Изобретение относится к соединениям формулы I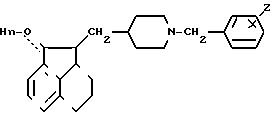 (I) где Z водород или галоген, а n 0 или 1. Эти соединения пригодны для облегчения различных дисфункций памяти, например болезни Альцгеймера.
(I) где Z водород или галоген, а n 0 или 1. Эти соединения пригодны для облегчения различных дисфункций памяти, например болезни Альцгеймера.
Если не указано особо, в описании и приложенной формуле изобретения будут применяться следующие определения.
Термин галоген будет означать фтор, хлор, бром или йод.
В описании данная формула или название будут заключать в себе все стерео- и таутомерные изомеры в тех случаях, где такие изомеры существуют, пунктирные линии будут означать возможные связи.
Соединения настоящего изобретения получают согласно следующей схеме синтеза.
В описании синтеза Z будет иметь приведенные значения, если не указано особо.
Соединение формулы VII дают реагировать с соединением формулы VIII, получая соединение формулы I, где n 1. Эту реакцию обычно осуществляют при помощи соответствующего основания, например карбоната калия или гидроксида калия, предпочтительно измельченного карбоната калия в соответствующем растворителе, например 2-бутаноне, при температуре 0-50оС, предпочтительно 10-40оС.
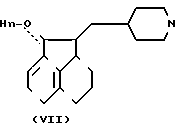
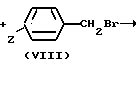
Hn-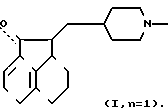 CH
CH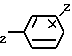
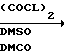
Соединению I дают реагировать с соответствующим окисляющим средством, например смесью хлорангидрида щавелевой кислоты и диметилсульфоксида, получая соединение формулы I, где n 0. Эту реакцию обычно осуществляют в соответствующем растворителе, например хлористом метилене или хлористом метилене/диметилсульфоксиле при температуре от -80 до -20оС, предпочтительно от -70 до -40оС.
 CH
CH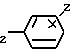
Соединения формулы I изобретения пригодны для лечения различных дисфункций памяти, например, болезни Альцгеймера. Активность в облегчении подобных дисфункций памяти проявляется благодаря способности этих соединений ингибировать фермент ацетилхолинэстеразы и повышать, тем самым, содержание холина в головном мозге.
Анализ на ингибирование холинэстеразы.
Холинэстеразы обнаруживают во всем теле, как в головном мозге, так и в сыворотке. Однако только распределение ацетилхолинэстеразы головного мозга (AChE) коррелируется с центральной холинергической иннервацией. Полагают, что та же самая иннервация ослабляется у больных болезнью Альцгеймера. Определили in vitro ингибирование активности ацетилхолинэстеразы в неостриатуме крысы.
in vitro ингибирование активности ацетилхолинэстеразы в неостриатуме крысы.
Ацетилхолинэстеразу (AChE), которую иногда называют истинной или особой холинэстеразой, обнаруживают в нервных клетках, скелетной мышце, гладкой мышце, различных железах и красных кровяных клетках AChE можно отличить от других холинэстераз по специфичности субстрата и ингибитора и по региональному распределению. Ее распределение в головном мозге приблизительно коррелируется с холинергической иннервацией, а субфракционирование показывает самое высокое содержание в нервных окончаниях.
Является общепринятым, что физиологическая роль AChE заключается в быстром гидролизе и инактивации ацетилхолина. Ингибиторы AChE оказывают значительное холиномиметическое действие в холинергически-иннервированных эффекторных органах и использовались терапевтически при лечении глаукомы, миастении и паралитической непроходимости кишечника. Однако недавние исследования навели на мысль, что ингибиторы AChE могут также благоприятствовать лечению болезни Альцгеймера.
Известный способ использовали в изобретении для анализирования активности холинэстеразы. Он является модификацией способа Ellman et al. Biochem. Pharmacol, 7,88, 1961.
Методика.
А. Реагенты.
1. Фосфатный буфер, 0,05 моль/л рН 7,2.
(а) 6,85 г NaH2PO4 ˙H2O/100 мл дистиллированной Н2О,
(b) 13,40 г Na2HPO4 ˙7H2O/100 мл дистиллированной Н2О,
(с) добавляют (а) к (b) до тех пор, пока рН не достигнет 7,2,
(d) разбавляют 1:10.
2. Субстрат в буфере.
(а) 198 мг ацетилтиохолинхлорида (10 ммоль/л),
(b) доводят до 100 мл 0,05 моль/л фосфатным буфером, рН 7,2 (реагент 1).
3. ДТНВ в буфере.
(а) 19,8 мг 5,5-дитиобиснитробензойной кислоты (ДТНВ) (0,5 ммоль/л),
(b) доводят до 100 мл 0,05 моль/л фосфатным буфером, рН 7,2 (реагент 1).
4. Раствор штамма испытуемого лекарства 2 ммоль/л готовят в соответствующем растворителе и доводят до объема посредством 0,5 ммоль/л ДТНВ (реагент 3). Лекарства последовательно разбавляют (1:10) таким образом, что конечная концентрация (в кювете) составляет 10-4 моль/л и проверяют на активность. При наличии активности определяют величины IC50на основании ингибирующей активности при последовательных концентрациях.
В. Приготовление тканей.
Wistar крыс мужского пола обезглавливали, быстро удаляли головной мозг, освобождали, иссекая неостриатумы, взвешивали и гомогенизировали в 19 объемах (приблизительно 7 мг белка/мл) фосфатного буфера 0,05 моль/л, рН 7,2, используя гомогенизатор Potter-Elvehjem 25 мкл, аликвоту гомогенного раствора добавляли к 1,0 мл разбавителя или различным концентрациям испытываемого лекарства и предварительно инкубировали в течение 10 мин при 37оС.
С. Анализ.
Ферментную активность измеряют при помощи спектрофотометра Beckman DU-50. Этот способ можно использовать для определения IC50 и для измерения кинетических констант.
Настройка измерительного прибора.
Программный модуль кинетики 598273(10) Программа 6. Кинетические данные. Источник vis. Длина волны 412 нм. Дозатор отсутствует. Кюветы 2 мл кюветы с использованием авто-6 пробоотборника.
Пустые 1 для каждой концентрации субстрата.
Время паузы 15 с (15 или 30 с для кинетики). Суммарное время 5 мин (5 или 10 мин для кинетики). Диаграмма да. Диапазон автошкала, Наклон возрастающий. Результаты да (дает наклон). Фактор-1.
Реагенты добавляют в пустую кювету и кювету для образца следующим образом: Пустая 0,8 мл фосфатного буфера (ДТНВ)
0,8 мл буфера/cубстрат Контрольная 0,8 мл фосфатного буфера (ДТНВ) фермент
0,8 мл фосфатного буфера/субстрат Лекарство 0,8 мл фосфатного буфера (ДТНВ/лекарство/фермент
0,8 мл фосфатного буфера/субстрат
Для каждого опыта определяют значения в пустых кюветах для контроля за неферментным гидролизом субстрата, и эти значения автоматически вычитаются программой кинетических данных на программный модуль кинетики. Эта программа также рассчитывает скорость изменения оптической плотности для каждой кюветы.
Для определения IC50:
Концентрация субстрата составляет 10 ммоль в анализе, субстрат разбавляют 1: 2, получая конечную концентрацию, равную 5 ммоль. Концентрация ДТНВ составляет 0,5 ммоль, давая конечную концентрацию 0,25 ммоль.
Ингибирования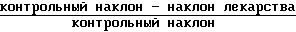 x100
x100
Результаты этого анализа для некоторых соединений изобретения и физостигмина (контрольное соединение) представлены в табл. 1.
Эта полезность дополнительно демонстрируется способностью этих соединений восстанавливать холинергически недостаточную память в нижеописанном анализе на избегание темноты.
Анализ на избежание темноты.
В этом анализе мышей испытывают на их способность запоминать неприятный раздражитель на промежуток времени, равный 24 ч. Мышь помещают в камеру, имеющую темное отделение, сильный свет от ламп накаливания загоняет ее в темное отделение, в котором посредством металлических пластин на полу осуществляют электрический удар. Животное удаляют из испытательного прибора и спустя 24 ч снова испытывают на способность запоминать электрический удар.
Если скополамин антихолинергическое средство, которое, как известно, вызывает недостаточность памяти, применяют до начального подвергания животного действию испытательной камеры, животное повторно входит в темное отделение после смещения в испытательную камеру 24 ч спустя. Это действие скополамина блокируется активным испытуемым соединением, приводя к большему промежутку времени до повторного входа в темное отделение.
Результаты для активного соединения выражаются в виде процента от группы животных, у которых действие скополамина блокируется, что проявляется в увеличенном промежутке времени между помещением в испытательную камеру и повторным входом в темное отделение.
Результаты этого анализа для некоторых соединений изобретения и результаты для такрина и пилокарпина (контрольные соединения) представлены в табл. 2.
Эффективные количества соединений изобретения могут применяться к больному посредством любого из различных способов, например перрорально в виде капсул или таблеток, парентерально в виде стерильных растворов или суспензий и в некоторых случаях внутривенно в виде стерильных растворов. Конечные продукты, представляющие собой свободные основания, будучи эффективными сами по себе, могут приготавливаться и применяться в виде их фармацевтически приемлемых солей, образованных присоединением кислоты, для целей стабильности, удобства кристаллизации, повышенной растворимости и т.п.
Кислоты, пригодные для приготовления фармацевтически приемлемых солей, образованных присоединением кислоты изобретения, включают неорганические кислоты, например соляную, бромистоводородную, серную, азотную, фосфорную и хлорную кислоты, а также органические кислоты, например винную, лимонную, уксусную, янтарную, малеиновую, фумаровую, 2-нафталинсульфоновую и щавелевую кислоты.
Активные соединения изобретения могут применяться перрорально, например, с инертным разбавителем или со съедобным носителем, или они могут быть заключены в желатиновые капсулы, или они могут быть спрессованы в таблетки. Для целей перрорального терапевтического применения активные соединения изобретения могут быть объединены с наполнителями и использоваться в виде таблеток, пастилок, капсул, эликсиров, суспензий, сиропов, облаток, жевательной резинки и т. п. Эти препараты должны содержать, по крайней мере, 0,5% активных соединений, но могут быть изменены, в зависимости от конкретной формы и могут удобным образом составлять от 4 до около 70% массы единицы лекарственной формы. Количество активного соединения в подобных композициях таково, что будет получена соответствующая доза. Предпочтительные композиции и препараты в соответствии с изобретением приготовляют таким образом, что единичная перроральная лекарственная форма содержит между 1,0-300 мг активного соединения.
Таблетки, пилюли, капсулы, пастилки и т.п. могут также содержать следующие компоненты: связующее вещество, например микрокристаллическую целлюлозу, трагант или желатин, наполнитель, например крахмал или лактозу, расщепляющее средство, например альгиновую кислоту. Примогель, кукурузный крахмал и т.п. смазывающее вещество, например стеарат магния или Sterotex, вещество, увеличивающее скольжение, например коллоидный диоксид кремния, и подсластитель, например могут быть добавлены сахароза или сахарин или средство, улучшающее вкус и запах лекарственного препарата (корригент), например вода перечной мяты, метилсалицилат или апельсиновая отдушка. Когда единичной лекарственной формой является капсула, она может содержать, кроме веществ указанного типа, жидкий носитель, например жирное масло. Другие единичные лекарственные формы могут содержать другие различные вещества, модифицирующие физическую форму лекарственной единицы, например, в качестве покрытий. Таким образом, таблетки или пилюли могут быть покрыты сахаром, шеллаком или другими желудочными покрывающими средствами. Сироп может содержать, кроме активных веществ, сахарозу, в качестве подсластителя, и некоторые консерванты, красители и корригенты. Вещества, используемые в приготовлении различных композиций, должны быть фармацевтически чистыми и нетоксичными в используемых количествах.
Для цели парентерального терапевтического применения активные соединения изобретения могут быть включены в раствор или суспензию. Эти препараты должны содержать, по крайней мере, 0,1% активного соединения, но это количество может изменяться между от 0,5 до около 30% от массы препарата. Количество активного соединения в таких композициях таково, что будет получена соответствующая доза. Предпочтительные композиции и препараты в соответствии с изобретением готовят таким образом, что парентеральная лекарственная форма содержит между 0,5-100 мг активного соединения.
Растворы или суспензии могут также включать следующие компоненты: стерильный разбавитель, например воду, для инъекций, соляной раствор, нелетучие масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители, антибактериальные средства, например бензиловый спирт или метилпарааминобензойные кислоты, антиоксиданты, например аскорбиновую кислоту или бисульфит натрия, хелатирующие средства, например этилендиаминтетрауксусную кислоту, буферы, например ацетаты, цитраты или фосфаты, и средства для регулирования тонуса, например хлорид натрия или виноградный сахар. Парентеральный препарат может быть заключен в одноразовые шприцы или флаконы с многократной дозой, сделанные из стекла или пластика.
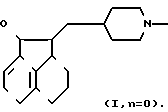
Примеры соединений изобретения включают: 2-[(N-Бензилпиперидин-4-ил/метил]-1,2,2a,3,4,5-гексагидроаценафтилен-1-ол,
2-[[N-(4-фторбензил)пиперидин-4-ил] метил]-1,2,2a,3,4,5- гексагидроаценафтилен-1-ол,
2-[(N-Бензилпиперидин-4-ил)метил] 2a,3,4,5-тетрагидро-1 (2Н)-аценафтилен-1-он,
2-[[N-(3-Фторбензил)пиперидин-4-ил] метил] -1,2,2a,3,4,5- гексагидроаценафтилен-1-ол,
2-[[N-(3-Фторбензил)пиперидин-4-ил] метил] -2a,3,4,5-тетрагидро-1(2Н)- аценафтилен-1-он,
2-[[N-Бензилпиперидин-4-ил/метил] -6- метокси-2а, 3,4,5-тетрагидро-1 (2Н)-аценафтилен-1-он,
2-[[N-(3-Фторбензил/пиперидин-4-ил] метил]-6-метокси-2а,3,4,5-тетрагидро- 1(2Н)-аценафтилен-1-он, 2-[(N-Бензилпиперидин-4-ил)метил]-6,7-диметокси-2а,3,4,5-тетрагидро-1(2Н)- аценафтилен-1-он,
2-[[N-(3-Фторбензил)пиперидин-4-ил] метил]-6,7-диметокси-2а,3,4,5-тетрагидро- 1(2Н)-аценафтилен-1-он, 2-[[N-(Бензилпиперидин-4-ил]метил]-6,7-диэтокси-2а, 3,4,5-тетрагидро-1(2Н)- аценафтилен-1-он, 2-[N-Бензилпиперидин-4-ил]метил]-6,7-диметокси-1(2Н)-аценафтилен-1-он,
2-[[N-(3-Нитробензил)пиперидин-4-ил] метил]-2a,3,4,5-тетрагидро-1-(2Н)- аценафтилен-1-он и
2-[(N-Бензилпиперидин-4-ил)метил] -6,7- метилендиокси-2а, 3,4,5-тетрагидро- 1(2Н)-аценафтилен-1-он.
П р и м е р 1. 7,8-Диметокси-2а,3,4,5-тетрагидро-1(2Н)-аценафтилен-1-он.
К перемешиваемому раствору 135 г полифосфорной кислоты при 90оС добавляли 18,9 г 6,7-диметокси-1,2,3,4-тетрагидро-1-нафталин уксусной кислоты. Смесь перемешивали 4-5 мин при 85-90оС, причем источник тепла удаляли на первые несколько минут, следующие за добавлением. К перемешиваемой смеси затем добавляли дополнительные 100 г полифосфорной кислоты при 80оС. Смесь перемешивали при 75-85оС в течение еще 5 мин, давали охладиться до 50оС и обрабатывали 350 г ледяной воды. Смесь перемешивали в течение 20 мин, охлаждали до 20-25оС, дважды экстрагировали эфиром, промывали последовательно водой, дважды 5% -ным гидроксидом натрия, водой, 3%-ной уксусной кислотой, бикарбонатом натрия и водой. Получающийся раствор сушили, фильтровали и концентрировали до масла, которое кристаллизовалось при стоянии. Вещество растворяли в небольшом объеме этилацетата и фильтровали через силикагель в тонком слое, элюируя смесью 15% этилацетат-гексан, с получением 12,6 г 7,8-диметокси-2а,3,4,5-тетрагидро-1(2Н)-аценафтилен-1-она, Т.пл. 80-84оС.
Вычислено для С14Н16О3, 72,39 C; 6,94 H
Найдено, 72,57 C; 6,93 H.
П р и м е р 2. 7,8-Диметокси-1(2Н)-аценафтилен-1-он.
Смесь 3,0 г 7,8-диметокси-2а,3,4,5-тетрагидро-1(2Н)-аценафтилен-1-она, 3 г ДДХ (2,3-дихлор-5,6-дициано-1,4-бензохинон) и 24 мл азеотропно высушенного бензола перемешивали при 75-85оС в течение 45 мин и давали охладиться до комнатной температуры. К смеси добавляли дополнительные 3 г ДДХ. Смесь перемешивали при 75-85оС в течение 1,25 ч, давали охладиться до комнатной температуры, разбавляли бензолом, и твердый осадок промывали бензолом. Фильтрат подвергали тонкослойной хроматографии на силикагеле, элюируя хлористым метиленом с получением 1,36 г 7,8-диметокси-1(2Н)-аценафтилен-1-она. Вещество дополнительно очищали тонкослойной хроматографией на силикагеле, элюируя смесью 25% этилацетат-гексан, с получением 1,17 г аналитически чистого твердого вещества, Т.пл. 81-83оС.
Вычислено для С14Н12О3, 73,67 C; 5,30 H
Найдено, 73,64 C; 5,33 H
П р и м е р 3. транс-7,8-Диметокси-2-[(пиридин-4-ил)метиленил]-2a, 3,4,5-тетрагидро-1(2Н)- аценафтилен-1-он.
К раствору 12,1 г литий бис-(триметилсилил)амида в 200 мл сухого ТГФ при от -65 до -70оС по каплям добавляли раствор 16,8 г 7,8-диметокси-2а,3,4,5-тетрагидро-1(2Н)-аценафтилен-1-она в 325 мл сухого ТГФ. Раствор перемешивали при от -70 до -75оС в течение одного часа. К раствору по каплям добавляли раствор 6,9 мл (7,73 г) пиридин-4-карбоксальдегида в 300 мл сухого ТГФ. Смесь перемешивали 1,5 ч при от -70 до -75оС и затем 4 ч при 0оС. Раствор выливали в ледяную воду, дважды экстрагировали этилацетатом, и органический экстракт последовательно промывали водой и насыщенным NaCl, после чего сушили Na2SO4-, фильтровали и концентрировали до масла. Тонкослойная хроматография на силикагеле, элюирование смесями 60% 70% и 100% этилацетат-гексан, давали после испарения растворителя 1,8 г чистого транс-7,8-диметокси-2-[(пиридин-4-ил)метиленил]-2a,3,4,5-тетрагидро-1(2Н)- аценафтилен-1-она, помимо 4,95 г транс-вещества, загрязненного небольшим количеством цис-соединения. Суммарный выход: 6,75 г
Вычислено для С20Н19NO3, 74,75 C; 5,96 H; 4,36 N.
Найдено, 74,76 C; 5,90 H; 4,34 N.
П р и м е р 4. 2-[(N-Бензилпиперидин-4-ил)метил]-1,2,2a,3,4,5-гексагидроаценафтилен-1-ол.
Смесь 4,08 г 2-[(пиридин-4-ил)метиленил]-2a,3,4,5-тетрагидро-1(2Н)-аценафтилен-1-она, растворенного в 64 мл уксусной кислоты и 0,81 г PtO2 (смоченного уксусной кислотой) встряхивали в среде Н2 (10 до 20 пси) при комнатной температуре в течение 24 ч. Смесь фильтровали через целит, и фильтрат концентрировали при высоком вакууме. Масло затем растворяли в 120 мл 2-бутанона. К раствору добавляли 12,0 г измельченного КОН, а затем 1,4 мл бензилбромида. Реакционную смесь перемешивали в течение 0,5 ч при комнатной температуре, затем фильтровали и концентрировали. Вещество очищали тонкослойной хроматографией, применяя смесь 20% ацетон-гексан. Фракции, содержащие продукт, соединяли и концентрировали. Остаток перекристаллизовывали из циклогексана и сушили при 110оС в течение 3 ч, получая 1,6 г 2-[(N-бензилпиперидин-4-ил)метил] -1,2,2a,3,4,5-гексагидро- аценафтилен-1-ола, Т.пл. 145-147оС.
Вычислено для С25Н32NO, 83,06 C; 8,64 H; 3,87 N.
Найдено, 82,96 C; 8,63 H; 3,92 N.
П р и м е р 5. 2-[[N-(4-Фторбензил)пиперидин-4-ил]метил]-1,2,2a,3,4,5- гексагидроаценафтилен-1-ол гидрохлорид моногидрат.
Смесь 1,25 г оксида платины (IV) 6,27 г 2-[(пиридин-4-ил)метиленил]-2a, 3,4,5-тетрагидро-1(2Н)-аценафтилен-1-она и 100 мл уксусной кислоты встряхивали в атмосфере Н2 (15 пси) в течение 24 ч. Катализатор удаляли фильтрованием, и фильтрат концентрировали при высоком вакууме. Остаточное масло растворяли в 187 мл 2-бутанона. К раствору добавляли 17 г измельченного карбоната калия, а затем 2,37 мл 4-фторбензилбромида. Реакционную смесь перемешивали при комнатной температуре в среде N2 в течение 24 ч. Реакционную смесь фильтровали, осадок промывали 2-бутаноном, и фильтрат концентрировали. Остаточное масло очищали посредством ГПЖХ, элюируя смесью 20% ацетон-гексан. Фракции, содержащие продукт, соединяли и концентрировали. Остаток растворяли в метаноле и добавляли эфирную HCl до тех пор, пока раствор не становился кислым. Растворитель удаляли в вакууме, а остаток растирали в порошок с эфиром и сушили при высоком вакууме сначала при комнатной температуре, а затем при 110оС в течение 24 ч, получая 2,1 г 2-[[N-(4-фторбензил)пиперидин-4-ил] метил] -1,2,2a,3,4,5- гексагидроаценафтилен-1-ол гидрохлорид моногидрата, Т.пл. 138-148оС.
Вычислено для С25Н33ClFNO2, 69,18 c; 7,60 H; 3,23 N.
Найдено, 69,20 C; 7,28 H; 3,23 N.
П р и м е р 6. 2-[(N-Бензилпиперидин-4-ил)метил]-2a,3,4,5-тетрагидро-1(2Н)-аценафтилен-1-он гидрохлорид.
К перемешиваемому раствору 0,76 мл хлорангидрида щавелевой кислоты в 20 мл ДХМ (хлористый метилен) при от -60 до -70оС по каплям добавляли раствор 1,30 мл ДМСО (диметилсульфоксид) в 10 мл ДХМ. Раствор перемешивали при от -60 до -70оС течение 4 мин. К раствору добавляли смесь 2,78 г 2-[(N-бензилпиперидин-4-ил)метил] -1,2,2a,3,4,5-гексагидроаценафтилен-1-ола 10 мл ДХМ и 8 мл ДМСО. Получающуюся смесь перемешивали в течение 15 мин при от -70 до -60оС. К раствору затем добавляли 5,6 мл триэтиламина, и затем смесь перемешивали 5 мин при от -70 до -60оС, давали нагреться до комнатной температуры, разбавляли ДХМ, последовательно промывали холодным карбонатом натрия, водой и рассолом, сушили (сульфат натрия) и концентрировали в масло. Масло затем растворяли в эфире, промывали водой, и водные промывки экстрагировали эфиром. Смешанные эфирные экстракты дважды промывали водой и рассолом, сушили (NaSO4) и концентрировали в масло. Масло очищали посредством тонкослойной хроматографии на силикагеле, элюируя смесью 40% этилацетат-гексан, а затем 50% этилацетат-гексан. Фракции, содержащие чистый продукт, соединяли и концентрировали, получая масло, которое растворяли в эфире и осаждали добавлением эфирного хлористого водорода. Фильтрование с последующим высушиванием при 113оС (2 мм) давало 1,72 г аналитически чистого 2-[(N-бензилпиперидин-4-ил)метил]-2a,3,4,5-тетрагидро-1(2Н)-аценафтилен-1-он гидрохлорида. Перекристаллизация из этанола давала бесцветные пластинки, Т.пл. 245-251оС.
Вычислено для С25Н30ClNO, 75,83 C; 7,64 H; 3,54 N.
Найдено, 76,01 C; 7,54 H; 3,51 N.
П р и м е р 7. 2-[[N-(3-Фторбензил)пиперидин-4-ил]метил]-1,2,2a,3,4,5-гексагидро- аценафтилен-1-ол гидрохлорид. Суспензию 5,03 г 2-[(пиридин-4-ил)метиленил-2а,3,4,5-тетрагидро-1(2Н)-аценафтилен-1-она, 80 мл уксусной кислоты и 1,0 г PtO2 встряхивали при 20 пси в атмосфере Н2 в течение 30 мин, 20-10 пси в течение 1 ч и 10 пси в течение 12 ч. Суспензию фильтровали, и фильтрат концентрировали в масло, которое затем дополнительно сушили перегонкой в азеотропной смеси с толуолом. Остаток растворяли в 150 мл 2-бутанона, к которому добавляли 15 г измельченного карбоната калия, а затем 1,9 мл 95% мета-фторбензилбромида. Суспензию перемешивали в течение 3 ч при комнатной температуре, фильтровали и промывали 2-бутаноном. Фильтрат подвергали тонкослойной хроматографии на силикагеле, элюируя смесью 40% этилацетат-гексан, а затем 50% этилацетат-гексан. Фракции, содержащие чистый продукт, соединяли и концентрировали, получая более твердое вещество (3,22 г). Вещество перекристаллизовывали из циклогексана, сушили, растворяли в эфире и осаждали в виде белого твердого вещества посредством эфирной HCl. Твердое вещество выделяли фильтрованием и сушили при 110оС в течение 1,5 ч, получая аналитический чистый 2-[[N-(3-фторбензил)пиперидин-4]-ил]метил-1,2,2а,3,4,5- гексагидроаце- нафтилен-1-ол гидрохлорид, Т.пл. 118-168оС.
Вычислено для С25Н31ClFNO, 72,19 C; 7,51 H; 3,37 N.
Найдено, 71,98 C; 7,54 H; 3,33 N.
П р и м е р 8. 2-[[N-(3-Фторбензил)пиперидин-4-ил]метил]-2a,3,4,5-тетрагидро-1(2Н)- аценафтилен-1-он гидрохлорид.
К перемешиваемому раствору 0,96 мл хлорангидрида щавелевой кислоты в 20 мл хлористого метилена при от -50 до -60оС по каплям добавляли раствор 1,63 мл ДМСО и 0,96 мл хлористого метилена. Раствор перемешивали в течение 4 мин, после чего по каплям добавляли раствор 3,70 г 2-[[N-(3-фторбензил)пиперидин-4-ил] метил] -1,2,2a,3,4,5- гексагидроаценафтилен-1-ола, растворенного в 13 мл хлористого метилена и 10 мл ДМСО. Реакционную смесь перемешивали в течение дополнительных 15 мин, после чего добавляли 7,10 мл Et3N. Реакционную смесь перемешивали в течение 5 мин. Реакционную смесь затем выливали в смесь хлористого метилена и Na2CO3, экстрагировали хлористым метиленом, последовательно промывали водой и рассолом, сушили с Na2SO4, фильтровали и концентрировали. Остаток очищали посредством ГПЖХ, элюируя смесь 30% EtOAc-гексан. Фракции, содержащие продукт, соединяли и концентрировали. Остаток растворяли в метаноле и добавляли эфирную HCl до тех пор, пока раствор не становился кислым. Растворитель удаляли в вакууме, а остаток растирали в порошок с эфиром и сушили в высоком вакууме при комнатной температуре и затем при 110оС в течение 2 ч, получая 2,2 г 2-[[N-(3-фторбензил)-пиперидин-4-ил] метил] -2a, 3,4,5-тетрагидро-1(2Н)- аценафтилен-1-он гидрохлорида, Т.пл. 220-222оС.
Вычислено для С25Н29СlFNO3, 72,54 C; 7,06 H; 3,38 N.
Найдено, 72,39 C; 7,09 H; 3,36 N.

Сущность изобретения: соединения ф-лы 1, где X-водород, низший алкил, низший алкокси, гидрокси или нитро, Y водород или низший алкокси, или же X и Y, соединенные между собой, образуют группу -OCH О-, в этом случае положения X и Y в половине бензольного кольца должны быть примыкающими друг к другу, Z водород, или галоген и их кислотно-аддитивные соли, а n 0 или 1, эти соединения обладают активностью по ингибированию ацетилхолинэстеразы и пригодны для облегчения различных дисфункций памяти, характеризуемых холинергической недостаточностью, например, болезнью Альцгеймера. Структура соединения ф-лы I: 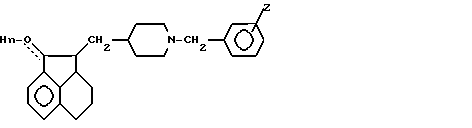 2 с.п. ф-лы, 6 з.п. ф-лы, 2 табл.
2 с.п. ф-лы, 6 з.п. ф-лы, 2 табл.
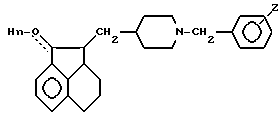
где Z водород или галоген;
n 1 или 0,
или их кислотно-аддитивные соли.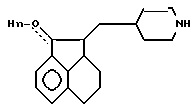
где n 1,
с соединением формулы III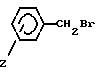
где Z имеет указанные значения,
для получения соединения формулы I, где n 1, Z имеет указанное значение,
и, необязательно, окисляют полученное соединение с образованием соединения формулы 1, в котором n 0.
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Патент США N 4246268, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
