Изобретение относится к усовершенствованному способу синтеза галоидопроизводственных олефиновых соединений, являющихся синтонами при получении биологически активных соединений, в частности, в синтезе витамина А [1]
Известен способ получения 4-галоген-3-метил-3-оксибутена (галогенгидрина изопрена) в результате реакции винилмагнийбромида с эфирным (тетрагидрофуран) раствором хлорацетона при 0оС [2] Выход хлоргидрина изопрена при этом достигает 42% Способ недостаточно технологичен вследствие необходимости использования труднодоступного хлорацетона, являющегося также лакриматором, взрывоопасного тетрагидрофурана, значительной продолжительности процесса и невысокого выхода хлоргидрина изопрена.
Другой подход к синтезу галогенгидрина изопрена отражен в работах (3-6). В основе способов, представленных в данных работах, лежат реакции присоединения элементов гипогалогенкислот к диеновым углеводородам, в частности к изопрену, т.е. реакции оксигалогенирования изопрена. В результате оксигалогенирования наряду с основным продуктом 1,2-присоединения отмечалось образование продукта 1,4-присоединения (4-6), дибромгидрина изопрена [3] Однако способы достижения селективности процесса и увеличения выхода целевого продукта оказались недостаточно эффективными (например, добавление тетрагидрофурана, значительный избыток изопрена или гипогалогенкислот), поэтому выход галогенгидрина изопрена в этих работах не превышает 50%
Наиболее близким по технической сущности и значительным по выходу галогенхлоргидрина изопрена является способ, основой которого также служит реакция оксигалогенирования изопрена элементами хлорноватистой кислоты. Метод заключается в том, что к водному раствору гипохлорита кальция прибавляется избыток изопрена при -5оС. Одновременно через реакционную смесь в течение 2 ч пропускают газообразную углекислоту. Выход изопренхлоргидрина при этом достигает 70% (7). При этом существенными недостатками метода являются:
низкая эффективность процесса вследствие наличия трех фаз (твердой, жидкой и газообразной) и слабого в этой связи межфазного взаимодействия;
недостаточно высокий выход хлоргидрина изопрена;
взрывоопасность процесса из-за склонности гипохлорита кальция к взрыву в присутствии органических соединений.
Целью заявляемого способа является повышение выхода 4-галоген-3-метил-4-оксибутена, упрощение технологического процесса и улучшение его безопасности.
Поставленная цель достигается тем, что согласно предлагаемому способу изопрен вводят во взаимодействие с галогенимидами карбоновых кислот в водной среде при добавлении апротонного биполярного растворителя, хорошо смешивающегося с водой. Реакция проходит при комнатной температуре (20-25оС) по схеме: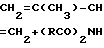 =CH2+(RCO)2NX+H2O ___→ CH2(X)-C(CH3)(OH)-CH=
=CH2+(RCO)2NX+H2O ___→ CH2(X)-C(CH3)(OH)-CH=
Cопоставление экспериментальных данных по выбору донора галоген-катиона показало, что наилучшие результаты и с химической, и с технологической точки зрения дает использование N-бром и N-хлорсукцинимидов. При этом изопрен берется в 20%-ном избытке по отношению к галогенимидам, что ниже, чем в аналогах. Образующийся в процессе синтеза галогенгидрина изопрена сукцинимид, по мере накопления, может быть возвращен в цикл после галогенирования, что является также несомненным достоинством предлагаемого метода. По данному способу продукт 1,4-присоединения не обнаружен.
Отличительной особенностью заявляемого способа является увеличение селективности процесса. Повышение селективности процесса, а следовательно, выхода целевого продукта, связано в данном случае с использованием апротонных биполярных растворителей, хорошо смешивающихся с водой. Роль апротонных биполярных растворителей в данной реакции заключается, вероятно, в разделении ионных пар, в частности, 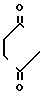 N-Br+, что увеличивает скорость реакции и активизирует процесс в целом. При этом достигается высокая селективность (массовая доля основного продукта составляет 93-95%), что обеспечивает высокий выход галогенгидрина изопрена (до 90%). В качестве апротонного биполярного растворителя могут быть использованы ацетонитрил, диметилсульфоксид, диметилформамид, гексаметилфосфортриамид. При промышленной реализации процесса предпочтительным является ацетонитрил ( ε=37,5), как наиболее доступный, дешевый и достаточно эффективный растворитель. Замена, например, ацетонитрила на диэтиловый эфир приводит к искомому продукту с выходом не превышающим 50% В отсутствие растворителя реакция в наших условиях практически не идет.
N-Br+, что увеличивает скорость реакции и активизирует процесс в целом. При этом достигается высокая селективность (массовая доля основного продукта составляет 93-95%), что обеспечивает высокий выход галогенгидрина изопрена (до 90%). В качестве апротонного биполярного растворителя могут быть использованы ацетонитрил, диметилсульфоксид, диметилформамид, гексаметилфосфортриамид. При промышленной реализации процесса предпочтительным является ацетонитрил ( ε=37,5), как наиболее доступный, дешевый и достаточно эффективный растворитель. Замена, например, ацетонитрила на диэтиловый эфир приводит к искомому продукту с выходом не превышающим 50% В отсутствие растворителя реакция в наших условиях практически не идет.
Существенным достоинством предлагаемого способа по сравнению с прототипом является упрощение процесса, повышение его технологичности. Отсутствие трехфазной системы, проведение процесса при температуре, не требующей дополнительных энергозатрат и позволяющей проводить реакцию менее, чем за час, возврат в цикл образовавшегося сукцинимида все это делает процесс эффективным и простым при его практической реализации.
Улучшение безопасности процесса связано с отсутствием веществ, склонных к взрываемости при наличии других органических соединений, как это имеет место в прототипе, при использовании гипохлорита кальция. Применение четырехлористого углерода в качестве экстрагента и для промывания осадка также снижает пожароопасность процесса.
Заявляемое техническое решение иллюстрируется примерами конкретного выполнения. Образование бромгидрина изопрена доказано ПМР и ИК-спектрами.
П р и м е р 1. В четырехгорлую колбу, снабженную мешалкой, обратным холодильником, термометром и капельной воронкой, загружают 5 г (0,028 г-моля) N-бромсукцинимида, 5 мл воды, 3 мл ацетонитрила. К этой смеси при комнатной температуре и перемешивании добавляют постепенно из капельной воронки 3,4 мл (0,036 г-моля) изопрена. Реакционную массу перемешивают до отрицательной реакции на активный катион галогена (проба с подкисленным раствором йодистого калия должна оставаться бесцветной), после чего реакционную смесь охлаждают, перемешивают при 0оС в течение часа. Выпавший сукцинимид отфильтровывают, промывают 10 мл четыреххлористого углерода, сушат. Выход возвратного сукцинимида 58% (0,8 г). В реакционную смесь добавляют промывной четыреххлористый углерод (ССl4), 20 мл насыщенного раствора хлористого натрия (NaCl) и 10 мл ССl4.
После деления слоев органическую часть промывают 20 мл 5%-ного раствора NaCl. Cушат над безводным сульфатом натрия. Затем отгоняют растворитель и получают 4,45 г бромидрина изопрена. Массовая доля основного вещества 93,6% (ГЖХ-анализ); выход 90,0% Полученный бромгидринизопрена далее может быть использован без дополнительной очистки. Т.кип. 49-51оС (10 мм рт.ст. nD20= 1,4897. ГЖХ-анализ проводили на хроматографе "Хром-5". Колонка стеклянная: l= 1,2 м, d=3 мм. Сорбент: 1% ПЕГ 2000 на хроматоне N-супер (0,16-0,20 мм). Тд=105оС, Тисп=190оС. Время удержания 5,5 мин.
ИК-спектр (в тонкой пленке) -λmax(см-1), 650 (С-Вr), 925 (-CH=CH2); 1370 (-OH); 1640 (-CН=CH2); 3090 (=C-H); 3420 (-OH).
Cпектры ЯМР записаны на спектрометре "Tesla" BS 567A (100,25 МГц) в СДС3 относительно ГМДС.
ЯМР 1Н, δм.д. 1,40(CH3), 3,43(CH2B), 5,15(=CH2), 5,32(=CH), 5,92(CH=).
ЯМР13С, δм.д. 114,81(C1), 141,79(C2), 72,14(C3), 44,31(C4), 26,11(C5).
П р и м е р 2. Выполняется аналогично примеру 1, но вместо ацетонитрила используется 3 мл гексаметилфосфортриамида (ГМФА). Выход возвратного сукцинимида 0,79 г (57,5%). После выделения и отгонки растворителя получают 4,3 г бромгидрина, массовая доля, согласно данным ГЖХ, 94,0% выход 87,3% Данные ЯМР и ИК-спектров согласуются с примером 1.
П р и м е р 3. Выполняется аналогично примеру 1, но вместо ацетонитрила используется 3 мл диэтилового эфира. После отгонки растворителя получают 2,2 г бромгидрилизопрена; массовая доля 93,3% выход 44,3% Все физико-химические данные согласуются с примером 1.
П р и м е р 4. Выполняется аналогично примеру 1, но вместо N-бромсукцинимида используется N-хлорсукцинимид (0,028 моля), выход возвратного сукцинимида 0,68 г (49,8% ). Получают 2,9 г хлоргидрина изопрена, массовая доля 93,5% выход 80,5% Т.кип. 48-49оС при 15 мм рт.ст. nD20=1,4588. Время удерживания по ГЖХ практически не изменилось; в ИК-спектре вместо λmax 650 cм-1 появилась λmax 680 cм-1; в ПМР спектре -δм.д. 3,46(С) (СH2Cl). Остальные показатели имеют в спектрах несущественные изменения.
П р и м е р 5. Выполняется аналогично примеру 1, но без добавления ацетонитрила. В отличие от предыдущих примеров внешних изменений ни при добавлении изопрена, ни при нагреве не наблюдалось. Проба с подкисленным раствором йодистого калия в течение 6 ч не обесцвечивалась. Реакция практически не идет.
Рассмотрение приведенных примеров позволяет заключить, что предлагаемый способ получения галогенгидрина изопрена обеспечивает существенное увеличение селективности процесса, и, как следствие, повышение выхода целевого продукта по сравнению с прототипом наряду с упрощением процесса и улучшением его безопасности.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения 5-ацетокси-3-бромпентанона-2 | 1989 |
|
SU1659397A1 |
| СПОСОБ ПЕРЕРАБОТКИ НЕПРОРЕАГИРОВАВШИХ ПРОДУКТОВ В ПРОЦЕССЕ СИНТЕЗА ВИТАМИНА B | 1991 |
|
RU2006496C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2,3,4,6-ДИ-О-ИЗОПРОПИЛИДЕН- α -L-СОРБОФУРАНОЗЫ | 1990 |
|
SU1788715A1 |
| СПОСОБ КОНЦЕНТРИРОВАНИЯ ВОДНОГО РАСТВОРА МЕДИЦИНСКОГО ВИТАМИНА С | 1992 |
|
RU2071818C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ПРОЦЕНТНОГО СОДЕРЖАНИЯ МЕДИЦИНСКОЙ АСКОРБИНОВОЙ КИСЛОТЫ В КРИСТАЛЛИЧЕСКОМ ПРЕПАРАТЕ | 1992 |
|
RU2072512C1 |
| СПОСОБ СТАБИЛИЗАЦИИ КОНЦЕНТРАЦИИ АСКОРБИНОВОЙ КИСЛОТЫ | 1990 |
|
RU2027164C1 |
| СПОСОБ ИЗМЕРЕНИЯ КОНЦЕНТРАЦИИ ВОДНОГО РАСТВОРА АСКОРБИНОВОЙ КИСЛОТЫ | 1991 |
|
RU2006838C1 |
| СПОСОБ ПОЛУЧЕНИЯ ВОДОРАСТВОРИМОГО БЕНЗАФЛАВИНА | 1988 |
|
SU1638845A1 |
| Способ получения 4-галоген-3-метил-2-бутениловых эфиров карбоновых кислот | 1985 |
|
SU1245565A1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИЭФИРНЫХ ТЕРМОЭЛАСТОПЛАСТОВ | 1993 |
|
RU2045543C1 |
Использование: в качестве синтона при получении биологически активных соединений, в синтезе витамина А. Сущность изобретения: реагент 1 изопрен. Реагент 2: N-галогенсукцинамид. Условия реакции: в растворителе, выбранном из группы: ацетонитрил, гексаметилфосфортриамид, диметилформамид, температура 20-25°С.
СПОСОБ ПОЛУЧЕНИЯ 4-ГАЛОГЕН-3-МЕТИЛ-3-ОКСИБУТЕНА оксигалогенированием изопрена в водной среде с выделением целевого продукта, отличающийся тем, что оксигалогенирование ведут N-галогенсукцинамидом в среде, содержащей апротонный биполярный растворитель, выбранный из группы ацетонитрил, диметилсульфоксид, диметилформамид, гексаметилфосфортриамид, при 20 25oС.
| Михантьев Б.Б | |||
| Сб | |||
| Мономеры и высокомолекулярные соединения, Воронеж, Госуниверситет, 1973, с.71. |
