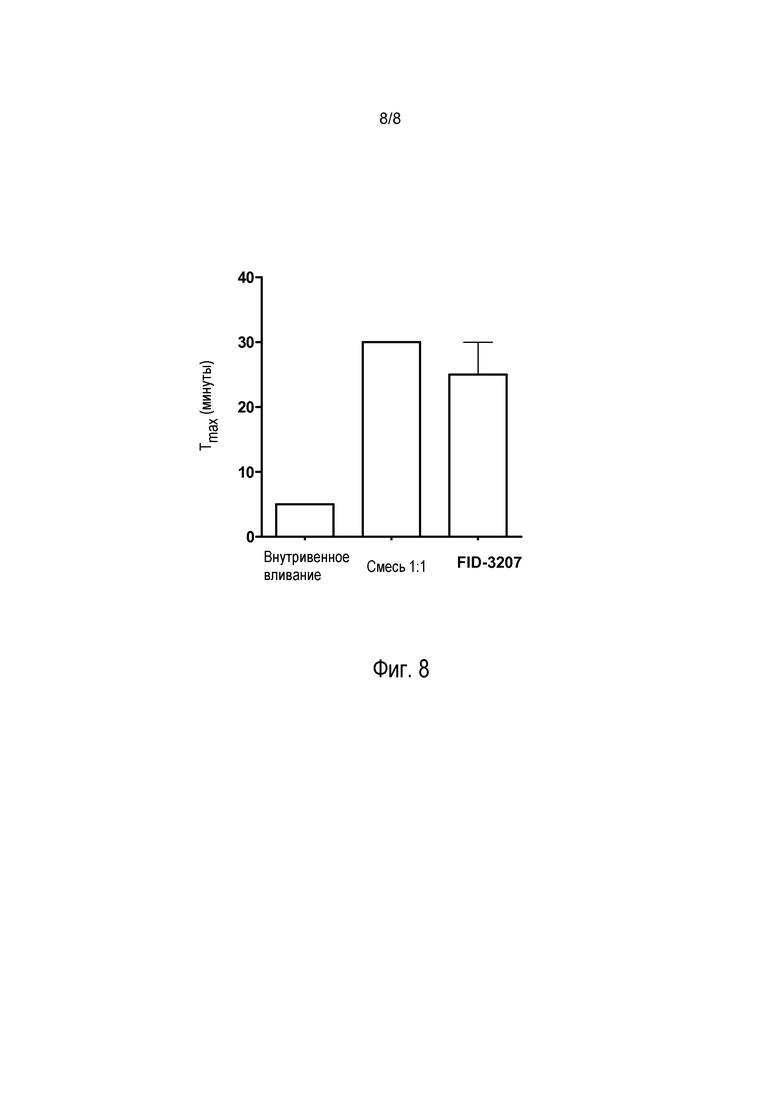
Группа изобретений относится к области медицины и фармацевтики. Первый объект представляет собой сухую фармацевтическую композицию, содержащую сухие частицы, которые включают частицы фармацевтически активного агента, фармацевтически приемлемое связующее вещество и фармацевтически приемлемый носитель. Второй и третий объекты – способ получения сухой фармацевтической композиции, включающий высушивание водной суспензии распылением, и композицию, полученную этим способом. Остальные объекты – способы введения активного агента, включающие введение сухой фармацевтической композиции, и применение сухой фармацевтической композиции в способе введения и в приготовлении лекарственного средства. Технический результат заключается в быстрой доставке активного агента, биоадгезии, лучшей устойчивости при хранении сухой фармацевтической композиции, предназначенной для введения через слизистую оболочку и содержащей вышеуказанные сухие частицы. 8 н. и 16 з.п. ф-лы, 8 ил., 14 табл., 7 пр.
1. Сухая фармацевтическая композиция, содержащая сухие частицы, включающие:
частицы фармацевтически активного агента, где указанные частицы активного агента имеют средний диаметр частиц от 0,01 до 1 мкм;
фармацевтически приемлемое связующее вещество и
фармацевтически приемлемый носитель, состоящий из материала, выбранного из группы, состоящей из микрокристаллической целлюлозы, кристаллической целлюлозы, целлюлозы, α-целлюлозы, перекрестносшитой натрий-карбоксиметилцеллюлозы, перекрестносшитого винилполивинилпирролидона, перекрестносшитого карбоксивинильного полимера или его солей, перекрестносшитого поливинилового спирта, перекрестносшитого полигидроксиэтилметакрилата, гидроксипропилового крахмала, карбоксиметилового крахмала, перекрестносшитого крахмала, амилазы, амилопектина, пектина, желатина, казеина, аравийской камеди, траганта и глюкоманнана;
в которой частицы фармацевтически активного агента связаны с частицами носителя.
2. Сухая фармацевтическая композиция по п.1, в которой сухие частицы являются высушенными распылением частицами, включающими частицы фармацевтически активного агента, фармацевтически приемлемое связующее вещество и фармацевтически приемлемый носитель, при этом:
частицы активного агента имеют средний диаметр до сушки распылением менее 1 мкм;
до 10% высушенных распылением частиц имеют размер менее 10 мкм, по меньшей мере 50% высушенных распылением частиц имеют размер, равный по меньшей мере 15 мкм, и по меньшей мере 90% высушенных распылением частиц имеют размер до 55 мкм.
3. Фармацевтическая композиция по п.1 или 2, в которой активный агент является прогестероном, его метаболитом, производным или пролекарством.
4. Фармацевтическая композиция по п.1 или 2, в которой активный агент является мометазоном или фуроатом мометазона.
5. Фармацевтическая композиция по п.1 или 2, в которой носитель представляет собой микрокристаллическую целлюлозу.
6. Фармацевтическая композиция по п.1 или 2, в которой фармацевтически приемлемое связующее вещество выбрано из группы, состоящей из гидроксипропилметилцеллюлозы (НРМС), натрий-карбоксиметилцеллюлозы (NaCMC), полиакрилатов и их солей, низших алкиловых эфиров целлюлозы, таких как кальций-карбоксиметилцеллюлоза, гидроксипропилцеллюлозы (НРС, НРС-SL и HPC-L), гидроксипропилметилцеллюлозы (НРМС), гипромеллозы 2910, натрий-карбоксиметилцеллюлозы (NaCМC), метилцеллюлозы, гидроксиэтилцеллюлозы, гидроксипропилцеллюлозы, фталата гидроксипропилметилцеллюлозы, некристаллической целлюлозы, олеиновой кислоты, глицерина, белого вазелина, сложного эфира глицерина гидрированной канифоли, полиэтиленгликолей, пропиленгликоля и полиэтиленгликоля 400.
7. Фармацевтическая композиция по п.1 или 2, которая дополнительно содержит интенсификатор размола, необязательно выбранный из группы, состоящей из полисорбата 20, полисорбата 80 и хлорида бензалкония.
8. Фармацевтическая композиция по п.1 или 2, в которой активный агент составляет от 10 до 75 мас.%, от 40 до 75 мас.%, от 25 до 50 мас.%, от 1,0 до 25,0 мас.% или от 5% до 10 мас.% частиц.
9. Фармацевтическая композиция по п.1 или 2, в которой фармацевтически приемлемое связующее вещество составляет от 1 до 15 мас.% или 10,0-50,0 мас.% высушенных распылением частиц.
10. Фармацевтическая композиция по п.7, в которой интенсификатор размола составляет от 0,1 до 5 мас.% высушенных распылением частиц.
11. Фармацевтическая композиция по п.1 или 2, в которой частицы фармацевтически активного агента состоят из активного агента и необязательно интенсификатора размола.
12. Фармацевтическая композиция по п.1 или 2, которая дополнительно включает один или несколько фармацевтически приемлемых наполнителей.
13. Фармацевтическая композиция по п.1 или 2, которая далее включает 5,0-30,0% мас./мас. микрокристаллической целлюлозы со средним диаметром частиц от 30 до 150 мкм, крахмал или их смесь и 0,1-10% мас./мас. трехосновного фосфата кальция.
14. Фармацевтическая композиция по п.1 или 2, в которой более 90% активного агента высвобождается из композиции в течение 60 минут при исследовании методом растворения.
15. Способ получения сухой фармацевтической композиции, как определена в п.1, где способ включает этапы комбинирования частиц фармацевтически активного агента, фармацевтически приемлемого связующего вещества и фармацевтически приемлемого носителя с получением водной суспензии, и высушивание водной суспензии распылением, с получением частиц, содержащих частицы фармацевтически активного агента, связанные с частицами носителя.
16. Способ по п.15, в котором
частицы активного агента имеют средний диаметр до сушки распылением менее 1 мкм;
до 10% высушенных распылением частиц имеют размер менее 10 мкм, по меньшей мере 50% высушенных распылением частиц имеют размер, равный по меньшей мере 15 мкм, и по меньшей мере 90% высушенных распылением частиц имеют размер до 55 мкм.
17. Способ по п.15 или 16, в котором активный агент является прогестероном, его метаболитом, производным или пролекарством, мометазоном или фуроатом мометазона, носитель включает микрокристаллическую целлюлозу (МСС), и фармацевтически приемлемое связующее вещество выбрано из группы, состоящей из гидроксипропилметилцеллюлозы (НРМС) и натрий-карбоксиметилцеллюлозы (NaCMC).
18. Сухая фармацевтическая композиция, полученная способом по любому из пп.15-17.
19. Способ введения активного агента, включающий введение нуждающемуся субъекту сухой фармацевтической композиции по любому из пп.1-14.
20. Способ введения активного агента, включающий введение нуждающемуся субъекту сухой фармацевтической композиции по любому из пп.1-14 через слизистую оболочку.
21. Применение сухой фармацевтической композиции по любому из пп.1-14 в способе введения активного агента нуждающемуся субъекту.
22. Применение по п.21, где указанный агент вводится через слизистую оболочку.
23. Применение сухой фармацевтической композиции по любому из пп.1-14 в приготовлении лекарственного средства, предназначенного для использования в способе введения активного агента нуждающемуся субъекту.
24. Применение сухой фармацевтической композиции по любому из пп.1-14 в приготовлении лекарственного средства, предназначенного для использования в способе введения активного агента нуждающемуся субъекту через слизистую оболочку.
| WO 2008075102 A1, 26.06.2008 | |||
| WO 2011133956 A1, 27.10.2011 | |||
| СПОСОБ УПРАВЛЕНИЯ ПЕРЕДАЧЕЙ ОБСЛУЖИВАНИЯ, ПОЛЬЗОВАТЕЛЬСКОЕ УСТРОЙСТВО, БАЗОВАЯ СТАНЦИЯ И СИСТЕМА РАДИОСВЯЗИ | 2011 |
|
RU2540281C2 |
| WO 91006292 A1, 16.05.1991 | |||
| US 2007031490 A1, 08.02.2007 | |||
| WO 2010138862 A2, 02.12.2010 | |||
| RU 94046097 A, 20.09.1996. | |||

