Изобретение относится к новым производным аминокислоты, их фармацевтически приемлемым солям присоединения кислот, способам их получения и фармацевтической композиции, обладающей антивирусной активностью на их основе.
Предлагаемые новые соединения ингибируют аспартилпротеазы вирусной природы и их можно использовать для профилактики и лечения вирусных инфекций, особенно инфекций, вызванных ВИЧ и другими ретровирусами, и для получения лекарственных средств для использования по указанному назначению.
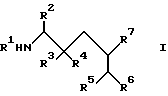
Согласно изобретению предлагаются производные аминокислоты общей формулы I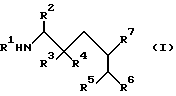
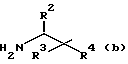
где R1 означает C1-C8-алкоксикарбонил или группу формулы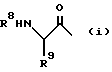
где R2 означает фенил- C1-C8-алкил, R3 означает водород, R4 означает гидроксильную группу, или R3 и R4, взятые вместе, означают оксогруппу, R5 означает C1-C8-алкоксикарбонил или C1-C8-алкилкарбамоил, R6 и R7, взятые вместе, означают триметилен или тетраметилен, R8 означает фенил-1C1-C8-алкоксикарбонил, хинолилкарбонил, и R9 означает C1-C8-алкил, фенил C1-C8-алкил, циано- C1-C8-алкил, карбамоил- C1-C8-алкил, алкилтио- C1-C8-алкил, или C1-C8-алкоксикарбонил- C1-C8-алкил, и фармацевтически приемлемые соли присоединения кислот указанных соединений формулы I, которые являются основными.
Предпочтительны соединения I, где R2 означает бензил, R3 означает водород, R4 означает гидроксильную группу, R5 означает C1-C8-алкилкарбамоил или R5 означает трет-бутилкарбамоил, либо R6 и R7 вместе означают незамещенный тетраметилен, либо соединения формулы I, где R1 означает трет-бутоксикарбонил или группу формулы (i), где R8 означает бензилоксикарбонил, а R9 означает карбамоилметил, R2 означает бензил, R3 означает водород, R4 означает гидроксильную группу, R5 означает трет-бутилкарбамоил, а R6 и R7 вместе означают незамещенный тетраметилен.
Наиболее предпочтительными являются соединения, выбранные из группы:
2(S)-[3(S)-(трет-бутоксиформамидо)-2(R)-гидрокси-4-фенилбутил] -N-трет-бутил-1(R)-циклогексанкарбоксамид;
2(R)-[3(S)-(трет-бутоксиформамидо)-2(R)-гидрокси-4-фенилбутил] -N-трет-бутил-1(R)-циклогексанкарбоксамид;
2(S)-[3(S)-[[N-(бензилоксикарбонил)-L-аспарагинил] -амино] -2(R)- гидрокси-4-фенилбутил]-N-трет-бутил-1(R)-циклогексанкарбоксамид;
2(R)-[3(S)-[[N-(бензилоксикарбонил)-L-аспарагинил]-амино]- 2(R)-гидрокси-4-фенилбутил]-N-трет-бутил-1(R)-циклогексанкарбоксамид.
Изобретение относится также к соединениям общей формулы A: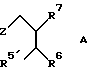
где Z означает группу формулы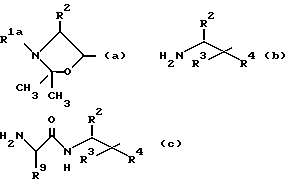
где R1a означает С1-C8-алкоксикарбонил, R5' означает C1-C8-алкоксикарбонил, C1-C8-алкилкарбамоил, винил или карбоксил и R2, R3, R4, R6, R7 и R9 имеют указанные значения.
Среди известных противовирусных средств отсутствуют сведения об аналогах новых производных аминокислоты. Указанные новые соединения можно отнести к категории нетоксичных соединений.
Используемый в данном описании термин "алкил" индивидуально либо в комбинации означает алкильную группу с прямой или разветвленной цепью, содержащую максимум до 8, предпочтительно до 4, атомов углерода, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, н-гексил и т.д. Термин "алкокси" отдельно или в комбинации означает алкильную группу, в которой "алкил" имеет указанные значения, например, метокси-, этокси-, н-пропокси-, изопропокси-, н-бутокси-, изобутокси-, втор-бутокси-, трет-бутокси- и т.д. Термин "фенилалкил" означает алкильную группу, имеющую указанные значения, в которой один атом водорода замещен фенильной группой, возможно имеющий один или несколько заместителей, выбранных из алкила, алкоксила, галогена, трифторметила, гидроксила, нитро-, аминогруппы и т.д. К примерам фенилалкильных групп относятся бензил, 4-хлорбензил, 4-метоксибензил, 2-фенилэтил. Термин "фенилалкоксикарбонил" означает группу формулы -C(O)-O-алкилфенил, где "алкил" имеет указанные значения.
Фармацевтически приемлемые соли присоединения кислот образуются в результате взаимодействия основных производных формулы I с неорганическими кислотами, например соляной и бромистоводородной кислотой, серной, азотной, фосфорной и другими кислотами, либо с органическими кислотами, например уксусной, лимонной, малеиновой, фумаровой, винной кислотами, метансульфоксилотой, пара-толуолсульфокислотой и т.д.
Соединения формулы I содержат по крайней мере 3 асимметрических атома углерода и, таким образом, могут быть в виде оптически чистых диастереоизомеров, их смесей, диастереоизомерных рацематов или смесей из диастереоизомерных рацематов. Притязания настоящего изобретения включают все эти формы.
Изобретение также относится к способам получения производных аминокислоты, указанным выше.
a) Для получения соединения формулы I, где R1 означает C1-C8-алкоксикарбонил, R3 означает водород, а R4 означает гидроксильную группу (cпособ A), соединение общей формулы II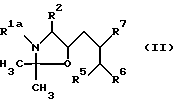
где R1a означает C1-C8-алкоксикарбонил, а R2, R5 и R7 имеют указанные значения обрабатывают кислотой и
б) при желании, превращают основное соединение формулы I в его фармацевтически приемлемую соль присоединения кислоты.
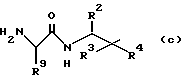
Изобретение относится к способу получения производных аминокислоты формул I и A (способ B), заключающийся в том, что:
а) осуществляют взаимодействие соединения общей формулы III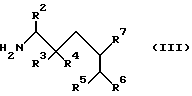
где R2, R3, R4, R5, R6 и R7 имеют указанные значения с ацидирующим агентом, который вводит группу R1, имеющую указанные значения; и
б) для получения соединения формулы I, где R3 и R4 вместе означают оксогруппу, окисляют соединение формулы I, где R3 означает водород, а R4 означает гидроксильную группу, и/или
в) при желании, превращают основное соединение формулы I в его фармацевтически приемлемую соль присоединения кислоты.
Другой способ получения соединения формулы I, где R1 означает группу формулы I (способ C), заключается в том, что осуществляют взаимодействие соединения общей формулы IV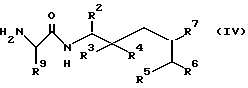
где R2, R3, R4, R5, R6, R7, R9 имеют указанные значения, с ацилирующим агентом, который вводит группу R8, имеющую значения, указанные выше, и
б) для получения соединения формулы I, где R3 и R4 вместе означают оксогруппу, окисляют соединение формулы I, где R3 означает водород, а R4 означает гидроксильную группу, и/или
в) при желании превращают основное соединение формулы I в его фармацевтически приемлемую соль присоединения кислоты.
При обработке соединения формулы II кислотой в соответствии с вариантом осуществления способа A получают соединение формулы I, где R1 означает алкоксикарбонил, R3 означает водород, а R4 означает гидроксильную группу. Используемый в данном варианте тип кислоты зависит от природы заместителя радикала R1a, присутствующего в соединении формулы II в качестве исходного материала. В случае, когда R1a означает алкоксикарбонил, например трет-бутоксикарбонил, такую обработку предпочтительно проводят при использовании сильной органической кислоты, особенно органической сульфокислоты, например алкансульфокислоты, например, метансульфокислоты, или ароматической сульфокислоты, например бензолсульфокислоты, паратолуолсульфокислоты, мезитиленсульфокислоты и т.д., и в присутствии любого органического растворителя, который инертен в реакционных условиях, например алканола, такого как метанол, этанол и т. д. Однако можно использовать вместо органической сульфокислоты галоидзамещенную кислоту, например трифторуксусную кислоту.
Взаимодействие соединения формулы III с ацилирующим агентом в соответствии с вариантом осуществления предлагаемого способа B можно проводить любым известным методом, в частности, при использовании в качестве ацилирующего агента соответствующей кислоты, или ее реакционноспособного производного. К подходящим реакционноспособным производным относятся галоидангидриды, например хлорангидриды, ангидриды кислот, смешанные ангидриды, активированные сложные эфиры и т.д. Если в качестве вещества для ацилирования используют реагент, который вводит группу формулы (i), то указанную реакцию целесообразно проводить при использовании кислоты в присутствии конденсирующего агента, например дициклогексилкарбодиимида или бензтриазол-1-илокси-трис(диметиламино)фосфония гексафторфосфат, и в присутствии основания, например триэтиламина, этилдиизопропиламина и т.п. Реакцию предпочтительно проводят при температуре примерно от 0oC до примерно комнатной температуры, предпочтительно при комнатной температуре.
Взаимодействие соединения формулы IV с ацилирующим агентом в соответствии с вариантом осуществления предлагаемого способа (C) можно проводить любым известным методом, в частности, с использованием в качестве ацилирующего агента соответствующей кислоты или ее реакционноспособного производного. К подходящим реакционноспособным производным относятся галоидангидриды, например хлорангидриды, ангидриды кислот, смешанные ангидриды, активированные сложные эфиры и т.д. Реакцию ацилирования целесообразно проводить при температуре в интервале примерно от 0oC до комнатной температуры, предпочтительно при комнатной температуре.
Реакцию окисления в соответствии со способом (б) предлагаемого способа можно проводить известным способом, используемым для окисления вторичных спиртов до образования кетонов. Так, например, реакцию окисления можно осуществлять при использовании бихромата пиридиния в диметилформамиде, хлорхромата пиридиния в дихлорметане, комплекса серного ангидрида с пиридином в диметилсульфоксиде, хлористого оксалила и триэтиламина в диметилсульфоксиде, дициклогексилкарбодиимида и любой органической кислоты, например такой, как дихлоруксусная или трифторуксусная кислоты в диметилсульфоксиде.
Разделение полученных продуктов в соответствии с вариантами осуществления предлагаемого способа можно проводить общеизвестными методами, например колоночной хроматографией, тонкослойной хроматографией, жидкостной хроматографией высокого давления и т.д.
Превращение основного соединения формулы I в фармацевтически приемлемую соль согласно осуществлению предлагаемого способа можно осуществлять путем обработки его традиционным методом при использовании либо неорганической кислоты, например галоидводородной кислоты, такой как соляная или бромистоводородная кислота, серная, азотная, фосфорная кислота и т.д., либо органической кислоты, например уксусной, лимонной, малеиновой, фумаровой, винной, метансульфоксилоты, пара-толуолсульфокислоты и других кислот.
Соединения формулы II, используемые в качестве исходного вещества по способу A осуществления способа, являются новыми и составляют еще один предмет данного изобретения. Их можно получить в соответствии с реакционной схемой, приведенной на чертеже, где R1a, R2, R6 и R7 имеют указанные значения.
Обращаясь к указанной схеме реакции, на первой стадии соединения формулы V взаимодействуют с соединением формулы VI с образованием соединения формулы VII. Реакцию проводят в условиях, известных для реакции Гриньяра, например, в органическом растворителе, инертном для указанных реакционных условий, таком как простой эфир, например диэтиловый эфир, при температуре в интервале примерно от 0oC до примерно 40oC, предпочтительно при комнатной температуре.
На следующей стадии соединение формулы VII превращают в соединение формулы VIII путем взаимодействия его с 2,2-диметоксипропаном в присутствии сильной органической кислоты, предпочтительно органической сульфокислоты, например пара-толуолсульфоксилоты. Для удобства указанную реакцию проводят примерно при комнатной температуре.
После этого соединение формулы VII окисляют до получения соединения формулы IX. Реакцию окисления предпочтительно проводить при использовании перманганата щелочного металла, например перманганата калия, примерно при комнатной температуре. Для удобства реакцию окисления проводят в системе растворителей из смеси воды, алканкарбоновой кислоты, например ледяной уксусной кислоты, и инертного органического несмешивающегося растворителя, например ароматического углеводорода, такого как бензол, толуол и тому подобное, в присутствии катализатора фазового переноса.
И наконец, соединение формулы IX превращают в соединение формулы II этерификацией или амидированием. Реакцию этерификации или амидирования осуществляют путем взаимодействия соединения формулы IX с соответствующим алканолом или амином известными методами.
Соединения формул V и VI, используемые для получения соединений формулы II, являются известными соединениями или их аналогами, которые можно получить методом, аналогичным для известных соединений. Более того, в приводимых далее примерах дана подробная информация относительно получения некоторых соединений формулы VI. Соединения формул VII, VIII и IX с другой стороны являются новыми соединениями и каждое представляет предмет данного изобретения.
Соединения формулы II, используемые в качестве исходных веществ в способе A, являются новыми и охватываются данным изобретением. Их можно получить путем удаления алкоксикарбонильной или аралкоксикарбонильной группы из соединения формулы I, в котором R1 означает алкоксикарбонил или аралкоксикарбонил. Такое удаление можно осуществлять известными методами, например, при использовании сильной неорганической кислоты, например, такой как галоидводородная кислота, или сильной органической кислоты, например, трифторуксусной кислоты, предпочтительно при температуре от примерно 0oC до примерно комнатной температуры. В альтернативном варианте аралкоксикарбонильную группу можно отщепить при использовании водорода в присутствии катализатора на основе благородных металлов, например палладиевой черни, в любом из органических носителей, инертном в условиях реакции, например алканоле, таком как этанол и т. д. , либо в эфире алканкарбоновой кислоты, например этилацетате, целесообразно при комнатной температуре.
Соединения формулы IV, используемые в качестве исходных веществ в способе C, являются новыми и охватываются изобретением. Их можно получить путем удаления алкоксикарбонильной или аралкоксикарбонильной группы из соединения формулы I, где R1 означает группу формулы (i), а R8 означает алкоксикарбонил или аралкоксикарбонил. Такое отщепление можно осуществлять известными методами, например, при использовании сильной неорганической кислоты, такой как галоидводородная кислота, или сильной органической кислоты, например трифторуксусной кислоты, предпочтительно при температуре примерно от 0oC до примерно комнатной температуры. В альтернативном варианте аралкоксикарбонильную группу можно отщеплять при использовании водорода в присутствии катализатора на основе благородных металлов, например палладиевой черни, в органическом инертном растворителе, например алканоле, таком как этанол и т. п., либо в эфире алканкарбоновой кислоты, например, этилацетате, целесообразно примерно при комнатной температуре.
Соединения формулы V и VI, используемые для получения соединений формулы II, являются известными соединениями или их аналогами, получаемыми аналогично известным соединениям. Более того, приводимые в описании примеры содержат подробную информацию относительно получения некоторых соединений формулы VI. Соединения формул VII, VIII и IX, с другой стороны, являются новыми и охватываются настоящим изобретением.
Как указывалось ранее, соединения формулы I и их фармацевтически приемлемые соли присоединения кислот к этим соединениям, которые являются основными, ингибируют аспартилпротеазы вирусной природы и пригодны для лечения и профилактики вирусных инфекций, вызванных вирусом иммунодефицита человека (ВИЧ) и другими ретровирусами.
Объектом изобретения является фармацевтическая композиция, обладающая антивирусной активностью, содержащая активные вещества и при необходимости фармацевтически приемлемый носитель. В качестве активного вещества композиция содержит производные аминокислоты формул I и A в эффективном количестве.
Использование общепринятых в фармацевтической практике наполнителей, разбавителей и иных добавок для приготовления композиций, содержащих эффективное количество активного ингредиента, дает возможность специалисту в данной области создавать различные лекарственные формы.
In vitro ингибирование ВИЧ-протеазы предлагаемыми изобретением производными аминокислоты можно продемонстрировать с помощью следующего испытания.
ВИЧ-протеазу экспрессировали в E.coli и частично очищали от растворимых экстрактов указанного микроорганизма методом фракционирования сульфатом аммония (0 - 30%). Протеазную активность анализировали при использовании защищенного гептапептидом сукцинил-Val-Ser-Gln-Asn-Phe-Pro-lle-изобутиламида в качестве субстрата. Степень гидролиза субстрата определяли по продуцированию H-Pro-lle изобутиламида спектрофотометрическим анализом N-концевого пролина.
1,25 ммоль субстрата растворяли в 125 ммоль цитратного буфера (pH 5,5), содержащего 0,125 мн/мл Твин-20, 10 мкл раствора с различными концентрациями испытуемого соединения (растворенного в метаноле или диметилсульфоксиде и разведенного водой, содержащей 0,1% Твин-20) и 10 мкл протеазы добавляли к 80 мкл указанного буферного субстрата. Гидролиз проводили при 37oC в течение заданного промежутка времени с последующим добавлением 1 мкл окрашивающего агента (30 мкг/мл изатина и 1,5 мг/мл 2- (4-хлорбензоил) бензойной кислоты в 10%-ном ацетоне, растворенном в этаноле, об/об). Полученный раствор нагревали в водяной бане, а затем окрашенные осадки растворяли в 1 мл 1%-ного пирогаллола в 33%-ном водном растворе ацетона (мас/об/об). Оптическую плотность раствора определяли спектрофотометрически на длине волны 599 нм. Полученный H-Pro-lle-изобутиламид в присутствии испытуемого соединения сравнивали с контрольными образцами и концентрацию испытуемого соединения, необходимую для 50% ингибирования (ИК50) устанавливали методом графопостроения испытуемого соединения, используемого в различных концентрациях.
В указанном испытании для определения ингибирования ВИЧ протеазы, ИК50 2(S)-[3(S)-[[N-бензилоксикарбонил)-1-аспарагинил]амино]- 2(R)-гидрокси-4-фенилбутил] -N-трет-бутил-1(R)-циклогексанкарбоксамид составляет 2 нмоль, а ИК50 2(R)-[[N-бензилоксикарбонил)-1-аспарагинил] амино] -2(R)-гидрокси-4- фенилбутил]-N-трет-бутил-1(R)-циклогексанкарбоксамида составляет 57 нмоль.
Антивирусную активность in vitro соединений формулы I можно продемонстрировать описанным анализом.
В этом анализе используют вирус T-клеточного лейкоза человека HTL V - III (штамм RF), выращенный в клетках C8166 (CD4+ T -лимфобластной линии) при использовании культуральной среды PPMI 1640 с бикарбонатным буфером, антибиотиками и 10% фетальной бычьей сыворотки.
Суспензию клеток инфицируют десятикратной ИД50 вирусных Т-клеток, а затем выдерживают в течение 90 мин при 37oC для поглощения клеток. После этого клетки промывают трижды при использовании указанной среды. Испытание проводят в 6 мл пробирках для тканевых культур, в которых содержалось 2•105 инфицированных клеток в 1,5 мл указанной среды. Испытуемые соединения растворяют либо в водной среде, либо в диметилсульфоксиде в соответствии со способностью к растворению и затем добавляют 15 мл раствора испытуемого соединения в указанные пробирки. Полученные культуры инкубируют в течение 72 ч при 37oC в увлажненной атмосфере, содержащей 5% двуокиси углерода в воздухе. Затем культуры центрифугируют, аликвоту супернатанта растворяют при использовании Nonidet P40 и подвергают иммуноанализу с захватом антигена при использовании первичной антисыворотки со специфической реакционной способностью против антигена вирусного p24, и системы детектирования на основе пероксидазы хрена обыкновенного. Цветной показатель определяют спектрофотометрическим методом, вычерчивают график зависимости от концентрации испытуемого соединения и устанавливают концентрацию, создающую 50% защиту (ИК50).
В указанном испытании для определения антивирусной активности in vitro ИК50 2(S)-[3(S)-[[N-(бензилоксикарбонил)-L-аспарагил] амино] -2(R)-гидрокси-4-фенилбутил] -N-трет-бутил-[1(R)- циклогексанкарбоксамида составляет 46 нмоль, а соединения 2(R)-[3(S)-[[N-(бензилоксикарбонил)-L-аспарагинил]амино] -2(R)- гидрокси-4-фенилбутил[-N-трет-бутил-1(R)-циклогексанкарбоксамида - 1000 нмоль.
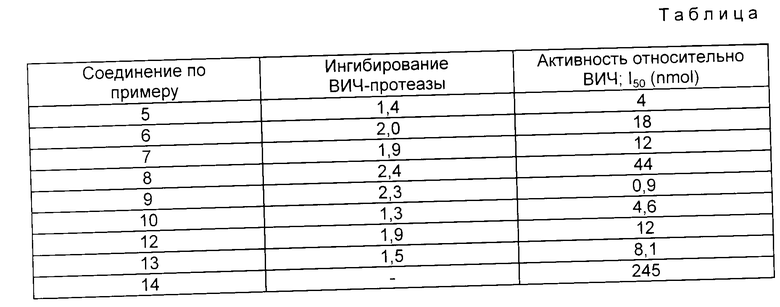
В таблице приведены данные по биологической активности соединений, полученные в соответствии с приведенными методиками.
Соединения формулы I и фармацевтически приемлемые соли указанных соединений, которые являются основными, можно использовать в качестве лекарственных средств, например в форме фармацевтических препаратов. Предлагаемые фармацевтические препараты можно вводить энтерально, например перорально, в виде таблеток, таблеток с покрытием, драже, твердых и мягких желатиновых капсул, растворов, эмульсий или суспензий, назально, аэрозолей для введения в нос, ректально, например в виде суппозиториев, или парентерально, например внутримышечно или внутривенно, например в виде растворов для инъекций.
Для получения фармацевтических препаратов указанные соединения и их соли можно использовать в сочетании с терапевтически инертными неорганическими или органическими наполнителями. В качестве таких наполнителей для таблеток, покрытых таблеток и твердых желатиновых капсул можно использовать, например, лактозу, кукурузный крахмал или их производные, тальк, стеариновую кислоту или ее соли. В качестве подходящих наполнителей для мягких желатиновых капсул можно использовать, например, растительные масла, воски, жиры, полутвердые или жидкие полиолы и тому подобное. В зависимости от характера активного вещества никаких наполнителей обычно не требуется для мягких желатиновых капсул. К подходящим наполнителям для получения растворов и сиропов относятся, например, вода, полиолы, сахароза, инвертированный сахар, глюкоза и тому подобное. В качестве подходящих носителей для получения инъекционных растворов можно использовать, например, воду, спирты, полиолы, глицерин, растительные масла и тому подобное. Натуральные и отвержденные масла, воски, жиры, полужидкие полиолы и тому подобное пригодны в качестве наполнителей для получения суппозиториев.
Фармацевтические препараты могут также содержать консерванты, стабилизаторы, смачивающие агенты, эмульгаторы, подслащивающие вещества, отдушки, окрашивающие вещества, соли для регулирования осмотического давления, буферные растворы, покрывающие вещества и противоокислители. Они могут также содержать другие терапевтически активные вещества.
Лекарственные препараты, содержащие соединение формулы I или соль основного соединения формулы I и фармацевтически инертный наполнитель, а также способ получения указанных лекарственных препаратов входят также в объем предлагаемого изобретения. Указанный способ включает приведение соединения формулы I или его соли в форму галеновых препаратов вместе с терапевтически инертным наполнителем, а при желании с одним или несколькими другими терапевтическими активными веществами.
Как указывалось ранее, соединения формулы I и их указанные соли можно использовать для лечения и предупреждения болезней, особенно в профилактике или лечении вирусных инфекций, в частности ретровирусных инфекций. Дозировка может варьироваться в широких пределах и, разумеется, должна устанавливаться с учетом индивидуальных особенностей в каждом конкретном случае. Обычно в случае перорального приема суточная доза составляет от примерно 3 мг до примерно 3 г, предпочтительно примерно от 10 мг до 1 г, хотя верхний предел может быть увеличен, если сочтут это целесообразным. Суточную дозировку можно вводить в виде однократной дозы или дробными дозами.
Пример 1. Раствор, содержащий 100 мг (0,20 ммоль) 2(S)-[[4(S)-бензил-3-(трет-бутоксикарбонил)-2,2-диметил-5(R)-оксазолидинил] метил]-N-трет-бутил-1(R)-циклогексанкарбоксамида и 3,3 мг (0,017 ммоль) пара-толуолсульфокислоты в 3 мл метанола выдерживают при комнатной температуре в течение 40 ч. Растворитель удаляют упариванием, и остаток разделяют на фазы при обработке 10 мл дихлорметана и 2 мл насыщенного раствора бикарбоната натрия. Органический слой сушат на безводном сульфате магния и после упаривания получают 90 мг смолы. Неочищенный продукт очищают колоночной хроматографией на силикагеле при использовании для элюирования 33% этилацетата в гексане. Получают 45 мг 2(S)-[3(S)-(трет-бутоксиформамидо)-2(R)-гидрокси-4-фенилбутил]- N-трет-бутил-1(R)-циклогексанкарбоксамида. Масс-спектр, м/e 447 [M+H]+.
2(S)-[[4(S)-бензил-3-(трет. -бутоксикарбонил)-2,2-диметил-5(R)- оксазолидинил]метил]-N-трет.-бутил-1(R)-циклогексанкарбоксамид, используемый в качестве исходного материала, получают по следующей методике.
(I) Раствор, содержащий 4,375 г (31 ммоль) транс-4-циклогексен-1(R), 2(R)-диметанола и 4,66 г (31 ммоль) трет- бутилдиметилсилилхлорида в 16 мл диметилформамида перемешивают в атмосфере азота и охлаждают до 0oC. Затем добавляют 5,30 г имидазола и полученную смесь постепенно нагревают до комнатной температуры, а затем перемешивают в течение ночи. Затем добавляют воду и смесь экстрагируют трижды порциями по 50 мл диметилового эфира. Объединенные экстракты промывают 100 мл воды, сушат на безводном сульфате магния и после упаривания получают 6,50 г масла. Это масло очищают колоночной хроматографией на силикагеле, используя для элюирования 10%-ный этилацетат, растворенный в гексане. В результате получают 2,80 г транс-1(R), 2(R)-бис [[(трет-бутил)(диметил)силилокси] метил] -4-циклогексана в виде бесцветного масла. После дополнительного элюирования колонки получают 2,576 г транс-6(R)-[[(трет-бутил)(диметил)силилокси] метил]-3-циклогексен- 1(R)-метанола в виде бесцветного масла. Масс-спектр, м/e 257 [M+H]+.
(II) Раствор, содержащий 2,57 г (10 ммоль) продукта, полученного на стадии (I), в 50 мл метанола гидрогенизируют на 130 мг катализатора, в качестве которого используют 10% палладиевую чернь при комнатной температуре и атмосферном давлении в течение 1 ч. Катализатор удаляют фильтрацией и после упаривания фильтрата получают 2,30 г транс-2(R)-[[(трет-бутил)(диметил)силилокси] метил] -1(R)- циклогексанметанола в виде бесцветного масла. Масс-спектр, м/e 259 [M+H]+.
(III) Раствор, содержащий 1,17 г (9,2 ммоль) хлористого оксалила в 23 мл дихлорметана, перемешивают в атмосфере азота и охлаждают до -78oC. Затем в течение 6 мин добавляют раствор, содержащий 1,3 мл (1,43 г, 18 ммоль) безводного диметилсульфоксида в 5 мл дихлорметана. Полученную смесь перемешивают еще в течение 2 мин, а затем в течение 9 мин вводят продукт, полученный в соответствии со стадией (II), растворенный в 10 мл дихлорметана. Полученную смесь перемешивают при -78oC в течение 20 мин, а затем в течение 6 мин добавляют 5,5 мл (3,99 г, 39 ммоль) триэтиламина. Затем полученную смесь нагревают постепенно до комнатной температуры и добавляют 40 мл воды. Образовавшиеся фазы разделяют и водный раствор экстрагируют дважды 50 мл порциями дихлорметана. Объединенные органические фазы сушат на безводном сульфате магния и после упаривания получают 2,45 г бесцветного масла. Полученное масло очищают колоночной хроматографией на силикагеле, используя для элюирования 2,5% этилацетата в гексане. В результате получают 1,53 г транс-2(R)-[[(трет-бутил)(диметил)силилокси] метил]-1(R)- циклогексанкарбоксальдегида в виде бесцветной смолы. Масс-спектр, м/e [M+H]+.
(IV) 3,46 мл (8,6 ммоль) 2,5 М раствора, содержащего н-бутиллитий в гексане, добавляют в течение 5 мин к перемешанной суспензии, содержащей 3,08 г (8,6 ммоль) метилтрифенилфосфонийбромида в 23 мл диэтилового эфира. Полученную смесь перемешивают при комнатной температуре в течение 85 мин и затем раствор 1,53 г (6,0 ммоль) продукта из стадии (III) в 10 мл диэтилового эфира добавляют в течение 5 мин. Смесь перемешивают при комнатной температуре в течение 140 мин, а затем добавляют 20 мл воды и образовавшиеся фазы разделяют. Водный раствор экстрагируют дважды порциями по 20 мл диэтилового эфира, а объединенные органические слои промывают 20 мл воды, сушат на безводном сульфате магния и после упаривания получают 1,85 г масла. Полученное масло очищают колоночной хроматографией на силикагеле и после элюирования при использовании 1%-ного этилацетата в гексане получают 0,870 г транс-1(R)-[[(трет-бутил)(диметил)силилокси] метил] -2(S)-винилциклогексана в виде бесцветного масла. Масс-спектр, м/e 239 [M-CH3]+.
(V) Раствор, содержащий 870 мг (3,4 ммоль) продукта, полученного на стадии (IV), растворенного в 6 мл 1M раствора тетрабутиламмония в тетрагидрофуране, выдерживают при комнатной температуре в течение 3,5 ч. Растворитель отгоняют, а осадок растворяют в 30 мл воды, а затем экстрагируют тремя порциями по 20 мл диэтилового эфира. Объединенные экстракты сушат на безводном сульфате магния и после упаривания получают 910 мг бесцветного масла. Это масло очищают колоночной хроматографией на силикагеле с использованием для элюирования 20%-ного этилацетата в гексане. В результате получают 435 г транс-2(S)-винил-1(R)-циклогексанметанола в виде бесцветного масла. Масс-спектр, м/e 141 [M+H]+.
(VI) Раствор, содержащий 435 мг (3,1 ммоль) продукта, полученного на стадии (V), в 10 мл пиридина перемешивают в атмосфере азота и охлаждают на ледяной бане. Затем добавляют 265 мкл (391 мг, 3,4 ммоль) хлористого метансульфонила, ледяную баню удаляют и смесь перемешивают в течение 4 ч. Растворитель отгоняют упариванием и остаток разделяют на фазы при использовании 30 мл 2M соляной кислоты и 30 мл диэтилового эфира. Водную фазу экстрагируют двумя порциями по 30 мл диэтилового эфира, и объединенные экстракты промывают 30 мл 2M соляной кислоты, 30 мл насыщенного раствора бикарбоната натрия и 30 мл воды, сушат на безводном сульфате магния, и после упаривания получают 615 мг 1(R)-[метан- сульфонилокси)-метил]-2(S)-винилциклогексана в виде бесцветного масла. Масс-спектр, м/e 219 [M+H]+.
(VII) Смесь, состоящую из 1,55 г (7,1 ммоль) продукта, полученного на стадии (VI), и 935 мг (10,7 ммоль) литийбромида, в 20 мл диметилформамида перемешивают при 60oC в атмосфере азота в течение 50 ч. Затем указанную смесь выливают в 250 мл воды и экстрагируют тремя порциями диэтилового эфира по 100 мл. Объединенные экстракты промывают 200 мл воды, сушат на безводном сульфате магния и после упаривания получают 1,312 г транс-1(R)-бромметил-2(S)-винилциклогексана в виде бесцветной жидкости. Масс-спектр, м/e 123 [M-Br]+.
(VIII) Смесь, состоящую из 172 мг (7,1 ммоль) магниевой стружки и кристаллического иода, в 2 мл тетрагидрофурана перемешивают в атмосфере аргона и добавляют 1,113 г (5,5 ммоль) продукта, полученного на стадии (VII), в течение 3 мин. Полученную смесь перемешивают и кипятят с обратным холодильником в течение 1 ч, а затем охлаждают до комнатной температуры. Раствор, содержащий 683 мг (2,74 ммоль) трет-бутил (L-α-формилфенетил)карбамата, добавляют в течение 10 мин и полученную смесь перемешивают еще 2,5 ч при комнатной температуре. Затем добавляют 30 мл 10%-ного раствора хлористого аммония, доводят ее до pH 2 при введении 2M соляной кислоты и после этого экстрагируют тремя порциями по 25 мл этилацетата. Объединенные экстракты промывают 25 мл насыщенного раствора бикарбоната натрия и 20 мл воды, сушат на безводном сульфате натрия и после упаривания получают 1,19 г масла. Это масло очищают колоночной хроматографией на силикагеле с использованием для элюирования 20% этилацетата в гексане. В результате получают 630 мг смеси из 1(S)-[3(S)-(трет-бутоксиформамидо)-2(S)-гидрокси-4-фенилбутил] -2(S)-винил циклогексана и 1(S)-[3(S)-(трет-бутоксиформамидо)-2(R)-гидрокси-4-фенилбутил] -2(S)-винилциклогексана в виде белого кристаллического вещества. Масс-спектр, м/e [M+H]+. Эту смесь диастереоизомеров используют на следующей стадии без последующей очистки.
(IX) Раствор, содержащий 630 мг (1,7 ммоль) продукта, полученного на стадии (VIII), в 20 мл диметилформамида перемешивают и охлаждают до 0oC. Затем добавляют 4,48 г (12 ммоль) бихромата пиридиния и полученную смесь перемешивают при 0oC в течение 10 мин. После этого смесь перемешивают при комнатной температуре в течение 6 ч, а затем ее выливают в 170 мл воды. Полученную смесь экстрагируют тремя порциями по 80 мл этилацетата и объединенные экстракты промывают 100 мл воды, сушат на безводном сульфате магния и после упаривания получают 640 мг 1(S)-[3(S)-(трет-бутоксиформамидо)-2-оксо-4-фенилбутил]-2(S)- винилциклогексана в виде бесцветного масла, кристаллизуемого при выдерживании. Полученный продукт используют на следующей стадии без последующей очистки.
(X) Раствор, содержащий 640 мг (1,7 ммоль) продукта, полученного на стадии (VIII), в 35 мл этанола перемешивают при 0oC, а затем добавляют 368 мг (9,7 ммоль) боргидрида натрия. Полученную смесь перемешивают при 0oC в течение 2,5 ч и растворитель затем отгоняют. Остаток разделяют на фазы между 100 мл воды и 100 мл этилацетата. Полученные фазы разделяют и водный слой экстрагируют отдельно порциями этилацетата по 100 и 50 мл. Объединенные экстракты сушат на безводном сульфате магния и после упаривания получают 595 мг белого маслянистого твердого продукта. Указанный продукт очищают колоночной хроматографией на силикагеле с использованием для элюирования 20%-ного этилацетата в гексане. В результате получают 85 мл 1(S)-3[(S)-(трет-бутоксиформамидо)-2(S)-гидрокси-4-фенилбутил] -2(S)- винилциклогексана в виде белого воскообразного твердого вещества. Масс-спектр, м/e [M+H]+. В результате дополнительного элюирования получают 263 мг 1(S)-[3(S)-(трет-бутоксиформамидо)-2(R)-гидрокси-4- фенилбутил] -2(S)-винилциклогексана в виде белого твердого продукта. Масс-спектр, м/e 374 [M+H]+.
(XI) Раствор, содержащий 370 мг (0,99 ммоль) продукта, полученного на стадии (X), и 225 мг (1,2 ммоль) моногидрата пара-толуолсульфокислоты в 7 мл 2,2-диметоксипропана, выдерживают при комнатной температуре в течение ночи. Раствор разбавляют 45 мл диэтилового эфира и промывают дважды порциями по 40 мл раствора бикарбоната натрия, сушат на безводном магнии и после упаривания получают 530 мг желтого масла. Полученное масло очищают колоночной хроматографией на силикагеле при использовании для элюирования 5%-ного этилацетата в гексане. В результате получают 230 мг 1(S)-[4(S)-бензил-3-(трет-бутоксикарбонил)-2,2-диметил-5(R)- оксазолидинилметил]-2(S)-винилциклогексана в виде бледно-желтого масла. Масс-спектр, м/e 414 [M+H]+.
(XII) Раствор, содержащий 670 мг (4,2 ммоль) перманганата калия в 7 мл воды, добавляют к смеси, состоящей из 230 мг (0,56 ммоль) продукта, полученного на стадии (XI), 123 Аликвата 336 и 0,65 мл ледяной уксусной кислоты в 8 мл бензола. Полученную смесь интенсивно перемешивают при комнатной температуре в течение ночи, а затем добавляют 428 мг метабисульфита натрия. После этого добавляют 24 мл диэтилового эфира и 2,5 мл 2M лимонной кислоты. Полученную смесь перемешивают в течение 10 мин, а затем фильтруют. Образовавшиеся фазы разделяют, и водный слой экстрагируют двумя порциями по 10 мл диэтилового эфира. Объединенные экстракты промывают дважды 10 мл воды, а затем сушат на безводном сульфате магния. После упаривания получают 340 мг масла. Полученное масло очищают колоночной хроматографией на силикагеле с использованием для элюирования 5%-ного метанола в дихлорметане. В результате получают 210 мг 2(S)-[[4(S)-бензил-3-(трет-бутоксикарбонил)-2,2-диметил-5(R)- оксазолидинил] метил]-1(R)-циклогексанкарбоновой кислоты в виде бесцветной смолы, которую используют на следующей стадии без последующей очистки.
(XIII) Смесь, состоящую из 210 мг (0,49 ммоль) продукта, полученного на стадии (XII), 67 мг (0,49 ммоль) гидрата 1-гидроксибензтриазола, 101 мг (0,49 ммоль) дициклогексилкарбодиимида и 52 мкл (36 мг, 0,49 ммоль) трет-бутиламина в 2 мл диметилформамида, перемешивают при комнатной температуре в атмосфере азота в течение 20 ч. Полученную смесь фильтруют и образовавшийся твердый продукт промывают 2 мл этилацетата. Объединенные фильтраты упаривают, остаток разделяют на фазы 10 мл этилацетата и 10 мл насыщенного раствора бикарбоната натрия. Фазы разделяют и водный слой экстрагируют двумя порциями по 10 мл этилацетата. Объединенные экстракты промывают 10 мл насыщенного раствора хлористого натрия, сушат на безводном сульфате магния и после упаривания получают 280 мг маслянистого твердого продукта. Этот продукт экстрагируют дважды порциями по 1 мл диэтилового эфира и сливают. Эфирные растворы упаривают с получением 260 мг неочищенного продукта. Этот продукт очищают колоночной хроматографией на силикагеле с использованием для элюирования 22% этилацетата в гексане. В результате получают 109 мг 2(S)-[[4(S)-бензил-3-(трет-бутоксикарбонил)-2,2-диметил-5(R)- оксазолидинил] метил] -N-трет-бутил-1(R)-циклогексанкарбоксамида в виде белого твердого вещества. Масс-спектр, м/e 487 [M+H]+.
Пример 2. Смесь, содержащую 35 мг (0,1 ммоль) 2(S)-[3(S)-2(R)-гидрокси-4-фенилбутил] -N-трет-бутил-1(R)- циклогексанкарбоксамида и 44 мг (0,1 ммоль) пентафторфенилового эфира N-бензилоксикарбонил-L-аспарагина в 1 мл диоксана, перемешивают при комнатной температуре в атмосфере азота в течение 16 ч. Растворитель упаривают, осадок растворяют в 10 мл этилацетата, полученный раствор промывают дважды 3 мл 2M соляной кислоты, 3 мл 2M NaOH и 3 мл насыщенного раствора хлористого натрия, сушат на безводном сульфате магния и упаривают. Полученный осадок хроматографируют на колонке с силикагелем при использовании в качестве элюента 6%-ного метанола в дихлорметане. В результате получают 34 мг белого твердого вещества. Это вещество перекристаллизовывают из смеси дихлорметан/гексан с получением 14 мг 2(S)-[3(S)-[[N-(бензилоксикарбонил)-L-аспарагинил] амино] -2(R)- гидрокси-фенилбутил]-N-трет-бутил-1(R)-циклогексанкарбоксамида в виде белого твердого вещества с т.пл. 195-199oC. Масс-спектр, м/e 595 [M+H]+.
2(S)-[3(S)-амино-2(R)-гидрокси-4-фенилбутил] -N-трет-бутил-1(R)- циклогексанкарбоксамид, используемый в качестве исходного материала, получают по следующей методике:
45 мг (0,1 ммоль) 2(S)-[3(S)-трет-бутоксиформамидо)-2(R)-гидрокси-4- фенилбутил]-N-трет-бутил-1(R)-циклогексанкарбоксамида (полученного по методике, описанной на стадии (I) примера 1), растворяют в 3 мл этилацетата и полученный раствор охлаждают до 0oC. Затем добавляют 0,2 мл насыщенного раствора хлористого водорода в этилацетате и полученную смесь выдерживают при комнатной температуре в течение ночи. Затем добавляют еще 1,0 мл хлористого водорода в этилацетате и реакционную смесь перемешивают в течение 4 ч при комнатной температуре, а затем упаривают досуха. Остаток растворяют в 10 мл дихлорметана и полученный раствор промывают 2 мл насыщенного раствора бикарбоната натрия, сушат на безводном сульфате магния и после упаривания получают 36 мг 2(S)-[3(S)-амино-2(R)-гидрокси-4-фенилбутил]-N-трет-бутил-1(R)- циклогексанкарбоксамида в виде бесцветной смолы. Масс-спектр, м/e 346 [M]+, который используют без последующей очистки.
Пример 3. В результате обработки 75 мг (0,15 ммоль) 2(R)-[[4(S)-бензил-3-трет-бутоксикарбонил)-2,2-диметил-5(R)- оксазолидинил]метил]-N-трет-бутил-1(R)-циклогексанкарбоксамида пара-толуолсульфокислотой в метаноле по методике, аналогичной описанной на стадии (I) примера 1, получают 20 мг 2(R)-[3(S)-трет-бутоксиформамидо)-2(R)-гидрокси-4-фенилбутил] -N-трет- бутил-1(R)-циклогексанкарбоксамид. Масс-спектр, м/e 469 [M+N]+, 447 [M+H]+.
2(R)-[[4(S)-бензил-3-трет-бутоксикарбонил)-2,2-диметил-5(R)- оксазолидинил] метил]-N-трет-бутил-1(R)-циклогексанкарбоксамид, используемый в качестве исходного материала, получают по следующей методике.
(I) Раствор, содержащий 18,40 г (0,10 ммоль) цис-4(R)-ацетоксиметил-5(S)-гидроксиметил)циклогексена и 16,50 г (0,11 моль) трет-бутилдиметилсилилхлорида в 45 мл диметилформамида перемешивают и охлаждают до 0oC. Затем добавляют 17,00 г (0,25 ммоль) имидазола и полученную смесь перемешивают при 0oC в течение 1 ч с последующим постепенным нагреванием до комнатной температуры и перемешиванием еще в течение 160 мин. Смесь затем выливают в 400 мл воды и экстрагируют двумя порциями диэтилового эфира по 100 мл. Объединенные экстракты промывают 200 мл воды, сушат на безводном сульфате магния и после упаривания получают 29,69 г жидкости соломенного цвета. Неочищенный продукт хроматографируют на колонке с силикагелем и при использовании для элюирования 10%-ного этилацетата в гексане получают 20,85 г цис-4(R)-(ацетоксиметил)-5(S)- [[(трет-бутил)(диметил)силилокси]метил]циклогексена в виде бесцветного масла. Масс-спектр, м/e 299 [M+H]+.
(II) 32,5 мл 2M раствора NaOH добавляют к раствору, содержащему 26,94 г (0,090 моль) продукта, полученного на стадии (I), растворенного в смеси из 90 мл этанола и 90 мл тетрагидрофурана. Полученную смесь перемешивают при комнатной температуре в течение 80 мин. Летучие растворители затем отгоняют, остаток разбавляют 520 мл воды и экстрагируют тремя порциями по 150 мл диэтилового эфира. Объединенные экстракты промывают последовательно 350 мл и 100 мл воды, сушат на безводном сульфате магния и после упаривания получают 22,77 г цис-6(S)-[[трет-бутил)(диметил)силилокси] метил] -3-циклогексен-1(R)-метанола в виде бесцветной жидкости. Масс-спектр, м/e 257 [M+H]+.
(III) По аналогичной методике, описанной в примере 1(II), 22,77 г (0,089 моль) продукта, полученного на стадии (II), гидрогенизируют с получением 19,85 г цис-2(S)-[[(трет-бутил)(диметил)силилокси]метил]-1(R)- циклогексанметанола в виде бесцветной жидкости. Масс-спектр, м/e 259 [M+H]+.
(IV) По аналогичной методике, описанной в примере 1(III), в результате окисления по методу Сверна 23,15 г (0,90 моль ) продукта, полученного на стадии (III), получают 22,78 г цис-2(S)-[[трет -бутил)(диметил)силилокси] метил] -1(R)-циклогексанкарбоксальдегида в виде бесцветного масла. Масс-спектр, м/e 257 [M+H]+.
(V) По методике, описанной в примере 1(IV), в результате обработки 17,70 г (0,069 моль) продукта, полученного на стадии (IV), бромистым метилтрифенилфосфонием и н-бутиллитием получают 14,51 г цис-1(S)-[[(трет-бутил)(диметил)силилокси] метил] -2(S)- -винилциклогексана в виде бесцветного масла. Масс-спектр, м/e 255 [M+H]+.
(VI) По аналогичной методике, описанной в примере 1(V), в результате обработки 12,7 г (0,049 моль) продукта, полученного на стадии (V), фтористым тетрабутиламмонием, получают 6,617 г цис-2(S)-винил-1(S)-циклогексанметанола в виде бесцветного масла. Масс-спектр, м/e 141 [M+H]+.
(VII) Раствор, содержащий 6,56 г (347 ммоль) продукта, полученного на стадии (VI), в 30 мл толуола, добавляют в течение 4 мин к перемешиваемой суспензии, содержащей 22,56 г (53 ммоль) дибромтрифенилфосфорана в 120 мл толуола. Полученную смесь перемешивают в атмосфере азота при комнатной температуре в течение 20 ч, затем фильтруют и остаток промывают 200 мл гексана. Объединенные фильтраты промывают двумя порциями по 200 мл насыщенного раствора бикарбоната натрия и 200 мл воды, сушат на безводном сульфате магния и после упаривания получают 7,898 г цис-1(S)-бромметил-2(S)-винилциклогексана в виде бесцветного масла. Масс-спектр, м/e 205, 203 [M+H] +.
(VIII) По аналогичной методике, описанной в примере 1(VIII), в результате обработки 7,766 г (38 ммоль) продукта, полученного на стадии (VII), магнием с последующим взаимодействием с трет-бутиловым эфиром L-α-формилфенетилкарбаминовой кислоты получают 3,196 г 1(R)-[3(S)-(трет-бутоксиформамидо)-2(S)-гидрокси-4-фенилбутил] -2(S)- -винилциклогексана, содержащего 10% (R)-спирта. Масс-спектр, м/e 374 [M+H]+.
(IX) По аналогичной методике, описанной в примере 1(IX), при окислении 2,81 г (7,5 ммоль) продукта, полученного на стадии (VIII), бихроматом пиридиния, получают 1,61 г (R)-[3(S)-(трет -бутоксиформамидо)-2-оксо-4-фенилбутил]-2(S)-винилциклогексана в виде бесцветного масла, которое кристаллизуется при стоянии. Масс-спектр, м/e 372 [M+H]+.
(X) По аналогичной методике, описанной в примере 1(X), в результате реакции восстановления 1,44 г продукта, полученного на стадии (IX), боргидридом натрия, получают 1,20 г 1(R)-[3(S)-(трет -бутоксиформамидо)-2(R)-гидрокси-4-фенилбутил]-2(S)- -винилциклогексана в виде бесцветной смолы, кристаллизующейся при хранении. Масс-спектр, м/e 373 [M]+.
(XI) По аналогичной методике, описанной в примере 1(XI), в результате обработки 1,20 г (3,2 ммоль) продукта, полученного на стадии (X), 2,2-диметоксипропаном получают 1,25 г 1(R)-[4(S)-бензил-3-(трет-бутоксикарбонил)-2,2-диметил-5(R)- -оксазолидинилметил] -2(S)-венилциклогексана в виде бледно-желтой смолы. Масс-спектр, м/e 414 [M+H]+.
(XII) По аналогичной методике, описанной в примере 1(XII), при обработке 850 мг (2,06 ммоль) продукта, полученного на стадии (XI), перманганатом калия получают 185 мг 2(R)-[4(S)-бензил-3-(трет -бутоксикарбонил)-2,2-диметил-5(R)-оксазолидинилметил] -1(R)-циклогексанкарбоновой кислоты в виде бесцветной смолы. Масс-спектр, м/e 432 [M+H]+.
(XIII) По аналогичной методике, описанной в примере 1(XIII), при взаимодействии 165 мг (0,38 ммоль) продукта, полученного на стадии (XII), с трет-бутиламином получают 85 мг 2(R)-[[4(S)-бензил-3-трет -бутоксикарбонил)-2,2-диметил-5(R)-оксазолидинил]метил]-N-трет- бутил-1(R)-циклогексанкарбоксамида в виде белого твердого продукта. Масс-спектр, м/e 487 [M+H]+.
Пример 4. По аналогичной методике, описанной на стадии (I) примера 2, из 2(R)-[3(S)-амино-2(R)-гидрокси-4-фенилбутил]-N-трет-бутил-1(R)-циклогексанкарбоксамида и N-бензилоксикарбонил-1- -аспарагинпентафторфенилового эфира получают 2(R)-[3(S)-[[N- -(бензилоксикарбонил)-L-аспарагинил]амино]-2(R)-гидрокси-4-фенилбутил] -N-трет-бутил-1(R)-циклогексанкарбоксамид в виде белого твердого продукта с точкой плавления 166,5-168oC. Масс-спектр м/e 617 [M+H] +.
2(R)-[3(S)-амино-2(R)-гидрокси-4-фенилбутил]-N-трет-бутил-1(R)-циклогексанкарбоксамид, используемый в качестве исходного материала, получают по аналогичной методике, описанной в примере 2(II) из 2(R)-[3(S)-(трет-бутоксиформамидо)-2(R)-гидрокси-4-фенилбутил] -N-трет-бутил-1(R)-циклогексанкарбоксамида (полученного по методике, описанной в примере 3(I)), и полученный продукт используют без последующей очистки.
Пример 5. Смесь, содержащая 65 мг (0,13 ммоля) 2(S)-[(3S)-[[L-аспарагинил] амино]-2(R)-окси-4-фенилбутил]-N-трет-бутил-1(R)-циклогексанкарбоксамида и 35 мг (0,13 ммоля) N-оксисукцинимидного эфира хинолинкарбоновой кислоты в 2 мл тетрагидрофурана перемешивают при комнатной температуре в атмосфере азота в течение 18 ч. Растворитель удаляют упариванием и остаток разделяют на фазы при обработке 10 мл этилацетата и 10 мл 10%-ного раствора карбоната натрия. Органический слой промывают 10 мл воды, сушат безводным сульфатом магния и упаривают. Полученный неочищенный продукт очищают колоночной хроматографией на силикагеле с использованием для элюирования 7%-ного метанола в дихлорметане. Получают 80 мг 2(S)-[[3(S)-[N-(2- -хинолилкарбонил)-L-аспарагинил] амино]-2(R)-гидрокси-4-фенилбутил]- -N-трет-бутил-1(R)-циклогексанкарбоксамида в виде белого твердого вещества. Масс-спектр, м/e 616 [M+H]+.
2(S)-[(3S)-[[L-аспарагинил] амино] -2(R)-окси-4-фенилбутил] -N-трет-бутил-1(R)-циклогексанкарбоксамид, используемый в качестве исходного материала, получают по следующей методике.
Раствор, содержащий 80 мг (0,13 ммоль) 2(S)-[3(S)-[[N-бензилоксикарбонил)-L-аспарагинил] амино]-2(R)-гидрокси-4- -фенилбутил]-N-трет-бутил-1(R)-циклогексанкарбоксамида (полученного по методике, описанной на стадии I примера 2) в 10 мл этанола гидрируют на 10 мг катализатора, в качестве которого используют 10% палладиевую чернь, в течение 4 ч. Катализатор удаляют фильтрацией и фильтрат упаривают. Остаток растворяют в толуоле и раствор упаривают. Этот способ повторяют еще один раз и получают 65 мг 2(S)-[3(S)-[[L-аспарагинил] амино-2(R)-гидрокси-4-фенилбутил] -N-трет-бутил-1(R)-циклогексанкарбоксамид в виде бесцветной пены. Полученный продукт используют без дальнейшей очистки.
Пример 6. Раствор, содержащий 90 мг (0,26 ммоля) 2(S)-[3(S)-амино-2(R)-гидрокси-4-фенилбутил] -N-трет-бутил-1(R)- -циклогексанкарбоксамид в 4 мл дихлорметана добавляют к раствору, содержащему 65 мг (0,26 ммоля) N-бензилоксикарбонил-циано-L-аланина, 35 мг (0,23 ммоля) гидрата оксибензотриазола и 54 мг (0,26 ммоля) дициклогексилкарбодиимида в 2 мл диметилформамида. Смесь перемешивают в атмосфере азота при комнатной температуре в течение 18 ч и затем фильтруют. Фильтрат упаривают и остаток разделяют на фазы обработкой 25 мл этилового ацетата и 10 мл насыщенного водного раствора бикарбоната натрия. Органический слой сушат над безводным сульфатом магния и упаривают. Неочищенный продукт очищают колоночной хроматографией на силикагеле при использовании для элюирования 3%-ного метанола в дихлорметане и полученный продукт дальше очищают кристаллизацией из смеси, содержащей 1 мл дихлорметана и 10 мл гексана. Получают 68 мг 2(S)-[(S)-[N-(бензилоксикарбонил)-3-циано-L-аланил] амино-2(R)-окси- 4-фенилбутил] -N-трет-бутил-1(R)-циклогексанкарбоксамида в виде белого твердого продукта. Масс-спектр, м/e 577 [M+H]+.
Пример 7. По аналогичной методике, описанной в примере 6, 73 мг (0,29 ммоля) N-бензилоксикарбонил-L-валина соединяют с 100 мг (0,29 ммоля) 2(S)-[3(S)-амино-2(R)-окси-4-фенилбутил]-N-трет-бутил-1-(R)- циклогексанкарбоксамида и получают 80 мг 2(S)-[3(S)-[N-(бензилоксикарбонил)L-валил]амино]-2(R)-окси-4- фенилбутил]-N-трет-бутил-1(R)-циклогексанкарбоксамида и получают 80 мг в виде белого твердого вещества. Масс-спектр, м/e 580 [M+H]+.
Пример 8. По аналогичной методике, описанной в примере 6, 78 мг (0,26 ммоля) N-бензилоксикарбонил-L-фенилаланина соединяют с 90 мг (0,26 моля) 2(S)-[3(S)-амино-2(R)-окси-4-фенилбутил]-N-трет-бутил-1(R)- циклогесанкарбоксамида. Получают 72 мг 2(S)-[3(S)-[[N-(бензилоксикарбонил)-L-фенилалаланил] амино] -2(R)-окси- 4-фенилбутил]-N-трет-бутил-1(R)-циклогексанкарбоксамида в виде белого твердого продукта. Масс-спектр, м/e 628 [M+H]+.
Пример 9. По аналогичной методике, описанной в примере 5, 95 мг (0,16 ммоль) 2(S)-[3(S)-[[N-(бензилоксикарболнил)-L-валил]амино]-2(R)-окси-4- фенилбутил] -N-трет-бутил-1(R)-циклогексанкарбоксамида гидрируют на катализаторе, в качестве которого используют 10% палладиевую чернь, и полученный продукт подвергают реакции с N-оксисукцинимидным эфиром хинолинкарбоновой кислоты и получают 50 мг 2(S)-[3(S)-[[N-(2- хинолилкарбонил)-L-валил]амино]-2(R)-окси-4-фенилбутил] -N-трет-бутил- 1(R)-циклогексанкарбоксамида в виде белого твердого вещества. Масс-спектр, м/e 601 [M+H]+.
Пример 10. По аналогичной методике, описанной в примере 6, 70 мг (0,26 ммоля) N-бензилдоксикарбонил-S-метил-L-цистеина соединяют с 90 мг (0,26 ммоля) 2(S)-[3(S)-амино-2(R)-окси-4-фенилбутил] -N-трет-бутил-1-(R)- циклогексанкарбоксамида. Получают 30 мг 2(S)-[3(S)-[[N-(бензилоксикарбонил)-S-метил-L-цистеинил]амино]- 2(R)-окси-4-фенилбутил]-N-трет-бутил-1(R)-циклогексанкарбоксамида в виде белого твердого вещества с т.пл. 143-144oC. Масс-спектр, м/e 598 [M+H]+.
Пример 11. 220 мг (0,59 ммоля) пиридиний дихромата добавляют к раствору, содержащему 48 мг (0,08 ммоля) 2(S)-[3(S)-[[N-(бензилоксикарбонил)-L-валил] -амино] -2-гидрокси-4- фенилбутил]-N-трет-бутил-1(R)-циклогексанкарбоксамида в 1 мл диметилформамида и смесь перемешивают при комнатной температуре в течение 18 ч. Добавляют 10 мл воды и смесь экстрагируют при использовании двух порций по 10 мл этилацетата. Объединенные экстракты промывают 10 мл воды, сушат безводным сульфатом магния и упаривают. Неочищенный продукт очищают колоночной хроматографией на силикагеле и после элюирования при использовании 4%-ного метанола в дихлорметане получают 31 мг 2(S)-[3(S)-[[N-(бензилоксикарбонил)L-валил] амино]-2-оксо-4-фенилбутил] -N-трет-бутил-1(R)-циклогексанкарбоксамида в виде белого твердого продукта с т.пл. 186-188oC. Масс-спектр, м/e 758 [M+H]+.
Пример 12. По аналогичной методике, описанной в примере 6, 72 мг (0,26 ммоля) β-метилового эфира N-бензилоксикарбонил-L-аспартагиновой кислоты соединяют с 90 мг (0,26 ммоля) 2(S)-[3-(S)-амино-2(R)-окси-4-фенилбутил]-N-трет-бутил-1(R)- циклогексанкарбоксамида и получают 90 мг 2(S)-[3(S)-[[N-(бензилоксикарбонил)-O-метил-L-аспартал] амино]-2(R) -окси-4-фенилбутил] -N-трет-бутил-1(R)-циклогексанкарбоксамида в виде бесцветной смолы. Масс-спектр, м/e 610 [M+H]+.
Пример 13. По аналогичной методике, описанной в примере 6, 68 мг (0,26 ммоля) N-бензилоксикарбонил-3-метил-L-валина соединяют с 90 г (0,26 ммоля) 2(S)-[3(S)-амино-2(R)-окси-4-фенилбутил] -N-трет-бутил-1(R)- циклогексанкарбоксамида и получают 40 мл 2(S)-[3(S)-[[N-(бензилоксикарбонил)-3-метил-L-валил] амино] -2(R)- окси-4-фенилбутил]-N-трет-бутил-1(R)-циклогексанкарбоксамида в виде белого твердого продукта с т.пл. 90oC (разложение). Масс-спектр, м/e 594 [M+H]+.
Пример 14. Раствор, содержащий 100 мг (0,23 ммоля) 2(S)-[[4(S)-бензил-3-(трет-бутоксикарбонил)-2,2-диметил-5(R)- оксазолидинил]метил]-1(R)-циклогексанкарбоновой кислоты (получение описано в примере 1) в 2 мл диэтилового эфир добавляют к 5 мл 0,3M раствора диазометана в диэтиловом эфире. Смесь выдерживают при комнатной температуре в течение ночи и затем упаривают. Остаток растворяют в 3 мл метанола и добавляют 4 мг n-толуол-сульфоновой кислоты. Смесь выдерживают в течение ночи при комнатной температуре и затем упаривают досуха. Остаток разделяют на фазы обработкой 10 мл дихлорметана и 3 мл насыщенного водного раствора бикарбоната натрия. Органический слой сушат безводным сульфатом магния и после упаривания получают 90 мг смолы. Неочищенный продукт очищают колоночной хроматографией на силикагеле с использованием для элюирования гексан/этилового ацетата (2:1, об/об) и получают метил 2(S)-[(S)-(трет-бутоксиформамидо)-2(R)-окси-4-фенилбутил]-1(R)- циклогексанкарбоксилата в виде белого твердого вещества с т.пл. 127-128oC. Масс-спектр, м/e 406 [M+H]+.
Следующий пример иллюстрирует получение типичного фармацевтического препарата, содержащего соединение формулы I или фармацевтически приемлемую соль присоединения кислот любого основного соединения формулы I в качестве действующего начала.
Пример A. Водный раствор, содержащий активное вещество, фильтруют с соблюдением стерильности и смешивают со стерильным раствором желатины, содержащим фенол в качестве консерванта, при этом компоненты берутся в таком количестве, что 1 мл конечного раствора содержит 3 мг активного вещества, 150 мг желатины, 4,7 фенола и остальное - дистиллированная вода. Полученную смесь заполняют во флаконы емкостью 1 мл с соблюдением стерильности.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ АМИНОКИСЛОТ И ИХ КИСЛОТНО-АДДИТИВНЫЕ СОЛИ | 1990 |
|
RU2071470C1 |
| ПРОИЗВОДНЫЕ 5-АРОИЛНАФТАЛИНА | 1998 |
|
RU2188192C2 |
| ПРОИЗВОДНЫЕ УКСУСНОЙ КИСЛОТЫ ИЛИ ИХ ГИДРАТЫ, ИЛИ ИХ СОЛЬВАТЫ, ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1991 |
|
RU2072359C1 |
| ЗАМЕЩЕННЫЕ ПИРРОЛЫ | 1994 |
|
RU2141960C1 |
| ПРОИЗВОДНЫЕ НИТРИЛОВ β-АМИНОКИСЛОТ | 2001 |
|
RU2245871C2 |
| ПРОИЗВОДНЫЕ ЦЕФАЛОСПОРИНА И ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ | 1994 |
|
RU2130939C1 |
| ПРОИЗВОДНЫЕ 1,2-ДИАМИНОПЕНТАНА КАК АНТАГОНИСТЫ РЕЦЕПТОРА CCR-3 И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2286339C2 |
| АРОМАТИЧЕСКИЕ ПРОИЗВОДНЫЕ КАРБОНОВОЙ КИСЛОТЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2130009C1 |
| ИЗОХИНОЛИНКАРБОКСАМИДЫ И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ ДЛЯ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕАЗЫ ВИЧ | 2001 |
|
RU2265016C2 |
| ПРОИЗВОДНЫЕ УКСУСНОЙ КИСЛОТЫ | 1994 |
|
RU2151768C1 |
Сущность: производные аминокислоты формулы I: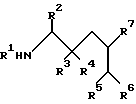
где R1 означает С1-С8- алкоксикарбонил или группу формулы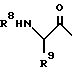
где R2 означает фенил-С1-С8- алкил; R3 означает водород, а R4 означает гидроксильную группу либо R3 и R4 в комбинации означают оксогруппу; R5 означает С1-С8- алкоксикарбонил или С1-С8- алкилкарбамоил; R6 и R7 в комбинации означают триметилен или тетраметилен; R8 означает фенил-С1-С8- алкоксикарбонил, хинолилкарбонил; R9 означает С1-С8-алкил, фенил-С1-С8-алкил, циано-С1-С8-алкил, карбамоил-С1-С8-алкил, С1-С8-алкилтил С1-С8-алкил, карбамоил-С1-С8-алкил или их фармацевтически приемлемые соли, которые являются основными, которые проявляют антивирусную активность, и могут быть использованы в форме лекарственных препаратов для профилактики или лечения вирусных инфекций. Их можно получить общеизвестными способами. 6 с. и 12 з. п.ф-лы, 1 табл.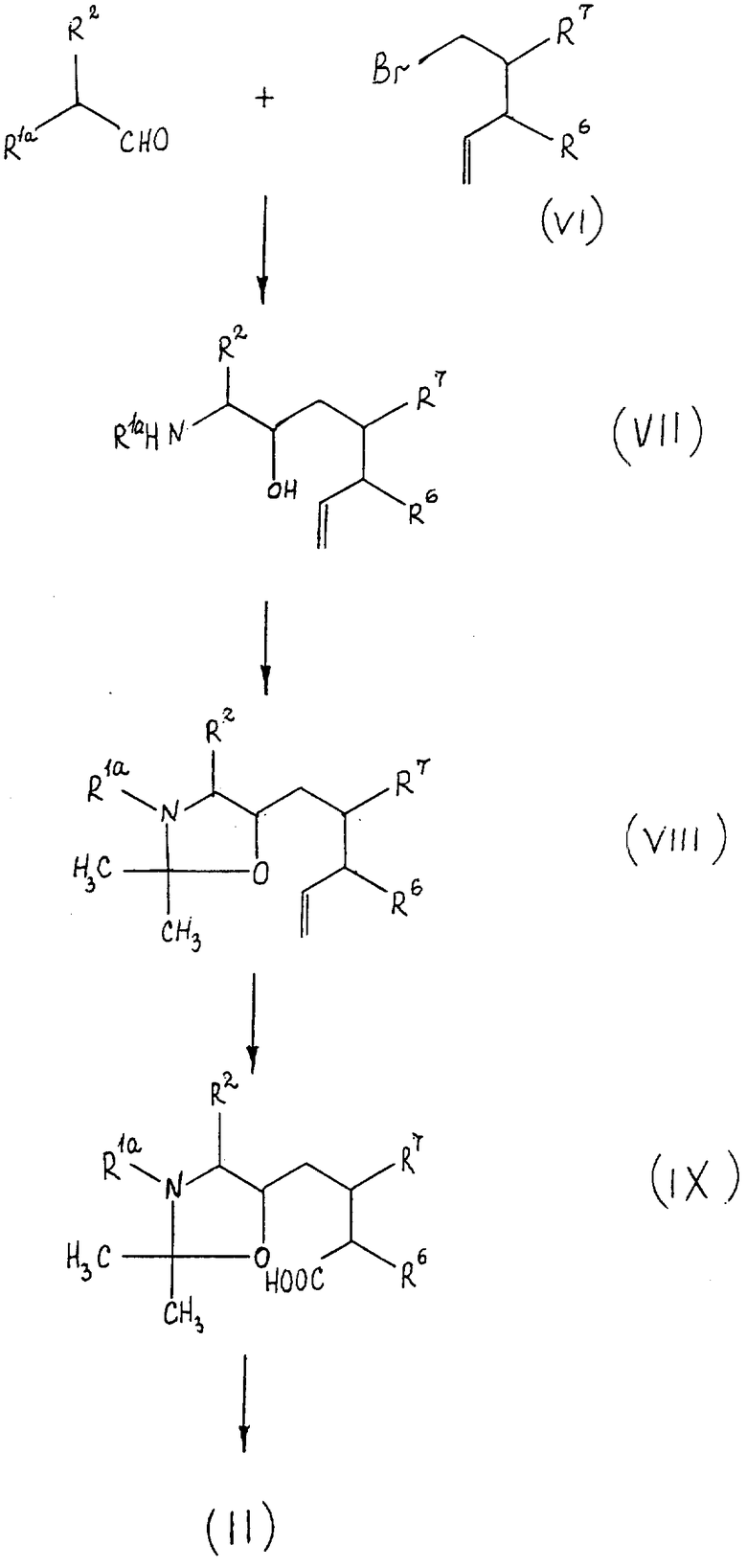
где R1 - С1 - С8-алкоксикарбонил, группа формулы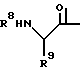
R2 - фенил-С1 - С8-алкил;
R3 - водород;
R4 - гидроксил или R3 и R4, взятые вместе, образуют оксогруппу;
R5 - С1 - С8-алкоксикарбонил, С1 - С8-алкилкарбамоил;
R6 и R7, вместе взятые, образуют триметилен или тетраметилен;
R8 - фенил-С1 - С8-алкоксикарбонил, хинолилкарбонил;
R9 - карбамоил-С1 - С8-алкил, циано-С1 - С8-алкил, С1 - С8-алкил, фенил-С1 - С8-алкил, С1 - С8-алкилтио-С1 - С8-алкил, С1 - С8-алкоксикарбонил-С1 - С8-алкил,
или их фармацевтически приемлемые соли, которые являются основными.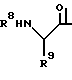
где R8 - бензилоксикарбонил;
R9 - карбамоилметил;
R2 - бензил,
R3 - водород, R4 - гидроксил, R5 - трет.-бутилкарбамоил, R6 и R7, вместе взятые, образуют незамещенный тетраметилен.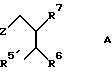
где Z - группа формул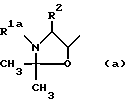
где R1 a - С1 - С8-алкоксикарбонил;
R2 - фенил-С1 - С8-алкил;
R3 - водород;
R4 - гидроксил или R3 и R4, вместе взятые, образуют оксогруппу;
R5' - С1 - С8-алкилкарбамоил, винил, карбоксил, С1 - С8-алкоксикарбонил;
R6 и R7, взятые вместе, образуют триметилен, тетраметилен;
R9 - карбамоил-С1 - С8-алкил, циано-С1 - С8-алкил, С1 - С8-алкил, фенил-С1 - С8-алкил, С1 - С8-алкилтио-С1 - С8-алкил, С1 - С8-алкоксикарбонил-С1 - С8-алкил.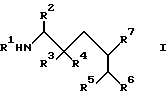
где R1 - С1 - С8-алкоксикарбонил;
R2 - фенил-С1 - С8-алкил;
R3 - водород;
R4 - гидроксил;
R5 - С1 - С8-алкоксикарбонил, С1 - С8-алкилкарбамоил;
R6 и R7, вместе взятые, образуют триметилен или тетраметилен,
отличающийся тем, что соединение формулы II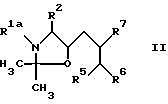
где R1 a - С1 - С8-алкоксикарбонил;
R2, R5 - R7 имеют указанные значения,
обрабатывают кислотой и полученный продукт выделяют в свободном виде или в виде фармацевтически приемлемой соли.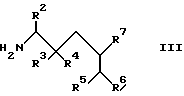
где R2 - R7 имеют указанные в п.1 значения,
обрабатывают ацилирующим агентом и в полученном продукте при необходимости окисляют соединение формулы I, где R3 - водород, а R4 - гидроксильная группа, и полученный продукт выделяют в свободном виде или переводят его в фармацевтически приемлемую соль.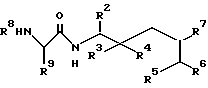
где R2 - R9 имеют значения, указанные в п.1,
отличающийся тем, что соединение общей формулы IV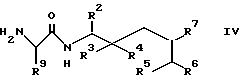
где R2 - R7 и R9 имеют указанные значения,
обрабатывают ацилирующим агентом с последующим окислением в случае необходимости соединения, у которого R3 - водород, а R4 - оксигруппа, и с выделением целевого продукта в свободном виде или в виде его фармацевтически приемлемой соли.
| ЕР, патент, 367601, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
