Изобретение относится к производным оксазолидинона. В частности, оно относится к производным оксазолидин-2-она общей формулы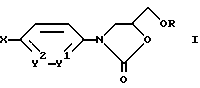
где R обозначает низший алкил;
X обозначает циклоалкенил; бицикло[2,2,1]гепт-2-ил, необязательно замещенный фенил-2-оксо-5-метоксиметилоксазолидинилом; бицикло[2,2,1]гепт-5-ен-2-ил; адамантил; или циклоалкил либо пиперидин, необязательно одно- или многократно замещенные галогеном, амино, низшим алкилом, нитрилом, оксогруппой, гидроксиимино, этилендиоксигруппой или -OR1,
где R обозначает -CH(C6H5)2, -(CH2)nC6H5, низший алкил, водород, -(CH2)nNHCOCH3, -(CH2)nNH2 -(CH2)nSOCH3,-(CH2)nCN, -(CH2)nSCH3, -(CH2)nSO2CH3, -CO-низший алкил, -COC6H5, необязательно замещенный оксазолидиниловой группой;
или замещенные на =CR2R3, где R2 обозначает водород или низший алкил; R3 обозначает водород, нитрил, низший алкил, фенил или COO-низший алкил;
или замещенные на -(CH2)nR4, где R4 обозначает нитрил, амино, -NHCOCH3, -COC6H5гал, фенил или гидроксильную группу;
или замещенные на -COR5, где R5 обозначает низший алкил, -CH=CH-C6H5-C6H5CF3, -O-C(CH3)3 или -O-низший алкил;
или замещенные на -NR6R7, где R6 обозначает водород или -COCH3; R7 обозначает -COCH3, бензил или -(CH2)nNHCOC6H4гал;
n обозначает 1-3;
Y1 обозначает -CH= или -N=;
Y2 обозначает -CH=, -C(OH)=, -C(NO2=), -C(NH2)=, -C(гал)= или -N=,
а также к фармацевтически приемлемым солям основных соединений формулы I с кислотами.
Эти соединения и соли являются новыми и отличаются ценными фармакологическими свойствами, так как обладают способностью ингибировать моноаминооксидазу (МАО). Благодаря такой активности соединения формулы I и их фармацевтически приемлемые соли могут применяться для лечения состояний депрессии, состояний паники и страха, нарушения умственных функций головного мозга и заболеваний нервно-дегенеративного характера, как болезнь Паркинсона и болезнь Альцгеймера.
Настоящее изобретение также направлено на получение соединений общей формулы I и их фармацевтически приемлемых солей как таковых и в качестве фармацевтических активных веществ, на разработку способа получения этих соединений и солей, на разработку содержащих их лекарственных средств для борьбы и соответственно предупреждения состояний депрессии, состояний паники и страха, заболеваний нервно-дегенеративного характера, как болезнь Паркинсона и болезнь Альцгеймера, и на изготовление таких лекарственных средств, на применение соединений общей формулы I и их фармацевтически приемлемых солей для борьбы или предупреждения болезней, соответственно для улучшения здоровья, а также на применение соединений общей формулы I и их фармацевтически приемлемых солей для изготовления лекарственных средств для борьбы и соответственно предупреждения состояний депрессии, состояний паники и страха, заболеваний нервно-дегенеративного характера, как болезнь Паркинсона и болезнь Альцгеймера.
Употребляемое в описании данной заявки понятие "низший алкил" обозначает линейные либо разветвленные насыщенные углеводородные радикалы, как метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор.-бутил, трет.-бутил и т. п.
Понятие "циклоалкил" обозначает насыщенные циклические углеводороды с числом атомов углерода в цикле предпочтительно 3-7, как, например, циклопропан, циклобутан, циклопентан, циклогексан или циклогептан.
Галоген обозначает фтор, хлор, бром или йод.
Понятие "уходящая группа" обозначает в рамках настоящего изобретения такие известные группы, как галоген, предпочтительно бром, арилсульфонилокси, алкилсульфонилокси и т.п.
Под реагентами, из которых получают карбонил, в рамках данной заявки могут пониматься, например, диэтилкарбонат, фосген или их аналоги.
Понятие "фармацевтически приемлемые соли" включает в себя соли неорганических и органических кислот, как соляная кислота, бромистоводородная кислота, серная кислота, фосфорная кислота, лимонная кислота, муравьиная кислота, малеиновая кислота, уксусная кислота, янтарная кислота, винная кислота, метансульфокислота, паратолуолсульфокислота и т.п.
К предпочтительным соединениям формулы I относятся те из них, в которых X представляет собой циклогексил или замещенный циклогексил, R обозначает метил, и Y1 и Y2 обозначают СН. Предпочтительными заместителями циклогексила являются такие, как оксогруппа, гидроксильная группа, с гидроксиимино, метокси, а также -O-(CH2)2-R8, где R8 обозначает -CN или -CH2NH2. Таковыми соединениями являются, например, следующие:
(RS)-3-(4-циклогексилфенил)-5-гидроксиметилоксазолидин-2-он;
(RS)-3-(4-циклогексилфенил)-5-метоксиметилоксазолидин-2-он;
(R)-3-(4-циклогексилфенил)-5-метоксиметилоксазолидин-2-он;
(RS)-3-[4-(4- оксоциклогексил)фенил]-5-метоксиметилоксазолидин-2-он;
(RS)-3-[4- (транс-4-гидроксициклогексил)-фенил]-5-метоксиметил- оксазолидин-2-он;
(RS)-3-[4-(4- гидроксииминоциклогексил)фенил] -5-метоксиметил-оксазолидин-2-он;
(R)-3-[4-транс-4-гидроксициклогексил)-фенил]-5-метоксиметил- оксазолидин-2-он;
(RS)-3-[4-(транс-4-метоксициклогексил)-фенил] -5- метоксиметил-оксазолидин-2-он;
(R)-3-[4-(4-оксоциклогексил)-фенил] -5-метоксиметилоксазолидин-2-он;
(RS)-3-[4-[транс-4-(2- цианоэтокси)циклогексил] фенил]-5-метоксиметилоксазолидин-2-он;
(RS) -3-[4-(транс-4-ацетоксициклогексил)фенил] -5-метоксиметилокса- золидин-2-он;
(RS)-3-[4-цис- и транс-4-гидроксиметилциклогексил)фенил]- 5-метоксиметилоксазолидин-2-он; и
(RS)-3-[4-цис- либо транс-4- гидрокси-4-метилциклогексил)фенил]-5-метоксиметил-оксазолидин-2-он.
Предпочтительными являются далее такие соединения, где Х обозначает бицикло[2,2,1] гепт-2-ил или бицикло[2,2,1]гепт-5-ен-2-ил, например следующие соединения:
3-[(1RS, 2RS, 4SR)-4-бициклo[2,2,1] гeпт-2-илфeнил]-5- метоксиметилоксазолидин-2-он и
3-[1RS, 2SR,4RS)-4-бицикло [2,2,1] гепт- 5-ен-2-ил-фенил]-5-метоксиметилоксазолидин-2-он.
Также к предпочтительным относятся такие соединения, в которых X представляет собой циклогексил или пиперидин, замещенный на -CH2CN, -COCH=CH-C6H5, -O(CH2)NH2 или -OCH2CN, R обозначает метил и Y1 и Y2 обозначают CH-группу, например следующие соединения:
(RS)-3-[4- (транс-4-цианометилциклогексил) фенил]-5- метоксиметилоксазолидин -2-он
(R)-3-[4-(транс-4-цианометилциклогексил) фенил] -5-метоксиметилоксазолидин -2-он
(E)-(RS)-5-метоксиметил-3-[4-[1-(3- фенилакрилоил)пиперидин-4-ил]-фенил] оксазолидин-2-он
(R)-3-[4- (транс-4-цианометоксициклогексил) фенил] -5- метоксиметилоксазолидин- 2-он
(RS)-3-[4-(транс-4- цианометоксициклогексил) фенил]-5- метоксиметилоксазолидин-2-он
гидрохлорид (RS)-3-[4-[транс-4-(2- аминоэтокси)циклогексил]фенил]-5-метоксиметилоксазолидин-2-она (1:1)
(R)-3-[4-[транс-4-(3-аминопропокси) циклогексил] фенил]-5- метоксиметилоксазолидин-2-он.
Соединения общей формулы I и их фармацевтически приемлемые соли могут быть получены согласно изобретению следующим образом:
а) соединение формулы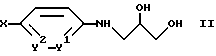
где X, Y1 и Y2 имеют указанные выше значения, циклизуют с помощью реагента, из которого выделяют карбонил, и при определенных условиях алкилируют, или
б) соединение формулы I, где R обозначает водород, алкилируют, или
в) соединение формулы I, где R обозначает бензил, подвергают каталитическому гидрированию, или
г) соединение формулы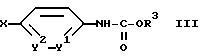
где X, Y1 и Y2 имеют указанные выше значения и R3 обозначает низший алкил или бензил, подвергают взаимодействию с соединением формулы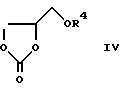
где R4 обозначает алкил или бензил, или
д) соединение общей формулы
где R, Y1 и Y2 имеют указанные выше значения и A обозначает уходящую группу, обрабатывают реагентом, из которого выделяют заместитель X, или
е) соединение формулы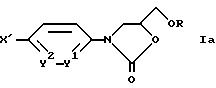
где X' обозначает (C5-C7)-циклоалкенильный остаток, который, как это расшифровано в формуле I для циклоалкила или пиперидина, может быть замещен,
и Y1, Y2 и R имеют указанные выше значения,
подвергают каталитическому гидрированию до соответствующего насыщенного циклоалкильного соединения, или
ж) соединение формулы I, где X обозначает замещенный на этилендиокси циклоалкил, гидрируют, или
з) соединение формулы I, где X обозначает циклоалкильный либо пиперидиновый остаток, замещенные -O-(CH2)2-CN, =CH-CN или -CH2CN, гидрируют до аминосоединения, или
и) соединение формулы I, где X обозначает замещенные оксогруппой циклоалкильный либо пиперидиновый остаток, трансформируют, а именно:
переводят оксогруппу в гидроксииминогруппу, или
восстанавливают ее до гидроксильной группы, или
переводят ее с помощью соответственно замещенных аминов в -NH(CH2)nNHCOC6H4R3-группу, где R3 и n имеют указанные выше значения, или
переводят ее в метиленовую группу, бензилиденовую группу, диметилметиленовую группу, метиленнитриловую группу либо метоксикарбонилметиленовую группу, или
переводят ее в этилендиоксигруппу, или
переводят ее в аминогруппу, или
переводят ее с помощью соответствующего фосфоната в =CH-CN-группу, или
к) соединение формулы I, где X обозначает замещенный гидроксильной группой циклоалкильный либо пиперидиновый остаток,
переводят гидроксильную группу с помощью акрилонитрила в -O(CH2)2CN-группу, или
алкилируют либо ацилируют ее с помощью соответствующих алкил-, арил- или ацилгалогенидов, или
галогенируют ее соответствующими галогенирующими средствами, или
трансформируют до соответствующих сульфаниловых соединений и при определенных условиях окисляют затем эти последние до сульфиниловых либо сульфониловых соединений, или
л) в соединении формулы I, где X обозначает замещенный на гидроксильную группу циклоалкильный остаток, дегидрируют этот последний до циклоалкенильного остатка, или
м) в соединении формулы I, где X обозначает замещенный на -O(CH2)n-SOCH3-группу циклоалкильный либо пиперидиновый остаток,
переводят эту группу в -O(CH2)nCN-группу, или
н) соединение формулы I, где X обозначает циклоалкильный остаток, пиперидиновый остаток либо защищенный пиперидиновый остаток, не связанный по атому азота,
ацилируют с помощью соответствующих ацилирующих средств, или
о) в соединении формулы I, где X обозначает замещенный на метиленовую группу циклоалкильный либо пиперидиновый остаток,
переводят эту группу в гидроксиметиловую группу или переводят ее в 4-гидрокси-4-метиловую группу, и
п) при необходимости основное соединение формулы I переводят с помощью соответствующей кислоты в фармацевтически приемлемую соль.
Соединение формулы II согласно варианту а) способа по изобретению может быть циклизовано в соединение формулы I путем обработки реагентом, из которого выделяют карбонил. Предпочтительно циклизацию осуществляют по известной методике с помощью диэтилкарбоната. При этом работают следующим образом: соединение формулы II растворяют в растворителе, например толуоле, обрабатывают диэтилкарбонатом и после добавки метанолового раствора метилата натрия перемешивают при нагреве в течение нескольких часов. Условия проведения реакции, а именно температура, продолжительность, растворители и т.д., могут варьироваться в зависимости от особенностей используемого реакционноспособного производного.
Согласно варианту б) способа по изобретению алкилируют гидроксильную группу. Это алкилирование может осуществляться по общеизвестным методам. Для метилирования целесообразно применять в качестве алкилирующего средства диметилсульфат. При этом можно работать таким образом, чтобы алкилируемое соединение сначала растворять в соответствующем растворителе, например толуоле, а затем обрабатывать диметилсульфатом, гидросульфатом тетрабутиламмония и раствором гидроксида натрия при интенсивном перемешивании. Условия реакции можно варьировать в зависимости от особенностей алкилирующего средства, соответственно алкилируемого соединения.
Если R представляет собой бензил, то каталитическое гидрирование можно осуществлять до R, обозначающего водород (вариант в) способа). В качестве катализатора могут применяться никель, платина или палладий. В качестве растворителей при каталитическом гидрировании пригодны, например, вода, спирт, метанол, уксусный эфир, ледяная уксусная кислота либо смеси этих растворителей. Предпочтительно простой бензиловый эфир растворяют в спирте и гидрируют при комнатной температуре с помощью палладиевого катализатора. Гидрирование осуществляют по известной методике в аппаратуре для встряхивания или автоклаве.
Согласно варианту г) способа по изобретению соединения формулы I могут быть получены также взаимодействием соединений формулы III и формулы IV.
Целесообразно при этом обрабатывать алкиловый эфир карбаминовой кислоты формулы III 4-(алкоксиметил)-1,3-диоксолан-2-оном формулы IV и перемешивать в течение нескольких часов при нагреве. Другая возможность получения соединений формулы I представлена вариантом д) способа. В этом случае соединения формулы V обрабатывают реагентом, из которого выделяют заместитель X. Если A в формуле V в качестве отщепляемой группы обозначает атом галогена, в частности бром или йод, то обменную реакцию целесообразно осуществлять следующим образом: оксазолидинон формулы V в течение нескольких часов подвергают обменной реакции в диметилформамиде (ДМФ) с добавками трифенилфосфина, дихлорида бис(трифенилфосфин) палладия (II), хлорида лития, 2,6-ди- трет.-бутил-пара-крезола и трибутилциклоалкенилстаннана, после чего проводят обработку с помощью известных методов.
Производные циклоалкила формулы I можно получать также, если соответствующие циклоалкенильные соединения формулы Iа подвергать каталитическому гидрированию (вариант е) способа). Пригодными для этой цели являются прежде всего 5-7-членные циклические системы, которые можно гидрировать до циклогексиловых, циклопентиловых или циклогептиловых соединений. Гидрирование осуществляют с помощью известных методов, предпочтительно с использованием палладиевого катализатора при комнатной температуре и при нормальном давлении.
Согласно варианту ж) способа замещенное на оксогруппу производное циклогексила формулы I получают путем гидролиза соединений формулы I, где X обозначает замещенный на этилендиокси циклогексил, проводимого по известной методике. Предпочтительно при этом готовят раствор в тетрагидрофуране, перемешивают в течение нескольких часов с соляной кислотой и обрабатывают натровым щелоком.
Соединение формулы I, где X представляет собой замещенный на -O-(CH2)2-CN-циклоалкил либо пиперидин, с помощью известных методов может гидрироваться в аминосоединение (вариант з) способа). Предпочтительно суспензию из борогидрида натрия, тетрагидрофурана (ТГФ) и трифторуксусной кислоты обрабатывают при комнатной температуре соответствующим соединением формулы I и перемешивают в течение нескольких часов. Затем следует переработка по обычной методике.
Если X обозначает замещенный на ацетонитриловую группу циклоалкил, то гидрирование до аминоэтиловой группы может проводиться в метаноловом аммиаке в присутствии никеля Ренея. Другой вариант, в котором получают аминосоединение формулы I, заключается в том, что сначала замещенный на =CH-CN-группу циклоалкил гидрируют с помощью Pd на угле, а затем, как это описано выше, преобразуют в аминосоединение.
Если X в формуле I обозначает замещенный на оксогруппу циклоалкильный либо пиперидиновый остаток, то эти соединения могут использоваться в качестве исходных веществ для получения других соединений формулы I (вариант и) способа).
Если X представляет собой циклоалканон, то оксогруппа может быть переведена в гидроксииминогруппу. Этот перевод осуществляют предпочтительно с помощью известных методов следующим образом: в соответствующее соединение формулы I при перемешивании и охлаждении добавляют водный раствор гидроксиламина. Соответствующее гироксииминосоединение после добавки водного раствора карбоната натрия выпадает в виде кристаллов.
Далее OH-замещенные производные формулы I могут быть получены путем восстановления оксогруппы. Это может осуществляться с помощью комплексного гидрида, как, например, борогидрида натрия или лития, в инертном в данных условиях реакции органическом растворителе, как метанол, этанол и т.п. Восстановление проводят преимущественно при комнатной температуре.
Замещенные оксогруппой производные циклоалкила либо пиперидина формулы I могут подвергаться взаимодействию также с аминами, например с N-(2-аминоэтил)-4-хлорбензамидом. Предпочтительно оба реагента обрабатывают при этом моногидратом пара-толуолсульфокислоты и растворяют в смеси толуола и ДМФ. Затем перемешивают в течение нескольких часов при нагреве и после охлаждения обрабатывают борогидридом натрия и соляной кислотой.
Далее согласно варианту и) соединения формулы I, где X обозначает замещенный на оксогруппу циклоалкильный либо пиперидиновый остаток, могут переводиться в метилен-, бензилиден-, диметилметилен-, метиленнитрил- или метоксикарбонилметилензамещенные производные циклоалкила либо пиперидина.
Целесообразно работать при этом следующим образом: в атмосфере аргона, например смеси из бромида метилтрифенилфосфония и амида натрия, или смеси из бромида изопропилтрифенилфосфония и амида натрия, или смеси из бромида бензилтрифенилфосфония и амида натрия или смеси бромида метоксикарбонилметилентрифенилфосфония и амида натрия суспендируют в ТГФ и перемешивают. Затем добавляют соответствующее оксоциклоалкильное либо оксопиперидиновое соединение формулы I и перемешивают в течение нескольких часов при нагреве. Последующую переработку проводят по обычной методике. Метиленовую группу при определенных условиях можно после этого повторно гидрировать.
Также согласно варианту и) способа можно получать соединения формулы I, где X обозначает замещенный на этилендиоксигруппу циклоалкильный либо пиперидиновый остаток. В этом случае соответственно замещенное оксоциклоалкильное или оксопиперидиновое соединение формулы I подвергают взаимодействию с этиленгликолем в присутствии моногидрата пара-толуолсульфокислоты в толуоле. Конечные соединения получают после перемешивания в течение нескольких часов при нагреве и обычной переработки.
Замещенный на оксогруппу циклоалкильный либо пиперидиновый остаток согласно варианту и) может переводиться в аминогруппу взаимодействием соответствующего оксосоединения предпочтительно с бензиламином и толуолсульфокислотой и последующим гидрированием в течение нескольких часов борогидридом натрия при комнатной температуре.
Оксоциклогексильная группа согласно варианту и) может быть переведена также в циклогексилиденацетонитриловую группу благодаря тому, что предпочтительно натрий растворяют в этаноле и этот раствор обрабатывают этаноловым раствором диэтилцианометилфосфоната, после чего осуществляют трансформацию с помощью соответствующего оксоциклогексильного соединения формулы I в конечное соединение.
Если X в формуле I обозначает замещенный на гидроксильную группу циклоалкильный либо пиперидиновый остаток, то также и эти соединения могут применяться в качестве исходных веществ для получения других соединений формулы I (вариант к) способа). Нитрилзамещенные соединения можно получать, например, взаимодействием производных гидроксициклоалкила либо пиперидина формулы I с акрилонитрилом в присутствии трет.-бутилата калия.
Полученные таким образом нитрилзамещенные соединения формулы I при определенных условиях могут трансформироваться в соответствующие аминосоединения путем их обработки сухим газообразным аммиаком, растворенным в метаноле, и гидрирования с помощью никеля Ренея.
Соединения формулы I, содержащие гидроксильную группу, с помощью хлоридов кислот могут быть ацилированы в соответствующие карбонилоксисоединения. Предпочтительно при этом замещенное гидроксильной группой циклоалкильное либо пиперидиновое соединение формулы I растворяют в растворителе, например в хлористом метилене, обрабатывают этот раствор пиридином и хлоридом кислоты, например ацетилхлоридом или бензоилхлоридом, и перемешивают в течение нескольких часов при комнатной температуре. Дальнейшую переработку проводят по известной методике.
Алкилирование, соответственно ацилирование соединений формулы I, содержащих гидроксильную группу, может осуществляться с помощью соответствующих алкил- или ацилгалогенидов, например, с помощью метилиодида, бензилбромида или хлордифенилметана. Эту трансформацию проводят по известным методам.
Галогенирование соединений формулы I, содержащих гидроксильную группу, также может проводиться по известной методике с помощью соответствующего галогенирующего средства. Так, например, фторциклоалкильное соединение формулы I образуется в результате взаимодействия гидроксициклоалкильного соединения со смесью из диэтиламинотрифторида серы и хлористого метилена после перемешивания при комнатой температуре.
Также согласно варианту к) способа соединения формулы I, содержащие гидроксильную группу, могут быть трансформированы в соответствующие сульфаниловые соединения. Эта трансформация может осуществляться так, что сначала гидроксильное соединение растворяют в диметилсульфоксиде, а затем обрабатывают ледяной уксусной кислотой и ангидридом уксусной кислоты и перемешивают в течение нескольких часов при комнатной температуре. Другие возможные варианты способа описаны в "Chemistry letters", стр.1277-1278, опубликованных Японским химическим обществом.
Соответствующее сульфиниловое соединение может быть получено при определенных условиях после осуществления описанного выше варианта путем окисления сульфанилового соединения. В качестве окислителя для этой цели особенно пригоден периодат натрия.
Соответствующее сульфониловое соединение может быть получено путем окисления сульфанилового соединения. Предпочтительно при этом в качестве окислителя используют хлорпербензойную кислоту. Пригодным для этой цели растворителем является, например, хлористый метилен.
Согласно варианту л) способа соединения формулы I, где X обозначает замещенный на гидроксильную группу циклоалкильный остаток, дегидрируют до циклоалкенильного остатка. Эту реакцию осуществляют предпочтительно взаимодействием гидроксильного соединения с трифенилфосфином и бензойной кислотой в тетрагидрофуране при комнатной температуре и последующей обработкой диэтиловым эфиром азодикарбоновой кислоты.
Согласно варианту м) способа, где X представляет собой замещенный на -O(CH2)n-SO-CH3-группу циклоалкильный либо пиперидиновый остаток, эту группу переводят в группу -O(CH2)nCN за счет того, что соответствующее сульфиниловое соединение растворяют в растворителе, например тетрагидрофуране, и преобразуют далее в присутствии иодида цинка и триметилсилилцианида. Эта реакция протекает в течение нескольких часов при комнатной температуре.
Соединение формулы I, где X представляет собой пиперидиновый или замещенный пиперидиновый остаток, не связанный по атому азота, либо циклоалкильный остаток, может быть ацилировано согласно варианту н) способа. Это ацилирование осуществляют предпочтительно с помощью соответствующих ацилирующих средств, например с помощью бензилбромида, ω-хлор-4-фторбутирофенона, трифторметилбензоилхлорида, ангидрида уксусной кислоты или хлорида коричной кислоты, по известной методике. В качестве растворителя при этом особенно пригодны диметилформамид или хлористый метилен.
Соединения формулы I, содержащие метиленовую группу в качестве заместителя циклоалкила либо пиперидина, также могут применяться как исходные вещества для получения других соединений формулы I (вариант о) способа).
Соответствующее соединение формулы I, где X обозначает замещенный метиленовой группой циклоалкильный либо пиперидиновый остаток, может переводиться в гидроксиметилзамещенное соединение путем гидроборирования этого соединения, растворенного в растворителе, например в ТГФ, борогидридом натрия в присутствии диметилсульфата и окисления промежуточного продукта с помощью H2O2.
4-метилензамещенные циклоалкильные соединения формулы I также могут быть трансформированы в соответствующие 4-гидрокси-4-метилзамещенные производные циклоалкила либо пиперидина. С этой целью ацетат ртути растворяют в ТГФ и обрабатывают 4-метилензамещенным циклоалкильным соединением формулы I. Затем перемешивают при комнатной температуре, обрабатывают натровым щелоком и подвергают взаимодействию с борогидридом натрия. Дальнейшую переработку проводят по обычной методике.
Соединение формулы I, где R обозначает бензил, может быть получено взаимодействием соответственно замещенного циклоалкил- либо пиперидинфенилэтоксикарбаминового эфира с бензилоксиметил-1,3-диоксолан-2- оном с помощью известных методов.
Фармацевтически приемлемые соли с учетом уровня техники и особенностей переводимого в соль соединения могут быть получены с помощью известных методов.
Применяемые согласно варианту а) способа в качестве исходных веществ соединения формулы II могут быть получены следующим путем: 4-циклогексиланилин или же другие соответственно замещенные анилины и (RS)-2,2-диметил-1,3- диоксолан-4-илметиловый эфир метансульфокислоты (описание см. Journ. Med. Chem. 27, 1176 [1934]) выдерживают в триэтиламине в течение нескольких часов при температуре порядка 140oС в тугоплавкой трубке. Затем после удаления растворителя остаток перемешивают с соляной кислотой, подщелачивают и перерабатывают далее по обычной методике.
Другая возможность состоит в проведении обменной реакции между 4-циклогексиланилином или другими соответственно замещенными анилинами и [(R)-2,2-диметил-1,3-диоксолан-4-метанолтолуол-4 -сульфонатом] в триэтиламине при температуре порядка 140oС. Далее перемешивают в течение нескольких часов и затем полученный продукт обрабатывают сначала HCl, а потом после перемешивания в течение приблизительно 1 ч подщелачивают натровым щелоком. Последующую переработку проводят по обычной и известной методике.
Используемое согласно варианту г) способа в качестве исходного вещества соединение формулы III известно и может быть получено следующим образом.
4-Циклоалкиланилин или другие соответственно замещенные анилины растворяют в растворителе, например в ТГФ и воде, обрабатывают бикарбонатом натрия, после чего подвергают обменной реакции с этиловым эфиром хлормуравьиной кислоты. Температура реакции не должна превышать при этом 20oС.
Соединения формулы IV известны и могут быть получены по способам, известным из существующих публикаций.
Требуемое для осуществления варианта д) способа соединение формулы V предпочтительно может быть получено следующим путем, причем в приведенном ниже примере Y2 обозначает азот.
Смесь из этилового эфира пиридин-3-илкарбаминовой кислоты, метоксиметил- 1,3-диоксолан-2-она и карбоната калия нагревают и перемешивают в течение нескольких часов. Образующееся соединение обрабатывают окислителем, предпочтительно 3-хлорпербензойной кислотой, после чего полученное соединение 5-метоксиметил-3-(1-оксипиридин-3-ил) оксазолидин-2-он бромируют и затем с помощью восстановителя, например трибромида фосфора, переводят в соединение формулы V. Это соединение может затем, как это описано выше, подвергаться взаимодействию с реагентом, из которого выделяют заместитель X. Пригодными для этой цели являются такие средства, которые в качестве радикала содержат, например, трибутилстанниловую группу. Получение названных соединений осуществляют с помощью известных методов.
Замещенные циклоалкильные соединения формулы Iа могут быть получены путем обработки соединений формулы V, где A обозначает отщепляемую группу, например галоген, реагентом, из которого выделяют заместитель X', где X' обозначает (C5-C7)-циклоалкенильный остаток, который, как это расшифровано в формуле I для циклоалкила, может быть замещен. Реактивная группа может представлять собой, например, триметилстанниловую группу. Предпочтительно обменную реакцию проводят в атмосфере аргона в присутствии дихлорида бис(трифенилфосфин) палладия (II) в ТГФ с помощью известных методов. Соединения, применяемые в вариантах б), в), е), ж), з), и), к), л), м), н), о) и п) в качестве исходных веществ, подпадают под формулу I и в соответствии с этим могут быть получены по методам, описанным для получения этих соединений согласно вариантам а), г) и д).
Соединения формулы I и их фармацевтически приемлемые соли обладают, как уже отмечалось выше, активностью, ингибирующей моноаминооксидазу (МАО). Благодаря этой активности соединения формулы I и их фармацевтически приемлемые соли могут применяться при лечении состояний депрессии, состояний паники и страха, нарушения умственных функций головного мозга и заболеваний нервно-дегенеративного характера. Примерами таких заболеваний являются гипомнезия, связанная с болезнью Паркинсона, первичная и вторичная деменция, например деменция типа болезни Альцгеймера или деменция, обусловленная неоднократным инфарктом, церебрально-сосудистые заболевания и последствия черепно-мозговых травм.
Ингибирующая МАО активность соединений по изобретению может определяться с помощью стандартных методов. Так испытуемые препараты подвергались описанному ниже тесту in vitro, по методу, аналогично описанному R.J. Wurtmann и J.A.Axelrod в Biochem. Pharmacol. 12, 1439- 1441 (1963).
Выделенный мозг крыс гомогенизировали в соотношении 1:9 (вес/объем) в 0,1 молярном буферном растворе фосфата калия (pH 7,4), после чего гомогенаты разбавляли в соотношении 1:4 (объем/объем) в том же самом буферном растворе и выдерживали при температуре -20oC. Для инкубации применяли смесь следующего состава:
100 мкл 1М фосфатного буфера (pH 7,4);
100 мкл солюбилизата испытуемой субстанции в воде или только в диметилсульфоксиде;
50 мкл гомогената мозга крыс;
и в качестве субстрата 50 мкл 14C-серотонина (5-HT), соответственно 14C-фенилэтиламина (ФЭА), соответственно по 100000 распадов в мин в соответствии с конечной концентрацией 2•10-4 мол/л, соответственно 2•10-5 мол/л.
Перед добавкой субстрата в течение 30 мин проводят предварительную инкубацию при 37oC. Инкубацию (в присутствии субстрата) осуществляют также при 37oC и она продолжается в течение 10 мин. Реакцию прекращают добавкой 200 мкл 2н. соляной кислоты. Для экстракции деаминированных продуктов, в зависимости от применения 5-HT или ФЭА в качестве субстрата встряхивают в течение 10 мин с 5 мл диэтилового эфира, соответственно с 5 мл н-гептана, после чего центрифугируют, водную фазу удаляют вымораживанием в ванне из сухого льда и органическую фазу декантируют в счетных стаканах.
На основании полученных с помощью счетчиков β-значений определяют активность МАО в сравнении с контрольными гомогенатами.
В условиях проводившихся опытов активность имеет линейный характер по отношению ко времени и концентрации гомогенизата. Значения IC50 определяли графическим путем как log кривой концентрация/активность.
В качестве IC50 определяли такую концентрацию испытуемой субстанции, которая снижала активность МАО при использовании субстрата 5HT, соответственно ФЭА на 50%.
Выявленная таким путем активность некоторых соединений по изобретению представлена в нижеследующей таблице 1, где приводятся значения IC50.
Соединения формулы I, а также их фармацевтически приемлемые аддитивные соли кислот могут находить применение в качестве лекарственных средств, например в виде фармацевтических препаратов. Фармацевтические препараты могут вводиться орально, например в форме таблеток, лаковых таблеток, драже, твердожелатиновых и мягкожелатиновых капсул, растворов, эмульсий или суспензий. Введение препаратов может осуществляться также ректально, например в форме суппозиториев, или же парэнтерально, например в форме растворов для инъекций.
Для изготовления таблеток, лаковых таблеток, драже и твердожелатиновых капсул в соединения формулы I и их фармацевтически приемлемые кислотно-аддитивные соли могут добавляться фармацевтически инертные, неорганические либо органические эксципиенты. В качестве таковых, например, для таблеток, драже и твердожелатиновых капсул могут использоваться лактоза, кукурузный крахмал или его производные, тальк, стеариновая кислота или ее соли и т.д.
Для использования в качестве эксципиентов для мягкожелатиновых капсул пригодны, например, растительные масла, воск, жиры, полутвердые и жидкие полиолы и т.д.
При изготовлении растворов и сиропов в качестве эксципиентов можно использовать воду, полиолы, сахарозу, инвертный сахар, глюкозу и т.д.
В качестве эксципиентов для инъекционных растворов пригодны, например, вода, спирты, полиолы, глицерин, растительные масла и т.д.
В качестве эксципиентов для суппозиториев пригодны, например, природные или отвержденные масла, воск, жиры, полужидкие или жидкие полиолы и т.д.
Фармацевтические препараты могут содержать, кроме того, еще и консерванты, растворители, стабилизаторы, смачивающие агенты, эмульгаторы, вещества, улучшающие вкус, красители, ароматизирующие средства, соли для изменения осмотического давления, буферы, покрытия или антиокислители. Они могут содержать также другие ценные в терапевтическом отношении вещества.
Согласно изобретению соединения общей формулы I и их фармацевтически приемлемые аддитивные соли кислот могут применяться для борьбы и соответственно предупреждения состояний депрессии, нарушения умственных функций головного мозга и заболеваний нервно-дегенеративного характера, как болезнь Паркинсона и болезнь Альцгеймера. Дозировку можно варьировать в широких пределах, естественно в зависимости в каждом отдельном случае от индивидуальных особенностей пациента. При оральном введении суточная доза может назначаться в основном из расчета от приблизительно 10 до 100 мг соединения общей формулы I, причем указанная верхняя граница отнюдь не является предельной, если это окажется целесообразным.
Предлагаемое изобретение поясняется подробнее на нижеследующих примерах, которые никоим образом не ограничивают его. Все температуры в примерах указаны в градусах Цельсия.
Пример 1
(RS)-3-(4-пиклогексилфенил)-5-гидроксиметилоксазолидин-2-он
2,4 г (9,62 ммолей) (RS)-3-(4- циклогексил-фениламино)-пропан-1,2-диола растворяли в 50 мл толуола, обрабатывали 1,25 г (10,6 ммолей) диэтилкарбоната и 0,4 мл 1М раствора метилата натрия в метаноле и перемешивали в течение ночи при температуре масляной бани 110oC. Растворитель отгоняли в водоструйном насосе, остаток обрабатывали водой и 1н. соляной кислотой и экстрагировали с помощью уксусного эфира. Органическую фазу промывали водой, сушили над сульфатом магния и растворитель отгоняли. Из уксусного эфира/гексана получали 1,4 г продукта в виде кристаллов желтоватого цвета. Далее из маточного раствора получали еще 0,9 г чистого продукта. Т.пл.: 167-167,5o.
Пример 2
(RS) -3-(4-циклогексилфенил)-5-метоксиметилоксазолидин-2-он
1,5 г (5,45 ммолей) (RS)-3-(4-циклогексилфенил)-5-гидроксиметил-оксазолидин-2-она обрабатывали 15 мл толуола, 1,6 мл диметилсульфата (16,3 ммолей), 185 мг гидросульфата тетрабутиламмония и раствором из 1,09 г (27,2 ммолей) гидроксида натрия в 1,3 мл воды и интенсивно перемешивали в течение 30 мин. Затем реакционную смесь сливали на ледяную воду и экстрагировали с помощью уксусного эфира. Органическую фазу промывали водой, сушили над сульфатом магния и растворитель отгоняли. В результате получали 1,6 г желтого масла. После обработки уксусным эфиром/гексаном получали 0,7 г бесцветных кристаллов. Т.пл.: 69-70o.
Пример 3
(S)-3-(4-циклогексилфенил)-5-гидроксиметилоксазолидин-2-он
Раствор из 1,40 г (5,6 ммолей) (S)-3-(4-циклогексил-фениламино)-пропан-1,2-диола в 50 мл толуола обрабатывали 0,73 г (6,2 ммолей) диэтилкарбоната и 0,5 мл 1М раствора метилата натрия и перемешивали в течение ночи при температуре масляной бани 110o. После отгонки растворителя обрабатывали водой и 5 мл 1н. соляной кислоты, после чего экстрагировали с помощью уксусного эфира. Органическую фазу промывали раствором из поваренной соли, сушили над сульфатом магния и растворитель отгоняли. Из уксусного эфира/гексана получали 1,35 г (S) -3-(4-циклогексилфенил)-5-гидроксиметилоксазолидин-2-она в виде бесцветных кристаллов. Т.пл.: 166-168o. [α]D= +51,25o (с=0,8/CH3OH).
Пример 4
(S)-3-(4-циклогексилфенил)-5-метоксиметилоксазолидин-2-он
В раствор из 1,0 г (3,63 ммоля) (S)-3-(4-циклогексилфенил)-5- гидроксиметилоксазолидин-2-она в 10 мл толуола добавляли 250 мг гидросульфата тетрабутиламмония, раствор из 0,73 г гидроксида натрия (18,2 ммолей) в 1 мл воды и 1,1 мл диметилсульфата (10,9 ммолей) и перемешивали в течение 1 ч при 110o. После охлаждения обрабатывали 10 мл воды и экстрагировали с помощью уксусного эфира. После концентрирования растворителя получали 1,2 г кристаллической смеси, которую хроматографировали на 30-кратном количестве силикагеля. Однородные согласно тонкослойной хроматограмме (ТСХ) фракции уксусный эфир/гексан (1: 1) соединяли и растворитель отгоняли. В результате получали 0,85 г (S)-3-(4-циклогексилфенил)-5-метоксиметилоксазолидин-2-она в виде бесцветных кристаллов. Т.пл.: 90-92o. [α]D=39,7o (с=0,7/CHCl3).
Пример 5
(R)-3-(4-циклогексилфенил)-5- гидроксиметилоксазолидин-2-он
6,2 г (24,8 ммоля) (R)-3-(4- циклогексилфениламино)-пропан-1,2-диола преобразовывали и перерабатывали аналогично тому, как это описано в примере 3. В результате получали 6,3 г (93%) кристаллов бежевого цвета. Т.пл.: 166-168o. [α]D=-48,9o (с=0,7/CH3OH).
Пример 6
(R)-3-(4-циклогексилфенил)-5-метоксиметилоксазолидин-2-он
2,0 г (7,26 ммолей) (R)-3-(4-циклогексилфенил)- 5-гидроксиметилоксазолидин- 2-она преобразовывали и перерабатывали аналогично тому, как это описано в примере 4. В результате получали 1,91 г (91%) (R)-3-(4- циклогексилфенил)-5-метоксиметилоксазолидин-2-она в виде бесцветных кристаллов. Т. пл.: 86-88o. [α]D=-37,7o (с=0,3/CHCl3).
Пример 7
(RS)-3-[4-(4-оксоциклогексил)фенил]-5-метоксиметилоксазолидин-2-он
10 г (38,3 ммолей) [4-(4-оксоциклогексил)фенил] этоксикарбаминового эфира, 10 г (75 ммолей) (RS)-4-(метоксиметил)-1,3-диоксолан-2-она и 0,5 г карбоната калия интенсивно перемешивали в течение 4 ч при температуре масляной бани более 160o. После охлаждения обрабатывали 50 мл воды и экстрагировали с помощью уксусного эфира. Полученное после отгонки растворителя желтое масло (15,5 г) хроматографировали на 30-кратном количестве силикагеля. Однородные согласно ТСХ (уксусный эфир/гексан 7:3) фракции хлористый метилен/уксусный эфир (15:1) соединяли и растворитель отгоняли. В результате получали 5,54 г (48%) бесцветного продукта. Т.пл.: 114-116o.
Пример 8
(RS)-3-[4-(транс- 4-гидроксициклогексил)-фенил] -5-метоксиметил- оксазолидин-2-он
5,0 г (16,44 ммолей) (RS)-5-метоксиметил-3-[4-(4-оксоциклогексил)- фенил] оксазолидин-2-она растворяли в 150 мл этанола в условиях тепла и после охлаждения при перемешивании обрабатывали 620 мг (16,4 ммолей) борогидрида натрия, охлаждали в течение ночи и затем отгоняли растворитель. Полученный маслянистый остаток обрабатывали водой и 1н. соляной кислотой, после чего продукт реакции растворяли в уксусном эфире. Органическую фазу промывали водой, сушили над сульфатом магния и растворитель отгоняли. Полученное бесцветное масло (5,3 г) растворяли в уксусном эфире, обрабатывали гексаном до помутнения и выпавший осадок через 1 ч отсасывали. В результате получали 3,3 г однородного согласно ЯМР-спектру спирта. Т.пл.: 109-110o.
Пример 9
(RS)-3-[4- (4-гидроксииминоциклогексил)фенил] -5-метоксиметил-оксазолидин-2-он
Приготовленный обычным путем из кетона оксим перекристаллизовывали из уксусного эфира. Т.пл.: 146-147o.
Пример 10
(RS)-5-бензилоксиметил-3-[4-(4-оксоциклогексил)фенил]-оксазолидин-2-он
20 г (76,5 ммолей) [4- (4-оксоциклогексил)фенилэтоксикарбаминового эфира, 23,8 г (RS)-4- (бензилоксиметил)-1,3-диоксолан-2-она и 0,25 г карбоната калия интенсивно перемешивали в течение 3 ч, после чего перерабатывали и хроматографировали аналогично тому, как это описано в примере 7. Полученный бензиловый эфир перерастворяли из трет.-бутилметилового эфира/гексана. Т.пл. : 126,5-128o.
Пример 11
(RS)-5-гидроксиметил- 3-[4-(4-оксоциклогексил)фенил]-ксазолидин-2-он
4,9 г (12,9 ммолей) (RS)-5-бензилокси-3-[4-(4-оксоциклогексил)-фенил] оксазолидин-2-она растворяли в 500 мл этанола в условиях тепла и после добавки Pd/C 5% гидрировали при комнатной температуре. После отделения катализатора растворитель отгоняли и полученное масло хроматографировали на 30-кратном количестве силикагеля. Элюаты хлористый метилен/уксусный эфир (1:4) соединяли и растворитель отгоняли. Из уксусного эфира/гексана получали 3,2 г продукта. Т.пл.: 151,5-152,5o.
Пример 12
(RS)-3-[4-(транс-гидроксициклогексил)фенил] -5- гидроксиметил-оксазолидин-2-он
0,5 г (1,73 ммоля) (RS)-5- гидроксиметил-3-[4-(4-оксоциклогексил)фенил] оксазолидин-2-она растворяли в 500 мл этанола и восстанавливали и обрабатывали 65 мг (1,73 ммоля) борогидрида натрия аналогично тому, как это описано в примере 8. В результате получали кристаллический продукт (0,38 г). Т.пл.: 180-182o.
Пример 13
(RS)-3-(4-циклопропилфенил)-5-метоксиметилоксазолидин-2-он
3,0 г (10,48 ммолей) (RS)-5-метоксиметил- 3-(4-бромфенил)-оксазолидин-2-она, 6,29 г (6,29 ммолей) трифенилфосфина, 0,88 г (1,25 ммоля) дихлорида бис(трифенилфосфин)палладия (II), 3,73 г хлорида лития, небольшое количество (на кончике шпателя) 2,6-ди-трет.-бутил-пара-крезола и 6,94 г трибутилциклопропилстаннана в 50 мл ДМФ перемешивали в течение 6 ч при 120o. Реакционную смесь обрабатывали водой и 1н. натровым щелоком и экстрагировали с помощью простого эфира. Полученный сырой продукт хроматографировали на 30-кратном количестве силикагеля. Однородные согласно ТСХ (силикагель, уксусный эфир/гексан 1:1) экстракты соединяли и растворитель отгоняли. Из трет.-бутилметилового эфира/гексана получали 0,3 г бесцветных кристаллов. Т.пл.: 58-60o.
Пример 14
(R)-3-[4-(4-оксоциклогексил)фенил]-5- метоксиметилоксазолидин-2-он
5,0 г (19,13 ммолей) [4-(4- оксоциклогексил)фенил] этоксикарбаминового эфира подвергали обменной реакции с 3,0 г (S)-4-метоксиметил-1,3-диоксолан-2-она и перерабатывали аналогично тому, как это описано в примере 7. Из трет.- бутилметилового эфира/гексана получали 1,25 г продукта в виде кристаллов желтоватого цвета. Т.пл.: 92-93o, [α]D=-38,1o (с=1, CHCl3)
Пример 15
(R)-3-[4-(транс-4-гидроксициклогексил)фенил] - 5-метоксиметил-оксазолидин-2-он
1,0 г кетона аналогично тому, как это описано в примере 8, восстанавливали с помощью борогидрида натрия в этаноле и перерабатывали. Из уксусного эфира/гексана выделяли 0,7 г бесцветных кристаллов. Т.пл.: 133,5-134,5o. [α] D=-38,6o (с=0,7, CHCl3).
Пример 16
(RS)-3-[4-(транс-4-метоксициклогексил)фенил] -5- метоксиметил-оксазолидин-2-он
0,5 г (1,64 ммоля) (RS)-3-[4-(транс-4- гидроксициклогексил)фенил]-5-метоксиметилоксазолидин-2-она растворяли в 5 мл диметилформамида, обрабатывали 0,51 мл (8,2 ммолей) метилиодида и 107 мг (2,46 ммоля) дисперсии гидрида натрия (55%) и перемешивали в течение ночи при 40o. Затем реакционную смесь сливали на ледяную воду и экстрагировали с помощью простого эфира. Органические фазы промывали водой, сушили над сульфатом магния и растворитель отгоняли. Полученный гель хроматографировали на 30-кратном количестве силикагеля с помощью хлористого метилена/уксусного эфира (1:4). После обработки трет.- бутилметиловым эфиром/гексаном выпадали кристаллы. В результате получали 0,33 г продукта. Т.пл.: 82-83o.
Пример 17
3-[(1RS, 2RS,4RS)-4-бицикло[2.2.1]-гепт-2-илфенил]-5-метоксиметил- оксазолидин-2-он (R:S=1:1)
Раствор из 56 мг диацетата бис(трифенилфосфин)палладия (II), 0,5 г (RS)-3-(4-иодфенил)-5- метоксиметилоксазолидин-2-она, 0,16 г бицикло [2,2,1] гепт-2-ена, 0,5 мл пиперидина, 1 мл диметилформамида и 0,15 мл муравьиной кислоты перемешивали в течение 3 ч в атмосфере аргона. Реакционную смесь разбавляли 50 мл уксусного эфира и выпавший осадок отделяли. Фильтрат дважды экстрагировали соответственно порциями по 20 мл воды, органическую фазу сушили над сульфатом магния и растворитель отгоняли. Полученное коричневое масло хроматографировали на 30-кратном количестве силикагеля с помощью уксусного эфира/гексана (7: 3). В результате после кристаллизации в уксусном эфире/гексане в условиях холода получали 0,43 г однородного продукта. Рацемическая смесь диастереомеров (1:1) имела т.пл. 83-84o.
Пример 18
3-[(1RS,2SR,4RS)-4-бицикло [2.2.1] гепт-5-ен-2-ил-фенил]-5-метоксиметил-оксазолидин-2-он (R:S = 1:1)
1,0 г (RS)-3-(4-иодфенил)-5-метоксиметил-оксазолидин-2-она трансформировали и перерабатывали аналогично тому, как это описано в примере 17. В результате получали 0,37 г рацемической смеси диастереомеров (1:1) в виде кристаллического продукта. Т.пл.: 46-48o.
Пример 19
Смесь из (RS)- и (SR)-5-метоксиметил-3- [4-[(RS)-3-оксоциклопентил] фенил] оксазолидин-2-она
Суспензию из 0,44 г (RS)-3-(4- иодфенил)-5-метоксиметил-2-оксазолидинона и 15 мг тетракис(трифенилфосфин)палладия в 1,1 мл 2-циклопентенона и 1,8 мл триэтиламина перемешивали в атмосфере аргона в течение 24 ч при 80o. Затем реакционную смесь охлаждали и после добавки 100 мл 2н. соляной кислоты экстрагировали с помощью этилового эфира уксусной кислоты. Органическую фазу сушили над сульфатом магния и концентрировали. Остаток (0,6 г коричневого масла) хроматографировали на силикагеле 60 с помощью смесей этилового эфира уксусной кислоты/гексана (1: 3-1: 1), после чего получали 0,26 г (RS)- и (SR)-5-метоксиметил-3-[4-[(RS)-3- оксоциклопентил] фенил] оксазолидин-2-она в виде бесцветного масла. 1H-ЯМР (CDCl3) част/млн: 7,53 (d, 2H), 7,26 (d, 2H), 4,76 (m, 1H), 4,06 (t, 1H), 3,93 (t, 1H), 3,65 (d, 2H), 3,44 (s, 3H), 3,40 (m, 1H). 2.65 (m. 1H), 2,36 (m, 4H), 1,98 (m, 1H).
Пример 20
(RS)-5-метоксиметил-3-[4-(4-оксопиперидин-1-ил)-фенил] -оксазолидин-2-он
В атмосфере аргона в течение 3 ч нагревали до 160o смесь из 6,0 г этилового эфира 4-(4-оксопиперидин-1-ил)- фенилкарбаминовой кислоты, 5,0 г 4-метоксиметил-1,3-диоксолан-2-она и 0,6 г карбоната калия. Затем реакционную смесь охлаждали, обрабатывали хлористым метиленом и водой и фазы разделяли. Органическую фазу сушили над сульфатом магния и концентрировали. Остаток (7,8 г) хроматографировали с помощью этилового эфира уксусной кислоты в качестве растворителя над силикагелем 60. В результате получали 0,6 г (RS)-5-метоксиметил-3-[4- (4-оксопиперидин-1-ил)фенил] оксазолидин-2-она. 1H-ЯМР (CDCl3) част/млн: 7,46 (d, 2H), 6,98 (d, 2H), 4,76 (m, 1H), 3,99 (t, 1H), 3,88 (t, 1H), 3,64 (d, 2H), 3,57 (t, 4H), 3,44 (s, 3H), 2,56 (t, 4H).
Пример 21
(RS)-3-[4-(4-гидроксипиперидин-1-ил)фенил] -5- метоксиметил-2-оксазолидин-2-он
В раствор из 0,6 г (RS)-5- метоксиметил-3-[4-(4-оксопиперидин-1-ил)-фенил] оксазолидин-2-она в 25 мл метанола и 2,5 мл воды добавляли 0,3 г борогидрида натрия. Эту смесь перемешивали в течение 24 ч при комнатной температуре и затем концентрировали. Остаток растворяли в хлористом метилене и промывали 10%-ным водным раствором аммиака. Органическую фазу сушили над сульфатом магния и концентрировали. Остаток (0,6 г) хроматографировали на силикагеле 60 с помощью смесей этилового эфира уксусной кислоты/гексана (1: 1-2: 1). После кристаллизации из этилового эфира уксусной кислоты получали 0,1 г (RS)-3-[4-(4-гидроксипиперидин-1- ил)фенил]-5-метоксиметил-2-оксазолидин-2-она. Т.пл.: 149-152o.
Пример 22
(RS)-3-[5-(1,4-диоксаспиро [4,5] декан-8-ил)-пиридин-2- ил]-5-метоксиметилоксазолидин-2-он
Раствор из 0,9 г (RS)-3-[5-(1,4- диоксаспиро[4,5]дец-7-ен-8-ил)-пиридин-2-ил] -5- метоксиметилоксазолидин-2-она и 0,4 г палладия на угле (10%) в 150 мл метанола гидрировали при комнатной температуре и при нормальном давлении. После отфильтровывания катализатора концентрировали и остаток перекристаллизовывали из диэтилового эфира. В результате получали 0,35 г (RS)-3-[5-(1,4-диоксаспиро [4,5] декан-8-ил)-пиридин- 2-ил]-5-метоксиметилоксазолидин-2-она. Т.пл. 117-121o.
Пример 23
(RS)-5-метоксиметил-3-[5-(4-оксоциклогексил)-пиридин-2-ил] - оксазолидин-2-он
Раствор из 0,5 г (RS)-3-[5-(1,4- диоксаспиро[4,5]декан-8-ил)-пиридин-2-ил] -5-метоксиметил-оксазолидин- 2-она в 20 мл тетрагидрофурана и 5 мл 2н. соляной кислоты перемешивали в течение 4 ч при комнатной температуре. Затем добавляли 10 мл 2н. натрового щелока и несколько раз экстрагировали с помощью этилового эфира уксусной кислоты. Органические фазы соединяли, сушили над сульфатом магния и концентрировали. После хроматографии с помощью этилового эфира уксусной кислоты на силикагеле получали 0,4 г (RS)-5- метоксиметил-3-[5-(4-оксоциклогексил) пиридин-2-ил] -оксазолидин- 2-она. 1H-ЯМР (CDCl3) част/млн: 8,22 (s, 1H), 8,18 (d, 1H), 7,58 (d, 1H), 4,78 (m, 1H), 4,27 (t, 1H), 4,09 (t, 1H), 3,64 (m, 2H), 3,43 (s, 3H), 3,08 (m, 1H), 2,51 (m, 4H), 2,04 (шир., 4H).
Пример 24
(RS)-3-[5-транс-4-гидроксициклогексил)пиридин-2-ил]-5- метоксиметилоксазолидин-2-он
Раствор из 0,4 г (RS)-5-метоксиметил- 3-[5-(4-оксо-циклогексил)-пиридин-2-ил] -оксазолидин-2-она в 25 мл метанола и 2,5 мл воды обрабатывали 0,2 г борогидрида натрия и перемешивали в течение 16 ч при комнатной температуре. Затем концентрировали, остаток растворяли в хлористом метилене и промывали водой. Водную фазу экстрагировали с помощью хлористого метилена, органические фазы соединяли, сушили над сульфатом магния и концентрировали. Остаток (0,4 г) перекристаллизовывали из уксусного эфира и в результате получали 0,2 г (RS)-3-[5-(транс-4- гидроксициклогексил)пиридин-2-ил]-5-метоксиметилоксазолидин-2-она. Т.пл.: 142-143o.
Пример 25
(RS)-3-[6-(1.4-диоксаспиро [4.5] декан- 8-ил)пиридин-3-ил]-5-метоксиметилоксазолидин-2-он
Раствор из 0.8 г (RS)-3-[6-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)-пиридин-3-ил] -5- метоксиметилоксазолидин-2-она и 1,1 г палладия на угле (10%) в 250 мл метанола гидрировали при комнатной температуре и при нормальном давлении. После отфильтровывания катализатора концентрировали. В результате получали 1,2 г (RS)-3-[6-(1,4-диоксаспиро [4,5]декан-8-ил)-пиридин-3-ил]- 5- метоксиметилоксазолидин-2-она. Далее на силикагеле 60 с помощью этилового эфира уксусной кислоты в качестве растворителя продолжали очищать пробу, которую затем кипятили с диэтиловым эфиром. Т.пл.: 93-94o.
Пример 26
(RS)-5-метоксиметил- 3-[6-(4-оксоциклогексил)-пиридин-3-ил] -оксазолидин-2-он
Раствор из 0,8 г (RS)-3-[6-(1,4-диоксаспиро[4,5]декан-8-ил-пиридин-3-ил] -5- метоксиметил-оксазолидин-2-она в 32 мл тетрагидрофурана и 8 мл 2н. соляной кислоты перемешивали в течение 4 ч при комнатной температуре. Затем добавляли 15 мл 2н. натрового щелока и несколько раз экстрагировали с помощью этилового эфира уксусной кислоты. Органические фазы соединяли, сушили над сульфатом магния и концентрировали. В результате получали 0,7 г (RS)-5-метоксиметил-3- [6-(4-оксо-циклогексил)-пиридин-3-ил] -оксазолидин-2-она. 1H-ЯМР (CDCl3) част/млн: 8,49 (s, 1H), 8,19 (d, 1H), 7,23 (d, 1H). 4,81 (m, 1H), 4,09 (t, 1H), 3,98 (t, 1H), 3,66 (m, 2H), 3,44 (s, 3H), 3,19 (m, 1H), 2,52 (m, 4H), 2,28 (m, 2H), 2,06 (m, 2H).
Пример 27
(RS)- 3-[6-транс-(4-гидроксициклогексил)-пиридин-3-ил]-оксазолидин-2-он
Раствор из 0,7 г (RS)-5-метоксиметил-3-[6-(4-оксоциклогексил)пиридин- 3-ил] -оксазолидин-2-она в 40 мл метанола и 4 мл воды обрабатывали 0,3 г борогидрида натрия и перемешивали в течение 16 ч при комнатной температуре. Затем концентрировали, остаток растворяли в хлористом метилене и промывали водой. Водную фазу сушили над сульфатом магния и концентрировали. Остаток (0,4 г) хроматографировали с помощью хлористого метилена/метанола (50:1) на силикагеле 60, после чего кипятили с гексаном. В результате получали 0,2 г (RS)-3-[6-транс- (4-гидроксициклогексил)пиридин-3-ил]-оксазолидин-2-она. Т. пл.: 134- 136o.
Пример 28
(RS)-3-[(4-циклогептил)фенил]-5-метоксиметилоксазолидин-2-он
4,18 г (16,0 ммолей) этилового эфира 4-циклогептилфенилкарбамидовой кислоты обрабатывали 1,3 г (16,0 ммолей) пиридина и 18 г (0,136 моля) (RS)-4-метоксиметил-1,3-диоксолан-2-она и перемешивали при температуре ванны 160o в течение 20 ч. После охлаждения реакционную смесь хроматографировали на 400 г силикагеля 60 с помощью простого эфира. После перекристаллизации из изопропилового эфира получали 2,05 г (RS)- 3-[(4-циклогептил)фенил]-5-метоксиметилоксазолидин-2-она в виде белого кристаллизата. Т.пл.: 70-72o.
Пример 29
(RS)-3-[(4-адамантан-1- ил) фенил]-5-метоксиметилоксазолидин-2-он
5,42 г (18,1 ммолей) этилового эфира 4-адамантан-1-ил-фенилкарбаминовой кислоты обрабатывали 1,43 г (18,1 ммолей) пиридина и 18,0 г (0,136 моля) (RS)- 4-метоксиметил-1,3-диоксолан-2-она и при температуре ванны 160o перемешивали в течение 20 ч. После охлаждения реакционную смесь хроматографировали на 500 г силикагеля 60 с помощью простого эфира. После перекристаллизации из хлористого метилена/простого эфира получали 1,4 г (RS)-3-[(4-адамантан-1-ил)фенил] -5-метоксиметил- оксазолидин-2-она в виде белого кристаллизата. Т.пл.: 145-147o.
Пример 30
(RS)-3-[4-[транс-4-(2-цианоэтокси)циклогексил] фенил] -5-метоксиметилоксазолидин-2-он
3,05 г (10,0 ммолей) (RS)-3-[4-транс-4- гидроксициклогексил)фенил]-5-метоксиметилоксазолидин-2-она растворяли в 150 мл хлористого метилена и последовательно обрабатывали 0,64 г (12,0 ммолей) акрилонитрила и 1,35 г (12,0 ммолей) трет.-бутилата калия. Реакционную смесь перемешивали в течение 18 ч при комнатной температуре, после чего ее промывали водой, органическую фазу сушили над сульфатом натрия, фильтровали и упаривали. Остаток (4,0 г) хроматографировали на 120 г силикагеля 60 с помощью уксусного эфира/н-гексана (6: 4). После перекристаллизации из уксусного эфира получали 1,8 г (RS)-3-[4-[транс-4-(2-цианоэтокси)циклогексил]фенил]-5- метоксиметилоксазолидин-2-она. Т.пл.: 82-84o.
Пример 31
Гидрохлорид (RS)-3-[4-[транс-4-(3- аминопропокси)циклогексил]-фенил]-5-метоксиметилоксазолидин-2-она (1:1)
545,3 мг (14,4 ммолей) борогидрида натрия, суспендированного в 20 мл абсолютного тетрагидрофурана, обрабатывали 1,64 г (14,4 ммолей) трифторуксусной кислоты в течение 5 мин при комнатной температуре. Затем в течение 15 мин при комнатной температуре добавляли по каплям 1,29 г (3,6 ммоля) (RS)-3-[транс-4-[4-(5-метоксиметил-2-оксо- оксазолидин-3-ил)-фенил] -циклогексилокси] -пропионитрила, растворенного в 20 мл абсолютного тетрагидрофурана. После перемешивания в течение 18 ч реакционную смесь упаривали, остаток распределяли на воду и хлористый метилен, органическую фазу промывали водой, сушили над сульфатом натрия, фильтровали и упаривали. Остаток (1,4 г) хроматографировали на 39 г силикагеля 60 с помощью хлористого метилена (насыщ. NH3)/метанола (98: 2). Продукт промывали в хлористом метилене водой, сушили над сульфатом натрия, фильтровали и фильтрат подкисляли эфирной соляной кислотой. После перекристаллизации из хлористого метилена/простого эфира получали 1,13 г гидрохлорида (RS)- 3-[4-[транс-4-(3-аминопропокси)циклогексил] фенил]-5- метоксиметилоксазолидин-2-она (1:1). Т.пл.: 173-175o.
Пример 32
(RS)-3-[4-[транс-4-[2-(4,5-дигидрооксазол-2-ил)бензоилокси] - циклогексил] фенил]-5-метоксиметилоксазолидин-2-он
В суспензию из 690 мг (15,8 ммолей) 55 %-ной дисперсии гидрида натрия в 20 мл абсолютного диметилформамида в атмосфере аргона в течение 25 мин по каплям добавляли раствор из 4,58 г (15,0 ммолей) (RS)-3-[4-транс-4- гидроксициклогексил)фенил] -5-метоксиметилоксазолидин-2-она в 30 мл абсолютного диметилформамида и перемешивали в течение 3 ч при комнатной температуре. Затем добавляли по каплям раствор из 3,81 г (15,0 ммолей) N-(2-брометил)фталимида в 20 мл абсолютного диметилформамида и перемешивали при комнатной температуре в течение ночи. Далее реакционную смесь обрабатывали 0,9 г (15,0 ммолей) ледяной уксусной кислоты и упаривали. Остаток растворяли в ледяной уксусной кислоте, промывали водой, сушили над сульфатом натрия, фильтровали и упаривали. После перекристаллизации из уксусного эфира получали 4,9 г (RS)-3-[4-[транс- 4-[2-(4,5-дигидрооксазол-2-ил)бензоилокси]-циклогексил] фенил]-5- метоксиметилоксазолидин-2-она. Т.пл.: 182-184o.
Пример 33
(RS)-3- [4-(транс-4-ацетоксициклогексил)фенил] -5-метоксиметилоксазолидин- 2-он
В раствор из 0,5 г (1,639 ммоля) (RS)-3-[4-транс-4-гидроксициклогексил) фенил] -5-метоксиметилоксазолидин-2-она в 20 мл хлористого метилена и 5 мл пиридина добавляли по каплям при перемешивании раствор из 0,58 мл ацетилхлорида (8,195 ммолей) в 10 мл хлористого метилена и продолжали перемешивать в течение 3 ч при комнатной температуре. Затем реакционную смесь сливали на ледяную воду, с помощью 3н. соляной кислоты подкисляли и экстрагировали с помощью хлористого метилена. Органическую фазу промывали водой, сушили над сульфатом магния и растворитель отгоняли. Ацетат кристаллизовался при обработке трет. - бутилметиловым эфиром. В результате получали 0,35 г (62%) (RS)-3-[4- (транс-4-ацетоксициклогексил)фенил] - 5- метоксиметилоксазолидин-2-она. Т.пл.: 78-79o.
Пример 34
(RS)-3-[4-(транс-4- бензоилоксициклогексил)фенил] -5-метоксиметилоксазолидин- 2-он
Раствор из 0,5 г (1,639 ммоля) (RS)-3-[4-(транс-4-гидрокси- циклогексил)фенил] -5-метоксиметилоксазолидин-2-она в 20 мл хлористого метилена и 5 мл пиридина обрабатывали по каплям раствором из 0,95 мл (8,195 ммолей) бензоилхлорида в 10 мл хлористого метилена и продолжали перемешивать в течение 3 ч при комнатной температуре. Затем реакционную смесь сливали на ледяную воду, с помощью 3н. соляной кислоты подкисляли и экстрагировали с помощью хлористого метилена. Органическую фазу промывали водой, сушили над сульфатом магния и растворитель отгоняли. Бензоат кристаллизовался при обработке трет. - бутилметиловым эфиром. В результате получали 0,52 г (77%) (RS)-3-[4- (транс-4-бензоилоксициклогексил)фенил] -5-метоксиметилоксазолидин-2- она. Т. пл.: 104-105o.
Пример 35
5-метокси-3-[4-[5-экзо-[4-(5-метоксиметил-2- оксооксазолидин-3-ил)-фенил]-бицикло [2.2.1] гепт-2-экзоил] фенил]- оксазолидин-2-он
При получении соединения из примера 18 в качестве побочного продукта образуется вышеназванное соединение, которое выделяли посредством хроматографии (силикагель, уксусный эфир/гексан (1:1)). Т.пл.: 152-154o.
Пример 36
Гидрохлорид (RS)-3-[4-[транс- 4-[2-(4-хлоробензиламино)этиламино]-циклогексил] фенил]-5- метоксиметилоксазолидин-2-она (1:1)
Раствор из 1,31 г (4,3 ммоля) (RS)-3-[4-(4-оксоциклогексил)фенил]-5-метоксиметилоксазолидин-2-она, 2,0 г (10 ммолей) N-(2-аминоэтил)-4-хлорбензамида и 37 мг (0,2 ммоля) моногидрата пара-толуолсульфокислоты в 20 мл толуола и 6 мл диметилформамида кипятили в течение 7 ч в водоотделителе. При перемешивании при комнатной температуре добавляли сначала 330 мг (8,7 ммолей) борогидрида натрия и через 30 мин 10 мл 1н. соляной кислоты. Органическую фазу отделяли. Водную фазу дважды экстрагировали с помощью хлористого метилена. Затем органические фазы сушили над сульфатом натрия, концентрировали и хроматографировали на силикагеле с помощью хлористого метилена/метанола (9: 1). Полученное твердое вещество перекристаллизовывали из этанола/простого эфира. В результате получали 200 мг белых кристаллов. Т.пл.: 233-235o.
Пример 37
(RS)- 5-метоксиметил-3-[4-(4-метиленциклогексил)фенил]-оксазолидин-2-он
В атмосфере аргона 5,76 г (13,8 ммолей) смесь из бромида метилтрифенилфосфония и амида натрия в 170 мл тетрагидрофурана перемешивали в течение 1 ч при комнатной температуре. Затем в течение 5 мин по каплям добавляли раствор из 4,20 г (13,8 ммолей) (RS)-3-[4-(4- оксоциклогексил)фенил]-5-метоксиметилоксазолидин-2-она. После кипячения с обратным холодильником в течение ночи сливали на воду и экстрагировали с помощью хлористого метилена. После сушки над сульфатом натрия и концентрирования хроматографировали с помощью уксусного эфира/н-гексана (1:2) на силикагеле. В результате получали 3,47 г (83%) твердого вещества. Затем небольшую пробу перекристаллизовывали из гексана. Т.пл.: 63-65o.
Пример 38
(RS)-3- [4-(цис- и транс-4-гидроксиметилциклогексил)фенил]-5- метоксиметилоксазолидин-2-он
В суспензию из 1,50 г (5,0 ммолей) (RS)- метоксиметил-3-[4-(4-метиленциклогексил)фенил]оксазолидин-2-она в 250 мл тетрагидрофурана добавляли 0,19 г (5,0 ммолей) борогидрида натрия и по каплям добавляли 0,46 мл (5,0 ммолей) диметилсульфата. После перемешивания в течение 4 ч при температуре приблизительно 40o охлаждали до 5o и добавляли 1,5 мл 2н. натрового щелока. Далее медленно добавляли по каплям 0,77 мл 30%-ного H2O2 (7,5 ммолей). Затем сливали на 100 мл насыщенного раствора поваренной соли, отделяли органическую фазу и водную фазу экстрагировали с помощью хлористого метилена. Органические фазы сушили над сульфатом натрия, концентрировали и хроматографировали на силикагеле с помощью хлористого метилена/метанола. В результате получали 350 мг бесцветного масла. МС: m/e (% основного пика): 319 (М+, 73), 246 (16), 233 (16), 170 (30), 158 (16), 144 (86), 118 (60), 95 (19), 91 (25), 77 (24), 71 (60), 45 (100).
Пример 39
(RS)-3-[4-(цис- либо транс-4-гидрокси- 4-метилциклогексил)фенил]-5-метоксиметил-осазолидин-2-он
Раствор из 525 мг (1,6 ммоля) ацетата ртути в 10 мл воды обрабатывали 10 мл тетрагидрофурана. После перемешивания в течение 15 мин в эту суспензию добавляли по каплям раствор из 500 мг (1,7 ммоля) (RS)-5-метоксиметил- 3-[4-(4-метиленциклогексил)фенил] оксазолидин-2-она в 10 мл тетрагидрофурана. Затем перемешивали в течение 1,5 ч при комнатной температуре. Далее добавляли сначала 10 мл 2н. натрового щелока и после перемешивания в течение 10 мин добавляли раствор из 200 мл (5,3 ммолей) борогидрида натрия в 10 мл 2н. натрового щелока. После перемешивания в течение 30 мин экстрагировали с помощью хлористого метилена, органическую фазу промывали насыщенным раствором поваренной соли и сушили над сульфатом натрия. После отгонки растворителя хроматографировали на силикагеле с помощью уксусного эфира/гексана (1:2). В результате получали 150 мг белых кристаллов. Т.пл.: 118-119o.
Пример 40
(RS)-3-[4-(1,4-диоксаспиро [4.5] дец-8- ил)-фенил]-5-метоксиметил]-оксазолидин-2-он
Раствор из 0,60 г (2 ммоля) (RS)-3-[4-(4-оксоциклогексил)фенил]-5- метоксиметилоксазолидинона, 0,15 мг (2,4 ммоля) этиленгликоля и 30 мг моногидрата пара-толуолсульфокислоты в 20 мл толуола кипятили в течение 7 ч в водоотделителе. Затем толуол отфильтровывали, остаток суспендировали в 10 мл 2н. натрового щелока и экстрагировали с помощью хлористого метилена. После сушки над сульфатом натрия растворитель отгоняли. После хроматографии на силикагеле с помощью простого эфира и перекристаллизации с помощью простого эфира получали 0,4 г белых кристаллов. Т.пл.: 120-123o.
Пример 41
(RS)-3-[4-(цис- 4-фторциклогексил)фенил]-5-метоксиметил-оксазолидин-2-он
0,5 г (RS)- 3-[4-(транс-4-гидроксициклогексил)фенил]-5-метоксиметил-оксазолидин- 2-она растворяли в 10 мл хлористого метилена и при комнатной температуре в течение 30 мин обрабатывали смесью из 2,2 мл диэтиламино-трифторида серы и 5 мл хлористого метилена. После перемешивания в течение 2 ч при комнатной температуре смесь сливали на 50 мл ледяной воды и дважды экстрагировали с помощью хлористого метилена. Органическую фазу промывали раствором хлорида натрия, раствором гидрокарбоната натрия и повторно раствором хлорида натрия, после чего сушили над сульфатом магния и концентрировали. Остаток, 0,6 г желтого меда, хроматографировали на 30-кратном количестве силикагеля с помощью хлористого метилена/уксусного эфира (19:1) и в результате получали 80 мг (RS)-3-[4-(цис-4-фторциклогексил)фенил]-5- метоксиметилоксазолидин-2-она в виде бесцветного масла. 1H-ЯМР (CDCl3) част./млн: 7,47 (d, 2H), 7,24 (d, 2H), 4,88 (шир., 1H), 4,74 (m, 1H), 4,04 (t, 1H), 3,91 (m, 1H), 3,64 (d, 2H), 3,43 (s, 3H), 2,53 (m, 1H), 2,16 (m, 2H), 1,72 (m, 6H).
Пример 42
Гидрохлорид (RS)-3-[4-(транс-4-аминоциклогексил)фенил]-5- метоксиметилоксазолидин-2-она (1:1)
а) Смесь из 1,0 г (RS)-3-[4-(4- оксоциклогексил)фенил]-5-метоксиметил-оксазолидин-2-она, 0,1 г пара- толуолсульфокислоты и 0,43 мл бензиламина в 100 мл толуола кипятили в течение 3 ч в водоотделителе. Растворитель удаляли, остаток растворяли в 50 мл метанола и перемешивали со 150 мг борогидрида натрия в течение ночи при комнатной температуре. Растворитель удаляли, остаток обрабатывали 1н. соляной кислотой и промывали уксусным эфиром. Водную фазу подщелачивали натровым щелоком и экстрагировали с помощью хлористого метилена. Хлористометиленовую фазу сушили над сульфатом магния и концентрировали. После кристаллизации остатка из уксусного эфира/гексана получали 0,6 г окрашенных в бежевый цвет кристаллов смеси (1:1) из (RS)-3-[4-цис- и (RS)-3-[4-(транс- бензиламиноциклогексил)фенил]-5-метоксиметилоксазолидин-2-она. Т.пл. 93- 94o.
б) 0,5 г смеси из (RS)-3-[4-цис- и (RS)-3-[4-(транс- бензиламиноциклогексил)фенил] - 5-метоксиметилоксазолидин- 2-она растворяли в 50 мл этанола при добавке одного эквивалента 1н. соляной кислоты и гидрировали в присутствии палладия на угле (10%) в качестве катализатора при комнатной температуре и нормальном давлении. После добавки диэтилового эфира выпадали 0,3 г гидрохлорида (RS)-3-[4- (транс-4-аминоциклогексил)фенил]-5-метоксиметилоксазолидин-2-она (1: 1) в виде бесцветных кристаллов. Т.пл.: > 250o. 1H-ЯМР (ДМСО) част./млн: 8,12 (шир., 3H), 7,47 (d, 2H), 7,25 (d, 2H), 4,81 (m, 1H), 4,09 (t, 1H), 3,77 (m, 1H), 3,58 (m, 2H), 3,32 (s, 3H), 3,03 (m, 1H), 2,47 (m, 1H), 2,06 (m, 2H), 1,80 (m, 2H), 1,50 (m, 4H).
Пример 43
(RS)-3-[4-(транс-4- бензгидрилоксициклогексил)фенил] -5-метоксиметил-оксазолидин-2-он
0,5 г (RS)-3-[4-(транс-4-гидрокси-циклогексил)-фенил] -5- метоксиметилоксазолидин-2-она нагревали в 5 мл этилдиизопропиламина с 0,35 мл хлордифенилметана до 100o и перемешивали в течение 7 ч при этой температуре. Затем реакционную смесь охлаждали, подкисляли 1н. соляной кислотой и экстрагировали с помощью уксусного эфира. Органическую фазу нейтрализовали промыванием водой, сушили над сульфатом магния и концентрировали. Остаток хроматографировали на 30-кратном количестве силикагеля с помощью смесей хлористого метилена/этилового эфира уксусной кислоты. В результате получали 0,3 г (RS)-3-[4-(транс-4-бензгидрилокси-циклогексил)фенил] -5- метоксиметилоксазолидин-2-она в виде бесцветных кристаллов. Т.пл.: 146- 148o.
Пример 44
Смесь (9:1) из (RS)-3-[4-(транс- и цис-4- бензилоксициклогексил)-фенил] -5-метоксиметилоксазолидин-2-она
1,0 г (RS)-3-[4-(гидроксициклогексил)фенил]- 5-метоксиметилоксазолидин- 2-она обрабатывали в 10 мл диметилформамида 0,21 г гидрида натрия и при комнатной температуре. После добавки 50 мл этилового эфира уксусной кислоты трижды промывали водой. Водные фазы экстрагировали с помощью этилового эфира уксусной кислоты. Органические фазы соединяли, сушили над сульфатом магния и концентрировали. После хроматографии остатка (1,6 г) на 30-кратном количестве силикагеля с помощью смесей хлористого метилена/уксусного эфира получали 0,8 г желтого масла, которое кристаллизовалось из трет.-бутилметилового эфира. В результате получали 0,6 г смеси (9:1) (RS)-3-[4-(транс- и цис-4- бензилоксициклогексил)фенил] -5-метоксиметил-оксазолидин-2-она в виде бесцветных кристаллов. Т. пл.: 82-84o. 1H-ЯМР (CDCl3) част./млн: 7,46 (d,2H), 7,35 (m, 5H), 7,23 (d, 2H), 4,74 (m, 1H), 4,60 (s, 2H), 3,99 (t, 1H), 3,90 (m, 1H), 3,63 (d, 2H), 3,43 (s, 3H), 3,40 (m, 1H), 2,50 (m, 1H), 2,22 (m, 2H), 1,91 (m, 2H), 1,46 (m, 4H).
Пример 45
(R)-3-[4-(транс-4-метоксициклогексил)фенил] -5- метоксиметил-оксазолидин-2-он
1,4 г (R)-3-[4-(транс-4- гидроксициклогексил)фенил]-5-метоксиметил-оксазолидин-2-она обрабатывали в 14 мл диметилформамида, 0,3 г гидрида натрия и 1,43 мл метилиодида. Реакционную смесь перемешивали в течение ночи при 40o. После добавки 100 мл диэтилового эфира трижды промывали водой. Водные фазы экстрагировали с помощью диэтилового эфира. Органические фазы соединяли, сушили над сульфатом магния и концентрировали. После хроматографирования остатка (1,5 г) на 30-кратном количестве силикагеля с помощью смесей хлористого метилена/уксусного эфира получали 1,3 г бесцветного масла, которое кристаллизовалось из трет.-бутилметилового эфира. В результате получали 1,1 г (R)-3-[4-(транс-4- метоксициклогексил)фенил]-5-метоксиметилоксазолидин-2-она в виде бесцветных кристаллов. Т.пл.: 73-74o.
Пример 46
(RS)-3-(4- циклогекс-1-енилфенил)-5-метоксиметилоксазолидин-2-он
Раствор из 410 мг диацетата бис(трифенилфосфин)палладия (II), 2,0 г (RS)-3-(4- иодфенил)-5-метоксиметилоксазолидин-2-она и 1,8 мл 1-трибутил-станнил- 1-циклогексена в 10 мл диметилформамида перемешивали в течение 18 ч при 80o. Затем реакционную смесь сливали на 100 мл воды и трижды экстрагировали с помощью этилового эфира уксусной кислоты. Органическую фазу промывали насыщенным раствором фторида калия, насыщенным раствором хлорида натрия и водой, сушили над сульфатом магния и концентрировали. После хроматографии остатка (2,0 г) на 30-кратном количестве силикагеля с помощью этилового эфира уксусной кислоты/гексана (1:2) получали 0,7 г твердого вещества, которое кристаллизовалось из этилового эфира уксусной кислоты/гексана. В результате получали 0,3 г (RS)-3-(4-циклогекс-1-енил-фенил)-5- метоксиметилоксазолидин-2-она в виде бесцветных кристаллов. Т.пл.: 106- 108o.
Пример 47
Смесь (RS)-и (SR)-3-[(RS)-4-циклогекс-3-енил- фенил]-5-метоксиметилоксазолидин-2-она
Раствор из 2,0 г (RS)-3- [4-(гидроксициклогексил)фенил]-5-метоксиметилоксазолидин-2-она, 2,1 г трифенилфосфина и 1,8 г бензойной кислоты в 80 мл тетрагидрофурана обрабатывали при комнатной температуре в течение 15 мин 1,2 мл диэтилового эфира азодикарбоновой кислоты в 20 мл тетрагидрофурана и перемешивали в течение 22 ч при комнатной температуре. Затем реакционную смесь концентрировали, остаток растворяли в 100 мл уксусного эфира, дважды промывали 10%-ным раствором карбоната натрия и дважды насыщенным раствором хлорида натрия, сушили над сульфатом магния и концентрировали. После хроматографирования остатка (7,0 г) на 30-кратном количестве силикагеля с помощью смесей хлористого метилена/гексана в качестве побочного продукта получали 0,6 г масла, которое кристаллизовалось из этилового эфира уксусной кислоты/гексана. В результате получали 0,4 г смеси (RS)- и (SR)-3-[(RS)-4-циклогекс-3- енилфенил] - 5-метоксиметилоксазолидин- 2-она в виде бесцветных кристаллов. Т.пл.: 74-75o. 1H-ЯМР (CDCl3) част./млн: 7,48 (d, 2H), 7,23 (d, 2H), 5,75 (m, 2H), 4,75 (m, 1H), 4,04 (t, 1H), 3,91 (m, 1H), 3,64 (d, 2H), 3,43 (s, 3H), 2,78 (m, 1H), 2,16 (m, 4H), 1,88 (m, 1H), 1,75 (m, 1H).
Пример 48
(RS)-3-[4-(4-цианометиленциклогексил)-фенил] -5-метоксиметилоксазолидин-2-он
0,46 г натрия растворяли в атмосфере аргона в 50 мл этанола. Затем при 20o добавляли 3,5 г (=3,1 мл) диэтилцианометилфосфоната, растворенного в 10 мл этанола. Затем продолжали перемешивать еще в течение 30 мин при комнатной температуре, после чего добавляли 2,0 г (RS)-5-метоксиметил-3-[4-(4- оксоциклогексил)фенил]-оксазолидин-2-она. Далее перемешивали еще в течение 1 ч при нагреве с обратным холодильником, после чего охлаждали до комнатной температуры, с помощью приблизительно 0,7 мл уксусного эфира устанавливали pH 6 и реакционную смесь упаривали в вакууме. Сырую смесь (≈4,2 г) хроматографировали на 200 г силикагеля. С помощью дихлорметана/этилацетата (9:1) элюировали 1,65 г (RS)-3- [4-(4-цианометиленциклогексил) -фенил]-5- метоксиметилоксазолидин- 2-она в виде белых кристаллов. После перекристаллизации пробы из этилацетата/гексана получали белые кристаллы с т.пл. 121-125o.
Пример 49
(RS)-3-[4-(транс-4-цианометилциклогексил)-фенил] -5- метоксиметилок-сазолидин-2-он
1,65 г смеси из a) (R)-[4-[4-[(R)-, б) (R)-[4-[4-[(S)-, в) (S)-[4-[4-[(R)- и г) (S)-[4-[4-[(8)-5- метоксиметил-2-оксооксазолидин-3-ил]фенил]- циклогексилиден] ацетонитрила (соотношение а:б=в:г=1:1) гидрировали в 175 мл ледяной уксусной кислоты и 0,3 г палладия на угле (10%) при нормальном давлении и при комнатной температуре до теоретического поглощения (в течение примерно 16 ч). Затем отфильтровывали на нутче от катализатора и упаривали в вакууме. Остаток (1,6 г) растворяли в этилацетате и промывали насыщенным 10%-ным раствором гидрокарбоната натрия, а также раствором хлорида натрия. После сушки и упаривания получали 1,6 г кристаллов, которые для очистки хроматографировали на 30 г силикагеля. Элюирование проводили с помощью дихлорметана/этилацетата в соотношении 9: 1. После соединения полученных согласно тонкослойной хроматограмме чистых фракций и перекристаллизации из этилацетата/гексана получали 0,72 г (RS)-3-[4-(транс-4- цианометилциклогексил)-фенил] -5-метоксиметилоксазолидин-2-она в виде белых кристаллов с т. пл. 118-120o.
Пример 50
(R)-3-[4-(транс-4- цианометилциклогексил) -фенил] -5-метоксиметилоксазолидин- 2-он
1,1 г (R)-[4-[4-(5-метоксиметил-2-оксо-оксазолидин-3-ил)-фенил]- циклогексилиден]ацетонитрила растворяли в 50 мл метанола, обрабатывали 0,2 г Pd/C (10%) и гидрировали при комнатной температуре и при нормальном давлении до теоретического поглощения водорода. Затем отфильтровывали на нутче от катализатора и упаривали в вакууме. Маслянистый остаток хроматографировали на 15 г силикагеля. Элюирование проводили с помощью дихлорметана/этилацетата (9: 1). Полученные согласно тонкослойной хроматографии чистые фракции соединяли и кристаллизовали из этилацетата/гексана. В результате получали 0,64 г (R)-3-[4-(транс-4- цианометилцикло-гексил)-фенил] -5-метоксиметилоксазолидин-2-она в виде белых кристаллов с т.пл. 90-91o.

Пример 51
Смесь (1: 1) гидрохлорида цис- и гидрохлорида транс-(RS)-3-[4-[4-(2-аминоэтил)циклогексил] фенил]-5- метоксиметилоксазолидин-2-она
2,1 г смеси из a) (R)-[4-[4-[(R)-, б) (R)-[4-[4-[(S)-, в) (S)-[4-[4-[(R)- и г) (S)-[4-[4-[(8)-5- метоксиметил-2-оксооксазолидин-3-ил]-фенил]- циклогексилиден] ацетонитрила (соотношение а:б=в:г=1:1) гидрировали в 200 мл метанолового аммиака (20% (вес/объем)) и 3,0 г никеля Ренея в течение приблизительно 40 ч при нормальном давлении и комнатной температуре до теоретического поглощения водорода. Затем отфильтровывали на нутче от катализатора и упаривали в вакууме. Сырой продукт (≈1,8 г) хроматографировали на 15 г силикагеля. Далее элюировали с помощью дихлорметана/метанола/аммиака (25% (250:5:1)) и дихлорметана, насыщенного аммиаком (25%) и метанолом (5%). Полученные согласно тонкослойной хроматографии чистые фракции соединяли и упаривали. В результате получали 1,4 г основания в виде мутного масла. 0,7 г этого масла переводили в гидрохлорид (этаноловая соляная кислота/простой эфир). Таким путем получали 0,5 г смеси (1:1) из гидрохлорида цис- и гидрохлорида транс-(RS)-3-[4-[4-(2-аминоэтил)- циклогексил] фенил]-5-метоксиметилоксазолидин-2-она в виде белых кристаллов с т.пл. 203-207o.
Пример 52
(RS)-3-[4-[4-(2-ацетиламиноэтил) циклогексил] фенил] -5- метоксиметилоксазолидин-2-он
0,57 г смеси (1:1) цис- и транс-(RS)-3-[4-[4-(2-аминоэтил)- циклогексил] фенил]-5-метоксиметилоксазолидин-2-она растворяли в 2 мл пиридина и обрабатывали 0,21 г ангидрида уксусной кислоты. Затем перемешивали в течение 3,5 ч при комнатной температуре, после чего реакционную смесь растворяли в дихлорметане. Органическую фазу промывали 1н. соляной кислотой, водой, гидрокарбонатом натрия и насыщенным раствором поваренной соли, затем сушили и упаривали. В качестве остатка получали 0,75 г мутного масла, которое кристаллизовали из этилацетата/метил-трет. -бутилового эфира. В результате получали 0,25 г (RS)-3-[4-[4-(2-ацетиламиноэтил) циклогексил]фенил]-5-метоксиметилоксазолидин-2-она в виде белых кристаллов с т.пл. 120-122o.
Пример 53
(RS)-3-[4-(1-трет. - бутоксикарбонилпиперидин-4-ил)фенил]-5-метоксиметилоксазолидин-2-он
5,5 г (15,0 ммолей) трет.-бутилового эфира 4-(4-этоксикарбониламино-фенил)пиперидин-1-карбоновой кислоты и 20 г (0,15 моля) (RS)-4-(метоксиметил)-1,3-диоксолан-2-она интенсивно перемешивали в присутствии 2,37 г (30 ммолей) пиридина в течение 18 ч при температуре масляной ванны 160o. После охлаждения реакционную смесь хроматографировали на 840 г силикагеля 60 с помощью простого эфира. В результате получали 3,4 г (RS)-3-[4-(1-трет. -бутоксикарбонилпиперидин- 4-ил)фенил]-5-метоксиметилоксазолидин-2-она в виде желтоватого масла. 1H-ЯМР (CDCl3) част./млн: 7,48 (d, 2H), 7,22 (d, 2H), 4,76 (m, 1H), 4,25 (шир., 2H), 4,05 (t, 1H), 3,92 (t, 1H), 3,64 (d, 2H), 3,43 (s, 3H), 2,80 (t, 2H), 2.64 (m, 1H), 1,78 (шир., 2H), 1,61 (m, 2H), 1,49 (s, 9H).
Пример 54
Гидрохлорид (RS)-5-метоксиметил-3-(4-пиперидин-4-ил-фенил)-оксазолидин-2-она (1:1)
2,51 г (6,4 ммолей) трет.-бутилового эфира (RS)-4-[4-(5-метоксиметил-2-оксооксазолидин-3- ил)фенил]пиперидин-1-карбоновой кислоты растворяли в 40 мл уксусного эфира и при перемешивании обрабатывали при комнатной температуре 40 мл 2,2 М насыщенного хлористым водородом раствора уксусного эфира. Через 90 мин суспензию упаривали и отгоняли с помощью толуола. В результате получали 1,47 г гидрохлорида (RS)-5-метоксиметил-3-(4- пиперидин-4-ил-фенил)-оксазолидин-2-она (1:1). Т.пл.: 161-163o. 1H-ЯМР (ДМСО) част./млн: 8,98 (шир., 2H), 7,52 (d, 2H), 7,24 (d, 2H), 4,81 (m, 1H), 4,10 (t, 1H), 3,79 (t, 1H), 3,58 (m, 2H), 3.35 (m. 2H), 3,32 (s, 3H), 2,97 (шир., 2H), 2,80 (m, 1H), 1,87 (шир., 4H).
Пример 55
Гидрохлорид (RS)-3-[4-(1-бензилпиперидин-4-ил)-фенил] -5- метоксиметилоксазолидин-2-она (1:1)
290,4 мг (1,0 ммоль) (RS)-4-[4- (5-метоксиметил-2-оксо-оксазолидин-3-ил) фенил] пиперидина растворяли в 5 мл абсолютного диметилформамида и обрабатывали в присутствии 404,8 мг (4,0 ммоля) триэтиламина 171,0 мг (1,0 ммоль) бензилбромида. Реакционную смесь перемешивали в течение 90 мин при 60o. После этого смесь упаривали, остаток распределяли на уксусный эфир и 3н. раствор аммиака, органическую фазу промывали водой, сушили над сульфатом натрия, фильтровали и упаривали. Основание растворяли в этаноле, подкисляли эфирной соляной кислотой и повторно отгоняли с помощью толуола. После перекристаллизации из этанола/простого эфира получали 291,2 мг гидрохлорида (RS)-3-[4-(1-бензилпиперидин-4-ил)-фенил] -5- метоксиметилоксазолидин-2-она (1:1). Т.пл.: 175-177o.
Пример 56
Гидрохлорид (RS)-3-[4-[1-[4-(4-фторофенил)-4-оксобутил]-пиперидин- 4-ил] -фенил]-5-метоксиметилоксазолидин-2-она (1:1)
253,0 мг (0,87 ммоля) (RS)-4-[4-(5-метоксиметил-2-оксооксазолидин-3-ил) фенил] пиперидина растворяли в 15 мл абсолютного диметилформамида и обрабатывали в присутствии 352,7 мг (3,485 ммоля) триэтиламина 174,8 мг (0,87 ммоля) ω-хлор-4-фторбутирофенона. Реакционную смесь перемешивали в течение 5 ч при 100o. Затем смесь упаривали, остаток распределяли на уксусный эфир и 3н. раствор аммиака, органическую фазу промывали водой, сушили над сульфатом натрия, фильтровали и упаривали. Остаток растворяли в этаноле и подкисляли эфирной соляной кислотой. После перекристаллизации из спирта/простого эфира получали 135,9 мг гидрохлорида (RS)-3-[4-[1-[4-(4-фторофенил)-4-оксобутил]- пиперидин-4-ил] -фенил] -5-метоксиметилоксазолидин-2-она (1:1). Т.пл.: 182-184o.
Пример 57
(RS)-5-метоксиметил-3- [4-[1-(4- трифторометилбензоил) -пиперидин-4-ил] -фенил]- оксазолидин-2-он
273,1 мг (0,94 ммоля) гидрохлорида (RS)-4-[4-(5-метоксиметил-2- оксооксазолидин-3-ил)фенил] -пиперидина растворяли в 20 мл хлористого метилена и обрабатывали в присутствии 380,7 мг (3,76 ммоля) триэтиламина 196,2 мг (0,94 ммоля) 4-трифторметилбензоилхлорида. Реакционную смесь перемешивали в течение 17 ч при комнатной температуре. Затем смесь обрабатывали уксусным эфиром, промывали 1H соляной кислотой и водой и сушили над сульфатом натрия. После перекристаллизации из уксусного эфира/простого эфира получали 356,3 мг (RS)-5-метоксиметил-3-[4-[1-(4-трифторометилбензоил)-пиперидин-4-ил]- фенил] -оксазолидин-2-она. Т.пл.: 164-166o.
Пример 58
(RS)-3-[4- (1-ацетилпиперидин-4-ил)- фенил]-5-метоксиметилоксазолидин- 2-он
177,1 мг (0,61 ммоля) (RS)-4-[4-(5-метоксиметил-2-оксо-оксазолидин-3-ил)-фенил] -пиперидина растворяли в 20 мл хлористого метилена и обрабатывали в присутствии 53,1 мг (0,67 ммоля) пиридина 68,5 мг (0,67 ммоля) ангидрида уксусной кислоты. После перемешивания в течение 18 ч при комнатной температуре промывали водой и органическую фазу сушили над сульфатом натрия. После фильтрации и упаривания фильтрата продукт сушили в высоком вакууме. В результате получали 197,3 мг (RS)-3-[4-(1- ацетилпиперидин-4-ил)-фенил]-5-метоксиметилоксазолидин-2-она в виде желтоватого масла. 1H-ЯМР (CDCl3) част. /млн: 7,49 (d, 2H), 7,20 (d, 2H), 4,80 (m, 2H), 4,05 (t, 1H), 3,92 (m, 2H), 3,64 (d, 2H), 3,43 (s, 3H), 3,21 (m, 1H), 2,68 (m, 2H), 2,14 (s, 3H), 1,85 (m, 2H), 1,60 (m, 2H).
Пример 59
(E)-(RS)-5-метоксиметил-3-[4-[1-(3- фенилакрилоил)пиперидин-4-ил]-фенил] оксазолидин-2-он
201,7 мг (0,695 ммоля) гидрохлорида (RS)-4-[4-(5-метоксиметил-2-оксооксазолидин-3- ил)фенил] пиперидина растворяли в 15 мл хлористого метилена и в присутствии 281,2 мг (2,78 ммоля) триэтиламина при перемешивании при комнатной температуре обрабатывали по каплям раствором из 115,7 мг (0,695 ммоля) хлорида коричной кислоты в 5 мл хлористого метилена. Через 17 ч разбавляли уксусным эфиром и органическую фазу промывали 1н. соляной кислотой и водой. После перекристаллизации из уксусного эфира/простого эфира получали 228,9 мг (Е)-(RS)-5-метоксиметил-3-[4-[1-(3-фенилакрилоил)пиперидин-4-ил] фенил] оксазолидин-2-она. Т.пл.: 136-138o.
Пример 60
(RS)-3-[4-(1-этоксикарбонилпиперидин-4-ил)фенил] -5- метоксиметилоксазолидин-2-он
1,12 г (3,5 ммоля) этилового эфира 4-(4-этосикарбониламино-фенил)-пиперидин-1-карбоновой кислоты обрабатывали в присутствии 276,8 мг (3,5 ммоля) пиридина, 4,0 г (30,28 ммолей) (RS)-4-(метоксиметил)-1,3- диоксолан-2-она и в течение 18 ч интенсивно перемешивали при температуре масляной ванны 160o. После охлаждения реакционную смесь хроматографировали на 125 г силикагеля 60 с помощью простого эфира. После перекристаллизации из хлористого метилена/изопропилового эфира получали 976 мг (RS)-3-[4-(1-этоксикарбонилпиперидин-4-ил) фенил]-5 -метоксиметилоксазолидин-2-она. Т.пл.: 86-88o.
Пример 61
(R)- 5-метоксиметил-3-[4-(транс-4-метилсульфанилметокси- циклогексил)фенил]оксазолидин-2-он
9,5 г (31,11 ммоля) (R)-3-[транс- (4-гидроксициклогексил)фенил]-5-метоксиметилоксазолидин-2-она растворяли в 120 мл диметилсульфоксида и последовательно обрабатывали 2 мл воды, 25 мл уксусного эфира и 80 мл ангидрида уксусной кислоты. Реакционную смесь перемешивали в течение 22 ч при комнатной температуре. Затем смесь добавляли порциями в охлажденный льдом раствор из 130 г карбоната натрия в 2 л воды. После окончания добавления смесь перемешивали еще в течение 1 ч. Далее продукт экстрагировали с помощью хлористого метилена, органическую фазу промывали водой, сушили над сульфатом натрия, фильтровали и упаривали. В результате получали 11,2 г желтоватого масла, из которого обработкой простым эфиром/н-гексаном получали кристаллический (R)-5- метоксиметил-3-[4-(транс-4-метилсульфанилметоксициклогексил)фенил] оксазолидин-2-он. Т.пл.: 54-56o, [α]D= -30,8o (с=1,0, CHCl3). 1H-ЯМР (COCl3) част. /млн: 7,47 (d, 2H), 7,21 (d, 2H), 4,70 (m, 1H), 4,04 (t, 1H), 3,93 (t, 1H), 3,70 (m, 1H), 3,63 (d, 2H), 3,43 (s, 3H). 2.50 (m. 1H). 2,18 (s. 3H). 2.12 (d. 2H), 1.90 (d, 2H), 1,56 (шир., 4H).
Пример 62
(R)-3- [4-(транс-4-метансульфинилметоксициклогексил)фенил]-5- метоксиметилоксазолидин-2-он (S-оксид R:S=1:1, смесь диастереомеров)
10,7 г (29,28 ммолей) (R)-5-метоксиметил-3-[4-(транс-4-метилсульфанилметоксициклогексил) фенил] оксазолидин-2-она растворяли в 500 мл метанола, охлаждали до 0o и в течение 90 мин при перемешивании обрабатывали по каплям раствором из 4,7 г (22,0 ммолей) периодата натрия в 150 мл воды. Реакционную смесь перемешивали в холодильнике при температуре 4-5o в течение 19 ч. Затем смесь разбавляли 1 л воды и экстрагировали с помощью хлористого метилена. Органическую фазу промывали водой, сушили над сульфатом натрия и фильтровали. Фильтрат фильтровали через 400 г силикагеля 60 в качестве буфера и последовательно, раздельно промывали уксусным эфиром (800 мл), метанолом (1 л) и тетрагидрофураном (800 мл). После упаривания уксусно-эфирного элюата получали еще 3,16 г эдукта. После упаривания метаноловых и тетрагидрофурановых элюатов получали 8,11 г (R)-3-[4- (транс-4-метансульфинилметоксициклогексил) -фенил] -5-метоксиметил- оказолидин-2-она (S-оксид R:S=1:1, смесь диастереомеров) в виде желтого масла. Т.кип.: 205o/0,02 мбар, [α]D= -32,2o (с=1,0%, ДМСО).
Пример 63
(R)-3-[4-(транс-4-цианометоксициклогексил) фенил] -5- метоксиметилоксазолидин-2-он
7,7 г (20,2 ммолей) (R)-3-[4-(транс-4- метансульфинилметоксициклогексил) фенил] -5- метоксиметилоксазолидин- 2-она (S-оксид, RS=1:1, смесь диастереомеров) растворяли в 200 мл абсолютного тетрагидрофурана и в присутствии 420 мг (1,32 ммоля) иодида цинка при перемешивании в течение 30 мин обрабатывали по каплям раствором из 17,73 г (0,179 моля) триметилсилилхлорида в 40 мл абсолютного тетрагидрофурана. Реакционную смесь перемешивали в течение 24 ч при комнатной температуре. Затем смесь упаривали, остаток распределяли на уксусный эфир и воду, органическую фазу промывали водой, сушили над сульфатом натрия, фильтровали и упаривали. Остаток (7,75 г) хроматографировали на 320 г силикагеля 60 с помощью уксусного эфира/н-гексана (1:1). В результате получали 2,64 г (R)-3-[4-( транс-4- цианометоксициклогексил)фенил]-5-метоксиметилоксазолидин-2-она в виде бесцветного масла; Т.кип.: 210o/0,02 мбар;] [α]D= -32,2o (с = 1,0% в CHCl3). 1H-ЯМР (COCl3) част./млн: 7,48 (d, 2H), 7,20 (d, 2H), 4,75 (m, 1H), 4,32 (d, 2H), 4,02 (t, 1H), 3,93 (t, 1H), 3,64 (d, 2H), 3,61 (m, 1H), 3,43 (s, 3H), 2,55 (m, 1H), 2,39 (m, 2H), 1,95 (m, 2H), 1,50 (шир., 4H).
После перекристаллизации из простого эфира/гексана получали белые кристаллы с т.пл. 43-45o.
Пример 64
(RS)-5-метоксиметил-3- [4-(транс-4-метилсульфанилметоксициклогексил) фенил]оксазолидин-2- он
305,4 мг (1,0 ммоль) (RS)-3-[транс-4- гидроксициклогексил)фенил]-5-метоксиметилоксазолидин-2-она вводили в 35 мл циклогексана и при комнатной температуре при перемешивании обрабатывали 186,9 мг (1,1 ммоля) нитрата серебра, 121,4 мг (1,2 ммоля) триэтиламина и 115,9 мг (1,2 ммоля) хлорметилметилсульфида. Реакционную смесь перемешивали в течение 20 ч. Затем смесь отфильтровывали от хлорида серебра через дикалит, фильтрат промывали насыщенным гидрокарбонатом натрия, органическую фазу сушили над сульфатом натрия, фильтровали и упаривали. В результате получали 354,5 мг (RS)-5-метоксиметил-3-[4-(транс-4- метилсульфанилметоксициклогексил)-фенил]оксазолидин-2-она в виде масла желтоватого цвета.
Пример 65
(RS)-3-[4-(транс-4- метансульфинилметоксициклогексил)фенил]-5-метоксиметилоксазолидин- 2-он (смесь диастереомеров)
1,71 г (4,68 ммоля) (RS)-5-метоксиметил-3-[4-(транс-4- метилсульфанилметоксициклогексил) фенил] оксазолидин-2-она растворяли в 100 мл метанола, охлаждали до 0o и при перемешивании в течение 40 мин обрабатывали по каплям раствором из 1,0 г (4,68 ммоля) метапериодата натрия, растворенного в 40 мл воды. Реакционную смесь выдерживали в течение 24 ч при температуре примерно 4oC. Затем смесь разбавляли водой и продукт экстрагировали с помощью хлористого метилена. Органическую фазу промывали водой, сушили над сульфатом натрия и фильтровали. Фильтрат фильтровали через 50 г силикагеля 60 и раздельно промывали метанолом. После упаривания метанолового элюата смесь диастереомеров (RS)-3-[4-(транс-4-метансульфинилметоксициклогексил) фенил]-5-метоксиметилоксазолидин-2-она кристаллизовали из бензола. Т.пл.: 98-100o.
Пример 66
(RS)-3-[4-(транс-4- цианометоксициклогексил)фенил] -5-метоксиметилоксазолидин-2-он
536,3 мг (1,40 ммоля) (RS)-3-[4-(транс-4- метансульфинилметоксициклогексил) фенил] -5-метоксиметилоксазолидин- 2-она (смесь диастереомеров) растворяли в 25 мл абсолютного тетрагидрофурана и при перемешивании обрабатывали 50 мг иодида цинка и 465 мг (2,81 ммоля) триметилсилилцианида. После перемешивания в течение 21 ч при комнатной температуре реакционную смесь разбавляли 40 мл хлористого метилена и 40 мл воды, органическую фазу промывали водой, сушили над сульфатом натрия, фильтровали и упаривали. Остаток (517,5 мг) хроматографировали на 25 г силикагеля 60 с помощью уксусного эфира/н-гексана (1: 1). В результате получали 90 мг (RS)-3-[4-(транс-4- цианометоксициклогексил) фенил]-5-метоксиметилоксазолидин-2-она в виде бесцветного масла. 1H-ЯМР (CDCl3) част./млн: 7,48 (d, 2H), 7,20 (d, 2H), 4,75 (m, 1H), 4,32 (d, 2H), 4,05 (t, 1H), 3,92 (t, 1H), 3,65 (d, 2H), 3,60 (m, 1H), 3,43 (s, 3H), 2,50 (m, 1H), 2,20 (d, 2H), 1,95 (d, 2H), 1,47 (шир., 4H).
Пример 67
(RS)-3-[4-(транс-4-метансульфонилметоксициклогексил)фенил] -5-метоксиметилоксазолидин-2-он
320,1 мг (0,88 ммоля) (RS)-5-метоксиметил-3-[4-(транс-4- метилсульфанилметоксициклогексил) фенил] оксазолидин-2-она растворяли в 15 мл хлористого метилена и обрабатывали 226,9 мг (0,97 ммоля) мета- хлорпербензойной кислоты. После перемешивания в течение 3 ч при комнатной температуре реакционную смесь промывали раствором гидрокарбоната натрия и водой, органическую фазу сушили над сульфатом натрия, фильтровали и упаривали. Остаток (343,2 мг) хроматографировали на 20 г силикагеля 60 с помощью уксусного эфира. После перекристаллизации из хлористого метилена/простого эфира получали 136,7 мг (RS)-3-[4-(транс-4-метансульфонилметоксициклогексил)фенил] - 5-метоксиметилоксазолидин-2-она в виде белого кристаллизата с т.пл. 145-147o.
Пример 68
Гидрохлорид (RS)-3-[4-[транс-4-(2- аминоэтокси)циклогексил] фенил]-5-метоксиметилоксазолидин-2-она (1:1)
189,7 мг (0,5508 ммоля) (RS)-[транс-4-[4-(5-метоксиметил-2- оксо-оксазолидин-3-ил)фенил]циклогексилокси] ацетонитрила растворяли в 15 мл насыщенного сухим аммиаком метанола и гидрировали в присутствии 300 мг никеля Ренея (тип В 113 W Degussa). Через 30 мин отфильтровывали от катализатора и фильтрат упаривали. Остаток растворяли в хлористом метилене, промывали водой, сушили над сульфатом натрия, фильтровали и упаривали. Основание растворяли в спирте и подкисляли эфирной соляной кислотой. После перекристаллизации из спирта/простого эфира получали 186,0 мг гидрохлорида (RS)-3-[4- [транс-4-(2-аминоэтокси)циклогексил]фенил]-5-метоксиметилоксазолидин- 2-она (1:1). Т. пл.: 204-206o.
Пример 69
(RS)-3-[4- [транс-4-(3-ацетиламинопропокси)циклогексил] фенил] -5- метоксиметилоксазолидин-2-он
199,5 мг (0,5 ммоля) (RS)-3-[транс-4- [4-(5-метоксиметил-2-оксооксазолидин-3- ил)фенил]циклогексилокси] пропилацетамида растворяли в 5 мл хлористого метилена и обрабатывали в присутствии 87,01 мг (1,1 ммоля) пиридина и 43,2 мг (0,55 ммоля) ацетилхлорида. Через 3 ч промывали водой, органическую фазу сушили над сульфатом натрия, фильтровали и упаривали. После перекристаллизации из уксусного эфира/простого эфира получали 175,8 мг (RS)-3-[4-[транс-4-(3- ацетиламинопропокси) циклогексил] фенил]-5- метоксиметилоксазолидин-2- она. Т.пл.: 72-74o.
Пример 70
(R)-3-[4-[транс-4-(2- цианоэтокси)циклогексил] фенил]-5-метоксиметилоксазолидин-2-он
4,58 г (15,0 ммолей) (R)-3-[транс-(4-гидроксициклогексил)фенил]-5- метоксиметилоксазолидин-2-она растворяли в 400 мл хлористого метилена и последовательно обрабатывали 0,96 г (18,0 ммолей) акрилонитрила и 2,02 г (18,0 ммолей) трет.-бутилата калия. Реакционную смесь перемешивали в течение 16 ч при комнатной температуре, после чего обрабатывали ее водой, отфильтровывали от нерастворившихся компонентов через дикалит, органическую фазу промывали водой, сушили над сульфатом натрия, фильтровали и упаривали. Остаток (5,1 г) хроматографировали на 204 г силикагеля 60 с помощью уксусного эфира/н-гексана (6: 4). После перекристаллизации из трет.-бутилметилового эфира получали 1,6 г (R)- 3-[4-[транс-4-(2-цианоэтокси)циклогексил] фенил]-5- метоксиметилоксазолидин- 2-она. Т.пл.: 57-59o. [α]D= -30,0o (с = 0,7 в CHCl3).
Пример 71
Гидрохлорид (R)-3-[транс-4-[3-(аминопропокси)циклогексил]фенил] -5-метоксиметилоксазолидин-2-она
932 мг (2,6 ммоля) (R)-3-[транс-4-[4-(5-метоксиметил-2-оксо- оксазолидин-3-ил)фенил] циклогексилокси]пропионитрила растворяли в 60 мл насыщенного сухим аммиаком метанола и гидрировали в присутствии 0,9 г никеля Ренея (тип B 113 W Degussa). Через 3 ч отфильтровывали на нутче от катализатора и фильтрат упаривали. Остаток растворяли в хлористом метилене, промывали водой, сушили над сульфатом натрия, фильтровали и упаривали. Основание растворяли в спирте и подкисляли эфирной соляной кислотой. После перекристаллизации из спирта/простого эфира получали 825,2 мг гидрохлорида (R)-3-[транс-4-[3- (аминопропокси)циклогексил] фенил] -5-метоксиметилоксазолидин-2-она. Т.пл.: 178-180o. [α]D= -29,78o (с=1%, в CHCl3).
Пример 72
(R)-5-метоксиметил-3-[4-(4- метиленциклогексил)фенил]оксазолидин-2-он
2,0 г (7,7 ммолей) [4-(4- метиленциклогексил)фенил]этоксикарбаминового эфира перемешивали в течение ночи при 160o с 1,02 г (7,7 ммолей) (S)-4-метоксиметил-1,3-диоксолан-2-она и 170 мг карбоната калия. Остаток растворяли в 50 мл дихлорметана и экстрагировали с помощью 50 мл воды. Органическую фазу сушили над сульфатом натрия и концентрировали. После хроматографии на силикагеле с помощью уксусного эфира/гексана (1:99) и перекристаллизации из гексана получали 1,04 г (45%) (R)-5- метоксиметил-3-[4-(4-метиленциклогексил)фенил]оксазолидин-2-она в виде белых кристаллов с т.пл. 64o.
Пример 73
(R)-3-[4-(цис- или транс-4- гидрокси-4-метилциклогексил)фенил]-5-метоксиметилоксазолидин-2-он
Раствор из 945 мг (3 ммоля) ацетата ртути в 18 мл воды обрабатывали 18 мл тетрагидрофурана. После перемешивания в течение 30 мин при комнатной температуре в эту суспензию по каплям добавляли раствор из 900 мг (3 ммоля) (R)-5-метоксиметил-3-[4-(4- метиленциклогексил)фенил]оксазолидин-2-она в 18 мл тетрагидрофурана. После выдерживания в холодильном шкафу в течение нескольких дней добавляли сначала 18 мл 2н. натрового щелока, а затем после перемешивания в течение 30 мин раствор из 360 мг (9,5 ммолей) борогидрида натрия в 12 мл 2н. натрового щелока. После перемешивания в течение 30 мин экстрагировали с помощью хлористого метилена, органическую фазу промывали водой и сушили над сульфатом натрия. После отгонки растворителя хроматографировали на силикагеле с помощью уксусного эфира/дихлорметана (1:1). В результате получали 200 мг (21%) (R)-3-[4-(цис-4-гидрокси-4-метил-циклогексил)фенил] -5- метоксиметилоксазолидин-2-она в виде белых кристаллов с т.пл. 122-123o и 500 мг смеси цис- и трансизомеров.
Пример 74
(R)-транс-5-метоксиметил-3-[4-(4-цианоциклогексил)фенил] -оксазолидин-2-он
Суспензию из 500 мг (1,84 ммоля) транс-[4-(4-цианоциклогексил)фенил] этоксикарбаминовой кислоты, 365 мг (2,8 ммоля) (S)-4-метоксиметил-1,3-диоксолан-2-она и 185 мг карбоната калия перемешивали в течение 16 ч при 160o. Реакционную смесь хроматографировали на силикагеле с помощью уксусного эфира/гексана (1:1), после чего полученные желтые кристаллы перекристаллизовывали из уксусного эфира. В результате получали 165 мг (29%) (R)-транс-5-метоксиметил-3-[4-(4-цианоциклогексил)фенил]- оксазолидин-2-она в виде белых кристаллов с т.пл. 174o.
Пример 75
(RS)-5-метоксиметил-3-[4-(4-диметилметиленциклогексил)-енил] - оксазолидин-2-он
1,60 г (3,3 ммоля) смеси из бромида изопропилтрифенилфосфония и амида натрия в 30 мл тетрагидрофурана перемешивали в течение 1 ч при комнатной температуре. Затем по каплям добавляли раствор из 1,0 г (3,3 ммоля) (RS)-3-[4-(4- оксоциклогексил)фенил] - 5-метоксиметилоксазолидин- 2-она в 25 мл тетрагидрофурана. После кипячения с обратным холодильником в течение уикэнда желировали в 100 мл воды и трижды экстрагировали соответственно порциями по 50 мл дихлорметана. После хроматографии на силикагеле с помощью уксусного эфира/гексана (1:2) и перекристаллизации из н-гексана получали 120 мг (11%) (RS)-5- метоксиметил-3- [4-(4-диметилметилен-циклогексил)фенил] оксазолидин-2- она в виде белых кристаллов с т.пл. 89-91o.
Пример 76
Смесь из (RS)- и (SR)-3-[4-[(RS)-4-бензилиденциклогексил]фенил]-5-метоксиметил оксазолидин-2-она
5,0 г (10 ммолей) смеси из бромида бензилтрифенилфосфония и амида натрия в 100 мл тетрагидрофурана перемешивали в течение 1 ч при комнатной температуре. Затем по каплям добавляли раствор из 304 г (10 ммолей) (RS)-3-[4-(4- оксоциклогексил)фенил] -5- метоксиметилоксазолидин-2-она в 150 мл тетрагидрофурана и в течение 20 ч кипятили с обратным холодильником. После отгонки растворителя растворяли в 150 мл воды и четырежды экстрагировали соответственно порциями по 80 мл дихлорметана. После хроматографии на силикагеле с помощью уксусного эфира/гексана (1:2) и перекристаллизации из уксусного эфира/гексана получали 250 мг (7%) белых кристаллов с т.пл. 65-69o.
Пример 77
(RS)-3-[4-(цис- и транс-4-бензилциклогексил)фенил]-5-метоксиметил- оксазолидин-2-он
290 мг маточного раствора в 40 мл этанола обрабатывали 100 мг 10%-ного палладия на угле и гидрировали в течение 20 ч при комнатной температуре. После хроматографии на силикагеле с помощью уксусного эфира/гексана (1:2) получали 110 мг смолы. МС: m/e (% основного пика): 379 (М+, 100), 246 (23), 144 (31), 118 (26), 91 (55), 71 (26), 45 (28).
Пример 78
(RS)-3-[4-(4- метоксикарбонилметиленциклогексил)фенил] -5-метоксиметилоксазолидин- 2-он
10,0 г (33 ммоля) (RS)-3-[4-(4-оксоциклогексил)-фенил]-5- метоксиметилоксазолидин-2-она и 11,0 г (33 ммоля) метоксикарбонилметилен-трифенилфосфорана в 500 мл бензола кипятили в течение 24 ч с обратным холодильником. После отгонки растворителя в вакууме хроматографировали на силикагеле с помощью уксусного эфира/гексана (1:1). В результате получали 3,6 г (43%) белых кристаллов с т.пл. 62-64o.
Пример 79
(RS)-3-(4-циклогексил-3- нитрофенил)-5-метоксиметилоксазолидин-2-он
2,44 г (8,4 ммолей) (4- циклогексил-3-нитрофенил)этоксикарбаминового эфира и 1,10 г (8,4 ммолей) (RS)-4-метоксиметил-1,3-диоксолан-2-она и 200 мг карбоната калия перемешивали в течение 1 дня при 160o. После хроматографии на силикагеле с помощью уксусного эфира/гексана (2:3) и перекристаллизации из уксусного эфира/простого эфира получали 0,94 г (34%) кристаллов бежевого цвета с т.пл. 105-106o.
Пример 80
(RS)-3-(3-амино-4-циклогексилфенил)-5- метоксиметилоксазолидин-2-он
0,5 г (1,5 ммоля) (RS)-3-(4-циклогексил- 3-нитрофенил)-5-метоксиметилоксазолидин-2-она (из маточного раствора в предыдущей реакции) в 75 мл этанола обрабатывали 100 мг 10%-ного палладия на угле и гидрировали в течение 5 ч при комнатной температуре. После фильтрации, концентрирования и перекристаллизации из уксусного эфира получали 150 мг белых кристаллов с т. пл. 130-132o.
Пример 81
(RS)-3-(4-циклогексил-3-иодфенил)-5-метоксиметилоксазолидин- 2-он
180 мг (0,6 ммоля) (RS)-3-(3-амино-4-циклогексил-енил)-5- метоксиметилоксазолидин-2-она (из маточного раствора в предыдущей реакции) суспендировали в смеси из 0,32 мл воды, 0,32 г 37%-ной соляной кислоты и 0,64 г льда и обрабатывали при температуре 0-5o раствором из 41,4 мг (0,6 ммоля) нитрита натрия в 0,3 мл воды. После перемешивания в течение 30 мин при этой температуре добавляли раствор из 0,1 г (0,6 ммоля) иодида калия в 0,3 мл воды. Затем перемешивали в течение ночи при комнатной температуре, экстрагировали на силикагеле с помощью уксусного эфира/гексана (3:1) и в результате получали 100 мг масла. МС: m/e (% основного пика): 415 (М+, 100), 372 (5), 346 (9), 289 (9), 245 (12), 143 (15), 71 (22).
Пример 82
(RS)-3-(4- циклогексил-3-гидроксифенил)-5-метоксиметилоксазолидин-2-он
200 мг (0,7 ммоля) (RS)-3-(3-амино-4-циклогексилфенил)-5- метоксиметилоксазолидин-2-она суспендировали в смеси из 0,86 мл воды, 0,72 г льда и 0,235 мл концентрированной серной кислоты и затем обрабатывали при температуре 0-5o раствором из 45 мг (0,66 ммоля) нитрита натрия в 0,4 мл воды. После кипячения с обратным холодильником в течение 3 ч экстрагировали с помощью дихлорметана, сушили над сульфатом натрия и отгоняли растворитель. После хроматографии на силикагеле с помощью уксусного эфира/гексана (1:2) получали 120 мг (60%) белых кристаллов с т.пл. 207-208o.
Пример 83
(RS)-3-(3-хлор-4-циклогексилфенил)-5-метоксиметилоксазолидин-2- он
0,45 г (1,6 ммоля) 3-хлор-4-циклогексилфенил)этоксикарбаминового эфира и 0,84 г (6,4 ммолей) (RS)-4-метоксиметил-1,3-диоксолан-2-она и 200 мг карбоната калия перемешивали в течение 24 ч при 160o. После хроматографии на силикагеле с помощью дихлорметана получали 0,57 г масла, содержавшего еще некоторое количество диоксолана. Этот диоксолан удаляли посредством молекулярной перегонки при 100o/0,2 мбар. В результате получали 0,38 г (73%) смолы. МС: m/e (% основного пика): 323 (М+, 100), 280 (13), 254 (13), 178 (25), 152 (18), 71 (36).
Получение промежуточных продуктов
Пример 84
(RS)-3-(4-циклогексилфениламино)-пропан-1,2-диол
8,05 г (44,6 ммолей) 4-циклогексиланилина, 12,0 г (57 ммолей) (RS)-2,2-диметил-1,3-диоксолана- 4-ил-метилового эфира метансульфокислоты и 11,2 мл (80 ммолей) триэтиламина выдерживали в течение 4 ч в тугоплавкой трубке при 140o. Растворитель отгоняли в водоструйном насосе, маслянистый остаток коричневого цвета обрабатывали 50 мл 3н. соляной кислоты и перемешивали в течение 1 ч при 50o. После охлаждения дважды экстрагировали соответственно порциями по 250 мл простого эфира, водную фазу подщелачивали с помощью 2н. натрового щелока и продукт растворяли в уксусном эфире. Органическую фазу промывали водой, сушили над сульфатом магния и растворитель отгоняли. Маслянистый остаток кристаллизовался при обработке уксусным эфиром/гексаном. В результате получали 2,2 г кристаллического (RS)-3-(4-циклогексилфениламино)- пропан-1,2-диола. Т.пл.: 121-122 (разложение).
Из маточного раствора дополнительно получали 0,65 г кристаллического продукта.
Пример 85
а) (S)-3-(4-циклогексилфениламинометил)-4-(2,2-диметил-1,3-диоксолан)
5,0 г (28,5 ммолей) 4-циклогексиланилина, 10,0 г (34,9 ммоля) (R)-2,2-диметил-1,3-диоксолан-4-метанолтолуол-4-сульфоната и 10 мл триэтиламина перемешивали в течение 3 ч при температуре масляной ванны 140o. Коричневое масло, полученное после отгонки легколетучих компонентов в водоструйном насосе, обрабатывали насыщенным раствором карбоната натрия и экстрагировали с помощью уксусного эфира. Органическую фазу промывали раствором поваренной соли, сушили над сульфатом магния и растворитель отгоняли. В результате получали 9,5 г коричневого масла, которое хроматографировали на 30-кратном количестве силикагеля. Полученные после элюирования с помощью уксусного эфира/гексана (1: 3) посредством ТСХ (уксусный эфир/гексан 1:2) однородные фракции соединяли и растворитель отгоняли. В результате получали 9,4 г масла желтоватого цвета. Т.кип.: 140o/0,01 мбар. [α]D= +1,43 (с=1, CHCl3).
б) (S)-3-(4-циклогексилфениламино)пропан-1,2-диол
4,5 г (S)-3-(4- циклогексилфениламинометил)-4-(2,2-диметил-1,3-диоксолана) обрабатывали 50 мл 3н. соляной кислоты и перемешивали в течение 1 ч при 50o. После охлаждения с помощью 3н. натрового щелока при охлаждении льдом подщелачивали, выпавшее в осадок масло растворяли в уксусном эфире, промывали раствором поваренной соли, сушили над сульфатом магния и растворитель отгоняли. Полученное светло-коричневое масло (4,1 г) хроматографировали на 30-кратном количестве силикагеля. Метиленовые фракции удаляли и уксуксноэфирные и хлористометиленовые фракции (1:1) соединяли. В результате получали 1,5 г продукта в виде кристаллов бежевого цвета. Т.пл.: 116o. [α]D= -12,5o, (с=0,6, CHCl3).
Пример 86
а) (R)-3-(4-циклогексилфениламинометил)-4-(2,2-диметил-1,3-диоксолан)
10,0 г (57 ммолей) 4-циклогексиланилина, аналогично тому, как это описано в примере 42a), трансформировали и обрабатывали 20,0 г (70 ммолей) (S)-2,2-диметил-1,3-диоксолан-4- метанолтолуол-4-сульфоната. В результате получали 11,7 г (71%) желтоватого масла. [α]D= -2,4o (с=1,0, CHCl3).
б) (R)-3-(4-циклогексилфениламино) пропан-1,2-диол
11,5 г (R)-3-(4-циклогексилфениламино-метил)-4-(2,2-диметил-1,2- диоксолана), аналогично тому, как это описано в примере 42б), трансформировали и обрабатывали 100 мл 3н. соляной кислоты. В результате получали 6,25 г продукта в виде кристаллов бежевого цвета. Т.пл.: 118-120o.  -12,75o, (с= 0,4, CHCl3).
-12,75o, (с= 0,4, CHCl3).
Пример 87
[4-(4-оксоциклогексил)фенил]этоксикарбаминовый эфир
10,0 г (52,8 ммоля) 4-(4-аминофенил)циклогексанона растворяли в 150 мл тетрагидрофурана. После добавки 10 мл воды и 6,65 г (79,2 ммолей) карбоната натрия интенсивно перемешивали в течение 15 мин, после чего в течение 20 мин по каплям добавляли раствор из 5,5 мл этилового эфира хлормуравьиной кислоты (58,1 ммолей) в 25 мл тетрагидрофурана, причем температура не должна была превышать 20o. Через 2 ч выпавший осадок отсасывали и растворитель отгоняли в водоструйном насосе. Маслянистый остаток обрабатывали водой и продукт реакции экстрагировали с помощью уксусного эфира. Органическую фазу промывали водой, сушили над сульфатом магния и растворитель отгоняли. В результате получали 13,8 г (100%) карбамата. Т.пл.: 160-162o.
Пример 88
а) 1-(4-аминофенил)-4-пиперидинон
5,6 г 1-(4-нитрофенил)-4-пиперидона суспендировали в атмосфере аргона в 60 мл диоксана, обрабатывали 2,6 мл триэтиламина и 0,6 г палладия на угле (5%) и гидрировали при комнатной температуре и при нормальном давлении. После фильтрации и концентрирования получали 5,0 г 1-(4-аминофенил)-4-пиперидинона.
б) Этиловый эфир 4-(4- оксопиперидин-1-ил) фенилкарбаминовой кислоты
4,9 г 1-(4-аминофенил)- 4-пиперидинона суспендировали в атмосфере аргона в смеси из 30 мл тетрагидрофурана и 9 мл воды и обрабатывали 3,3 г гидрокарбоната натрия. Затем при энергичном перемешивании при комнатной температуре добавляли по каплям раствор из 2,7 мл этилового эфира хлормуравьиной кислоты в 20 мл тетрагидрофурана и после окончания добавления продолжали перемешивать еще в течение 1 ч. После добавления воды и этилового эфира уксусной кислоты фазы разделяли и водную фазу экстрагировали с помощью этилового эфира уксусной кислоты. Органические фазы соединяли, сушили над сульфатом магния и концентрировали. Остаток (6,6 г) хроматографировали на силикагеле 60 с помощью смеси из этилового эфира уксусной кислоты и гексана (1:2- 1:0). После кристаллизации из диэтилового эфира получали 3,8 г этилового эфира 4-(4-оксопиперидин-1-ил)фенилкарбаминовой кислоты в виде серых кристаллов. Т.пл.: 145-146o.
Пример 89
а) (RS)-3-(5-бромпиридин-2-ил)-5-метоксиметилоксазолидин-2-он
Смесь из 9,3 г этилового эфира 5-бромпиридин-2-ил-карбаминовой кислоты, 7,5 г 4-метоксиметил-1,3-диоксолан-2-она, 1,0 г карбоната калия и 10 мл декагидронафталина нагревали в течение 6 ч до 160o. Затем смесь охлаждали и обрабатывали этиловым эфиром уксусной кислоты и водой. Фазы разделяли, водную фазу экстрагировали с помощью этилового эфира уксусной кислоты и органические фазы промывали насыщенным раствором хлорида натрия. Органические фазы соединяли, сушили над сульфатом магния и затем концентрировали. После хроматографии остатка (20,2 г) с помощью смесей этилового эфира уксусной кислоты/гексана (1: 2-1: 1) на силикагеле получали 6,4 г (RS)-3-(5-бромпиридин-2-ил)-5- метоксиметилоксазолидин-2-она, который перекристаллизовывали из диэтилового эфира. Т.пл.: 90-92o.
б) (RS)-3-[5-(1,4-диоксаспиро[4,5]дец- 7-ен-8-ил)-пиридин-2-ил]-5-метоксиметилоксазолидин-2-он
В атмосфере аргона в течение 18 ч при 70o перемешивали суспензию из 1,4 г 3-(5-бромпиридин-2-ил)-5-метоксиметилоксазолидин-2-она, 1,4 г 8- триметилстаннил-1,4-диоксаспиро[4,5] дец-7-ена и 0,3 г дихлорида бис(трифенилфосфин) палладия (II) в 20 мл тетрагидрофурана. Затем суспензию охлаждали, обрабатывали 150 мл этилового эфира уксусной кислоты и 150 мл насыщенного раствора фторида калия и перемешивали в течение 30 мин. Смесь фильтровали через Celite®-буфер, фазы разделяли и водную фазу экстрагировали с помощью этилового эфира уксусной кислоты. Органические фазы соединяли, сушили над сульфатом магния, концентрировали и хроматографировали с помощью диэтилового эфира в качестве растворителя на силикагеле 60. В результате получали 0,6 г (RS)-3-[5-(1,4- диоксаспиро[4,5]дец-7-ен-8-ил)-пиридин-2-ил]-5- метоксиметилоксазолидин-2-она. 1H-ЯМР (CDCl3) част./млн: 8,41 (s, 1H), 8,03 (d, 1H), 7,89 (d, 1H), 6,08 (t, 1H), 4,82 (m, 1H), 4,21 (t, 1H), 3,91 (t, 1H), 3,92 (s, 4H), 3,60 (m, 2H), 3,23 (s, 3H), 2,54 (m, 2H), 2,38 (m, 2H), 1,82 (t, 2H).
Пример 90
а) 1.4-диоксаспиро[4.5]дец-7-ен-8-иловый эфир трифторметансульфокислоты.
Из 48 мл диизопропиламина и 210 мл 1,6М раствора n-бутиллития в гексане приготавливали в 1 л абсолютного тетрагидрофурана в атмосфере аргона раствор диизопропиламида лития. В этот раствор при -70o добавляли по каплям раствор из 50 г 1,4-циклогександионмоноэтиленкеталя в 200 мл тетрагидрофурана. После удаления охлаждающей ванны температура смеси повышалась до -20o, после чего ее повторно понижали до -70o и быстро обрабатывали 120 г N-фенилбис(трифторметансульфонимида) в 500 мл тетрагидрофурана. После удаления охлаждающей ванны температура смеси поднималась до комнатной температуры и смесь концентрировали. Остаток (215 г) фильтровали с помощью этилового эфира уксусной кислоты/гексана (1:9) через силикагель 60. В результате получали с количественным выходом 1,4-диоксаспиро[4,5]дец-7-ен-8-илового эфира трифторметансульфокислоты. 1H-ЯМР (CDCl3) част./млн: 5,66 (m, 1H), 3,99 (s, 4H), 2,53 (m, 2H), 2,42 (m, 2H), 1,90 (t, 2H).
б) 8-трибутилстаннил-1.4-диокса-спиро[4,5]дец-7-ен
Из 13 мл диизопропиламина и 59 мл 1,6М раствора н-бутиллития в гексане приготавливали в 400 мл абсолютного тетрагидрофурана в атмосфере аргона раствор диизопропиламида лития. При -70o в этот раствор добавляли по каплям 21 мл гидрида трибутилолова и перемешивали в течение 2 ч. После добавки 3,5 г цианида меди (I) смеси давали нагреться до -50o и затем быстро добавляли по каплям раствор из 10,4 г 1,4-диоксаспиро[4,5]дец-7-ен-8-илового эфира трифторметансульфокислоты в 80 мл тетрагидрофурана. При перемешивании в течение 2 ч при -30o реакционная смесь медленно окрашивалась в красный цвет. Затем удаляли охлаждающую ванну и смесь обрабатывали насыщенным раствором хлористого аммиака и гексаном. После фильтрации через Celite®-буфер водную фазу отделяли, а органическую фазу промывали последовательно водой и насыщенным раствором хлорида натрия. Затем органическую фазу сушили над сульфатом магния и концентрировали. Остаток растворяли в 300 мл этилового эфира уксусной кислоты, обрабатывали 18 г ацетата серебра и перемешивали на воздухе. Далее смесь фильтровали через Celite®\-буфер и фильтрат промывали водой и насыщенным раствором хлорида натрия. Органическую фазу сушили над сульфатом магния, концентрировали и хроматографировали на силикагеле 60 (дезактивация с помощью триэтиламина). В результате получали 8,9 г 8-трибутилстаннил-1,4-диокса-спиро[4,5] дец-7-ена. 1H-ЯМР (CDCl3) част./млн: 5,70 (m, 1H), 3,98 (s, 4H), 2,36 (шир., 4H), 1,74 (t, 2H), 1,45 (m, 6H), 1,30 (m, 6H), 0,88 (m, 9H).
Пример 91
a) (RS)-5-метоксиметил-3-пиридин-3-илоксазолидин -2-он
Смесь из 16,8 г этилового эфира пиридин-3-илкарбаминовой кислоты, 20 г 4-метоксиметил-1,3-диоксолан-2-она и 2,8 г карбоната калия нагревали в течение 3 ч до 160o. Затем смесь охлаждали и обрабатывали этиловым эфиром уксусной кислоты и водой. Фазы разделяли, после чего водную фазу экстрагировали с помощью этилового эфира уксусной кислоты, а органические фазы промывали насыщенным раствором хлорида натрия. Органические фазы соединяли, сушили над сульфатом магния и концентрировали. После хроматографии остатка (21 г) с помощью смесей этилового эфира уксусной кислоты/метанола (1:0-20:1) на силикагеле получали 17,6 г (RS)-5-метоксиметил-3- пиридин-3-ил-оксазолидин-2-она, который перекристаллизовывали из диэтилового эфира. Т.пл.: 65-66o.
б) (RS)-5- метоксиметил-3-(1-оксипиридин-3-ил)оксазолидин-2-он
В атмосфере аргона в раствор из 16,5 г (RS)-5-метоксиметил-3-пиридин-3- илоксазолидин-2-она в 250 мл хлористого метилена добавляли 33,4 г 3-хлорпербензойной кислоты и смесь перемешивали в течение 4 ч при комнатной температуре. Затем смесь промывали водой и водную фазу экстрагировали с помощью хлористого метилена. Органические фазы соединяли, сушили над сульфатом магния, концентрировали и хроматографировали на силикагеле с помощью смесей хлористого метилена/метанола (50: 1-20:1). Продукт, 15,9 г (RS)-5-метоксиметил-3-(1-оксипиридин-3-ил)оксазолидин- 2-она, кипятили для последующей очистки с диэтиловым эфиром. Т.пл.: 82- 83o.
в) (RS)-3-(6-бром-1-оксипиридин-3-ил)-5-метоксиметилоксазолидин-2-он
При комнатной температуре в раствор из 25,5 г (RS)-5- метоксиметил-3-(1-оксипиридин-3-ил)-оксазолидин-2-она в 300 мл хлористого метилена в атмосфере аргона добавляли по каплям 5,9 мл брома, растворенного в 35 мл хлористого метилена. Смесь продолжали перемешивать еще в течение 3 ч, затем обрабатывали 350 мл гидрокарбоната натрия, после чего фазы разделяли. Водную фазу еще несколько раз экстрагировали с помощью хлористого метилена. Органические фазы соединяли, сушили над сульфатом магния, концентрировали и хроматографировали на силикагеле 60 с помощью смесей хлористого метилена/метанола (50: 1-20: 1). После хроматографии получали 22,5 г (RS)-3-(6-бpoм-1-oкcипиpидин-3-ил)-5-мeтoкcимeтилoкcaзoлидин-2- oнa. 1H-ЯМР (CDCl3) част. /млн: 8,54 (s, 1H), 7,74 (d, 1H), 7,59 (d, 1H), 4,82 (m, 1H), 4,00 (t, 1H), 3,90 (t, 1H), 3,66 (m, 2H), 3,43 (s, 3H).
г) (RS)-3-(6-бромпиридин-3-ил)-5-метоксиметилоксазолидин-2-он
В кипящий раствор из 22,5 г (RS)-3-(6-бром-1-оксипиридин-3-ил)-5- метоксиметилоксазолидин-2-она в хлористом метилене в атмосфере аргона добавляли по каплям 10,5 мл трибромида фосфора и смесь кипятили в течение ночи. Затем смесь охлаждали и при энергичном перемешивании сливали на 300 мл 2н. натрового щелока. Водную фазу несколько раз экстрагировали с помощью хлористого метилена, органические фазы соединяли, сушили над сульфатом магния и концентрировали. Остаток (11,3 г) хроматографировали на силикагеле 60 с помощью хлористого метилена в качестве растворителя. После хроматографии получали 9,9 г (RS)-3-(6-бромпиридин-3-ил)-5-метоксиметилоксазолидин-2-она, который кипятили для последующей очистки с диэтиловым эфиром. Т.пл.: 108-110o.
Пример 92
(RS)-3-[6-(1,4-диоксаспиро [4.5]дец-7-ен-8-ил)пиридин-3-ил]-5-метоксиметилоксазолидин-2-он
В атмосфере аргона в течение 22 ч при 85o перемешивали суспензию из 8,9 г (RS)-3-(6-бромпиридин-3-ил)-5-метоксиметилоксазолидин-2-она, 6,5 г 8-трибутилстаннил-1,4-диокса-спиро[4,5]дец-7-ена и 1,4 г дихлорида бис (трифенилфосфин) палладия (II) в 40 мл диметилформамида. Затем смесь охлаждали, фильтровали через Celite®-буфер, фильтровальный остаток промывали большим количеством этилового эфира уксусной кислоты и органическую фазу несколько раз промывали полунасыщенным раствором хлорида натрия. Водные фазы экстрагировали с помощью этилового эфира уксусной кислоты, органические фазы соединяли, концентрировали до объема 250 мл, обрабатывали 250 мл насыщенного раствора фторида калия и перемешивали в течение 30 мин. Затем смесь повторно фильтровали через Celite®-буфер, фазы разделяли и водную фазу экстрагировали с помощью этилового эфира уксусной кислоты. Органические фазы соединяли, сушили над сульфатом магния, концентрировали и хроматографировали на силикагеле 60 с помощью диэтилового эфира в качестве растворителя. В результате получали 1,2 г (RS)-3-[6-(1,4-диокса-спиро[4,5]дец-7-ен-8-ил)-пиридин- 3-ил] -5-метоксиметилоксазолидин-2-она. 1H-ЯМР (CDCl3) част./млн: 8,48 (s, 1H), 8,19 (d, 1H), 7,56 (d, 1H), 6,50 (m, 1H), 4,79 (m, 1H), 4,12 (t, 1H), 4,02 (s, 4H), 3.98 (t, 1H), 3,64 (m, 2H), 3,44 (s, 3H), 2,80 (m, 2H), 2,52 (m, 2H), 1,96 (t, 2H).
Пример 93
Этиловый эфир 4-циклогептилфенилкарбаминовой кислоты
6,81 г (36,0 ммолей) 4-аминофенилциклогептана растворяли в 60 мл хлористого метилена и обрабатывали 3,13 г (39,6 ммолей) пиридина. Реакционную смесь при перемешивании при комнатной температуре в течение 10 мин обрабатывали по каплям раствором из 4,3 г (39,6 ммолей) этилового эфира хлормуравьиной кислоты в 15 мл хлористого метилена. Через 2 ч промывали водой, органическую фазу сушили над сульфатом натрия, фильтровали и упаривали. Этиловый эфир 4-циклогептилфенилкарбаминовой кислоты получали путем перегонки при 165-170o и давлении 0,3 мбар.
Пример 94
Этиловый эфир 4-(адамантан-1-ил)-фенилкарбаминовой кислоты
5,28 г (20 ммолей) гидрохлорида 4-адамантан-1-илфениламина помещали в 100 мл хлористого метилена и обрабатывали 3,22 г (42,0 ммоля) пиридина. В эту суспензию при перемешивании при комнатной температуре в течение 15 мин добавляли по каплям раствор из 2,39 г (22,0 ммоля) этилового эфира хлормуравьиной кислоты в 15 мл хлористого метилена. Реакционную смесь перемешивали в течение 2 ч при комнатной температуре. Затем промывали водой, органическую фазу сушили над сульфатом натрия, фильтровали и упаривали. Остаток перекристаллизовывали из хлористого метилена/н-гексана. В результате получали 5,6 г этилового эфира 4-адамантан-1- илфенилкарбаминовой кислоты в виде белого кристаллизата. Т.пл.: 132- 134o.
Пример 95
Смесь (R)- и (S)-[4-[4-[(R)-5-метоксиметил-2- оксооксазолидин-3-ил]-фенил]циклогексилиден]ацетонитрила
0,66 г натрия растворяли в атмосфере аргона в 70 мл этанола. Затем при 20o добавляли 5,08 г (=4,51 мл) диэтилцианометилфосфоната, растворенного в 20 мл этанола. Затем перемешивали в течение 30 мин при комнатной температуре, после чего добавляли 2,9 г (R)-5-метоксиметил-3-[4-(4-оксо-циклогексил) фенил] оксазолидин-2-она. Далее перемешивали еще в течение 1 ч при нагреве с обратным холодильником, охлаждали до комнатной температуры, устанавливали с помощью примерно 1 мл уксусного эфира pH 6 и реакционную смесь упаривали в вакууме. Остаток растворяли в дихлорметане и трижды промывали водой, после чего сушили и упаривали. Избыточный фосфорный эфир, еще имевшийся в остатке, отгоняли при температуре ванны ≈100o и вакууме 0,03 мм (Т. кип. 0,03 34o). Образовавшийся остаток (3,2 г) хроматографировали на 250 г силикагеля при среднем давлении. Элюирование проводили с помощью дихлорметана и дихлорметана/этилацетата (20: 1). Полученные согласно тонкослойной хроматограмме чистые фракции соединяли и концентрировали в вакууме. В результате получали 2,6 г смеси из (R)- и (S)-[4-[4-[ (R)-5-метоксиметил-2-оксооксазолидин-3-ил)] фенил]циклогексилиден] ацетонитрила (примерно 1:2 E/Z или Z/E) в виде слегка замутненного масла, которое непосредственно использовали для восстановления.
Пример 96
Трет.-бутиловый эфир 4-(4-нитрофенил)пиперидин-1-карбоновой кислоты
7,36 г (37,0 ммолей) 4-(4- нитрофенил)пиперидина и 8,88 г (40,7 ммолей) ди-трет.-бутилдикарбоната перемешивали в 150 мл хлороформа в течение 4 ч при комнатной температуре. Затем промывали водой, органическую фазу сушили над сульфатом натрия, фильтровали и упаривали. После перекристаллизации из н-гексана получали 11,2 г трет.-бутилового эфира 4-(4-нитрофенил)- пиперидин-1-карбоновой кислоты. Т.пл.: 60-62o.
Пример 97
Трет.-бутиловый эфир 4-(4-аминофенил) \ пиперидин-1-карбоновой кислоты
11,03 г (36,0 ммолей) трет.-бутилового эфира 4-(4-нитрофенил)-пиперидин-1- карбоновой кислоты растворяли в 200 мл спирта и гидрировали в присутствии 1 г палладия на угле (10%) при комнатной температуре и при нормальном давлении. После отфильтровывания катализатора фильтрат концентрировали и остаток (10,0 г) промывали в хлористом метилене водой. После перекристаллизации из простого эфира/н-гексана получали 8,0 г трет.-бутилового эфира 4-(4-аминофенил)пиперидин-1-карбоновой кислоты. Т.пл.: 114-116o.
Пример 98
Трет. -бутиловый эфир 4-(4- этоксикарбониламинофенил)пиперидин-1-карбоновой кислоты
8,84 г (32,0 ммоля) трет.-бутилового эфира 4-(4-аминофенил)пиперидин-1-карбоновой кислоты растворяли в 150 мл хлористого метилена и обрабатывали в присутствии 2,78 г (35,2 ммолей) пиридина и 3,82 г (35,2 ммолей) этилового эфира хлормуравьиной кислоты. Реакционную смесь перемешивали в течение 16 ч при комнатной температуре, после чего промывали водой, органическую фазу сушили над сульфатом натрия, фильтровали и упаривали. Остаток (11,3 г) хроматографировали на 95 г силикагеля 60 с помощью простого эфира/n-гексана (4: 6). После перекристаллизации из простого эфира/н-гексана получали 6,15 г трет.-бутилового эфира 4-(4- этоксикарбониламинофенил)пиперидин-1-карбоновой кислоты. Т.пл.: 126- 128o.
Пример 99
Этиловый эфир 4-(4-этоксикарбониламинофенил) пиперидин-1- карбоновой кислоты
3,01 г (14,6 ммолей) 4-(4-аминофенил)пиперидина растворяли в 100 мл хлористого метилена и при перемешивании обрабатывали в присутствии 11,55 г (146 ммолей) пиридина и 1,74 г (16,06 ммолей) этилового эфира хлормуравьиной кислоты. Через 3 ч смесь упаривали, остаток распределяли на уксусный эфир/воду, органическую фазу промывали водой, сушили над сульфатом натрия, фильтровали и упаривали. Остаток (2,7 г) хроматографировали на 85 г силикагеля 60 с помощью хлористого метилена/метанола (98:2). После перекристаллизации из простого эфира/н-гексана получали 0,9 г этилового эфира 4-(4- этоксикарбониламинофенил)пиперидин-1-карбоновой кислоты. Т.пл.: 105- 107o.
Пример 100
[4-(4-метиленциклогексил)фенил]этоксикарбаминовый эфир
Суспензию из 15,95 г смеси бромида метилтрифенилфосфония и амида натрия (38,3 ммолей бромида метилтрифенилфосфония) в 300 мл перемешивали в течение 1 ч при комнатной температуре. Затем добавляли раствор из 10,0 г (38,3 ммолей) [4-(4-оксоциклогексил) фенил] этоксикарбаминового эфира в 120 мл тетрагидрофурана и кипятили в течение ночи с обратным холодильником. Растворитель отгоняли и остаток хроматографировали на силикагеле с помощью уксусного эфира/гексана (1: 3). В результате получали 6,1 г (61%) [4-(4-метиленциклогексил)фенил]этоксикарбаминового эфира в виде белых кристаллов с т.пл. 98o.
Пример 101
Цис- и транс-4-(4-пианоциклогексил)анилин
В суспензию из 278 мг (1,5 ммоля) 4-(4- оксоциклогексил)анилина и 370 мг (1,9 ммоля) толуол-4- сульфонилметилизоцианида в 5 мл 1,2-диметоксиэтана и 0,15 мл этанола при -10oC порциями добавляли 392 мг (3,5 ммоля) трет.-бутилата калия. После перемешивания в течение 1 ч при -5oC и в течение последующих 70 ч при комнатной температуре добавляли 25 мл воды и 10 мл насыщенного раствора поваренной соли и трижды экстрагировали соответственно порциями по 40 мл дихлорметана. Органическую фазу сушили над сульфатом натрия и концентрировали. После хроматографии на силикагеле с помощью уксусного эфира/гексана (1: 2) получали 165 мг (56%) 4-(4-цианоциклогексил)-анилина с т. пл. 132-140o.
Пример 102
Цис-[4-(4-цианоциклогексил)фенил] этоксикарбаминовый эфир и транс-[4-(4-цианоциклогексил)фенил] этоксикарбаминовый эфир
В раствор из 2,75 г (14 ммолей) цис- и транс-4-(4-цианоциклогексил)-анилина в 50 мл тетрагидрофурана и 4 мл воды добавляли сначала 1,74 г (21 ммоль) гидрокарбоната натрия, а затем через 15 мин 1,46 мл (15,3 ммолей) этилового эфира хлормуравьиной кислоты, растворенного в 5 мл тетрагидрофурана. После перемешивания в течение 4 дней при комнатной температуре растворитель отгоняли, остаток суспендировали в 100 мл дихлорметана и экстрагировали с помощью воды и насыщенного раствора поваренной соли. Органическую фазу сушили над сульфатом натрия и концентрировали. После хроматографии остатка на силикагеле с помощью уксусного эфира/гексана (1:2) получали 630 мг (17%) транс-[4-(4- цианоциклогексил)фенил]-этоксикарбаминового эфира в виде белых кристаллов с т.пл. 145o и 80 мг (2%) цис-[4-(4-цианоциклогексил) фенил]этоксикарбаминового эфира в виде белых кристаллов с т.пл. 110o, а также 2,5 г (67%) смешанной цис-транс-фракции.
Пример 103
4-циклогексил-3-нитрофенилэтоксикарбаминовый эфир
Смесь из 1,97 г (8,9 ммолей) 4-циклогексил-3-нитроанилина и 1,13 г (13,4 ммолей) гидрокарбоната натрия в 25 мл тетрагидрофурана и 1,7 мл воды обрабатывали 1,07 г (9,9 ммолей) этилового эфира хлормуравьиной кислоты, растворенного в 5 мл тетрагидрофурана. После перемешивания в течение ночи при комнатной температуре растворитель отгоняли и остаток хроматографировали на силикагеле с помощью уксусного эфира/гексана. В результате получали 2,44 г (93%) желтого масла. МС: m/e (% основного пика):292 (М+, 7), 275 (100), 247 (14), 229 (27), 201 (18), 41 (26).
Пример 104
3-хлор-4-циклогексиланилин
Смесь из 1,0 г (5,0 ммолей) 4-циклогексилнитробензола и 0,1 г хлорида железа (III) нагревали до 60o. После расплавления циклогексилнитробензола в хорошо перемешанную смесь подавали в течение 15 мин газообразный хлор, после чего добавляли 20 мл ледяной воды и экстрагировали с помощью простого 5 эфира. Затем эфир отгоняли и остаток растворяли в 60 мл метанола. Далее обрабатывали 2,5 г (25 ммолей) хлорида меди (I) и порциями 1,6 г (29 ммолей) борогидрида калия. После перемешивания в течение 30 мин желировали в 0,5 г воды и экстрагировали с помощью простого эфира. После сушки над сульфатом натрия, отгонки эфира и хроматографии на силикагеле с помощью гексана/уксусного эфира (9:1) получали 0,68 г масла. МС: m/e (% основного пика) = 209 (М+, 75), 166 (100), 140 (78), 131 (62).
Пример 105
(3-хлор-4-циклогексилфенил) этоксикарбаминовый эфир
Смесь из 0,59 г (2,8 ммоля) 3-хлор-4-циклогексиланилина и 0,36 г (4,2 ммоля) гидрокарбоната натрия в 10 мл тетрагидрофурана и 0,75 мл воды обрабатывали 0,34 г (3,1 ммоля) этилового эфира хлормуравьиной кислоты, растворенного в 1 мл тетрагидрофурана. После 5-часового перемешивания при комнатной температуре сливали на 25 мл воды и экстрагировали с помощью дихлорметана. После хроматографии на силикагеле с помощью гексана/дихлорметана (2: 1) получали 0,47 г (59%) масла. МС: m/e (% основного пика) = 281 (М+, 100), 238 (18), 225 (12), 212 (34), 192 (23), 166 (19), 140 (20).
Пример A
(RS)-3-(4-циклогексилфенил)-5-гидроксиметилоксазолидин-2-он может применяться в качестве активного вещества при изготовлении по известным методам фармацевтических препаратов следующего состава:
1. Капсулы по 100 мг
Активное вещество - 100,0 мг
Лактоза порошкообр. - 104,7 мг
Тальк - 12,0 мг
Стеарат магния - 3,0 мг
Кукурузный крахмал - 70,0 мг
Гидроксипропилметилцеллюлоза - 10,0 мг
Сульфосукцинат диоктилнатрия - 0,3 мг - _________ - 300 мг
2. Таблетки по 50 мг
Активное вещество - 50,0 мг
Лактоза порошкообр. - 50,0 мг
Микрокристаллическая целлюлоза - 82,0 мг
Na-карбоксиметилированный крахмал - 15,0 мг
Стеарат магния - 3,0 мг - _________ - 200 мг
3. Суппозитории по 500 мг
Активное вещество - 500 мг
Масса суппозитория - До 2000 мг
4. Суппозитории по 500 мг
Активное вещество - 500 мг
Масса суппозитория - До 2000 мг
5. Мягкожелатиновые капсулы по 100 мг
Активное вещество - 100 мг
Триглицерид - 300 мг - _________ - 400 мге
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ИНДОЛА И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ КИСЛОТНО-АДДИТИВНЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ НА ИХ ОСНОВЕ | 1994 |
|
RU2136662C1 |
| ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ПИРАЗОЛА | 1995 |
|
RU2152388C1 |
| ПРОИЗВОДНЫЕ КАРБАМИНОВОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 1999 |
|
RU2179969C2 |
| ПРОИЗВОДНЫЕ ЦЕФАЛОСПОРИНА И ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ | 1994 |
|
RU2130939C1 |
| ИМИДАЗОДИАЗЕПИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 1995 |
|
RU2139873C1 |
| БЕНЗОЛСУЛЬФОНОВЫЕ ПРОИЗВОДНЫЕ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 1999 |
|
RU2201922C2 |
| ЗАМЕЩЕННЫЕ ПИРРОЛЫ | 1994 |
|
RU2141960C1 |
| ТРИЦИКЛИЧЕСКИЕ ДИКАРБОНИЛЬНЫЕ ПРОИЗВОДНЫЕ И ЛЕКАРСТВЕННЫЙ ПРЕПАРАТ НА ИХ ОСНОВЕ | 1995 |
|
RU2145606C1 |
| О-АРИЛЬНЫЕ ЭФИРЫ МОРФИНАНОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2123000C1 |
| ПРОИЗВОДНЫЕ АМИНОКИСЛОТ И ИХ КИСЛОТНО-АДДИТИВНЫЕ СОЛИ | 1990 |
|
RU2071470C1 |


Описываются новые производные оксазолидин-2-она общей формулы I, где R обозначает низший алкил; Х обозначает циклоалкенил; бицикло (2,2,1)гепт-2-ил, необязательно замещенный фенил-2-оксо-5-метоксиметилоксазолидинилом; бицикло (2,2,1)гепт-5-ен-2-ил; адамантил или циклоалкил либо пиперидин, необязательно одно- или многократно замещенный галогеном, амино, низшим алкилом, нитрилом, оксогруппой, гидроксиимино, этилендиоксигруппой или OR1, где R1 обозначает -СН(С6Н5)2, -(CН2)nС6Н5, низший алкил, водород, -(СН2)nNНСОСН3-(СН2)nNH2, -(CH2)nSOCH3, -(CH2)nСN, -(CH2)nSCH3,
-(CH2)nSO2CH3-СО-низший алкил, -СОС6Н5, необязательно замещенный оксазолидиниловой группой, или замещенный =CR2R3, где R2 обозначает водород или низший алкил; R3 обозначает водород, нитрил, низший алкил, фенил или СОО-низший алкил, или замещенный -(СН2)nR4, где R4 обозначает нитрил, амино, -NНСОСН3-, -СОС6Н4гал, фенил или гидроксильную группу; или замещенный -COR5, где R5 обозначает низший алкил, -СН=СН-С6Н5, -С6Н5СF3, -О-С/СН3/3 или -0-низший алкил; или замещенный -NR6R7, где R6 обозначает водород или -СОСН3; R7 обозначает -СОСН3 бензил или -(СН2)nNНСОС6Н4гал; n-1-3; Y1- -СН = или -N= ; Y2-СН= , -С(ОН)=, -С(NO2)=, -С(NH2)=, -С (гал) - или -N=, а также фармацевтически приемлемые соли основных соединений формулы I с кислотами. Эти соединения обладают способностью ингибировать моноаминооксидазу. Описывается лекарственное средство на основе соединений формулы I, которое может применяться для лечения состояний депрессии, состояний паники и страха. 2 с. и 21 з.п.ф-лы.
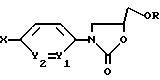
где R обозначает низший алкил;
X обозначает циклоалкенил; бицикло[2,2,1]гепт-2-ил, не обязательно замещенный фенил-2-оксо-5-метоксиметилоксазолидинилом; бицикло[2,2,1]гепт-5-ен-2-ил; адамантил или циклоалкил либо пиперидин, не обязательно одно- или многократно замещенный галогеном, амино, низшим алкилом, нитрилом, оксогруппой, гидроксиимино, этилендиоксигруппой или -OR1, где R1 обозначает -CH(C6H5)2, -(CH2)nC6H5, низший алкил, водород, -(CH2)nNHCOCH3, -(CH2)nNH2,
-(CH2)nSOCH3, -(CH2)nCN, -(CH2)nSCH3, -(CH2)nSO2CH3, -CO-низший алкил, -COC6H5, не обязательно замещенный оксазолидиниловой группой или замещенный =СR2R3, где R2 обозначает водород или низший алкил; R3 обозначает водород, нитрил, низший алкил, фенил или -COO-низший алкил; или замещенный -(CH2)nR4, где R4 обозначает нитрил, амино, -NHCOCH3, -COC6H4гал, фенил или гидроксильную группу; или замещенный -COR5, где R5 обозначает низший алкил, -CH= CH-C6H5, -C6H5CF3, -O-C(CH3)3 или -O-низший алкил; или замещенный -NR6R7, где R6 обозначает водород или -COCH3; R7 обозначает -COCH3, бензил или -(CH2)nNHCOC6H4гал;
n обозначает 1 - 3;
Y1 обозначает -CH= или -N=;
Y2 обозначает -CH=, -C(OH)=, -C(NO2)=, -C(NH2)=, -C(гал)= или -N=,
а также фармацевтически приемлемые соли основных соединений формулы I с кислотами.
(RS)-3-[4-(4-оксоциклогексил)-фенил]-5-метоксиметилоксазолидин-2-он;
(RS)-3-[4-(транс-4-гидроксициклогексил)-фенил] -5-метоксиметилоксазолидин-2-он;
(RS)-3-[4-(4-гидроксииминоциклогексил)-фенил] -5-метоксиметилоксазолидин-2-он;
(R)-3-[4-(транс-4-гидроксициклогексил)-фенил] -5-метоксиметилоксазолидин-2-он;
(RS)-3-[4-(транс-4-метоксициклогексил)-фенил] -5-метоксиметилоксазолидин-2-он;
(RS)-3-[4-(транс-4-ацетоксициклогексил)-фенил] -5-метоксиметилоксазолидин-2-он.
Приоритет по признакам:
13.12.93 - при R - низший алкил; X - бицикло[2,2,1]гепт-2-ил, не обязательно замещенный фенил-2-оксо-5-метоксиметилоксазолидинилом; бицикло[2,2,1]гепт-5-ен-2-ил; адамантил; или циклоалкил либо пиперидин, не обязательно одно- или многократно замещенный галогеном, низшим алкилом, оксогруппой, гидроксиимино, этилендиоксигруппой или -OR1, где R1 - низший алкил, водород, -(CH2)3NH2, -(CH2)2CN, -CO-низший алкил, -COC6H5, необязательно замещенный оксазолидиниловой группой; или замещенный = СR2R3, где R2 и R3 обозначают водород; или замещенный группой -CH2OH; или замещенный -NR6R7, где R6 обозначает водород; R7 обозначает -(CH2)nNHCOC6H4гал; n = 1 - 3; Y1 обозначает -CH= или -N=; Y2 обозначает -CH= или -N=, а также фармацевтически приемлемые соли основных соединений формулы I с кислотами;
27.09.94 - при R - низший алкил; X - циклоалкенил; или циклоалкил либо пиперидин, необязательно одно- или многократно замещенный амино, нитрилом или -OR1, где R1 обозначает -CH(C6H5)2, -(CH2)nC6H5, где n = 1 - 3, -(CH2)nNHCOCH3, где n = 1 - 3, -(CH2)nNH2, где n = 1, 2; -(CH2)nSOCH3, где n = 1 - 3, -(CH2)nCN, где n = 1, 3; -(CH2)nSCH3, где n = 1 - 3; -(CH2)nSO2CH3, где n = 1 - 3; или замещенный =СR2R3, где R2 обозначает водород или низший алкил; R3 обозначает нитрил, низший алкил, фенил или -COO-низший алкил; или замещенный -(CH2)nR4, где R4 обозначает нитрил, амино, -NHCOCH3, -COC6H5гал, фенил, при n = 1 - 3, или R4 - гидроксильная группа при n = 2, 3; или замещенный -COR5, где R5 обозначает низший алкил, -CH=CH-C6H5, -C6H5CF3, -O-C(CH3)3 или -O-низший алкил; или замещенный -NR6R7, где R6 обозначает водород или -COCH3; R7 обозначает -COCH3 или бензил; Y1 обозначает -CH= или -N= ; Y2 обозначает -C(OH)=; -C(NO2)=; -C(NH2)=; -C(гал)=, а также фармацевтически приемлемые соли основных соединений формулы I с кислотами.
| СЫВОРОТОЧНЫЙ ИММУНОБИОЛОГИЧЕСКИЙ ПРЕПАРАТ ДЛЯ ЭКСТРЕННОЙ ПРОФИЛАКТИКИ И ЛЕЧЕНИЯ СИБИРСКОЙ ЯЗВЫ | 2008 |
|
RU2381037C1 |
| Способ получения 2-оксооксазолидинилбензолсульфонамидов | 1982 |
|
SU1194274A3 |
| GB 2003151 A, 1979 | |||
| РЕМЕНЬ БЕЗОПАСНОСТИ ТРАНСПОРТНОГО СРЕДСТВА | 1994 |
|
RU2076813C1 |
| Гидроразбиватель для первичной обработки макулатуры | 1972 |
|
SU511031A3 |
| УСТРОЙСТВО ДЛЯ ИНДИКАЦИИ | 1973 |
|
SU428421A1 |

