Настоящее изобретение относится к новым биологическим активным соединениям. В частности, изобретение относится к N-замещенным гетероциклическим производным, к производным пиримидина и имидазолина - промежуточным соединениям для получения замещенных гетероциклических производных, и к фармацевтической композиции на основе замещенных гетероциклических производных.
Соединения в соответствии с изобретением обладают антагонистическим действием по отношению к ангиотензину II, который является пептидным гормоном формулы:
H-Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-OH
Ангиотензин II является мощным вазопрессором, и представляет собой биологически активный продукт в системе ренин-ангиотензин; ренин воздействует на ангиотензиноген плазмы с получением ангиотензина I, который превращается в ангиотензин II под действием фермента, осуществляющего конверсию ангиотензина I.
Соединения согласно изобретению являются непептидными соединениями. Ингибируя действие ангиотензина II на соответствующие рецепторы, соединения в соответствии с изобретением препятствуют, в частности, увеличению кровяного давления, создаваемого в результате взаимодействия гормон -рецептор, они обладают также другими физиологическими действиями на уровне центральной нервной системы.
Таким образом, новые соединения могут быть полезны при лечении таких сердечно-сосудистых заболеваний, как гипертензия, сердечная недостаточность, а также при лечении заболеваний центральной нервной системы, при лечении глаукомы и диабетической ретинопатии.
Предметом настоящего изобретения являются соединения формулы: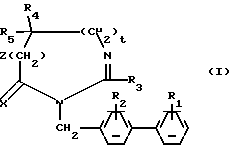
в которой R1 и R2 одинаковые или различные и представляют каждый независимо друг от друга: водород или группу, выбираемую из карбокси, алкоксикарбонила (C1-C4), циано, тетразолила, метилтетразолила, метилсульфониламино, трифторметилсульфониламино, трифторметилсульфониламинометила, N -цианоацетамида, N-гидроксиацетатамида, N -[(4-карбокси)-1,3-тиазол-2-ил)] -ацетамида, уреидо, 2 -цианогуанидинкарбонила, 2 -цианогуанидинметила, 1 -имидазолилкарбонила, 3-циано-2 -метилизотиоуреидометила, при условии, что по меньшей мере один из заместителей R1 или R2 отличается от водорода;
R3 водород, алкил (C1-C6), незамещенный или замещенный одним или несколькими атомами галогена, алкенил (C2-C6), циклоалкил (C3-C7), фенил, фенилалкил (C1-C3), причем названные фенильные группы не замещены;
R4 и R5 представляет каждый независимо друг от друга алкил (C1-C6), фенил, причем алкил или фенил не замещены или замещены одним или несколькими атомами галогена или группой перфторалкила (C1-C4);
или R4 и R5 образуют вместе группу формулыCR7R8, в которой R7 представляет водород и R8 представляет фенил;
или же R4 и R5 представляют вместе группу (CH2)n, в которой n=3-11, кроме того, если эта группа представляет циклогексан, она может быть замещена алкилом (C1-C4) или фенилом; или группу (CH2)p Y (CH2)q, в которой p=q=2:
Y NR6, где R6 ацетил, COF3, COC6H5, остаток аргинина, аспарагиновой кислоты, цистеина или изолейцина;
или же R4 и R5 вместе с атомом углерода, с которым они соединены, образуют индан или адамантан;
X представляет атом кислорода или атом серы;
Z и t представляют нули или один представляет нуль, а другой 1, и их соли.
Если соединение по изобретению имеет асимметрический углерод, то изобретение включает 2 оптических изомера этого соединения.
Соли соединений формулы (I) включают соли с минеральными или органическими кислотами, которые позволяют выделять или кристаллизовать соединения формулы (I), с такими как пикриновая, щавелевая кислота или оптически активная кислота, например, миндальная или камфорсульфокислота, или которые образуют фармацевтически приемлемые соли, такие как хлоргидраты, бромгидраты, сульфаты, гидросульфаты, дигидрофосфаты, метансульфонаты, метилсульфаты, малеаты, фумараты, 2 -нафталинсульфонаты.
Соли соединений формулы (I) включают также соли с органическими и минеральными основаниями, например, соли щелочных или щелочноземельных металлов, такие как соли натрия, калия, кальция, причем соли натрия и калия предпочтительны, или с третичным амином, таким как трометамол, или же соли аргинина, лизина или любого физиологически приемлемого амина.
В настоящем описании под атомом галогена понимают атом брома, хлора или фтора; под N-защитной группой (обозначаемой также как Pг) понимают группу, используемую обычно в химии пептидов для обеспечения временной защиты аминогруппы, такую, как например, группу БОК, Z, ФМОК (см. ниже) или бензильную группу, под этерифицированной карбоксигруппой понимают сложный эфир, неустойчивый в соответствующих условиях, как например, метиловый, этиловый, бензиловый или третбутиловый сложный эфир. "Алкилом" обозначают остатки алифатических насыщенных углеводородов, линейных или разветвленных.
Предпочтительными соединениями являются:
соединения формулы (I), в которой R1 находится в ортоположении и представляет собой карбоксигруппу или тетразолил, а R2 является водородом;
соединения формулы (I), в которых R4 и R5 образуют вместе с углеродом, с которым они связаны, циклопентан или циклогексан;
соединения формулы (I), в которых R3 представляет собой линейную алкильную группу C1-C6;
соединения формулы (I,) в которых X представляет собой атом кислорода;
соединения формулы (I), в которых Z t 0.
В описании и в примерах используются следующие сокращения:
Et этил
н-Bu, т-Bu н-бутил, трет-бутил
ДМФА диметилформамид
ТГФ тетрагидрофуран
ДХМ дихлорметан
NБС N-бромсукцинимид
ДЦК дициклогексилкарбодиимид
ДИПЭА диизопропилэтиламин
Эфир этиловый эфир
ТФК трифторуксусная кислота
Z бензилоксикарбонил
БОК трет-бутоксикарбонил
БОФ гексафторфосфат бензотриазолилокситрисдиметиламинофосфония
ФМОК флуоренилметилоксикарбонил
Соединения формулы I могут быть получены согласно одному из нижеописанных способов, называемому cпособ 1, cпособ 2 и cпособ 3.
Способ 1.
а) Осуществляют реакцию гетероциклического производного формулы:
в которой Z, t, R3, R4 и R5 имеют значения, указанные выше для соединения формулы (I) с производным (4-бифенилил) метила формулы:
в которой Гал представляет собой атом галогена, а  и
и  представляют собой соответственно либо R1 и R2, описанные выше, либо группу -предшественник для R1 и R2;
представляют собой соответственно либо R1 и R2, описанные выше, либо группу -предшественник для R1 и R2;
б1) в случае необходимости, полученное таким образом соединение формулы: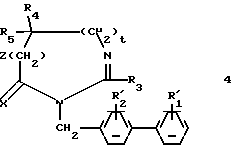
обрабатывают реактивом Лавессона; 2,4-бис-(4-метоксифенил)-1,3-дитиа-2,4 -дифосфэтан-2,4-дисульфидом;
в1) соединение, полученное на стадиях а1 или б1 формулы: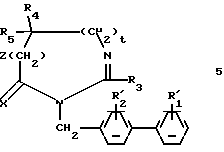
в которой X представляет собой атом кислорода или атом серы, обрабатывают с целью получения соединения (I) путем превращения групп  и/или
и/или 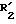 в соответственно группы R1 и/или R2.
в соответственно группы R1 и/или R2.
Среди соединений 2 соединения формулы (II), описанные ниже, являются новыми.
Таким образом, изобретение относится также к соединениям формулы II: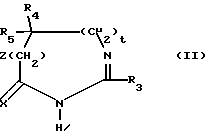
в которой; R3 представляет водород, алкил (C1-C6), незамещенный одним или несколькими атомами галогена, алкенил (C2-C6), циклоалкил (C3-C7), фенил, фенилалкил (C1-C3), причем названные фенильные группы не замещены,
R4 и R5 представляют каждый независимо друг от друга алкил (C1-C6), фенил, алкил или фенил, не замещенные или замещенные одним или несколькими атомами галогена или группой перфторалкила (C1-C4);
или R4 и R5 образуют вместе группу формулыCR7R8, в которой R7 представляет водород и R8 представляет фенил;
или же R4 и R5 представляют вместе группу (CH2)n, в которой n= 3 11, причем, если эта группа представляет циклогексан, она может быть замещена алкилом (C1-C4) или фенилом; или группу (CH2)pY (CH2)q, в которой p q 2;
Y NR6, где R6 ацетил, COF3, COC6H5, остаток аргинина, аспарагиновой кислоты, цистеина или изолейцина;
или же R4 и R5 вместе с атомом углерода, с которым они соединены, образуют индан или адамантан;
X представляет атом кислорода или атом серы;
Z и t представляют нули или один представляет нуль, а другой представляет 1, и их соли.
Среди производных формулы (II) соединения, в которых Z t О, а R4 и R5 вместе с углеродом, с которым они связаны, образуют циклопентан, являются предпочтительными соединениями. Эти соединения отвечают формуле:
в которой X представляет собой атом кислорода или атом серы, а R3 представляет собой водород, алкил C1-C6, незамещенный или замещенный одним или несколькими атомами галогена; алкенил C2 -C6, циклоалкил C3-C7, фенил, фенилалкил, с алкильной частью C1-C3, причем указанные фенильные группы являются незамещенными при условии, что R3 отличается от замещенного фенила, если X означает кислород.
Соединения (II), в которых Z 0 и t 1, формулы:
в которой R3, R4, R5 и X имеют значения, данные выше для соединения (II), являются также предпочтительными соединениями.
Наконец, соединения (II), в которых Z=1 и t=0, формулы
в которой: R3 представляет собой водород, алкил C1-C6, незамещенный или замещенный одним или несколькими атомами галогена, алкенил C2-C6, циклоалкил C3-C7, причем указанные фенильные группы являются незамещенными;
R4 и R5 представляют собой каждый независимо друг от друга алкил C1-C6, фенил, причем указанные алкильная или фенильная группы являются незамещенными или замещенными одним или несколькими атомами галогена или одной группой перфторалкила C1-C4;
или R4 и R5 вместе образуют группу формулыCR7R8, в которой R7 представляет собой водород, а R8 представляет собой фенил;
или же R4 и R5 представляют вместе группу (CH2)n в которой n 3-11, причем, если эта группа представляет циклогексан, то она может быть замещена алкилом (C1-C4) или фенилом; или группу (CH2)pY /CH2)q в которой p=q=2;
Y NR6, где R6 ацетил, COF3, COC6H5, остаток аргинина, аспарагиновой кислоты, цистеина или изолейцина;
или же R4 и R5 вместе с атомом углерода, с которым они соединены, образуют индан или адамантан,
X означает атом кислорода или серы.
Производные 2 получаются по известным методам. Например, можно использовать метод, описанный Jacquier et al. (Bull. Chim. Soc. France. 1971, 3, 1040-1051) или Brunken et Bach (Chem. Ber. 1956, 89, 1363-1373), и подействовать алкилимидатом на аминокислоту или ее сложный эфир в соответствии со следующей реакционной схемой: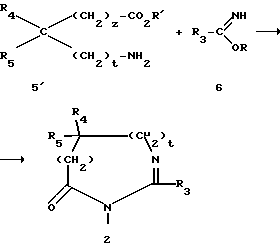
в которой R представляет собой алкил C1-C4, R' представляет собой водород или алкил C1-C4, а R3, R4, R5, Z и t являются такими, как определено выше для соединения (I).
Эта реакция осуществляется в кислой среде при нагревании в инертном растворителе, таком как ксилол или толуол.
Производное (4-бифенилил)-метила (3) получается в соответствии со способом, описанным в заявке на европейский патент ЕП 324 377
Превращение группы, 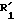 и/или
и/или 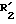 в группу R1 и/или R2 осуществляется при помощи методов, хорошо известных специалисту. Так, когда соединение (I), которое нужно получить, имеет группу R1 и/или R2 карбоксигруппа, то
в группу R1 и/или R2 осуществляется при помощи методов, хорошо известных специалисту. Так, когда соединение (I), которое нужно получить, имеет группу R1 и/или R2 карбоксигруппа, то  и/или
и/или 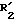 представляют собой этерифицированную карбокси группу. Когда получаемое соединение (I) имеет группу R1, и/или R2 тетразолил, то
представляют собой этерифицированную карбокси группу. Когда получаемое соединение (I) имеет группу R1, и/или R2 тетразолил, то 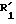 или
или  могут представлять собой либо тетразолил, защищенный, например, тритильной группой, либо цианогруппу, которая затем замещается тетразолильной группой, в случае необходимости, защищенной тритилом. Превращение цианогруппы в тетразолил может быть осуществлено под действием азида, например, азида трибутилолова или под действием азида натрия.
могут представлять собой либо тетразолил, защищенный, например, тритильной группой, либо цианогруппу, которая затем замещается тетразолильной группой, в случае необходимости, защищенной тритилом. Превращение цианогруппы в тетразолил может быть осуществлено под действием азида, например, азида трибутилолова или под действием азида натрия.
Стадия а1 осуществляется в инертном растворителе, таком как ДМФА, ДМСО или ТГФ, в щелочной среде, например в присутствии гидроксида калия, алкоголята металла, гидрида металла, карбоната кальция или триэтиламина.
Стадия б1 осуществляется при нагревании под азотом в таком растворителе, как толуол, по методу, описанному M.P.Cava et al. Tetrahedron, 1985, 41, 22, 5061.
С другой стороны, соединения (I) могут быть получены по другому способу, который называется способ 2. Этот способ заключается в том, что:
а2) проводят реакцию аминокислоты формулы: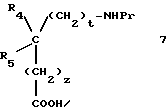
в которой Z, t, R4 и R5 имеют значения, указанные выше для соединения (1), и в которой аминогруппа защищена группой Pг, с производным (4-бифенилил) -метиламина, имеющим формулу:
в которой  представляют собой соответственно либо R1 и R2, либо группировку -предшественник для R1 и R2;
представляют собой соответственно либо R1 и R2, либо группировку -предшественник для R1 и R2;
б2) после снятия защиты с аминогруппы полученное соединение формулы:
обрабатывается затем алкиловым эфиром ортокислоты формулы R3C(OR)3 (10), в которой R3 имеет значение, указанное выше для соединения (I), а R является алкилом C1-C4;
в2) в случае необходимости, полученное соединение формулы:
обрабатывается реактивом Лавессона,
г2) соединение, полученное указанным образом на стадиях б2 или в2, формулы: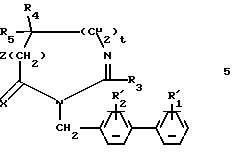
обрабатывается затем в условиях, подходящих для получения соединения (I) путем превращения групп  и/или
и/или 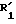 в соответственно группы R2 и/или R1.
в соответственно группы R2 и/или R1.
Соединения 7 являются известными или получаются при помощи известных методов (Chemistry of the Amino Acids, Greenstein and Winitz, John Wiley ed. 1961, т. 1, с. 697).
Соединения 8 получаются в соответствии с заявкой на европейский патент ЕП 324 377. Стадия а2 осуществляется в условиях, обычных для присоединения кислоты к амину, например, в присутствии БОФ и ДИПЭА.
Стадия б2, которая является циклизацией соединения 9 в присутствии соединения 10, осуществляется в соответствии с Jacquier et al. (Bull. Soc. Chim. France, 1971, 3, 1040-1051) и в соответствии с Brunken et Bach (Chem. Ber. 1956, 89, 1363-1373).
В соответствии с одним вариантом способа 2 на стадии б2 можно, в случае необходимости, выделять промежуточное соединение 9' формулы: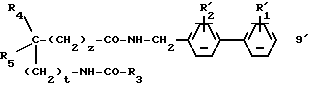
а затем получать соединение 4 в результате циклизации в кислой среде.
В соответствии с другим вариантом способа 2 для получения соединения (I) в котором R4R5 представляют собой группуCR7R8, можно проводить реакцию в кислой среде между аминокислотой формулы: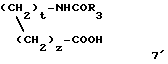
и альдегидом или кетоном с формулой:
R7COR8,
в которой R7 и R8 имеют значения, данные выше для соединения (I), а затем в результате действия соединения 8 получают соединения формулы:
Циклизация этого соединения в кислой среде приводит к получению соединения 4.
В этом способе для получения соединения (I), у которого R1 и/или R2 являются карбоксигруппой, заместители  и/или
и/или  представляют собой предпочтительно третбутоксикарбонил.
представляют собой предпочтительно третбутоксикарбонил.
Наконец, другой альтернативой для получения соединений (I) по изобретению, у которых z, и t равны 0, является способ фотоокисления, называемый способ 3.
Этот способ заключается в том, что:
а3) на производное имидазола формулы: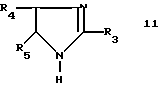
в которой R3, R4, R5 имеют значения, указанные выше для соединения (I), действуют производным (4-бифенилил) -метила формулы: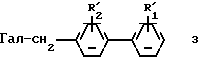
в которой Гал представляет собой атом галогена, а  и
и  представляют собой соответственно либо R1 и R2, либо группировку-предшественник для R1 и R2, в присутствии кислорода и при действии УФ-облучения в щелочной среде;
представляют собой соответственно либо R1 и R2, либо группировку-предшественник для R1 и R2, в присутствии кислорода и при действии УФ-облучения в щелочной среде;
б3) в случае необходимости, полученное соединение формулы: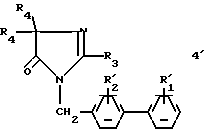
обрабатывают реактивом Лавессона: 2,4-бис-(4-метоксифенил)-1,3-дитиа-2,4 -дифосфенат-2,4-дисульфидом;
в3) соединение, полученное на стадиях б3 или в3 формулы: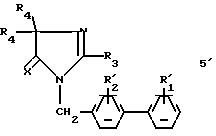
обрабатывают затем в условиях, подходящих для получения соединения (I) путем превращения групп  и/или
и/или  в соответственно группы R1 и/или R2.
в соответственно группы R1 и/или R2.
Производное имидазола 11 является либо коммерческим, либо получаемым по известным методам, указанным выше для получения соединений 2.
Стадия а3 осуществляется в инертном растворителе, таком как ДМФА, например: для облегчения реакции можно добавлять фотосенсибилизирующее соединение, как, например, метиленовая синь.
Соединения в соответствии с изобретением, у которых R4 и R5, соединенные вместе, представляют собой группу с формулой (CH2)pY(CH2)q, в которой Y является группой NH, могут быть получены в результате каталитического гидролиза соответствующего соединения (I), в котором Y является группой N-R6, причем R6 является бензилом.
Сродство продуктов в соответствии с изобретением по отношению к рецепторам ангиотензина II изучалось при помощи теста на связывание ангиотензина II, меченного при помощи иода-125, с клеточными рецепторами печени крыс. Использованный метод является методом, описанным S.Keppens et al. Biochem. J. 1982, 208, 809-817.
Измеряют Cl50: концентрация, которая дает 50% замещения меченного ангиотензина II, специфически связанного с рецептором. Значение Cl50 для соединений в соответствии с изобретением составляет менее 10-6 М. Результаты приведены в прилагаемой табл. 2.
Кроме того, антагонистическое действие по отношению к ангиотензину II для продуктов в соответствии с изобретением было установлено для различных видов животных, у которых предварительно была активирована система ренин-ангиотензин (C. Lacouret al. J. Hypertension, 1989, 7, suppl. S33-S35). Протокол испытаний и результаты представлены ниже.
Соединения в соответствии с изобретением сохраняют активность при введении различными путями, в частности, оральным путем.
Не обнаружено никакого признака токсичности для этих соединений при фармакологически активных дозах. Соединения могут использоваться при лечении различных сердечно-сосудистых заболеваний, в частности, гипертензии, сердечной недостаточности, венозной недостаточности, а также при лечении глаукомы, диабетических ретинопатий и различных заболеваний центральной нервной системы, например, беспокойства, депрессии, амнезии или болезни Альцгеймера.
Изобретение относится также к фармацевтической композиции, содержащей эффективную дозу соединения в соответствии с изобретением или его фармацевтически приемлемую соль и подходящие инертные вещества. Инертные вещества выбираются в зависимости от фармацевтической формы и от желаемого способа введения.
В фармацевтических композициях для орального, подъязычного, подкожного, внутримышечного, внутривенного, местного, внутритрахейного, интраназального, трансдермического или ректального введения активные компоненты формулы (I) или их возможные соли могут вводиться в виде единичных форм введения в смеси с классическими фармацевтическими носителями. Надлежащие единичные формы введения включают оральные формы, такие как таблетки, желатиновые капсулы, порошки, гранулы и оральные растворы или суспензии, формы подъязычного, защечного, внутритрахейного, интраназального введения формы подкожного, внутримышечного или внутривенного введения и формы ректального введения. Для местного применения можно использовать кремы, мази или лосьоны.
Доза активного компонента может варьироваться между 0,01 и 50 мг на кг веса тела в день.
Каждая единичная доза может содержать от 0,1 до 1000 мг, предпочтительно от 1 до 500 мг активного ингредиента в комбинации с фармацевтическим носителем.
Когда готовят твердую композицию в виде таблеток, то смешивают основной активный компонент с фармацевтическим носителем, таким как желатина, крахмал, лактоза, стеарат магния, тальк, гуммиарабик или аналогичные. На таблетки можно наносить покрытие из сахарозы, производного целлюлозы или других соответствующих веществ или же можно обрабатывать их таким образом, чтобы они имели активность продленного действия или действия с запаздыванием и чтобы они непрерывно высвобождали предопределенное количество активного компонента.
Препарат в желатиновых капсулах получают, смешивая активный ингредиент с разбавителем и заливая полученную смесь в мягкие или твердые желатиновые капсулы.
Препарат в виде сиропа или эликсира или для введения в виде капель может содержать активный компонент одновременно с подсластителем, предпочтительно бескалорийным, метилпарабеном и пропилпарабеном в качестве антисептиков, а также с придающим вкус веществом и с соответствующим красителем.
Порошки и гранулы, диспергируемые в воде, могут содержать активный ингредиент в смеси с диспергирующими агентами или смачивающими агентами или агентами для перевода в суспензию, как, например, поливинилпирролидон, а также с подсластителями или с корректорами вкуса.
Для ректального введения используют свечи, которые приготавливаются со связующими, плавящимися при ректальной температуре, например, масло, какао или полиэтиленгликоли.
Для парентерального введения используют водные суспензии, солевые изотонические растворы или стерильные растворы, пригодные для впрыскивания, которые содержат фармакологически совместимые диспергирующие и/или смачивающие агенты, например, пропиленгликоль или бутиленгликоль.
Активный компонент может быть введен в рецептуру также в виде микрокапсул, в случае необходимости, с одним или несколькими носителями или дополнительными веществами.
Последующие примеры иллюстрируют изобретение, вместе с тем не ограничивая его. В этих примерах используют следующие сокращения: d означает плотность, КТ означает комнатную температуру, KHSO4 K2SO4 означает водный раствор, содержащий 16,6 г бисульфата калия и 33,3 г сульфата калия в 1 л.
Температуры плавления (Тпл) приводятся в градусах Цельсия; за исключением особо оговоренных случаев, они были измерены без перекристаллизации продукта.
Чистота продуктов проверяется методом тонкослойной хроматографии (ТСХ) или методом высокоэффективной жидкостной хроматографии (ВЭЖХ). Продукты характеризуются по их спектру ЯМР, зарегистрированному при 200 МГц в дейтерированном ДМСО, причем внутренним стандартом является тетраметилсилан.
Для интерпретации спектров ЯМР используют следующие обозначения:
с для синглета,
ш.с. для широкого синглета,
д для дублета,
т для триплета,
к для квадруплета,
кв для квинтета,
сек для секстета,
м для массива или мультиплета.
Кроме того, им означает имидазол.
Классическим образом атомы водорода на бифенилиле нумеруются, как изображено на следующей формуле: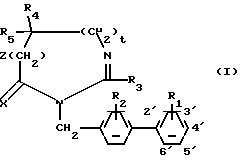
В последующих примерах z и t равны нулю, за исключением случая, когда полученное соединение является пиримидином.
Пример 1.
2-н-бутил-4 -спироциклопентан-1 -[(2'-трет -бутоксикарбонил-4-бифенилил) метил]-2-имидазолин-5-он и
трифторацетат 2-н-бутил-1-[(2' -карбокси-4-бифенилил)-метил] -4 - спироциклопентан-2-имидазолин-5-она. (Способ 2).
А) N-ФМОК-1 -аминоциклопентанкарбоновая кислота получается в соответствии со способом, описанным Chi Den Chang et al. (Int. J.Peptide Protein. Res. 1980, 15, 59 -66).
Тпл 89-91oC.
В) N-[2'-трет-бутоксикарбонил-4 -бифенилилметил]-1-[N-ФМОК-амино]-1 -циклопентанкарбоксамид.
Растворяют 700 мг продукта, полученного на предыдущей стадии, в 8 мл ДМФА и прибавляют последовательно 576 мг 4-аминометил-[2'-трет-бутоксикарбонил] -бифенила, 970 мг БОФ и количество ДИПЭА, достаточное для доведения до pH 6.
После перемешивания в течение 1 ч реакционную среду разбавляют посредством 100 мл этилацетата и 20 мл воды; органическую фазу промывают последовательно насыщенным раствором бикарбоната натрия, затем раствором KHSO4-K2SO4 и, наконец, насыщенным раствором хлорида натрия. После сушки на сульфате натрия раствор выпаривают досуха. Получают жидкое масло с массой m, равной 1,2 г.
С) N-[2'-трет-бутоксикарбонил-4 -бифенилилметил]-[1-амино]-1- циклопентанкарбоксамид.
Растворяют продукт, полученный на предыдущей стадии, в 10 мл ДМФА, затем прибавляют 1 мл диэтиламина и перемешивают в течение 1 ч 15 мин при КТ. Реакционную среду извлекают посредством 100 мл этилацетата и 20 мл воды, затем органическую фазу промывают 1 раз в воде, 1 раз насыщенным раствором хлорида натрия, затем сушат на сульфате натрия и выпаривают досуха.
Остаток хроматографируется на силикагеле при элюировании смесью этилацетат/метанол/аммиак с концентрацией 30% (99/1/0,5 по об.). Получают 600 мг ожидаемого соединения.
ИК (HCl3):
3350 см-1: H (амид и амин),
1700 см-1: C O (CO2 тBu),
1650 см-1: C O (CONH).
Спектр ЯМР:
1,25 ppm: с: 9 H: тBu,
2,15 1,40 ppm: с: 10 H: (C5H8, NH2),
4,40 ppm: д: 2H: CH2- NH,
7,15 7,75 ppm: м: 8H: бифенил,
8,60 ppm: т: 1 H: NH CH2.
D) 2-н-бутил-4-спироциклопентан-1 -[(2'-трет-бутоксикарбонил-4- бифенилил) -метил]-2-имидазодин-5-он.
Смешивают 394 мг продукта, полученного на предыдущей стадии, и 250 мг этилортовалерата в 2 мл ДХМ. Прибавляют 1 каплю уксусной кислоты, затем нагревают до температуры 90oC, позволяя испаряться ДХМ. Спустя 1 ч 15 мин реакционную смесь извлекают в 50 мл этилацетата, 10 мл воды и 1 мл насыщенного раствора бикарбоната натрия. Затем органическую фазу промывают насыщенным раствором хлорида натрия, потом сушат на сульфате натрия и выпаривают досуха. Остаток хроматографируется на силикагеле при элюировании смесью этилацетат/толуол (1/2 по об.). Получают 390 мг ожидаемого продукта, который кристаллизируется.
Тпл 63-65oC.
ИК(CHCl3):
1710-1720 см-1: C O, C=O (сложный эфир и имидазолин);
1625 см-1: C=N.
Спектр ЯМР:
0,88 ppm: т: 3 H: CH3 (нBu),
1,20 ppm: с: 9 Н: тBu,
1,35 ppm: сек: 2 H: CH3-CH2 -,
1,58 ppm: кв: 2 H: CH3-CH2CH2-,
1,95-1,65 ppm: с: 8 Н: циклопентан,
2,42 ppm: т: 2 H: CH3-CH2-CH3-CH2-,
4,78 ppm: с: 2 H: CH2-C6H4-,
7,20-7,80 ppm: м: 8 H: ароматические H.
Масс-спектр: MH+: 461.
Е) трифторацетат 2-н-бутил-1-[(2'-карбокси-4-бифенилил)-метил]-4 - спироциклопентан-2-имидазодин-5-она.
Обрабатывают 180 мг продукта, полученного на предыдущей стадии, посредством 3 мл ДХМ и 4 мл ТФК в течение 45 мин. После выпаривания под вакуумом остаток извлекают эфиром. Получают твердое вещество белого цвета, которое фильтруется, промывается в эфире, затем сушится под вакуумом
m 155 мг.
Тпл 176-178oC.
Спектр ЯМР:
0,78 ppm: т: 3 H: CH3 (нBu),
1,25 ppm: сек: 2 H: CH3-CH2,
1,50 ppm: кв: 2 H: CH3-CH2-CH2,
1,75-2,00 ppm: м: 8 H: циклопентан,
2,65 ppm: т: 2 H: CH3-CH2-CH2-CH2-,
4,83 ppm: с: 2 H: CH2-C6H4-,
7,20-7,75 ppm: м: 8 H: ароматические.
Масс-спектр: MH+: 405.
Пример 2.
Трифторацетат 2-н-бутил-1 -[(2' -карбокси-4-бифенилил)-метил]-4 - спироциклопентан-2-имидазолин-5-она. (Способ 1).
А) 2-н-бутил-4-спироциклопентан-2 -имидазолин-5-он.
Этиловый эфир 1-аминоциклопентанкарбоновой кислоты получается в соответствии с методом Adkins et Billica (J. Amer. Chem. Soc. 1948, 70, 3121).
Этилвалеримидат получается в соответствии с методом Mac. Elvain (J. Amer. Chem Soc. 1942, 64, 1825-1827), затем выделяется из своего хлоргидрата под действием карбоната калия и экстракции посредством ДХМ.
Этиловый эфир 1-аминоциклопентанкарбоновой кислоты (1,57 г) и этилвалеримидат (1,56 г) растворяют в 12 мл ксилола, содержащего 6 капель уксусной кислоты. После нагревания в течение шести с половиной часов с обратным холодильником реакционную среду концентрируют под вакуумом, затем остаток хроматографируют на силикагеле при элюировании смесью: хлороформ/метанол/уксусная кислота (94/4/2: об/об/об). Фракцию, содержащую ожидаемый продукт, выпаривают несколько раз в присутствии ксилола, а затем бензола, чтобы удалить уксусную кислоту. Получают 1,91 г продукта в виде густого жидкого масла.
ИК(CHCl3):
1720 см-1: C O,
1635 см-1: C N.
Замечание: тот факт, что не видна полоса между 1500 и 1600 см-1 указывает, что в растворе хлороформа продукт представляет собой имидазолинон-5.
Спектр ЯМР:
0,92 ppm: т: 3 H: CH3 (нBu),
1,35 ppm: сек: 2 H: CH3-CH2-,
1,50-1,93 ppm: м: 10 H: CH3-CH2-CH2 и циклопентан,
2,33 ppm: т: 2 H: CH3-CH2-CH2-CH2-,
10,7 ppm: м: NH.
Масс-спектр: MH+: 195.
Полученный на стадии А 2-н-бутил-4 -спироциклопентан-2 имидазолин-5-он также может быть получен в соответствии с другой методикой, описанной ниже, используя в качестве исходного реагента циклопентанон.
а) 1-аминоциклопентаннитрил.
Эта стадия осуществляется в соответствии с методикой A.Strecker (Org. Synth 1955, 3).
В колбе растворяют 1,97 г цианида натрия в 3,9 мл воды и добавляют раствор, содержащий 2,33 г хлорида аммония в 5,9 мл воды и 3,5 мл 20% аммиака, наконец, прибавляют в колбу 3 г циклопентанона в 3,8 мл метанола. После перемешивания в течение 1 ч 30 мин выдерживают при температуре 60oC в течение 45 мин, затем прекращают нагревание, поддерживают перемешивание в течение 45 мин, затем охлаждают до температуры 25oC. Экстрагируют несколько раз метиленхлоридом. Сушат на сульфате натрия, фильтруют и концентрируют под вакуумом. Получают 4 г ожидаемого продукта в виде маслянистой жидкости.
Полученный 1-аминоциклопентаннитрил растворяют в 300 мл ацетона и прибавляют при перемешивании раствор, содержащий 2,25 г дигидратированной щавелевой кислоты в 200 мл ацетона. Образованный осадок центрифугируют, промывают ацетоном, затем сушат.
m 4,71 г, Тпл 220oC.
Это соединение является гемиоксалатом 1-аминоциклопентаннитрила.
b) 1-аминоциклопентанацетамид.
Эта стадия осуществляется в соответствии с методикой J. Zabichy (The Chemistry of Amides, Intersciences, New York, 1970, 119).
Обрабатывают 5,1 г оксалата, полученного на предыдущей стадии, в течение 45 мин при перемешивании с 7,65 мл концентрированной серной кислоты (d 1,84). Наблюдают газовыделение, температура поднимается до 100oC. Охлаждают до температуры 35oC и приливают к смеси лед-концентрированный аммиак (10 г/ 2,8 мл). Образуемую суспензию экстрагируют 6 раз подряд при помощи хлороформа, содержащего 5% метанола. Добавляют 3 мл раствора аммиака (d0,92) к водной фазе и снова экстрагируют хлороформом, содержащим метанол (1/0,5 по об. ). Объединенные органические фазы сушат на сульфате натрия, фильтруют и концентрируют. Ожидаемый продукт получается в виде твердого вещества белого цвета.
m 3,79 г, Тпл 95oC.
Результаты анализа и спектр МК позволяют подтвердить структуру.
с) 2-н-бутил-4-спироциклопентан-2 -имидазолин-5-он.
Эта стадия осуществляется в соответствии с методикой H.Takenaka et al. Heterocycles, 1989, 29 (6), 1185 -89.
Помещают 3 г соединения, полученного на предыдущей стадии, в 70 мл безводного ТГФ и 3,3 мл триэтиламина и прибавляют при перемешивании 3 мл валерилхлорида в 10 мл безводного ТГФ. Образуется суспензия белого цвета. Полученное промежуточное соединение, однако не выделяемое в чистом виде, является (N-валерил)-1 -аминоциклопентанкарбоксамидом. Прибавляют 6 г гидроксида калия в виде таблеток, 7 мл воды и 16 мл метанола. Нагревают с обратным холодильником в течение 2 ч 30 мин, затем прибавляют 9 г хлорида аммония. После перемешивания в течение 15 мин концентрируют под вакуумом. Полученный остаток извлекают посредством 40 мл воды и экстрагируется при помощи 10 мл этилацетата, а затем 2 раза по 5 мл этилацетата. Объединенные органические фазы сушат на сульфате натрия и фильтруют. Фильтрат концентрируют досуха. Получают 4,85 г ожидаемого продукта. Спектр ЯМР подобен спектру, описанному перед этим. Можно получить хлоргидрат этого соединения в результате прибавления концентрированной хлороводородной кислоты. Хлоргидрат плавится при температуре 240oC, возгоняясь при этом.
В) 2-н-бутил-4-спироциклопентан-1 -[2'-трет(бутоксикарбонил-4- бифенилил) -метил]-2-имидазолин-5-он.
Растворяют 970 мг продукта, полученного на стадии А, в 10 мл ДМФА. Прибавляют 270 мг метилата натрия и оставляют при перемешивании в течение 15 мин при КТ. Добавляют 2,08 г 4-бромметил -(2'-трет-бутоксикарбонил)-бифенила к суспензии, затем спустя 30 мин нагревают при температуре 40oC в атмосфере азота в течение 3 ч 30 мин. Реакционную среду извлекают смесью, содержащей 100 мл этилацетата, 10 мл воды и 1 мл насыщенного раствора бикарбоната натрия. Органическую фазу промывают насыщенным раствором хлорида натрия, затем сушат на сульфате натрия и выпаривают досуха. Остаток хроматографируют на силикагеле при элюировании смесью: этилацетат/толуол (1/2: об/об). Получают 1,25 г ожидаемого продукта, который кристаллизуется.
Тпл 63-66oC.
Спектры ИК, ЯМР и масс-спектр, а также Rf. являются идентичными тем, которые получены на стадии D примера 1.
С) Трифторацетат 2-н-бутил-1-[(2' -карбокси-4-бифенилил)-метил]-4- спироциклопентан-2-имидазолин-5-она.
Перемешивают 1,22 г продукта, полученного на предыдущей стадии, в течение 40 мин в растворе, содержащем 6 мл ДХМ и 8 мл ТФК. После концентрирования под вакуумом остаток извлекают этиловым эфиром; образованный осадок белого цвета фильтруют, промывают эфиром, затем сушат под вакуумом. Получают 1,15 г ожидаемого продукта.
Тпл 176-178oC.
Спектры ИК, ЯМР и масс-спектр являются идентичными тем, которые получены в примере 1Е; аналогично Rf, наблюдаемое в ТСХ, является идентичным.
Пример 3.
Трифторацетат 2-н-бутил-1 -[(2'-карбокси-4-бифенилил)-метил] 4 -спироциклопентан-2-имидазолин-5-она. (Способ 3).
А) Получают 2-н-бутилбензимидазол в соответствии с методикой W.O. Pool (J. Amer. Chem. Soc. 1937, 59, 178] затем получают 2-н-бутил-4,5,6,7 -тетрагидробензимидазол в соответствии с методикой M.Hartmann et L Panizzon (Helv. Chim. Acta, 1938, 21, 1692-1694).
Тпл 145oC.
Спектр ЯМР:
0,82 ppm: т: 3 H: CH3 (нBu),
1,23 ppm: сек: 2 H: CH3-CH2-,
1,50 ppm: кв: 2 H: CH3-CH2-CH2-,
1,65 ppm: с: 4 Н; H5, H6 (тетрагидробензимидазол),
2,35 ppm: с: 4 H: H4, H7 (тетрагидробензимидазол),
2,45 ppm: т: 2 H: CH3-CH2-CH2-CH2-,
11,1 ppm: м: NH.
Масс-спектр: M+: 178.
В) 2-н-бутил-4-спироциклопентан-1 -[(2'-трет-бутоксикарбонил-4- бифенилил -метил]-2-имидазолин-5-он.
Растворяют 1 г продукта, полученного на предыдущей стадии, в 45 мл ДМФА с 303 мг метилата натрия и несколькими мг метиленовой сини. Барботируют кислород в реакционную среду, которая освещается УФ-лампой. Спустя 15 мин прибавляют 2,14 г 4 -бромметил-(2'-трет-бутоксикарбонил) -бифенила, затем спустя 1 ч реакционную среду извлекают 300 мл этилацетата с добавкой 50 мл воды и 5 мл насыщенного раствора бикарбоната натрия. Потом органическую фазу промывают насыщенным раствором хлорида натрия, затем сушат на сульфате натрия и выпаривают досуха. Остаток хроматографируют на силикагеле при элюировании смесью: этилацетат/толуол (1/2: об/об) Получают 610 мг ожидаемого продукта, который кристаллизуется.
Тпл 62-65oC.
Спектры ИК, ЯМР, масс-спектр, а также Rf являются идентичными тем, которые получены ранее для того же самого соединения.
С) Трифторацетат 2-н-бутил-1-[(2'-карбокси-4-бифенилил)-метил]-4 - спироциклопентан-2-имидазолин-5-она.
Это соединение получается путем обработки в кислой среде, как описано на последней стадии примера 1 и примера 2. Физико-химические данные являются идентичными тем, которые были получены для того же самого соединения, приготовленного по способам 1 или 2.
Пример 4.
2-н-бутил-4,4-диметил-1 -[(2'-трет -бутоксикарбонил-4- бифенилил) -метил]-2 -имидазолин-5-он и
трифторацетат 2-н-бутил-1-[(2' -карбокси-4-бифенилил)-метил]-4,4 - диметил-2-имидазолин-5-она. (Способ 1).
А) 2-н-(бутил-4,4-диметил-2 -имидазолин-5-он.
Получают этиловый эфир альфа-аминоизомасляной кислоты в соответствии с методикой R. Jacquier et al. (Bull. Soc. Chim. France, 1971, 3, 1040-1051). Растворяют 650 мг этого соединения и 780 мг этилвалеримидата в 8 мл ксилола, содержащего 4 капли уксусной кислоты, и нагревают с обратным холодильником в течение 7 ч. Затем реакционную среду концентрируют под вакуумом, а остаток хроматографируют на силикагеле при элюировании смесью: хлороформ/метанол /уксусная кислота (95/5/2 по об.). После нескольких выпариваний с ксилолом, а затем с бензилом для удаления уксусной кислоты получают 560 мг ожидаемого продукта, который кристаллизуется.
Тпл 35-38oC.
ИК (CHCl3):
1725 см-1 C O,
1635 см-1: C N.
Замечание: отсутствие сигнала между 1500 и 1600 см-1 подтверждает, что соединение, находящееся в растворе хлороформа, является 2-имидазолин-5 -оном.
Спектр ЯМР:
0,92 ppm: т: 3 H: CH3 (нBu),
1,20 ppm: с: 6 H: C(CH3)2,
1,38 ppm: сек: 2 H: CH3-CH2,
1,63 ppm: кв: 2 H, CH3-CH2-CH2-,
2,38 ppm: т: 2 H: CH3-CH2-CH2-CH2-,
10,7 ppm: м: 1 H: N-H.
Масс-спектр: MH+: 169.
В) 2-н-бутил-4,4-диметил-1-[(2' -трет-бутоксикарбонил-4-бифенилил)-метил] 2-имидазолин-5-он.
Растворяют 520 мг продукта, полученного на предыдущей стадии, в 10 мл ДМФА. Прибавляют 167 мг метилата натрия и перемешивают в атмосфере азота в течение 15 мин. Затем прибавляют 1,25 г 4-бромметил-(2'-трет-бутоксикарбонил) -бифенила и оставляют при перемешивании в течение 3 ч 30 мин при температуре 40oC. Реакционную среду извлекают с помощью 150 мл этилацетат, затем 20 мл воды и 2 мл насыщенного раствора бикарбоната натрия. Органическую фазу промывают насыщенным раствором хлорида натрия, сушат на сульфате натрия и выпаривают досуха. Остаток хроматографируют на силикагеле при элюировании смесью: этилацетат/толуол (1,2/2:об/об). Получают 570 мг ожидаемого продукта, который кристаллизуется.
Тпл 98-100oC.
ИК (CHCl3):
1710-1720 см-1: C O, C O (имидазолинон, сложный эфир);
1625 см-1: C N
Спектр ЯМР:
0,78 ppm: т; 3 H: CH3 (нBu),
1,08 ppm: с: 9 H: C(CH3)3,
1,15 ppm: с: C(CH3)2 и 1,20 ppm: сек: CH3-CH2-: 8 H,
1,45 ppm: кв: 2 H: CH3-CH2-CH2-:
2,30 ppm: т: 2 H: CH3-CH2-CH2-CH2-,
4,65 ppm: с: 2 H: CH2-C6H4-,
7,15-7,65 ppm: м: 8 H: ароматические H.
Изучение Я.Э.О. (Ядерный Эффект Оверхаузера) подтверждает положение заместителей 5-он и 4,4-диметил на имидазолиноне.
Масс-спектр: MH+: 435.
С) Трифторацетат 2-н-бутил-1-[(2' -карбокси-4-бифенилил)-метил]-4,4 - диметил-2-имидазолин-5-она.
Обрабатывают 460 мг продукта, полученного на предыдущей стадии, посредством 3 мл ДХМ и 4 мл ТФК в течение 45 мин. После концентрирования под вакуумом остаток извлекается эфиром, а образованный осадок фильтруют, промывают в эфире, затем сушат под вакуумом. Получают 450 мг ожидаемого продукта в виде твердого вещества белого цвета.
Тпл 168-171oC.
Спектр ЯМР:
0,82 ppm: т: 3 H: CH3 (нBu),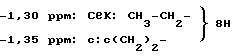
1,55 ppm: кв; 2 H: CH3-CH2-CH2-,
2,62 ppm; т: 2 H: CH3-CH2-CH2-CH2-,
4,82 ppm: с: 2 H: CH2-C6H4 -
7,20-7,75 ppm: м: 8 H: ароматические.
Масс-спектр: MH+; 379.
Пример 5.
1-[(2'-циано-4-бифенилил) -метил] -2 -н-бутил-4-спироциклопентан-2 - имидазолин-5-он и
2-н-бутил-4-спироциклопентан-1-[(2' -(5-тетразолил)-4-бифенилил)- метил] -2 -имидазолин-5-он. (Способ 1).
А) 1-[(2'-циано-4-бифенилил)-метил] -2-н-бутил-4-спироциклопентан-2 - имидазолин-5-он.
В атмосфере азота получают смесь, содержащую 250 мг гидрида натрия (диспергированного в минеральном масле с содержанием 80%) и 5 мл ДМФА, и прибавляют по капле раствор, содержащий 0,97 г 2-н-бутил-4-спироциклопентан-2-имидазолин-5-она (полученного в примере 2 на стадии А) в 10 мл ДМФА. Перемешивают в течение 30 мин при КТ, затем прибавляют раствор, содержащий 1,5 г 4-бромметил-2'-цианобифенила в 10 мл ДМФА. После перемешивания в течение 1 ч при КТ выпаривают ДМФА при пониженном давлении, затем извлекают остаток этилацетатом, промывают органическую фазу водой, потом сушат на сульфате натрия, фильтруют и выпаривают. Остаток хроматографируется на силикагеле при элюировании смесью: ДХМ/этилацетат (9/1: об/об). Выделяют 1,68 г ожидаемого продукта.
Тпл 92-93oC,
В) 2-н-бутил-1-спироциклопентан-1 -[2'-(5-трифенилметилтетразолил)-4 - бифенилил-метил]-2-имидазолин-5-он.
Нагревают с обратным холодильником 1,56 г полученного выше продукта, 2,6 г азида трибутилолова и 30 мл ксилола в течение 66 ч. Затем ксилол выпаривают, а остаток растворяют в 20 мл ДХМ и 5 мл ТГФ, прибавляя 0,8 мл 10н гидроксида натрия и после перемешивания в течение 30 мин 2,5 г тритилхлорида, и оставляют при перемешивании в течение 26 ч. После выпаривания растворителей остаток извлекают этилацетатом и промывают водой, затем 3% раствором гидросульфата калия и водой. Сушат и выпаривают. Остаток хроматографируется на глиноземе при элюировании смесью: гексан/этилацетат (9/1: об/об). Получают 1,97 г ожидаемого продукта.
Тпл 150-152oC
С) 2-н-бутил-4-спироциклопентан-1 -[(2'-(5-тетразолил)-4-бифенилил)- метил] -2-имидазолин-5-он.
Растворяют 1,96 г продукта, полученного на предыдущей стадии, в 10 мл метанола и 10 мл ТГФ. После охлаждения реакционной среды до температуры 5oC прибавляют 1,5 мл 4н хлороводородной кислоты и перемешивают в течение 3 ч при КТ и в течение 1 ч при температуре 30oC. После выпаривания растворителей остаток извлекают водой и доводят до значения pH 12 путем прибавления 10н гидроксида натрия. Водную фазу экстрагируют эфиром, толуолом и снова эфиром. Подкисляют водную фазу до значения pH 2 путем прибавления 1н хлороводородной кислоты, затем экстрагируют этилацетатом, с ушат и выпаривают. Полученное твердое вещество белого цвета сушится при температуре 50oC и давлении 0,05 мм ртутного столба. Получают 840 мг ожидаемого продукта.
Тпл180-181oC.
Спектр ЯМР:
0,75 ppm: т: 3 H: CH3 (нBu),
1,10 ppm: сек: 2 H: CH3-CH2-,
1,20 ppm: кв: 2 H: CH3-CH2-CH2-,
165 2 ppm: м: 8 H: -C5H8,
2,2 ppm: т: 2 H: CH3-CH2-CH2-CH2-,
4,6 ppm: с: 2 H: CH2C6H4-,
7 ppm: с: 4 Н: CH2-C6H4-,
7,35-7,7 ppm: м: 4 H: H3 4', 5', 6' ароматические.
Изучение Я. Э. О. Подтверждает положение заместителя 5-он на имидазолиноне.
D) Калиевая соль 2-н-бутил-4 -спироциклопентан-1-[2'-(5-тетразолин)-4 - бифенилил)-метил]-2-имидазолин-5-она.
Растворяют 970 мг соединения, полученного на предыдущей стадии, в 40 мл смеси изопропанол-метанол (1/1: об/об), устанавливают значение pH 12 путем прибавления 85% раствора гидроксида калия к смеси метанол-вода (20/1: об/об). Выпаривают, извлекают остаток изопропанолом и выпаривают снова. Остаток растворяют в 20 мл изопропанола, слегка нагревая, затем дают вернуться в комнатной температуре. Декантируют, выпаривают фильтрат, затем извлекают остаток гептаном. После порошкования продукт уплотняют; фильтруют его, затем снова промывают гептаном и сушат под вакуумом. Получают 945 мг ожидаемой калиевой соли.
Тпл=142-144oC.
Элементный анализ: C25H27KH6O•H2O
Вычислено: C: 61,95; H: 6,03; N: 17,34
Найдено, С: 62,02; H: 6,13; N: 17,14
Пример 6.
Трифторацетат 2-н-бутил-1 -[(2'-карбокси-4-бифенилил)-метил]-4-(4 - спиротетрагидропиран)-2-имидазолин-5-она и
2-н-бутил-4-(4 -спиротетрагидропиран)-1-[(2'-трет -бутоксикарбонил-4- бифенилил)-метил]-2 -имидазолин-5-он. (Способ 2).
А) 4-амино-4 -тетрагидропиранкарбоновая кислота получается, исходя из 4 -тетрагидропиранона в соответствии с методом, описанным в германском патенте DE 2215721.
В) 4-(N-бензилоксикарбониламино)-4 -карбокситетрагидропиран.
Помещают 1,015 г соединения со стадии А в 12 мл воды и обрабатывают при температуре 10oC посредством 1,22 мл диизопропилэтиламина, затем 3,33 г N -(бензилоксикарбонилокси)-сукцинимида, растворенного в 12 мл ацетонитрила. Спустя 1 ч 15 мин реакционную среду разбавляют посредством 70 мл этилацетата и 10 мл воды, затем доводят до значения pH 2 путем прибавления насыщенного раствора бисульфата калия. После декантации органическую фазу промывают насыщенным раствором хлорида натрия, сушат на сульфате натрия, затем выпаривают под вакуумом. Остаток разбавляют в 60 мл эфира, затем прибавляют 7 ммоль дициклогексиламина. Образованный осадок фильтруют и промывают эфирам; затем его растворяют в смеси этилацетат-вода и доводят раствор до значения pH 1,5 путем прибавления насыщенного раствора бисульфата калия. Органическую фазу декантируют, промывают насыщенным раствором хлорида натрия, выпаривают под вакуумом и получают 1,9 г твердого вещества белого цвета.
Тпл=110-115oC.
С) N-(2'-трет-бутоксикарбонил-4 -бифенилилметил)-4-(N - бензилоксикарбониламино)-4 -тетрагидропиранкарбоксамид.
Растворяют 850 мг соединения, полученного на стадии В, в 15 мл ДМФА и прибавляют эквимолярные количества 4 -аминометил-(2'-трет-бутоксикарбонил) -бифенила, ДИПЭА, а затем БОФ (с 10% избытком). Спустя 40 мин реакционную среду извлекают посредством 200 мл этилацетата и 200 мл воды. Органическую фазу декантируют, затем промывают 2 раза насыщенным раствором бикарбоната натрия, 2 раза 5% раствором бисульфата натрия, затем 1 раз насыщенным раствором хлорида натрия. После сушки на сульфате натрия органическую фазу выпаривают досуха. Получают 1,8 г ожидаемого продукта.
D) N-(2'-трет-бутоксикарбонил-4 -бифенилил-метил)-4-амино-4 - тетрагидропиранкарбоксамид.
Продукт, полученный на стадии С, растворяют в 30 мл метанола. Прибавляют 400 мг палладия с содержанием 10% на угле и гидрируют при атмосферном давлении. Спустя 1 ч катализатор отфильтровывают, затем фильтрат концентрируют под вакуумом. Остаток хроматографируют на кремнеземе при элюировании смесью: этилацетат-метанол -раствор аммиака с концентрацией 33% (99/1/0,5: об/об/об). Получают 0,93 г ожидаемого продукта в виде твердого вещества белого цвета.
Тпл 125-127oC.
Спектр ЯМР:
8,50 ppm: т: 1 H: амидный H,
7,60-7,05 ppm: м: 8 H: ароматические H,
4,25 ppm: д: 2 H: CH2-C6H4,
3,70-3,50 ppm: м: 4 H: CH2 в положениях 2 и 6 тетрагидропирана,
2,00-1,80 ppm: м: 4 H: CH2 в положениях 3 и 5 тетрагидропирана,
1,05 ppm: с: 9 H: тBu.
Е) 2-н-бутил-4-(4 -спиротетрагидропиран)-1-[(2' -третбутоксикарбонил-4- бифенилил)-метил] -2-имидазолин-5-он.
Нагревают в течение 3 ч при температуре 110oC смесь, содержащую 0,9 г соединения, полученного на стадии D, 327 мг метилортовалерата и 2 капли уксусной кислоты. Реакционную среду извлекают посредством 100 мл этилацетата, затем промывают насыщенным раствором бикарбоната натрия, насыщенным раствором хлорида натрия, потом сушат на сульфате натрия и выпаривают этилацетат. Полученный остаток хроматографируют на кремнеземе при элюировании смесью: этилацетат/толуол (2/1: об/об) Получают 550 мг ожидаемого продукта в виде воска.
Спектр ЯМР:
7,05-7,60 ppm: м: 8 H: ароматические H,
4,63 ppm: с: 2 H: CH2-C6H4-,
3,85-3,55 ppm: м: 4 H: CH2 в положениях 2 и 6 тетрагидропирана,
2,30 ppm: т: 2 H: CH2-C3H7,
1,05-1,80 ppm: м: 8 H: CH2-CH2-CH2 -CH3 и CH2 в положениях 3 и 5 тетрагидропирана,
1,03 ppm: с: 9 H: тBu,
0,75 ppm: т: 3 H: (CH2)3-CH3.
ИК (CHCl3):
1710-1720 см-1: C O, C O,
1625 см-1: CN.
F) Трифторацетат 2-н-бутил-4-(4 -спиротетрагидропиран)-1-[(2'-трет- бутоксикарбонил-1-бифенилил)-метил]-2 -имидазолин-5-она.
Обрабатывают 530 мг продукта, полученного на предыдущей стадии, посредством 4 мл дихлорметана и 5 мл ТФК в течение 45 мин. После выпаривания под вакуумом остаток извлекают эфиром, образованный осадок фильтруют, промывают в эфире, затем сушат под вакуумом. Получают 510 мг ожидаемого продукта.
Тпл 159-162oC.
Спектр ЯМР:
7,80-7,10 ppm: м: 8 H: ароматические H,
4,80 ppm: с: 2 И: CH2-C6H4-,
4,00-3,75 ppm: м: 4 H: CH2 в положениях 2 и 6 тетрагидропирана,
2,60 ppm: т: 2 H: CH2-C3H7,
1,45-2,00 ppm: м: 6 H: CH2-CH2-CH2 -CH3 и CH2 в положениях 3 и 5 тетрагидропирана,
1,30 ppm: сек: 2 H: CH2CH2-CH2-CH3,
0,80 ppm: т: 3 H: (CH2)3-CH3.
Пример 7.
Трифторацетат 2-н-бутил-1 -[(2'-карбокси-4-бифенилил)-метил]-4 -[спиро- (1-бензил-4-пиперидин)]-2 -имидазолин-5-она и
2-н-бутил-4-[спиро-(1-бензил-4 -пиперидин)]-1-[(2'-третбутоксикарбонил - 4-бифенилил)-метил]-2-имидазолин-5-он. (Способ 1).
А) 4-амино-1-бензил-4 -пиперидинкарбоновая кислота получается, исходя из N-бензил-4-пиперидона в соответствии с методом, описанным в германском патенте DE 2 215 721.
В) 4-амино-1-бензил-4 -этилпиперидинкарбоксилат. К раствору, содержащему 13 г хлороводородной кислоты в 50 мл этанола, прибавляют 3,80 г соединения, полученного на стадии А, при температуре 0oC, затем выдерживают с обратным холодильником в течение 5 ч. После концентрирования под вакуумом остаток промывают в эфире, затем растворяют в смеси эфир-вода, к которой прибавляют насыщенный раствор карбоната калия для достижения значения pH 9. Эфирную фазу декантируют, промывают насыщенным раствором хлорида натрия, сушат на сульфате натрия, затем выпаривают досуха. Получают 3,50 г ожидаемого продукта в виде жидкого масла.
Спектр ЯМР:
7,20-7,40 -ppm: м: 5 H: ароматические H,
4,10 ppm: к: 2 H: CH2-CH3,
3,45 ppm: с: 2 H: CH2 в бензиле,
2,25-2,60 ppm: м: 4 H: CH2 в положениях 2 и 6 пиперидина,
1,12 ppm: т: 3 H: CH3 CH2-.
С) 2-н-бутил-4-[спиро-(1-бензил-4 -пиперидин)]-2-имидазолин-5-он.
Этилвалеримидат получается, как в примере 2 на стадии А. Смешивают 2,06 г этилвалеримидата, 3,40 г соединения, полученного на стадии В, и 8 капель уксусной кислоты с 15 мл ксилола и нагревают с обратным холодильником в течение 6 ч. После концентрирования под вакуумом остаток хроматографируют на силикагеле при элюировании смесью: хлороформ/метанол/уксусная кислота (82/15/3: об/об/об) Получают 2,80 г ожидаемого продукта в результате экстракции хлороформом при pH 9, чтобы удалить уксусную кислоту.
Тпл 170-172oC.
ИК (хлороформ):
1725 см-1: C O,
1640 см-1: C N
Спектр ЯМР:
7,10-7,30 ppm: м: 5 H: ароматические H,
3,45 ppm: с: 2 H: -CH2-C6H5,
1,10-2,75 ppm: 5 M: 14 H: CH2 в положениях 2, 3, 5, 6 пиперидина и (CH2)3-CH3,
0,80 ppm: т: 3 H: (CH2)3-CH3.
D) 2-н-бутил-4-[спиро-(1-бензил-4 -пиперидин)] -1-[(2'-трет- бутоксикарбонил-4-бифенилил)-метил]-2-имидазолин-5-он.
К 2,78 г соединения, полученного на стадии С и растворенного в 25 мл ДМФА, прибавляют 513 мг метилата натрия и спустя 15 мин 4,16 г 4-бромметил-(2'-трет-бутоксикарбонил)-бифенила. Нагревают при температура 40oC в течение 5 ч, затем реакционную смесь извлекают посредством 300 мл этилацетата, 50 мл воды и 5 мл насыщенного раствора бикарбоната натрия. Органическую фазу декантируют, промывают один раз насыщенным раствором хлорида натрия, сушат на сульфате натрия и выпаривают в вакууме. Остаток хроматографируют на кремнеземе при элюировании смесью: этилацетат/метанол (95/5 по об.). Получают 0,98 г ожидаемого продукта.
Тпл=103-106oC.
ИК (CHCl3):
1710-1725 см-1: C O, C O (имидазолин, сложный эфир);
1630 см-1: C N.
Спектр ЯМР:
7,70-7,10 ppm: м: 13 H: ароматические H,
4,70 ppm: с: 2 H: CH2-C6H4-,
3,55 ppm: с: 2 H: CH2-C6H5,
1,20-2,75 ppm: 5 M: 14 H: CH2 в положениях 2, 3, 5, 6 пиперидина и (CH2)3-CH3,
1,15 ppm: с: 9 H: тBu,
0,85 ppm: т: 3 H: (CH2)3-CH3.
Е) Трифторацетат 2-н-бутил-1-[(2'-карбокси-4-бифенилил)-метил]-4-[спиро
(1-бензил-4-пиперидин)]-2-имидазолин-5 -она.
Растворяют 350 мг соединения, полученного на стадии D, в 4 мл дихлорметана и 5 мл ТФК. Спустя 45 мин реакционную среду концентрируют под вакуумом, затем остаток извлекают смесью эфир-гексан, образованный осадок фильтруют, промывают в эфире и сушат под вакуумом. Получают 350 мг ожидаемого продукта.
Тпл 198-200oC.
Спектр ЯМР:
7,05-7,75 ppm: м: 13 H: ароматические H,
4,75 ppm: с: 2 Н: CH2-C6H4-,
4,40 ppm: с: 2 H: CH2-C6H5,
3,20-3,60 ppm: м: 4 H: CH2 в положениях 2 и 6 пиперидина,
2,35 ppm: т: 2 H: CH2-CH2-CH2-CH3,
2,20-1,40 ppm: 3 массива: CH2 в положениях 3 и 5 пиперидина и CH2-CH2-CH2 -CH3,
1,25 ppm: сек: 2 H: CH2-CH2-CH2-CH3,
0,80 ppm: т: 3 И: (CH2)3-CH3
Пример 8.
Дитрифторацетат 2-н-бутил-1-[(2'-карбокси-4-бифенилил)-метил] -4-(4 - спиропиперидин)-2-имидазолин-5-она и
2-н-бутил-4-(4-спиропиперидин)-1 -[(2'-трет-бутоксикарбонил-4- бифенилил) -метил]-2-имидазолин-5-он.
А) 2-н-бутил-4-(4-спиропиперидин)-1 -[(2'-трет-бутоксикарбонил-4- бифенилил) -метил]-2-имидазолин-5-он.
Растворяют 300 мг соединения, полученного в примере 7 на стадии D, в 10 мл метанола. Прибавляют 180 мг палладия с содержанием 10% на угле и гидрируют в течение 3 ч под атмосферным давлением. Отфильтровывают катализатор и концентрируют фильтрат под вакуумом. Получают 200 мг ожидаемого продукта.
Спектр ЯМР:
7,20 7,75 ppm: м: 8 H: ароматические H,
4,75 ppm: с: 2 H: CH2 C6H4-,
3,00-1,70 ppm: 3 массива для 4 CH2 -групп пиперидина,
2,40 ppm: т: 2 H: CH2-CH2-CH3,
1,60 ppm: кв: 2 H: CH2-CH2-CH2-CH3,
1,35 ppm: сек: 2 H: CH2-CH2-CH2-CH3,
1,20 ppm: с: 9 H: тBu,
0,90 ppm: т: 3 H: (CH2)3-CH3.
В) Дитрифторацетат 2-н-бутил-1-[(2'-карбокси-4-бифенилил)-метил]-4- (4 -спиропиперидин)-2-имидазолин-5-она.
Перемешивают 160 мг продукта, полученного на стадии А, в 3 мл дихлорметана и 4 мл трифторуксусной кислоты в течение 45 мин. Концентрируют под вакуумом, остаток извлекают эфиром. Получают каучукоподобное вещество, а затем после сушки под вакуумом пеноподобное вещество.
m 150 мг,
Тпл 80-85oC.
Спектр ЯМР:
7,15-7,80 ppm: м: 8 H: ароматические H,
4,75 ppm: с: 2 H: CH2-C6H4-,
3,20-1,60 ppm: 3 массива: 4 CH2 -группы пиперидина,
2,40 ppm: т: 2 H: CH2-CH2-CH2-CH3,
1,50 ppm: кв: 2 H: CH2-CH2-CH2-CH3,
1,30 ppm: сек: 2 H: CH2-CH2-CH2-CH3,
0,80 ppm: т: 3 H: (CH2)3-CH3.
Пример 9.
Трифторацетат 2-н-бутил-1-[(2'-карбокси-4-бифенилил)-метил]-4,4-дифенил
2-имидазолин-5-она и
2-н-бутил-4,4-дифенил-1-[(2'-трет-бутоксикарбонил-4-бифенилил)-метил] - 2-имидазолан-5-он. (Способ 1).
А) Хлоргидрат валерамидина.
Прибавляют 6 г хлоргидрата этилвалеримидата в растворе 6,75 г аммиака в 80 мл метанола, при температуре 0oC. Спустя 18 ч реакционную среду концентрируют под вакуумом, и получают ожидаемый продукт в виде твердого вещества белого цвета.
В) 2-н-бутил-4,4-дифенил-2 -имидазолин-5-он.
Это соединение получается в соответствии с методикой, описанной J. Nyitrai, K. Lempert, Tetrahedron, 1969, 25, 4265-4275, исходя из дибензоила и хлоргидрата валерамидина.
Тпл 135oC.
ИК (CHCl3):
1725 см-1: C O,
1640 см-1: C N
Спектр ЯМР:
7,20-7,50 ppm: м: 10 H: ароматические H,
2,50 ppm: т: 2 H: CH2-CH2-CH2-CH3,
1,65 ppm: кв: 2 H: CH2-CH2-CH2-CH3,
1,35 ppm: сек: 2 H: CH2-CH2-CH2-CH3,
0,90 ppm: т: 3 H: CH2-CH2-CH2-CH3,
11 ppm: ш. С. NH.
С) 2-н-бутил-4,4-дифенил-1-[(2'-трет-бутоксикарбонил-4-бифенилил)- метил]-2-имидазолин-5-он.
Это соединение получается в соответствии с обычным способом при действии на соединение, полученное на стадии В, 4-бромметил-(2'-трет-бутоксикарбонил)-бифенилом в присутствии метилата натрия в ДМФА.
ИК (CHCl3):
1715-1725 см-1: C O, C O (сложный эфир, имидазолинон),
1635 см-1: C N.
Спектр ЯМР:
7,25-7,80 ppm: м: 18 H: ароматические H,
4,85 ppm: с: 2 H: N-CH2-C6H4-,
2,60 ppm: т: 2 H: CH2-CH2-CH2-CH3,
1,75 ppm: кв: 2 H: CH2-CH2-CH2-CH3,
1,40 ppm: сек: 2 H: CH2-CH2-CH2-CH3,
1,15 ppm: с: 9 H: тBu,
0,90 ppm: т: 3 H: CH3 н-бутила.
D) Трифторацетат 2-н-бутил-1-[(2' -карбокси-4-бифенилил)-метил]-4,4 - дифенил-2-имидазолин-5-она.
Обрабатывают 500 мг продукта, полученного на стадии С, посредством 2,5 мл дихлорметана и 2,5 мл трифторуксусной кислоты при температуре 20oC в течение 40 мин. После концентрирования под вакуумом остаток извлекают смесью эфир -гексан, образованный осадок фильтруют, промывают в гексане и сушат. Получают 440 мг ожидаемого продукта.
Тпл 55-60oC.
Спектр ЯМР:
7,15-7,80 ppm: м: 18 H: ароматические H,
4,85 ppm: с: 2 H: N- CH2-C6H4 -
2,60 ppm: т: 2 H: CH2-CH2-CH2-CH3,
1,70 ppm: кв: 2 H: CH2-CH2-CH2-CH3,
1,40 ppm: сек: 2 H: CH2-CH2-CH2-CH3,
0,90 ppm: т: 3 H: CH3 бутила.
Пример 10.
Трифторацетат 2-н-бутил -3-[(2'-карбокси-4-бифенилил)-метил]-6 - спироциклопентан-5,6-дигидро-1Н-4 -пиримидинона.
А) (1-аминоциклопентил)-уксусная кислота. Циклопентилиденуксусная кислота получается в соответствии с методикой G.A.R.Kon, R.P.Linstead, J. Chem. Soc. 1925, 127, 616. В автоклав помещают 740 мг этой кислоты и 5 мл 20% раствора аммиака и нагревают при 150oC в течение 24 ч. После выпаривания растворителей хроматографируют остаток на колонке с кремнеземом при элюировании смесью; ДХМ -метанол-20% водный раствор аммиака (70/30/1: об/об/об). Получают 330 мг ожидаемой кислоты.
В) (1-аминоциклопентил)-этилацетат.
Растворяют 330 мг кислоты в 10 мл этанола. Охлаждают в ледяной бане и насыщают газообразной хлороводородной кислотой. После выдерживания в течение 24 ч при температуре образования флегмы выпаривают реакционную среду, извлекают остаток раствором карбоната натрия и экстрагируют этилацетатом, затем сушат на сульфате натрия, фильтруют и выпаривают. Получают 312 мг ожидаемого сложного эфира.
С) 2-н-бутил-6-спироциклопентан-5,6 -дигидро-1Н-4-пиримидинон.
Доводят до образования флегмы смесь, содержащую 310 мг соединения, полученного на стадии В, 348 мг этилвалеримидата, 10 мл ксилола и 6 капель уксусной кислоты. Прибавляют снова спустя 2 ч и 18 ч 348 мг этилвалеримидата и после выдерживания в течение 24 ч при температуре образования флегмы всей смеси выпаривают реакционную среду, затем хроматографируют на кремнеземе при элюировании смесью; ДХМ -метанол (97/3: об/об). Получают 153 мг ожидаемого продукта.
D) 2-н-бутил-3-[(2'-трет -бутоксикарбонил-4-бифенилил)-метил]-5,6 - дигидро-1Н-4-пиримидинон.
В атмосфере азота приготавливают смесь, содержащую 10 мл ДМФА и 40 мг гидрида натрия с концентрацией 80% в жидком масле. Прибавляют по капле при комнатной температуре 144 мг соединения, полученного на стадии С и растворенного в 5 мл ДМФА. После перемешивания в течение 30 мин прибавляют 288 мг 4 -бромметил-2'-трет-бутоксикарбонилбифенила, растворенного в 5 мл ДМФА. Оставляют на 2 ч при перемешивании, затем выпаривают, извлекают остаток водой и экстрагируют этилацетатом. Сушат на сульфате натрия, фильтруют и выпаривают, затем очищают методом хроматографии на колонке при элюировании смесью гексан-этилацетат (85/5: об/об). Получают 174 мг ожидаемого продукта.
E) Охлаждают 10 мл трифторуксусной кислоты в водоледяной бане и прибавляют 161 мг соединения, полученного на стадии Д. Оставляют при перемешивании в течение 30 мин, затем выпаривают. Извлекают остаток этиловым эфиром, затем снова выпаривают. Эта операция повторяется, потом сушат остаток под вакуумом. Получают 140 мг ожидаемого соединения в виде аморфного порошка.
Тпл 108-115oC.
Спектр ЯМР:
0,9 ppm: т: 3 H: (CH2)3-CH3,
1,1 -2,1 ppm: м: 12 H: циклопентан и CH2-CH2- CH2-CH3,
2,7 ppm: т: 2Н: CH2-CH2-CH3,
3,1 ppm: с: 2 H: -CH2-CO,
5,1 ppm: с: 2 H: N-CH2-C6H5,
7,2- 7,8 ppm: м: 8 H: ароматические H.
Пример 11.
2-н-бутил-4 -спироциклопентан-1 -[(2'-трет-бутоксикарбонил-4-бифенилил)
метил]-2 -имидазолин-5-тион и
трифторацетат 2-н-бутил-1-[(2'-карбокси -4-бифенилил)-метил]-4 - спироциклопентан -2-имидазолин-5-тиона.
А) 2-н-бутил-4-спироциклопентан-1 -[(2'-трет-бутоксикарбонил-4- бифенилил) -метил]-2-имидазолин-5-тион.
Растворяют 5,63 г соединения, полученного на стадии D Д примера 1, в 40 мл безводного толуола и обрабатывают при температуре 80oC в атмосфере азота посредством 3 г реактива Лавессона. Спустя 6 ч реакционную среду фильтруют и концентрируют. Хроматографируют на кремнеземе при элюировании смесью: ДХМ -этилацетат (95/5: об/об). Получают ожидаемый продукт в виде жидкого масла, которое кристаллизуется на холоду.
m 4,5 г, Тпл 77-79oC.
Спектр ЯМР:
0,90 ppm: т: 3 H: CH3 (нBu),
1,20 ppm: с: 9 H: тBu,
1,35 ppm: сек: 2 H: CH3-CH2-,
1,60 ppm: кв: 2 H: CH3-CH2-CH2-,
1,80-2,10 ppm: м: 8 H: циклопентан,
2,60 ppm: т: 2 H: CH3-CH2-CH2-CH2,
5,35 ppm: с: 2 H: CH2-C6H4-,
7,25-7,80 ppm: м: 8 H: ароматические H.
В) Трифторацетат 2-н-бутил-1-[(2' -карбокси-4-бифенилил)-метил] -4 - спироциклопентан-2-имидазолин-5-тиона.
Обрабатывают 225 мг соединения, полученного на стадии А, посредством 5 мл ДХМ и 5 мл ТФК в течение 30 мин. После концентрирования остаток извлекают эфиром. Ожидаемое соединение получается в виде порошка желтого цвета, который споласкивается гексаном.
m 160 мг, Тпл 185-190oC.
Масс-спектр: MH+: 421.
Спектр ЯМР:
0,78 ppm: т: 3 H: CH3 (нBu),
1,20 ppm: сек: 2 H: CH3-CH2,
1,50 ppm: кв: 2 H: CH3-CH2-CH2-,
1,75-2,00 ppm: м: 8 H: циклопентан,
2,40 ppm: т: 2 H: CH3-CH2-CH2-CH3,
5,20 ppm: с: 2 H: CH2-C6H4-,
7,00-7,65 ppm: м: 8 H: ароматические H.
Пример 12.
2-н-бутил-4-(2 -спироиндан)-1-[(2'-трет-бутоксикарбонил -4-бифенилил) - метил]-2-имидазолин-5-он и
2- н-бутил-1-[(2'- карбокси-4 -бифенилил)-метил]-4-(2-спироиндан)-2 - имидазолин-5-он. (Способ 1).
А) 2-амино-2-инданкарбоновая кислота получается в соответствии с методикой R. M.Pinder, J. Med. Chem. 1971, 14, 9, 892, а соответствующий этиловый сложный эфир получается затем в соответствии с методикой Adkins (ссылка, указанная в примере 2А).
В) 2-н-бутил-4-(2-спироиндан)-2 -имидазолин-5-он.
Растворяют 2,78 г этилового сложного эфира, полученного на стадии А, и 2,5 г этилвалеримидата в 20 мл ксилола в присутствии 60 мкл уксусной кислоты и выдерживают при температуре образования флегмы в течение 3 ч. Снова прибавляют 500 мг этилвалеримидата и выдерживают при температуре образования флегмы в течение дополнительных 3 ч. Реакционную среду концентрируют, затем хроматографируют на кремнеземе при элюировании смесью: гексан-этилацетат -уксусная кислота (3/8/0,3: об/об/об). Чистые фракции объединяются и выпариваются с толуолом. Получают 3,07 г ожидаемого продукта в виде твердого вещества белого цвета.
Тпл 148-150oC.
Спектр ЯМР:
0,90 ppm: т: 3 H: CH3 (нBu),
1,2-1,7 ppm: м: 4 H: CH2-CH2-CH3,
2,4 ppm: т: 2 H: CH2-(CH2)2-CH3,
2,8-3,2 ppm: к: 4 H: 2CH2 (индан),
4,90 ppm: с: 2 H: CH2-C6H4-,
7,2 ppm: м: 4 Н: ароматические H.
С) 2-н-бутил-4-(2-спироиндан)-1 -[(2'-трет-бутоксикарбонил-4-бифенилил)
метил]-2-имидазолин-5-он.
Соединение, полученное на предыдущей стадии, растворяют в 20 мл безводного ДМФА и обрабатывают посредством 450 мл метилата натрия в атмосфере азота. Спустя 20 мин при комнатной температуре прибавляют 3,6 г 4 -бромметил-(2'-трет-бутоксикарбонил) -бифенила и оставляют при перемешивании в течение 6 ч при температуре 40oC. Реакционную среду концентрируют, затем проводят обычные промывания и хроматографируют на кремнеземе при элюировании смесью: дихлорметан -этилацетат (95/5: об/об) получают ожидаемое соединение в виде пенообразного вещества (m 1,84 г).
Спектр ЯМР:
0,80 ppm: т: 3 H: CH3 в нBu,
1,20 ppm: с: 9 H: тBu,
1,20-1,60 ppm: м: 4 Н: CH2-CH2-CH3,
2,40 ppm: т: 2 H: CH2-(CH2)2-CH3,
2,9-3,3 ppm: к: 4 Н: 2CH2 (индан),
4,80 ppm: с: 2 H: N-CH2-C6H4,
7,20-7,80 ppm: м: 12 H: ароматические H.
D) 2-н-бутил-1-[(2'-карбокси-4 -бифенилил)-метил]-4-(2-спироиндан)-2 - имидазалии-5-он.
Растворяют 1,71 г соединения, полученного на предыдущей стадии, в 15 мл ДХМ и обрабатывают посредством 20 мл ТФК. Спустя 30 мин реакционную среду концентрируют, затем извлекают эфиром. После порошкообразования полученное твердое вещество отжимают, споласкивают эфиром и сушат. Получают 1,42 г ожидаемого продукта.
Тпл 217-218oC.
Спектр ЯМР:
0,70 ppm: т: 3 H: CH3 (нBu),
1,10-1,50 ppm: м: 4 Н: CH2-CH2-CH3,
2,30 ppm: т: 2 H: CH2-(CH2)2 -CH3,
2,8-3,3 ppm: к: 4 Н: 2CH2 (индан),
4,70 ppm: с: 2 H: N-CH2-C6H4-,
7,1-7,7 ppm: м: 12 H: ароматические H.
Другие соединения в соответствии с изобретением были получены по одному из способов, описанных выше. Они сведены в табл. 1. Структура каждого из этих соединений подтверждается анализом их спектра ЯМР.
Пример 13.
2-н-бутил-1-[(2'-(1 -имидазолилкарбонил)-4-бифенилил)-метил] - 4-спироциклопентан-2-имидазолин-5-он.

Перемешивают при комнатной температуре в течение 72 ч смесь, содержащую 404 мг соединения, полученного в примере 1 на стадии Е, 15 мл ТГФ и 260 мг карбонилдиимидазола. Реакционную смесь выпаривают, извлекают этилацетатом, промывают водой, затем раствором хлорида натрия, при этом получают 420 мг продукта, который очищается методом хроматографии на кремнеземе при элюировании смесью: ДХМ -этилацетат (70/30: об/об), чтобы получить ожидаемое соединение.
m 230 мг, Тпл 120oC.
Пример 14.
2-н-бутил-1-[(2'-(3 -циано-2 -метилизотиоуреидометил)-4 -бифенилил) -метил] 4-спироциклопентан-2 -имидазолин -5-он.

А) 1-[(2'-аминометил-4-бифенилил) -метил]-2-н-бутил-4-спироциклопентан- 2 -имидазолин-5-он.
Это соединение получается путем гидрирования соединения, полученного в примере 5.
Помещают 1 г соединения, полученного в примере 5 на стадии А, в 15 мл абсолютного метанола и 2,3 мл этанола в присутствии 0,5 г палладия с содержанием 5% на угле и гидрируют при комнатной температуре в течение 24 ч. После обработки получают 730 мг ожидаемого продукта в виде маслянистой жидкости.
B) Выдерживают при температуре образования флегмы в течение 24 ч смесь, содержащую 300 мг соединения, полученного на предыдущей стадии, и 113 мг N-цианимидо-S, S-диметилдитиокарбоната в 3 мл этанола. После обычной обработки реакционную среду очищают методом хроматографии на кремнеземе при элюировании смесью: ДХМ-этилацетат (50/50: об/об) Ожидаемый продукт выделяется в виде твердого вещества белого цвета.
m 307 мг, Тпл 83oC.
Пример 15.
2- н-бутил-1-[(2'-(2 -цианогуанидинметил)-4-бифенилил)-метил] -4-спироциклопентан-2-имидазолин-5-он.
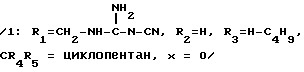
Это соединение получается, исходя из соединения, полученного в предыдущем примере. Помещают 200 мг этого соединения в 10 мл абсолютного этанола, насыщают аммиаком при температуре около 10oC, затем нагревают при температуре 80oC в автоклаве в течение ночи. После концентрирования досуха реакционной среды проводят хроматографирование на кремнеземе при элюировании смесью: ДХМ -метанол (95/5: об/об). Получают 130 мг ожидаемого продукта.
Тпл 100oC.
Пример 16.
Трифторметилсульфонат 2 -н-бутил-4 -спироциклопентан-1-[(2'- трифторметилсульфониламино-4-бифенилил) -метил]-2-имидазолин-5-она.
/1 R1 -NHSO2CF3, R2 H, R3 н -C4H9, CR4R5 циклопентан, X O/,
А) 4-метил-2'-нитробифенил.
Смешивают 11,2 г 2-нитробромбензола и 15 г 4-иодтолуола, нагревают до температуры 195oC и оставляют при перемешивании при этой температуре в течение 3 ч 30 мин. После возвращения к комнатной температуре извлекают посредством ДХМ, доводят до образования флегмы и фильтруют горячий раствор на целите R, затем выпаривают ДХМ.
m 6, 5 г.
Температура кипения Ткип 80-120oC при давлении 0,2 мм ртутного столба.
n
В) 4-бромметил-2'-нитробифенил.
Нагревают с обратным холодильником в течение 5 ч смесь, содержащую 6,5 г 4 -метил-2'-нитробифенила, 5,42 г NБС, 118 мг азобисизобутиронитрила и 500 мл четыреххлористого углерода. Охлаждают до температуры 0oC, центрифугируют и концентрируют фильтрат, в результате чего получают 9 г маслянистого продукта, используемого в таком виде на следующей стадии.
С) 2-н-бутил-1-[(2'-нитро-4 -бифенилил)-метил]-4-спироциклопентан-2 -имидазолин-5-он.
Приготавливают смесь, содержащую 260 мг гидрида натрия с концентрацией 80% в 5 мл ДМФА, и прибавляют при комнатной температуре в атмосфере азота 500 мг 2-н-бутил-4-спироциклопентан-2 -имидазолин-5-она, полученного в примере 2 на стадии А. После перемешивания в течение 15 мин прибавляют 901 мг 4 -бромметил-2'-нитробифенила в 5 мл ДМФА и выдерживают при перемешивании в течение 24 ч. Реакционную среду концентрируют досуха, извлекают смесью вода -этилацетат. Органическую фазу декантируют, сушат на сульфате натрия и фильтруют, затем выпаривают этилацетат. Полученный продукт хроматографируется на кремнеземе при элюировании смесью ДХМ-этилацетат (9/1: об/об). Получают 500 мг ожидаемого продукта.
D) 1-[(2'-амино-4-бифенилил)-метил] -2-н-бутил-4-спироциклопентан-2 - имидазолин-5-он.
Помещают 450 мг продукта, полученного на предыдущей стадии, в 10 мл метанола в присутствии палладия с содержанием 5% на угле при комнатной температуре для проведения гидрирования. После фильтрования катализатора и выпаривания получают 240 мг ожидаемого продукта.
E) В 4 мл ДХМ смешивают 225 мг продукта, полученного на предыдущей стадии, 0,1 мл триэтиламина и прибавляют в атмосфере аргона при температуре -78oC 0,2 мл ангидрида трифторметилсульфокислоты, затем дают вернуться к комнатной температуре. Реакционную среду промывают водой, раствором гидрокарбоната натрия, затем сушат и концентрируют. Получают 150 мг аморфного твердого вещества белого цвета.
Спектр ЯМР:
0,4-1,3 ppm: м: 7 Н: CH3-CH2CH2-,
1,4-2,3 ppm: м: 10 H: CH3-CH2-CH2 -CH2 и циклопентан,
4-4,8 ppm: система АВ: 2 H: N-CH2 -C6H4-,
7 7,6 ppm: м: 8 H: ароматические,
8,3 ppm: с: 1 Н: -NH,
10 ppm: ш.с. 1 Н: CF3SO3H.
Пример 17.
Трифторметилсульфонат 2 -н-бутил-4 -спироциклопентан-1-[(2' - трифторметилсульфониламинометил-4 -бифенил ил)-метил]-2-имидазолин-5-она.
/1 R1 CH2NHSO2CF3, R2 H, R3 н-C4H9, CR4R5 циклопентан, X O/. Синтез осуществляется, исходя из 1-[(2' -аминометил-4бифенилил)-метил]-2- н-бутил -4-спироциклопентан-2-имидазолин-5-она, полученного в примере 14 на стадии А. Помещают 322 мг этого соединения и 0,122 мл триэтиламина в 3,4 мл ДХМ при температуре -70oC и прибавляют 0,294 мл ангидрида трифторметилсульфокислоты. Дают вернуться к комнатной температуре, приливают к разбавленной уксусной кислоте, экстрагируют посредством ДХМ, сушат на сульфате натрия и выпаривают ДХМ. Остаток хроматографируют 2 раза на кремнеземе при элюировании смесью: ДХМ -этилацетат (95/5: об/об), затем (99,5/0,5: об/об).
Получают m 90 мг, Тпл 90oC.
Спектр ЯМР:
0,4-1,2 ppm: м: 7 Н: -CH2-CH2-CH3
1,3-2,45 ppm: м: 10 H: CH2-CH2-CH2 -CH3 и циклопентан,
4,1-5 ppm: м: 4 Н: N-CH2-C6H4 и NH -CH2-C6H4
7,1-7,7 ppm: м: 8 H: ароматические H,
8,4 ppm: с: 1 Н: NH.
Пример 18.
2-н-бутил-1-[(2'-(N -гидроксиацетамид)-4-бифенилил)-метил]-4 - спироциклопентан-2-имидазолин-5-он.
/1 R1 -CO-NHOH, R2 H, R3 н-C4H9, CR4R5 циклопентан, X O/.
Соединение, полученное в примере 2, высвобождается из его соли с трифторуксусной кислотой, извлекая это соединение смесью этилацетат-вода и доводя значение pH раствора до 6 путем прибавления насыщенного раствора гидрокарбоната натрия. Органическую фазу промывают насыщенным раствором хлорида натрия, сушат на сульфате натрия, фильтруют и концентрируют с образованием свободного основания в виде твердого вещества белого цвета.
Растворяют 450 мг этого соединения в хлороформе, прибавляют 860 мл тионилхлорида при температуре 0oC и оставляют при перемешивании в течение 2 ч при комнатной температуре. Раствор концентрируют, а следы тионилхлорида удаляют в результате азеотропной перегонки с толуолом. Полученный таким образом хлорангидрид кислоты прибавляют по каплям в виде раствора в ДМФ к раствору, содержащему 200 мг хлоргидрата гидроксиламина и 700 мл ДИПЭА в 10 мл ДМФ. Спустя 2 ч при температуре 0oC реакционную среду концентрируют, извлекают посредством 100 мл ДХМ и 50 мл воды. Доводят до значения pH 7, экстрагируют органическую фазу, затем сушат на сульфате натрия. После фильтрования раствор концентрируется. Полученный продукт перекристаллизовывается в смеси этилацетат-этиловый эфир-гексан.
m 360 мг, Тпл 85oC.
Пример 19.
2-н-бутил-4 -спироциклопентан-1 -[(2'-уреидо-4 -бифенилил)-метил]-2 - имидазолин-5-он.
/1 R1 NHCONH2, R2 H, R3 н-C4H9, CR4R5 циклопентан, X O/.
Это соединение получается при использовании метода, описанного B.B.Kobu et al. Org. Synth. 1957, 37, 52, исходя из 1-[(2'-амино-4-бифенилил) -метил] -2-н-бутил-4-спироциклопентан-2 -имидазолин- 5-она, полученного в примере 14 на стадии А. Растворяют 1 г последнего соединения в 50 мл 6н хлороводородной кислоты и обрабатывают изоцианатом калия в течение 1 ч при температуре 5oC. Реакционную среду концентрируют, извлекают этилацетатом, промывают гидрокарбонатом натрия, затем насыщенным раствором хлорида натрия. После сушки на сульфате натрия и фильтрования раствор концентрируют, а полученное жидкое масло очищают методом хроматографии на кремнеземе при элюировании смесью: ДХМ-метанол (9/1: об/об)
m 600 мг,
Спектр ЯМР:
0,85 ppm: т: 3 H: CH2-CH3,
1,35 ppm: сек: 2 Н; CH2-CH3,
1,66 ppm: кв: 2 H: CH2-CH2-CH3,
1,7-2 ppm: м: 8 H: циклопентан,
2,45 ppm: т: 2 H: CH2-CH2-CH2-CH3,
4,8 ppm: с: 2 H: -CH2-C6H4,
6,05 ppm: с: 2 H: NH2,
7-8 ppm: м: 9 H: 8 H ароматические + NHCO
Примеры 20 и 21.
1-[(2'- карбокси-4 -бифенилил) -метил]-2-н-пропил-4 -спироциклогексан-2
имидазолин-5-он и
1-[(2'-N-цианокарбоксамид-4 -бифенилил) -метил]-2-н-пропил-4 - спироциклогексан-2 -имилазолин-5-он.
/1 R1 CO-NH-CN, R2 H, R3 н -C3H7, CR4R5 циклогексан, X O/.
А) Хлоргидрат этилбутиримидата:
Это соединение получается в соответствии с методикой Mc Elvain (J. Amer. Chem. Soc. 1942, 64, 1825-1827).
К раствору 10,6 г газообразного хлористого водорода в 20 мл безводного этанола, прибавляют при температуре 0oC 23 мл бутиронитрила, затем после хранения реакционной среды в течение 4 суток при температуре 0oC приливают ее при перемешивании к 200 мл безводного эфира при 0oC; образованный осадок фильтруют, промывают в эфире, затем сушат под вакуумом. Получают 25,8 г ожидаемого продукта.
В) Этилбутиримидат.
Растворяют 16 г имидата, полученного на стадии А, в 100 мл дихлорметана и 50 мл воды и прибавляют 15 г карбоната калия. После декантирования дихлорметан сушат на карбонате калия, затем выпаривают досуха без нагревания.
С) Этиловый эфир 1 -аминоциклогексанкарбоновой кислоты.
1 Аминоциклогексанкарбоновая кислота является коммерческим продуктом. При температуре 0oC 15 г этой аминокислоты прибавляют к раствору, содержащему 23 г газообразного хлористого водорода в 150 мл безводного этанола. Нагревают с обратным холодильником в течение 5 ч, затем концентрируют досуха реакционную среду и извлекают ее эфиром. Полученное твердое вещество белого цвета фильтруют, промывают эфиром, затем растворяют в смеси, состоящей из 300 мл эфира и 100 мл воды. Доводят до значения pH 9 путем прибавления раствора карбоната калия. Органическую фазу декантируют, промывают насыщенным раствором хлорида натрия, сушат на сульфате натрия, затем выпаривают досуха. Получают 14 г ожидаемого продукта в виде жидкого масла.
D) 2-н-пропил-4-спироциклогексан-2 -имидазолин-5-он.
Растворяют 14 г продукта, полученного на стадии С, в 200 мл ксилола, содержащего 0,6 мл уксусной кислоты. Прибавляют половину имидата, полученного на стадии В, и нагревают до образования флегмы. Спустя 1 ч 30 мин прибавляют половину от оставшегося имидата, а затем спустя 4 ч последнюю четверть. После выдерживания при температуре образования флегмы в течение 7 ч в целом реакционную среду выпаривают досуха. Полученное твердое вещество извлекают гексаном, фильтруют, промывают в эфире, затем сушат. Получают 10,3 г ожидаемого имидазолинона.
Тпл 124-125oC.
ИК (CHCl3):
1715 см-1: C 0,
1635 см-1: C N,
Примечание: Соединение, находящееся в растворе, является 5-имидазолиноном согласно значениям полос ИК-спектра.
Е) 2-н-пропил-4-спироциклогексан-1 -[(2'-трет-бутоксикарбонил-4- бифенилил) -метил]-2-имидазолин-5-он.
К суспензии, содержащей в 10 мл диметилформамида 0,24 г гидрида натрия с концентрацией 80% в жидком масле, прибавляют 970 мг имидазолинона, полученного на стадии D. После перемешивания в течение 20 мин в атмосфере азота прибавляют за 5 мин 1,91 г 4-бромметил-2'-трет -бутоксикарбонилбифенила, полученного в соответствии с заявкой на европейский патент 324 377. После перемешивания в течение 1 ч реакционную среду концентрируют наполовину под вакуумом и извлекают посредством 100 мл этилацетата, затем 20 мл воды. Органическую фазу декантируют, промывают насыщенным раствором хлорида натрия, сушат на сульфате натрия, затем концентрируют под вакуумом. Остатки хроматографируют на кремнеземе при элюировании смесью: этилацетат-толуол. Получают 2,10 г ожидаемого продукта в виде воскообразного вещества.
ИК (CHCl3):
1705 1715 см-1: C O, C O (сложный эфир, имидазолинон),
1635 см-1: C N.
Анализ спектра ЯМР подтверждает структуру.
F) 1-[(2'-карбокси-4-бифенилил) -метил]-2-н-пропил-4-спироциклогексан-2
имидазолин-5-он. (Пример 20).
В течение 45 мин перемешивают 1,25 г трет-бутилового сложного эфира, полученного на стадии Е, в смеси, содержащей 11 мл дихлорметана и 15 мл трифторуксусной кислоты. После концентрирования под вакуумом остаток извлекают эфиром. Образованное твердое вещество фильтруют, промывают в эфире, затем сушат. Получают 1,04 г твердого вещества белого цвета.
Тпл 170-172oC.
Спектр ЯМР:
7,10-7,80 ppm: м: 8Н: ароматические,
4,90 ppm: с: 2 H: N- CH2-C6H4-,
2,45 ppm: т: 2 H: CH3-CH2-CH2-,
1,40-1,80 ppm: м: 12 H: CH3-CH2-CH2 и спироциклогексан,
0,90 ppm: т: 3 H: CH3-CH2-CH2 -.
Растворяют 1,60 г полученного выше трифторацетата в 150 мл этилацетата и 20 мл воды. Прибавляют 1н гидроксид натрия для получения значения pH 5,0. Органическую фазу декантируют, промывают насыщенным раствором хлорида натрия, сушат на сульфате натрия, затем выпаривают досуха. Твердый остаток извлекают этиловым эфиром, фильтруют и сушат.
m 1, 1 4 г, Тпл 208-210oC.
G) 1-[(2'-N-цианокарбоксамид-4 -бифенилил)-метил]-2-пропил-4 - спироциклогексан-2-имидазолин-5-он. (Пример 21).
К суспензии, содержащей в 5 мл ДХМ 300 мг соединения, полученного на предыдущей стадии, прибавляют 0,54 мл тионилхлорида. Спустя 1 ч 30 мин реакционную среду концентрируют под вакуумом, затем выпаривают 2 раза с бензолом. Полученный таким образом хлорангидрид кислоты растворяют в 2 мл диоксана и прибавляют к 42 мг цианамида, растворенного в 1 мл диоксана, содержащего 0,2 мл 10н гидроксида натрия. Спустя 1 ч 30 мин реакционную среду разбавляют посредством 150 мл этилацетата, 20 мл воды и доводят до значения pH 5 путем прибавления уксусной кислоты, органическую фазу декантируют, промывают насыщенным раствором хлорида натрия, сушат на сульфате натрия и выпаривают досуха. Остаток хроматографируют на кремнеземе при элюировании смесью; хлороформ/метанол/уксусная кислота (90/8/2: об/об/об). Получают 160 мг ожидаемого продукта в виде твердого вещества.
ИК (KBr):
2150 см-1: C ≡ N.
Масс-спектр: MH+: 429.
Спектр ЯМР:
7,20-7,70 ppm: м: 8 H: ароматические,
4,75 ppm: с: 2 H: N-CH2-C6H4,
2,40 ppm: т: 2 H: CH3-CH2-CH2-,
1,30-1,80 ppm: м: 12 H: CH3-CH2-CH2 и спироциклогексан,
0,85 ppm: т: 3 H: CH3-CH2-CH2 -.
Пример 22.
1-{[2'-N-(4-карбокси-1,3 -тиазол-2 -илацетамид)-4-бифенилил]-метил} -2-н пропил-4-спироциклогексан-2 -имидазолин -5-он.

CR4R5 циклогексан, X O).
Это соединение получается, исходя из соединения, полученного в примере 20.
2-Амино-4-этоксикарбонил-1,3-тиазол получается в соответствии с методикой B.Plouvier et al. J. Heterocycl. Chem. 1989, 26 (6), 1646.
А) 1-{[2'-N-(4-карбэтокси-1,3 -тиазол-2-ил-ацетамид)4-бифенилил]метил} - 2-н-пропил-4-спироциклогексан-2 -имидазолин-5-он.
К раствору, содержащему 404 мг соединения, полученного в примере 20, и 190 мг производного тиазола в 4 мл ДХМ и 1 мл ДМФА, прибавляют 500 мг БОФ и 0,14 мл триэтиламина. Перемешивают в течение 40 ч при комнатной температуре, затем в течение 7 ч при температуре 50oC. Реакционную смесь извлекают с помощью 50 мл этилацетата и промывают 2 раза раствором KHSO4-K2SO4, затем 2 раза насыщенным раствором бикарбоната натрия, потом 1 раз насыщенным раствором хлорида натрия. После сушки на сульфате натрия органическую фазу концентрируют под вакуумом и остаток хроматографируется на кремнеземе при элюировании смесью: этилацетат-толуол. Получают 120 мг ожидаемого продукта.
Тпл 96-98oC.
В) К 110 мг продукта, полученного на предыдущей стадии и растворенного в 1 мл метанола и 1 мл диоксана, прибавляют 0,5 мл 2н гидроксида натрия. После перемешивания в течение 35 мин реакционную среду разбавляют при помощи 10 мл воды и 60 мл этилацетата и доводят до значения pH 5 путем прибавление 1н хлороводородной кислоты. Органическую фазу декантируют, промывают насыщенным раствором хлорида натрия, сушат на сульфате натрия, затем концентрируют. Остаток извлекают эфиром, фильтруют и сушат.
m 100 мг, Тпл 145-148oC.
Спектр ЯМР:
8,0 ppm: с: 1 Н: H в положении 5 тиазола,
7,1-7,7 ppm: м: 8 H: ароматические H,
4,7 ppm: с: 2 H: N-CH2-C6H4-,
2,25 ppm: т: 2 H: CH2-CH2-CH3,
1,2-1,8 ppm: м: 12 H: CH2-CH2-CH3 и циклогексан,
0,85 ppm; т: 3 H: CH2-CH2-CH3.
Пример 23.
2-н-бутил-1-{ [2'-(2 -цианогуанидинкарбонил)-4-бифенилил] -метил} -4- спироциклопент ан-2-имидазолин -5-он.
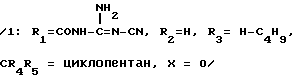
Получают хлорангидрид соединения, синтезированного в примере 2, следующим образом: помещают 1 г этого соединения в 20 мл ДХМ в присутствии 1,8 мл тионилхлорида и перемешивают при комнатной температуре в течение 2 ч. После концентрирования реакционной среды проводят извлечение бензолом, затем снова концентрируют. Выделенный неочищенный продукт используется дальше. Его смешивают с 417 мг дицианодиамида, 0,5 мл 10 н гидроксида натрия, 0,5 мл воды и 10 мл диоксана, затем оставляют при перемешивании в течение 5 ч. Реакционную среду извлекают водой и этилацетатом, прибавляют карбонат калия, затем концентрируют. Полученный остаток хроматографируют на кремнеземе при элюировании смесью: ДХМ-метанол (95/5: об/об). Выделяют 100 мг ожидаемого продукта.
Тпл 105oC.
Пример 24.
Трифторацетат 4 -бензилиден-2-н -бутил-1-[(2'-карбокси)4 -бифенилил - метил]-2-имидазолин-5-она.
/1 R1 CO2H, R2 H, R3 н-C4H9, R4R5 CH-C6H5, X O/.
А) 4-(1-бензилиден-1-валериламинометиламидометил)-2'-бифенил -трет- бутилкарбоксилат.

Исходя из N-БОК- α -дегидро-(L) -фенилаланина, получают N -карбоксиангидрид a -дегидро-(L) -фенилаланина в соответствии с методикой R. Jacquier et al. Tetrahedron Lett. 1984, 25 (26), р. 2775. К 430 мг этого соединения, растворенного в 5 мл ТГФ, прибавляют 644 мг 4-аминометил-2'бифенил -трет-бутилкарбоксилата, перемешивают в течение 2 ч при комнатной температуре, затем прибавляют 1 мл метилортовалерата и выпаривают досуха под вакуумом без нагревания. Остаток нагревается в течение 3 ч при температуре 100oC, концентрируется под вакуумом, затем хроматографируется на кремнеземе при элюировании смесью; гексан-этилацетат (4/1: об/об) Получают 580 мг твердого вещества белого цвета.
Тпл 154oC.
Спектр ЯМР:
1,3 ppm: с: 9 H: тBu,
0,65 ppm: т: 3 Н; CH3 (нBu),
2 ppm: т: 2 Н CH3-CH2-CH2-CH2-CO,
4,4 ppm: д; 1 H: CH2 NH,
6,8 ppm: с: 1 И: CH (=CH-C6H5).
В) 4-бензилиден-2-н-бутил-1-[(2'-трет-бутоксикарбонил-4-бифенилил)- метил]-2- имидазолин-5-он.
Растворяют 440 мг соединения, полученного на стадии А, в 1 мл уксусной кислоты и нагревают в течение 30 мин при температуре 100oC.
Выпаривают досуха под вакуумом и хроматографируют остаток на кремнеземе при элюировании смесью: гексан-этилацетат (4/1: об/об) Получают 130 мг ожидаемого продукта в виде маслянистой жидкости.
Спектр ЯМР:
4,9 ppm: с: 2 H: CH2 (N-CH2-C6H5-).
C) Растворяют 100 мг соединения, полученного на предыдущей стадии, в 1 мл ДХМ и прибавляют 1 мл трифторуксусной кислоты, затем оставляют при перемешивании в течение 40 мин при комнатной температуре и выпаривают под вакуумом. Извлекают несколько раз посредством ДХМ, затем выпаривают. В результате прибавления этилового эфира выпадает твердый осадок белого цвета.
m 101 мг, Тпл 85oC.
Масс-спектр; MH+: 439.
Спектр ЯМР:
0,82 ppm: т: 3Н: CH3 (нBu),
1,3 ppm: сек: 2 H: CH3- CH2-,
1,6 ppm: м: 2 H: CH3-CH2-CH2-,
2,6 ppm: т: 2 H: CH3-CH2-CH2-CH2-,
4,82 ppm: с: 2 H: CH2-C6H4-,
7,05 ppm: с: 1 H:CH-C6H4-,
7,2-8,2 ppm: м: 13 H: ароматические.
Пример 25.
4-бензилиден-1-[(2'-карбокси)-4 -дифенилил-метил]-2-фенил-2 -имидазолин
5-он.
/1 R1 CO2H, R2 H, R3 C6H5, R4R5 CH-C6H5, X O/,
A) 4-бензилиден-2-фенил-5 -оксазолон.
Растворяют 1,8 г гиппуровой кислоты и 0,4 г бикарбоната калия в 4 мл уксусного ангидрида, нагревают в течение нескольких минут при температуре 50oC, затем охлаждают до комнатной температуры и прибавляют 1,49 г бензальдегида. Спустя 1 ч при комнатной температуре прибавляют 20 мл дистиллированной воды при температуре 80oC. Твердое вещество, которое выпадает в осадок, отжимают, промывают водой, этанолом, затем сушат. Получают 1,24 г ожидаемого продукта в виде твердого вещества желтого цвета.
Тпл 215oC.
Спектр ЯМР:
7,4 ppm: с 11 Н:CH-C6H5,
8,1-8,4 ppm: с: 10 Н: ароматические.
В) 4-(1-бензоиламино-1 -бензилиденметиламидометил)-2'-бифенил -трет- бутилкарбоксилат.
Нагревают при температуре 110oC в течение 3 ч смесь, содержащую 500 мг соединения, полученного на предыдущей стадии, 570 мг 4-аминометил-2'-бифенил -трет-бутилкарбоксилата и 10 мл пиридина. Выпаривают под вакуумом, извлекают хлороформом, затем снова выпаривают. Остаток хроматографируют на кремнеземе при элюировании смесью: гексан-этилацетат (3/1, затем 2/1: об/об). Получают 106 мг ожидаемого продукта в виде твердого вещества желтого цвета.
Спектра ЯМР:
1,1 ppm: с: 9 H: тBu,
4,35 ppm: т: 2 H: -CH2-NH -
7,05-7,06 ppm: м: 19 H: ароматические H + C6H5-CH=
8,65 ppm: т: 1 Н: NH-CH2,
9,9 ppm: с: 1 Н: NH-CH.
C) Нагревают в течение 6 ч с обратным холодильником смесь, содержащую в 5 мл уксусной кислоты 1,2 г соединения, полученного на предыдущей стадии, и 1,1 г свежерасплавленного ацетата натрия. Дают охладиться, затем путем прибавления хлороформа осаждают нерастворимое вещество. Фильтрат выпаривается, а остаток хроматографируется на кремнеземе при элюировании смесью: хлороформ-метанол (98/2:об/об). Полученное твердое вещество перекристаллизовывают из этилового эфира.
m= 692 мг, Тпл 120oC.
Спектр ЯМР;
4,95 ppm: с: 2 Н; CH2-C6H4-,
7,1-8,3 ppm: м: 19 H: ароматические H +CH-C6H5.
Примеры 26 и 27.
2-н-бутил-1-[(2'- (2-метил-5 -тетразолил)-4-бифенилил) -метил]-4 - спироциклопентан-2-имидазолин -5-он (Пример 26) и
2-н-бутил-1-[(2'-(1 -метил-5-тетразолил)-4-бифенилил)-метил] -4- спироциклопентан-2-имидазолин-5-он (Пример 27).
В 10 мл ДМФА смешивают 500 мг соединения, полученного в примере 5, и 58 мг гидрида натрия, перемешивают в течение 30 мин, затем прибавляют 179 мг метилиодида и 2 мл ДМФА и оставляют при перемешивании в течение 4 ч при комнатной температуре. Концентрируют реакционную среду, извлекают водой, затем экстрагируют этилацетатом. Сушат на сульфате натрия, фильтруют и выпаривают растворитель. Остаток хроматографируется на кремнеземе при элюировании смесью: гексан-этилацетат: (6/4: об/об). Выделяют 2 фракции:
90 мг соединения по примеру 26,
184 мг соединения по примеру 27.
Спектр ЯМР:
Пример 26;
0,7 ppm: т: 3 H: CH3 (нBu)
1,2 ppm: сек: 2 H: CH3-CH2-,
1,4 ppm: кв: 2 H: CH3-CH2-CH2-,
1,5-1,9 ppm: м: 8 H: циклопентан,
2,25 ppm: т: 2 H: CH3-CH2-CH2-CH2-,
4,15 ppm: с: 3 H: N-CH3,
4,6 ppm: с: 2 H: -N-CH2-C6H4-,
7 ppm: система АА' ВВ': 4 Н: CH2 -C6H4-,)
7,3-7,75 ppm: м: 4 Н: CH2-C6H4-C6H4-.
Пример 27.
0,7 ppm: т: 3 H: CH3 (нBu),
1,15 ppm: сек: 2 H: CH3 CH2-,
1,38 ppm: кв: 2 H: CH3-CH2-CH2,
1,5-1,9 ppm: м: 8 H: циклопентан,
2,2 ppm: т: 2 Н; CH3-CH2-CH2-CH2-,
3,35 ppm: с: 3 H: N-CH3,
4,6 ppm; с: 2 H: N-CH2-C6H4,
7 ppm: система АА', ВВ': 4 Н: N-CH2 -C6H4-,
7,4-7,8 ppm: м: 4 Н: CH2-C6H4-C6H4-.
Пример 28.
2-н-бутил-6-спироциклопентан-3-[(2' -(5-тетразолил)-4 -бифенилил)- метил]-4 -(1H)-5,6-дигидро-4 -пиримидинон.
А) Этилциклопентилиденацетат.
В 40 мл бензола помещают 6 г 80% гидрида натрия и прибавляют по капле 57,1 мл триэтилфосфоноацетата этила при температуре ниже 35oC. После выдерживания в течение 1 ч при комнатной температуре прибавляют по капле 24,3 мл циклопентанона. Нагревают при температуре 65oC в течение 15 мин, затем охлаждают до комнатной температуры и декантируют плавающую на поверхности жидкость. Прибавляют 25 мл бензола, нагревают при температуре 65oC в течение 15 мин, охлаждают, декантируют, затем выделяют плавающую на поверхности жидкость. Операцию повторяют еще 1 раз. В результате выпаривания выделенной жидкости получают 42 г ожидаемого продукта, который перегоняется.
Тпл 102oC при 11 мм рт.ст. m 22,8 г.
В) (1-аминоциклопентил)-ацетамид.
Прибавляют 150 мл газообразного аммиака к 20 г этил - циклопентилиденацетата, полученного перед этим, и нагревают при температуре 150oC в течение 72 ч. Продукт, полученный после выпаривания, очищают методом хроматографии на кремнеземе при элюировании смесью: ДХМ-метанол-20 раствор аммиака (90/10/1: об/об/об). Полученный продукт растворяют в ДХМ, сушат на сульфате натрия. Фильтруют и выпаривают ДХМ, в результате чего получают 7,2 г ожидаемого продукта.
С) 2-н-бутил-6-спироциклопентан-4 -(1Н)-5,6-дигидро-4-пиримидинон.
Нагревают при температуре 100oC в течение 18 ч смесь, содержащую 4,57 г (1 -аминоциклопентил)-ацетамида, полученного выше, 25 мл метилортовалерата и несколько капель уксусной кислоты. После выпаривания избытка ортовалерата остаток извлекают смесью этилацетат-бикарбонат натрия, затем промывают водным раствором хлорида натрия, сушат на сульфате натрия, затем очищают методом хроматографии на кремнеземе при элюировании смесью: ДХМ-метанол (98/2: об/об).
m 5 г.
Спектр ЯМР:
0,75 ppm: т: 3 H: CH3 (нBu),
1,2 ppm: сек; 2 H: CH3-CH2-,
1,3-1,8 ppm: м: 10 Н: CH3-CH2-CH2 и циклопентан,
2 ppm: т: 2 H: CH3-CH2-CH2-CH2-,
2,15 ppm: с: 2 H: CH2-CO,
9,95 ppm: ш.с. 1 Н: NH.
Это соединение является таким, которое получено в примере 10 на стадии С.
D) 2-н-бутил-4-спироциклопентан-1 -[2'-(трифенилметил-5-тетразолил)- 4 -бифенилил-метил]-6 -пиримидинон.
Смешивают в течение 30 мин в атмосфере азота 327 г 80% гидрида натрия в 30 мл ДМФА с 1,5 г пиримидинона, полученного перед этим, и прибавляют 5,27 г 4-бромметил-2'-[трифенилметил-5 -тетразолил]-бифенила. После перемешивания в течение 4 ч при комнатной температуре выпаривают растворители, извлекают этилацетатом и водой, сушат на сульфате натрия и концентрируют. Полученный продукт очищают методом хроматографии на кремнеземе при элюировании смесью: этилацетат/гексан (3/7: об/об).
m 3,2 г.
Е) Помещают 3 г соединения, полученного на предыдущей стадии, в 15 мл метанола и охлаждают при помощи водо-ледяной бани, прибавляют 2,2 мл 4н HCl и оставляют при перемешивании в течение 5 ч при комнатной температуре. После выпаривания извлекают этилацетатом и водой, затем прибавляют гидроксид натрия для создания щелочной среды (pH 11). Декантируют, промывают водную фазу этиловым эфиром и толуолом, затем снова эфиром. Доводят эту водную фазу до значения pH 5 путем прибавления разбавленной хлороводородной кислоты, затем экстрагируют этилацетатом, сушат и концентрируют. Полученный продукт очищается на кремнеземе при элюировании смесью ДХМ-метанол (95/5: об/об) Получают 800 мг ожидаемого продукта.
Спектр ЯМР:
0,85 ppm: т: 3 H: CH3 (нBu),
1,30 ppm: сек: 2 H: CH3-CH2-,
1,40-1,95 ppm: м: 10 Н: CH2-CH2-CH2 -CH3 и циклопентан
2,30 ppm: т: 2 H: CH2-CH2-CH2-CH3,
2,55 ppm: с: 2 H: CH2-CO,
4,95 ppm: с: 2 H: N-CH2-C6H4,
7,05 ppm: м: 4 Н: CH2-C6H4 -
7,55-7,82 ppm: м: 4 Н: CH2-C6H4-C6H4 -.
Пример 29.
Трифторацетат 2-н-бутил -3-[(2'-карбокси-4-бифенилил)-метил]-5 - спироциклопентан-5-(1Н)-5,6-дигидро-4 -пиримидинона.
А) 1-цианоциклопентенкарбоксилат этила.
Это соединение получается в соответствии с методикой Helv. Chim. Acta, 1952, 35 (7), 2561.
Растворяют 9,2 г натрия в 200 мл абсолютного этанола. Половину образованного раствора этилацетата натрия приливается в колбу. К оставшейся половине прибавляют 24,88 г этилцианоацетата и нагревают до образования флегмы. В другую колбу приливают 43,19 г 1,4-дибромбутана, прибавляют по капле в реакционную среду одновременно раствор этилацетата натрия и 1,4-дибромбутана. По окончании прибавления поддерживают орошение флегмой в течение 2 ч. Выпаривают, извлекают смесью этиловый эфир-вода, промывают насыщенным раствором хлорида натрия, потом сушат. Полученный продукт перегоняют при температуре 115-120oC под давлением 11 мм ртутного столба.
m 24 г.
В) 1 -аминометилциклопентанкарбоксилат этила.
Это соединение синтезируется путем каталитического гидрирования 1 -цианоциклопентанкарбоксилата этила.
Помещают 20 г 1-цианоциклопентанкарбоксилата этила в 200 мл этанола с 10% аммиака и гидрируют при температуре 60oC под давлением 100 бар в присутствии родня на оксиде алюминия в течение 72 ч. После фильтрования на целите R и выпаривания остаток хроматографируют на кремнеземе при элюировании смесью: ДХМ-метанол-20% раствор аммиака (98/2/0,5: об/об/об).
m 12,8 г.
С) 2-н-бутил-5-спироциклопентан-4 -(1Н)-5,6-дигидро-4-пиримидинон.
Нагревают с обратным холодильником в течение 13 ч смесь, содержащую 13,12 г соединения, полученного на предыдущей стадии, 13,5 г этилвалеримида в 100 мл ксилола, содержащего несколько капель уксусной кислоты. Выпаривают реакционную среду, извлекают этилацетатом и 10% раствором карбоната натрия, затем сушат и концентрируют.
m=14 г.
Tпл=89 91oC.
Спектр ЯМР:
0,80 ppm т 3Н CH3 (нBu),
1,10 1,80 ppm м 12Н CH3-CH2 -CH2- и циклопентан,
2,05 ppm т 2Н CH3-CH2-CH2-CH2 -,
3,20 ppm с 2Н CH2 (пиримидинон).
10 ppm с 1Н NH-CO.
D) 2-н-бутил-5-спироциклопентан-3 -[(2'-трет-бутоксикарбонил-4-бифенилил) метил]-4-(1Н)-5,6-дигидро-4-пиримидинон.
Помещают 500 мг продукта, полученного на предыдущей стадии, в 40 мл ДМФА в присутствии 115 г гидрида натрия с содержанием 80% в жидком масле в атмосфере аргона и перемешивают при комнатной температуре в течение получаса. Прибавляют 1,08 г 4-бромметил -2'-трет-бутоксикарбонилбифенила и выдерживают в течение 2 ч при перемешивании. После выпаривания остаток извлекают смесью этилацетат-вода, промывается насыщенным раствором хлорида натрия, затем сушится, концентрируется и хроматографируется на кремнеземе при элюировании смесью: этилацетат-гексан (3/7: об/об).
m 280 мг.
Растворяют 250 мг трет-бутилового сложного эфира, полученного на предыдущей стадии, в 10 мл ДХМ. Охлаждают в водоледяной бане, затем прибавляют 5 мл холодной трифторуксусной кислоты и оставляют на холоду при перемешивании в течение 1 ч, затем в течение 1 ч при комнатной температуре. Выпаривают при пониженном давлении. Остаток извлекают этиловым эфиром, затем выпаривают. Операцию повторяют 3 раза, затем извлекают остаток после выпаривания гексаном, тритируют, затем декантируют гексан. Извлекают этиловым эфиром и отфильтровывают осадок.
m 190 мг.
Тпл 153-155oC.
Спектр ЯМР:
0,85 ppm: т: 3 H: CH3 (нBu),
1,35 ppm: сек: 2 H: CH3-CH2-,
1,45-2,20 ppm: м: 10 Н: CH3-CH2-CH2- и циклопентан,
2,80 ppm: т: 2 H: CH3-CH2-CH2-CH2-,
3,80 ppm: с: 2 H: CH3 (пиримидинон),
5,15 ppm: с: 2 H: N-CH2-,
7,25 ppm: м: 8 H: ароматические.
Примеры 30-68 см. в табл.1.
Заявитель исследовал ряд новых соединений с целью изучения их антагонистической активности к ангиотензину II на живом организме (in vivo). Протокол испытаний.
Антагонистический эффект соединений показан с помощью теста на ингибирование изменения давления, вызванного ангиотензином II у нормальных крыс (n 6 животных на группу), осуществленного по методу П.К. Вонга (J.Pharmacol. Exp. Ther. 252(2), 726-732, 1990). Ангиотензин 11 вводили путем внутривенной инъекции, с дозой, равной 40 мг/кг, что вызвало подъем систолического артериального давления примерно на 40 мм рт. ст. Соединения вводили перорально с дозой 3 или 10 мг/кг в виде 6%-ного раствора гуммиарабика. Ниже приводятся результаты в процентах ингибирования изменения давления, вызванного ангиотензином II.
Пример ингибирования (доза)
1 второе соединение 63 (10)
4 50(10)
5 второе соединение 65(3)
10 37(1)
20 35(10)
28 68(3)
30 66 (10)
31 20(10)
32 45(10)
34 60(10)
42 40(10)
44 50(10)
49 45(10).
Сродство ангиотензина II к рецепторам мембраны печени крыс показано в табл.2.
Соединения согласно изобретению могут входить в составы следующих композиций, представленные ниже в качестве иллюстрации:
Таблетки
Соединение (пример 5) 75,00 мг
Моногидратная лактоза 15,37 мг
Модифицированный кукурузный крахмал 22,50 мг
Структурированная натрийсодержащая карбоксиметилцеллюлоза 3,75 мг
Полоксамер (Плуроник Ф-68) 4,50 мг
Безводный коллоидный двуоксид кремния 3,00 мг
Чистая вода
Для таблетки 150 мг
Таблетки
Соединение из примера 5 150,00 мг
Моногидратная лактоза 30,75 мг
Модифицированный кукурузный крахмал 45,00 мг
Структурированная натрийсодержащая карбоксиметилцеллюлоза 7,50 мг
Полоксамер (Плуроник Ф-68) 9,00 мг
Безводный коллоидный двуоксид кремния 6,00 мг
Чистая вода
Для таблетки 300 г
Таблетки
Соединение из примера 5 300,00 мг
Моногидратная лактоза 61,50 мг
Модифицированный кукурузный крахмал 90,00 мг
Структурированная натрийсодержащая карбоксиметилцеллюлоза 15,00 мг
Полоксамер (Плуроник Ф-68) 18,00 мг
Безводный коллоидный двуоксид кремния 12,00 мг
Чистая вода
Для таблетки 600 мг
Желатиновая капсула
Соединение примера 5 5,00 мг
Лактоза 276,50 мг
Модифицированный кукурузный крахмал 50,00 мг
Тальк 25,00 мг
Безводный коллоидный двуоксид кремния 0,50 мг
Стеарат магния 1,00 мг
Для заполненной желатиновой капсулы 358 мг
Желатиновая капсула
Соединение примера 5 12,50 мг
Лактоза 235,00 мг
Модифицированный кукурузный крахмал 50,00 мг
Тальк 25,00 мг
Безводный коллоидный двуоксид кремния 0,50 мг
Стеарат магния 1,00 мг
Для заполненной желатиновой капсулы 324 мг
Желатиновая капсула
Соединение примера 5 25,00 мг
Лактоза 171,50 мг
Модифицированный кукурузный крахмал 50,00 мг
Тальк 25,00 мг
Безводный коллоидный двуоксид кремния 0,50 мг
Стеарат магния 1,00 мг
Для заполненной желатиновой капсулы 273 мгю
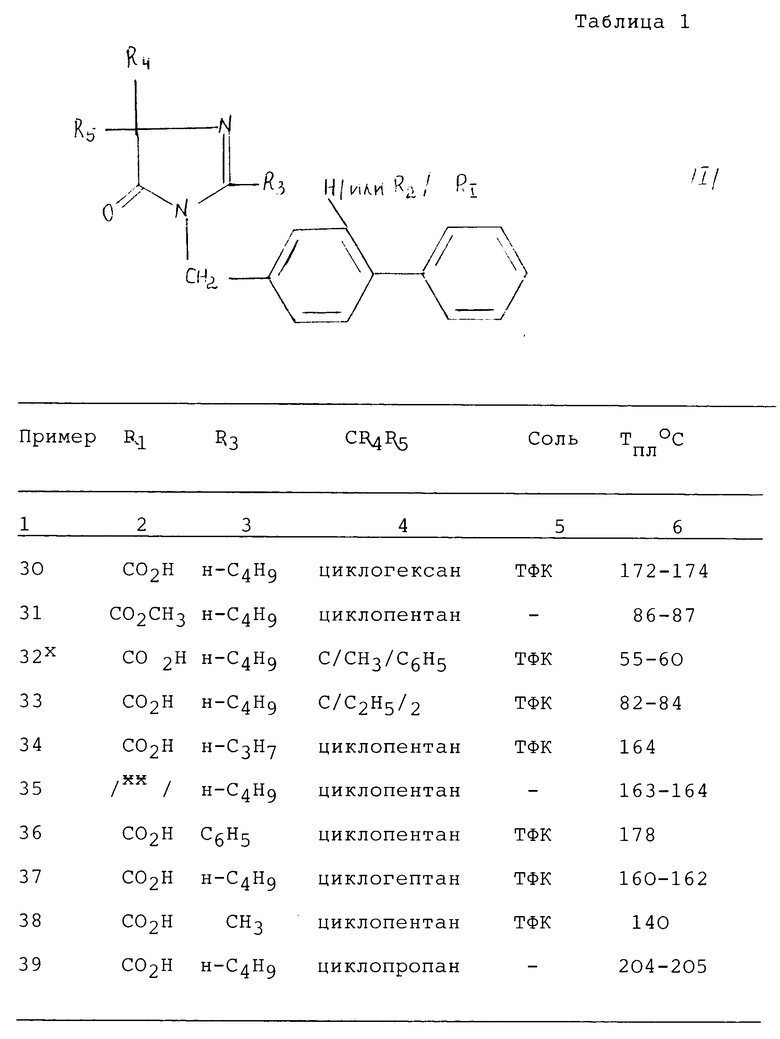

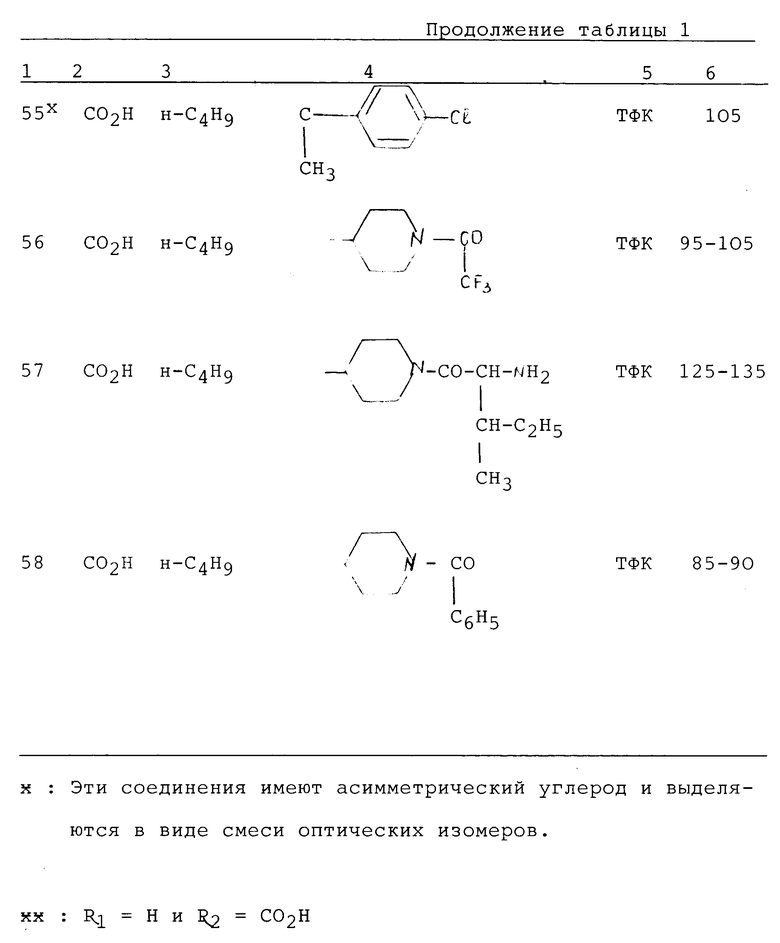



Использование: в медицине в качестве соединений, обладающих антагонистическим действием по отношению к ангиотензину II, могут найти применение при лечении сердечно -сосудистых заболеваний. Сущность: N -замещенные гетероциклические производные формулы I, указанной в описании, в которой R1 и R2 являются одинаковыми или различными и представляют собой каждый независимо друг от друга водород или группу, выбираемую среди алкила C1-C4, циано, тетразолила, метилтетразолила, метилсульфониламино, трифторметилсульфониламино, трифторметилсульфониламинометила, N -цианоацетамида, N-гидроксиацетамида, N -[(4-карбокси)-1,3-тиазол-2-ил] -ацетамида, уреидо, 2 -цианогуанидинкарбонила, 2 -цианогуанидинметила, 1 -имидазолилкарбонила, 3-циано-2 -метилизотиоуреидометила, при условии, что по меньшей мере один из заместителей R1 или R2 отличается от водорода; R3 представляют собой водород, алкил (C1-C6), незамещенный или замещенный одним или несколькими атомами галогена, алкенил (C2-C6), циклоалкил (C3-C7), фенил, фенилалкил (C1-C3), R4 и R5 представляют собой каждый независимо друг от друга алкил (C1-C6), фенил, причем алкил или фенил не замещены или замещены одним или несколькими атомами галогена или одной группой, перфторалкила (C1-C4); R4 и R5 вместе образуют группу формулы =CR7R8, в которой R7 представляет собой водород, а R8 = фенил; или же R4R5, соединенные вместе, представляют собой либо группу с формулой (CH2)n, в которой n = 3-11, если эта группа представляет собой циклогексан, она может быть замещена алкилом (C1-C4) или фенилом; или группу (CH2)pY(CH2)q, где p = q = 2; - Y - = HR6, R6 = ацетил, COF3, COC6H5, остаток Arg, Asp, Cys или Ile, или R4 и R5, соединенные вместе с атомом углерода, с которым они связаны, образуют индан или адамантан; X представляет собой атом кислорода или серы; Z и t равны нулю или один из этих индексов равен нулю, а другой представляет собой единицу; или их соли, а также промежуточные продукты - производные пиримидина и имидазолина и фармацевтическая композиция, содержащая в качестве активного начала соединение I в эффективном количестве. 3 с. и 8 з.п. ф-лы.
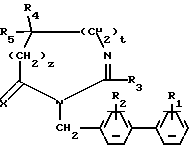
в которой R1 и R2 одинаковые или различные и представляют каждый независимо друг от друга водород или группу, выбираемую из карбокси, алкоксикарбонила (C1 C4), циано, тетразолила, метилтетразолила, метилсульфониламино, трифторметилсульфониламино, трифторметилсульфониламинометила, N-цианоацетамида, N-гидроксиацетамида, N-/(4-карбокси)-1,3-тиазол 2-ил)/-ацетамида, уреидо, 2-цианогуанидинкарбонила, 2-цианогоуанидинметила, 1-имидазолилкарбонила, 3-циано-2- метилизотиоуреидометила, при условии, что по меньшей мере один из заместителей R1 и R2 отличается от водорода;
R3 водород, алкил (C1 C6), незамещенный или замещенный одним или несколькими атомами галогена, алкенил (C2 - C6), циклоалкил (C3 C7), фенил, фенилалкил (C1 - C3), причем названные фенильные группы незамещены;
R4 и R5 представляют каждый независимо друг от друга алкил (C1 C6), фенил, причем алкил или фенил незамещены или замещены одним или несколькими атомами галогена или группой перфторалкила (C1 - C4);
или R4 и R5 образуют вместе группу формулы CR7R8, в которой R7 представляет водород и R8 представляет фенил;
или же R4 и R5 представляют вместе группу (CH2)n, в которой n 3 11, кроме того, если эта группа представляет циклогексан, она может быть замещена алкилом (C1 - C4), или фенилом; или группу (CH2)p Y(CH2)q, в которой p q 2;
Y NR6; R6 ацетил, COF3, COC6H5, остаток аргинина, аспарагиновой кислоты, цистеина или изолейцина;
или же R4 и R5 вместе с атомом углерода, с которым они соединены, образуют индан или адамантан;
X атом кислорода или атом серы;
Z и t 0 или один равен 0, а другой 1,
или их соли.
в которой R3 водород или алкил C1 C6, незамещенный или замещенный одним или несколькими атомами галогена, алкенил C2 C6, циклоалкил C3 C7, фенил, фенилалкил, в котором алкил C1 C3, причем указанные фенильные группы незамещены;
R4 и R5 представляют собой, каждый, независимо друг от друга, алкил C1 C6, фенил, причем алкильные и фенильные группы незамещены или замещены одним или несколькими атомами галогена или группой перфторалкил C1 C4;
или R4 и R5 вместе образуют группу формулы CR7R8, в которой R7 водород, и R8 фенил;
или R4 и R5, связанные друг с другом, представляют собой либо группу формулы (CH2)n, в которой n 3 11, причем если эта группа означает циклогексан, то она может быть замещена алкилом C1 C4 или фенилом, либо группу (CH)p Y(CH2)q, в которой p q 2.
Y NR6, где R6 ацетил, COF3, COC6H5, остаток аргинина, аспарагиновой кислоты, цистеина или изолейцина;
или R4 и R5, связанные вместе с атомом углерода, с которым они соединены, образуют индан или адамантан;
X атом кислорода или серы;
Z и t 0 или один из них равен 0, а другой 1, и их соли,
в качестве промежуточных соединений в синтезе замещенных гетероциклических производных формулы I по п.1.
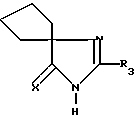
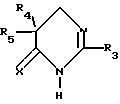

Приоритет по признакам:
20.03.90 при t 0; Z 0; R1 и R2 одинаковые или различные и представляют каждый, независимо друг от друга, водород или группу, выбираемую из карбокси, алкоксикарбонила (C1 C4), циано, тетразолила, метилсульфониламино, трифторметилсульфониламино при условии, что по меньшей мере один из заместителей R1 или R2 не является водородом; R3 алкил (C1 C6), незамещенный алкенил (C2 C6), фенил; фенилалкил (C1 C3), - R4 и R5 представляют каждый независимо друг от друга алкил (C1 C6), фенил, причем алкил или фенил незамещены или замещены одним или несколькими атомами галогена; или же R4 и R5 представляют вместе группу (CH2)n, в которой n 3 6; X представляет атом кислорода.
| EP, заявка N 0051829, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| DE, патент N 3042466, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| US, патент N 4349684, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| EP, заявка N 0226947, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

