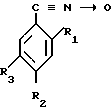
Изобретение относится к химии высокомолекулярных соединений, в частности к производным ароматических нитрилов ди-N-оксидам динитрилов диалкилбензолов формулы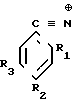

 где R1,2=C2H5; R3=C ≡
где R1,2=C2H5; R3=C ≡  _→
_→ 
R1,2=CH3; R3=C ≡  _→
_→ 
R1,3=C2H5; R2=C ≡  _→
_→ 
R1,3=CH3; R2=C ≡  _→
_→  низкотемпературным отвердителям каучуков с малой непредельностью.
низкотемпературным отвердителям каучуков с малой непредельностью.
Изобретение относится также к способу получения указанных веществ.
Изобретение может быть использовано при производстве эластичных каучуков с широким спектром эксплуатационных характеристик.
Одним из важных показателей физико-химических свойств и химического состава известных видов натуральных и синтетических каучуков, наряду с плотностью, температурой стеклования, средневязкостной массой, содержанием примесей и другими, является показатель процентного содержания непредельных связей. По этому параметру каучуки можно условно разделить на три группы: 1-я группа каучуки, имеющие высокий (до 48 мас. >С=С< связей) процент непредельности, например, натуральные каучуки (Смокед-шитс), изопреновые (СКИ-3), бутадиеновые (СКБ) и др.
2-я группа каучуки, обладающие малой непредельностью (0,2-1,0 мас. >С=С< связей), например, бутилкаучуки (БК-0845ТД, БК-2045Н) и др.
3-я группа каучуки с полностью насыщенной основной цепью, к которым можно отнести полиизобутилены (П-155), эпихлоргидриновые каучуки (СКЭХГ-200), фторкаучуки (СКФ-260) и др. [1]
Только на основе каучуков последних двух групп были получены эластичные полимеры, отвечающие современным требованиям по отношению к окислителям (озон, кислород), действию тепла, с высокой газонепроницаемостью и стойкостью к набуханию в различных маслах.
Для получения эластомеров, как с указанными выше, так и другими специальными свойствами, в процессе вулканизации каучуков применяют разнообразные химические добавки (ингредиенты). К их числу можно отнести вулканизирующие вещества, ускорители, активаторы, замедлители вулканизации, противостарители, наполнители и др. Особую роль в этом ряду занимают вулканизирующие вещества, или отвердители полимеров, так как они, в основном, определяют технологические параметры структурирования полимеров, в том числе температурные режимы.
Одним из недостатком известных отвердителей каучуков, например, серы, органических перекисей является необходимость применения в процессе отверждения повышенных температур, как правило, превышающих 100оС [2] Это приводит к существенным энергозатратам при переработке каучуков с малой непредельностью, так как объемы их производства составляют десятки тысяч тонн в год.
Наиболее старый, но широкоприменяемый до сих пор вулканизатор сера для отверждения каучука требует затрат большого количества энергии, так как только при высокой температуре (выше 150оС) происходит радикальный распад восьмичленного цикла серы, т. е. ее активация, с последующим очень медленным химическим структурированием каучука. Сложность этого процесса возрастает при получении вулканизатов на основе каучуков с малой непредельностью, так как экспериментально установлено, что даже незначительное снижение непредельности с 2% до 0,8% (в от непредельности натуральных каучуков) снижает скорость сшивки почти в четыре раза [3] Применение повышенных температур (в соответствии с аррениусовской зависимостью) не всегда дает положительных результатов из-за увеличения вклада побочных реакций окисления и пиролиза.
Поскольку вулканизация каучука только элементарной серой является весьма длительным высокотемпературным процессом для его интенсификации используют ускорители вулканизации, в том числе, различного вида карбаматы (МН, ЭН, МЦ, БЦ, ЭФЦ, ДМЦ, ПМП, ЭЭА), тиурамы (ММ, Д, Е, МТ), каптакс, цинкапт, альтакс, сульфенамиды (Ц, 2Ц, М, ТМ, ДТМ, БТ, Ф) и ряд других, а для полимеров с малой непредельностью суперускорители [4] Действие подавляющего числа перечисленных выше ускорителей сводится, главным образом, к сокращению времени процесса вулканизации и не меняет его температурные параметры.
Так, ультраускоритель вулканизации резиновых смесей пентаметилендитиокарбамат пиперидиния, например Карбамат ПМП (СССР), Naugatex 144 (US), Pip-Pip (GB), Vulkacit (Ger), позволяет проводить вулканизацию при температуре не ниже 100-140оС.
Для вулканизации каучуков различной природы широкое применение получили органические перекиси: перекись ди-трет-бутила (Пербут), перекись дикумила (Перкум), перекись бензоила (Пербенил). Так, для вулканизации каучуков с небольшой непредельностью, например, кремнийорганических: (СКТВ) достаточно эффективна перекись ди-трет-бутила. Полученные вулканизаты обладают высокой твердостью и прочностью на разрыв. Однако во всех случаях температура вулканизации лежит в интервале 120-170оС, причем процесс ведут в течение длительного времени под давлением, так как температура ее кипения (110оС) ниже температуры ее распада для начала структурирования [1]
Известно также, что для вулканизации непредельных каучуков, а особенно каучуков с малой непредельностью (бутилкаучук, СКЭПТ), используют также фенолформальдегидные смолы, например, n-трет-Бутилфенолформальдегидную смолу (Фенофор Б), n-трет-Октилфенолформальдегидную смолу (Фенофор О), бромметилированную n-трет-бутилфенолформальдегидную смолу (Фенофор ББ). Но и в этом варианте отверждения необходимым условием проведения процесса является температура 140-180оС. Причем по данным работы [1] сложно получить хорошие характеристики вулканизатов при непредельности исходного каучука ниже 1,8% в том числе из-за низкой скорости сшивки и длительности процесса.
Известна низкотемпературная система отверждения каучуков на основе полухлористой серы. Однако при переработке каучуков в присутствии полухлористой серы уже в первые 1-2 мин наблюдается "скорчинг" смеси, что делает их непригодными для дальнейшего использования с целью формования, прессования и не позволяет получать равномерно сшитые эластомеры. Кроме этого, по данным работы [5] низкотемпературное отверждение допустимо только в отношении натуральных, т.е. имеющих максимальный процент непредельности каучуков, причем существенным недостатком этой системы отверждения является то, что в процессе происходит выделение соляной кислоты. Специфика отверждения полухлористой серой заключается также в том, что на ее основе можно получать лишь тонкостенные изделия из-за действия в парах или в бензиновом растворе.
К числу вулканизирующих агентов относятся производные хинонов, например, n-хинондиоксим (ПХДО), превращающийся при действии окислителей, например оксидов свинца (IУ), в n-динитрозобензол, который и обеспечивает поперечное сшивание при 80-100оС [5] Известно, что эти агенты являются взрывоопасными компонентами и повышают склонность смесей к подвулканизации, и сейчас вытесняются фенолформальдегидными смолами. Следует отметить высокую токсичность как свинца, так и n-динитрозобензола [6]
Существует возможность использования при отверждении каучуков с малой непредельностью хиноловых эфиров продуктов взаимодействия n-бензохинондиоксима с 2,4,6-сильнозатрудненными фенолами. Именно при применении таких вулканизирующих агентов было достигнуто существенное улучшение как технологических, так и эксплуатационных свойств резин на основе малонепредельного бутилкаучука [7] Несмотря на удовлетворительные результаты по свойствам конечных эластомеров и в этом варианте отверждения необходимо использовать повышенные температуры для процесса структурирования в интервале 100-143оС [8]
Низкотемпературные отвердители каучуков с малой непредельностью (до 1,0 мас.) неизвестны.
Не обнаружено сведений о ди-N-оксидах 1,5-динитрил-2,4-диэтилбензола, 1,5-динитрил-2,4-диметилбензола, 1,4-динитрил-2,5-диэтилбензола, 1,4-динитрил-2,5-диметилбензола и их специфических свойствах.
Известен способ получения производных ароматических нитрилов полифункциональных N-оксидов (терефтало-бис-нитрил-N-оксида, окси-бис-4-бензоилкарбонитрил-N-оксида, адипобискарбонитрил-N-оксида) [9] Так, в суспензию 19,8 г оксима терефталевого альдегида в 357 г эфира пропускают NOCl при 0оС до исчезновения зеленой окраски. После удаления эфира и перекристаллизации получают 5,9 г терефтало-бис-оксимоилхлорида с т.пл. 177,5-179оС. К раствору 5 г последнего в 66 г метилового спирта прибавляют при 0оС в течение 1 ч раствор 4,2 г триэтиламина в 1,2 г метилового спирта. Твердый продукт промывают дважды метиловым спиртом и получают 3,17 г терефтало-бис-нитрил-N-оксида. Выход целевого продукта в расчете на исходный оксим составляет около 19,0 мас. (от теор. ). Следует отметить, что исходный оксим требует отдельного синтеза, что, безусловно, означает дальнейшее снижение выхода конечного соединения. Низкий выход N-оксидов и абсолютные условия получения являются препятствием на пути использования известного способа синтеза производных ароматических нитрилов на практике.
Полученные известным способом полифункциональные N-оксиды были испытаны в качестве низкотемпературных (25оС) отвердителей цис-1,4-полибутадиена, каучука с высокой непредельностью (48 мас.  C=C
C=C связей). Действие отвердителей не распространяется на каучуки с малой непредельностью. Кроме того, область потенциального использования этих отвердителей ограничена тем, что процесс отверждения протекает в присутствии растворителя (бензола) и поэтому проведение структурирования в "массе" невозможно.
связей). Действие отвердителей не распространяется на каучуки с малой непредельностью. Кроме того, область потенциального использования этих отвердителей ограничена тем, что процесс отверждения протекает в присутствии растворителя (бензола) и поэтому проведение структурирования в "массе" невозможно.
Известен также способ получения производных ароматических нитрилов-полифункциональных N-оксидов из ароматических альдегидов производных мезитилена, дурола и 1,3,5-триэтилбензола [10]
Синтез N-оксидов включает стадии оксимирования диальдегидов и последующего окисления диоксимов в целевой продукт.
Так, 2,4,6-триэтилизофталодинитрилоксид получен по следующей схеме:
H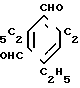 H5
H5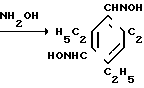 H5
H5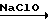
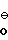

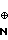 H
H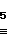 C
C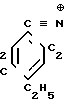 H
H
К раствору 0,1 моля диальдегида в 300-400 мл этилового спирта прибавляли последовательно при 50оС растворы 0,4 моля NaOH в 20 мл H2О и 0,4 моля NH2OHxHCl в 50 мл Н2О. Реакционную массу кипятили 4 ч. растворитель отгоняли, остаток обрабатывали 50-100 мл СHCl3 в 100 мл Н2О, отделяли органический слой и промывали его водой (3х200 мл). После отгонки СНСl3 остаток перекристаллизовывали из гексана. Т.пл. 122-123,5оС. Выход 74% (от теор.).
К энергично перемешиваемому и охлажденному до 5-7оС водному раствору 0,5 моля 10-15% гипохлорита натрия, содержащему 8-10% NaOH, за 5-10 мин прибавляли раствор 0,1 моля диоксима в 150-250 мл СН2Сl3. Смесь энергично перемешивали 1 ч при постепенном повышении температуры до 15оС, органический слой отделяли, а водный экстрагировали СН2Cl3 (2х50 мл). Объединенный органический слой промывали водой (4х100 мл) и после отгонки растворителя в вакууме остаток перекристаллизовывали из гексана. Т.пл. 93-96оС. Выход диоксида 75% (от теор.). (В расчете на оксим или около 38% в расчете на промежуточный (дихлорметил)-формил триэтилбензол [10, 11]).
Исходные соединения для этого синтеза диальдегиды получали формилированием в абсолютных условиях соответствующих алкилбензолов (дихлорметил)-метиловым эфиром в присутствии AlCl3 или TiCl4 [11]
В результате реализации известного способа получены N-оксиды, способные вступать в реакцию 1,3-диполярного циклоприсоединения с веществами, содержащими кратные связи, т.е. потенциально способные выступать в качестве отвердителей каучуков. Однако в анализируемом источнике [10] отсутствуют какие-либо конкретные сведения об отвердителях на основе полученных N-оксидов.
Недостатками известного способа являются многостадийность получения целевого продукта, что снижает его выход в расчете на исходные мезитилен, дурол, 1,3,5-триэтилбензол и, в конечном счете, делает его бесперспективным с практической точки зрения. Кроме того, реагенты синтеза труднодоступны и непрактичны с экологической точки зрения. Продукты, получаемые на каждой стадии, требуют тщательной очистки.
Цель изобретения уменьшение температуры процесса вулканизации и, следовательно, снижение уровня энергозатрат при производстве каучуков с малой непредельностью.
Сущность изобретения состоит в том, что предложены новые вещества ди-N-оксиды динитрилов диалкилбензолов формулы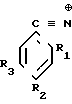

 где R1,2=C2H5; R3=C ≡
где R1,2=C2H5; R3=C ≡  _→
_→ 
R1,3=CH3; R2=C ≡  _→
_→ 
R1,3=C2H5; R2=C  N
N 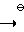 O
O
R1,3= CH3; R2=C ≡  _→
_→  в качестве низкотемпературных отвердителей каучуков с малой непредельностью и специально разработанный способ их получения, который реализуется следующей совокупностью существенных признаков:
в качестве низкотемпературных отвердителей каучуков с малой непредельностью и специально разработанный способ их получения, который реализуется следующей совокупностью существенных признаков:
на первой стадии синтеза проводят реакцию селективного бис-хлорметилирования исходного диалкилбензола в кислой среде, содержащей формальдегид, соляную кислоту, серную кислоту, уксусную кислоту, взятых в мольных соотношениях диалкилбензол: формальдегид соляная кислота серная кислота уксусная кислота 1,0 (2,5-3,2) (2,2-3,2) (6,0-14,0) (2,0-4,5) при 70-85оС в течение 2-5 ч;
в качестве диалкилбензола используют 2,4-диэтилбензол, 2,4-диметилбензол, 2,5-диэтилбензол, 2.5-диметилбензол;
на второй стадии последовательно обрабатывают полученный бис(хлорметил)-диалкилбензол 10-80% -ным водным раствором гексаметилентетрамина, взятого в мольном соотношении бис(хлорметил)-диалкилбензол гексаметилентетрамин 1: (2-4) в течение 1-2 ч при температуре кипения и 10-80%-ной водной уксусной кислотой в течение 2-5 ч при нагревании до кипения, с последующей экстракцией продуктов реакции органическим растворителем из ряда хлоралканов и обработкой полученного экстракта гидроксиламином, взятом в мольном соотношении бис(хлорметил)-диалкилбензол гидроксиламин 1:(2-3) при рН 6-8 в течение 0,5-1 ч при 25-50оС, добавлением водной натриевой щелочи для получения рН 12-14, отделением органического растворителя с последующей нейтрализацией водного раствора 6-7 и выделением соответствующего диоксима диальдегида диалкилбензола;
на третьей стадии проводят окислительное хлорирование диоксима 5-15%-ным водным раствором гипохлорита натрия в течение 0,5-1 ч при рН 6-10 и температуре (-10)-(+10)оС с получением целевого соединения.
П р и м е р 1. К 33,7 г (30,9 мл; 392 ммоль) 35%-ного водного формалина при комнатной температуре и перемешивании прибавляют последовательно 40,9 г (34,9 мл; 392 ммоль) 36,5%-ной соляной кислоты и 16,5 г (15,7 мл; 275 ммоль) концентрированной уксусной кислоты. Далее к полученной кислотной смеси при указанной температуре приливают 17,6 г (20,4 мл; 131 ммоль) 2,4-диэтилбензола и в течение 25-30 мин дозируют 90,2 г (50 мл; 920 ммоль) 94,5%-ной серной кислоты (соотношения реагентов приведены в табл.2). Нагревают реакционную смесь до 70оС и выдерживают при перемешивании в течение 3 ч. После окончания выдержки продукты реакции охлаждают до 15-25оС, выпавшие кристаллы 1,5-бис(хлорметил)-2,4-диэтилбензола отфильтровывают, промывают на фильтре водой (2х100 мл), 3% -ным раствором бикарбоната натрия (2х50 мл) до рН промывных вод 6-7, водой (2х100 мл) и высушивают до постоянной массы. Получают 29,7 г (98% от теор. ) бис-хлорметильного производного 2,4-диэтилбензола, который используется без дополнительной обработки для последующего синтеза. Для того, чтобы охарактеризовать полученное соединение кристаллизацией из спирта получен образец с т.пл. 63-66оС.
Вычислено, С 62,33; Н 6,93; Cl 30,74.
Найдено, С 62,54; 62,26; Н 7,02; 7,12; Cl 30,46; 30,93;
Мол.м. (масс-спектрометрически): 231.
ИК-спектр содержит полосы поглощения, характерные для хлорметильной группы, см-1: С-Cl 1250; 650.
Спектр ПМР (хлороформ -d3);
Триплет 1,07; 1,14; 1,22 (6Н, СН3);
Квартет 2,48; 2,56; 2,62; 2,70 (4Н, СН2);
Синглеты 4,38 (4Н, СН2Cl);
6,88; 7,08 (2Н, ароматические).
К раствору 25,2 г (180 ммоль) гексаметилентетрамина в 20 мл воды прибавляют 13,9 г (60 ммоль) 1,5-бис(хлорметил)-2,4-диэтилбензола (соотношения реагентов приведены в табл. 3). Реакционную массу нагревают до 95-100оС и выдерживают в течение 1 ч, а затем прибавляют 45 мл концентрированной уксусной кислоты и выдерживают при 95-100оС в течение 3 ч. Из реакционной массы экстрагируют органические продукты четыреххлористым углеродом (2х35 мл), промывают экстракт 100 мл воды. К промытому экстракту приливают 30 мл воды. При перемешивании добавляют 10,5 г (80 ммоль) гидроксиламина гидрохлорида. Нейтрализуют смесь раствором 4 г (100 моль) NaОН в 10 мл воды до рН 7-9 и выдерживают при 25-35оС в течение 30 мин. Далее прибавляют раствор 14 г (350 ммоль) NaOH в 40 мл воды (рН > 12) и делают выдержку 30 мин при 25-35оС. Отделяют органический слой. Водный слой охлаждают до 20оС и нейтрализуют соляной кислотой до рН 7. Выделившийся осадок диоксима 1,5-диальдегид-2,4-диэтилбензола отфильтровывают, промывают на фильтре водой (2х50 мл) и высушивают на воздухе до постоянной массы. Получают 7,5 г (57%) диоксима 1,5-диальдегид-2,4-диэтилбензола с т. пл. 156-160оС. Без дополнительной очистки диоксим может быть использован для получения N-оксида.
Вычислено, С 65,45; Н 7,27; N 12,73.
Найдено, С 65,32; 65,21; Н 7,53; 7,70; N 12,38; 12,65.
Мол.м. (масс-спектрометрически): 220.
ИК-спектр содержит полосы поглощения, характерные для оксимной группы, см-1: 3300 (ОН); 1610 (С=N); 950, 970 (N-ОН).
Спектр ПМР (хлороформ -d3):
Триплет 1,07; 1,14; 1,22 (6Н, СН3);
Квартет 2,58; 2,66; 2,73; 2,81 (4Н, СН2);
Синглеты 6,93 (2Н, СНN);
7,86; 8,16 (2Н, ароматические).
К 60 мл 100% -ного водного раствора гипохлорита натрия (81 ммоль) при перемешивании прибавляют концентрированную соляную кислоту до рН 7-9, а затем 7,9 г (39 ммоль) мелкоизмельченного диоксима 1,5-диальдегид-2,4-диэтилбензола при (-10)-0оС в течение 40 мин. Реакционную массу выдерживают при этой температуре 35 мин. Выпавший кристаллический продукт отфильтровывают, промывают на воронке водой (3х50 мл) и высушивают на воздухе до постоянного веса. Получают 7 г (выход 90%) ди-N-оксида 1,5-динитрил-2,4-диэтилбензола. Тн.и.р. 85-89оС (по данным дериватографии).
Вычислено, С 66,67; Н 5,56; N 12,96.
Найдено, С 66,78; 66,85; Н 5,42; 5,39; N 13,05; 13,10.
Мол.м. (масс-спектрометрически): 216.
ИК-спектр содержит полосы, характерные для нитрилоксидной группы, см-1: 2310, 1340.
Спектр ПМР (хлороформ -d3):
Триплет 1,07; 1,14; 1,22 (6Н, СН3);
Квартет 2,66; 2,72; 2,87; 2,90 (4Н, СН2);
Синглеты 7,96; 8,86 (2Н, ароматические).
Ди-N-оксиды 1,5-динитрил-2,4-диметилбензола, 1,4-динитрил-2,5-диэтилбензола, 1,4-динитрил-2,5-диметилбензола получены аналогично примеру.
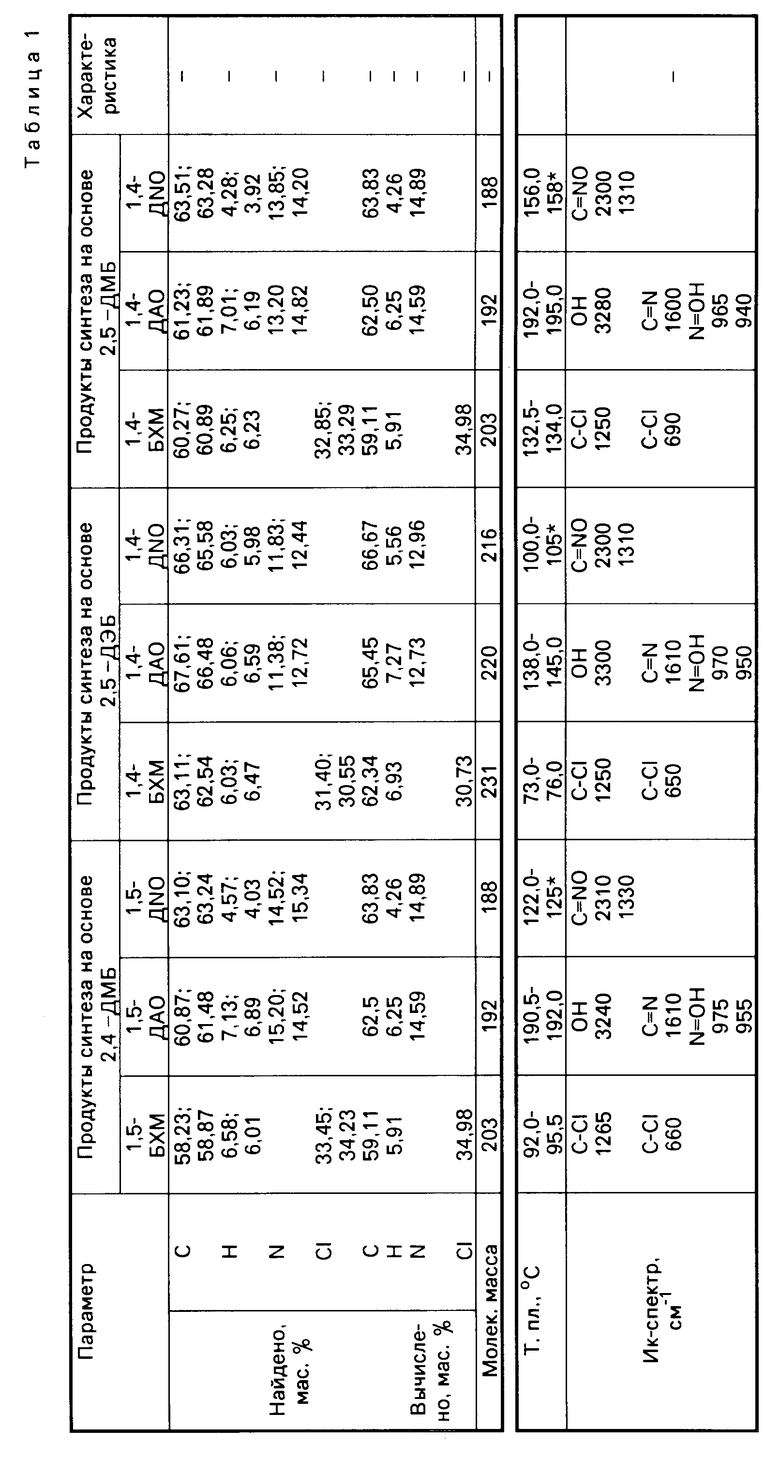
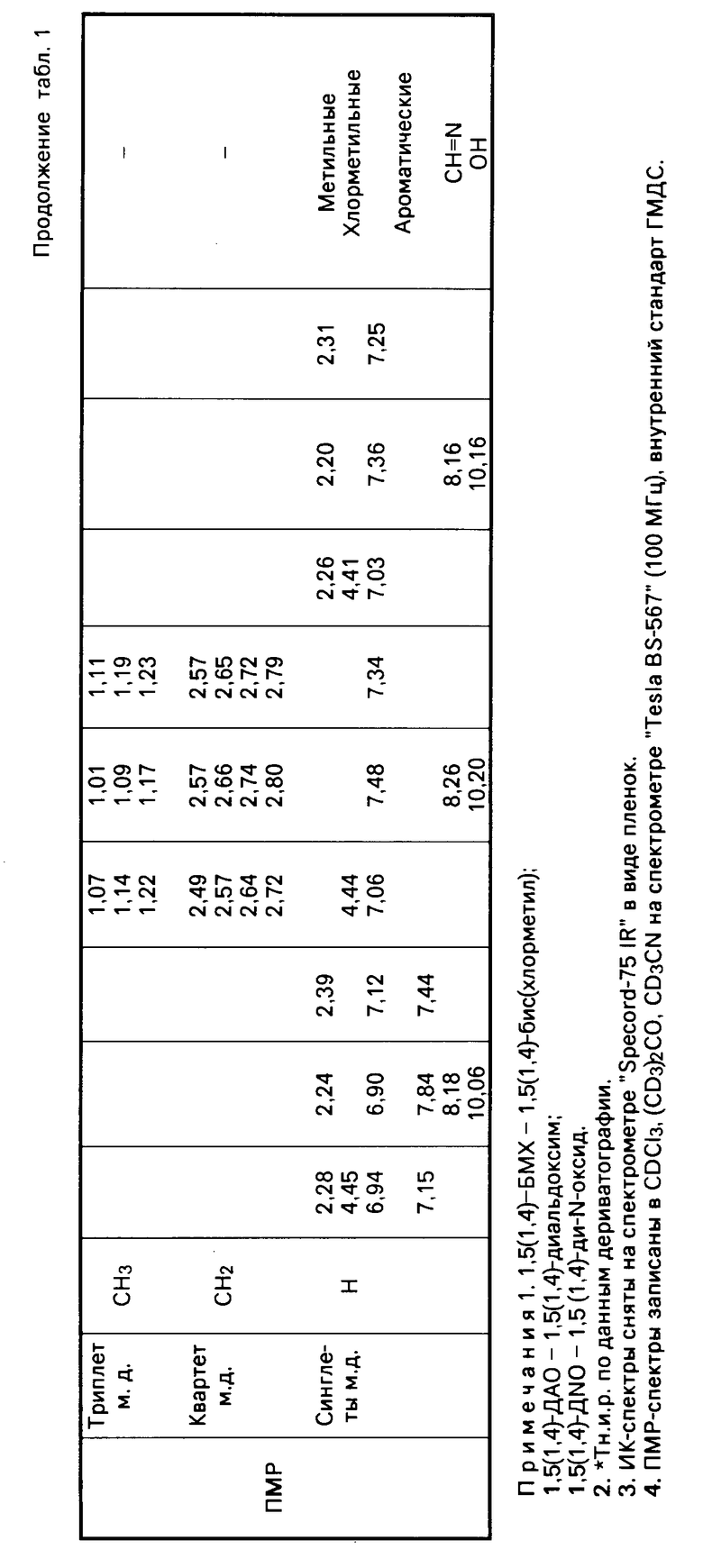
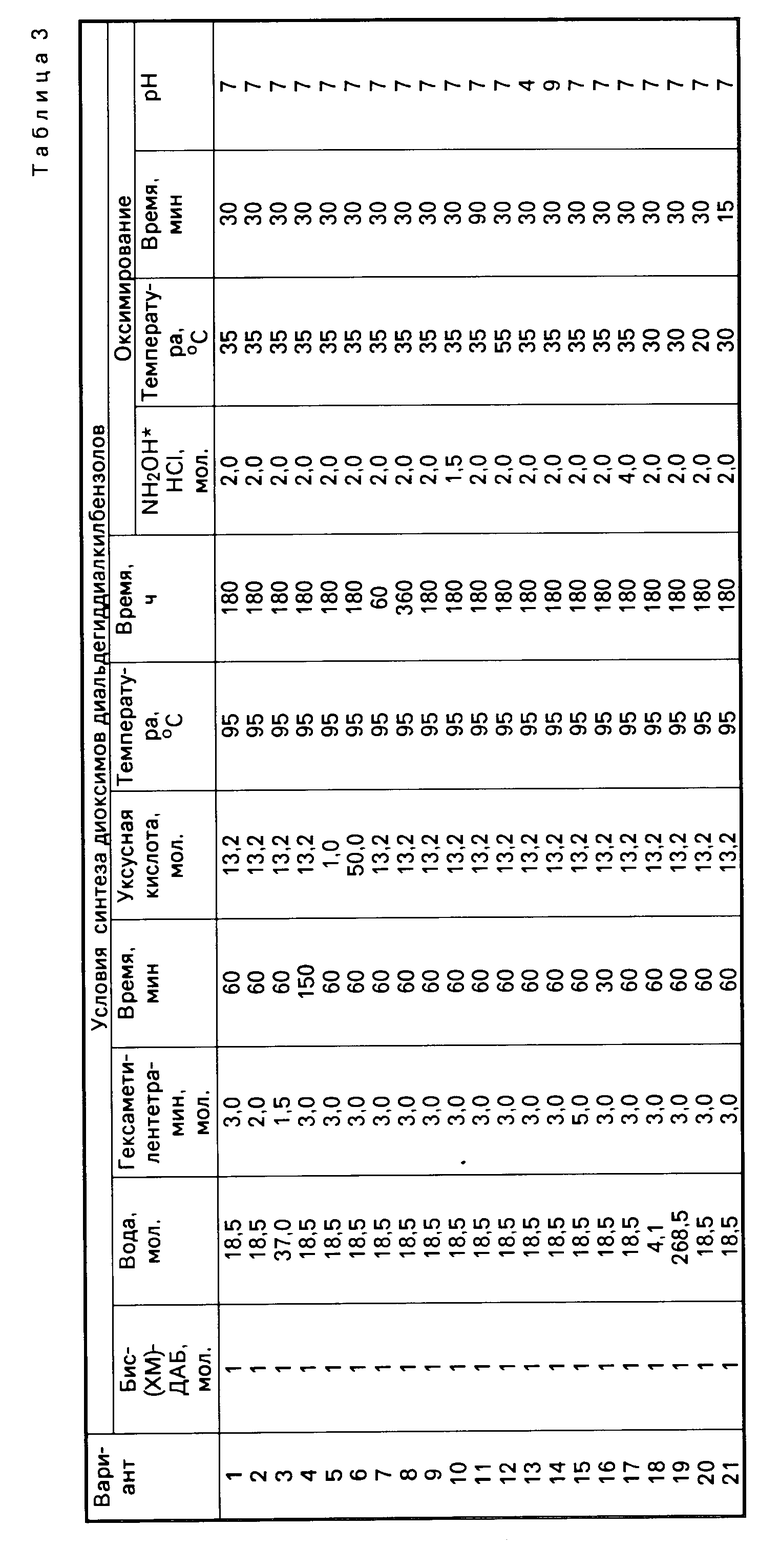
Свойства указанных динитрилоксидов сведены в табл. 1, которая характеризует физико-химические свойства промежуточных и конечных продуктов синтеза.
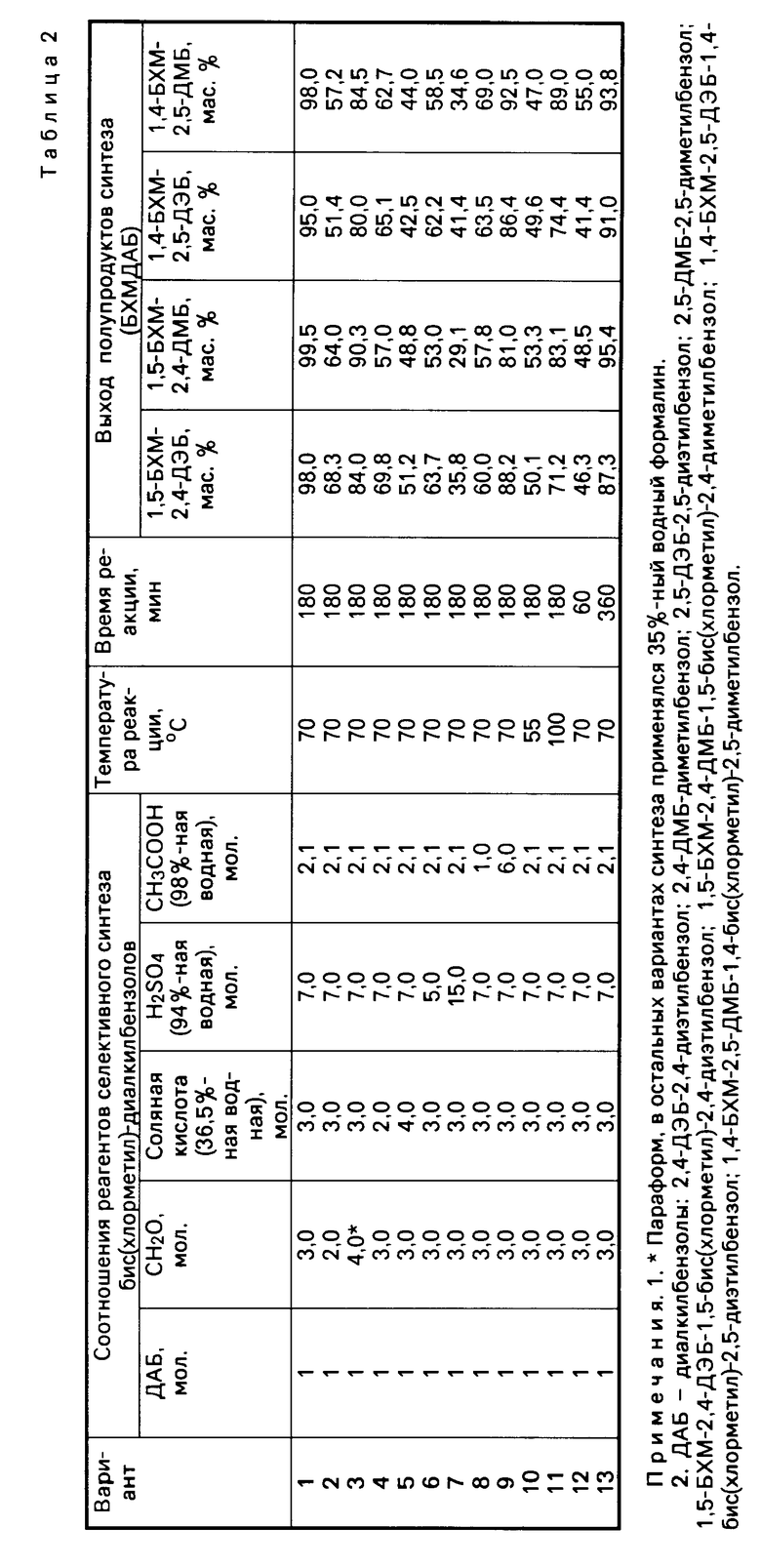
Табл.2 иллюстрирует влияние отдельных параметров синтеза на выход промежуточных бис(хлорметил)-диалкилбензолов.
Как видно из данных табл.2, отклонение от указанных в формуле изобретения оптимальных параметров селективного бис-хлорметилирования приводит к снижению выхода промежуточных бис(хлорметил)-диалкилбензолов, что подтверждается, например, результатами, полученными как при снижении, так и при увеличении мольного соотношения ДАБ: формалин:HCl:H2SO4:CH3COOH (см. варианты 2,3; 4,5; 6,7; 8,9).
Табл.3 иллюстрирует влияние отдельных параметров синтеза на выход промежуточных диоксимов диальдегиддиалкилбензолов.
Как видно из данных табл.3, отклонение от оптимальных параметров второй стадии процесса приводит к снижению выхода промежуточных диоксимов диальдегиддиалкилбензолов.
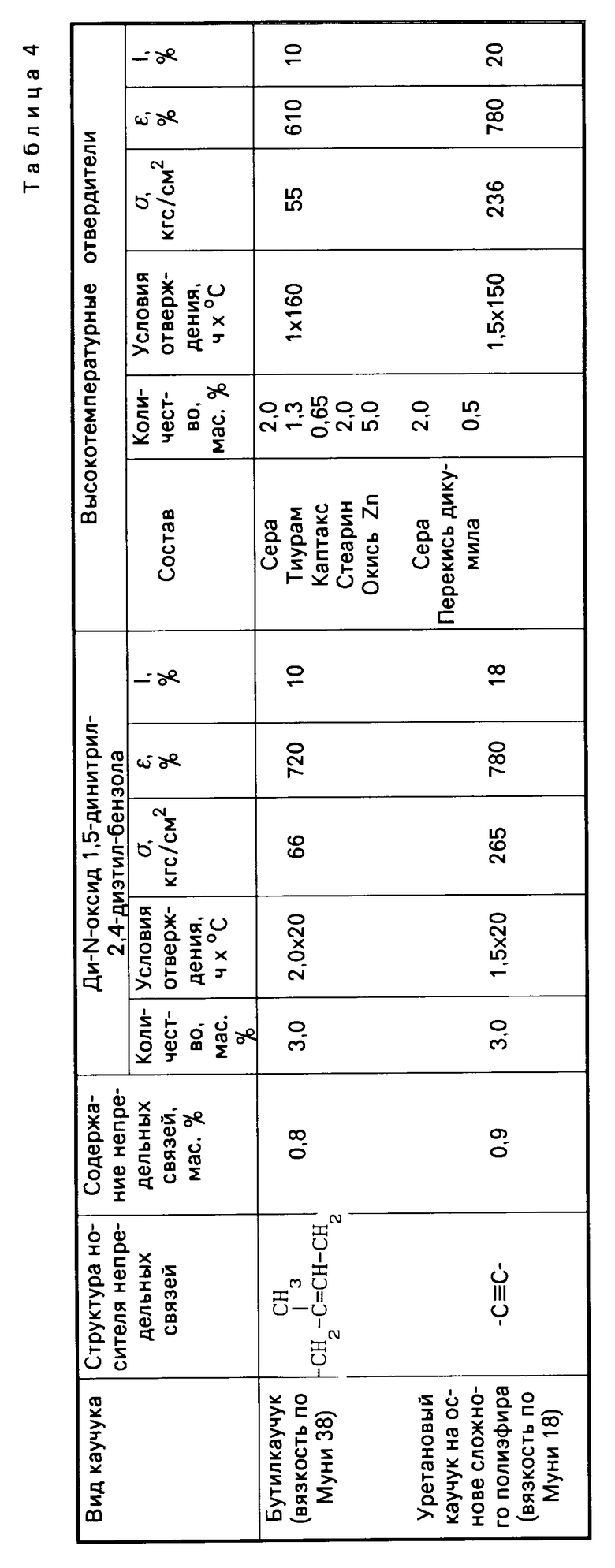
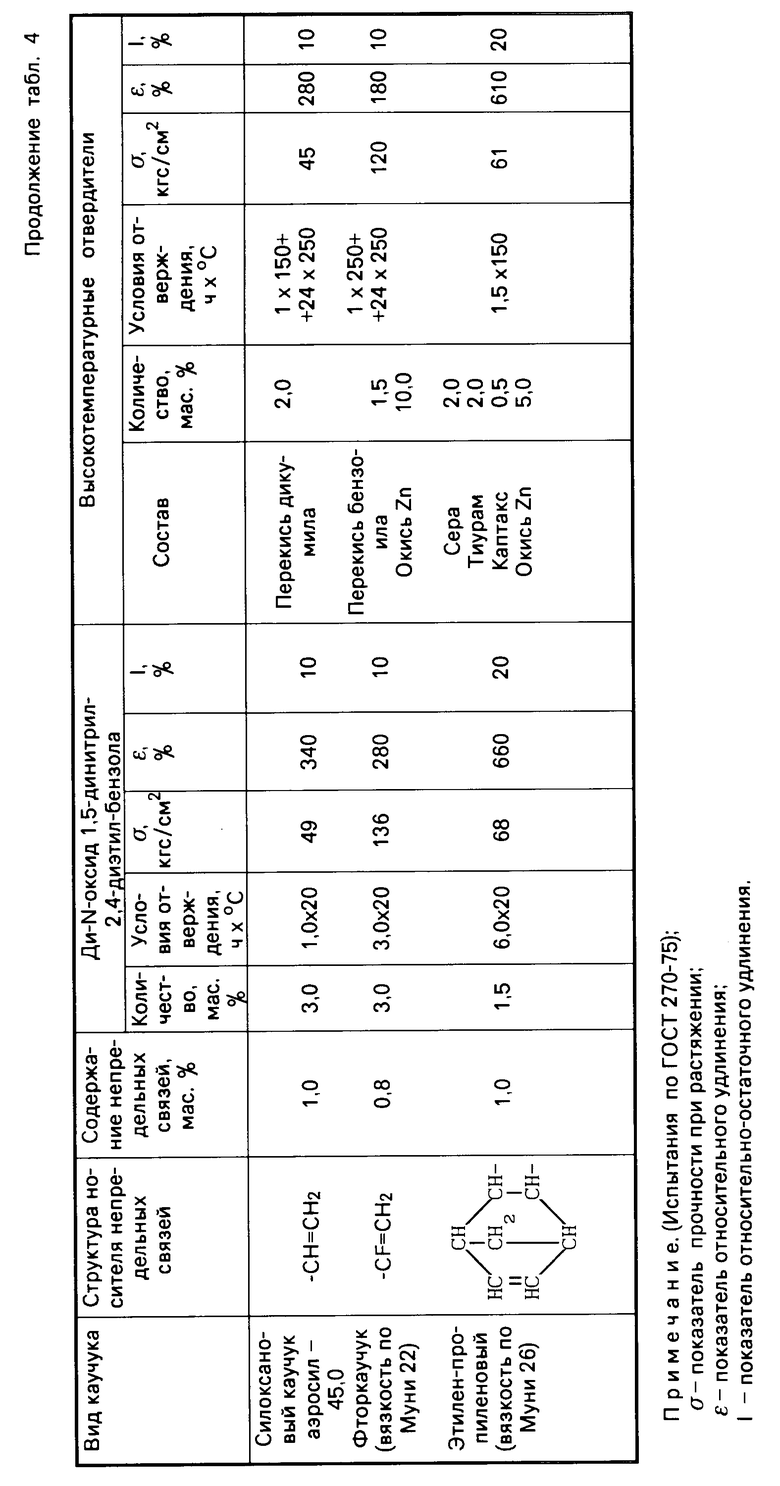
Табл. 4 иллюстрирует результаты сравнительных испытаний отверждающего действия ди-N-оксида 1,5-динитрил-2,4-диэтилбензола и традиционных систем отверждения по отношению к различным типам каучуков с малой непредельностью
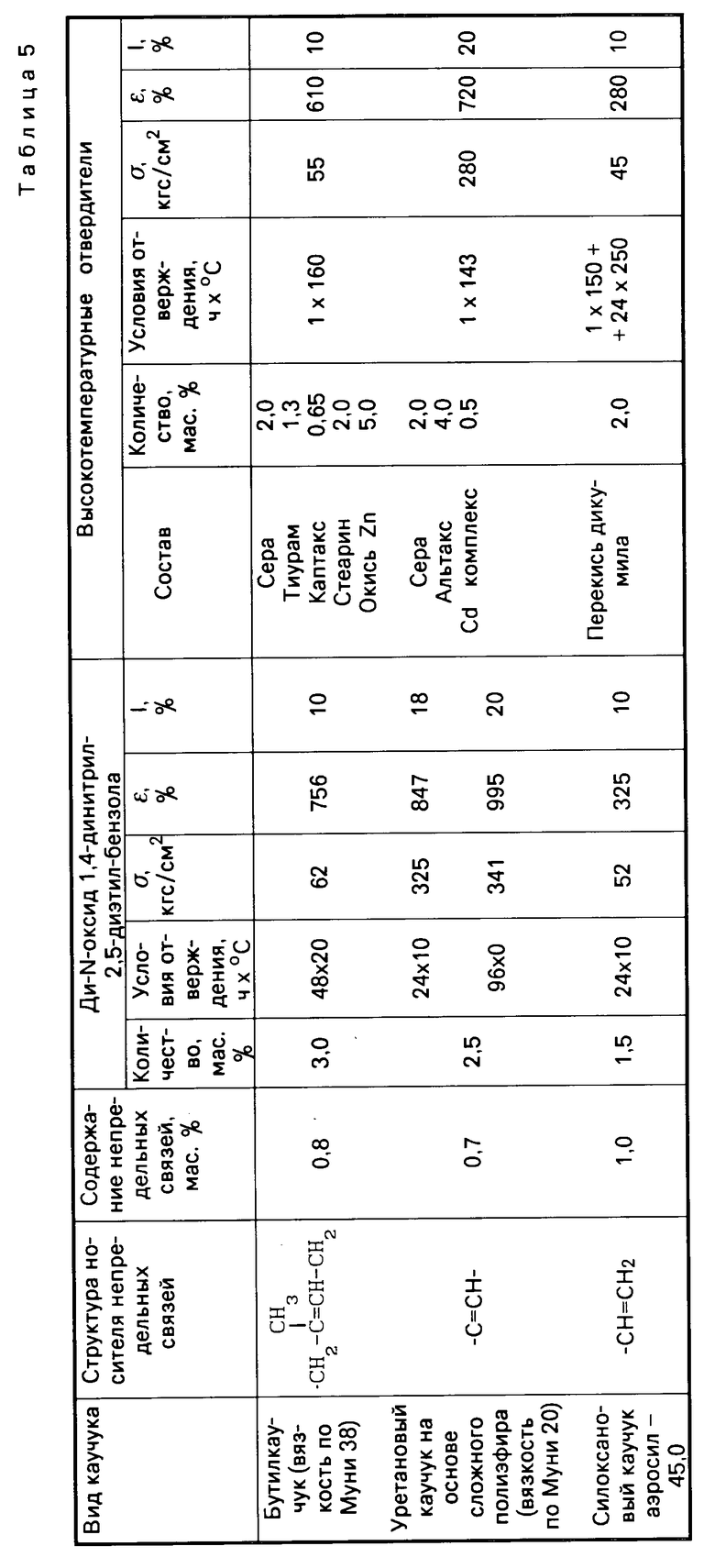
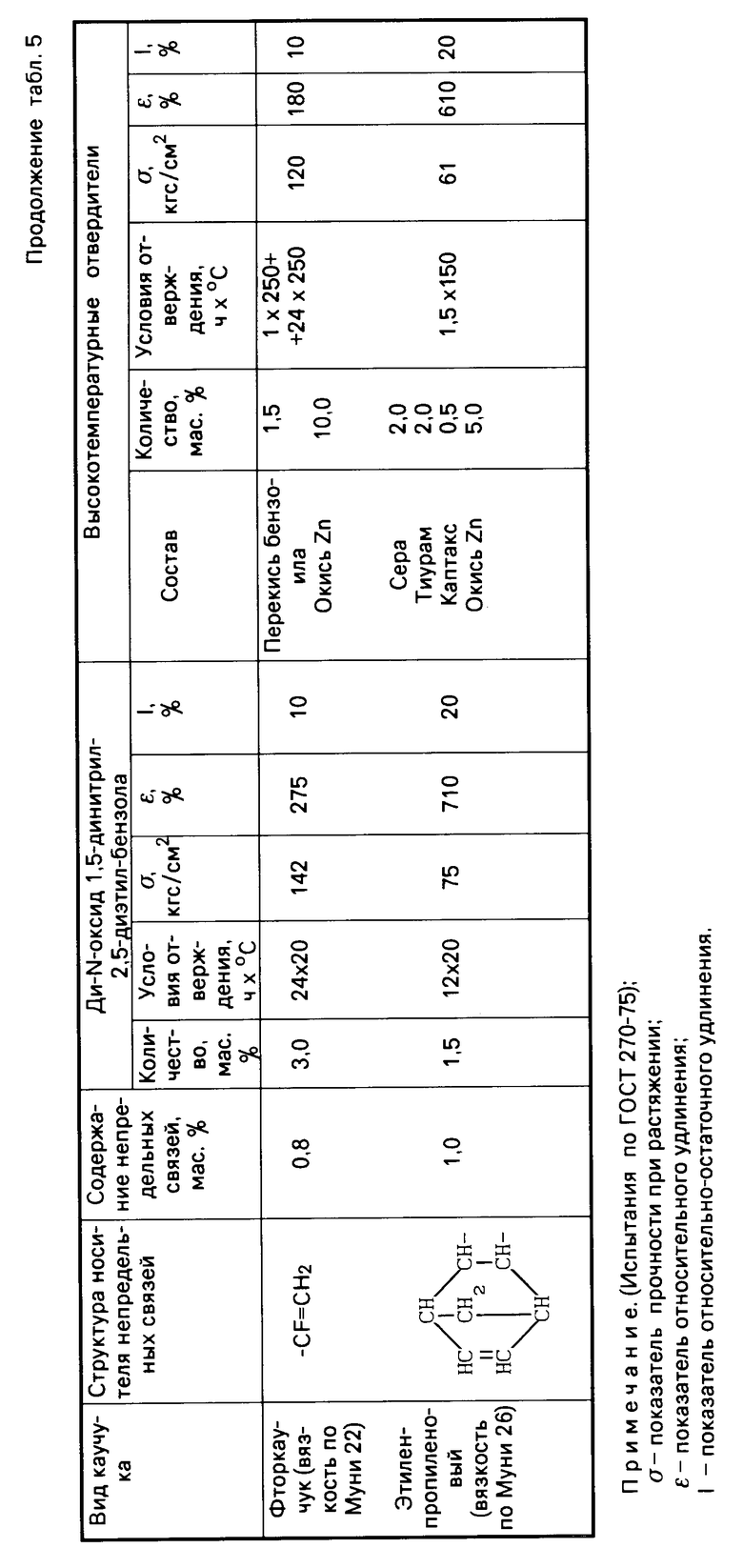
Табл. 5 иллюстрирует результаты сравнительных испытаний отверждающего действия ди-N-оксида 1,4-динитрил-2,5-диэтилбензола и традиционных систем отверждения по отношению к различным типам каучуков с малой непредельностью.
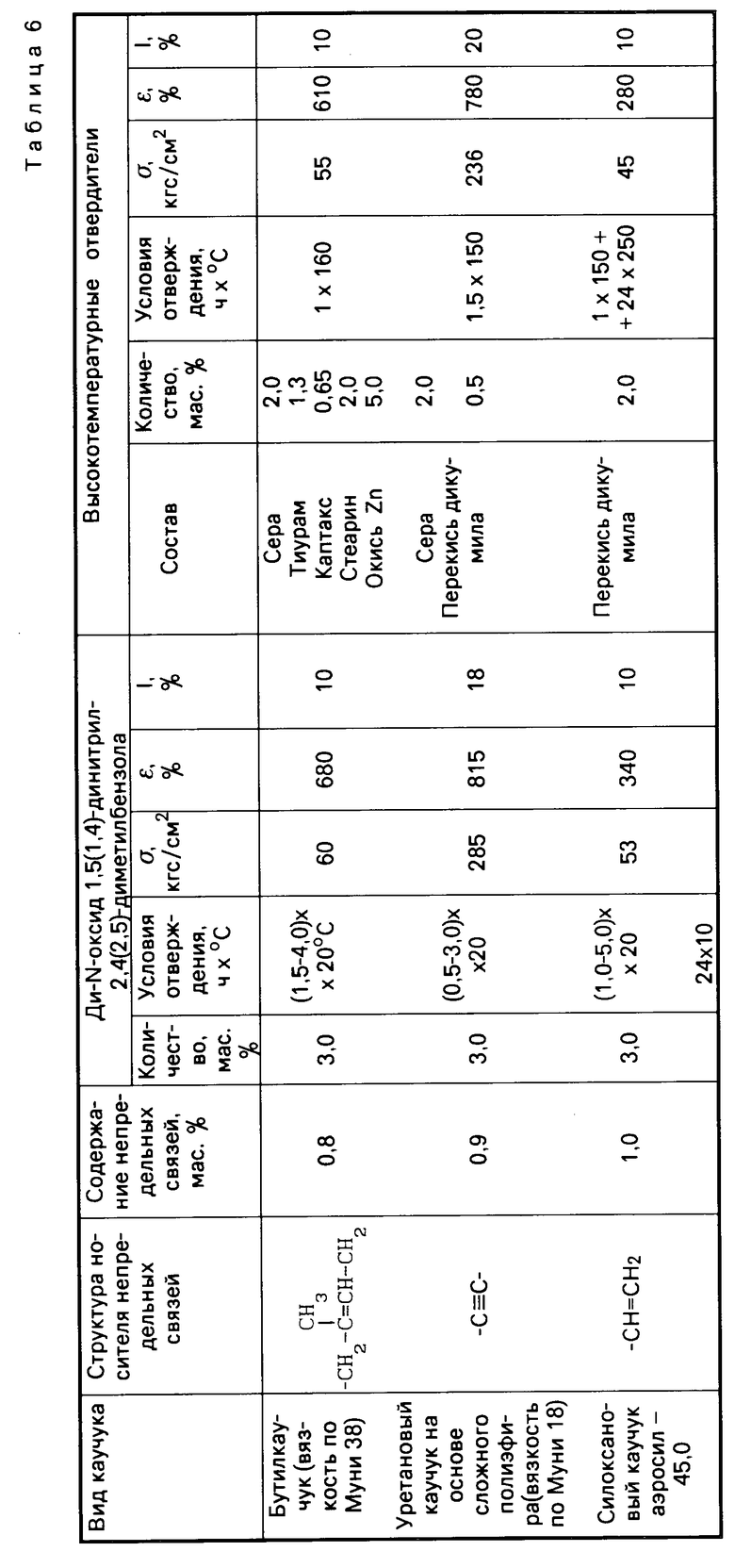
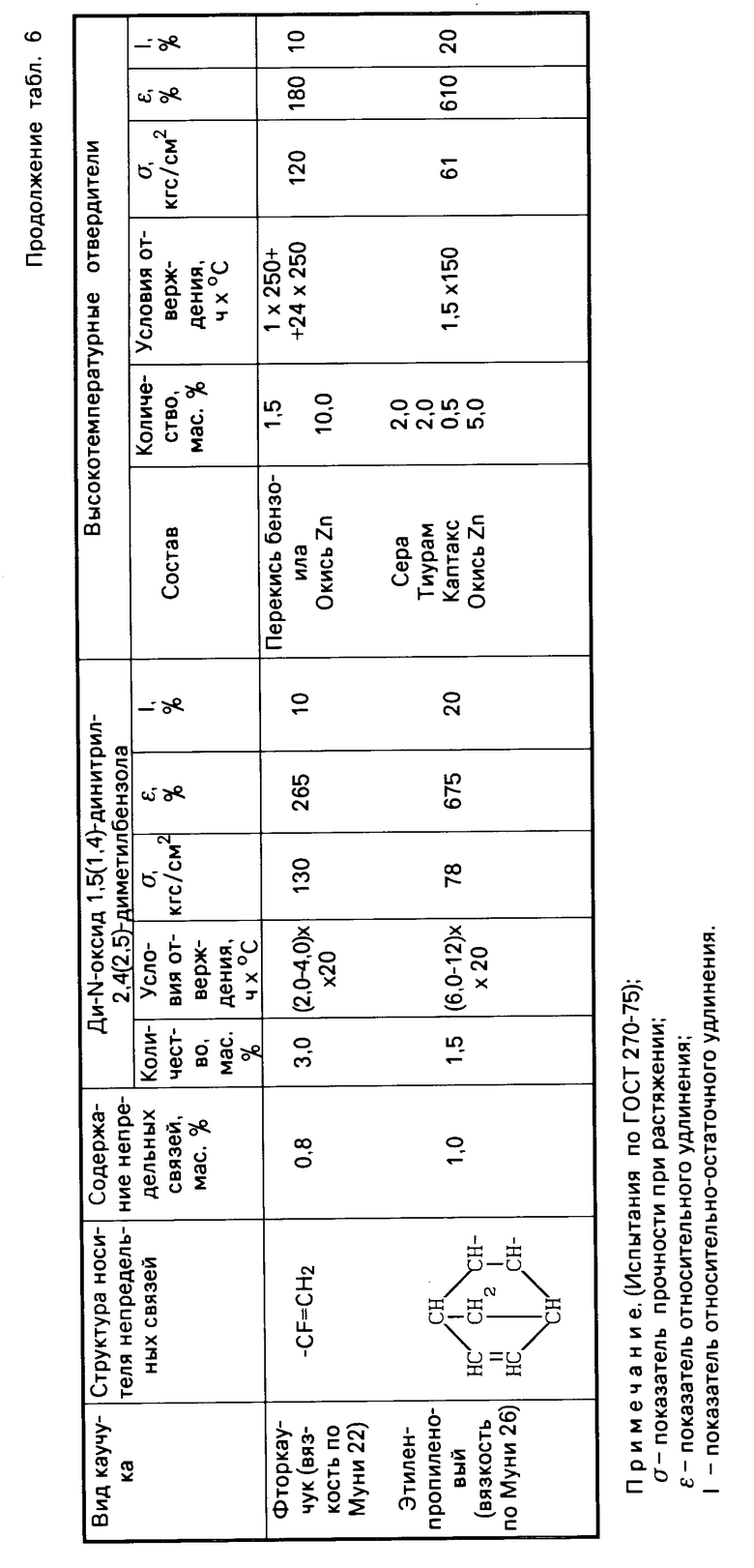
Табл.6 иллюстрирует результаты сравнительных испытаний отверждающего действия ди-N-оксидов 1,5-динитрил-2,4-диметилбензола и 1,4-динитрил-2,5-диметилбензола, и традиционных систем отверждения по отношению к различным типам каучуков с малой непредельностью.
Как видно из табл.4, 5 и 6, изобретение позволяет решить проблему уменьшения температуры процесса вулканизации и, следовательно, снижения энергозатрат при производстве каучуков за счет использования новых низкотемпературных отвердителей ди-N-оксидов динитрилов диалкилбензолов, полученных способом, пригодным для реализации в промышленности. По уровню физико-механических свойств каучуки, полученные с использованием новых низкотемпературных отвердителей, не уступают, а по эластичности превосходят каучуки, полученные с использованием традиционных многокомпонентных высокотемпературных отвердителей.
Предлагаемый способ получения ди-N-оксидов динитрилов диалкилэтилбензолов соответствует требованиям химической промышленности к новым технологиям.
В отличие от аналога и прототипа вещества, используемые в синтезе исходные диалкилбензолы, водный формалин, кислоты, уротропин, четыреххлористый углерод, являются техническими продуктами, широко производимыми химической промышленностью.
Предлагаемый метод синтеза превосходит аналог и прототип по выходу конечного продукта.
Выход N-оксида следующий: аналог менее 19 мас. (в расчете на исходный оксим, с учетом его синтеза выход конечного продукта значительно уменьшится);
прототип около 38 мас. (в расчете на промежуточный (дихлорметил) формилтриэтилбензол [4, 5]
расчет на алкилбензол приведет к дальнейшему уменьшению выхода соответствующего N-оксида);
изобретение 50 мас. (в расчете на исходный алкилбензол).
Учитывая тот факт, что вопросы экологии и утилизации отходов производства имеют в настоящее время приоритетное значение, предлагаемый метод синтеза позволяет производить полный рецикл применяемой в процессе уксусной кислоты и органического растворителя традиционными приемами химической технологии (простой перегонкой или ректификацией). Тем самым создаются предпосылки для создания полностью замкнутого технологического процесса при условии решения вопроса регенерации отработанной кислотной смеси на стадии селективного бис-хлорметилирования, что может быть достигнуто насыщением ее доступным серным ангидридом. Избытки реагентов близки к теоретическим, что определяет экономичность метода синтеза.
Интервалы варьирования температуры (-10)-(+105)оС и времени процесса по отдельным стадиям (3 ч, 10 ч, 1 ч) и в целом (14 ч) являются обычными и не требуют специального технологического оформления.
С точки зрения химика-технолога важно то, что предлагаемый метод синтеза не только позволяет достичь высокого качества полу- и целевого продуктов, но и максимально увязывает технологические параметры отдельных стадий (в ходе осуществления синтеза нет необходимости в проведении нерациональной промежуточной сушки или дополнительной непрактичной перекристаллизации).
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения ди-N,N'-оксидов динитрилов 2,4,6-триалкилбензол-1,3-дикарбоновых кислот | 2018 |
|
RU2694261C1 |
| ГИДРОКСИЗАМЕЩЕННЫЕ СТЕРИЧЕСКИ ЗАТРУДНЕННЫЕ N-АЛКОКСИАМИНЫ | 2000 |
|
RU2243216C2 |

Использование: в качестве низкотемпературных отвердителей каучуков с малой непредельностью. Сущность изобретения: новые ди-N-оксиды динитрилов диалкилбензолов и способ их получения. Реагент 1: соответствующий диалкилбензол. Реагент 2: формальдегид, соляная кислота, серная кислота, уксусная кислота. Условия реакции: реагент 1 обрабатывают реагентом 2 в кислой среде при 70 85°С в течение 5 -6 ч, с последующей обработкой полученного продукта 10 80%-ным водным раствором гексаметилентетрамина при определенных условиях. Затем проводят экстракцию, нейтрализацию и окисление полученного продукта. 2 с. и 1 з.п. ф-лы, 6 табл.
1. R1, R2=C2H5; R3=C ≡ N _→ O;
2. R1, R2=CH3; R3=C ≡ N _→ O;
3. R1, R2=C2H5; R2=C ≡ N _→ O;
4. R1, R3=CH3; R2=C ≡ N _→ O,
в качестве низкотемпературных отвердителей каучуков с малой непредельностью.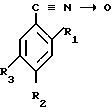
где 1. R1, R2=C2H5; R3=C ≡ N _→ O;
2. R1, R2=CH3; R3=C ≡ N _→ O;
3. R1, R3=; R2=C ≡ N _→ O;
4. R1, R3=CH3; R2=C ≡ N _→ 0,
отличающийся тем, что соответствующий диалкилбензол подвергают селективному бис-хлорметилированию в кислой среде, содержащей формальдегид, соляную кислоту, серную кислоту, уксусную кислоту при молярном соотношении диалкилбензол: формальдегид: соляная кислота: серная кислота:уксусная кислота, равном 1,0 2,5 + 3,2 2,2 + 3,2 6,0 14,0 2,0 4,5, при 70 + 85oС в течение 2 5 ч, полученный при этом соответствующий бис(хлорметил) диалкилбензол обрабатывают 10 80% -ным водным раствором гексаметилентетрамина при молярном соотношении бис(хлорметил)диалкилбензол, гексаметилентетрамин 1,0 2 4 в течение 1 2 ч при температуре кипения и 20 80%-ной водной уксусной кислотой в течение 2 5 ч при кипячении, полученный при этом продукт экстрагируют органическим растворителем, выбранным из ряда хлоралканов, экстракт обрабатывают гидроксиламином при молярном соотношении бис-(хлорметил)диалкилбензол:гидроксиламин соответственно, равном 1 2 3, при pH 6 8 в течение 0,5 1 ч при 25 + 50oС, добавляют водный раствор едкого натра для получения pH 12 14, отделяют органический растворитель, нейтрализуют водный раствор кислотой до pH 6 8 с получением соответствующего диоксима, который подвергают окислению гипохлоритом натрия в щелочной среде при температуре 10 10 oС.
| Походная разборная печь для варки пищи и печения хлеба | 1920 |
|
SU11A1 |
| Якубов А.П., Цыганов В.Д.;Беленький Л.И., Краюшкин М.М., Синтез стерически затрудненных альдегидов ароматического ряда | |||
| Изв.АН СССР | |||
| Сер | |||
| Химия | |||
| Циркуль-угломер | 1920 |
|
SU1991A1 |
