Область техники
Настоящее изобретение относится к новым ингибиторам рецептора эпидермального роста фактора (EGFR), а также к их фармацевтически приемлемым солям, сольватам или стереоизомерам, фармацевтическим композициям, содержащим данные соединения, к способам лечения и применению данных соединений в качестве фармацевтических препаратов для лечения заболеваний или нарушений.
Уровень техники
Рецептор эпидермального фактора роста (EGFR) представляет собой трансмембранный гликопротеин - тирозинкиназу, являющуюся членом семейства erbB рецептора. EGFR состоит из гликозилированного наружного лиганд-связывающего домена (621 остаток) и цитоплазматического домена (542 остатка), связанных коротким трансмембранным линкером из 23 аминокислот. Внеклеточная часть EGFR содержит 25 дисульфидных мостиков, 12 N-связанных сайтов гликозилирования и обычно считается состоящей из четырех субдоменов. Рентгеновские кристаллические структуры EGFR позволяют полагать, что рецептор может принимать как аутоингибированную связанную конформацию, которая неспособна связывать эпидермальный фактор роста (EGF) (Ferguson et al., Mol Cell, 2003, vol 11: 507-517), так и активную конформацию, которая может опосредовать связывание лиганда EGF и димеризацию рецептора (Garrett et al., Cell 2002, vol 110:763-773; Ogiso et al., Cell, 2002, vol 110: 775-787). При связывании лиганда фактора роста, такого как EGF, рецептор может гомодимеризоваться с другой молекулой EGFR или гетеродимеризоваться с другим членом семейства, таким как erbB2 (HER2), erbB3 (HER3) или erbB4 (HER4). Гомо- и/или гетеро-димеризация erbB рецепторов приводит к фосфорилированию ключевых тирозиновых остатков во внутриклеточном домене и ведет к стимуляции огромного количества внутриклеточных путей передачи сигнала, вовлеченных в клеточную пролиферацию и выживание. Подробное описание передачи сигнала erbB рецептора и его вовлеченности в онкогенез представлено в статьях Ciardiello F.N. Engl J Med 2008; 358:1160-1174 и Robert Roskoski Jr. Biochemical and Biophysical Research Communications 319 (2004) 1-11.
Связь EGFR с онкологическими заболеваниями впервые была признана, когда трансформирующий онкоген v-ErbB вируса птичьего эритроблатоза был признан мутантным гомологом EGFR человека (Downward J. Nature. 1984; 307:521-527). Было обнаружено, что онкоген v-ErbB содержит рекомбинации трансмембранных и цитоплазматических доменов EGFR (Olofsson В. Eur. J. Biochem. 1986; 160:261-266), что приводит к онкогенным аберрациям EGFR. В дополнение к мутациям была определена повышенная экспрессия EGFR, способствующая прогрессированию ряда злокачественных опухолей (Gusterson В. Cell Biol. Int. Rep. 1984; 8:649-658), в том числе сарком (Gusterson В. Int. J. Cancer. 1985; 36:689-693), немелкоклеточного рака легкого (НМРЛ) (Veale D. Br. J. Cancer. 1987; 55:513-516) и злокачественных глиом (Wong A.J. Proc. Natl. Acad. Sci. USA. 1987; 84:6899-6903).
В настоящее время известно, что EGFR регулирует многочисленные процессы в клетке посредством путей сигнальной трансдукции, опосредуемых тирозинкиназой, включая, но не ограничиваясь контролем клеточной пролиферации, дифференцировки, выживаемости, апоптоза, опухолевого ангиогенеза, митогенеза и метастазирования (Atalay et al., Ann. Oncology 14: 1346-1363 [2003]; Herbst R.S. Cancer. 2002; 94: 1593-1611; Modjtahedi et al., Br. J. Cancer. 1996; 73: 228-235). Гиперэкспрессия EGFR подтверждена при многочисленных злокачественных опухолях у человека, включая рак мочевого пузыря, головного мозга, головы и шеи, поджелудочной железы, легкого, молочной железы, яичников, толстой кишки, простаты и почек (Atalay et al., Ann. Oncology 14: 1346-1363 [2003]; Herbst R.S. Cancer. 2002; 94: 1593-1611; Modjtahedi et al., Br. J. Cancer. 1996; 73: 228-235). EGFR экспрессируется также в клетках нормальных тканей, особенного эпителиальных тканей кожи, печени и желудочно-кишечного тракта, хотя обычно на более низких уровнях, чем в злокачественных клетках (Herbst R.S. Cancer. 2002; 94: 1593-1611).
Известно, что низкомолекулярные ингибиторы тирозинкиназы EGFR применяются для лечения онкологических заболеваний, например, для лечения немелкоклеточного рака легкого, рака поджелудочной железы, антитела к EGFR применяются при лечении колоректального рака и рака головы и шеи (Ping Wee. Cancers (Basel). 2017 May; 9(5): 52).
При множестве онкологических заболеваний наблюдаются частые мутации и гиперэкспрессия EGFR, поэтому сохраняется актуальность разработки новых эффективных и безопасных препаратов, направленных на ингибирование активности EGFR.
Описание изобретения
Ниже приведены определения терминов, которые использованы в описании этого изобретения.
Необязательно замещенный в одном, двух, трех или нескольких положениях обозначает, что описанная группа может быть замещена в одном, двух, трех или от одного до шести положениях любым одним или любой комбинацией радикалов.
«Алкил» означает алифатическую углеводородную линейную или разветвленную группу с 1-12 атомами углерода в цепи, более предпочтительно с 1-6 атомами углерода в цепи. Разветвленная означает, что алкильная цепь имеет один или несколько «низших алкильных» заместителей. Примеры алкильных групп включают, но не ограничиваются ими, метил, этил, н-пропил, изо-пропил, н-бутил, изо-бутил, втор-бутил, трет-бутил, н-пентил, 2-пентил, 3-пентил, нео-пентил, н-гексил. Алкил может иметь заместители, которые могут быть одинаковыми или разными.
«Циклоалкил» означает полностью насыщенное карбоциклическое кольцо, содержащее 3-10 атомов углерода в цикле. Примеры циклоалкильных групп включают, но не ограничиваются ими, моноциклические группы, такие как циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклононил или циклодецил, бициклические группы, такие как бициклогептил или бициклооктил. Циклоалкил может иметь заместители, которые могут быть одинаковыми или разными.
«Алкенил» означает углеводородную линейную или разветвленную группу с 2-12 атомами углерода в цепи, более предпочтительно с 2-6 атомами углерода в цепи, которая содержит одну или несколько двойных связей углерод-углерод. Алкенил может иметь заместители, которые могут быть одинаковыми или разными.
«Алкинил» означает углеводородную линейную или разветвленную группу с 2-12 атомами углерода в цепи, более предпочтительно с 2-6 атомами углерода в цепи, которая содержит одну или несколько тройных связей углерод-углерод. Алкинил может иметь заместители, которые могут быть одинаковыми или разными.
«Арил» означает ароматическую моноциклическую или полициклическую систему, включающую от 6 до 14 атомов углерода, преимущественно от 6 до 10 атомов углерода. Примеры арильных групп включают, но не ограничиваются ими, фенил, фенилен, бензолтриил, инданил, нафтил, нафтилен, нафталентриил и антрилен. Арил может иметь заместители циклической системы, которые могут быть одинаковыми или разными. Арил может быть аннелирован с неароматической циклической системой или гетероциклом.
«Алкилокси», «Алкокси» или «алкилокси-группа» означает алкил-О-группу, в которой алкил определен в данном разделе. Примеры алкокси групп включают, но не ограничиваются ими, метокси, этокси, н-пропокси, изо-пропокси, н-бутокси, трет-бутокси, изо-бутокси.
«Арилокси» или «арилокси-группа» означает арил-О- группу, в которой арил определен в данном разделе. Примером арилокси-группой является, но не ограничивается, фенокси группа.
«Циклоалкилокси» или «циклоалкилокси-группа» означает циклоаклкил-О- группу, в которой циклоалкил определен в данном разделе. Примеры циклоалкилокси групп включают, но не ограничиваются ими, циклогексилокси, циклопентилокси, циклобутилокси или циклопропилокси.
«Аминогруппа» означает R'R''N- группу.
«Аминокарбонил» означает -C(=O)NR'R'' группу.
Примеры R' и R'' включают, но не ограничиваются ими, заместители, выбранные из группы, содержащей водород, алкил, алкенил, алкинил, циклоалкил, арил, гетероциклил, гетероарил, или R' и R'' совместно с атомом азота, к которому они присоединены, могут образовывать 4-7-членный гетероциклил или гетероарил.
«Низший алкил» означает линейный или разветвленный алкил с 1-4 атомами углерода.
«Гало» или «Галоген» (Hal) означает фтор, хлор, бром или йод.
«Гетероцикл», «гетероциклил», «гетероциклическое кольцо» означает моноциклическую или полициклическую систему, включающую от 3 до 11 атомов углерода, в которой один или несколько атомов углерода заменены на гетероатом, такой как азот, кислород, сера. Гетероцикл может быть конденсирован с арилом или гетероарилом. Гетероцикл может иметь один или несколько заместителей, которые могут быть одинаковыми или разными. Атомы азота и серы, находящиеся в гетероцикле могут быть окислены до N-оксида, S-оксида или S-диоксида. Гетероцикл может быть насыщенным, частично ненасыщенным или ненасыщенным. Примеры гетероциклов включают, но не ограничиваются ими, азетидин, пирролидин, пиперидин, 2,8-диазаспиро[4.5]декан, пиперазин, морфолин и др.
«Гетероарил» означает ароматическую моноциклическую или полициклическую систему, включающую от 5 до 11 атомов углерода, предпочтительно от 5 до 10, в которой один или несколько атомов углерода замещены на гетероатом, такой как азот, сера или кислород. Атом азота, находящийся в гетероариле, может быть окислен до N-оксида. Гетероарил может иметь один или несколько заместителей, которые могут быть одинаковыми или разными. Представителями гетероарилов являются пирролил, фуранил, тиенил, пиридил, пиразинил, пиримидинил, пиридазинил, изооксазолил, изотиазолил, тетразолил, оксазолил, тиазолил, пиразолил, фуразанил, триазолил, 1,2,4-тиадиазолил, хиноксалинил, фталазинил, имидазо[1,2-а]пиридинил, имидазо[2,1-b]тиазолил, бензофуразанил, индолил, азаиндолил, бензимидазолил, бензотиазенил, хинолинил, имидазолил, пиразолил, тиенопиридил, хиназолинил, нафтиридинил, тиенопиримидинил, пирролопиридинил, имидазопиридил, изохинолинил, бензоазаиндолил, 1,2,4-триазинил, тиенопирролил, фуропирролил и др.
«Частично ненасыщенный» означает кольцевую систему, которая включает по меньшей мере одну двойную или тройную связь. Термин «частично ненасыщенный» относится к кольцам, имеющим множество сайтов для насыщения, но не включает арильные и гетероарильные системы, как они определены выше.
Термин «оксо», используемый в настоящем документе, относится к радикалу = O.
«Заместитель» означает химический радикал, который присоединяется к молекулярному остову (скэффолду, фрагменту).
«Сольват» означает молекулярный комплекс соединения по настоящему изобретению, включая его фармацевтически приемлемые соли, с одной или более молекулами растворителя. Такие молекулы растворителя представляют собой молекулы, обычно используемые в фармацевтике, которые известны как безвредные для реципиента, например, воду, этанол, этиленгликоль и подобные. Другие растворители можно использовать как промежуточные сольваты в получении более желательных сольватов, такие как метанол, метил-трет-бутиловый эфир, этилацетат, метилацетат, (S)-пропиленгликоль, (R)-пропиленгликоль, 1,4-бутандиол и подобные.
Термин «гидрат» относится к комплексу, в котором молекула растворителя представляет собой воду.
Сольваты и/или гидраты предпочтительно существуют в кристаллической форме.
Термин «связь», «химическая связь» или «одинарная связь» относится к химической связи между двумя атомами или двумя группировками (группами, фрагментами), если два атома, соединенные связью, рассматриваются как часть более крупной субструктуры.
Термин "стереоизомеры" относится к соединениям, которые имеют идентичный химический состав и одинаковое строение, но отличаются пространственным расположением атомов или групп. Стереоизомеры могут включать в себя геометрические изомеры, энантиомеры, диастереомеры.
Термин "защитная группа" относится к группам, которые применяются для блокирования реакционной способности функциональных групп, таких как аминогруппа, карбоксильная группа или гидроксигруппа. Примерами, без ограничения, защитных групп являются трет-бутоксикарбонил (Boc), бензилоксикарбонил (Cbz), 2-(триметилсилил)этокси) метилацеталь (SEM), триалкилсилил, алкил(диарил)силил или алкил.
Термин «эксципиент» используется в данном документе для описания любого ингредиента, отличающегося от соединения(-ий) по данному изобретению.
«Фармацевтическая композиция» обозначает композицию, включающую в себя соединение согласно изобретению и, по крайней мере, один эксципиент. Эксципиент может быть выбран из группы, состоящей из фармацевтически приемлемых и фармакологически совместимых наполнителей, растворителей, разбавителей, носителей, вспомогательных, распределяющих и воспринимающих средств, средств доставки, таких как консерванты, стабилизаторы, наполнители, измельчители, увлажнители, эмульгаторы, суспендирующие агенты, загустители, подсластители, отдушки, ароматизаторы, антибактериальные агенты, фунгициды, лубриканты, регуляторы пролонгированной доставки, выбор и соотношение которых зависит от природы и способа назначения и дозировки. Примерами суспендирующих агентов являются этоксилированный изостеариловый спирт, полиоксиэтилен, сорбитол и сорбитовый эфир, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, а также смеси этих веществ. Защита от действия микроорганизмов может быть обеспечена с помощью разнообразных антибактериальных и противогрибковых агентов, например, таких как парабены, хлорбутанол, сорбиновая кислота и подобные им соединения. Композиция может включать также изотонические агенты, например, сахара, хлористый натрий и им подобные. Пролонгированное действие композиции может быть обеспечено с помощью агентов, замедляющих абсорбцию активного начала, например, моностеарат алюминия и желатин. Примерами подходящих носителей, растворителей, разбавителей и средств доставки являются вода, этанол, полиспирты, а также их смеси, растительные масла (такие, как оливковое масло) и инъекционные органические сложные эфиры (такие, как этилолеат). Примерами наполнителей являются лактоза, молочный сахар, цитрат натрия, карбонат кальция, фосфат кальция и им подобные. Примерами измельчителей и распределяющих средств являются крахмал, альгиновая кислота и ее соли, силикаты и им подобные. Примерами лубрикантов являются стеарат магния, лаурилсульфат натрия, тальк, а также полиэтиленгликоль с высоким молекулярным весом. Фармацевтическая композиция для перорального, сублингвального, трансдермального, внутримышечного, внутривенного, подкожного, местного или ректального введения активного начала, одного или в комбинации с другим активным началом, может быть введена животным и людям в стандартной форме введения в виде смеси с традиционными фармацевтическими носителями.
Пригодные стандартные формы введения включают пероральные формы, такие как таблетки, желатиновые капсулы, пилюли, порошки, гранулы, жевательные резинки и пероральные растворы или суспензии, сублингвальные и трансбуккальные формы введения, аэрозоли, имплантаты, местные, трансдермальные, подкожные, внутримышечные, внутривенные, интраназальные или внутриглазные формы введения и ректальные формы введения.
«Фармацевтически приемлемая соль» означает относительно нетоксичные органические и неорганические соли кислот и оснований, заявленных в настоящем изобретении. Эти соли могут быть получены in situ в процессе синтеза, выделения или очистки соединений, или приготовлены специально. В частности, соли оснований могут быть получены специально, исходя из очищенного свободного основания заявленного соединения и подходящей органической или неорганической кислоты. Примерами полученных таким образом солей являются гидрохлориды, гидробромиды, сульфаты, бисульфаты, фосфаты, нитраты, ацетаты, оксалаты, валериаты, олеаты, пальмитаты, стеараты, лаураты, бораты, бензоаты, лактаты, тозилаты, цитраты, малеаты, фумараты, сукцинаты, тартраты, мезилаты, малонаты, салицилаты, пропионаты, этансульфонаты, бензолсульфонаты, сульфаматы и им подобные (Подробное описание свойств таких солей дано в Berge S.M., et al., "Pharmaceutical Salts" J. Pharm. Sci. 1977, 66: 1-19). Соли заявленных кислот также могут быть специально получены реакцией очищенной кислоты с подходящим основанием, при этом могут быть синтезированы соли металлов и аминов. К солям металлов относятся соли натрия, калия, кальция, бария, цинка, магния, лития и алюминия, наиболее желательными из которых, являются соли натрия и калия. Подходящими неорганическими основаниями, из которых могут быть получены соли металлов, являются гидроксид, карбонат, бикарбонат и гидрид натрия, гидроксид и бикарбонат калия, гидроксид лития, гидроксид кальция, гидроксид магния, гидроксид цинка. В качестве органических оснований, из которых могут быть получены соли заявленных кислот, выбраны амины и аминокислоты, обладающие достаточной основностью, чтобы образовать устойчивую соль, и пригодные для использования в медицинских целях (в частности, они должны обладать низкой токсичностью). К таким аминам относятся аммиак, метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, бензиламин, дибензиламин, дициклогексиламин, пиперазин, этилпиперидин, трис(гидроксиметил)аминометан и подобные им. Кроме того, для солеобразования могут быть использованы гидроокиси тетраалкиламмония, например, такие как холин, тетраметиламмоний, тетраэтиламмоний и им подобные. В качестве аминокислот могут быть использованы основные аминокислоты - лизин, орнитин и аргинин.
«Лекарственное средство (препарат)» - вещество (или смесь веществ в виде фармацевтической композиции) в виде таблеток, гранул, капсул, инъекций, мазей и др. готовых форм, предназначенное для восстановления, исправления или изменения физиологических функций у человека и животных, а также для лечения и профилактики болезней, диагностики, анестезии, контрацепции, косметологии и прочего.
«Лечить», «лечение» и «терапия» относятся к методу смягчения или устранения биологического расстройства и/или по меньшей мере одного из сопутствующих ему симптомов. Термин «облегчить» болезнь, заболевание или состояние, означает уменьшение тяжести и/или частоты возникновения симптомов заболевания, расстройства или состояния. Кроме того, содержащиеся в данном документе ссылки на «лечение» включают ссылки на лечебную, паллиативную терапию.
«Профилактика», «профилактическая терапия» относится к комплексу мероприятий, направленных на предупреждение возникновения, устранение факторов риска или на ранее выявление заболевания или нарушения, их обострения, рецидивов, осложнений или других последствий.
В одном аспекте пациент или субъект лечения, или профилактики является млекопитающим, предпочтительно человеческим субъектом.
Вышеупомянутый субъект может быть мужского или женского пола любого возраста.
Термин "нарушение" означает любое состояние, которое можно улучшить в результате лечения по настоящему изобретению. В определение данного термина входят хронические и острые заболевания или патологические состояния, которые вызывают предрасположенность млекопитающего к возникновению этих заболеваний. Неограничивающие примеры подлежащих лечению заболеваний включают в себя доброкачественные и злокачественные новообразования или новообразования неуточненной природы, в том числе опухоли, исходящие из клеток крови и лимфоидных клеток. Примером могут быть: рак мочевого пузыря, рак яичника, рак шейки матки, колоректальный рак, рак молочной железы, рак поджелудочной железы, рак головы и шеи, глиома, глиобластома, меланома, рак предстательной железы, лейкоз, лимфома, неходжкинская лимфома, лимфома Ходжкина, рак легкого, гепатоцеллюлярный рак, рак пищевода, рак желудка, стромальная опухоль желудочно-кишечного тракта, рак щитовидной железы, рак желчных протоков, рак эндометрия, рак почки, рак печени, анапластическая крупноклеточная лимфома, острый миелоидный лейкоз, множественная миелома, меланома, мезотелиома, гематологические злокачественные опухоли.
«Терапевтически эффективным количеством» считается количество вводимого в процессе лечения терапевтического агента, которое избавит в определенной степени от одного или нескольких симптомов заболевания, по поводу которого проводится лечение.
В настоящем описании и в последующей формуле изобретения, если контекстом не предусмотрено иное, слова «иметь», «включать» и «содержать» или их вариации, такие как «имеет», «имеющий», «включает», «включающий», «содержит» или «содержащий», следует понимать, как включение указанного целого или группы целых, но не исключение любого другого целого или группы целых.
Подробное описание изобретения
В одном варианте осуществления, настоящее изобретение относится к соединению формулы I:

или его фармацевтически приемлемой соли, сольвату или стереоизомеру,
где L представляет собой -С(О)- или -СНОН-;
X1 представляет собой СН или N;
А представляет собой
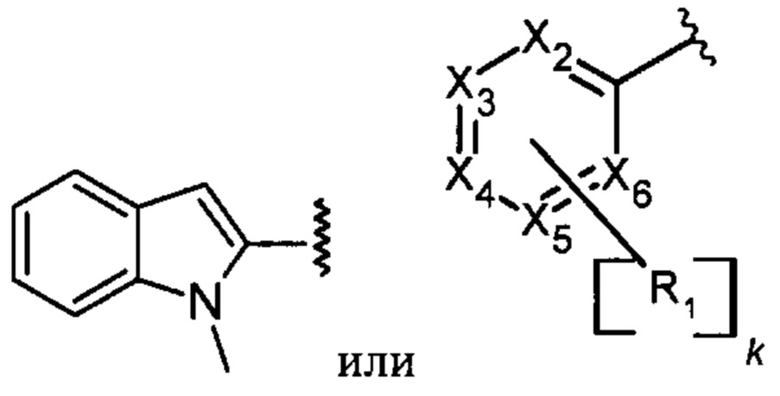
Х2, Х3, Х4, Х5, Х6 каждый независимо представляют собой С, СН или N;
R1 каждый независимо представляет собой водород; Hal; циано; нитро; гидроксигруппу; C1-С6 алкил, незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, гидроксигруппы, NR2R3; C1-С6 алкилокси-группу, незамещенную или замещенную одним или несколькими радикалами, выбранными из Hal, NR2R3, гидроксигруппы, C1-С6 алкилокси, арила, незамещенного или замещенного одним или несколькими радикалами, выбранными из Hal, гидроксигруппы, NR2R3; арилокси, незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, C1-С6 алкила, гидроксигруппы, NR2R3; С3-С6 циклоалкилокси, незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, гидроксигруппы, NR2R3; C1-С6 алкилокси C1-С6 алкил; NR2R3; арил, незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, гидроксигруппы, NR2R3; 5-6 членный гетероарил с 1-2 гетероатомами, выбранными из N и/или О, незамещенный или замещенный одним или несколькими заместителями, выбранными из Hal, циано, C1-С6 алкила, гидроксигруппы, C1-С6 алкилокси, NR2R3; 4-7 членный гетероциклил с 1-2 гетероатомами, выбранными из N и/или О, незамещенный или замещенный одним или несколькими заместителями, выбранными из Hal, циано, гидроксигруппы, оксо, C1-С6 алкила, C1-С6 алкилокси, NR2R3; R2 или R3 каждый независимо представляет собой водород; C1-С6 алкил, незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, гидроксигруппы, C1-С6 алкилокси; к представляет собой 0, 1, 2 или 3;
Hal представляет собой атом фтора, брома, хлора или йода.
В еще одном варианте осуществления настоящее изобретение относится к соединению формулы I, где фрагмент

выбран из группы, включающей:

где R1 каждый независимо представляет собой водород; Hal; циано; нитро; гидроксигруппу; C1-С6 алкил, незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, гидроксигруппы, -NR2R3; C1-С6 алкилокси-группу, незамещенную или замещенную одним или несколькими радикалами, выбранными из Hal, -NR2R3, гидроксигруппы, C1-С6 алкилокси, арила, незамещенного или замещенного одним или несколькими радикалами, выбранными из Hal, гидроксигруппы, -NR2R3; арилокси, незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, C1-С6 алкила, гидроксигруппы, -NR2R3; С3-С6 циклоалкилокси, незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, гидроксигруппы, -NR2R3; C1-С6 алкилокси C1-С6 алкил; -NR2R3; арил, незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, гидроксигруппы, -NR2R3; 5-6 членный гетероарил с 1-2 гетероатомами, выбранными из N и/или О, незамещенный или замещенный одним или несколькими заместителями, выбранными из Hal, циано, C1-С6 алкила, гидроксигруппы, C1-С6 алкилокси, -NR2R3; 4-7 членный гетероциклил с 1-2 гетероатомами, выбранными из N и/или О, незамещенный или замещенный одним или несколькими заместителями, выбранными из Hal, циано, гидроксигруппы, оксо, C1-С6 алкила, C1-С6 алкилокси, -NR2R3;
R2 или R3 каждый независимо представляет собой водород, C1-С6 алкил незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, гидроксигруппы, C1-С6 алкилокси; к представляет собой 0, 1, 2 или 3;
Hal представляет собой атом фтора, брома, хлора или йода.
В еще одном варианте осуществления настоящее изобретение относится к соединению формулы I, где фрагмент

выбран из группы, включающей:
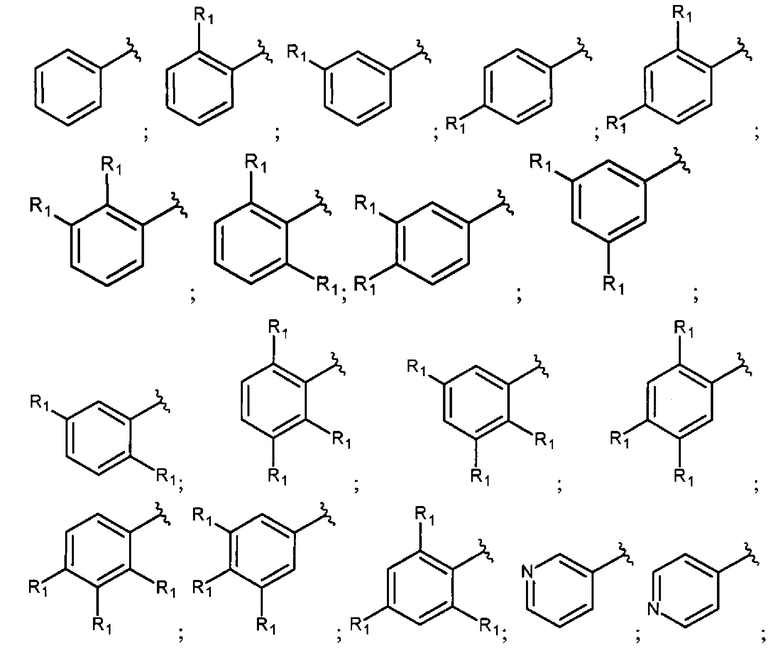

где R1 каждый независимо представляет собой водород; Hal; циано; нитро; гидроксигруппу; C1-С6 алкил, незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, гидроксигруппы, -NR2R3; С1-С6 алкилокси-группу, незамещенную или замещенную одним или несколькими радикалами, выбранными из Hal, -NR2R3, гидроксигруппы, C1-С6 алкилокси, арила, незамещенного или замещенного одним или несколькими радикалами, выбранными из Hal, гидроксигруппы, -NR2R3; арилокси, незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, C1-С6 алкила, гидроксигруппы, -NR2R3; С3-C6 циклоалкилокси, незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, гидроксигруппы, -NR2R3; C1-С6 алкилокси C1-С6 алкил; -NR2R3; арил, незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, гидроксигруппы, -NR2R3; 5-6 членный гетероарил с 1-2 гетероатомами, выбранными из N и/или О, незамещенный или замещенный одним или несколькими заместителями, выбранными из Hal, циано, C1-С6 алкила, гидроксигруппы, C1-С6 алкилокси, -NR2R3; 4-7 членный гетероциклил с 1-2 гетероатомами, выбранными из N и/или О, незамещенный или замещенный одним или несколькими заместителями, выбранными из Hal, циано, гидроксигруппы, оксо, C1-С6 алкила, C1-С6 алкилокси, -NR2R3;
R2 или R3 каждый независимо представляет собой водород, C1-С6 алкил незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, гидроксигруппы, C1-С6 алкилокси; к представляет собой 0, 1, 2 или 3;
Hal представляет собой атом фтора, брома, хлора или йода.
В еще одном варианте осуществления настоящее изобретение относится к соединению формулы I, где фрагмент

выбран из группы, включающей:

где R1 каждый независимо представляет собой водород; Hal; циано; нитро; гидроксигруппу; C1-С6 алкил, незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, гидроксигруппы, -NR2R3; C1-С6 алкилокси-группу, незамещенную или замещенную одним или несколькими радикалами, выбранными из Hal, -NR2R3, гидроксигруппы, C1-С6 алкилокси, арила, незамещенного или замещенного одним или несколькими радикалами, выбранными из Hal, гидроксигруппы, -NR2R3; арилокси, незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, C1-С6 алкила, гидроксигруппы, -NR2R3; С3-С6 циклоалкилокси, незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, гидроксигруппы, -NR2R3; C1-С6 алкилокси C1-С6 алкил; -NR2R3; арил, незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, гидроксигруппы, -NR2R3; 5-6 членный гетероарил с 1-2 гетероатомами, выбранными из N и/или О, незамещенный или замещенный одним или несколькими заместителями, выбранными из Hal, циано, C1-С6 алкила, гидроксигруппы, C1-С6 алкилокси, -NR2R3; 4-7 членный гетероциклил с 1-2 гетероатомами, выбранными из N и/или О, незамещенный или замещенный одним или несколькими заместителями, выбранными из Hal, циано, гидроксигруппы, оксо, C1-С6 алкила, C1-С6 алкилокси, -NR2R3;
R2 или R3 каждый независимо представляет собой водород, C1-С6 алкил незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, гидроксигруппы, C1-С6 алкилокси;
k представляет собой 0, 1, 2 или 3;
Hal представляет собой атом фтора, брома или хлора.
В еще одном варианте осуществления настоящее изобретение относится к соединению формулы I, где фрагмент

выбран из группы, включающей:

где R1 каждый независимо представляет собой водород; Hal; циано; нитро; гидроксигруппу; C1-С6 алкил, незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, гидроксигруппы, -NR2R3; C1-С6 алкилокси-группу, незамещенную или замещенную одним или несколькими радикалами, выбранными из Hal, -NR2R3, гидроксигруппы, C1-С6 алкилокси, арила, незамещенного или замещенного одним или несколькими радикалами, выбранными из Hal, гидроксигруппы, -NR2R3; арилокси, незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, C1-С6 алкила, гидроксигруппы, -NR2R3; С3-С6 циклоалкилокси, незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, гидроксигруппы, -NR2R3; C1-С6 алкилокси C1-С6 алкил; -NR2R3; арил, незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, гидроксигруппы, -NR2R3; 5-6 членный гетероарил с 1-2 гетероатомами, выбранными из N и/или О, незамещенный или замещенный одним или несколькими заместителями, выбранными из Hal, циано, C1-С6 алкила, гидроксигруппы, C1-С6 алкилокси, -NR2R3; 4-7 членный гетероциклил с 1-2 гетероатомами, выбранными из N и/или О, незамещенный или замещенный одним или несколькими заместителями, выбранными из Hal, циано, гидроксигруппы, оксо, C1-С6 алкила, C1-С6 алкилокси, -NR2R3;
R2 или R3 каждый независимо представляет собой водород, C1-С6 алкил незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, гидроксигруппы, C1-С6 алкилокси;
Hal представляет собой атом фтора, брома или хлора.
Соединения, описанные в настоящем изобретении, могут быть получены в виде и/или их можно применять в виде фармацевтически приемлемых солей. Типы фармацевтически приемлемых солей включают следующие, но не ограничены ими: соли присоединения кислот, образованные при взаимодействии соединения в форме свободного основания с фармацевтически приемлемой неорганической кислотой, такой как соляная, бромистоводородная, серная, азотная, фосфорная, метафосфорная кислоты и т.п.; или с органической кислотой, такой как уксусная, пропионовая, капроновая, циклопентанпропионовая, гликолевая, пировиноградная, молочная, малоновая, янтарная, яблочная, малеиновая, фумаровая, трифторуксусная, винная, лимонная, бензойная, 3-(4-гидроксибензоил)бензойная, коричная, миндальная кислоты, метансульфокислота, этансульфокислота, 1,2-этандисульфокислота, 2-гидроксиэтандисульфокислота, бензолсульфокислота, толуолсульфокислота, 2-нафталинсульфокислота, 4-метилбицикло [2.2.2] окт-2-ен-1-карбоновая, глюкогептоновая, 4,4'-метилен-бис-3-гидрокси-2-ен-1-карбоновая, 3-фенилпропионовая, триметилуксусная, третбутилуксусная, лаурилсерная, глюконовая, глутаминовая, гидроксинафтойная, салициловая, стеариновая, муконовая кислоты и т.п.
Соответствующие противоионы фармацевтически приемлемых солей можно исследовать и идентифицировать с использованием различных методов, включая перечисленные, но не ограничиваясь ими: ионнообменную хроматографию, ионную хроматографию, капиллярный электрофорез, индукционное связывание плазмы, атомно-абсорбционную спектроскопию, масс-спектрометрию или любую их комбинацию.
Соли восстанавливают с применением по меньшей мере одной из следующих методик: фильтрация, осаждение с осадителем с последующей фильтрацией, выпариванием растворителя или в случае водных растворов лиофилизацией. Следует понимать, что упоминание фармацевтически приемлемой соли включает формы аддитивной соли с растворителем или их кристаллические формы, в частности сольваты или полиморфы. Сольваты содержат стехиометрическое или нестехиометрическое количество растворителя и могут быть образованы в ходе процесса кристаллизации с фармацевтически приемлемыми растворителями, такими как вода, этанол и т.п. Гидраты образуются в случае, если растворителем является вода, а алкоголяты образуются в случае, когда растворителем является спирт. Сольваты соединений, описанных в настоящем патенте, могут быть легко получены или образованы в способах, описанных в настоящем изобретении. Кроме того, соединения, предусмотренные настоящим изобретением, могут существовать в несольватированной, а также в сольватированной формах. В целом, сольватированные формы рассматриваются как эквивалент несольватированных форм при описании соединений и способов, предусмотренных настоящим изобретением.
Соединения, описанные в настоящем изобретении, могут быть представлены в различных формах, включая перечисленные, но не ограничиваясь ими: бесструктурные формы, молотые формы и наночастицы. Кроме того, описанные в настоящем изобретении соединения включают кристаллические формы, также известные как полиморфы. Полиморфы включают кристаллы с различной структурой одинакового элементного состава соединения. Полиморфы, как правило, имеют различный характер рентгеновской дифракции, различные инфракрасные спектры, температуру плавления, различную плотность, твердость, кристаллическую форму, оптические и электрические свойства, стабильность и растворимость. Различные факторы, такие как растворитель для рекристаллизации, степень кристаллизации и температура хранения, могут обусловливать доминирование одной кристаллической формы.
Скрининг и определение характеристик фармацевтически приемлемых солей, полиморфов и/или сольватов можно осуществлять рядом методов, включая перечисленные, но не ограничиваясь ими: термический анализ, рентгено-дифракционный метод, спектроскопию, сорбцию пара и микроскопию. Термические методы анализа направлены на исследование термохимического разложения или термофизических процессов, включая, но не ограничиваясь, полиморфные переходы, и такие методы применяют для анализа связи между полиморфными формами, определения потери в массе, для нахождения температуры стеклования или исследования совместимости с наполнителем. Такие способы включают, без ограничения, дифференциальную сканирующую калориметрию (ДСК), модулирующую дифференциальную сканирующую калориметрию (МДСК), термогравиметрический анализ (ТГА), термогравиметрический и инфракрасный анализ (ТГ/ИК). Кристаллографические методы включают перечисленные, но не ограничиваются ими: монокристаллические и порошковые дифрактометры и синхротронные источники. Различные используемые спектроскопические методы включают перечисленные, но не ограничены ими: определение спектра Рамана (комбинационного рассеяния), FTIR, UVIS и ЯМР (жидкого и твердого состояния). Различные методы микроскопии включают перечисленные, но не ограничены ими: микроскопию в поляризованном свете, сканирующую электронную микроскопию (СЭМ) с рентгеновским анализом методом энергетической дисперсии (EDX), сканирующую электронную микроскопию в режиме естественной среды с EDX (в атмосфере газа или водяного пара), ИК-микроскопию и микроскопию комбинационного рассеяния.
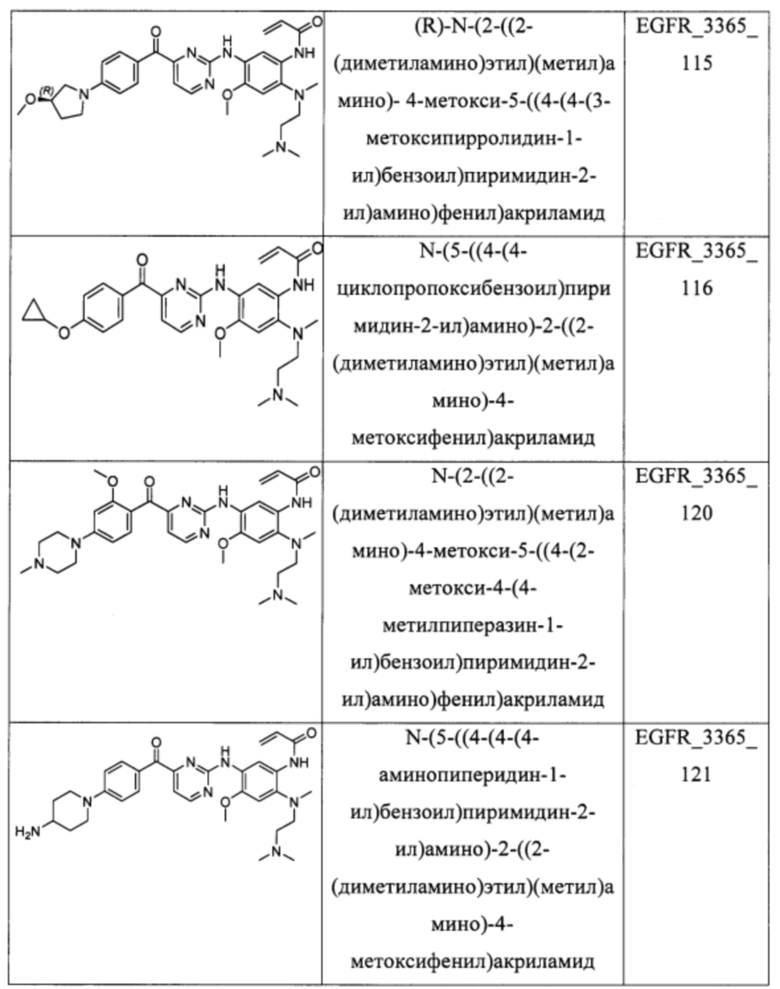
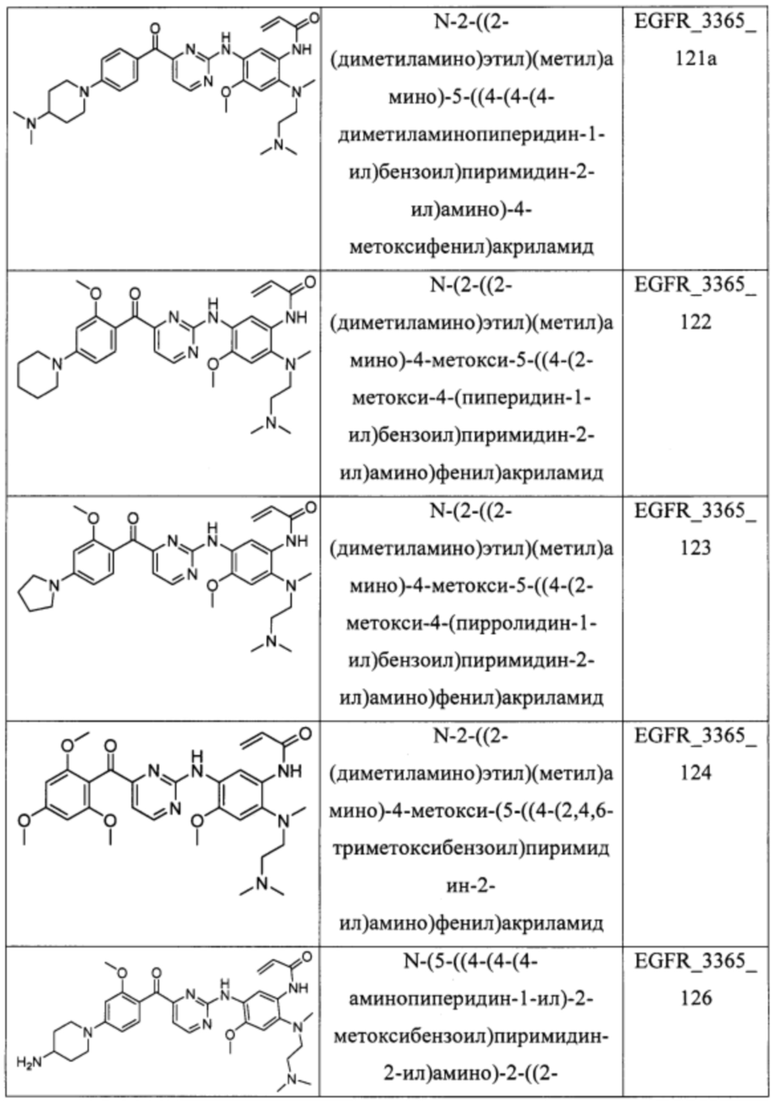
В еще одном варианте осуществления настоящее изобретение относится к соединениям, выбранным из группы, включающей:
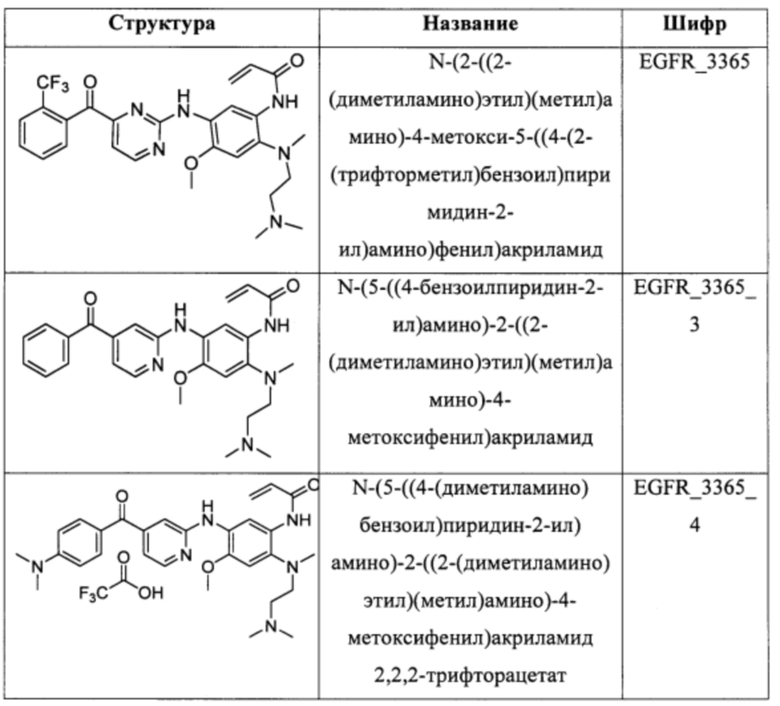


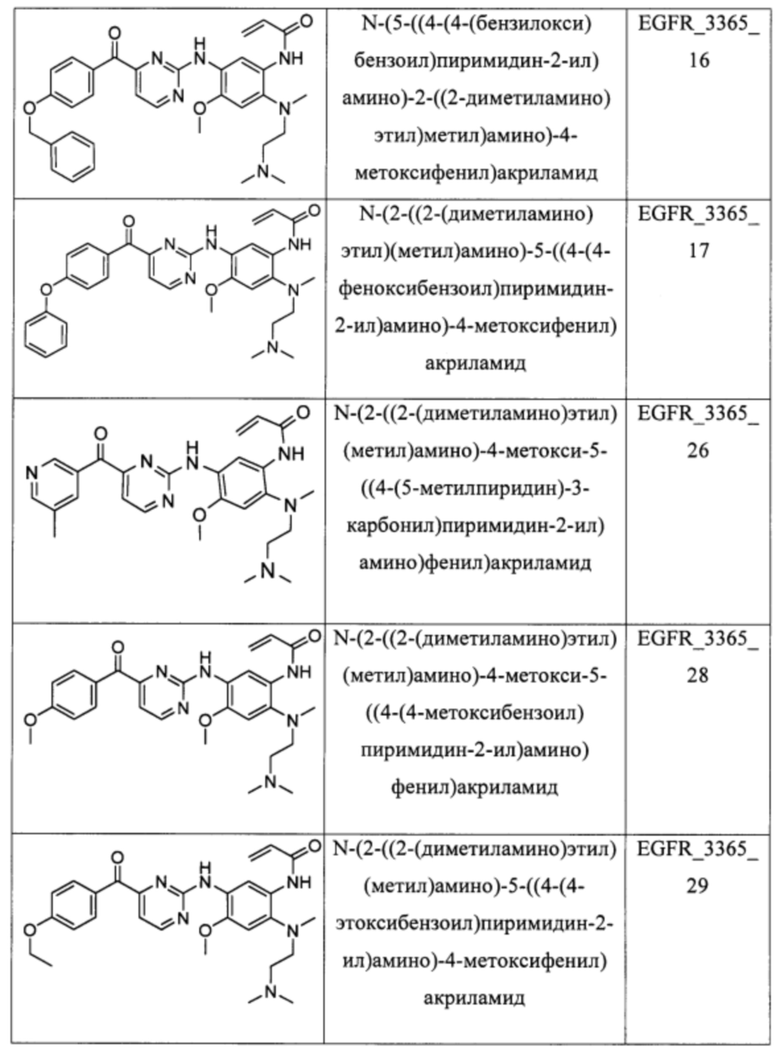
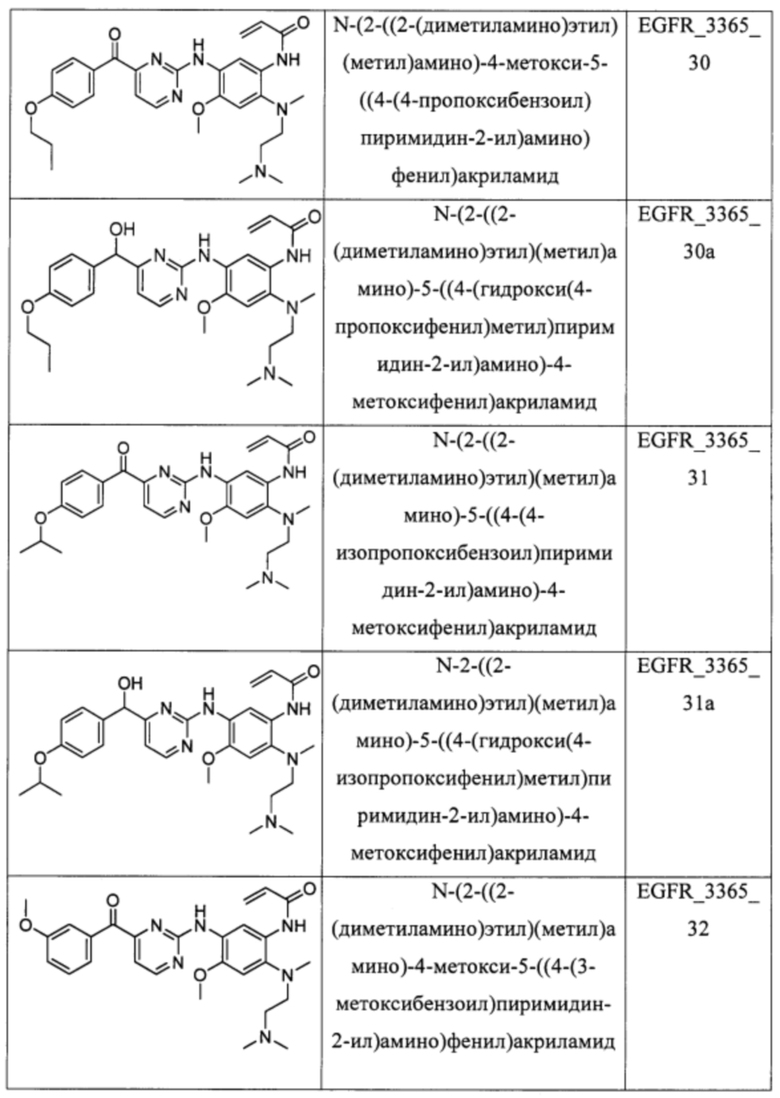



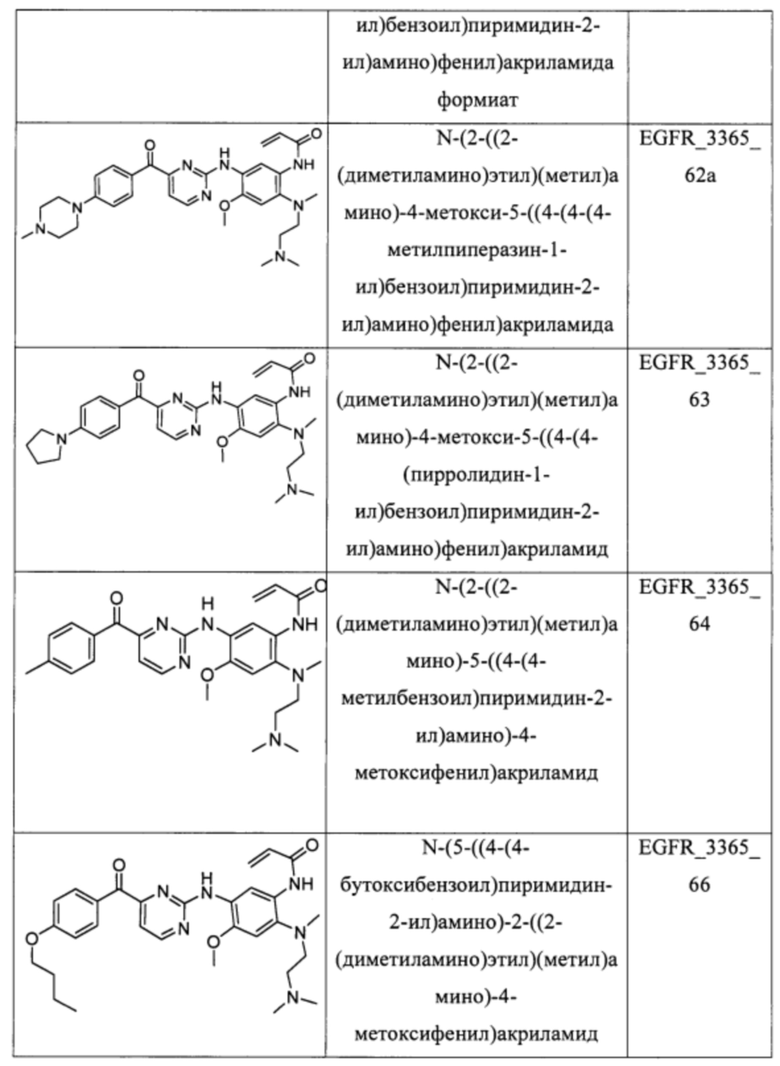
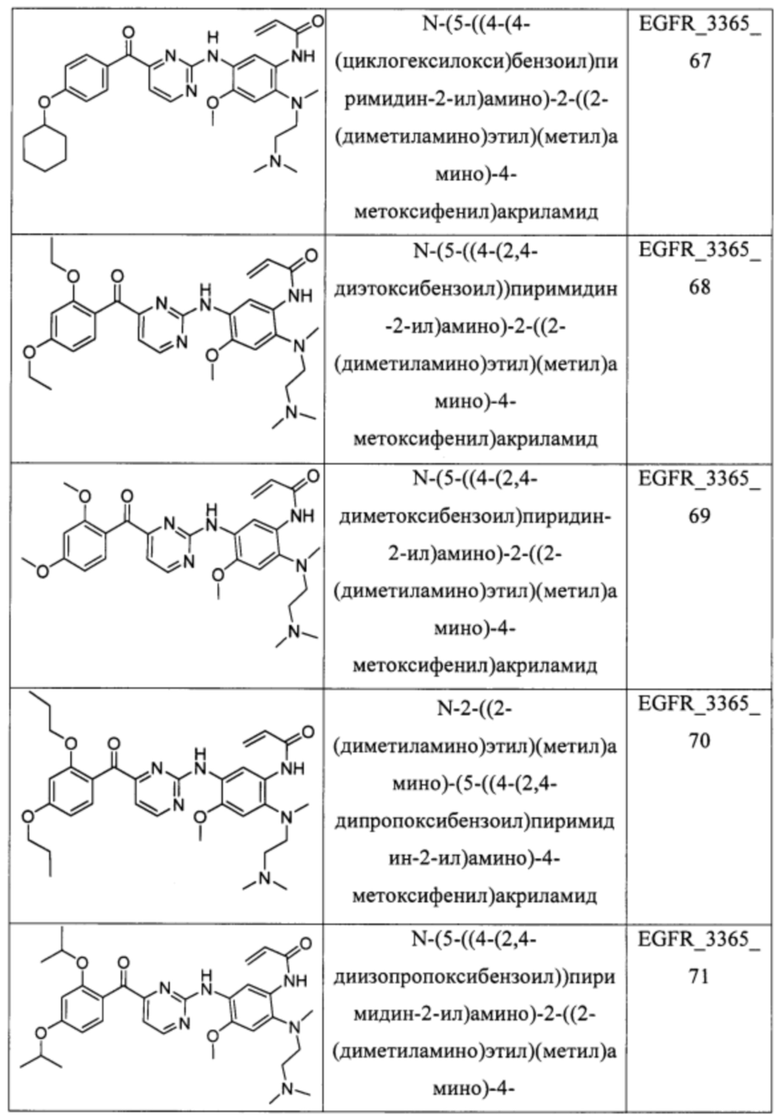
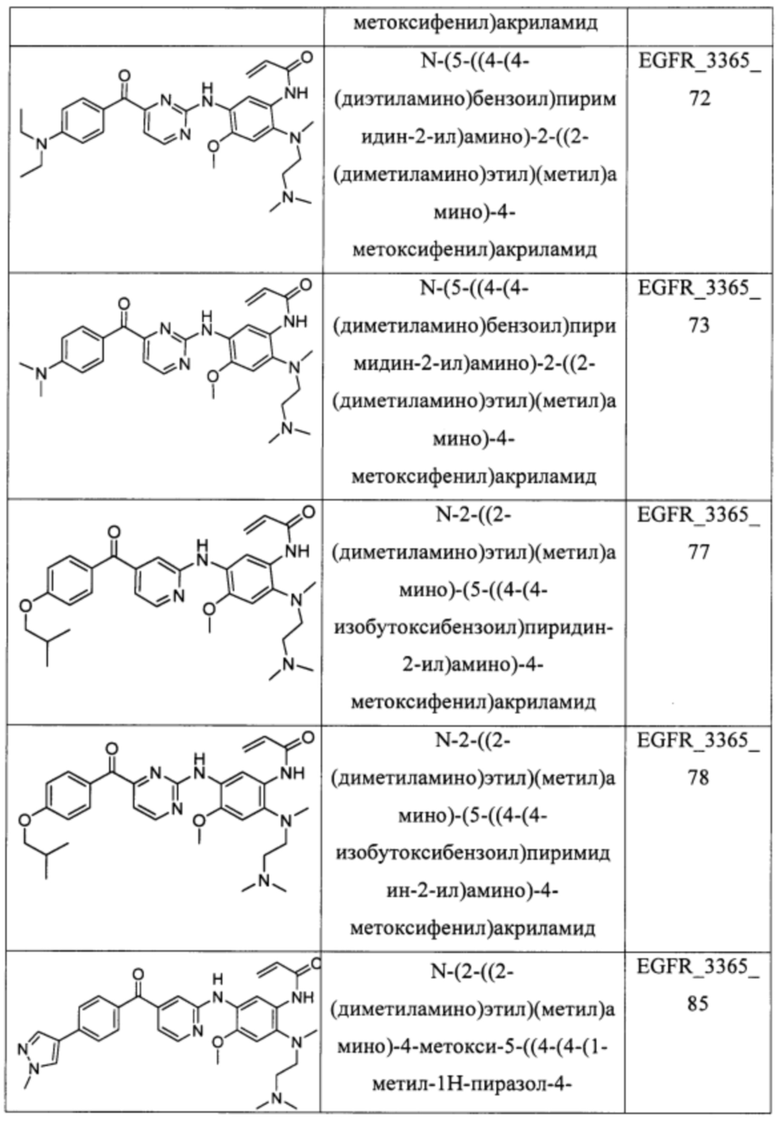
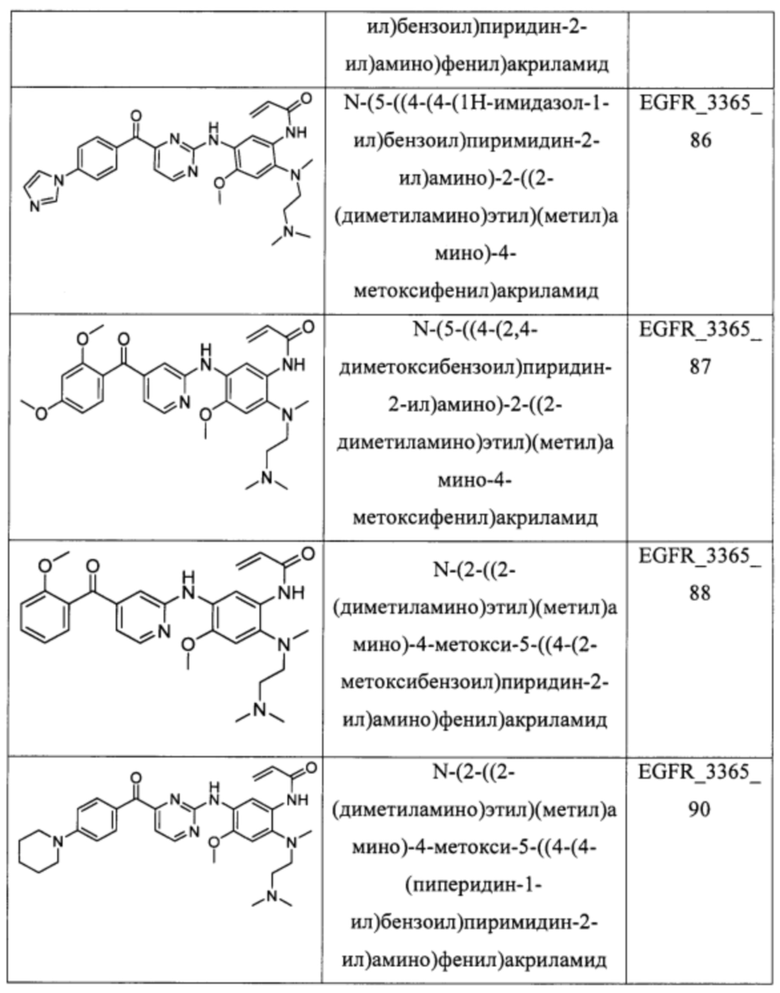

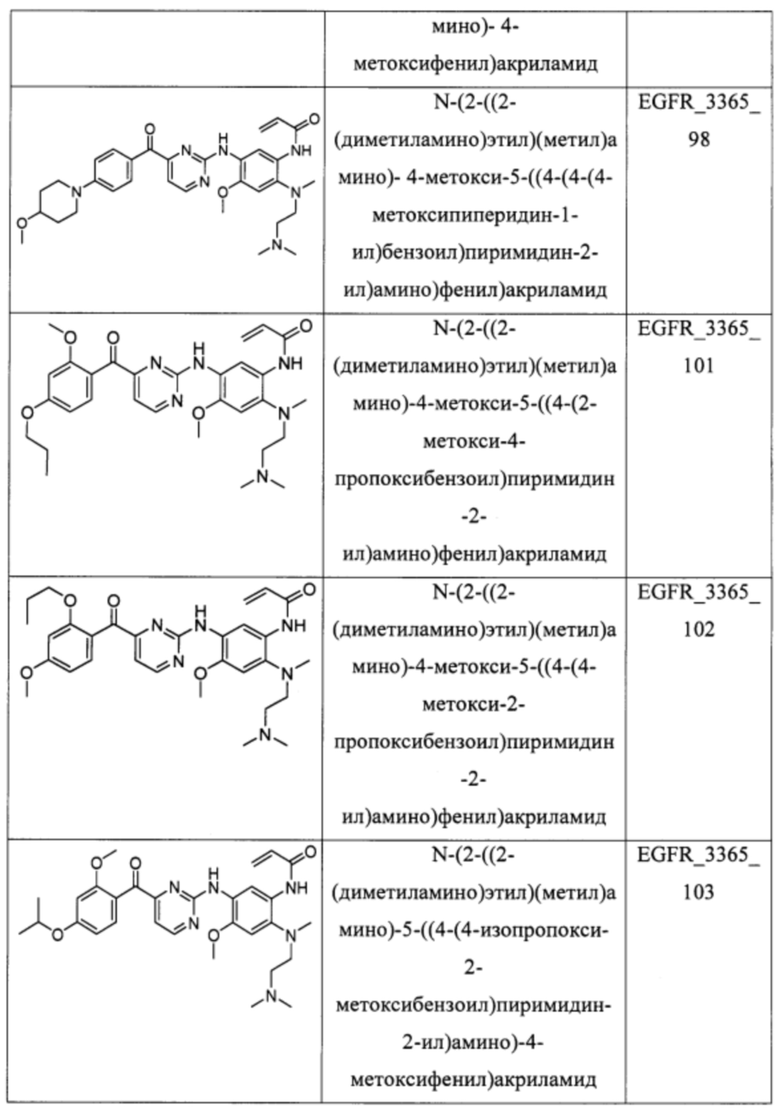
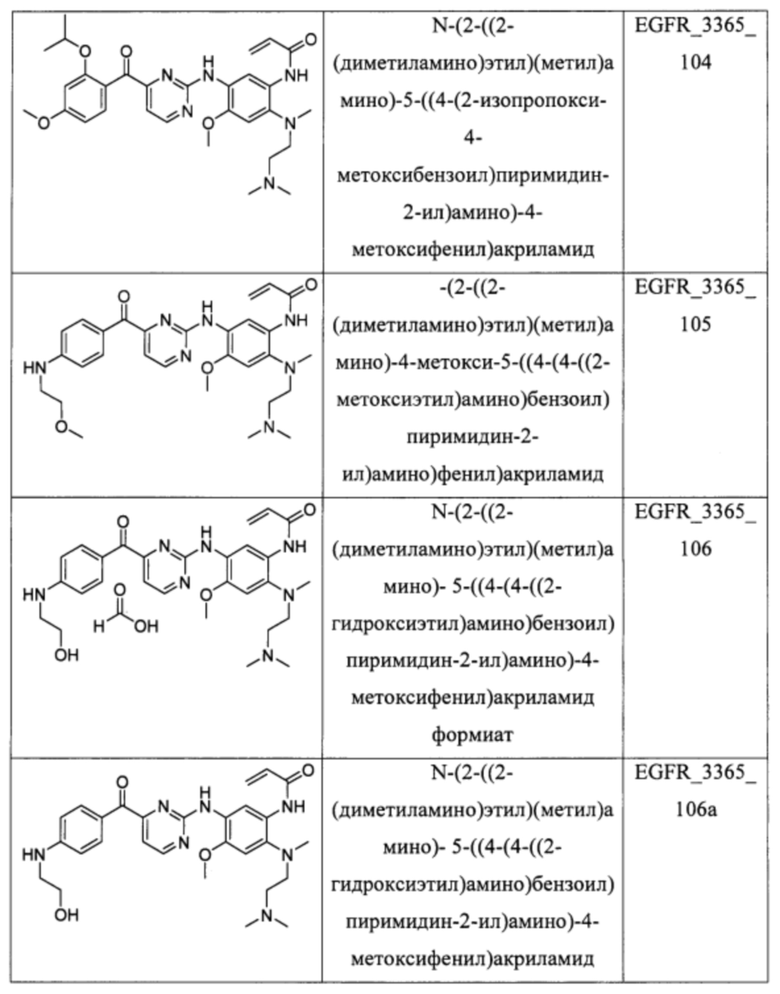

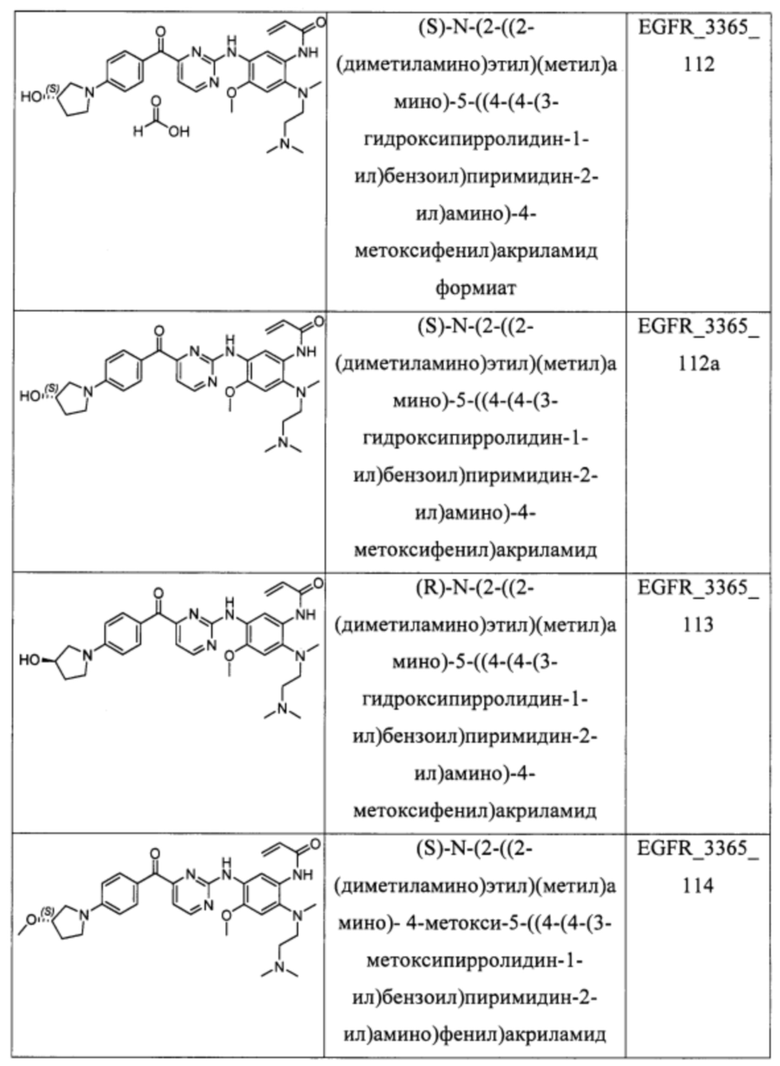

Настоящее изобретение также относится к способу ингибирования у субъекта биологической активности EGFR, заключающемуся в контактировании EGFR с соединением, описанном в настоящем документе.
Соединения, ингибирующие EGFR, можно применять для производства лекарственных препаратов для лечения любого из описанных в настоящем документе патологических состояний, например, соединения формулы I, фармацевтически приемлемые соли, сольваты или стереоизомеры будут полезны в лечении заболеваний или медицинских состояний, опосредованных исключительно или отчасти активностью EGFR, например, онкологических заболеваний. Примеры онкологических заболеваний, которые могут поддаваться лечению с использованием вышеуказанных соединений, включают рак мочевого пузыря, рак яичника, рак шейки матки, колоректальный рак, рак молочной железы, рак поджелудочной железы, рак головы и шеи, глиому, глиобластому, меланому, рак предстательной железы, лейкоз, лимфому, неходжкинскую лимфому, лимфому Ходжкина, рак легкого (например, немелкоклеточный рак легкого), гепатоцеллюлярный рак, рак пищевода, рак желудка, стромальную опухоль желудочно-кишечного тракта, рак щитовидной железы, рак желчных протоков, рак эндометрия, рак почки, рак печени, анапластическую крупноклеточную лимфому, острый миелоидный лейкоз, множественную миелому, меланому, мезотелиому, гематологические злокачественные опухоли, но не ограничены ими.
В одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей в терапевтически эффективном количестве по меньшей мере одно из соединений, описанных в настоящем документе, или его фармацевтически приемлемую соль, сольват, стереоизомер и один или несколько фармацевтически приемлемых эксципиентов. В еще одном варианте настоящего изобретения фармацевтическая композиция, содержащая соединения по данному изобретению, предназначена для профилактики или лечения заболевания, или нарушения, опосредованного активностью EGFR.
В еще одном варианте настоящего изобретения фармацевтическая композиция, содержащая соединения по настоящему изобретению, предназначена для профилактики или лечения заболевания или нарушения, опосредованного активностью EGFR с мутацией L858R и/или мутацией Т790М и/или делецией в экзоне 19 и/или мутацией C797S.
В еще одном варианте настоящего изобретения фармацевтическая композиция, содержащая соединения по настоящему изобретению, предназначена для профилактики или лечения онкологических заболеваний, включающих рак мочевого пузыря, рак яичника, рак шейки матки, колоректальный рак, рак молочной железы, рак поджелудочной железы, рак головы и шеи, глиому, глиобластому, меланому, рак предстательной железы, лейкоз, лимфому, неходжкинскую лимфому, лимфому Ходжкина, рак легкого (например, немелкоклеточный рак легкого), гепатоцеллюлярный рак, рак пищевода, рак желудка, стромальную опухоль желудочно-кишечного тракта, рак щитовидной железы, рак желчных протоков, рак эндометрия, рак почки, рак печени, анапластическую крупноклеточную лимфому, острый миелоидный лейкоз, множественную миелому, меланому, мезотелиому, гематологические злокачественные опухоли.
В еще одном варианте настоящего изобретения фармацевтическая композиция, содержащая соединения по настоящему изобретению, предназначена для профилактики или лечения онкологического заболевания, которое представляет собой немелкоклеточный рак легкого
Фармацевтическая композиция по настоящему изобретению содержит, например, от приблизительно 5% до приблизительно 100% мас. активных ингредиентов, предпочтительно от приблизительно 10% мас. до приблизительно 60% мас. активных ингредиентов. Подразумевается, что содержание активного ингредиента или ингредиентов в индивидуальной дозе каждой лекарственной формы не обязательно составляет эффективное количество, поскольку необходимое эффективное количество может достигаться при введении нескольких стандартных лекарственных форм.
Типичную композицию получают посредством смешивания соединения по настоящему изобретению и носителя, разбавителя, или эксципиента. Подходящие носители, разбавители и эксципиенты хорошо известны специалистам в данной области и включают такие вещества, как углеводы, воска, водорастворимые и/или набухающие полимеры, гидрофильные или гидрофобные вещества, желатин, масла, растворители, воду и подобное. Конкретный используемый носитель, разбавитель или эксципиент будет зависеть от средств и цели, для которой применяют соединение по настоящему изобретению. Растворители в общем случае выбирают на основании растворителей, признанных специалистами в данной области техники безопасными (GRAS) для введения млекопитающему. В общем случае безопасные растворители представляют собой нетоксичные водные растворители, такие как вода и другие нетоксичные растворители, которые растворимы в воде или смешиваются с водой. Подходящие водные растворители включают воду, этанол, пропиленгликоль, полиэтиленгликоли (например, PEG400, PEG300) и т.д. и их смеси. Композиции также могут включать один или более буферов, стабилизирующих агентов, поверхностно-активных веществ, увлажняющих агентов, смазывающих агентов, эмульгаторов, суспендирующих агентов, консервантов, антиокислителей, матирующих агентов, скользящих веществ, технологических добавок, красителей, подсластителей, отдушек, ароматизаторов и других известных добавок для получения хорошего внешнего вида лекарственного средства (т.е. соединения по настоящему изобретению или его фармацевтической композиции) или чтобы способствовать изготовлению фармацевтического продукта (т.е. лекарственного средства). Производство фармацевтических композиций предпочтительно должно соответствовать требованиям GMP (надлежащей производственной практики).
Фармацевтические композиции также включают сольваты и гидраты соединений по настоящему изобретению, или стабилизированную форму соединения (например, комплекс с производным циклодекстрина или другим известным агентом комплексообразования).
Фармацевтические композиции по настоящему изобретению, как правило, пригодны для перорального введения. Пероральное введение может включать глотание, так что соединение поступает в желудочно-кишечный тракт и/или буккально, лингвально или сублингвально поступает в кровоток непосредственно из полости рта.
Лекарственные формы, пригодные для перорального введения, включают твердые, полутвердые и жидкие системы, такие как таблетки; мягкие или твердые капсулы, содержащие мульти- или наночастицы, жидкости или порошки; гранулы; пастилки (включая заполненные жидкостью); жевательные формы; гели; быстро растворимые лекарственные формы; пленки; суппозитории; спреи; и щечные/мукоадгезивные пластыри. Более предпочтительными лекарственными формами для перорального введения являются таблетки, гранулы и капсулы.
Жидкие лекарственные формы включают суспензии, растворы, сиропы и эликсиры. Такие лекарственные формы могут быть использованы как наполнители в мягких или жестких капсулах (например, из желатина или гидроксипропилметилцеллюлозы) и обычно содержат носитель, например, воду, этанол, полиэтиленгликоль, пропиленгликоль, метилцеллюлозу или подходящее масло и один или более эмульгаторов и/или суспендирующих агентов. Жидкие лекарственные формы могут быть также изготовлены путем восстановления твердого вещества, например, из саше.
Соединения по настоящему изобретению могут также вводиться парентерально. Используемый в данном документе термин «парентеральное введение» фармацевтической композиции включает любой способ введения, для которого характерно физическое нарушение целостности ткани субъекта и введение фармацевтической композиции через нарушение в ткани, что обычно приводит к прямому попаданию в кровоток, в мышцу или во внутренний орган. Таким образом, парентеральное введение включает, помимо прочего, введение фармацевтической композиции путем инъекции композиции посредством введения композиции через хирургический разрез, путем нанесения композиции с помощью проникающей в ткани нехирургической раны и т.п. В частности, предполагается, что парентеральное введение включает, помимо прочего, подкожную, внутрибрюшинную, внутримышечную, внутривенную, внутриартериальную, интратекальную, внутрижелудочковую, интрауретральную, внутричерепную, внутрисуставнную инъекцию или инфузии; и почечные диализные инфузионные методики. Внутриопухолевая доставка, например, внутриопухолевая инъекция, также может оказаться полезной. Также предусмотрена региональная перфузия.
Лекарственные формы фармацевтических композиций, подходящие для парентерального введения, обычно содержат активный ингредиент в сочетании с фармацевтически приемлемым носителем, например, стерильной водой или стерильным изотоническим раствором. Такие лекарственные формы могут быть изготовлены, упакованы или проданы в форме, подходящей для болюсного введения или для непрерывного введения. Инъекционные лекарственные формы могут быть изготовлены, упакованы или проданы в стандартной лекарственной форме, например, в ампулах, или в многодозовых контейнерах, содержащих консервант. Лекарственные формы для парентерального введения включают, помимо прочего, суспензии, растворы, эмульсии в масляных или водных основах, пасты и тому подобное.
Соединения по настоящему изобретению могут также вводиться интраназально или ингаляционно, обычно в форме сухого порошка (самостоятельно, в виде смеси или в виде частиц со смешанными компонентами, например, смешанными с подходящим фармацевтически приемлемым наполнителем) из ингалятора с сухим порошком, такого как аэрозольный контейнер под давлением, помпа, спрей, распылитель (предпочтительно распылитель, в котором использован принцип электрогидродинамики для получения мелкодисперсного тумана) или небулайзер, в котором используется или не используется подходящий пропеллент, или в виде назальных капель.
Контейнер под давлением, помпа, спрей, распылитель или небулайзер обычно содержит раствор или суспензию соединения по данному изобретению, включая, например, подходящее вещество для диспергирования, растворения или продления высвобождения активного вещества, пропеллент в качестве растворителя.
До использования в виде сухого порошка или суспензии, лекарственный препарат обычно микронизируют до размера, подходящего для доставки путем ингаляции (обычно менее 5 микрон). Этого можно достичь любым подходящим способом измельчения, таким как размол на спиральной струйной мельнице, размол на струйной мельнице с кипящим слоем, сверхкритическая очистка жидкости для формирования наночастиц, гомогенизация высоким давлением или распылительная сушка.
Капсулы, блистеры и картриджи для использования в ингаляторе или инсуфляторе могут быть выполнены так, чтобы содержать порошковую смесь соединения по данному изобретению, подходящей порошковой основы и модификатор активности.
Подходящая формула раствора для использования в распылителе, в котором использован принцип электрогидродинамики для получения мелкодисперсного тумана, может содержать подходящую дозу соединения по данному изобретению на одно нажатие, и объем на нажатие может варьироваться, например, от 1 мкл до 100 мкл.
В лекарственные формы по данному изобретения, предназначенные для ингаляции/интраназального введения, могут быть добавлены подходящие ароматизаторы, такие как ментол и левоментол, или подсластители, такие как сахарин или натрия сахарин.
Лекарственные формы могут быть выполнены для немедленного и/или модифицированного высвобождения. Лекарственные формы с модифицированным высвобождением включают отсроченное, замедленное, пульсирующее, контролируемое, нацеленное и программируемое высвобождение.
В одном варианте настоящее изобретение относится к способу лечения заболевания или нарушения, опосредованного активностью EGFR, который включает введение в терапевтически эффективном количестве любого соединения, описанного выше, или фармацевтической композиции по данному изобретению субъекту, нуждающемуся в таком лечении.
Еще в одном варианте изобретение относится к способу лечения, описанному выше, где указанное заболевание или нарушение представляет собой заболевание или нарушение, опосредованное активностью EGFR с мутацией L858R и/или мутацией Т790М и/или делецией в экзоне 19 и/или мутацией C797S.
В еще одном варианте изобретение относится к способу лечения, описанному выше, где заболевание или нарушение, опосредованное активностью EGFR, представляет собой онкологические заболевания. В еще одном варианте изобретение относится к способу лечения, описанному выше, где онкологические заболевания выбирают из группы, включающей рак мочевого пузыря, рак яичника, рак шейки матки, колоректальный рак, рак молочной железы, рак поджелудочной железы, рак головы и шеи, глиому, глиобластому, меланому, рак предстательной железы, лейкоз, лимфому, неходжкинскую лимфому, лимфому Ходжкина, рак легкого (например, немелкоклеточный рак легкого), гепатоцеллюлярный рак, рак пищевода, рак желудка, стромальную опухоль желудочно-кишечного тракта, рак щитовидной железы, рак желчных протоков, рак эндометрия, рак почки, рак печени, анапластическую крупноклеточную лимфому, острый миелоидный лейкоз, множественную миелому, меланому, мезотелиому, гематологические злокачественные опухоли.
В еще одном варианте изобретение относится к способу лечения, описанному выше, где онкологическое заболевание представляет собой немелкоклеточный рак легкого.
В одном варианте настоящее изобретение относится к применению соединения по настоящему изобретению или фармацевтической композиции, описанной выше, для лечения заболевания или нарушения, опосредованного активностью EGFR у субъекта, нуждающегося в таком лечении.
В еще одном варианте настоящее изобретение относится к применению, описанному выше, где указанное заболевание или нарушение представляет собой заболевание или нарушение, опосредованное активностью EGFR с мутацией L858R и/или мутацией Т790М и/или делецией в экзоне 19 и/или мутацией C797S.
В еще одном варианте настоящее изобретение относится к применению, описанному выше, где заболевание или нарушение, опосредованное активностью EGFR, представляет собой онкологические заболевания. В еще одном варианте изобретение относится к применению, описанному выше, где онкологические заболевания выбирают из группы, включающей рак мочевого пузыря, рак яичника, рак шейки матки, колоректальный рак, рак молочной железы, рак поджелудочной железы, рак головы и шеи, глиому, глиобластому, меланому, рак предстательной железы, лейкоз, лимфому, неходжкинскую лимфому, лимфому Ходжкина, рак легкого (например, немелкоклеточный рак легкого), гепатоцеллюлярный рак, рак пищевода, рак желудка, стромальную опухоль желудочно-кишечного тракта, рак щитовидной железы, рак желчных протоков, рак эндометрия, рак почки, рак печени, анапластическую крупноклеточную лимфому, острый миелоидный лейкоз, множественную миелому, меланому, мезотелиому, гематологические злокачественные опухоли.
В еще одном варианте изобретение относится к применению, описанному выше, где онкологическое заболевание представляет собой немелкоклеточный рак легкого.
Соединения по настоящему изобретению могут назначаться отдельно или в сочетании с одним или более другими препаратами или антителами (или в любой их комбинации). Таким образом, фармацевтические композиции, способы и использование изобретения также охватывает варианты осуществления комбинаций (совместное назначение) с другими активными агентами.
Используемые в данном документе термины «совместное назначение», «совместно назначенный» и «в сочетании с» относящиеся к данным соединениям с одним или более другими терапевтическими агентами, как предполагается, означают, ссылаются или включают:
- одновременное введение такой комбинации соединения по данному изобретению и терапевтического агента пациенту, который нуждается в лечении, когда такие компоненты сформулированы вместе в одной лекарственной форме, из которой указанные компоненты высвобождаются практически одновременно указанному пациенту,
- одновременное введение такой комбинации соединения по данному изобретению и терапевтического агента пациенту, который нуждается в лечении, когда такие компоненты сформулированы отдельно в разных лекарственных формах, введение которых происходит практически в одно и то же время указанному пациенту, после чего указанные компоненты высвобождаются практически одновременно указанному пациенту,
- последовательное введение такой комбинации соединения по данному изобретению и терапевтического агента пациенту, который нуждается в лечении, когда такие компоненты сформулированы отдельно друг от друга в отдельных лекарственных формах, которые принимаются в последовательно по времени указанным пациентом со значимым временным интервалом между каждым введением, после чего указанные компоненты высвобождаются в практически разное время указанному пациенту; а также
- последовательное введение такой комбинации соединения по данному изобретению и терапевтического агента пациенту, который нуждается в лечении, когда такие компоненты сформулированы вместе в единый лекарственной форме, из которой высвобождение указанных компонентов происходит контролируемым образом, после чего они одновременно, последовательно или совместно высвобождаются в одно и то же время и/или разное время указанному пациенту, где каждая часть может быть введена одним или разными путями.
Специалистам в данной области известно, что терапевтически эффективные дозировки могут меняться при применении препаратов в комбинированном лечении. Способы для экспериментального определения терапевтически эффективных дозировок препаратов и других агентов для применения в режимах комбинированного лечения описаны в литературе. Например, применение равномерного дозирования, т.е. введение более частых и меньших доз для минимизации токсичных побочных эффектов, описано в литературе. Комбинированное лечение, кроме того, включает периодическое лечение, которое начинается и останавливается в различное время в соответствии с планом лечения пациента. В комбинированной терапии, описанной в настоящем патенте, дозировки совместно вводимых соединений, несомненно, меняются в зависимости от типа применяемого вспомогательного лекарственного средства, специфики применяемого лекарственного средства, болезни или состояния, подвергаемого лечению, и т.д.
Противоопухолевое лечение, описанное выше, можно применять в качестве единственной терапии, либо в сочетании с хирургическим вмешательством, или лучевой терапией, или лекарственной терапией. Такая терапия может быть введена параллельно, одновременно, последовательно или раздельно с лечением соединением по изобретению и может включать один или более агентов из следующих категорий противоопухолевых агентов: антипролиферативные/ противоопухолевые лекарственные средства и их комбинации, применяемые в медицинской онкологии, такие как алкилирующие агенты (например, цисплатина, оксалиплатин, карбоплатин, циклофосфамид, хлорметин, мелфалан, хлорамбуцил, бусульфан, треосульфан, темозоломид, бендамустин, проспидин, спиробромин, преднимустин, эстрамустин, пафенцил, лофенал, ифосфамид, мафосфамид, трофосфамид, глюфосфамид и препараты нитрозомочевины, в т.ч. кармустин, ломустин, нимустин, фотемустин, араноза, стрептозоцин); антиметаболиты (например, гемцитабин, фторурацил, флоксуридин, тегафур, ралтитрексид, метотрексат, триметрексат, пеметрексед, пралатрексат, кальция левофолинат, цитозинарабинозид, гидроксимочевина, азатиоприн, кладрибин, флударабин, пентостатин, меркаптопурин, неларабин, тиогуанин, фопурин, азацитидин, капецитабин, флударабин, кладрибин, неларабин, азатиоприн, клофарабин, цитарабин, эноцитабин, кармофур, гемцитабин, сапацитабин, элацитарабин, доксифлуридин); противоопухолевые антибиотики (например, блеомицин, доксорубицин, дауномицин, эпирубицин, идарубицин, митомицина, дактиномицин, митрамицин, даунорубицин, карубицин, эпирубицин, валрубицин, зорубицин, акларубицин, пирарубицин, неморубицин, амрубицин, зиностатин, стрептозоцин, митоксантрон); антимитотические агенты (например, винкаалкалоиды типа винкристина, винбластина, винфлунина, виндезина и винорелбина, таксоиды типа паклитаксела и доцетаксела, кабазитаксела, тезетаксела, ингибиторы полокиназы); и ингибиторы топоизомеразы (например, эпиподофиллотоксины типа этопозида и тенипозида, амсакрина, топотекана, иринотекан, белотекан, ворелоксин, амонафид и камптотецин); цитостатические агенты, такие как антиэстрогены (например, тамоксифен, клостилбегит, фулвестрант, торемифен, ралоксифен, дролоксифен и йодоксифен), антиандрогены (например, бикалутамид, флутамид, нилутамид, топилутамид, энзалутамид и ципротерона ацетат, хлормадинон), антагонисты рилизинг-фактора лютеинизирующего гормона (LHRH) или агонисты LHRH (например, гозерелин, лейпрорелин и бусерелин), прогестагены (например, хлормадинон, гестонорона капроат, медроксипрогестерон, мегестрола ацетат), ингибиторы ароматазы (например, анастрозол, летрозол, воразол и эксеместан) и ингибиторы 5α-редуктазы (например, финастерид, дутастерид, эпристерид); антиинвазивные агенты (например, ингибиторы семейства с-Src-киназ (например, саракатиниб, дазатиниб и босутиниб), ингибиторы металлопротеиназы (например, маримастат), ингибиторы функции урокиназных рецепторов активаторов (например, плазминоген или антитела к гепараназе); ингибиторы функции фактора роста: например, такие ингибиторы включают антитела к факторам роста и антитела к рецепторам фактора роста (например, трастузумаб, панитумумаб, цетуксимаб и любые антитела к факторам роста и рецепторам фактора роста, раскрытые Stern et al. Critical reviews in oncology/haematology, 2005, Vol. 54, p. 11-29); такие ингибиторы также включают ингибиторы тирозинкиназы, в том числе ингибиторы семейства эпидермального фактора роста (например, ингибиторы тирозинкиназы EGFR, такие как гефитиниб, эрлотиниб, канертиниб (CI 1033), афатиниб, осимертиниб, роцилетиниб, икотиниб, дакомитиниб; ингибиторы erbB2 тирозинкиназы, такие как лапатиниб); ингибиторы семейства факторов роста гепатоцитов; ингибиторы семейства инсулиноподобных факторов роста; ингибиторы семейства тромбоцитарных факторов роста, такие как иматиниб, нилотиниб; ингибиторы серин/треонинкиназ (например, ингибиторы передачи сигнала Ras/Raf, такие как ингибиторы фарнезилтрансферазы, например сорафениб, типифарниб и лонафарниб), ингибиторы передачи сигнала через МЕК- и/или АКТ-киназы, ингибиторы c-kit, ингибиторы abl-киназы, ингибиторы PI3-киназы, ингибиторы Plt3-киназы, ингибиторы CSF-1R-киназы, ингибиторы IGF рецепторной (инсулиноподобный фактор роста) киназы; ингибиторы aurora-киназы (например, barasertib (AZD1152), danusertib (РНА-739358), tozasertib (VX-680), MLN8054, R763, МР235, МР529, VX-528 и AX39459) и ингибиторы циклинзависимой киназы, такие как CDK2 и/или CDK4 ингибиторы; антиангиогенные агенты, такие как агенты, которые ингибируют эффекты сосудистого эндотелиального фактора роста (например, бевацизумаб, вандетаниб, ваталаниб, сунитиниб, акситиниб, пазопаниб, кризотиниб и цедираниб (AZD2171), линомид, ингибиторы функции avp3 интегрина, ангиостатин, эндостатин, талидомид, эверолимус, сиролимус, итраконазол, сурамин, semaxanib, тромбоспондин, рамуцирумаб, tasquinimod, ранибизумаб, сорафениб, соединения, раскрытые в публикациях международных заявок WO 97/22596, WO 97/30035, WO 97/32856 и WO 98/13354); сосудоповреждающие агенты (например, комбретастатин А4, омбрабулин и соединения, раскрытые в публикациях международных заявок WO 99/02166, WO 00/40529, WO 00/41669, WO 01/92224, WO 02/04434 и WO 02/08213; антагонист эндотелиновых рецепторов (например, бозентан, ситаксентан, амбризентан, BQ-123, BQ-788, мацитентан, tezosentan, зиботентан, атрасентан); антисмысловые терапии (например, которые направлены на мишени, перечисленные выше, такие как ISIS 2503, антисмысловая anti-ras, антисмысловая анти-EGFR, custirsen, apatorsen, ISIS-STAT3Rx (ISIS 481464/ AZD9150), ISIS-ARRx (AZD5312), Trabedersen (AP 12009), EZN-2968, LErafAON-ETU); подходы генной терапии, включая, например, подходы для замещения аберрантных генов (например, аберрантный р53 или аберрантный BRCA1 или BRCA2), подходы GDEPT (ген-направленная фермент-пролекарственная терапия, например, подходы, использующие цитозиндеаминазу, тимидинкиназу или бактериальный фермент нитроредуктазу), и подходы увеличения толерантности пациента к химиотерапии или радиотерапии (например, генная терапия множественной лекарственной устойчивости); и иммуннотерапевтические подходы, включая, например, ингибиторы контрольных точек, таких как PD-1/PD-L1 (ниволумаб, пембролизумаб, атезолизумаб, дурвалумаб, авелумаб, пидилизумаб и другие), а также препараты, воздействующие на CTLA-4 (в том числе ипилимумаб, тремелимумаб), ОХ-40, VISTA, ICOS, TIGIT, LAG-3, 4-1ВВ, GITR, CD40, CCR4 и другие; другие ex-vivo и in-vivo подходы для увеличения иммуногенности опухолевых клеток пациента, такие как трансфекция цитокинами, такими как интерлейкин 2, интерлейкин 4, интерлейкин 15 или гранулоцитарно-макрофагеальный колониестимулирующий фактор, подходы для уменьшения анергии Т-клеток, подходы, использующие трансфицированные иммунные клетки, такие как цитокин-трансфицированные дендритные клетки, подходы, использующие цитокинтрансфицированные опухолевые клеточные линии, подходы, использующие анти-идиотипические антитела, подходы к снижению функции иммуносупрессивных клеток, таких как регуляторные Т-клетки, миелоидные супрессорные клетки или IDO (индоламин-2,3,-дезоксигеназа)-экспрессирующие дендритные клетки, и подходы, использующие вакцины против рака, состоящие из белков или пептидов, имеющих происхождение из опухолеассоциированных антигенов, таких как NY-ESO-1, MAGE-3, WT1 или Her2/neu.
Таким образом, в еще одном варианте изобретения описан фармацевтический продукт, содержащий соединение формулы I или его фармацевтически приемлемую соль, сольват или стереоизомер, как определено выше, в сочетании с противоопухолевым средством, как определено выше, для совместного лечения рака.
Схемы приема лекарственных средств можно регулировать, чтобы обеспечить оптимальный желаемый ответ. Например, может быть введена одна доза, несколько разделенных доз могут быть введены в течение некоторого времени, или доза может быть пропорционально уменьшена или увеличена в зависимости от остроты терапевтической ситуации. Особенно полезным является изготовление пероральных композиций в стандартной лекарственной форме для простоты введения и однородности дозирования. Стандартная лекарственная форма при использовании в данном документе, относится к физически дискретным единицам, пригодным в качестве единичных доз для пациентов/субъектов, подлежащих лечению; каждая единица содержит заданное количество активного соединения, рассчитанное для получения желаемого терапевтического эффекта в сочетании с требуемым фармацевтическим носителем. Спецификация для стандартных лекарственных форм по настоящему изобретению, как правило, диктуется и непосредственно зависит от (а) уникальных характеристик терапевтического агента и конкретного терапевтического или профилактического эффекта, которые должны быть достигнуты, и (b) ограничений, присущих в технике компаундирования такого активного соединения для лечения чувствительности у субъектов.
Таким образом, квалифицированным специалистам понятно, исходя из раскрытия, представленного в данном документе, что дозы и режим дозирования корректируются в соответствии со способами, хорошо известными в терапевтической области. Это означает, что может быть легко установлена максимально переносимая доза и может быть также определено эффективное количество, обеспечивающее обнаруживаемый терапевтический эффект для пациента, так же как и требования к времени введения каждого агента для достижения видимого терапевтического эффекта для пациента. Таким образом, хотя некоторые дозы и схемы режима введения приведены в качестве примеров в данном документе, эти примеры никоим образом не ограничивают дозы и режимы введения, которые могут понадобиться для пациента в практике применения настоящего изобретения.
Следует отметить, что значения дозировки могут изменяться, в зависимости от типа и тяжести состояния, которое следует облегчить, и может включать одну или более доз. Кроме того, необходимо понимать, что для любого конкретного пациента, конкретные схемы введения должны быть скорректированы через некоторое время согласно индивидуальной потребности и на усмотрение медицинского работника, который осуществляет введение или контролирует введение композиций, и что диапазоны концентрации, приведенные в данном описании, приведены только в качестве примера и не предназначены для ограничения объема или практики заявленных композиций. Кроме того, режим дозирования с композициями по данному изобретению может быть основан на различных факторах, включая тип заболевания, возраст, вес, пол, состояния здоровья пациента, тяжесть состояния, путь введения и конкретное используемое соединение по настоящему изобретению. Таким образом, режим дозирования может широко варьироваться, но может определяться регулярно с помощью стандартных методов. Например, дозы могут быть скорректированы на основе фармакокинетических и фармакодинамических параметров, которые могут включать клинические эффекты, такие как токсические эффекты или лабораторные значения. Таким образом, настоящее изобретение охватывает индивидуальное повышение дозы, которое определяется квалифицированным специалистом. Определение необходимой дозы и режимы хорошо известны в соответствующей области техники и будут понятны специалисту в данной области после ознакомления с идеями, раскрытыми в данном документе.
Как правило, дозы, применяемые для лечения взрослого человека, обычно находятся в диапазоне 0,02-5000 мг в день или приблизительно от 1-1500 мг в день.
При улучшении состояния пациента вводится поддерживающая доза, если это необходимо. Впоследствии, дозировка или частота введения, или то и другое могут быть уменьшены, в зависимости от симптомов, до уровня, при котором поддерживается облегченное состояние болезни, нарушения или состояния. Пациентам может, однако, потребоваться периодическое лечение в течение долгого времени при любом рецидиве симптомов.
Вышеизложенный спектр является только предположительным, поскольку количество переменных в отношении индивидуального режима лечения велико, и значительные отклонения от этих рекомендованных значений являются весьма обычными. Эти дозировки могут быть изменены в зависимости от множества переменных, не ограниченных активностью применяемого соединения, болезни или состояния, подвергаемого лечению, способа введения, потребности индивидуального субъекта, тяжести болезни или состояния, подвергаемого лечению, и мнения лечащего врача.
Для наилучшего понимания изобретения приводятся следующие примеры. Эти примеры приведены только в иллюстративных целях и не должны толковаться как ограничивающие сферу применения изобретения в любой форме.
Все публикации, патенты и патентные заявки, указанные в этой спецификации включены в данный документ путем отсылки. Хотя вышеупомянутое изобретение было довольно подробно описано путем иллюстрации и примера в целях исключения двусмысленного толкования, специалистам в данной области на основе идей, раскрытых в данном изобретении, будет вполне понятно, что могут быть внесены определенные изменения и модификации без отклонения от сущности и объема прилагаемых вариантов осуществления изобретения.
Примеры
Пример 1. Способ получения соединения 4b.

Стадия 1. Получение соединения 4_2.
К раствору 4-фтор-2-метокси-5-нитроанилина 4_1 (10.0 г, 53.7 ммоль) и DMAP (0.33 г, 2.66 ммоль) в дихлорметане (100 мл) при 0°С по каплям в течение 30 мин добавили раствор ди-трет-бутилдикарбоната (16.2 г, 74.3 ммоль) в 30 мл дихлорметана. Реакционную смесь довели до комнатной температуры и перемешивали в течение 24 ч. Полученную смесь сконцентрировали в вакууме, продукт выделили с помощью колоночной хроматографии на силикагеле с использованием элюента дихлорметан/гексан (градиент от 1:1 до 1:0). Выход соединения 4_2 составил 6.40 г (42%).
Стадия 2. Получение соединения 4_3.
К раствору анилина 4_2 (6.10 г, 20.9 ммоль) в ДМФА (10 мл) при комнатной температуре добавили N,N,N'-триметилэтилендиамин (3.59 мл, 27.2 ммоль) и DIPEA (5.24 мл, 31.3 ммоль). Реакционную смесь перемешивали 2 ч при 60°С. Полученную смесь вылили в воду, продукт экстрагировали этилацетатом. Объединенные органические фракции промыли водой и насыщенным раствором NaCl, высушили над Na2SO4, профильтровали и сконцентрировали в вакууме. Выход соединения 4_3 составил 7.62 г (99%).
Стадия 3. Получение соединения 4_4.
К раствору соединения 4_3 (7.50 г, 20.2 ммоль) в метаноле (90 мл) добавили Pd/C (1.30 г, 3.02 ммоль) и восстанавливали под давлением водорода (2 атм.) в течение 1 ч. Реакционную смесь профильтровали и сконцентрировали в вакууме. Выход соединения 4_4 составил 6.69 г (98%).
Стадия 4. Получение соединения 4_5.
К смеси соединения 4_4 (6.69 г, 19.8 ммоль) и DIPEA (3.59 мл, 27.2 ммоль) в дихлорметане (150 мл) при 0°С по каплям (в течение 30 мин) добавили раствор акролоил хлорида (3.59 мл, 27.2 ммоль) в дихлорметане (80 мл). Полученную смесь перемешивали при комнатной температуре. Через 3.5 ч добавили дополнительные количества акролоил хлорида (0.38 мл, 4.60 ммоль) и DIPEA (0.80 мл, 4.60 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. К полученной смеси добавили насыщенный раствор Na2CO3, органический слой промыли насыщенным раствором NaCl, высушили над Na2SO4, профильтровали и сконцентрировали в вакууме. Продукт выделилули с помощью колоночной хроматографии на силикагеле с использованием элюента этилацетат/гексан/триэтиламин (градиент от 8:2:0 до 8:2:0.05). Выход соединения 4_5 составил 5.04 г (65%).
Стадия 5. Получение соединения 4b.
Соединение 4_5 (5.04 г, 12.8 ммоль) растворили в трифторуксусной кислоте (30 мл) и перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь по каплям добавили к насыщенному раствору Na2CO3, продукт экстрагировали этилацетатом. Объединенные органические фракции промыли водой и насыщенным раствором NaCl, высушили над Na2SO4, профильтровали и сконцентрировали в вакууме. Продукт выделили с помощью колоночной хроматографии на силикагеле с использованием элюента этилацетат/гексан/триэтиламин (градиент от 8:2:0 до 8:2:0.07). Выход соединения 4b составил 3.23 г (85%).
Пример 2. Способ получения соединений EGFR_3365_3, EGFR_3365_4, EGFR_3365_50, EGFR_3365_51, EGFR_3365_52, EGFR_3365_54, EGFR_3365_56, EGFR_3365_57, EGFR_3365_77, EGFR_3365_85, EGFR_3365_87, EGFR_3365_88, EGFR_3365_93.
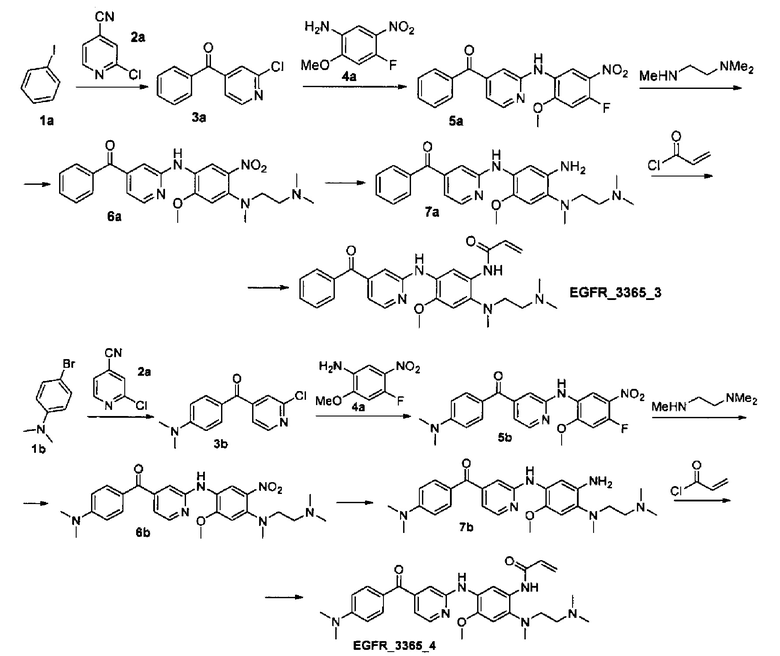
Стадия 1. Получение соединения 3а.
К раствору йодбензола 1а (8.35 г, 39.7 ммоль) в 350 мл диэтилового эфира добавили по каплям 2.5 М раствор н-бутиллития в гексане (15.9 мл, 39.7 ммоль) за 15 мин при -78°С в атмосфере азота. Затем температуру реакционной смеси довели до 0°С и перемешивали при данной температуре в течение 30 мин. Далее реакционную смесь вновь охладили до -78°С и медленно добавили раствор нитрила 2а (5.00 г, 36.1 ммоль) в 50 мл диэтилового эфира. Полученную смесь перемешивали 1 час при -78°С, затем температуру смеси довели до -30°С и добавили 100 мл 2М HCl. Реакционную смесь перемешивали в течение 1 ч, затем нейтрализовали 1М раствором NaOH, продукт экстрагировали этилацетатом. Объединенные органические фракции высушили над Na2SO4, профильтровали и сконцентрировали в вакууме. Продукт выделили с помощью колоночной хроматографии на силикагеле с использованием элюента этилацетат/гексан (1:4). Выход соединения 3а составил 7.04 г (90%).
Аналогично получено соединение 3b из соответствующего исходного реагента 1b.
Стадия 2. Получение соединения 5а.
К раствору соединения 3а (0.100 г, 0.46 ммоль) в 2 мл 1,4-диоксана добавили Cs2CO3 (0.300 г, 0.92 ммоль), BINAP (0.057 г, 0.09 ммоль) и анилин 4а (0.094 г, 0.51 ммоль) при комнатной температуре. Полученный раствор дегазировали азотом в течение 10 мин. Затем к реакционной смеси при перемешивании добавили Pd(OAc)2 (0.010 г, 0.05 ммоль). Полученную смесь кипятили 3 ч в атмосфере азота. Затем смесь разбавили 10 мл дихлорметана и профильтровали через целит. Фильтрат сконцентрировали в вакууме, продукт выделили с помощью колоночной хроматографии на силикагеле с использованием дихлорметана в качестве элюента. Выход соединения 5а составил 0.112 г (66%).
Аналогично получено соединение 5b из соответствующего промежуточного 3b.
Стадия 3. Получение соединения 6а.
К раствору соединения 5а (0.110 г, 0.3 ммоль) в 3 мл диметилформамида добавили N,N,N'-триметилэтан-1,2-диамин (0.034 г, 0.33 ммоль) и DIPEA (0.077 г, 0.6 ммоль). Реакционную смесь перемешивали 12 часов при комнатной температуре. Полученную смесь сконцентрировали, продукт выделили при помощи колоночной хроматографии на силикагеле с использованием элюента дихлорметан/этилацетат (8:1) с градиентом триэтиламина (от 0%об. до 10%об.). Выход соединения 6а составил 0.127 г (95%).
Аналогично получено соединение 6b из соответствующего промежуточного 5b.
Стадия 4. Получение соединения 7а.
К раствору соединения 6а (0.127 г, 0.28 ммоль) в 9 мл смеси тетрагидрофуран/метанол/вода (3:1:5) добавили NaHCO3 (0.214 г, 2.54 ммоль). К полученной смеси добавили Na2S2O4 (0.442 г, 2.54 ммоль) по частям в течение 30 мин при 0°С. Полученную смесь перемешивали 15 мин при комнатной температуре. Далее смесь разбавили водой, продукт экстрагировали этилацетатом. Объединенные органические фракции высушили над Na2SO4, профильтровали и сконцентрировали в вакууме. Продукт выделили с помощью колоночной хроматографии на силикагеле с использованием элюента дихлорметан/этилацетат (3:1) с градиентом триэтиламина (от 0.5%об. до 5%об.). Выход соединения 7а составил 0.107 г (90%).
Аналогично получено соединение 7b из соответствующего промежуточного 6b.
Стадия 5. Получение соединения EGFR_3365_3.
К раствору соединения 7а (0.107 г, 0.26 ммоль) в 5 мл дихлорметана добавили DIPEA (0.035 г, 0.27 ммоль). К полученной смеси добавили по каплям раствор акрилоил хлорида (0.024 г, 0.26 ммоль) в 3 мл дихлорметана при -70°С за 1 ч. Температуру реакционной смеси довели до -30°С и перемешивали при данной температуре в течение 30 мин. К полученной смеси добавили насыщенный раствор NaHCO3, продукт экстрагировали дихлорметаном. Объединенные органические фракции промыли насыщенным раствором NaCl, высушили над Na2SO4, профильтровали и сконцентрировали в вакууме. Продукт выделили с помощью колоночной хроматографии на силикагеле с использованием элюента дихлорметан/гексан/этилацетат (3:1:1) с градиентом триэтиламина (от 0.5%об. до 3%об.). Выход соединения EGFR_3365_3 составил 0.055 г (45%).
Аналогично получено соединение EGFR_3365_4 из соответствующего промежуточного 7b (продукт был дополнительно очищен с помощью препаративной хроматографии). Соединение EGFR_3365_4a было получено с помощью повторной лиофилизации соединения EGFR_3365_4.
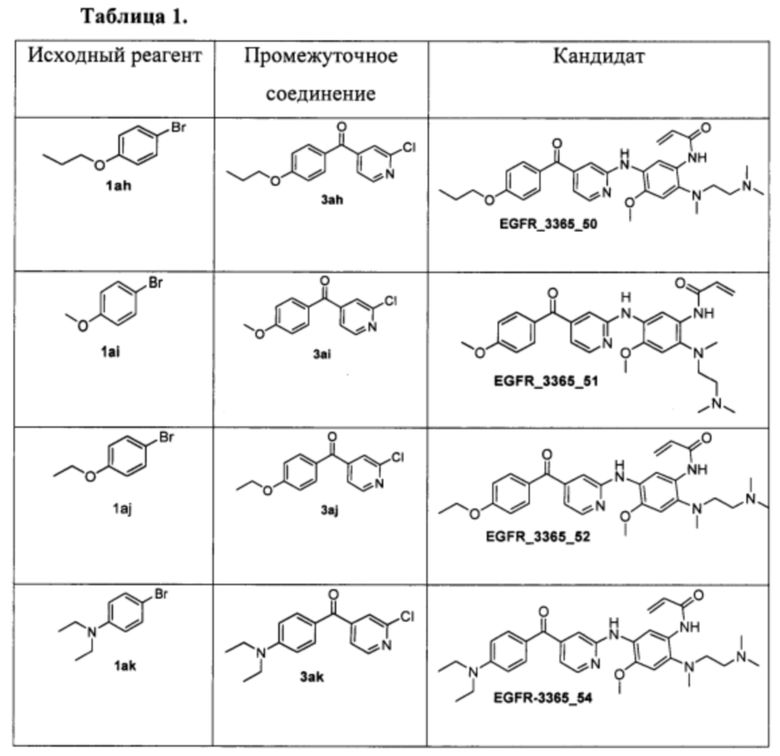
Аналогично соединению 5а получены кандидаты EGFR_3365_50, EGFR_3365_51, EGFR_3365_52, EGFR_3365_54, EGFR_3365_56, EGFR_3365_57, EGFR_3365_77, EGFR_3365_85, EGFR_3365_87, EGFR_3365_88, EGFR_3365_93 с использованием анилина 4b вместо 4a из соответствующих исходных реагентов через соответствующие промежуточные соединения, указанные в таблице 1.

Пример 3. Способ получения соединения EGFR_3365_5, EGFR_3365_15, EGFR_3365_26, EGFR_3365_73, EGFR_3365_101, EGFR_3365_102, EGFR_3365_103, EGFR_3365_104, EGFR_3365_116, EGFR_3365_124.
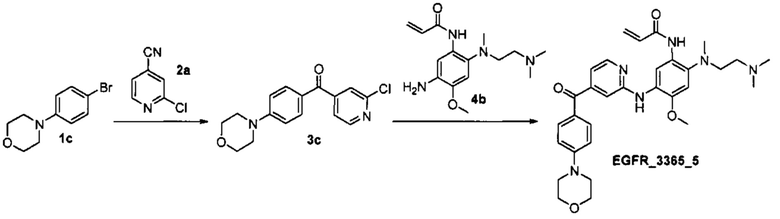
Стадия 1. Получение соединения 3с.
К суспензии магния (0.061 г, 2.52 ммоль) в 20 мл тетрагидрофурана добавили арилбромид 1с (0.5 г, 2.06 ммоль) и каталитическое количество дибромэтана. Полученную суспензию кипятили в течение 2.5 ч, затем охладили до комнатной температуры и добавили нитрил 2а (0.277 г, 2.0 ммоль). Реакционную смесь перемешивали при комнатной температуре двое суток, затем разбавили насыщенным раствором NH4Cl, органический слой отделили и сконцентрировали. Остаток растворили в 10 мл диэтилового эфира, к раствору добавили 40 мл 1М HCl, полученную смесь перемешивали в течение 30 минут при комнатной температуре. Водный слой отделили и нейтрализовали насыщенным раствором NaHCO3, продукт экстрагировали дихлорметаном, объединенные органические фракции промыли насыщенным раствором NaCl, высушили над Na2SO4, профильтровали и сконцентрировали в вакууме. Продукт выделили с помощью колоночной хроматографии на силикагеле с использованием элюента этилацетат/гексан (1:4). Выход соединения 3 с составил 0.407 г (65%).
Стадия 2. Получение соединения EGFR_3365_5.
Соединение EGFR_3365_5 было получено аналогично соединению 5а (стадия 2), используя соединение 3с вместо соединения 3а и анилин 4b вместо анилина 4а. Соединение EGFR_3365_5a было получено с помощью повторной лиофилизации соединения EGFR_3365_5.
Аналогично были получены соединения EGFR_3365_15, EGFR_3365_26, EGFR_3365_73, EGFR_3365_101, EGFR_3365_102, EGFR_3365_103, EGFR_3365_104, EGFR_3365_116, EGFR_3365_124 из соответствующих исходных реагентов через соответствующие промежуточные соединения, указанные в таблице 2.


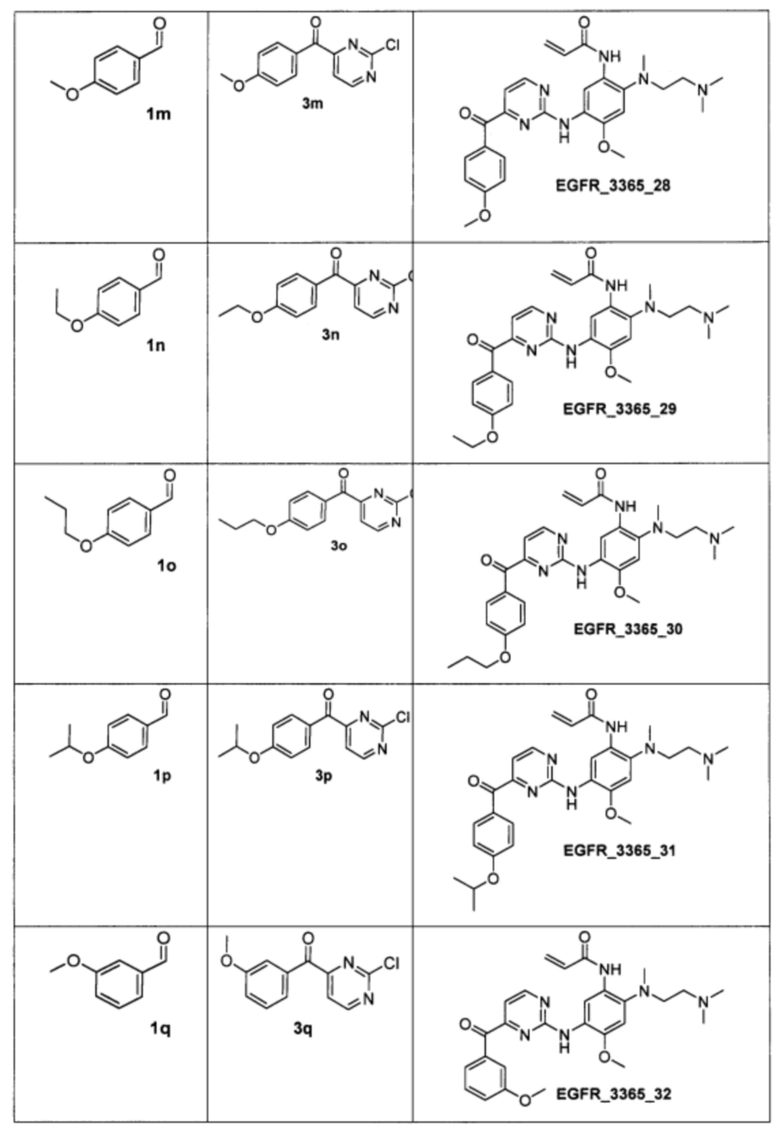
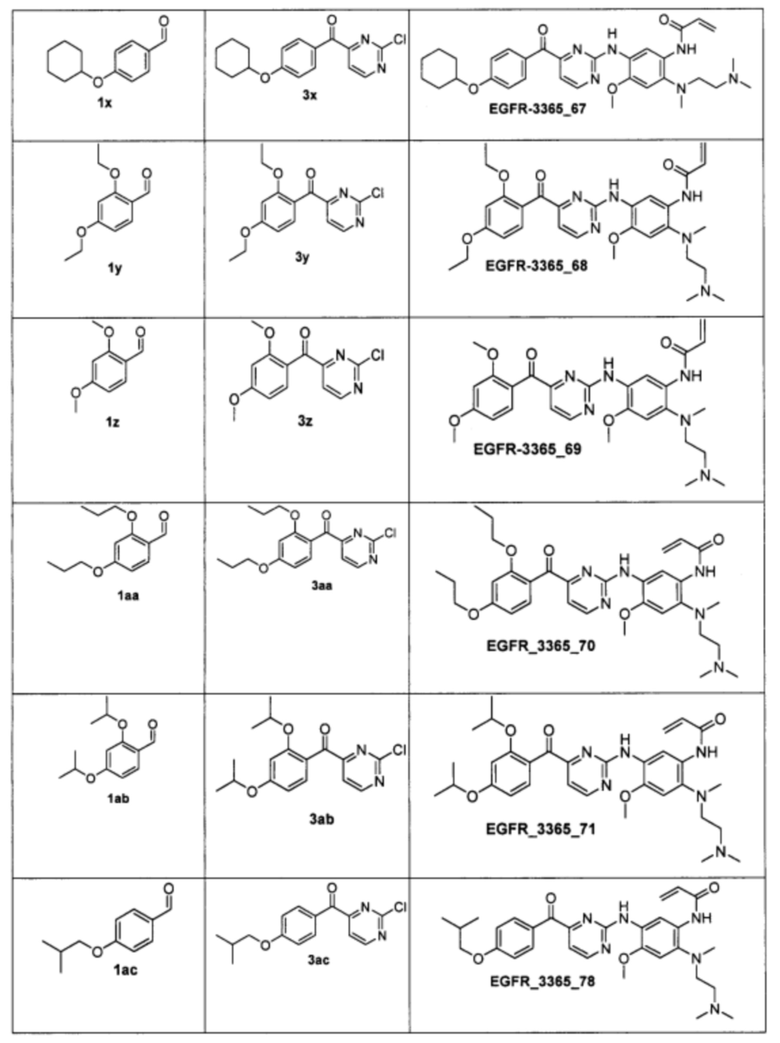
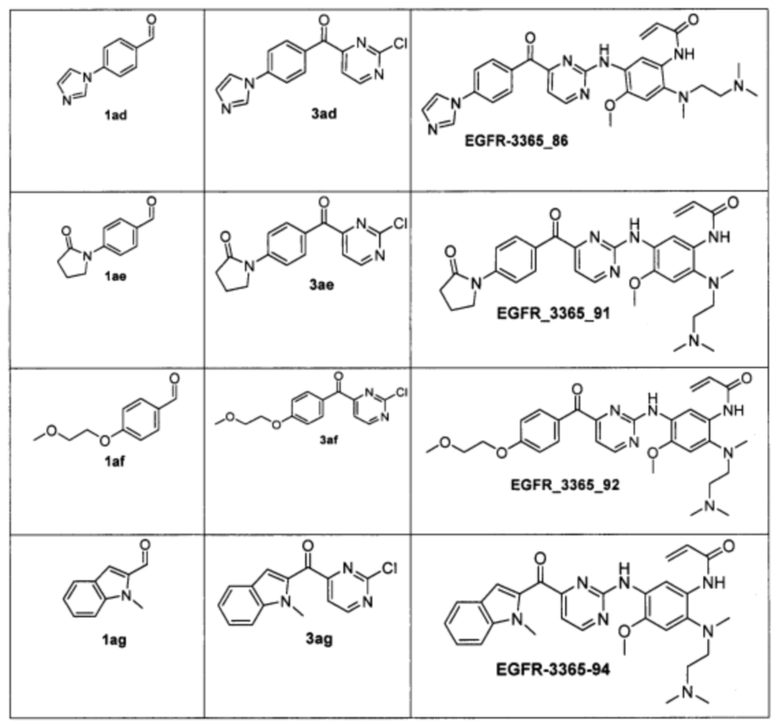
Пример 4. Способ получения соединения EGFR_3365_10, EGFR_3365_11, EGFR_3365_12, EGFR_3365_13, EGFR_3365_14, EGFR_3365_14a, EGFR_3365_16, EGFR_3365_17, EGFR_3365_28, EGFR_3365_29, EGFR_3365_30, EGFR_3365_31, EGFR_3365_32, EGFR_3365_33, EGFR_3365_34, EGFR_3365_36, EGFR_3365, EGFR_3365_64, EGFR_3365_66, EGFR_3365_67, EGFR_3365_68, EGFR_3365_69, EGFR_3365_70, EGFR_3365_71, EGFR_3365_78, EGFR_3365_86, EGFR_3365_91, EGFR_3365_92, EGFR_3365_94.
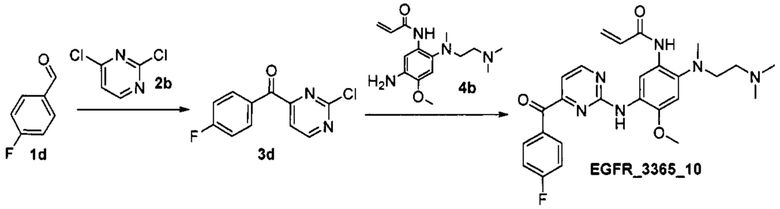
Стадия 1. Получение соединения 3d.
К раствору альдегида 1d (0.478 г, 3.85 ммоль) в 10 мл ДМФА добавили 2,4-дихлорпиримидин 2b (0.500 г, 3.35 ммоль) и N,N'-диметилимидазолий йодид (0.375 г, 1.67 ммоль) при комнатной температуре. Полученный раствор дегазировали азотом в течение 10 мин, затем добавили NaH (60% масс., 0.201 г, 5.02 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Полученную смесь вылили в холодную воду, продукт экстрагировали этилацетатом. Объединенные органические фракции промыли водой и насыщенным раствором NaCl, высушили над Na2SO4, профильтровали и сконцентрировали в вакууме. Продукт выделили с помощью колоночной хроматографии на силикагеле с использованием элюента этилацетат/гексан (1:4). Выход соединения 3d составил 0.166 г (21%).
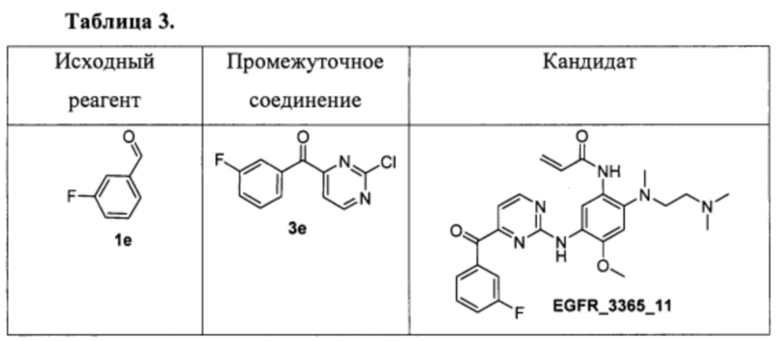
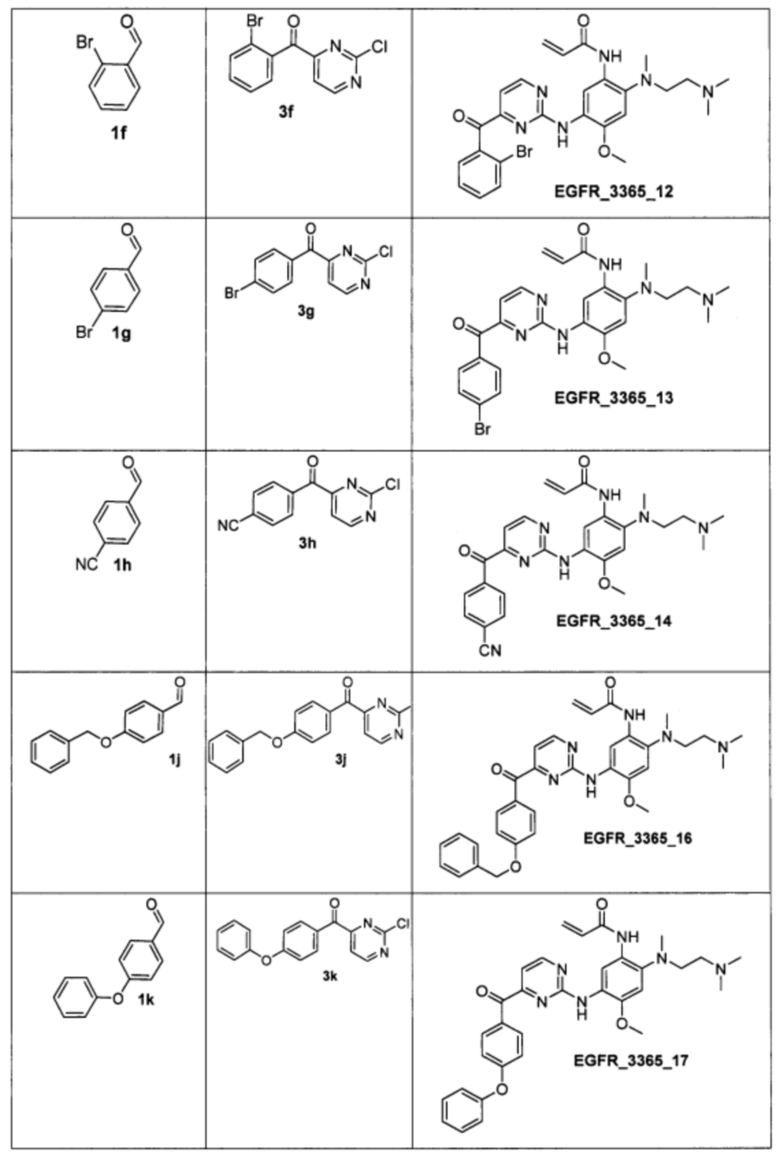
Аналогично были получены соединения 3е - 3u (приведены в таблице 2). Для синтеза соединений 3f и 3g в качестве растворителя использовалась смесь 1,4-диоксана и ДМСО в соотношении 10:1.
Стадия 2. Получение соединения EGFR_3365_10.
К раствору соединения 3d (0.114 г, 0.48 ммоль) в 2 мл изопропилового спирта добавили анилин 4b (0.115 г, 0.53 ммоль) и трифторуксусную кислоту (0.185 мл, 2.64 ммоль) при комнатной температуре. Реакционную смесь кипятили в течение 24 часов. К полученной смеси добавили насыщенный раствор NaHCO3 и дихлорметан. Органический слой промыли насыщенным раствором NaCl, высушили над Na2SO4, профильтровали и сконцентрировали в вакууме. Продукт выделили с помощью колоночной хроматографии на силикагеле с использованием элюента дихлорметан/гексан/этилацетат (3:1:1) с градиентом триэтиламина (от 0.5%об. до 3%об.). Выход соединения EGFR_3365_10 составил 54 мг (23%). Продукт был дополнительно очищен с помощью препаративной хроматографии.
Аналогично были получены соединения EGFR_3365_11, EGFR_3365_12, EGFR_3365_13, EGFR_3365_14, EGFR_3365_14a, EGFR_3365_16, EGFR_3365_17, EGFR_3365_28, EGFR_3365_29, EGFR_3365_30, EGFR_3365_31, EGFR_3365_32, EGFR_3365_33, EGFR_3365_34, EGFR_3365_36, EGFR_3365, EGFR_3365_64, EGFR_3365_66, EGFR_3365_67, EGFR_3365_68, EGFR_3365_69, EGFR_3365_70, EGFR_3365_71, EGFR_3365_78, EGFR_3365_86, EGFR_3365_91, EGFR_3365_92, EGFR_3365_94 из соответствующих исходных реагентов через соответствующие промежуточные соединения, указанные в таблице 3.

Пример 5. Способ получения соединения EGFR_3365_31a, EGFR_3365_30a.

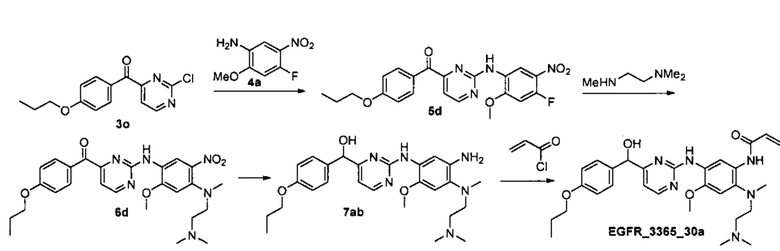
Стадия 1. Получение соединения 5с.
Соединение 5с было получено аналогично соединению EGFR_3365_10, используя соединение 3p вместо соединения 3d и анилин 4а вместо анилина 4b.
Аналогично было получено соединение 5d из соответствующего исходного 3о.
Стадия 2. Получение соединения 6с.
Соединение 6с было получено аналогично соединению 6а, используя соединение 5с вместо соединения 5а.
Аналогично было получено соединение 6d из соответствующего промежуточного 5d.
Стадия 3. Получение соединения 7аа.
К раствору соединения 6с (4.1 г, 7.82 ммоль) в 120 мл смеси метанол/вода (1:2) добавили Na2S2O4 (13.75 г, 78.2 ммоль). Реакционную смесь перемешивали 1 час при 40°C. Полученную смесь сконцентрировали, продукт экстрагировали дихлорметаном, объединенные органические фракции высушили над Na2SO4, профильтровали и сконцентрировали в вакууме. Продукт выделили с помощью колоночной хроматографии на силикагеле с использованием элюента дихлорметан/метанол (градиент от 9:1 до 1:1). Выход соединения 7аа составил 3.6 г (96%), коричневое маслообразное вещество.
Аналогично было получено соединение 7ab из соответствующего промежуточного 6d.
Стадия 4. Способ получения соединения EGFR_3365_31a.
К раствору соединения 7аа (2.0 г, 4.03 ммоль) в 40 мл дихлорметана при -70°C в атмосфере азота добавили по каплям раствор N,N-диизопропилэтиламина (0.63 г, 4.84 ммоль) в 5 мл дихлорметана, затем раствор акрилоилхлорида (0.33 г, 3.63 ммоль) в 5 мл дихлорметана. Реакционную смесь перемешивали в течение 2 ч при -40°C, затем при этой же температуре добавили воду, продукт экстрагировали дихлорметаном, объединенные органические фракции высушили над Na2SO4, профильтровали и сконцентрировали в вакууме. Продукт выделили с помощью колоночной хроматографии на силикагеле с использованием элюента дихлорметан/метанол (градиент от 99:1 до 90:1). Выход соединения EGFR_3365_31a составил 0.81 г (37%), белый порошок.
Аналогично было получено соединение EGFR_3365_30a из соответствующего промежуточного 7ab.
Пример 6. Способ получения соединения EGFR_3365_63, EGFR_3365_58, EGFR_3365_61, EGFR_3365_62, EGFR_3365_62a, EGFR_3365_72, EGFR_3365_90, EGFR_3365_97, EGFR_3365_98, EGFR_3365_105, EGFR_3365_106, EGFR_3365_106a, EGFR_3365_108, EGFR_3365_109, EGFR_3365_110, EGFR_3365_111, EGFR_3365_112, EGFR_3365_112a, EGFR_3365_113, EGFR_3365_114, EGFR_3365_115, EGFR_3365_121a.
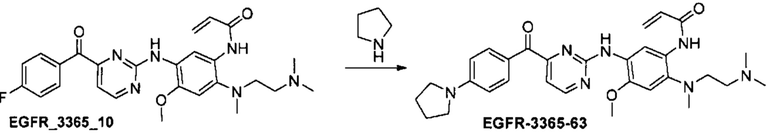
К раствору соединения EGFR_3365_10 (102 мг, 0.21 ммоль) и пирролидина (17 мкл, 0.21 ммоль, 1 экв.) в ДМФА (1 мл) добавили K2CO3 (36 мг, 0.258 ммоль, 1.25 экв.). Полученную суспензию перемешивали при 80°C в течение 12 часов. Реакционную массу вылили в воду, продукт экстрагировали этилацетатом. Объединенные органические фракции высушили над Na2SO4, профильтровали и сконцентрировали в вакууме. Продукт выделили с помощью колоночной хроматографии на силикагеле с использованием элюента этилацетат/триэтиламин (1/0.075). Выход соединения EGFR_3365_63 составил 72 мг. Продукт был дополнительно очищен с помощью препаративной ВЭЖХ, в результате было получено 48 мг (43%).
Аналогично были получены соединения EGFR_3365_58, EGFR_3365_61, EGFR_3365_62, EGFR_3365_62a, EGFR_3365_72, EGFR_3365_90, EGFR_3365_97, EGFR_3365_98, EGFR_3365_105, EGFR_3365_106, EGFR_3365_106a, EGFR_3365_108, EGFR_3365_109, EGFR_3365_110, EGFR_3365_111, EGFR_3365_112, EGFR_3365_112a, EGFR_3365_113, EGFR_3365_114, EGFR_3365_115, EGFR_3365_121a из соответствующих исходных реагентов, указанных в таблице 4.



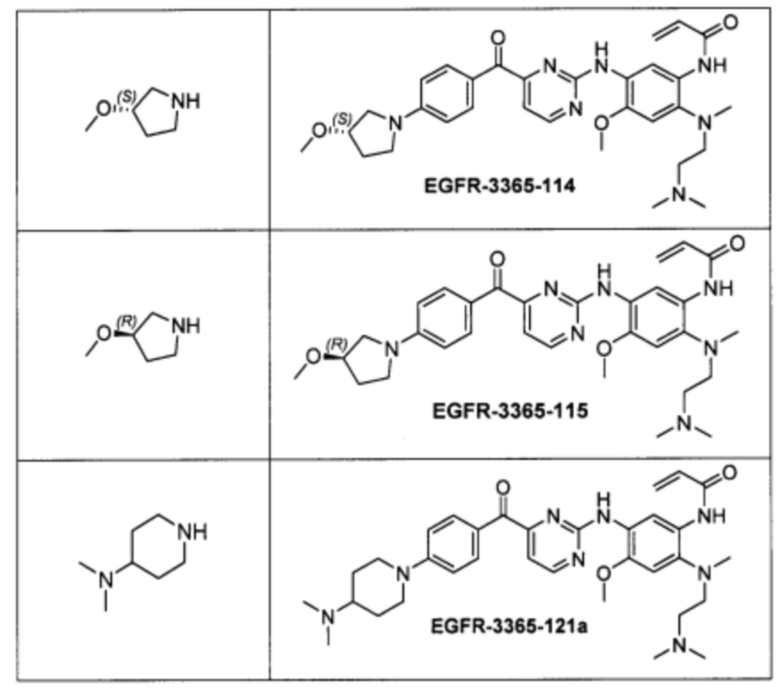
Пример 7. Способ получения соединения EGFR_3365_120, EGFR_3365_122, EGFR_3365_123, EGFR_3365_127.
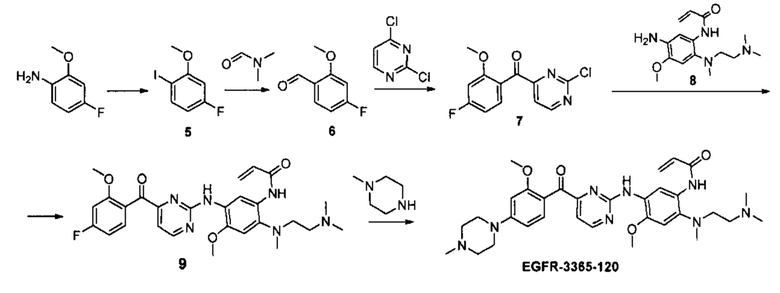
Стадия 1. Получение соединения 5.
4-Фтор-2-метоксианилин (2.50 г, 17.5 ммоль) растворили в 14 мл HClконц., охладили до 0°C, затем прибавили при перемешивании раствор NaNO2 (1.45 г, 21.0 ммоль) в 13 мл воды. Перемешивание продолжали в течение 40 мин, полученный раствор прибавили по каплям к раствору KI (8.73 г, 52.6 ммоль) в 30 мл воды при комнатной температуре. Полученную смесь перемешивали при 35-40°C в течение 1 ч, продукт экстрагировали этилацетатом (3×70 мл), экстракт промыли раствором Na2S2O3, высушили над Na2SO4. Продукт был выделен при помощи хроматографии на силикагеле с использованием гексана в качестве элюента. Получили 3.46 г (78%) 2-иод-5-фторанизола 5 в виде твердой бесцветной массы.
Стадия 2. Получение соединения 6.
2-Иод-5-фторанизол 5 (3.46 г, 13.0 ммоль) растворили в 70 мл ТГФ, охладили реакционную массу в атмосфере азота до -10°C, прибавили 2 М раствор изопропилмагний хлорида в ТГФ (8.50 мл, 17.0 моль), продолжали перемешивание 30 мин, прибавили ДМФ (2.89 г, 39.1 ммоль), продолжали перемешивание в течение 30 мин, нагрели реакционную массу до комнатной температуры, прибавили 20 мл насыщенного водного раствора NH4Cl. Полученную смесь экстрагировали этилацетатом (3×70 мл), экстракт промывали водой (5×20 мл), высушили над Na2SO4. Продукт был выделен при помощи хроматографии на силикагеле, элюент этилацетат-гексан (1:9). Получили 1.9 г (94%) 2-метокси-4-фторбензальдегида 6.
Стадия 3. Получение соединения 7.
2-Метокси-4-фторбензальдегид (0.40 г, 2.60 ммоль), 2,4-дихлорпиримидин (0.59 г, 3.89 ммоль), 1,3-диметилимидазолий иодид (0.31 г, 1.30 ммоль) растворили в 9 мл ДМФ, пропускали ток азота через реакционную массу в течение 2 мин, прибавили NaH в виде 60% суспензии в парафине (0.13 г, 2.24 ммоль), полученную смесь перемешивали в атмосфере азота при 75°C в течение 4 ч. Затем прибавили к реакционной массе 2,4-дихлорпиримидин (0.3 г, 1.95 ммоль), 1,3-диметилимидазолий иодид (0.16 г, 0.65 ммоль) и NaH (0.065 г, 1.12 ммоль) и продолжали нагревание при перемешивании в течение 1 ч, прибавили NaH (0.065 г, 1.12 ммоль) и продолжали нагревание еще 2 ч. К полученной смеси прибавили 20 мл воды, продукт экстрагировали этилацетатом (3×50 мл), экстракт промыли водой (4×10 мл), высушили над Na2SO4. Продукт был выделен при помощи хроматографии на силикагеле, элюент этилацетат-гексан (1:9). Получили 0.21 г (30%) соединения 7.
Стадия 4. Получение соединения 9.
Соединение 7 (0.12 г, 0.45 ммоль), анилин 8 (0.28 г, 0.90 ммоль), BINAP (0.057 г, 0.09 ммоль), Cs2CO3 (0.44 г, 1.35 ммоль) и Pd(OAc)2 (10 мг, 0.05 ммоль, 10 мольн. %) растворили в 3.6 мл диоксана и поместили в сосуд с завинчивающейся крышкой. Реакционную массу дегазировали током азота в течение 5 мин, перемешивали в атмосфере азота при 90°C в течение 4 ч. Продукт был выделен при помощи хроматографии на силикагеле, элюент триэтиламин-этилацетат-гексан (2:40:10). Получили 0.18 г (69%) соединения 9.
Стадия 5. Получение соединения EGFR_3365_120.
В сосуд с завинчивающейся крышкой поместили соединение 9 (0.10 г, 0.17 ммоль), K2CO3 (0.029 г, 0.21 ммоль), 1 мл ДМСО и N-метилпиперазин (0.019 г, 0.19 ммоль) в указанной последовательности. Реакционную массу перемешивали при 80°C в течение 5 ч. К полученной смеси прибавили 5 мл воды, продукт экстрагировали этилацетатом (3×30 мл), экстракт промыли водой (3×5 мл), высушили над Na2SO4. Продукт был выделен при помощи хроматографии на силикагеле, элюент - метанол, затем метанол-триэтиламин (100:1). Получили 0.054 г (52%) соединения EGFR_3365_120.
Аналогично были получены соединения EGFR_3365_122, EGFR_3365_123 и EGFR_3365_127 из соответствующих исходных реагентов, указанных в таблице 5.


Пример 8. Способ получения соединения EGFR_3365_121, EGFR_3365_126.
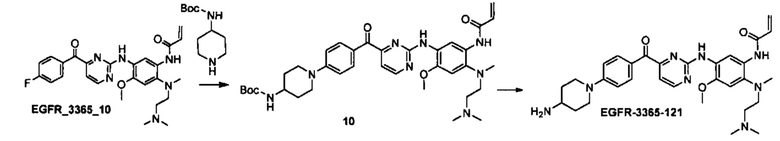
Стадия 1. Получение соединения 10.
Соединение 10 было получено аналогично соединению EGFR_3365_63, используя соединение трет-бутил-пиперидин-4-илкарбамат вместо пирролидина.
Стадия 2. Получение соединения EGFR_3365_121.
К раствору соединения 9 (0.051 г, 0.07 ммоль) в дихлорметане (7 мл) при комнатной температуре добавили трифторуксусную кислоту (1.8 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Полученную смесь аккуратно вылили в насыщенный раствор Na2CO3 (дегазированный). Продукт экстрагировали этилацетатом (3×15 мл), объединенные органические фракции промыли водой (2×10 мл), насыщенным раствором NaCl (1×10 мл), высушили над Na2SO4, профильтровали и сконцентрировали в вакууме. Полученную субстанцию (0.032 г, 80%) очистили с помощью препаративной ВЭЖХ. Полученный образец обработали насыщенным раствором Na2CO3 (дегазирован) до слабощелочной среды, продукт экстрагировали этилацетатом (дегазирован) (3×15 мл). Объединенные органические фракции промыли насыщенным раствором соли (1×10 мл). Полученный экстракт высушили над Na2SO4, профильтровали и сконцентрировали в вакууме. После лиофилизации получили 0.03 г (75%) кристаллического вещества желтого цвета.
Аналогично было получено соединение EGFR_3365_126 из соответствующего промежуточного соединения 9 и трет-бутил-пиперидин-4-илкарбамата.
Пример 9. Способ получения соединения EGFR_3365_53.
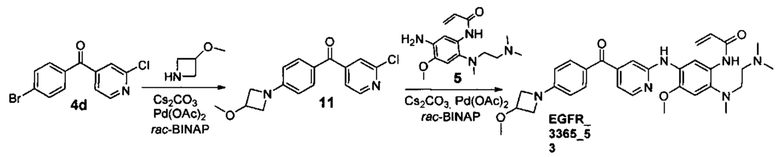
Стадия 1. Получение соединения 11.
К смеси, содержащей кетон 4d (200 мг, 0.65 ммоль), 3-метоксиазетидин гидрохлорид (74 мг, 0.58 ммоль, 0.9 экв), rac-BINAP (82 мг, 0.013 ммоль, 0.2 экв), Cs2CO3 (646 мг, 1.96 ммоль, 3 экв) в сухом 1,4-диоксане (14 мл) под током аргона добавили Pd(OAc)2 (15 мг, 0.06 ммоль, 0.1 экв). Реакционную смесь перемешивали в течение 3 часов при 90°C. К полученной смеси добавили воду (45 мл), продукт экстрагировали этилацетатом (3×15 мл). Объединенные органические фракции высушили над Na2SO4, профильтровали и сконцентрировали в вакууме. Продукт выделили с помощью колоночной хроматографии на силикагеле с использованием элюента этилацетат/гексан (10/90). Выход соединения 7 составил 110 мг (45%), желтый порошок.
Стадия 2. Получение соединения EGFR_3365_53.
К смеси, содержащей соединение 11 (100 мг, 0.29 ммоль), блок 5 (79 мг, 0.27 ммоль, 0.9 экв), rac-BINAP (37 мг, 0.059 ммоль, 0.2 экв), Cs2CO3 (293 мг, 0.89 ммоль, 3 экв) в сухом 1,4-диоксане (7 мл) под током аргона добавили Pd(OAc)2 (7 мг, 0.03 ммоль, 0.1 экв). Реакционную смесь перемешивали в течение 4 часов при 90°C. К полученной смеси добавили воду (30 мл), продукт экстрагировали этилацетатом (3×10 мл). Объединенные органические фракции высушили над Na2SO4, профильтровали и сконцентрировали в вакууме. Продукт выделили с помощью колоночной хроматографии на силикагеле с использованием элюента этилацетат/триэтиламин (98/2). Выход EGFR_3365_53 составил 40 мг (24%), желтый порошок.
Пример 10. Анализ полученных соединений.
Чистота и строение полученных соединений была подтверждена методом хромато-масс-спектрометрии LC/MS и спектроскопии 1Н ЯМР (Табл. 6).
Данные на оборудование:





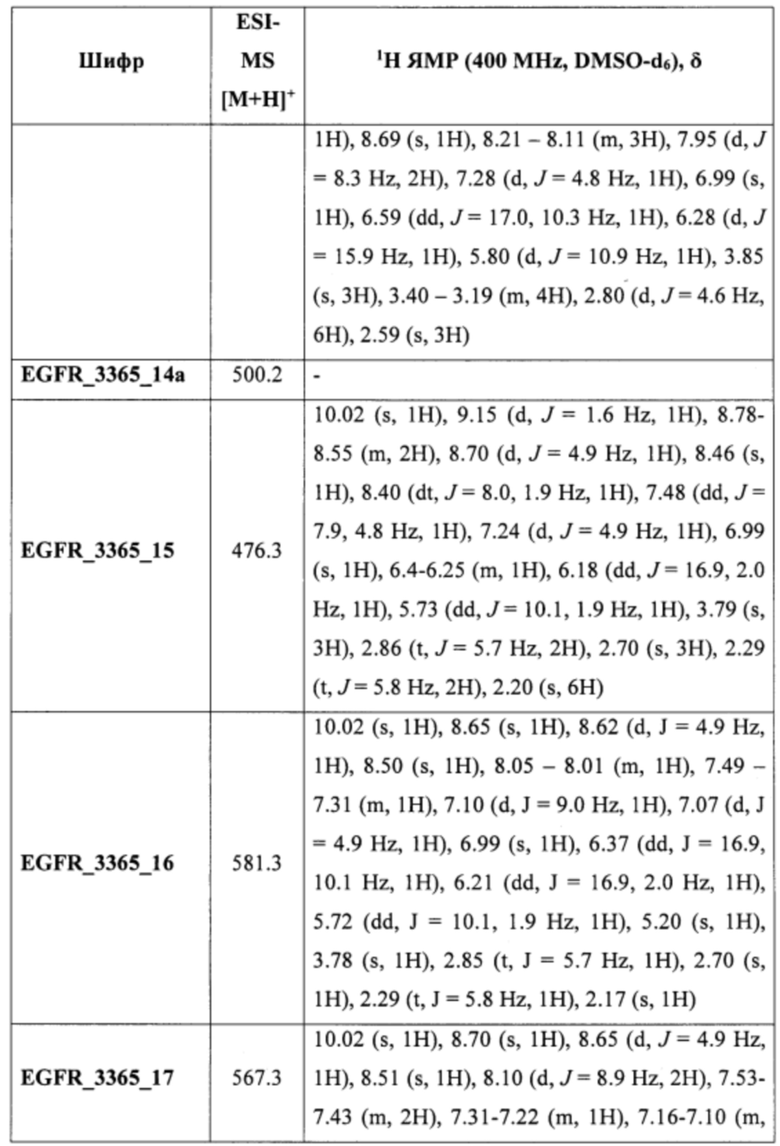
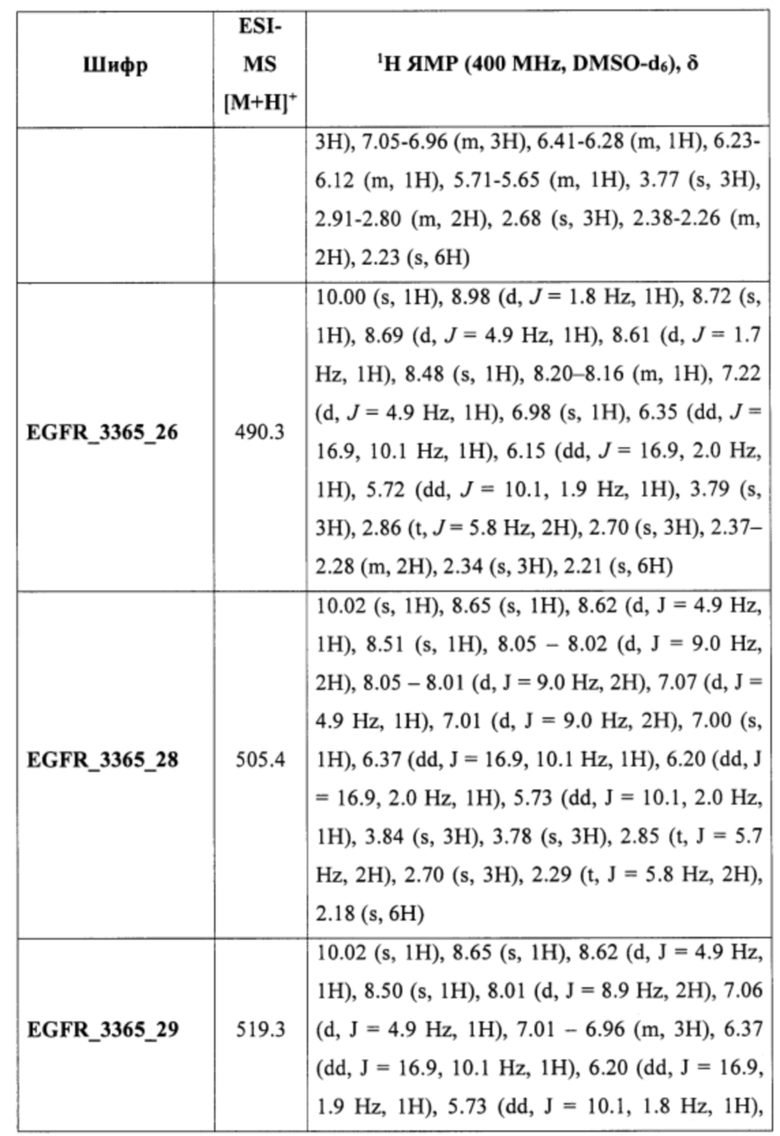













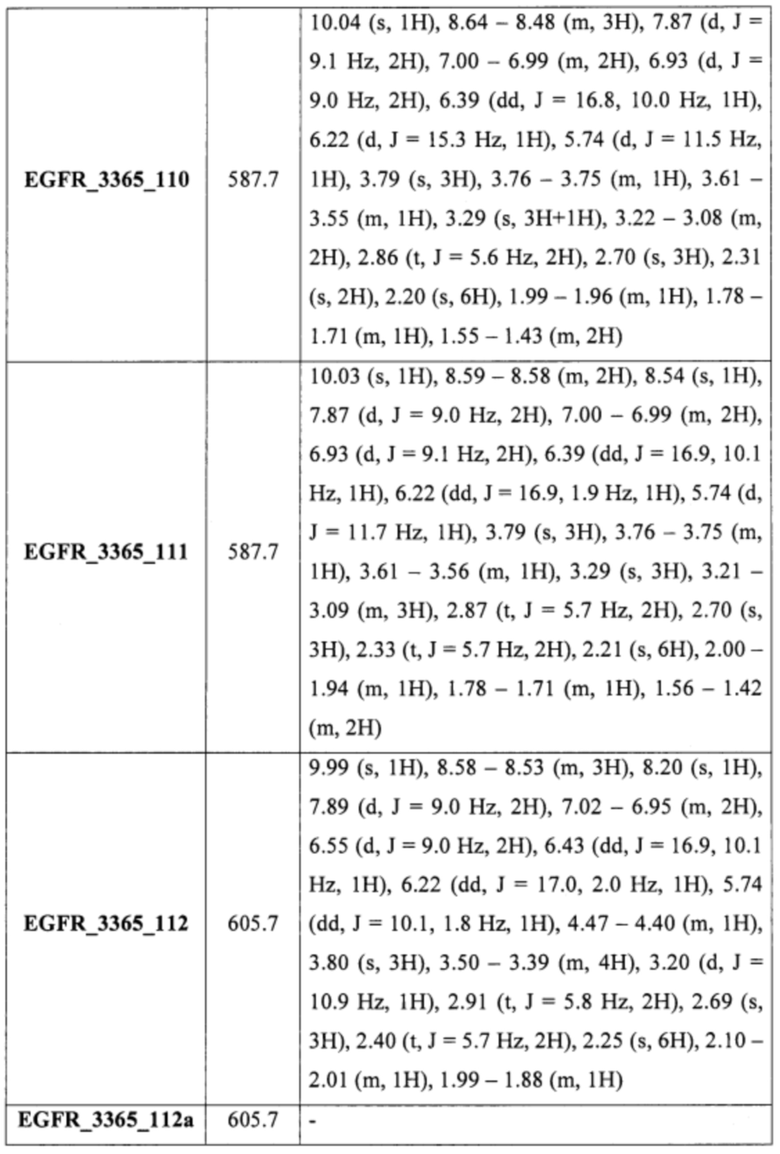
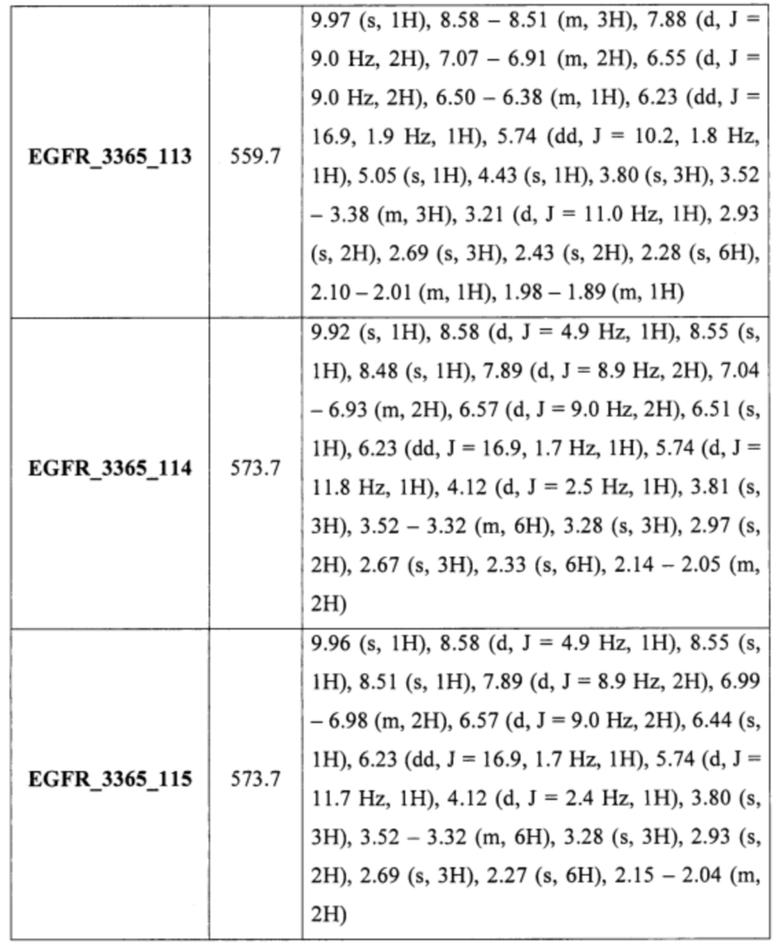
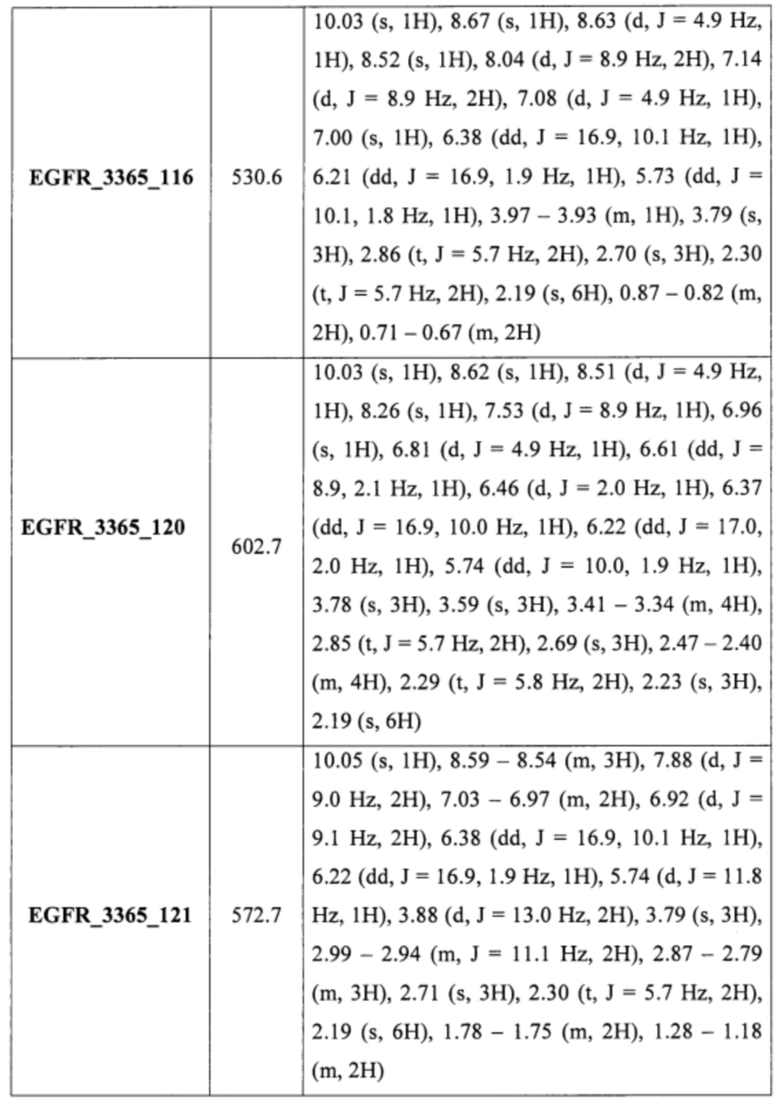
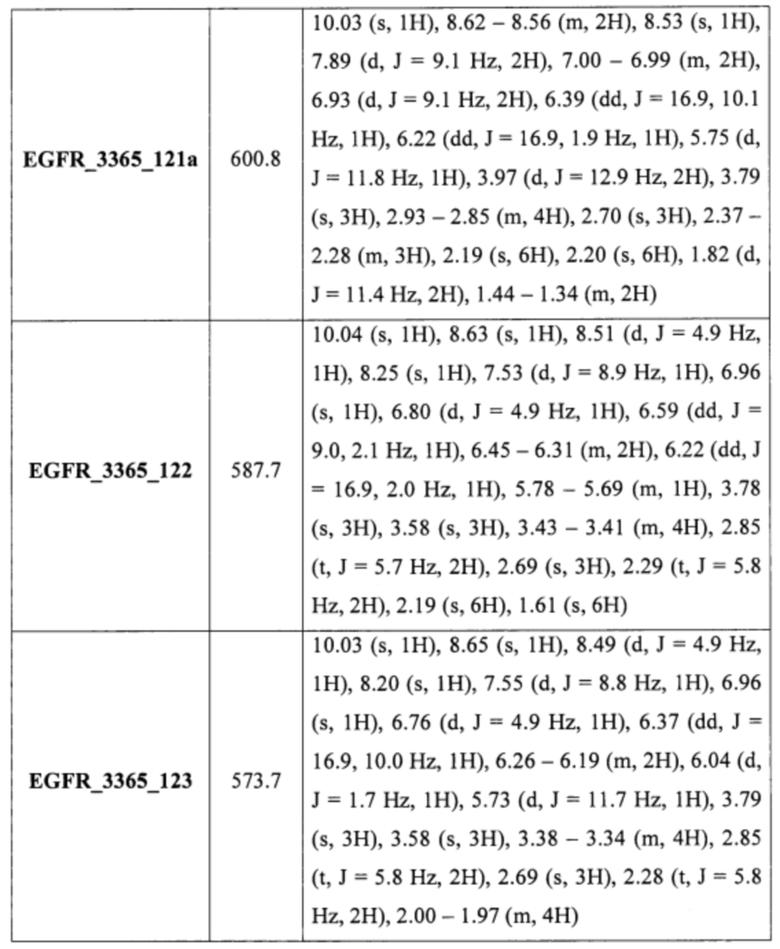

Пример 11. Определение химической стабильности в искусственных биологических средах.
1) Определение химической стабильности в искусственном кишечном соке, искусственном желудочном соке и плазме крови человека.
В качестве искусственного желудочного сока использовали SGF без ферментов концентрат, рН=1,4 (Sigma Ireland, cat#01651). Исходный раствор кандидата (10 мМ в ДМСО) разводили рабочим раствором SGF до концентрации 10 мкМ (испытуемый раствор). Испытуемый раствор выдерживали в твердотельном термостате 2 часа при температуре 37°C. В качестве искусственного кишечного сока использовали SIF без ферментов концентрат, рН=6.5 (Sigma Ireland, cat#55331). Исходный раствор кандидата (10 мМ в ДМСО) разводили рабочим раствором SIF до концентрации 10 мкМ (испытуемый раствор). Испытуемый раствор выдерживали в твердотельном термостате 2 часа при температуре 37°C. Методом ВЭЖХ с использованием хроматографа Agilent1200 (Agilent, США) определяли площади пиков соединений в испытуемых образцах, соответствующие начальному времени испытания (до выдерживания) и конечному времени испытания (после выдерживании в твердотельном термостате 2 часа при температуре 37°C). Хроматографический анализ проводили в градиентном режиме элюирования при скорости потока 1 мл/мин. Определяли количество вещества в образце в % после термостатирования.
Оценивали стабильность соединений. Соединения, описанные в настоящем изобретении, имели значения химической стабильности в искусственных биологических средах более 80% (см. табл. 9), т.е. являются химически стабильными в кислой среде искусственного желудочного сока и слабокислой среде искусственного кишечного сока.
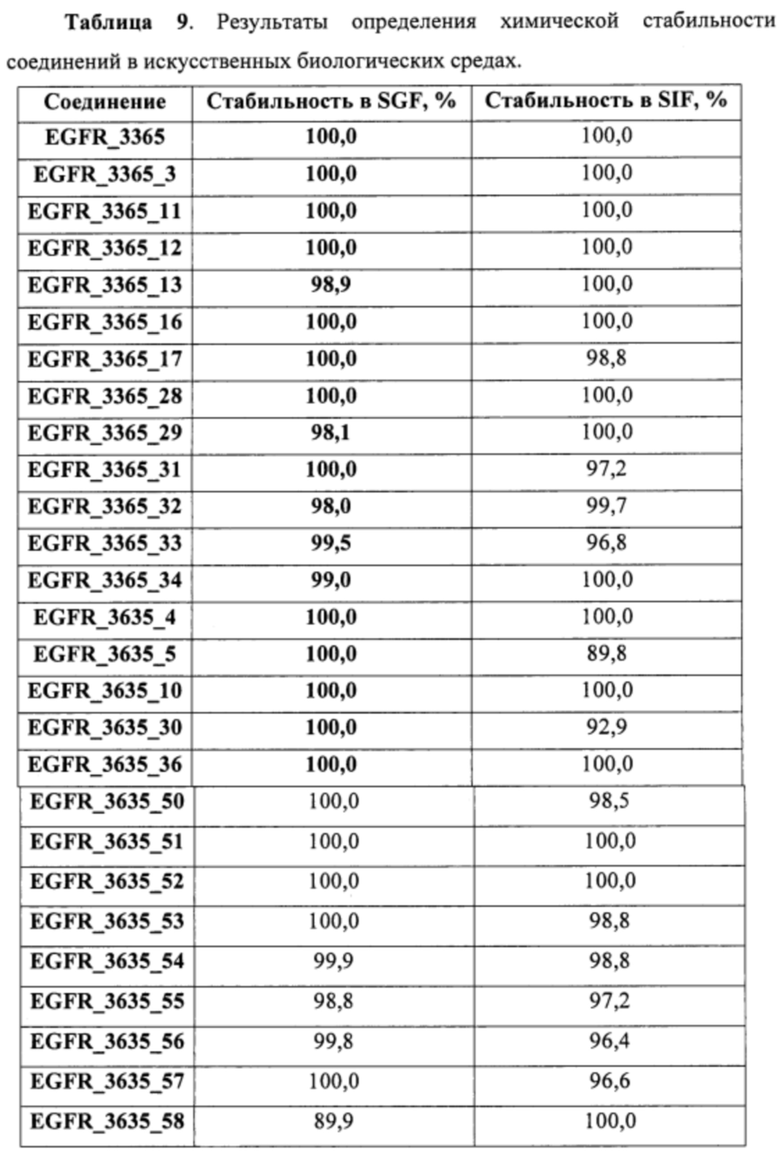
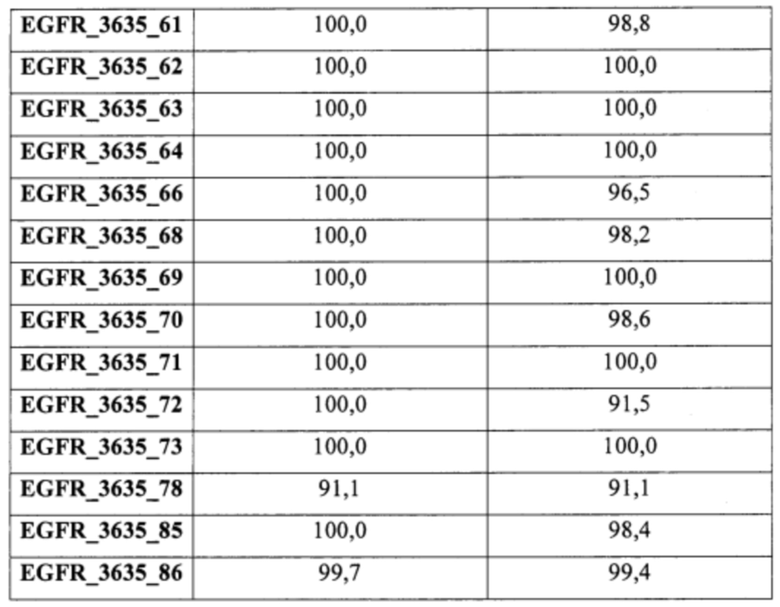
2) Определение химической стабильности в плазме крови человека.
Определение стабильности в плазме крови человека приводили с использованием пулированной плазмы крови человека, взятой у десяти здоровых доноров. Исходный раствор кандидата (10 мМ в ДМСО) разводили пулированной плазмой крови до концентрации 10 мкМ (испытуемый раствор). Испытуемый раствор выдерживали в твердотельном термостате 4 часа при температуре 37°C. Методом ВЭЖХ с использованием хроматографа Agilent1200 (Agilent, США) определяли площади пиков соединений в испытуемых образцах, соответствующие начальному времени испытания (до выдерживания) и конечному времени испытания (после выдерживании в твердотельном термостате 4 часа при температуре 37°C) с предварительным осаждением белков ацетонитрилом. Хроматографический анализ проводили в градиентном режиме элюирования при скорости потока 1 мл/мин. Определяли количество вещества в образце в % после термостатирования.
Оценивали стабильность соединений. Соединения, описанные в настоящем изобретении, имели значения химической стабильности в плазме крови человека более 60% (см. табл. 10).
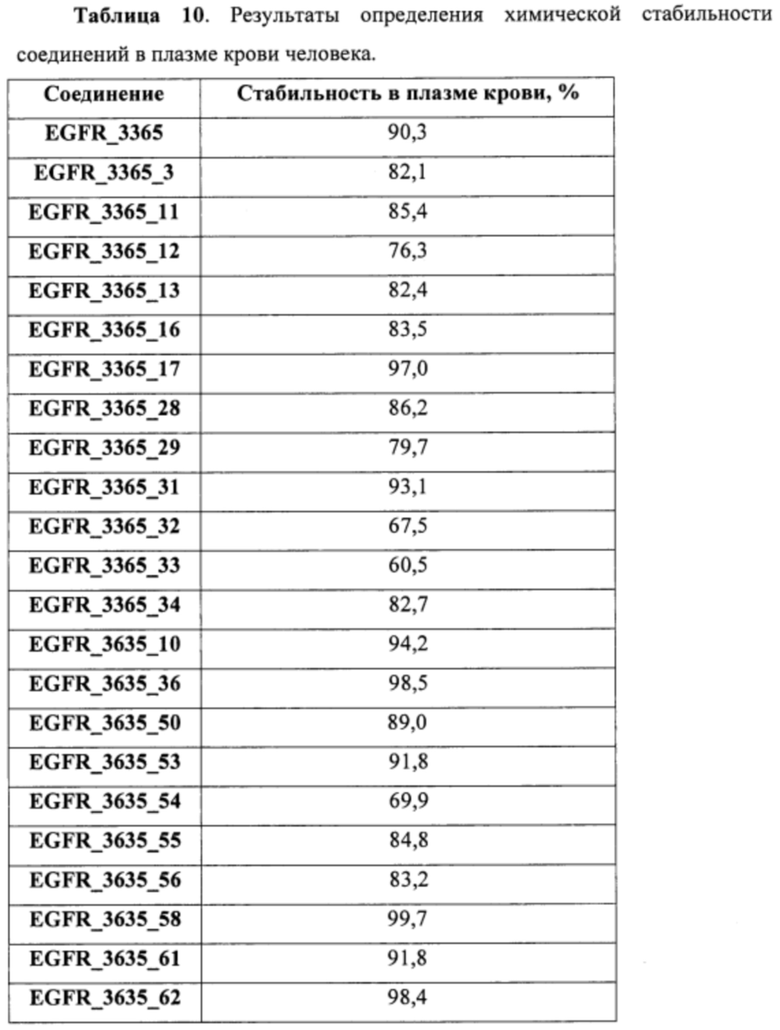

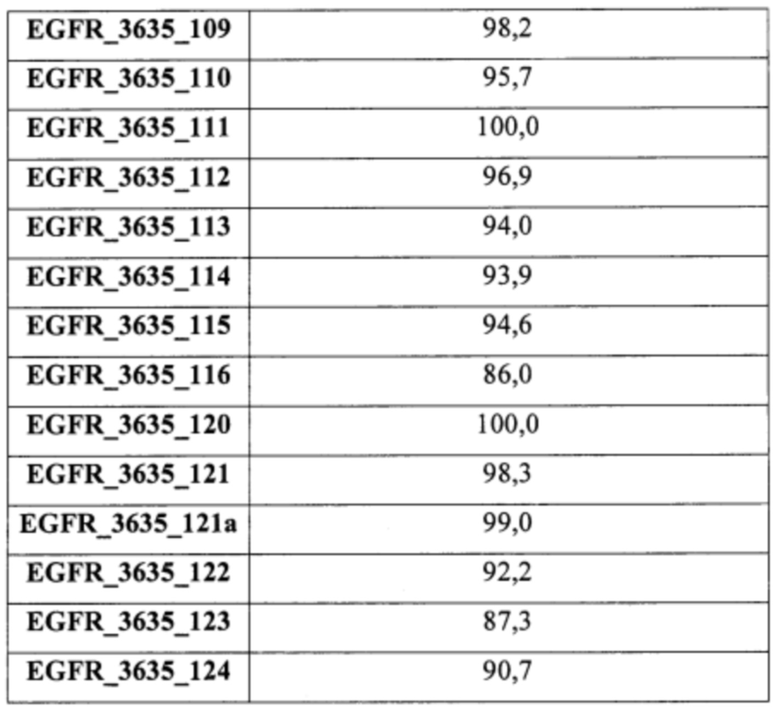
Таким образом, из примера 11 видно, что заявляемые соединения формулы I являются достаточно стабильными в кислой среде искусственного желудочного сока, в слабокислой среде искусственного кишечного сока и в плазме крови человека.
Пример 12. Определение ферментативной стабильности.
Определение ферментативной стабильности кандидатов позволило оценить устойчивость соединений к действию ферментов печени.
Скорость ферментативного разложения соединения определяли путем выдерживания в твердотельном термостате при температуре 37°C реакционной смеси, содержащей 0,5 мг/мл пулированных S9 фракций печени человека (XenoTech, США, cat# Н0610), 10 мкм соединения, 2 мМ β-никотинамидадениндинуклеотида (Carbosynth, UK, cat#NN10871) и 4 мМ хлорида магния в 0,1М натрий-фосфатном буфере рН=7,4. Реакцию останавливали ацетонитрилом из расчета 100 мкл ацетонитрила на 100 мкл реакционной смеси. После остановки реакции образцы центрифугировали 10 минут при 10000 об/мин. Надосадочную жидкость анализировали хроматографическим методом с использованием хроматографа Agilent1200 (Agilent, США). Хроматографический анализ проводили в градиентном режиме элюирования при скорости потока 1 мл/мин. Строили график зависимости логарифма площади пика вещества от времени. Зависимый коэффициент этой прямой соответствовал константе элиминирования k, на основе которой рассчитывали период полураспада препарата Т1/2 и скорость разложения CLint:
Elimination rate constant (k)=(-gradient)
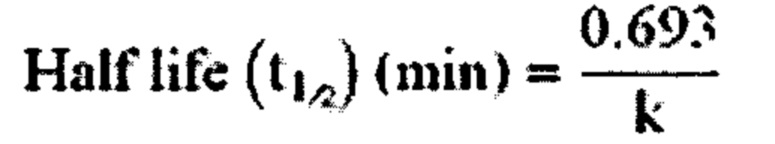

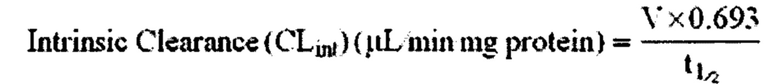
На основе полученных данных делали вывод о ферментативной стабильности кандидатов в S9 фракциях печени человека (см. табл. 11).
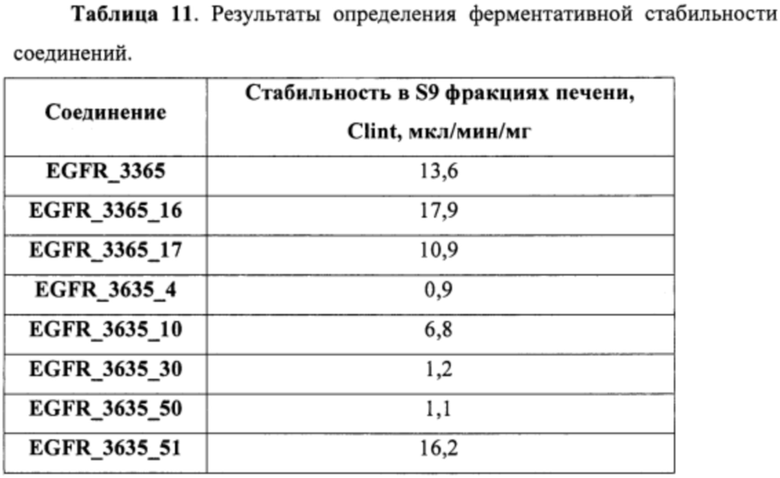

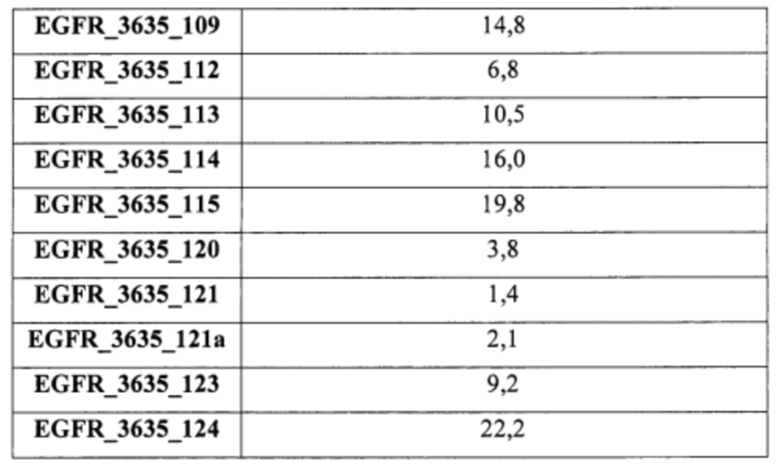
Таким образом, из примера 12 видно, что заявляемые соединения формулы I показали достаточную устойчивость к действию ферментов печени и имели скорость ферментативного разложения Clint менее 24 мкл/мин/мг.
Пример 13. Определение проницаемости соединений.
1) Определение пассивной проницаемости через искусственную мембрану.
Пассивную проницаемость определяли при помощи искусственной мембраны, где роль липидного бислоя выполняет мембрана из L-α-фосфотидилхолина.
5 мкл раствора L-α-фосфотидилхолина соевого (Sigma, Ирландия, cat#8002-43-5) в концентрации 20 мг/мл в ДМСО наносили на мембрану акцепторного фильтрующего планшета, в лунки акцепторного фильтрующего планшета вносили по 150 мкл 0,01М натрий-фосфатного буфера рН=7,4. В лунки донорного планшета вносили по 300 мкл испытуемого раствора соединения (10 мМ в ДМСО). Собранную систему выдерживали 20 часов при комнатной температуре. Образцы из донорного и акцепторного планшетов анализировали хроматографическим методом с использованием хроматографа Agilent1200 (Agilent, США). Хроматографический анализ проводили в градиентном режиме элюирования при скорости потока 1 мл/мин. На хроматограммах определяли площади пиков аналитов в доноре и акцепторе, рассчитывали концентрации соединения. Пассивную проницаемость через искусственную мембрану Ре рассчитывали по формуле:

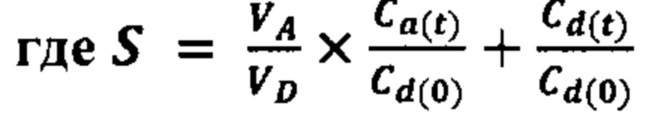
Ре - эффективная константа проницаемости, м/с
VD - объем раствора донора (0,3 мл), мл
VA - объем раствора акцептора (0,15 мл), мл
Area - площадь поверхности мембраны (0,24 см2), см2
t1 - время выдерживания (72000 сек), сек
t0 - время необходимое на заполнение мембраны (1140 сек), сек
Cd(0) - концентрация раствора акцептора в начальный момент времени, мкМ
Cd(0) - концентрация раствора донора в начальный момент времени, мкМ
Cd(t) - концентрация раствора акцептора после 20 часов, мкМ
Cd(t) - концентрация раствора донора после 20 часов, мкМ
Соединения показали высокую скорость пассивного транспорта (см. табл. 12), т.е. соединения способны проникать внутрь клетки через мембрану.
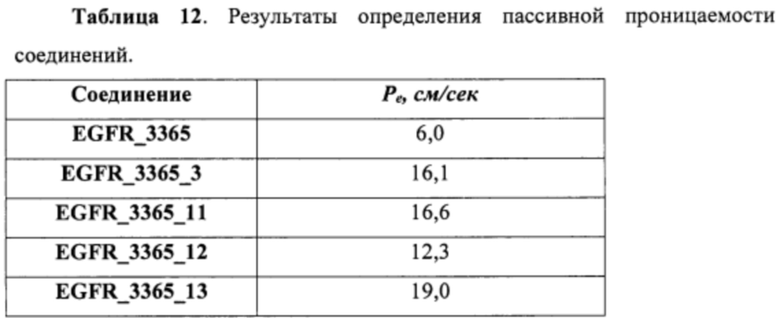
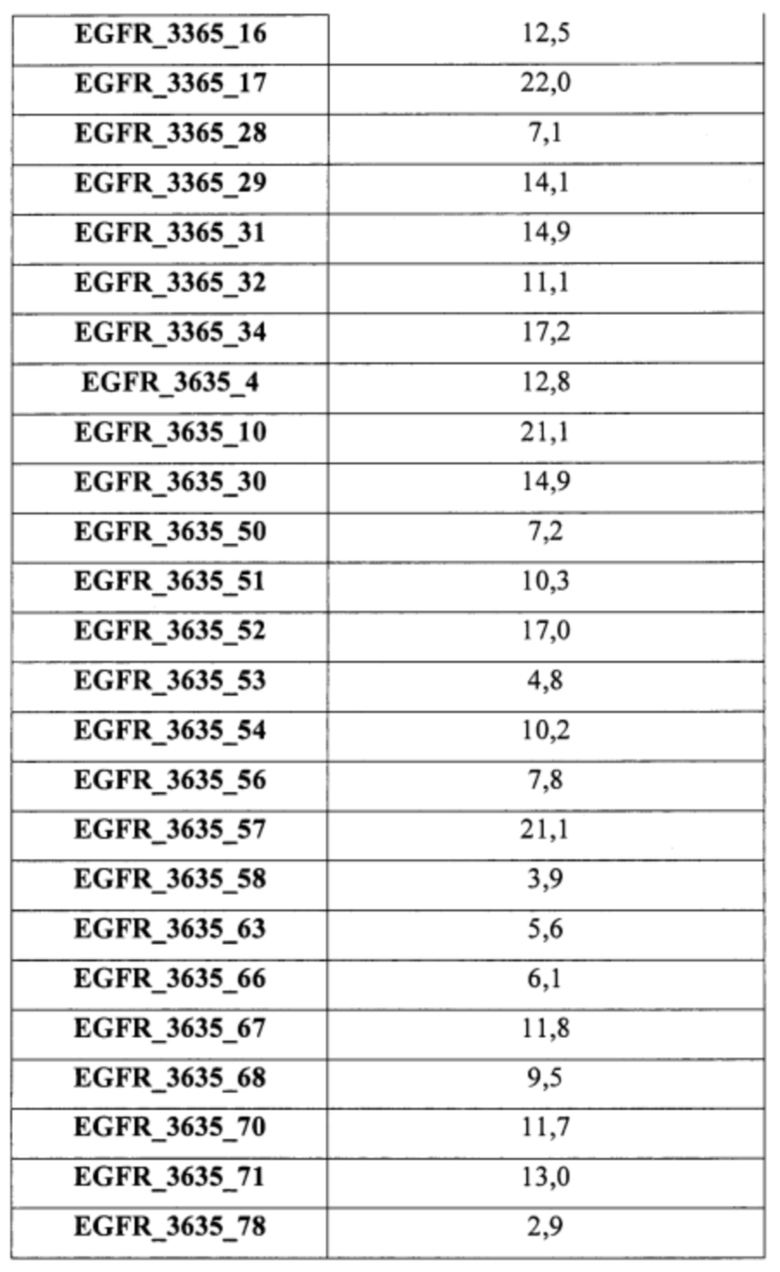
2) Определение проницаемости через монослой клеток Caco-2.
Определение проницаемости через монослой клеток Caco-2 позволило оценить способность веществ проникать через биологические мембраны как посредством активного, так и пассивного транспорта.
Клетки кишечного эпителия Caco-2 культивировали во вставках с фильтрами (с порами 0,4 мкм, BD Falcon with High Density) в течение 21 дня, после чего проверяли целостность монослоя с помощью красителя Lucifer Yellow (Sigma-Aldrich, США) по стандартному протоколу. При постановке переноса А->В (перенос «просвет кишечника» - «кровоток»), растворы исследуемых веществ вносили в буфере рН 6.5 (HBSS, 10 мМ HEPES, 15 мМ p-p Глюкозы) в концентрации 10 мкМ в верхнюю камеру; нижнюю камеру при этом заполняли буфером с рН 7.4 (HBSS, 10 мМ HEPES, 15 мМ p-p Глюкозы, 1% BSA). При постановке переноса В->А (перенос «кровоток»-«просвет кишечника»), верхнюю камеру заполняли буфером рН 6.5, а растворы исследуемых веществ вносили в буфере рН 7.4 в концентрации 10 мкМ в нижнюю камеру. В качестве контроля использовали пропранолол, как вещество высокой проницаемости.
После инкубации в течение 2 ч при 37°C в атмосфере с 5% СО2, определяли количества исследуемых веществ в верхних и нижних камерах методом ВЭЖХ с использованием хроматографа Agilent 1200 (Agilent, США) с предварительным осаждением белков ацетонитрилом. Хроматографический анализ проводили в градиентном режиме элюирования при скорости потока 1 мл/мин. На хроматограммах определяли площади пиков, соответствующие соединениям. На основе значений площади пиков соединения в калибровочных стандартах, определяли концентрацию соединения в исходном растворе и в образцах из лунок верхней и нижней камер.
Проницаемость через слой клеток Рарр рассчитывали по формуле:
Papp=(C(t)*V)/(C(0)*t*Area), где
Papp - эффективная константа проницаемости, м/с
V - объем раствора (в тесте А->В - 0,8 мл, в тесте В->А - 0,2 мл), мл
Area - площадь поверхности мембраны (0,33 см2), см2
t - время выдерживания (7200 сек), сек
C(0) - концентрация исходного раствора, мкМ
C(t) - концентрация раствора после 2 часов (в тесте А->В - концентрация в образце из лунки нижней камеры; в тесте В->А - концентрация в образце из лунки верхней камеры), мкМ
Коэффициент эффлюкса показал способность клеток элиминировать вещество из кровотока. Значение рассчитывали по формуле:
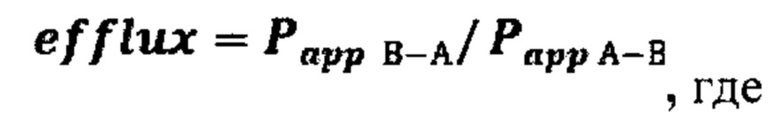
Papp A-B - значение проницаемости прямого анализа А->В;
Papp B-A - значение проницаемости обратного анализа В->А.
Соединения показали высокую скорость прямого транспорта «просвет кишечника» - «кровоток», при этом коэффициент эффлюкса не превышал 2 (см. табл. 13), что указывает на то, что транспортер Pgp не накладывает ограничения на биодоступность испытуемых соединений.
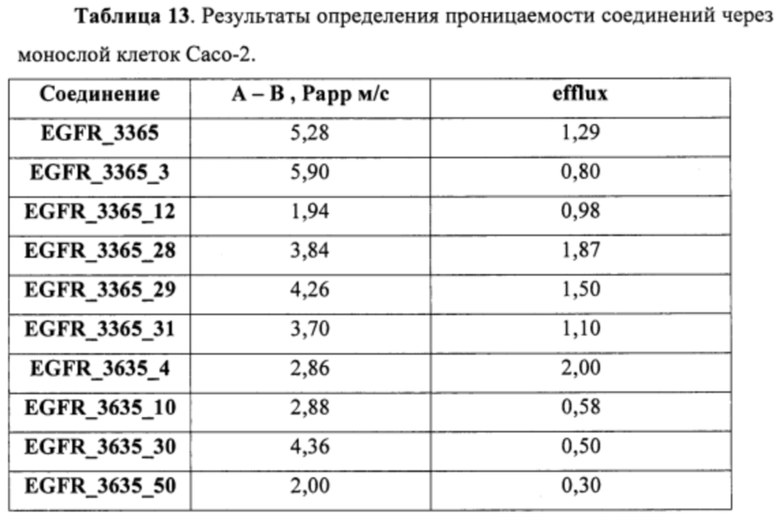

Таким образом, из примера 13 видно, что заявляемые соединения формулы I имеют достаточно высокую скорость пассивного и активного транспорта, из чего следует, что соединения по настоящему изобретению потенциально имеют хорошую биодоступность.
Пример 14. Ингибирующая активность в отношении EGFR in vitro.
Значения IC50 соединений, описанных в настоящем изобретении, определяли с помощью биохимического теста на ингибирование киназной активности в неклеточной системе и с помощью функционального клеточного антипролиферативного теста.
Ингибирование активности киназ WT EGFR (Wild-TypeEGFR), DM EGFR(DoubleMutantEGFR, L828R/T790M) определяли с помощью киназной системы SignalChem и набора для детекции ADP-Glo™ Kinase Assay (V9102, Promega).
Измерения проводили в 96-луночном формате в реакционном объеме 25 мкл. Фермент и ингибитор прединкубировали в течение 10 минут в реакционном буфере, содержащем 25 мМ MOPS, рН 7.2, 12.5 мМ B-глицерол-фосфата, 27 мМ MgCl2, 2 мМ MnCl2 5 мМ EGTA, 2 мМ EDTA, 0.3 мМ DTT, 1,2 мг/мл бычьего сывороточного альбумина. В качестве контрольного ингибитора использовали стауроспорин (S4400, Sigma), в качестве отрицательного контроля - 0,1% раствор ДМСО в реакционном буфере. Добавляли раствор пептидного субстрата 0,5 мг/мл и 50 мкМ АТФ в том же буфере, инкубировали 180 минут при 37°C, детектировали количество израсходованной в киназной реакции АТФ с помощью системы детекции ADP-Glo™ (V9102, Promega). Измерение сигнала люминесценции проводили на планшетном спектрофотометре Infinite M200Pro (Tecan, Швейцария). Величину IC50 определяли с использованием программы Magellan 7.2 (Tecan, Швейцария), аппроксимируя экспериментальные точки по четырехпараметрической модели с оптимизацией по Левенбергу-Маркарту (таблица 14).
Антипролиферативную активность ингибиторов EGFR (таблицы 15 и 16) измеряли в клеточном тесте на перевиваемых культурах эпителиальных клеток А549 (аденокарцинома легкого, АТСС® CRM-CCL-185™ - WT), НСС827 (аденокарцинома легкого, АТСС® CRL-2868™ - SM (SingleMutantEGFR, exon19delE746-A750)) и Н1975 (аденокарцинома легкого, АТСС® CRL-5908™ - DM (DoubleMutantEGFR, L828R/T790M)) с помощью прижизненного красителя Alamar Blue (ThermoFisher, #DAL1100). Клетки субкультивировали в среде RPMI-1640 (ПанЭко, С330п) с добавлением 10% FBS (Gibco, #16140071) не менее 1 пассажа после разморозки, промывали и повторно высевали в 96-луночные культуральные планшеты (3599, Corning) в питательную среду RPMI-1640 с добавлением 2% FBS в количестве 5*103 клеток/лунку для А549, 10*103 клеток/лунку для НСС827 и 15*103 клеток/лунку для H1975 в 100 мкл. Инкубировали 16-18 часов в СО2 инкубаторе (Thermo Forma, США) при 37°C с атмосферой 5% CO2 для прикрепления клеток.
Исследуемые соединения растворяли в ДМСО в выбранных концентрационных диапазонах и переносили в среду RPMI-1640 (ПанЭко, С330п) с добавлением 2% FBS. После добавления 50 мкл приготовленных разведений к клеткам, инкубационная смесь содержала конечные концентрации исследуемых веществ и не более 1% ДМСО. Содержимое планшетов инкубировали при 37°C в течение 72 ч, после чего в лунки добавляли по 15 мкл витального красителя Alamar Blue (ThermoFisher, #DAL1100), перемешивали на орбитальном шейкере (Biosan, Латвия) и дополнительно инкубировали 3-5 часов при 37°C в СО2-инкубаторе (Thermo Forma, США). Количество живых клеток детектировали на планшетном спектрофотометре Infinite M200Pro (Tecan, Швейцария), измеряя флуоресцентный сигнал при длине волны возбуждения (λЕх) 540 нм и длине волны детектирования (ΔEm) 590 нм.
Величину IC50 определяли с использованием программы Magellan 7.2 (Tecan, Швейцария), аппроксимируя экспериментальные точки по четырехпараметрической модели с оптимизацией по Левенбергу-Маркарту.
Общую цитотоксичность (СС50) определяли в тесте на клетках HepG2 (гепатоцеллюлярная карцинома, АТСС® НВ-8065™) (таблица 16). Клетки субкультивировали в среде DMEM (ПанЭко, С420п) с добавлением 10% FBS (Gibco, #16140071) не менее 1 пассажа после разморозки, промывали и повторно высевали в 96-луночные культуральные планшеты (3599, Corning) в концентрации 2*104 клеток / 100 мкл на лунку. Инкубировали 16-18 часов. Исследуемые вещества титровали в ДМСО и переносили в среду DMEM (ПанЭко, С420п) с 2% FBS, добавляли к клеткам и инкубировали при 37°C в течение 72 часов, после чего проводили детекцию жизнеспособности клеток с помощью витального красителя Alamar Blue (ThermoFisher, #DAL1100). Величину CC50 определяли аналогично IC50.
Терапевтический индекс (Therapeutic Index - TI) определяли как отношение СС50 на линии HepG2 к IC50 на клеточной линии HI975:
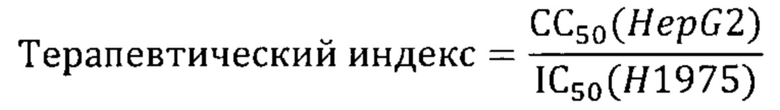
Индекс селективности (Selectivity Index - SI) определяли как отношение IC50 для линий, содержащих EGFR дикого типа (А549) к IC50 для линии с целевой мутацией L828R/T790M (HI975):

Соединения по настоящему изобретению продемонстрировали эффективное ингибирование активности киназы с целевой мутацией EGFR (L828R/T790M), а также показали низкую активность к EGFR дикого типа. Соединения по настоящему изобретению показали высокую селективность в отношении EGFR с мутациями.
В клеточных тестах соединения по настоящему изобретению показали высокую антипролиферативную активность в отношении целевых клеточных линий (EGFR с мутацией L828R/T790M и с делецией в экзоне 19).
По результатам теста на общую цитотоксичность соединения по настоящему изобретению проявили низкую токсичность.

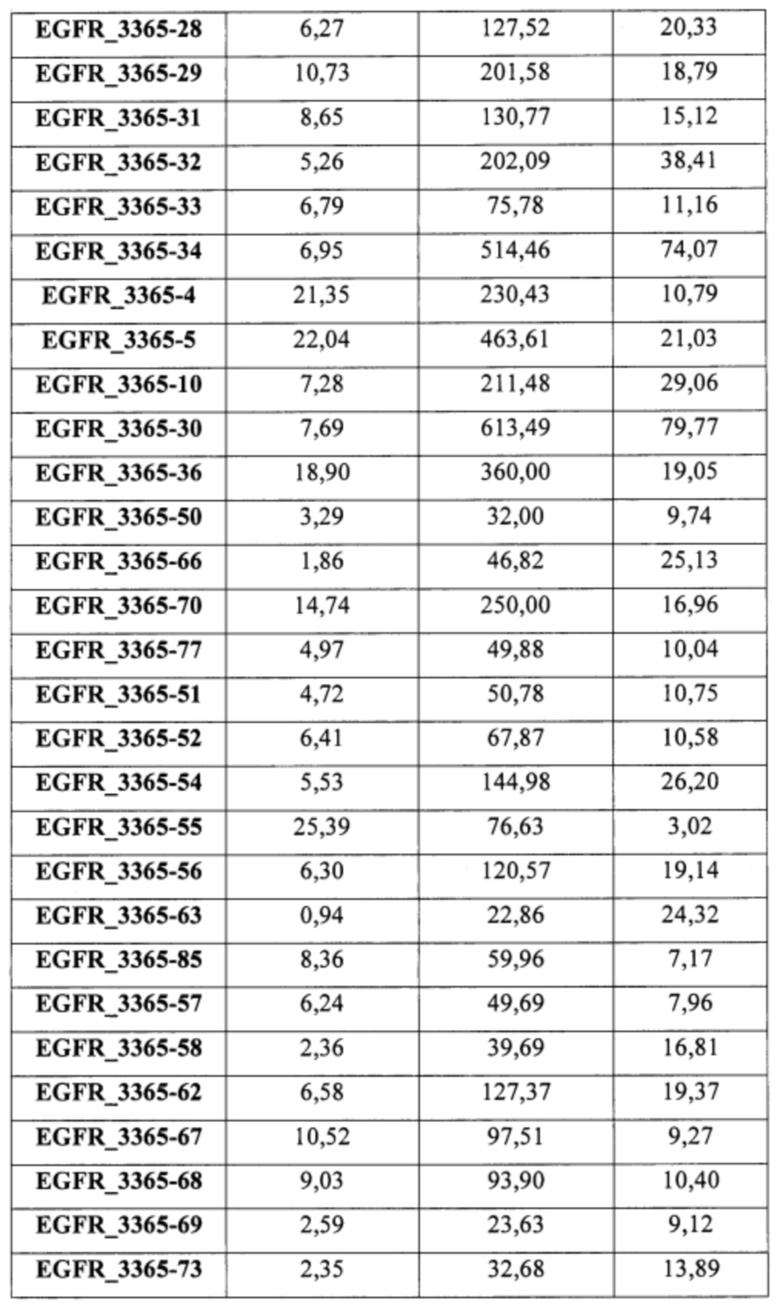
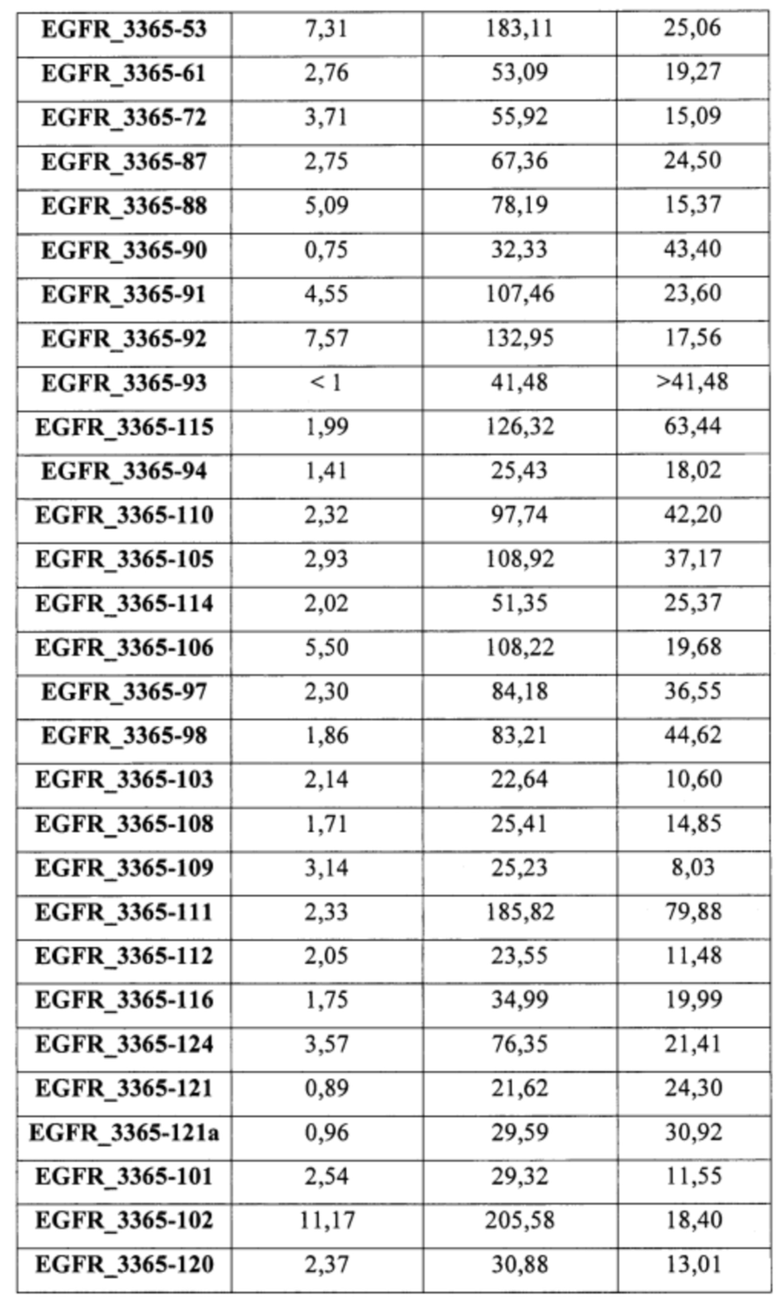

* После знака «>» и «<» приводятся значения IC50, которые находятся выше предела диапазона исследуемых концентраций.
** После знака «<» и «>»приводятся ориентировочные значения, рассчитанные на основании значения IC50, находящегося вне диапазона исследуемых концентраций.
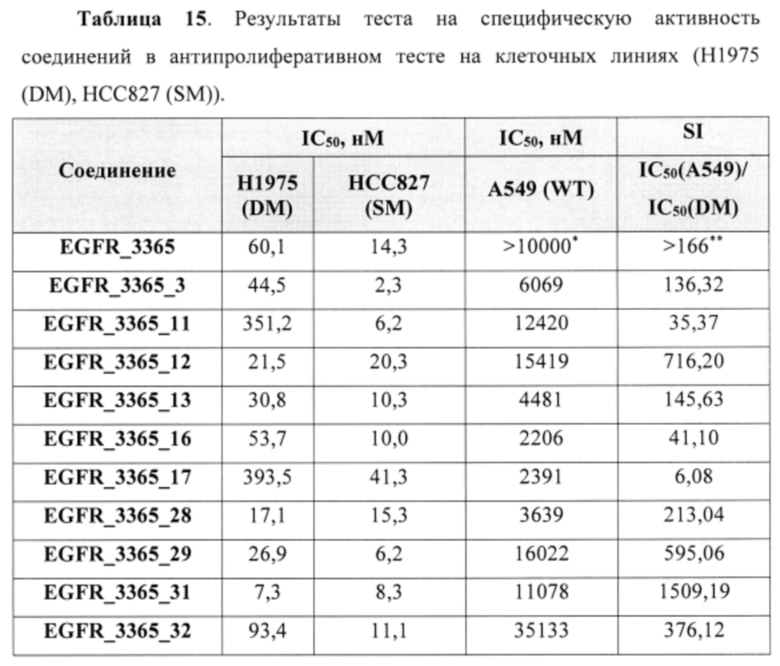

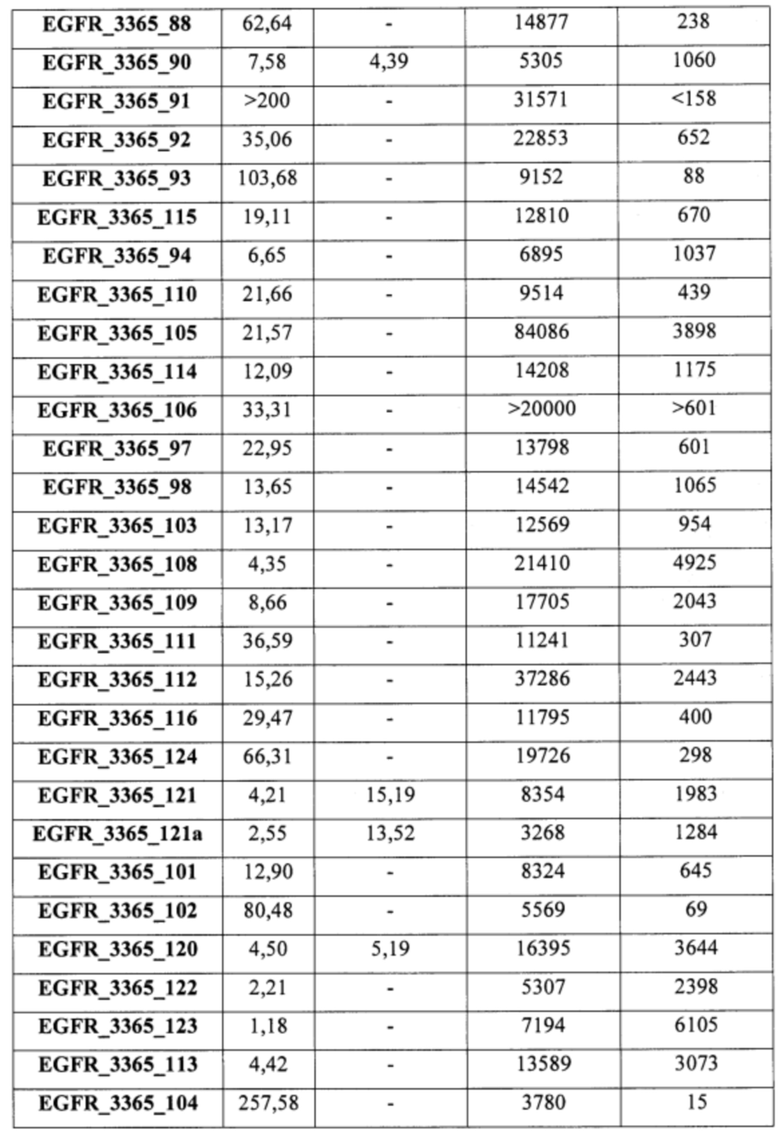

* После знака «>» приводятся значения IC50, которые находятся выше предела диапазона исследуемых концентраций.
** После знака «>» приводятся ориентировочные значения, рассчитанные на основании значения IC50, находящегося вне диапазона исследуемых концентраций.


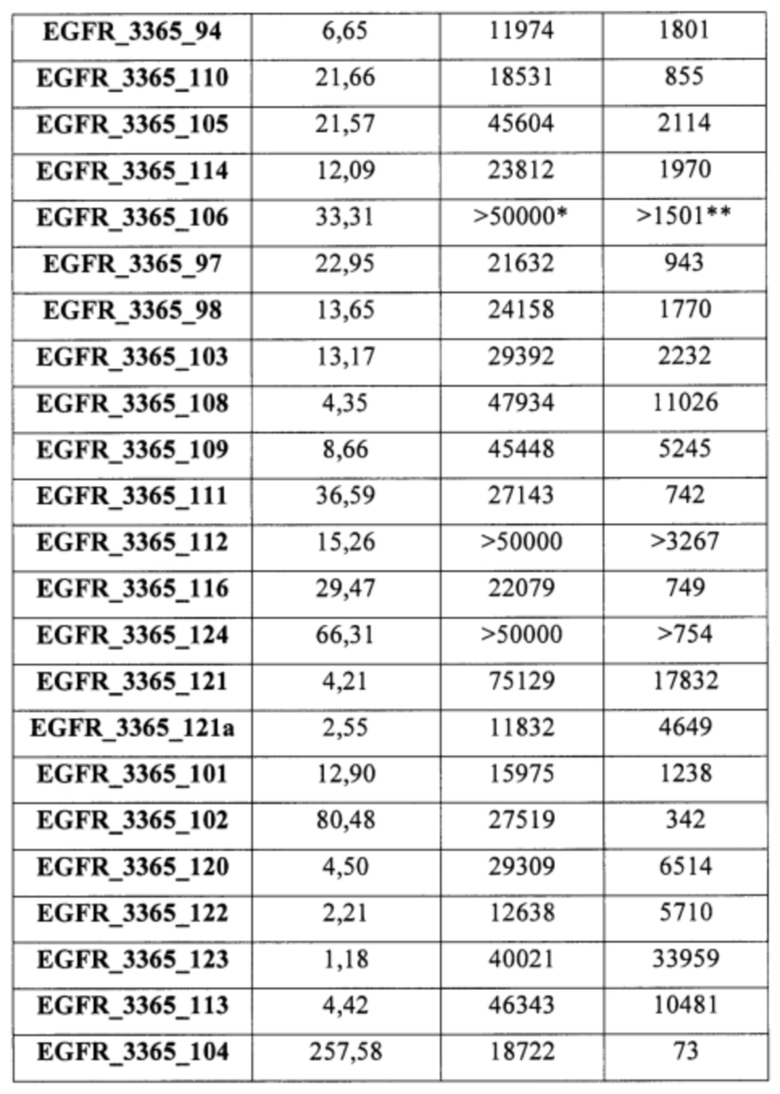
* После знака «>» приводятся значения IC50, которые находятся выше предела диапазона исследуемых концентраций.
** После знака «>» приводятся ориентировочные значения, рассчитанные на основании значения IC50, находящегося вне диапазона исследуемых концентраций.
| название | год | авторы | номер документа |
|---|---|---|---|
| Новые гетероциклические соединения как ингибиторы CDK8/19 | 2018 |
|
RU2739489C2 |
| Ингибиторы рецептора эпидермального фактора роста | 2022 |
|
RU2821531C2 |
| НОВЫЕ ИНГИБИТОРЫ CDK8/19 | 2019 |
|
RU2754441C2 |
| ПРОИЗВОДНОЕ АКРИЛАНИЛИДА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЯ В ФАРМАКОЛОГИИ | 2016 |
|
RU2742372C2 |
| МОДУЛЯТОРЫ ПРОТЕИН-ТИРОЗИНКИНАЗЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2014 |
|
RU2656591C2 |
| СОЕДИНЕНИЯ-ИНГИБИТОРЫ EGFR | 2017 |
|
RU2751341C2 |
| СОЕДИНЕНИЯ ДЛЯ ИНГИБИРОВАНИЯ КИНАЗЫ EGFR, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2020 |
|
RU2832925C2 |
| СОЕДИНЕНИЕ 2-АМИНОПИРИМИДИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ ДАННОГО СОЕДИНЕНИЯ | 2015 |
|
RU2704129C2 |
| НЕКОТОРЫЕ ХИМИЧЕСКИЕ СОЕДИНЕНИЯ, КОМПОЗИЦИИ И СПОСОБЫ | 2014 |
|
RU2718876C2 |
| ИНГИБИТОР EGFR И ЕГО ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2015 |
|
RU2702631C2 |
Настоящая группа изобретений относится к новым соединениям формулы I
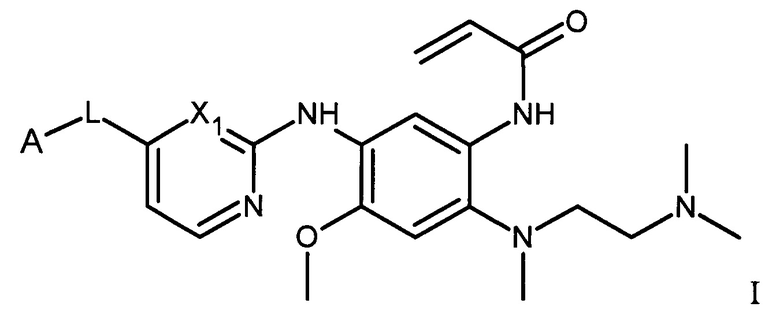 ,
,
их фармацевтически приемлемым солям, а также к фармацевтической композиции, способу ингибирования биологической активности рецептора эпидермального фактора роста (EFGR), способу лечения заболеваний или нарушений, опосредованных активностью EFGR, и применению заявляемых соединений или указанной композиции для лечения заболевания или нарушения, опосредованного активностью EFGR. 5 н. и 11 з.п. ф-лы, 14 пр., 16 табл.
1. Соединение формулы I:
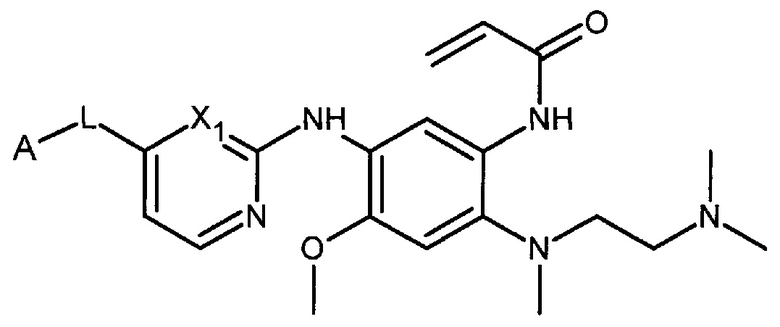
или его фармацевтически приемлемая соль,
где L представляет собой -С(О)-;
X1 представляет собой СН или N;
А представляет собой
 где
где
Х2, Х3, Х4, Х5, Х6 каждый независимо представляет собой С, СН или N,
R1 каждый независимо представляет собой водород; Hal; циано; нитро; гидроксигруппу; C1-С6 алкил, незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, гидроксигруппы; C1-С6 алкилоксигруппу, незамещенную или замещенную одним или несколькими радикалами, выбранными из гидроксигруппы, C1-С6 алкилокси, C6-С10 арила; фенокси; C3-С6 циклоалкилокси; -NR2R3; 5-6-членный гетероарил с 1-2 гетероатомами, выбранными из N и/или О, незамещенный или замещенный одним или несколькими заместителями, выбранными из C1-С6 алкила; 4-7-членный гетероциклил с 1-2 гетероатомами, выбранными из N и/или О, незамещенный или замещенный одним или несколькими заместителями, выбранными из циано, гидроксигруппы, оксо, С1-С6 алкила, C1-С6 алкилокси, -NR2R3;
R2 или R3 - каждый независимо представляет собой водород, C1-С6 алкил, незамещенный или замещенный одним или несколькими радикалами, выбранными из гидроксигруппы, C1-С6 алкилокси;
k представляет собой 0, 1, 2 или 3;
Hal представляет собой атом фтора, брома или хлора.
2. Соединение по п. 1, где фрагмент

выбран из группы, включающей:

где R1 каждый независимо представляет собой Hal; циано; нитро; гидроксигруппу; C1-С6 алкил, незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, гидроксигруппы; C1-С6 алкилоксигруппу, незамещенную или замещенную одним или несколькими радикалами, выбранными из гидроксигруппы, C1-С6 алкилокси, C6-С10 арила; фенокси; С3-С6 циклоалкилокси; -NR2R3; 5-6-членный гетероарил с 1-2 гетероатомами, выбранными из N и/или О, незамещенный или замещенный одним или несколькими заместителями, выбранными из C1-С6 алкила; 4-7-членный гетероциклил с 1-2 гетероатомами, выбранными из N и/или О, незамещенный или замещенный одним или несколькими заместителями, выбранными из циано, гидроксигруппы, оксо, C1-С6 алкила, C1-С6 алкилокси, -NR2R3;
R2 или R3 - каждый независимо представляет собой водород, C1-С6 алкил, незамещенный или замещенный одним или несколькими радикалами, выбранными из гидроксигруппы, C1-С6 алкилокси;
k представляет собой 0, 1, 2 или 3;
Hal представляет собой атом фтора, брома или хлора.
3. Соединение по п. 1, где фрагмент

выбран из группы, включающей:
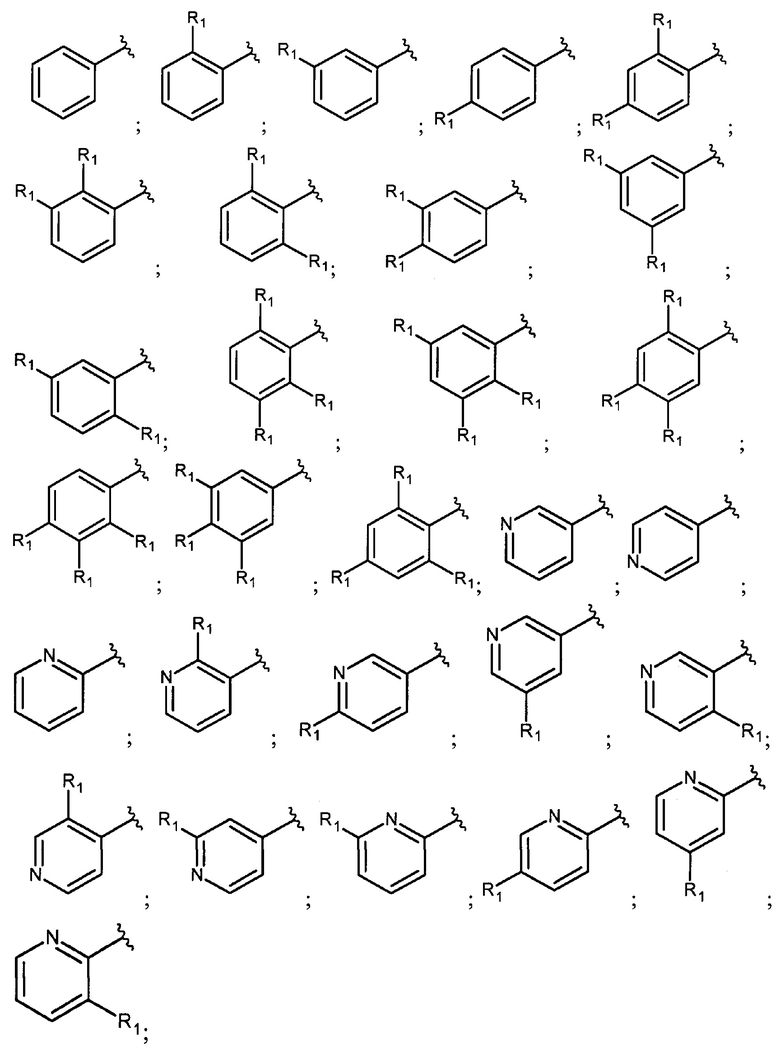
где R1 каждый независимо представляет собой Hal; циано; нитро; гидроксигруппу; C1-С6 алкил, незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, гидроксигруппы; C1-С6 алкилоксигруппу, незамещенную или замещенную одним или несколькими радикалами, выбранными из Hal, C1-С6 алкилокси, C6-С10 арила; фенокси; С3-С6 циклоалкилокси; -NR2R3; 5-6-членный гетероарил с 1-2 гетероатомами, выбранными из N и/или О, незамещенный или замещенный одним или несколькими заместителями, выбранными из C1-С6 алкила; 4-7-членный гетероциклил с 1-2 гетероатомами, выбранными из N и/или О, незамещенный или замещенный одним или несколькими заместителями, выбранными из циано, гидроксигруппы, оксо, C1-С6 алкила, C1-С6 алкилокси, -NR2R3;
R2 или R3 - каждый независимо представляет собой водород, C1-С6 алкил, незамещенный или замещенный одним или несколькими радикалами, выбранными из гидроксигруппы, C1-С6 алкилокси;
k представляет собой 0, 1, 2 или 3;
Hal представляет собой атом фтора, брома или хлора.
4. Соединение по п. 1, где фрагмент

выбран из группы, включающей:
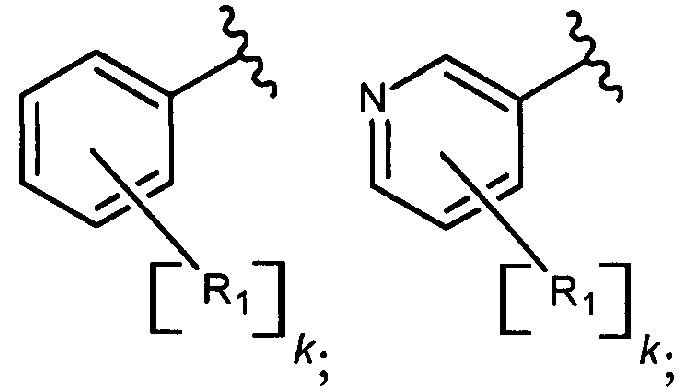
где R1 каждый независимо представляет собой Hal; циано; нитро; гидроксигруппу; C1-С6 алкил, незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, гидроксигруппы; C1-С6 алкилоксигруппу, незамещенную или замещенную одним или несколькими радикалами, выбранными из гидроксигруппы, C1-С6 алкилокси, C6-С10 арила; фенокси; С3-С6 циклоалкилокси; -NR2R3; 5-6-членный гетероарил с 1-2 гетероатомами, выбранными из N и/или О, незамещенный или замещенный одним или несколькими заместителями, выбранными из C1-С6 алкила; 4-7-членный гетероциклил с 1-2 гетероатомами, выбранными из N и/или О, незамещенный или замещенный одним или несколькими заместителями, выбранными из циано, гидроксигруппы, оксо, C1-С6 алкила, C1-С6 алкилокси, -NR2R3;
R2 или R3 - каждый независимо представляет собой водород, C1-С6 алкил, незамещенный или замещенный одним или несколькими радикалами, выбранными из гидроксигруппы, C1-С6 алкилокси;
k представляет собой 0, 1, 2 или 3;
Hal представляет собой атом фтора, брома или хлора.
5. Соединение по п. 1, где фрагмент

выбран из группы, включающей:
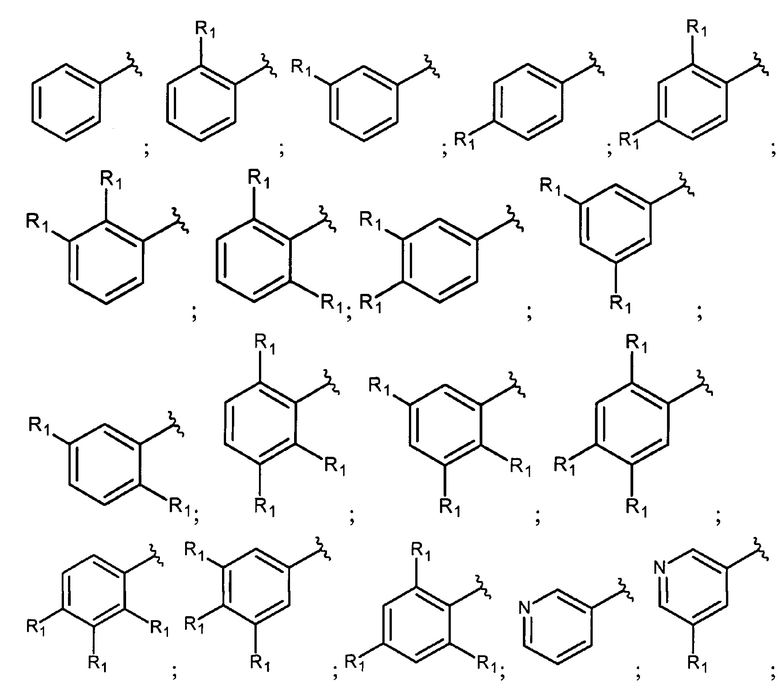
где R1 каждый независимо представляет собой Hal; циано; нитро; гидроксигруппу; C1-С6 алкил, незамещенный или замещенный одним или несколькими радикалами, выбранными из Hal, гидроксигруппы; C1-С6 алкилоксигруппу, незамещенную или замещенную одним или несколькими радикалами, выбранными из гидроксигруппы, C1-С6 алкилокси, C6-С10 арила; фенокси; С3-С6 циклоалкилокси; -NR2R3; 5-6-членный гетероарил с 1-2 гетероатомами, выбранными из N и/или О, незамещенный или замещенный одним или несколькими заместителями, выбранными из C1-С6 алкила; 4-7-членный гетероциклил с 1-2 гетероатомами, выбранными из N и/или О, незамещенный или замещенный одним или несколькими заместителями, выбранными из циано, гидроксигруппы, оксо, C1-С6 алкила, C1-С6 алкилокси, -NR2R3;
R2 или R3 - каждый независимо представляет собой водород, C1-С6 алкил, незамещенный или замещенный одним или несколькими радикалами, выбранными из гидроксигруппы, C1-С6 алкилокси;
Hal представляет собой атом фтора, брома или хлора.
6. Соединения по пп. 1-5, представляющие собой:
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(2-(трифторметил)бензоил)пиримидин-2-ил)амино)фенил)акриламид (EGFR_3365);
N-(5-((4-бензоилпиридин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид (EGFR_3365_3);
N-(5-((4-(диметиламино)бензоил)пиридин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид 2,2,2-трифторацетат (EGFR_3365_4);
N-(5-((4-(диметиламино)бензоил)пиридин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид (EGFR_3365_4a);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-морфолинобензоил)пиридин-2-ил)амино)фенил)акриламид 2,2,2-трифторацетат (EGFR_3365_5);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-морфолинобензоил) пиридин-2-ил)амино)фенил) акриламид (EGFR_3365_5a);
N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(4-фторбензоил)пиримидин-2-ил)амино)-4-метоксифенил)акриламид (EGFR_3365_10);
N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(3-фторбензоил)пиримидин-2-ил)амино)-4-метоксифенил)акриламид (EGFR_3365_11);
N-(5-((4-(2-бромбензоил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)проп-2-енамид (EGFR_3365_12);
N-(5-((4-(4-бромбензоил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)проп-2-енамид (EGFR_3365_13);
N-(5-((4-(4-цианобензоил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)проп-2-енамид 2,2,2-трифторацетат (EGFR_3365_14);
N-(5-((4-(4-цианобензоил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метокси фенил)проп-2-енамид (EGFR_3365_14a);
N-(2-((2-(диметиламино)этил)(метил)амино)амино)-4-метокси-5-((4-(пиридин-3-карбонил)пиримидин-2-ил)амино)фенил)проп-2-енамид (EGFR_3365_15);
N-(5-((4-(4-(бензилокси)бензоил)пиримидин-2-ил)амино)-2-((2-диметил амино)этил)метил)амино)-4-метоксифенил)акриламид (EGFR_3365_16);
N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(4-феноксибензоил)пиримидин-2-ил)амино)-4-метоксифенил)акриламид (EGFR_3365_17);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(5-метилпиридин)-3-карбонил)пиримидин-2-ил)амино)фенил)проп-2-енамид (EGFR_3365_26);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-метоксибензоил)пиримидин-2-ил)амино)фенил)акриламид (EGFR_3365_28);
N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(4-этоксибензоил)пиримидин-2-ил)амино)-4-метоксифенил)акриламид (EGFR_3365_29);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-пропоксибензоил)пиримидин-2-ил)амино)фенил)акриламид (EGFR_3365_30);
N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(гидрокси(4-пропоксифенил)метил)пиримидин-2-ил)амино)-4-метоксифенил)акриламид (EGFR_3365_30a);
N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(4-изопропоксибензоил)пиримидин-2-ил)амино)-4-метоксифенил)акриламид (EGFR_3365_31);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(3-метоксибензоил)пиримидин-2-ил)амино)фенил)акриламид (EGFR_3365_32);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(2-метоксибензоил)пиримидин-2-ил)амино)фенил)акриламид (EGFR_3365_33);
N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(3-нитробензоил)пиримидин-2-ил)амино)-4-метоксифенил)акриламид (EGFR_3365_34);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(2-нитробензоил)пиримидин-2-ил)амино)фенил)акриламид (EGFR_3365_36);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-пропоксибензоил)пиридин-2-ил)амино)фенил)акриламид (EGFR_3365_50);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-метоксибензоил)пиридин-2-ил)амино)фенил)акриламид (EGFR_3365_51);
N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(4-этоксибензоил)пиридин-2-ил)амино)-4-метоксифенил)акриламид (EGFR_3365_52);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-(3-метоксиазетидин-1-ил)бензоил)пиридин-2-ил)амино)фенил)акриламид (EGFR_3365_53);
N-(5-((4-(4-диэтиламино)бензоил)пиридин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид (EGFR_3365_54);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-(4-метилпиперазин-1-ил)бензоил)пиридин-2-ил)амино)фенил)акриламид (EGFR_3365_55);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-(пирролидин-1-ил)бензоил)пиридин-2-ил)амино)фенил)акриламид (EGFR_3365_56);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-метилбензоил)пиридин-2-ил)амино)фенил)акриламид (EGFR_3365_57);
N-(5-((4-(4-(азетидин-1-ил)бензоил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид (EGFR_3365_58);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-(3-метоксиазетидин-1-ил)бензоил)пиримидин-2-ил)амино)фенил)акриламид (EGFR_3365_61);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-(4-метилпиперазин-1-ил)бензоил)пиримидин-2-ил)амино)фенил)акриламид (EGFR_3365_62);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-(4-метилпиперазин-1-ил)бензоил)пиримидин-2-ил)амино)фенил)акриламида (EGFR_3365_62a);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-(пирролидин-1-ил)бензоил)пиримидин-2-ил)амино)фенил)акриламид (EGFR_3365_63);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-метилбензоил)пиримидин-2-ил)амино)фенил)акриламид (EGFR_3365_64);
N-(5-((4-(4-бутоксибензоил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид (EGFR_3365_66);
N-(5-((4-(4-(циклогексилокси)бензоил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид (EGFR_3365_67);
N-(5-((4-(2,4-диэтоксибензоил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид (EGFR_3365_68);
N-(5-((4-(2,4-диметоксибензоил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид (EGFR_3365_69);
N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(2,4-дипропоксибензоил)пиримидин-2-ил)амино)-4-метоксифенил)акриламид (EGFR_3365_70);
N-(5-((4-(2,4-диизопропоксибензоил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид (EGFR_3365_71);
N-(5-((4-(4-(диэтиламино)бензоил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид (EGFR_3365_72);
N-(5-((4-(4-(диметиламино)бензоил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид (EGFR_3365_73);
N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(4-изобутоксибензоил)пиридин-2-ил)амино)-4-метоксифенил)акриламид (EGFR_3365_77);
N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(4-изобутоксибензоил)пиримидин-2-ил)амино)-4-метоксифенил)акриламид (EGFR_3365_78);
N-2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-(1-метил-1Н-пиразол-4-ил)бензоил)пиридин-2-ил)амино)фенил)акриламид (EGFR_3365_85);
N-(5-((4-(4-(1Н-имидазол-1-ил)бензоил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид (EGFR_3365_86);
N-(5-((4-(2,4-диметоксибензоил)пиридин-2-ил)амино)-2-((2-диметиламино)этил)(метил)амино-4-метоксифенил)акриламид (EGFR_3365_87);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(2-метоксибензоил)пиридин-2-ил)амино)фенил)акриламид (EGFR_3365_88);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-(пиперидин-1-ил)бензоил)пиримидин-2-ил)амино)фенил)акриламид (EGFR_3365_90);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-(2-оксопирролидин-1-ил)бензоил)пиримидин-2-ил)амино)фенил)акриламид (EGFR_3365_91);
N-[2-{[2-(диметиламино)этил)(метил)амино)-4-метокси-5-({4-[4-(2-метоксиэтокси)бензоил]пиридин-2-ил}амино)фенил]проп-2-енамид (EGFR_3365_92);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-(2-метоксиэтокси)бензоил)пиридин-2-ил)амино)фенил)акриламид (EGFR_3365_93);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(1-метил-1Н-индол-2-карбонил)пиримидин-2-ил)амино)фенил)акриламид (EGFR_3365_94);
N-(5-((4-(4-(4-цианопиперидин-1-ил)бензоил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид (EGFR_3365_97);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-(4-метоксипиперидин-1-ил)бензоил)пиримидин-2-ил)амино)фенил)акриламид (EGFR_3365_98);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(2-метокси-4-пропоксибензоил)пиримидин-2-ил)амино)фенил)акриламид (EGFR_3365_101);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-метокси-2-пропоксибензоил)пиримидин-2-ил)амино)фенил)акриламид (EGFR_3365_102);
N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(4-изопропокси-2-метоксибензоил)пиримидин-2-ил)амино)-4-метоксифенил)акриламид (EGFR_3365_103);
N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(2-изопропокси-4-метоксибензоил)пиримидин-2-ил)амино)-4-метоксифенил)акриламид (EGFR_3365_104);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-((2-метоксиэтил)амино)бензоил)пиримидин-2-ил)амино)фенил)акриламид (EGFR_3365_105);
N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(4-((2-гидроксиэтил)амино)бензоил)пиримидин-2-ил)амино)-4-метоксифенил)акриламидформиат (EGFR_3365_106);
N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(4-((2-гидроксиэтил)амино)бензоил)пиримидин-2-ил)амино)-4-метоксифенил)акриламид (EGFR_3365_106a);
(S)-N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(4-(3-гидроксипиперидин-1-ил)бензоил)пиримидин-2-ил)амино)-4-метоксифенил)акриламид (EGFR_3365_108);
(R)-N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(4-(3-гидроксипиперидин-1-ил)бензоил)пиримидин-2-ил)амино)-4-метоксифенил)акриламид (EGFR_3365_109);
(S)-N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-(3-метоксипиперидин-1-ил)бензоил)пиримидин-2-ил)амино)фенил)акриламид (EGFR_3365_110);
(R)-N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-(3-метоксипиперидин-1-ил)бензоил)пиримидин-2-ил)амино)фенил)акриламид (EGFR_3365_111);
(S)-N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(4-(3-гидроксипирролидин-1-ил)бензоил)пиримидин-2-ил)амино)-4-метоксифенил)акриламид формиат (EGFR_3365_112);
(S)-N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(4-(3-гидроксипирролидин-1-ил)бензоил)пиримидин-2-ил)амино)-4-метоксифенил)акриламид (EGFR_3365_112а);
(R)-N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(4-(3-гидроксипирролидин-1-ил)бензоил)пиримидин-2-ил)амино)-4-метоксифенил)акриламид (EGFR_3365_113);
(S)-N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-(3-метоксипирролидин-1-ил)бензоил)пиримидин-2-ил)амино)фенил)акриламид (EGFR_3365_114);
(R)-N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-(3-метоксипирролидин-1-ил)бензоил)пиримидин-2-ил)амино)фенил)акриламид (EGFR_3365_115);
N-(5-((4-(4-циклопропоксибензоил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид (EGFR_3365_116);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(2-метокси-4-(4-метилпиперазин-1-ил)бензоил)пиримидин-2-ил)амино)фенил)акриламид (EGFR_3365_120);
N-(5-((4-(4-(4-аминопиперидин-1-ил)бензоил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид (EGFR_3365_121);
N-2-((2-(диметиламино)этил)(метил)амино)-5-((4-(4-(4-диметиламинопиперидин-1-ил)бензоил)пиримидин-2-ил)амино)-4-метоксифенил)акриламид (EGFR_3365_121a);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(2-метокси-4-(пиперидин-1-ил)бензоил)пиримидин-2-ил)амино)фенил)акриламид (EGFR_3365_122);
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(2-метокси-4-(пирролидин-1-ил)бензоил)пиримидин-2-ил)амино)фенил)акриламид (EGFR_3365_123);
N-2-((2-(диметиламино)этил)(метил)амино)-4-метокси-(5-((4-(2,4,6-триметоксибензоил)пиримидин-2-ил)амино)фенил)акриламид (EGFR_3365_124);
N-(5-((4-(4-(4-аминопиперидин-1-ил)-2-метоксибензоил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид (EGFR_3365_126);
N-2-((2-(диметиламино)этил)(метил)амино)-5-((4-(4-(4-диметиламинопиперидин-1-ил)-2-метоксибензоил)пиримидин-2-ил)амино)-4-метоксифенил)акриламид (EGFR_3365_127).
7. Способ ингибирования биологической активности рецептора эпидермального фактора роста (EGFR) у субъекта, заключающийся в контактировании EGFR с соединением по любому из пп. 1-6.
8. Фармацевтическая композиция, содержащая терапевтически эффективное количество соединения по любому из пп. 1-6 и один или несколько фармацевтически приемлемых эксципиентов и предназначенная для профилактики или лечения заболевания, или нарушения, опосредованного активностью EGFR с мутацией L858R и/или мутацией Т790М и/или делецией в экзоне 19 и/или мутацией C797S.
9. Способ лечения заболевания или нарушения, опосредованного активностью EGFR, включающий введение в терапевтически эффективном количестве соединения по любому из пп. 1-6 или фармацевтической композиции по п. 8 субъекту, нуждающемуся в таком лечении, где указанное заболевание или нарушение представляет собой заболевание или нарушение, опосредованное активностью EGFR с мутацией L858R и/или мутацией Т790М и/или делецией в экзоне 19 и/или мутацией C797S.
10. Способ по п. 9, где заболевание или нарушение, опосредованное активностью EGFR, представляет собой онкологическое заболевание.
11. Способ по п. 10, где заболевание или нарушение представляет собой рак мочевого пузыря, рак яичника, рак шейки матки, колоректальный рак, рак молочной железы, рак поджелудочной железы, рак головы и шеи, глиому, глиобластому, меланому, рак предстательной железы, лейкоз, лимфому, неходжкинскую лимфому, лимфому Ходжкина, рак легкого, гепатоцеллюлярный рак, рак пищевода, рак желудка, стромальную опухоль желудочно-кишечного тракта, рак щитовидной железы, рак желчных протоков, рак эндометрия, рак почки, рак печени, анапластическую крупноклеточную лимфому, острый миелоидный лейкоз, множественную миелому, меланому, мезотелиому, гематологические злокачественные опухоли.
12. Способ по п. 11, где онкологическое заболевание представляет собой немелкоклеточный рак легкого.
13. Применение соединения по любому из пп. 1-6 или фармацевтической композиции по п. 8 для лечения заболевания или нарушения, опосредованного активностью EGFR у субъекта, нуждающегося в таком лечении, где указанное заболевание или нарушение представляет собой заболевание или нарушение, опосредованное активностью EGFR с мутацией L858R и/или мутацией Т790М и/или делецией в экзоне 19 и/или мутацией C797S.
14. Применение по п. 13, где заболевание или нарушение, опосредованное активностью EGFR, представляет собой онкологическое заболевание.
15. Применение по п. 14, где заболевание или нарушение представляет собой рак мочевого пузыря, рак яичника, рак шейки матки, колоректальный рак, рак молочной железы, рак поджелудочной железы, рак головы и шеи, глиому, глиобластому, меланому, рак предстательной железы, лейкоз, лимфому, неходжкинскую лимфому, лимфому Ходжкина, рак легкого, гепатоцеллюлярный рак, рак пищевода, рак желудка, стромальную опухоль желудочно-кишечного тракта, рак щитовидной железы, рак желчных протоков, рак эндометрия, рак почки, рак печени, анапластическую крупноклеточную лимфому, острый миелоидный лейкоз, множественную миелому, меланому, мезотелиому, гематологические злокачественные опухоли.
16. Применение по п. 15, где онкологическое заболевание представляет собой немелкоклеточный рак легкого.
| WO 2016054987 A1, 14.04.2016 | |||
| EP 3181559 A1, 21.06.2017 | |||
| WO 2011140338 A1, 10.11.2011 | |||
| US 2011207736 A1, 25.08.2011 | |||
| Anne Marie Healy et al | |||
| "Pharmaceutical solvates, hydrates and amorphous forms: A special emphasis on cocrystals", Advanced Drug Delivery Reviews, march 2017, P | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| интернет-источник | |||

