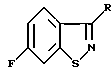
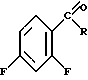
Изобретение относится к способу получения 6-фтор-1,2-бензизотиазолов формулы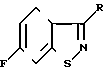 (I) где R - атом водорода, низший алкил или группа формулы
(I) где R - атом водорода, низший алкил или группа формулы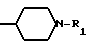 или
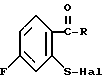
или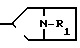 где R1 означает -СНО или - CN, заключающемуся в том, что о-галогенфенациловое производное формулы:
где R1 означает -СНО или - CN, заключающемуся в том, что о-галогенфенациловое производное формулы: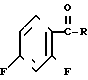
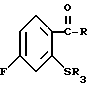
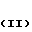 где R имеет указанные значения, подвергают взаимодействию с R3SH, где R3- бензил, в среде апротонного органического растворителя, с образованием соединения формулы
где R имеет указанные значения, подвергают взаимодействию с R3SH, где R3- бензил, в среде апротонного органического растворителя, с образованием соединения формулы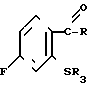 (III) где R и R3 имеют указанные значения, которое подвергают взаимодействию с галогенирующим агентом для получения соответствующего сульфенилгалоида формулы
(III) где R и R3 имеют указанные значения, которое подвергают взаимодействию с галогенирующим агентом для получения соответствующего сульфенилгалоида формулы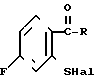 (IV) Подвергают взаимодействию полученный сульфенилгалоид с аммиаком с образованием в результате соединения формулы
(IV) Подвергают взаимодействию полученный сульфенилгалоид с аммиаком с образованием в результате соединения формулы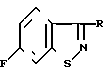 (I)
(I)
Полученные в результате 1,2-бензизотиазолы формулы I можно использовать в качестве промежуточных продуктов для получения фармацевтически активных соединений, которые могут использоваться, например, в качестве антипсихотических агентов, а также в качестве ингибиторов повторного поглощения серотонина.
Настоящее изобретение, кроме того, относится к соединениям формулы (III) где R и R3 имеют вышеуказанные значения,
(III) где R и R3 имеют вышеуказанные значения,
которые могут использоваться в качестве промежуточных продуктов для получения соединений формулы IV.
Если это специально не оговорено, под выражением "низший алкил" имеется ввиду прямой или разветвленный алкил с 1-6 атомами углерода. Примерами такого алкила являются метил, этил, н-пропил, изобутил, пентил и гексил.
Если это специально не оговорено, под выражением "галоген" имеются ввиду атомы фтора, хлора, брома или йода.
Известны способы получения 1,2-бензизотиазолов, исходя из о-галогенфенациловых соединений. Примером одного из таких способов является обработка о-галогенфенациловых соединений в автоклаве при высоких температурах молекулярной серой и аммиаком. Таким способом нельзя, однако, получить 6-фтор-1,2-бензизотиазолы с высоким выходом.
Предлагаемый в соответствии с настоящим изобретением способ дает возможность получать 6-фтор-1,2-бензизотиазолы из о-галогенфенациловых соединений с высоким выходом, не прибегая к высоким температурам и без использования автоклава.
Было установлено, что при протекании вышеуказанной реакции с образованием соединения III замещение нуклеофильной группой R3SH происходит селективно у атома фтора, соседнего с карбонильной функциональной группой фенильного кольца. Реакцию обычно проводят в среде органического растворителя, например тетрагидрофурана, в присутствии основания например трет-бутоксида калия, при температуре ≈ 0-150оС, предпочтительно при ≈10-100оС, наиболее предпочтительно ≈15-80оС. Предпочтительные R3SH - соединения включают бензилмеркаптаны.
Для получения сульфенилгалогенида IV соединение III подвергают взаимодействию с галогенирующим агентом, например сульфурилхлоридом, сульфурилбромидом, бромом или хлором, наиболее предпочтительно с сульфурилхлоридом. Реакцию обычно проводят в среде органического растворителя, например дихлорэтана или дихлорметана, при температуре ≈0-100оС предпочтительно при ≈15-80оС.
Образующееся соединение IV обычно выделяют из реакционной смеси и без дополнительной очистки подвергают взаимодействию с аммиаком, получая в результате целевой бензизотиазол. Реакцию обычно проводят в среде органического растворителя, например тетрагидрофурана, при температуре ≈0-100оС, предпочтительно при ≈15-80оС наиболее предпочтительно при температуре окружающей среды.
Исходные дифторкетоны, у которых R означает гетероциклоалкил, например пиперидинил, получают, например, путем ацилирования 1,3-дифторбензола по реакции Фриделя-Крафтса известным способом, используя соответствующий галоидангидрид.
Исходные дифторкетоны, у которых R означает атом водорода, низший алкил, получают известными способами.
Исходные дифторкетоны, у которых R означает бициклогетероалкил, например тропанил, получают из диалкилацеталя 2,4-дифторбензальдегида, который подвергают взаимодействию с фосфорилирующим агентом, например триэтилфосфитом и бортрифторидэтератом, в присутствии подходящего растворителя, например дихлорметана, с образованием в результате соединения формулы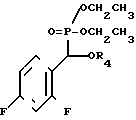 (V) где R4 означает низший алкил. Эту реакцию обычно проводят при температуре от -25оС до комнатной температуры, в течение 10-30 ч.
(V) где R4 означает низший алкил. Эту реакцию обычно проводят при температуре от -25оС до комнатной температуры, в течение 10-30 ч.
Полученное соединение V подвергают затем взаимодействию с н-бутиллитием или другим подходящим реагентом, например литийдиизопропиламидом, и тропиноном формулы с образованием в результате соединения формулы
с образованием в результате соединения формулы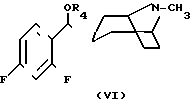 Реакцию обычно проводят в среде подходящего растворителя, например тетрагидрофурана, при температуре от -78оС до комнатной, в течение 10-30 ч.
Реакцию обычно проводят в среде подходящего растворителя, например тетрагидрофурана, при температуре от -78оС до комнатной, в течение 10-30 ч.
Соединение VI подвергают затем взаимодействию с водным раствором HCl в среде ацетона при температуре кипения, в течение 2-8 ч с образованием в результате соединения
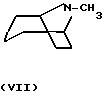
Полученное соединение VII переводят в соединение II, где R1означает CN или CO2R2 известными способами, например путем взаимодействия с бромцианом или хлорформиатом, в присутствии основания, например карбоната калия, с образованием в результате соединения II, где R1 означает CN, соответственно CO2R2.
Нижеследующие примеры приведены исключительно для иллюстрации изобретения и их нельзя рассматривать как ограничивающие объем его защиты. Все температуры, если это специально не оговорено, приведены в градусах Цельсия.
П р и м е р 1. 4-Фтор-2-(фенилметилтио)бензальдегид
Бензилмеркаптан (1,74 г) в 10 мл тетрагидрофурана (ТГФ) добавляли по каплям к раствору трет-бутоксида калия (1,58 г) в 60 мл ТГФ. Полученную суспензию перемешивали при температуре окружающей среды в течение 5 мин, затем добавляли к ней 2,4-дифторбензальдегид (2,0 г) и перемешивали реакционную смесь в течение получаса при комнатной температуре. После этого добавляли к ней насыщенный раствор хлорида аммония и подвергали экстракции этилацетатом (EtOAc). Объединенные органические слои промывали рассолом, высушивали над MgSO4, фильтровали и упаривали, получая 3,11 г сырого продукта. После кристаллизации из метанола получали твердое вещество с т.пл. 81-82оС.
Вычислено, %: С 68,27; Н 14,50
С14H11FOS
Найдено, %: С 68,31; Н 4,37
В. 6-Фтор-1,2-бензиизотиазол
Сульфурилхлорид (1,04 г) добавляли по каплям к суспензии 4-фтор-2-(фенилметилтио)бензальдегида (2,0 г) в 23 мл дихлорэтана при комнатной температуре. Образующийся раствор перемешивали в течение 0,5 ч, после чего растворитель удаляли в вакууме, а остаток суспендировали в 10 мл ТГФ и обрабатывали при комнатной температуре 10 мл этанола (EtOH), насыщенного аммиаком (NH3). Полученную смесь перемешивали в течение получаса, добавляли к ней воду и образующийся продукт экстрагирования EtOAc. Объединенные органические слои промывали рассолом, высушивали над MgSO4, фильтровали и упаривали, получая в результате 1,0 г сырого продукта. В результате очистки с помощью ускоренной препаративной тонкослойной хроматографии (элюирование смесью этилацетата и гексана) получали 0,66 г жидкости бледно-желтого цвета.
П р и м е р 2. А. 4-Фтор-2-(фенилметилтио)ацетофенон
Бензилмеркаптан (1,59 г) в 10 мл ТГФ добавляли по каплям к раствору третбутоксида калия (1,4 г) в 55 мл ТГФ. Полученную суспензию перемешивали в течение 5 мин при температуре окружающей среды, после чего добавляли к ней 2,4-дифторацетофенон (2,0 г). Реакционную смесь перемешивали в течение получаса при комнатной температуре, добавляли к ней насыщенный раствор хлорида аммония, после чего подвергали экстракции EtOAc. Объединенные органические слои промывали рассолом, высушивали над MgSO4, фильтровали и упаривали, получая в результате 3,2 г сырого продукта. После перекристаллизации из метанола получали 2,1 г (63%) продукта в виде твердого вещества с т.пл. 111-112оС.
Расчитано, %: C 69,21; Н 5,03
C15H13FOS
Найдено, %: С 69,42; Н 5,11
В. 6-Фтор-3-метил-1,2-бензизотиазол
Сульфурилхлорид (0,52 г) добавляли по каплям к суспензии 4-фтор-2-(фенилметилтио)ацетофенона (1,0 г) в 12 мл дихлорэтана при комнатной температуре. Полученный раствор перемешивали в течение получаса, после чего растворитель удаляли в вакууме, а остаток суспендировали в 12 мл ТГФ и обрабатывали при комнатной температуре 12 мл ЕtOH, насыщенного NH3. Полученную смесь перемешивали в течение получаса, после чего добавляли к ней воду и образующийся продукт экстрагировали EtOAc. Объединенные органические слои промывали рассолом, высушивали над MgSO4, фильтровали и упаривали, получая в результате 0,6 г сырого продукта. В результате очистки с помощью ускоренной препаративной тонкослойной хроматографии (элюирование смесью этилацетата и гексана) получали 0,37 г (58%) твердого вещества бледно-желтого цвета с т.пл 50-57оС.
П р и м е р 3. А. 4-[4-Фтор-2-(фенилметилтио)бензоил]-1-пиперидинкарбоксаль-дегид
К перемешиваемому раствору трет-бутоксида калия (4,6 г) в ТГФ (125 мл) добавляли по каплям бензилмеркаптан (5,1 г) в ТГФ (60 мл). После перемешивания в течение получаса при температуре окружающей среды добавляли порциями 4-(2,4-ди-фторбензоил)-1-пиперидинкарбоксальдегид (10,3 г). Присутствующие в реакционной смеси белое хлопьевидное твердое вещество переходило в раствор и перемешивание при температуре окружающей среды продолжали в течение 2 ч. Реакцию затем прекращали, выливая реакционную смесь в воду, и водную суспензию подвергали экстракции EtOAc. Органический экстракт промывали (водой), высушивали (MgSO4) и растворитель отгоняли, получая 14,0 г вязкой маслянистой жидкости. Полученную маслянистую жидкость смешивали с порцией из другой партии (14,7 г) и весь продукт растирали с EtO2, получая в результате 20,4 г (63%) твердого вещества с темпер. плавления 96-98оС. После перекристаллизации из смеси толуола и гексана получали не совсем белый твердый продукт с темп. плавл. 101-103оС.
Рассчитано, %: С 67,20; Н 5,64; N 3,92
C20H20FNO2S
Найдено, %: С 67,42; Н 5,96; N 3,90
В. 4-(6-Фтор-1,2-бензизотиазол-3-ил)-1-пиперидинкарбоксальдегид
К перемешиваемому раствору 4-[4-фтор-2-(фенилметилтио)]-1-пиперидинкар- боксальдегида (40 г) в метиленхлориде (СН2Сl2) (400 мл) добавляли по каплям сульфурилхлорид (8,9 мл) в СН2Сl2 (28 мл). По окончании добавления реакционную смесь перемешивали в течение 1 ч при температуре окружающей среды. После этого ее фильтровали для удаления небольших количеств нерастворившегося материала и упаривали, получая в результате 40,7 г сульфенилхлорида в виде густой маслянистой жидкости, которая отверждалась при стоянии. Этот сырой сульфенилхлорид растворяли в ТГФ (500 мл) и к полученному раствору добавляли по каплям насыщенный раствор NH3 в ЕtOH (180 мл). Реакционную смесь перемешивали в течение 2 ч при комнатной температуре и затем оставляли стоять при этой температуре в течение 16 ч. После этого ее выливали в воду и водную суспензию подвергали экстракции EtOAc. Органический экстракт промывали (водой), высушивали (MgSO4) и растворитель отгоняли, получая в результате 29,6 г густой маслянистой жидкости, которую подвергали очистке с помощью препаративной высокопроизводительной жидкостной хроматографии на заполненной силикагелем колонке, осуществляя элюирование 3% Et2NH - EtOAc. Выход целевого соединения, представляющего собой маслянистую жидкость, составлял 12,2 г (42%). Путем растирания с изопропиловым эфиром и поцарапывания получали твердое вещество, которое перекристаллизовывали из смеси EtOAc и гексана. Температура плавления полученного твердого продукта равнялась 104-106оС.
Рассчитано, %: С 59,07; Н 4,96; N 10,60
C13H13FN2OS
Найдено, %: С 59,17; Н 5,07; N 10,48
П р и м е р 4. А. Диэтил-1-(2,4-дифторфенил)-1-метоксиметанфосфонат
Бортрифторидэтерат (86 г) добавляли по каплям к раствору диметилацеталя 2,4-дифторбензальдегида (114 г) и триэтилфосфита (101 г) в 1,2 л СН2Cl2 при -25оС. Полученному раствору давали нагреться до комнатной температуры и перемешивали его в течение 20 ч. После этого к нему добавляли воду и энергично перемешивали смесь в течение 10 мин. Органический слой отделяли, промывали рассолом, высушивали над MgSO4, фильтровали и концентрировали, получая в качестве остатка жидкое вещество. В результате очистки с помощью высокопроизводительной жидкостной хроматографии на колонке с силикагелем (элюирование CH2Cl2 и затем 5% EtOAc - СН2Сl2) получали 136 г диэтил-1-(2,4-дифторфенил)-1-метоксиметанфосфоната в виде жидкости.
В. (2,4-дифторфенил)(8-метил-8-азабицикло-[3.2.1]-октан-3-ил)метанонгидрохло-ри д
Диэтил-1-(2,4-дифторфенил)-1-метокси- метанфосфонат (76 г) растворяли в ТГФ (1600 мл). Раствор охлаждали до -78оС и добавляли к нему по каплям н-бутиллитий (103,4 мл 2,5М раствора в гексане) с такой скоростью, чтобы температура не поднималась выше -65оС. Полученный раствор перемешивали в течение 1 ч, после чего добавляли к реакционной смеси медленно по каплям тропинон (32,7 г), растворенный в ТГФ (100 мл). По окончании добавления смеси медленно давали нагреться до комнатной температуры и перемешивали в течение ночи. Затем к ней добавляли насыщенный раствор NaCl (1,5 л), слои разделяли, органические слои собирали и высушивали над MgSO4 и растворитель отгоняли, получая в результате маслянистую жидкость (73 г). Эту маслянистую жидкость (38 г) растворяли в ацетоне (2 л), медленно добавляли к полученному раствору воду (35 мл) и концентрированную HСl (182 мл) и кипятили смесь в течение 3 ч с обратным холодильником. После этого ацетон отгоняли и остаток в виде водного раствора подвергали экстракции EtOAc, подщелоченный К2СО3, затем экстракции CH2Cl2 и высушивали над MgSO4. После отгонки растворителя получали 31,3 г маслянистой жидкости, которая отверждалась. Часть этого твердого вещества (3 г) растворяли в EtOH (75 мл), подкисляли полученный раствор этанольным раствором НСl и добавляли к нему этиловый эфир (75 мл), в результате чего из раствора выпадал осадок продукта в виде соли (2,65 г), т. пл. 224-225оС.
Рассчитано, %: С 59,70; Н 6,01; N 4,64
С15Н18ClF2NO
Найдено, %: С 59,52; Н 5,87; N 4,55
с. 3-(2,4-Дифторбензоил)-8-азабицикло[3.2.1]-октан-8-карбонитрил
Бромциан (4,0 г) в один прием добавляли к суспензии (2,4-дифтоторфенил)8-метил-8-азабицикло[3.2.1] окстан-3-ил/-метанона (5,0 г) и карбоната калия (5,2 г) в 80 мл диметилформамида при комнатной температуре. Смесь перемешивали в течение 2 ч и затем разбавляли водой и EtOAc. Слои разделяли и водную фазу подвергали экстракции EtOAc. Объединенные органические слои промывали водой и рассолом, высушивали над MgSO4, фильтровали и концентрировали, получая в результате 3,5 г твердого вещества, которое перекристаллизовывали из смеси EtOAc и гептана. Температура плавления 162-164оС.
Рассчитано, %: С 65,21; Н 5,11; N 10,14
С15Н14F2N2O
Найдено, %: С 65,16; Н 5,10; N 10,08
d. 3-[4-Фтор-2(фенилметилтио)бензил]-8-азабицикло[3.2.1]октан-8-карбонитрил
Бензилмеркаптан (0,67 г) в 5 мл ТГФ добавляли по каплям к раствору третбутоксида калия (0,6 г) в 15 мл ТГФ. Образующуюся суспензию перемешивали при температуре окружающей среды в течение 5 мин, после чего добавляли к ней (2,4-дифторбензoил)-8-азабицикло[3.2.1] октан-8-карбонитрил (1,5 г). Реакционную смесь перемешивали в течение получаса при комнатной температуре, затем добавляли к ней насыщенный раствор хлорида аммония и образующийся продукт экстрагировали EtOAc. Объединенные органические слои промывали рассолом, высушивали над MgSO4, фильтровали и концентрировали, получая в результате 1,7 г сырого продукта. После перекристаллизации из метанола получали 0,87 г продукта в виде твердого вещества с т.пл. 166-167оС.
Рассчитано, %: С 69,45; Н 5,56; N 7,36
C22H21FN2OS
Найдено, %: С 69,37; Н 5,69; N 7,15.
е. 3-(6-Фтор-1,2-бензизотиазол-3-ил)-8-азабицикло[3.2.1] октан-8-карбонитрил
Сульфурилхлорид (5,33 г) добавляли по каплям при комнатной температуре к суспензии 3-[4-фтор-2-(фенилметилтио)бензоил] -8-азабицикло-[3.2.1] октан-8-карбонитрила (15 г) в 160 мл дихлорэтана. Образующийся раствор перемешивали в течение 1,25 ч, после чего растворитель отгоняли в вакууме и остаток суспендировали в 160 мл ТГФ и обрабатывали при комнатной температуре 160 мл этанола, насыщенного аммиаком. Образующуюся смесь перемешивали в течение 1,5 ч, после чего растворитель удаляли в вакууме и остаток распределяли между водой и CH2Cl2. Объединенные органические слои промывали рассолом, высушивали над MgSO4, фильтровали и упаривали, получая в результате 10,7 г твердого вещества, которое фильтровали через силикагель (элюирование СН2Сl2). Твердое вещество, полученное после упаривания фильтрата (6,82 г) перекристаллизовывли из EtOAc, получая 4,1 г белого твердого вещества с т. пл. 193-195оС.
Рассчитано, %: С 62,70; Н 4,91; N 14,62
С15Н14FN3S
Найдено, %: С 62,63; Н 4,90; N 14,73
Следует отметить, что возможны изменения и различные варианты в рамках объема изобретения, определяемого прилагаемой формулой изобретения.
Использование: в качестве промежуточных продуктов для получения фармацевтически активных соединений. Сущность изобретения: продукт 1 - 6-фтор-1,2-бензоизотиазолы ф-лы 1, где R - водород, низший алкил или группа ф-лы 2 или 3, где R1CHO или -CN. Реагент 1: соединение ф-лы 4, где R указано выше; R3 -бензил Реагент 2: галогенирующий агент, лучше сульфурилхлорид. Реагенты 1 и 2 при взаимодействии дают соединение ф-лы 5, которое подвергают взаимодействию с аммиаком, лучше при комнатной температуре. Соединение ф-лы 4 получают из соединения ф-лы 6 и бензилмеркаптана в среде апротонного органического растворителя. 3с. и 2 з.п. ф-лы. Структура формул 1, 2, 3, 4, 5 и 6 (см. рис.).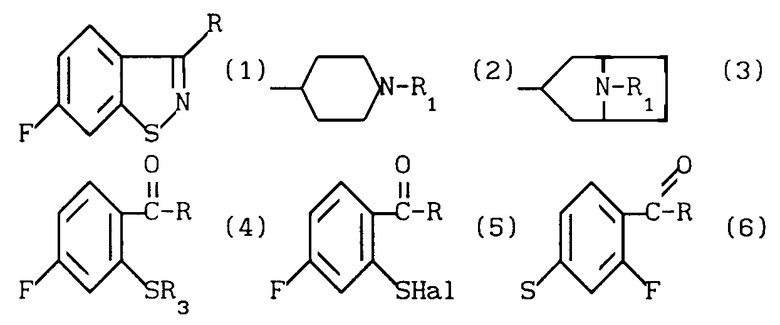
СПОСОБ ПОЛУЧЕНИЯ 6-ФТОР-1,2-БЕНЗИЗОТИАЗОЛОВ, ОРТОЗАМЕЩЕННОЕ ФЕНАЦИЛОВОЕ ПРОИЗВОДНОЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ.
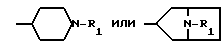


| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| US N 4458076, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Колосниковая решетка с чередующимися неподвижными и движущимися возвратно-поступательно колосниками | 1917 |
|
SU1984A1 |

