Релевантные заявки
По данной непредварительной заявке, поданной согласно 37 CFR § 1.53(b) испрашивается приоритет согласно 35 USC § 119(e) предварительной заявки на патент США № 61/037410, поданной 18 марта 2008 г., которая полностью включена в данный документ посредством ссылки.
Область техники, к которой относится изобретение
Изобретение главным образом относится к фармацевтическим комбинациям соединений, обладающим активностью в отношении гиперпролиферативных нарушений, таких как злокачественная опухоль. Также изобретение относится к способам применения комбинаций соединений для диагностики или лечения клеток млекопитающих в условиях in vitro, in situ и in vivo, или ассоциированных патологических состояний.
Уровень техники
HER2 (ErbB2) рецептор тирозинкиназы является членом семейства рецепторов эпидермальных факторов роста (EGFR) трансмембранных рецепторов. Сверхэкспрессию HER2 отмечают примерно в 20% злокачественных опухолей молочной железы человека, и она ассоциируется с агрессивным ростом и неблагоприятным прогнозом для пациенток с данными опухолями (Slamon et al., 1987, Science, 235:177-182).
Трастузумаб (номер по CAS 180288-69-1, ГЕРЦЕПТИН®, huMAb4D5-8, rhuMAb HER2, Genentech) представляет собой вариант рекомбинантного, полученного из ДНК гуманизированного, IgG1 каппа, моноклонального антитела мышиного антитела к HER2, которое избирательно связывается с высокой аффинностью в клеточном тесте (Kd=5 нМ) с внеклеточным доменом белка рецептора 2 человеческого эпидермального фактора роста, HER2 (ErbB2) (патент США № 5677171; патент США № 5821337; патент США № 6054297; патент США № 6165464; патент США № 6339142; патент США № 6407213; патент США № 6639055; патент США № 6719971; патент США № 6800738; патент США № 7074404; Coussens et al., 1985, Science, 230:1132-1139; Slamon et al., 1989, Science, 244:707-712; Slamon et al., 2001, New Engl. J. Med., 344:783-792). Трастузумаб содержит человеческие каркасные области с определяющими комплементарность областями мышиного антитела (4D5), которое связывается с HER2. Трастузумаб связывается с антигеном HER2 и таким образом ингибирует рост опухолевых клеток. В тестах в условиях in vitro и на животных было показано, что трастузумаб ингибирует пролиферацию человеческих опухолевых клеток со сверхэкспрессией HER2 (Hudziak et al., 1989, Mol. Cell Biol., 9:1165-1172; Lewis et al., 1993, Cancer Immunol. Immunother: 37:255-63; Baselga et al., 1998, Cancer Res., 58:2825-2831). Трастузумаб является медиатором антителозависимой клеточной цитотоксичности, ADCC (Lewis et al., 1993, Cancer Immunol. Immunother., 37(4):255-263; Hotaling et al., 1996, [abstract]. Proc. Annual Meeting Am. Assoc. Cancer Res., 37:471; Pegram M.D. et al., 1997, [abstract]. Proc. Annual Meeting Am. Assoc. Cancer Res., 38:602; Sliwkowski M. et al., 1999, Seminars in Oncology, 26(4), Suppl. 12:60-70; Yarden Y. and Sliwkowski M., 2001, Nature Reviews: Molecular Cell Biology, Macmillan Magazines, Ltd., Vol. 2:127-137).
Герцептин® был разрешен к применению в 1998 для лечения пациенток с метастатическими злокачественными опухолями молочной железы со сверхэкспрессией ErbB2 (Baselga et al., 1996, J. Clin. Oncol., 14:737-744), которые до этого получали интенсивную противоопухолевую терапию, и с того времени его применили на более чем 300000 пациентках (Slamon D.J. et al., N. Engl. J. Med., 2001, 344:783-92; Vogel C.L. et al., J. Clin. Oncol., 2002, 20:719-26; Marty M. et al., J. Clin. Oncol., 2005, 23:4265-74; Romond E.H. et al., Т N. Engl. J. Med., 2005, 353:1673-84; Piccart-Gebhart M.J. et al., N. Engl. J. Med., 2005, 353:1659-72; Slamon D. et al., [abstract]. Breast Cancer Res. Treat., 2006, 100 (Suppl. 1):52). В 2006 г. FDA разрешила герцептин® (трастузумаб, Genentech Inc.) к применению в качестве составного компонента в схеме лечения, содержащей доксорубицин, циклофосфамид и паклитаксел, для адъювантного лечения пациенток с HER2-положительной, метастатической злокачественной опухолью молочной железы. Несмотря на то, что герцептин®, разработанный для пациенток с HER2-положительными опухолями, обеспечивал более высокий терапевтический эффект по сравнению с одной химиотерапией, в конечном итоге у всех пациенток с HER2-положительной метастатической злокачественной опухолью молочной железы (MBC) имело место прогрессирование заболевания на доступных лекарственных средствах. Оставались возможности для улучшения эффективности в лечении пациенток с МВС. Несмотря на различные механизмы действия трастузумаба, у ряда пациенток, подвергшихся лечению трастузумабом, не было ответа на лечение или ответ был остановлен после положительного периода лечения. Некоторые HER2+ (HER2-положительные) опухоли не отвечали на лечение герцептином® и у большинства пациенток, у которых наблюдали ответ опухолей, в конечном итоге имело место прогрессирование заболевания. В клинике имеется существенная потребность в разработке дополнительных, направленных на HER2 противоопухолевых средств для лечения пациентов с опухолями, сверхэкспрессирующими HER2, или с другими заболеваниями, ассоциированными с экспрессией HER2, которые не отвечают или слабо отвечают на лечение герцептином®.
Альтернативным подходом для антитело-направленной терапии является применение антител для доставки цитотоксических лекарственных средств специфически в антиген-экспрессирующие опухолевые клетки. Майтансиноиды, производные антимитотического средства майтансина, связываются с микротрубочками аналогично лекарственным средствам на основе алкалоидов барвинка (Issell B.F. et al., 1978, Cancer Treat. Rev., 5:199-207; Cabanillas F. et al., 1979, Cancer Treat. Rep., 63:507-509). Конъюгаты антитело-лекарственное средство (ADC), состоящие из майтансиноида DM1, связанного с трастузумабом, проявляют высокую противоопухолевую активность на HER2-экспрессирующих чувствительных к трастузумабу и резистентных к трастузумабу линиях опухолевых клеток и на моделях ксенотрансплантатов злокачественной опухоли молочной железы человека. Эффективность конъюгата майтансиноидов, связанных с мышиным антителом против HER2, ТА.1 через линкер МСС, в 200 раз выше по сравнению с соответствующим конъюгатом с дисульфидным линкером (Chari et al., 1992, Cancer Res., 127-133). Конъюгаты антитело-лекарственное средство (ADC), состоящие из майтансиноида, DM1, связанного с трастузумабом, проявляют высокую противоопухолевую активность на чувствительных к трастузумабу и резистентных к трастузумабу линиях опухолевых клеток и на моделях ксенотрансплантатов злокачественной опухоли молочной железы человека. В настоящее время конъюгат трастузумаб-MCC-DM1 (T-DM1) проходит фазу II клинических испытаний у пациентов, которые устойчивы к лечению препаратами, направленными на HER2 (Beeram et al., 2007, «A phase I study of trastuzumab-MCC-DM1 (T-DM1), a first-in-class HER2 antibody-drug conjugate (ADC) in patients (pts) with HER2+ metastatic breast cancer (BC)», American Society of Clinical Oncology 43rd:June 02 (Abs 1042; Krop et al., European Cancer Conference ECCO, Poster 2118, September 23-27, 2007, Barcelona; патент США № 7097840; заявка на патент США № 2005/0276812; заявка на патент США № 2005/0166993).
Комбинированная терапия, в которой используют два или несколько лекарственных средств по определенной схеме или в определенной форме введения, как правило, имеет одну или несколько целей: (i) снижение частоты возникновения приобретенной резистентности объединением лекарственных средств с минимальной перекрестной резистентностью, (ii) снижение дозировок лекарственных средств, которые не являются токсичными, и имеют аналогичный терапевтический профиль для достижения эффективности с проявлением меньших побочных эффектов, т.е. увеличением терапевтического индекса, (iii) сенсибилизация клеток к воздействию одного лекарственного средства через действие другого лекарственного средства, посредством изменения стадии клеточного цикла или способности к росту и (iv) достижение повышенной эффективности при использовании аддитивного действия или несколько чем аддитивного действия, эффектов в биологической активности двух лекарственных средств (Pegram M. et al., 1999, Oncogene, 18:2241-2251; Konecny G. et al., 2001, Breast Cancer Res. and Treatment, 67:223-233; Pegram M. et al., 2004, J. Nat. Cancer Inst., 96(10):739-749; Fitzgarald et al., 2006, Nature Chem. Biol., 2(9):458-466; Borisy et al., 2003, Proc. Natl. Acad. Sci., 100(13):7977-7982).
Аддитивность Леве (Chou T.C. and Talalay P., 1977, J. Biol. Chem., 252:6438-6442; Chou T.C. and Talalay P., 1984, Adv. Enzyme Regul., 22:27-55; Berenbaum M.C., 1989, Pharmacol. Rev., 41:93-141) и независимость/синергия Блисса (Bliss C.I., 1956, Bacteriol. Rev., 20:243-258; Greco et al., 1995, Pharmacol. Rev., 47:331-385) являются методами, применяемыми для расчета предполагаемого взаимоотношения доза-эффект при комбинированной терапии по сравнению с монотерапией, основанные на таких параметрах, как IC50, доза лекарственного средства, необходимая для ингибирования 50% мишени и равная Ki в наиболее простом случае.
Сообщалось о применении антител-ингибиторов димеризации HER2 и ингибиторов EGFR для комбинированной терапии рака (заявка на патент США № 2007/0020261). Трастузумаб-MCC-DM1 (T-DM1) и пертузумаб по отдельности проявили эффективность на пациентках с MBC, и было показано, что комбинация пертузумаба и трастузумаба является активной у пациенток с HER-положительными MBC (Baselga J. et al., «A Phase II trial of trastuzumab and pertuzumab in patients with HER2-positive metastatic breast cancer that had progressed during trastuzumab therapy: full response data», European Society of Medical Oncology, Stockholm, Sweden, September 12-16, 2008).
Сущность изобретения
Изобретение главным образом относится к конъюгату анти-HER2-антитело-лекарственное средство, трастузумабу-МСС-DM1, который вводят в комбинации с одним или несколькими химиотерапевтическими средствами, для подавления роста опухолевых клеток. Некоторые комбинации трастузумаба-МСС-DM1 и химиотерапевтического средства проявляют синергическое действие в подавлении роста опухолевых клеток в условиях in vitro и in vivo. Комбинации и способы по изобретению могут быть пригодными при лечении гиперпролиферативных нарушений, таких как злокачественная опухоль. Комбинации могут ингибировать рост опухолей у млекопитающих и могут быть пригодными для лечения людей со злокачественными опухолями.
В одном аспекте изобретение относится к способу лечения гиперпролиферативного нарушения, включающему введение терапевтической комбинации в виде комбинированной композиции или поочередно млекопитающему, в котором терапевтическая комбинация содержит терапевтически эффективное количество трастузумаба-МСС-DM1 и терапевтически эффективное количество химиотерапевтического средства, выбранного из антитела-ингибитора димеризации HER2, антитела к VEGF, 5-FU, карбоплатина, лапатиниба, ABT-869, доцетаксела, GDC-0941 и GNE-390.
Терапевтически эффективное количество трастузумаба-МСС-DM1 и терапевтически эффективное количество химиотерапевтического средства можно вводить в виде комбинированной композиции или поочередно.
Также изобретение относится к способам применения композиций для диагностики или лечения клеток млекопитающих, организмов в условиях in vitro, in situ и in vivo, или ассоциированных патологических состояний.
Также изобретение относится к способам, в которых введение терапевтической комбинации приводит к синергическому эффекту.
Еще один аспект изобретения относится к фармацевтическим композициям, содержащим трастузумаб-МСС-DM1, химиотерапевтическое средство, выбранное из антитела-ингибитора димеризации HER2, антитела к VEGF, 5-FU, карбоплатина, лапатиниба, ABT-869, доцетаксела, GDC-0941 и GNE-390; и один или несколько фармацевтически приемлемых носителей, регуляторов сыпучести, разбавителей или наполнителей.
Еще один аспект изобретения относится к способам лечения гиперпролиферативного заболевания или нарушения, включающим введение млекопитающему, нуждающемуся в таком лечении, терапевтически эффективных количеств трастузумаба-МСС-DM1 и химиотерапевтического средства. Трастузумаб-МСС-DM1 и химиотерапевтическое средство можно формулировать совместно для введения в комбинации в виде фармацевтической композиции или их можно вводить поочередно (чередующиеся, последовательные введения) в виде терапевтической комбинации. В одном варианте осуществления T-DM1 вводят инфузией, и химиотерапевтическое средство вводят перорально.
Еще один аспект изобретения относится к способам прогноза эффективных лекарственных комбинаций в отношении эффективности в условиях in vivo, где комбинации содержат трастузумаб-МСС-DM1 и противоопухолевое, широко применяемое химиотерапевтическое средство. Данные по эффективности, полученные в опытах по оценке пролиферации клеток в условиях in vitro и на опухолевых ксенотрансплантатах в условиях in vivo, подвергают качественному и количественному анализу. Методы количественного анализа могут быть основаны на принципе медианного эффекта Chou&Talalay и изоболограммах, с помощью которых определяют значение комбинационного индекса (CI) для установления синергизма, антагонизма или аддитивного эффекта, или они могут быть основаны на независимости отклонения Блисса.
Еще один аспект изобретения относится к способу применения терапевтической комбинации по изобретению для лечения у млекопитающего заболевания или состояния, такого как злокачественная опухоль, включающего заболевание, которое модулируется HER2 или KDR9 (рецептором 1 VEGF).
Еще один аспект изобретения относится к применению терапевтической комбинации по изобретению в производстве лекарственного средства для лечения у млекопитающего заболевания или состояния, такого как злокачественная опухоль, включающего заболевание, которое модулируется HER2 или KDR9 (рецептором 1 VEGF).
Еще один аспект изобретения относится к изделиям или наборам, содержащим трастузумаб-МСС-DM1, химиотерапевтическое средство, контейнер и необязательно вкладыш в упаковке или этикетку, с описанием лечения.
Еще один аспект изобретения относится к способу определения соединений для применения в комбинации для лечения злокачественной опухоли, включающему: (а) введение терапевтической комбинации трастузумаба-МСС-DM1 и химиотерапевтического средства, выбранного из антитела-ингибитора димеризации HER2, антитела к VEGF, 5-FU, карбоплатина, лапатиниба, ABT-869, доцетаксела, GDC-0941 и GNE-390 в опухолевую клеточную линию в условиях in vitro и (b) определение синергического или не синергического эффекта.
Дополнительные преимущества и новые признаки данного изобретения будут частично приведены в последующем описании, и частично станут понятными специалистам в данной области при ознакомлении с последующей заявкой или изучены при практическом применении изобретения. Преимущества изобретения могут быть реализованы и достигнуты посредством инструментального исполнения, комбинаций, композиций и способов, заявленных в прилагаемой формуле изобретения.
Краткое описание фигур
На фиг.1 приведен график зависимости жизнеспособности клеток SK-BR-3 в условиях in vitro на 3 сутки от кратных концентраций IC50 трастузумаба, трастузумаба-МСС-DM1 (T-DM1) и комбинации трастузумаба и T-DM1.
На фиг.2 приведен график зависимости жизнеспособности клеток BT-474 EEI в условиях in vitro на 3 сутки от кратных концентраций IC50 трастузумаба, трастузумаба-МСС-DM1 (T-DM1) и комбинации трастузумаба и T-DM1.
На фиг.3 приведен график зависимости жизнеспособности клеток MDA-MB-175 в условиях in vitro на 5 сутки от кратных концентраций IC50 пертузумаба, трастузумаба-МСС-DM1 (T-DM1) и комбинации пертузумаба и T-DM1.
На фиг.3а приведен график зависимости жизнеспособности клеток MDA-MB-175 в условиях in vitro на 5 сутки от кратных концентраций IC50 пертузумаба, трастузумаба-МСС-DM1 (T-DM1) и комбинации пертузумаба и T-DM1.
На фиг.4 приведен график зависимости жизнеспособности клеток BT-474 в условиях in vitro на 5 сутки от различных фиксированных концентраций пертузумаба в комбинации с концентрацией ответной реакции трастузумаба-МСС-DM1 (T-DM1) и различных концентраций одного T-DM1.
На фиг.5 приведен график зависимости жизнеспособности клеток BT-474 в условиях in vitro на 5 сутки от различных фиксированных концентраций трастузумаба-МСС-DM1 (T-DM1) в комбинации с концентрацией ответной реакции пертузумаба и различных концентраций одного пертузумаба.
На фиг.6 приведен график зависимости жизнеспособности клеток ВТ-474 в условиях in vitro на 5 сутки от кратных концентраций IC50 пертузумаба, трастузумаба-МСС-DM1 (T-DM1) и комбинации пертузумаба и T-DM1.
На фиг.7 приведен график зависимости жизнеспособности клеток SK-BR-3 в условиях in vitro на 3 сутки от различных концентраций T-DM1 в комбинации с фиксированными концентрациями лапатиниба (4,5 нМ; 14 нМ; 41 нМ; 123 нМ) и различных концентраций одного T-DM1 (0-1000 нг/мл).
На фиг.7а приведен график зависимости жизнеспособности клеток SK-BR-3 в условиях in vitro на 3 сутки от кратных концентраций IC50 T-DM1, лапатиниба и комбинаций с соотношением фиксированных концентраций T-DM1 и лапатиниба.
На фиг.8а приведен график зависимости жизнеспособности клеток BT-474 в условиях in vitro на 3 сутки от кратных концентраций IC50 T-DM1, лапатиниба и комбинаций с соотношением фиксированных концентраций T-DM1 и лапатиниба.
На фиг.8 приведен график зависимости жизнеспособности клеток BT-474 в условиях in vitro на 3 сутки от различных концентраций T-DM1 в комбинации с фиксированными концентрациями лапатиниба (1,5 нМ; 4,5 нМ; 14 нМ; 41 нМ; 123 нМ) и различных концентраций одного T-DM1 (0-1000 нг/мл).
На фиг.9 приведен график зависимости жизнеспособности клеток BT-474-EEI в условиях in vitro на 3 сутки от различных концентраций T-DM1 в комбинации с фиксированными концентрациями лапатиниба (14 нМ; 41 нМ; 123 нМ; 370 нМ; 1111 нМ) и различных концентраций одного T-DM1 (0-1000 нг/мл).
На фиг.10 приведен график изменения среднего объема опухолей в течение времени в условиях in vivo для опухолей KPL-4, имплантированных в жировую подушку молочной железы иммунодефицитных мышей SCID (3 миллиона клеток в матригеле на мышь) после введения: (1) буфера ADC; (2) пертузумаба в дозе 15 мг/кг; (3) T-DM1 в дозе 0,3 мг/кг; (4) T-DM1 в дозе 1 мг/кг; (5) T-DM1 в дозе 3 мг/кг, (6) пертузумаба в дозе 15 мг/кг + T-DM1 в дозе 0,3 мг/кг; (7) пертузумаба в дозе 15 мг/кг + T-DM1 в дозе 1 мг/кг; (8) пертузумаба в дозе 15 мг/кг + T-DM1 в дозе 3 мг/кг. Буфер ADC и T-DM1 вводили один раз на 0 сутки. Претузумаб вводили на 0; 7 и 14 сутки.
На фиг.11 приведен график изменения среднего объема опухолей в течение времени в условиях in vivo для опухолей KPL-4, имплантированных в жировую подушку молочной железы иммунодефицитных мышей SCID (3 миллиона клеток в матригеле на мышь) после введения: (1) буфера ADC; (2) 5-FU в дозе 100 мг/кг; (3) пертузумаба в дозе 40 мг/кг, первая доза пертузумаба (группы 5, 7 и 9) была 2× нагрузочной дозой; (4) B20-4.1 в дозе 5 мг/кг; (5) T-DM1 в дозе 5 мг/кг; (6) 5-FU в дозе 100 мг/кг + T-DM1 в дозе 5 мг/кг; (7) пертузумаба в дозе 40 мг/кг + T-DM1 в дозе 5 мг/кг; (8) B20-4.1 в дозе 5 мг/кг + T-DM1 в дозе 5 мг/кг; (9) B20-4.1 в дозе 5 мг/кг + пертузумаб в дозе 40 мг/кг. Буфер ADC и T-DM1 вводили внутривенно один раз на 0 сутки. Претузумаб вводили на 0; 7 и 14 сутки (один раз в неделю ×4). 5FU вводили на 0; 7 14 и 21 сутки (один раз в неделю ×3). В20-4.1 вводили на 0; 3; 7; 10; 14; 17; 21 и 24 сутки (2×/неделю ×8 в целом).
На фиг.12 приведен график изменения среднего объема опухолей в течение времени в условиях in vivo для трансгенных опухолей молочной железы MMTV-Her2 Fo5, имплантированных в жировую подушку молочной железы мышей CRL nu/nu, после введения: (1) растворителя (буфера ADC); (2) B20-4.1 в дозе 5 мг/кг; (3) T-DM1 в дозе 3 мг/кг; (4) T-DM1 в дозе 5 мг/кг; (5) T-DM1 в дозе 10 мг/кг; (6) B20-4.1 в дозе 5 мг/кг + T-DM1 в дозе 3 мг/кг; (7) B20-4.1 в дозе 5 мг/кг + T-DM1 в дозе 5 мг/кг; (8) B20-4.1 в дозе 5 мг/кг + T-DM1 в дозе 10 мг/кг. Буфер ADC и T-DM1 вводили на 0 и 21 сутки. B20-4.1 вводили на 0; 3; 7; 10; 14; 17; 21 и 24 сутки (2×/неделю ×4, 8 в целом).
На фиг.13 приведен график изменения среднего объема опухолей в течение времени в условиях in vivo для трансгенных опухолей молочной железы MMTV-Her2 Fo5, имплантированных в жировую подушку молочной железы мышей CRL nu/nu, после введения: (1) растворителя (буфера ADC); (2) T-DM1 в дозе 10 мг/кг; (3) 5-FU в дозе 100 мг/кг; (4) гемцитабина в дозе 120 мг/кг; (5) карбоплатина в дозе 100 мг/кг; (6) 5-FU в дозе 100 мг/кг + T-DM1 в дозе 10 мг/кг; (7) гемцитабина в дозе 120 мг/кг + T-DM1 в дозе 10 мг/кг; (8) карбоплатина в дозе 100 мг/кг + T-DM1 в дозе 10 мг/кг. Буфер ADC, T-DM1 и карбоплатин вводили однократно на 0 сутки. 5-FU вводили однократно на 0; 7 и 14 сутки (один раз в неделю ×3). Гемцитабин вводили на 0; 3; 6 и 9 сутки (каждые 3 дня ×4).
На фиг.14 приведен график изменения среднего объема опухолей в течение времени в условиях in vivo для трансгенных опухолей молочной железы MMTV-Her2 Fo5, имплантированных в жировую подушку молочной железы бестимусных мышей Harlan, после введения: (1) растворителя (буфера PBS) в/в, один раз в неделю ×4; (2) лапатиниба в дозе 101 мг/кг, перорально, два раза день ×21; (3) пертузумаба в дозе 40 мг/кг, в/в, один раз в неделю ×4; (4) B20-4.1 в дозе 5 мг/кг, в/б, 2×/в неделю ×4; (5) T-DM1 в дозе 15 мг/кг, в/в, один раз в 3 недели до конца; (6) лапатиниба в дозе 101 мг/кг перорально, два раза в день ×21 + T-DM1 в дозе 15 мг/кг, в/в, один раз в 3 недели до конца; (7) пертузумаба в дозе 40 мг/кг в/в, один раз в неделю ×4 + T-DM1 в дозе 15 мг/кг, в/в, один раз в 3 недели до конца; (8) B20-4.1 в дозе 5 мг/кг, в/б, 2×/в неделю ×4 + T-DM1 в дозе 15 мг/кг, в/в, один раз в 3 недели до конца.
На фиг.15 приведен график изменения среднего объема опухолей в течение времени в условиях in vivo для трансгенных опухолей молочной железы MMTV-Her2 Fo5, имплантированных в жировую подушку молочной железы бестимусных мышей Harlan, после введения: (1) растворителя (буфера PBS) перорально, два раза в день ×21; (2) T-DM1 в дозе 7,5 мг/кг, в/в, один раз в день ×1; (3) T-DM1 в дозе 15 мг/кг, в/в, один раз в день ×1; (4) ABT-869 в дозе 5 мг/кг, перорально, два раза в день ×21; (5) ABT-86 в дозе 15 мг/кг, перорально, два раза в день ×21; (6) T-DM1 в дозе 7,5 мг/кг в/в, один раз в день ×1 + ABT-869 в дозе 5 мг/кг, перорально, два раза в день ×21; (7) T-DM1 в дозе 7,5 мг/кг в/в, один раз в день ×1 + ABT-869 в дозе 15 мг/кг, перорально, два раза в день ×21; (8) T-DM1 в дозе 15 мг/кг, в/в, один раз в день ×1 + ABT-869 в дозе 5 мг/кг, перорально, два раза в день ×21; (9) T-DM1 в дозе 15 мг/кг, в/в, один раз в день ×1 + ABT-869 в дозе 15 мг/кг, перорально, два раза в день ×21.
На фиг.16 приведен график изменения среднего объема опухолей в течение времени в условиях in vivo для трансгенных опухолей молочной железы MMTV-Her2 Fo5, имплантированных в жировую подушку молочной железы бестимусных мышей Harlan, после введения: (1) растворителя в/в, один раз в неделю ×3; (2) T-DM1 в дозе 7,5 мг/кг в/в, один раз в 3 недели ×2; (3) T-DM1 в дозе 15 мг/кг в/в, один раз в 3 недели ×2; (4) доцетаксела в дозе 30 мг/кг в/в, один раз в неделю ×3; (5) T-DM1 в дозе 7,5 мг/кг в/в, один раз в 3 недели ×2 + доцетаксела в дозе 30 мг/кг в/в, один раз в неделю ×3; (6) T-DM1 в дозе 15 мг/кг в/в, один раз в 3 недели ×2 + доцетаксела в дозе 30 мг/кг в/в, один раз в неделю ×3.
На фиг.17 приведен график изменения среднего объема опухолей в условиях in vivo для трансгенных опухолей MMTV-Her2 Fo5, имплантированных в жировую подушку молочной железы бестимусных мышей Harlan, после введения: (1) растворителя перорально, один раз в день ×21; (2) T-DM1 в дозе 7,5 мг/кг в/в, один раз в 3 недели ×2; (3) T-DM1 в дозе 15 мг/кг в/в, один раз в 3 недели ×2; (4) лапатиниба в дозе 100 мг/кг перорально, два раза в день ×21; (5) T-DM1 в дозе 7,5 мг/кг в/в, один раз в 3 недели ×2 + лапатиниба в дозе 100 мг/кг перорально, два раза в день ×21; (6) T-DM1 в дозе 15 мг/кг в/в, один раз в 3 недели ×2 + лапатиниба в дозе 100 мг/кг перорально, два раза в день ×21.
На фиг.18 приведен график зависимости жизнеспособности клеток SK-BR-3 в условиях in vitro на 3 сутки от кратных концентраций IC50 5-FU, трастузумаба-MCC-DM1 (T-DM1) и комбинаций с соотношением фиксированных концентраций 5-FU и T-DM1.
На фиг.19 приведен график зависимости жизнеспособности клеток BT-474 в условиях in vitro на 3 сутки от кратных концентраций IC50 5-FU, трастузумаба-MCC-DM1 (T-DM1) и комбинаций с соотношением фиксированных концентраций 5-FU и T-DM1.
На фиг.20 приведен график зависимости жизнеспособности клеток SK-BR-3 в условиях in vitro на 3 сутки от кратных концентраций IC50 гемцитабина, трастузумаба-MCC-DM1 (T-DM1) и комбинаций с соотношением фиксированных концентраций гемцитабина и T-DM1.
На фиг.21 приведен график зависимости жизнеспособности клеток MDA-MD-361 в условиях in vitro на 3 сутки от кратных концентраций IC50 гемцитабина, трастузумаба-MCC-DM1 (T-DM1) и комбинаций с соотношением фиксированных концентраций гемцитабина и T-DM1.
На фиг.22 приведен график жизнеспособности (пролиферации) клеток KPL4 в условиях in vitro на 3 сутки после обработки T-DM1, GDC-0941 и комбинациями с соотношением фиксированных концентраций 1:10 T-DM1 и GDC-0941 (от 62,5 нМ до 1 мкМ) при кратных концентрациях IC50 от 0,25× до 4×. Строили график прогноза аддитивного действия Блисса в виде пунктирной линии.
На фиг.23 приведен график жизнеспособности (пролиферации) клеток KPL4 в условиях in vitro на 3 сутки после обработки T-DM1, GDC-0941 и комбинациями с соотношением фиксированных концентраций 1:25 (от 1,25 до 80 нг/мл) и GDC-0941 (от 31,25 нМ до 2 мкМ) при кратных концентрациях IC50 от 0,0625× до 16×. Строили график прогноза аддитивного действия Блисса в виде пунктирной линии.
На фиг.24 приведен график жизнеспособности (пролиферации) клеток KPL4 с амплификацией Her2, резистентных к герцептину®, мутантных по PIK3CA (H1047R) в условиях in vitro на 3 сутки после обработки T-DM1, PI103, GDC-0941 и комбинациями с соотношением фиксированных концентраций T-DM1+PI103 и T-DM1+GDC-0941 при кратных концентрациях IC50 от 0 до 16×.
На фиг.25 приведен график жизнеспособности (пролиферации) клеток KPL4 каспаза 3/7 в условиях in vitro через 24 ч после обработки T-DM1, GDC-0941 и комбинациями с соотношением фиксированных концентраций T-DM1 и GDC-0941 при концентрациях T-DM1 до 160 нг/мл.
На фиг.26 приведен график жизнеспособности (пролиферации) клеток KPL4 в условиях in vitro через 3 суток после обработки T-DM1, GDC-0941 и комбинациями с соотношением фиксированных концентраций T-DM1 и GDC-0941 при концентрациях T-DM1 от 0 до 200 нг/мл.
На фиг.27 приведен график жизнеспособности (пролиферации) клеток MDA-0MB-361 в условиях in vitro на 3 сутки после обработки T-DM1, GDC-0941 и комбинациями с соотношением фиксированных концентраций 1:20 T-DM1 (от 3,125 до 50 нг/мл) и GDC-0941 (от 62,5 нМ до 1 мкМ) при кратных концентрациях IC50 от 0,125× до 8×. Строили график прогноза аддитивного действия Блисса в виде пунктирной линии.
На фиг.28 приведен график жизнеспособности (пролиферации) клеток MDA-0MB-361 в условиях in vitro на 3 сутки после обработки T-DM1, GDC-0941 и комбинациями с соотношением фиксированных концентраций 1:20 T-DM1 (от 3,125 до 100 нг/мл) и GDC-0941 (от 62,5 нМ до 2 мкМ) при кратных концентрациях IC50 от 0,125× до 8×. Строили график прогноза аддитивного действия Блисса в виде пунктирной линии.
На фиг.29 приведен график жизнеспособности (пролиферации) клеток BT-474 в условиях in vitro на 3 сутки после обработки T-DM1, GDC-0941 и комбинациями с соотношением фиксированных концентраций 1:10 T-DM1 (от 3,125 до 100 нг/мл) и GDC-0941 (от 31,25 нМ до 1 мкМ) при кратных концентрациях IC50 от 0,125× до 4×. Строили график прогноза аддитивного действия Блисса в виде пунктирной линии.
На фиг.30 приведен график жизнеспособности (пролиферации) клеток BT-474 в условиях in vitro на 3 сутки после обработки T-DM1, GDC-0941 и комбинациями с соотношением фиксированных концентраций 1:10 T-DM1 (от 6,25 до 100 нг/мл) и GDC-0941 (от 62,5 нМ до 1 мкМ) при кратных концентрациях IC50 от 0,25× до 4×. Строили график прогноза аддитивного действия Блисса в виде пунктирной линии.
На фиг.31 приведен график жизнеспособности (пролиферации) клеток AU565 с амплификацией Her2, мутантных без PIK3 в условиях in vitro на 3 сутки после обработки T-DM1, PI103, GDC-0941 и комбинациями с соотношением фиксированных концентраций T-DM1+PI103 и T-DM1+GDC-0941 при кратных концентрациях IC50 от 0 до 16×.
На фиг.32 приведен график жизнеспособности (пролиферации) клеток EFM192А с амплификацией Her2, мутантных по PIK3СА (C420R) в условиях in vitro на 3 сутки после обработки T-DM1, PI103, GDC-0941 и комбинациями с соотношением фиксированных концентраций T-DM1+PI103 и T-DM1+GDC-0941 при кратных концентрациях IC50 от 0 до 16×.
На фиг.33 приведен график жизнеспособности (пролиферации) клеток HCC1954 с амплификацией Her2, резистентных к герцептину®, мутантных по PIK3CA (H1047R) в условиях in vitro после обработки T-DM1, PI103, GDC-0941 и комбинациями с соотношением фиксированных концентраций T-DM1+PI103 и T-DM1+GDC-0941 при кратных концентрациях IC50 от 0 до 16×.
На фиг.34 приведен график изменения среднего объема опухолей в течение времени в условиях in vivo для трансгенных опухолей MMTV-Her2 Fo5, имплантированных в жировую подушку молочной железы мышей CRL nu/nu, после введения: (1) растворителя перорально, один раз в день ×21; (2) T-DM1 в дозе 10 мг/кг в/в, один раз в 3 недели; (3) 5-FU в дозе 100 мг/кг перорально, один раз в неделю ×2; (4) T-DM1 в дозе 5 мг/кг в/в, один раз в 3 недели + 5-FU в дозе 100 мг/кг перорально, один раз в неделю ×2.
На фиг.35 приведен график изменения среднего объема опухолей в течение времени в условиях in vivo для трансгенных опухолей MMTV-Her2 Fo5, имплантированных в жировую подушку молочной железы мышей CRL nu/nu, после введения: (1) растворителя перорально, один раз в день ×21; (2) T-DM1 в дозе 5 мг/кг в/в, один раз в день ×1; (3) GDC-0941 в дозе 100 мг/кг перорально, один раз в день ×21; (4) GDC-0152 дозе 50 мг/кг перорально, один раз в неделю ×3; (5) T-DM1 в дозе 5 мг/кг в/в, один раз в день ×1 + GDC-0941 в дозе 100 мг/кг перорально, один раз в день ×21; (6) T-DM1 в дозе 5 мг/кг в/в, один раз в день ×1 + GDC-0152 в дозе 50 мг/кг перорально, один раз в неделю ×3.
На фиг.36 приведен график изменения среднего объема опухолей в течение времени в условиях in vivo для опухолей MDA-MB-361.1, имплантированных в жировую подушку молочной железы мышей CRL nu/nu, после введения: (1) растворителя перорально, один раз в день ×21; (2) GDC-0941 в дозе 25 мг/кг, перорально, один раз в день ×21; (3) GDC-0941 в дозе 50 мг/кг, перорально, один раз в день ×21; (4) GDC-0941 в дозе 100 мг/кг, перорально, один раз в день ×21; (5) T-DM1 в дозе 3 мг/кг в/в, один раз в день ×1; (6) T-DM1 в дозе 10 мг/кг в/в, один раз в день ×1; (7) GDC-0941 в дозе 25 мг/кг перорально, один раз в день ×21 + T-DM1 в дозе 3 мг/кг в/в, один раз в день ×1; (8) GDC-0941 в дозе 50 мг/кг перорально, один раз в день ×21 + T-DM1 в дозе 3 мг/кг в/в, один раз в день ×1; (9) GDC-0941 в дозе 100 мг/кг перорально, один раз в день ×21 + T-DM1 в дозе 3 мг/кг в/в, один раз в день ×1; (10) GDC-0941 в дозе 25 мг/кг перорально, один раз в день ×21 + T-DM1 в дозе 10 мг/кг в/в, один раз в день ×1; (11) GDC-0941 в дозе 50 мг/кг перорально, один раз в день ×21 + T-DM1 в дозе 10 мг/кг в/в, один раз в день ×1; (12) GDC-0941 в дозе 100 мг/кг перорально, один раз в день ×21 + T-DM1 в дозе 10 мг/кг в/в, один раз в день ×1.
На фиг.37 приведен график изменения среднего объема опухолей в течение времени в условиях in vivo для опухолей MDA-MB-361.1, имплантированных в жировую подушку молочной железы мышей CRL nu/nu, после введения: (1) растворителей [MCT (0,5% метилцеллюлоза/0,2% твин 80) + сукцинатный буфер (100 мМ сукцината натрия, 100 мг/мл трегалозы, 0,1% твина 80, рН 5,0)] перорально+в/в, один раз в день ×21 и один раз в день; (2) GNE-390 в дозе 1,0 мг/кг, перорально, один раз в день ×21; (3) GNE-390 в дозе 2,5 мг/кг, перорально, один раз в день ×21; (4) T-DM1 в дозе 3 мг/кг в/в, один раз в день; (5) GNE-390 в дозе 1,0 мг/кг перорально, один раз в день ×21 + T-DM1 в дозе 3 мг/кг в/в, один раз в день; (6) GNE-390 в дозе 2,5 мг/кг перорально, один раз в день ×21 + T-DM1 в дозе 3 мг/кг в/в, один раз в день.
Подробное описание предпочтительных вариантов осуществления
Далее будут подробно описаны некоторые варианты осуществления изобретения, примеры которых приведены с прилагаемыми структурами и формулами. Несмотря на то, что изобретение будет описано в сочетании с перечисленными вариантами осуществления, очевидно, понятно, что они не предназначены для ограничения изобретения данными вариантами осуществления. Напротив изобретение предназначено для включения всех альтернатив, модификаций и эквивалентных вариантов, которые могут быть включены в объем настоящего изобретения, определенного формулой изобретения. Специалистам в данной области, очевидно, понятно, что существует много способов и веществ, аналогичных или эквивалентных описанным в данном документе, которые можно использовать в практике настоящего изобретения. Настоящее изобретение никоим образом не ограничивается описанными способами и веществами. В случае, когда один или несколько включенных источников литературы, патентов и аналогичных материалов отличается или противоречит данной заявке, включая, не ограничиваясь этим, определение терминов, использование терминов, описание методик или тому подобное, то данная заявка контролирует это.
Определения
Выражения «содержит», «содержащий», «включает», «включающий» и «включает» при использовании в настоящей заявке и формуле изобретения, предназначаются для определения наличия указанных признаков, целых чисел, компонентов или стадий, но они не препятствуют наличию или добавлению одного или несколько признаков, целых чисел, компонентов или стадий или их групп.
Термины «лечить» и «лечение» относятся к лечебным и профилактическим или превентивным мероприятиям, целью которых является предупреждение или замедление (ослабление) нежелательного физиологического изменения или нарушения, такого как рост, развитие или распространение гиперпролиферативного состояния, такого как злокачественная опухоль. Для целей настоящего изобретения положительные или желательные клинические результаты включают, не ограничиваясь этим, ослабление симптомов, снижение степени выраженности заболевания, обеспечение стабильного (т.е., отсутствие ухудшения) состояния заболевания, снижение или замедление прогрессирования заболевания, ослабление или смягчение болезненного состояния и ремиссия (частичная или полная), независимо от того, являются они детектируемыми или не детектируемыми. Также термин «лечение» может означать пролонгированную выживаемость по сравнению с предполагаемой выживаемостью по сравнению с ситуацией при отсутствии лечения. Субъекты, нуждающиеся в лечении, включают пациентов уже с имеющимся состоянием или нарушением, а также субъектов склонных к развитию состояния или нарушения, и субъектов, у которых состояние или нарушение следует предупредить.
Выражение «терапевтически эффективное количество» означает количество соединения по настоящему изобретению, с помощью которого можно (i) лечить конкретное заболевание, состояние или нарушение, (ii) ослаблять, облегчать или элиминировать один или несколько симптомов конкретного заболевания, состояния или нарушения и (iii) предупреждать или замедлять начало развития одного или несколько симптомов конкретного заболевания, состояния или нарушения, описанного в данном документе. В случае злокачественной опухоли терапевтически эффективное количество лекарственного средства может привести к уменьшению количества опухолевых клеток, уменьшению размера опухоли, подавлению (т.е. замедлению до определенной степени и предпочтительно остановке) инфильтрации опухолевых клеток в периферические органы, подавлению (т.е. замедлению до определенной степени и предпочтительно остановке) метастазирования опухоли, подавлению до определенной степени роста опухоли и/или ослаблению до определенной степени одного или несколько симптомов, ассоциированных со злокачественной опухолью. Лекарственное средство может до определенной степени предупредить рост опухоли и/или привести к гибели имеющихся опухолевых клеток, оно может быть цитостатическим и/или цитотоксическим. В отношении терапии злокачественной опухоли, то эффективность можно оценить, например, по времени прогрессирования заболевания (ТТР) и/или по скорости проявления ответной реакции (RR).
Термин «гиперпролиферативное нарушение» относится к опухолям, раку и новообразованиям, включая презлокачественные и не-злокачественные стадии, а также он включает псориаз, эндометриоз, полипы и фиброаденому.
Термины «рак» и «злокачественная опухоль» относятся к или описывают физиологическое состояние у млекопитающих, которое характеризуется неконтролируемым ростом клеток. «Опухоль» содержит одну или несколько опухолевых клеток. Примеры злокачественной опухоли включают, не ограничиваясь этим, карциному, лимфому, бластому, саркому и лейкоз или злокачественные опухоли лимфоидной ткани. Более конкретные примеры таких злокачественных опухолей включают плоскоклеточный рак (например, эпителиальный плоскоклеточный рак), рак легких, в том числе, мелкоклеточную карциному легких, немелкоклеточную карциному легких («NSCLC»), аденокарциному легких и плоскоклеточную карциному легких, рак брюшины, гепатоцеллюлярную карциному, рак желудка, в том числе, злокачественные опухоли органов пищеварительного тракта, рак поджелудочной железы, глиобластому, рак шейки матки, рак яичников, печени, мочевого пузыря, гепатому, рак молочной железы, ободочной кишки, прямой кишки, колоректальный рак, рак эндометрия или матки, рак слюнных желез, почек или органов выделительной системы, вульвы, щитовидной железы, карциному печени, рак анального отверстия, рак полового члена, а также злокачественные опухоли головы и шеи.
Термин «химиотерапевтическое средство» представляет собой химическое соединение, пригодное для лечения рака, независимо от механизма действия. Группы химиотерапевтических средств включают, не ограничиваясь этим: алкилирующие агенты, антиметаболиты, растительные алкалоиды, представляющие собой яды митотического веретена, цитотоксические/противоопухолевые антибиотики, ингибиторы топоизомеразы, антитела, фотосенсибилизаторы и ингибиторы киназы. Химиотерапевтические средства включают соединения, применяемые в «целенаправленной терапии» и обычной химиотерапии. Примеры химиотерапевтических средств включают: эрлотиниб (тарцева®, Genentech/OSI Pharm.), доцетаксел (таксотер®, Sanofi-Aventis), 5-FU (фторурацил, 5-фторурацил, номер по CAS 51-21-8), гемцитабин (гемзар®, Lilly), PD-0325901 (номер по CAS 391210-10-9, Pfizer), цисплатин (цис-диамин дихлорплатины (II), номер по CAS 15663-27-1), карбоплатин (номер по CAS 41575-94-4), паклитаксел (таксол®, Bristol-Myers Squibb Oncology, Princeton, N.J.), трастузумаб (герцептин®, Genentech), темозоломид (4-метил-5-оксо-2,3,4,6,8-пентабицикло[4.3.0]нона-2,7,9-триен-9-карбоксамид, номер по CAS 85622-93-1, темодар®, темодал®, Shering Plough), тамоксифен ((Z)-2-[4-(1,2-дифенилбут-1-енил)фенокси]-N,N-диметилэтанамин, нольвадекс®, истубал®, валодекс®) и доксорубицин (адриамицин®), акти-1/2, HPPD и рапамицин.
Другие примеры химиотерапевтических средств включают: оксалиплатин (элоксатин®, Sanofi), бортезомиб (велкаде®, Millennium Pharm.), сутент (сунитиниб®, SU11248, Pfizer), летрозол (фемара®, Novartis), иматиниб мезилат (гливек®, Novartis), XL-518 (ингибитор МЕК, Exelixis, международная заявка WO 2007/044515), ARRY-886 (ингибитор МЕК, AZD6244, Array BioPharma, Astra Zeneca), SF-1126 (ингибитор PI3K, Semafore Pharmaceuticals), BEZ-235 (ингибитор PI3K, Novartis), XL-147 (ингибитор PI3K, Exelixis), PTK787/ZK 222584 (Novartis), фулвестрант (фазлодекс®, AstraZeneca), лейковорин (фолиновая кислота), рапамицин (сиролимус, рапамун®, Wyeth), лапатиниб (тикерб®, GSK572016, Glaxo Smith Kline), лонафарниб (сарасарТМ, SCH 66336, Shering Plough), сорафениб (нексавар®, BAY43-9006, Bayer Labs), гефитиниб (иресса®, AstraZeneca), иринотекан (камтосар®, CPT-11, Pfizer), типифарниб (зарнестраТМ, Johnson&Johnson), абраксанТМ (не содержащий кремофора), наночастицы на основе альбумина с паклитакселом (American Pharmaceutical Partners, Schaumberg, Il), вандетаниб (rINN, ZD6474, зактима®, AstraZeneca), хлорамбуцил, AG1478, AG1571 (SU 5271; Sugen), темсиролимус (торисел®, Wyeth), пазопаниб (GlaxoSmithKline), канфосфамид (телсита®, Telik), тиотеру и циклофосфамид (цитоксан®, неосар®); алкисульфонаты, такие как бусульфан, импросульфан и пипосульфан; азиридины, такие как бензодопа, карбоквон, метуредопа и уредопа; этиленимины и метиламеламины, включая альтретамин; триэтиленамин, триэтиленфосфорамид, триэтилентиофосфорамид и триметиломеламин; ацетогенины (в частности, буллатацин и буллатацинон); камптотецин (включая синтетический аналог топотекан); бриостатин; каллистатин; СС-1065 (включая его синтетические аналоги адозелезин, карзелезин и бизелезин); криптофицины (в частности, криптофицин 1 и криптофицин 8); доластатин; дуокармицин (включая синтетические аналоги KW-2189 и CB1-TM1); элеутеробин; панкратистатин; саркодиктиин; спонгистатин; азотистые иприты, такие как хлорамбуцил, хлорнафазин, хлорфосфамид, эстрамустин, ифосфамид, мехлоретамин, мехлортамина оксид гидрохлорид, мелфалан, новембихин, фенэстрин, преднимустин, трофосфамид, урациловый иприт; нитрозомочевины, такие как кармустин, хлорзотоцин, фотемустин, ломустин, нимустин и ранимнустин; антибиотики, такие как энедииновые антибиотики (например, калихеамицин, калихеамицин гамма 1I, калихеамицин омегаI1 (Angew Chem. Intl. Ed. Engl., 1994, 33:183-186); динемицин, динемицин А; бифосфонаты, такие как клодронат; эсперамицин; а также неокарзиностатин хромофор и близкие хромопротеиновые энедииновые антибиотики хромофоры), аклациномизины, актиномицин, аутрамицин, азасерин, блеомицины, кактиномицин, карабицин, карминомицин, карзинофилин, хромомицины, дактиномицин, даунорубицин, деторубицин, 6-диазо-5-оксо-L-норлейцин, морфолино-доксорубицин, цианоморфолино-доксорубицин, 2-пирролино-доксорубицин и дезоксидоксорубицин), эпирубицин, эзорубицин, идарубицин, марцелломицин, митомицины, такие как митомицин С, микофеноловую кислоту, ногаламицин, оливомицины, пепломицин, порфиромицин, пуромицин, квеламицин, родорубицин, стрептонигрин, стрептозоцин, туберцидин, убенимес, зиностатин, зорубицин; антиметаболиты, такие как метотрексат и 5-фторурацил (5-FU); аналоги фолиевой кислоты, такие как деноптерин, метотрексат, птероптерин, триметрексат; аналоги пуринов, такие как флударабин, 6-меркаптопурин, тиамиприн, тиогуанин; аналоги пиримидинов, такие как анцитабин, азацитидин, 6-азауридин, кармофур, цитарабин, дидезоксиуридин, доксифлуридин, эноцитабин, флоксуридин; андрогены, такие как калустерон, дромостанолон пропионат, эпитиостанол, мепитиостан, тестолактон; ингибиторы надпочечников, такие как аминоглутетимид, митотан, трилостан; пополнители фолиевой кислоты, такие как фролиновая кислота; ацеглатон; альдофосфамид гликозид; аминолевулиновую кислоту; энилурацил; амсакрин; бестрабуцил; бисантрен; эдатраксат; дефофамин; демеколцин; диазиквон; элфорнитрин; эллиптиний ацетат; эпотилон; этоглюцид; нитрат галлия; гидроксимочевину; лентинан; лонидаинин; майтансиноиды, такие как майтансин и ансамитоцины; митогуазон; митоксантрон; мопиданмол; нитаэрин; пентостатин; фенамет; парарубицин; лозоксантрон; подофилловую кислоту; 2-этилгидразид; прокарбазин; полисахаридный комплекс PSK® (JHS Natural Products, Eugene, OR); разоксан; ризоксин; сизофиран; спирогерманий; тенуазоновую кислоту; триазиквон; 2,2’,2”-трихлорэтиламин; трихотецены (Т-2 токсин, верракурин А, роридин А и ангуидин); уретан; виндезин; дакарбазин; манномустин; митобронитол; митолактол; пипоброман; гацитозин; арабинозид (Ара-С); циклофосфамид; тиотепа; 6-тиогуанин; меркаптопурин; метотрексат; аналоги платины, такие как цисплатин и карбоплатин; винбластин; этопозид (VP-16); ифосфамид; митоксантрон; винкристин; винорельбин (навелбин®); новантрон; тенипозид; этатрексат; дауномицин; аминоптерин; капецитабин (кселода®, Roche); ибандронат; СРТ-11; ингибитор топоизомеразы RFS 2000; дифторметилорнитин (DMFO); ретиноиды, такие как ретиноевая кислота, и фармацевтически приемлемые соли, кислоты и производные любого из перечисленных выше продуктов.
Также в понятие «химиотерапевтическое средство» входят: (i) антигормональные средства, регулирующие или ингибирующие действие гормонов на опухоли, такие как антиэстрогены и селективные модуляторы рецепторов эстрогенов (SERM), включая, например, тамоксифен (нольвадекс®; тамоксифена цитрат), ралоксифен, дролоксифен, 4-гидрокситамоксифен, триоксифен, кеоксифен, LY117018, онапристон и фарестон® (торемифин цитрат); (ii) ингибиторы ароматазы, ингибирующие фермент ароматазу, которая регулирует продукцию эстрогенов в надпочечниках, например, такие как 4(5)-имидазолы, аминоглютетимид, мегаза® (мегестрол ацетат), аромазин® (эксеместан, Pfizer), форместан, фадрозол, ривизор® (ворозол), фемара® (летрозол, Novartis) и аримидекс® (анастрозол, AstraZeneca); (iii) антиандрогены, такие как флутамид, нилутамид, бикалутамид, лейпролид и гозерелин, а также троксацитабин (аналог нуклеозида цитозина 1,3-диоксолан); (iv) ингибиторы протеинкиназы, такие как ингибиторы MEK (международная заявка WO 2007/044515); (v) ингибиторы липидкиназы; (vi) антисмысловые олигонуклеотиды, в частности, которые ингибируют экспрессию генов в сигнальных путях, принимающих участие в аномальной пролиферации клеток, например, PKC-альфа, Raf и H-Ras, такие как облимерсен (генасенс®, Genta Inc.); (vii) рибозимы, такие как ингибиторы экспрессии VEGF (например, ангиозим®) и ингибиторы экспрессии HER2; (viii) вакцины, такие как вакцины на основе генной терапии, например, алловектин®, лейвектин® и ваксид®; пролейкин® rIL-2; ингибиторы топоизомеразы 1, такие как луртотекан®; абареликс® rmRH; (ix) антиангиогенные средства, такие как бевацизумаб (авастин®, Genentech), и фармацевтически приемлемые соли, кислоты и производные любого из перечисленных выше продуктов.
Также в понятие «химиотерапевтическое средство» входят терапевтические антитела, такие как алемтузумаб (Campath), бевацизумаб (авастин®, Genentech); цетуксимаб (эрбитукс®, Imclone); панитумумаб (вектибикс®, Amgen), ритуксимаб (ритуксан®, Genentech/Biogen Idec), пертузумаб (омнитаргТМ, 2С4, Genentech), трастузумаб (герцептин®, Genentech), тозитумомаб (Bexxar, Corixia) и конъюгат антитело-лекарственное средство, гемтузумаб озогамицин (милотарг®, Wyeth).
Гуманизированные моноклональные антитела с лечебной активностью в виде химиотерапевтических средств в комбинации с трастузумабом-MCC-DM1 включают: алемтузумаб, аплизумаб, азелизумаб, атлизумаб, бапиньюзумаб, бевацизумаб, биватузумаб мертанзин, кантузумаб мертанзин, цеделизумаб, сертолизумаб пегол, цитфуситузумаб, цидтузумаб, даклизумаб, экулизумаб, эфализумаб, эптатузумаб, эрлизумаб, фелвизумаб, фонтолизумаб, гемтузумаб озогамицин, инотузумаб озогамицин, ипилимумаб, лабетузумаб, линтузумаб, матузумаб, меполизумаб, мотавизумаб, мотовизумаб, натализумаб, нимоиузумаб, ноловизумаб, нумавизумаб, окрелизумаб, омализумаб, паливизумаб, пасколизумаб, пекфуситузумаб, пектузумаб, пертузумаб, пекселизумаб, раливизумаб, ранибизумаб, ресливизумаб, реслизумаб, резивизумаб, ровелизумаб, руплизумаб, сибротузумаб, сиплизумаб, сонтузумаб, такатузумаб тетраксетан, тадоцизумаб, тализумаб, тефибазумаб, тоцилизумаб, торазумаб, трастузумаб, тукотузумаб целмолейкин, тукузитузумаб, умавизумаб, уртоксазумаб и визилизумаб.
Термин «метаболит» означает продукт, синтезированный в результате метаболизма определенного соединения или его соли в организме. Метаболиты соединения можно идентифицировать с использованием обычных методов, известных в данной области, и их активность оценить с использованием тестов, описанных в данном документе. Такие продукты могут образоваться, например, в результате окисления, восстановления, гидролиза, амидирования, деамидирования, этерификации, деэтерификации, ферментативного расщепления и тому подобное, вводимого соединения. Следовательно, изобретение включает метаболиты соединений по изобретению, в том числе, соединения, продуцированные способом, включающим контактирование соединения по настоящему изобретению с млекопитающим в течение периода времени, достаточного для образования продукта его метаболизма.
В том смысле, в котором термин «вкладыш в упаковке» используется в данном документе, он относится к инструкциям, которые обычно включают в промышленные упаковки лекарственных продуктов, содержащие информацию о показаниях, применении, дозировке, введении, противопоказаниях и/или предостережениях, касающихся применения таких лекарственных продуктов.
В том смысле, в котором термин «фармацевтически приемлемая соль» используется в данном документе, он относится к фармацевтически приемлемым органическим или неорганическим солям соединения по изобретению. Приведенные в качестве примера соли включают такие соли, не ограничиваясь этим, как сульфат, цитрат, ацетат, оксалат, хлорид, бромид, иодид, нитрат, бисульфат, фосфат, гидрофосфат, изоникотинат, лактат, салицилат, гидроцитрат, тартрат, олеат, таннат, пантотенат, битартрат, аскорбат, сукцинат, малеат, гентизинат, фумарат, глюконат, глюкуронат, сахарат, формиат, бензоат, глутамат, метансульфонат, «мезилат», этансульфонат, бензолсульфонат, п-толуолсульфонат и памоат (т.е. 1,1’-метилен-бис-(2-гидрокси-3-нафтоат)). Фармацевтически приемлемые соли могут содержать включение другой молекулы, такой как ион ацетат, ион сукцинат или другой противоион. Противоион может представлять собой любую органическую или неорганическую группу, которая стабилизирует заряд в исходном соединении. Кроме того, фармацевтически приемлемые соли могут содержать в своей структуре более чем один заряженный атом. В тех случаях, когда многочисленные заряженные атомы являются частью фармацевтически приемлемой соли, то они могут содержать многочисленные противоионы. Следовательно, фармацевтически приемлемая соль может содержать один или несколько заряженных атомов и/или один или несколько противоионов.
В том случае, когда соединение по изобретению является основанием, то желаемую фармацевтически приемлемую соль можно получить с помощью любого подходящего метода, известного в данной области, например, обработкой свободного основания неорганической кислотой, такой как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, метансульфоновая кислота, фосфорная кислота и тому подобное, или органической кислотой, такой как уксусная кислота, малеиновая кислота, янтарная кислота, миндальная кислота, фумаровая кислота, малоновая кислота, пировиноградная кислота, щавелевая кислота, гликолевая кислота, салициловая кислота, пиранозидильная кислота, такая как глюкуроновая кислота или галактуроновая кислота, альфа-оксикислота, такая как лимонная кислота или винная кислота, аминокислота, такая как аспарагиновая кислота или глютаминовая кислота, ароматическая кислота, такая как бензойная кислота или циннамовая кислота, сульфоновая кислота, такая как п-толуолсульфоновая кислота или этансульфоновая кислота, или тому подобное. Общие аспекты по кислотам, подходящим для получения фармацевтически пригодных или приемлемых солей из основных фармацевтических соединений, обсуждаются, например, P. Stahl et al., Camille G. (eds.) Handbook of Pharmaceutical Salts. Properties, Selection and Use, 2002, Zurich: Wiley-VCH; S. Berge et al., Journal of Pharmaceutical Sciences, 1977, 66(1), 119; P. Gould, International J. of Pharmaceutics, 1986, 33:201-217; Anderson et al., the Practice of Medicinal Chemistry, 1996, Academic Press, New York; Remington’s Pharmaceutical Sciences, 18th ed., 1995, Mack Publishing Co., Easton P.A.; и в The Orange Book (Food&Drug Administration, Washington, D.C. на их сайте). Указанные источники включены в данный документ для сведения.
В том случае, когда соединение по изобретению является кислотой, то желаемую фармацевтически приемлемую соль можно получить с помощью любого подходящего метода, например, обработкой свободной кислоты неорганическим или органическим основанием, таким как амин (первичный, вторичный или третичный), гидроксид щелочного металла или гидроксид щелочно-земельного металла или тому подобное. Показательные примеры подходящих солей включают, не ограничиваясь этим, органические соли, полученные из аминокислот, таких как глицин и аргинин, аммиака, первичных, вторичных и третичных аминов и циклических аминов, таких как пиперидин, морфолин и пиперазин, и неорганические соли, полученные из натрия, кальция, калия, магния, марганца, железа, меди, цинка, алюминия и лития.
Выражение «фармацевтически приемлемые» указывает на то, что соединение или композиция могут быть совместимыми химически и/или с точки зрения токсикологии с другими ингредиентами, входящими в состав композиции, или с млекопитающим, которое подвергается лечению ими.
Термин «сольват» относится к физической ассоциации или комплексу одной или несколько молекул растворителя и соединения по изобретению. Соединения по изобретению могут находиться как в несольватированной форме, так и сольватированной форме. Примеры растворителей, которые образуют сольваты, включают, не ограничиваясь этим, воду, изопропанол, этанол, метанол, ДМСО, этилацетат, уксусную кислоту и этаноламин. Термин «гидрат» относится к комплексу, в котором молекула растворителя представляет собой воду. Данная физическая ассоциация имеет различные степени ионного и ковалентного связывания, в том числе, водородные связи. В некоторых случаях сольват можно выделить, например, когда одна или несколько молекул растворителя входят в кристаллическую решетку кристаллического твердого вещества. В общем получение сольватов известно, например, оно описано M. Caira et al., J. Pharmaceutical Sci., 93(3), 601-611, 2004. Аналогичные способы получения сольватов, гемисольватов, гидратов и тому подобное описаны E.C. van Tonder et al., AAPS Pharm. Sci. Tech., 5(1), article 12, 2004 и A.L. Bingham et al., Chem. Commun., 603-604, 2001. Типичный, не ограничивающий способ включает растворение соединения по изобретению в желаемом количестве желаемого растворителя (органического растворителя или воды, или их смесей) при более высокой, чем комнатная температура, и охлаждение раствора со скоростью, достаточной для образования кристаллов, которые затем выделяют обычными методами. С помощью аналитических методов, например, таких как ИК-спектроскопия, определяют присутствие растворителя (или воды) в кристаллах в виде сольвата (или гидрата).
В том смысле, в котором в данном документе используется термин «синергический», он относится к терапевтической комбинации, которая является более эффективной по сравнению с аддитивным действием двух или несколько одних агентов. Определение синергического взаимодействия между трастузумабом-MCC-DM1 и одним или несколько химиотерапевтическим средством может быть основано на результатах тестов, описанных в данном документе. Результаты данных тестов анализируют с использованием комбинационного метода Chou и Talalay и анализа доза-эффект с использованием программы CalcuSyn для определения комбинационного индекса «CI» (Chou and Talalay, 1984, Adv. Enzyme Regul., 22:27-55). Комбинации по настоящему изобретению были оценены в нескольких аналитических системах и полученные данные можно подвергнуть обработке с использованием стандартной программы для количественного определения синергизма, аддитивного эффекта и антагонизма у противоопухолевых средств. Предпочтительно используют программу, описанную Chou and Talalay в «New Avenues in Developmental Cancer Chemotherapy», Academic Press, 1987, chapter 2. Значения комбинационного индекса (CI) ниже 0,8 указывают на наличие синергизма, значения выше 1,2 указывают на антагонизм и значения в пределах от 0,8 до 1,2 указывают на аддитивный эффект. Комбинированная терапия может обеспечивать «синергию» и быть «синергической», т.е. когда достигаемый эффект при совместном использовании активных ингредиентов выше по сравнению с суммой эффектов, обеспечиваемых с использованием соединений по отдельности. Синергическое действие можно обеспечить, когда активные ингредиенты: (1) формулируют вместе и вводят или доставляют одновременно в комбинированной разовой лекарственной форме; (2) вводят по очередности в виде отдельных лекарственных форм или (3) вводят по другой схеме. При введении по очередности синергическое действие можно обеспечить, когда соединения вводят или доставляют последовательно, например, с помощью разных инъекций в отдельных шприцах. В основном во время «поочередной» терапии эффективную дозу каждого активного ингредиента вводят последовательно, например, поочередно во времени.
Трастузумаб-MCC-DM1
Настоящее изобретение относится к терапевтическим комбинациям, содержащим трастузумаб-MCC-DM1(T-DM1), конъюгат антитело-лекарственное средство (номер по CAS № 139504-50-0), имеющий формулу:
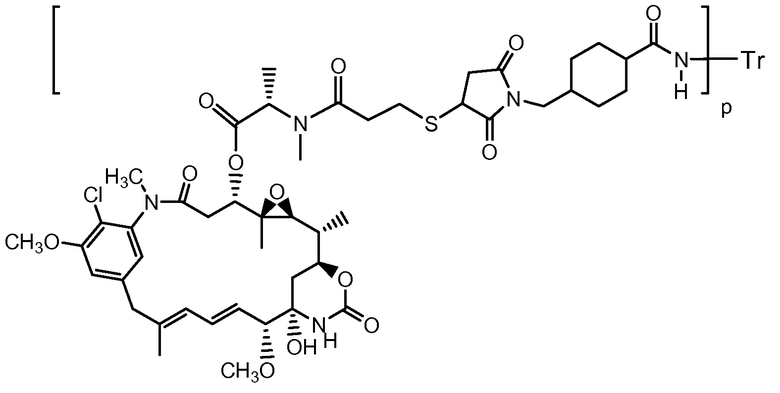
где Tr представляет трастузумаб, связанный через линкер МСС, с группой лекарственного средства майтансиноида, DM1 (патент США № 5208020; патент США № 6441163). Соотношение лекарственного средства и антитела или нагрузка лекарственным средством представлена показателем «р» в приведенной выше структуре трастузумаба-MCC-DM1 и пределы целых значений составляют от 1 до примерно 8. Значение нагрузки лекарственным средством «р» составляет 1-8. Трастузумаб-MCC-DM1 содержит все смеси различно нагруженных и присоединенных конъюгатов антитело-лекарственное средство, где 1, 2, 3, 4, 5, 6, 7 и 8 групп лекарственного средства ковалентно присоединены к антителу трастузумаб (патент США № 7097840; заявка на патент США № 2005/0276812; заявка на патент США № 2005/0166993). Конъюгат трастузумаб-MCC-DM1 можно приготовить, следуя способу, описанному в примере 1.
Трастузумаб продуцируется в суспензионной культуре клеток млекопитающих (клеток яичника китайского хомячка, СНО). Прото-онкоген HER2 (или c-erbB2) кодирует трансмембранный белок рецептора массой 185 kDa, который в структурном отношении близок к рецептору эпидермального фактора роста. Сверхэкспрессию белка HER2 отмечают в 25-30% первичных злокачественных опухолей молочной железы и ее можно детектировать с использованием иммуногистохимического метода, основанного на анализе фиксированных опухолевых блоков (Press M.F. et al., 1993, Cancer Res., 53:4960-70). Трастузумаб представляет собой антитело, содержащее антигенсвязывающие остатки или полученное из мышиного антитела 4D5 (ATCC CRL 10463, которое депозировано в Американской коллекции типовых культур, 12301 Parklawn Drive, Rockville, Md. 20852 по Будапештскому соглашению от 24 мая 1990). Приводимые в качестве примера гуманизированные антитела 4D5 включают huMAb4D5-1, huMAb4D5-2, huMAb4D5-3, huMAb4D5-4, huMAb4D5-5, huMAb4D5-6, huMAb4D5-7 и huMAb4D5-8 (герцептин®), описанные в патенте США № 5821337.
В фазе I клинических испытаний было установлено, что максимально переносимая доза (MTD) T-DM1, при внутривенной инфузии каждые 3 недели, составляет 3,6 мг/кг. Значение DLT (ограничивающая дозы токсичность) по тромбоцитопении степени 4 у 2 из 3 пациентов равняется 4,8 мг/кг. Побочные эффекты степени ≥2, наблюдаемые в дозе 3,6 мг/кг, проявлялись редко и были незначительными. Данная схема лечения хорошо переносилась больными и обеспечивала хороший клинический эффект, как описано ранее. В фазе II клинических испытаний было показано, что доза 3,6 мг/кг хорошо переносилась при введении каждые 3 недели при небольшом проценте пациентов (3 из 112 пациентов), для которых требовалось снижение дозировки средства. Таким образом, дозу T-DM1, равную 3,6 мг/кг каждые 3 недели, выбрали для тестирования в настоящем исследовании, основываясь на следующем: (1) была показана эффективность и безопасность T-DM1 в дозе 3,6 мг/кг каждые 3 недели и (2) для данной группы пациентов удобна схема один раз в 3 недели.
Химиотерапевтические средства
Было показано, что некоторые химиотерапевтические средства проявляют удивительные и неожиданные свойства в комбинации с трастузумабом-МСС-DM1 в подавлении клеточной пролиферации в условиях in vitro и in vivo. Такие химиотерапевтические средства включают антитело-ингибитор димеризации HER2, антитело к VEGF, 5-FU, карбоплатин, лапатиниб, АВТ-869, доцетаксел, GDC-0941 и GNE-390.
Пертузумаб (номер по CAS 380610-27-5, омнитарг®, 2С4, Genentech) представляет рекомбинантное, гуманизированное моноклональное антитело, которое ингибирует димеризацию HER2 (патент США № 6054297; патент США № 6407213; патент США № 6800738; патент США № 6949245; патент США № 7041292). Пертузумаб и трастузумаб направлены на различные внеклеточные области рецептора тирозинкиназы HER-2 (Nahta et al., 2004, Cancer Res., 64:2343-2346). Гибридомная клеточная линия, экспрессирующая 2С4 (пертузумаб) депозирована в Американской коллекции типовых культур (АТСС), 10801 University Boulevard, Manassas, Va. 20110-2209, USA как ATCC HB-12697 8 апреля 1999. Пертузумаб блокирует способность рецептора HER2 соединяться с другими членами семейства рецепторов HER2, т.е. HER1/EGFR, HER3 и HER4 (Agus et al., 2002, Cancer Cell, 2:127-137; Jackson et al., 2004, Cancer Res., 64:2601-2609; Takai et al., 2005, Cancer, 104:2701-2708; патент США № 6949245). В опухолевых клетках нарушение способности HER2 к взаимодействию с другими членами семейства рецепторов HER приводит к блокированию передачи клеточных сигналов и в конечном итоге может привести к подавлению роста опухолевых клеток и гибели опухолевых клеток. HDI за счет уникального механизма их действия обладают способностью функционировать в широком ряде опухолей, включая опухоли без сверхэкспрессии HER2 (Mullen et al., 2007, Molecular Cancer Therapeutics, 6:93-100).
Пертузумаб основан на последовательностях каркасной области человеческого IgG1 (κ). Он состоит из двух тяжелых цепей и двух легких цепей. Подобно трастузумабу пертузумаб направлен на внеклеточный домен HER2. Однако он отличается от трастузумаба по эпитоп-связывающим областям легкой цепи и тяжелой цепи. В результате пертузумаб связывается с эпитопом внутри, который известен как субдомен 2 HER2, в то время как эпитоп из трастузумаба находится в субдомене 4 (Cho et al., 2003; Franklin et al., 2004). Пертузумаб функционирует посредством блокирования ассоциации HER2 с другими членами семейства HER, включая HER1 (рецептор эпидермального фактора роста; EGFR), HER3 и HER4. Данная ассоциация необходима для передачи сигналов в присутствии лиганда через МАР-киназу и PI3-киназу. В результате пертузумаб ингибирует инициированную лигандом внутриклеточную передачу сигналов. Ингибирование данных сигнальных путей может привести к соответственно к остановке роста и апоптозу (Hanahan and Weinberg, 2000). В результате того, что пертузумаб и трастузумаб связываются с различными эпитопами в рецепторе HER2, активированная лигандом передача сигналов ниже блокируется пертузумабом, но не под действием трастузумаба. Следовательно, для пертузумаба не требуется сверхэкспрессия HER2 для проявления его активности в качестве противоопухолевого средства. Кроме того, в результате дополняющих механизмов их действия комбинация пертузумаба и T-DM1, обладает потенциальной возможностью применяться в лечении заболеваний, при которых имеется сверхэкспрессия HER2.
Пертузумаб оценивали в качестве препарата для монотерапии в пяти клинических испытаниях в фазе II, проведенных на различных злокачественных опухолях, включая MBC с низким уровнем экспрессии HER2, немелкоклеточную карциному легких, гормон-устойчивую злокачественную опухоль предстательной железы и злокачественную опухоль яичников. В фазе II клинических испытаний оценивали эффективность пертузумаба в качестве самостоятельного препарата при второй или третьей линии химиотерапии у пациенток с метастатическим раком молочной железы (МВС) с нормальной экспрессией HER2 (Cortes et al., 2005, J. Clin. Oncol., 23:3068). Оценивали эффективность пертузумаба в фазе II клинических испытаний в комбинации с трастузумабом (Baselga J. et al., «A Phase II trial of trastuzumab and pertuzumab in patients with HER-2-positive metastatic breast cancer that had progressed during trastuzumab therapy: full response data», European Society of Medical Oncology, Stockholm, Sweden, September 12-16, 2008; Gelmon et al., 2008, J. Clin. Oncol., 26:1026). В первом исследовании участвовало 11 пациенток с HER-положительной МВС, которые ранее получали до трех курсов лечения с включением трастузумаба (Portera et al., 2007).
Бевацизумаб (номер по CAS 216974-75-3, авастин®, Genentech) представляет собой моноклональное антитело к VEGF, к васкулярному эндотелиальному фактору роста (патент США № 7227004; патент США № 6884879; патент США № 7060269; патент США № 7169901; патент США № 7297334), препарат применяют для лечения злокачественных опухолей, где он подавляет их рост, блокируя образование новых кровеносных сосудов. Бевацизумаб был первым применяемым в клинике ингибитором ангиогенеза в США, разрешенным FDA в 2004 г., для применения в комбинации с обычной химиотерапией для лечения метастатического рака ободочной кишки и большинства форм метастатической немелкоклеточной карциномы легких. Было проведено несколько завершающих стадий клинических испытаний для оценки его безопасности и эффективности у пациентов с адъювантным/неметастатическим раком ободочной кишки, метастатическим раком молочной железы, метастатическим раком почки, метастатической мультиформной глиобластомой, метастатическим раком яичников, метастатическим гормон-устойчивым раком предстательной железы и метастатическим или неоперабельным местно-распространенным раком поджелудочной железы.
Обычно анти-VEGF-антитело не будет связываться с другими гомологами VEGF, такими как VEGF-В или VEGF-С, и с другими факторами роста, такими как PIGF, PDGF или bFGF. Предпочтительные антитела к VEGF включают моноклональное антитело, которое связывается с тем же эпитопом, что и моноклональное анти-VEGF-антитело А4.6.1, продуцированное гибридомой АТСС НВ 10709; рекомбинантное гуманизированное моноклональное антитело к VEGF, полученное, как описано Presta et al. 1997, Cancer Res., 57:4593-4599, включая, не ограничиваясь этим, бевацизумаб. Бевацизумаб содержит мутантные каркасные области человеческого IgG1 и антигенсвязывающие определяющие комплементарность области из мышиного моноклонального анти-hVEGF-антитела А4.6.1, которое блокирует связывание человеческого VEGF с его рецепторами. Примерно 93% аминокислотной последовательности бевацизумаба, включая большую часть каркасных областей, происходит из человеческого IgG1 и примерно 7% последовательности происходит из мышиного антитела А4.6.1. Бевацизумаб имеет молекулярную массу, составляющую примерно 149000 дальтон, и он гликозилирован. Бевацизумаб и другие гуманизированные анти-VEGF-антитела подробно описаны в патенте США № 6884879. Дополнительные анти-VEGF-антитела включают антитела серий G6 и В20 (например, G6-31, В20-4.1), как показано на фиг.27-29 в международной заявке WO2005/012359. В одном варианте осуществления антитело серии В20 связывается с функциональным эпитопом в человеческом VEGF, содержащем остатки F17, M18, D19, Y21, Y25, Q89, I91, K101, E103 и С104.
Гибридомные линии, экспрессирующие анти-VEGF-антитела А4.6.1 (АТСС НВ 10709) и В2.6.2 (АТСС НВ 10710), депозированы и хранятся в Американской коллекции типовых культур (АТСС), 10801 University Boulevard, Manassas, VА 20110-2209, USA. Клон, экспрессирующий полипептид VEGF-E (патент США № 6391311), кодированный инсертом нуклеотидной последовательности депозита АТСС под номером DNA29101-1276, депозирован 5 марта 1998 и хранится под номером АТСС 209653 в Американской коллекции типовых культур (АТСС), 10801 University Boulevard, Manassas, Va. 20110-2209, USA.
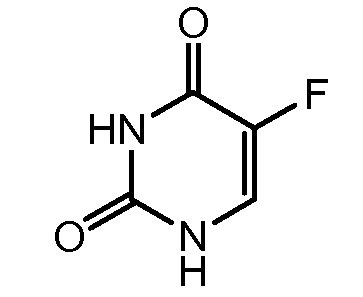
5-FU (фторурацил, 5-фторурацил, номер по CAS № 51-21-8) представляет собой ингибитор тимидилатсинтазы, и в течение десятилетий его применяли для лечения рака, включая коллоректальный рак и рак поджелудочной (патент США № 2802005; патент США № 2885396; Barton et al., 1972, Jour. Org. Chem., 37:329; Hansen R.M., 1991, Cancer Invest., 9:637-642). 5-FU представляет 5-фтор-1Н-пиримидин-2,4-дион и он имеет формулу:
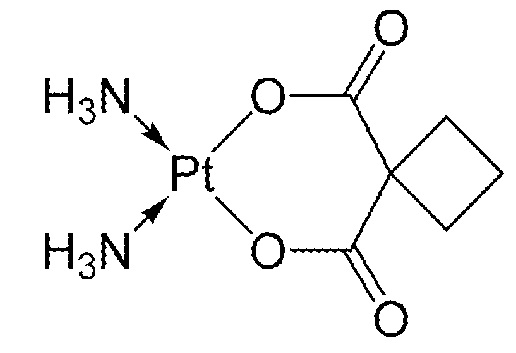
Карбоплатин (номер по CAS № 41575-94-4) представляет собой химиотерапевтическое средство, которое применяют для лечения злокачественных опухолей яичников, легких, головы и шеи (патент США № 4140707). Карбоплатин представляет азанид; циклобутан-1,1-дикарбоновой кислоты платину и имеет формулу:
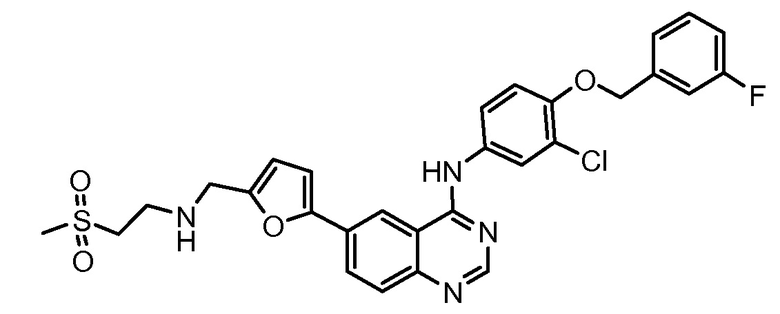
Лапатиниб (номер по CAS № 388082-78-8, тикерб®, GW572016, Glaxo SmithKline) разрешен для применения в комбинации с капецитабином (кселода®, Roche) для лечения пациенток с местно-распространенной или метастатической злокачественной опухолью молочной железы со сверхэкспрессией HER2 (ErbB2) и пациенток, ранее получавших терапию антрациклином, таксаном и трастузумабом. Лапатиниб является АТФ-конкурентным эпидермальным фактором роста (EGFR) и двойным ингибитором Her2/neu (ErbB-2) тирозинкиназы (патент США № 6727256; патент США № 6713485; патент США № 7109333; патент США № 6933299; патент США № 7084147; патент США № 7157466; патент США № 7141576), который ингибирует аутофосфорилирование и активирование рецепторов посредством связывания с АТФ-связывающим участком домена EGFR/HER2 протеинкиназы. Лапатиниб представляет N-(3-хлор-4-(3-фторбензилокси)фенил)-6-(5-((2-(метилсульфонил)этиламино)метил)фуран-2-ил)хиназолин-4-амин и он имеет формулу:
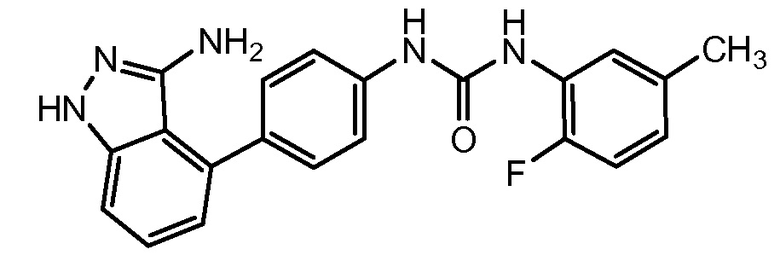
АВТ-869 (Abbott and Genentech) является многомишеневым ингибитором семейства рецепторов тирозинкиназ VEGF и PDGF для потенциального лечения рака при приеме препарата перорально (патент США № 7297709; заявка на патент США № 2004/235892; заявка на патент США № 2007/104780). Начаты клинические испытания препарата при лечении немелкоклеточного рака легких (NSCLC), гепатоцеллюлярной карциномы (НСС) и почечно-клеточной карциномы (RCC). В химическом отношении АВТ-869 представляет 1-(4-(3-амино-1Н-индазол-4-ил)фенил)-3-(2-фтор-5-метилфенил)мочевину (номер по CAS № 796967-16-3) и имеет формулу:
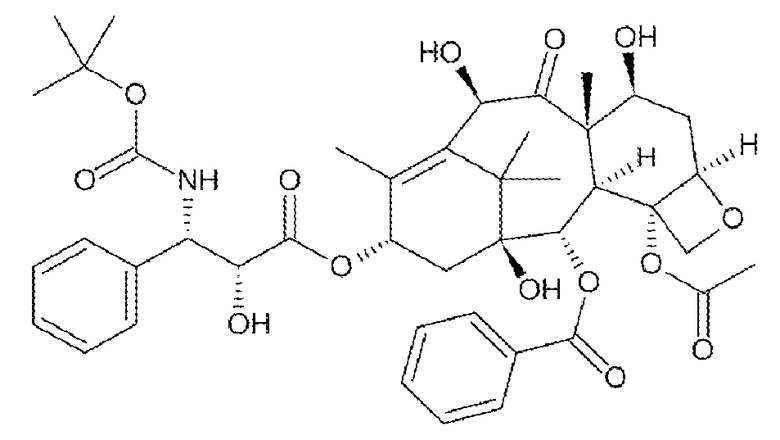
Доцетаксел (таксотер®, Sanofi-Aventis) применяют для лечения рака молочной железы, яичников и NSCLC (патент США № 4814470; патент США № 5438072; патент США № 5968582; патент США № 5714512; патент США № 5750561). Доцетаксел представляет N-трет-бутиловый эфир (2R,3S)-N-карбокси-3-фенилизосерина, 13-эфир 5,20-эпокси-1,2,4,7,10,13-гексагидрокситакс-11-ен-9-она 4-ацетат-2-бензоата тригидрата (патент США № 4814470; Европейский патент 253738; номер по CAS 114977-28-5) и имеет формулу:
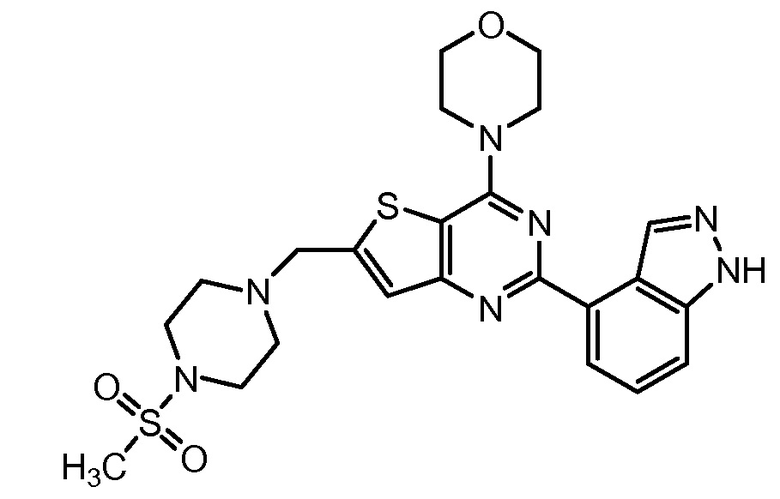
GDC-0941 (Genentech Inc.) представляет собой селективный, обладающий хорошей биодоступностью при пероральном приеме тиенопиримидиновый ингибитор PI3K с обещающими фармакокинетическими и фармацевтическими свойствами (Folkes et al., 2008, J. Med. Chem., 51(18):5522-5532; заявка на патент США № 2008/0076768; заявка на патент США № 2008/0207611; Belvin et al., American Association for Cancer Research Annual Meeting 2008, 99th:April 15, Abstract 4004; Folkes et al., American Association for Cancer Research Annual Meeting 2008, 99th:April 15, Abstract LB-146; Friedman et al., American Association for Cancer Research Annual Meeting 2008, 99th:April 14, Abstract LВ-110). GDC-0941 демонстрирует синергическую активность в условиях in vitro и in vivo в комбинации с некоторыми химиотерапевтическими средствами на клеточных линиях солидных опухолей (заявка на патент США № 12/208227, Belvin et al. «Combinations of phosphoinositide 3-kinase inhibitor compounds and chemotherapeutic agents and methods of use», поданная 10 сентября 2008). В химическом отношении GDC-0941 представляет 4-(2-(1Н-индазол-4-ил)-6-((4-(метилсульфонил)пиперазин-1-ил)метил)тиено[3,2-d]пиримидин-4-ил)морфолин (номер по CAS 957054-30-7) и имеет формулу:
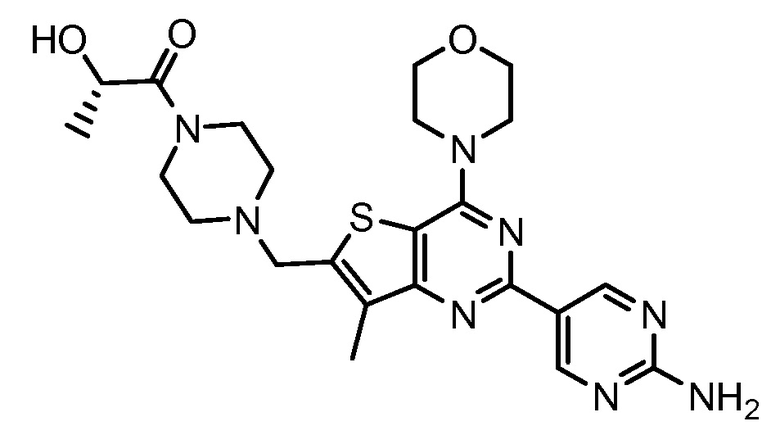
GNE-390 (Genentech Inc.) представляет собой селективный, обладающий хорошей биодоступностью при пероральном приеме тиенопиримидиновый ингибитор PI3K с обещающими фармакокинетическими и фармацевтическими свойствами (заявка на патент США № 2008/0242665; международная заявка WO 2008/070740). GNE-390 демонстрирует синергическую активность в условиях in vitro и in vivo в комбинации с некоторыми химиотерапевтическими средствами на клеточных линиях солидных опухолей (заявка на патент США № 12/208227, Belvin et al. «Combinations of phosphoinositide 3-kinase inhibitor compounds and chemotherapeutic agents and methods of use», поданная 10 сентября 2008). GDC-390 представляет (S)-1-(4-((2-(2-аминопиримидин-5-ил)-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-ил)метил)пиперазин-1-ил)-2-гидроксипропан-1-он и имеет формулу:
Оценка биологической активности
Исследования на клеточных культурах в условиях in vitro с использованием трастузумаба-MCC-DM1(T-DM1) в комбинации с различными химиотерапевтическими средствами или биологически нацеленными средствами проводили на ряде клеточных линий с амплификацией HER2. Данные анализировали с использованием метода Chou&Talalay для определения комбинационного индекса (CI) для каждой комбинации при кратных значениях IC50 для каждого лекарственного средства. Значения CI ниже 0,7 означают наличие синергии; CI в пределах 0,7-1,3 - аддитивный эффект и значения CI выше 1,3 - антагонизм. В отношении комбинаций с химиотерапевтическими средствами, T-DM1 в комбинации с доцетакселем или 5-FU приводили к обеспечению аддитивной или синергической антипролиферативной активности, в то время как комбинации с гемцитабином или карбоплатином были не активны или оказывали антагонистическое действие по отношению к T-DM1. В опытах с мышиными ксенотрансплантатами были получены аналогичные результаты, где T-DM1 в комбинации с доцетакселем или 5-FU обеспечивал существенно более высокую противоопухолевую активность по сравнению с этими средствами по отдельности. T-DM1 в комбинации с карбоплатином обеспечивал более высокую эффективность по сравнению с одними препаратами, в то время как комбинация T-DM1 с гемцитабином не была более эффективной по сравнению с одним T-DM1. T-DM1 в сочетании с пертузумабом, лапатинибом или GDC-0941 приводил к проявлению аддитивного или синергического антипролиферативного действия на клеточных культурах, и противоопухолевая активность существенно повышалась в условиях in vivo по сравнению с лечением препаратами по отдельности. В противоположность неконъюгированный трастузумаб проявлял антагонистическое действие в отношении T-DM1 за счет связывания с одним и тем же эпитопом на HER2. В экспериментах в условиях in vivo с T-DM1 в комбинации с антиангиогенными средствами, такими как антитело В20-4.1 или ингибитор на основе небольшой молекулы АВТ-869, было показано усиление противоопухолевой эффективности со всеми тестированными комбинациями, за исключением наиболее высокой дозы T-DM1 (10 или 15 мг/кг) в сочетании с В20-4.1.
Оценивали эффективность комбинаций трастузумаба-MCC-DM1(T-DM1) и многочисленных противоопухолевых средств по определению их антипролиферативной активности на моделях в условиях in vitro на клетках опухоли молочной железы со сверхэкспрессией HER2 и по определению противоопухолевой эффективности в условиях in vivo на ксенотрансплантатах злокачественной опухоли молочной железы. В данных опытах трастузумаб-MCC-DM1 добавляли к цитотоксическим химиотерапевтическим средствам, антителам или ингибиторам киназы на основе небольшой молекулы.
Комбинация мышиного анти-VEGF-антитела В20-4.1 (Liang et al., 2006, J. Biol. Chem., 281:951-961), аналога бевацизумаба, и трастузумаба-MCC-DM1 при испытании на модели ксенотрансплантатов злокачественной опухоли молочной железы на мышах проявляла более высокую противоопухолевую активность по сравнению с одним В20-4.1. Результаты данных исследований обеспечивают прогностическую основу синергических эффектов и обоснование для будущих клинических испытаний лечебных схем, включающих трастузумаб-MCC-DM1 с различными противоопухолевыми средствами при HER-2-положительных злокачественных опухолях молочной железы.
Синергическое действие наблюдали с комбинациями средств, направленных на HER2, таких как трастузумаб-DM1 плюс лапатиниб или трастузумаб-DM1 плюс антитело к HER2 пертузумаб (ингибитор димеризации HER2).
Трастузумаб-МСС-DM1 в сочетании с карбоплатином или 5-FU показывал повышенную активность по сравнению с лечением одними этими средствами, в то время как комбинированное лечение с гемцитабином не приводило к усилению противоопухолевой активности.
Блокада пути с участием PI3-киназы с использованием GDC-0941, ингибитора киназы с небольшой молекулой р110 изоформ (международная заявка WO 2007/129161), усиливала активность трастузумаба-DM1.
Использование T-DM1 в сочетании с ингибитором PI3K GDC-0941 приводило к усилению противоопухолевой активности на клеточных линиях HER2-экспрессирующей злокачественной опухоли молочной железы с мутантной PI3K: BT-474 (K111N), MDA-361.1 (E545K) и KPL4 (H1047R). Комбинированная обработка в условиях in vitro приводила к аддитивному или синергическому ингибированию пролиферации клеток, а также повышенному апоптозу. Аналогично эффективность в условиях in vivo усиливалась при комбинированной обработке лекарственными средствами по сравнению с активностью одних препаратов на моделях MDA-MB-361.1 и ксенотрансплантатах Fo5 с амплификацией HER2. Результаты биохимического определения биомаркеров при испытании каждого препарата показывали, что имело место ингибирование фосфо-Akt и фосфо-ERK под действием обоих T-DM1 и GDC-0941, снижение фосфорилирования Rb и PRAS40 под действием GDC-0941 и повышение уровня митотических маркеров фосфогистона Н3 и циклина В1 после обработки T-DM1. Кроме того, введение T-DM1 приводило к апоптозу клеток на данных моделях рака молочной железы по данным определения фрагмента расщепления PARP массой 23 kDa, снижения уровня Bcl-XL, а также активации каспаз 3 и 7. Добавление GDC-0941 к T-DM1 дополнительно индуцировало усиление апоптоза. В данных исследованиях была получена доказательная основа для применения рациональных комбинаций лекарственных средств при лечении злокачественной опухоли молочной железы с амплификацией HER2 и предложений по дополнительным терапевтическим подходам для пациентов, у которых имеет место прогрессирование заболевания при терапии трастузумабом или лапатинибом.
Тесты оценки пролиферации клеток в условиях in vitro
Активность комбинаций трастузумаба-MCC-DM1 с химиотерапевтическими средствами оценивали в условиях in vitro с использованием теста оценки пролиферации клеток, описанного в примере 2; промышленно доступный тест CellTiter-Glo® Luminescent Cell Viability от Promega Corp., Madison, WI. Данный гомогенный тест основан на рекомбинантной экспрессии люциферазы Coleoptera (патент США № 5583024; патент США № 5674713; патент США № 5700670) и определении количества жизнеспособных клеток в культуре по количеству присутствующей АТФ, индикатора метаболически активных клеток (Crouch et al., 1993, J. Immunol. Meth., 160:81-88; патент США № 6602677). Тест CellTiter-Glo® проводили в формате 96- или 384-луночных планшетов, сделав их пригодными для автоматического высокопропускного скрининга (HTS) (Cree et al., 1995, AntiCancer Drugs, 6:398-404). Гомогенный анализ включает добавление одного реагента (реагента CellTiter-Glo®) непосредственно к клеткам, культивированным в среде с добавлением сыворотки. Отмывка клеток, удаление среды и многочисленные стадии пипетирования не требуются. С помощью данной системы можно детектировать такое небольшое количество клеток, как 15 клеток/лунку в 384-луночном планшете в течение 10 мин после внесения реагента и перемешивания.
Гомогенный формат «добавление-перемешивание-определение» приводит к лизису клеток и генерации люминесцентного сигнала, прямо пропорционального количеству присутствующей АТФ. Количество АТФ прямо пропорционально количеству клеток, находящихся в культуре. Тест CellTiter-Glo® воспроизводит люминесцентный сигнал типа свечения, образующийся в реакции с участием люциферазы, который имеет период полураспада более чем 5 ч, в зависимости от используемого типа клеток и среды. Жизнеспособные клетки отражаются в относительных единицах люминесценции (RLU). Субстрат люциферин жуков подвергается окислительному декарбоксилированию, которое катализируется рекомбинантной люциферазой светляков, с одновременным превращением АТФ в АМФ и генерацией фотонов. В результате большого периода полураспада устраняется необходимость в использовании инжекторов, и обеспечивается гибкость для непрерывного или периодического способа обработки множества планшетов. Данный тест оценки пролиферации клеток можно использовать в различных многолуночных форматах, например, 96- или 384-луночном формате. Данные можно регистрировать с помощью люминометра или визуализирующего устройства с CCD камерой. Выход люминесценции выражают в относительных световых единицах (RLU) в течение времени.
Антипролиферативное действие трастузумаба-MCC-DM1 и комбинаций с химиотерапевтическими средствами оценивали с использованием теста CellTiter-Glo® (пример 2) на опухолевых клеточных линиях, результаты представлены на фиг. 1-9 и 18-33.
Приведенные в качестве примера варианты осуществления включают способ идентификации соединений для применения в комбинации для лечения рака, включающий: (а) введение терапевтической комбинации трастузумаба-MCC-DM1 (T-DM1) и химиотерапевтического средства в опухолевую клеточную линию в условиях in vitro и (b) определение синергического или несинергического эффекта. Значение комбинационного индекса (CI) выше 1,3 означает антагонизм; значение CI в пределах от 0,7 до 1,3 означает аддитивный эффект и значение CI ниже 0,7 означает синергическое взаимодействие препаратов.
На фиг.1 показано антагонистическое действие трастузумаба в комбинации с трастузумабом-MCC-DM1 (T-DM1) в различных концентрациях при кратных отдельных значениях IC50 (таблица 1) на клетках SK-BR-3, которые являются чувствительными к трастузумабу. Строили график зависимости количества жизнеспособных клеток против кратных значений IC50. Комбинационный индекс (CI) в интервале от IC10 до IC90 для каждой комбинации выше 2, что указывает на наличие антагонистического действия в условиях in vitro. Однако комбинация T-DM1+трастузумаб не показала антагонистического действия в условиях in vivo.
На фиг.2 показано антагонистическое действие трастузумаба в комбинации с трастузумабом-MCC-DM1 (T-DM1) в различных концентрациях при кратных отдельных значениях IC50 (таблица 2) на клетках BT-474 EEI, которые являются резистентными к трастузумабу. Строили график зависимости количества жизнеспособных клеток от кратных значений IC50. Комбинационный индекс (CI) в интервале от IC10 до IC90 для каждой комбинации выше 2, что указывает на антагонистическое действие.
На фиг.3 показано синергическое действие пертузумаба в комбинации с трастузумабом-MCC-DM1 (T-DM1) в различных концентрациях при кратных отдельных значениях IC50 (таблица 3) на клетках MDA-MB-175. Строили график зависимости количества жизнеспособных клеток от кратных значений IC50. Комбинационный индекс (CI) в интервале от IC10 до IC90 для каждой комбинации ниже 1, что указывает на наличие синергического действия.
На фиг.3а приведен график зависимости жизнеспособности клеток в условиях in vitro на 5 сутки от кратных концентраций IC50 пертузумаба, трастузумаба-MCC-DM1 (T-DM1) и комбинации пертузумаба и T-DM1. Строили график зависимости количества жизнеспособных клеток от кратных значений IC50. Комбинационный индекс (CI) в интервале от IC10 до IC90 для каждой комбинации ниже 1 со средним значением CI=0,096, что указывает на синергическое действие (Таблица 3а).
На фиг.4 приведен график зависимости жизнеспособности клеток BT-474 в условиях in vitro на 5 сутки от различных фиксированных концентраций пертузумаба в комбинации с концентрацией ответной реакции трастузумаба-МСС-DM1 (T-DM1) и различных концентраций одного T-DM1. На фиг.4 показано влияние фиксированных концентраций T-DM1 в комбинации с различными концентрациями пертузумаба. Добавление пертузумаба к T-DM1 приводит к обеспечению несколько более высокой антипролиферативной активности по сравнению с одним T-DM1.
На фиг.5 приведен график зависимости жизнеспособности клеток BT-474 в условиях in vitro на 5 сутки от различных фиксированных концентраций трастузумаба-МСС-DM1 (T-DM1) в комбинации с концентрацией ответной реакции пертузумаба и различных концентраций одного пертузумаба. На фиг.5 показано влияние фиксированных концентраций пертузумаба в комбинации с различными концентрациями T-DM1 на пролиферацию клеток BT-474. Добавление T-DM1 к пертузумабу приводит к усилению действия одного пертузумаба.
На фиг.6 показано синергическое действие пертузумаба в комбинации с трастузумабом-МСС-DM1 в различных концентрациях при кратных отдельных значениях IC50 (таблица 4) на клетках BT-474. Строили график зависимости количества жизнеспособных клеток от кратных значений IC50. Комбинационный индекс (CI) в интервале от IC10 до IC90 находится в пределах от 0,198 до 1,328. Среднее значение CI для данного предела = 0,658, что указывает на синергическое действие.
На фиг.7 приведен график зависимости жизнеспособности клеток SK-BR-3 в условиях in vitro на 3 сутки от различных концентраций T-DM1 в комбинации с фиксированными концентрациями лапатиниба (4,5 нМ; 14 нМ; 41 нМ; 123 нМ) и различных концентраций одного T-DM1 (0-1000 нг/мл). Добавление T-DM1 к лапатинибу приводит к некоторому усилению антипролиферативного действия по сравнению с одним T-DM1.
На фиг.7а приведен график зависимости жизнеспособности клеток SK-BR-3 в условиях in vitro на 3 сутки от кратных концентраций IC50 T-DM1, лапатиниба и комбинаций с соотношением фиксированных концентраций T-DM1 и лапатиниба, как показано в таблице 7а. Среднее значение CI в интервале от IC10 до IC90=0,793, что указывает на наличие аддитивного действия.
На фиг.8а приведен график зависимости жизнеспособности клеток BT-474 в условиях in vitro на 3 сутки от кратных концентраций IC50 T-DM1, лапатиниба и комбинаций с соотношением фиксированных концентраций T-DM1 и лапатиниба, как показано в таблице 8а. Среднее значение CI в интервале от IC10 до IC90=0,403, что указывает на наличие синергии.
На фиг.8 приведен график зависимости жизнеспособности клеток BT-474 в условиях in vitro на 3 сутки от различных концентраций T-DM1 в комбинации с фиксированными концентрациями лапатиниба (1,5 нМ; 4,5 нМ; 14 нМ; 41 нМ; 123 нМ) и различных концентраций одного T-DM1 (0-1000 нг/мл). Добавление лапатиниба к T-DM1 приводит к усилению антипролиферативного действия по сравнению с одним препаратом.
На фиг.9 приведен график зависимости жизнеспособности клеток BT-474-EEI в условиях in vitro на 3 сутки от различных концентраций T-DM1 в комбинации с фиксированными концентрациями лапатиниба (14 нМ; 41 нМ; 123 нМ; 370 нМ; 1111 нМ) и различных концентраций одного T-DM1 (0-1000 нг/мл). Добавление лапатиниба к T-DM1 приводит к усилению антипролиферативного действия по сравнению с одним препаратом.
На фиг.18 приведен график зависимости жизнеспособности клеток SK-BR-3 в условиях in vitro на 3 сутки от кратных концентраций IC50 5-FU, трастузумаба-MCC-DM1 (T-DM1) и комбинаций с соотношением фиксированных концентраций 5-FU и T-DM1 (таблица 18). Комбинация 5-FU и T-DM1 оказывает аддитивное действие на клетки SK-BR-3 при среднем значении CI в интервале от IC10 до IC90=0,952.
На фиг.19 приведен график зависимости жизнеспособности клеток BT-474 в условиях in vitro на 3 сутки от кратных концентраций IC50 5-FU, трастузумаба-MCC-DM1 (T-DM1) и комбинаций с соотношением фиксированных концентраций 5-FU и T-DM1 (таблица 19). Комбинация 5-FU и T-DM1 оказывает синергическое действие на клетки BT-474 со средним значением CI=0,623.
На фиг.20 приведен график зависимости жизнеспособности клеток SK-BR-3 в условиях in vitro на 3 сутки от кратных концентраций IC50 гемцитабина, трастузумаба-MCC-DM1 (T-DM1) и комбинаций с соотношением фиксированных концентраций гемцитабина и T-DM1 (таблица 20). Гемцитабин в комбинации с T-DM1 приводит к антагонистическому взаимодействию лекарственных средств при среднем значении CI>1,3 при всех тестированных комбинациях.
На фиг.21 приведен график зависимости жизнеспособности клеток MDA-MD-361 в условиях in vitro на 3 сутки от кратных концентраций IC50 гемцитабина, трастузумаба-MCC-DM1 (T-DM1) и комбинаций с соотношением фиксированных концентраций гемцитабина и T-DM1 (таблица 21). Комбинация препаратов дает антагонистический эффект при среднем значении CI=1,706.
На фиг.22 приведен график жизнеспособности (пролиферации) клеток KPL4 в условиях in vitro на 3 сутки после обработки T-DM1, GDC-0941 и комбинациями с соотношением фиксированных концентраций T-DM1 (от 6,25 до 100 нг/мл) и GDC-0941 (от 62,5 нМ до 1 мкМ) при кратных концентрациях IC50 от 0,25× до 4×. В таблице 22 показано действие в интервале ингибирования 10-90% с расчетными значениями CI и средним значением CI, равным 1,111.
Строили график прогноза аддитивного действия Блисса в виде пунктирной линии на фиг.22. График независимости Блисса показывает рассчитанное аддитивное действие комбинации двух отдельных соединений.
На фиг.23 приведен график жизнеспособности (пролиферации) клеток KPL4 в условиях in vitro на 3 сутки после обработки T-DM1, GDC-0941 и комбинациями с соотношением фиксированных концентраций T-DM1 (от 1,25 до 80 нг/мл) и GDC-0941 (от 31,25 нМ до 2 мкМ) при кратных концентрациях IC50 от 0,0625× до 16×. Строили график прогноза аддитивного действия Блисса в виде пунктирной линии. В таблице 23 показано действие в интервале ингибирования 10-90% с расчетными значениями CI и средним значением CI, равным 0,802. Комбинация T-DM1 и GDC-0941 проявляет аддитивный эффект на клеточной линии KPL4.
На фиг.24 приведен график жизнеспособности (пролиферации) клеток KPL4 с амплификацией Her2, резистентных к герцептину®, мутантных по PIK3CA (H1047R) в условиях in vitro через 3 суток после обработки T-DM1, PI103, GDC-0941 и комбинациями с соотношением фиксированных концентраций T-DM1+PI103 и T-DM1+GDC-0941 при кратных концентрациях IC50 от 0 до 16×. В таблице 24 приведены значения комбинационного индекса. На основе полученных результатов можно предположить, что между T-DM1 и GDC-0941 имеет место синергия в условиях in vitro, поскольку значения CI находятся в пределах от 0,5 до 1, и между T-DM1 и PI103 имеет место аддитивный эффект, т.к. значения CI близки к 1.
PI3K представляет избирательный ингибитор PI103 (Hayakawa et al., 2007, Bioorg. Med. Chem. Lett., 17:2438-2442; Raynaud et al., 2007, Cancer Res., 67:5850; Fan et al., 2006, Cancer Cell, 9:341-349; патент США № 6608053) и имеет формулу:
На фиг.25 приведен график апоптоза (запрограммированной клеточной гибели) клеток KPL4 каспаза 3/7 в условиях in vitro через 24 ч после обработки T-DM1, GDC-0941 и комбинациями с соотношением фиксированных концентраций T-DM1 и GDC-0941. Комбинация T-DM1 и GDC-0941 приводит к существенному усилению апоптоза по сравнению с одним препаратом.
На фиг.26 приведен график апоптоза (запрограммированной клеточной гибели) клеток KPL4 в условиях in vitro на 3 сутки после обработки T-DM1, GDC-0941 и комбинациями с соотношением фиксированных концентраций T-DM1 и GDC-0941 в соотношениях фиксированных концентраций. Комбинация T-DM1 и GDC-0941 приводит к существенному увеличению апоптоза по сравнению с одним препаратом.
На фиг.27 приведен график жизнеспособности (пролиферации) клеток MDA-MB-361 в условиях in vitro на 3 сутки после обработки T-DM1, GDC-0941 и комбинациями с соотношением фиксированных концентраций T-DM1 (от 3,125 до 50 нг/мл) и GDC-0941 (от 62,5 нМ до 1 мкМ) при кратных концентрациях IC50 от 0,125× до 8×. Строили график прогноза аддитивного действия Блисса в виде пунктирной линии. В таблице 27 показано действие в интервале ингибирования 10-90% с расчетными значениями CI и средним значением CI, равным 0,888. Комбинация T-DM1 и GDC-0941 приводит к аддитивному антипролиферативному эффекту на клетках MDA-MB-361 со средним значением CI=0,889.
На фиг.28 приведен график жизнеспособности (пролиферации) клеток MDA-MB-361 в условиях in vitro на 3 сутки после обработки T-DM1, GDC-0941 и комбинациями с соотношением фиксированных концентраций T-DM1 (от 3,125 до 100 нг/мл) и GDC-0941 (от 62,5 нМ до 2 мкМ) при кратных концентрациях IC50 от 0,125× до 8×. Строили график прогноза аддитивного действия Блисса в виде пунктирной линии. В таблице 28 показано действие в интервале ингибирования 10-90% с расчетными значениями CI и средним значением CI, равным 0,813. T-DM1 в комбинации с GDC-0941 приводит к проявлению аддитивного антипролиферативного эффекта на клетках MDA-MB-361 при среднем значении CI=0,813.
На фиг.29 приведен график жизнеспособности (пролиферации) клеток BT-474 в условиях in vitro на 3 сутки после обработки T-DM1, GDC-0941 и комбинациями с соотношением фиксированных концентраций T-DM1 (от 3,125 до 100 нг/мл) и GDC-0941 (от 31,25 нМ до 1 мкМ) при кратных концентрациях IC50 от 0,125× до 4×. Строили график прогноза аддитивного действия Блисса в виде пунктирной линии. В таблице 29 показано действие в интервале ингибирования 10-90% с расчетными значениями CI и средним значением CI, равным 1,2122. В использованных соотношениях концентраций GDC-0941 в комбинации с T-DM1 не проявляют комбинированного эффекта на клетках BT-474.
На фиг.30 приведен график жизнеспособности (пролиферации) клеток BT-474 в условиях in vitro на 3 сутки после обработки T-DM1, GDC-0941 и комбинациями с соотношением фиксированных концентраций T-DM1 (от 6,25 до 100 нг/мл) и GDC-0941 (от 62,5 нМ до 1 мкМ) при кратных концентрациях IC50 от 0,25× до 4×. Строили график прогноза аддитивного действия Блисса в виде пунктирной линии. В таблице 30 показано действие в интервале ингибирования 10-90% с расчетными значениями CI и средним значением CI, равным 0,997, что указывает на аддитивное действие.
На фиг.31 приведен график жизнеспособности (пролиферации) клеток AU565 с амплификацией Her2, мутантных без PI3K в условиях in vitro на 3 сутки после обработки T-DM1, PI103, GDC-0941 и комбинациями с соотношением фиксированных концентраций T-DM1+PI103 и T-DM1+GDC-0941 при кратных концентрациях IC50 от 0 до 16×. В таблице 31 приведены значения комбинационного индекса. На основе полученных результатов можно предположить, что между T-DM1 и GDC-0941 имеет место антагонизм в условиях in vitro, поскольку значения CI составляют >1, и между T-DM1 и PI103 существует аддитивный эффект или незначительный антагонизм, т.к. значения CI близки или несколько выше 1.
На фиг.32 приведен график жизнеспособности (пролиферации) клеток EFM192А с амплификацией Her2, мутантных по PIK3CA (C420R) в условиях in vitro на 3 сутки после обработки T-DM1, PI103, GDC-0941 и комбинациями с соотношением фиксированных концентраций T-DM1+PI103 и T-DM1+GDC-0941 при кратных концентрациях IC50 от 0 до 16×. В таблице 32 приведены значения комбинационного индекса. На основе полученных результатов можно предположить о наличии умеренной синергии между T-DM1 и GDC-0941 в условиях in vitro, поскольку значения CI находятся в пределах от 0,5 до 1, и синергии между T-DM1 и PI103, т.к. значения CI близки к 0,5.
На фиг.33 приведен график жизнеспособности (пролиферации) клеток HCC1954 с амплификацией Her2, резистентных к герцептину®, мутантных по PIK3CA (H1047R) в условиях in vitro на 3 сутки после обработки T-DM1, PI103, GDC-0941 и комбинациями с соотношением фиксированных концентраций T-DM1+PI103 и T-DM1+GDC-0941 при кратных концентрациях IC50 от 0 до 16×. В таблице 33 приведены значения комбинационного индекса. На основе полученных результатов можно предположить о наличии аддитивного эффекта или незначительной синергии между T-DM1 и GDC-0941 в условиях in vitro, поскольку значения CI близки к 1, и имеется незначительная синергия между T-DM1 и PI103, т.к. значения CI <1.
Эффективность на опухолевых ксенотрансплантатах
в условиях in vivo
Эффективность комбинаций по изобретению можно оценить в условиях in vivo при имплантации аллотрансплантатов или ксенотрансплантатов опухолевых клеток на грызунах и обработкой опухолей комбинациями. Предполагается получение различных результатов в зависимости от клеточной линии, наличия или отсутствия определенных мутаций в опухолевых клетках, последовательности введения трастузумаба-MCC-DM1 и химиотерапевтического средства, схемы введения и других факторов. Мышей обрабатывали лекарственным средством(ми) или контрольным раствором (растворителем) и наблюдали в течение нескольких недель или несколько для оценки периода времени, необходимого для двукратного увеличения опухоли, log гибели клеток и подавления роста опухолей (пример 3). На фиг.10-17 и 34-37 показана эффективность трастузумаба-MCC-DM1 в комбинации с химиотерапевтическими средствами в отношении подавления роста опухолевых ксенотрансплантатов у мышей.
На фиг.10 приведен график изменения среднего объема опухолей в течение времени в условиях in vivo для опухолей KPL-4, имплантированных в жировую подушку молочной железы иммунодефицитных мышей SCID, после введения: (1) буфера ADC; (2) пертузумаба в дозе 15 мг/кг; (3) T-DM1 в дозе 0,3 мг/кг; (4) T-DM1 в дозе 1 мг/кг; (5) T-DM1 в дозе 3 мг/кг, (6) пертузумаба в дозе 15 мг/кг + T-DM1 в дозе 0,3 мг/кг; (7) пертузумаба в дозе 15 мг/кг + T-DM1 в дозе 1 мг/кг; (8) пертузумаба в дозе 15 мг/кг + T-DM1 в дозе 3 мг/кг. У животных, которым вводили буфер ADC (1) было 0 PR и 0 CR. У животных, которым вводили пертузумаб в дозе 15 мг/кг (2), было 0 PR и 0 CR. У животных, которым вводили один T-DM1 в дозе 0,3 мг/кг (3) было 0 PR и 0 CR. У животных, которым вводили один T-DM1 в дозе 1 мг/кг (4) было 1 PR и 0 CR. У животных, которым вводили один T-DM1 в дозе 3 мг/кг (5) было 7 PR и 0 CR. У животных, которым вводили комбинацию пертузумаба в дозе 15 мг/кг и T-DM1 в дозе 0,3 мг/кг (6) было 5 PR и 0 CR. Результат у животных, которым вводили комбинацию пертузумаба в дозе 15 мг/кг и T-DM1 в дозе 1 мг/кг (7), составил 8 PR и 0 CR. У животных, которым вводили комбинацию пертузумаба в дозе 15 мг/кг и T-DM1 в дозе 3 мг/кг (8) было 8 PR и 0 CR. Комбинация пертузумаба и T-DM1 обеспечивает более высокую противоопухолевую активность на ксенотрансплантатах KPL4 по сравнению с одним препаратом.
На фиг.11 приведен график изменения среднего объема опухолей в течение времени в условиях in vivo для опухолей KPL-4, имплантированных в жировую подушку молочной железы иммунодефицитных мышей SCID, после введения: (1) буфера ADC; (2) 5-FU в дозе 100 мг/кг; (3) пертузумаба в дозе 40 мг/кг; (4) B20-4.1 в дозе 5 мг/кг; (5) T-DM1 в дозе 5 мг/кг; (6) 5-FU в дозе 100 мг/кг + T-DM1 в дозе 5 мг/кг; (7) пертузумаба в дозе 40 мг/кг + T-DM1 в дозе 5 мг/кг; (8) B20-4.1 в дозе 5 мг/кг + T-DM1 в дозе 5 мг/кг; (9) B20-4.1 в дозе 5 мг/кг + пертузумаба в дозе 40 мг/кг. В конце опыта проводили гистологическое исследование всех оставшихся опухолей, которые имели объем менее чем 50 мм3, и установили, что в 8 образцах опухолей от мышей, получавших один T-DM1 в дозе 5 мг/кг (5), 5 образцах от мышей в группе, получавшей комбинацию 5-FU в дозе 100 мг/кг + T-DM1 в дозе 5 мг/кг (6) и 8 образцах от мышей, получавших комбинацию пертузумаба в дозе 40 мг/кг + T-DM1 в дозе 5 мг/кг (7), отсутствовали жизнеспособные опухолевые клетки.
На фиг.12 приведен график изменения среднего объема опухолей в течение времени в условиях in vivo для трансгенных опухолей молочной железы MMTV-Her2 Fo5, имплантированных в жировую подушку молочной железы мышей CRL nu/nu, после введения: (1) растворителя (буфера ADC); (2) B20-4.1 в дозе 5 мг/кг; (3) T-DM1 в дозе 3 мг/кг; (4) T-DM1 в дозе 5 мг/кг; (5) T-DM1 в дозе 10 мг/кг; (6) B20-4.1 в дозе 5 мг/кг + T-DM1 в дозе 3 мг/кг; (7) B20-4.1 в дозе 5 мг/кг + T-DM1 в дозе 5 мг/кг; (8) B20-4.1 в дозе 5 мг/кг + T-DM1 в дозе 10 мг/кг. Комбинация T-DM1 и B20-4.1 обеспечивает усиленное ингибирование роста опухолей при дозе T-DM1 3 и 5 мг/кг, но не 10 мг/кг.
На фиг.13 приведен график изменения среднего объема опухолей в течение времени в условиях in vivo для трансгенных опухолей молочной железы MMTV-Her2 Fo5, имплантированных в жировую подушку молочной железы мышей CRL nu/nu, после введения: (1) растворителя (буфера ADC); (2) T-DM1 в дозе 10 мг/кг; (3) 5-FU в дозе 100 мг/кг; (4) гемцитабина в дозе 120 мг/кг; (5) карбоплатина в дозе 100 мг/кг; (6) 5-FU в дозе 100 мг/кг + T-DM1 в дозе 10 мг/кг; (7) гемцитабина в дозе 120 мг/кг + T-DM1 в дозе 10 мг/кг; (8) карбоплатина в дозе 100 мг/кг + T-DM1 в дозе 10 мг/кг. Комбинация T-DM1 с 5-FU, карбоплатином или гемцитабином обеспечивает более высокую противоопухолевую активность по сравнению с обработкой одним препаратом.
На фиг.14 приведен график изменения среднего объема опухолей в течение времени в условиях in vivo для ксенотрансплантатов трансгенных опухолей молочной железы MMTV-Her2 Fo5, имплантированных в жировую подушку молочной железы бестимусных мышей Harlan, после введения: (1) растворителя (буфера PBS) в/в, один раз в неделю ×4; (2) лапатиниба в дозе 101 мг/кг, перорально, два раза день ×21; (3) пертузумаба в дозе 40 мг/кг, в/в, один раз в неделю ×4; (4) B20-4.1 в дозе 5 мг/кг, в/б, 2×/в неделю ×4; (5) T-DM1 в дозе 15 мг/кг, в/в, один раз в 3 недели до конца; (6) лапатиниба в дозе 101 мг/кг перорально, два раза в день ×21 + T-DM1 в дозе 15 мг/кг, в/в, один раз в 3 недели до конца; (7) пертузумаба в дозе 40 мг/кг в/в, один раз в неделю ×4 + T-DM1 в дозе 15 мг/кг, в/в, один раз в 3 недели до конца; (8) B20-4.1 в дозе 5 мг/кг, перорально, 2×/в неделю ×4 + T-DM1 в дозе 15 мг/кг, в/б, один раз в 3 недели до конца.
Один T-DM1 в дозе 15 мг/кг (5) по эффективности не отличается достоверно от комбинации T-DM1 в дозе 15 мг/кг и B20-4.1 в дозе 5 мг/кг (8). В настоящем опыте лапатиниб и пертузумаб не отличались от растворителя. Для B20-4.1 была характерна тенденция к повышению эффективности по сравнению с контролем. T-DM1 был эффективен в виде одного препарата (р<0,01). Комбинация T-DM1 с лапатинибом была достоверно эффективнее по сравнению с одним лапатинибом (р<0,01), но ее эффективность не различалась по сравнению с одним T-DM1. Эффективность комбинации T-DM1 с пертузумабом была достоверно выше по сравнению с одним пертузумабом (р<0,01), но не отличалась по сравнению с одним T-DM1. Комбинация T-DM1 с B20-4.1 была статистически достоверно эффективнее по сравнению с одним B20-4.1 (р<0,01), но не отличалась от этого показателя для одного T-DM1.
На фиг.15 приведен график изменения среднего объема опухолей в течение времени в условиях in vivo для ксенотрансплантатов трансгенных опухолей молочной железы MMTV-Her2 Fo5, имплантированных в жировую подушку молочной железы бестимусных мышей Harlan, после введения: (1) растворителя (буфера PBS) перорально, два раза в день ×21; (2) T-DM1 в дозе 7,5 мг/кг, в/в, один раз в день ×1; (3) T-DM1 в дозе 15 мг/кг, в/в, один раз в день ×1; (4) ABT-869 в дозе 5 мг/кг, перорально, два раза в день ×21; (5) ABT-869 в дозе 15 мг/кг, перорально, два раза в день ×21; (6) T-DM1 в дозе 7,5 мг/кг в/в, один раз в день ×1 + ABT-869 в дозе 5 мг/кг, перорально, два раза в день ×21; (7) T-DM1 в дозе 7,5 мг/кг в/в, один раз в день ×21 + ABT-869 в дозе 5 мг/кг, перорально, два раза в день ×21; (8) T-DM1 в дозе 15 мг/кг, в/в, один раз в день ×1 + ABT-869 в дозе 5 мг/кг, перорально, два раза в день ×21; (9) T-DM1 в дозе 15 мг/кг, в/в, один раз в день ×1 + ABT-869 в дозе 15 мг/кг, перорально, два раза в день ×21.
Комбинация T-DM1 и ABT-869 в дозе 5 мг/кг показывала два частичных ответа (8) и не была статистически достоверно эффективнее по сравнению с одним ABT-869 в дозе 5 мг/кг (4). Комбинация T-DM1 и ABT-869 в дозе 15 мг/кг (9) является несколько более эффективной по сравнению с одним ABT-869 в дозе 15 мг/кг (5). ABT-869 в дозе 5 мг/кг был достоверно эффективнее по сравнению с растворителем до времени конечной точки (р<0,01), но его эффективность не отличалась ко времени удвоения опухоли. Эффективность ABT-869 в дозе 15 мг/кг и T-DM1 в дозе 7,5 или 15 мг/кг была статистически достоверно выше по сравнению с растворителем ко времени удвоения объема опухоли и ко времени конечной точки опухоли (р<0,01). Комбинация T-DM1 в дозе 7,5 мг/кг и ABT-869 в дозе 5 мг/кг достоверно не отличалась по эффективности в сравнении с одним T-DM1 в дозе 7,5 мг/кг. Эффективность комбинации T-DM1 в дозе 7,5 мг/кг и ABT-869 в дозе 5 мг/кг была достоверно выше по сравнению с одним ABT-869 в дозе 5 мг/кг ко времени удвоения объема опухоли (р<0,01), но не отличалась ко времени конечной точки. Комбинация T-DM1 в дозе 7,5 мг/кг и ABT-869 в дозе 15 мг/кг была достоверно лучше по эффективности по сравнению с каждым одним препаратом (р<0,01). Эффективность комбинации T-DM1 в дозе 15 мг/кг + ABT-869 в дозе 5 мг/кг не различалась с одним T-DM1 в дозе 15 мг/кг. По сравнению с одним ABT-869 в дозе 5 мг/кг эффективность комбинации T-DM1 в дозе 15 мг/кг + ABT-869 в дозе 5 мг/кг не отличалась ко времени конечной точки, но на время удвоения объема опухоли была достоверно выше (р<0,01). Эффективность комбинации T-DM1 в дозе 15 мг/кг + ABT-869 в дозе 15 мг/кг была достоверно выше по сравнению с одним ABT-869 в дозе 15 мг/кг и была выше по сравнению с одним T-DM1 в дозе 15 мг/кг ко времени удвоения объема опухоли (р<0,01). На время конечной точки показатели противоопухолевой активности T-DM1 в дозе 15 мг/кг и T-DM1 в дозе 15 мг/кг + ABT-869 в дозе 15 мг/кг не различались между собой.
На фиг.16 приведен график изменения среднего объема опухолей в течение времени в условиях in vivo для ксенотрансплантатов трансгенных опухолей молочной железы MMTV-Her2 Fo5, имплантированных в жировую подушку молочной железы бестимусных мышей Harlan, после введения: (1) растворителя в/в, один раз в неделю ×3; (2) T-DM1 в дозе 7,5 мг/кг в/в, один раз в 3 недели ×2; (3) T-DM1 в дозе 15 мг/кг в/в, один раз в 3 недели ×2; (4) доцетаксела в дозе 30 мг/кг в/в, один раз в неделю ×3; (5) T-DM1 в дозе 7,5 мг/кг в/в, один раз в 3 недели ×2 + доцетаксела в дозе 30 мг/кг в/в, один раз в неделю ×3; (6) T-DM1 в дозе 15 мг/кг в/в, один раз в 3 недели ×2 + доцетаксела в дозе 30 мг/кг в/в, один раз в неделю ×3.
У животных, которым вводили один T-DM1 в дозе 15 мг/кг (3) было 6 частичных ответов (PR) и 1 полный ответ (CR). У животных, которым вводили один доцетаксел в дозе 30 мг/кг (4) было 2 PR. У животных, которым вводили комбинацию T-DM1 в дозе 7,5 мг/кг и доцетаксела в дозе 30 мг/кг (5), было 10 PR. У животных, которым вводили комбинацию T-DM1 в дозе 15 мг/кг и доцетаксела в дозе 30 мг/кг (6) было 7 PR и 3 CR. Все группы с введением одних препаратов достоверно отличались от результатов, полученных на животных, которым вводили растворитель (р<0,01). Эффективность комбинации T-DM1 в дозе 7,5 мг/кг и доцетаксела была достоверно выше по сравнению с каждым одним препаратом на время удвоения объема опухоли и на время конечной точки (р<0,01). Объективные ответы отсутствовали в группе мышей, получавших T-DM1 в дозе 7,5 мг/кг, и имело место 2 частичных ответа (PR) в группе с введением одного доцетаксела. Введение комбинации T-DM1 в дозе 7,5 мг/кг и доцетаксела приводило к 9 PR и 1 одному полному ответу (CR). Эффективность комбинации T-DM1 в дозе 15 мг/кг + доцетаксела была достоверно выше по сравнению с каждым одним препаратом ко времени удвоения объема опухоли и ко времени конечной точки (р<0,01). Введение одного T-DM1 в дозе 15 мг/кг давало 5 PR и 2 CR. Комбинированное введение T-DM1 в дозе 15 мг/кг + доцетаксела повышало количество объективных ответов на лечение до 7 PR и 3 CR. Все мыши в данной группе с комбинированной схемой имели объективный ответ на лечение.
На фиг.17 приведен график изменения среднего объема опухолей в условиях in vivo для ксенотрансплантатов трансгенных опухолей MMTV-Her2 Fo5, имплантированных в жировую подушку молочной железы бестимусных мышей Harlan, после введения: (1) растворителя перорально, один раз в день ×21; (2) T-DM1 в дозе 7,5 мг/кг в/в, один раз в 3 недели ×2; (3) T-DM1 в дозе 15 мг/кг в/в, один раз в 3 недели ×2; (4) лапатиниба в дозе 100 мг/кг перорально, два раза в день ×21; (5) T-DM1 в дозе 7,5 мг/кг в/в, один раз в 3 недели ×2 + лапатиниба в дозе 100 мг/кг перорально, два раза в день ×21; (6) T-DM1 в дозе 15 мг/кг в/в, один раз в 3 недели ×2 + лапатиниба в дозе 100 мг/кг перорально, два раза в день ×21.
У животных, которым вводили один T-DM1 в дозе 15 мг/кг (3) было 6 частичных ответов (PR) и 3 полных ответа (CR). У животных, которым вводили комбинацию T-DM1 в дозе 7,5 мг/кг и лапатиниба в дозе 100 мг/кг (5), имело место 4 PR и 5 CR. У животных, которым вводили комбинацию T-DM1 в дозе 15 мг/кг и лапатиниба в дозе 100 мг/кг (6), ответная реакция на лечение выразилась в 8 CR. Все группы с одним препаратом достоверно отличались от результатов, полученных на животных, которым вводили растворитель (р<0,01) на время удвоения объема опухоли и на время конечной точки. Эффективность комбинации T-DM1 в дозе 7,5 мг/кг с лапатинибом была достоверно выше по сравнению с лапатинибом и T-DM1 в дозе 7,5 мг/кг при введении в виде одного препарата (р<0,01). Эффективность T-DM1 в дозе 15 мг/кг в комбинации с лапатинибом была достоверно выше по сравнению с одним лапатинибом (р<0,01). Данная комбинация не отличалась по эффективности от одного T-DM1 в дозе 15 мг/кг.
Время удвоения объема опухоли определяли с помощью статистического анализа Каплана-Мейера в виде 2 ×Vo. Время удвоения объема опухоли и анализ выживаемости количественно анализировали по значениям log-рангов-р. Время прогрессирования определяли в виде времени, прошедшего до достижения объема опухоли 1000 мм3 или времени выживаемости, если объем опухоли 1000 мм3 не достигался. T-DM1 в комбинации с лапатинибом обеспечивал существенно более высокую противоопухолевую эффективность по сравнению с лечением одним препаратом.
На фиг.34 приведен график изменения среднего объема опухолей в течение времени в условиях in vivo для ксенотрансплантатов трансгенных опухолей MMTV-Her2 Fo5, имплантированных в жировую подушку молочной железы мышей CRL nu/nu, после введения: (1) растворителя перорально, один раз в день ×21; (2) T-DM1 в дозе 10 мг/кг в/в, один раз в 3 недели; (3) 5-FU в дозе 100 мг/кг перорально, один раз в неделю ×2; (4) (5) T-DM1 в дозе 5 мг/кг в/в, один раз в 3 недели + 5-FU в дозе 100 мг/кг перорально, один раз в неделю ×2. У животных, которым вводили растворитель, имело место 0 частичных ответов (PR) и 0 полных ответов (CR). У животных, которым вводили один T-DM1, детектировали 1 PR и 0 CR. У животных, получавших один 5-FU, имело место 0 PR и 0 CR. У животных, которым вводили комбинацию T-DM1 и 5-FU, регистрировали 3 PR и 0 CR на временную точку 42 суток. Обработка T-DM1 и 5-FU приводила к обеспечению повышенной противоопухолевой активности по сравнению с каждым одним препаратом.
На фиг.35 приведен график изменения среднего объема опухолей в течение времени в условиях in vivo для ксенотрансплантатов трансгенных опухолей MMTV-Her2 Fo5, имплантированных в жировую подушку молочной железы мышей CRL nu/nu, после введения: (1) растворителя перорально, один раз в день ×21; (2) T-DM1 в дозе 5 мг/кг в/в, один раз в 3 недели; (3) GDC-0941 в дозе 100 мг/кг перорально, два раза в день ×21; (4) GDC-0152 дозе 50 мг/кг перорально, один раз в неделю ×2; (5) T-DM1 в дозе 5 мг/кг в/в, один раз в 3 недели + GDC-0941 в дозе 100 мг/кг перорально, два раза в день ×21; (6) T-DM1 в дозе 5 мг/кг в/в, один раз в 3 недели + GDC-0152 в дозе 50 мг/кг перорально, один раз в неделю ×2. Обработка T-DM1 и GDC-0941 приводит к обеспечению повышенной противоопухолевой активности по сравнению с лечением одним препаратом, в то время как комбинация T-DM1 и GDC-0152 не была более эффективной по сравнению с одним T-DM1.
GDC-0152 является ингибитором каспаз, которые подавляют белки апоптоза (Call et al., 2008, The Lancet Oncology, 9(10):1002-1011; Deveraux et al., 1999, J. Clin. Immunol., 19:388-398).
На фиг.36 приведен график изменения среднего объема опухолей в течение времени в условиях in vivo для опухолей MDA-MB-361.1, имплантированных в жировую подушку молочной железы мышей CRL nu/nu, после введения: (1) растворителя перорально, один раз в день ×21; (2) GDC-0941 в дозе 25 мг/кг, перорально, один раз в день ×21; (3) GDC-0941 в дозе 50 мг/кг, перорально, один раз в день ×21; (4) GDC-0941 в дозе 100 мг/кг, перорально, один раз в день ×21; (5) T-DM1 в дозе 3 мг/кг в/в, один раз в 3 недели; (6) T-DM1 в дозе 10 мг/кг в/в, один раз в 3 недели; (7) GDC-0941 в дозе 25 мг/кг перорально, один раз в день ×21 + T-DM1 в дозе 3 мг/кг в/в, один раз в 3 недели; (8) GDC-0941 в дозе 50 мг/кг перорально, один раз в день ×21 + T-DM1 в дозе 3 мг/кг в/в, один раз в 3 недели; (9) GDC-0941 в дозе 100 мг/кг перорально, один раз в день ×21 + T-DM1 в дозе 3 мг/кг в/в, один раз в 3 недели; (10) GDC-0941 в дозе 25 мг/кг перорально, один раз в день ×21 + T-DM1 в дозе 10 мг/кг в/в, один раз в 3 недели; (11) GDC-0941 в дозе 50 мг/кг перорально, один раз в день ×21 + T-DM1 в дозе 10 мг/кг в/в, один раз в 3 недели; (12) GDC-0941 в дозе 100 мг/кг перорально, один раз в день ×21 + T-DM1 в дозе 10 мг/кг в/в, один раз в 3 недели.
У животных, которым вводили растворитель (1) имело место 0 частичных ответов (PR) и 0 полных ответов (CR). У животных, которым вводили один GDC-0941 в дозе 25 мг/кг (2) было 0 PR и 0 CR. У животных, которым вводили один GDC-0941 в дозе 50 мг/кг (3) был 1 PR и 0 CR. Эффективность на животных, которым вводили один GDC-0941 в дозе 100 мг/кг (4), выразилась в 0 PR и 0 CR. У животных, которым вводили один T-DM1 в дозе 3 мг/кг (5), было 1 PR и 1 CR. У животных, которым вводили один T-DM1 в дозе 10 мг/кг (6), регистрировали 8 PR и 1 CR. У животных, которым вводили комбинацию T-DM1 в дозе 3 мг/кг и GDC-0941 в дозе 25 мг/кг (7), имело место 5 PR и 0 CR. У животных, которым вводили комбинацию T-DM1 в дозе 3 мг/кг и GDC-0941 в дозе 50 мг/кг (8), имело место 3 PR и 0 CR. У животных, которым вводили комбинацию T-DM1 в дозе 3 мг/кг и GDC-0941 в дозе 100 мг/кг (9), имело место 3 PR и 1 CR. У животных, которым вводили комбинацию T-DM1 в дозе 10 мг/кг и GDC-0941 в дозе 50 мг/кг (10) было 9 PR и 0 CR. У животных, которым вводили комбинацию T-DM1 в дозе 10 мг/кг и GDC-0941 в дозе 50 мг/кг (11), было 7 PR и 2 CR. На животных, которым вводили комбинацию T-DM1 в дозе 10 мг/кг и GDC-0941 в дозе 100 мг/кг (12), ее эффективность составила 9 PR и 1 CR.
На фиг.37 приведен график изменения среднего объема опухолей в течение времени в условиях in vivo для опухолей MDA-MB-361.1, имплантированных в жировую подушку молочной железы мышей CRL nu/nu, после введения: (1) растворителей [MCT (0,5% метилцеллюлоза/0,2% твин 80) + сукцинатный буфер (100 мМ сукцината натрия, 100 мг/мл трегалозы, 0,1% твина 80, рН 5,0)] перорально+в/в, один раз в день ×21 и один раз в день; (2) GNE-390 в дозе 1,0 мг/кг, перорально, один раз в день ×21; (3) GNE-390 в дозе 2,5 мг/кг, перорально, один раз в день ×21; (4) T-DM1 в дозе 3 мг/кг в/в, один раз в день; (5) GNE-390 в дозе 1,0 мг/кг перорально, один раз в день ×21 + T-DM1 в дозе 3 мг/кг в/в, один раз в день; (6) GNE-390 в дозе 2,5 мг/кг перорально, один раз в день ×21 + T-DM1 в дозе 3 мг/кг в/в, один раз в день.
У животных, которым вводили растворитель (1), имело место 0 частичных ответов (PR) и 0 полных ответов (CR). У животных, которым вводили один GNE-390 в дозе 1,0 мг/кг (2), было 0 PR и 0 CR. Эффективность на животных, получавших один GNE-390 в дозе 2,5 мг/кг (3), составила 1 PR и 0 CR. У животных, которым вводили один T-DM1 в дозе 3 мг/кг (5), регистрировали 1 PR и 1 CR. У животных, которым вводили один T-DM1 в дозе 3 мг/кг (4), было 0 PR и 0 CR. У животных, которым вводили комбинацию T-DM1 в дозе 3 мг/кг и GNE-390 в дозе 25 мг/кг (5), имело место 3 PR и 0 CR. Эффективность комбинации T-DM1 в дозе 3 мг/кг и GNE-390 в дозе 2,5 мг/кг (6) составила 5 PR и 1 CR. Комбинация GNE-390 с T-DM1 приводила к достоверному увеличению противоопухолевой активности по сравнению с одними GNE-390 или T-DM1 на модели ксенотрансплантатов злокачественной опухоли молочной железы MB-361.1.
Фармацевтические композиции
Фармацевтические композиции или лекарственные формы по настоящему изобретению содержат комбинации трастузумаба-MCC-DM1, химиотерапевтического средства, и один или несколько фармацевтически приемлемых носителей, регуляторов сыпучести, разбавителей или наполнителей.
Трастузумаб-MCC-DM1 и химиотерапевтические средства по настоящему изобретению можно использовать в несольватированной форме, а также в сольватированных формах с фармацевтически приемлемыми растворителями, такими как вода, этанол и тому подобное, и предполагается, что изобретение включает сольватированные и несольватированные формы.
Трастузумаб-MCC-DM1 и химиотерапевтические средства по настоящему изобретению могут находиться в различных таутомерных формах, и все такие формы входят в объем изобретения. Термины «таутомер» или «таутомерная форма» относятся к структурным изомерам с различной энергией, которые превращаются друг в друга через низкоэнергетический барьер. Например, протонные таутомеры (также известные как прототропные таутомеры) включают взаимопревращения посредством миграции протона, такие как изомеризации кето-енол и имин-енамин. Валентные таутомеры включают взаимопревращения, происходящие посредством реорганизации некоторых участвующих в образовании связей электронов.
Фармацевтические композиции включают насыпные композиции и отдельные разовые лекарственные формы, содержащие более чем одно (например, два) фармацевтически активных веществ, в том числе, трастузумаб-MCC-DM1 и химиотерапевтическое средство, выбранное из перечня дополнительных лекарственных средств, описанных в данном документе, наряду с любыми фармацевтически неактивными наполнителями, разбавителями, носителями или регуляторами сыпучести. Насыпная композиция и каждая отдельная разовая лекарственная форма может содержать фиксированные количества вышеуказанных фармацевтически активных веществ. Насыпная композиция представляет вещество, которое не было формулировано в отдельные разовые лекарственные формы. Показательной лекарственной формой является лекарственная форма для приема перорально, такая как таблетки, пилюли, капсулы и тому подобное. Аналогично описанный в данном документе способ лечения пациента введением фармацевтической композиции также предназначен для включения введения насыпной композиции и отдельных разовых лекарственных форм.
Фармацевтические композиции также включают меченные изотопами соединения по настоящему изобретению, которые аналогичны уже описанным в данном документе, но отличаются тем, что один или несколько атомов замещены атомом, имеющим атомную массу или массовое число, отличающиеся от атомной массы или массового числа, которые обычно имеются в природе. Все изотопы любого конкретного атома или элемента, как уже упомянуто, включаются в объем соединений по изобретению и их применений. Приведенные в качестве примера изотопы, которые можно включить в соединения по изобретению, включают водород, углерод, азот, кислород, фосфор, серу, фтор, хлор и иод, такие как 2H, 3H, 11C, 13C, 14C, 13N, 15N, 15O, 17O, 18O, 32P, 33P, 35S, 18F, 36Cl, 123I и 125I. Некоторые меченные изотопами соединения по изобретению (например, меченные 3Н и 14С) являются пригодными для постановки анализа распределения соединения и/или субстрата в тканях. Изотопы тритий (3Н) и углерод-14 (14С) подходят для простого получения и детектирования соединений. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий (2Н), может придать некоторые терапевтические преимущества в результате более высокой метаболической стабильности (например, повышенный период полураспада или несколько редкая необходимость во введении) и следовательно, в некоторых ситуациях могут быть предпочтительными. Испускающие позитроны изотопы, такие как 15О, 13N, 11C и 18F, являются пригодными для исследований с позитронно-эмиссионной томографией (PET) для изучения занятости рецептора субстратом. Как правило, меченные изотопами соединения по настоящему изобретению можно получить с использованием следующих методов, аналогичных описанным на схемах и/или в примерах, приведенных в данном документе ниже, замещением немеченного изотопами реагента на меченный изотопами реагент.
Трастузумаб-MCC-DM1 и химиотерапевтические средства можно формулировать согласно обычной фармацевтической практике для применения в терапевтической комбинации для медикаментозного лечения (включая профилактическое лечение) гиперпролиферативных нарушений у млекопитающих, в том числе, людей. Изобретение относится к фармацевтической композиции, содержащей трастузумаб-MCC-DM1 в ассоциации с одним или несколькими фармацевтически приемлемыми носителями, регуляторами сыпучести, разбавителями или наполнителями.
Подходящие носители, разбавители и наполнители хорошо известны специалистам в данной области, и они включают вещества, такие как углеводы, воска, водорастворимые и/или набухаемые полимеры, гидрофильные или гидрофобные вещества, желатин, масла, растворители, воду и тому подобное. Конкретный носитель, разбавитель и наполнитель для использования будет зависеть от способов и цели, для которых применяют соединение по настоящему изобретению. Как правило, растворители, которые выбирают, представляют собой растворители, известные специалистам в данной области как безопасные (GRAS) для введения млекопитающему. В основном, безопасными растворителями являются нетоксичные водные растворители, такие как вода, и другие нетоксичные растворители, которые растворимы или смешиваются с водой. Подходящие растворители включают воду, этанол, пропиленгликоль, полиэтиленгликоли (например, ПЭГ 400, ПЭГ 300) и т.д. и их смеси. Также композиции могут содержать одно или несколько из буферов, стабилизаторов, поверхностно-активных веществ, смачивающих веществ, смазывающих веществ, эмульгаторов, суспендирующих веществ, консервантов, антиоксидантов, замутняющих веществ, регуляторов сыпучести, средств для обработки, красителей, подсластителей, ароматизаторов, вкусовых веществ и других известных добавок для обеспечения элегантной формы представления лекарственного средства (т.е. соединения по настоящему изобретению или его фармацевтической композиции) или вспомогательных, применяющихся в производстве фармацевтического продукта (т.е. лекарственного средства).
Композиции можно приготовить с использованием обычного растворения и смешивания. Например, насыпное лекарственное средство (т.е. соединение по настоящему изобретению или стабилизированная форма соединения) (например, комплекс с циклодекстрином или другим комплексообразующим агентом) растворяют в подходящем растворителе в присутствии одного или несколько наполнителей, указанных выше. Как правило, соединение по настоящему изобретению формулируют в виде фармацевтических лекарственных форм для обеспечения простого контролированного введения лекарственного средства и в целях удобства для пациента по прописанной схеме.
Фармацевтическую композицию (или лекарственную форму) для применения можно упаковать различными путями в зависимости от используемого способа введения лекарственного средства. Как правило, предмет для введения включает контейнер, содержащий находящуюся в нем фармацевтическую композицию в соответствующей форме. Подходящие контейнеры хорошо известны специалистам в данной области и включают материалы, такие как флаконы (из пластика или стекла), саше, ампулы, пластиковые мешки, металлические цилиндры и тому подобное. Также контейнер может включать надежное соединение для предупреждения нечаянного доступа к содержимому упаковки. Кроме того, контейнер также имеет находящуюся на нем этикетку, на которой приводится описание содержимого контейнера. Также на этикетке могут находиться соответствующие предостережения.
Фармацевтические композиции соединений по настоящему изобретению можно приготовить для различных путей и типов введения с фармацевтически приемлемыми разбавителями, носителями, наполнителями или стабилизаторами (Remington’s Pharmaceutical Sciences, 1995, 18th edition, Mack Publ. Co., Easton, PA) в виде лиофилизованной композиции, измельченного порошка или водного раствора. Композицию можно приготовить смешением при комнатной температуре, при соответствующем значении рН и при желаемой степени чистоты с физиологически приемлемыми носителями, т.е. носителями, которые не являются токсичными для реципиентов в используемых дозах и концентрациях. Значение рН композиции в основном зависит от конкретного применения и концентрации соединения, но может находиться в пределах примерно от 3 до примерно 8.
Предпочтительно фармацевтическая композиция является стерильной. В частности, композиции для применения при введении в условиях in vivo должны быть стерильными. Такую стерилизацию легко провести фильтрованием через стерильные мембранные фильтры.
Обычно фармацевтическую композицию можно хранить в виде твердой композиции, лиофилизованной композиции или в виде водного раствора.
Фармацевтические композиции по изобретению следует дозировать и вводить таким образом, т.е. в количествах, концентрациях, схемах, курсе, растворителях и путях введения, соответствующих доброкачественной медицинской практике. Факторы, которые следует учитывать в этом отношении, включают конкретное нарушение, которое лечат, конкретное млекопитающее, которое подвергается лечению, клиническое состояние отдельного пациента, причину нарушения, место введения препарата, способ введения, схему введения и другие факторы, известные практикующим врачам. При определении «терапевтически эффективного количества» соединения по изобретению для введения следует учитывать вышеуказанные факторы, и оно является минимальным количеством, необходимым для предупреждения, ослабления или лечения нарушения, опосредованного фактором коагуляции. Предпочтительно такое количество ниже, количества, которое является токсичным для хозяина или делает хозяина существенно более чувствительным к кровотечению.
В качестве общего положения первоначальное фармацевтически эффективное количество трастузумаба-MCC-DM1, вводимое на дозу, находится в пределах примерно 0,01-1000 мг/кг, точнее примерно от 0,1 до 20 мг/кг массы тела пациента в день, с типичными начальными пределами используемого соединения от 0,3 до 15 мг/кг.
Приемлемые разбавители, носители, наполнители и стабилизаторы являются нетоксичными для реципиентов в используемых дозах и концентрациях, и они включают буферы, такие как фосфатный, цитратный и другие органические кислоты; антиоксиданты, в том числе аскорбиновую кислоту и метионин; консерванты (такие как октадецилдиметилбензиламмония хлорид; гексаметония хлорид; бензалкония хлорид; бензетония хлорид; фенол, бутилэтанол или бензиловый спирт; алкилпарабены, такие как метил- или пропилпарабен; катехол; резорцин; циклогексанол; 3-пентанол и м-крезол); низкомолекулярные полипептиды (длиной примерно менее из 10 остатков); белки, такие как сывороточный альбумин, желатин или иммуноглобулин; гидрофильные полимеры, такие как поливинилпирролидон; аминокислоты, такие как глицин, глутамин, аспарагин, гистидин, аргинин или лизин; моносахариды, дисахариды и другие углеводы, включая глюкозу, маннозу или декстрины; комплексообразующие агенты, такие как ЭДТА; сахара, такие как сахароза, маннит, трегалоза или сорбит; солеобразующие противоионы, такие как натрий; комплексы металлов (например, комплексы Zn-протеин) и/или неионогенные поверхностно-активные вещества, такие как твинТМ, включая твин 80, плуроникиТМ или полиэтиленгликоль (ПЭГ), в том чисел, ПЭГ400. Активные фармацевтические ингредиенты также можно включить в микрокапсулы, приготовленные, например, коацервацией или межфазной полимеризацией, например, микрокапсулы соответственно из гидроксиметилцеллюлозы или желатина и поли(метилметакрилата), в коллоидные системы для доставки лекарственных средств (например, липосомы, альбуминовые микросферы, микроэмульсии, наночастицы и нанокапсулы) или в макроэмульсии. Такие методы раскрыты в Remington’s Pharmaceutical Sciences 18th edition, 1995, Mack Publ. Co., Easton, PA.
Фармацевтические композиции включают композиции, подходящие для введения путями, подробно описанными в данном документе. Композиции можно соответственно представить в разовой лекарственной форме и можно приготовить любым из методов, хорошо известных в области фармации. Общее описание методов и композиций можно найти в Remington’s Pharmaceutical Sciences 18th edition, 1995, Mack Publ. Co., Easton, PA. Такие методы включают стадию приведения в контактирование активного ингредиента с носителем, который представляет одно или несколько вспомогательных веществ. В целом композиции готовят однородным и тщательным приведением в ассоциацию активного ингредиента с жидкими носителями или мелко измельченными твердыми носителями или обоими видами носителей и затем при необходимости приданием формы продукту.
Композиции химиотерапевтического средства, подходящие для приема перорально, можно приготовить в виде дискретных разовых лекарственных форм, таких как пилюли, твердые или мягкие, например, желатиновые капсулы, облатки, пастилки, лекарственные леденцы, водные или масляные суспензии, диспергируемые порошки или гранулы, эмульсии, сиропы или эликсиры, каждая содержащая заранее определенное количество соединения трастузумаба-MCC-DM1 и/или химиотерапевтического средства. Такие композиции можно приготовить с использованием любого известного в данной области метода, используемого для производства фармацевтических композиций, и такие композиции могут содержать один или несколько добавок, включающих подсластители, вкусовые вещества, красители и консерванты, для обеспечения приятного на вкус и запах препарата. Прессованные таблетки можно приготовить прессованием, с использованием подходящего устройства, активного ингредиента в сыпучей форме, такой как порошок или гранулы, необязательно в смеси со связующим веществом, регулятором сыпучести, инертным разбавителем, консервантом, поверхностно-активным или диспергирующим веществом. Формованные таблетки можно приготовить формованием на подходящем устройстве смеси порошкообразного активного ингредиента, увлажненного инертным жидким разбавителем. Таблетки можно необязательно покрыть оболочкой или сделать насечки, и необязательно формулировать с обеспечением замедленного или контролируемого высвобождения из них активного ингредиента.
Наполнители в таблетках фармацевтической композиции по изобретению могут включать: наполнитель (или разбавитель) для увеличения объема порошкообразного препарата для приготовления таблетки; разрыхлители для обеспечения разрушения таблетки на небольшие фрагменты при пероральном приеме, в идеале отдельные частицы лекарственного средства, и быстрого растворения и всасывания лекарственного средства; связующее вещество для получения гранул и таблетки с необходимой механической силой и поддержания целостности таблеток после прессования, предупреждения ее разрушения на составляющие порошки во время упаковки, транспортировки и обычного обращения препарата; регулятор сыпучести для обеспечения сыпучести порошка во время производства таблеток; смазывающее вещество для обеспечения того, чтобы порошок для приготовления таблеток не прилипал к оборудованию, которое используют для прессования таблеток в производстве. Эти вещества повышают сыпучесть порошкообразных смесей при прохождении через прессы и сводят до минимума истираемость и ломкость во время извлечения конечных таблеток из оборудования; антиприлипатель с функцией, аналогичной регулятору сыпучести, который снижает адгезию между порошком для приготовления таблеток и аппаратом, который используют для штамповки таблеток в производстве; вкусовое вещество, которое включают в таблетки для придания им более приятного вкуса или маскировки неприятного вкуса, и краситель для обеспечения идентификации и удобства для пациента.
Таблетки, содержащие активный ингредиент в смеси с нетоксичным фармацевтически приемлемым наполнителем, который является пригодным для производства таблеток, являются приемлемыми. Данные наполнители могут, например, представлять собой инертные разбавители, такие как карбонат кальция или натрия, лактозу, фосфат кальция или натрия; вспомогательные средства для гранулирования и разрыхления, такие как кукурузный крахмал или альгиновая кислота; связывающие вещества, такие как крахмал, желатин или камедь; и смазывающие вещества, такие как стеарат магния, стеариновая кислота или тальк. Таблетки могут быть без оболочки или их можно покрыть оболочкой с использованием известных методов, включающих микрокапсулирование для замедления разрыхления и всасывания в желудочно-кишечном тракте и тем самым обеспечения замедленного действия в течение более длительного периода времени. Например, можно использовать вещество для замедления всасывания во времени, такое как глицерилмоностеарат или глицерилдистеарат одни или с воском.
Для лечения глаз или других наружных тканей, например, ротовой полости и кожи, композиции предпочтительно наносят в виде мази или крема для наружного применения, содержащих активный ингредиент(ы) в количестве, например, от 0,075 до 20% мас./мас. При формуляции в виде мази активные ингредиенты можно использовать с парафиновой или смешивающейся с водой основой для мази. Альтернативно активные ингредиенты можно формулировать в креме с основой для крема масло-в-воде.
Если желательно, то водная фаза основы для крема может содержать многоатомный спирт, т.е. спирт, имеющий две или несколько гидроксильные группы, такой как пропиленгликоль, бутан-1,3-диол, маннит, сорбит, глицерин и полиэтиленгликоль (включая ПЭГ 400) и их смеси. Для композиций для наружного применения может быть желательным включить в их состав соединение, которое усиливает всасывание или проникновение активного ингредиента через кожу или другие пораженные области. Примеры таких веществ, усиливающих проникновение через кожу, включают диметилсульфоксид и близкие аналоги.
Масляная фаза эмульсий по данному изобретению может состоять из известных компонентов в известном соотношении, включая смесь, по меньшей мере, одного эмульгатора с жиром или маслом, или с жиром или маслом вместе. Предпочтительно гидрофильный эмульгатор включают вместе с липофильным эмульгатором, который функционирует в качестве стабилизатора. Эмульгатор(ы) с или без стабилизатора(ов) обеспечивают эмульгирующий воск, и воск вместе с маслом и жиром создает эмульгирующую основу мази, которая образует масляную диспергированную фазу кремовых композиций. Эмульгаторы и стабилизаторы эмульсии, подходящие для применения в композиции по изобретению, включают твин®60, спан® 80, цетостеариловый спирт, бензиловый спирт, миристиловый спирт, глицерилмоностеарат и лаурилсульфат натрия.
Водные суспензии фармацевтических композиций по изобретению содержат активные вещества в смеси с наполнителями, подходящими для производства водных суспензий. Подобные наполнители включают суспендирующее вещество, такое как натриевая соль карбоксиметилцеллюлозы, кроскармелозу, повидон, метилцеллюлозу, гидроксипропилметилцеллюлозу, альгинат натрия, поливинилпирролидон, трагакантовую камедь и аравийскую камедь, и диспергирующие или смачивающие вещества, такие как природный фосфатид (например, лецитин), продукт конденсации алкиленоксида с жирной кислотой (например, полиэтиленстеарат), продукт конденсации этиленоксида с длинноцепочечным алифатическим спиртом (например, гептанэтиленоксицетанол), продукт конденсации этиленоксида с неполным эфиром, полученным из жирной кислоты и ангидрида гекситола (например, полиоксиэтилен сорбитанмоноолеата). Водная суспензия также может содержать один или несколько консервантов, таких как этил- или н-пропил-п-гидроксибензоат, один или несколько красителей, одно или несколько вкусовых веществ и один или несколько подсластителей, таких как сахароза или сахарин.
Фармацевтические композиции могут находиться в виде стерильного инъекционного препарата, такого как стерильная водная или масляная суспензия для инъекций. Данную суспензию можно формулировать согласно способам, известным в данной области, с использованием подходящих диспергирующих или смачивающих средств и суспендирующих веществ, указанных выше. Стерильный инъекционный препарат может представлять собой раствор или суспензию в нетоксичном, приемлемом для парентерального введения разбавителе или растворителе, такой как раствор в 1,3-бутандиоле, или приготовленный из лиофилизованного порошка. Среди приемлемых носителей или растворителей, которые можно использовать, можно упомянуть воду, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, можно использовать стерильные нелетучие масла, обычно применяемые в качестве растворителя или суспендирующей среды. Для данной цели можно использовать любое мягкое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, для приготовления инъекционных растворов также можно использовать жирные кислоты, такие как олеиновая кислота.
Количество активного ингредиента, которое можно объединить с носителем для приготовления разовой лекарственной формы, может варьировать в зависимости от хозяина, который подвергается лечению и конкретного пути введения. Например, лекарственная форма с замедленным высвобождением, предназначенная для перорального введения людям, может содержать примерно от 1 до 1000 мг активного вещества, формулированного с соответствующим и подходящим количеством носителя, содержание которого может находиться в пределах примерно от 5 до примерно 95% к массе общей композиции (масса:массе). Фармацевтическую композицию можно приготовить таким образом, чтобы можно было легко определять количество для введения. Например, водный раствор для внутривенной инфузии, может содержать примерно от 3 до 500 мкг активного ингредиента на миллилитр раствора для обеспечения подходящего объема для инфузии со скоростью введения примерно 30 мл/ч.
Композиции, подходящие для парентерального введения, включают водные или неводные стерильные инъекционные растворы, которые могут содержать антиоксиданты, буферы, бактериостатики и растворенные вещества, которые придают композиции изотоничность с кровью предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут включать суспендирующие вещества и загустители.
Композиции, подходящие для местного применения для глаз, включают глазные капли, в которых активный ингредиент растворен или суспендирован в подходящем носителе, в частности, водном растворителе для активного ингредиента. Предпочтительно активный ингредиент находится в таких композициях в концентрации примерно от 0,5 до 20% мас./мас., например, примерно от 0,5 до 10% мас./мас., например, она составляет 1,5% мас./мас.
Композиции, подходящие для местного применения в ротовой полости, включают лечебные леденцы, содержащие активный ингредиент в основе с вкусовым веществом, обычно сахарозой и аравийской камедью или трагакантом; пастилки, содержащие активный ингредиент в инертной основе, такой как желатин и глицерин, или сахароза и аравийская камедь; и жидкости для полоскания ротовой полости, содержащие активный ингредиент в подходящем жидком носителе.
Композиции для ректального введения могут быть представлены в виде суппозитория с подходящей основой, содержащей, например, какао-масло или салицилат.
Композиции, подходящие для интрапульмонарного или назального введения, имеют размер частиц, например, в пределах от 0,1 до 500 микрон (включая размер частиц в пределах от 0,1 до 500 микрон с приростом в микронах, таком как 0,5; 1; 30; 35 микрон и т.д.), которые вводят быстрой ингаляцией через носовые ходы или ингаляцией через рот таким образом, чтобы достичь альвеолярных мешков. Подходящие композиции включают водные или масляные растворы активного ингредиента. Композиции, подходящие для введения в виде аэрозоля или сухого порошка, можно приготовить с использованием обычных методов и их можно вводить с другими лекарственными средствами, такими как соединения, применяемые при лечении или профилактике заболеваний, указанных в данном документе.
Композиции, подходящие для интравагинального введения, могут быть представлены в виде вагинальных свечей, тампонов, кремов, гелей, паст, пен или спреев, содержащих в дополнении к активному ингредиенту подходящие носители, которые известны в данной области.
Композиции можно упаковать в виде разовых лекарственных форм или мультидозовых контейнеров, например, запаянных ампул и флаконов, и их можно хранить в высушенном вымораживанием (лиофилизованном) состоянии, когда требуется только добавить стерильный жидкий носитель, например, воду для инъекций, непосредственно перед использованием. Приготавливаемые экстемпоро инъекционные растворы и суспензии готовят из стерильных порошков, гранул и таблеток описанного ранее типа. Предпочтительными разовыми лекарственными формами являются формы, содержащие суточную дозу или неполную суточную дозу активного ингредиента, указанные выше, или их соответствующую фракцию.
Также изобретение относится к ветеринарным композициям, содержащим, по меньшей мере, один активный ингредиент, указанный выше, вместе с носителем для него, применяемым в ветеринарии. Носители, применяемые в ветеринарии, представляют вещества, пригодные для введения композиции, и они могут быть твердыми, жидкими или газообразными веществами, которые являются инертными или приемлемыми для применения в ветеринарии и совместимыми с активным ингредиентом. Данные ветеринарные композиции можно вводить парентерально, перорально или любым другим путем.
Комбинированная терапия
Трастузумаб-MCC-DM1 можно использовать в комбинации с другими химиотерапевтическими средствами для лечения гиперпролиферативного заболевания или нарушения, включая опухоли, злокачественные опухоли и неопластическую ткань, наряду с презлокачественными и не неопластическими или доброкачественными гиперпролиферативными заболеваниями. В некоторых вариантах осуществления трастузумаб-МСС-DM1 комбинируют в фармацевтической комбинированной композиции или в схеме введения для комбинированной терапии со вторым соединением, обладающим антигиперпролиферативными свойствами, или которое является пригодным для лечения гиперпролиферативного нарушения. Второе соединение в фармацевтической комбинированной композиции или в схеме введения предпочтительно обладает комплементарными активностями для трастузумаба-MCC-DM1, и таким образом они не оказывают отрицательного воздействия друг на друга. Такие соединения соответственно находятся в комбинации в количествах, которые являются эффективными для предназначенной цели. В одном варианте осуществления композиция по данному изобретению содержит трастузумаб-MCC-DM1 в комбинации с химиотерапевтическим средством, описанным в данном документе. В примерах 4 и 5 приводятся клинические протоколы соответственно для T-DM1 + пертузумаба и T-DM1 + GDC-0941.
Терапевтические комбинации по изобретению включают композицию, схему введения или иной курс лечения, включающие введение трастузумаба-MCC-DM1 и химиотерапевтического средства, выбранного из антитела-ингибитора димеризации HER2, антитела к VEGF, 5-FU, карбоплатина, лапатиниба, ABT-869 и доцетаксела, в виде комбинированного препарата для раздельного, одновременного или последовательного применения в лечении гиперпролиферативного нарушения.
Комбинированную терапию можно проводить в виде одновременной или последовательной схемы. При последовательном введении комбинацию можно вводить в два или несколько приемов. Комбинированное введение включает совместное введение, применение раздельных лекарственных форм или одной фармацевтической формы, и последовательное введение в любом порядке, где предпочтительно имеется период времени, когда оба (или все) активные соединения одновременно проявляют свою биологическую активность.
Подходящие дозы любого из указанных выше препаратов для совместного введения представляют собой применяемые в настоящее время и которые можно снизить за счет комбинированного действия (синергии) вновь идентифицированного препарата и других химиотерапевтических средств или видов лечения.
В конкретном варианте осуществления противоопухолевой терапии трастузумаб-MCC-DM1 можно комбинировать с химиотерапевтическим средством, в том числе, гормональными средствами или антителами, такими как описаны выше, а также объединить с хирургическим лечением и лучевой терапией. Количества трастузумаба-MCC-DM1 и другого фармацевтически активного химиотерапевтического средства(в) и относительное время введения выбирают для достижения желаемого комбинированного лечебного эффекта.
Введение фармацевтических композиций
Соединения по изобретению можно вводить любым путем, который соответствует состоянию, которое подвергается лечению. Подходящие пути включают пероральный, парентеральный (в том числе, подкожный, внутримышечный, внутривенный, внутриартериальный, ингаляционный, внутрикожный, интратекальный, эпидуральный и инфузионный), трансдермальный, ректальный, назальный, местный (включая буккальный или сублингвальный), вагинальный, внутрибрюшинный, интрапульмонарный и интраназальный. Местное введение также может включать применение трансдермального введения с помощью трансдермальных пластырей или устройств для ионофореза. Формуляция лекарственных средств обсуждается в Remington’s Pharmaceutical Sciences, 1995, 18th edition, Mack Publ. Co., Easton, PA. Другие примеры лекарственных форм можно найти у Liberman H.A. and Lachman L., Eds., Pharmaceutical Dosage Forms, Marcel Decker, Vol. 3, 2nd Ed., New York, NY. Для местной иммуносупрессорной терапии соединения можно вводить в область поражения, включая перфузию или иное контактирование трансплантата с супрессором перед трансплантацией. Специалисты в данной области, очевидно, понимают, что предпочтительный путь может варьировать в зависимости, например, от состояния реципиента. В том случае, когда соединение вводят перорально, то его можно формулировать в виде пилюли, капсулы, таблетки и т.д. с фармацевтически приемлемым носителем, регулятором сыпучести или наполнителем. В том случае, когда соединение вводят парентерально, то его можно формулировать с фармацевтически приемлемым растворителем или наполнителем для парентерального введения и в разовой лекарственной инъекционной форме, как подробно описано ниже.
Доза трастузумаба-MCC-DM1 для лечения людей может находиться в пределах примерно от 100 мг до примерно 500 мг. Дозу трастузумаба-MCC-DM1 можно вводить один раз каждые 6 недель, один раз каждые 3 недели, один раз в неделю или чаще в зависимости от фармакокинетических (PK) и фармакодинамических (PD) свойств препарата, включая всасывание, распределение, метаболизм и выделение. Доза химиотерапевтического средства, используемого в комбинации с трастузумабом-MCC-DM1, может находиться в пределах примерно от 10 мг до примерно 1000 мг. Химиотерапевтическое средство можно вводить один раз каждые 6 недель, один раз каждые 3 недели, один раз в неделю или чаще, например, один или два раза в день. Кроме того, на дозу и схему введения могут оказывать влияние токсические факторы. Для введения перорально пилюлю, капсулу или таблетку можно принимать ежедневно или реже в течение определенного периода времени. Схему можно повторять в течение ряда терапевтических курсов.
Способы лечения
Терапевтические комбинации: (1) трастузумаб-MCC-DM1 и (2) химиотерапевтическое средство являются пригодными для лечения заболеваний, состояний и/или нарушений, включая, не ограничиваясь этим, заболевания, для которых характерна активация пути HER2. Следовательно, другой аспект данного изобретения относится к способам лечения заболеваний или состояний, которые можно лечить направленным воздействием на HER2 или рецептор 1 VEGR. Терапевтические комбинации: (1) трастузумаб-MCC-DM1 и (2) химиотерапевтического средства можно использовать для лечения гиперпролиферативного заболевания или нарушения, включая опухоли, злокачественные опухоли и неопластическую ткань, наряду с презлокачественными и не неопластическими или доброкачественными гиперпролиферативными заболеваниями.
Злокачественные опухоли, которые можно лечить способами по данному изобретению, включают, не ограничиваясь этим, рак молочной железы, яичников, шейки матки, предстательной железы, яичка, органов мочеполовой системы, пищевода, гортани, глиобластому, нейробластому, рак желудка, кожи, кератоакантому, рак легких, плоскоклеточную карциному, крупноклеточную карциному, немелкоклеточную карциному легких (NSCLC), мелкоклеточную карциному, аденокарциному легких, рак кости, ободочной кишки, аденому, рак поджелудочной железы, аденокарциному, рак щитовидной железы, фолликулярную карциному, недифференцированную карциному, папиллярную карциному, семиному, меланому, саркому, карциному мочевого пузыря, карциному печени и желчных протоков, карциному почек, миелоидные нарушения, лимфоидные нарушения, рак волосистых клеток, рак щечного кармана и глотки (ротовой полости), рак губ, языка, ротовой полости, глотки, тонкого кишечника, рак ободочной-прямой кишки, толстого кишечника, прямой кишки, мозга и центральной нервной системы, ходжкинскую лимфому и лейкоз.
Другой аспект данного изобретения относится к фармацевтической композиции или терапевтической комбинации для применения в лечении заболеваний или состояний, указанных в данном документе, у млекопитающего, например, человека, страдающего таким заболеванием или состоянием. Также обеспечивается применение фармацевтической композиции в производстве лекарственного средства для лечения заболеваний и состояний, указанных в данном документе, у теплокровного животного, такого как млекопитающее, например, человека, страдающего таким нарушением.
Изделия
В другом варианте осуществления изобретения обеспечивается изделие или «набор», содержащий трастузумаб-MCC-DM1, пригодный для лечения заболеваний или нарушений, описанных выше. В одном варианте осуществления набор включает контейнер, содержащий трастузумаб-MCC-DM1. Набор может дополнительно содержать этикетку или вкладыш в упаковке на или связанные с контейнером. Термин «вкладыш в упаковке» используется в отношении инструкций, обычно вкладываемых в промышленные упаковки терапевтических продуктов, которые содержат информацию о показаниях, применении, дозировке, введении, противопоказаниях и/или предостережениях, касающихся применения таких терапевтических продуктов. Подходящие контейнеры включают, например, бутыли, флаконы, шприцы, блистерные упаковки и т.д. Контейнер может быть изготовлен из различных материалов, таких как стекло или пластик. Контейнер может содержать трастузумаб-MCC-DM1 или его композицию, которая является эффективной для лечения состояния и может иметь стерильное входное отверстие (например, контейнер может представлять мешок с раствором для внутривенного введения или флакон с пробкой, проткнутой иглой для внутрикожных инъекций). По меньшей мере, одно активное соединение в композиции представляет собой трастузумаб-MCC-DM1. На этикетке или вкладыше в упаковке указывается, что композиция используется для лечения состояния выбора, такого как рак. В одном варианте осуществления на этикетке или вкладыше в упаковке указывается, что композицию, содержащую трастузумаб-MCC-DM1, можно использовать для лечения нарушения, возникающего в результате аномального роста клеток. Также на этикетке или вкладыше в упаковке может указываться, что композицию можно использовать для лечения других нарушений. Альтернативно или дополнительно изделие может также содержать второй контейнер, содержащий фармацевтически приемлемый буфер, такой как бактериостатическая вода для инъекций (BWFI), забуференный фосфатом физиологический раствор, раствор Рингера и раствор декстрозы. Дополнительно он может содержать другие вещества, желательные с коммерческой точки зрения и с точки зрения потребителя, включая буферы, разбавители, фильтры, иглы и шприцы.
Набор может дополнительно содержать инструкции по введению трастузумаба-MCC-DM1 и, если он присутствует, второй фармацевтический композиции. Например, если набор включает первую композицию, содержащую трастузумаб-MCC-DM1, и вторую фармацевтическую композицию, то набор может дополнительно содержать инструкции по одновременному, последовательному или раздельному введению первой и второй фармацевтической композиций пациенту, нуждающемуся в этом.
В другом варианте осуществления наборы подходят для введения твердых лекарственных форм трастузумаба-MCC-DM1 для приема перорально, таких как таблетки или капсулы. Предпочтительно такой набор содержит ряд разовых лекарственных форм. Такие наборы могут включать карточку с дозами, расположенными в порядке для предназначенного использования. Примером такого набора является «блистерная упаковка». Блистерные упаковки хорошо известны в упаковочной промышленности, и они широко используются для упаковки фармацевтических разовых лекарственных форм. Если желательно, то можно обеспечить набор памяткой, например, в форме чисел, букв или других пометок или календарем-вкладышем, на котором приведена схема лечения в днях, по которой можно вводить дозы.
По одному варианту осуществления набор содержит (а) первый контейнер с трастузумабом-MCC-DM1 в нем и необязательно (b) второй контейнер со второй фармацевтической композицией в нем, где вторая фармацевтическая композиция содержит второе соединение, обладающее антигиперпролиферативной активностью. Альтернативно или дополнительно набор также может включать третий контейнер, содержащий фармацевтически приемлемый буфер, такой как бактериостатическая вода для инъекций (BWFI), забуференный фосфатом физиологический раствор, раствор Рингера и раствор декстрозы. Дополнительно он может содержать другие вещества, желательные с коммерческой точки зрения и с точки зрения потребителя, включая буферы, разбавители, фильтры, иглы и шприцы.
В том случае, если набор содержит композицию трастузумаба-MCC-DM1 и второго лекарственного средства, т.е. химиотерапевтического средства, то набор может содержать контейнер для отдельных композиций, такой как разделенная бутыль или разделенная упаковка из фольги, однако, отдельные композиции также могут находиться в одном, неразделенном контейнере. Как правило, набор содержит инструкции для введения отдельных компонентов. Форма набора является особенно преимущественной, когда отдельные компоненты предпочтительно вводят в различных лекарственных формах (например, перорально и парентерально), вводят при различных интервалах введения или когда для врача желательно провести титрование отдельных компонентов комбинации.
Примеры
Для иллюстрации изобретения приводятся следующие примеры. Однако, очевидно, понятно, что данные примеры не ограничивают изобретение и только означают предложения для способа применения изобретения на практике.
Пример 1. Получение трастузумаба-MCC-DM1
Трастузумаб выделяли из герцептина® буфер-обменом в концентрации 20 мг/мл в буфере, состоящем из 50 мМ фосфата калия/50 мМ хлорида натрия/2 мМ ЭДТА, рН 6,5 и обрабатывали 7,5-10 моль-экв сукцинимидил-4-(N-малеимидометил)циклогексан-1-карбоксилата (SMCC, Pierce Biotechnology, Inc), 20 мМ в ДМСО или DMA (диметилацетамиде), 6,7 мг/мл (заявка на патент США 2005/0169933; заявка на патент США 2005/0276812). После перемешивания в течение 2-4 ч в атмосфере аргона при комнатной температуре реакционную смесь пропускали через колонку с сефадексом G25, уравновешенную буфером из 50 мМ фосфата калия/50 мМ хлорида натрия/2 мМ ЭДТА, рН 6,5. Альтернативно реакционную смесь фильтровали через гель с 30 мМ цитратом и 150 мМ хлоридом натрия при рН 6. Фракции, содержащие антитело, объединяли и анализировали. Выделение трастузумаба-SMCC составляло 88%.
Промежуточное соединение лекарственное средство-линкер, трастузумаб-MCC, указанный выше, разводили буфером из 50 мМ фосфата калия/50 мМ хлорида натрия/2 мМ ЭДТА, рН 6,5 до конечной концентрации 10 мг/мл и подвергали взаимодействию с 10 мМ раствором DM1 (1,7 экв, приняв соотношение 5 SMCC/трастузумаб, 7,37 мг/мл) в диметилацетамиде. DM1 можно приготовить из продукта ферментации ансамитоцина (патент США № 6790954; патент США № 7432088) и дериватизировали для конъюгации (патент США № 6333410; RE 39151). Реакционную смесь перемешивали при комнатной температуре в атмосфере аргона в течение примерно 4-16 ч. Реакционную смесь для конъюгирования пропускали через колонку для фильтрования с сефадексом G25 (1,5×4,9) 1× PBS с рН 6,5. Альтернативно реакционную смесь пропускали через гель с 10 мМ цитратом и 150 мМ хлоридом натрия при рН 5. Соотношение DMI/трастузумаб (р) составляло 3,1 по данным определения поглощения при длинах волн 252 нм и 280 нм. Соотношение лекарственного средства к антителу (р) можно определить с использованием масс-спектрометрии. Также ход конъюгирования можно проследить с использованием электрофореза в SDS-полиакриламидном геле. Агрегацию можно оценить анализом рассеивания лазерного пучка света.
Альтернативно трастузумаб-MCC-DM1 можно получить приготовлением реагента MCC-DM1 линкер-лекарственное средство и затем взаимодействием с трастузумабом.
Как правило, реакция конъюгации трастузумаба-MCC с DM1 приводит к образованию гетерогенной смеси, содержащей антитела с различным количеством присоединенного конъюгированного лекарственного средства DM1, т.е. с различной нагрузкой лекарственным средством, где р представляет распределение от 1 до примерно 8. Имеется дополнительное измерение гетерогенности с различными сайтами присоединения SMCC к трастузумабу, где множество различных нуклеофилов в трастузумабе, например, концевые аминогруппы лизина, могут взаимодействовать с SMCC. Таким образом, трастузумаб-MCC-DM1 включает выделенные, очищенные молекулы, а также смеси со средней нагрузкой лекарственным средством от 1 до 8, и где MCC-DM1 присоединен через любой сайт антитела трастузумаба.
Среднее количество групп лекарственного средства DM1 на антитело трастузумаб в препаратах трастузумаба-MCC-DM1, полученных реакцией конъюгации, можно определить обычными методами, такими как масс-спектроскопия, ELISA, электрофорез и ВЭЖХ. Также можно определить количественное распределение трастузумаба-MCC-DM1 по значению р. С помощью ELISA можно определить среднее значение р в конкретном препарате ADC (Hamblett et al., 2004, Clinical Cancer Res., 10:7063-7070; Sanderson et al., 2005; Clinical Cancer Res., 11:843-852). Однако распределение значений р (лекарственного средства) не различимо по связыванию антитело-антиген и с учетом предела детектирования ELISA. Также с помощью ELISA для конъюгатов антитело-лекарственное средство невозможно установить, в какой области группы лекарственного средства присоединяются к антителу, например, к фрагментам тяжелой цепи или легкой цепи, или к конкретным аминокислотным остаткам. В некоторых случаях можно провести выделение, очистку и характеристику гомогенного трастузумаба-MCC-DM1, где р имеет определенные значения, из трастузумаба-MCC-DM1 с нагрузкой другими лекарственными средствами обратнофазовой ВЭЖХ или электрофорезом.
Пример 2. Тест оценки пролиферации клеток в условиях in vitro
Эффективность комбинаций по изобретению можно определить тестом оценки пролиферации клеток с использованием следующего протокола (Promega Corp. Technical Bulletin TB288; Mendoza et al., 2002, Cancer Res., 62:5485-5488). Реагенты и протокол для постановки анализа Cell-Titer Glo являются промышленно доступными (Promega). С помощью данного теста можно оценить способность соединений проникать в клетки и воздействовать на пролиферацию клеток. Принцип метода заключается в определении количества имеющихся жизнеспособных клеток по количественному анализу АТФ. Cell-Titer Glo представляет собой реагент, используемый для количественного анализа. Это гомогенный анализ, при котором добавление реагента Cell-Titer Glo приводит к лизису клеток и генерации люминесцентного сигнала в реакции с люциферазой. Сигнал люминесценции прямо пропорционален количеству присутствующей АТФ.
Планшеты для ДМСО и сред: 96-луночные полипропиленовые планшеты производства Nunc (cat.#249946).
Планшеты для клеток: 384-луночные черные, с прозрачным дном (microclear) планшеты ТС с крышкой производства Falcon (353962).
Культуральная клеточная среда: RPMI или DMEM с высоким содержанием глюкозы; среда Ham’s F-12 (50:50), 10% фетальной бычьей сыворотки, 2 мМ L-глутамина.
Cell-Titer Glo: Promega (Cat.#G7572).
Протокол метода:
1 сутки – клетки высевают в планшеты для клеток, собирают клетки, высевают клетки с титром 1000-2000 клеток в 54 мкл на лунку в 384-луночные планшеты для клеток на 3 суток. Инкубируют в течение ночи (примерно 16 ч) при 37°С, в атмосфере с 5% СО2.
2 сутки – к клеткам прибавляют лекарственное средство, соединение в разведении, планшеты для ДМСО (серийные разведения 1:2 на 9 точек). Вносят 20 мкл соединений (10 мМ маточный раствор лекарственных средств с небольшой молекулой) во второй ряд 96-луночного планшета. Проводят серийные разведения 1:2 по планшету (10 мкл +10 мкл 100% ДМСО) в целом на 9 точек с использованием планшетов Precision Media (разведение 1:50). Вносят 147 мкл среды во все лунки отдельного 96-луночного планшета для сред. Из каждой лунки в планшете для ДМСО переносят 3 мкл ДМСО+соединение в соответствующую лунку в планшете для сред с использованием Rapidplate. В опытах с комбинацией двух лекарственных средств одно лекарственное средство в 1,5 мкл ДМСО+соединение из каждой лунки в планшете для ДМСО переносят в каждую соответствующую лунку в планшете для сред с использованием Rapidplate. Затем переносят 1,5 мкл другого препарата в планшет для сред.
К клеткам прибавляют лекарственные средства, планшет для клеток (разведение 1:10), добавляют 6 мкл среда+соединение непосредственно к клеткам (54 мкл среды уже на клетках). Инкубируют в течение 3 суток в термостате при 37°С, в атмосфере с 5% СО2, который не должен часто открываться.
5 сутки – развивают планшеты, размораживают буфер для Cell-Titer Glo при комнатной температуре. Вынимают планшеты для клеток из термостата с температурой 37°С и уравновешивают до комнатной температуры в течение примерно 30 мин. К субстрату Cell- Titer Glo добавляют буфер Cell-Titer Glo (бутыль на бутыль). Добавляют 30 мкл реагента Cell Titer-Glo в каждую лунку с клетками. Помещают на шейкер для планшетов примерно на 30 мин. Определяют люминесценцию на ридере для планшетов PerkinElmer Envision (0,1 сек на лунку) или Analyst HT (полсекунды на лунку).
Анализ жизнеспособности клеток и комбинационные тесты: клетки высевают с титром 1000-2000/лунку в 384-луночные планшеты на 16 ч. На 2 сутки проводят девять серийных разведений 1:2 соединения в ДМСО в 96-луночном планшете. Соединения затем разводят в культуральной среде с использованием автоматического устройства Rapidplate (Zymark Corp., Hopkinton, MA). Затем разведенные соединения вносят в четыре лунки в 384-луночных планшетах для клеток и инкубируют при 37°С и в атмосфере с 5% СО2. Через 4 суток определяют относительные количества жизнеспособных клеток по люминесценции с использованием Cell-Titer Glo (Promega), следуя инструкциям производителя, и сигнал регистрируют на ридере Envision или Wallac Multilabel (PerkinElmer, Foster City). Рассчитывают ЕС50 с использованием программного обеспечения Kaleidagraph 4.0 (Synergy Software) или Prism 4.0 (GraphPad, San Diego). Лекарственные средства в комбинационных тестах дозируют, начиная с концентрации 8× ЕС50. В тех случаях, когда значение ЕС50 лекарственного средства составляло >2,5 мкМ, то использовали наиболее высокую концентрацию 10 мкМ. Во всех тестах трастузумаб-MCC-DM1 и химиотерапевтические средства добавляли одновременно или раздельно с интервалом 4 ч (один перед другим).
В дополнительном приведенном в качестве примера описании теста оценки пролиферации клеток в условиях in vitro анализ включает следующие стадии:
1. Аликвотную порцию клеточной культуры объемом 100 мкл, содержащую примерно 104 клеток (см. на фиг.1 клеточные линии и тип опухолей) в среде помещают в каждую лунку 384-луночного планшета с непрозрачными стенками.
2. Готовят контрольные лунки, содержащие среду и без клеток.
3. В опытные лунки добавляют соединение и инкубируют в течение 3-5 суток.
4. Планшеты уравновешивают до комнатной температуры в течение примерно 30 мин.
5. В каждую лунку вносят реагент Cell-Titer Glo в объеме, равном объему культуральной клеточной среды, находящейся в каждой лунке.
6. Содержимое перемешивают в течение 2 мин на орбитальном шейкере для индукции лизиса клеток.
7. Планшет инкубируют при комнатной температуре в течение 10 мин для стабилизации сигнала люминесценции.
8. Регистрируют люминесценцию и строят графики в RLU=относительных единицах люминесценции.
Альтернативно клетки высевали с оптимальной плотностью в 96-луночном планшете и инкубировали в течение 4 суток в присутствии испытуемого соединения. Затем вносили аламар синийТМ в среду для постановки теста и клетки инкубировали в течение 6 ч перед регистрацией сигнала при длине волны возбуждения 544 нм и длине эмиссии 590 нм. Значения ЕС50 рассчитывали с использованием сигмоидальной кривой зависимости доза-эффект.
Пример 3
Опухолевые ксенотрансплантаты в условиях in vivo
Животных, подходящих для трансгенных опытов, можно получить из обычных коммерческих питомников. Иммунодефицитным мышам-самкам CB-17 SCID (Charles River Laboratory) имплантировали 3 млн. клеток KPL-4 (со сверхэкспрессией Her2) злокачественной опухоли молочной железы в матригеле в жировую подушку молочной железы. Бестимусным мышам-самкам nude (Charles River Laboratory или Harlan) имплантировали фрагменты трансгенных опухолей 2х2 мм3 молочной железы MMTV-Her2 Fo5 в жировую подушку молочной железы. На 0 сутки в ксенотрансплантаты у мышей вводили лекарственное средство, комбинацию лекарственных средств или носитель по схеме, соответствующей каждой опухолевой модели. 5-FU, гемцитабин, карбоплатин и B20-4.1 вводили внутрибрюшинно, пертузумаб вводили внутривенно или внутрибрюшинно, как это соответствует, трастузумаб-MCC-DM1 и доцетаксел вводили внутривенно, лапатиниб, GDC-0941 и АВТ-869 вводили перорально с помощью желудочного зонда. Размеры опухолей определяли дважды в неделю в течение всего опыта. Также дважды в неделю определяли массу мышей и регулярно проводили наблюдение за состоянием животных. Объем опухолей определяли в двух измерениях (по длине и ширине) с использованием штангенциркуля Ultra Cal IV (Model 54-10-111; Fred V. Fowler Co., Inc.; Newton, MA) и анализировали с использованием программы Excel v. 11.2 (Microsoft Corporation; Redmond, WA). Графики ингибирования опухолей строили с использованием программы KaleidaGraph 3,6 (Synergy Software; Reading, PA). Объем опухолей рассчитывали по формуле: размер опухолей (мм3) = (более длинное измерение × более короткое измерение2) ×0,5.
Массу животных анализировали с использованием программы Adventurera Pro AV812 (Ohaus Corporation; Pine Brook, NJ). Графики строили с использованием программы KaleidaGraph Version 3.6. Изменение массы тела в процентах рассчитывали с использованием формулы: изменение массы по группе в процентах = (1-(исходная масса/новая масса)) × 100.
Мышей с объемом опухолей, превышающем 2000 мм3, или с потерей масса тела, составляющей более 20% от исходной, подвергали эвтаназии согласно принятой практике.
Замедление роста опухолей в процентах (% TGD) в конце опыта (EOS) рассчитывали с использованием формулы: % TGD=100 × (среднее время до конечной точки в опытной группе – среднее время до конечной точки в контрольной группе)/среднее время до конечной точки в контрольной группе.
Частоту опухолей (TI) определяли, основываясь на количестве определяемых опухолей, остающихся в каждой группе в конце опыта. Частичный ответ (PR) определяли как более чем 50%, но менее чем 100% снижение объема опухолей по сравнению с исходным объемом опухолей для трех последовательных измерений. Полный ответ (CR) определяли как 100% снижение объема опухолей по сравнению с исходным объемом опухолей для трех последовательных измерений. Данные анализировали и р-значения определяли с использованием t-теста Даннетта со статистической программой JMP, версия 5.1.2 (SAS Institute; Cary, NC). Значения объема отдельных опухолей в конце опыта и среднего объема опухолей±SEM обрабатывали с использованием статистической программы JMP, версия 5.1.2. Строили график по данным определения массы тела на основе среднего процента изменения по сравнению со значениями исходной массы тела±SEM.
Пример 4. Клиническое изучение трастузумаба-MCC-DM1
(T-DM1) в комбинации с пертузумабом
Проводят фазу 1b/II открытого клинического изучения безопасности, переносимости и эффективности трастузумаба-MCC-DM1 (T-DM1) в комбинации с пертузумабом при внутривенном введении пациенткам с HER2-положительным местно-распространенным или метастатическим раком молочной железы для оценки безопасности и переносимости комбинации. Комбинацию вводят один раз в 3 недели пациенткам с HER2-положительным местно-распространенным или метастатическим раком молочной железы, которые ранее получали трастузумаб в любой линии терапии, получали химиотерапию в комбинации с HER2-направленней терапией для заболевания на поздних стадиях или у которых имело место прогрессирование заболевания при получении последней терапии. Другой целью является оценка фармакокинетики T-DM1 при введении комбинации T-DM1 и пертузумаба по данной схеме. Еще одной целью исследований является предварительная оценка эффективности комбинации T-DM1 и пертузумаба при введении по данной схеме по результатам объективных ответов, основанных на оценке исследователей, с использованием критериев оценки ответов на терапию при солидных опухолях (RECIST), версия 1.0. Вторичными целями данного исследования являются следующие: (1) установление выживаемости без прогрессирования заболевания (PFS) у пациенток, получавших комбинацию T-DM1 и пертузумаба при введении по данной схеме; (2) оценка продолжительности ответа на комбинацию T-DM1 и пертузумаба при введении по данной схеме и (3) оценка выработки антител к T-DM1.
T-DM1 вводят внутривенной (в/в) инфузией в комбинации с пертузумабом, который также вводят внутривенной (в/в) инфузией, пациенткам с HER2-положительным местно-распространенным или метастатическим раком молочной железы, которые ранее получали трастузумаб и у которых наблюдали прогрессирование заболевания после или во время последней терапии. Пациентки получают комбинацию T-DM1 и пертузумаба в повторных курсах с минимальным интервалом, составляющим 3 недели.
Проводят наблюдение за состоянием пациенток при введении данной дозы на DLT (ограничивающую дозирование токсичность) во время периода наблюдений за DLT (21 сутки со времени первого введения T-DM1) после получения первых доз испытуемых средств перед введением более высоких доз любой пациентке. В том случае, если у данных пациенток не наблюдают DLT во время периода наблюдений за DLT, то следующую дозу можно увеличить.
DLT определяют как проявление любого из токсических эффектов, связанных с лечением, в период наблюдений за DLT: (1) побочный эффект степени ≥3, не связанный с гематологическими показателями, который проявляется не в результате прогрессирования заболевания или по иной четко определяемой причине, за исключением алопеции любой степени; (2) диарея степени ≥3, которая соответствует стандарту лечения; (3) тошнота или рвота степени 3 при отсутствии премедикации, которые соответствуют стандарту лечения; (4) повышение уровня билирубина, активности трансаминаз печени (АЛТ или АСТ) или щелочной фосфатазы (ЩФ) в сыворотке крови степени ≥3, продолжающееся в течение 72 ч, за исключением пациенток с активностью трансаминаз печени и ЩФ степени 2 на исходном уровне (≤5 верхняя граница нормы [ULN] в результате наличия метастазов в печени и костях. Активность трансаминаз или ЩФ степени ≥10 ULN будет рассматриваться в качестве проявления DLT; (5) тромбоцитопения степени ≥4, продолжающаяся 24 ч; (6) нейтропения степени ≥4 (абсолютное количество нейтрофилов <500 клеток/мм3), продолжающаяся в течение 4 суток или сопровождающаяся лихорадкой (температура при измерении в ротовой полости и тимпаническая температура 100,4°F или 38°С); (7) любой субъективно оцениваемый побочный эффект, отмечаемый исследователем, связанный с введением каждого испытуемого соединения; (8) любое проявление токсичности, связанное с лечением, препятствующее началу второго курса лечения.
После принятия решения о продолжении лечения в следующей более высокой дозе, то разрешается повышение дозы для пациенток; пациентки, принимающие участие в данном исследовании, первоначально получают пониженную дозу T-DM1 (3,0 мг/кг) вместе с полной дозой пертузумаба. Этим пациенткам разрешается повысить дозы обоих препаратов до полных при дальнейших курсах, если они прошли период наблюдений за DLT. Однако безопасность дозировки 3,6 мг/кг будет основываться на оценке DLT. Пациентки (включая пациенток, принимающих участие в исследовании во время фазы повышения дозы в исследовании) считаются оцениваемыми в отношении эффективности, если они остаются в исследовании до первой последующей оценки опухоли. В конце первого курса и затем каждые три курса во время лечения следует провести эхокардиографию (ECHO) или MUGA-сканирование (многовходную артериографию).
Составление лекарственной формы T-DM1
T-DM1 можно формулировать в виде лиофилизованной композиции для однократного применения в стеклянном флаконе емкостью 20 мл типа I по фармакопее США/Европейской фармакопее с фторрезиновой ламинированной пробкой размером 20 мм и закатанном алюминиевым колпачком с темно-серой съемной пластиковой крышкой. После восстановления 8,0 мл стерильной воды для инъекций (SWFI) полученный продукт содержит T-DM1 в концентрации 20 мг/мл в 10 мМ сукцинате натрия, рН 5,0, 6% (мас./об.) сахарозы и 0,02% (мас./об.) полисорбата 20. Каждый флакон емкостью 20 мл содержит примерно 172 мг T-DM1, что позволяет вводить 160 мг T-DM1. Указанный объем раствора T-DM1 извлекают из флакона(в) и переносят в мешок для внутривенного введения лекарственных средств. Восстановленный T-DM1 разводят в поливинилхлоридных (PVC) или не содержащих латекс, не содержащих PVC полиолефиновых мешках (РО) с 0,45% или 0,9% хлоридом натрия для инъекций (минимальный объем 250 мл). Предпочтительным является применение мешков из PVC или РО, содержащих 0,45% хлорид натрия. В случае применения мешков из PVC или РО, содержащих 0,9% хлорид натрия, рекомендуется использовать встроенные 0,22 мкм фильтры. Мешок осторожно переворачивают для перемешивания раствора. Раствор T-DM1 для инфузий, разведенный в поливинилхлоридных (PVC) или несодержащих латекс, не содержащих PVC полиолефиновых мешках (РО) с 0,9% или 0,45% раствором хлорида натрия для инъекций USP, можно хранить при 2°-8°С (36°-46°F) в течение короткого периода времени.
Составление лекарственной формы пертузумаба
Пертузумаб можно формулировать в виде композиции для одноразового применения, содержащей пертузумаб в концентрации 30 мг/мл в буфере из 20 мМ L-гистидина (рН 6,0), 120 мМ сахарозы и 0,02% полисорбата 20. Каждый флакон емкостью 20 см3 содержит примерно 420 мг пертузумаба (14,0 мл/флакон). Указанный объем раствор пертузумаба извлекают из флаконов и переносят в мешок для внутривенного введения емкостью 250 см3 с 0,9% раствором хлорида натрия для инъекций. Мешок осторожно переворачивают для перемешивания раствора и визуально осматривают на наличие частиц и обесцвечивания перед введением. Раствор пертузумаба для инфузий, разведенный в полиэтиленовых или несодержащих PVC полиолефиновых мешках с 0,9% раствором хлорида натрия для инъекций, можно хранить при 2°-8°С (36°-46°F) в течение короткого периода времени.
Определение показателей безопасности
Безопасность и переносимость T-DM1 и пертузумаба оценивают по следующим основным показателям безопасности: (1) частота, происхождение и тяжесть побочных эффектов; (2) побочные эффекты или изменения в физическом состоянии и результатах клинических лабораторных анализов во время и после введения испытуемых средств, которые приводят к изменению дозы, снижению дозы или прекращению лечения T-DM1 и пертузумабом и (3) изменения в функции сердца (т.е. фракции выброса левого желудочка [LVEF], аномалий сегментной стенки), включая данные ECHO или MUGA.
Определение фармакокинетических и
фармакодинамических параметров
У всех пациенток, получающих испытуемое лечение, определяют фармакокинетические параметры T-DM1 и пертузумаба, с использованием некомпартментного и/или популяционного методов, когда они соответствуют, в виде следующих параметров: (1) концентрация T-DM1 (конъюгата) в сыворотке крови, трастузумаба в целом (свободного и конъюгированного с DM1); (2) концентрация свободного DM1 в плазме крови; (3) общее воздействие (площадь под кривой концентрация-время [AUC]; (4) максимальная концентрация в сыворотке крови (Cmax); (5) минимальная концентрация (Cmin); (6) клиренс; (7) объем распределения; (8) конечный период полураспада; (9) антитела к T-DM1.
Определение эффективности
В качестве данных оценки эффективности определяют объективные ответы с использованием программы RECIST v. 1.0. В данном исследовании эффективность оценивают по следующим вторичным показателям: (1) PFS, определяемое как время от начала испытуемого лечения до первого проявления прогрессирования заболевания или смерти в период проведения исследования (в течение 30 суток после введения последней дозы при испытуемом лечении) по любой причине по данным отчета оценки опухолей с использованием RECIST v. 1.0 и (2) продолжительность ответа, определяемой в виде первого проявления документированного объективного ответа до времени начала прогрессирования заболевания по данным отчета оценки опухолей с использованием модифицированной RECIST v. 1.0 или смерти в период проведения исследования (в течение 30 суток после введения последней дозы при испытуемом лечении) по любой причине.
Испытуемое лечение
T-DM1 вводят не чаще, чем один раз в 3 недели в дозах 2,4; 3,0 или 3,6 мг/кг внутривенно. Любую пациентку можно перевести на более низкую дозу T-DM1, равную 2,4 мг/кг. В зависимости от проявляемой токсичности, наблюдаемой в группе пациенток, которые начали получать терапию в дозе 3,0 мг/кг, и если подтверждается, что T-DM1 хорошо переносится в дозе 3,0 мг/кг, то в последующих курсах пациенток можно перевести на более высокую дозу 3,6 мг/кг в/в один раз каждые 3 недели. Пертузумаб вводят в нагрузочной дозе 840 мг в/в на 1 сутки, 1 курс, затем 420 мг в/в каждые 3 недели в последующих циклах.
Статистические методы
Конечной точкой определения первичной эффективности в данном исследовании является оцениваемый исследователем объективный ответ, определяемый в виде полного или неполного ответа в двух последовательных обследованиях с интервалом ≥4 недели. Значение частоты проявления основного ответа получают с использованием компьютерной программы, а также соответствующий 95% доверительный интервал. В отношении объективного ответа, то пациенток без достоверной оценки опухоли на исходном уровне, рассматривают в качестве пациенток, не давших ответ на лечение. В отношении продолжительности ответа и PFS, то данные от пациенток, потерявшихся к последующему осмотру, рассматриваются на последнее время, когда имелись сведения об отсутствии прогрессирования заболевания. Данные для пациенток без оценки опухолей после лечения или на момент смерти рассматриваются на время начала лечения плюс 1 сутки.
Пример 5. Клиническое изучение трастузумаба-MCC-DM1
(T-DM1) в комбинации с GDC-0941
Проводят фазу Ib открытого клинического изучения комбинации Т-DM1, который вводят в/в, и GDC-0941, который вводят перорально, пациенткам с HER2-положительным местно-распространенным или метастатическим раком молочной железы, у которых имело место прогрессирование заболевания на предшествующей терапии трастузумабом, для оценки безопасности, переносимости, фармакокинетики и эффективности комбинации. Основными целями данного исследования являются: оценка безопасности и переносимости GDC-0941 при введении в комбинации с Т-DM1; определение MTD GDC-0941 при введении в комбинации с Т-DM1; идентификация рекомендованной в фазе II дозы для GDC-0941 в комбинации с Т-DM1 и оценка любой наблюдаемой противоопухолевой активности GDC-0941 при введении в комбинации с Т-DM1. Целями в отношении фармакокинетики являются: характеристика фармакокинетики GDC-0941 при отсутствии и в присутствии Т-DM1 и характеристика фармакокинетики Т-DM1 в относительном отсутствии и в присутствии GDC-0941.
Составление лекарственной формы GDC-0941
GDC-0941 представляет собой сухой порошок, предназначенный для перорального введения. Приготовленный лекарственный продукт помещают в твердые желатиновые капсулы с двумя силами действия (15 и 50 мг), которые инкапсулируют в оболочки 0 размера и дифференцируют цветом. Наполнителями, включенными в капсулы, являются микрокристаллическая целлюлоза NF/EP, лаурилсульфат натрия NF/DP (только в капсулах с силой действия 50 мг), лимонная кислота безводная USP/EP, натриевая соль кроскармелозы NF/EP, коллоидный диоксид кремния NF/EP и стеарат магния (не бычий) NF/EP. Капсулы с GDC-0941 следует хранить при охлаждении при температуре между 36°F и 46°F (2°С и 8°С). Пациенток следует инструктировать в отношении требуемого режима хранения лекарственного средства при охлаждении при температуре между 36°F и 46°F (2°С и 8°С).
Определение эффективности
Определяют и оценивают параметры безопасности, фармакокинетики, фармакодинамики и эффективности, включая статистические методы, как описано в примере 4.
Испытуемое лечение
Испытуемое лечение проводят 3-недельными курсами. Пациентки, у которых наблюдается клинический эффект в результате испытуемого лечения, могут иметь возможность пройти большее число курсов терапии, которые могут иметь место в отдельном исследовании, в зависимости от стадии разработки препарата, доступности лекарственного средства и других факторов.
На стадии повышения дозировки в исследовании пациентки, принимающие участие в исследовании, получают однократную дозу GDC-0941 на 1 день 1 курса натощак для отбора проб до и после введения препарата для проведения фармакокинетических исследований и наблюдений в отношении вариабельности среди пациенток. Исходная доза GDC-0941 составляет 60 мг один раз в день, которая, как было определено в фазе I клинического изучения, является безопасной при монотерапии препаратом без проявления ограничивающей дозировку токсичности. На 2 день 1 курса вводят полную дозу T-DM1, составляющую 3,6 мг/кг в/в в течение 90 мин без нагрузочной дозы. Затем следует введение дозы GDC-0941. За состоянием пациенток наблюдают в течение 90 мин после первой инфузии T-DM1. Затем GDC-0941 вводят один раз в день в целом в течение 14 суток с последующим интервалом в 1 неделю после первого цикла.
Повышение дозы GDC-0941 у последующих пациенток продолжают до прогрессирования заболевания или проявления непереносимости препарата. Продолжительность последующих курсов испытуемого лечения будет составлять 3 недели при дозе T-DM1, равной 3,6 мг/кг в/в в течение 30 мин на 1 день каждого курса, и после инфузии T-DM1 вводят GDC-0941 и лечение продолжают 2 недели и делают интервал в 1 неделю. Введение продолжают до прогрессирования заболевания или проявления непереносимости препарата. T-DM1 следует вводить в течение 30-90 (±10) мин в/в инфузией в зависимости от того, как переносится T-DM1 в первоначальном исследовании. Если инфузия продолжительностью 90 мин переносится хорошо, то последующие инфузии можно проводить в течение 30 (±10) мин.
Приведенное выше описание следует рассматривать только в качестве иллюстративного в отношении принципов изобретения. Кроме того, поскольку для специалистов в данной области очевидны многочисленные модификации и изменения, то не желательно ограничивать изобретение точным конструированием и способом, описанными в данном документе. Следовательно, все подходящие модификации и эквивалентные варианты входят в объем изобретения, определяемый формулой изобретения, которая следует.
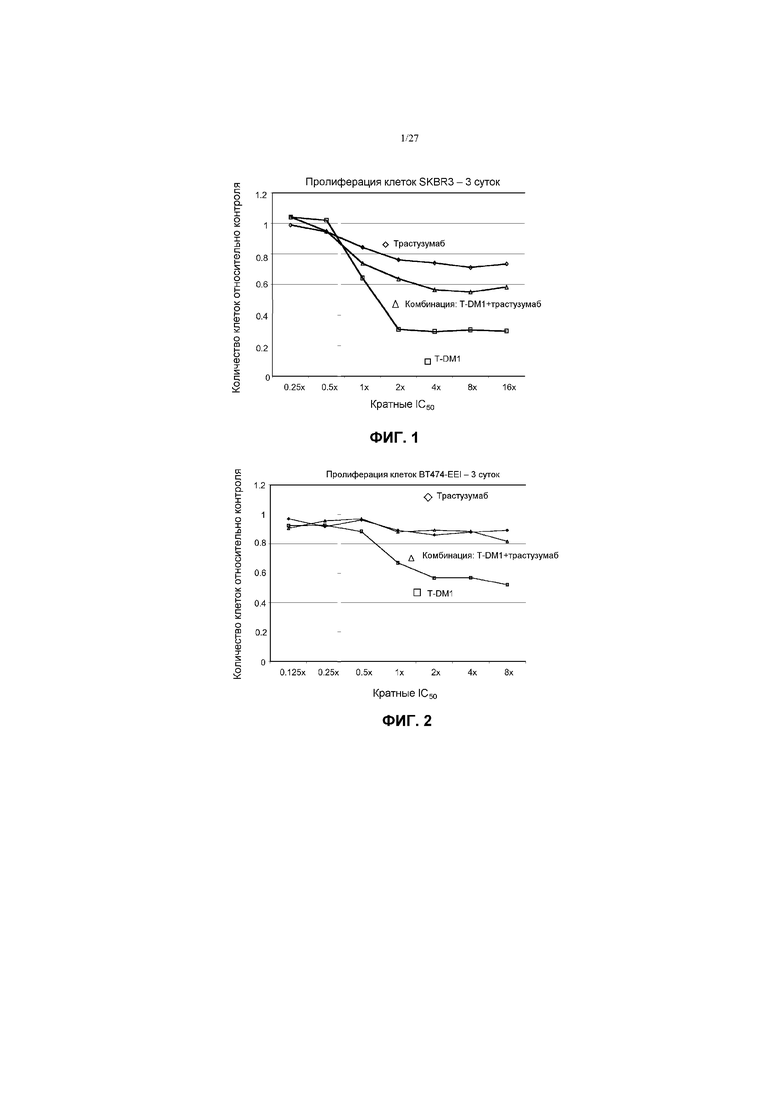
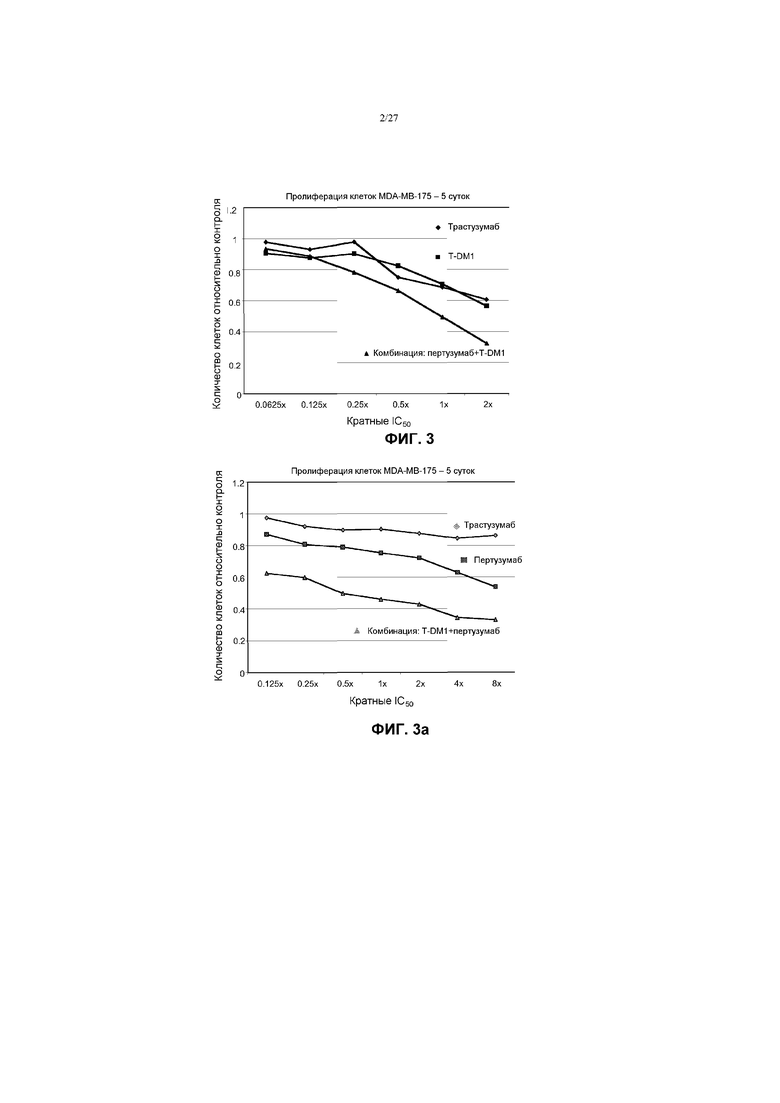
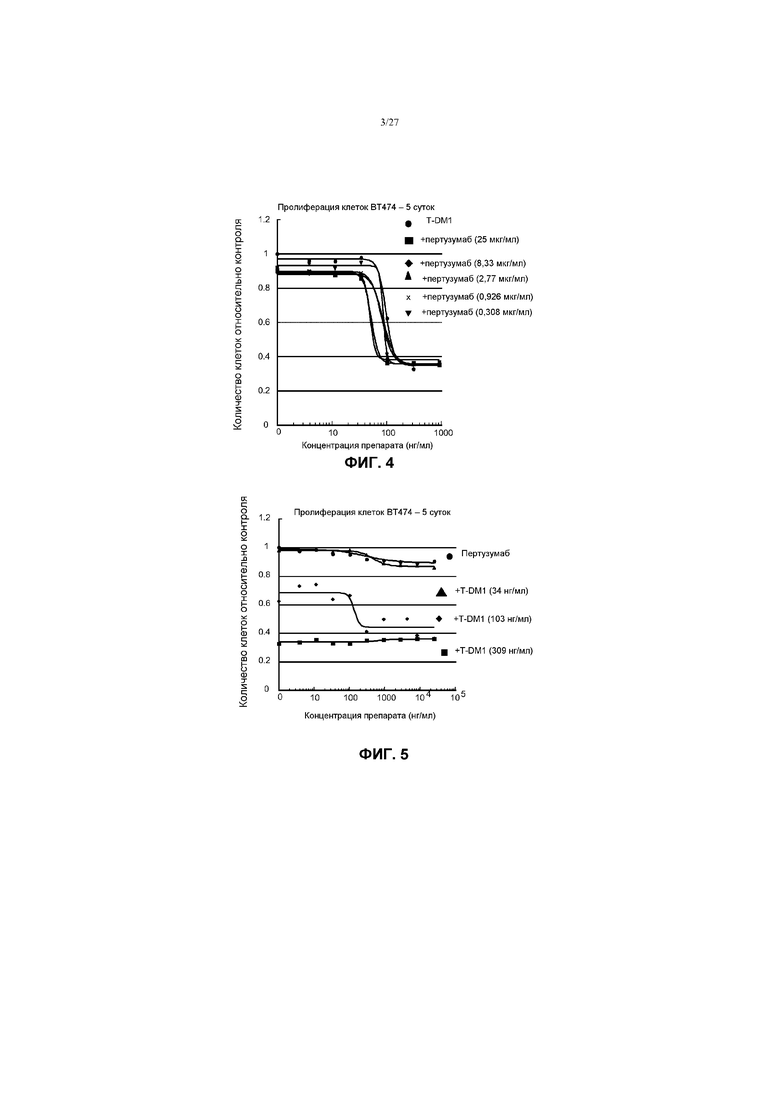
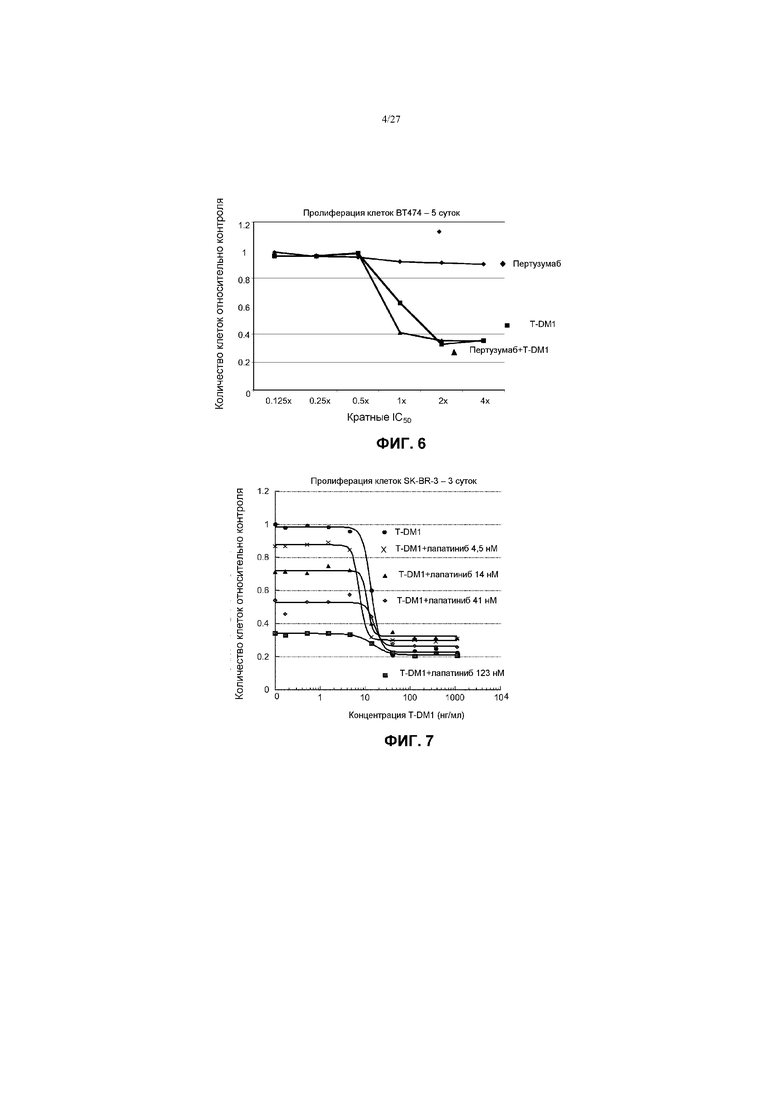
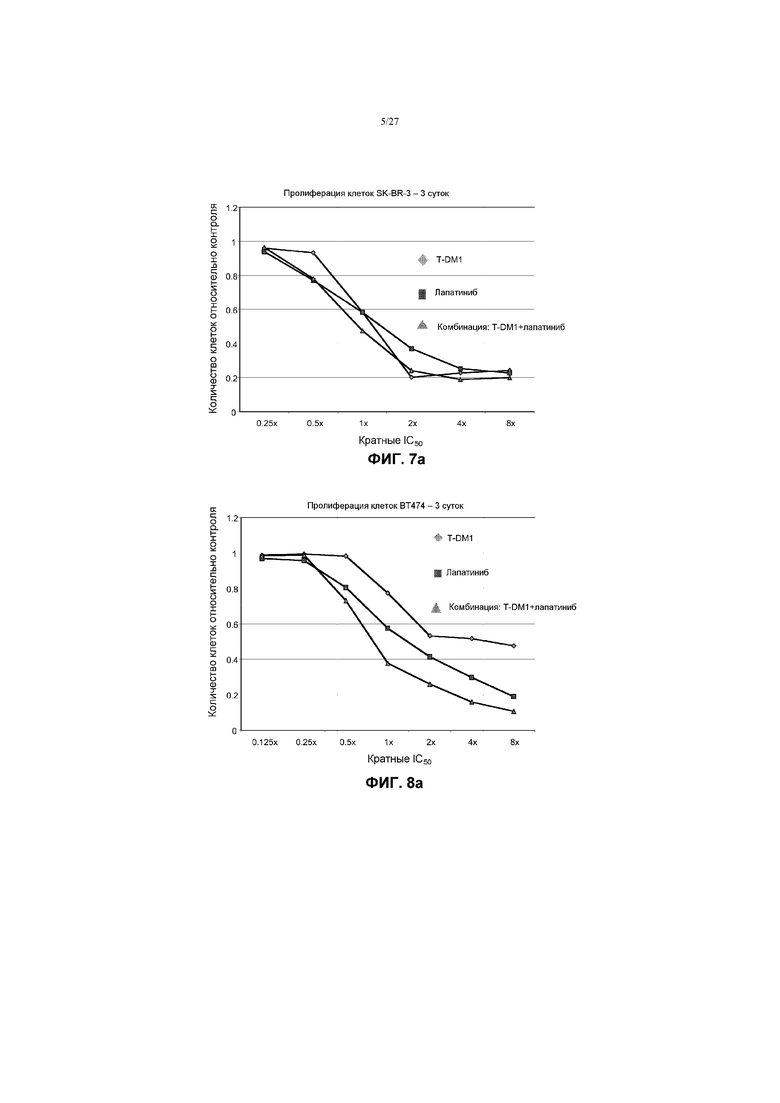
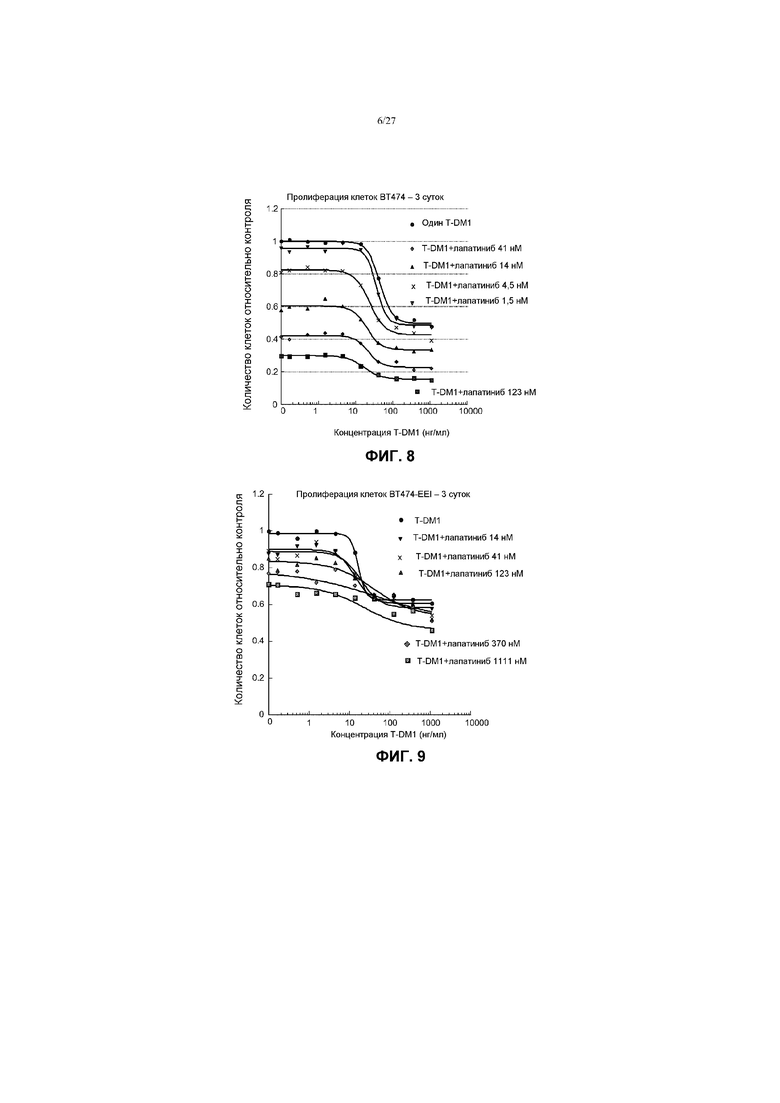
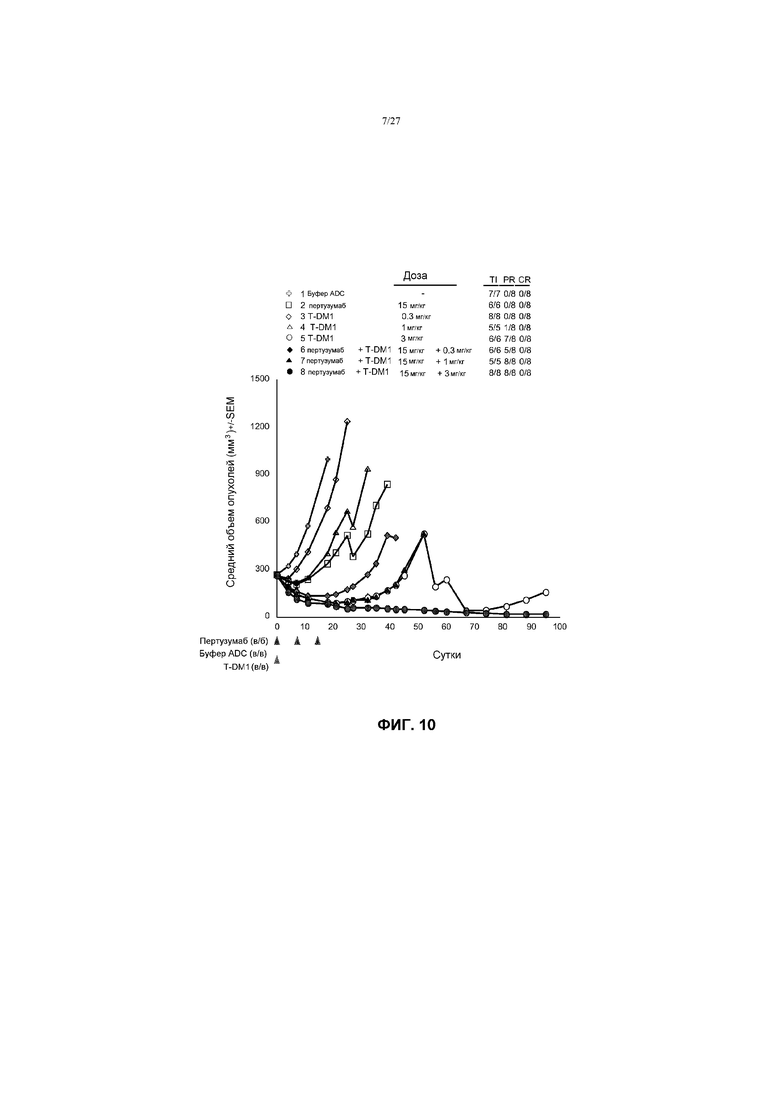
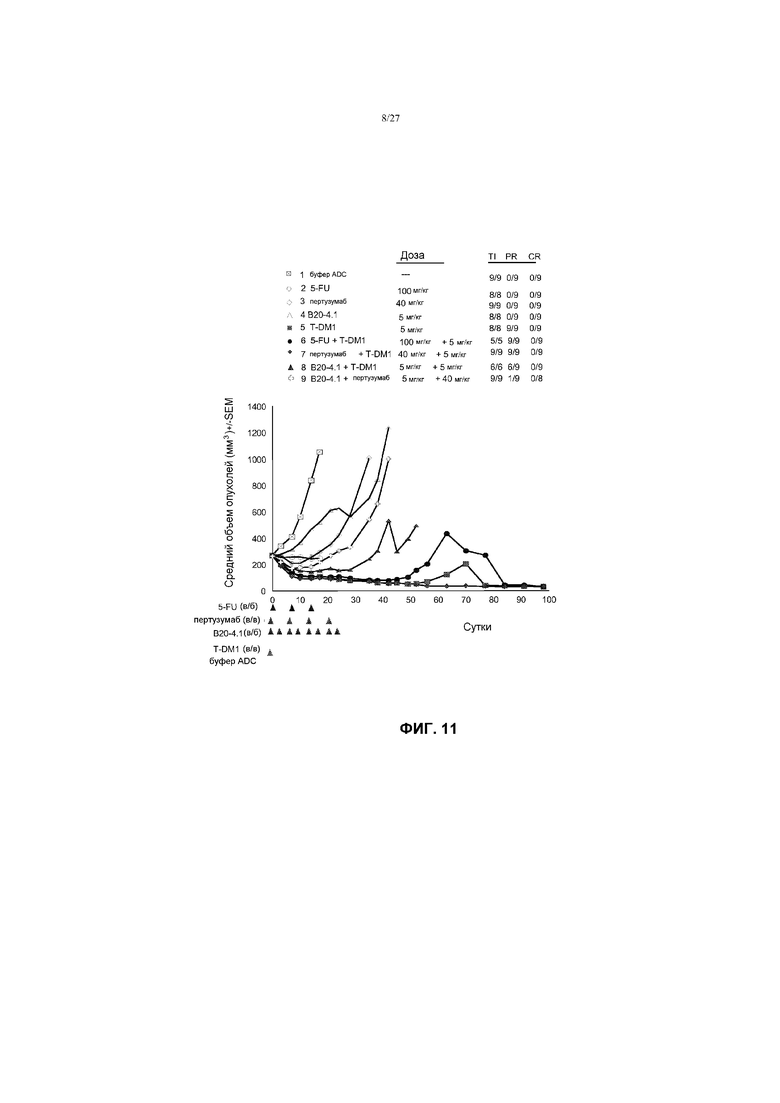
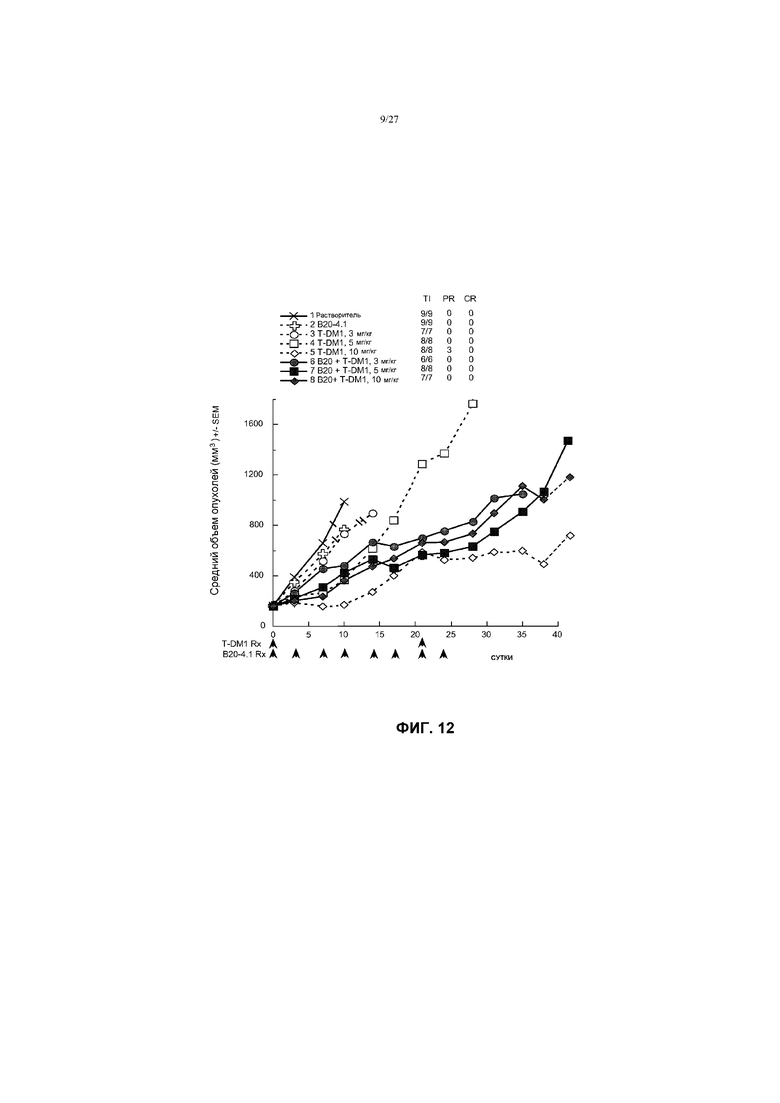
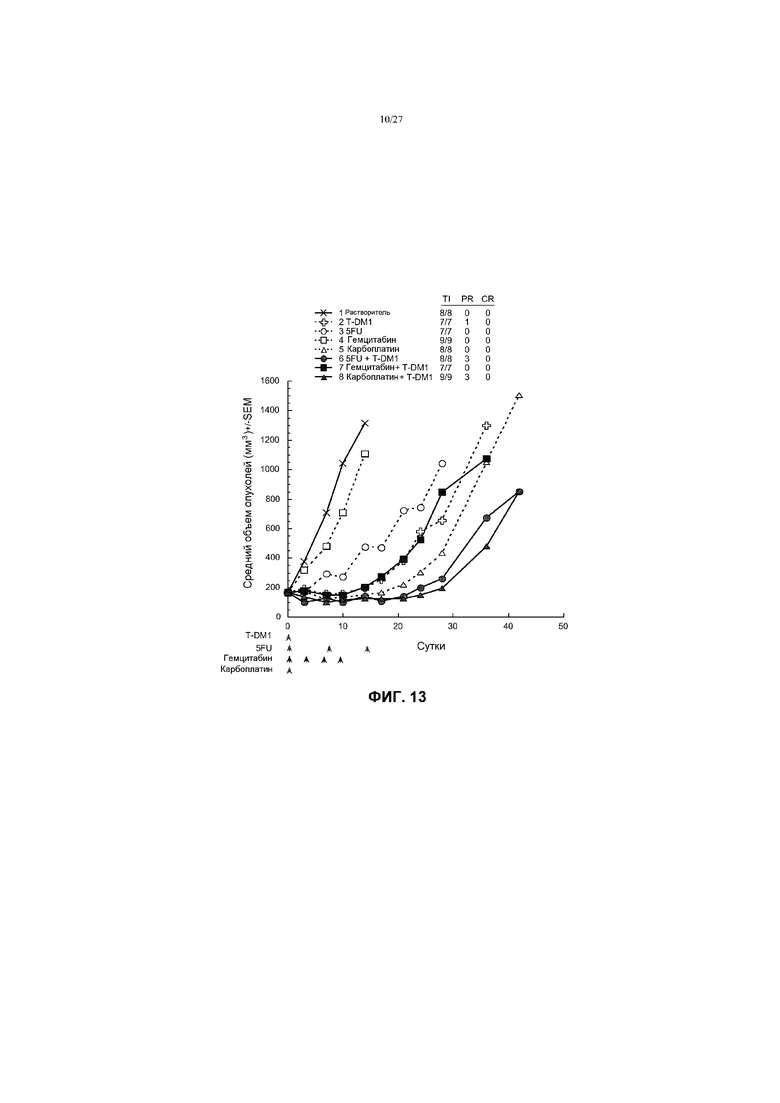

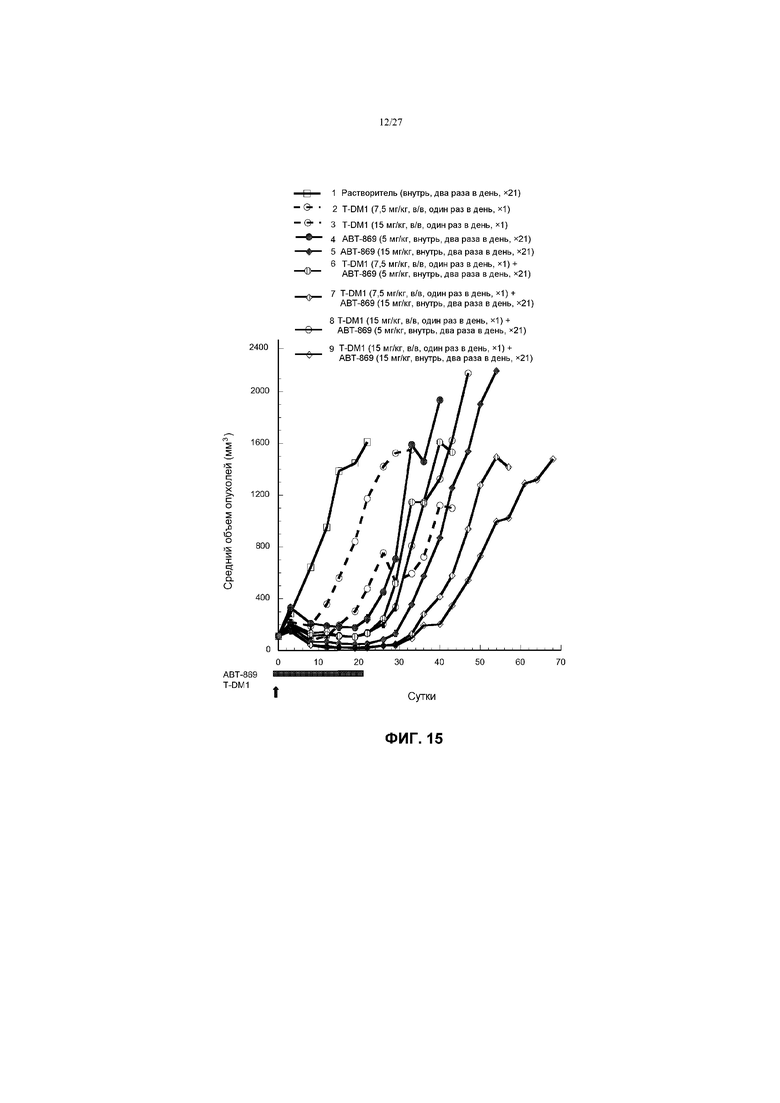

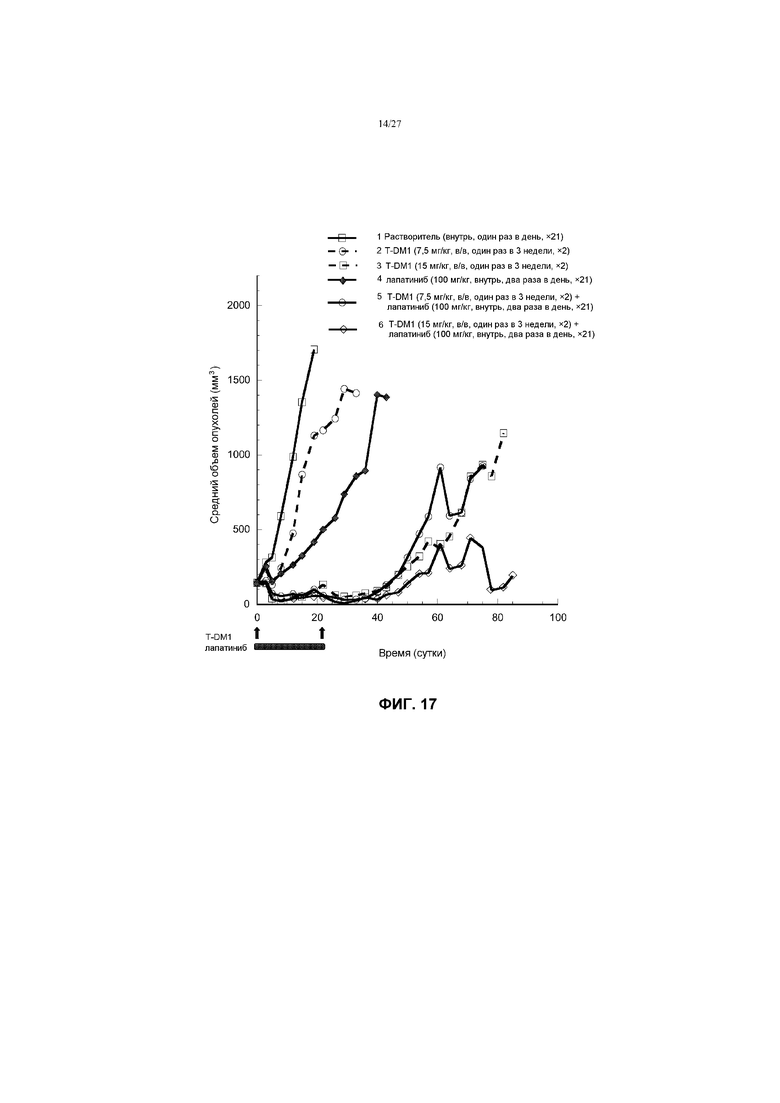
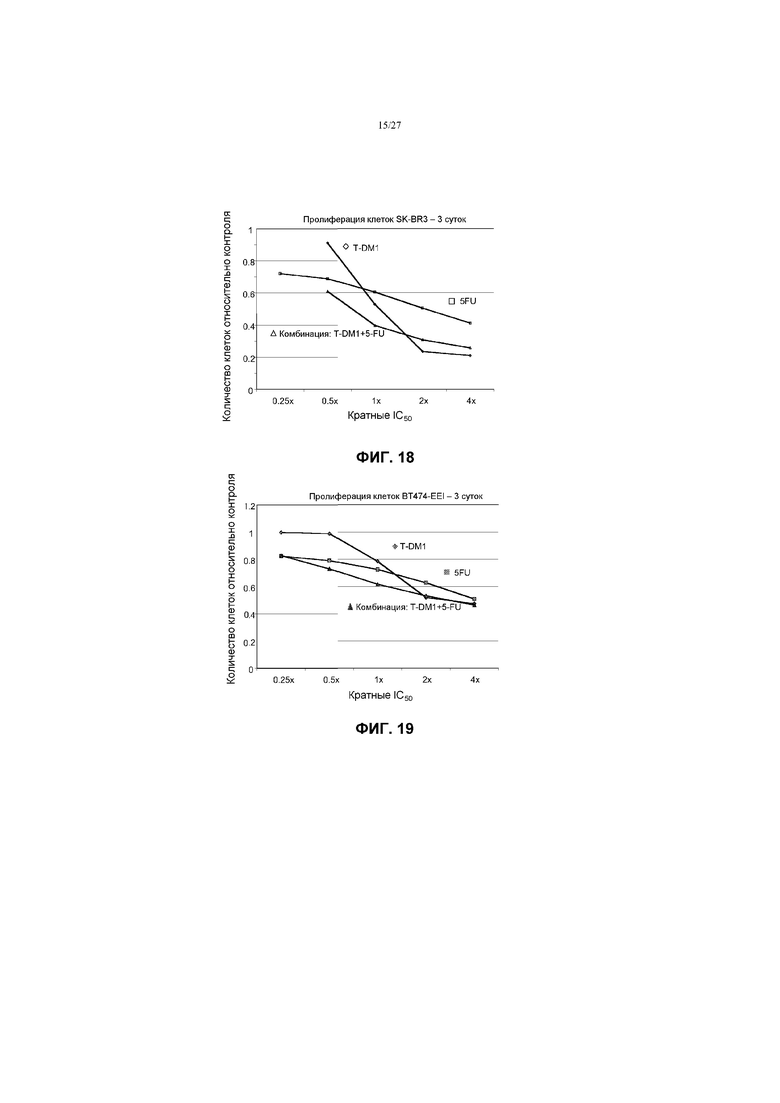
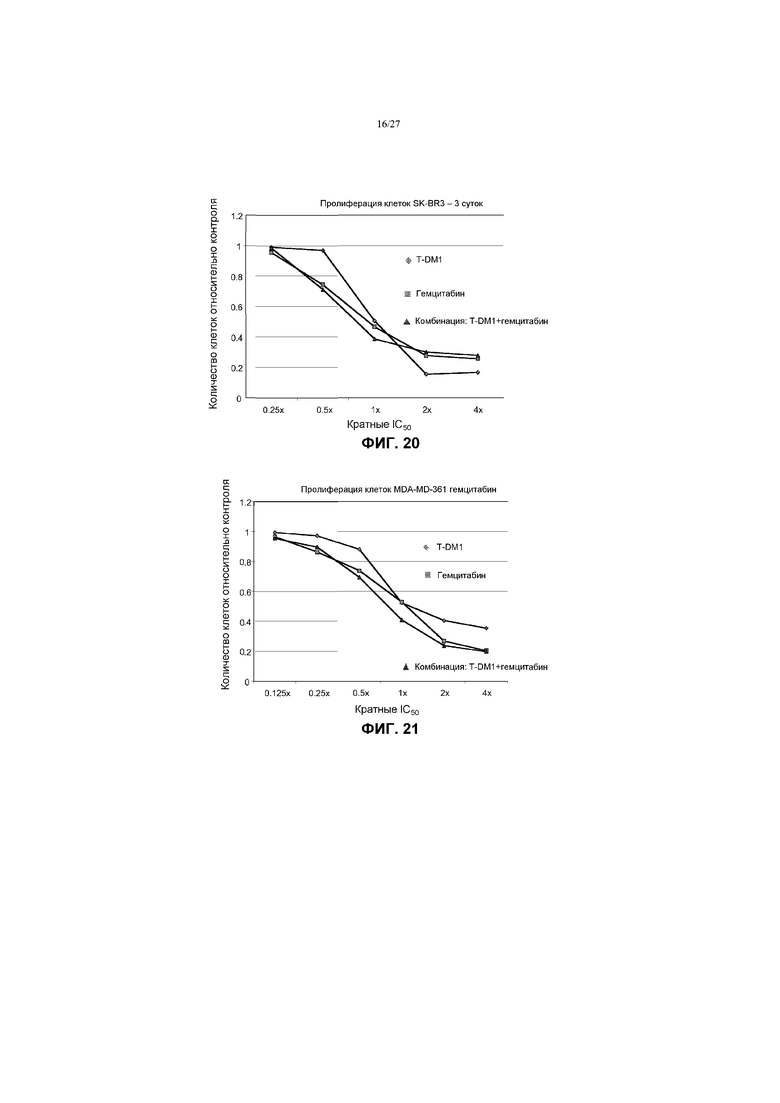
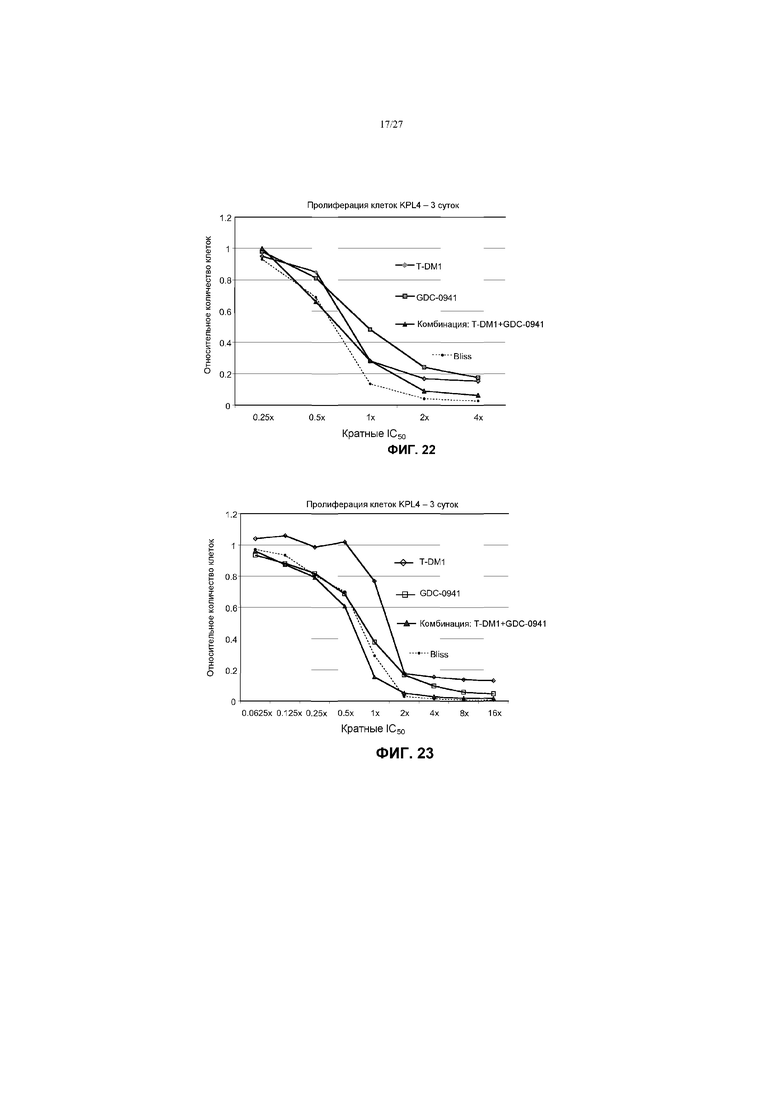
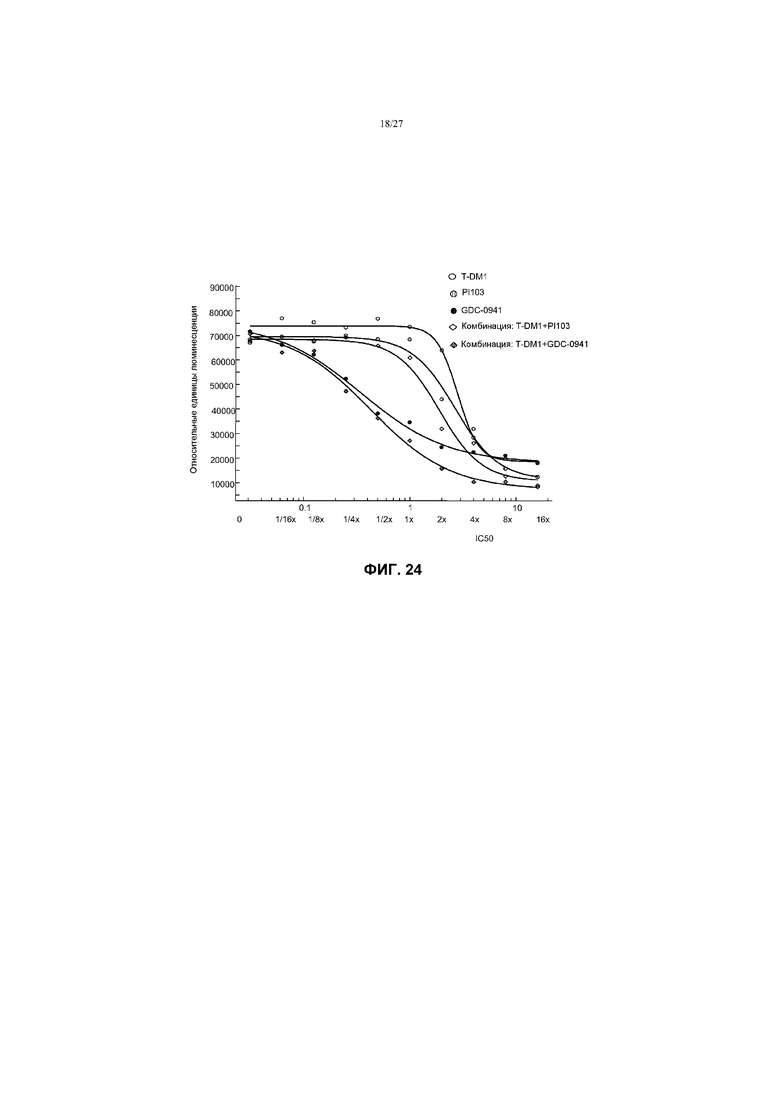
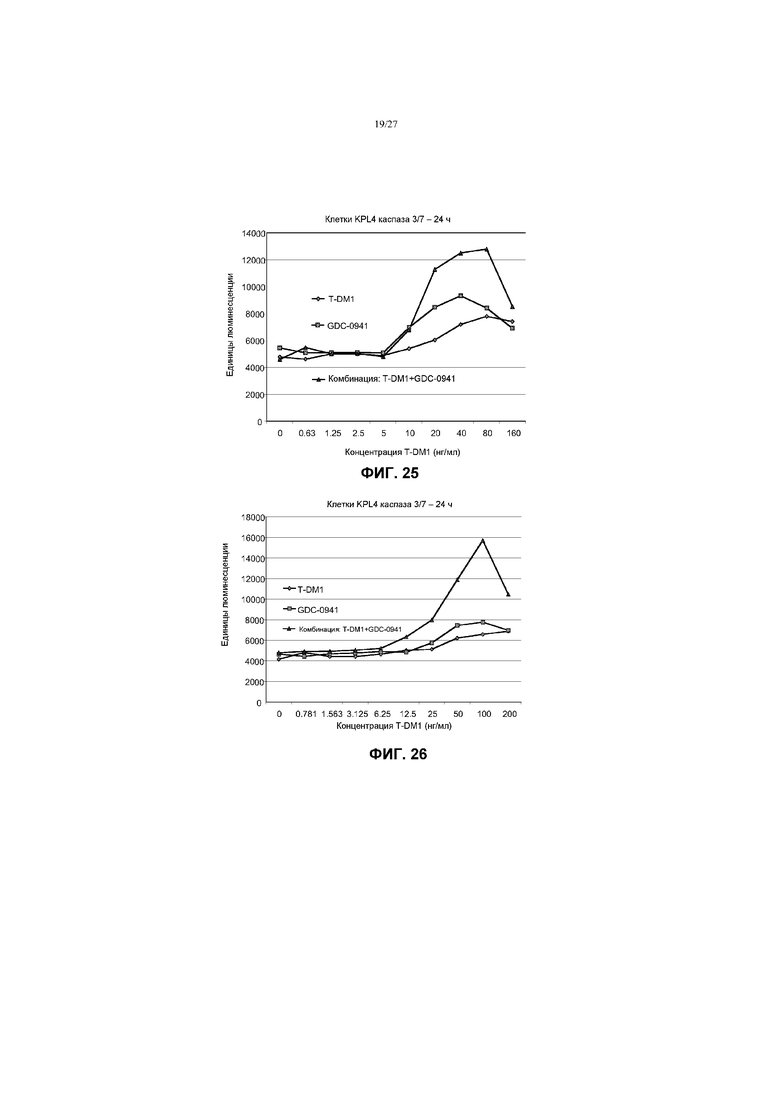
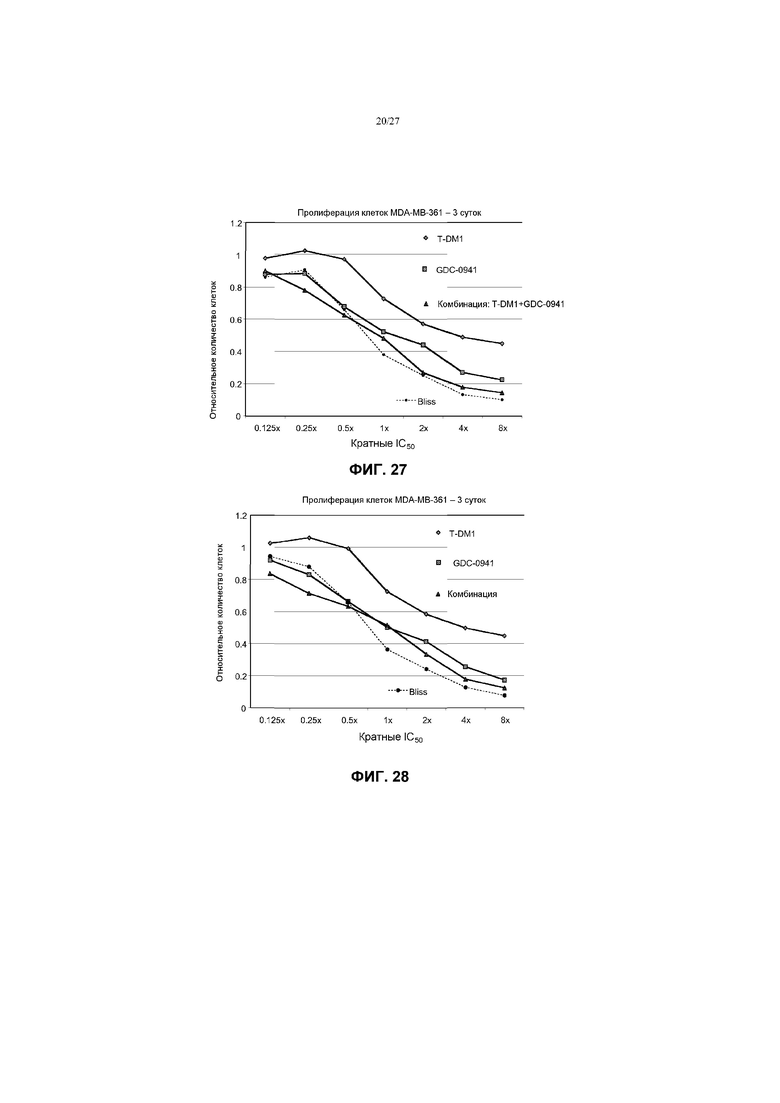
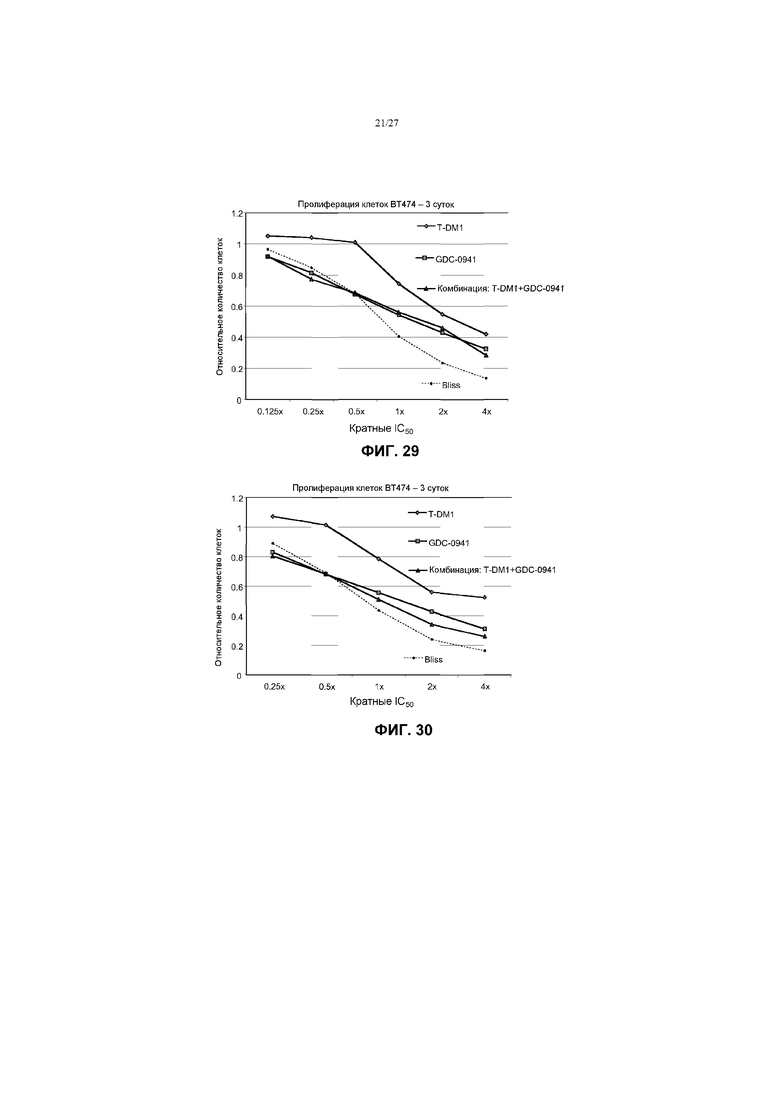
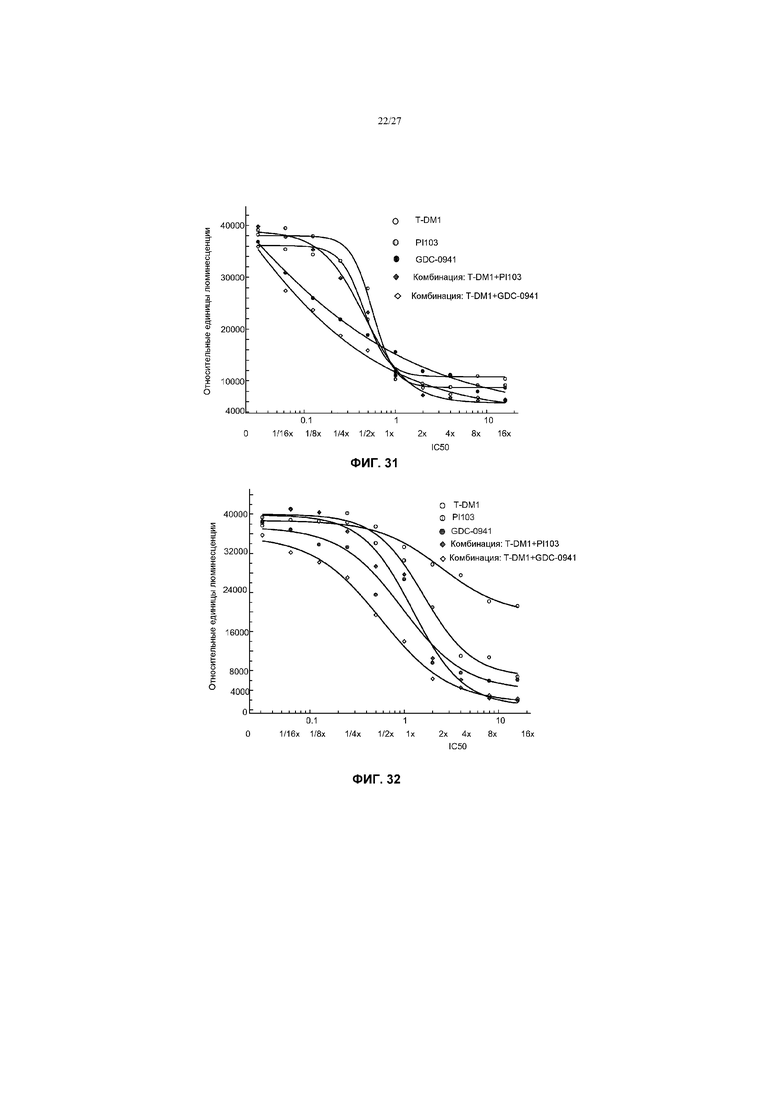
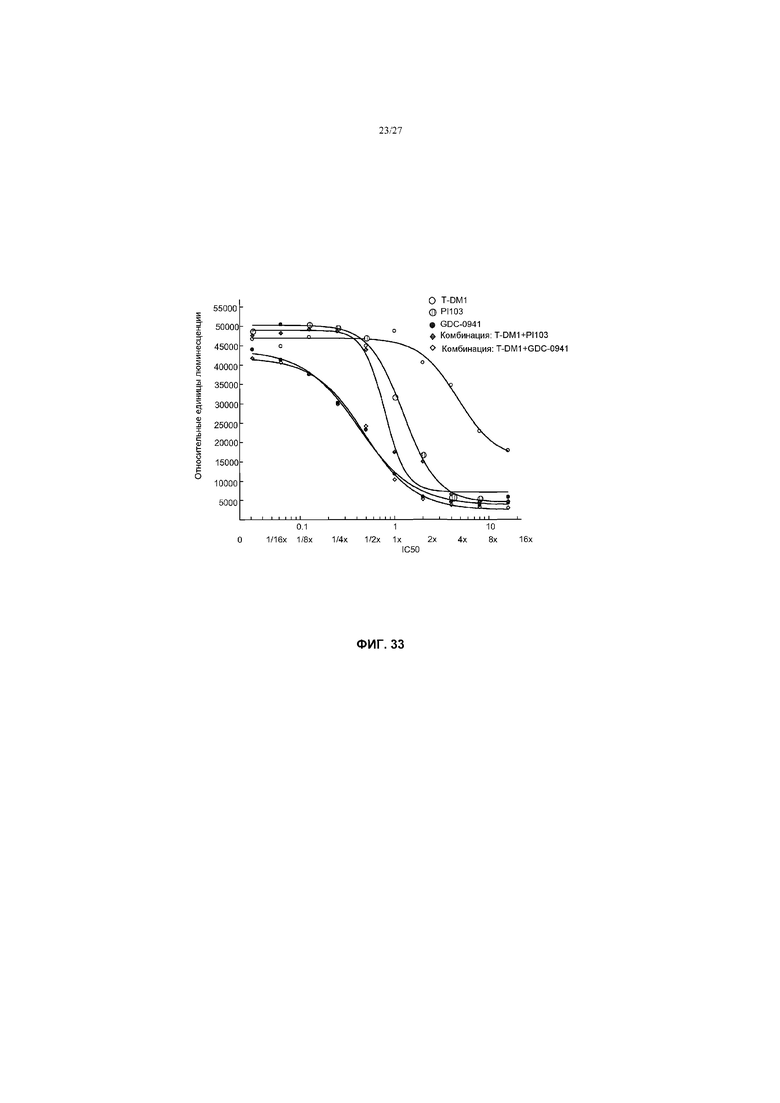
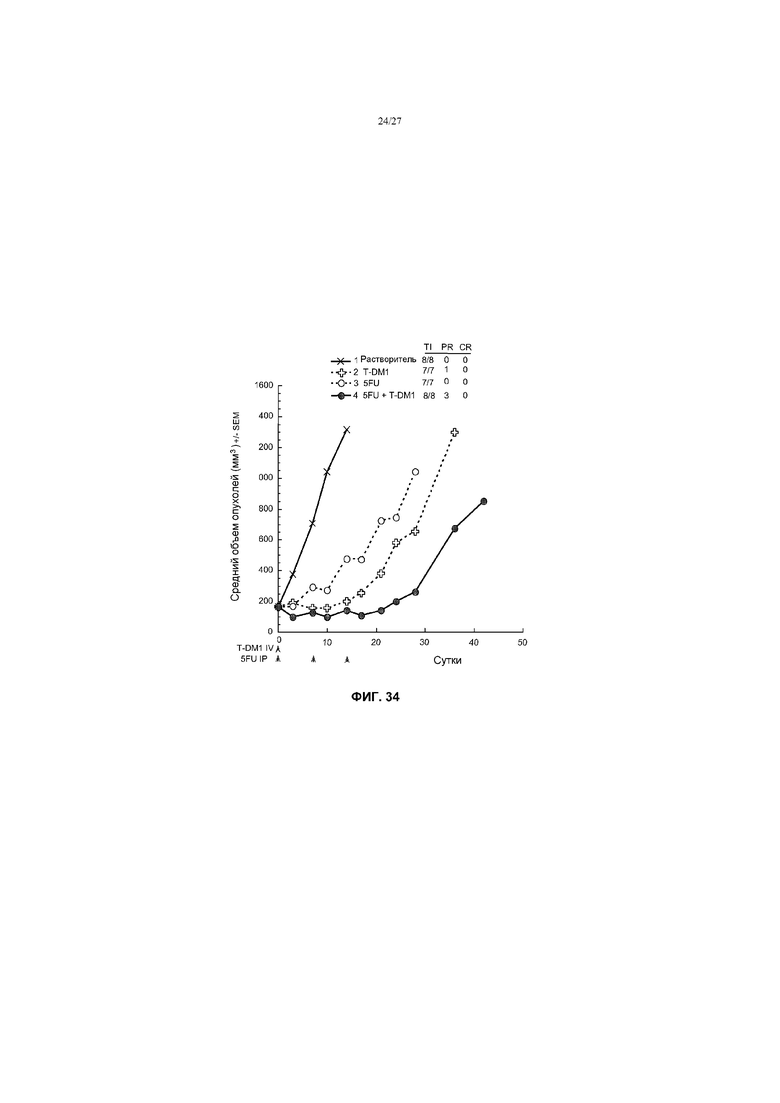

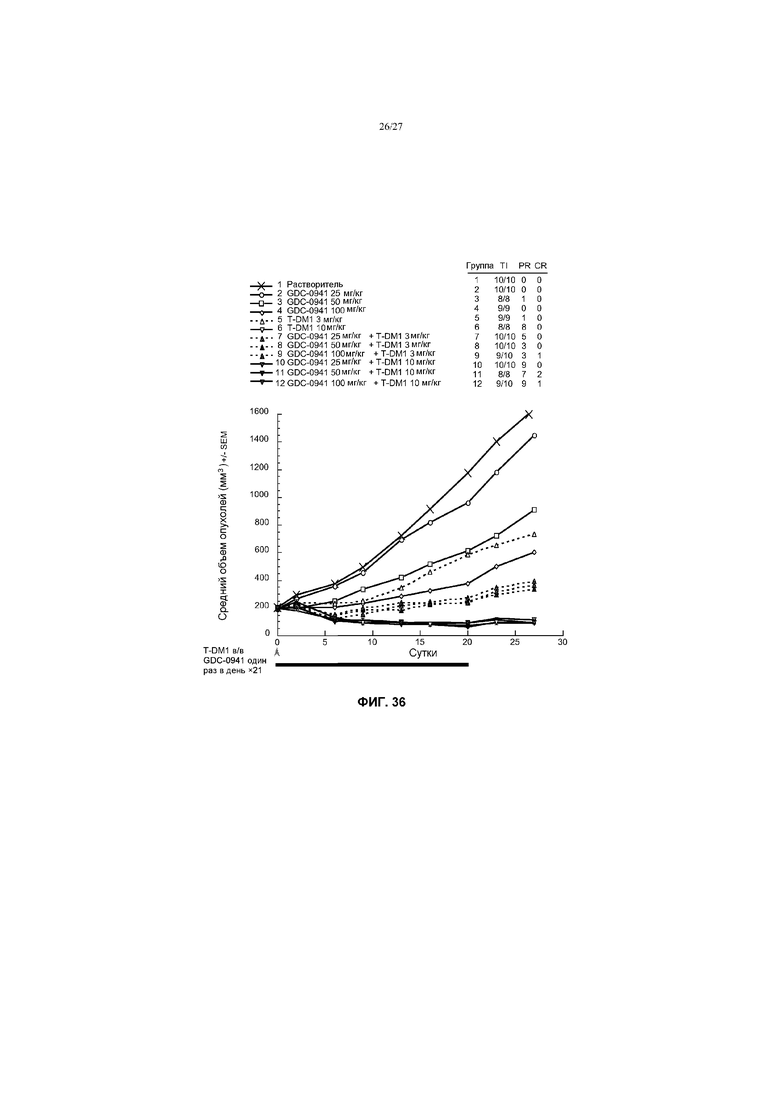

Изобретение относится к способу лечения злокачественной опухоли, экспрессирующей ErbB2, включающий введение терапевтической комбинации в виде комбинированной композиции или поочередно млекопитающему, где терапевтическая комбинация содержит трастузумаб-МСС-DM1 и доцетаксел, где введение терапевтической комбинации приводит к аддитивному эффекту по сравнению с введением трастузумаба-МСС-DM1 или доцетаксела в виде отдельных средств, где количество трастузумаба-МСС-DM1 составляет от 0,3 до 15 мг/кг, и где количество доцетаксела составляет приблизительно 30 мг/кг. 9 з.п. ф-лы, 37 ил., 21 табл., 5 пр.
1. Способ лечения злокачественной опухоли, экспрессирующей ErbB2, включающий введение терапевтической комбинации в виде комбинированной композиции или поочередно млекопитающему, где терапевтическая комбинация содержит трастузумаб-МСС-DM1 и доцетаксел, где введение терапевтической комбинации приводит к аддитивному эффекту по сравнению с введением трастузумаба-МСС-DM1 или доцетаксела в виде отдельных средств,
где количество трастузумаба-МСС-DM1 составляет от 0,3 до 15 мг/кг, и где количество доцетаксела составляет приблизительно 30 мг/кг.
2. Способ по п. 1, где трастузумаб-МСС-DM1 и доцетаксел вводят в виде комбинированной композиции.
3. Способ по п. 1, где трастузумаб-МСС-DM1 и доцетаксел вводят поочередно.
4. Способ по п. 3, где млекопитающему вводят доцетаксел и затем впоследствии вводят трастузумаб-MCC-DM1.
5. Способ по любому из пп. 1-4, где терапевтическую комбинацию вводят человеку, страдающему злокачественной опухолью, экспрессирующей ErbB2, с интервалами приблизительно три недели.
6. Способ по п. 3, где трастузумаб-MCC-DM1 вводят человеку, страдающему злокачественной опухолью, экспрессирующей ErbB2, с интервалами от приблизительно одной недели до трех недель.
7. Способ по п. 3, где трастузумаб-MCC-DM1 вводят не чаще, чем каждые 3 недели при дозировке 2,4, 3,0 или 3,6 мг/кг внутривенно.
8. Способ по любому из пп. 1-7, где количество трастузумаба-MCC-DM1 и количество доцетаксела составляет для каждого от приблизительно 1 мг до приблизительно 1000 мг, и количество трастузумаба-MCC-DM1 и количество доцетаксела находятся в соотношении от приблизительно 1:10 до приблизительно 10:1 по массе.
9. Способ по п. 1, где млекопитающим является пациент с положительными реакциями на HER2.
10. Способ по п. 9, где пациент с положительными реакциями на HER2 получал терапию трастузумабом или лапатинибом.
| US 20050276812 A1, 15.12.2005 | |||
| US 7097840 B2, 29.08.2006. |

