ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка испрашивает приоритет по предварительной заявке США № 61/659211 (поданной 13 июня 2012 г.) и предварительной заявке США № 61/784382 (поданной 14 марта 2013 г.), каждая из которых включена в настоящее описание посредством ссылки.
УРОВЕНЬ ТЕХНИКИ
Несмотря на прогресс в лечении рака, сохраняется острая необходимость дальнейшего совершенствования способов лечения для продления жизни пациентов при сохранении качества жизни, особенно в случае поздних стадий рака, например, рака поджелудочной железы, которые часто являются устойчивыми или приобретают устойчивость к текущим терапевтическим процедурам.
Заболеваемость раком поджелудочной железы заметно возросла за последние несколько десятилетий. В настоящее время он занимает четвертое место среди основных причин смерти от рака в США. Высокая смертность от рака поджелудочной железы обусловлена недостатком эффективных способов лечения и полным отсутствием надежных способов лечения. Из-за расположения поджелудочной железы рак поджелудочной железы, как правило, не диагностируется, пока опухоль не становится достаточно большой для появления системных симптомов. Это, в сочетании с отсутствием хороших инструментов для скрининга и ограниченным пониманием факторов риска, приводит к тому, что на момент постановки диагноза у пациентов, как правило, имеют место поздние (часто метастазирующие) стадии рака. Прогноз при метастазирующем раке поджелудочной железы неблагоприятен и почти всегда фатален, общая пятилетняя выживаемость составляет менее 4%.
Показано, что химиотерапия с применением одного или более из 5-фторурацила (5-FU) и гемцитабина продлевает выживаемость при раке поджелудочной железы. Комбинированная терапия, включающая применение фолиновой кислоты (лейковорина или левулолейковорина), 5-фторурацила и иринотекана (FOLFIRI), фолиновой кислоты, 5-фторурацила, иринотекана и оксалиплатина (FOLFIRINOX) или, реже, комбинации фолиновой кислоты, 5-фторурацила и оксалиплатина (FOLFOX) также используется для лечения некоторых вариантов рака поджелудочной железы. Иринотекан представляет собой 7-этил-10-[4-(1-пиперидин)-1-пиперидин]карбонилоксикапотецин, название по ИЮПАК (S)-4,11-диэтил-3,4,12,14-тетрагидро-4-гидрокси-3,14-диоксо-1H-пиран-[3',4':6,7]-индолизин-[1,2-b]-хинолин-9-ил-[1,4'-бипиперидин]-1'-карбоксилат. Иринотекан является членом класса лекарственных веществ-ингибиторов топоизомеразы I и представляет собой полусинтетический водорастворимый аналог природного алкалоида - камптотецина. Известный также как СРТ-11, иринотекан в настоящее время продается на рынке в виде водного раствора как Camptosar® (иринотекана гидрохлорид для инъекций). Ингибиторы топоизомеразы I, например, иринотекан, останавливают неконтролируемый рост клеток путем ингибирования раскручивания ДНК, тем самым предотвращая репликацию ДНК.
Иринотекан характеризуется сложной фармакологией; активация, инактивация и выведение данного лекарства включают интенсивные метаболические преобразования. Иринотекан является пролекарством, которое преобразуется неспецифическими карбоксилэстеразами в 100-1000-кратно более активный метаболит, SN-38. SN-38 не распознается P-гликопротеином, переносчиком лекарственных веществ, который играет важную роль в приобретенной лекарственной резистентности за счет выведения некоторых лекарственных веществ из клетки, поэтому иринотекан вероятно будет активным в опухолях, устойчивых к другим стандартным химиотерапевтическим средствам. SN-38 выводится из организма за счет глюкуронизации, для которой описаны основные варианты фармакогенетической изменчивости, и экскреции с желчью. Указанные свойства данного лекарственного вещества вносят вклад в заметную неоднородность эффективности и токсичности иринотекана, наблюдаемую в клинических условиях. Применение инъекций иринотекана гидрохлорида одобрено в США для лечения метастазирующего рака толстой кишки или почек; указанное средство также применяют для лечения рака ободочной и прямой кишки, желудка, легких, матки, шейки матки и яичников.
Существует несколько утвержденных вариантов лечения поздних или метастазирующих стадий рака поджелудочной железы, особенно экзокринного происхождения. Монотерапия с применением гемцитабина является текущим стандартом лечения первой линии поздних и метастазирующих стадий аденокарциномы поджелудочной железы. В клинических исследованиях монотерапия с применением гемцитабина неизменно демонстрировала среднее увеличение продолжительности жизни на 5-6 месяцев и выживаемость в течение 1 года приблизительно 20%. Использование монотерапии с применением гемцитабина также утверждено в качестве лечения второй линии для пациентов, ранее получавших, но уже не реагирующих на 5-фторурацил; при этом медианное общее увеличение продолжительности жизни составляло 3,9 месяцев.
На основании знаний о биологии рака поджелудочной железы была произведена оценка ряда агентов, оказывающих направленной воздействие, однако для использования в качестве терапевтического средства первой линии при поздних стадиях рака поджелудочной железы одобрен только эрлотиниб, ингибитор протеинтирозинкиназы, мишенью которого является EGFR, и данное одобрение дано только для применения в комбинации с гемцитабином. Совместное введение эрлотиниба с гемцитабином приводило к статистически значимому благоприятному действию на выживаемости и улучшению медианной продолжительности жизни (6,4 месяца по сравнению с 5,9 месяца) и выживаемости в течение 1 года (24% по сравнению с 17%) при сравнении с монотерапией с применением гемцитабина. Клинические исследования по оценке других агентов, оказывающих адресное воздействие, в том числе исследования антител бевацизумаб и цетуксимаб, дали неутешительный отрицательный результат. Таким образом, существует настоятельная потребность в усовершенствовании и эффективной альтернативе для текущих способов лечения рака поджелудочной железы. Описанное изобретение удовлетворяет эту потребность и обеспечивает другие преимущества.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Представлены способы лечения рака поджелудочной железы у пациента (т.е. пациента-человека), включающие введение пациенту липосомального иринотекана (например, инъекции липосомального препарата октасульфатной соли иринотекана - сахарозы, также называемой ММ-398) отдельно или в комбинации с 5-фторурацилом (5-FU) и лейковорином (вместе обозначаемым как 5-FU/LV) в соответствии с конкретной клинической схемой введения. Кроме того, предложены композиции, адаптированные для использования в рамках таких способов.
В одном из аспектов представлен способ лечения (например, эффективного лечения) рака поджелудочной железы у пациента, включающий: введение пациенту эффективного количества липосомального иринотекана, причем указанный способ включает по меньшей мере один цикл, где цикл представляет собой период продолжительностью 3 недели, причем в течение каждого цикла липосомальный иринотекан вводят в 1 день цикла в дозе 120 мг/м2, за исключением случая, если пациент является гомозиготным по аллелю UGT1A1*28; в этом случае липосомальный иринотекан вводят в 1 день 1 цикла в дозе 80 мг/м2. В одном варианте реализации дозу липосомального иринотекана, вводимого пациенту, гомозиготному по аллелю UGT1A1*28, повышают после одного с шагом 20 мг/м2, но не более 120 мг/м2.
В еще одном аспекте способ лечения рака поджелудочной железы у пациента включает совместное введение пациенту эффективного количества липосомального иринотекана, 5-фторурацила (5-FU) и лейковорина, причем указанный способ включает по меньшей мере один цикл введения, равный 2-недельному периоду, причем для каждого цикла:
(a) липосомальный иринотекан вводят пациентам, не гомозиготным по аллелю UGT1A1*28, в 1 день каждого цикла в дозе 80 мг/м2, а пациентам, гомозиготным по аллелю UGT1A1*28, в 1 день 1 цикла в дозе 60 мг/м2 и в 1 день каждого последующего цикла в дозе от 60 мг/м2 до 80 мг/м2 (например, 60 мг/м2 или 70 мг/м2 или 80 мг/м2);
(b) 5-FU вводят в дозе 2400 мг/м2; и
(с) лейковорин вводят в дозе 200 мг/м2 (l-формы, или леволейковорина) или 400 мг/м2 (l+d-рацемической формы).
В одном варианте реализации дозу липосомального иринотекана, вводимую пациенту, гомозиготному по аллелю UGT1A1*28, увеличивают после одного цикла до 80 мг/м2. В одном варианте реализации в каждом цикле липосомальный иринотекан вводят перед лейковорином, а лейковорин - перед 5-FU.
В еще одном варианте реализации липосомальный иринотекан вводят внутривенно в течение 90 минут.
В еще одном варианте реализации 5-FU вводят внутривенно в течение 46 часов.
В еще одном варианте реализации лейковорин вводят внутривенно в течение 30 минут.
В еще одном варианте реализации перед каждым введением липосомального иринотекана осуществляют премедикацию пациента дексаметазоном и/или антагонистом 5-HT3 или другим противорвотным средством.
В еще одном варианте реализации рак поджелудочной железы представляет собой экзокринный рак поджелудочной железы, выбранный из группы, состоящей из ацинарно-клеточной карциномы, аденокарциномы, железисто-плоскоклеточной карциномы, гигантоклеточной опухоли, внутрипротокового папиллярного муцинозного новообразования (IPMN), муцинозной цистаденокарциномы, панкреатобластомы, серозной цистаденокарциномы и солидных и псевдопапиллярных опухолей.
В одном варианте реализации лечение пациента приводит к положительному результату, причем положительный результат представляет собой полный патологический ответ (pCR), полный ответ (CR), частичный ответ (PR) или стабильное заболевание (SD). В еще одном варианте реализации комбинированная терапия с применением липосомального иринотекана, 5-FU и лейковорина приводит к терапевтическому синергизму.
В еще одном варианте реализации липосомальный иринотекан изготовлен в инъекционной липосомальной форме иринотекана - сахарозы октасульфата (ММ-398). Инъекционную липосомальную форму иринотекана - сахарозы октасульфата также можно называть инъекционной липосомальной формой иринотекана-HCl, поскольку иринотекан-HCl является активным фармацевтическим ингредиентом, используемым для загрузки иринотекана в липосомы, содержащие триэтиламмонийсахарозы октасульфат, для получения липосом ММ-398. Эту номенклатуру можно использовать даже при том, что гидрохлорид-ион иринотекана-HCl реагирует с триэтиламмоний-ионом триэтиламмонийсахарозы октасульфата с образованием хлорида триэтиламмония (гидрохлорида триэтиламина), в результате чего иринотекана - сахарозы октасульфат остается в качестве фармацевтического средства в составе липосом ММ-398. В еще одном аспекте представлены наборы для лечения рака поджелудочной железы у пациента, содержащие дозу липосомального иринотекана и инструкции по применению липосомального иринотекана, как описано в настоящем документе.
В еще одном аспекте представлены наборы для лечения рака поджелудочной железы у пациента, содержащие дозу липосомального иринотекана, 5-фторурацила (5-FU) и лейковорина, а также инструкции по применению липосомального иринотекана, 5-FU и лейковорина, как описано в настоящем документе.
В одном варианте реализации набор охватывает лечение экзокринного рака поджелудочной железы, выбранного из группы, состоящей из ацинарно-клеточной карциномы, аденокарциномы, железисто-плоскоклеточной карциномы, гигантоклеточной опухоли, внутрипротокового папиллярного муцинозного новообразования (IPMN), муцинозной цистаденокарциномы, панкреатобластомы, серозной цистаденокарциномы и солидных и псевдопапиллярных опухолей.
В одном варианте реализации липосомальный иринотекан представляет собой инъекционную липосомальную форму иринотекана - сахарозы октасульфата (ММ-398).
В еще одном аспекте представлен состав липосомального иринотекана для совместного введения с 5-фторурацилом (5-FU) и лейковорином в течение по меньшей мере одного цикла, причем цикл представляет собой период продолжительностью 2 недели, состав иринотекана - липосомальнй состав иринотекана, и:
(a) липосомальный иринотекан вводят пациентам, не гомозиготным по аллелю UGT1A1*28, в 1 день каждого цикла в дозе 80 мг/м2, а пациентам, гомозиготным по аллелю UGT1A1*28, в 1 день 1 цикла в дозе 60 мг/м2 и в 1 день каждого последующего цикла в дозе от 60 мг/м2 или 80 мг/м2;
(b) 5-FU вводят в дозе 2400 мг/м2; и
(с) лейковорин вводят в дозе 200 мг/м2 (l-формы, или леволейковорина) или 400 мг/м2 (l+d-рацемической формы).
В одном варианте реализации дозу липосомального иринотекана, вводимую пациенту, гомозиготному по аллелю UGT1A1*28, увеличивают после 1 цикла до 80 мг/м2. В еще одном варианте реализации липосомальный иринотекан вводят внутривенно в течение 90 минут.
В еще одном варианте реализации 5-FU вводят внутривенно в течение 46 часов.
В еще одном варианте реализации лейковорин вводят внутривенно в течение 30 минут.
В еще одном варианте реализации перед каждым введением липосомального иринотекана осуществляют премедикацию пациента дексаметазоном и/или антагонистом 5-HT3 или другим противорвотным средством.
В еще одном варианте реализации рак поджелудочной железы представляет собой экзокринный рак поджелудочной железы, выбранный из группы, состоящей из ацинарно-клеточной карциномы, аденокарциномы, железисто-плоскоклеточной карциномы, гигантоклеточной опухоли, внутрипротокового папиллярного муцинозного новообразования (IPMN), муцинозной цистаденокарциномы, панкреатобластомы, серозной цистаденокарциномы и солидных и псевдопапиллярных опухолей.
В еще одном варианте реализации липосомальный состав иринотекана представляет собой инъекционную липосомальную форму иринотекана - сахарозы октасульфата.
В еще одном аспекте представлен способ улучшения результатов химиотерапии за счет увеличения васкуляризации опухоли, включающий введение пациенту с опухолью инъекции липосомального препарата октасульфатной соли иринотекана - сахарозы в количестве, эффективном для увеличения васкуляризации опухоли, и одновременное введение пациенту эффективного количества химиотерапевтического агента, не являющегося иринотеканом.
В еще одном аспекте представлена инъекционная липосомальная форма иринотекана - сахарозы октасульфата для совместного введения пациенту с опухолью: 1) инъекционной липосомальной формы иринотекана - сахарозы октасульфата в количестве, эффективном для увеличения васкуляризации опухоли, и 2) эффективного количества химиотерапевтического агента, не являющегося иринотеканом.
Далее, изобретение включает способ лечения экзокринного рака поджелудочной железы, включающий внутривенное применение противоопухолевой терапии один раз каждые две недели к пациенту-человеку, имеющему экзокринный рак поджелудочной железы, при этом противоопухолевая терапия состоит из:
a) введения дозы 60-80 мг/м2 композиции липосомального иринотекана в разбавленном инъекционном составе иринотекана пациенту-человеку в однократной инфузии в течение приблизительно 90 минут, причем разбавленный инъекционный состав иринотекана содержит липосомы иринотекана в 500 мл инъецируемой жидкости и объем 5 мг/мл липосомальной композиции иринотекана эффективен для доставки дозы липосомального иринотекана, в комбинации с
b) введением терапевтически эффективного количества лейковорина и 5-фторурацила,
для лечения экзокринного рака поджелудочной железы у пациента, где липосомы иринотекана состоят из фосфатидилхолина, холестерина и фосфатидилэтаноламина, модифицированного полиэтиленгликолем, и имеют диаметр приблизительно 80-140 нм.
В предпочтительном варианте, фосфатидилэтаноламин, модифицированный полиэтиленгликолем, присутствует в количестве приблизительно одна молекула полиэтиленгликоля на 200 молекул фосфолипидов в липосомах иринотекана.
В одном из вариантов изобретения, липосомы иринотекана представляют собой моноламеллярные липидные двуслойные везикулы, инкапсулирующие соль иринотекана с октасульфатом сахарозы.
Изобретение также относится к случаям, когда экзокринный рак поджелудочной железы прогрессировал на фоне терапии с применением гемцитабина до введения липосом иринотекана.
Предпочтительно, терапевтически эффективное количество лейковорина составляет 200 мг/м2 (l) формы лейковорина, необязательно введенного как 400 мг/м2 (l+d) рацемической формы лейковорина, и терапевтически эффективное количество 5-фторурацила составляет 2400 мг/м2 на дозу.
В одном из вариантов, разбавленный инъекционный состав иринотекана, лейковорин и 5-фторурацил вводят пациенту последовательно, начиная с первого дня двухнедельного цикла, что приводит к одновременному присутствию липосом иринотекана, лейковорина и 5-фторурацила в крови пациента.
Предпочтительно, разбавленный инъекционный состав иринотекана вводят пациенту до 5-фторурацила.
Далее, согласно изобретению:
a) пациента предварительно лечили гемцитабином;
b) лейковорин вводится как 400 мг/м2 (l+d) рацемической формы лейковорина;
c) экзокринный рак поджелудочной железы прогрессировал на фоне терапии с применением гемцитабина до введения разбавленного инъекционного состава иринотекана;
d) липосомы иринотекана представляют собой моноламеллярные липидные двуслойные везикулы, инкапсулирующие соль иринотекана с октасульфатом сахарозы, и
e) фосфатидилэтаноламин, модифицированный полиэтиленгликолем, присутствует в количестве приблизительно одна молекула полиэтиленгликоля на 200 молекул фосфолипидов в липосомах иринотекана.
Изобретение может дополнительно включать хранение разбавленного инъекционного состава иринотекана при температуре приблизительно 2-8°C в течение не более 24 часов до введения.
Предпочтительно, липосомы иринотекана представляют собой моноламеллярные липидные двуслойные везикулы, которые инкапсулируют водное пространство, содержащее иринотекан в гелеобразном или осажденном состоянии в виде соли с октасульфатом сахарозы.
В предпочтительном варианте осуществления изобретения, инъецируемая жидкость представляет собой 5% инъекционный раствор декстрозы.
Далее, изобретение включает способ лечения экзокринного рака поджелудочной железы у пациента-человека, которого предварительно лечили гемцитабином, включающий внутривенное применение к пациенту противоопухолевой терапии, состоящей из:
a) дозы 60-80 мг/м2 композиции липосомального иринотекана, содержащей липосомы иринотекана в разбавленном инъекционном составе иринотекана, при этом разбавленный инъекционный состав получен посредством разбавления объема композиции липосомального иринотекана, содержащей 5 мг/мл липосомального иринотекана в 500 мл инъецируемой жидкости, в комбинации с
b) дозой 200 мг/м2 (l) формы лейковорина и дозой 2400 мг/м2 5-фторурацила,
для лечения экзокринного рака поджелудочной железы у пациента, где липосомальная композиция иринотекана содержит соль иринотекана с октасульфатом сахарозы, инкапсулированную в липосомах иринотекана, состоящих из фосфатидилхолина, холестерина и фосфатидилэтаноламина, модифицированного полиэтиленгликолем, и имеющих диаметр приблизительно 80-140 нм, при этом разбавленный инъекционный состав иринотекана, лейковорин и 5-фторурацил вводят пациенту последовательно, начиная с первого дня двухнедельного цикла.
Изобретение дополнительно может включать хранение разбавленного инъекционного состава иринотекана при температуре приблизительно 2-8°C в течение не более 24 часов до введения.
В предпочтительном варианте, фосфатидилэтаноламин, модифицированный полиэтиленгликолем, присутствует в липосомах иринотекана в количестве приблизительно одна молекула полиэтиленгликоля на каждые 200 молекул фосфолипидов.
В одном из вариантов осуществления изобретения, пациент ранее безуспешно получал лечение с применением гемцитабина или приобрел устойчивость к гемцитабину до введения разбавленного инъекционного состава иринотекана.
Согласно одному из вариантов изобретения, доза 200 мг/м2 (l) формы лейковорина обеспечивается посредством введения дозы 400 мг/м2 (l+d) формы лейковорина.
Согласно изобретению, доза композиции липосомального иринотекана составляет 60 мг/м2, 70 мг/м2 или 80 мг/м2, предпочтительно, доза композиции липосомального иринотекана составляет 80 мг/м2, и пациент-человек не является гомозиготным по аллелю UGT1A1*28.
В одном из вариантов осуществления изобретения, композицию липосомального иринотекана вводят в виде разбавленного инъекционного состава иринотекана в качестве 90-минутной инфузии, лейковорин вводят после иринотекана в течение 30 минут, и 5-фторурацил вводят в течение 46 часов после лейковорина.
Предпочтительно, липосомы иринотекана представляют собой моноламеллярные липидные двуслойные везикулы, которые инкапсулируют водное пространство, содержащее иринотекан в гелеобразном или осажденном состоянии в виде соли с октасульфатом сахарозы.
Предпочтительно, инъецируемая жидкость представляет собой 5% инъекционный раствор декстрозы.
Кроме того, изобретение относится к способу лечения рака поджелудочной железы у пациента-человека, у которого рак прогрессировал после терапии с применением гемцитабина, включающему применение противоопухолевой терапии к пациенту-человеку один раз каждые две недели, при этом противоопухолевая терапия состоит из внутривенного введения пациенту-человеку: разбавленного инъекционного состава иринотекана, обеспечивающего дозу 60, 70 или 80 мг/м2 липосомального иринотекана, в комбинации с дозой 200 мг/м2 (l) формы лейковорина и дозой 2400 мг/м2 5-фторурацила для лечения рака поджелудочной железы у пациента-человека, где
a) липосомы в разбавленном инъекционном составе иринотекана имеют диаметр приблизительно 80-140 нм, и состоят из фосфатидилхолина, холестерина и фосфатидилэтаноламина, модифицированного полиэтиленгликолем; и
b) разбавленный инъекционный состав иринотекана содержит липосомы иринотекана в 500 мл инъецируемой жидкости, хранящейся при температуре приблизительно 2-8°C в течение не более 24 часов до каждого введения.
Согласно вышеприведенному способу, дозу липосомального иринотекана 80 мг/м2 вводят пациенту-человеку, который не является гомозиготным по аллелю UGT1A1*28, и доза 200 мг/м2 (l) формы лейковорина обеспечивается введением дозы 400 мг/м2 (l+d) формы лейковорина.
Указанный способ может дополнительно включать введение противорвотного средства пациенту перед применением противоопухолевой терапии; при этом липосомальный иринотекан вводят в составе разбавленного инъекционного состава иринотекана в качестве одной 90-минутной инфузии, дозу лейковорина вводят после липосомального иринотекана в течение 30 минут, и дозу 5-фторурацила вводят в течение 46 часов после лейковорина.
Предпочтительно, липосомы иринотекана представляют собой моноламеллярные липидные двуслойные везикулы, которые инкапсулируют водное пространство, содержащее иринотекан в гелеобразном или осажденном состоянии в виде соли с октасульфатом сахарозы.
При этом, инъецируемая жидкость представляет собой 5% инъекционный раствор декстрозы.
Далее, изобретение включает способ лечения метастатической аденокарциномы поджелудочной железы после прогрессирования заболевания после терапии с применением гемцитабина у пациента-человека, который не является гомозиготным по аллелю UGT1A1*28, включающий следующие этапы, осуществляемые однократно каждые две недели:
a) разбавление объема 5 мг/мл липосомальной композиции иринотекана в 500 мл инъецируемой жидкости, с получением разбавленного инъекционного состава иринотекана; затем
b) необязательно, хранение разбавленного инъекционного состава иринотекана при температуре приблизительно 2-8°C в течение не более 24 часов до введения; затем
c) применение противоопухолевой терапии к пациенту-человеку, при этом противоопухолевая терапия состоит из внутривенного введения пациенту-человеку: разбавленного инъекционного состава иринотекана, обеспечивающего дозу 80 мг/м2 липосомального иринотекана, в комбинации с дозой 200 мг/м2 (l) формы лейковорина и дозой 2400 мг/м2 5-фторурацила, для лечения рака поджелудочной железы у пациента-человека;
где липосомы в липосомальной композиции иринотекана имеют диаметр приблизительно 80-140 нм, и содержат моноламеллярную липидную двуслойную везикулу, которая инкапсулирует водное пространство, содержащее иринотекан в гелеобразном или осажденном состоянии в виде соли с октасульфатом сахарозы, где моноламеллярный липидные бислой состоит из фосфатидилхолина, холестерина и фосфатидилэтаноламина, модифицированного полиэтиленгликолем.
В предпочтительном варианте осуществления, инъецируемая жидкость представляет собой 5% инъекционный раствор декстрозы.
Дополнительно, изобретение представляет собой инъекционную липосомальную форму иринотекана - сахарозы октасульфата для совместного введения пациенту с опухолью: 1) количества инъекционной липосомальной формы препарата иринотекана - сахарозы октасульфата каждые две недели, эффективного для увеличения васкуляризации опухоли, и 2) эффективного количества по меньшей мере одного противоракового агента, не являющегося иринотеканом.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фигура 1 представляет собой график, на котором показана противоопухолевая активность ММ-398 в ортотопической модели опухоли поджелудочной железы, экспрессирующей люциферазу (L3.6pl).
Фигура 2 представляет собой график, на котором показано накопление SN-38 в опухоли после лечения с применением свободного иринотекана или липосомального иринотекана (ММ-398).
Фигура 3 представляет собой график, на котором показано действие ММ-398 на окрашивание карбоангидразы IX в модели ксенотрансплантата НТ29.
На фигуре 4 показано влияние ММ-398 на перфузию низкомолекулярного красителя Хехста.
На фигуре 5 приведены сводные данные по фармакокинетике ММ-398 в q3w (иринотекан, липосомальный + свободное лекарственное вещество).
На фигуре 6 приведены сводные данные по фармакокинетике ММ-398 в q3w.
Фигура 7 представляет собой схематическое изображение схемы исследования 3 фазы.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
I. Определения
В настоящем документе термин «субъект» или «пациент» представляет собой человека, больного раком.
В настоящем документе «эффективное лечение» относится к лечению, дающему положительный эффект, например, улучшение по меньшей мере одного симптома заболевания или расстройства. Положительный эффект может принимать форму улучшения по сравнению с исходным уровнем, т.е. улучшения по сравнению с измерением или наблюдением, сделанным до начала лечения в соответствии с указанным способом. Положительный эффект также может принимать форму остановки, замедления, задержки или стабилизация вредоносного прогрессирования маркера рака. Эффективное лечение может относиться к облегчению по меньшей мере одного симптома рака. Такое эффективное лечение может, например, уменьшить боль пациента, уменьшить размер и/или количество очагов, может снизить или предотвратить метастазирование раковой опухоли и/или может замедлить рост раковой опухоли.
Термин "эффективное количество" относится к количеству агента, которое обеспечивает желательный биологический, терапевтический и/или профилактический результат. Этим результатом может быть снижение, улучшение, облегчение, уменьшение, замедление и/или смягчение одного или более признаков, симптомов или причин заболевания, или любое другое желаемое изменение биологической системы. Применительно к раку, эффективное количество включает количество, достаточное, чтобы вызвать уменьшение опухоли и/или уменьшить скорость роста опухоли (например, подавить рост опухоли) или предотвратить или задержать другую нежелательную пролиферацию клеток. В некоторых вариантах реализации эффективное количество представляет собой количество, достаточное для задержки развития опухоли. В некоторых вариантах реализации эффективное количество представляет собой количество, достаточное для предупреждения или отсрочки рецидива опухоли. Эффективное количество может быть введено за один или более приемов. Эффективное количество лекарства или композиции может: (i) снижать количество раковых клеток; (ii) уменьшать размер опухоли; (iii) ингибировать, задерживать, замедлять до некоторой степени и останавливать инфильтрацию раковых клеток в периферические органы; (iv) ингибировать (то есть замедлять до некоторой степени и останавливать) метастазирование опухоли; (v) ингибировать рост опухоли; (vi) предупреждать или вызывать отсрочку возникновения и/или рецидива опухоли; и/или (vii) облегчать до некоторой степени один или более симптомов, связанных с раком.
Термины "комбинированная терапия", "совместное введение", "совместно введенные" или "одновременное введение" (или указанные термины с незначительными вариациями) включают одновременное введение по меньшей мере двух терапевтических агентов пациенту или их последовательное введение в течение периода времени, когда первый вводимый терапевтический агент все еще присутствует в организме пациента при введении второго вводимого терапевтического агента.
Термин "монотерапия" относится к введению единственного лекарственного средства для лечения заболевания или расстройства при отсутствии совместного введения любого другого терапевтического агента для лечения того же заболевания или расстройства.
"Дозировка" относится к параметрам введения лекарственного средства в определенных количествах за единицу времени (например, в час, день, неделю, месяц и т.д.) пациенту. Такие параметры включают, например, размер каждой дозы. Такие параметры также включают конфигурацию каждой дозы, которую можно вводить в виде одной или нескольких единиц, например, принимаемых за одно введение, например, перорально (например, в виде одной, двух, трех или более таблеток, капсул, и т.д.) или путем инъекции (например, в виде болюса). Размеры дозировки могут быть связаны с дозами, вводимыми непрерывно (например, в виде внутривенного инфузии в течение нескольких минут или часов). Такие параметры дополнительно включают частоту введения отдельных доз, которая может меняться с течением времени.
"Доза" относится к количеству лекарственного средства, принимаемому при однократном введении.
В настоящем документе "рак" относится к состоянию, характеризующихся аномальным, нерегулируемым, злокачественным ростом клеток. В одном варианте реализации рак представляет собой экзокринный рак поджелудочной железы. В еще одном варианте реализации экзокринный рак поджелудочной железы выбран из группы, состоящей из ацинарно-клеточной карциномы, аденокарциномы, железисто-плоскоклеточной карциномы, гигантоклеточной опухоли, внутрипротокового папиллярного муцинозного новообразования (IPMN), муцинозной цистаденокарциномы, панкреатобластомы, серозной цистаденокарциномы и солидных и псевдопапиллярных опухолей.
Термины "резистентный" и "устойчивый" относятся к опухолевым клеткам, которые выживают при лечении с применением терапевтического агента. Такие клетки могут вначале реагировать на терапевтический агент, но впоследствии демонстрировать снижение реакции во время лечения, или не демонстрировать адекватную реакцию на терапевтический агент в том смысле, что указанные клетки продолжают пролиферировать в процессе лечения с применением данного агента.
II. Инъекционная липосомальная форма иринотекана - сахарозы сульфата (ММ-398; PEP02)
В настоящем документе иринотекан вводят в стабильном липосомальном составе в инъекционной липосомальной форме иринотекана - сахарозы сульфата (иначе называемой "инъекционной липосомальной формой иринотекана – сахарозы окстасульфата" или "инъекционная липосомальная форма иринотекана сахарософата"), причем указанный состав в настоящем документе называют "ММ-398" (а также PEP02, см. US 8147867). ММ-398 может быть представлен в виде стерильной жидкости для внутривенной инъекции. Необходимое количество ММ-398 можно разбавить, например, в 500 мл 5% раствора декстрозы для инъекций класса USP, и вводить путем инфузии в течение 90 минут.
Липосома MM-398 представляет собой моноламеллярную липидную двуслойную везикулу диаметром приблизительно 80-140 нм, в которой закапсулировано водное пространство, содержащее комплекс иринотекана в гелеобразном или осажденном состоянии в виде соли с октасульфатом сахарозы. Липидная мембрана липосомы состоит из фосфатидилхолина, холестерина и фосфатидилэтаноламина, модифицированного полиэтиленгликолем в количестве приблизительно одна молекула полиэтиленгликоля (ПЭГ) на 200 молекул фосфолипидов.
Указанный стабильный липосомальный состав иринотекана обладает несколькими свойствами, которые могут обеспечить улучшенный терапевтический индекс. Контролируемое и замедленное высвобождение улучшает активность указанного лекарственного средства в зависимости от расписания приема за счет увеличения длительности воздействия лекарственного вещества на ткань опухоли; данное свойство позволяет ему присутствовать в большем процентном количестве клеток во время S-фазы клеточного цикла, когда требуется раскручивание ДНК на предварительном этапе репликации ДНК. Фармакокинетика при длительном нахождении в кровотоке и высокое внутрисосудистое удерживание лекарственного вещества в липосомах может способствовать эффекту повышенной проницаемости и удерживания (EPR). EPR позволяет липосомам накапливаться в таких участках, как злокачественные опухоли, где нарушена нормальная целостность сосудов (в частности, капилляров), что приводит к утечке таких частиц, как липосомы, из просвета капилляра. EPR может, таким образом, способствовать сайт-специфической доставке липосом в солидные опухоли. EPR ММ-398, может привести к последующему эффекту депо, при котором липосомы накапливаются в макрофагах, ассоциированных с опухолью (TAM), которые метаболизируют иринотекан, локально преобразуя его в существенно более токсичный SN-38. Считается, что эта локальная биоактивация приводит к уменьшению воздействия лекарственного вещества на потенциальные сайты токсичности и усилению воздействия на раковые клетки в опухоли.
Фармакогенетика глюкуронидации иринотекана
Фермент, продуцируемый геном UGT1A1, UDP-глюкуронозилтрансфераза 1, отвечает за метаболизм билирубина, а также опосредует глюкуронидацию SN-38, являющуюся первым этапом преобладающего пути метаболического клиренса этого активного метаболита иринотекана. Кроме противоопухолевой активности, SN-38 также отвечает за высокую токсичность, иногда ассоциируемую с терапией с применением иринотекана. Таким образом, глюкуронидация SN-38 до неактивной формы, глюкуронида SN-38, является важным этапом регуляции токсичности иринотекана.
Описан мутационный полиморфизм промотора гена UGT1A1, при котором существует переменное количество тимин-адениновых (ta) повторов. Обнаружено, что промоторы, содержащие семь тимин-адениновых (ta) повторов (в аллеле UGT1A1*28), были менее активны, чем промоторы дикого типа, содержащие шесть повторов, что приводило к пониженной экспрессии UDP-глюкуронилтрансферазы 1. Пациенты, несущие две дефектных аллеля UGT1A1, демонстрируют пониженную глюкуронизацию SN-38. В некоторых случаях сообщается, что лица, гомозиготные по аллелю UGT1A1*28 (называемые лицами с генотипом UGT1A1 7/7, поскольку оба аллеля представляют собой аллели UGT1A1*28, содержащие 7 ta-повторов, в отличие от генотипа UGT1A1 6/6 (дикого типа), при котором оба аллеля содержат 6 ta-повторов) и характеризующиеся нестабильным повышением билирубина в сыворотке крови (например, пациенты с синдромом Жильбера), могут подвергаться большему риску токсичности при получении стандартных доз иринотекана. Это указывает на наличие связи между гомозиготностью по аллелю UGT1A1*28, уровнями билирубина и токсичностью иринотекана.
Метаболическая трансформация ММ-398 в SN-38 (например, в плазме) включает два важнейших этапа: (1) высвобождение иринотекана из липосом и (2) преобразование свободного иринотекана в SN-38. Без учета теоретических аспектов считается, что как только иринотекан покидает липосомы, он подвергается катаболизму по тем же метаболическим путям, что и обычный (свободный) иринотекан. Поэтому подобные генетические полиморфизмы человека, являющиеся прогностическим фактором токсичности и эффективности, можно считать аналогичными для иринотекана и ММ-398. Тем не менее, из-за меньшего распределения в тканях, сниженного выведения, более высокого системного воздействия и более длительного периода полувыведения SN-38 в составе ММ-398 по сравнению со свободным иринотеканом, дефектные генетические полиморфизмы могут демонстрировать большую ассоциацию с тяжелыми нежелательными явлениями и/или эффективностью.
Пациенты с пониженной активностью UGT1A1
Показано, что лица, гомозиготные по аллелю UGT1A1*28 (генотип UGT1A1 7/7), характеризуются повышенным риском нейтропении после начала лечения с применением иринотекана. Согласно инструкции по применению иринотекана (Camptosar®), в исследовании с участием 66 пациентов, получавших иринотекан в качестве монотерапии (350 мг/м2 раз в 3 недели), заболеваемость нейтропенией 4 степени у пациентов, гомозиготных по аллелю UGT1A1*28, составляла 50%, а у пациентов, гетерозиготных по этому аллелю (генотип UGT1A1 6/7) заболеваемость составляла 12,5%. Важно, что нейтропения 4 степени не наблюдалась у пациентов, гомозиготных по аллелю дикого типа (генотип UGT1A1 6/6). В других исследованиях описана более низкая распространенность нейтропении, опасной для жизни. По этой причине у пациентов, набранных в исследование 3 фазы, описанное в разделе "Примеры" настоящего документа, и гомозиготных по аллелю UGT1A1*28 (генотип UGT1A1 7/7), лечение с применением ММ-398 начинали с более низкой дозы, чем у пациентов с одним (например, UGT1A1 6/7) или двумя (UGT1A1 6/6) аллелями дикого типа.
Дополнительные генотипические модификаторы метаболизма иринотекана
Хотя аллель UGT1A1*28 относительно часто встречается у европеоидов (по оценкам, у 10%), ее распространенность варьирует у других этнических групп. Кроме того, обнаружены дополнительные генотипы UGT1A1 с более высокой распространенностью, например, в популяциях монголоидов; это может иметь важное значение для метаболизма иринотекана в этих популяциях. Например, аллель UGT1A1*6 более распространена среди монголоидов. Эта аллель связана не с ta-повторами, а с мутацией Gly71Arg, которая снижает активность фермента. В более ранних и текущих исследованиях ММ-398 были собраны фармакогенетические данные на набранных пациентов. В исследовании под названием PEP0203 четкая взаимосвязь генетического полиморфизма семейства UGT1A и DPYD (дигидропиримидиндегидрогеназы, фермента, связанного с катаболизмом 5-FU) с фармакокинетическими параметрами и токсичностью ММ-398 не была установлена вследствие малого размера выборки оцениваемых субъектов. Вместе с тем было отмечено, что у пациентов с комбинированным полиморфизмом UGT1A1*6/*28 была повышенная ППК SN-38, нормированная по дозе, и перенесенная DLT.
III. 5-фторурацил (5-FU) и лейковорин
5-фторурацил является пиримидиновым антагонистом, который влияет на биосинтез нуклеиновых кислот. Дезоксирибонуклеотид указанного лекарственного вещества ингибирует тимидилатсинтетазу, таким образом подавляя образование тимидиловой кислоты из дезоксиуридиловой кислоты и мешая синтезу ДНК. Он также препятствует синтезу РНК.
Лейковорин (также называемый фолиновой кислотой) действует как биохимический кофактор реакций переноса 1-углерода при синтезе пуринов и пиримидинов. Лейковорин не требует фермента дигидрофолатредуктазы (ДГФР) для преобразования в тетрагидрофолиевую кислоту. Лейковорин ингибирует эффекты метотрексата и других антагонистов ДГФР. Лейковорин может усиливать цитотоксические эффекты фторированных пиримидинов (т.е. фторурацила и флоксуридина). После того, как 5-FU активируется в клетке, он сопровождается фолатным кофактором и ингибирует фермент тимидилатсинтетазу, таким образом подавляя синтез пиримидинов. Лейковорин увеличивает пул фолата, тем самым усиливая связывание фолатного кофактора и активного 5-FU с тимидилатсинтетазой.
Лейковорин имеет декстро- и лево-изомеры, причем только последний из них пригоден для фармакологических целей. Поэтому применение биоактивного лево-изомера («леволейковорина») также было одобрено FDA для лечения рака. Дозировка леволейковорина обычно составляет половину от дозировки рацемической смеси, содержащей декстро- (d) и лево- (l) изомеры.
С FU и лейковорином следует обращаться и хранить их в соответствии с вкладышем в упаковку, специальным для каждой страны.
IV. Введение
Липосомальный иринотекан вводят внутривенно, по отдельности или в комбинации с 5-фторурацилом (5-FU) и/или лейковорином. В одном варианте реализации липосомальный иринотекан вводят до 5-FU и лейковорина. В еще одном варианте реализации лейковорин вводят до 5-FU. В еще одном варианте реализации липосомальный иринотекан вводят внутривенно в течение 90 минут. В еще одном варианте реализации 5-FU вводят внутривенно в течение 46 часов. В еще одном варианте реализации лейковорин вводят внутривенно в течение 30 минут. В различных вариантах реализации липосомальный иринотекан представляет собой MM-398.
V. Популяции пациентов
В одном варианте реализации пациент, получающий лечение с применением способов и композиций, описанных в настоящем документе, демонстрирует признаки рецидивирующего или рецидивирующего рака поджелудочной железы после первичной химиотерапии.
В еще одном варианте реализации пациент имеет историю безуспешного лечения основного или рецидивирующего заболевания с применением по меньшей мере одной схемы химиотерапии на основе соединений платины, например, схемы химиотерапии, включающей карбоплатин, цисплатин или другое органическое соединение платины.
В дополнительном варианте реализации пациент ранее безуспешно получал лечение с применением гемцитабина или приобрел устойчивость к гемцитабину.
В одном варианте реализации резистентная или устойчивая опухоль является опухолью, при которой интервал без лечения для пациентов с опухолью составляет менее 6 месяцев (например, из-за рецидива рака) или при которой опухоль прогрессирует во время курса лечения.
В еще одном варианте реализации рак поджелудочной железы у пациентов, которых лечат, представляет собой позднюю стадию рака поджелудочной железы, являющегося опухолью поджелудочной железы, демонстрирующей отдаленные метастазы и/или перипанкреатическое разрастание опухоли.
Композиции и способы, описанные в настоящем документе, можно применять для лечения всех видов рака поджелудочной железы, в том числе рака поджелудочной железой, резистентного или устойчивого к другим видам противоракового лечения.
VI. Комбинированная терапия
В одном варианте реализации липосомальный иринотекан вводят пациентам с раком поджелудочной железы совместно с 5-фторурацилом (5-FU) и лейковорином, согласно определенной схеме клинической дозировки, например, описанной в настоящем документе. В одном варианте реализации липосомальный иринотекан представляет собой MM-398.
В настоящем документе дополнительное или комбинированное введение (совместное введение) включает одновременное введение соединений в одной или разных лекарственных формах, или раздельное введение соединений (например, последовательное введение). Например, липосомальный иринотекан можно вводить одновременно с 5-FU и лейковорином. В альтернативном варианте липосомальный иринотекан можно вводить в комбинации с 5-FU и лейковорином, причем липосомальный иринотекан, 5-FU и лейковорин изготовлены в форме для раздельного введения и вводятся одновременно или последовательно. Например, вначале можно ввести липосомальный иринотекан, а затем (например, сразу после этого) ввести 5-FU и лейковорин. Такое параллельное или последовательное введение дает то преимущество, что липосомальный иринотекан, 5-FU и лейковорин одновременно присутствуют в организме пациентов, получающих лечение. В конкретном варианте реализации липосомальный иринотекан вводят до 5-FU и лейковорина. В еще одном конкретном варианте реализации лейковорин вводят до 5-FU.
В еще одном варианте реализации липосомальный иринотекан, 5-FU и лейковорин изготовлены в форме для внутривенного введения. В конкретном варианте реализации пациенту вводят эффективное количество каждого из следующих веществ: липосомального иринотекана, 5-фторурацила (5-FU) и лейковорина, причем лечение включает по меньшей мере один цикл, где цикл представляет собой период продолжительностью 2 недели, и для каждого цикла: (a) липосомальный иринотекан вводят в 1 день цикла в дозе 80 мг/м2, за исключением случаев, когда пациент гомозиготен по аллелю UGT1A1*28; в таком случае липосомальный иринотекан вводят в 1 день 1 цикла в дозе 60 мг/м2; (b) 5-FU вводят в дозе 2400 мг/м2; и (c) лейковорин вводят в дозе 200 мг/м2 (l-форма) или 400 мг/м2 (l+d-рацемическая форма). В конкретном варианте реализации дозу липосомального иринотекана, вводимую пациенту, гомозиготному по аллелю UGT1A1*28, увеличивают после одного цикла до 80 мг/м2.
В одном варианте реализации липосомальный иринотекан можно вначале вводить в высокой дозе и снижать дозу с течением времени. В еще одном варианте реализации липосомальный иринотекан вначале вводят в низкой дозе и повышают дозу с течением времени. В одном варианте реализации липосомальный иринотекан вводят в форме монотерапии.
В еще одном варианте реализации дозу 5-FU меняют со временем. Например, 5-FU можно вначале вводить в высокой дозе и снижать дозу с течением времени. В еще одном варианте реализации 5-FU вначале вводят в низкой дозе и повышают дозу с течением времени.
В еще одном варианте реализации дозу лейковорина меняют со временем. Например, лейковорин можно вначале вводить в высокой дозе и снижать дозу с течением времени. В еще одном варианте реализации лейоворин вначале вводят в низкой дозе и повышают дозу с течением времени.
VII. Протоколы лечения
Подходящие протоколы лечения включают, например, протоколы, где пациенту вводят эффективное количество липосомального иринотекана, причем лечение включает по меньшей мере один цикл, равный 3-недельномк периоду, и в каждом цикле липосомальный иринотекан вводят в первый день цикла в дозе 120 мг/м2, за исключением случая, если пациент гомозиготен по аллелю UGT1A1*28; в таком случае липосомальный иринотекан вводят в 1 день 1 цикла в дозе 80 мг/м2. В одном варианте реализации дозу липосомального иринотекана, вводимого пациенту, гомозиготному по аллелю UGT1A1*28, повышают после одного с шагом 20 мг/м2, но не более 120 мг/м2.
В еще одном варианте реализации протокол лечения включает введение пациенту эффективного количества каждого из следующих средств: липосомального иринотекана, 5-фторурацила (5-FU) и лейковорина, причем лечение включает по меньшей мере один цикл, где цикл представляет собой период продолжительностью 2 недели, и для каждого цикла: (a) липосомальный иринотекан вводят в 1 день цикла в дозе 80 мг/м2, за исключением случаев, когда пациент гомозиготен по аллелю UGT1A1*28; в таком случае липосомальный иринотекан вводят в 1 день 1 цикла в дозе 60 мг/м2; (b) 5-FU вводят в дозе 2400 мг/м2; и (c) лейковорин вводят в дозе 200 мг/м2 (l-форма) или 400 мг/м2 (l+d-рацемическая форма). В конкретном варианте реализации дозу липосомального иринотекана, вводимую пациенту, гомозиготному по аллелю UGT1A1*28, увеличивают после одного цикла до 80 мг/м2.
VIII. Результаты
Настоящее изобретение обеспечивает способы лечения рака поджелудочной железы у пациента, включающие введение пациенту липосомального иринотекана (ММ-398) отдельно или в комбинации с 5-фторурацилом (5-FU) и лейковорином, в соответствии с конкретной схемой клинического приема.
Предпочтительно, при комбинированной терапии с применением липосомального иринотекана с 5-FU и лейковорином наблюдается терапевтический синергизм.
«Терапевтический синергизм» относится к явлению, при котором лечение больных с применением комбинации терапевтических агентов приводит к терапевтически улучшенным результатам по сравнению с результатами, достигаемыми за счет каждого отдельного компонента комбинации, применяемого в оптимальной дозе (T. H. Corbett et al., 1982, Cancer Treatment Reports, 66, 1187). В указанном контексте терапевтически улучшенный результат представляет собой результат, при котором пациенты: а) демонстрируют меньшую частоту неблагоприятных явлений при получении терапевтического благоприятного эффекта, который равен или превышает эффект, получаемый при введении каждого из отдельных компонентов комбинации в качестве монотерапия в той же дозе, что и в комбинации, или b) не демонстрируют дозолимитирующей токсичности при получении терапевтического благоприятного эффекта, превышающего эффект, получаемый при введении каждого отдельного компонента комбинации в тех же дозах, что и в комбинации(ях). В моделях ксенотрансплантата при применении комбинации, используемой в максимально переносимой дозе, причем каждый из компонентов присутствует в дозе, как правило, не превышающей его собственной максимально переносимой дозы, наблюдается терапевтический синергизм, если за счет введения комбинации достигается снижение роста опухоли, превышающее наилучшее значение снижение роста опухоли при введении отдельных компонентов.
Таким образом, в комбинации компоненты таких комбинаций оказывают аддитивное или сверхаддитивное воздействие на подавление роста опухоли поджелудочной железы по сравнению с монотерапией с применением только иринотекана, инкапсулированного в липосомах, или лечением с применением химиотерапевтического(их) агента(ов) в отсутствие липосомального иринотекана. Под термином «аддитивный» подразумевается результат, превышающий (например, по степени снижения митотического индекса опухоли или роста опухоли или степени сокращения размеров опухоли или частоты и/или продолжительности бессимптомных периодов или периодов с уменьшением симптомов) лучший отдельный результат, достигнутый за счет монотерапии с применением каждого отдельного компонента, а термин «супераддитивный» используется для обозначения результата, превышающего сумму таких отдельных результатов. В одном варианте реализации аддитивный эффект измеряют как замедление или остановку роста опухоли поджелудочной железы. Аддитивный эффект также можно измерять как, например, снижение размера опухоли поджелудочной железы, снижение митотического индекса опухоли, снижение количества метастазов со временем, повышение общей скорости реакции, повышение медианной или общей выживаемости.
Один из неограничивающих примеров возможного количественного показателя эффективности терапевтического лечения - расчет десятичного логарифма уничтожения клеток, определяемого по следующей формуле:
lg уничтожения клеток = TC(дней)/3,32×Td
где TC - задержка роста клеток, т.е. среднее время (в сутках), за которое опухоли в группе, получавшей лечение (T), и опухоли в контрольной группе (C) достигают заданного значения (например, 1 г, или 10 мл), а Td - время (в сутках), необходимое для удвоения объема опухоли у контрольных животных. При применении данного показателя продукт считают активным, если lg уничтожения клеток больше или равен 0,7, и очень активным - если lg уничтожения клеток превышает 2,8. При применении данного показателя для комбинации, используемой в максимально переносимой дозе, причем каждый из компонентов присутствует в дозе, обычно не превышающей его собственной максимально переносимой дозы, наблюдается терапевтический синергизм, если 10 lg уничтожения клеток превышает наилучшее значение 10 lg уничтожения клеток при введении отдельных компонентов. В типичном случае 10 lg уничтожения клеток для комбинации превышает наилучшее значение 10 lg уничтожения клеток при введении отдельного компонента комбинации по меньшей мере на 0,1 lg уничтожения клеток, по меньшей мере на 0,5 lg уничтожения клеток или по меньшей мере на 1,0 lg уничтожения клеток.
Реакция на лечение может включать:
Полный патологический ответ (pCR): отсутствие инвазивного рака молочной железы и лимфатических узлов после основного системного лечения.
полный ответ (CR): исчезновение всех очагов-мишеней. Любые патологические лимфатические узлы (являющиеся или не являющиеся мишенями), уменьшившиеся по короткой оси на <10 мм;
частичный ответ (PR): по меньшей мере 30% снижение суммы размеров очагов-мишеней по сравнению с исходной суммой диаметров;
стабильное заболевание (SD): отсутствие как значимого снижения размеров, соответствующего частичной реакции, так и значимого увеличения, соответствующего прогрессирующему заболеванию, по сравнению с наименьшей суммой диаметров, зарегистрированной в процессе исследования; либо
при этом не CR/не PD означает сохранение одного или более очагов, не являющихся мишенями, и/или поддержание уровня маркера опухоли выше нормы.
Прогрессирующее заболевание (PD) обозначает увеличение суммы размеров очагов-мишеней по меньшей мере на 20% по сравнению с наименьшей суммой, зарегистрированной в ходе исследования (включая исходную сумму, если она является наименьшей в ходе исследования). Помимо относительного увеличения на 20%, должно произойти абсолютное увеличение суммы на 5 мм. Появление одного или нескольких новых очагов также считается прогрессированием.
При типичном результате пациенты, получающие лечение в соответствии со способами, описанными в настоящем документе, могут испытывать улучшение по меньшей мере одного из признаков рака поджелудочной железы.
В одном варианте реализации у пациента, получающего такое лечение, наблюдается pCR, CR, PR или SD.
В еще одном варианте реализации пациент, получающий такое лечение, испытывает сокращение размеров опухоли и/или снижение скорости роста, т.е. подавление роста опухоли. В еще одном варианте реализации происходит снижение или ингибирование нежелательной пролиферации клеток. В еще одном варианте реализации может произойти одно или более из следующих явлений: может снизиться количество раковых клеток; может уменьшиться размер опухоли; может произойти ингибирование, задержка, замедление или остановка проникновения раковых клеток в периферические органы; может произойти замедление или ингибирование метастазирования опухоли; может произойти ингибирование роста опухоли; может достигаться предотвращение или задержка рецидива опухоли; может произойти некоторое облегчение одного или более симптомов, связанных с раком.
В других вариантах реализации такое улучшение измеряется по уменьшению количества и/или размера измеримых опухолевых очагов. Измеримые очаги определяются как очаги, которые можно точно измерить по меньшей мере по одному измерению (при этом регистрируют максимальный диаметр) как >10 мм согласно компьютерной томографии (с использованием КТ-среза не толще 5 мм), 10 мм согласно измерению штангенциркулем при клиническом обследовании или >20 мм согласно рентгенографии грудной клетки. В рамках улучшения также можно измерять размер очагов, не являющихся мишенями, например, патологических лимфатических узлов. В одном варианте реализации очаги можно измерить на рентгенограммах грудной клетки или КТ- или МРТ-пленках.
В других вариантах реализации для оценки реакции на лечение можно использовать цитологию и гистологию. При дифференцировке реакции или стабильного заболевания от прогрессирующего заболевания необходимо учитывать цитологическое подтверждение неопластического происхождения любого выделения, появляющегося или ухудшающегося во время лечения, если измеримые опухоли соответствуют критериям реакции или стабильного заболевания (выделение может быть побочным эффектом лечения).
В некоторых вариантах реализации введение эффективного количества липосомального иринотекана, 5-FU и лейковорина согласно любому из способов, предоставленных в настоящем документе, приводит к по меньшей мере одному терапевтическому эффекту, выбранному из группы, состоящей из снижения размеров опухоли молочной железы, сокращения количества метастатических очагов, возникающих с течением времени, полной ремиссии, частичной ремиссии, стабильного заболевания, увеличения скорости общей реакции, или патологической завершенной реакции. В некоторых вариантах реализации предложенные способы лечения приводят к сопоставимой степени клинического благоприятного эффекта (CBR=CR+PR+SD ≥6 месяцев), лучшей, чем степень, достигаемая за счет этой же комбинации противораковых агентов, вводимой без сопутствующего введения ММ-398. В других вариантах реализации улучшение степени клинического благоприятного эффекта составляет приблизительно 20%, 20%, 30%, 40%, 50%, 60%, 70%, 80% или более по сравнению с этими же комбинациями противораковых агентов, вводимых без сопутствующего введения ММ-398.
Следующие примеры являются иллюстративными и никоим образом не должны рассматриваться в качестве ограничения объема настоящего описания; для специалистов в данной области техники после почтения настоящего описания станут очевидны различные изменения и эквивалентные варианты описанного.
ПРИМЕРЫ
Пример 1: Активность ММ-398 в модели ортотопической опухоли поджелудочной железы, экспрессирующей люциферазу (L3.6pl)
Противоопухолевую активность ММ-398 оценивали на ортотопической модели рака поджелудочной железы (L3.6pl), высокогипоксической доклинической модели опухоли. Примерно 2,5×10-5 клеток опухоли поджелудочной железы L3.6pl имплантировали путем прямой инъекции в поджелудочную железу. Опухолевую нагрузку/количественную оценку отслеживали со временем с помощью биолюминесцентной визуализации (BLI). ММ-398 и свободный иринотекан вводили в дозе 20 мг/кг/неделю в течение трех недель. Как показано на фигуре 1, ММ-398 (липосомальный CPT11) обладал значительной противоопухолевой активностью по сравнению с контролем (HBS) и свободным CPT11.
Пример 2: Накопление SN-38 в опухолях после лечения с применением свободного иринотекана или липосомального иринотекана (ММ-398)
Было высказано предположение, что противоопухолевая активность, наблюдавшаяся в ортотопической модели рака поджелудочной железы, была обусловлена местным действием макрофагов по преобразованию иринотекана в более активный SN-38. Для проверки указанного предположения клетки рака толстой кишки человека (HT-29) подкожно вводили SCID-мышам, после достижения размера опухолей 1000 мм3 внутривенно вводили 40 мг/кг свободного иринотекана или ММ-398. Мышей с опухолями умерщвляли в разные моменты времени, извлекали опухоли из животных обеих групп и измеряли концентрации SN-38.
Как показано на фигуре 2, в опухоли наблюдалось 20-кратное увеличение ППКSN-38 для ММ-398 по сравнению со свободным иринотеканом. Длительность воздействия позволяла оказывать продолжительное воздействие активного метаболита на медленно пролиферирующие раковые клетки на протяжении клеточного цикла. Кроме того, было высказано предположение, что указанная активность обусловлена также снижением гипоксии внутри опухоли и последующим подавляющим воздействием на ангиогенез, метастазирование и иммунодепрессивные условия в опухоли.
Пример 3: Влияние ММ-398 на окрашивание карбоангидразы IX в модели ксенотрансплантата HT29
Для проверки действия ММ-398 по снижению маркеров гипоксии выполнили эксперименты на модели клеток рака толстой кишки человека (HT-29). Конкретно, клетки HT-29 подкожно вводили бестимусным голым мышам, на 13 день внутривенно вводили PBS-контроль или 1,25, 2,5, 5, 10 или 20 мг/кг ММ-398. MM-398 вводили раз в неделю в течение 4 недель в назначенных дозах. Опухоли из обеих групп животных (n=5) извлекали через 24 часа после введения последней дозы. Срезы замороженных опухолей использовали для иммуногистохимического окрашивания карбоангидразы IX (CAIX). Количественную оценку окрашивания CAIX выполняли с помощью программного обеспечения Definiens® (Definiens AG, Мюнхен).
Как показано на фигуре 3, ММ-398 снижал уровни маркеров гипоксии. В частности, на графиках на фигуре 3 показан процент клеток со средней (средняя треть) или высокой (верхняя треть) интенсивностью окрашивания CAIX. Показаны типичные образцы из каждой группы, а также среднее по группе (среднее +/- стандартное отклонение). Лечение с применением ММ-398 изменяло микроокружение опухоли, уменьшая процент средне- и высокоположительных по CAIX клеток в зависимости от дозы. Поскольку гипоксия является отличительным признаком устойчивого и агрессивного заболевания; ожидается, что снижение гипоксии повышает чувствительность опухолевых клеток к химиотерапии.
Пример 4: ММ-398 увеличивает перфузию красителя Хехста
Помимо изменения чувствительности опухолевых клеток к химиотерапии путем модификации микроокружения опухоли, снижение гипоксии может указывать на улучшение васкуляризации опухоли, что может облегчить доставку низкомолекулярных терапевтических средств. Лечение с применением ММ-398 привело к повышению микрососудистой плотности через 6 дней после лечения согласно измерению окрашивания CD31 (молекулы адгезии эндотелиальных клеток к тромбоцитам) в исследовании ксенотрансплантата HT29. Для дальнейшей оценки воздействия ММ-398 на низкомолекулярную васкуляризацию опухоли выполнили эксперимент с перфузией красителя Hoechst 33342. В частности, в организме мыши NOD-SCID выращивали первичную опухоль поджелудочной железы и вводили однократную дозу ММ-398 (20 мг/кг). Через 24 часа вводили краситель Hoechst 33342, и через 20 минут умерщвляли животное. Как показано на фигуре 4, увеличение интенсивности окрашивания у обработанных мышей было статистически значимым, p<0,001. Указанные данные показывают, что ММ-398 изменял микроокружение опухоли, повышая восприимчивость опухоли к таким агентам, как 5-FU/LV посредством снижения гипоксии опухоли и увеличения перфузии низкомолекулярных соединений.
Пример 5: Фармакокинетика ММ-398 в организме человека (I фаза)
Фармакокинетический профиль ММ-398 при применении в качестве монотерапии изучали в клиническом исследовании I фазы (PEP0201) у пациентов при уровне дозы 60, 120 или 180 мг/м2 и в клиническом исследовании II фазы у пациентов с раком желудка (PEP0206) при 120 мг/м2. В указанных исследованиях измеряли уровни общего иринотекана, SN-38 и инкапсулированного иринотекана в плазме.
Пиковые концентрации общего иринотекана в сыворотке (Cмакс) для 120 мг/м2 ММ-398 находились в диапазоне 48-79 мкг/мл, что было приблизительно в 50 раз выше, чем для 125 мг/м2 свободного иринотекана. Период полувыведения общего иринотекана (t1/2) для ММ-398 находился в диапазоне от 21 до 48 часов, что было приблизительно в 2-3 раза выше, чем для 125 мг/м2 свободного иринотекана. В целом, воздействие общего иринотекана за одну неделю (ППК 0-T) при дозе 120 мг/м2 ММ-398 колебалось в диапазоне 1200-3000 (мкг*ч/мл), что было приблизительно в 50-100 раз выше, чем для 300 мг/м2 свободного иринотекана. В противоположность этому, уровни Cмакс SN38 при 120 мг/м2 ММ-398 колебались от 9 до 17 нг/мл, что было приблизительно на 50% меньше, чем при 125 мг/м2 свободного иринотекана. В целом, воздействие SN38 за одну неделю (ППК 0-T) варьировалось от 474 до 997 нг/мл и было лишь в 1-2 раза выше, чем при применении 300 мг/м2 свободного иринотекана. Как для SN38, так и для общего иринотекана, ППК возрастала менее чем пропорционально дозе ММ-398. ФК параметры инкапсулированного иринотекана почти совпадают с параметрами общего иринотекана, что указывает на то, что большая часть иринотекана оставалась капсулированной в липосомах при циркуляции. ФК параметры ММ-398 не подвергались существенным изменениям в комбинации с 5-FU/LV. На фигурах 5 и 6 приведена сводная информация по ФК результатам предыдущих исследований ММ-398.
Пример 6: Исследование 1 фазы с эскалацией дозы
Схему с применением комбинации фторурацила, лейковорина и ММ-398 изучили в исследовании 1 фазы солидных опухолей у 16 субъектов, из которых 5 являлись пациентами с раком поджелудочной железы. Объективная частота реакции опухоли, продолжительность реакции и частота контроля заболевания являлись конечными показателями эффективности в данном исследовании. Среди 15 пациентов, у которых была возможна оценка эффективности, у 2 (13,3%) подтвердилась PR, у 9 (60,0%) наблюдали SD и у 4 (26,7%) - PD. Общая частота контроля заболевания составила 73,3%. Частичную реакцию наблюдали у одного пациента с раком желудка (при уровне дозы 80 мг/м2) и одного пациента с раком молочной железы (при уровне дозы 100 мг/м2) при продолжительности реакции 142 и 76 дней, соответственно. Среди 6 пациентов, получивших MTD-дозу 80 мг/м2, наблюдали 1 PR, 4 SD и 1 PD. Частота реакции опухоли и частота контроля заболевания составили 16,7% и 83,3%, соответственно. Основные DLT представляли собой диарею 3 степени, лейкопению, нейтропению и лихорадочную нейтропению. MTD для ММ-398 составляла 80 мг/м2.
В исследовании 1 фазы с эскалацией дозы ММ-398 в комбинации с 5-FU/LV при поздних стадиях развития солидных опухолей (PEP0203) сообщалось в общей сложности в 401 эпизоде НЯ у 16 субъектов, получавших лечение (популяция для исследования безопасности), из которых 74 (18,4%) были 3 категории CTC или выше. Среди всех НЯ, по мнению исследователей, 231 НЯ (57,6%) было связано с лечением. Наиболее распространенные НЯ, связанные с лечением, включали тошноту (81,3%), диарею (75,0%), рвоту (68,8%), утомляемость (43,8%), мукозит (43,8%), лейкопению (37,5%), нейтропению (37,5%), потерю массы тела (37,5%), анемию (31,3%) и алопецию (31,3%). Острая холинергическая диарея наблюдалась редко. В таблице 1 представлена частота нежелательных явлений, возникших при лечении, в зависимости от максимальной категории CTC и причинно-следственной связи (частота встречаемости ≥ 20%), согласно исследованию PEP0203. В таблице 2 представлена частота нежелательных явлений 3 или более высокой категории, возникших при лечении, наблюдавшихся у 5 пациентов с раком поджелудочной железы, получавших лечение в рамках исследования PEP0203.
Частота нежелательных явлений, возникших при лечении, в зависимости от максимальной категории CTC и причинно-следственной связи (частота≥20%) в исследовании PEP0203
Предпочтительный термин
(N=16)
1: При классификации тяжести используется максимальная степень, когда-либо зарегистрированная для каждого субъекта, сообщавшего о таком нежелательном явлении
2: Определяется как субъект, когда-либо испытывавший НЯ, связанный с исследуемым препаратом, или нет
Частота нежелательных явлений 3 или более высокой категории, возникших при лечении у больных раком поджелудочной железы в исследовании PEP0203
Пример 7: Исследование 3 фазы
Многообещающие данные по эффективности и безопасности, полученные в ходе исследования 1 фазы (описанного выше) обеспечили дальнейшее изучение комбинации ММ-398, 5-FU и лейковорина в исследовании 3 фазы.
А. Цели
Основная цель исследования 3 фазы заключается в сравнении общей выживаемости после лечения с применением ММ-398 в комбинации с 5-фторурацилом и лейковорином или без них по сравнению с применением 5-фторурацила и лейковорина у пациентов с метастазирующим раком поджелудочной железы, прогрессирующим на фоне терапии с применением гемцитабина. Дополнительные цели включали:
сравнить конечные показатели времени до наступления явления в экспериментальной и контрольной группах (т.е. выживаемость без прогрессирования (PFS) и время до констатации отсутствия эффекта лечения (TTF));
- сравнить частоту объективной реакции (ORR) в группах, получавших лечение;
- сравнить реакцию опухолевого маркера CA 19-9 в группах, получавших лечение;
- сравнить частоту клинической эффективности (CBR) в группах, получавших лечение;
- оценить результаты, регистрируемые пациентами (PRO) в группах, получавших лечение, с помощью основной анкеты для оценки качества жизни Европейской организации исследования и лечения рака (EORTC) (EORTC-QLQ-C30);
- сравнить профиль безопасности и нежелательных явлений в группах, получавших лечение; и
- определить фармакокинетические свойства ММ-398 при применении в качестве монотерапии и в комбинации с 5-FU и лейковорином.
Ключевая цель настоящего исследования заключалась в изучении биомаркеров, связанных с токсичностью и эффективностью лечения с применением ММ-398 и ММ-398 в комбинации с 5-FU и лейковорином.
В. Схема исследования
Данное исследование представляет собой открытое рандомизированное исследование 3 фазы ММ-398 в комбинации с 5-FU и лейковорином или без них по сравнению с 5-фторурацилом (5-FU) и лейковорином (также известным как фолиновая кислота) в трех группах пациентов с метастазирующим раком поджелудочной железы, прогрессировавшим на фоне предшествующей терапии с применением гемцитабина.
В данном глобальном исследовании будут участвовать приблизительно 405 подходящих пациентов согласно протоколу версии 2 или более поздней версии. Все пациенты примут участие скрининге продолжительностью до 28 дней, в ходе которого будет выполнена оценка их пригодности и скрининг аллеля UGT1A1*28. Пригодные пациенты будут рандомизированы в соотношении 1:1:1 в одну из следующих групп, получающих лечение:
5-FU и лейковорин
Леволейковорин в дозе 200 мг/м2 или лейковорин (l+d-рацемическая смесь) в дозе 400 мг/м2 вводили в/в в течение 30 минут раз в неделю на протяжении 4 недель (в дни 1, 8, 15 и 22) с последующими 2 неделями отдыха в рамках 6-недельного цикла.
5-FU 2400 мг/м2 в/в в течение 46-часов, раз в 2 недели.
Леволейковорин в дозе 200 мг/м2 или l+d-рацемическая смесь в дозе 400 мг/м2 в/в в течение 30 минут раз в 2 недели.
ММ-398 следует вводить перед 5-FU и лейковорином; лейковорин всегда следует вводить перед 5-FU. Не следует вводить другие лекарственные средства в комбинации, если необходимо приостановить прием ММ-398 или 5-FU/лейковорина.
Пациенты будут равномерно рандомизированы в группы, получающие лечение, с помощью централизованной интерактивной системы с доступом через Интернет (IWRS). Рандомизацию стратифицируют на основании следующих прогностических факторов:
- исходного уровня альбумина (≥4,0 г/дл по сравнению с <4,0 г/дл);
- KPS (70 и 80 по сравнению с ≥90);
- этнической принадлежности (европеоиды по сравнению с выходцами из Восточной Азии по сравнению со всеми остальными).
Лекарственное средство будут вводить циклами. Пациентов будут лечить до появления признаков прогрессирования заболевания (радиологического или клинического ухудшения), непереносимой токсичности или других оснований для прекращения исследования. Реакцию опухоли будут оценивать с использованием руководящих принципов RECIST (Eisenhauer, E.A., et al., “New response evaluation criteria in solid tumors: Revised RECIST guideline (version 1.1). European Journal of Cancer, 2009. 45:pp. 228-247) каждые 6 недель или раньше при очевидном прогрессировании заболевания на основании клинических признаков и симптомов. Снимки для измерения опухоли будут собирать и хранить для всех пациентов на протяжении всего исследования. Вместе с тем, все решения о лечении будут основываться на оценке статуса заболевания местным рентгенологом или руководителем исследования. При необходимости независимого анализа ORR и/или PFS можно выполнить независимый обзор изображений.
Через 30 дней после прекращения лечения требуется посещение врача в рамках последующего наблюдения. Впоследствии все пациенты будут обследоваться раз в месяц на предмет оценки общей выживаемости (по телефону или посредством визита в исследовательский центр) до смерти или завершения исследования, в зависимости от того, какое из этих событий наступит первым. Пациентов, прекративших прием исследуемого средства по причинам, не связанным с объективным прогрессированием заболевания (в том числе пациентов, досрочно прекративших участие в исследовании вследствие ухудшения симптомов), следует продолжать обследовать каждые 6 недель в период последующего наблюдения на предмет радиологического прогрессирования.
Всем пациентам будет предложено заполнить дневник оценки боли и приема анальгетиков на протяжении всего периода их участия в исследовании, в котором пациент будет документировать оценку интенсивности боли и ежедневное потребление анальгетиков. Ответы пациента будут использоваться для оценки клинической благоприятной реакции наряду с другими параметрами. От всех пациентов также потребуется заполнить анкету EORTC-QLQ-C30 для оценки качества жизни.
Для решения задач этого исследования от всех центров будет требоваться участие в протоколе дополнительного трансляционного исследования (TR) (ММ-398-07-03-01.TR), если это не запрещено местным законодательством. Участие в этом исследовании будет носить факультативный характер для пациентов; они будут обязаны предоставить отдельное согласие для трансляционного исследования.
Первичный анализ общей выживаемости (OS) будет осуществляться после смерти по меньшей мере 305 пациентов, зачисленных в исследование в рамках протокола версии 2 или более поздней версии. Пациенты, получающие исследуемое лечение во время основного анализа общей выживаемости, будут продолжать получать лечение до выполнения одного из критериев прекращения. В ходе этого исследования независимый комитет по мониторингу данных (DSMB) будет проводить регулярный обзор данных о безопасности. На фигуре 7 показана схема исследования.
С. Отбор пациентов и досрочное прекращение участия
В данном глобальном исследовании будут участвовать приблизительно 405 пациентов согласно протоколу версии 2 или более поздней версии. Для включения в исследование пациенты должны иметь/быть:
1. Гистологически или цитологически подтвержденную экзокринную аденокарциному поджелудочной железы.
2. Документально зарегистрированную метастазирующую опухоль; статус заболевания может поддаваться или не поддаваться измерению в соответствии с руководящими принципами RECIST вер. 1.1.
3. Документально зарегистрированное прогрессирование заболевания на фоне предшествующего приема гемцитабина или гемцитабин-содержащего лекарственного средства при локально распространенном или метастазирующем раке. Примеры допустимых лекарственных средств включают, но не ограничиваются следующими:
- монотерапию с применением гемцитабина;
- любую схему лечения на основе гемцитабина, с поддерживающим лечением на основе гемцитабина или без него;
- монотерапию с применением гемцитабина, к которой впоследствии добавили агент на основе платины, фторпиримидин или эрлотиниб;
- гемцитабин, вводившийся в комбинации с адъювантом в случае рецидива заболевания в течение 6 месяцев после завершения адъювантной терапии.
4. Индекс общего состояния по Карнофски (KPS)>70.
5. Адекватные резервы костного мозга, подтверждаемые следующими параметрами:
- АЧН>1500 клеток/мкл без применения гемопоэтических факторов роста;
- количество тромбоцитов >100000 клеток/мкл; и
- гемоглобин >9 г/дл (для пациентов с уровнем гемоглобина ниже 9 г/дл допускается переливание крови).
6. Адекватную функцию печени, подтверждаемую следующими параметрами:
- общий билирубин в сыворотке в пределах нормального диапазона для организации (при обструкции желчных протоков допускается их дренаж);
- уровень альбумина ≥3,0 г/дл;
- уровень аспартатаминотрансферазы (АСТ) и аланинаминотрансферазы (АЛТ)≤2,5×ВПН (при наличии метастазов в печения приемлемым является уровень ≤ 5×ВПН)
7. Адекватную функцию почек, подтверждаемую уровнем креатинина в сыворотке ≤1,5×ВПН.
8. Нормальную ЭКГ или ЭКГ без клинически значимых отклонений.
9. Оправиться от последствий предшествующей хирургической операции, лучевой терапии или другой противораковой терапии.
10. Возраст по меньшей мере 18 лет.
11. Способен понимать и подписать информированное согласие (или иметь законного представителя, который в состоянии сделать это).
Пациенты должны соответствовать всем критериям включения, перечисленным выше, и не соответствовать ни одному из следующих критериев исключения:
1. Активные метастазы в ЦНС (проявляющиеся клиническими симптомами, отеком головного мозга, необходимостью приема стероидов или прогрессирующим заболеванием).
2. Клинически значимые нарушения желудочно-кишечного тракта, в том числе расстройства печени, кровотечение, воспаление, непроходимость или диарею >1 степени.
3. Любое другое злокачественное новообразование в анамнезе в последние 5 лет; субъекты с предшествующим анамнезом рака in situ или базального или плоскоклеточного рака кожи допускаются к участию. Субъекты с другими злокачественными новообразованиями допускаются к участию, если признаки заболевания совершенно не проявлялись у них в последние 5 лет.
4. Тяжелая артериальная тромбоэмболия (инфаркт миокарда, нестабильная стенокардия, инсульт) менее чем за 6 месяцев до включения.
5. Застойная сердечная недостаточность III или IV класса по критериям NYHA, желудочковые аритмии или неконтролируемое давление крови.
6. Активная инфекция или необъяснимая лихорадка >38,5°C во время скрининговых посещений или в первый день запланированного приема препарата (пациенты с опухолевой лихорадкой могут допускаться к зачислению на усмотрение исследователя), которая, по мнению исследователя, может поставить под угрозу участие пациента в исследовании или влияет на результат исследования
7. Известная гиперчувствительность к любому из компонентов ММ-398, другим липосомальным продуктам, фторпиримидинам или лейковорину.
8. Исследуемое лекарственное средство, принимаемое в течение 4 недель или менее чем 5-кратного времени полувыведения исследуемого агента до дня первого запланированного приема лекарства в данном исследовании, в зависимости от того, что из вышеперечисленного продолжается дольше.
9. Другие медицинские или социальные состояния, которые, по мнению исследователя, могут мешать способности пациента подписать информированное согласие, сотрудничать и участвовать в исследовании, или мешать интерпретации результатов.
10. Беременность или кормление грудью; у женщин, способных к деторождению, на момент включения в исследование должен быть отрицательный тест на беременность, основанный на анализе мочи или сыворотки крови. Мужчины и женщины, способные к деторождению, должны согласиться на использование надежного метода контроля рождаемости во время исследования и до 3 месяцев после приема последней дозы исследуемого препарата.
Критерии зачисления должны прямо соблюдаться. Прекращение приема исследуемого препарата предусмотрено для пациентов в следующих случаях:
- у пациента есть признаки прогрессирования заболевания на основании критериев RECIST v1.1;
- у пациента наблюдается ухудшение симптомов;
- пациент испытывает непереносимое токсическое действие или нежелательное явление, которое требует:
третьего снижения дозы;
приостановки лечения более чем на 21 день от начала следующего цикла, если, по мнению исследователя, пациент не получает благоприятного действия от исследуемого препарата;
- пациент, по оценке руководителя исследования, не придерживается процедур согласно исследованию;
- пациент или лечащий врач пациента просит пациента воздержаться от приема исследуемого препарата;
- исследователь или спонсор по какой-либо причине, принимая во внимание права, безопасность и благополучие пациентов и в соответствии с руководящими принципами ICH/GCP и местным законодательством, прекращает исследование или участие пациента в исследовании.
Если пациент потерян для последующего наблюдения или воздерживается от приема исследуемого препарата, следует попытаться связаться с пациентом для выяснения причины прекращения участия. Если пациент потерян для последующего наблюдения, следует предпринять по меньшей мере 3 документально подтвержденные попытки связи с пациентом, в том числе одну - заказным письмом, прежде чем считать пациента потерянным для последующего наблюдения. Если пациент прекращает прием исследуемого препарата по причинам, не связанным с объективным прогрессированием, следует продолжать рентгенологическую оценку заболевания у пациента каждые 6 недель до обнаружения объективного прогрессирования.
Все пациенты, прекратившие прием исследуемого препарата, должны оставаться на последующем наблюдении, как требует протокол. Единственным обстоятельством, при котором у пациента не следует отслеживать конечные показатели исследования, является отзыв согласия пациента. Отзыв согласия должен быть решением самого пациента и должен означать не только желание пациента прекратить прием исследуемого препарата и визиты в рамках последующего наблюдения, но и то, что исследователь не вправе прилагать дальнейшие усилия для контакта с пациентом, включая попытки определения его статуса выживания.
D. Метод распределения пациентов в группы лечения
После завершения всех обследований в рамках скрининга и получения результатов анализа UGT1A1*28 пациентов следует рандомизировать с использованием компьютеризированной интерактивной системы с доступом через Интернет (IWRS) в соотношении 1:1:1 в одну из следующих групп, получающих лечение:
- группа A (экспериментальная группа): ММ-398;
- группа B (контрольная группа): 5-FU и лейковорин;
- группа C (экспериментальная группа): ММ-398, 5-FU и лейковорин.
Рандомизация должна произойти в течение 7 дней до планируемого приема препаратов. Рандомизацию стратифицируют на основании следующих прогностических факторов:
- исходного уровня альбумина (≥ 4,0 г/дл по сравнению с <4,0 г/дл);
- KPS (70 и 80 по сравнению с ≥90);
- этнической принадлежности (европеоиды/восточные азиаты/все остальные).
Е. Описание MM-398
ММ-398 представляет собой иринотекан (также известный как CPT-11), инкапсулированный в систему липосомальной доставки лекарственных веществ. Он будет поставляться в стерильных, одноразовых флаконах, содержащих 9,5 мл ММ-398 в концентрации 5 мг/мл. Флаконы будут содяржать дополнительные 0,5 мл для облегчения извлечения указанного количества из каждого 10-мл флакона.
ММ-398 необходимо хранить в холодильнике при температуре 2-8°C с защитой от света. Во время инфузии защита от света не требуется. ММ-398 нельзя замораживать. Ответственные лица должны проверять содержимое флакона на наличие твердых частиц до и после отбора лекарственного препарата из флакона в шприц.
ММ-398 необходимо разбавлять перед введением. Разбавленный раствор физически и химически стабилен в течение 6 часов при комнатной температуре (15-30°C), но предпочтительно должен храниться при температуре холодильника (2-8°C) без доступа света. Разбавленный раствор нельзя замораживать. Из-за возможного микробного загрязнения во время разбавления желательно использовать разбавленный раствор в течение 24 часов при хранении в холодильнике (2-8°C) и в течение 6 часов при хранении при комнатной температуре (15-30°C).
Двадцать флаконов ММ-398 будут упакованы в картонный контейнер. Отдельные флаконы, а также наружная картонная тара, будут маркированы в соответствии с местными нормативными требованиями.
ММ-398 будут вводить следующим образом. Будет выполнен исходный скрининг всех пациентов на аллель UGT1A1*28.
- Пациенты, гомозиготные по UGT1A1*28, получат первый цикл лечения при пониженной дозе 80 мг/м2. Если пациент не испытывает токсического действия лекарственного вещества после первого приема ММ-398, то со 2 цикла и далее можно увеличивать их дозу с шагом 20 мг/м2 до максимального значения 120 мг/м2.
- Пациенты, гомозиготные по аллелю UGT1A1*28 и рандомизированные в группу C, получат первый цикл лечения при пониженной дозе 60 мг/м2. Если пациент не испытывает токсического действия лекарственного вещества после первого приема ММ-398, то со 2 цикла и далее можно увеличить дозу до 80 мг/м2.
- ММ-398 следует вводить перед введением 5-FU и лейковорина.
В группе A ММ-398 будут вводить путем в/в инфузии в течение 90 минут в первый день каждого 3-недельного цикла в исследовательском центре. В группе C ММ-398 будут вводить путем в/в инфузии в течение 90 минут во время первого цикла; время инфузии может быть сокращено до 60 минут, начиная со 2 цикла, если в ходе 1 цикла не возникало острой реакции на инфузию. Продолжительность цикла составляет 3 недели для группы A и 2 недели для группы C. Первый день 1 цикла - фиксированный день; последующие дозы следует вводить в первый день каждого цикла +/-3 дня.
Перед введением соответствующую дозу ММ-398 необходимо разбавить 5% раствором декстрозы для инъекции (D5W) до окончательного объема 500 мл. Следует соблюдать внимательность и не использовать встроенные фильтры или растворители, кроме D5W. ММ-398 можно вводить с использованием стандартных ПВХ-содержащих пакетов и трубок для внутривенного введения.
Фактическую вводимую дозу ММ-398 определят путем вычисления площади поверхности тела пациента в начале каждого цикла. Для удобства введения будет допускаться +/-5% разброс рассчитанной общей дозы. Поскольку флаконы с ММ-398 одноразовые, сотрудники центра не должны хранить неиспользованную часть флакона для будущего использования; неиспользованную часть продукта необходимо выбросить.
Перед инфузией ММ-398 все пациенты должны получить стандартные дозы дексаметазона и антагониста 5-HT3 или другого противорвотного средства в соответствии со стандартной ведомственной практикой введения иринотекана. Для пациентов, перенесших острые холинэргические симптомы в предыдущих циклах, можно назначить атропин в профилактических целях.
F. Описание 5-FU и лейковорина
5-фторурацил является пиримидиновым антагонистом, который влияет на биосинтез нуклеиновых кислот. Дезоксирибонуклеотид указанного лекарственного вещества ингибирует тимидилатсинтетазу, таким образом подавляя образование тимидиловой кислоты из дезоксиуридиловой кислоты и мешая синтезу ДНК. Он также препятствует синтезу РНК.
Лейковорин действует как биохимический кофактор реакций переноса 1-углерода при синтезе пуринов и пиримидинов. Лейковорин не требует фермента дигидрофолатредуктазы (ДГФР) для преобразования в тетрагидрофолиевую кислоту. Лейковорин ингибирует эффекты метотрексата и других антагонистов ДГФР. Лейковорин может усиливать цитотоксические эффекты фторированных пиримидинов (т.е. фторурацила и флоксуридина). После того, как 5-FU активируется в клетке, он сопровождается фолатным кофактором и ингибирует фермент тимидилатсинтетазу, таким образом подавляя синтез пиримидинов. Лейковорин увеличивает пул фолата, тем самым усиливая связывание фолатного кофактора и активного 5-FU с тимидилатсинтетазой.
С FU и лейковорином следует обращаться и хранить их в соответствии с вкладышем в упаковку, специфичным по отношению к стране. Доступные для приобретения 5-FU и лейковорин будут предоставляться всем пациентам в исследовании, рандомизированным в группы B и C.
5-FU и лейковорин будут вводить следующим образом.
- Лейковорин будут вводить в дозе 200 мг/м2 (l-форма) или 400 мг/м2 (l+d-рацемическая форма) в виде в/в инфузии в течение 30 минут раз в неделю в течение 4 недель (в дни 1, 8, 15 и 22) с последующими 2 неделями отдыха в рамках 6-недельного цикла
- Лейковорин будут вводить в дозе 200 мг/м2 (l-форма) или 400 мг/м2(l+d-рацемическая форма) в виде инфузии в течение 30 минут каждые 2 недели
Лейковорин следует растворить согласно инструкции на вкладыше в упаковку или стандартному ведомственному руководству по растворению лейковорина. Лейковорин следует вводить перед инфузией 5-FU.
Фактическую вводимую дозу 5-FU и лейковорина будут определять путем вычисления площади поверхности тела пациента до каждого цикла. Для удобства введения будет допускаться +/-5% разброс рассчитанной общей дозы.
После цикла 1 для начала каждого нового цикла будет допускаться "окно" продолжительностью +/-3 дня, а для инфузий в 8, 15 и 22 дни - "окно" продолжительностью +/-1 день.
Все пациенты до инфузии 5-FU и лейковорина должны получить стандартные дозы дексаметазона, прохлорперазин или другого эквивалентного противорвотного средства согласно стандартной ведомственной практике введения 5-FU.
G. Важные соображения для лечения с применением ММ-398
Данные предыдущих исследований ММ-398 не выявили неожиданных токсических эффектов по сравнению с подробно изученным активным ингредиентом - иринотеканом. Предупреждения и меры предосторожности при использовании иринотекана и процедуры лечения указанных токсических эффектов приведены ниже.
Диарея
Иринотекан может вызывать ранние и поздние формы диареи, которые, по-видимому, обусловлены различными механизмами. Ранняя диарея (возникающая во время или вскоре после инфузии иринотекана) холинергическая по природе. Она обычно носит временный характер и редко бывает тяжелой. Она может сопровождаться симптомами ринита, увеличением слюноотделения, миозом, слезотечением, потоотделением, гиперемией и усиленной перистальтикой кишечника, что может вызвать спастические боли в животе. Для пациентов, испытывавших ранние холинергические симптомы во время предыдущего цикла приема ММ-398, в профилактических целях по усмотрению исследователя будут вводить атропин.
Поздняя диарея (как правило, возникающая спустя более чем 24 часа после введения иринотекана) может быть опасной для жизни, так как она может быть продолжительной и привести к обезвоживанию, электролитному дисбалансу или сепсису. Позднюю диарею необходимо своевременно должны лечить с применением лоперамида, а если диарея сохраняется после лоперамида, следует рассмотреть возможность приема октреотида. Потеря жидкости и электролитов, связанная со стойкой или тяжелой диареей, может привести к опасному для жизни обезвоживанию, почечной недостаточности и электролитному дисбалансу и вносить вклад в заболевания сердечно-сосудистой системы. Повышается риск инфекционных осложнений, которые могут привести к сепсису у больных с нейтропенией, вызванной химиотерапией. Пациенты с диареей должны находиться под тщательным контролем, при начале обезвоживания необходима замена жидкости и электролитов, а при развитии кишечной непроходимости, лихорадки или тяжелой нейтропении - поддержка антибиотиками.
Нейтропения
У пациентов, получавших иринотекан, были зарегистрированы случаи смерти в результате сепсиса после тяжелой нейтропении. Нейтропенические осложнения следует своевременно лечить с применением поддержки антибиотиками. Для лечения нейтропении по усмотрению исследователя можно применять Г-КСФ. Пациенты, испытавшие нейтропению 3 или 4 степени при предшествующей противоопухолевой терапии, нуждаются в тщательном мониторинге и лечении.
Гиперчувствительность
Наблюдались реакции гиперчувствительности, в том числе тяжелые анафилактические или анафилактоидные реакции. При возникновении гиперчувствительности следует немедленно приостановить прием подозрительного лекарственного средства и начать инвазивную терапию.
Колит/кишечная непроходимость
Наблюдались случаи колита, осложненного изъязвлением, кровотечением, кишечной непроходимостью и инфекцией. Пациенты, испытывающие кишечную непроходимость, должны получать своевременную поддержку антибиотиками.
Тромбоэмболия
Тромбоэмболические явления отмечались у пациентов со схемой лечения, включавшей прием иринотекана; конкретные причины этих явлений не определялись.
Беременность
Иринотекан относится к категории D безопасности применения при беременности. Женщинам, способным к деторождению, следует рекомендовать избегать беременности во время лечения с применением иринотекана. При сообщении о беременности лечение следует прекратить. Пациентку следует вывести из исследования и отслеживать беременность до тех пор, пока не станет известен ее результат.
Уход за областью внутривенного введения
Следует избегать кровоизлияний; необходимо контролироваться наличие признаков воспаления в области инфузии. При возникновении кровоизлияния рекомендуется промыть область стерильным физиологическим раствором и приложить лед.
Пациенты из группы особого риска
В клинических исследованиях с недельным графиком приема иринотекана было отмечено, что пациенты с умеренно повышенным исходным уровнем общего билирубина в сыворотке (1,0-2,0 мг/дл) характеризовались значительно большей вероятностью возникновения нейтропении 3 или 4 степени во время первого цикла, чем пациенты с уровнем билирубина менее 1,0 мг/дл (50,0% [19/38] по сравнению с 17,7% [47/226]; p<0,001). Пациенты с аномалиями глюкуронидации билирубина, например, с синдромом Жильбера, также могут подвергаться повышенному риску миелосупрессии при лечении с применением иринотекана.
Острые реакции, ассоциированные с инфузией
Острые реакции, ассоциированные с инфузией и характеризующиеся гиперемией, одышкой, отеком лица, головной болью, ознобом, болью в спине, чувством стеснения в груди или глотке и гипотензией, отмечались у небольшого количества пациентов, получавших липосомальные лекарственные средства. У большинства пациентов указанные реакции проходили в течение 24 часов после прекращения инфузии. У некоторых пациентов эти реакции проходили при снижении скорости инфузии. Большинство пациентов, испытывавших реакции, ассоциированные с острым инфузией липосомальных лекарственных средств, могли переносить дальнейшие инфузии без осложнений.
Другие возможные токсические эффекты
MM-398, новый липосомальный состав иринотекана, отличается от иринотекана в некапсулированном составе, и поэтому существует возможность токсических эффектов, не вызываемых свободным иринотеканом. Необходимо осуществлять тщательный мониторинг признаков и симптомов токсического действия лекарственного вещества у всех пациентов, особенно в начале лечения.
Н. Требования к изменению дозы
Прием можно приостановить на срок до 3 недель от запланированной даты для восстановления пациента после токсических эффектов, связанных с исследуемым препаратом. Если на восстановление после токсического эффекта требуется более 3 недель, следует прекратить участие пациента в исследовании, при условии, что пациент не получает благоприятного действия от исследуемого препарата; в противном случае исследователь и спонсор или его представитель должны обсудить возможность продолжения участия пациента с учетом риска и выгоды от продолжения.
При снижении дозы для пациента во время исследования вследствие токсичности она должна оставаться пониженной в течение всего исследования; повторное увеличение дозы до предшествующего уровня не допускается. Если для пациента требуется снижение дозы в третий раз в связи с продолжением нежелательного явления после двух снижений дозы, следует прекратить участие пациента в исследовании.
Следует контролировать реакции на инфузию. Реакции на инфузию будут оценивать согласно определению национального института рака CTCAE (версия 4.0) для аллергических реакций/реакций на инфузию и анафилаксии, приведенному ниже:
При лечении реакций на инфузию следует использовать правила, принятые в исследовательском центре, или следующие руководящие принципы.
- Снизить скорость инфузии на 50%
- Отслеживать состояние пациента каждые 15 минут на предмет ухудшения
- Остановить инфузию
- Ввести 50 мг дифенгидрамина гидрохлорида в/в, 650 мг ацетаминофена п/о, дать кислород
- Возобновить инфузию при 50% скорости после купирования реакции на инфузию
- Отслеживать состояние пациента каждые 15 минут на предмет ухудшения
- Перед всеми последующими инфузиими вводить 25-50 мг дифенгидрамина гидрохлорида в/в
- Остановить инфузию и отсоединить трубки для инфузии от пациента
- Ввести 50 мг дифенгидрамина гидрохлорида в/в, 10 мг дексаметазона в/в, бронходилататоры при бронхоспазме и другие лекарства или кислород по необходимости
- Дальнейшее лечение с применением ММ-398 не допускается
- Остановить инфузию и отсоединить трубки для инфузии от пациента
- Ввести эпинефрин, бронходилататоры или кислород, как показано при бронхоспазме
- Ввести 50 мг дифенгидрамина гидрохлорида в/в, 10 мг дексаметазона в/в
- Рассмотреть возможность госпитализации для наблюдения
- Дальнейшее лечение с применением ММ-398 не допускается
Для пациентов, которые испытывают реакции на инфузию 1 или 2 степени, последующие инфузии по усмотрению исследователя можно осуществлять при сниженной скорости (в течение 120 минут).
Для пациентов, которые испытывают реакции на инфузию 1 или 2 степени во второй раз, следует ввести 10 мг дексаметазона в/в. Перед всеми последующими инфузиями следует вводить 50 мг дифенгидрамина гидрохлорида в/в, 10 мг дексаметазона в/в и 650 мг ацетаминофена п/о.
I. Изменение дозы ММ-398 при гематологической токсичности
Перед началом нового цикла терапии у пациентов должны быть следующие показатели:
- АЧН>1500/мм3;
- количество тромбоцитов >100000/мм3.
Следует отложить лечение, чтобы дать достаточно времени для восстановления и после восстановления; лечение следует осуществлять в соответствии с руководящими принципами, приведенными ниже в таблицах. При лихорадочной нейтропении АЧН следует восстановить до ≥1500/мм3, а пациент должен оправиться от инфекции.
Изменение дозы ММ-398 при различном количестве нейтрофилов
Группа С: Пациенты, не гомозиготные по UGT1A1*28
(3/4 степень) или лихорадочная нейтропения
a Все изменения дозы должны основываться на наихудшей предшествующей токсичности.
b Пациентов, для которых требуется дальнейшее снижение дозы менее 80 мг/м2, необходимо исключить из исследования.
c Пациентов, для которых требуется дальнейшее снижение дозы менее 50 мг/м2, необходимо исключить из исследования.
d Для пациентов, гомозиготных по UGT1A1*28, у которых повышали дозу, следует снизить дозу в соответствии с руководством для пациентов, не гомозиготных по UGT1A1*28.
е Пациентов, для которых требуется дальнейшее снижение дозы менее 40 мг/м2, необходимо исключить из исследования.
Изменение дозы ММ-398 при других видах гематологической токсичности
Группа С: Пациенты, не гомозиготные по UGT1A1*28
a Все изменения дозы должны основываться на наихудшей предшествующей токсичности.
b Пациентов, для которых требуется дальнейшее снижение дозы менее 80 мг/м2, необходимо исключить из исследования.
с Пациентов, для которых требуется дальнейшее снижение дозы менее 50 мг/м2, необходимо исключить из исследования.
d Для пациентов, гомозиготных по UGT1A1*28, у которых повышали дозу, следует снизить дозу в соответствии с руководством для пациентов, не гомозиготных по UGT1A1*28.
е Пациентов, для которых требуется дальнейшее снижение дозы менее 40 мг/м2, необходимо исключить из исследования.
J. Изменение дозы ММ-398 при негематологической токсичности
Следует отложить лечение до коррекции диареи до ≤1 степени, а для других негематологических токсических эффектов 3 или 4 степени - до их коррекции до 1 степени или исходного уровня. Руководство по коррекции дозы MM-398 при медикаментозной диарее и других негематологических токсических эффектах 3 или 4 степени представлено ниже. При реакциях на инфузию следует действовать, как описано выше.
Изменение дозы ММ-398 при диарее
Группа С: Пациенты, не гомозиготные по UGT1A1*28
a Все изменения дозы должны основываться на наихудшей предшествующей токсичности.
b Пациентов, для которых требуется дальнейшее снижение дозы менее 80 мг/м2, необходимо исключить из исследования.
c Пациентов, для которых требуется дальнейшее снижение дозы менее 50 мг/м2, необходимо исключить из исследования.
d Для пациентов, гомозиготных по UGT1A1*28, у которых повышали дозу, следует снизить дозу в соответствии с руководством для пациентов, не гомозиготных по UGT1A1*28.
е Пациентов, для которых требуется дальнейшее снижение дозы менее 40 мг/м2, необходимо исключить из исследования.
Изменение дозы ММ-398 при негематологической токсичности, не связанной с диареей, астенией и анорексией 3 степениd
Группа С: Пациенты, не гомозиготные по UGT1A1*28
a Все изменения дозы должны основываться на наихудшей предшествующей токсичности.
b Пациентов, для которых требуется дальнейшее снижение дозы менее 80 мг/м2, необходимо исключить из исследования.
c Пациентов, для которых требуется дальнейшее снижение дозы менее 50 мг/м2, необходимо исключить из исследования.
d При астении и анорексии 3 степени изменение дозы не требуется.
е Для пациентов, гомозиготных по UGT1A1*28, у которых повышали дозу, следует снизить дозу в соответствии с руководством для пациентов, не гомозиготных по UGT1A1*28.
f Пациентов, для которых требуется дальнейшее снижение дозы менее 40 мг/м2, необходимо исключить из исследования.
K. Изменение дозы 5-FU и лейковорина (группы B и C)
Ниже приводятся рекомендации по изменению дозы 5-FU. Корректировка дозы лейковорина из-за токсичности не требуется. Лейковорин необходимо вводить непосредственно перед каждой дозы 5-FU; следовательно, при отсрочке введения дозы 5-FU введение дозы лейковорина также нужно отложить. В случае, если пациент испытывает реакцию на инфузию, следует использовать ведомственное руководство или руководство для лечения реакций на инфузию ММ-398.
L. Изменение дозы 5-FU при гематологической токсичности
Перед введением следующей дозы в цикле или началом нового цикла лечения у пациентов должны быть следующие показатели:
- АЧН≥1500/мм3
- лейкоциты ≥3500/мм3;
- количество тромбоцитов ≥75000/мм3 (согласно Европейской сводке характеристик продукта для 5-FU, до начала лечения тромбоциты должны восстановиться до уровня ≥100000/мм3).
Следует отложить лечение, чтобы дать достаточно времени для восстановления и после восстановления; лечение следует осуществлять в соответствии с руководящими принципами, приведенными ниже в таблице. Продолжительность циклов фиксирована и составляет 6 недель, и если пациент не может получить дозу D8, D15 или D22 из-за токсичности, эта доза будет считаться пропущенной.
Изменение дозы 5-FU при гематологической токсичности
(группа В и С)
(клеток/мм3)
a Все изменения дозы должны основываться на наихудшей предшествующей токсичности.
b Пациентов, которым требуется более 2 снижений дозы, необходимо исключить из исследования.
M. Изменение дозы 5-FU при негематологической токсичности
Лечение необходимо отложить до коррекции всех негематологических токсических эффектов 3 или 4 степени до 1 степени или исходного уровня. Руководящие принципы коррекции дозы 5-FU из-за токсичности представлены ниже. Продолжительность циклов фиксирована и составляет 6 недель, и если пациент не может получить дозу D8, D15 или D22 из-за токсичности, эта доза будет считаться пропущенной.
Изменение дозы 5-FU при негематологической токсичности, не связанной с астенией и анорексией 3-й степени с (группа В и С)
a Все изменения дозы должны основываться на наихудшей предшествующей токсичности.
b Пациентов, которым требуется более 2 снижений дозы, необходимо исключить из исследования.
с При астении и анорексии 3 степени изменение дозы не требуется.
N. Другие токсические эффекты, требующие особого внимания
Для групп, получавших 5-FU и MM-398, следует лечить удлинение QTc, происходящее на фоне электролитного дисбаланса, вызванного диареей, с применением насыщения соответствующим электролитом. После коррекции основной аномалии и аномалий ЭКГ можно возобновить лечение при условии тщательного мониторинга и соответствующего изменения дозы при диарее, как описано выше.
O. Сопутствующая терапия
Все параллельные медицинские состояния и осложнения основного злокачественного заболевания следует лечить по усмотрению исследователя в соответствии с приемлемыми местными стандартами медицинской помощи. Пациенты по мере необходимости должны получать анальгетики, противорвотные средства, антибиотики, жаропонижающие средства и препараты крови. Хотя терапия с применением антикоагулянтов типа варфарина допускается, необходим тщательный мониторинг параметров свертывания во избежание осложнений возможных межлекарственных взаимодействий. Все сопутствующие медицинские препараты, в том числе переливание препаратов крови, следует регистрировать в соответствующей индивидуальной регистрационной карте.
Руководящие принципы лечения некоторых заболеваний обсуждаются ниже; вместе с тем, также можно использовать ведомственные принципы лечения этих состояний. Сопутствующие лекарственные средства, требующие особого внимания, обсуждаются ниже.
Противорвотные средства
Дексаметазон и блокатор 5-НТ3 (например, ондансетрон или гранисетрон) можно вводить всем больным в рамках медикаментозной подготовки при отсутствии индивидуальных противопоказаний. Противорвотные средства также будут выписываться по клиническим показаниям в течение периода исследования.
Колониестимулирующие факторы
Применение гранулоцитарного колониестимулирующего фактора (Г-КСФ) допускается для лечения пациентов с нейтропенией или лихорадочной нейтропенией; профилактическое применение Г-КСФ допускается только для пациентов, у которых был по меньшей мере один эпизод нейтропении 3 или 4 степени или лихорадочной нейтропении при получении исследуемого препарата или документально подтвержденная нейтропения 3 или 4 степени или лихорадочная нейтропения при получении противоопухолевых лекарственных средств в прошлом.
Лечение диареи
Острая диарея и спастическая боль в животе, развивающаяся во время или в течение 24 часов после введения ММ-398, могут являться частью холинергического синдрома. Этот синдром следует лечить с применением атропина. Для пациентов, испытывающих холинергические симптомы во время исследования, следует рассмотреть необходимость профилактического или терапевтического введения атропина
Диарея может быть изнурительной и в редких случаях - потенциально опасной для жизни. Руководящие принципы, разработанные группой ASCO для лечения диареи, вызванной химиотерапией, кратко изложены ниже.
Рекомендации по лечению диареи, вызванной химиотерапией
При контроле диареи, вызванной химиотерапией на основе фторпиримидинов, показана эффективность синтетического октапептида октреотида при введении в повышающихся дозах путем непрерывного инфузии или подкожной инъекции. Октреотид можно вводить в дозах от 100 мкг дважды в день до 500 мкг три раза в день при максимально переносимой дозе 2000 мкг три раза в день в ходе 5-дневного курса. Пациентам следует рекомендовать обильное питье на протяжении всего лечения.
Другие средства лечения
Симптоматическое лечение других токсических эффектов следует проводить согласно ведомственным руководствам. Допускается профилактика алопеции с помощью охлаждающей шапочки или стоматита с помощью ледяных полосканий рта.
P. Запрещенные терапевтические средства
В инструкции по применению иринотекана указано, что с иринотеканом взаимодействуют следующие лекарственные средства: зверобой, CYP3A4-индуцирующие противосудорожные препараты (фенитоин, фенобарбитал, карбамазепин), кетоконазол, итраконазол, тролеандомицин, эритромицин, дилтиазем и верапамил. Следует по возможности избегать лечения с применением этих и других агентов, которые взаимодействуют с иринотеканом. Поскольку 5-FU взаимодействует с варфарином, следует проявлять осторожность при необходимости их одновременного применения. Информацию о других межлекарственных взаимодействиях см. во вкладышах в упаковку 5-FU и лейковорина для конкретной страны.
В ходе исследования не допускается применение следующих терапевтических средств:
- других противоопухолевых терапевтических средств, в том числе цитотоксических, агентов адресного действия, гормональных терапевтических средств или других антител;
- потенциально лечебной лучевой терапии; допускается применение паллиативной лучевой терапии; и
- применение любых других исследуемых препаратов не допускаются.
Q. Лабораторные процедуры
Общий анализ крови
Общий анализ крови (ОАК) будет выполняться на местной основе и должен включать оценку количества лейкоцитов (лейкоциты), определение лейкоцитарной формулы, гемоглобина, гематокрита и количества тромбоцитов.
Биохимический анализ сыворотки
Панель биохимического анализа сыворотки будет осуществляться централизованно. Кроме того, биохимический анализ можно выполнять на местной основе и использовать результаты, полученные в местной лаборатории, для зачисления и принятия решений о лечении при недоступности результатов из центральной лаборатории. Если результаты местной лаборатории используются для зачисления, то результаты местной лаборатории следует использовать при принятии всех последующих решений о лечении. Биохимический анализ сыворотки должен включать определение электролитов (натрия, калия, хлорида и бикарбоната), АМК, креатинина в сыворотке крови, глюкозы, прямого и общего билирубина, АСТ, АЛТ, щелочной фосфатазы, ЛДГ, мочевой кислоты, общего белка, альбумина, кальция, магния и фосфата.
CA 19-9
Уровни CA 19-9 будут измерять централизованно для всех пациентов.
Тест на беременность
Все женщины, способные к деторождения, должны пройти тест на беременность с использованием сыворотки или мочи.
Аллель UGT1A1*28
Образцы цельной крови будут собраны у всех пациентов в начале исследования и отправлены в центральную лабораторию для проверки статуса аллеля UGT1A1*28. Результаты местной лаборатории можно использовать при недоступности результатов центральной лаборатории на момент рандомизации.
Фармакокинетические оценки
ФК-анализ будет осуществляться централизованно. Образцы плазмы для оценки ФК будут собраны в ходе первого цикла у всех пациентов, рандомизированных в данном исследовании, в следующие моменты времени:
- группа А: непосредственно перед инфузией, во время инфузии (от 80 до 90 минут после начала инфузии), между 2 с половиной и четырьмя часами после начала инфузии и в C1D8;
- группа В: один образец в конце инфузии 5-FU (C1D2);
- группа C: непосредственно перед инфузией ММ-398, во время инфузии ММ-398 (от 80 до 90 минут после начала инфузии), между 2 с половиной и четырьмя часами после начала инфузии ММ-398, в конце инфузии 5-FU и в C1D8.
Кроме того, образец для оценки ФК будет отобран в ходе первого цикла, в любое время между 8 и 72 ч после введения ММ-398 у пациентов, рандомизированных в группы А и C, предоставивших дополнительное согласие на отбор данного образца.
R. Оценка боли и потребления анальгетиков
Пациентам будут предоставлены дневники оценки боли и потребления анальгетиков с целью ежедневной записи интенсивности боли по визуальной аналоговой шкале и документирования ежедневного потребления анальгетиков.
S. EORTC-QLQ-C30
Качество жизни будет оцениваться с помощью инструмента EORTC-QLQ-C30. EORTC-QLQ-C30 является надежным и объективным показателем качества жизни онкологических пациентов при межнациональных клинических исследованиях. Он включает девять многоэлементных шкал: пять функциональных шкал (физическую, ролевую, познавательную, эмоциональную и социальную); три шкалы симптомов (усталости, боли и тошноты и рвоты); и шкалу общего состояния здоровья и качества жизни. Кроме того, в него входят несколько одноэлементных показателей симптомов.
От пациентов требуется заполнить анкету EORTC-QLQ-C30 в моменты времени, указанные в графике оценки. В дни, когда пациент должен получить исследуемый препарат, оценки следует выполнить до начала введения исследуемого препарата. Анкету должны заполнять только те пациенты, для которых доступны проверенные переводы анкеты EORTC-QLQ-C30.
T. Общая выживаемость/последующее наблюдение после исследования
Данные по общей выживаемости будут собирать после выполнения пациентом контрольного посещения на 30 день, каждый месяц (+/-1 неделя) после даты контрольного посещения на 30 день. Данные, собираемые после прекращения, должны включать: дату прогрессирования заболевания (если она еще не документирована; если пациент досрочно прекратил прием исследуемого препарата по причинам, не связанным с объективным прогрессированием заболевания, пациент должен продолжать подвергаться оценке опухоли каждые 6 недель до начала новой противоопухолевой терапии или прогрессирования заболевания); документацию о противораковом лечении, получаемом пациентом, в том числе даты системной терапии, лучевой терапии или хирургического вмешательства после прекращения участия в исследовании; и дату смерти. Все пациенты должны состоять на учете вплоть до смерти или закрытия исследования, в зависимости от того, какое из этих событий наступит раньше.
U. Определение тяжести и связи нежелательных явлений
Категорию каждого нежелательного явления будут определять в соответствии с NCI CTCAE вер. 4.0, находящемся на http://ctep.cАЧНer.gov/reporting/ctc.html. Явления, не перечисленные в CTCAE, будут обозначаться как легкие, умеренные, тяжелые, опасные для жизни или фатальные, что соответствует категориям NCI CTCAE 1, 2, 3, 4 и 5, соответственно, и следующим определениям:
- легкое: явление, не приводящее к нетрудоспособности или инвалидности, и устраняющееся без вмешательства;
- умеренное: явление, не приводящее к нетрудоспособности или инвалидности, но требующее вмешательства;
- тяжелое: явление, приводящее к временной нетрудоспособности или инвалидности и требующее вмешательства;
- опасное для жизни: явление, при котором пациент подвергается риску смерти во время явления
- фатальное: явление, которое приводит к смерти пациента
Исследователь должен попытаться определить наличие обоснованной вероятности того, что нежелательное явление связано с применением исследуемого препарата. Эту взаимосвязь следует описывать как "связано" или "не связано".
V. Анализ общей выживаемости
Общая выживаемость (OS) представляет собой основной конечный показатель исследования. Общая выживаемость определяется как время с даты рандомизации пациентов до даты смерти или даты последней информации о том, что субъект жив. Для каждого пациента, в отношении которого неизвестно, скончался ли он на момент завершения сбора данных, общая выживаемость будет пересматриваться с учетом анализа на момент последнего контакта до завершения сбора данных.
Первичный анализ исследования будет включать два попарных сравнения выживаемости при приеме исследуемых препаратов в популяции ITT с использованием нестратифицированного логарифмического рангового критерия. Тестирование будет проводиться в соответствии с процедурой Бонферрони-Холма, которая жестко контролирует частоту ошибок с поправкой на эффект множественных сравнений на уровне 0,05 (двустороннем) [25]:
Отклонить 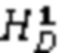
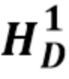 :SA(t)=SB(t), т.е. монотерапия с применением ММ-398 не оказывает эффекта по сравнению с контролем, если логарифмическое ранговое значение р для этого теста составляет не менее 0,025, или если логарифмическое ранговое значение р для этого теста менее 0,05 и логарифмическое ранговое значение р для сравнения групп B и С менее 0,025.
:SA(t)=SB(t), т.е. монотерапия с применением ММ-398 не оказывает эффекта по сравнению с контролем, если логарифмическое ранговое значение р для этого теста составляет не менее 0,025, или если логарифмическое ранговое значение р для этого теста менее 0,05 и логарифмическое ранговое значение р для сравнения групп B и С менее 0,025.
Отклонить 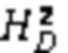
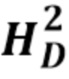 :SC(t)=SB(t), т.е. комбинированная терапия с применением ММ-398 не оказывает действия по сравнению с контролем, если логарифмическое ранговое значение р для этого теста составляет не менее 0,025, или если логарифмическое ранговое значение р для этого теста менее 0,05 и логарифмическое ранговое значение р для сравнения групп А и В менее 0,025.
:SC(t)=SB(t), т.е. комбинированная терапия с применением ММ-398 не оказывает действия по сравнению с контролем, если логарифмическое ранговое значение р для этого теста составляет не менее 0,025, или если логарифмическое ранговое значение р для этого теста менее 0,05 и логарифмическое ранговое значение р для сравнения групп А и В менее 0,025.
Для каждой группы, получающей лечение, будет выполнен анализ Каплана-Мейера с целью получения непараметрических оценок функции выживаемости и медианного времени выживаемости. Будут рассчитаны соответствующие 95% доверительные интервалы с использованием двойного логарифмического способа. Моделирование пропорциональных рисков Кокса будет использоваться для оценки соотношения рисков и соответствующих 95% доверительных интервалов.
Для общей выживаемости в популяции ITT (за исключением указанного) будут выполнены следующие дополнительные анализы чувствительности с целью оценки надежности результатов первичного анализа:
логарифмическое ранговое сравнение лечения в популяции PP;
стратифицированные логарифмические ранговые анализы с использованием факторов стратификации рандомизации [с параметрами соотношения угроз, рассчитанными при стратифицированном моделировании Кокса];
сравнение лечения по Уилкоксону;
модель регрессии Кокса с поэтапным отбором (вводимое значение p<0,25, остаточное значение p<0,15) параметров модели, где лечение и прогностические факторы (отмеченные ниже) являются кандидатами для включения;
одномерный анализ с целью оценки потенциальных независимых прогностических факторов с помощью регрессии Кокса;
анализ подгрупп для проверки различий в эффектах лечения в различных сегментах исследуемой популяции.
Повторить все анализы (первичные и анализы чувствительности) с участием только пациентов, зачисленных согласно протоколу версии 2 (и более поздней)
Проверяемые прогностические факторы включают: исходный KPS, исходный альбумин, этническую принадлежность, географическое положение, стадию заболевания на момент постановки диагноза, исходное местоположение опухоли, количество предшествующих курсов химиотерапии, предшествующую лучевую терапию, предшествующие хирургические операции, время с момента последнего лечения, лучшую реакцию на предшествующее лечение, исходный CA 19-9, пол и возраст.
W. Дополнительные анализы эффективности
Выживаемость без прогрессирования
PFS определяют как количество месяцев с даты рандомизации до даты смерти или прогрессирования, в зависимости от того, какое из этих событий наступит ранее (согласно RECIST 1.1). Если во время исследования не наблюдается ни смерти, ни прогрессирования, данные по PFS будут пересмотрены на момент последней достоверной оценки опухоли.
Сравнение PFS между группами, получающими лечение, будет выполнено с помощью парного нестратифицированного логарифмического рангового критерия. Кривые PFS будут оценивать с использованием оценок по Каплану-Мейеру. С помощью модели пропорциональных рисков Кокса будут получены оценки соотношения угроз и соответствующие 95% доверительные интервалы. Кроме того, будет проведен стратифицированный анализ с использованием факторов стратификации рандомизации. Будут изучены эффекты лечения с поправками на переменные стратификации и другие прогностические независимые переменные. Кроме того, для выполнения анализов чувствительности PFS можно использовать различные способы отбора и подстановки пропущенных данных. Методология анализа чувствительности будет полностью описана в плане статистического анализа.
Анализы будут выполнены для популяций ITT, РР и EP.
Время до констатации отсутствия эффекта лечения
Время до констатации отсутствия эффекта лечения определяется как время от рандомизации до прогрессирования заболевания, смерти или досрочного прекращения участия в исследовании ввиду токсичности. Для времени до констатации отсутствия эффекта лечения будут выполнены анализы Каплана-Мейера, как указано для анализа выживаемости без прогрессирования.
Анализы будут выполнены для популяций ITT, РР и EP.
Частота объективных реакций
Оценки опухоли, связанные с ORR, будут определять с помощью RECIST v1.1. Если спонсор требует независимого обзора радиологических оценок для поддержки нового применения лекарственного средства или по любой другой причине, статус реакции всех пациентов может рассматриваться независимой группой клиницистов и спонсором или его представителем. В случае расхождения между оценкой независимой группы и оценкой исследователя оценка независимой группы будет иметь приоритет.
Частоту объективных реакций (ORR) для каждой группы, получающей лечение, будут рассчитывать, складывая количество пациентов с наилучшей общей реакцией подтвержденных CR или PR согласно RECIST. ORR представляет собой наилучшую реакцию, зарегистрированную от рандомизации до прогрессирования или до окончания исследования. Будет представлено количество и процент пациентов, испытывающих объективную реакцию (подтвержденные CR+PR) во время анализа и рассчитан 95% доверительный интервал для доли. Сравнение частоты объективных реакций в группах, получающих лечение, будет выполнено с помощью попарного точного критерия Фишера. Анализы будут выполнены для популяций ITT, РР и EP.
Анализ реакции маркера опухоли
Уровни CA 19-9 в сыворотке будут оценивать в течение 7 дней до начала лечения (исходный уровень) и далее через каждые 6 недель. Реакцию маркера опухоли CA19-9 будут оценивать по изменению уровня CA19-9 в сыворотке. Реакция определяется как по меньшей мере однократное снижение CA 19-9 на 50% по сравнению с исходным уровнем за период лечения. Расчет частоты реакции маркера опухоли будут выполнять только для пациентов с повышенным исходным уровнем CA 19-9 (>30 ед/мл).
Анализ результатов согласно сообщениям пациентов
Анализ анкет EORTC-QLQ-C30 будут выполнять в соответствии с руководящими принципами EORTC [22].
Анализ безопасности
Нежелательные явления, возникающие при лечении, будут представлены в зависимости от группы, получающей лечение, пациента, категории NCI CTCAE и класса системы органов (SOC) по MedDRA. Для общих нежелательных явлений, серьезных нежелательных явлений, нежелательных явлений, связанных с исследуемыми препаратами, и нежелательных явлений 3 и 4 степени будут представлены отдельные списки. Лабораторные данные будут представлены в зависимости от группы, получающей лечение, и визита. Аномальные лабораторные значения по возможности будут оценивать в соответствии с категорией NCI CTCAE. Оценка QTc будет выполнена на основе способа коррекции по формуле Фридеричиа. К QTcF будут применять критерии CTCAE (т.е. 3 категория = QTc>500 мс). Все анализы безопасности при необходимости будут выполнять в зависимости от группы, получающей лечение, цикла лечения и недели. Общую безопасность также будут оценивать в зависимости от категории на протяжении нескольких циклов, SOC и степени воздействия. Кроме того, анализ безопасности будет включать сравнение между группами, получающими лечение, для всех пациентов в популяции для оценки безопасности:
- требуемое количество переливаний крови;
- доля пациентов, для которых требуется Г-КСФ;
- нежелательные явления, приведшие к задержке или изменению дозы.
Анализ фармакокинетики
Фармакокинетические данные будут собраны для всех пациентов, рандомизированных в любую из групп, получающих ММ-398. Данные о концентрации в плазме в зависимости от времени будут анализировать с помощью популяционных фармакокинетических способов. Фармакокинетические параметры будут оценивать путем нелинейного моделирования со смешанными эффектами с использованием NONMEM® версии 7, уровня 1.0 (ICON Development Solutions, Дублин, Ирландия). ФК параметры будут включать Cмакс, Тмакс, ППК (площадь под кривой концентрации) в плазме, клиренс, объем распределения и терминальный период полувыведения. Будет оцениваться влияние пациент-специфических факторов (возраста, расы, пола, массы тела, показателей функции печени и почек, значение ECOG и т.д.) на фармакокинетические параметры. Для оценки взаимосвязи между воздействием лекарственных средств и параметрами эффективности и/или токсичности (например, нейтропенией, диареей) будут использоваться популяционные ФК/ФД-методики. Для прояснения вопросов безопасности, эффективности или ФК, связанных с ММ-398, которые возникают в ходе исследования, можно выполнить дополнительный эксплораторный анализ ФК-образцов. Уровни концентрации 5-FU будут обобщаться описательно.
Примечания
Хотя настоящее изобретение описано в связи с конкретными вариантами его реализации, следует понимать, что возможны их дальнейшие изменения, и подразумевается, что настоящая заявка охватывает любые изменения, варианты применения или модификации настоящего изобретения, в целом соответствующие сущности изобретения и включая такие отступления от настоящего описания, которые возникают в пределах известной или общепринятой практической деятельности в рамках области техники, к которой относится изобретение, и могут применяться к основным функциям, изложенным в настоящем документе. Описание всевозможных патентов США, международных или иных патентов или патентных заявок или публикаций, упомянутых в настоящем документе, включено в настоящий документ посредством ссылки в полном объеме.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ЛЕЧЕНИЯ РАКА ПОДЖЕЛУДОЧНОЙ ЖЕЛЕЗЫ С ПРИМЕНЕНИЕМ КОМБИНИРОВАННОЙ ТЕРАПИИ, ВКЛЮЧАЮЩЕЙ ЛИПОСОМНЫЙ ИРИНОТЕКАН | 2013 |
|
RU2663450C2 |
| ЛЕЧЕНИЕ РАКА ЖЕЛУДКА С ПРИМЕНЕНИЕМ КОМБИНАЦИОННЫХ ВИДОВ ТЕРАПИИ, СОДЕРЖАЩИХ ЛИПОСОМАЛЬНЫЙ ИРИНОТЕКАН, ОКСАЛИПЛАТИН, 5-ФТОРУРАЦИЛ (И ЛЕЙКОВОРИН) | 2017 |
|
RU2761953C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ С ПРИМЕНЕНИЕМ ЛИПОСОМАЛЬНОГО ИРИНОТЕКАНА И ИНГИБИТОРА PARP ДЛЯ ЛЕЧЕНИЯ РАКА | 2016 |
|
RU2760185C2 |
| ОПТИМИЗАЦИЯ ИНДИВИДУАЛЬНЫХ ДОЗ 5-ФТОРУРАЦИЛА ПРИ РЕЖИМЕ FOLFIRI | 2008 |
|
RU2496499C2 |
| ВВЕДЕНИЕ МНОЖЕСТВЕННЫХ БОЛЮСОВ [6R]-MTHF В ХИМИОТЕРАПИИ НА ОСНОВЕ 5-ФТОРУРАЦИЛА | 2018 |
|
RU2762596C2 |
| ЛЕЧЕНИЕ РАКА С ПРИМЕНЕНИЕМ ВИРУСОВ, ФТОРПИРИМИДИНОВ И КАМПТОТЕЦИНОВ | 2006 |
|
RU2435586C2 |
| [6R]-MTHF - ЭФФЕКТИВНАЯ ФОЛАТНАЯ АЛЬТЕРНАТИВА В ХИМИОТЕРАПИИ НА ОСНОВЕ 5-ФТОРУРАЦИЛА | 2018 |
|
RU2763934C2 |
| СТАБИЛИЗИРУЮЩИЕ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ КАМПТОТЕЦИНА | 2016 |
|
RU2833053C2 |
| СТАБИЛИЗИРОВАННЫЕ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ КАМПТОТЕЦИНА | 2016 |
|
RU2732567C2 |
| СПОСОБЫ ЛЕЧЕНИЯ КОЛОРЕКТАЛЬНОГО И МЕТАСТАТИЧЕСКОГО КОЛОРЕКТАЛЬНОГО РАКА | 2019 |
|
RU2779535C2 |
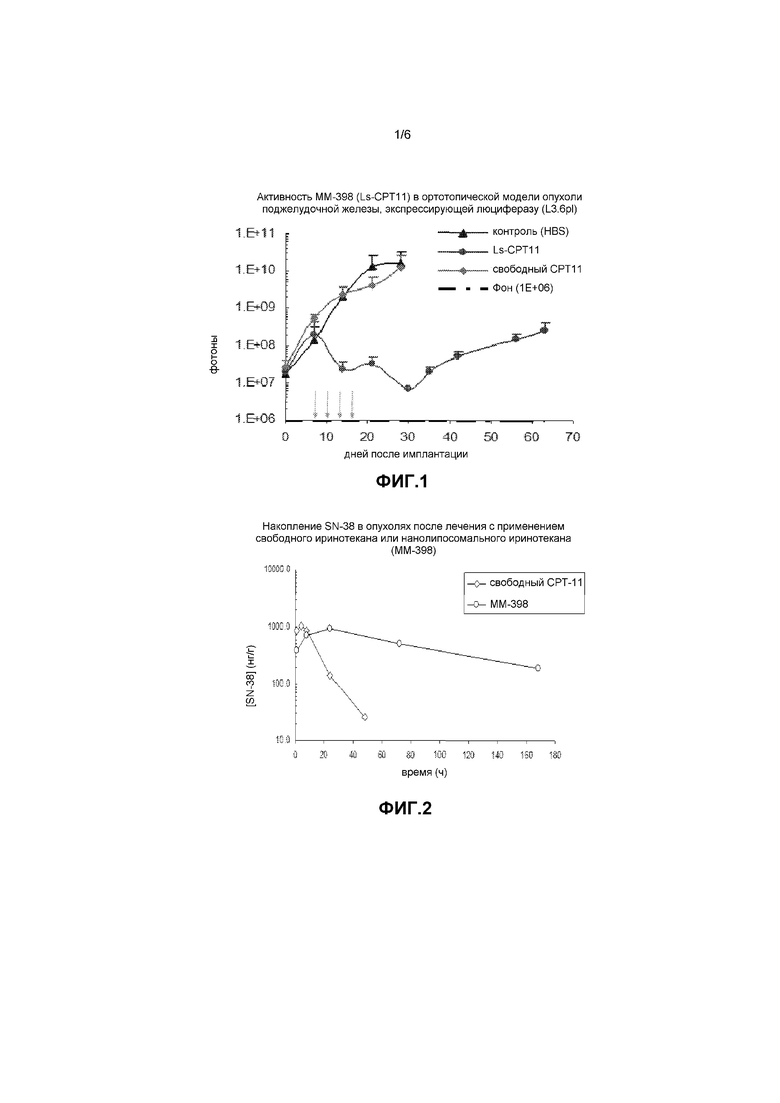
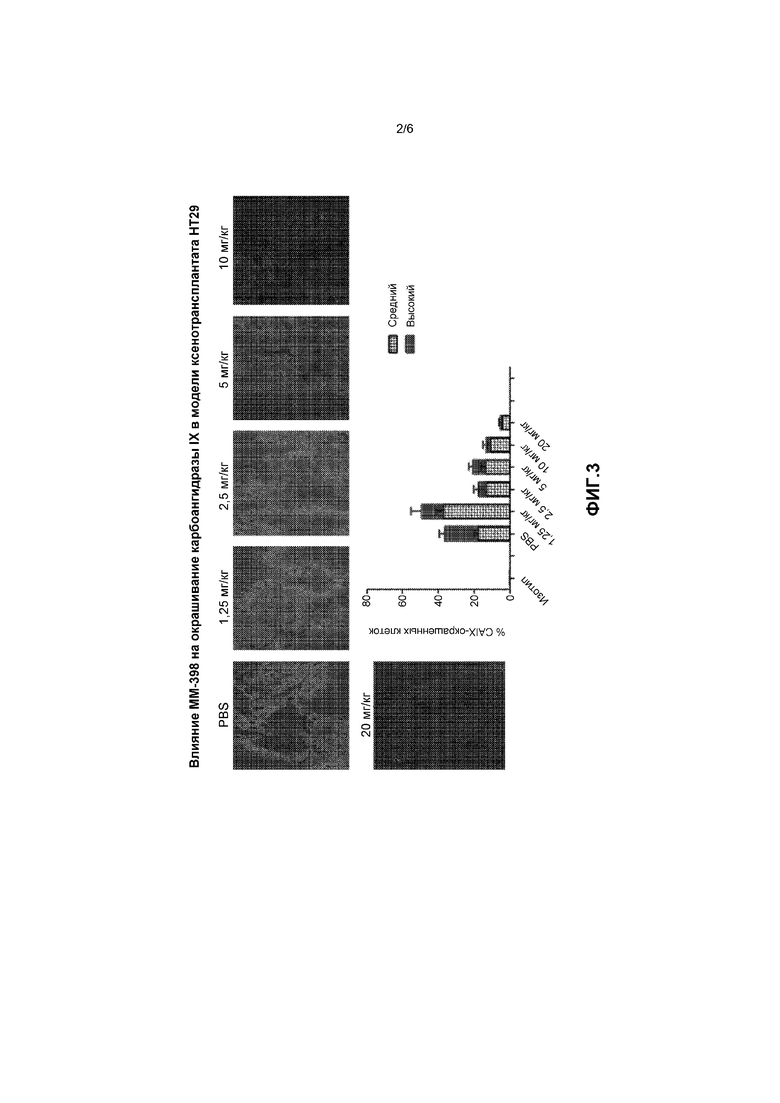
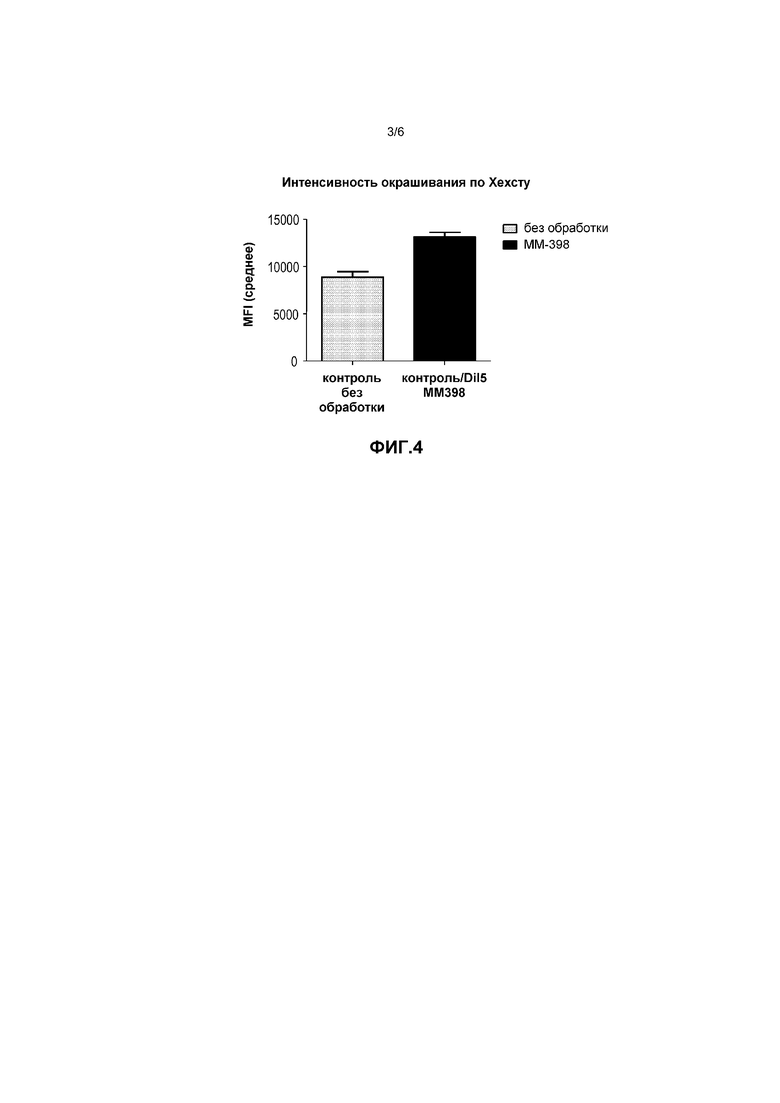
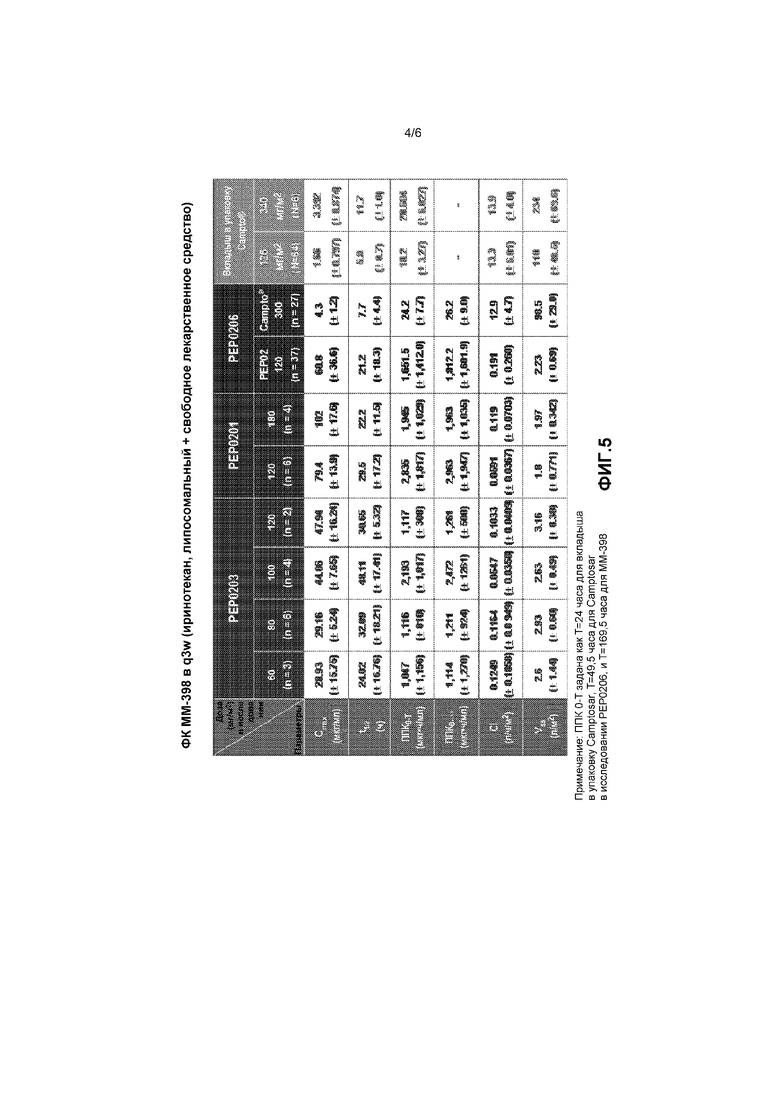
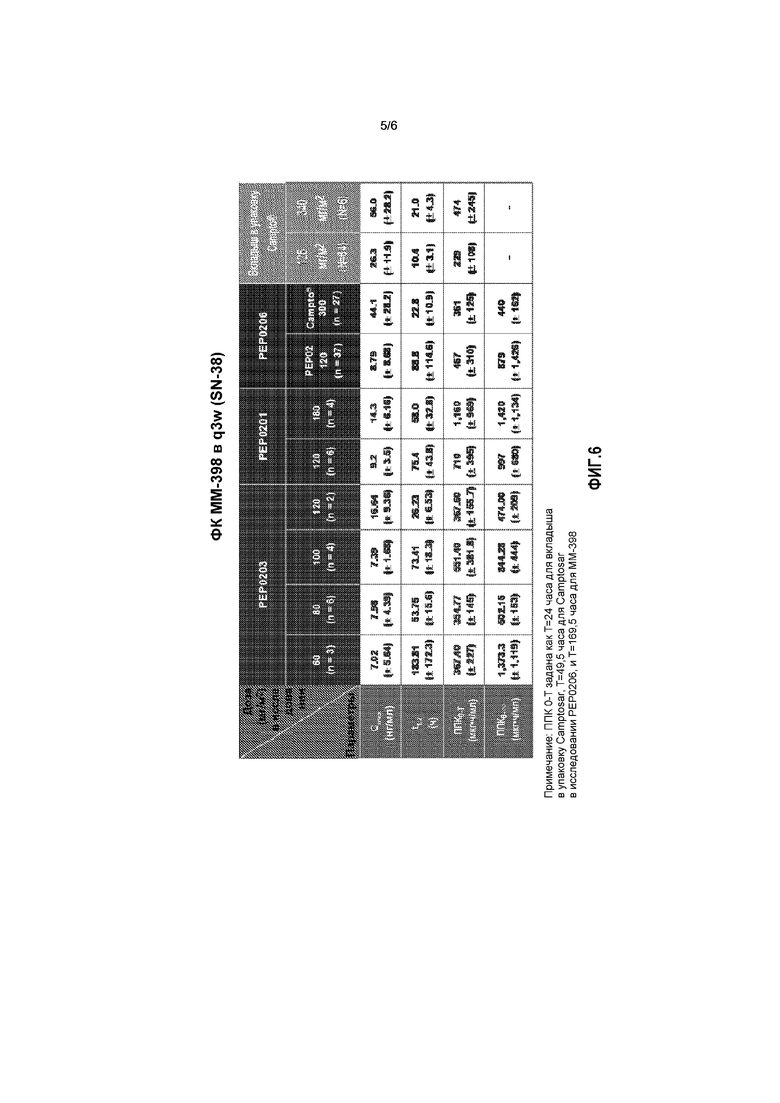
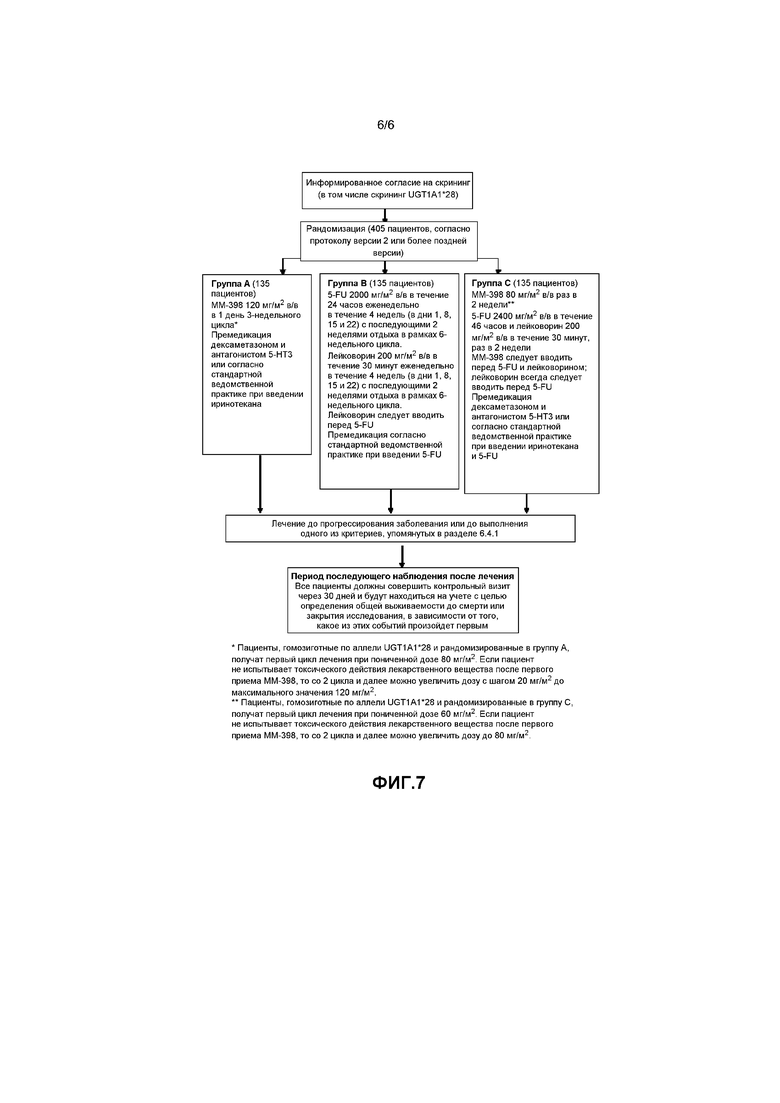
Настоящее изобретение относится к способу лечения экзокринного рака поджелудочной железы у пациента-человека, которого предварительно лечили гемцитабином, включающему внутривенное применение к пациенту противоопухолевой терапии, состоящей из: a) дозы 60, 70 или 80 мг/м2 композиции липосомального иринотекана, содержащей липосомы иринотекана в разбавленном инъекционном составе иринотекана, при этом разбавленный инъекционный состав иринотекана получен посредством разбавления объема композиции липосомального иринотекана, содержащей 5 мг/мл липосомального иринотекана в 500 мл 5% раствора декстрозы для инъекций, в комбинации с b) дозой 200 мг/м2 (l) формы лейковорина и дозой 2400 мг/м2 5-фторурацила, для лечения экзокринного рака поджелудочной железы у пациента, где липосомальная композиция иринотекана содержит соль иринотекана с октасульфатом сахарозы, инкапсулированную в липосомах иринотекана, состоящих из фосфатидилхолина, холестерина и фосфатидилэтаноламина, модифицированного полиэтиленгликолем, и имеющих диаметр приблизительно 80-140 нм, при этом разбавленный инъекционный состав иринотекана, лейковорин и 5-фторурацил вводят пациенту последовательно, начиная с первого дня двухнедельного цикла. Настоящее изобретение обеспечивает терапевтический синергизм при комбинированной терапии с применением липосомального иринотекана с 5-FU и лейковорином. 11 з.п. ф-лы, 7 пр., 7 ил., 14 табл.
1. Способ лечения экзокринного рака поджелудочной железы у пациента-человека, которого предварительно лечили гемцитабином, включающий внутривенное применение к пациенту противоопухолевой терапии, состоящей из:
a) дозы 60, 70 или 80 мг/м2 композиции липосомального иринотекана, содержащей липосомы иринотекана в разбавленном инъекционном составе иринотекана, при этом разбавленный инъекционный состав иринотекана получен посредством разбавления объема композиции липосомального иринотекана, содержащей 5 мг/мл липосомального иринотекана в 500 мл 5% раствора декстрозы для инъекций, в комбинации с
b) дозой 200 мг/м2 (l) формы лейковорина и дозой 2400 мг/м2 5-фторурацила,
для лечения экзокринного рака поджелудочной железы у пациента, где липосомальная композиция иринотекана содержит соль иринотекана с октасульфатом сахарозы, инкапсулированную в липосомах иринотекана, состоящих из фосфатидилхолина, холестерина и фосфатидилэтаноламина, модифицированного полиэтиленгликолем, и имеющих диаметр приблизительно 80-140 нм, при этом разбавленный инъекционный состав иринотекана, лейковорин и 5-фторурацил вводят пациенту последовательно, начиная с первого дня двухнедельного цикла.
2. Способ по п.1, где фосфатидилэтаноламин, модифицированный полиэтиленгликолем, присутствует в количестве приблизительно одна молекула полиэтиленгликоля (ПЭГ) на 200 молекул фосфолипидов в липосомах иринотекана.
3. Способ по п.1, где липосомы иринотекана представляют собой моноламеллярные липидные двуслойные везикулы, инкапсулирующие соль иринотекана с октасульфатом сахарозы.
4. Способ по п.1, в котором экзокринный рак поджелудочной железы прогрессировал на фоне терапии с применением гемцитабина до введения липосом иринотекана.
5. Способ по п.1, в котором 200 мг/м2 (l) формы лейковорина, введены как 400 мг/м2 (l+d) рацемической формы лейковорина.
6. Способ по п.1, в котором разбавленный инъекционный состав иринотекана вводят пациенту до 5-фторурацила.
7. Способ по п.6, в котором:
a) лейковорин вводится как 400 мг/м2 (l+d) рацемической формы лейковорина;
b) экзокринный рак поджелудочной железы прогрессировал на фоне терапии с применением гемцитабина до введения разбавленного инъекционного состава иринотекана;
c) липосомы иринотекана представляют собой моноламеллярные липидные двуслойные везикулы, инкапсулирующие соль иринотекана с октасульфатом сахарозы, и
d) фосфатидилэтаноламин, модифицированный полиэтиленгликолем, присутствует в количестве приблизительно одна молекула полиэтиленгликоля на 200 молекул фосфолипидов в липосомах иринотекана.
8. Способ по п.1, дополнительно включающий хранение разбавленного инъекционного состава иринотекана при температуре приблизительно 2-8°C в течение не более 24 часов до введения.
9. Способ по п.1, где липосомы иринотекана представляют собой моноламеллярные липидные двуслойные везикулы, которые инкапсулируют водное пространство, содержащее иринотекан в гелеобразном или осажденном состоянии в виде соли с октасульфатом сахарозы.
10. Способ по п.1, где пациент ранее безуспешно получал лечение с применением гемцитабина или приобрел устойчивость к гемцитабину до введения разбавленного инъекционного состава иринотекана.
11. Способ по п.1, в котором доза композиции липосомального иринотекана составляет 80 мг/м2, и пациент-человек не является гомозиготным по аллелю UGT1A1*28.
12. Способ по п.11, где композицию липосомального иринотекана вводят в виде разбавленного инъекционного состава иринотекана в качестве 90-минутной инфузии, лейковорин вводят после иринотекана в течение 30 минут, и 5-фторурацил вводят в течение 46 часов после лейковорина.
| C Yoo et al., A randomised phase II study of modified FOLFIRI.3 vs modified FOLFOX as second-line therapy in patients with gemcitabine-refractory advanced pancreatic cancer / British Journal of Cancer, 2009, Vol | |||
| Приспособление для записи звуковых явлений на светочувствительной поверхности | 1919 |
|
SU101A1 |
| Металлический ключ для пчеловодов | 1924 |
|
SU1658A1 |
| L | |||
| Chen et al., Phase I study of liposome irinotecan (PEP02) in combination with weekly infusion of 5-FU/LV in | |||
