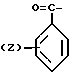
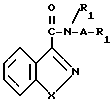
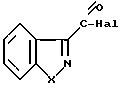
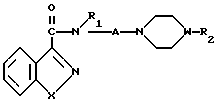
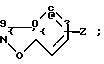
Изобретение касается бензизотиазол- и бензоизооксазол-3-карбоксамидов формулы I (где) R1 водород или низший алкил; R2 низший алкил или группа одной из формул
(где) R1 водород или низший алкил; R2 низший алкил или группа одной из формул (Z),
(Z), 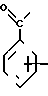 (Z), N
(Z), N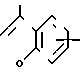 (Z)
(Z)
(Z)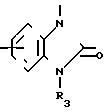
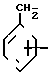 (Z), N
(Z), N (Z)
(Z)
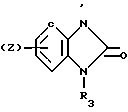
 (Z) где R3 водород; А низший алкилен, группа формулы -СНR4CH=CHR4- или -СНR4C≡ CHR4-; R4 водород; Х кислород или сера; W азот или СН; Z водород, низший алкил, низшая алкоксигруппа или галоген, где сплошная черта (---) указывает место присоединения группы к обозначенному члену формулы; а также оно касается геометрических и оптических изомеров упомянутых соединений или же их фармацевтически приемлемых солей, предназначенных для лечения психических расстройств как самостоятельно, так и в сочетании с инертными адъювантами.
(Z) где R3 водород; А низший алкилен, группа формулы -СНR4CH=CHR4- или -СНR4C≡ CHR4-; R4 водород; Х кислород или сера; W азот или СН; Z водород, низший алкил, низшая алкоксигруппа или галоген, где сплошная черта (---) указывает место присоединения группы к обозначенному члену формулы; а также оно касается геометрических и оптических изомеров упомянутых соединений или же их фармацевтически приемлемых солей, предназначенных для лечения психических расстройств как самостоятельно, так и в сочетании с инертными адъювантами.
Бензизотиазол- и бензизоксазол-3-карбоксамиды, отвечающие изобретению, порождают также ряд производных от них соединений, где а. R2 группа одной из формул: (Z),
(Z),  (Z),
(Z),  (Z) b. R2 группа одной из формул:
(Z) b. R2 группа одной из формул:
N (Z), N
(Z), N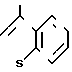 или
или 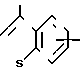 (Z) с. R2 группа формулы:
(Z) с. R2 группа формулы:
(Z)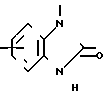 d. R2 группа формулы
d. R2 группа формулы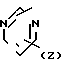
По всему тексту описания изобретения и в приложенных к нему пунктах формулы изобретения термин "алкил" означает углеводородный радикал с прямой или разветвленной цепью, не содержащий ненасыщенных структурных элементов и включающий 1-7 атомов углерода, такой как метил, этил, 1-пропил, 2-пропил, 1-бутил, 1-пентил, 2-пентил, 3-гексил, 4-гептил и т.п. термин "алкоксигруппа" означает одновалентный заместитель, содержащий группу алкила, присоединенную через кислород простого эфира, имеющий свободную валентную связь от кислорода этой эфирной группы и представляющий собой метокси-, этокси-, пропокси-, бутокси-, 1,1-диметилэтокси-, пентокси-, 3-метил-пентокси-, 2-этилпентокси-группу и т.п. термин "галоген" относится к семейству, в которое входят хлор, фтор, бром и йод. Термин "низший" относится к любой из упомянутых групп и означает группу, содержащую до 6 атомов углерода включительно.
Соединения, отвечающие изобретению, по своей молекулярной структуре несимметричны и существуют в виде оптических антиподов или их рацемических форм. Оптический антипод может быть получен из соответствующих рацемических форм по стандартной методике оптического разделения, включающей, например, разделение диастереомерических солей указанных соединений, характеризующееся присутствием основной аминогруппы и оптически активной кислоты, или же синтезом из оптически активных веществ-предшественников.
Изобретение охватывает все оптические изомеры и их рацемические формы. Формулы таких соединений приведены для того, чтобы специалист мог ориентироваться во всевозможных оптических изомерах этих соединений, определенных указанным образом.
Обозначения "Е" и "L" указывают на то или иное расположение заместителей, связанных с группировкой С=С 1-пиперазинил-2-бутенов согласно изобретению. В бутенах "Е" атомы водорода расположены по обе стороны от двойной связи, т.е. транс-позиции по отношению друг к другу. В бутенах "Z" атомы водорода расположены по одну сторону от двойной связи, т.е. в цис-позиции по отношению друг к другу.
Бензоизотиазол- и бензоизоксанол-3-карбоксамиды, отвечающие изобретению, предпочтительно получают алкилированием галоалкиламида формулы I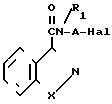 где R1, А, Х имеют указанные значения; Наl хлор, бром или йод, со вторичным амином формулы II
где R1, А, Х имеют указанные значения; Наl хлор, бром или йод, со вторичным амином формулы II
HN W-R2 где R и W имеют указанные значения.
W-R2 где R и W имеют указанные значения.
В N-метилпирролидоне, диполярном апротонном растворителе, при повышенных температурах в интервале 50-200оС, предпочтительно в температурном интервале 120-190оС. Помимо N-метилпирролидона можно использовать и другие диполярные апротонные растворители, такие как, например, диметилацетамид, диметилформамид, гексаметилфосфорамид и диметилсульфоксид при температуре конденсации, обеспечивающие удовлетворительную скорость реакции в температурном интервале, когда они совместимы с растворителем.
Можно использовать также основание, а именно карбонат или бикарбонат щелочного или щелочно-земельного металла, например, карбонат или бикарбонат лития, натрия или калия как самостоятельно, так и в сочетании с промотором алкилирования: йодидом щелочного металла, а именно, йодидом лития, натрия или калия. Предпочтительным основанием и предпочтительным промотором конденсирования являются соответственно карбонат калия и йодид натрия.
Если алкилирование протекает под действием основания и промотора алкилирования, то растворителем может служить ацетонитрил, причем алкилирование проводят в температурном интервале от 50оС до температуры обратной перегонки реакционной среды, предпочтительно в температурном интервале от 75оС до температуры обратной перегонки.
По другому варианту бензоизотиазол- и бензоизоксазол-3-карбоксамиды, отвечающие настоящему изобретению, получают конденсированием кислого галида формулы III где Х имеет приведенные значения, Наl представляет собой хлор или бром, с гетероциклическим амином формулы II
где Х имеет приведенные значения, Наl представляет собой хлор или бром, с гетероциклическим амином формулы II
H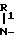 A-N
A-N W-R2 где R1, R2, А и W имеют приведенные значения.
W-R2 где R1, R2, А и W имеют приведенные значения.
В присутствии кислого акцептора: третичного амина, такого как триалкиламин, а именно триметиламин, триэтиламин или трипропиламин, или же гетероциклического амина, а именно пиридина, пиколина, лутидина или коллидина, в подходящем растворителе. К подходящим растворителям относятся, например, дихлорметан, трихлорометан. 1,1- и 1,2-дихлороэтан, причем предпочтителен дихлорометан; а также ароматические растворители, например, бензол, толуол и ксилол, причем предпочтителен толуол. Предпочтительными кислыми акцепторами являются триалкиламины. Наиболее предпочтительным акцептором является триэтиламин.
Бензоизотиазол- и бензоизоксазол-3-карбоксамиды, отвечающие изобретению, получают также аминированием сложного эфира бензоизотиазол- или бензоизоксазол-3-карбоновой кислоты, имеющего формулу IY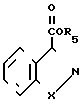 где R5 алкил; Х имеет приведенные значения, аминоалканом формулы Y
где R5 алкил; Х имеет приведенные значения, аминоалканом формулы Y
H OH где А и R1 имеют приведенные значения, с образованием гидроксилалкиламинокарбоксамида формулы YI
OH где А и R1 имеют приведенные значения, с образованием гидроксилалкиламинокарбоксамида формулы YI который переводят в отвечающие изобретению бензоизотиазол- и бензоизоксазол-карбоксамиды. Предпочтительно проводить аминирование в ароматическом растворителе, например, в бензоле, толуоле, ксилоле или метилоле или же в спирте, например, этиловом, 2-пропиловом или 1-бутиловом, при температурах 100-140оС. Предпочтительным растворителем является толуол. Если реакцию аминирования проводят при комнатной температуре, то для этого лучше использовать реактор, работающий под давлением.
который переводят в отвечающие изобретению бензоизотиазол- и бензоизоксазол-карбоксамиды. Предпочтительно проводить аминирование в ароматическом растворителе, например, в бензоле, толуоле, ксилоле или метилоле или же в спирте, например, этиловом, 2-пропиловом или 1-бутиловом, при температурах 100-140оС. Предпочтительным растворителем является толуол. Если реакцию аминирования проводят при комнатной температуре, то для этого лучше использовать реактор, работающий под давлением.
Гидроксилалкиламинокарбоксамид YI превращают в конечный продукт, а именно в бензоизотиазол- или бензоизоксазол посредством обработки гидроксилалкилкарбоксамида YI сульфонилхлоридом формулы YII
R6SO2Hal где R6 алкил, фенил или толуол; Наl хлор или бром, в присутствии кислотного акцептора, например, триалкиламина или триэтиламина, в ароматическом растворителе; бензоле, толуоле или ксилоле, при температуре реакции в интервале 0-25оС с образованием сульфоната формулы YIII гидроксилалкилкарбоксамида YI: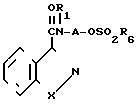 где А, R1, R6, Х определены выше,
где А, R1, R6, Х определены выше,
обрабатываемого (лучше без выделения в чистом виде) пиперидином или пиперазином II, взятым в том виде, как он есть или в виде раствора в эфирном растворителе, таком как 1,2-диметоксиэтан, 2-метоксиэтиловый эфир, тетрагидрофуран или диоксан, при температуре реакции от 25оС до температуры обратной перегонки реакционной среды. Предпочтительным эфирным растворителем является тетрагидрофуран, а предпочтительной температурой проведения реакции температура обратной перегонки.
На стадии сульфонирования можно использовать дополнительный растворитель, такой как тетрагидрофуран.
Предпочтительными сульфонирующими реагентами 7 являются алкилсульфонилхлориды, из которых наилучший метансульфонилхлорид.
В качестве промежуточных продуктов, предназначенных для приготовления искомых бензоизотиазол- и бензоизоксазол-3-карбоксамидов, были получены следующие сульфонаты N-(гидроксиалкил)-бензоизоксазол- и бензоизотиазол-3-карбоксамида:
а. Метаносульфонат N-метил-N-(2-гидроксиэтил)-1,2-бензоизотиазол-3-карбоксамида;
b. Метаносульфонат N-метил-N-(2-гидроксиэтил)-1,2-бензоизоксазол-3-карбоксамида;
с. Метаносульфонат N-(1-метилэтил)-N-(2-гидроксиэтил)-1,2-бензоизоксазол-3-карбоксамида;
d. Метаносульфонат N-(1-метилэтил)-N-(3-гидроксипропил)-1,2-бензоизоксазол-3-карбоксамида.
Исходные материалы и вспомогательные вещества для синтеза бензоизотиазол- и бензоизоксазол-3-карбоксамида согласно изобретению либо имеются в продаже, либо их можно получить методами, хорошо известными специалистам в данной области. Например, 3-галокарбонилбензоизотиазолы III и 3-галоалкиламинокарбонил-бензоизотиазолы I получают способом, который описали Аморетти и сотр. см. Amoretti L. et al. IL FARМACO (Ed. Sc.), 1972, V. 27, р. 855. Соответствующие бензоизоксазолы, например, исходные вещества формул I и III, где Х кислород, получают адаптированием методик, которые описали Аморетти и сотр. см. там же, р. 859-861, или раскрыли Сато и Хираиин, см. патент США N 4758503, 1988.
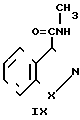
В частности, 3-галоалкиламинокарбонильный бензоизотиазол или бензоизоксазол II, где R1 метил, А алкил с 2-4 атомами углерода, а Х, Y, Hal и n определены выше, получают алкилированием бензоизотиазол- или бензоизоксазол-3-карбоксамида IX, в котором R1 метил, а Х, Y и n определены выше, дигалоалканом Х формулы НаlAHal, где A и радикалы Наl- те, что описаны выше, в присутствии гидрида щелочного металла, например, гидрида натрия или его масляной эмульсии, в диполярном апротонном растворителе, например, в диметилформамиде, при температуре реакции, находящейся в интервале 0-25оС.

Необходимые реагенты имеются в свободной продаже и указаны в каталогах или же их можно приготовить обычными методами. Так, например, 4-(4-фторобензоил)пиперидин, реагент для получения отвечающих изобретению бензоизотиазол- и бензоизоксазол-3-карбоксамидов, где R2имеет формулу где W представляет собой СН, описан в патенте США N 3576810, 1971;
где W представляет собой СН, описан в патенте США N 3576810, 1971;
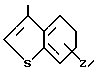
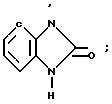
4-(6-Хлоро-1,2-бензоизоксазол-3-ил)пиперидин, служащий субстратом при получении конечных искомых соединений согласно изобретению, т.е. соединений, в которых R2 имеет формулу
N (Z) а W представляет собой СН, рассмотрен в патенте США N 4327103, 1982;
(Z) а W представляет собой СН, рассмотрен в патенте США N 4327103, 1982;
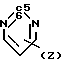
4-(Бензоизотиазол-3-ил)пиперазины, а именно пиперазиновые субстраты для синтеза карбоксамидов, содержащие радикал R2 формулы
N (Z) и радикал W, представляющий собой водород, описан в патенте США N 4452799, 1984.
(Z) и радикал W, представляющий собой водород, описан в патенте США N 4452799, 1984.
Реагенты, необходимые для построения молекулярных структур бензоизотиазол- и бензоизоксазол-3-карбоксамидов, имеющие радикал А формулы -СН2СН= СНСН2- или -СН2С ≡ССН2-, а именно, амины формул
H H2CH=CHCH2N
H2CH=CHCH2N W-R2
W-R2
H H2C
H2C CCH2N
CCH2N W-R2 где R1, R2, W и m определены выше, упомянуты в кассете патентных заявок США, под номером НR 1155.
W-R2 где R1, R2, W и m определены выше, упомянуты в кассете патентных заявок США, под номером НR 1155.
Бензоизотиазол- и бензоизоксазол-3-карбоксамиды, отвечающие изобретению, полезны для лечения психических расстройств благодаря способности блокировать возбудимость, вызванную апоморфином, у млекопитающих.
Антиневротическую активность определяют в ходе т.н. "испытания на вскарабкивание мышей" (Сlimbing mice assay) по методике, похожей на ту, что описали Протэ и сотр. см. Protais P. et al. Psychopharmacol. 1976, V. 50, р. 1, а также Косталь, см. Costal B. Eur. J. Pharmacol. 1978, V. 50, р. 39.
Объектами испытаний служили самцы мышей линии СК-1 с массой тела 23-27 г, разбитые на группы согласно стандартной методике лабораторных испытаний. Мышей помещали в проволочные клетки размерами 4 х 4 х 10 дюймов и выдерживали в течение 1 с для адаптирования и привыкания к новой окружающей обстановке. Затем мышам вводили инъекцией подкожно апоморфин при дозировке 1,5 мг на 1 кг живой массы, так что всех подопытных животных эта доза заставляла на 30 мин вскарабкиваться (climb) на стенки клетки. Соединения, подлежащие испытанию на антиневротическую активность, вводили инъекцией в брюшную полость за 30 мин до количественной оценки действия апоморфина при дозировке 10 мг/кг.
Для количественной оценки "вскарабкивания на стенку клетки" брали по 3 значения (для 10, 20 и 30 мин после назначения апоморфина) согласно приведенной шкале.
Мышей, постоянно вскарабкивающихся на стенку еще до инъекции апоморфина, в результатах эксперимента не учитывали.
Развитая форма вскарабкивания на стенку под действием апоморфина выражалась в длительном повисании животных на стенках клеток по большей части в неподвижном состоянии. По контрасту с этим вскарабкивание, вызванное просто двигательным стимулированием, обычно длится лишь несколько секунд. Поведение мышей Баллы вскарабкивающихся на стенку клетки опирается всеми 4-мя лапами на пол 0 ( вскарабкивания на стенку клетки не наблюдается); цепляется 2-мя передними лапами за стенку клетки 1 ( стоит на задних лапах); цепляется за стенку клетки всеми 4-мя лапами 2 (" лезет на стену") и висит
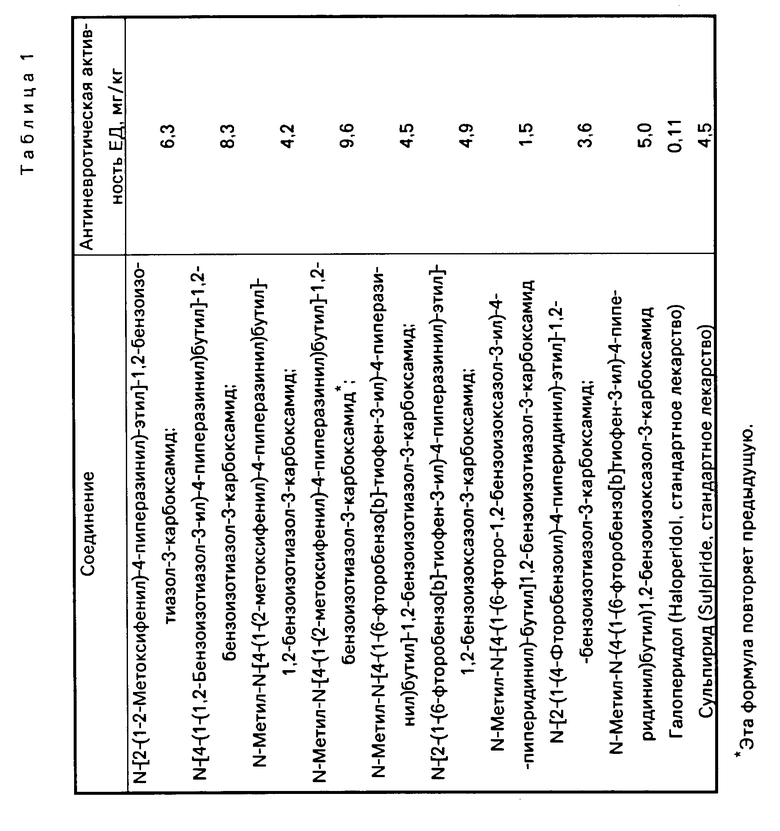
Баллы оценки вскарабкивания были суммированы по отдельности (максимальная сумма баллов: 6 на одну мышь для трех считываний). За 100% принимали сумму баллов для контрольной группы, животным которой в брюшную полость вводили разбавитель, а подкожно апоморфин. Значения ЕD50 вычисляли методом линейного регрессионного анализа с 95%-ной достоверностью. В табл. 1 представленa антиневротическая активность, выраженная через показатель ЕD50 для указанных в табл. 1 бензоизотиазоил- и бензоизоксазол-3-карбоксамидов, а также для двух стандартных антиневротиков.
Антиневротическая активность достигается, когда рассматриваемые бензоизотиазол- и бензоизоксазол-3-карбоксамиды назначают подопытным животным, нуждающимся в таком лечении, в виде эффективных дозировок 0,01-50 мг на 1 кг живой массы в сутки, через полость рта, а также посредством внутривенной инъекции в ткани извне. В особенности эффективна дозировка 25 мг на 1 кг живой массы в сутки. Следует иметь в виду, что каждое подопытное животное требует регулирования индивидуального режима дозирования (с учетом конкретных особенностей организма) квалифицированным специалистом, который вводит животным указанное соединение или наблюдает за правильностью введения последнего. Следует также иметь в виду, что указанные дозы приведены исключительно для примера и ни в малейшей степени не ограничивают область и практику применения изобретения.
Антиневротический характер рассматриваемых бензоизотиазол- и бензоизоксазол-3-карбоксамидов является следствием их парадоксально слабой способности вызывать нежелательные экстрапирамидальные (ехtra pyramidal) побочные эффекты. Степень нежелательной активности, обусловленной экстрапирамидальным побочным эффектом, устанавливают по подавлению стереотипного поведения, вызванного апоморфином, пользуясь методикой, которую описали Анден и сотр. см. Anden N.Е. et al. J.Pharma. Pharmacol. 1967, V. 19, р. 627, а также Эрнст и сотр. см. Ernst A.M. et al. Psychopharmacologia (Berl), 1967, V. 10, р. 316.
По этой методике брали самцов крыс линии Wistar массой тела 125-200 г, которых разбивали на группы с доступом к пище и воде ad libitum. Лекарства готовили на дистиллированной воде, в которую в случае нерастворимости компонентов добавляли поверхностно-активное вещество (ПАВ). Режим назначения можно было варьировать. Объемная дозировка составляла 10 мл на 1 кг массы тела. При первичной оценке численность животных в группе равнялась шести. Лекарство назначали за час до считывания показаний в баллах. Животных размещали по одному в пластмассовых "аквариумах" размерами 24 х 14 х 13 см. Животным контрольной группы вводили только растворитель без лекарства. Готовили раствор гидрохлорида апоморфина с концентрацией 15 мг указанного соединения в 10 мл 0,03% раствора аскорбиновой кислоты, содержащего 30 мкг аскорбиновой кислоты в 100 мл 1%-ного физиологического солевого раствора, что повышало стабильность гидрохлорида апоморфина в растворе. Раствор гидрохлоридa апоморфина назначали подкожно при дозировке 1,5 мг на 1 кг живой массы (объемная дозировка составляла 1 мл на 1 кг живой массы). Через 50 мин после введения лекарства отмечали стереотип поведения животного, стереотипную активность классифицировали как фырканье, облизывание и жевательные движения, повторяющиеся неоднократно в беспрерывном режиме. Если эти проявления иногда прерывались, то таких животных считали защищенными от невротического расстройства.
За показатель эффективности лекарства (в) принимали относительное число таких "защищенных" животных в каждой наблюдаемой группе. В том случае, когда в ходе данного испытания антиневротики показывали незначительный эффект, считали, что это свидетельствует об их слабой способности вызывать нежелательные экстрапирамидальные побочные эффекты и/или о наличии у данных животных дискинезии, замедляющей движения, см. работу Мура и Гершона (Moore N.C. Gershon S. Clinical neuropharmacology, 1989, V. 12, р. 167).
Реакцию на дозировку определяли так же, как и первичную оценку, за исключением того, что группа насчитывала 10 животных, и этим животным дозы лекарства вводили несистематизированно. Животным одной из групп вводили только растворитель. Показатель ЕD50 стереотипного поведения вычисляли методом пробит-анализа (рrobit analysis).
В табл. 2 приведены значения показателя (%) подавления стереотипного поведения, проявляющегося под действием апоморфина, для рассматриваемых бензоизотиазол- и бензоизоксазол-3-карбоксамидов, отвечающих изобретению, а также для двух стандартных лекарств.
Соединения, отвечающие изобретению, включают:
а. N-[2-((1-метил)-4-пиперазинил)этил]-6-метил-1,2-бензоизотиазол-3- карбоксамид;
b. 5-метокси-N-[2-((1-метил)-4-пиперазинил)этил] -1,2-бензоизотиазол-3- карбоксамид;
с. 5-гидрокси-N-[2-((1-метил)-4-пиперазинил)этил]-1,2-бензоизотиазол-3- карбоксамид;
d. 7-хлоро-N-[2-((1-метил)-4-пиперазинил)этил]-1,2-бензоизотиазол-3- карбоксамид;
е. N-2-[(1-метил)-4-пиперазинил)этил)] -6-трифторометил- 1,2-бензоизотиазол-3-карбоксамид;
f. 6,7-дихлоро-N-[2-((1-метил)-4-пиперазинил)этил]-6-метил-1,2- бензоизотиазол-3-карбоксамид;
g. N-[2-[1-(3-метилбензил)] -4-пиперазинилэтил] -1,2-бензоизотиазол-3- карбоксамид;
h. N-[2-[1-(3,4-дихлоробензил)] -4-пиперазинилэтил]-1,2-бензоизотиазол- 3-карбоксамид;
i. N-[2-[1-(2-гидроксибензил)]-4-пиперазинилэтил]-1,2-бензоизотиазол-3- карбоксамид;
j. N-[2-[1-(4-трифторометил)бензил]-4-пиперазинилэтил]-1,2-бензоизотиазол- 3-карбоксамид;
k. N-[4-(1-(2-метоксифенил)-4-пиперазинил)-2-бутенил]-1,2-бензоизотиазол- 3-карбоксамид;
l. N-[3-(1-(1,2-бензоизотиазол-3-ил)-4-пиперазинил)пропил]-1,2-бензоизотиазол- 3-карбоксамид;
m. N-метил-N-[2-(бензо[b]тиофен-3-ил)-4-пиперазинил)этил]-1,2-бензоизоксазол- 3-карбоксамид.
Эффективные количества соединений, отвечающих изобретению, можно назначать самыми различными методами, например, через желудочно-кишечный тракт в виде капсул или таблеток, инъекциями в ткани стерильных растворов или суспензий, а в некоторых случаях внутривенным вливанием стерильных растворов. Конечные соединения, представляющие собой свободные основания, эффективны и сами по себе, но их также можно вводить в рецептуры и назначать в виде их фармацевтически приемлемых аддитивных солей с целью улучшения стабильности, удобства кристаллизации, повышения растворимости и т.п.
Предпочтительные фармацевтически приемлемые аддитивные соли включают соли минеральных кислот, например, соляной, азотной, серной и т.п. соли одноосновных карбоновых кислот, таких как, например, уксусная, пропионовая и т. п. а также двухосновных карбоновых кислот, таких как, например, малеиновая, фумаровая, щавелевая и т.п. а кроме того, трехосновных карбоновых кислот, таких как, например, карбоксиянтарная, лимонная и т.п.
Для приема через полость рта активные соединения, отвечающие изобретению, можно назначать, например, в сочетании с инертным разбавителем или съедобным наполнителем. Эти вещества можно заключать в желатиновые капсулы или спрессовывать в таблетки. При терапевтическом назначении через полость рта данные вещества можно сочетать с эксципиентами и использовать в виде таблеток, облаток, капсул, эликсиров, суспензий, сиропов, вафель, жевательных резинок и т.п. Такие рецептуры должны содержать не менее 0,5% активного соединения, но это содержание может варьировать в зависимости от конкретной придаваемой лекарству формы и обычно составляет 4-75% от массы штучной формы. Количество предлагаемого активного соединения в подобном составе таково, что оно обеспечивает подходящую дозировку. Предпочтительные составы и рецептуры, отвечающие настоящему изобретению, готовят таким образом, что дозировка для приема штучной формы лекарства через полость рта составляет 1,0-300 мг активного соединения.
Таблетки, пилюли, капсулы, облатки и т.п. могут также содержать следующие ингредиенты: связующее, такое как микрокристаллическая целлюлоза, трагакантовая камедь или желатин; эксципиент, такой как крахмал или лактоза; размельчитель, такой как альгиновая кислота, "Примогель" (Primogel), кукурузный крахмал и т. п. смазывающее вещество, такое как стеарат магния или "Стеротес" (Sterotes); глидант (вещество, облегчающее скольжение), такой как коллоидный кремнезем; подслащивающую добавку, такую как сахароза или сахарин, или ароматизатор, такой как мята, метилсалицилат, апельсиновый ароматизатор. Если дозировочной штучной формой является капсула, то кроме веществ указанных выше типов она также содержит жидкий носитель, такой как жирная кислота. Прочие дозировочные штучные формы лекарства могут содержать различные иные вещества, видоизменяющие физическую природу дозировочной штучной формы, например, могут иметь покрытия. Так таблетки или пилюли могут иметь покрытия из сахара, шеллака или иных веществ, образующих оболочки. Сиропы помимо активных компонентов могут содержать сахарозу, являющуюся подслащивающим средством, а также те или иные консерванты, пигменты, красители и ароматизаторы. Вещества, использу- емые при приготовлении указанных составов, должны быть фармацевтически чистыми и в тех дозах, в которых они применяются, нетоксичными.
При терапевтическом назначении инъекциями в ткани организма извне активные соединения, отвечающие изобретению, можно включить в состав раствора или суспензии. Такие рецептуры должны содержать не менее 0,1% упомянутого активного соединения, но это содержание может варьировать в пределах 0,5-50% от массы раствора или суспензии. Содержание активного вещества в подобных составах таково, что оно обеспечивает подходящую дозировку. Предпочтительные составы и рецептуры, отвечающие настоящему изобретению, готовят таким образом, что дозировка для одной штучной формы лекарства, предназначенной для введения в ткани организма извне, составляет 0,5-100 мг активного соединения.
Растворы или суспензии могут также включать следующие компоненты: стерильный разбавитель, такой как вода для инъекции, физиологический солевой раствор, фиксированные масла, полиэтиленгликоль, глицерин, пропиленгликоль и иные синтетические растворители, бактерицидные средства, такие как бензиловый спирт или метилпарабены; противоокислители, такие как аскорбиновая кислота или бисульфит натрия; желатирующие агенты, такие как этилендиаминтетрауксусная кислота; буферные реагенты, такие как ацетаты, цитраты или фосфаты; а также регуляторы тоничности (tonicity), такие как хлорид натрия или декстроза.
Рецептурные составы для непосредственного введения в ткани извне, можно заключать в ампулы, рассасываемые швы или склянки из стекла или пластмассы, вмещающие по нескольку доз.
П р и м е р 1. Гидрохлорид N-метил-N-[4-(1-(4-фторобензоил)-4-пиперидинил)-бутил]-1,2-бензоизотиазол-3- карб
В течение 17 ч в газовой среде азота нагревали при 70оС смесь, содержащую 5,12 г N-метил-N-(4-бромобутил)-1,2-бензоизотиазол-3-карбоксамида, 3,75 г 4-фторобензоилпиперидина, 6,77 г карбоната калия, 0,350 г йодида натрия и 75 мл диметилформамида. Реакционную смесь разбавляли введением 300 мл воды и 20 мл 5%-ного раствора гидроксида натрия, после чего осуществляли экстрагирование эфиром. Объединенные вместе экстракты промывали водой и рассолом, высушивали над безводным сульфатом натрия и фильтровали с последующим выпариванием фильтрата при пониженном давлении. Остаток очищали методом хроматографии на силикагеле с использованием в качестве вымывающей жидкости раствора метилового спирта в дихлорометане. Подходящие фракции отбирали и выпаривали. Обработкой остатка раствором хлористого водорода в этиловом спирте получали хлористоводородную соль искомого соединения. Рекристаллизацией из дихлорометана и этилацетата было получено (при 45,9% выходе 3,15 г продукта с температурой плавления 148-150оС dec.
Вычислено, C 61,28; H 5,96; N 8,57.
C25H29ClFN3O2S
Найдено, C 61,22; H 5,98; N 8,54.
П р и м е р 2. N-[2-(1-(6-Хлоро-1,2-бензоизоксазол-3-ил)-4-пиперидинил)этил]-1,2- бензоизотиазол-3-карбоксамид.
В газовой среде азота при перемешивании нагревали до 180оС смесь, содержащую 2,9 г N-(2-хлороэтил)-1,2-бензоизотиазол-3-карбоксамида и 3,48 г 1-(6-хлоро-1,2-бензоиозоксазол-3-ил)пиперидина в 125 мл обезвоженного N-метилпирролидинона. Спустя 5 ч смеси давали остыть до комнатной температуры, разбавляли смесь водой и насыщенный водным раствором карбоната натрия сообщали этой смеси основность, после чего осуществляли экстрагирование этилацетатом. Органическую фазу промывали водой, обезвоживали над безводным сульфатом магния и фильтровали, после чего фильтрат концентрировали в вакууме. Получившийся остаток подвергали хроматографической очистке на силикагеле с использованием эфира в качестве вымывающей жидкости. Подходящие фракции собирали вместе и выпаривали. Образовавшийся остаток растирали в присутствии дихлорметана и эфира, в результате чего было получено (при 44,6% выходе) 2,38 г продукта с т.пл. 132-134оС.
Вычислено, C 59,93; H 4,80; N 12,71.
C22H21Cl4O2S
Найдено, C 59,98; H 4,91; N 12,65.
П р и м е р 3. N-[3-(1-(6-Хлоро-1,2-бензоизоксазол-3-ил)-4-пиперидинил)пропил] 1,2-бензоизотиазол-3-карбоксамид.
В газовой среде азота при перемешивании нагревали до 180оС смесь, содержащую 5,0 г N-(3-хлоропропил)-1,2-бензоизотиазол-3-карбоксамида, 5,40 г гидрохлорида 1-(6-хлоро-1,2-бензоизоксазол-3-ил)пиперидина, 5,40 г карбоната калия и 100 мг йодида натрия в 100 мл обезвоженного N-метилпирролидинона. Спустя 36 ч реакционной смеси давали остыть при комнатной температуре и проводили разделение фаз на этилацетатную и водную. Органическую фазу промывали водой, обезвоживали над безводным сульфатом магния и фильтровали с последующим концентрированием фильтрата в вакууме. Получившийся остаток подвергали хроматографической очистке на силикагеле с использованием этилацетата в качестве разбавителя. Подходящие фракции собирали вместе и концентрировали. Фильтрат обрабатывали раствором хлористого водорода в эфире. Смесь подвергали рекристаллизации из метилового спирта, дихлорометана и эфира, в результате чего было получено (при 29,4% выходе) 2,87 г продукта с температурой плавления 221-223оС.
Вычислено, C 56,21; H 4,92; N 11,40.
C23H24Cl2N4O2S
Найдено, C 55,85; H 4,95; N 11,19.
П р и м е р 4. Гидрохлорид N-метил-N-[4-(1-(6-фторо-1,2-бензоизоксазол-3-ил)-4-пиперидинил)бутил]-1,2-б ензоизотиазол-3-карбоксамида.
В течение 21 ч в газовой среде азота нагревали при 75оС смесь, содержащую 4,84 г N-метил-N-(4-бромобутил)-1,2-бензоизотиазол-3-карбоксамида, 3,15 г 1-(6-фторо-1,2-бензоизоксазол-3-ил)пиперидина, 4,50 г карбоната калия, 0,560 г йодида натрия и 200 мл ацетонитрила. Реакционную смесь фильтровали, отфильтрованный корж остатка промывали дихлорметаном, после чего фильтрат концентрировали при пониженном давлении. Полученный остаток переносили в дихлорометан, промывали 5%-ным водным раствором гидроксида натрия и водой, обезвоживали над безводным сульфатом натрия и фильтровали с последующим испарением фильтрата при пониженном давлении. Образовавшийся остаток подвергали хроматографической очистке на силикагеле с использованием 5-10% -ного раствора метилового спирта в дихлорометане в качестве вымывающей жидкости. Подходящие фракции собирали вместе и концентрировали. Остаток обрабатывали раствором хлористого водорода в этиловом спирте. Рекристаллизацией из дихлорометана и этилацетата было получено (при 39,7% выходе) 2,60 г продукта с т.пл. 204-205оС dec.
Вычислено, C 59,69; H 5,61; N 11,14.
C25H28ClFN4O2S
Найдено, C 59,37; H 5,57; N 11,06.
П р и м е р 5. N-[2-(1-(4-(2-Оксо-1-бензоимидазолинил)пиперидинил)этил] -1,2- бензоизотиазол-3-карбоксамид.
В газовой среде азота при перемешивании нагревали до 180оС смесь, содержащую 5,0 г 1,2-бензоизотиазол-3-[N-(2-хлороэтил)-карбоксамида и 5,87 г 4-(2-оксо-1-бензоимидазолинил)пиперидина в 150 мл 1-метил-2-пирролидинона. Спустя 18 ч реакционной смеси давали остыть до комнатной температуры, после чего выливали эту смесь в воду. Водную фазу экстрагировали этилацетатом. Соединенные вместе органические экстракты промывали водой, обезвоживали над безводным сульфатом магния и фильтровали с последующим концентрированием фильтрата в вакууме. Получившийся остаток подвергали рекристаллизации из этилацетата, в результате чего было получено (при 46,0% выходе) 4,03 г продукта с т.пл. 174-177оС.
Вычислено, C 62,69; H 5,50; N 16,61.
C17H24N4O2S
Найдено, C 62,54; H 5,43; N 16,39.
П р и м е р 6. Дигидрохлорид N-[2-(1-метил)-4-пиперазинил)этил]-1,2-бензоизотиазол-3-карбоксамида.
В газовой среде азота при перемешивании нагревали до 180оС смесь, содержащую 3,9 г N-(2-хлороэтил)-1,2-бензоизотиазол-3-карбоксамида и 3,6 мл 1-метилпиперазина в 150 мл обезвоженного N-метилпирролидинона. Спустя 18 ч реакционной смеси давали остыть до комнатной температуры, а затем разбавляли эту смесь водой с последующим экстрагированием этилацетатом. Органическую фазу промывали водой, обезвоживали над безводным сульфатом магния и фильтровали, после чего фильтрат собирали вместе и концентрировали в вакууме. Остаток подвергали хроматографической очистке на силикагеле с использованием смеси метилового спирта и этилацетата, взятых в соотношении 1:1, в качестве промывочной жидкости. Подходящие фракции собирали вместе и концентрировали. Остаток переносили в эфир. Туда же добавляли раствор хлористого водорода в эфире. Рекристаллизацией и осаждением из метилового спирта, дихлорметана и этилацетата было получено (при 37,9% выходе) 2,33 г соли с т.пл. 227-230оС.
Вычислено, C 47,75; H 5,88; N 14,85.
C15H22Cl2N4OS
Найдено, C 47,86; H 5,95; N 14,80.
П р и м е р 7. Полугидрат дигидрохлорида N-[2-(1-бензил)-4-пиперазинил)этил]-1,2-бензоизотиазол-3-карбоксамида.
В газовой среде азота при перемешивании нагревали до 180оС смесь, содержащую 3,0 г N-(2-хлороэтил)-1,2-бензоизотиазол-3-карбоксамида и 2,6 мл 1-бензилпиперазина в 100 мл обезвоженного N-метилпирролидинона. Спустя 5 ч реакционной смеси давали остыть при комнатной температуре, разбавляли ее водой и насыщенным водным раствором карбоната натрия сообщали этой смеси основность с последующим экстрагированием этилацетатом. Органическую фазу промывали водой, обезвоживали над безводным сульфатом магния и фильтровали с последующим концентрированием фильтрата в вакууме. Остаток подвергали хроматографической очистке на силикагеле с использованием этилацетата в качестве вымывающей жидкости. Подходящие фракции собирали вместе и выпаривали. Получившийся остаток растворяли в эфире. Туда же добавляли раствор хлористого водорода в эфире. Осадок подвергали рекристаллизации из метилового спирта, дихлорометана и эфира, в результате чего было получено (при 40,9% выходе) 2,36 г продукта с т.пл. 207-210оС.
Вычислено, C 54,53; N 5,88; N 12,11.
C21H27Cl2N4O1,5S
Найдено, C 54,86; H 5,69; N 12,36.
П р и м е р 8. Дигидрохлорид N-[2-(1-2-метоксифенил)-4-пиперазинил)этил] -бензоизотиазол-3- карбоксамида.
В газовой среде азота при перемешивании нагревали до 120оС смесь, содержащую 2,24 г 1,2-бензоизотиазол-3-[N-(2-хлороэтил) карбоксамида и 1,8 г 1-(2-метоксифенил)пиперазина в 100 г мл обезвоженного 1-метил-2-пирролидинона. Спустя 24 ч реакционную смесь охлаждали до комнатной температуры и выливали в насыщенный водный раствор карбоната натрия с последующим экстрагированием эфиром. Органические экстракты соединяли вместе, обезвоживали над безводным сульфатом магния и фильтровали с последующим концентрированием фильтрата в вакууме. Получившийся остаток подвергали хроматографированию на силикагеле с использованием эфира в качестве вымывающей жидкости. Подходящие фракции соединяли вместе и концентрировали. Образовавшийся остаток переносили в эфир. Туда же добавляли раствор хлористого водорода в эфире. Осадок подвергали рекристаллизации из эфира и дихлорометана, в результате чего было получено (при 30,6% выходе) 1,34 г продукта с температурой плавления 205-208оС.
Вычислено, C 53,73; H 5,58; N 11,93.
C21H24N4O2S.2HCl
Найдено, C 53,52; H 5,35; N 11,73.
П р и м е р 9. N-[2-(1-(3-Хлорофенил)-4-пиперазинил)этил]-1.2-бензоизотиазол-3- карбоксамид.
В газовой среде азота при перемешивании нагревали до 180оС смесь, содержащую 4,23 г 1,2-бензоизотиазол-3-[N-(2-хлороэтил)] карбоксамида и 4,15 г 1-(3-хлорофенил)пиперазина в 125 мл обезвоженного 1-метил-пирролидинона. Спустя 18 ч реакционную смесь охлаждали до комнатной температуры и выливали в насыщенный водный раствор карбоната натрия. Водную фазу экстрагировали эфиром, после чего объединенные вместе органические экстракты промывали водой, обезвоживали над безводным сульфатом магния и фильтровали с последующим концентрированием фильтрата в вакууме. Получившийся остаток подвергали хроматографированию на силикагеле с использованием эфира в качестве вымывающей жидкости. Подходящие фракции собирали вместе и концентрировали. Образовавшийся остаток переводили в твердое вещество. Рекристаллизацией из эфира и дихлорометана было получено (при 37,2% выходе) 2,62 г продукта с т.пл. 115-117оС.
Вычислено, C 59,92; H 5,28; N 13,97.
C20H21ClN4OS
Найдено, C 59,84; H 5,15; N 13,93.
П р и м е р 10. Дигидрохлорид N-[3-(1-(2-метоксифенил)-4-пиперазинил)пропил]-1,2-бензоизотиазол-3- карбоксамида.
В газовой среде азота при перемешивании нагревали до 180оС смесь, содержащую 4,2 г N-(3-хлоропропил)-1,2-бензоизотиазол-3-карбоксамида, 3,3 г 1-(2-метоксифенил)пиперазина, 4,55 г карбоната калия и 100 мг йодида натрия в 150 мл обезвоженного N-метилпирролидинона. Спустя 24 ч реакционной смеси давали остыть до комнатной температуры и осуществляли разделение фаз на эфирную и водную. Органическую фазу обезвоживали над безводным сульфатом магния и фильтровали с последующим концентрированием фильтрата в вакууме. Получившийся остаток подвергали хроматографированию на силикагеле с использованием эфира в качестве вымывающей жидкости. Подходящие фракции собирали вместе и выпаривали. Остаток переводили в эфир. Туда же добавляли раствор хлористого водорода в эфире. Рекристаллизацией осадка из метилового спирта, дихлорометана и эфира было получено (при 26,6% выходе) 2,12 г продукта с т.пл. 191-194оС.
Вычислено, C 54,66; H 5,84; N 11,59.
C22H28Cl2N4O2S
Найдено, C 54,60; H 5,75; N 11,51.
П р и м е р 11. Дигидрохлорид N-метил-N-[3-(1-(2-метоксифенил)-4-пиперазинил)-пропил]-1,2- бензоизотиазол-3-карбоксамида.
В газовой среде азота при перемешивании нагревали до 80оС смесь, содержащую 3,4 г N-метил-N-(3-бромопропил)-1,2-бензоизотиазол-3-карбоксамида, 2,09 г 1-(2-метоксифенил)пиперазина, 3,0 г карбоната калия и 100 мг йодида натрия в 125 мл обезвоженного ацетонитрила. Спустя 24 ч смеси давали остыть до комнатной температуры, после чего эту смесь выпаривали в вакууме. Далее проводили разделение фаз остатка на этилацетатную и водную. Органическую фазу обезвоживали над безводным сульфатом магния и фильтровали с последующим концентрированием фильтрата в вакууме. Полученный остаток подвергали хроматографированию на силикагеле с использованием этилацетата в качестве вымывающей жидкости. Подходящие фракции собирали вместе и концентрировали. Образовавшийся остаток переносили в эфир. Туда же добавляли раствор хлористого водорода в эфире. Осадок подвергали рекристаллизации из дихлорометана и этилацетата, в результате чего было получено (при 30,0% выходе) 1,62 г продукта с температурой плавления 166-169оС.
Вычислено, C 55,53; H 6,08; N 11,26.
C23H28N4O2S.HCl
Найдено, C 55,21; H 5,90; N 11,16.
П р и м е р 12. Дигидрохлорид N-[4-(1-(2-метоксифенил)-4-пиперазинил)бутил]-1,2-бензоизотиазол-3- карбоксамида.
С вечера до утра при перемешивании нагревали до 80оС смесь, содержащую 2,44 г хлорида 1,2-бензоизотиазол-3-карбоновой кислоты, 3,26 г 1-(2-метоксифенил)-4-(4-аминобутил)пиперазина и 6 мл триэтиламина в 100 мл обезвоженного "просеиванием" (sieve-dried) толуола. Спустя 24 ч реакционную смесь охлаждали до комнатной температуры, после чего выливали в воду. Далее отделяли органическую фазу, а водную фазу экстрагировали эфиром. Эфирные экстракты и толуоловую фазу объединяли вместе, обезвоживали над безводным сульфатом магния и фильтровали с последующим концентрированием фильтрата в вакууме. Полученный остаток подвергали хроматографированию на силикагеле с использованием этилацетата в качестве вымывающей жидкости. Подходящие фракции собирали вместе и концентрировали. Образовавшийся остаток переносили в эфир. Туда же добавляли раствор хлористого водорода в эфире. Осадок подвергали рекристаллизации из этилового спирта и этилацетата с последующим обезвоживанием при температуре обратной перегонки толуола в вакууме, составлявшем 0,1 мм рт. ст. в результате чего было получено (при 34,6% выходе) 2,13 г продукта с т.пл. 175-178оС.
Вычислено, C 55,53; H 6,08; N 11,26.
C23H28N4OS.HCl
Найдено, C 55,49; H 5,80; N 11,19.
П р и м е р 13. Полугидрат дигидрохлорида N-метил-N-[4-1-(2-метоксифенил)-4-пиперазинил)бутил]-1,2-бензоизотиазол- 3-карбоксамида.
В газовой среде азота при перемешивании нагревали до 80оС смесь, содержащую 3,42 г N-метил-N-(4-бромобутил)-1,2-бензоизотиазол-3-карбоксамида, 2,01 г 1-(2-метоксифенил)пепиразина, 2,9 г карбоната калия и 20 мг йодида натрия в 100 мл обезвоженного ацетонитрила. Спустя 18 ч реакционной смеси давали остыть до комнатной температуры, после чего эту смесь выпаривали в вакууме. Далее проводили разделение фаз остатка на ацетатную и водную. Органическую фазу обезвоживали над безводным сульфатом магния и фильтровали с последующим концентрированием фильтрата в вакууме. Полученный остаток переносили в эфир. Туда же добавляли раствор хлористого водорода в эфире. Осадок подвергали рекристаллизации из дихлорометана и эфира, в результате чего было получено (при 34,3% выходе) 1,87 г продукта с т.пл. 169-171оС.
Вычислено, C 55,37; H 6,38; N 10,76.
C24H30N4.O2S.2HCl.0,5H2O
Найдено, C 55,64; H 6,42; N 10,75.
П р и м е р 14. N-[2-(1-(1,2-бензоизотиазол-3-ил)-4-пиперазинил)этил]-1,2-бензоизотиазол- 3-карбоксамид.
В газовой среде азота при перемешивании нагревали до 190оС смесь, содержащую 1,2 г 1,2-бензоизотиазол-3-[N-(2-хлороэтил) карбоксамида и 1,3 г 1,2-бензоизотиазол-3-ил-пиперазина в 25 мл 1-метил-2-пирролидинона. Спустя 2 ч реакционную смесь охлаждали до комнатной температуры, после чего выливали в насыщенный водный раствор карбоната натрия. Водную фазу экстрагировали эфиром, после чего органические экстракты объединяли вместе, обезвоживали над безводным сульфатом магния и фильтровали с последующим концентрированием фильтрата в вакууме. Полученный остаток подвергали хроматографированию на силикагеле с использованием эфира в качестве вымывающей жидкости. Подходящие фракции собирали вместе и концентрировали. Оставшееся твердое вещество подвергали рекристаллизации из эфира, в результате чего было получено (при 62,8% выходе) 1,33 г продукта с т.пл. 160-163оС.
Вычислено, C 59,55; H 5,00; N 16,53.
C21H21N5OS2
Найдено, C 59,20; H 4,99; N 16,31.
П р и м е р 15. Гидрохлорид N-[4-(1-(1,2-бензоизотиазол-3-ил)4-пиперазинил)-бутил]-1,2-бензоизотиазол- 3-карбоксамид.
С вечера до утра при перемешивании нагревали до 80оС смесь, содержащую 2,6 г хлорида 1,2-бензоизотиазол-3-карбоновой кислоты, 3,45 г 1-(1,2-бензоизотиазол-3-ил)-4-(4-аминобутил)-пиперазина и 5 мл триэтиламина в 100 мл обезвоженного "просеиванием" (sieve-dried) толуола. Спустя 24 ч реакционную смесь охлаждали до комнатной температуры, после чего выливали в воду. Далее отделяли органическую фазу, а водную фазу экстрагировали этилацетатом. Этилацетатные экстракты и толуоловую фазу объединяли вместе и обезвоживали над безводным сульфатом магния. Органическую фазу фильтровали с последующим концентрированием фильтрата в вакууме. Полученный остаток подвергали хроматографированию на силикагеле с использованием этилацетата. Подходящие фракции собирали вместе и концентрировали в вакууме. Образовавшийся остаток переносили в эфир. Туда же добавляли раствор хлористого водорода в эфире. Осадок повергали рекристаллизации из дихлорометана и эфира и обезвоживали при температуре обратной перегонки изопропилового спирта в вакууме, составляющем 0,1 мм рт.ст. в результате чего было получено (при 27,1% выходе) 1,60 г продукта с температурой плавления 203-205оС.
Вычислено, C 55,57; H 5,47; N 14,09.
C23H23N5OS2.HCl.0,5H2O.
Найдено, C 55,32; H 5,20; N 13,86.
П р и м е р 16. Гидрохлорид N-метил-N-[4-(1-(6-фторобензо[b]тиофен-3-ил)-4-пиперазинил)бутил] 1,2-бензоизотиазол-3-карбоксамида.
В газовой среде азота при температуре обратной перегонки растворителя нагревали смесь, содержащую 4,0 г N-метил-N-(4-бромобутил)-1,2-бензоизотиазол-3-карбоксамида, 4,0 г 6-фторо-3-пиперазинилбензо[b]тиофена, 5,0 г карбоната калия, 350 мг йодида натрия и 200 мл ацетонитрила. Спустя 16 ч реакционную смесь фильтровали, отфильтрованный корж осадка промывали дихлорометаном, а фильтрат концентрировали при пониженном давлении. Полученный остаток переносили в дихлорометан, промывали 5%-ным раствором гидроксида натрия и водой, а затем обезвоживали над безводным сульфатом натрия и фильтровали с последующим концентрированием фильтрата. Образовавшийся остаток подвергали хроматографированию на силикагеле с протекающим по нему 7,5%-ным раствором метилового спирта в дихлорометане. Подходящие фракции собирали вместе и концентрировали. К остатку добавляли раствор хлористого водорода в эфире. Осадок подвергали рекристаллизации из дихлорометана и этилацетата, в результате чего было получено (при 44,2% выходе) 2,80 г продукта с т.пл. 183-185оС.
Вычислено, C 57,85; H 5,44; N 10,79.
C25H28ClFN4OS2
Найдено, C 57,66; H 5,21; N 10,61.
П р и м е р 17. Гидрохлорид N-метил-N-[4-(1-(1,2-бензоизотиазол-3-ил)-4-пиперанизил)бутил]-1,2- бензоизотиазол-3-карбоксамида.
В газовой среде азота при перемешивании нагревали до 80оС смесь, содержащую 4,0 г N-метил-N-(4-бромобутил)-1,2-бензоизотиазол-3-карбоксамида, 3,13 г гидрохлорида 1-(1,2-бензизотиазол-3-ил)пиперазина, 5,10 г карбоната калия и 20 мг йодида натрия в 100 мл обезвоженного ацетонитрила. Спустя 18 ч реакционной смеси давали остыть до комнатной температуры, после чего проводили ее концентрирование в вакууме. Далее проводили разделение фаз остатка на этилацетатную и водную. Органическую фазу обезвоживали над обезвоженным сульфатом магния и фильтровали с последующим концентрированием фильтрата в вакууме. Полученный остаток подвергали хроматографированию на силикагеле с использованием смеси, содержащей 10% метилового спирта и 90% этилацетата, в качестве вымывающей жидкости. Подходящие фракции собирали вместе и выпаривали. Образовавшийся остаток переносили в эфир. Туда же добавляли раствор хлористого водорода в эфире. Рекристаллизацией из дихлорометана и эфира было получено (при 55,7% выходе) 3,36 г продукта с т.пл. 210-211оС.
Вычислено, C 57,41; H 5,62; N 13,95.
C24H28ClN5OS2
Найдено, C 57,22; H 5,49; N 13,83.
П р и м е р 18. Дигидрохлорид N-[4-(1-(2-метоксифенил)-4-пиперазинил)-2-бутинил]-1,2-бензоизотиазол- 3-карбоксамида.
Смесь, содержащую 2,0 г хлорида 1,2-бензоизотиазол-3-карбоновой кислоты и 2,63 г 1-(2-метоксифенил)-4-(4-амино-2-бутинил)пиперазина в 100 мл дихлорометана, при перемешивании вводили по каплям 2,83 мл триэтиламина. Перемешивание продолжалось с вечера до утра. Через 24 ч реакционную смесь разбавляли водой и экстрагировали дихлорометаном. Органическую фазу обезвоживали над безводным сульфатом магния и фильтровали с последующим концентрированием фильтрата в вакууме. Остаток подвергали хроматографированию на силикагеле с использованием этилацетата в качестве вымывающей жидкости. Подходящие фракции собирали вместе и концентрировали. Оста- ток переносили в смесь эфира с дихлорометаном. Туда же добавляли раствор хлористого водорода в эфире. Остаток подвергали рекристаллизации из метилового спирта, дихлорометана и эфира, в результате чего было получено (при 45,7% выходе) 2,27 г продукта с т.пл. 162-164оС.
Вычислено, C 55,98; H 5,31; N 11,35.
C23H26Cl2N4O2S
Найдено, C 55,84; H 5,06; N 11,25.
П р и м е р 19. Дигидрохлорид Z-N-[4-(1-(2-метоксифенил)-4-пиперазинил)-2-бутенил]-1,2-бензоизотиазол- 3-карбоксамида.
В смесь, содержащую 1,77 г хлорида 1,2-бензоизотиазол-3-карбоновой кислоты и 2,34 г Z-1-(2-метоксифенил)-4-(4-амино-2-бутинил)пиперазина в 100 мл дихлорометана, при перемешивании вводили по каплям 2,51 мл триэтиламина. Перемешивание продолжалось с вечера до утра. Через 24 ч реакционную смесь разбавляли, добавляя к ней воду и насыщенный водный раствор карбоната натрия, после чего проводили экстрагирование дихлорометаном. Органическую фазу промывали водой, обезвоживали над безводным сульфатом магния и фильтровали с последующим концентрированием фильтрата в вакууме. Полученный остаток переносили в смесь дихлорометана с эфиром. Туда же добавляли раствор хлористого водорода в эфире. Осадок подвергали рекристаллизации из метилового спирта, дихлорометана и эфира, в результате чего было получено (при 51,7% выходе) 2,31 г продукта с т.пл. 184-187оС.
Вычислено, C 55,76; H 5,70; N 11,31.
C23H28Cl2N4O2S
Найдено, C 55,52; H 5,65; N 11,21.
П р и м е р 20. N-[4-(1-(2-Пиримидил)-4-пиперанизил)-2-бутинил]-1,2-бензоизотиазол-3 -карбоксамид.
В смесь, содержащую 2,55 г хлорида 1,2-бензоизотиазол-карбоновой кислоты и 3,0 г 1-(2-пиримидил)-4-(4-амино-2-бутинил)пиперазина в 100 мл дихлорометана, при перемешивании вводили по каплям 3,62 мл триэтиламина. Перемешивание продолжалось с вечера до утра. Через 24 реакционную смесь разбавляли водой, обезвоживали над безводным сульфатом магния и фильтровали с последующим концентрированием фильтрата в вакууме. Получившийся остаток подвергали рекристаллизации из дихлорометана, эфира и гексана, в результате чего было получено (при 72,3% выходе) 3,67 г продукта с т.пл. 102-104оС.
Вычислено, C 61,21; H 5,14; N 21,41.
C20H20N6OS
Найдено, C 61,10; H 4,92; N 21,25.
П р и м е р 21. Гидрохлорид N-метил-N-[4-(1-(6-фторобензо[b]тиофен-3-ил)-4-пиперазинил)бутил]-1,2 бензоизоксазол-3-карбоксамида.
В течение 17 ч в газовой среде азота нагревали при 75оС смесь, содержащую 4,22 г N-метил-N-(4-бромбутил)-1,2-бензоизоксазол-3-карбоксамида, 3,89 г 1-(6-фторобензо[b] тиофен-3-ил)пиперазина, 5,00 г карбоната калия, 0,80 г йодида натрия и 200 мл ацетонитрила. Затем реакционную смесь фильтровали, корж отфильтрованного остатка промывали дихлорометаном, а фильтрат выпаривали. Получившийся остаток переносили в дихлорометан, промывали 5%-ным раствором гидроксида натрия и водой, обезвоживали над безводным сульфатом натрия и фильтровали с последующим выпариванием фильтрата при пониженном давлении. Образовавшийся остаток подвергали хроматографической очистке на силикагеле с использованием 7,5%-ного раствора метилового спирта в этилацетате в качестве вымывающей жидкости. Подходящие фракции собирали вместе и выпаривали. Туда же добавляли раствор хлористого водорода в этиловом спирте. Рекристаллизацией осадка из этилового спирта и этилацетата было получено (при 30,8% выходе) 2,11 г продукта с т.пл. 145-147оС.
Вычислено, C 59,69; H 5,61; N 11,14.
C25H28ClFN4O2S
Найдено, C 59,72; H 5,72; N 11,15.
П р и м е р 22. N-[2-(1-(2-метоксифенил)-4-пиперазинил)этил]-1,2-бензоизотиазол-3- карбоксамид.
В течение 4,5 ч нагревали при 170оС смесь, содержащую 4,00 г N-(2-хлороэтил)-1,2-бензоизотиазол-3-карбоксамида, 4,15 г N-(2-метоксифенил)пиперазина и 100 мл N-метилпирролидинона. Реакционной смеси давали остыть, после чего разбавляли ее 400 мл 5%-ного раствора гидроксида натрия. Образовавшуюся водную смесь экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом, после чего обезвоживали над безводным сульфатом магния и фильтровали. Фильтрат концентрировали в вакууме. Получившийся остаток подвергали хроматографированию на силикагеле с использованием 5%-ного раствора метилового спирта в дихлорометане. Подходящие фракции собирали вместе и концентрировали. Рекристаллизацией образовавшегося остатка из эфира и гексанов было получено (при 32,8% выходе) 2,22 г продукта с температурой плавления 107-110оС.
Вычислено, C 66,30; H 6,36; N 14,73.
C21H24N4O3
Найдено, C 66,38; H 6,22; N 14,72.
П р и м е р 23. Гидрохлорид N-[2-(1-(6-фторобензо[b]тиофен-3-ил)-4-пиперазинил)этил]-1,2- бензоизотиазол-3-карбоксамида.
В течение 3,0 ч нагревали при 160оС смесь, содержащую 4,40 г N-(2-хлороэтил)-1,2-бензоизоксазол-3-карбоксамида, 5,90 г 6-фторо-3-(1-пиперазинил)бензо[b] тиофена и 100 мл N-метилпирролидинона. Реакционной смеси давали остыть, после чего разбавляли ее 350 мл воды и при помощи 25%-ного раствора гидроксида натрия сообщали ей основность (рН 8), после чего проводили экстрагирование эфиром. Объединенные экстракты промывали водой и рассолом, обезвоживали над безводным сульфатом магния и фильтровали. Фильтрат концентрировали в вакууме. Получившийся остаток подвергали хроматографированию на силикагеле с использованием 75%-ного раствора этилацетата в гексанах в качестве вымывающей жидкости. Подходящие фракции собирали вместе и концентрировали. К образовавшемуся остатку добавляли раствор хлористого водорода в этиловом спирте. Рекристаллизацией осадка из метилового и этилового спиртов было получено (при 23,6% выходе) 2,13 г продукта с т.пл. 225-228оС.
Вычислено, C 57,32; H 4,81; N 12,15.
C22H22ClFN4O2S
Найдено, C 57,24; H 4,45; N 12,07.
П р и м е р 24. Полугидрат гидрохлорида N-[2-(1-(4-фторобензоил)-4-пиперидинил)этил]-1,2-бензоизотиазол-3- карбоксамида.
В газовой среде азота при перемешивании нагревали до 180оС смесь, содержащую 3,5 г N-(2-хлороэтил)-1,2-бензоизотиазол-3-карбоксамида и 3,63 г 1-(4-фторобензоил)пиперидина в 125 мл обезвоженного 1-метил-2-пирролидинона. Спустя 3 ч реакционную смесь охлаждали до комнатной температуры, после чего выливали эту смесь в насыщенный водный раствор карбоната натрия. Водную фpазу экстрагировали этилацетатом, а объединенные органические экстракты промывали водой, обезвоживали над безводным сульфатом магния и фильтровали с последующим концентрированием фильтрата в вакууме. Получившийся остаток растирали с эфиром, фильтрат концентрировали. Образовавшийся остаток подвергали хроматографированию на силикагеле с использованием этилацетата в качестве вымывающей жидкости. Подходящие фракции объединяли вместе и концентрировали. Остаток переносили в эфир. Туда же добавляли раствор хлористого водорода в эфире. Остаток подвергали рекристаллизации из метилового спирта, дихлорометана и эфира, в результате чего было получено (при 18,1% выходе) 1,21 г продукта с температурой плавления 188-190оС.
Вычислено, C 57,82; H 5,29; N 9,19.
C22H24ClFN3O2,5S
Найдено, C 57,73; H 5,06; N 9,09.
П р и м е р 25. N-Mетил-N-(4-бромбутил)-1,2-бензоизоксазол-3-карбоксамид.
К охлажденной в ледяной ванне суспензии, содержащей 1,09 г гидроксила натрия в виде 60%-ной дисперсии в масле и 10 мл диметилформамида добавляли в виде раствора 4,38 г N-метил-1,2-бензоизотиазол-3-карбоксамида и 10 мл диметилформамида в условиях перемешивания с интенсивностью, обеспечивающей слабое выделение водорода. По завершении указанного добавления реакционную смесь перемешивали при температуре ледяной ванны в течение 10 мин. Затем ледяную ванну удаляли и полученную смесь вводили по каплям при перемешивании в раствор 7,8 мл 1,4-дибромобутана в 10 мл диметилформамида. Смесь перемешивали при комнатной температуре с вечера до утра, после чего добавляли к ней 250 мл воды. Затем смесь экстрагировали эфиром. Объединенные экстракты промывали водой и насыщенным раствором хлорида натрия, обезвоживали над безводным сульфатом натрия и фильтровали. Далее фильтрат концентрировали, а получившийся остаток подвергали хроматографированию на 500 г силикагеля. По колонке при этом пропускали 5%-ный раствор этилацетата в дихлорометане. Подходящие фракции собирали вместе и выпаривали, в результате чего было получено (при 55% выходе) 4,28 маслянистого продукта.
П р и м е р 26. Малеат 6-фторо-3-(1-пиперазинил)бензо[b]тиофена.
В течение 2 ч в газовой среде азота нагревали при 176оС смесь, содержащую 40,1 г 6-фторо-3-аминобензо[b]тиофен-2-карбоксилата, 13,1 г 1-метилпиперазина и 100 мл 1-метил-2-пирролидинона. Затем раствор разбавляли водой в количестве 400 мл и экстрагировали эфиром. Объединенные экстракты промывали водой и рассолом, обезвоживали над безводным сульфатом натрия и фильтровали с последующим концентрированием фильтрата при пониженном давлении, в результате чего было получено 9,32 г 3-амино-6-фторобензо[b]тиофена.
Далее в течение 14 ч в газовой среде азота нагревали при 192оС смесь, содержащую 9,32 г 3-амино-6-фторобензо[b]тиофена, 15,0 г пиперазина и 100 мл 1-метил-2-пирролидинона. После этого смесь охлаждали, разбавляли водой в количестве 500 мл и экстрагировали эфиром. Объединенные экстракты промывали водой и рассолом, обезвоживали над безводным карбонатом калия и фильтровали с последующим концентрированием фильтрата при пониженном давлении. Получившийся остаток подвергали хроматографированию на силикагеле с вымыванием 30% -ным раствором метилового спирта в дихлорометане, в результате чего было получено 3,03 г маслянистого вещества в виде свободного основания. К раствору, содержащему 3,03 г указанного свободного основания и 20 мл 2-пропилового спирта, добавляли раствор, содержащий 1,49 г малеиновой кислоты в 20 мл 2-пропилового спирта. Смесь концентрировали при пониженном давлении, а остаток подвергали рекристаллизации из метилового спирта и этилацетата, в результате чего было получено (при 8,80% выходе) 2,72 г продукта с т.пл. 173-175оС.
Вычислено, C 54,54; H 4,86; N 7,95.
C16H17FN
Найдено, C 54,53; H 4,69; N 8,01.
П р и м е р 27. N-(1-Метилэтил)-N-(2-гидроксиэтил)-1,2-бензоизоксазол-3-карбоксамид.
В течение 4 ч в герметичном контейнере нагревали до 140оС смесь, содержащую 10: 0 г этил-1,2-бензоизоксазол-3-карбоксилата, 16,1 г 2-(1-метилэтиламино)этанола и 80 мл толуола. Раствор разбавляли эфиром в количестве 50 мл, промывали 5%-ным раствором бикарбоната натрия, водой и рассолом, обезвоживали над безводным сульфатом натрия и концентрировали при пониженном давлении. Получившийся остаток подвергали хроматографированию на силикагеле при использовании в качестве вымывающей жидкости 60%-ного раствора этилацетата в гексанах, в результате чего было получено (при 81,4% выходе) 10,6 г продукта. Рекристаллизацией из дихлорометана и гексанов была получена анализируемая проба вещества с температурой плавления 92-94оС.
Вычислено, C 62,89; H 6,50; N 11,28.
C13H16N2O3
Найдено, C 63,00; H 6,51; N 11,24.
П р и м е р 28. Полугидрат гидрохлорида N-метил-N-[2-1-(6-фторо-1,2-бензизоксазол-3-ил)-4-пиперазинил)этил] -1,2-бензоизотиазол-3-карбоксамида.
В газовой среде азота при 0оС в условиях перемешивания к смеси, содержащей 4,00 г N-метил-N-(2-гидроксиэтил)-1,2-бензоизоксазол-3-карбоксамида и 2,48 мл триэтиламина в 100 мл толуола, обезвоженного "просеиванием" (sieve dried), по каплям вводили 1,34 мл метансульфонилхлорида. По окончании добавления этого реагента смесь перемешивали и медленно подогревали до комнатной температуры. Через 45 мин добавляли 3,72 г 4-(6-фторо-1,2-бензоизоксазол-3-ил)пиперидина. Полученную смесь при перемешивании нагревали в течение 18 ч при температуре обратной перегонки. Далее смеси давали остыть до комнатной температуры, после чего ее приливали к 2%-ному раствору гидроксида натрия. Затем отделяли органический слой, а водный раствор экстрагировали этилацетатом. Объединенные экстракты промывали водой, обезвоживали над безводным сульфатом магния и фильтровали с последующим концентрированием фильтрата в вакууме. Получившийся остаток подвергали хроматографированию на силикагеле с использованием этилацетата в качестве вымывающей жидкости. Подходящие фракции собирали вместе и выпаривали. Образовавшийся остаток растворяли в дихлорометане, в этот раствор добавляли также раствор хлористого водорода в этиловом спирте, а затем эфир. Образовавшуюся соль подвергали рекристаллизации из дихлорометана и эфира, в результате чего (при 14,0% выходе) было получено 1,45 г продукта с т.пл. 211-214оС.
Вычислено, C 57,07; H 5,21; N 11,58.
C23H23N4O2S.HCl 0,5H2O
Найдено, C 57,04; H 5,08; N 11,52.
П р и м е р 29. Полугидрат гидрохлорида N-метил-N[(2-(1-(4-фторобензоил)-4-пиперидинил)этил]-1,2-бензоизотиазол- 3-карбоксамида.
В газовой среде азота при 0оС к смеси, содержащей 3,43 г N-метил-N-(2-гидроксиэтил)-1,2-бензоизотиазол-3-карбоксамида и 2,13 мл триэтиламина в 100 мл толуола, обезвоженного "просеиванием" (sieve dried), добавляли по каплям 1,15 мл метансульфонилхлорида. После этого смеси давали нагреться до комнатной температуры при непрерывном перемешивании. Через 45 мин к смеси добавляли 3,01 г 1-(4-фторобензоил)пиперидина. Далее смесь в течение 18 ч при перемешивании нагревали при температуре обратной перегонки. Затем смеси давали остыть до комнатной температуры и приливали ее к 5%-ному водному раствору бикарбоната натрия. После этого органический слой отделяли, а водный слой экстрагировали этилацетатом. Объединенные вместе органические фракции промывали водой, обезвоживали над сульфатом магния и фильтровали. Фильтрат концентрировали в вакууме. Получившийся остаток подвергали хроматографированию на силикагеле с использованием этилацетата в качестве вымывающей жидкости. Подходящие фракции собирали вместе и концентрировали. Образовавшийся остаток растворяли в дихлорометане, в этот раствор добавляли раствор хлористого водорода в этиловом спирте, а затем эфире. Соль подвергли рекристаллизации из дихлорометана и этилацетата, в результате чего (при 22,6% выходе) было получено 1,54 г продукта с т.пл. 214-217оС.
Вычислено, C 58,65; H 5,56; N 8,92.
C23H24FN3O2S.HCl 0,5H2O
Найдено, C 58,28; H 5,68; N 8,78.
П р и м е р 30. Гидрохлорид N-метил-N-[(2-((1-(4-фторобензоил)-4-пиперидинил)-этил]-1,2- бензоизоксазол-3-карбоксамида.
К раствору 4,98 г N-метил-N-(2-гидроксиэтил)-1,2-бензоизоксазол-3-карбоксамида и 2,36 г триэтиламина в 55 мл толуоле при 0оС добавляли 2,60 г метансульфонилхлорида. Полученную смесь перемешивали при 0оС в течение 25 мин. Далее к этой смеси добавляли 6,48 г гидрохлорида 4-(4-фторобензоил)пиперидина и 6,46 г триэтиламина. Образовавшуюся реакционную смесь нагревали в течение 13 ч при температуре обратной перегонки. Эту смесь фильтровали с последующим концентрированием фильтрата при пониженном давлении. Образовавшийся остаток растворяли в 225 мл дихлорометана, промывали 10%-ным раствором гидроксида натрия и водой, обезвоживали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток подвергали хроматографированию на силикагеле с использованием 0-10%-ного раствора этилового спирта в этилацетате в качестве вымывающей жидкости. Подходящие фракции собирали вместе и концентрировали, в результате чего было получено 3,74 г продукта в виде свободного основания. Последнее переводили в гидрохлорид раствором хлористого водорода в этиловом спирте. Рекpисталлизацией из метилового спирта и этилацетата было получено (при 22,2% выходе) 2,24 г продукта с т.пл. 218-221оС.
Вычислено, C 61,95; H 5,65; N 9,42.
C23H25ClFN3O3
Найдено, C 61,82; H 5,57; N 9,37.
П р и м е р 31. N-[1-Метилэтил)-N-[(2-(1-(6-фторо-1,2-бензоизоксазол-3-ил)-4- пиперидинил)этил]-1,2-бензоизоксазол-3-карбоксамид.
В газовой среде азота при 0оС быстро вводили 3,70 г метансульфонилхлорида в раствор, содержащий 8,00 г N-(1-метилэтил)-N-(2-гидроксиэтил)-1,2-бензоизоксазол-3-карбоксамида, 3,27 г триэтиламина в 150 мл тетрагидрофурана. Полученную смесь перемешивали при 0оС в течение 30 мин. К этой смеси быстро добавляли суспензию, содержащую 7,80 г 4-(6-фторо-1,2-бензоизоксазол-3-ил)пиперидина и 6,53 г триэтиламина в 60 мл тетрагидрофурана. Полученную смесь нагревали в течение 12 ч при температуре обратной перегонки. Затем добавляли воду, после чего раствор концентрировали при пониженной температуре. Остаток растворяли в 150 мл дихлорометана, промывали 10%-ным раствором гидроксида натрия и водой, обезвоживали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Получившийся остаток подвергали хроматографированию на силикагеле с использованием 80%-ного раствора этилацетата в гексанах. Подходящие фракции собирали вместе и выпаривали, а остаток подвергали рекристаллизации из эфира, в результате чего (при 16,0% выходе) было получено 2,33 г продукта с т.пл. 118-120оС.
Вычислено, C 66,65; H 6,04; N 12,44.
C25H27FN4O3.
Найдено, C 66,63; H 6,04; N 12,41.
П р и м е р 32. N-Метил-N-[(2-(1-(6-фторо-1,2-бензоизоксазол-3-ил)-4-пиперидинил) этил]-1,2-бензоизоксазол-3-карбоксамида.
В газовой среде азота при 0оС быстро вводили 3,70 г метансульфонилхлорида в раствор, содержащий 7,10 г N-метил-N-(2-гидроксиэтил)-1,2-бензоизоксазол-3-карбоксамида и 3,27 г триэтиламина в 100 мл тетрагидрофурана. Смесь перемешивали при 0оС в течение 60 мин. К этой смеси быстро добавляли суспензию, содержащую 8,10 г 4-(6-фторо-1,2-бензоизоксазол-3-ил)пиперидина и 7,26 г триэтиламина в 80 мл тетрагидрофурана. Смесь нагревали в течение 16 ч при температуре обратной перегонки. Затем добавляли воду, после чего раствор концентрировали при пониженной температуре. Получившийся остаток разбавляли 10%-ным раствором гидроксида натрия и экстрагировали 50%-ным раствором эфира в толуоле. Объединенные вместе экстракты промывали водой и рассолом, обезвоживали над безводным карбонатом калия и фильтровали с последующим концентрированием фильтрата при пониженном давлении. Образовавшийся остаток подвергали хроматографированию на силикагеле с использованием этилацетата в качестве вымывающей жидкости. Подходящие фракции собирали вместе и выпаривали. Остаток подвергали рекристаллизации из эфира, в результате чего было получено (при 22,1% выходе) 3,01 г продукта с т.пл. 94-97оС.
Вычислено, C 65,39; H 5,49; N 13,26.
C23H23FN4O3
Найдено, C 65,30; H 5,51; N 13,18.
П р и м е р 33. Полугидрат гидрохлорида N-(1-метилэтил)-N-[(3-(1-(4-фторобензоил)-4-пиперидинил)пропил]-1,2 -бензоизоксазол-3-карбоксамида.
В газовой среде азота при 0оС быстро вводили 3,40 г метансульфонилхлорида в раствор, содержащий 7,80 г N-(1-метилэтил)-N-(3-гидроксипропил)-1,2-бензоизоксазол-3-карбоксамида и 3,05 г триэтиламина в 200 мл толуола. Образовавшуюся смесь перемешивали при 0оС. Через 45 мин в этот раствор вводили 7,95 г гидрохлорида 4-(4-фторобензоил)пиперидина и 8,71 г триэтиламина. Полученную смесь нагревали в течение 16 ч при температуре обратной перегонки. Далее реакционную смесь промывали 10%-ным раствором гидроксида натрия, водой и рассолом, обезвоживали над безводным сульфатом натрия и фильтровали с последующим концентрированием фильтрата. Получившийся остаток подвергали хроматографированию на силикагеле с использованием 5-10% раствора метилового спирта в этилацетате в качестве вымывающей жидкости. Подходящие фракции собирали вместе и выпаривали. Далее раствор этого остатка в метиловом спирте подкисляли раствором хлористого водорода в этиловом спирте и разбавляли эфиром. Смесь вымораживали и получали твердое вещество. Рекристаллизацией этого твердого вещества из этилового спирта и этилацетата было получено (при 10,2% выходе) 1,50 г продукта с т.пл. 177-179оС.
Вычислено, C 62,83; H 6,49; N 8,45.
C26H31ClFN3O3.0,5H2O
Найдено, C 62,86; H 6,28; N 8,30.
П р и м е р 34. Дигидрохлорид N-метил-N-[3-(1-(4-фторобензоил)пиперидинил)-пропил]-1,2-бензоизотиазол- 3-карбоксамида.
В газовой среде азота при перемешивании нагревали до 120оС смесь, содержащую 7,13 г N-метил-N-(3-хлоропропил)-1,2-бензоизотиазол-3-карбоксамида, 6,44 г 1-(4-фторобензоил)пиперидина, 7,33 г карбоната калия и 300 мг йодида натрия в 150 мл обезвоженного 1-метил-2-пирролидинона. Спустя 24 ч смеси давали остыть до комнатной температуры и проводили разделение фаз остатка на ацетатную и водную. Органическую фазу промывали водой, обезвоживали над безводным сульфатом магния и фильтровали с последующим концентрированием фильтрата в вакууме. Черновой остаток дважды подвергали хроматографированию на кремнеземе с использованием смеси, содержащей 10% метилового спирта и 90% этилацетата в качестве вымывающей жидкости. Подходящие фракции собирали вместе и выпаривали. Образовавшийся остаток растворяли в эфире. Туда же добавляли раствор хлористого водорода в этиловом спирте. Рекристаллизацией осадка из дихлорометана и эфира было получено (при 5,09% выходе) 0,642 г продукта с т.пл. 189-191оС.
Вычислено, C 60,56; H 5,72; N 8,83.
C24H27ClFN3O2S
Найдено, C 60,32; H 5,75; N 8,70.
П р и м е р 35. N-метил-N-3-(1-(4-фторобензоил)-4-пиперидинил)пропил)-1,2- бензоизоксазол-3-карбоксамид.
В газовой среде азота в течение 27,5 ч нагревали при 100оС смесь, содержащую 4,42 г N-метил-N-(3-хлоропропил)-1,2-бензоизоксазол-3-карбоксамида, 4,40 г гидрохлорида 4-фторобензоилпиперидина, 8,12 г карбоната калия и 0,400 г йодида натрия в 250 мл ацетонитрила. Реакционную смесь фильтровали, после чего отфильтрованный корж остатка промывали дихлорометаном. Фильтрат концентрировали при пониженном давлении. Получившийся остаток растворяли в дихлорометане, промывали 10%-ным раствором гидроксида натрия и водой, обезвоживали над безводным сульфатом натрия и фильтровали с последующим концентрированием фильтрата при пониженном давлении. Получившийся остаток подвергали хроматографированию на силикагеле с использованием 5%-ного раствора метилового спирта в дихлорометане в качестве вымывающей жидкости. Подходящие фракции собирали вместе и выпаривали. Остаток подвергали рекристаллизации из метилового спирта и гексана, в результате чего было получено (при 16,2% выходе) 1,55 г продукта с т.пл. 85-87оС.
Вычислено, C 68,07; H 6,19; N 9,92.
C24H26FN3O3
Найдено, C 68,02; H 6,14; N 9,89.
Ниже приводятся данные по токсичности для заявленных соединений, определенные в результате анализа РОЕ (Первичный явный эффект) и выраженные в виде острой летальной дозы 50.
Ниже приводится методика проведения испытаний и результаты.
Первичные явные эффекты у крыс.
Использовали группу из четырех самцов крыс Вистар (125-300 г). Перед испытанием животных помещали на 24 ч по крайней мере в помещение с контролируемым климатом, обеспечивая вдоволь пищи и воды. В день испытания животных извлекали из их обычных клеток, и помещали 4/клетку в белые полупрозрачные пластиковые боксы (45х25х29 см) с крышками из металлических прутков, и переносили в помещение для испытаний.
Соединения готовят с использованием дистиллированной воды, а если они нерастворимы, добавляют поверхностно-активное вещество и постоянно перемешивают полученную в результате суспензию.
Перед введением лекарства всех животных обследовали на наличие каких-либо аномальностей, которые могли впоследствии запутать действие лекарства. Они включают положение глаз, ясность глаз, кровь вокруг глаз или носа, необычность походки, аномальное поведение во время обращения с ними и аномальное поведение в пластиковых боксах. Затем определяют температуру тела (или ректально или интраперитонеально).
Затем животным вводят лекарство, контрольная группа получает носитель. Обычно объем дозы составляет 10 см3/кг, обычным путем введения является интраперитонеальный.
Животных наблюдают в пластиковых боксах непрерывно в течение часа после введения лекарства и отмечают какие-либо явные эффекты. Полное неврологическое обследование, детали которого приведены в приложении, приводится для каждого животного через 1, 2, 4 и 6 ч после введения лекарства (болевой ответ также измеряется через 1/2 ч) и результаты регистрируются. Во время испытания в помещении должно быть тихо. Явные эффекты, которые видны между этими промежутками времени, регистрируются. Животным дают пищу и воду после 6 ч и выдерживают до 24 ч, в то время как наблюдают их общее явное состояние. Все параметры приведены как максимально нормализованный процент, за исключением температуры тела, которая приводится как разница с контролем носителя. Результаты даны в табл. 3.
Нормализованный  × 100%
× 100%
Используя величины нормализованных процентов для различных параметров, выполнены следующие расчеты:
Нормализованный процент смертности х 0,8. Измеренные параметры: Смертность.
Отмечают время смерти для каждого животного и отмечают время, когда произошла первая и последняя смерть. Оценка:
Смерть +1
Нет смерти 0
Фармацевтические препараты Таблетка:
Ингредиенты В каждой
таблетке, мг
N-Метил-N[4-(1-(6-фтор-
1,2-бензизоксазол-3-ил)-
4-пиперидинил)-бутил]
1,2-бензизотиазол-3-кар-
боксамид 300 Поливинилпирролидон 22,5 Лактоза 61,75 Спирт 3А-200 крепость 4,5 Стеариновая кислота 9 Тальк 13,5 Кукурузный крахмал 43,25
Смешивают N-метил-N-[4-(1-(6-фтор-1,2-бензизоксазол-3-ил)-4-пиперидинил)бутил] 1,2-бензизотиазол-3-карбоксамид, поливинилпирролидон и лактозу вместе, пропускают через сито 40 меш. Добавляют медленно спирт и тщательно перемешивают. Пропускают сырую массу через сито 4 меш. Сушат гранулят при 50оС до утра. Пропускают сухой гранулят через сито 20 меш. Просеивают стеариновую кислоту, тальк и кукурузный крахмал через сито 60 меш. перед смешением в барабане с гранулятом. Таблетируют с использованием стандартного 7/16-дюймового пуансона, 10 таблеток имеют массу 4,5 г. Мыльце:
Ингредиенты В каждой
свече, мг
N-Метил-N-[4-(1-(6-фтор-
1,2-бензизоксазол-3-ил)-
4-пиперидинил)бутил] -1,2-бензизотиазол-3- карбоксамид 300 Глицерин 3000 Очищенная вода 200
Глицерин нагревают в подходящем сосуде до примерно 120оС. Лекарство растворяют с легким перемешиванием в нагретом глицерине, после чего добавляется очищенная вода, смешивается, и горячая смесь сразу выливается в соответствующую форму.
Эмульсия. Количество
Ингредиенты.
Желатин типа А (получен-
ный из кислотообработан-
ных предшественников; используется при рН ≈ 3,2) 4 г
N-Метил-N-[4-(1-(6-фтор-
1,2-бензизоксазол-3-ил)-4-
пиперидинил) бутил]-1,2-
бензизотиазол-3-карбокса- мид 360 мг
Ароматизирующее вещест- во, по желанию 30 мл Спирт 250 мл Масло 250 мл
Очищенная вода, добавля-
ется до 500 мл.
К примерно 300 мл очищенной воды добавляется желатин и лекарство, выстаивается несколько минут, нагревается до растворения желатина, затем поднимается температура до примерно 98оС и выдерживается при этой температуре примерно 20 мин. Охлаждают до 50оС и добавляют ароматизирующее вещество, спирт и достаточно очищенную воду до 500 мл. Добавляют масло, тщательно перемешивают смесь и пропускают ее через гомогенизатор или коллоидную мельницу до получения полной и однородной дисперсии масла.

Использование: в медицине, в качестве веществ, обладающих антипсихотической активностью. Сущность изобретения: продукт бензизотиазол- или бензизоксазол-3-карбоксамиды ф-лы I, где R1 -Н или низший алкил; R2 низший алкил или группа ф-лы II, III, IV, V, VI, VII, VIII, IX, где А низший алкилен, группа ф-лы -CH2CH=CH-CH2-CH2≡ CCH2-; Х-О или S; W-N или группа СН; Z-H, низшая алкоксигруппа или галоген, а сплошная линия указывает место присоединения группы к обозначенному члену ф-лы или их фармацевтически приемлемые соли и фармкомпозиции с подходящим носителем, содержащие 1 360 мг соединения I на единичную дозу. Соединения I получают взаимодействием соединения X, где R2-Cl, Br, I или группа OSO2R6 где R6 -алкил, фенил или топил, с соединением ф-лы XI или взаимодействием соединения ф-лы XII, где Hal-Cl или Br, с соединением ф-лы XIII. Структура ф-л I, II, III, IV, V, VI, VII, VIII, IX, X, XI, XII, XIII: 
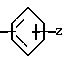
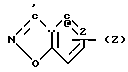



 3 с. и 2 з.п. ф-лы, 2 табл.
3 с. и 2 з.п. ф-лы, 2 табл.
где R1 водород или низший алкил;
R2 низший алкил или группа одной из общих формул

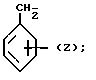
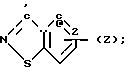


А низший алкилен, группа формулы CH2 CH=CHCH2 или -CH2 C≡ CCH2-;
X кислород или сера;
W азот или группа CH-;
Z водород, низшая алкоксигруппа или галоген;
сплошная линия (_____) указывает место присоединения группы к обозначенному члену формулы;
или их фармацевтически приемлемые соли.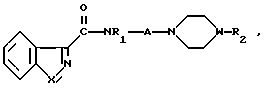
где R1 водород или низший алкил;
R2 низший алкил или группа одной из общих формул
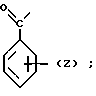




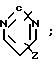

A низший алкилен, группа формулы -CH2-CH=CH-CH2- или -CH2 C≡ C-CH2-;
X кислород или сера;
W азот или группа -CH-;
Z водород, низшая алкоксигруппа или галоген;
сплошная линия указывает место присоединения группы к обозначенному члену формулы,
отличающийся тем, что соединения общей формулы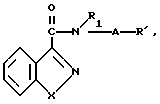
где R1,A,X имеют указанные значения;
R' хлор, бром, йод или группа OSO2R6, где R6 алкил, фенил или толил,
подвергают взаимодействию с соединением общей формулы
где R2 и W имеют указанные значения.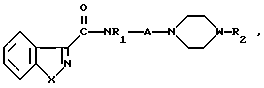
где R1 водород или низший алкил;
R2 низший алкил или группа одной из общих формул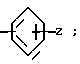



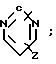

A низший алкилен, группа формулы -CH2-CH=CH-CH2- или
-CH2≡ CCH2-;
X кислород или сера;
W азот или группа CH-;
Z водород, низшая алкоксигруппа или галоген,
сплошная линия указывает место присоединения группы к обозначенному члену формулы,
отличающийся тем, что соединение общей формулы
где X имеет указанные значения;
Hal хлор или бром,
подвергают взаимодействию с соединением общей формулы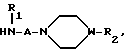
где A, R1, R2 и W имеют указанные значения.
| EP N 0378111, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
