В данной заявке заявлен приоритет по предварительной заявке на патент Китая № CN201910600229.1, поданной 4 июля 2019 года, полное содержание которой включено в данный документ посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ
Данное изобретение относится к области фармацевтических препаратов и, более конкретно, относится к серии ингибиторов EGFR, способам их получения и их применения.
УРОВЕНЬ ТЕХНИКИ
Рецептор эпидермального фактора роста (EGFR) представляет собой рецепторную тирозинкиназу семейства ErbB в цитоплазматической мембране. Другие члены семейства ErbB включают ERBB2 (HER2), ERBB3 (HER3) и ERBB4 (HER4). EGFR ускоряет клеточный рост посредством активации пути передачи сигналов MAPK и PI3K. EGFR, сверхактивированный вследствие мутации, амплификации или сверхэкспрессии, идентифицирован во многих солидных опухолях, особенно в раке легких.
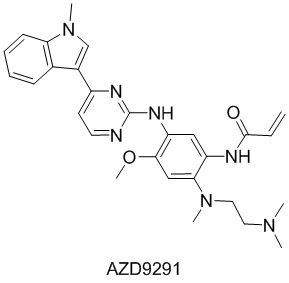
Распространенность мутации EGFR при немелкоклеточном раке легких (NSCLC) составляет 50% в Восточной Азии и 15% в Европе и Америке. Большинство мутаций EGFR встречаются с 18 экзона по 21 экзон. Первое поколение ингибиторов тирозинкиназы EGFR (TKI), включая гефитиниб и эрлотиниб, направленно воздействует, главным образом, на мутации в экзоне 18, 19 и 21. Однако в ходе лечения неизбежно развивается резистентность. На долю мутации EGFR-T790M приходится более 60% приобретенной резистентности к TKI первого поколения. Афатиниб, необратимый ингибитор EGFR второго поколения, активен против T790M, однако он ассоциируется с существенной токсичностью, включая сыпь и диарею, вследствие его активности против EGFR дикого типа. Третье поколение TKI EGFR TKI, AZD9291, специфически блокирует T790M и одобрено в качестве средства лечения пациентов с немелкоклеточным раком легких, положительным по мутации EGFR T790M.
Помимо вышеупомянутых классических мутаций EGFR, вставки в 20 экзон образуют третью наибольшую группу мутаций EGFR с встречаемостью 4-10% среди всех мутаций EGFR, которые более распространены среди женщин, некурящих, жителей Азии и пациентов с аденокарциномой, и ассоциируются с такими же клиническими характеристиками, как при классических мутациях.
Мутации в 20 экзоне сгруппированы между аминокислотами 762 и 823, и все они являются вставками, за исключением T790M. Помимо EGFR, около 2% пациентов с NSCLC несут мутации her2, 90% из которых являются вставками в 20 экзоне. Мутации со вставкой в 20 экзоне Her2 встречаются в положении, структурно аналогичном положению в EGFR с подобными молекулярными особенностями и чувствительностью к лекарственным средствам. В совокупности их широко классифицируют как вставки в 20 экзоне. До настоящего времени идентифицированы 122 подтипа вставок в 20 экзоне EGFR, из которых самой распространенной является Asp770_Asn771ins, за ней следуют Va1769_Asp770ins, A1a767_Va1769ins и Ser768_Asp770ins. В то же время наиболее распространенным вариантом мутации в 20 экзоне Her2 является A775_G776insYVM, на долю которой приходится 70% случаев. Все вставки в 20 экзоне EGR и Her2 ускоряют лиганд-независимую активацию.
Большинство вставок в 20 экзоне EGFR являются «наивными», и некоторые из них являются приобретенными. Помимо рака легких, вставки в 20 экзоне также обнаружены в редкой форме рака головы и шеи, известной как синоназальная плоскоклеточная карцинома. С учетом наличия вставок в 20 экзоне у существенного количества пациентов, агенты, ингибирующие EGFR, несущий вставки в 20 экзоне, могут быть особенно пригодны для данной группы пациентов. Однако во многих исследованиях показано, что вставки в 20 экзоне, в частности, вставки после 764 аминокислоты, не чувствительны к одобренным TKI, и доступно ограниченное количество терапевтических возможностей. В настоящее время проходят клинические исследования позиотиниба и мобоцертиниба, двух TKI против вставок в 20 экзоне. Из них позиотиниб ассоциируется с тяжелыми неблагоприятными эффектами, возможно вследствие одновременного ингибирования EGFR дикого типа. Таким образом, необходима разработка TKI с селективностью в отношении вставок в 20 экзоне по сравнению с EGFR дикого типа. Новое поколение TKI, описанное в данном патенте, демонстрирует превосходящую биохимическую и клеточную активность в отношении T790M и мутаций со вставкой в 20 экзоне по сравнению с EGFR дикого типа.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
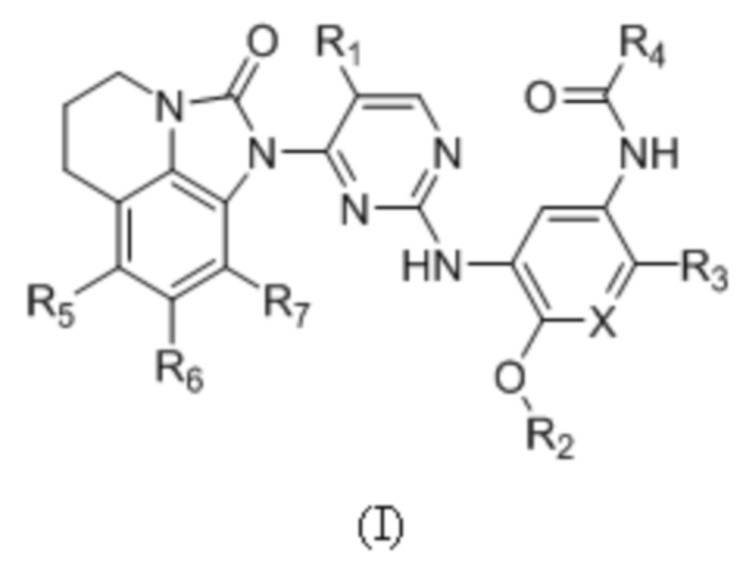
В данном изобретении предложено соединение общей формулы (I), его фармацевтически приемлемая соль:
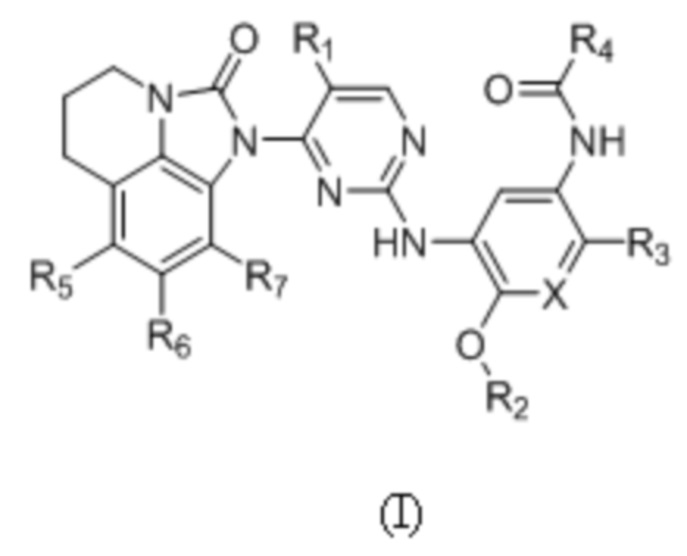
где:
X выбран из группы, состоящей из N и CH;
R1 выбран из группы, состоящей из водорода, галогена, C1-6 алкила, C3-6 циклоалкила, -C(O)OR8 и CN;
R2 выбран из группы, состоящей из C1-6 алкила, дейтерированного C1-6 алкила, C3-6 циклоалкила и C1-6 галогеналкила;
R3 выбран из группы, состоящей из -NR9(CH2)2NR9'R9'', 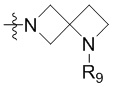 ,
, 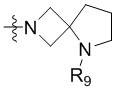 ,
, 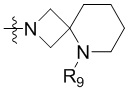 ,
,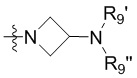 и
и 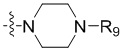 ;
;
R4 представляет собой 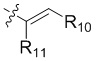 ;
;
R5, R6 и R7 независимо выбраны из группы, состоящей из водорода, галогена, C1-6 алкила, C1-6 галогеналкила, C1-6 алкокси и CN;
R8 выбран из группы, состоящей из водорода, C1-6 алкила и C1-6 галогеналкила;
R9 выбран из группы, состоящей из водорода, C1-6 алкила, дейтерированного C1-6 алкила и C1-6 галогеналкила;
R9' и R9'' независимо выбраны из группы, состоящей из водорода, C1-6 алкила, C3-6 циклоалкила, дейтерированного C1-6 алкила и C1-6 галогеналкила, или R9' и R9'' вместе с атомом азота, связанным с ними, образуют гетероцикл, указанный гетероцикл является незамещенным или необязательно замещен 1-3 группами, выбранными из группы, состоящей из галогена, C1-6 алкила, C1-3 алкокси, метилтио, метансульфонила и C1-6 галогеналкила;
R10 выбран из группы, состоящей из водорода, галогена, C1-6 алкила и -CH2NR12'R12'';
R11 выбран из группы, состоящей из водорода, галогена и C1-6 алкила; и
R12 и R12' независимо выбраны из группы, состоящей из водорода, C1-6 алкила и C1-6 галогеналкила, или R12' и R12'' вместе с атомом азота, связанным с ними, образуют гетероцикл, указанный гетероцикл является незамещенным или необязательно замещен 1-3 группами, выбранными из группы, состоящей из галогена, C1-6 алкила и C1-6 галогеналкила.
В общей формуле (I) R1 предпочтительно выбран из группы, состоящей из водорода, галогена, C1-6 алкила, -C(O)OR8 или CN; и R5, R6 и R7 предпочтительно независимо выбраны из группы, состоящей из водорода и галогена.
В данном изобретении предложены соединения формулы (I), способные ингибировать один или более EGFR-активируемых или лекарственно-устойчивых мутантов, например, лекарственно-устойчивый мутант T790M, мутант, активируемый вставкой в 20 экзоне, и, следовательно, такие соединения могут быть использованы в схемах лечения рака для пациентов с приобретенной лекарственной резистентностью к существующим терапиям на основе ингибиторов EGFR.
В данном изобретении предложены соединения общей формулы (I), которые обладают более эффективным ингибированием EGFR, образованного активированным или резистентным мутантом, чем EGFR дикого типа вследствие сниженной токсичности, ассоциированной с ингибированием EGFR дикого типа, поэтому указанные соединения являются более подходящими для применения в качестве терапевтических агентов, в частности, для лечения рака.
В данном изобретении предложен способ получения соединения общей формулы (I).
В данном изобретении предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, вспомогательное вещество или разбавитель.
В данном изобретении предложено применение соединения формулы (I) или его фармацевтически приемлемой соли для лечения заболеваний, опосредованных EGFR-активированными или лекарственно-устойчивыми мутантами, у млекопитающих, в частности, у людей и, в частности, для лечения рака.
В данном изобретении предложен способ лечения заболевания, в частности, рака, опосредованного EGFR-активированными или лекарственно-устойчивыми мутантами, у млекопитающих, в частности, у людей, включающий введение пациенту соединения формулы (I) или его фармацевтически приемлемой соли, или фармацевтической композиции, содержащей терапевтически эффективное количество соединения формулы (I) и фармацевтически приемлемый носитель, вспомогательное вещество или разбавитель.
В данном изобретении предложен способ селективного ингибирования EGFR-активированных или лекарственно-устойчивых мутантов по сравнению с EGFR дикого типа, включающий приведение в контакт биологического образца с соединением формулы (I) или его фармацевтически приемлемой солью, или с его фармацевтической композицией, или введение пациенту соединения формулы (I) или его фармацевтически приемлемой соли, или его фармацевтической композиции.
Рак, упомянутый в данном изобретении, может быть выбран из гепатоцеллюлярной карциномы, рака легких, рака поджелудочной железы, рака молочной железы, рака шейки матки, эндометриального рака, рака толстой и прямой кишок, рака желудка, рака носоглотки, рака яичника, рака предстательной железы, лейкоза, лимфомы, неходжкинской лимфомы, миеломы, глиомы, глиобластомы, меланомы, желудочно-кишечной стромальной опухоли (GIST), рака щитовидной железы, холангиокарциномы, рака почек, анапластической крупноклеточной лимфомы, острого миелоидного лейкоза (AML), множественной миеломы или мезотелиомы.
В данном изобретении особенно предпочтительные соединения формулы (I) или их фармацевтически приемлемые соли включают следующие:
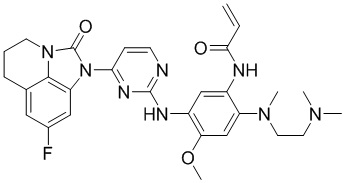
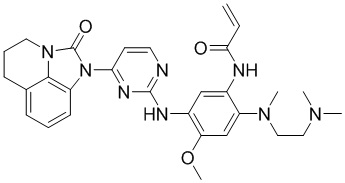
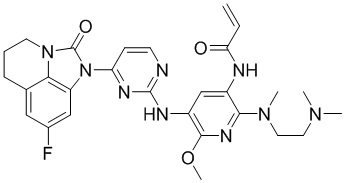
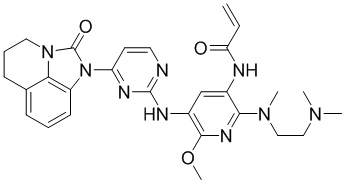
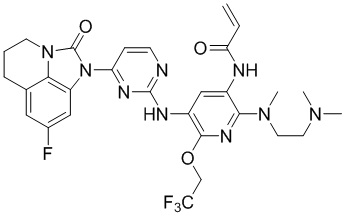
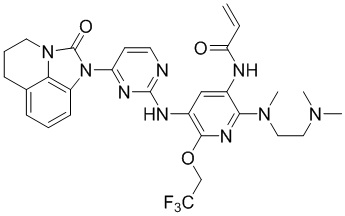
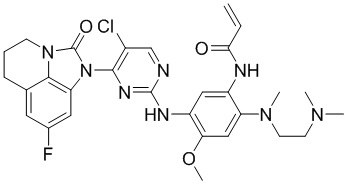
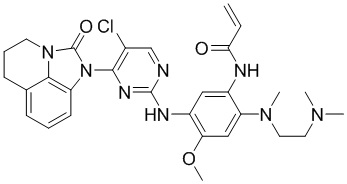
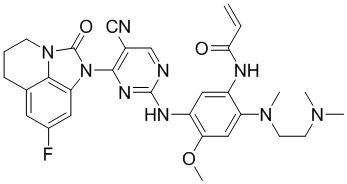
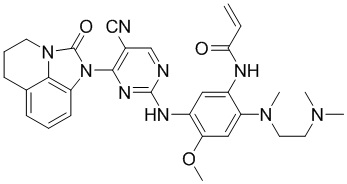
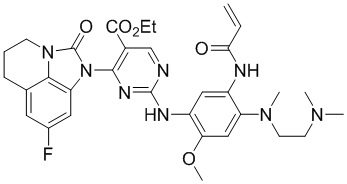
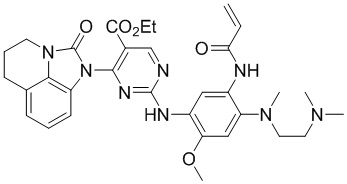
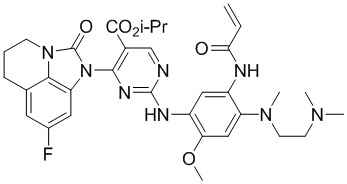
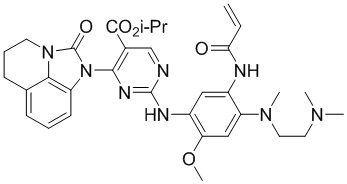
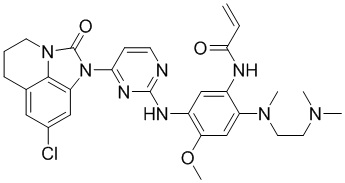
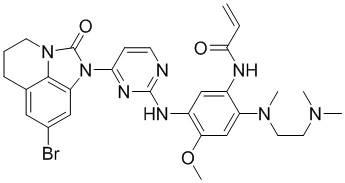
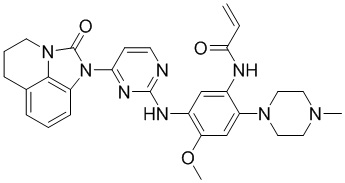
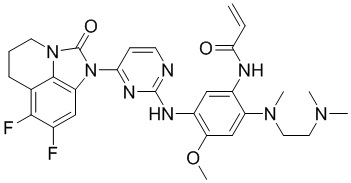
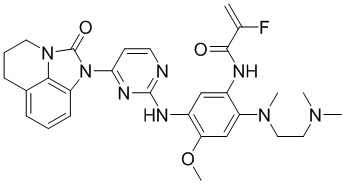
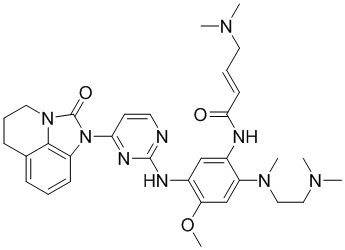
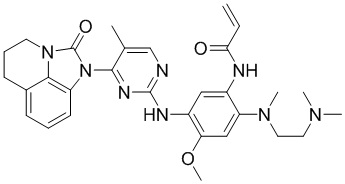
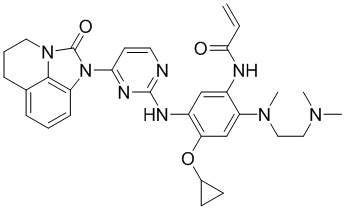
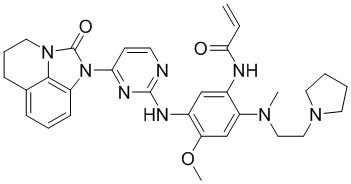
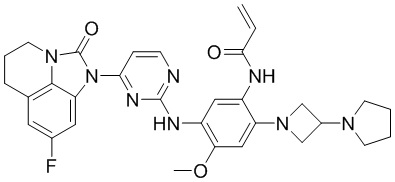
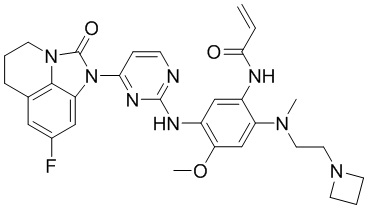
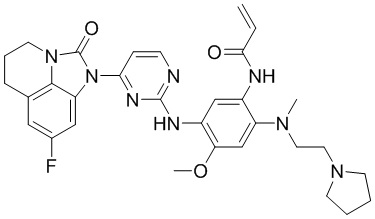
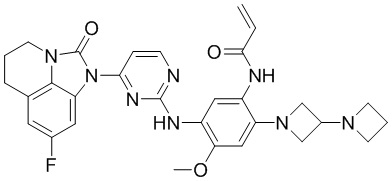
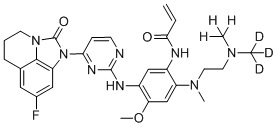
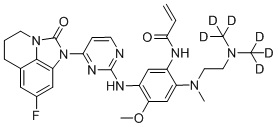
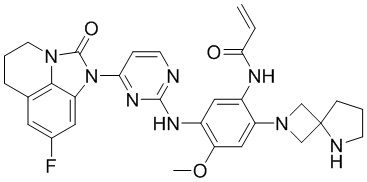
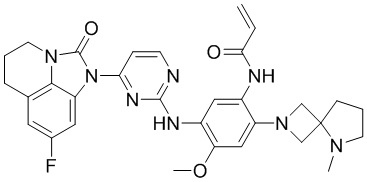
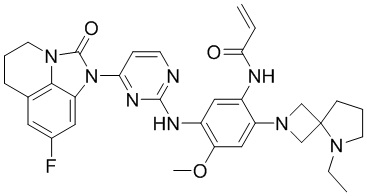
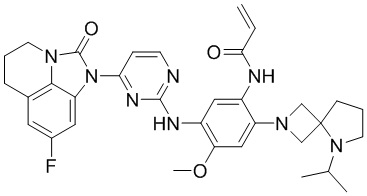
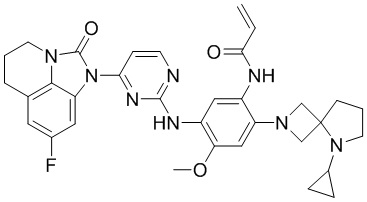
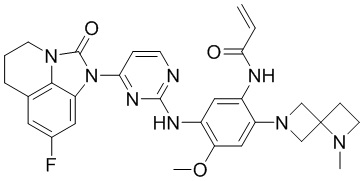
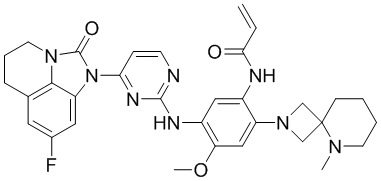
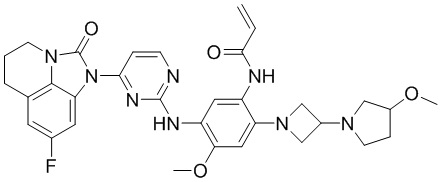
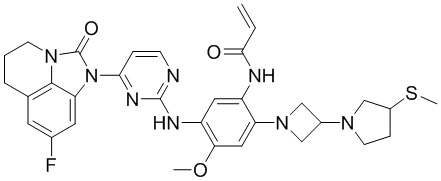
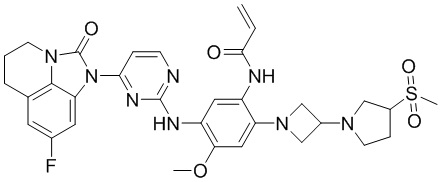
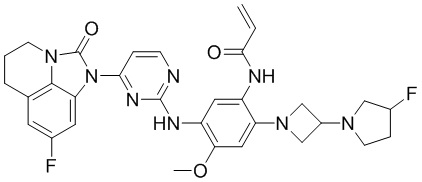
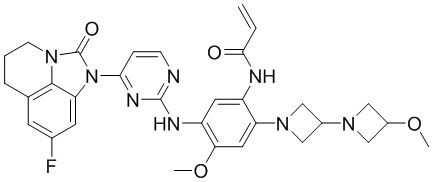
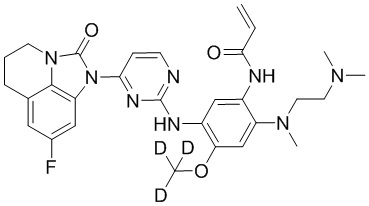
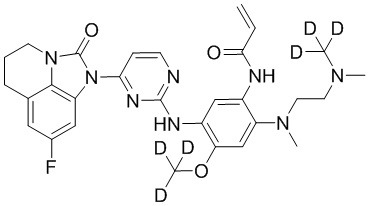
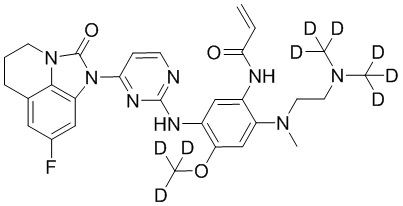
В данном изобретении предложен способ получения соединения формулы (I), включающий следующие стадии:
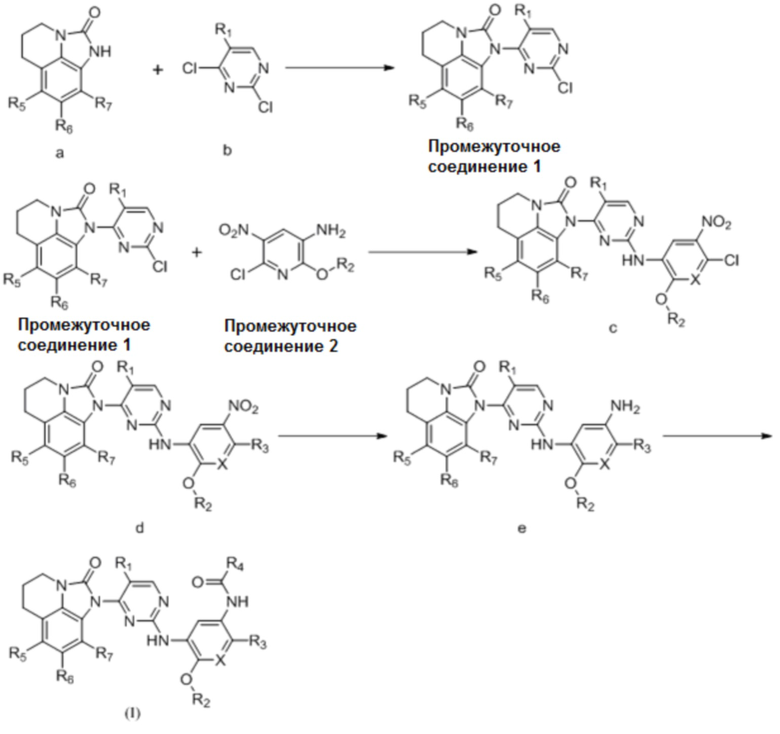
или
где R1, R2, R3, R4, R5, R6 и R7 имеют такие же значения, как указаны выше для общей формулы (I).
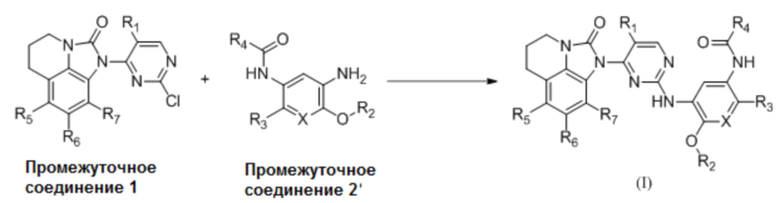
Используя соединения (a) и (b) в качестве исходных материалов, осуществляя реакцию замещения в основных условиях с получением промежуточного соединения 1, осуществляя реакцию замещения или конденсации с использованием промежуточного соединения 1 и промежуточного соединения 2 с получением соединения (c), соединение (c) подвергают нуклеофильному замещению с получением соединения (d), восстанавливают нитро-группу соединения (d) с получением соединения (e), затем соединение (e) подвергают ацилированию с получением соединения (I); или промежуточное соединение 1 и промежуточное соединение 2' напрямую подвергают реакции замещения или конденсации с получением соединения (I).
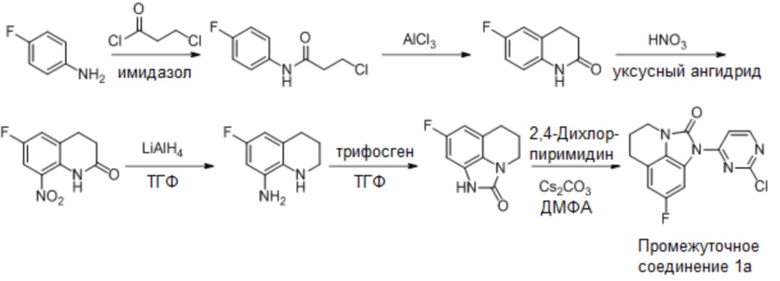
В одном варианте реализации, если промежуточное соединение 1 представляет собой промежуточное соединение 1a, соединение формулы (I) получают следующим образом:
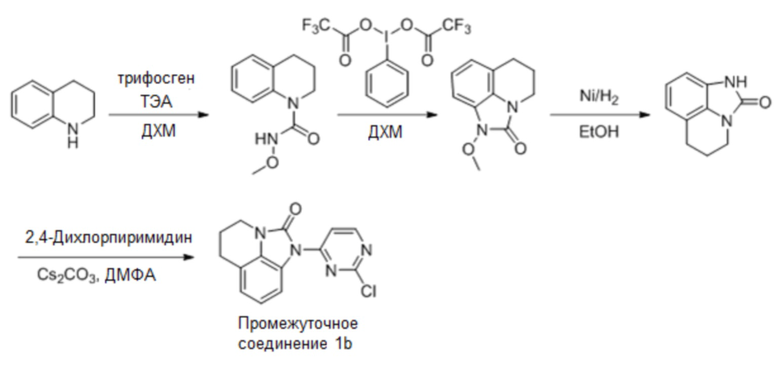
В одном варианте реализации, если промежуточное соединение 1 представляет собой промежуточное соединение 1b, соединение формулы (I) получают следующим образом:
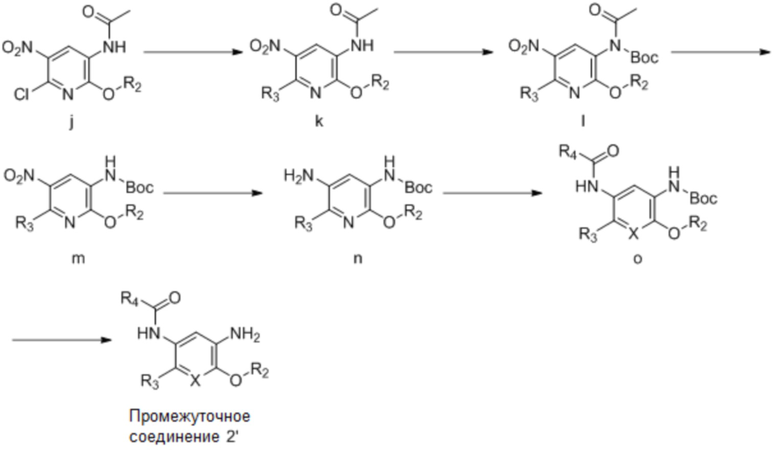
В одном варианте реализации настоящего изобретения, который относится к получению соединения формулы (I), способ получения промежуточного соединения 2, промежуточного соединения 2' включает следующие стадии:
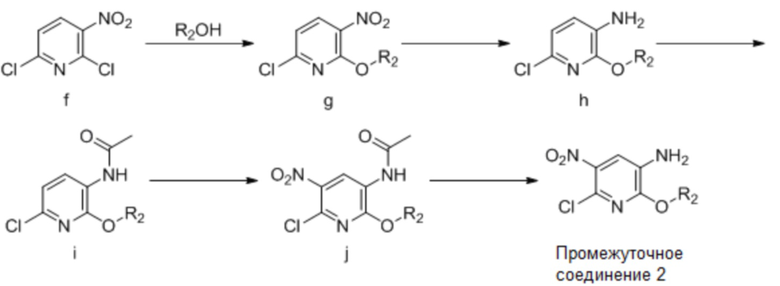
где R2, R3 и R4 имеют такие же значения, как указаны выше для общей формулы (I).
Используя 2,6-дихлор-3-нитропиридин в качестве исходного вещества, осуществляя реакцию этерификации с получением соединения (g), которое подвергают восстановлению нитро-группы соединения (g) с получением соединения (h), соединение (h) подвергают ацилированию с получением соединения (i), затем соединение (i) подвергают реакции нитрования с получением соединения (j), с которого затем снимают защиту с получением промежуточного соединения 2.
Соединение (j) подвергают взаимодействию с R3H по реакции замещения с получением соединения (k), в соединении (k) удаляют защитную Boc-группу с получением соединения (l), которое подвергают деацетилированию с введением защитной группы с получением соединения (m), нитро-группу соединения (m) восстанавливают с получением соединения (n), затем соединение (n) ацилируют с получением соединения (o) и, наконец, в соединении (o) удаляют защитную группу с получением промежуточного соединения.
В способе получения промежуточных соединений 2 и 2' реакцию этерификации осуществляют под действием сильного основания, причем сильное основание включает, но не ограничиваясь ими, гидрид натрия, гидрид калия, гидроксид натрия, гидроксид калия, этоксид натрия и метоксид натрия; в способах восстановления нитро-группы используют обычные восстановительные агенты, известные в данной области техники, включая, но не ограничиваясь ими, порошок железа, порошок цинка, сульфид натрия, H2/PtO2; внедрение защитной группы или удаление защитной группы осуществляют обычными способами, известными в данной области техники, в соответствующих кислотных или основных условиях.
«Галоген» (или «гало») относится к фтору, хлору, брому или йоду.
«C1-6 алкил» относится к линейной или разветвленной алкильной группе, содержащей от 1 до 6 атомов углерода, предпочтительно к линейной или разветвленной алкильной группе, содержащей от 1 до 4 атомов углерода. Разветвленная цепь означает, что одна или более алкильных групп, содержащих от 1 до 4 атомов углерода, таких как метил, этил или пропил и т.д., присоединены к линейной алкильной группе. Предпочтительные C1-6 алкильные группы включают, но не ограничиваясь ими, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил и т.п.
«Дейтерированный алкил» означает, что один или более атомов водорода в алкильной группе заменены на дейтерий. Например, все три атома водорода в метильной группе заменены на дейтерий с образованием дейтерированной метильной группы CD3.
«C1-6 галогеналкил» относится к C1-6 алкильной группе, определение которой приведено выше, содержащей один или более атомов галогена в качестве заместителей.
«C1-6 гетероалкил» означает, что C1-6 алкил, определение которого приведено выше, содержит один или более заместителей, выбранных из группы, состоящей из O, S, N, -(S=O)-, -(O=S=O)- и т.п.
«C3-6 циклоалкил» относится к неароматической моноциклической или полициклической группе, содержащей от 3 до 6 атомов углерода, предпочтительно от 3 до 6 атомов углерода. Предпочтительные моноциклические C3-6 циклоалкильные группы включают, но не ограничиваясь ими, циклопропил, циклопентил, циклогексил и т.п.
«C1-6 алкокси» относится к группе C1-6 алкил-O-, связанной с исходным фрагментом через атом кислорода, в которой C1-6 алкил является таким, как описано выше. Предпочтительные C1-6 алкокси-группы включают, но не ограничиваясь ими, метокси, этокси, н-пропокси, изопропокси и н-бутокси.
Любая из вышеупомянутых функциональных групп по данному изобретению может быть незамещенной или замещенной заместителями, описанными в данном документе. Термин «замещенная» (или замещение) относится к замене одного или более атомов водорода у указанного атома группой, выбранной из указанной группы, при условии, что не превышено нормальное валентное состояние указанного атома, и такое замещение приводит к образованию устойчивого соединения. Комбинации таких заместителей и/или переменных допустимы только в том случае, если такая комбинация приводит к образованию устойчивого соединения.
Данное изобретение также включает фармацевтически приемлемую соль соединения формулы (I). Термин «фармацевтически приемлемая соль» относится к относительно нетоксичной кислотно-аддитивной соли или основно-аддитивной соли соединения по данному изобретению. Кислотно-аддитивная соль представляет собой соль указанного соединения формулы (I) по данному изобретению с подходящей неорганической или органической кислотой, и указанная соль может быть получена при окончательном выделении и очистке соединения или может быть получена посредством взаимодействия очищенного соединения формулы (I) в форме его свободного основания с подходящей органической или неорганической кислотой. Иллюстративные кислотно-аддитивные соли включают гидробромид, гидрохлорид, сульфат, бисульфат, сульфит, ацетат, оксалат, валерат, олеат, пальмитат, стеарат, лаурат, борат, бензоат, лактат, фосфат, гидрофосфат, карбонат, бикарбонат, толуат, цитрат, малеат, фумарат, сукцинат, тартрат, бензоат, метансульфонат, п-толуолсульфонат, глюконат, лактат, лаурат и т.п. Основно-аддитивная соль представляет собой соль указанного соединения формулы (I) по данному изобретению с подходящим неорганическим или органическим основанием, включая, например, соль со щелочным металлом, щелочноземельными металлами, катионом четвертичного аммония, таким как натрий, литий, калий, кальций, магний, тетраметиламмоний, тетраэтиламмоний и т.п.; соль амина, включая соль, образованную с аммиаком (NH3), первичным амином, вторичным амином или третичным амином, такую как соль метиламина, соль диметиламина, соль триметиламина, соль триэтиламина, соль этиламина и т.п.
В анализах активности фермента показано, что соединение по данному изобретению обладает высокой активностью против мутанта вставки в 20 экзоне; в клеточных анализах, а именно в in vitro антипролиферативных анализах клеток с активированным мутантом, т.е. клеток с активированным мутантом по типу вставки в 20 экзон, лекарственно-устойчивых клеток опухоли и клеток кожи человека с EGFR дикого типа, показано, что предложенное соединение обладает высокой антипролиферативной активностью в отношении клеток с активированным мутантом или лекарственно-устойчивых мутантных опухолевых клеток, но слабой антипролиферативной активностью в отношении раковых клеток с EGFR дикого типа с высокой чувствительностью. Соединение по данному изобретению пригодно для лечения заболеваний или патологических состояний, опосредованных активностью EGFR-активированных или резистентных мутантов, в частности, для лечения рака. Такие раковые заболевания включают, но не ограничиваясь ими, гепатоцеллюлярную карциному, рак легких, рак головы и шеи, рак поджелудочной железы, рак молочной железы, рак шейки матки, эндометриальный рак, рак толстой и прямой кишок, рак желудка, рак легких, рак носоглотки, рак яичников, рак предстательной железы, лейкоз, лимфому, неходжкинскую лимфому, миелому, глиому, глиобластому, меланому, желудочно-кишечную стромальную опухоль (GIST), рак щитовидной железы, холангиокарциному, рак почек, анапластическую крупноклеточную лимфому, острый миелоидный лейкоз (AML), множественную миелому или мезотелиому, в частности, опухоль с мутацией 790 треонина в метионин в рецепторе эпидермального фактора роста (EGFR T790M) и опухоль с мутацией активированного типа, и более эффективным является применение для опухолей с активированной мутацией по типу вставки в 20 экзоне.
Следует понимать, что и изложенной выше общее описание, и следующее подробное описание данного изобретения являются иллюстративными и пояснительными, и они предназначены для дополнительного объяснения заявленного изобретения.
Следует понимать, что специалистами в данной области техники могут быть сделаны различные изменения или модификации без отступления от объема и сущности данного изобретения, и специалистам в данной области техники понятно, что такие эквиваленты также могут входить в объем данного изобретения, определенный прилагаемой формулой изобретения.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На Фиг. 1 показано влияние соединения 1 на степень выживания in-situ «голых» мышей с PC9 в головном мозге.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Далее представлено подробное описание вариантов реализации данного изобретения.
ПРИМЕРЫ
Данное изобретение дополнительно проиллюстрировано конкретными примерами. Следует понимать, что данные примеры представлены лишь для иллюстрации данного изобретения и не предназначены для ограничения объема данного изобретения, и данное изобретение не ограничено приведенными примерами. Специалистам в данной области техники понятно, что указанные соединения могут быть получены с использованием известных вариаций условий и приемов, описанных в следующих способах получения. При отсутствии специального описания, исходные материалы, использованные в данном изобретении, доступны в продаже.
Сокращения: комнатная температура (RT, комн. т-ра); водный раствор (водн.); петролейный эфир (PE); этилацетат (EA); дихлорметан (ДХМ); метанол (MeOH); этанол (EtOH); тетрагидрофуран (ТГФ); диметилформамид (ДМФА); диметилсульфоксид (ДМСО); триэтиламин (ТЭА), диизопропилэтиламин (DI(P)EA); 4-диметиламинопиридин (DMAP); палладий на углероде (Pd/C); эквивалент (экв.); грамм/миллиграмм (г/мг); моль/миллимоль (моль/ммоль); литр/миллилитр (л/мл); минута (секунда) (мин (с)); час (ч.); азот (N2); ядерный магнитный резонанс (ЯМР); тонкослойная хроматография (ТСХ).
Общий способ синтеза:
Если не указано иное, все реакции проводили в атмосфере инертного газа (например, аргона или азота), используя доступные в продаже реагенты и безводные растворители без дополнительной обработки.
Масс-спектры записывали на жидкостном хроматомасс-спектрометре (ЖХ-МС) (одноступенчатый и четырехступенчатый ЖХ-МС Agilent 6120B). Спектры ядерного магнитного резонанса (такие как спектры водорода (1H), углерода (13C), фосфора (31P) и фтора (19F)) записывали на ЯМР спектрометре BrukerAMX-400, Gemini-300 или AMX-600 в дейтерированном растворителе, таком как дейтерированных хлороформ, дейтерированный метанол, дейтерированная вода или дейтерированный диметилсульфоксид, с использованием пика дейтерированного растворителя в качестве эталонного стандарта. Химический сдвиг δ выражен в м.д., а константа связывания (J) выражена в герцах (Гц). Пики расщепления при связывании на спектре ЯМР записывали как широкий синглет (шс), синглет (с), дублет (д), дублет дублетов (дд), триплет (т), квадруплет (к) и мультиплет (м).
Подробное описание изобретения:
1. Примеры получения промежуточных соединений по данному изобретению
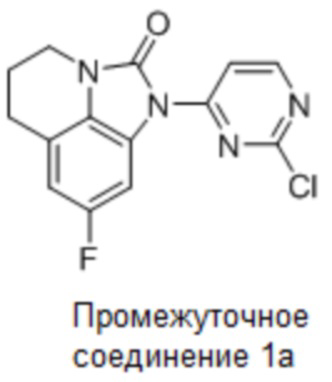
Промежуточное соединение 1a: Синтез 1-(2-хлорпиримидин-4-ил)-8-фтор-5,6-дигидро-4H-имидазо[4,5,1-ij]хинолин-2(1H)-она
Стадия 1: Синтез 3-хлор-N-(4-фторфенил)пропенамида
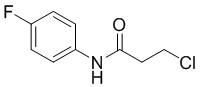
3-Хлорпропионилхлорид (653 г, 1 экв.) растворяли в 6,5 л дихлорметана и по каплям добавляли исходное вещество, 4-фторанилин (783,6 г, 1,05 экв.) при охлаждении на бане из сухого льда/этанола, поддерживая внутреннюю температуру от 0 до 10°С, в осадок выпало большое количество твердого вещества. После капельного добавления перемешивали смесь еще 0,5 часа и по частям добавляли имидазол (405 г, 1,01 экв.) (с заметным повышением температуры) для поддержания внутренней температуры от 0 до 10°С. Реакция была завершена после перемешивания в течение 1 часа, реакционный раствор выливали в разбавленную хлористоводородную кислоту, разделяли, концентрировали органическую фазу до осаждения большого количества твердого вещества, добавляли 800 мл PE/EA(5/1), перемешивали в течение ночи, фильтровали и промывали PE/EA (5/1) с получением 950 г 3-хлор-N-(4-фторфенил)пропенамида в виде белого твердого вещества. МС (ИЭР): m/z = 202 [M+H]+, 1H ЯМР (400 МГц, ДМСО-d6) δ 10,16 (с, 1H), 7,71-7,60 (м, 2H), 7,23-7,13 (м, 2H), 3,91 (т, J = 6,3 Гц, 2H), 2,84 (т, J = 6,3 Гц, 2H).
Стадия 2: Синтез 6-фтор-3,4-дигидрохинолин-2(1H)-она
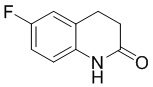
В трехгорлую колбу объемом 5 л добавляли 3-хлор-N-(4-фторфенил)пропионамид (820 г, 1 экв.), затем при перемешивании добавляли безводный трихлорид алюминия (1640 г, 3 экв.), затем три раза заменяли атмосферу на азот. Внешнюю температуру устанавливали на 60°С и перемешивали содержимое колбы до расплавленного состояния (внутренняя температура повысилась до 70°С). После снижения внутренней температуры нагревали колбу до 100°С (внутренняя температура составляла 97°С) и перемешивали смесь в течение 4 часов. ЖХМС показала степень превращения около 58%, добавляли 500 г трихлорида алюминия и перемешивали смесь еще 4 часа, ЖХМС показала степень превращения около 73%, и добавляли еще 200 г трихлорида алюминия и перемешивали в течение 4 часов, ЖХМС показала лишь небольшое количество непревращенного исходного вещества. После охлаждения смеси до 40°С в смеси добавляли ДХМ (2 л), затем в смесь по каплям добавляли ТГФ (6 л), наблюдая интенсивную экзотерму. Добавляли EA (3 л) и непрерывно добавляли воду для отделения большого количества осадка. Отделяли органическую фазу, концентрировали органическую фазу и фильтровали водную фазу. Объединенные продукты суспендировали, соответственно, с EA и водой с получением 600 г влажного продукта в виде белого твердого вещества. МС (ИЭР): m/z = 166 [M+H]+, 1H ЯМР (400 МГц, ДМСО-d6) δ 10,16 (с, 1H), 7,71-7,61 (м, 2H), 7,23-7,13 (м, 2H), 3,92 (т, J = 6,3 Гц, 2H), 2,85 (т, J = 6,3 Гц, 2H).
Стадия 3: Синтез 6-фтор-8-нитро-3,4-дигидрохинолин-2(1H)-она
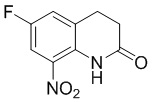
6-Фтор-3,4-дигидрохинолин-2(1H)-он (700 г, 1 экв.) добавляли в трехгорлую колбу объемом 5 л, затем добавляли 3,5 л уксусного ангидрида, регулируя внутреннюю температуру на уровне 15-20°С, по каплям медленно добавляли концентрированную азотную кислоту (485 г, 1,2 экв.), после добавления раствор стал прозрачным, затем перемешивали при 25°С в течение 30 минут, затем в осадок выпало большое количеств твердого вещества, выливали реакционный раствор в воду (20 л), перемешивали до завершения гидролиза, фильтровали, промывали осадок на фильтре водой, пока промывочный раствор не стал бесцветным, и сушили с получением 700 г требуемого промежуточного соединения в виде желтого твердого вещества; МС (ИЭР): m/z = 211 [M+H]+, H ЯМР: 1H ЯМР (400 МГц, ДМСО-d6) δ 9,84 (с, 1H), 7,91 (дд, J = 8,9, 2,9 Гц, 1H), 7,70 (дд, J = 8,2, 2,8 Гц, 1H), 3,15-3,04 (м, 2H), 2,63 (дд, J = 8,3, 6,7 Гц, 2H).
Стадия 4: Синтез 6-фтор-1,2,3,4-тетрагидрохинолин-8-амина
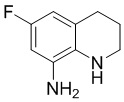
LiAlH4 (48 г, 1,27 моль) растворяли в ТГФ (1 л) и по частям добавляли суспензию 6-фтор-8-нитро-3,4-дигидрохинолин-2(1H)-она (89 г, 0,42 моль) в ТГФ (100 мл), поддерживая внутреннюю температуру от 5 до 10°С. После завершения капельного добавления смесь естественным образом возвращалась к температуре 12°С, и ее перемешивали в течение 0,5 часа. Затем смесь охлаждали до температуры ниже 0°С, последовательно гасили водой (48 мл), 15% NaOH (48 мл) и водой (144 мл), поддерживая внутреннюю температуру ниже 5°С, и добавляли диатомовую землю (90 г). После перемешивания в течение 30 минут при температуре ниже 5°С фильтровали смесь с диатомовой землей, промывали ТГФ, осадок на фильтре снова суспендировали с ТГФ, фильтровали и концентрировали органическую фазу. Остаток очищали колоночной хроматографией (подвижная фаза PE/EA, соотношение 1/10, 1/4 и 2/3 с содержанием 0,1% ТЭА) с получением 57 г требуемого промежуточного соединения в виде винно-красной маслянистой жидкости; МС (ИЭР): m/z = 167 [M+H]+.
Стадия 5: Синтез 8-фтор-5,6-дигидро-4H-имидазо[4,5,1-ij]хинолин-2(1H)-она
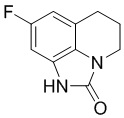
6-Фтор-8-амино-1,2,3,4-тетрагидрохинолин (166 г, 1 моль) растворяли в ТГФ (1 л) и по каплям добавляли суспензию трифосгена (118 г, 0,4 моль) в ТГФ (300 мл), поддерживая внутреннюю температуру от 5 до 10°С. После завершения добавления продолжали перемешивание в течение 0,5 часа, по каплям добавляли имидазол (160 г, 20 моль), поддерживали внутреннюю температуру от 10 до 20°С и продолжали перемешивание в течение 15 минут после возвращения температуры к комнатной температуре. После расходования исходных материалов, по данным ЖХМС, добавляли 1 л 13% раствора NaCl, затем добавляли ТГФ (1 л), отделяли органическую фазу, экстрагировали ТГФ (2 л*2), сушили и концентрировали, остаток суспендировали с EA в течение ночи и фильтровали с получением 168 г требуемого промежуточного соединения в виде светло-коричневого твердого вещества. МС (ИЭР): m/z = 193 [M+H].
Стадия 6: Синтез 1-(2-хлорпиримидин-4-ил)-8-фтор-5,6-дигидро-4H-имидазо-[4,5,1-ij]хинолин-2(1H)-она
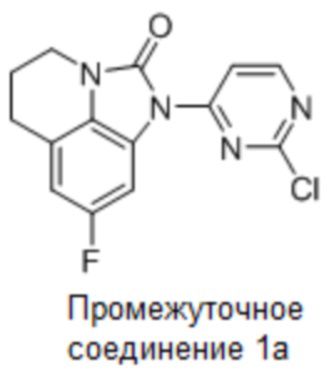
8-Фтор-5,6-дигидро-4H-имидазо[4,5,1-ij]хинолин-2(1H)-он (36 г, 0,19 моль) и 2,4-дихлорпиримидин (34 г, 0,23 моль) растворяли в ДМФА (400 мл), добавляли карбонат цезия (122 г, 0,37 моль) и перемешивали в течение 4 часов при комнатной температуре. По данным ЖХМС, реакция была завершена. Разбавляли смесь водой (250 мл), отфильтровывали твердое вещество и дополнительно очищали неочищенный образец колоночной хроматографией (ДХМ/EA, 100/1), концентрировали до около 50 мл, суспендировали с PE (200 мл) и фильтровали с получением 45 г требуемого промежуточного соединения в виде белого твердого вещества; МС (ИЭР): m/z = 305 [M+H]+, 1H ЯМР (400 МГц, ДМСО-d6) δ 8,81 (д, J = 5,7 Гц, 1H), 8,42 (д, J = 5,8 Гц, 1H), 7,75 (д, J = 9,7 Гц, 1H), 7,00 (д, J = 9,6 Гц, 1H), 3,82 (т, J = 5,5 Гц, 2H), 2,85 (т, J = 5,6 Гц, 2H), 2,15-2,01 (м, 2H).
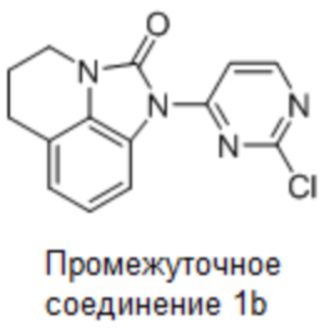
Промежуточное соединение 1b: Синтез 1-(2-хлорпиримидин-4-ил)-5,6-дигидро-4H-имидазо[4,5,1-ij]хинолин-2(1H)-она
Стадия 1: Синтез N-метокси-3,4-дигидрохинолин-1(2H)-карбоксамида
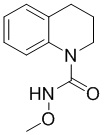
Растворяли трифосген (335 г, 1,13 моль) в ДХМ (3 л), по каплям добавляли раствор 1,2,3,4-тетрагидрохинолина (300 г, 2,26 моль) и триэтиламина (390 г, 3,86 ммоль) в ДХМ (2 л) в течение 1,5 часа при температуре от 0 до 5°С. После добавления перемешивали смесь при комнатной температуре в течение 1 часа. ТСХ (PE:EA = 5:1) показала, что израсходована большая часть 1,2,3,4-тетрагидрохинолина. Добавляли триэтиламин (800 г, 7,92 моль) и гидрохлорид метоксиамина (375 г, 4,52 моль) и перемешивали при комнатной температуре (15°С) еще 16 часов. ТСХ (PE:EA = 5:1) показала, что осталось небольшое количество (около 20%) исходного вещества, тогда реакционную смесь нагревали до 30°С (на водяной бане) еще 3 часа. По данным ТСХ (PE:EA = 5:1), реакция была завершена, реакционный раствор промывали хлористоводородной кислотой (2 M, 3 л), экстрагировали водную фазу ДХМ (1 л), объединяли органические фазы, промывали насыщенным раствором бикарбоната натрия (3 л) и насыщенным солевым раствором (2 л), сушили над безводным сульфатом натрия, фильтровали и сушили с получением требуемого промежуточного соединения (580 г) в виде желтого твердого вещества.
Стадия 2: Синтез 1-метокси-5,6-дигидро-4H-имидазо[4,5,1-ij]хинолин-2(1H)-она
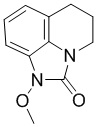
N-Метокси-3,4-дигидрохинолин-1(2H)-карбоксамид (неочищенный, 580 г, 1,13 моль) растворяли в ДХМ (500 мл), по каплям добавляли раствор бис(трифторацетокси)йодбензола (1250 г, 2,91 моль) в ДХМ (1,2 л) при температуре от -3°С до 2°С, после добавления естественным образом нагревали до комнатной температуры (15°С) и перемешивали еще 1 час. По данным ТСХ (PE:EA = 1:1), реакция была завершена, к смеси добавляли насыщенный раствор бикарбоната натрия (8 л), отделяли органическую фазу, концентрировали, очищали остаток колоночной хроматографией (PE:EA = от 5:1 до 1:1) с получением требуемого промежуточного соединения (205 г, выход 44,5%) в виде желтого твердого вещества.
Стадия 3: Синтез 5,6-дигидро-4H-имидазо[4,5,1-ij]хинолин-2(1H)-она
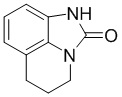
1-Метокси-5,6-дигидро-4H-имидазо[4,5,1-ij]хинолин-2(1H)-он (51,25 г, 251,22 моль) растворяли в этаноле (500 мл), добавляли никель Ренея (20 г) при комнатной температуре (15°С), затем повышали температуру до 50°С и дополнительно перемешивали смесь в течение 16 часов в атмосфере водорода из баллона. ТСХ (PE:EA = 1:1) показала, что осталось около 30% исходного вещества. Дополнительно перемешивали смесь при 50°С в течение 4 часов в атмосфере водорода из нового баллона, ТСХ (PE:EA = 1:1) показала, что осталось около 20% исходного вещества. Добавляли дополнительное количество никеля Ренея (8 г) при комнатной температуре и перемешивали смесь при 50°С в течение 16 часов в атмосфере водорода из нового баллона. По данным ТСХ (PE:EA = 1:1), реакция была завершена. Реакционный раствор охлаждали до комнатной температуры, фильтровали через целит, три раза промывали осадок на фильтре метанолом (150 мл) и концентрировали фильтрат. Неочищенный продукт (четыре объединенные партии) суспендировали с PE/EA (1:1, 800 мл), фильтровали с получением требуемого промежуточного соединения (155 г, выход 88,6%) в виде почти белого твердого вещества.
Стадия 4: Синтез 1-(2-хлорпиримидин-4-ил)-5,6-дигидро-4H-имидазо[4,5,1-ij]хинолин-2(1H)-она
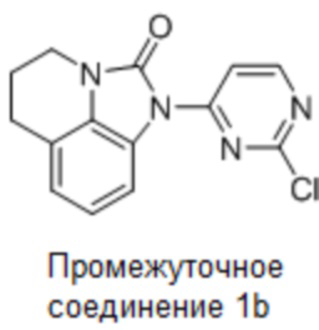
5,6-Дигидро-4H-имидазо[4,5,1-ij]хинолин-2(1H)-он (155 г, 890,80 моль) растворяли в ДМФА (1,5 л), добавляли 2,4-дихлорпиримидин (158 г, 1,06 моль) и карбонат цезия (580 г, 1,78 моль) при комнатной температуре (10°С), затем нагревали до 30°С и перемешивали еще 16 часов. По данным ТСХ (ДХМ:MeOH = 20:1), реакция была завершена, в реакционную смесь добавляли воду (3 л) и перемешивали еще 1 час. Фильтровали, промывали осадок на фильтре водой (1 л). Осадок на фильтре суспендировали с PE:EA (1:1, 1,5 л), фильтровали и сушили с получением требуемого промежуточного соединения (230 г, выход 90,2%) в виде почти белого твердого вещества.
Промежуточное соединение 2a: Синтез N-(5-амино-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида
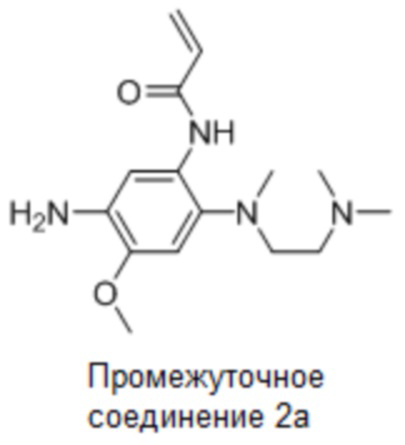
Стадия 1: Синтез N1-(2-(диметиламино)этил)-5-метокси-N1-метил-2-нитробензол-1,4-диамина
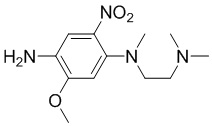
4-Фтор-2-метокси-5-нитроанилин (3 г, 16 ммоль) и N1,N1,N2-триметилэтан-1,2- диамин (2,47 г, 24 ммоль) растворяли в ДМФА (30 мл), добавляли карбонат калия (4,5 г, 32 ммоль), перемешивали при 80°С в течение 2 часов. По данным ЖХМС, реакция была завершена, реакционную смесь охлаждали до комнатной температуры, разбавляли смесь водой (60 мл), фильтровали, осадок на фильтре суспендировали с EtOH/H2O (1/1), фильтровали и сушили с получением требуемого промежуточного соединения (3,1 г) в виде желтого твердого вещества; МС (ИЭР): m/z = 269 [M+H]+.
Стадия 2: Синтез трет-бутил-(4-((2-(диметиламино)этил)(метил)амино)-2-метокси-5-нитрофенил)карбамата
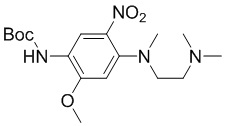
N 1-(2-(Диметиламино)этил)-5-метокси-N1-метил-2-нитробензол-1,4-диамин (3,1 г, 12 ммоль) растворяли в ТГФ (40 мл), добавляли ди-трет-бутилдикарбонат (3,8 г, 17 ммоль) и перемешивали смесь при 70°С в течение 6 часов до завершения реакции. Затем концентрировали, суспендировали остаток с EA/PE (1/5) с получением требуемого промежуточного соединения (3,8 г) в виде бледно-желтого твердого вещества, МС (ИЭР): m/z = 369 [M+H]+.
Стадия 3: Синтез трет-бутил-(5-амино-4-((2-(диметиламино)этил)(метил)амино)-2-метоксифенил)карбамата
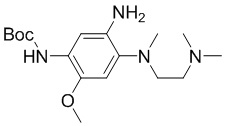
Трет-бутил-(4-((2-(диметиламино)этил)(метил)амино)-2-метокси-5-нитрофенил)карбамат (3,8 г, 10,3 ммоль) растворяли в MeOH (40 мл), три раза меняли атмосферу на азот, затем добавляли Pd/C (0,4 г) и три раза меняли атмосфера на водород. Затем перемешивали смесь при комнатной температуре в течение 4 часов. После завершения реакции фильтровали и концентрировали смесь, неочищенный продукт напрямую использовали на следующей стадии без дополнительной очистки. МС (ИЭР): m/z = 339 [M+H]+.
Стадия 4: Синтез трет-бутил-(5-акриламидо-4-((2-(диметиламино)этил)(метил)-амино)-2-метоксифенил)карбамата
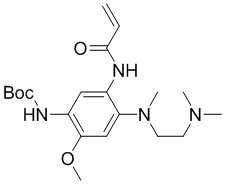
Трет-бутил-(5-амино-4-((2-(диметиламино)этил)(метил)амино)-2-метоксифенил)карбамат (10,3 ммоль) растворяли в ДХМ (50 мл), затем по каплям добавляли акрилоилхлорид (1,36 г, 15 ммоль) при охлаждении на ледяной бане, затем перемешивали в течение 0,5 часа при естественном нагревании до комнатной температуры. Доводили рН до 8, добавляя насыщенный раствор бикарбоната натрия, отделяли водную фазу и экстрагировали ДХМ (50 мл), объединяли органические фазы, сушили и концентрировали, очищали остаток колоночной хроматографией (MeOH/ДХМ = от 1/70 до 1/20) с получением требуемого промежуточного соединения (1,4 г) в виде серого твердого вещества; МС (ИЭР): m/z = 393 [M+H]+.
Стадия 5: Синтез N-(5-амино-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида
Трет-бутил-(5-акриламидо-4-((2-(диметиламино)этил)(метил)амино)-2-метокси-фенил)карбамат (392 мг, 1 ммоль) растворяли в ДХМ (5 мл), по каплям добавляли ТФФК (1 мл), и после перемешивания при комнатной температуре в течение 1 часа реакция была завершена. Доводили рН до 8, добавляя насыщенный раствор бикарбоната натрия при охлаждении на ледяной бане. Отделяли водную фазу, экстрагировали ДХМ (50 мл), сушили и концентрировали, остаток очищали колоночной хроматографией (MeOH/ДХМ = от 1/20 до 1/10) с получением требуемого промежуточного соединения (200 мг) в виде коричневого сиропообразного вещества; МС (ИЭР): m/z = 293 [M+H]+.
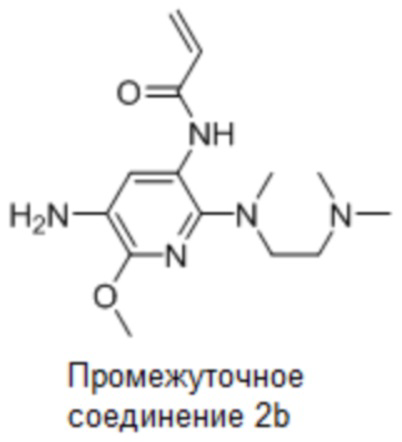
Промежуточное соединение 2b: Синтез N-(5-амино-2-((2-(диметиламино)этил)(метил)-амино)-6-метоксипиридин-3-ил)акриламида
Стадия 1: Синтез 6-хлор-3-нитро-2-(2,2,2-трифторэтокси)пиридина
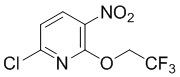
2,6-Дихлор-3-нитропиридин (500 г, 2,6 моль) растворяли в ТГФ (1 л), охлаждали до температуры ниже -10°С, добавляли гидрид натрия (104 г, 2,6 моль), по каплям добавляли трифторэтанол (260 г, 2,6 моль) при -15°С, после добавления нагревали до комнатной температуры и перемешивали в течение ночи. По данным ТСХ (PE/EA = 5/1), реакция была завершена, смесь выливали в ледяную воду (1 л), перемешивали и разделяли. Органическую фазу концентрировали до небольшого объема, дважды экстрагировали EA, объединяли органические фазы, промывали водой и насыщенным солевым раствором, сушили и концентрировали с получением требуемого промежуточного соединения (720 г) в виде желтого маслянисто-твердого вещества; МС (ИЭР): m/z = 257 [M+H]+.
Стадия 2: Синтез 6-хлор-2-(2,2,2-трифторэтокси)пиридин-3-амина
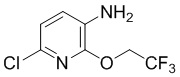
6-Хлор-3-нитро-2-(2,2,2-трифторэтокси)пиридин (150 г, 0,58 моль) растворяли в смеси этанол/вода (1,2 л/0,3 л), добавляли хлорид аммония (160 г, 2,9 моль). После повышения температуры до 50°С (внутренняя температура) медленно, по частям добавляли порошок железа (166 г, 2,9 моль), затем суспендировали при 80°С в течение 1 часа. По данным ТСХ (PE/EA = 5/1), реакция была завершена, температуру понижали до 40°С (внутренняя температура), добавляли карбонат натрия (160 г) и диатомит (160 г), затем перемешивали в течение 20 минут, фильтровали с помощью диатомита, осадок на фильтре суспендировали с ДХМ, маточный раствор водно-этанольный раствор концентрировали досуха, остаток дважды экстрагировали ДХМ, в котором суспендировали осадок на фильтре, объединяли органические фазы, промывали водой с насыщенным солевым раствором, сушили и концентрировали с получением требуемого промежуточного соединения (122 г) в виде черного маслянистого вещества; МС (ИЭР): m/z = 227 [M+H]+.
Стадия 3: Синтез N-(6-хлор-2-(2,2,2-трифторэтокси)пиридин-3-ил)ацетамида
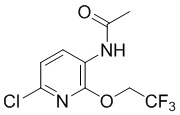
6-Хлор-2-трифторэтоксипиридин-3-амин (570 г, 2,5 моль) растворяли в ДХМ (4,5 л), добавляли DIPEA (540 мл, 3,8 моль). После понижения температуры до 0°С по каплям добавляли ацетилхлорид (200 мл, 3 моль) в течение 1 часа для поддержания температуры около 10°С, затем перемешивали в течение 30 минут, и ТСХ (PE/EA = 5/1) показала, что реакция завершена. Добавляли воду (2 л) при охлаждении на ледяной бане, отделяли органическую фазу и экстрагировали водную фазу ДХМ, объединяли органические фазы, промывали 1 М раствором хлористоводородной кислоты и насыщенным солевым раствором, сушили и концентрировали, остаток очищали колоночной хроматографией (PE/EA = 5/1) с получением требуемого промежуточного соединения (480 г) в виде желтой смеси твердого вещества и жидкости; МС (ИЭР): m/z = 269 [M+H]+.
Стадия 4: Синтез N-(6-хлор-5-нитро-2-(2,2,2-трифторэтокси)пиридин-3-ил)ацетамида
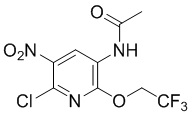
N-(6-хлор-2-(2,2,2-трифторэтокси)пиридин-3-ил)ацетамид (300 г, 1,1 моль) суспендировали в трифторуксусном ангидриде (1,5 л) и охлаждали до температуры ниже -5°С. По каплям добавляли концентрированную азотную кислоту (125 г, 1,2 моль) в течение 1 часа, затем перемешивали при -5°С в течение 4 часов, реакция была завершена, по данным ТСХ (PE/EA = 2/1), затем смесь добавляли в ледяную воду при перемешивании, затем некоторое время перемешивали, фильтровали, последовательно выщелачивали осадок на фильтре водой и PE, суспендировали влажный продукт (185 г) с PE/EA (400 мл) в течение ночи, фильтровали и снова суспендировали осадок на фильтре с PE/EA (5/1), фильтровали и сушили с получением требуемого промежуточного соединения (220 г) в виде желтого твердого вещества; МС (ИЭР): m/z = 314 [M+H]+.
Стадия 5: Синтез 6-хлор-5-нитро-2-(2,2,2-трифторэтокси)пиридин-3-амина
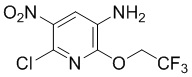
N-(6-Хлор-5-нитро-2-(2,2,2-трифторэтокси)пиридин-3-ил)ацетамид (220 г, 0,7 моль) суспендировали в смешанном растворителе из метанола/концентрированной хлористоводородной кислоты (900/220 мл), нагревали до 50°С для взаимодействия в течение около 4 часов, реакционная смесь становилась прозрачной. По данным ТСХ, реакция была завершена, реакционный раствор добавляли в воду при перемешивании, фильтровали, промывали осадок на фильтре водой, затем суспендировали с насыщенным раствором бикарбоната натрия, фильтровали и последовательно выщелачивали осадок на фильтре водой и PE, сушили с получением требуемого промежуточного соединения (175 г) в виде желтого твердого вещества; МС (ИЭР): m/z = 272 [M+H]+.
Стадия 6: Синтез N2-(2-(диметиламино)этил)-N2-метил-3-нитро-6-(2,2,2-трифторэтокси)пиридин-2,5-диамина
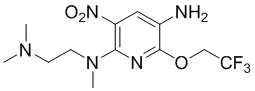
6-Хлор-5-нитро-2-(2,2,2-трифторэтокси)пиридин-3-амин (950 мг, 3,5 ммоль) растворяли в ацетонитриле (15 мл), добавляли K2CO3 (967 мг, 7 ммоль) и N,N,N'- триметилэтилендиамин (643 мг, 6,3 ммоль) при комнатной температуре, затем перемешивали реакционную смесь при 80°С в течение ночи. Реакционный раствор фильтровали, концентрировали фильтрат, очищали остаток колоночной хроматографией на силикагеле с получением требуемого промежуточного соединения (1,16 г) в виде красного маслянистого вещества; МС (ИЭР): m/z = 338,2 [M+H]+.
Стадия 7: Синтез N2-(2-(диметиламино)этил)-N2-метил-3-нитро-5-ди-трет-бутоксикарбониламино-6-(2,2,2-трифторэтокси)-2-амина
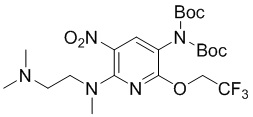
N 2-(2-(Диметиламино)этил)-N2-метил-3-нитро-6-(2,2,2-трифторэтокси)пиридин-2,5-диамин (1,01 г, 3,5 ммоль) и DMAP (110 мг, 0,9 ммоль) растворяли в 1,4-диоксане (30 мл), добавляли ди-трет-бутилдикарбонат (1,96 г, 10,5 ммоль), затем перемешивали на масляной бане при 100°С в течение 8 часов, концентрировали, очищали остаток колоночной хроматографией с получением требуемого промежуточного соединения (680 мг) в виде желтого маслянистого вещества; МС (ИЭР): m/z = 538 [M+H]+.
Стадия 8: Синтез N2-(2-(диметиламино)этил)-N2-метил-5-ди-трет-бутоксикарбониламино-6-(2,2,2- трифторэтокси)-2,3-диамина
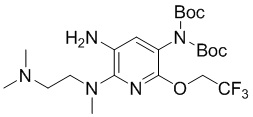
N 2-(2-(Диметиламино)этил)-N2-метил-3-нитро-5-ди-трет-бутоксикарбониламино-6-(2,2,2-трифторэтокси)-2-амин (680 мг, 1,3 ммоль) растворяли в MeOH (30 мл), добавляли 10% Pd-C (136 мг), три раза меняли атмосферу воздуха в колбе на водород, затем перемешивали при комнатной температуре в течение 1 часа. После завершения реакции фильтровали смесь через целит, концентрировали и очищали остаток колоночной хроматографией с получением требуемого промежуточного соединения (415 мг) в виде коричневого маслянистого вещества; МС (ИЭР): m/z = 508,3 [M+H]+.
Стадия 9: Синтез N-(5-ди-трет-бутоксикарбониламино-2-((2-(диметиламино)этил)(метил)амино)-6-(2,2,2- трифторэтокси)пиридин-3-ил)акриламида
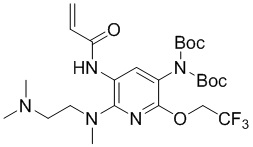
К N2-(2-(Диметиламино)этил)-N2-метил-5-ди-трет-бутоксикарбониламино-6-(2,2,2- трифторэтокси)-2,3-диамину (415 мг, 0,8 ммоль) в ДХМ (15 мл) добавляли триэтиламин (248 мг, 2,4 ммоль), перемешивали на ледяной бане, по каплям добавляли акрилоилхлорид (148 мг, 1,6 ммоль), затем нагревали до комнатной температуры, продолжали перемешивание в течение 10 минут, затем гасили водой, экстрагировали ДХМ (15 мл*3), сушили и концентрировали объединенные органические фазы, очищали остаток колоночной хроматографией с получением требуемого промежуточного соединения (318 мг) в виде коричневого маслянистого вещества; МС (ИЭР): m/z = 562,3 [M+H]+.
Стадия 10: Синтез N-(5-амино-2-((2-(диметиламино)этил)(метил)амино)-6-метоксипиридин-3-ил)акриламида
N-(5-Ди-трет-бутоксикарбониламино-2-((2-(диметиламино)этил)(метил)амино)-6-(2,2,2-трифторэтокси)пиридин-3-ил)акриламид (318 мг, 0,57 ммоль) растворяли в ДХМ (20 мл), по каплям добавляли метансульфоновую кислоту (1,63 г, 5,7 ммоль) при охлаждении на ледяной бане, затем продолжали перемешивание в течение 2,5 часа, после чего естественным образом повышали температуру до комнатной температуры. Постепенно доводили рН до 8, по каплям добавляя насыщенный раствор бикарбоната натрия при охлаждении на ледяной бане, экстрагировали ДХМ (25 мл*3), объединяли органические фазы, сушили и концентрировали, очищали остаток колоночной хроматографией с получением требуемого промежуточного соединения (176 мг) в виде бледного коричневато-зеленого твердого вещества; МС (ИЭР): m/z = 362,2 [M+H]+.
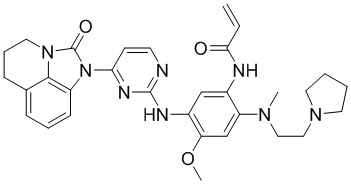
Пример 1: Синтез N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(8-фтор-2-оксо-5,6-дигидро-4H-имидазо[4,5,1-ij]хинолин-1(2H)-ил)пиримидин-2-ил)амино)-4-метоксифенил)акриламида
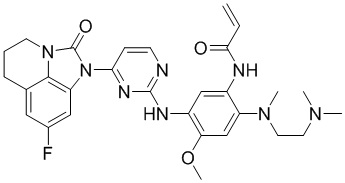
Промежуточное соединение 1a (152 мг, 0,2 ммоль), промежуточное соединение 2a (200 мг, 0,68 ммоль), ацетат палладия (45 мг, 0,2 ммоль), Xanphos (116 мг, 0,2 ммоль) и карбонат цезия (130 мг, 0,4 ммоль) добавляли в 1,4-диоксан (5 мл) при перемешивании при 90°С в течение 10 часов. После завершения реакции фильтровали смесь через целит, концентрировали и очищали остаток колоночной хроматографией (MeOH/ДХМ = 1/10) с получением требуемого соединения (41 мг) в виде бледно-коричневого твердого вещества.
Таким же способом синтезировали соединения, представленные в следующей таблице:
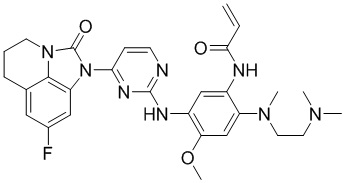
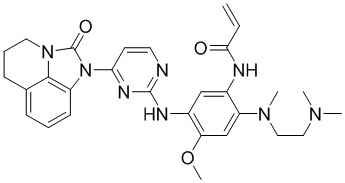
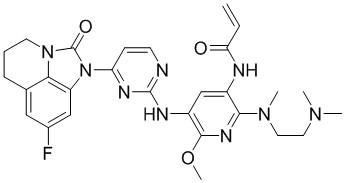
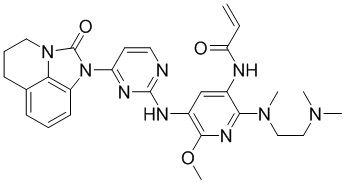
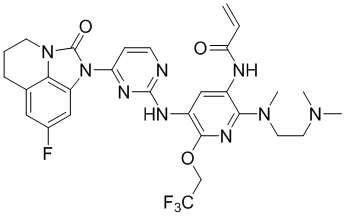
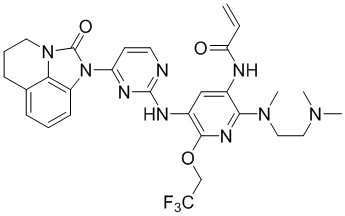
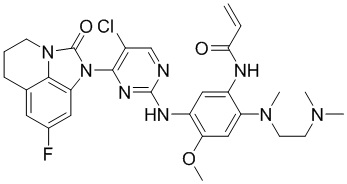
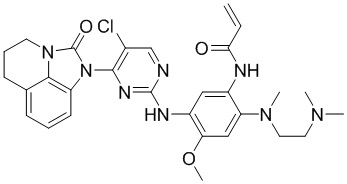
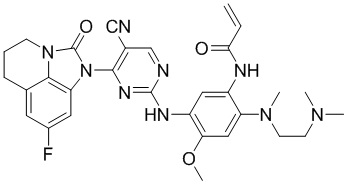
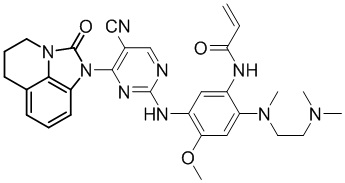
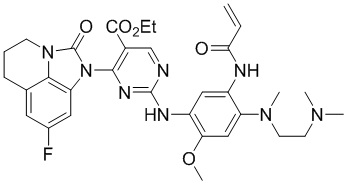
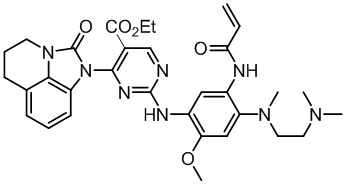
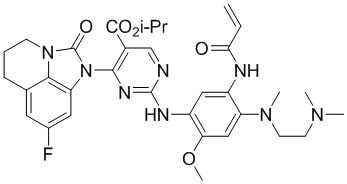
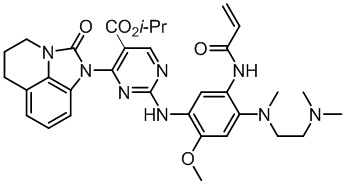
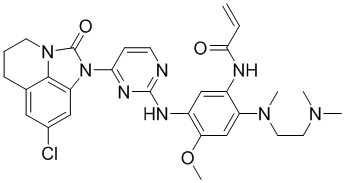
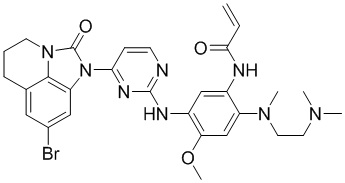
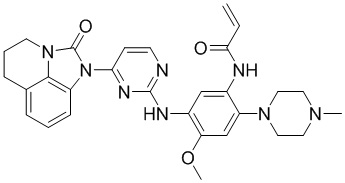
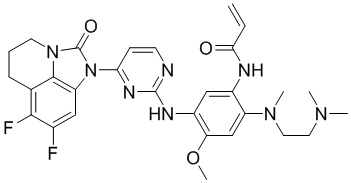
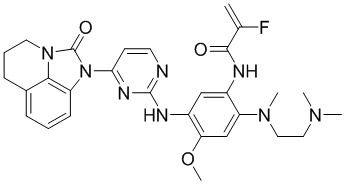
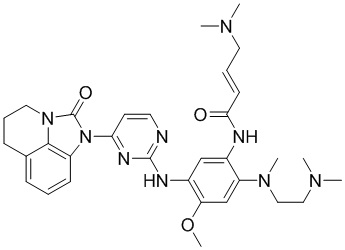
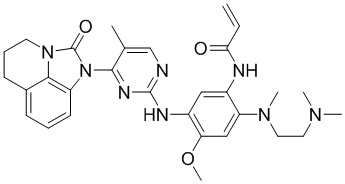
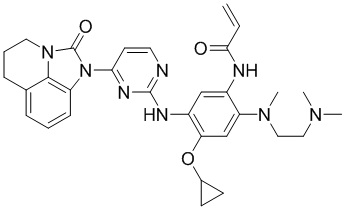
Ссылаясь на синтез соединения 1, получали соединения, представленные в следующей таблице:
Примеры биологических анализов соединений по данному изобретению
Анализ 1: анализ биохимической активности в отношении EGFR дикого типа, HER2 и HER4, и мутантного EGFR
10 нл серийно разбавленных соединений переносили на аналитические планшеты с помощью устройства Labcyte Echo 550, и последовательно вводили 5 мкл 2X растворов ферментов в аналитическом буфере. Аналитический планшет накрывали клейкой пленкой для планшетов и кратковременно вращали в течение 30 с при 1000 g. Добавляли 5 мкл 2X раствора TK-субстрат-биотина и АТФ, смешанных в аналитическом буфере.
Через 40 минут инкубации при комнатной температуре добавляли 10 мкл Sa-XL 665 и TK-антитело-криптата, смешанных в аналитическом буфере HTRF, для инициации связывания антитела.
Еще через 60 минут инкубации при комнатной температуре измеряли сигналы на приборе Envision 2104 при длине волны 615 нм (криптат) и 665 нм (XL665). Рассчитывали отношение сигналов при 665 нм к сигналам при 615 нм и использовали значения, полученные для отрицательного контрольного образца, для нормализации для расчета процентного ингибирования. Рассчитывали IC50 и анализировали с помощью 4-параметрической логистической модели.
IC50 (нМ)
IC50 (нМ)
IC50 (нМ)
IC50 (нМ)
IC50 (нМ)
insFHEA
insNPG T790M
D770-N771insNPG
Как показано в представленной выше таблице, соединения, описанные в данном патенте, демонстрируют более высокую активность против широкого спектра мутантов EGFR, включая вставки в 20 экзоне и точечные мутации, по сравнению с AZD9291. Для соединений, не показанных в таблице, также наблюдали превосходную активность.
Анализ 2: Клеточный анализ пролиферации A431 (EGFR дикого типа, рак кожи), H1975 (EGFR L858R/T790M, немелкоклеточный рак легких) и Ba/F3 (EGFR D770_N771insSVD или EGFR V769_D770insASV, pro-B)
Клетки A431, клетки H1975 и Ba/F3, экспрессирующие различные мутанты EGFR, собирали из культур на экспоненциальной фазе роста и высевали в 96-луночные планшеты при плотности клеток 3000 на лунку для клеток A431 и H1975, и 10000 на лунку для клеток Ba/F3. После прикрепления в течение ночи проводили 3-кратное серийное разбавление соединений и вводили их в клетки в концентрациях 30 мкМ, 10 мкМ, 3 мкМ, 1 мкМ, 0,3 мкМ, 0,1 мкМ, 0,03 мкМ и 0,01 мкМ в трех повторениях, и инкубировали в течение трех дней. Затем добавляли 20 мкл раствора MTT с концентрацией 5 мг/мл, после чего дополнительно добавляли 50 мкл 10% раствора SDS вместе с 5% изобутилового спирта в растворе HCl с концентрацией 0,01 моль/л. Инкубировали планшеты в течение ночи. Количественно измеряли абсорбцию (A) при длине волны 570 нм. Процент ингибирования использовали для расчета IC50 методом Блисса. Результаты представлены в таблице 5.
D770_N771insSVD
IC50 (нМ)
V769_D770insASV
IC50 (нМ)
IC50 (нМ)
IC50 (нМ)
По сравнению с AZD9291, соединения из таблицы 5 демонстрируют более высокую активность при ингибировании пролиферации клеток BaF3, содержащих EGFR D770_N771insSVD или EGFR V769_D770insAS, и сопоставимую активность в отношении H1975 и A431, что позволяет предположить, что соединения по данному описанию демонстрируют существенно улучшенную активность в отношении вставок в 20 экзон EGFR при сохранении высокой активности в отношении L858R/T790M EGFR наряду с высокой селективностью в отношении EGFR дикого типа. Другие примеры данной заявки, не перечисленные в указанной таблице, также показали профили активности, подобные описанным выше.
Анализ 3. In vivo исследования в мышиных моделях ксенотрансплантатов, полученных из клеточной линии (CSX) и полученных от пациентов (PDX)
Клетки (H1975) или кусочки ткани (LU0493 и LU0426) подкожно имплантировали в левую подмышку «голых» мышей. По достижении среднего объема опухоли 100-150 мм3 мышей рандомизировали по объему опухоли и вводили носитель, соединение 1 или позиотиниб, соответственно. Объем опухоли и массу тела измеряли два раза в неделю. Мышей усыпляли на 21 день или на 28 день и записывали объем опухоли и конечную массу тела. Рассчитывали относительный объем опухоли, значения процентного соотношения эксперимент/контроль и ингибирование роста опухоли, и осуществляли статистические расчеты.
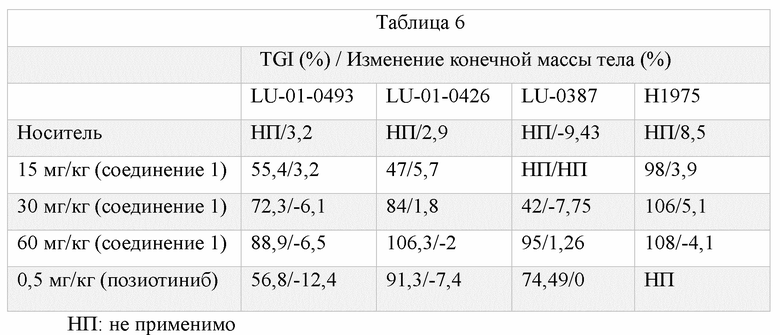
*: P < 0,05 относительно группы с носителем; D1: первый день лечения препаратом; RTV: относительный объем опухоли; RTV = Vt / V0; T/C (%) = TRTV / CRTV X 100; TRTV: RTV экспериментальной группы; CRTV : RTV группы с носителем; TGI (%): ингибирование роста опухоли (%); T/C (%) > 60: неэффективно; T/C (%) ≤ 60 и P < 0,05: эффективно. Изменение конечной массы тела рассчитывали как процент изменения массы тела с 1 дня по 21 день.
Как показано в представленной выше таблице, по сравнению с позиотинибом соединение 1 является более эффективным в отношении блокирования роста опухоли со вставками в 20 экзон EGFR и мутациями T790M при меньшем влиянии на массу тела, что свидетельствует об увеличении диапазона безопасности.
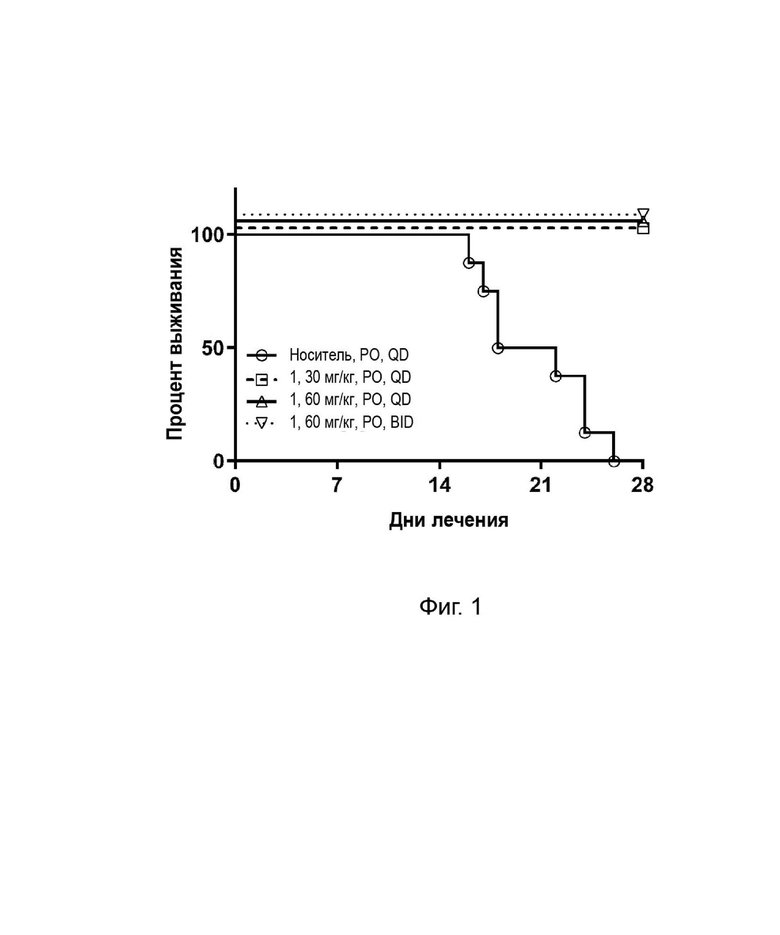
Анализ 4. In vivo мышиная модель ортотопического ксенотрансплантата PC9 в головном мозге
В головной мозг мышей вводили инъекцию 3x105 клеток PC9, экспрессирующих люциферазу. Мышей рандомизировали по интенсивности флуоресценции головного мозга и массе тела и перорально вводили носитель или соединение 1. Выживаемость и массу тела контролировали ежедневно, и усыпляли мышей с потерей массы тела более 20%.
Как показано в таблице 7 и на фиг. 1, все мыши в группе с носителем погибли в течение 28 дней после введения, при этом все мыши, получавшие соединение 1, выжили, что позволяет предположить, что соединение 1 может проникать в головной мозг и ингибировать рост опухоли, улучшая выживаемость.

Результаты анализов 1-4 демонстрируют, что соединение по данному описанию ингибирует активность мутанта EGFR со вставками в 20 экзоне и точечными мутациями, а также пролиферацию клеток Ba/F3, содержащих различные мутации EGFR, с высокой селективностью по сравнению с EGFR дикого типа. По сравнению с позиотинибом, соединение 1 демонстрирует более высокую in vivo эффективность в мышиных моделях PDX с улучшенным диапазоном безопасности. Оно также является активным в ортотопической модели PC9 головного мозга, что свидетельствует о высокой проницаемости в головной мозг. Другие соединения по данному описанию также являются эффективными in vivo в отношении блокирования роста опухоли.
Несмотря на то, что выше описаны конкретные варианты реализации данного изобретения, специалистам в данной области техники понятно, что они являются лишь примерами, и в отношении приведенных вариантов реализации могут быть сделаны различные изменения или модификации без отступления от принципов и сущности данного изобретения. Соответственно, объем данного изобретения определяется прилагаемой формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| МОДУЛЯТОРЫ МОНОАЦИЛГЛИЦЕРИНЛИПАЗЫ | 2019 |
|
RU2797323C2 |
| ЗАМЕЩЕННЫЕ 1,2-ДИГИДРО-3Н-ПИРАЗОЛО[3,4-D]ПИРИМИДИН-3-ОНЫ | 2019 |
|
RU2812726C2 |
| СОЕДИНЕНИЕ 2-АМИНОПИРИМИДИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ ДАННОГО СОЕДИНЕНИЯ | 2015 |
|
RU2704129C2 |
| ИНГИБИТОРЫ HPK1 И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2839132C2 |
| ПРОИЗВОДНЫЕ 1,5-НАФТИРИДИНА И ИНГИБИТОРЫ MELK, СОДЕРЖАЩИЕ ИХ | 2012 |
|
RU2645339C1 |
| ИНГИБИТОРЫ МУТАЦИИ HER2 | 2022 |
|
RU2834124C2 |
| ИМИДАЗОПИРИМИДИНЫ КАК ИНГИБИТОРЫ EED И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2836176C2 |
| БИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ЕГО ПРИМЕНЕНИЕ ДЛЯ ИНГИБИРОВАНИЯ SUV39H2 | 2016 |
|
RU2729187C1 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2716136C2 |
| ТРИЦИКЛИЧЕСКИЕ АЗОТСОДЕРЖАЩИЕ СОЕДИНЕНИЯ И ПРОИЗВОДНЫЕ ТЕТРАГИДРОХИНОЛИНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1991 |
|
RU2023712C1 |
Изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли. В формуле (I): X выбран из группы, состоящей из N и CH; R1 представляет собой водород; R2 выбран из группы, состоящей из C1-6 алкила, дейтерированного C1-6 алкила, C3-6 циклоалкила и C1-6 галогеналкила; R3 представляет собой -NR9(CH2)2NR9’R9’’; R4 представляет собой 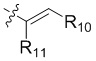 ; R5, R6 и R7 независимо выбраны из группы, состоящей из водорода и галогена; R8 выбран из группы, состоящей из водорода, C1-6 алкила и C1-6 галогеналкила; R9 выбран из группы, состоящей из водорода, C1-6 алкила, дейтерированного C1-6 алкила и C1-6 галогеналкила; R9’ и R9’’ независимо выбраны из группы, состоящей из водорода, C1-6 алкила, C3-6 циклоалкила, дейтерированного C1-6 алкила и C1-6 галогеналкила, или R9’ и R9’’ вместе с атомом азота, связанным с ними, образуют гетероцикл, указанный гетероцикл является незамещенным или необязательно замещен 1-3 группами, выбранными из группы, состоящей из галогена, C1-6 алкила, C1-3 алкокси, метилтио, метансульфонила и C1-6 галогеналкила; R10 представляет собой водород и R11 представляет собой водород. Предложенные соединения могут селективно ингибировать активность мутантов рецептора эпидермального фактора роста (EGFR), демонстрируют высокий ингибирующий эффект в отношении мутанта EGFR и антипролиферативную активность против раковых клеток и могут быть использованы для лечения рака. 1 з.п. ф-лы, 1 ил., 7 табл., 1 пр.
; R5, R6 и R7 независимо выбраны из группы, состоящей из водорода и галогена; R8 выбран из группы, состоящей из водорода, C1-6 алкила и C1-6 галогеналкила; R9 выбран из группы, состоящей из водорода, C1-6 алкила, дейтерированного C1-6 алкила и C1-6 галогеналкила; R9’ и R9’’ независимо выбраны из группы, состоящей из водорода, C1-6 алкила, C3-6 циклоалкила, дейтерированного C1-6 алкила и C1-6 галогеналкила, или R9’ и R9’’ вместе с атомом азота, связанным с ними, образуют гетероцикл, указанный гетероцикл является незамещенным или необязательно замещен 1-3 группами, выбранными из группы, состоящей из галогена, C1-6 алкила, C1-3 алкокси, метилтио, метансульфонила и C1-6 галогеналкила; R10 представляет собой водород и R11 представляет собой водород. Предложенные соединения могут селективно ингибировать активность мутантов рецептора эпидермального фактора роста (EGFR), демонстрируют высокий ингибирующий эффект в отношении мутанта EGFR и антипролиферативную активность против раковых клеток и могут быть использованы для лечения рака. 1 з.п. ф-лы, 1 ил., 7 табл., 1 пр.
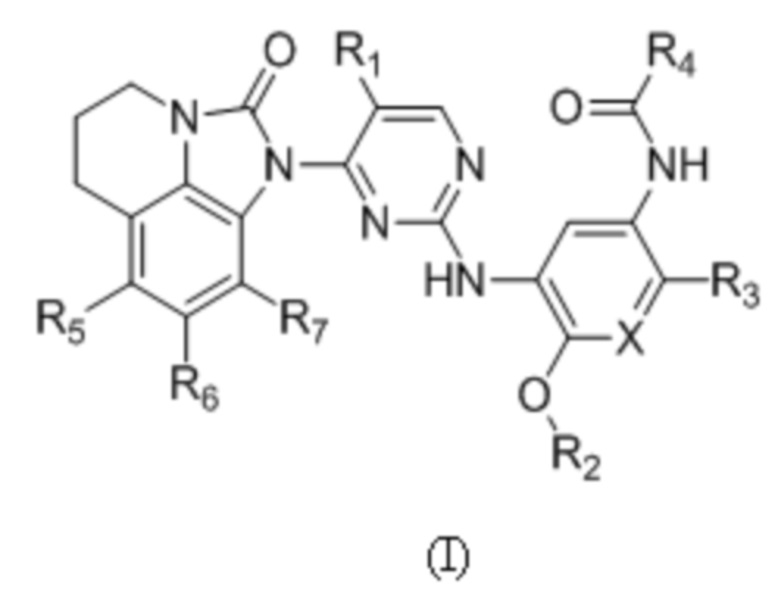
1. Соединение общей формулы (I) или его фармацевтически приемлемая соль
 ,
,
где X выбран из группы, состоящей из N и CH;
R1 представляет собой водород;
R2 выбран из группы, состоящей из C1-6 алкила, дейтерированного C1-6 алкила, C3-6 циклоалкила и C1-6 галогеналкила;
R3 представляет собой -NR9(CH2)2NR9’R9’’;
R4 представляет собой 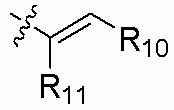 ;
;
R5, R6 и R7 независимо выбраны из группы, состоящей из водорода и галогена;
R8 выбран из группы, состоящей из водорода, C1-6 алкила и C1-6 галогеналкила;
R9 выбран из группы, состоящей из водорода, C1-6 алкила, дейтерированного C1-6 алкила и C1-6 галогеналкила;
R9’ и R9’’ независимо выбраны из группы, состоящей из водорода, C1-6 алкила, C3-6 циклоалкила, дейтерированного C1-6 алкила и C1-6 галогеналкила, или R9’ и R9’’ вместе с атомом азота, связанным с ними, образуют гетероцикл, указанный гетероцикл является незамещенным или необязательно замещен 1-3 группами, выбранными из группы, состоящей из галогена, C1-6 алкила, C1-3 алкокси, метилтио, метансульфонила и C1-6 галогеналкила;
R10 представляет собой водород; и
R11 представляет собой водород.
2. Соединение общей формулы (I) или его фармацевтически приемлемая соль по п. 1, отличающиеся тем, что указанное соединение представляет собой:
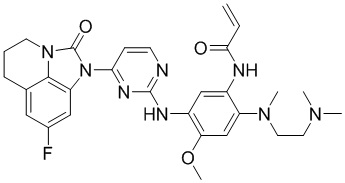 ,
, 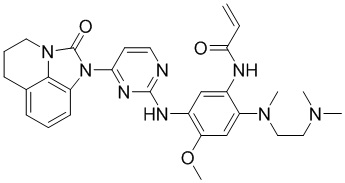 ,
,
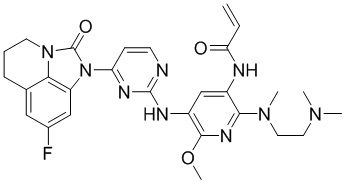 ,
, 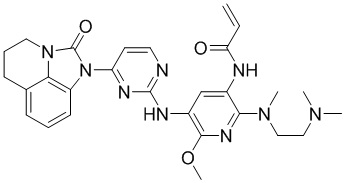 ,
,
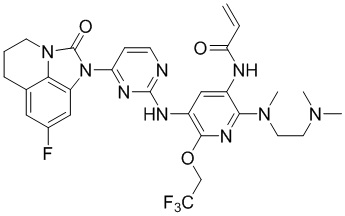 ,
, 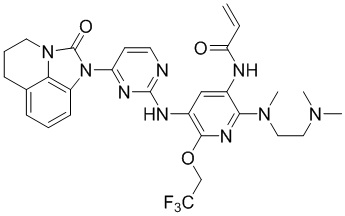 ,
,
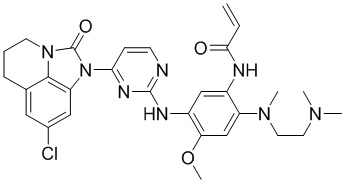 ,
, 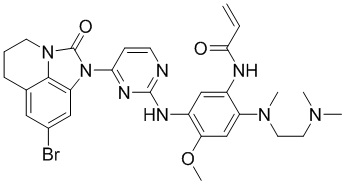 ,
,
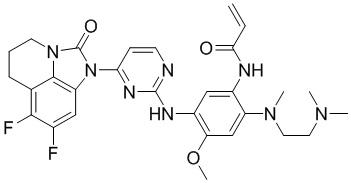 ,
, 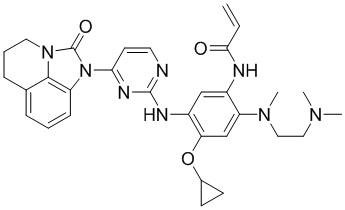 ,
,
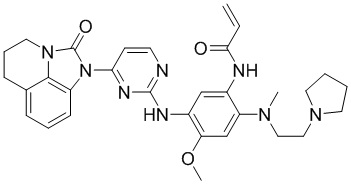 ,
,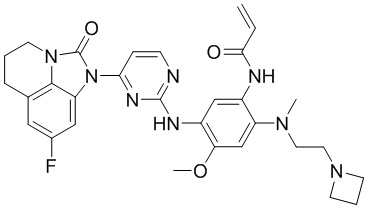 ,
, 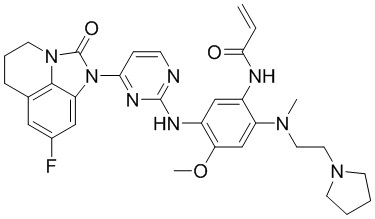 ,
, 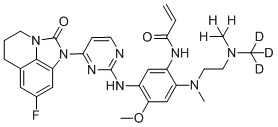 ,
, 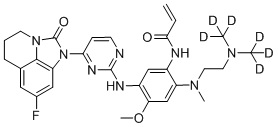 ,
,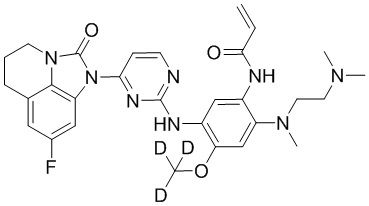 ,
,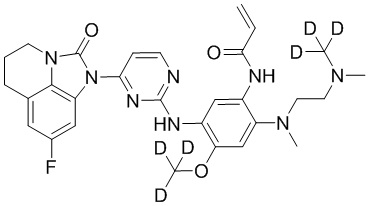 или
или 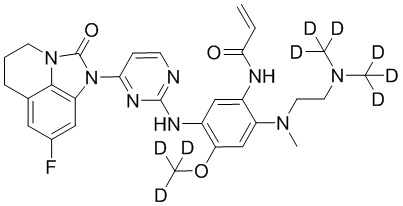 .
.
| WO 2017120429 A1, 13.07.2017 | |||
| US 20180208581 A1, 26.07.2018 | |||
| US 20190152954 A1, 23.05.2019 | |||
| EA 201391491 A1, 29.08.2014. |

