Изобретение относится к соединениям общей формулы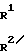 N-Q-CH
N-Q-CH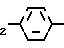 CO
CO (I) где R1 и R2 каждый означают водород, низший алкил или вместе образуют неразветвленный алкилен С2-4;
(I) где R1 и R2 каждый означают водород, низший алкил или вместе образуют неразветвленный алкилен С2-4;
Q алкилен С4-11 минимум с двумя атомами С между двумя свободными валентностями;
R означает, что кольцо не замещено или замещено галогеном, трифторметилом, нитрогруппой или низшим алкилом, а также к приемлемым для фармацевтических целей солям этих соединений с кислотами. Эти соединения обладают ценными фармакологическими свойствами, в частности имеют выраженные противогрибковое и синергическое действия в комбинации с известными противогрибковыми веществами, подавляющими биосинтез таких стеролов, как кетоцоназол и тербинафин. Поэтому соединения формулы (I) можно использовать в качестве лекарств, в частности для борьбы с топическими или системными заражениями, вызываемыми патогенными грибками, или для предупреждения подобного рода заражений.
Выражение "низший" обозначает остатки и соединения с числом атомов С не более семи, предпочтительно не более четырех.
Выражение "алкил" обозначает неразветвленные или разветвленные насыщенные углеводородные остатки, например метил, этил, пропил, изопропил, трет.-бутил.
Выражение "алкокси" обозначает присоединенные через атом кислорода алкилы, такие как метокси и этокси.
Выражение "алкилен" обозначает неразветвленные или разветвленные насыщенные углеводородные остатки с двумя свободными валентностями, такими как диметилен, триметилен, тетраметилен, пентаметилен, гексаметилен.
Выражение "галоген" обозначает четыре вида галогенов фтор, хлор, бром, йод.
В рамках этого аспекта изобретения предпочитаются следующие соединения формулы (I):
3-(4-хлорфенил)-4'-[3-(диметиламино) пропил]пропиофенон;
3-(2-метилфенил)-4'-[3-(диметиламино)пропил]пропиофенон;
(Е)-3-фенил-4'-[3-(1-пирролидинил)про- пил]акрилофенон.
Новые соединения формулы (I) и их приемлемые для фармацевтических целей соли с кислотами согласно изобретению получают тем, что
а) соединение общей формулы
X Q CH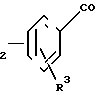
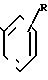 (II) где Х отщепляемая группа;
(II) где Х отщепляемая группа;
R3, Q, R имеют вышеуказанное значение, подвергают реакции взаимодействия с амином общей формулы
HNR1R2, где R1 и R2 имеют вышеуказанное значение,
б) соединение общей формулы N-Q-CH
N-Q-CH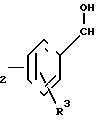
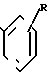 (III) где R1, R2, R3, Q и R имеют вышеуказанное значение, окисляют, или
(III) где R1, R2, R3, Q и R имеют вышеуказанное значение, окисляют, или
в) соединение общей формулы N-Q-CH
N-Q-CH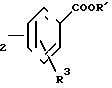 (IV) где R1 низший алкил;
(IV) где R1 низший алкил;
R1, R2, R3, Q имеют вышеуказанное значение, подвергают реакции взаимодействия с соединением общей формулы
 (V) где М -MgCl, -MgBr, -MgJ или -Li;
(V) где М -MgCl, -MgBr, -MgJ или -Li;
R имеет вышеуказанное значение, или
г) соединение общей формулы N-Q-CH
N-Q-CH (VI) где R'' и R''' каждый означают низший алкил или вместе образуют диметилен или триметилен;
(VI) где R'' и R''' каждый означают низший алкил или вместе образуют диметилен или триметилен;
R1, R2, R3, Q и R имеют вышеуказанное значение, обрабатывают водной кислотой, или
д) соединение общей формулы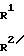 N-Q-CH
N-Q-CH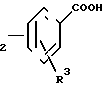 (VIIа)
(VIIа)
или
 (VIIб) в виде реакционноспособного производного в присутствии кислоты Льюиса подвергают реакции взаимодействия с соединением общей формулы
(VIIб) в виде реакционноспособного производного в присутствии кислоты Льюиса подвергают реакции взаимодействия с соединением общей формулы (VIIIа)
(VIIIа)
или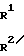 N-Q-CH
N-Q-CH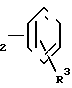 (VIIIб) где R1, R2, R3, Q и R имеют вышеуказанное значение, или
(VIIIб) где R1, R2, R3, Q и R имеют вышеуказанное значение, или
е) соединение общей формулы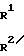 N-Q-CH
N-Q-CH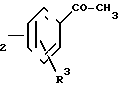 (IX) где R1, R2, R3, Q имеют вышеуказанное значение, в присутствии основания подвергают реакции взаимодействия с соединением общей формулы
(IX) где R1, R2, R3, Q имеют вышеуказанное значение, в присутствии основания подвергают реакции взаимодействия с соединением общей формулы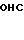
 (X) где R имеет вышеуказанное значение, или
(X) где R имеет вышеуказанное значение, или
ж) соединение общей формулы N-Q-CH
N-Q-CH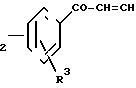
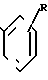 или
или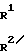 N-Q′‗‗ CH
N-Q′‗‗ CH 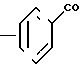
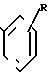 (XI) где Q' группа Q, уменьшенная на атом водорода;
(XI) где Q' группа Q, уменьшенная на атом водорода;
R1, R2, R3, Q и R имеют вышеуказанное значение, гидрируют и
з) полученное соединение формулы (I) при необходимости переводят в приемлемую для фармацевтических целей соль с кислотой.
Реакцию взаимодействия соединения формулы (II) с амином HNR1R2согласно варианту а) способа осуществляют известными специалисту методами. Ее, в частности, проводят в полярном растворителе и в присутствии основания в качестве акцептора кислоты в температурном интервале около 0-150оС. При этом пригодными растворителями являются, например, такие низшие спирты, как метанол и этанол, и такие низшие диалкилкетоны, как ацетон. Пригодными основаниями являются, например, амин формулы NHR1R2 в избытке, третичные амины, например, триэтиламин, и такие неорганические основания, как карбонаты, гидроокиси и алкоголяты щелочных металлов.
Реакцию окисления соединения (III) согласно варианту б) также осуществляют известными специалисту методами, в частности в присутствии инертного растворителя и в присутствии окислителя в приблизительном температурном интервале от 80оС до комнатной температуры. Пригодными растворителями являются, например, хлорированные низшие углеводороды, например, метиленхлорид и хлороформ. Пригодными окислителями являются, например, двуокись марганца или смеси ДМСО с оксалилхлоридом, дициклогексилкарбодиимидом или ацетангидридом и третичным амином, например триэтиламином.
Реакцию взаимодействия соединения (IV) с соединением (V) по варианту в) также осуществляют известными специалисту способами, в частности в инертном растворителе и в приблизительном температурном интервале от 80оС до комнатной температуры. Подходящими растворителями являются, например, такие нециклические и циклические простые эфиры, как диэтиловый эфир, метил-трет.-бутиловый эфир и тетрагидрофуран, и смеси этих соединений.
Для обработки соединения формулы VI водной кислотой согласно варианту г) применяют предпочтительно разбавленную водную минеральную кислоту, например разбавленную соляную, и проводят реакцию в температурном интервале примерно от 0оС до комнатной температуры.
Реакцию взаимодействия реакционноспособного производного соединения формулы (VIIa) с соединением формулы (VIIIa) или реакционноспособного соединения формулы (VIIб) с соединением формулы (VIIIб) согласно варианту д) способа осуществляют известными специалисту методами. Реакцию проводят предпочтительно в инертном растворителе и в присутствии кислоты Льюиса в температурном интервале примерно от 0оС до примерно 100оС. Подходящими растворителями являются, например, такие галогенированные низшие углеводороды, как метиленхлорид, хлороформ и этиленхлорид, нитробензол, сероуглерод и избыток соединения формулы (VIIIa). В качестве кислоты Льюиса применяют предпочтительно хлористый алюминий. Подходящими реакционноспособными производными соединений формулы (VIIa) или (VIIб) являются, например, хлориды карбоновых кислот.
Реакцию взаимодействия соединения формулы (IX) с соединением формулы (X) в присутствии основания согласно варианту е) способа осуществляют известными специалисту способами. Реакцию осуществляют преимущественно в полярном растворителе и в температурном интервале около 0-60оС. Подходящими растворителями являются, например, низшие спирты, такие как метанол и этанол, и смеси этих соединений с водой. В качестве оснований применяют предпочтительно карбонаты и гидроокиси щелочных металлов, такие как карбонат калия и гидроокись натрия.
Реакцию гидрирования соединения формулы (I) или (XI) согласно варианту ж) способа осуществляют известными специалисту методами, в частности в полярном растворителе с элементарным водородом в присутствии пригодного катализатора гидрирования и в приблизительном температурном интервале от 0оС до комнатной температуры. Пригодными растворителями являются также низшие спирты, как метанол и этанол. Пригодными катализаторами являются, например, палладий или платина на угле, окись платины или никель Ранея.
Получение приемлемых для фармацевтических целей солей соединений формулы (I) с кислотами согласно варианту з) способа осуществляют известными специалисту способами. Имеются в виду соли с приемлемыми для фармацевтических целей неорганическими или органическими кислотами. Предпочтительными солями с кислотами являются гидрохлориды, гидробромиды, сульфаты, нитраты, цитраты, ацетаты, сукцинаты, фумараты, метансульфонаты, п-толуолсульфонаты.
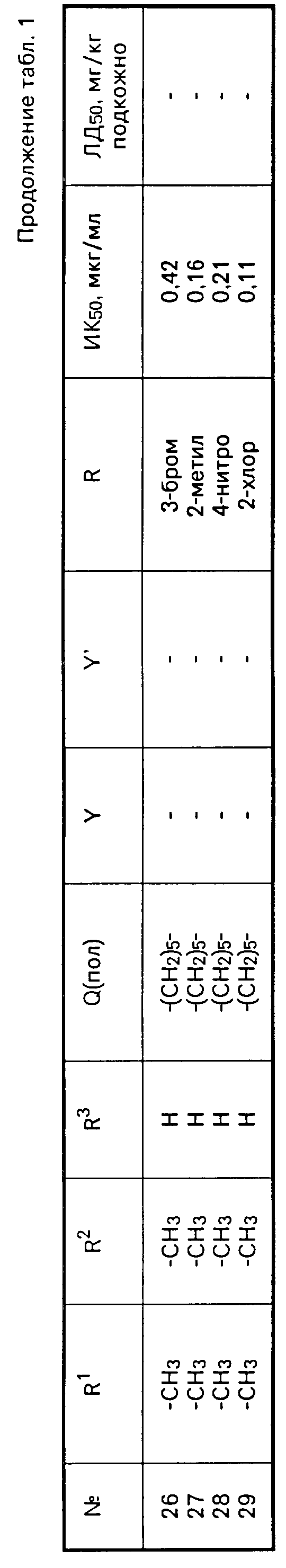
Соединения формулы (I) и их приемлемые для фармацевтических целей соли с кислотами обладают ценными противогрибковыми качествами. Они оказываются эффективными в борьбе со множеством патогенных грибков, вызывающих топические и системные заражения, например с такими видами грибков, как Candida albicans и Histoplasma capsulatum. Незаменимым энзимом в жизнедеятельности грибков, участвующим в биосинтезе стерола эукариотической клетки, является 2,3-эпоксисквален-ланостерол-циклаза. Так, штаммы S. cerevisiae, лишенные этого энзима, не жизнеспособны (см. F. Karst and F. Lacroute, Molec. Gen. Genet. 154, 269 (1977). Тормозящее действие соединений формулы (I) на вышеназванный энзим, содержащийся в грибке C. albicans, выбрали как критерий противогрибкового действия. Торможение можно измерить, например, нижеописанным методом.
Установление торможения активности 2,3-эпоксисквален-ланостерол-циклазы грибка Candida albicans определением значения IC50. Сначала собирают клетки культуры Candida albicans в конце логарифмической фазы роста и промывают их 100 мМ фосфатным буфером (рН 6,9) буфером для переваривания, и 50 мМ фосфатным буфером (рН 7,4), содержащим 1 М маннита и 5 мМ DTT.
1,0 г клеток взвешивают в 5 мл буфера для переваривания и во взвесь добавляют 1 мг зимолазы 100Т (ф-мы Seikagaku Kogyo, Япония) и 12,5 мкл β-меркаптоэтанола, после чего ее выдерживают в течение 30 мин при 30оС. Образовавшиеся за это время протопласты отделяют путем центрифугирования (10 мин при 2500 g) с последующим их разрывом путем добавления 2 мл 100 мМ фосфатного буфера (рН 6,9). В результате повторного центрифугирования (10 мин при 10000 g) получают надосадочную жидкость, представляющую собой не содержащий клеток экстракт (CFE). Последний разбавляют до содержания 10 мг белка/мл, после чего значение рН доводят до 6,9.
Активность 2,3-эпоксисквален-ланостерол-циклазы в CFE измеряют обработкой эпоксида сквалена -С14 в присутствии n-децилпентаоксиэтилена в качестве детергента. Титрацией подходящих количеств испытуемого вещества определяют значения IC50, т.е. концентрацию испытуемого вещества, уменьшающую активность энзима на половину.
Этот опыт проводят следующим образом. Обработкой ультразвуком сначала приготовляют 250 мкМ раствор эпоксида сквалена-С14 в 100 мМ фосфатном буфере (рН 6,9), содержащем 1% n-децилпентаоксиэтилена. К 100 мкл этого раствора добавляют 20 мкл раствора испытуемого вещества (или 20 мкл чистого диметилсульфоксида в качестве контроля). После добавления 880 мкл CFE хорошо смешанный раствор со встряхиванием выдерживают в течение 1 ч при 30оС. Затем добавлением 500 мкл 15%-ной гидроокиси калия в 90%-ном этаноле прекращают реакцию.
Полученную смесь два раза экстрагируют 1 мл n-гексана, после чего упаривают гексан и образовавшийся липидный остаток смешивают с 200 мкл диэтилового эфира. После тонкослойной хроматографии продукта на силикагеле с метиленхлоридом в качестве растворителя пластинки ТСХ исследуют с помощью сканирующего устройства для установления наличия радиоактивных веществ.
В вышеописанных условиях в качестве радиоактивного продукта обнаруживают только ланостерол. Количество последнего сравнивают с количеством радиоактивного ланостерола в контрольном веществе.
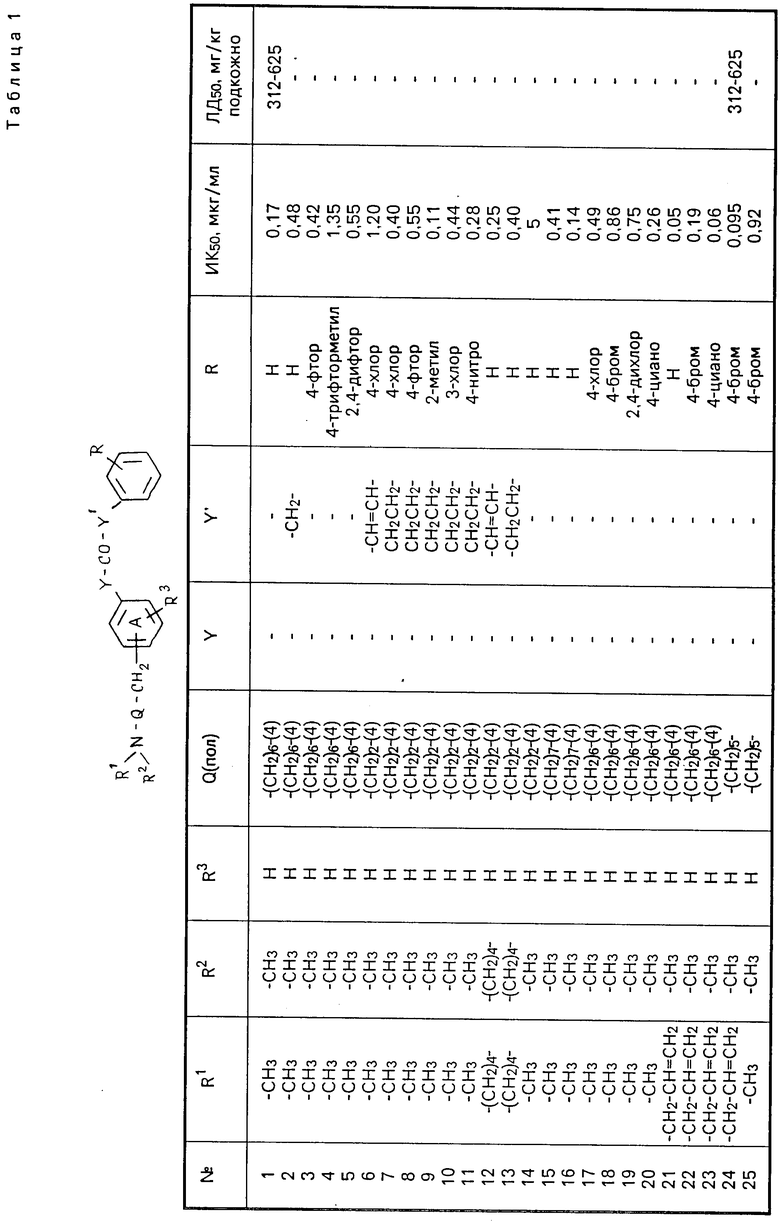
Значения IC50 определяют графическим способом, и их указывают в мкг испытуемого вещества на мл. В табл.1 приведены значения IC50 для показательных представителей соединений формулы (I), а также данные об острой токсичности при подкожном введении испытуемых соединений мышам (ЛД50 в мг/кг).
Вышеуказанное синергическое действие соединений формулы (I) и их фармацевтически приемлемых солей с кислотами в комбинации с ингибиторами биосинтеза стерола, например, кетоконазола, можно, например, показать методом серийного разведения на агаре. В этих целях используют казеиновый агар типа "Casitonagar" и инокуляты (10 клеток/мл) двухсуточных культур Candida albicans. Тест-вещества (соединения формулы (I), ТВ) наносят в концентрациях 80-1,25 мкг/мл, а ингибиторы биосинтеза стерола (ИБС) в концентрации 20-0,001 мкг/мл) с кратностью разведения 1:2. Культуры в течение 2 сут инкубируют при 37оС. Определяют минимальную ингибирующую концентрацию (МИК) отдельных действующих начал в чистом виде и в комбинации с другими веществами. Исходя из значений МИК, вычисляют фракционированную ингибирующую концентрацию (ФИК) по следующей формуле:
ФИК  +
+ 
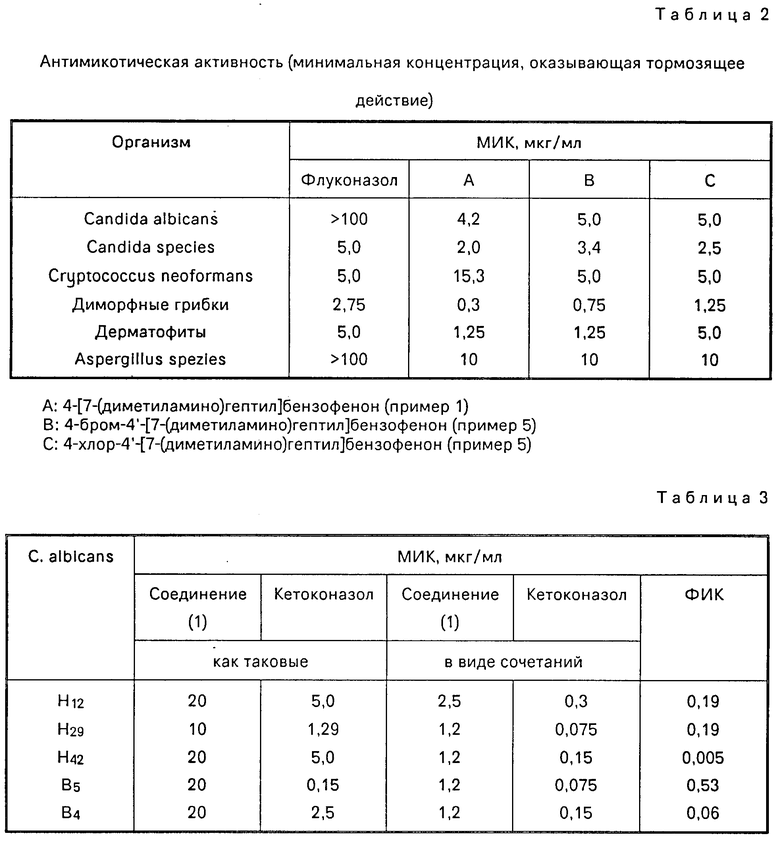
Соединения формулы (I) имеют синергическое действие при том условии, что значение ФИК < 0,5. Приведенные в табл.2 и 3 данные для соединения (I) по табл. 1, представляющего собой показательный представитель определенного формулой (I) класса соединений, в комбинации с кетоконазолом, представляющим собой показательный ингибитор биосинтеза стерола, являются доказательством синергического действия соединений.
Соединения формулы (I) и их приемлемые для фармацевтических целей соли с кислотами могут применяться как лекарства, например в виде фармацевтических препаратов для энтерального или парентерального введения или топического применения. Они могут быть введены орально, например в виде таблеток, таблеток с покрытием, драже, твердых и мягких желатиновых капсул, растворов, эмульсий или суспензий, ректально, например в виде суппозиториев, парентерально, например в виде растворов для инъекций или инфузий, или же топически, например в виде мазей, кремов или масел.
Получение фармацевтических препаратов осуществляют известным специалисту образцом, т.е. тем, что описанные соединения формулы (I) и их приемлемые для фармацевтических целей соли, если нужно, в сочетании с другими терапевтически ценными веществами, например с указанными ингибиторами биосинтеза стерола, вместе с подходящими, нетоксичными, инертными, терапевтически совместимыми твердыми или жидкими носителями и, если нужно, с обычными фармацевтическими вспомогательными веществами перерабатывают в галеновый препарат.
Пригодными для сочетания с соединениями формулы (I) ингибиторами биосинтеза стерола могут служить, например, системно действующие противогрибковые азолы типа мицоназола, такие как кетоконазол, итраконазол и флуконазол, а также системно действующие противогрибковые аллиламины типа нафтифина, такие как нафтифин и тербинафин.
Носителями могут служить как неорганические, так и органические носители. В качестве подобных носителей для таблеток с покрытием и без такового, а также для драже и твердых желатиновых капсул можно использовать, например, лактозу, кукурузный крахмал или его производные, тальк, стеариновую кислоту или ее соли. Пригодными носителями для мягких желатиновых капсул являются, например, растительные масла, воски, жиры, полутвердые и жидкие полиолы и т. д. (В зависимости от природы действующего начала применение носителей для получения мягких желатиновых капсул может оказаться и ненужным). Для получения растворов и сиропов пригодны такие носители, как вода, полиолы, сахароза, инвертный сахар, глюкоза и т.п. Пригодными носителями для впрыскиваемых растворов являются, например, вода, спирты, полиолы, глицерин, растительные масла и т.п. Подходящими носителями для суппозиториев являются, например, естественные или отвержденные масла, воски, жиры и полужидкие или жидкие полиолы. Подходящими носителями для топических препаратов являются глицериды, полусинтетические и синтетические глицериды, гидрированные масла, жидкие воски, жидкие парафины, жидкие спирты жирного ряда, стеролы, полиэтиленгликоли, производные целлюлозы.
В качестве вспомогательных фармацевтических веществ могут применяться обычные стабилизаторы, консерванты, смачиватели, эмульгаторы, средства для повышения консистентности, подслащивающие и ароматизирующие вещества, соли для изменения осмотического давления, буферы, агенты растворения, красители, покрывающие вещества и противоокислители.
В зависимости от вида патогенных грибков, возраста и состояния здоровья больного, а также от способа введения лекарств их дозировка может варьироваться в широких пределах. Ее следует приспособить к индивидуальным потребностям больного. В целях предупреждения тонических и системных заражений, вызываемых патогенными грибками, а также для борьбы с подобными заражениями при монотерапии больного пригодна суточная доза около 0,01-4 г, в частности около 0,05-2 г. В зависимости от дозировки целесообразно ввести суточную дозу в виде нескольких единичных доз. Пригодная суточная доза для комбинированной терапии составляет около 0,01-2 г, в частности около 0,02-1 г соединения формулы (I) и около 0,02-0,2 г ингибитора биосинтеза стерола.
Фармацевтические монопрепараты обычно содержат около 10-1000 мг, предпочтительно 50-500 мг соединения формулы (I), а комбинированные препараты около 10-500 мг, предпочтительно 20-250 мг соединения формулы (I) и около 50-100 мг ингибитора биосинтеза стерола.
Превосходство новых соединений по сравнению с наиболее близким прототипом вытекает из вышеприведенного отчета об опытах. Однако о прогрессивности новых соединений по сравнению с известным фунгицидом нельзя судить только на основании сравнения числовых значений концентраций, оказывающих тормозящее действие. Новые соединения являются ценными прежде всего потому, что они обладают другим механизмом действия, чем известные антимикотические средства. Известные антимикотические средства представляют собой либо ингибиторы деметилирования 4,4',14-триметилстеринов (к ним относятся азолы и подобные соединения), либо ингибиторы Δ8- Δ7 стерол-изомеразы (например, фенпропидин и фенпропиморф) или ингибиторы сквален-эпоксидазы (к ним относятся алкиламины и подобные соединения). В противоположность этому, новые соединения представляют собой ингибиторы 2,3-эпоксисквален-ланостерол-циклазы. Для решения проблем резистентности необходимо иметь в распоряжении соединения, оказывающие действие в других местах обмена веществ грибков, чем используемые до сих пор соединения. Таким образом, новые соединения обогащают ассортимент имеющихся антимикотических средств.
П р и м е р 1. а) 9,3 г 1,6-дибромгексана и 5 г трифенилфосфина перемешивают в атмосфере аргона при 100оС в течение 1 ч. Затем полученную реакционную массу охлаждают и хроматографируют на 90 г силикагеля смесью метиленхлорида и метанола (95: 5). К полученному 6-бромгексил-фосфонийбромиду (6,37 г, 65%) добавляют 35 мл 33%-ного раствора диметиламина в этаноле и перемешивают в течение 1 сут при комнатной температуре в атмосфере аргона. Раствор выпаривают и полученный остаток полностью высушивают в высоком вакууме. Полученный таким образом гидробромид 6-(диметиламино)гексил-трифенилфосфоний- бромида взвешивают в 50 мл ТГФ, после чего при 0оС по порциям добавляют 2,09 г трет.бутилата калия. Полученную смесь перемешивают в течение 15 мин при 0оС. Потом при 0оС добавляют раствор 1,86 г 4-бензоил-бензальдегида [Tetrahed. Let. 24, 4287 (1983)] в 10 мл ТГФ, после чего полученную смесь перемешивают при комнатной температуре в течение 18 ч. Затем выпаривают реакционную массу. Остаток смешивают со 100 мл 1 н. соляной кислоты и экстрагируют три раза 100 мл диэтилового эфира. Водную фазу с охлаждением льдом с помощью 2 н. раствора едкого натра доводят до щелочной реакции и три раза экстрагируют метиленхлоридом. Органические фазы высушивают сульфатом магния и выпаривают. Полученный сырой продукт хроматографируют на 100 г нейтрального оксида алюминия (степень активности III) смесью метиленхлорида и этилацетата (95:5). В результате выпаривания получают 1,98 г (31%) 4-[(Z)-7-(диметиламино)-1-гептенил]бензофенона в виде бесцветного масла.
Данные 1Н-ЯМР-анализа (CDCl3): 1,3-1,8 (м, 6Н); 2,22 (с, 6Н); 2,29 (т, J 8 Гц, 2Н); 2,35 (м, 2Н), 5,80 (дхт, J 12 Гц, J 7 Гц, 1 H); 6,48 (дхт, J 12 Гц, J 2 Гц, 1 H); 7,39 (д, J 8 Гц, 2 H); 7,4-7,8 (м, 7H) ppm.
б) 1,12 г 4-[(Z)-7-(диметиламино)-1-гептенил]бензофенона растворяют в 30 мл метанола. К раствору добавляют 20 мг 5%-ного палладия на угле и перемешивают взвесь в атмосфере водорода под нормальным давлением при комнатной температуре в течение 2,5 ч. Реакционную массу фильтруют через силикагель, а фильтрат упаривают. Получают 1,10 г (98%) 4-[7-(диметиламино)гептил]бензофенона в виде бесцветного масла.
Данные 1Н-ЯМР-анализа (CDCl3): 1,3-1,6 (м, 8Н); 1,67 (кв. J 7,5 Гц, 2 H); 2,23 (с, 6Н); 2,29 (т, J 8 Гц, 2); 2,69 (т, J 7,5 Гц, 2 H); 7,3-7,9 (м, 9H) ppm.
П р и м е р 2. а) К взвеси 5,06 г гидробромида 6-(диметиламино)гексилтрифенилфосфонийбромида (см. пример 1а) в 50 мл ТГФ при 0оС в атмосфере аргона добавляют 2,47 г трет.-бутилата калия. К этой взвеси в течение 15 мин по каплям добавляют раствор 1,64 г метилового эфира р-формилбензойной кислоты в 20 мл ТГФ. Реакционную массу перемешивают в течение 18 ч при комнатной температуре, а потом выпаривают. Остаток растворяют в 100 мл 1 н. соляной кислоты. Водную фазу промывают три раза 100 мл диэтилового эфира, затем с помощью твердого карбоната калия ее доводят до щелочной реакции и экстрагируют три раза 100 мл диэтилового эфира. Эфирные фазы высушивают сульфатом магния и выпаривают. Остаток хроматографируют на 100 г силикагеля смесью метиленхлорида, метанола и гидроокиси аммония (100:10:1). Получают 880 мг (32%) метилового эфира 4-[(Z)-7-(диметиламино)-1-гептенил] бензой- ной кислоты в виде бесцветного масла.
б) 0,88 г метилового эфира (4-[(Z)-7-(диметиламино)-1-гептенил]-бензойной кислоты растворяют в 20 мл этанола, к раствору добавляют 20 мг 5%-ного палладия на угле и перемешивают полученную взвесь в атмосфере водорода под нормальным давлением при комнатной температуре в течение 2 ч. Реакционную массу фильтруют через диатомовую землю, а фильтрат выпаривают. Получают 0,85 г (95%) метилового эфира 4-[7-(диметиламино)гептил]бензойной кислоты в виде бесцветного масла.
в) К раствору 0,85 г метилового эфира 4-[7-(диметиламино)гептил]бензойной кислоты в 20 мл ТГФ при -78оС в атмосфере аргона по каплям добавляют раствор бензилмагнийбромида (приготовленного из 74 мг магния и 524 мг бензилбромида) в 20 мл диэтилового эфира в течение 30 мин. Реакционную массу перемешивают при -78оС в течение 2 ч, затем ее выливают на 50 мл насыщенного раствора хлористого алюминия и экстрагируют три раза 50 мл этилацетата. Собранные органические фазы промывают 100 мл насыщенного раствора хлористого натрия, высушивают сульфатом магния и выпаривают. После хроматографии остатка на 100 г силикагеля смесью метиленхлорида, метанола и гидроокиси алюминия (100: 10: 1) получают 340 мг (35%) 4'-[7-(диметиламино)гептил]-2-фенилацето- фенона в виде желтоватого масла.
Данные 1Н-ЯМР-анализа (CDCl3): 1,3-1,7 (м, 10 H); 2,40 (с, 6Н); 2,52 (т, J 8 Гц, 2Н); 2,64 (т, J 7 Гц, 2Н); 4,26 (с, 2Н); 7,3-7,4 (м, 7Н); 7,93 (д, J 8 Гц, 2 Н) ppm.
П р и м е р 3. Аналогично примеру 2в) из метилового эфира 4-[7-(диметиламино)гептил] бензойной кислоты и а) фенэтилмагнийбромида получают 4'- [7-(диме- тиламино)-гептил]-3-фенилпропиофенон в виде бесцветного масла (выход 35%).
Данные 1Н-ЯМР-анализа (CDCl3): 1,3-1,7 (м, 10Н); 2,20 (с, 6Н); 2,1-2,3 (м, 2Н); 2,64 (т, J 8 Гц, 2Н); 3,0-3,4 (м, 4Н); 7,2-7,4 (м, 7Н); 7,88 (д, J 8 Гц, 2Н) ppm.
б) р-(трифторметил)-фенилмагнийбромида 4-[7-(диметиламино)гептил]-4'-(трифторметил)бензофенон в виде бесцветного масла (выход 40%).
Данные 1Н-ЯМР-анализа (CDCl3): 1,3-1,7 (м, 10Н); 2,24 (с, 6Н); 2,28 (т, J 8 Гц, 2Н); 2,69 (т, J 8 Гц, 2Н); 7,29 (д, J 9 Гц, 2Н); 7,7-7,9 (м, 6Н) ppm.
Данные МС-анализа: 391 (1,5% М+); 372 (1%); 235 (2,5%); 173 (3,8%); 145 (4,5%); 58 (100%).
П р и м е р 4. а) К 1,2 г метилового эфира 4-[7-(диметиламино)гептил]-бензойной кислоты добавляют 25 мл метанола и 10 мл 20%-ного раствора едкого натра в воде, после чего полученную смесь в течение 2 ч нагревают при температуре дефлегмации. После охлаждения до 0оС значение рН раствора добавлением уксусной кислоты доводят до 5. Выпавшее в результате вещество отсасывают на нутч-фильтре и высушивают в высоком вакууме под давлением около 6,7 Па в течение ночи. В результате получают 0,97 г (85%) 4-[7-(диметиламино)гептил] бензойной кислоты.
б) Полученное вещество взвешивают в 20 мл метиленхлорида, к полученной взвеси добавляют 1,2 мл оксалилхлорида, а полученную реакционную массу перемешивают при комнатной температуре в течение 2 ч и выпаривают. Сырой хлорангидрид кислоты высушивают в высоком вакууме, растворяют в 3 мл фторбензола, а раствор охлаждают до 0оС. К этому раствору добавляют 760 мг хлористого алюминия. Полученную массу перемешивают в течение 30 мин при 0оС и затем в течение 2 ч при комнатной температуре. Реакционную массу разбавляют 50 мл метиленхлорида и промывают два раза 50 мл 2 н. раствора едкого натра. Метиленхлоридную фазу высушивают сульфатом магния и выпаривают. Сырой продукт очищают хроматографией на 100 г силикагеля смесью метиленхлорида, метанола и гидроокиси алюминия (90:10:1). Получают 0,5 г (40%) 4-[7-(диметиламино)гептил]-4'-фторбензофенона в виде бесцветного масла.
Данные 1Н-ЯМР-анализа (CDCl3): 1,3-1,7 (м, 10Н); 2,23 (с, 6Н); 2,26 (т, J 7,5 Гц, 2Н); 2,68 (т, J 7,5 Гц, 2Н); 7,1-7,2 (м, 2Н); 7,28 (д, J 8 Гц, 2Н); 7,70 (д, J 8 Гц, 2Н); 7,7-7,9 (м, 2Н) ppm.
П р и м е р 5. Аналогично примеру 4в) из 4-[7-(диметиламино)гептил]-бензойной кислоты и 1,3 дифторбензола получают 2,4-дифтор-4'-[7-(диметиламино)гептил]бензо-фенон в виде бесцветного масла (выход 32%).
Данные 1Н-ЯМР-анализа (CDCl3): 1,3-1,7 (м, 10Н); 2,25 (с, 6Н); 2,28 (т, J 7 Гц, 2Н); 2,67 (т, J 8 Гц, 2Н); 6,8-7,8 (м, 7Н) ppm.
П р и м е р 6. К взвеси 486 мг магниевых стружек в 10 мл в атмосфере аргона в течение 30 мин по каплям добавляют раствор 6,1 г 2-(р-бромфенил)-2-фенил-1,3-диоксолана (по выкладке ФРГ N 2509474) в 50 мл ТГФ. Полученный коричневый раствор перемешивают при комнатной температуре в течение 1 ч, затем его охлаждают до 0оС и в течение 1 ч при 0оС по каплям к раствору добавляют 5,12 г дибромгексана и 0,1 ммоль дилитийтетрахлоркупрата (Synthesis, 1971, 303) в 10 мл ТГФ. Массу перемешивают в течение 18 ч при комнатной температуре, затем ее выпаривают. К остатку добавляют 100 мл насыщенного раствора хлористого аммония, после чего его экстрагируют три раза 100 мл диэтилового эфира. Органические фазы высушивают сульфатом магния и выпаривают. К остатку, содержащему 2-[4-(6-бромгексил)фенил]-2-фенил-1,3-диоксо-лан, добавляют 25 мл 33%-ного раствора диметиламина в этаноле. Полученную массу перемешивают в течение 24 ч при комнатной температуре. Раствор концентрируют, полученный остаток, содержащий 2-[4-[6-(диметиламино)гексил] фенил] -2-фенил-1,3-диоксолан, растворяют в 100 мл 1 н. соляной кислоты и промывают три раза 100 мл диэтилового эфира. Водную фазу, охлаждая ее льдом, с помощью 2 н. раствора едкого натра доводят до щелочной реакции и экстрагируют три раза 100 мл диэтилового эфира.
Органические фазы высушивают сульфатом магния и выпаривают. Сырой продукт очищают перегонкой в печи, снабженной трубкой с шаровым расширением, при 180-190оС и под давлением около 6,7 Па. Получают 2,55 г (41%) 4-[6-(диме- тиламино)гексил]бензофенона в виде желтоватого масла.
Это масло растворяют в 5 мл этанола и добавляют к горячему раствору 0,956 г фумаровой кислоты в 20 мл этанола. После добавления 50 мл диэтилового эфира и охлаждения до 0оС бесцветные кристаллы фильтруют на нутч-фильтре, промывают диэтиловым эфиром и высушивают. Получают 3 г (85%) 4-[6-(диметиламино)гексил] бензофенон-фумарата (1:1) с температурой плавления 87-89оС.
П р и м е р 7. а) 15 г 4-(3-бромпропил)ацетофенона и 10,8 мл пирролидина растворяют в 60 мл этанола и в течение 24 ч нагревают до 40оС. Реакционную массу затем выпаривают, а к остатку добавляют 250 мл этилацетата и 150 мл полунасыщенного раствора поваренной соли, после чего встряхивают массу. Водную фазу еще раз экстрагируют 250 мл этилацетата, а органические фазы еще раз промывают 150 мл полунасыщенного раствора поваренной соли. Собранные органические экстракты высушивают сульфатом магния и выпаривают. Получают 13,8 г (96% ) 4'-[3-(1-пирролидинил)пропил]ацетофенона в виде коричневатой жидкости; данные анализа масс-спектра m/e: М+ 431 (6,4), 84 (100), 42 (10,9).
б) 4 г 4'-[3-(1-пирролидинил)пропил] ацетофенона и 2 г бензальдегида растворяют в 80 мл метанола и, охлаждая раствор льдом, к нему в течение 15 мин добавляют раствор 9,55 г карбоната калия в 38 мл воды. Затем массе дают реагировать в течение 18 ч при комнатной температуре и в течение еще 18 ч при температуре около 40оС. Реакционную массу растворяют в 200 мл этилацетата и встряхивают 100 мл воды. Водную фазу еще раз экстрагируют 200 мл этилацетата, а органические фазы еще раз 100 мл полунасыщенного раствора поваренной соли. Собранные органические фазы высушивают сульфатом магния и выпаривают. Остаток хроматографируют на силикагеле ацетоном. Получают 4,5 г (81%) (Е)-3-фенил-4'-[3-(1-пирролидинил)-пропил] -акрилофенона в виде желтого масла; данные анализа масс-спектра m/e: M+ 319 (7,1), 131 (1,4), 84 (100), 42 (7,5). В целях получения гидрохлорида растворяют свободный амин в небольшом количестве этанола и затем к нему добавляют 10 М этанолического раствора соляной кислоты. После перекристаллизации из изопропанола получают чистый гидрохлорид; тпл. 211-213оС.
П р и м е р 8. 4,1 г 4-[3-(диметиламино)пропил]ацетофенона и 3,1 г 4-хлорбензальдегида растворяют в 80 мл метанола. К полученному раствору в течение 15 мин при комнатной температуре добавляют раствор 6,4 г гидроокиси натрия в 32 мл воды. Массу перемешивают в течение 48 ч. Для дальнейшей обработки массы ее выливают на 150 мл ледяной воды. Выпавшие кристаллы фильтруют на нутч-фильтре, промывают три раза 30 мл воды и высушивают при 40оС под пониженным давлением. Получают 6,36 г (97%) (Е)-3-(4-хлорфенил)-4'-[3-(диметиламино)пропил]акрилфенона; Tпл 83-86оС.
4-[3-(диметиламино)припил]ацетофе- нон можно получить аналогично примеру 7а).
П р и м е р 9. 3 г (Е)-3-(4-хлорфенил)-4'-[3-(диметиламино)пропил]-акрилофенона растворяют в 100 мл этанола. После добавления 300 мг 5%-ного палладия на угле гидрируют раствор. После поглощения теоретического количества воды отфильтровывают катализатор, а фильтрат выпаривают. Остаток растворяют в этаноле. Затем с помощью 10 М раствора соляной кислоты в этаноле его переводят в гидрохлорид, который осаждается после добавления гексана. Перекристаллизацией из смеси этилацетата и этанола (5:1) получают 1,44 г (43%) гидрохлорида 3-(4-хлорфенил)-4'-[3-(диметиламино)пропил] -пропиофенона; тпл 161-164оС.
П р и м е р 10. Аналогично примерам 7б и 8 можно получить нижеприведенные соединения: а) (Е)-3-(4-метилфенил)-4'-[3-(диметиламино]пропил]-акрилофенон; тпл76,5-77,5оС;
б) (Е)-3-(4-изопропилфенил)-4'-[3-(диметиламино)пропил]-акрилофенон; тпл 44-45оС;
в) (Е)-3-(4-метоксифенил)-4'-[3-(диметиламино)пропил]-акрилофенон в виде масла; 1Н-ЯМР (CDCl3, 400 МГц): 2,24 (с, 6Н); 3,86 (с, 3Н) ppm;
г) (Е)-3-[(4-трифторметил)фенил]-4'-[3-(диметиламино)-пропил]-акрилофенон; тпл= 71,5-73,5оС;
д) (Е)-3-(3-метилфенил)-4'-[3-(диметиламино)пропил] -акрилофенон в виде масла; 1Н-ЯМР (CDCl3, 400 МГц): 2,25 (с, 6Н); 2,40 (с, 3Н) ppm;
е) (Е)-3-(4-нитрофенил)-4'-[3-(диметиламино] пропил] -акрилофенон; тпл 97-99оС;
ж) (Е)-3-(3-хлорфенил)-4'-[3-(диметиламино] пропил] -акрилофенон в виде воска;
масс-спектр m/e: М+ 327 (1,1), 205 (0,8), 143 (0,9), 58 (100);
з) (Е)-3-фенил-4'-[3-(этилметиламино] пропил]-акрилофенон в виде масла; масс-спектр m/e: М+ 307 (4), 72 (100) (применяемый в качестве исходного вещества 4-[3-(этилметиламино)пропил] ацетофенон получают аналогично примеру 7а);
и) гидрохлорид (Е)-3-(4-трет.бутилфенил-4'-[3-(диметиламино]пропил]акрилофе- нона; тпл= 217-219оС (из этанола);
й) гидрохлорид (Е)-3-(3,5-дихлорфенил)-4'-[3-(диметиламино]пропил]акрилофенона; тпл > 230оС (с разл.);
к) гидрохлорид (Е)-3-(4-фторфенил)-4'-[3-(диметиламино]-пропил]акрилофенона; тпл= 219-220,5оС (из этанола);
л) гидрохлорид (Е)-3-(2-метилфенил)-4'-[3-(диметиламино]-пропил]акрилофенона; тпл 150,5-152оС (из смеси этанола и толуола).
П р и м е р 11. Аналогично примеру 9 можно получить нижеприведенные соединения: а) 3-(4-метилфенил)-4'-[3-(диметиламино)пропил] пропиофенон в виде масла; 1Н-ЯМР (CDCl3, 400 МГц): 2,22 (с, 6Н); 2,32 (с, 3Н) ppm;
б) 3-(4-изопропилфенил)-4'-[3-(диметиламино)пропил] -пропиофенон в виде масла; масс-спектр m/e: М+ 337 (5), 133 (4), 58 (100);
в) 3-(4-метоксифенил)-4'-[3-(диметиламино(пропил]пропиофенон в виде сиропа; масс-спектр m/e: М+ 325 (4), 121 (9), 58 (100);
г) 3-(4-трифторметилфенил)-4'-[3-(диметиламино)пропил]-пропиофенон в виде масла; масс-спектр m/e: М+ 363 (2), 159 (3), 58 (100);
д) 3-(4-метилфенил)-4'-[3-(диметиламино)пропил]пропиофенон в виде масла; масс-спектр m/e: М+ 309 (10,4), 145 (3,8), 105 (7,0), 58 (100);
е) 3-(3-хлорфенил)-4'-[3-(диметиламино)пропил]пропиофенон в виде масла; масс-спектр m/e: М+ 329 (1,0), 125 (2,5), 58 (100);
ж) 3-фенил-4'-[3-(этилметиламино)пропил]пропиофенон в виде сиропа; масс-спектр m/e: М+ 309 (6), 294 (1), 91 (6), 72 (100);
з) 3-(4-трет.бутилфенил)-4'-[3-(диметиламино)пропил]-пропиофенон в виде масла; масс-спектр m/e: М+ 351 (34,7), 131 (32,8), 91 (34,5), 58 (100);
и) 3-(4-фторфенил)-4'-[3-(диметиламино)пропил]пропиофенон в виде сиропа; масс-спектр m/e: М+ 313 (3), 109 (11), 58 (100);
й) 3-(2-метилфенил)-4'-[3-(диметиламино)пропил]пропиофенон; тпл145-147оС (из ТГФ);
к) 3-фенил-4'-[3-(1-пирролидинил)пропил]пропиофенон в виде сиропа; масс-спектр m/e: М+ 321 (7), 91 (6), 84 (100).
П р и м е р 12. Аналогично примеру 1 можно получить, например, следующие соединения: а) 4-[5-(диметиламино)пентил]бензофенон в виде бесцветного масла (выход 96%).
Масс-спектр m/e: М+ 295 (2% М+), 100 (3%), 58 (100%).
б) 4-[8-(диметиламино)октил] бензофенон в виде бесцветного масла (выход 88%).
Масс-спектр m/e: М+ 337 (2% М+), 149 (2,8%), 105 (3,3%), 58 (100%).
П р и м е р 13. а) К раствору 10 г 7-хлоргептанола [Rec. Trav. Chim. Pays-Bas, 99, 87 (1980)] и 18,7 мл триэтиламина в 100 мл метиленхлорида в атмосфере аргона при 0оС в течение 1 ч по каплям добавляют раствор 13,3 г р-толуолсульфонилхлорида в 100 мл метиленхлорида. Реакционную массу перемешивают в течение 6 ч при комнатной температуре, после чего ее промывают два раза 200 мл 2 н. соляной кислоты, два раза 200 мл насыщенного раствора бикарбоната натрия и один раз 200 мл насыщенного раствора хлорида натрия. Затем высушивают органическую фазу сульфатом магния и концентрируют ее. Полученный сырой 7-хлор-гептил-р-толуолсульфонат (19,5 г, 96%) растворяют в 40 мл ТГФ с 0,6 ммоль тетрахлоркупрата дилития. Этот раствор охлаждают до 0оС в атмосфере аргона. Затем в течение 2 ч по каплям добавляют раствор бромида фенилмагния, полученного из 3,1 г магниевой стружки и 20 г бромбензола в 80 мл ТГФ. Полученную реакционную массу перемешивают в течение 18 ч при комнатной температуре и выпаривают.
К остатку добавляют 200 мл насыщенного раствора хлористого аммония и три раза экстрагируют диэтиловым эфиром (по 200 мл). Органические фазы высушивают сульфатом магния и выпаривают. Остаток перегоняют в высоком вакууме при 100-110оС и под давлением около 13,4 Па. В результате получают 13 г (86%) 7-хлоргептилбензола в виде бесцветной жидкости.
б) 1,07 г 7-хлоргептилбензола и 0,89 г 4-хлорбензоилхлорида растворяют в 10 мл нитробензола, после чего в атмосфере аргона с охлаждением льдом добавляют 0,82 г хлористого алюминия. Полученную смесь перемешивают в течение 18 ч при комнатной температуре, после чего смесь смешивают со 100 мл ледяного 2 н. раствора соляной кислоты и три раза экстрагируют диэтиловым эфиром (по 50 мл). Органические фазы промывают сначала 50 мл 10%-ного раствора бикарбоната натрия и потом 50 мл насыщенного раствора хлорида натрия, после чего их высушивают сульфатом магния и концентрируют. Потом удаляют нитробензол, перегоняя массу при 80-100оС под давлением 20 Па. В результате перегонки при 240-250оС и под давлением около 13,4 Па получают 1,32 г (74%) 4-хлор-4'-(7-хлоргептил)бензофенона в виде желтоватого масла.
Данные 1Н-ЯМР-анализа (CDCl3): 1,2-1,9 (м, 9Н), 1,71 (т, J 7,5 Гц, 2Н), 3,53 (т, J 7,2 Гц, 2Н), 7,2-7,9 (м, 8Н) ppm.
в) 1,32 г 4-хлор-4'-(7-хлоргептил)бензофенона и 0,85 г йодида натрия нагревают при температуре дефлегмации в 20 мл этилметилкетона. Через 24 ч выпаривают реакционную массу. Остаток взвешивают в 50 мл воды и экстрагируют три раза этилацетатом (по 50 мл). Собранные органические фазы промывают 50 мл насыщенного раствора хлорида натрия, высушивают сульфатом магния и выпаривают. Получают 1,5 г (90%) сырого 4-хлор-4'-(7-йодгептил)бензофенона в виде желтоватого масла, которое без дальнейшей очистки применяют на следующей стадии способа.
г) К этому сырому маслу добавляют 10 мл 33%-ного раствора диметиламина в этаноле. Массу перемешивают в течение 24 ч при комнатной температуре, потом ее выпаривают. Остаток растворяют в 50 мл 1 н. соляной кислоты и промывают три раза диэтиловым эфиром (по 50 мл). Охлаждая водную фазу льдом, ее доводят до щелочной реакции с помощью 2 н. раствора едкого натра. После этого ее экстрагируют три раза диэтиловым эфиром (по 50 мл). Органические фазы высушивают сульфатом магния и выпаривают. Сырой продукт хроматографируют на силикагеле (100 г) смесью метиленхлорида, метанола и хлористого аммония (90: 10: 1). Получают 0,6 г (51%) 4-хлор-4'-[7-(диметиламино)гептил]бензофенона в виде бесцветного масла. Масс-спектр m/e: 357 (0,6% М+), 128 (4%), 58 (100%).
П р и м е р 14. Аналогично примеру 13 можно получить нижеприведенные соединения: а) 4-бром-4'-[7-(диметиламино)гептил]бензофенон в виде бесцветного масла (выход 38%). После добавления эфирной соляной кислоты получают соответствующий гидрохлорид с тпл 122-123оС (выход 85%).
б) 2,4-дихлор-4'-[7-(диметиламино)гептил] бензофенон в виде бесцветного масла (выход 73%).
Масс-спектр m/e: 391 (0,3% М+), 173 (1,3%), 128 (4,2%), 58 (100%).
в) 4-[4-(диметиламино)гептил] бензоил] бензонитрил в виде бесцветного масла (выход 68%).
Масс-спектр m/e: 348 (0,6% М+), 128 (2,7%), 58 (100%).
г) 4-[7-(аллилметиламино)гептил] бензофенон (выход 66%) в виде бесцветного масла.
Масс-спектр m/e: 349 (1,5% М+), 167 (4,3%), 84 (100%).
д) 4-[7-(аллилметиламино)гептил] -4'-бромбензофенон в виде желтоватого масла (выход 62%).
Масс-спектр m/e: 427 (0,7% М+), 400 (3,3%), 84 (100%).
е) 4-[4'-[7-(аллилметиламино)гептил] бензоил]бензонитрил в виде желтого масла (выход 46%).
Масс-спектр m/e: 374 (2% М+), 345 (8%), 84 (100%).
ж) Гидрохлорид 4-бром-4'-[6-(диметиламино)гексил]бензофенона (выход 72%) в виде бесцветных кристаллов с тпл 115-117оС.
з) 4-[6-(аллилметиламино)гексил] -4'-бромбензофенон (выход 77%) в виде бесцветного масла.
Масс-спектр m/e: 415 (0,8% М+), 413 (0,8%), 386 (2%), 84 (100%).
и) 3-бром-4'-[6-(диметиламино)гексил]бензофенон (выход 48%) в виде желтоватого масла.
Масс-спектр m/e: 387 (0,3% М+), 114 (7%), 58 (100%).
й) 4-[6-диметиламино)гексил]-2'-метилбензофенон (выход 36%) в виде желтоватого масла.
Масс-спектр m/e: 325 (0,9% М+), 114 (8%), 58 (100%).
к) 4-[6-(диметиламино)гексил] -4'-нитробензофенон (выход 80%) в виде желтого масла.
Масс-спектр m/e: 354 (0,4% М+), 114 (6%), 58 (100%).
л) 2-хлор-4'-[6-(диметиламино)гексил]бензофенон (выход 59%). Обработкой соединения эфирным раствором соляной кислоты получают соответствующий гидрохлорид с тпл 176-178оС (выход 86%).
м) 4-[7-(диметиламино)гептил]-2-метилбензофенон в виде бесцветного масла.
Масс-спектр m/e: 337 (1,5% М+), 128 (6%), 105 (4%), 77 (3%), 58 (100%).
П р и м е р 15. Соединение 4-[7-(диметиламино)гептил]бензофенон применяют в качестве действующего начала для получения таблеток при следующих соотношениях компонентов, мг/таблетку:
Действующее начало 200
Сахар молочный,
измельченный в порошок 100 Повидоне К 30 15 Крахмал карбокси-
метиловый натриевый 10 Тальк 3 Стеарат магния 2
Вес таблетки 330 мг. Действующее начало хорошо смешивают с измельченным в порошок молочным сахаром. Полученную массу смачивают водным раствором вещества Повидоне К 30 и замешивают. Полученную массу гранулируют, высушивают и пропускают через сито. Гранулят смешивают с остальными компонентами, а полученную массу спрессовывают в таблетки подходящей величины.
Использование: в медицине как обладающее микостатической активностью. Сущность изобретения: производные аминоалкилбензола формулы I: (см.рис.), где R1 и R2 - водород, низший алкил или вместе - неразветвленный алкилен C2-4 O - алкилен C4-11 минимум с двумя атомами углерода между двумя свободными валентностями; у - простая связь; R - водород, Cl, CF3 NO2 низший алкил или фармацевтически приемлемая соль с кислотой. 3 табл.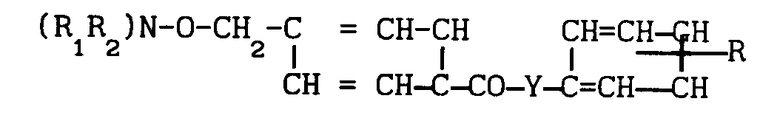
Производные аминоалкилбензола общей формулы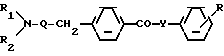
где R1 и R2 каждый водород, низший алкил или вместе образуют неразветвленный C2 C4-алкилен;
Q C4 C11-алкилен минимум с двумя атомами углерода между двумя свободными валентностями;
Y простая связь;
R обозначает, что кольцо незамещено или замещено галогеном, трифторметилом, нитрогруппой или низшим алкилом,
или их фармацевтичестки приемлемые соли с кислотами.
| Патент США 4072760, кл | |||
| Устройство для сортировки каменного угля | 1921 |
|
SU61A1 |
| Чугунный экономайзер с вертикально-расположенными трубами с поперечными ребрами | 1911 |
|
SU1978A1 |
