Изобретение относится к N-сульфонилиндолиновым производным, к способу их получения и к фармацевтическим композициям на их основе.
Известны некоторые производные N-сульфонилиндола[1], соответствующие формуле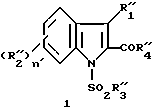
в которой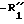 является водородом, алкилом или замещенным или незамещенным фенилом;
является водородом, алкилом или замещенным или незамещенным фенилом;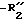 является галогеном, алкилом, -алкокси, нитро или трифторметилом;
является галогеном, алкилом, -алкокси, нитро или трифторметилом;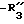 является алкилом, фенилом или алкилфенилом;
является алкилом, фенилом или алкилфенилом;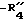 является алкилом, замещенным или незамещенным фенилом, алкокси или фенокси;
является алкилом, замещенным или незамещенным фенилом, алкокси или фенокси;
n' = 0, 1 или 2.
Эти соединения (1) представляют собой промежуточные соединения для получения индольных производных, являющихся активными по отношению к центральной нервной системе, имеющих формулу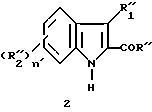
в которой
R'' является алкилом, замещенным или незамещенным фенилом или гидроксилом.
Индолиновые производные, соответствующие данному изобретению, обладают сродством по отношению к вазопрессиновым и оцитоциновым рецепторам.
Вазопрессин является известным гормоном антидиуретического действия, известна также его роль в регулировании артериального давления. Он стимулирует деятельность нескольких типов рецепторов
V1, V2, V1а, V1b и таким образом обладает сердечно-сосудистым, гепатическим, антидиуретическим, рвотным и агрегирующим действием, а также проявляет пролиферативный и митотический эффекты, особенно в сердечно-сосудистых и печеночных тканях. Антагонисты рецепторов вазопрессина могут воздействовать на регулирование центрального или периферического кровообращения, особенно коронарного, почечного и желудочного кровообращения, а также на метаболизм воды и выделение адренокортикотропного гормона (АКТГ). Вазопрессиновые рецепторы аналогично рецепторам оцитоцина найдены также в гладкой мышце матки. Оцитоцин имеет пептидную структуру, аналогичную структуре вазопрессина. Его рецепторы найдены также в миоэпителиальных клетках молочной железы и в центральной нервной системе (Presse Medicale 1987, 16 (10) 481-485, J.Lab. Chin. Med. .1989, 114 (6), 617-632 и Pharmacol^Rev. 1991, 43 (1), 73-108).
Таким образом, соединения, соответствующие изобретению, могут быть полезными, в частности, при лечении заболеваний центральной нервной системы, сердечно-сосудистой системы и в желудочной области людей и животных.
Изобретение относится к индолиновым производным формулы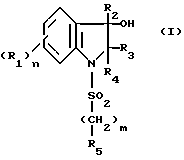
в которой
R1 - атом галогена, C1-C4-алкил, C1-C4- алкокси;
R2 C1-C6-алкил, C3-C7-циклоалкил, фенил, незамещенный или замещенный одной или несколькими группами C1-C4-алкил, или галогеном;
R3 - атом водорода;
R4- карбамоильная группа формулы CONR6R7;
R5-C1-C4; фенил, незамещенный или замещенный одним или несколькими заместителями, выбираемыми из галогена, C1-C4-алкила, C1-C4-алкокси;
R6- C1-C6-алкил, или R6 такой же как R7;
R7 - группа пиперидин-4-ил, незамещенная или замещенная на азоте C1-C4-алкилом, C1-C4 -алкоксикарбонилом; группа (CH2)r , которая сама замещена группой 2-, 3- или 4-пиридил или группой амино, свободной или замещенной одним или двумя C1-C4 -алкилами, карбоксильной группой, группой C1-C4 -алкоксикарбонил, карбамоильной группой, свободной или замещенной одним или двумя C1-C4-алкилами;
или R6 и R7 образуют вместе с атомом азота, с которым они связаны, гетероцикл, выбираемый из морфолина, тиоморфолина, тиазолидина или 2,2-диметилтиазолидина, пиперазина незамещенного или замещенного в положении 4 группой R''8 , ненасыщенного 5-членного цикла, содержащего единственный атом азота, замещенного радикалом R8 или насыщенного 3,4,5, или 6-членного цикла, содержащего единственный атом азота и замещенного радикалами R8 и R9;
R8 означает R1 8 или группу (CH2)r, которая сама замещена гидроксилом или амино-группой, свободной или замещенной одним или двумя C1-C4-алкилами;
R1 8 означает группу (CH2)g, которая сама замещена карбоксильной группой, C1-C4--алкоксикарбонильной группой, бензилоксикарбонильной группой, карбамоильной группой, свободной или замещенной гидроксилом или одним или двумя C1-C4-алкилами или аминокарботиоильной группой, свободной или замещенной одним или двумя C1-C4-алкилами;
R''8 означает R1 8или группу (CH2)2NH2, свободную или замещенную одним или двумя C1-C4- -алкилами;
R9 представляет водород, группу (CH2)rNR10R11, группу азидо или группу NH-бензилоксикарбонил;
R10 и R11 представляют независимо друг от друга водород или C1-C4-алкил;
n- 0,1 или 2;
m- 0,1 или 2;
q- 0,1,2 или 3;
r- представляет 0,1,2 или 3 при условии, что r не равно нулю, когда R8 или R9 находятся в альфа-положении по отношению к амидному межцикличному азоту, а также к их возможным солям.
Соли соединений формулы (1) включают соли с неорганическими или органическими кислотами, которые позволяют провести соответствующее выделение или кристаллизацию соединений формулы (1), такие, как пикриновая кислота, щавелевая кислота, или с оптически активными кислотами, например миндальная кислота или камфарсульфокислота, и с такими кислотами, которые образуют фармацевтически приемлемые соли, такие, как гидрохлориды, гидрогенсульфат, дигидрогенфосфат, метансульфонат, малеинат, фумарат или 2-нафталинсульфонат.
Соли соединений формулы (1) включают также соли с органическими или неорганическими основаниями, например соли щелочных или щелочно-земельных металлов, такие, как соли натрия, калия или кальция, предпочтительными являются соли натрия и калия, или с амином, такими, как трометомол, или соли аргинина, или лизина, или любого фармацевтически приемлемого амина.
Соединения (1) проявляют цис-трансизомерию по отношению к связи 2,3 индолина. Различные изомеры входят в объем изобретения.
Соединения (1), в которых R2 и R4 расположены по одну сторону кольца, называются цисизомерами.
Соединения (1), в которых R2 и R4 располагаются по разные стороны от кольца, называются трансизомерами.
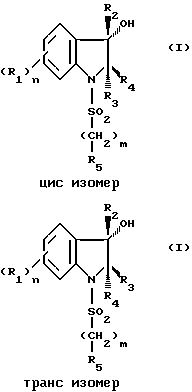
Кроме того, соединения, соответствующие изобретению, имеют 2 асимметричных углеродных атома или более, когда R4 содержит 1 или 2 асимметричных углерода. Оптические изомеры соединений (1) составляют часть изобретения.
В представленном описании под галогеном следует понимать атом фтора, хлора, брома или йода, под алкильной группой обозначаются линейный или разветвленные углеводородные группы.
Предпочтительными соединениями (1), соответствующими изобретению, являются такие, которые удовлетворяют следующим условиям:
R1 является атомом хлора или брома или метокси группой, а n = 1;
R2 представляет собой хлорфенил, метоксифенил или циклогексил;
R4 является группой CONR6R7, в которой R6 и R7 или NR6R7 имеют одно из следующих значений:
NR6R7 является пирролидино группой, которая является замещенной в положении 2 группой (CH2)q, которая сама является замещенной карбоксильной или карбамоильной группой с значением q= 0,1,2 или 3;
NR6R7 является пиперидино группой, которая в положении 4 замещается амино группой, C1-C4 -алкиламино или C1-C4-диалкиламино;
NR6R7 является триазолидино группой, которая замещается группой (CH2)q, которая в свою очередь замещается карбоксильной или карбамоильной группой с значением q= 0,1, 2 или 3.
NR6R7 является пирролидино группой, которая замещается в положении 2 группой (CH2)q, которая сама замещается карбоксильной или карбамоильной группой при значении q= 0, 1, 2 или 3 и которая в положении 4 замещается амино группой, C1-C4 -алкиламино или C1-C4 диалкиламино,
R6 представляет собой C1-C41-алкил, а R7 является группой (CH2)r, которая сама замещается карбоксильной группой или карбомоильной группой при значении r = 1, 2 или 3.
R5 является фенилом, замещенным в положении 3 и 4 или в положении 2 и 4 метокси группой, или R5 представляет собой фенил, замещенный в положении 4 метильной группой, -m = 0.
Наиболее предпочтительными являются соединения (1) в виде цисизомеров.
В описании и примерах используются следующие аббревиатуры.
ДХМ: дихлорметан
Эфир изо: изопропиловый эфир
AcOEt: этилацетат
MeOH: метанол
EtcOH: этанол
Эфир: этиловый эфир
ДМФ: диметилформамид
ТГФ: тетрагидрофуран
ТЭА: триэтиламин
ДМСО: диметилсульфоксид
ДИПЭА: диизопропилэтиламин
ДЦК: N,N' -дициклогексилкарбодиимид
ДБУ: 1,8-диазабицикло [5.4.0]ундек-7-ен
ТБД: 1,5,7-триазабицикло [4.4.0]дек-5-ен
ДБН: 1,5-диазабицикло [4.3.0]нон-5-ен
ДМАП: 4-диметиламинопиридин
ДМП: 3-диметил-2-оксогексагидропиримидинон
ТМЭДА: тетраметилэтилендиамин
ЛДА: литий диизопропиламид
ГМФА: гексаметилфосфорамид
ГОБТ: 1-гидроксибензотриазол гидрат
БОФ: бензотриазолилокситрисдиметиламинофосфоний гексафторфосфат
ТФУ: трифторуксусная кислота
Реактив Лавессона: 2,4-бис(4-метоксифенил)-1,3-дитиа2,4-дифосфэтан-2,4-дисульфид
Т.пл.: точка плавления
Соленой раствор: вода, насыщенная хлоридом натрия
Сухой лед: твердая двуокись углерода
ТСХ: тонкослойная хроматография
ВЭЖХ: высокоэффективная жидкостная хроматография
ЯМР: ядерный магнитный резонанс
с: синглет
м: мультиплет
ус: уширенный синглет
д: дублет
Гидрохлоридная вода: разбавленная хлористоводородная кислота концентрации около 1Н
80%-ный NaH: дисперсия гидрида натрия в минеральном масле
Me: метил
Et: этил; iPr: изопропил, Pr: пропил
iPentil: изопентил
iBu: изобутил
tBu: трет-бутил, Bu: бутил
Bz: бензил
Ph: фенил
Далее изобретение относится к способу получения соединений (1)
Способ получения соединений формулы (1) заключается в том, что подвергают взаимодействию 2 - аминофеноновое производное формулы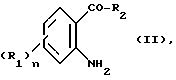
в которой
R1, R2 и n имеют значения, определенные выше, с сульфонильным производным формулы
Hal-SO2 -(CH2)m-R5 (III)
в которой Hal представляет собой галоген, предпочтительно хлор или бром,
m и R5 имеют значения, определенные выше, полученное соединение формулы IV: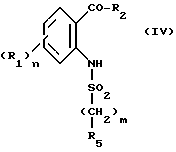
обрабатывают галогенированным производным формулы
Hal'-CH2COA (V)
в которой Hal' представляет собой галоген, предпочтительно бром, а A представляет собой или группу NR6R7 или группу OR, в которой R является третичным бутилом или бензилом с получением соединения формулы VI'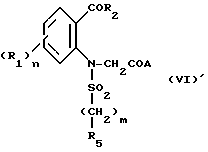
которое подвергают циклизации в основной среде, если A = NR6R7, с получением целевого продукта, либо в случае, когда A = OR, подвергают операции снятия защитной группы с получением соответствующей кислоты формулы VI''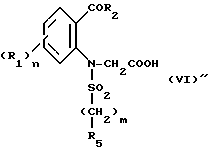
или ее хлорангидрида формулы VI'''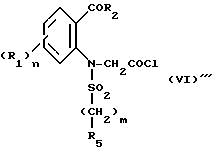
которые подвергают обработке амином NR6R7 для получения амидного производного формулы VI: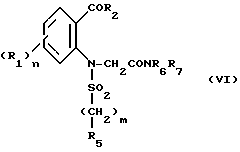
которое подвергают вышеуказанной циклизации с получением целевого продукта формулы 1, разделяют при необходимости полученный продукт на цис-и трансизомеры и выделяют энантиомеры.
2-аминофеноновые производные формулы (II) являются известными или получаются известными методами, аналогичными тем, которые описаны A.K.Singh и др. Synth Conunun 1986,16 (4), 485 и G.N.Walker, J. Org.Chem. 1962, 21 1929
2-амино-2'-трифторметилбензофеноны и другие трифторметилированные производные получаются в соответствии с методом, описанным в патенте США 3341592.
2,4-диметоксибензолсульфонилхлорид получается в соответствии с описанием в J. Am.Chem.Soc. 1952, 74, 2008.
Сульфонильные производные формулы (III) являются известными и получаются известными методами. Так, например, 4-диметиламинобензолсульфонилхлорид получается в соответствии с работой C.N.Sukenic и др. J. Am.Chem. Soc. 1977, 19,851-858, получение п-бензилоксибензолсульфонилхлорида описано в Европейской патентной заявке ЕР 229,566.
Алкоксибензолсульфонилхлорид получается из натрий -алкоксибензолсульфоната, который в свою очередь получается взаимодействием галоидного алкида с гидроксибензолсульфонатом натрия.
Галоидированные производные формулы (V) являются известными или получаются известными методами, такими, как описанные A.J.Vogel A.Text Book of Practical Organic Chemistry: Longman 3rd.,1956, p.383 или G.Kirchner и др. J. Am.Chem.Soc. 1985, 107, 14, 7072.
Взаимодействие соединения формулы II с соединением формулы III осуществляется в пиридине нагреванием при температуре между комнатной и точкой кипения растворителя в течение времени от нескольких часов до нескольких дней. Если нужно, реакцию можно проводить в присутствии диметиламинопиридина, который используется в каталитическом или стехиометрическом количестве.
Стадия обработки сульфонамида формулы (IV) соединением формулы (V) осуществляется при избытке галогенированного производного формулы (У) в растворителе, таком, как диметилформамид или диметилсульфоксид, в инертной атмосфере при температуре от 0oC до комнатной температуры, в течение периода времени от нескольких часов до 24 ч в присутствии гидрида натрия.
В том случае, когда группа -NR6R7 содержит вторичную амино-группу, то есть когда R6 и/или R7 являются замещенными амино группой, предпочитают использовать галогенированное производное (V) формулы Hal'-CH2-CO2R, в которой R представляет собой трет-бутил или бензил для того, чтобы получить промежуточные соединения формулы (VI') и затем (VI''). В этом случае этап образования кислоты формулы (VI'') осуществляется либо действием галогена в присутствии катализатора, такого, как палладий на угле, если R представляет собой бензил, либо в кислой среде, если R является трет-бутилом, например, в присутствии ТФУ-кислоты или в присутствии бромистоводородной кислоты в уксусной кислоте, или даже в присутствии ZnBr2 в дихлорметане.
Обработка соединения формулы (VI'') или (VI''') амином проводится в стандартных условиях амидного сочетания, например в присутствии БОФ или ГОБТ и ДЦК.
Соединения HNR6R7 являются известными или получаются известными методами. В качестве примера стереоспецифический синтез (R) и (S) 2-пирролидинилуксусных кислот проводится в соответствии с работой Н.Rueger и др. в Heterocycles, 1982, 19, (9), 1677 из пролинового производного соответствующей конфигурации. Получение метил N-Вос-3,4-дегидро-альфа-пролината осуществляется в соответствии с работой J.R. Dormoy Synthesis 1982, 753. Получение оптических чистых производных пипеколовой кислоты описывается, например в Tetrahedron 1992, 48, (3), 431-442 и Tetrahedron, 1991, 47, (24),4039-4062.
Получение производных азиридинкарбоксильной кислоты осуществляется в соответствии с работой K.Nakajima и др. в Bull. Chem.Soc. Jap. 1978, 51 (5), 1577.
Этап циклизации протекает по типу реакции альдолизации: метиленовая группа в альфа-положении амида депротонируется, а карбонильная группа фенона затем выступает в качестве внутреннего акцептора электронов, приводя к циклизации с появлением двух асимметричных углеродов (C*).
Реакция может быть проиллюстрирована следующей схемой: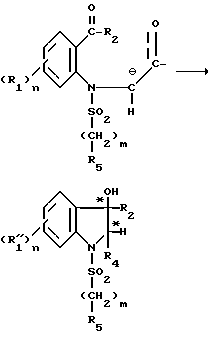
Принципы альдольной реакции присоединения рассмотрены С. H. Heathcock в Asymetric Synthesis, vol. 3 : Stereodifferentiating additions reactions, часть В, 111-112, Academic Press, 1984, издание J.D.Morrison.
Известно, что альдольная реакция ахиральных амидных анионов приводит к образованию двух рацемических диастереоизомеров бета-гидроксиламидов в соотношении, которое в значительной степени зависит от используемых условий эксперимента. Среди этих условий могут быть отмечены следующие: природа используемого неорганического или органического основания, природа катионов или противоионов, возможное присутствие добавок в реакционной среде, растворитель, температура реакции и структура соединения, участвующего в этой реакции.
В том случае, когда группы R6 и R7 не содержат группы, гидролизующейся в щелочной среде, можно использовать гидроокись натрия в воде в присутствии сорастворителя, с добавлением или без добавления катализатора фазового перехода, можно также использовать гидроокиси четвертичного аммония, например гидроокиси бензилтриметиламмония в метаноле.
Для проведения такой реакции адольдолизации возможно также использование органических оснований, например
гуанидинов, таких, как 1,5,7-триазабицикло[4.4.0]-дек-5-ен,
амидинов, таких, как 1,8-диaзaбициклo[5.4.0]ундeк-5-ен
или 1,5-диазабицикло[4.3.0]нон-5-ен
в растворителе или смеси растворителей, выбираемых, например, из бензола, ТГФ, дихлорметана, метанола и диметилформамида, реакция осуществляется в инертной атмосфере в интервале от -10oC до 110oC, количество использованного основания по меньшей мере является стехиометрическим, реакция может проводиться также и без растворителя при температуре бани.
Предпочтительно этап циклизации проводить в присутствии 1,8- диазабицикло[5.4.0] ундек-5-ена, (ДБУ) в растворителе, таком, как дихлорметан или метанол, при температуре в интервале от -10oC до температуры рефлюкса растворителя.
Возможно также использование алкоголята первичного, вторичного и третичного спирта с литием, натрием, калием, кальцием и магнием.
Алкоголят используется в каталитическом или стехиометрическом количестве в безводном растворителе, например, в спирте (если нужно в присутствии сорастворителя, такого, как ТГФ), или в стехиометрическом количестве в ТГФ, ДМФ или ДМСО, если необходимо в присутствии краун-эфиров, например, дициклогексил -18-краун-6, реакция проводится в интервале температур от -15 до 80oC.
Использование амида типа RR'NLr или RR'NMgBr, в котором R и R' одновалентные радикалы в качестве депротонирующего агента, является методом образования енолятов амидов, которые являются промежуточными соединениями в реакции альдолизации, этот метод был рассмотрен R.E. Ireland и др. J- Org.Chem., 1991, 56, 650. Растворителем реакции может быть бензол, гексан или ТГФ, используемые в безводной форме в инертной атмосфере. Могут быть добавлены адьюванты, такие, как LiF, LiCl, LiBr, LiI, LiBu
ТМЭДА, ДМП, ГМФА, или краун-эфир. (M.Murakate и др. J. Chem. Commen, 1990, 1657). В качестве примера можно отметить использование диизопропиламида лития в интервале температур -78oC и -80oC в безводном ТГФ в инертной атмосфере или ТГФ в присутствии добавок, таких, как, например, тетрамелиндиамин, ДМП или ГМФА. Примерами других известных амидов, которые могут быть использованы, являются циклогексиламид лития, и 2,2,6,6-тетраметилциклогексиламид лития. Возможно также получение других амидов взаимодействием требуемого количества бутиллития в гексане с линейными или циклическими вторичными аминами, при этом реакция протекает в одном из перечисленных выше растворителей.
Другую группу оснований, которые могут быть использованы, составляют силиламиды лития, натрия или калия, среди которых могут быть упомянуты (Me3Si)2NLi, (Me2PhSi)2 NLi, (Et3Si)2NLi, (Me3Si)2NK, (Me3Si)2NNa,
Возможно также использование смешанных амидов, например литиевой соли N-(триметилсилил)бензиламина или аналога, в котором бензиламин заменен хиральным первичным амином, таким, как (R)- или (S) альфа-метилбензиламин.
Когда соединение формулы (1), которое должно быть получено, имеет 2 асимметричных углеводородных атома, использование хиральных амидов или алкоголятов в этапе циклизации делает возможным энантиомерное обогащение каждым из цис- или трансстереоизомеров. Затем определяется пропорция каждого из энантиомеров методом высокоэффективной жидкостной хроматографии на хиральной колонке.
Когда соединение формулы (1), которое должно быть получено, имеет 3 или 4 асимметричных углеродных атома, этап циклизации соединение формулы (VI') может сопровождаться диастереоизомерным обогащением и использование соответствующего хирального основания делает возможным изменение диастереоизомерного обогащения.
На последнем этапе цис-и трансгеометрические изомеры соединения (1) экстрагируются по стандартным методам и разделяются хроматографическим методом или фракционной кристаллизацией.
Если необходимо, оптические изомеры каждого из цис-и трансизомеров разделяются, например, препаративной хроматографией на хиральной колонке, за которой следуют, если нужно, фракционная кристаллизация или образование оптически активной соли в присутствии соответственно выбранной хиральной кислоты или основания.
Таким образом, в том случае, если соединение, соответствующее изобретению, имеет 2 асимметричных углеродных атома, энантиомеры могут быть разделены хиральной ВЭЖХ.
Когда соединение, соответствующее изобретению, имеет 3 или 4 асимметричных атома углерода, диастереоизомеры могут быть выделены с использованием хроматографических методов и методов фракционной кристаллизации.
Для идентификации цисизомеров и трансизомеров соединений формулы (1) могут быть использованы несколько методов. В том случае, когда R3 является водородом, используется сравнительный анализ с применением высокой области ЯМР (250 МГц) в сочетании, например, с изучением эффекта Оверхаузера (N.O.E), например, между протоном индолина (R3=Н) и протоном гидроксила.
ИК-спектры цис-и трансизомеров в растворе ДХМ различны. Цисизомер в большинстве случаев имеет интенсивную четкую и симметричную полосу абсорбций в области 3550-3520 см-1, относящуюся к колебанию гидроксила, в то время как трансизомер не имеет разрешающей колебательной линии в этой области.
На основании обобщенных данных установлено, что цисизомер является в основном более подвижным в ТСХ на окисной алюминиевой пластинке (60F 254 нейтральная. Тип E Merok) при элюировании ДХМ, содержащим различные количества AcOEt. Аналогично при хроматографировании на колонке с алюминием (окись алюминия 90, размер частиц 0,063-0,200 мм), цисизомер в большинстве элюировался первым, при этом элюентом являлся ДХМ, содержащий различные количества AcOEt или MeOH.
Таким образом, цис-и трансизомерия соединения (1) в большинстве случаев может быть определена аналитическим методом. Возможно также использовать аналогию между сходными соединениями или между соединениями, полученными один из другого.
Абсолютная конфигурация некоторых соединений, соответствующих изобретению, была определена рентгенографическим анализом. Исходя из него, принимая во внимание значение вращения плоскости поляризации, можно также установить абсолютную конфигурацию других соединений, полученных аналогичным образом.
Из соединений (1), в которых заместители R6 и/или R7 или группа NR6R7 содержат C1-C4 -алкоксикарбонильную группу, можно получить путем гидролиза эфира соединения (1), в которых R6 и/или R7 или группа NR6R7 содержат карбоксильную группу, а другие заместители соединения (1) остаются неизменными. Кроме того, из соединений, в которых R6 и/или R7 или NR6R7 содержат карбоксильную группу, могут быть получены реакцией стандартного амидного сочетания соединения (1), в которых R6 и/или R7 или группа NR6R7 содержат карбамоильную группу, которая является свободной или замещена одним или двумя C1-C4-алкилами, а другие заместители являются идентичными.
Наконец, из соединений (1), в которых R6 и/или R7, или группа NR61R7 содержит карбамоильную группу, могут быть получены реакцией перегруппировки Гофмана соединения (1), в которых R6 и/или R7 или группа NR61R7 содержат аминогруппу, то другие заместители являются идентичными (J. Org. Chem. 1979, 41 (10) 1746).
Таким образом, в соответствии с изобретением способ получения соединений (1), в которых R6 и/или R7 или группа NR6R7 содержат аминогруппу, которая является свободной или замещена одним или двумя C1-C4-алкилами, может иметь два варианта
1) На этапе взаимодействия соединения формулы (IV) с соединением (V) осуществляют обработку соединения (IV) галогенированным производным формулы (V) Hal'-CH2CONR6R7, в котором R6 и/или R7 или группа NR61R7 содержат группу - предшественник аминогруппы, например карбоксиэфир, карбоксил или карбамоил, затем осуществляется этап циклизации и затем группу - предшественник аминогруппы превращают в аминогруппу, например гидролизуют карбоксиэфирную группу соединения (I) до карбоксильной группы, которая затем превращается в карбамоильную группу и затем в аминогруппу посредством перегруппировки Гофмана;
2) либо осуществляют обработку соединения (IV) галогенированным производным (V) формулы Hal'-CH2COOR, в котором R является бензилом или трет-бутилом; снимают защиту с эфиpa соединения (VI'), полученного таким образом, подвергая соответствующей обработке, затем осуществляют сочетание с соединением HNR6R7, в котором аминогруппа радикалов R6 и/или R7 может быть защищена, затем полученное таким образом соединение (VI) подвергают циклизации и, если необходимо, получают соединение (I), в котором аминогруппа является свободной, путем снятия защитных групп с амина.
Соединения формулы (VI), используемые в качестве промежуточных соединений для получения соединений (I), являются новыми и составляют часть изобретения.
Таким образом, представленное изобретение относится также к промежуточным соединениям общей формулы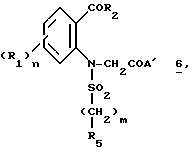
где A' представляет собой группу, выбираемую из HNR6R7;
-R1 является атомом галогена, C1-C4 -алкилом, C1-C4 -алкокси;
R5 - C1-C4-алкил, фенил, незамещенный или замещенный одним или несколькими заместителями, выбираемыми из галогена, C1-C4-алкила;
R6 - C1-C4 -алкил или R6 такой же, как R7;
R7 - группа пиперидин-4-ил, незамещенная или замещенная на азоте C1-C4 -алкилом, C1-C4 -алкоксикарбонилом; группа (CH2)r которая сама замещена группой 2-, 3- или 4-пиридил или группой амино, свободной или замещенной одним или двумя C1-C4-алкилами, карбоксильной группой, группой C1-C4-алкоксикарбонил, карбамоильной группой, свободной или замещенной одним или двумя C1-C4-алкилами;
или R6 и R7 образуют вместе с атомом азота, с которым они связаны, гетероцикл, выбираемый из морфолина, тиоморфолина, тиазолидина или 2,2-диметилтиазолидина, пиперазина незамещенного или замещенного в положении 4 группой R''8, ненасыщенного 5-членного цикла, содержащего единственный атом азота, замещенного радикалом R8, или ненасыщенного 3-; 4-; 5- или 6-членного цикла, содержащего единственный атом азота и замещенного радикалами R8 и R9;
R8 означает R1 8 или группу (CH2)r, которая сама замещена гидроксилом или аминогруппой, свободной или замещенной одним или двумя C1-C4-алкилами;
R1 8 означает группу (CH2)g, которая сама замещена карбоксильной группой, C1-C4-алкоксикарбонильной группой, бензилоксикарбонильной группой, карбамоильной группой, свободной или замещенной гидроксилом или одним или двумя C1-C4-алкилами;
R''8 означает R1 8 или группу (CH2)2NH2, свободную или замещенную одним или двумя C1-C4-алкилами;
R9 представляет водород, группу (CH2)r NR10R11, группу азидо или группу NH-бензилоксикарбонил;
R10 и R11 представляют независимо друг от друга водород или C1-C4-алкил;
n- 0, 1 или 2;
m- 0, 1 или 2;
q- 0, 1, 2 или 3;
r представляет 0,1,2 или 3 при условии, что r не равно нулю, когда R8 или R9 находятся в альфа-положении по отношению к амидному межцикличному азоту, или к их возможным солям.
Сродство соединений, соответствующих изобретению, к рецепторам вазопрессина было установлено in vitro с использованием метода, описанного в J. Biol.Chem., 1985, 260 (5), 2844-2851. Этот метод состоит в изучении смещения меченного тритием вазопрессина, связанного с участками V1- рецептора в мембранах печени крысы, 50%-ные концентрации ингибирования (IC50) для соединений, соответствующих изобретению, в отношении связывания меченного тритием вазопрессина, имеют низкие значения, достигающие 10-9 М.
Кроме того, на примере человеческой плазмы, богатой тромбоцитами, отмечалось ингибирование агрегирования тромбоцитов, наведенного вазопрессином, при использовании метода, описанного в работе Thrombosis Res. 1987, 45, 7-16. Соединения, соответствующие изобретению, ингибируют агрегирование, вызванное концентрациями вазопрессина от 50 до 100 нМ при низком значении ID50 (ингибирующая доза), подходящем до 10-9 М. Эти результаты показывают антагонистическую активность соединений, соответствующих изобретению, в отношении рецепторов V1.
Сродство соединений (1) к рецепторам V2 измерялось методом, предложенным P.Crause и др. Molecular and Cellular Endocrinology 1982, 28, 529-541.
Соединения, соответствующие изобретению и имеющие цисконфигурацию по отношению к связи 2,3 индолина, характеризуются заметной селективностью в отношении рецепторов V1.
Сродство соединений (1), соответствующие изобретению, к оцитоциновым рецепторам было определено in virto по смещению меченного тритием оцитоцина, связанного с рецепторами в мембранном препарате желез беременной крысы. Значения IC50 соединений, соответствующих изобретению, являются низкими и лежат между 10-5 М и 10-8 М.
Соединения, соответствующие изобретению, активны при различных путях приема, особенно при оральном введении.
У этих соединений не наблюдается симптомов токсичности при приеме фармакологически активных доз.
Следовательно, соединения изобретения могут быть использованы при лечении или профилактике различных, связанных с вазопрессином нарушений, особенно сердечно-сосудистых нарушений, таких, как артериальная гипертония, сердечная недостаточность, тромбоз или коронарный вазоспазм, в частности у курильщиков, нарушений центральной нервной системы, например при мозговых отеках, психотических состояниях, расстройствах аппетита или расстройствах памяти, нарушений почечной системы, таких, как почечный вазоспазм или некроз коркового вещества почек, расстройства желудочной системы, например язвы, или синдром несоответствующей секреции антидиуретического гормона.
Соединения, соответствующие изобретению, могут быть использованы также в качестве антирвотных средств, особенно при болезни укачивания, и в качестве антипролиферирующих агентов, например, при раке или атеросклерозе.
У женщин соединения, соответствующие изобретению, могут быть использованы также для лечения дисменореи или преждевременных родов.
Изобретение относится также к фармацевтической композиции, содержащей эффективную дозу соединения, соответствующего изобретению, или его фармацевтически приемлемой соли и фармацевтически приемлемые добавки. Названные добавки выбираются в соответствии с фармацевтической формой и необходимым способом приема.
В фармацевтических композициях, предназначенных для орального, подъязычного, подкожного, внутримышечного, внутривенного, местного, внутритрахеального, носового, чрезкожного или ректального приемов, содержание активного вещества формулы (1) или его солей соответствует единичной дозе приема и смешивается со стандартными фармацевтическими носителями.
Соответствующие единичные дозы выпускаются для орального приема в виде таблеток, желатиновых капсул, порошков, гранул и растворов или суспензий, или в соответствующих формах для подъязычного, защечного, внутритрахеального или носового приема, для подкожного, внутримышечного или внутривенного приема и для ректального приема. Для местного применения соединения могут быть использованы в кремах, мазях или лосьонах.
Для получения желаемого профилактического или терапевтического эффекта дозы активного содержимого могут варьироваться в интервале 0,01 - 50 мг на 1 кг веса тела в день.
Каждая единичная доза может содержать 0,5 - 1000 мг, предпочтительно 1 - 500 мг активного ингредиента в сочетании с фармацевтическим носителем. Такая единичная доза может приниматься от 1 до 5 раз в день так, чтобы ежедневная дозировка составила 0,5 - 5000 мг, предпочтительно 1 - 2 5000 мг.
Для получения твердой композиции в виде таблеток активный ингредиент смешивают с фармацевтическим наполнителем, таким, как желатина, крахмал, лактоза, стеарат магния, тальк, гуммиарабик или подобные им.
Таблетки могут быть покрыты сахарозой, целлюлозными производными или другими соответствующими веществами или они могут быть также обработаны для обеспечения пролонгирующего действия с тем, чтобы выделять непрерывно предварительно определенное количество активного компонента.
Препарат в форме желатиновых капсул готовится смешением активного ингредиента с разбавителем и разливкой образующейся смеси в мягкие или твердые желатиновые капсулы.
Препарат в форме сиропа или эликсира или для приема в форме капель может содержать активный ингредиент в сочетании с подслащивающим некалорийным веществом, предпочтительно и с метилпарабеном и пропилпарабеном в качестве антисептиков, а также агент для придания вкуса и соответствующий краситель.
Вододиспергируемые гранулы и порошки должны содержать активный ингредиент, смешанный с диспергаторами или увлажняющими агентами или с суспендирующими агентами, такими, как поливинилпирролидон, а также с подслаивающими веществами или с веществами, придающими вкус.
Для ректального приема используют свечи, которые готовят на основе связующих, плавящихся при ректальной температуре, например, с какао-маслом или полиэтиленгликолем.
Для парентерального приема используют водные суспензии, изотонические солевые растворы или стерильные растворы для инъекции, которые содержат фармакологически совместимые диспергаторы и/или увлажняющие агенты, например, пропиленгликоль или бутиленгликоль.
Активное соединение может быть использовано в виде макрокапсул, если необходимо с одним или несколькими носителями или добавками.
Помимо соединений формулы 1 и их фармацевтически приемлемых солей композиции могут содержать другие активно-действующие компоненты, которые могут быть использованы при лечении названных выше расстройств и болезней.
Соединения характеризуются их точкой плавления (Т.пл. oC) (или их точкой кипения Т.кип.) и/или их ЯМР спектром, снятым при 200 МГц в ДМСО и/или показателем вращения плоскости поляризации (альфаD), измеренной при 25oC (если нет других указаний).
Измеренное значение вращения плоскости поляризации зависит от количества остаточного растворителя, присутствующего в приготовленном продукте.
За исключением особо указанных случаев обозначение "цисизомер" или "трансизомер" означает, что выделенное соединение является смесью энантиомеров или цисконфигурацией, или трансконфигурацией.
Оптическая чистота соединений исследуется с использованием высокоэффективной жидкостной хроматографией (ВЭЖХ)
Пример 1. N-метил-N-метоксикарбонилметил-5-бром-3-(2-фторфенил) -1- (3,4-диметоксифенилсульфонил) -3-гидрокси-2-индолинкарбоксамид, цисизомер.
А) Метил- N-бромацетилсаркосинат
Это соединение получается в соответствии с Т.D.Harris и др. в J.Heterocyclic, Chem., 1981, 18 423.
В) 5-бром-2-(3,4-диметоксифенилсульфонамид) -2'-фторбензофенон.
20г 2-амино-5-бром-2'-фторбензофенона нагревают при 85oC в течение 48 ч в 120 мл сухого пиридина в присутствии 20 г 3,4-диметоксифенилсульфонилхлорида. Смесь охлаждают, выливают в охлажденную льдом воду, твердое вещество отфильтровывают, экстрагируют с помощью AcOEt, органическую фазу промывают водой, раствором хлористоводородной кислоты (1Н), водой и затем соленой водой. После высушивания над сульфатом магния и испарения растворителя в вакууме получают твердое вещество, которое перекристаллизовывают из смеси (ДХМ) изопропиловый эфир.
m 28 г
Т.пл. 125-128oC
С) 5-Бром-2-1[N-(3,4-диметоксифенилсульфонил)-N-(N'-метил- N'-метоксикарбонилкарбамоилметил)]-2-амино-2'-фторбензофенон.
3,5 г соединения, полученного на этапе В, растворяют в безводном ДМФ при 0oC в атмосфере аргона и добавляют 80%-ный гидрид натрия, через 15 мин добавляют 4,85 г соединения, полученного на этапе А, и смесь оставляют перемешиваться при комнатной температуре на 12 ч. Реакционную смесь выливают в воду, твердое вещество отфильтровывают, затем его растворяют в AcOEt, органическую фазу промывают водой, затем соленой водой, и растворитель выпаривают в вакууме. Полученное масло фильтруют на двуокиси кремния при элюировании смесью ДХМ/AcOEt (85/15, по объему)
m 3,2 г
Т.пл. 136-137oC
Д) 14 -метил- N -метоксикарбонилметил-5-бром-3- (2-фторфенил) 1- (3,4-диметоксифенилсульфонил)-3-гидрокси-2-индолинкарбоксамид, цисизомер.
3,2 г продукта, полученного на предшествующей стадии, растворяют в ДХМ (3 мл), добавляют 750 мг ДБУ, и смесь оставляют при перемешивании на 24 ч при комнатной температуре. Реакционную смесь выливают в силикагельную колонку, при элюировании смесью ДХМ/AcOEt (90/10 по объему), полученный продукт является смесью изомеров (цис и транс) целевого соединения. Этот продукт растирают в порошок в смеси гексан/изопропиловый эфир, и полученное твердое вещество фильтруют. Отфильтрованные жидкости хроматографируют на окисно-алюминиевой колонке, которая была предварительно уравновешена смесью ДХМ/AcOEt (70/30 по объему). Наименее полярное соединение элюируется смесью ДХМ/AcOEt (60/40 по объему) и затем перекристаллизовывается из смеси ДХМ/гексан/изопропиловый эфир.
Т.пл. 95oC с выделением газа
Примеры 2 и 3. [(4-Бензилоксикарбонил)-1-пиперазинил] 2-карбонил-5-хлор-3-(2-хлорфенил)-1-(3,4-диметоксифенилсульфонил)- 3-гидроксииндолин, цисизомер и трансизомер
А) 1-Бромацетил-4-(бензилоксикарбонил) пиперазин
Смесь из 22 г 4-бензилоксикарбонилпиперазина и 10,1 г триэтиламина в 200 мл эфира охлаждают до 0oC. В течение 30 мин добавляют 20,2 г бромацетилбромида в 100 мл эфира, и смесь доводят до комнатной температуры. Спустя 4 ч реакционную смесь промывают водой, сушат, концентрируют и затем хроматографируют на двуокиси кремния. Целевое соединение элюиругот смесью ДХМ/AcOEt (95/5 по объему) и перекристаллизовывают из ДХМ/изопропиловый эфир.
m 9г
Т.пл. 100-101oC
В) 2',5-Дихлор-2-(3,4-диметоксифенилсульфонамидо) бензофенон
5,6 г 2- амино-2',5-дихлорбензофенона и 5 г 3,4-диметоксифенилсульфонилахлорида нагревают в течение ночи в пиридине при 100oC.
Пиридин выпаривают до сухого состояния, добавляют воду и проводят экстракцию этилацетатом, содержащим небольшое количество ДХМ. После неоднократного промывания водой и высушивания над сульфатом натрия экстракт выпаривают в вакууме, и 7,7 г целевого продукта перекристаллизовывают из смеси ДХМ/AcOEt.
Т.пл. 164oC
С) 2', 5-Дихлор-2-1[N-(3,4-диметоксифенилсульфонил) -N(4-бензилоксикарбонил-1-пиперазинкарбонилметил)]-аминобензофенон
2,3 г бензофенона, полученного на этапе В, помещают в 10 мл ДМФ и обрабатывают 200 мг 80%-ного гидрида натрия в масле. Спустя 30 мин добавляют 5,3 г соединения, полученного на этапе А, и смесь перемешивают в течение 60 ч при комнатной температуре. Смесь выливают в воду, осадок фильтруют, растворяют в ДХМ, высушивают и затем концентрируют и хроматографируют на двуокиси кремния. Целевой продукт элюируют смесью ДХМ/AcOEt. (90/10, по объему) и кристаллизуют из смеси ДХМ/изопропиловый эфир.
m 2 г
Т.пл. 173-175oC
Д) [(4-Бензилоксикарбонил)-1-пиперазинил] 2-карбонил-5- хлор 3-(2-хлорфенил)-1-(3,4-диметоксифенилсульфонил)-3- гидроксииндолин, цисизомер и трансизомер.
1 г соединения, полученного на предыдущем этапе, суспендируют в 20 мл метанола и 20 мл ТГФ и подвергают действию 75 мг метилата натрия. Спустя 2 ч смесь нейтрализуют добавлением небольших количеств сухого льда, концентрируют до сухого состояния и затем поглощают водой, затем смесь экстрагируют с помощью ДХМ, экстракт высушивают и концентрируют. Неочищенный продукт подвергают хроматографии на колонке с окисью алюминия, при элюировании смесью ДХМ/AcOEt (80/20 по объему) получают последовательно два изомера.
Наименее полярный изомер перекристаллизовывают из смеси ДХМ/гексан. Это соединение представляет собой цисизомер.
m 262 мг
Т.пл. 169-179oC
Наиболее полярный изомер перекристаллизовывают из смеси ДХМ/изопропиловый эфир
m 200 мг
Т.пл. 209-211oC
Пример 4. 5-Хлор-3-(2-хлорфенил)-1-(3,4-диметоксифенилсульфонил) -3-гидрокси-2-(1-пиперазинилкарбонил)индолин, цисизомер
200 мг цисизомера, полученного в предыдущем примере, растворяют в 10 мл этанола и 5 мл ТГФ и гидрируют при комнатной температуре в присутствии 10%-ного палладия на угле. Через 30 мин смесь фильтруют через целит, отфильтрованные жидкости концентрируют и затем подвергают хроматографированию на двуокиси кремния. Целевой продукт подвергают элюированию смесью MeOH/ДХМ (10/90 по объему) и перекристаллизовывают из смеси ДХМ/изопропиловый эфир.
m 110 мг
Т.пл. 230-233oC
Примеры 5 и 6. 5-Хлор-3-(2-хлорфенил)-1- (3,4-диметоксифенилсульфонил)-3-гидрокси-2-морфолинокарбонилиндолин, цисизомер и трансизомер
А) 2',5-Дихлор[N-(3,4-диметоксифенилсульфонил) -N-(морфолинокарбонилметил)] 2-аминобензофенон
5 г 2',5-дихлор-2-(3,4-диметоксифенилсульфонамидо) бензофенон подвергают действию 350 мг 80%-ного гидрида натрия в 30 мл ДМФ при комнатной температуре в течение 20 мин. Добавляют 4,5 г морфолинобромацетамида и затем смесь перемешивают при комнатной температуре в течение 48 ч. Смесь выливают в воду, осадок отфильтровывают, растворяют в ДХМ, раствор сушат и концентрируют. Образованный продукт перекристаллизовывают из смеси ДХМ/изопропиловый эфир. Получают 3,4 г продукта.
Т.пл. 173-176oC
В) 5-Хлор-3- (2-хлорфенил) -1-(3,4-диметоксифенилсульфонил) - 3-гидрокси-2-морфолинокарбонилиндолин, цисизомер
1 г продукта, полученного на предыдущем этапе, растворяют в смеси метанола (10 мл) и ТГФ (20 мл), и на образующийся продукт действуют 92 мг метилата натрия при комнатной температуре в течение 1 ч. Смесь нейтрализуют сухим льдом, растворители частично испаряют, смесь растворяют в воде, экстрагируют с помощью ДХМ, а экстракт сушат, концентрируют и хроматографируют на окиси алюминия. Элюирование смесью ДХМ/AcOEt (70/30 по объему) дает наименее полярный изомер, который перекристаллизовывают из смеси ДХМ/изопропиловый эфир.
m 215 мг цисизомер
Т.пл. 260-264oC
C) 5-Хлор-3- (2-хлорфенил)-1-(3,4-диметоксифенилсульфонил)- 3-гидрокси-2-морфолинокарбонилиндолин, трансизомер
Хроматографированием продукта предыдущего этапа и элюированием смесью AcOEt/MeOH (90/10 по объему) получают наиболее полярный продукт. После перекристаллизации из смеси ДХМ/изопропиловый эфир получают
m 513 мг трансизомер
Т.пл. 240-241oC
Пример 7. N-метил-N-карбоксиметил-5-бром-3-(2-фторфенил) -1-(3,4-диметоксифенилсульфонил)-3-гидрокси-2-индолин-карбоксамид, цисизомер
200 мг соединения, полученного в примере 1, растворяют в 3 мл MeOH и 1 мг воды, содержащей 13 мг гидроокиси натрия. После перемешивания в течение 24 ч при комнатной температуре добавляют одну каплю концентрированного раствора гидроокиси натрия для завершения реакции, а затем спустя 15 мин смесь подкисляют до pH 3 добавлением раствора гидросульфата калия. Добавляют воду, смесь экстрагируют AcOEt и экстракт промывают водой и сушат над сульфатом магния, а растворитель выпаривают в вакууме. Полученный продукт перекристаллизовывают из смеси ДХМ/изопропиловый эфир.
Т.пл. 206-208oC
Примеры 8 и 9. 5-Хлор-3- (2-хлорфенил)-1-(3,4-диметоксифенилсульфонил)-3-гидрокси-2- (4-этилкарбоксилатпиперидинокарбонил) индолин, цисизомер, трансизомер.
А) Этил-N-бромацетил-4-пиперидинкарбоксилат
Этот продукт получают из этил-4-пиперидинкарбоксилата, который является коммерчески доступным.
B) 2',5-Дихлор-[N-(3,4-диметоксифенилсульфонил)-N (4-этилкарбоксилатпиперидинокарбонилметил)] 2-аминобензофенон
8 г 2',5-дихлор-2-(3,4-диметоксифенилсульфонамидо) бензофенона растворяют в 100 мл ДМФ и затем добавляют 541 мг гидрида натрия. После перемешивания в течение 30 мин добавляют 9,5 г соединения этапа А, смесь оставляют стоять при перемешивании при комнатной температуре на 18 ч. Смесь концентрируют в вакууме, растворяют в воде, экстрагируют этилацетатом и экстракт высушивают и концентрируют. Полученное масло хроматографируют на двуокиси кремния с использованием в качестве элюента смеси этилацетат ДХМ/гексан (40/10/50 по объему). Экстрагированный продукт кристаллизуют из эфира.
m 3,5 г
Т.пл. 128oC
C) 5 -Хлор-3-(2-хлорфенил)-1-(3,4-диметоксифенилсульфонил) -3-гидрокси-2-(4-этилкарбоксилатпиперидинокарбонил)индолин, цисизомер, трансизомер
Смесь, содержащую 3,4 г соединения, полученного на предыдущем этапе, и 869 мг ДБУ в 10 мл хлороформа в течение 18 ч нагревают до 60oC. Затем реакционную смесь фильтруют через колонку с окисью алюминия с использованием в качестве элюента смеси ДХМ/этилацетат (90/10 по объему) с целью получения цисизомера.
m 700 мг
Т.пл. 110oC
Чистый этилацетат элюирует трансизомер.
m 610 мг
Т.пл. 187oC
Примеры 10 и 11. N-метил-N- (2-пиридилэтил) -5-хлор-3- (2-хлорфенил) -1-(3,4-диметоксифенилсульфонил) -3-гидрокси-2-индолинкарбоксамид, цисизомер, трансизомер
A) N-[2-(2-хлорфенилкарбонил)-5-хлорфенил] N-(3,4-диметоксифенилсульфонил) аминоуксусная кислота
а) 2',5-дихлор-2- (3,4-диметоксифенилсульфонамидо) бензофенон.
Это соединение получается в примере 2-3, этап В.
в) 2'5-дихлор-[N-(3,4-диметоксифенилсульфонил) -N-бензилоксикарбонилметил]2-аминобензофенон.
172 г продукта, полученного на предшествующей стадии, растворяют в 800 мл ДХМ и охлаждают до 0oC. Постепенно в атмосфере азота добавляют 11,7 г 80%-ного гидрида натрия, а затем, спустя 30 мин, добавляют 256 г бензилбромацетата и смесь оставляют перемешиваться при комнатной температуре на 24 ч. ДМФ испаряют, остаток растворяют в воде, экстрагируют в ДХМ, и экстракт высушивают и концентрируют. Целевой продукт кристаллизуют из изопропилового эфира, а затем перекристаллизовывают из смеси ДХМ/изопропиловый эфир.
m 136,5 г
Т. пл. 102-104oC
с) N-[2-(2-Хлорфенилкарбонил)-5-хлорфенил] -N- (3,4-диметоксифенилсульфонил) аминоуксусная кислота
50 г бензилового эфира, полученного на предыдущем этапе, растворяют в 500 мл этилацетата и в атмосфере азота добавляют 2,5 г 5%-го палладия на угле. Раствор интенсивно перемешивают и через него пропускают в течение 5 ч ток водорода. В конце гидрогенерирования продукт кристаллизуют. Смесь фильтруют через целит, осадок обильно промывают горячим ДХМ и затем органическую фазу концентрируют. Целевой продукт кристаллизуют, а затем перекристаллизовывают из смеси ДХМ/изопропиловый эфир.
m 33,7 г
Т. пл. 177-178oC
В) 2',5-Дихлор-[N-(3,4-диметоксифенилсульфонил) -N(N'-2-пирид-2-ил-этил-N'-метил) карбамоилметил] 2-аминобензофенон
2 г кислоты, полученной на стадии А, вводят в 30 мл ДХМ и добавляют 1,13 г 2- (2-метиламиноэтил)-пиридина, затем 844 мг триэтиламина и, наконец, 1,92 г БОФ, после чего смесь оставляют стоять на 18 ч при комнатной температуре с перемешиванием. Смесь растворяют в воде, органическую фазу отделяют, промывают раствором карбоната натрия, высушивают и концентрируют. После хроматографирования на двуокиси кремния целевой продукт получают элюированием смесью ДХМ/MeOH (95/5 по объему)
m 2 г
Т.пл. 150oC
C) N-метил-N-(пирид-ил-этил)-5-хлор-3-(2-хлорфенил)-1- (3,4-диметоксифенилсульфонил)-3-гидрокси-2-индолинкарбоксамид.
Смесь, состоящую из 1,7 г продукта, полученного на предшествующем этапе, и 422 мг ДБУ в ДХМ нагревают в течение 18 ч при 55oC. Реакционную смесь хроматографируют на окиси алюминия. Элюирование смесью этилацетат/ДХМ (40/60, по объему) дает цисизомер
m 410 мг
Т.пл. 191oC
Чистый этилацетат элюирует трансизомер
m 790 мг
Т.пл. 154oC
Пример 12.
2-(4-Карбоксипиперидинкарбонил) -5-хлор-3- (2-хлорфенил) -1- (3,4-диметоксифенилсульфонил) -3-гидрокси-индолин, цисизомер
500 мг изомера, полученного в примере 9, помещают в 5 мл метанола в присутствии 48 мг гидроксида натрия в 1 мл воды. После перемешивания в течение 18 ч смесь выливают в воду, подкисляют разбавленной хлористоводородной кислотой, затем экстрагируют в ДХМ и экстракт высушивают и концентрируют. Полученное твердое вещество очищают хроматографией на двуокиси кремния, элюируют смесью ДХМ/MeOH (95/5 по объему) и полученный продукт затем кристаллизуют из смеси ДХМ/изопропиловый эфир.
m 250 мг
Т.пл. 150oC
Примеры 13 и 14.
N-Метил-N(1-метил-пиперидин-4-ил)-5-хлор-3-(2-хлорфенил) -1-(3,4-диметоксифенилсульфонил)-3-гидрокси-2-индолин-карбоксамид, цисизомер и трансизомер
А) 2',5-Дихлор-[N-(3,4-диметоксифенилсульфонил)-N- (N'-метил-N'-мeтил-пипepидин-4-ил)кapбaмoилмeтил] 2-аминобензофенон
2 г кислоты, приготовленной в примере 10-11, этап А, в 50 мл ДХМ смешивают с 650 мг 4-метиламино-1-метилпиперидина в присутствии 1,90 г БОФ. После перемешивания при комнатной температуре в течение 2 ч органическую фазу промывают карбонатной водой, высушивают и концентрируют. Затем остаток хроматографируют на двуокиси кремния с элюированием смесью ДХМ/MeOH (90/10 по объему). Получают 1,2 г целевого продукта.
Т.пл. 165-1ббoC
В) N-метил-N-(метил-пиперидин-4-ил)-5-хлор-(2-хлорфенил)-1- (3,4-диметоксифенилсульфонил) -3-гидрокси-2-индолинкарбоксамид, цисизомер и трансизомер
650 мг продукта, полученного на предыдущем этапе, обрабатывают в течение ночи 100 мг метилата натрия в 5 мл метанола. Добавляют сухой лед, растворитель выпаривают, остаток растворяют в карбонатной воде, экстрагируют в ДХМ и концентрируют, затем хроматографируют на двуокиси кремния. Смесь метанол/ДХМ (5/95 по объему) элюирует последовательно 2 изомера. Затем каждый изомер перекристаллизовывают из смеси ДХМ/изопропиловый эфир.
Трансизомер является в этих условиях наименее полярным
m 205 мг
Т.пл. 181oC
Цисизомер
m 150 мг
Т.пл. 97oC, содержит 0,25 М изопропилового эфира.
Примеры 15 и 16.
5-Хлор-3-(2-хлорфенил)-1-(3,4-диметоксифенилсульфонил) - 3-гидрокси-2[(4-метил)-пиперазин-1-илкарбонил]-индолин, цисизомер и трансизомер
А) 2'5-Дихлор-[N-(3,4-диметоксифенилсульфонил) -N-((4-метил-пиперазин-1-ил)карбамоилметил)-, 2-аминобензофенон
Это соединение получается действием N-метил-пиперазина на кислоту, полученную в примерах 10 и 11, этап А.
Т.пл. 165-167oC
В) 5-Хлор-3-(2-хлорфенил)-1-(3,4-диметоксифенилсульфонил) - 3-гидрокси-2-[(4-метил)пиперазин-1-илкарбонил]-индолин, цисизомер и трансизомер.
Соединение предшествующего этапа циклизуют согласно методике примеров 12-13. Два образованных изомера разделяют хроматографией на окиси алюминия. Смесь ДХМ/ этилацетат (75/25 по объему) элюирует наименее полярный продукт: цисизомер, который перекристаллизовывают из смеси ДХМ/ изопропиловый эфир.
Т. пл. 120oC: содержит 0,25 М изопропилового эфира. Смесь ДХМ/ MeOH элюирует наиболее полярное соединение, трансизомер, который затем перекристаллизовывают из метанола.
Т. пл. 189oC
Примеры 17 и 18. N-Изопропил-N-метоксикарбонилэтил-5-хлор-3-(2-хлорфенил) -1-(3,4-диметоксифенилсульфонил) -3-гидрокси-2-индолинкарбоксамид, цисизомер и трансизомер.
A) N -Изопропил- N -(метоксикарбонилэтил) бромацетамид.
90 г изопропиламина по каплям добавляют к 130 г раствора метилакрилата в 300 мл метанола, охлажденного до -10oC. После 72 ч выдержки при комнатной температуре смесь выпаривают, а затем остаток перегоняют. Образованное масло представляет собой метил-(N-изопропил) - 3-аминопропионат.
Т. кип. 73-78oC при 15 мм рт. ст.
29 г полученного соединения в 100 мл ДХМ смешивают с 20,2 бромацетилбромида в 100 мл ДХМ при ОoC. После выдерживания смеси при комнатной температуре в течение 12 ч растворитель выпаривают, остаток растворяют в воде, экстрагируют этилацетатом и экстракт сушат и концентрируют. Полученное масло используется как таковое на следующем этапе.
В) 2',5-Дихлор-N-(3,4-диметоксифенилсульфонил)-N-(N'- изопропил-N'-метоксикарбонилэтил)карбамоилметил] -2-аминобензофенон
Это соединение получается использованием следующих обычных методов, т.е. взаимодействием продукта, полученного на этапе А с 2',5-дихлор-2-(3,4-диметоксифенилсульфонамидо)-бензофеноном в присутствии гидрида натрия.
Т.пл. 135-137oC (перекристаллизация: ДХМ/ изопропиловый эфир).
C) N-Изопропил-N-Метоксикарбонилэтил-5-хлор-3-(2-хлорфенил) -1- (3,4-диметоксифенилсульфонил) -3-гидрокси-2-индолинкарбоксамид, цисизомер и трансизомер.
Этот продукт получается циклизацией соединения, полученного на этапе В в присутствии ДБУ. Цисизомер выделяется хроматографированием на окиси алюминия при элюировании смесью ДХМ/этилацетат (90/10 по объему). Затем продукт перекристаллизуют из смеси этилацетат/гексан.
Т. пл. 153-155oC
Трансизомер получается элюированием колонки из окиси алюминия этилацетатом. Затем продукт перекристаллизовывают из смеси метанол/изопропиловый эфир.
Т. пл. 182-185oC
Примеры 19 и 20. N-Метил-N-метоксикарбонилметил-5-хлор-3- (2-хлорфенил) -1- (3,4-диметоксифенилсульфонил)-3-гидрокси-2-индолинкарбоксамид, цисизомер и трансизомер.
2 изомера этого соединения получают в соответствии с методикой, описанной в примере 1. Они разделяются хроматографически на окиси алюминия. Смесь ДХМ/этилацетат (80/20 по объему) элюирует цисизомер. Его кристаллизуют из смеси ДХМ/изопропиловый эфир в виде белого порошка, содержащего 0,25 моль изопропилового эфира. Он превращается в пенящееся вещество при нагревании в вакууме.
ЯМР-спектр цисизомера (пример 19) дается на фиг.1.
Трансизомер элюируется чистым этилацетатом. Его перекристаллизовывают из смеси ДХМ/изопропиловый эфир.
Т. пл. 176-178oC
ЯМР- спектр трансизомера (пример 20) дается на фиг.2.
Примеры 21 и 22.
N-Метил-N-карбоксиметил-5-хлор-3- (2-хлорфенил) -1- (3,4-диметоксифенилсульфонил) -3-гидрокси-2-индолинкарбоксамид, цисизомер и трансизомер
Каждый из этих соединений получается из соединений, описанный в примерах 19 и 20 в соответствии с методикой, приведенной в примере 8.
Цисизомер: Т. пл. 220-222oC после перекристаллизации из смеси ДХМ/изопропиловый эфир/метиловый спирт
Трансизомер: Т. пл. 222-225oC после перекристаллизации из смеси ДХМ/изопропиловый эфир.
Примеры 23 и 24. N-Метил-N-карбамоилметил-5-хлор-3-(2-хлорфенил) -1-(3,4-диметоксифенилсульфонил) -3-гидрокси-2-индолинкарбоксамид, цисизомер и трансизомер
Каждый изомер получается из соответствующего изомера кислоты, приготовленного в примерах 21 и 22.
605 мг трансизомера кислоты, полученной в предшествующем примере, растворяют в 10 мл ДХМ и к ним добавляют 435 мг БОФ и 260 мг ДИПЭА. После выдерживания в течение 5 мин при комнатной температуре при интенсивном перемешивании добавляют 6 мл 20%-ного водного раствора аммиака и смесь перемешивают еще 4 ч. Добавляют раствор карбоната натрия, затем смесь экстрагируют в ДХМ. Органическую фазу последовательно промывают водой, раствором гидросульфата натрия, снова водой и затем сушат над сульфатом магния. После выпаривания остаток хроматографируют на силикагеле и элюируют смесью этилацетат/метанол (95/5 по объему).
Полученный продукт дважды кристаллизуют из смеси ДХМ/ метанол при 0oC.
Т. пл. 236oC
ЯМР-спектр трансизомера (пример 23) дается на фиг.3.
При использовании той же самой методики получается цисизомер.
Целевой продукт кристаллизуют из смеси ДХМ/изопропиловый эфир. Измельченное соединение, высушенное в вакууме при 70oC в течение 8 ч, содержит 0,25 моль изопропилового эфира.
Спектр ЯМР-цисизомера (пример 24) дается на фиг.4.
Примеры 25 и 26.
5-Хлор-З-(2-хлорфенил)-1-(3,4-диметоксифенилсульфонил)-3- гидрокси(4-гидрокси-пиперидин-1-ил)-1-карбонилиндолин, трансизомер
Это соединение получают из N-[2-(2-хлорфенилкарбонил) -5-хлорфенил]-N-(3,4-диметоксифенилсульфонил) аминоуксусной кислоты, получаемой в примерах 10 и 11, этап А. Затем процесс продолжают в соответствии с методикой примеров 11 и 12, стадия В, но используя 4-гидроксипиперидин в присутствии БОФ и триэтиламина. Затем полученный продукт циклизуют в соответствии с обычной методикой в присутствии ДБУ. Два изомера разделяют хроматографически на окиси алюминия. Смесь ДХМ/метанол (99/1 по объему) элюирует цисизомер.
Продукт кристаллизуют из смеси ДХМ/гексан/метанол и затем полученное твердое вещество растирают в порошок в смеси ДХМ/гексан, образуя аморфный порошок.
Цисизомер характеризуется по его спектру ЯМР при 388К.
1-1.8 м.д.: м: 4Н: CH2 в положениях 3 и 5 пиперидина 2.8- 3.65 м.д.: м: 5Н : CH2 в положениях 2 и 6 пиперидина и CH в положении 4.
3.75 м.д. : 2с:6H:20CH3
4.15 м.д. : д:1Н:ОН в пиперидине
5.45 м.д. : с:1Н:CH (индолин)
6.1 м.д.:с:1Н:ОН индилин
6.8-7.6 м.д.: м: 10Н:H ароматический
ДМСО: 2.4 м.д.
ДОН: 2.75 м.д.
Смесь ДХМ/метанол (97/3 по объему) элюирует трансизомер, который перекристаллизовывается из смеси ДХМ/изопропиловый эфир.
Т. пл. 232-234o
Синтез в (L)-пролиновой серии: примеры 27 - 30.
Примеры 27 и 27а. 5-Хлор-3- (2-хлорфенил) -1-(3,4-диметоксифенилсульфонил) -3-гидрокси-2-[(2S)-(2-метоксикарбонил)пирролидинокарбонил] индолин, (цисизомер: 2 соединения)
А) Метил (L)-N-(бромацетил)пролинат
20 г триэтиламина и 20 г бромацетилбромида в 30 мл ДХМ одновременно добавляют к раствору, содержащему 16,7 г метил-(L)-пролинат гидрохлорида в 20 мл ДХМ при поддержании температуры при -5oC, после чего смесь перемешивают в течение 24 ч при комнатной температуре. Добавляют воду, и смесь промывают раствором KHSO4, водой, раствором бикарбоната натрия, снова водой, после чего высушивают над сульфатом магния. После выпаривания получают масло, которое сушат в вакууме. Это масло, очищенное с помощью ТСХ, используется фактически на следующем этапе.
В) 2', 5-Дихлор-[N-(3,4-диметоксифенилсульфонил)-N-((2S)- (2-метоксикарбонил) пирролидинокарбонилметил)] 2-аминобензофенон.
4,66 г 2',5-дихлор-2-(3,4-диметоксифенилсульфонамидо)бензофенона растворяют в 40 мл безводного ДМФ в атмосфере аргона при 0oC, добавляют 340 мг 80%-ного гидрида натрия и затем, спустя 30 мин, 6,5 г соединения, полученного на этапе А. После выдерживания в течение 4 дней при комнатной температуре смесь выливают в воду, экстрагируют этилацетатом, экстракт промывают водой, соленой водой и затем сушат над сульфатом магния и выпаривают в вакууме. Твердое вещество, содержащее небольшое количество исходного бромированного производного, элюируют смесью ДХМ/этилацетат (85/15 по объему) хроматографированием на силикагеле. Образец перекристаллизовывают из смеси ДХМ/изопропиловый эфир.
m = 1,2 г
Т. пл. 141-142oC
α
Анализ
Рассчитано,%: С 54,81; H 4,44; N 4,41
Установлено,%: C 54,40; H 4,54; N 4,55
С) 5-Хлор-3-(2-хлорфенил)-1-(3,4-диметоксифенилсульфонил) - 3-гидрокси-2- [(2S)-(2-метоксикарбонил)пирролидинокарбонил] индолин (цисизомер)
1,1 соединения, полученного на предыдущем этапе, нагревают в 4 мл метиленхлорида в течение 24 ч с эквивалентным количеством ДБУ. Анализ аликвотной доли, выполненный с использованием ВЭЖХ, выявляет наличие 4 целевых изомеров. Через 24 ч реакционную смесь выливают в колонку с окисью алюминия, предварительно уравновешенную смесью ДХМ/этилацетат (90/10 по объему), и элюируют смесью ДХМ/ этилацетат (от 90/10 по объему до 70/30 по объему), получают 510 мг смеси 2-х наименее полярных соединений при соотношении 4/1 (измеренном с помощью ВЭЖХ).
1o) Две последовательных кристаллизации из смеси ДХМ/ изопропиловый эфир в холодном состоянии дает соединение, находящееся в наибольшем количестве.
m 180 мг
α
Т. пл. 187-190oC
2o) Кристаллизационные маточные растворы полученного соединения подвергают хроматографированию с использованием окиси алюминия при элюировании смесью ДХМ/этилацетат (85/15 по объему). Таким образом полученное выше соединение отделяется от второго, и второе соединение растворяют в минимальном количестве ДХМ и затем осаждают добавлением минимального количества гексана.
α
Пример 28. 2-((2S)-2-Карбоксипирролидинкарбонил) -5-хлор-3- (2-хлорфенил) -1- (3,4-диметоксифенилсульфонил) -3-гидроксииндрлин, цисизомер
430 мг соединения, полученного в примере 27, растворяют в 6 мл метанола, добавляют 41 мг гидроокиси натрия в 1 мл воды, и смесь перемешивают при комнатной температуре в течение 24 ч. Смесь подкисляют до pH 3 несколькими каплями раствора гидросульфата калия и экстрагируют этилацетатом. Экстракт промывают водой, затем высушивают над сульфатом магния. Проводится хроматографирование на колонке с двуокисью кремния, обработанной смесью ДХМ/пентан (80/20 по объему). Непрореагировавший эфир элюируют смесью ДХМ/AcOEt (70/30 об). Смесь AcOEt/MeOH (80/20 по объему) элюирует целевую кислоту, которую перекристаллизовывают из смеси ДХМ/изопропиловый эфир.
Т. пл. 232-234oC
α
Примеры 29 и 29а. 2-((2S)-2-Карбамоилпирролидинкарбонил) -5-хлор-3- (2-хлорфенил) -1-(3,4-диметоксифенилсульфонил)-3-гидроксииндолин, (цисизомер: два соединения)
230 мг соединения, полученного в примере 28, растворяют в 5 мл ДХМ, добавляют 50 мг ДИПЭА и затем 165 мг БОФ, и смесь оставляют при комнатной температуре на 5 мин. Смесь охлаждают на ледяной бане и через нее пропускают ток газообразного аммиака в течение 1 мин, а спустя 15 мин еще в течение 1 мин. Добавляют воду, а затем большой объем этилацетата - для получения двух фаз. Органический раствор промывают раствором карбоната натрия, водой, раствором гидросульфата калия, водой, а затем соленой водой. После сушки остаток подвергают хроматографированию на двуокиси кремния с элюированием смесью ДХМ/метанол (93/7 по объему). Полученный продукт растирают в порошок в смеси ДХМ/изопропиловый эфир/гексан. Он содержит 1/3 моль изопропилового эфира.
α
Соединение примера 29 может быть получено с использованием другой методики.
А) 2', 5-Дихлор-[N-(3,4-диметоксифенилсульфонил) -N-((2S)- 2-карбамоилпирролидинкарбонилметил)] 2-аминобензофенон
33,9 г кислоты, полученной в примере 10-11, этап А, растворяют в 300 мл хлороформа. Добавляют 15 г тионилхлорида, и смесь в течение 1,5 ч выдерживают при рефлюксе. Смесь выпаривают до сухого состояния, затем остаток растворяют в ДХМ, доводят до 0oC и к ней добавляют 10,5 г хлоргидрата (L) - пролинамида, а затем 18 г ДИПЭА в 20 мл ДХМ - последнее добавляют медленно с тем, чтобы температура реакционной смеси не превысила 3oC. После выдерживания в течение ночи при комнатной температуре реакционную смесь промывают бикарбонатом натрия (дважды), а затем - гидросульфатом калия (дважды), реакционную смесь сушат и концентрируют. Полученный неочищенный продукт растворяют в минимальном количестве ДХМ и по каплям добавляют к изопропиловому эфиру (1,2 л) при перемешивании. После перемешивания в течение 2 ч полученный осадок фильтруют и затем высушивают в вакууме при 60oC в течение 6 ч. Получают 42 г продукта.
α
В) 2-((2S)2-карбамоилпирролидинокарбонил)-5-хлор-3-(2-хлорфенил) -1-(3,4-диметоксифенилсульфонил) -3-гидроксииндолин), (цисизомеры: 2 соединения)
5 г продукта, приготовленного на предшествующей стадии, растворяют в 50 мл метанола. Раствор охлаждают до -10oC, добавляют 1,35 г ДБУ, и смесь выдерживают в течение 60 ч при -10oC. Соединение кристаллизуется, его отфильтровывают (циссоединение 1). Кристаллизационные маточные растворы нейтрализуют KHSO4, и смесь выпаривают до сухого состояния. Ее растворяют в воде, дважды экстрагируют ДХМ, а экстракт сушат и концентрируют. Полученный неочищенный продукт хроматографируют на двуокиси кремния с использованием в качестве элюента смеси этилацетата/ДХМ (28/72 по объему). Полученную смесь в горячем виде растворяют в минимальном количестве метанола, нерастворившееся вещество отфильтровывают, жидкости затем выдерживают в течение ночи при -4oC,и циссоединение 2 кристаллизуется.
m = 25 г
α
Анализ спектра ЯМР обнаруживает присутствие одного моля метанола в моле продукта. Перекристаллизация продукта из этанола дает возможность удалить растворитель из кристаллов.
Т. пл. 154-162oC
α
α
Это соединение является идентичным, исключая растворитель, соединению, полученному согласно первой методике данного примера.
Соединение, которое кристаллизуется на этапе В) выше, названное циссоединение 1, перекристаллизовывают из метанола.
Т. пл. 190oC
αD = +115o (с = 0,3, хлороформ)
Пример 30.
5-Хлор-3- (2-хлорфенил)-1-(3,4-диметоксифенилсульфонил) -3-гидрокси-2 -[(2S)(2-гидроксиметил)пирролидинкарбонил] индолин, цисизомеры.
А) 2', 5-Дихлор-[N-(3,4-диметоксифенилсульфонил) -N((2-гидроксиметил)пирролидинкарбонилметил)] -2-аминобензофенон.
Это соединение получают взаимодействием (L)-пролинола с кислотой, полученной в примерах 10 и 11, этап А, согласно следующей стандартной методике.
В) 5-Хлор-3- (2-хлорфенил) -1- (3,4-диметоксифенилсульфонил) -3-гидрокси-2-[(2S)(2-гидроксиметил)пирролидинокарбонил] индолин, цисизомер.
1,5 г соединения предыдущего этапа циклизуют в присутствии 380 мг ДБУ в 2 мл ДХМ. После выдерживания при комнатной температуре в течение 3 дней добавляют 1 мл ДХМ, и затем смесь нагревают в течение ночи при 40oC. При применении ТСХ на двуокиси кремния (элюант этилацетат) обнаруживается образование трех соединений, находящихся в наибольшем количестве.
Наименее полярная фракция элюируется при хроматографировании на двуокиси кремния с использованием смеси ДХМ/ этилацетат от 60/40 до 80/20 по объему. Затем осуществляют хроматографирование на окиси алюминия с элюированием ДХМ/метанол (99/1 по объему). Полученная фракция является гомогенной по данным ТСХ. Продукт перекристаллизовывают три раза из смеси ДХМ/изопропиловый эфир. Целевой продукт получают с использованием ВЭЖХ с чистотою более чем 99%.
m 155 мг
Т.пл. 194-197oC
α
Синтез с (D) - Пролиновыми фракциями.
Пример 31.
5-Хлор-3- (2-хлорфенил) -1-(3,4-диметоксифенилсульфонил) -3-гидрокси-2-х(2R)(2-метоксикарбонил)пирролидинкарбонил]индолин, цисизомер.
A) 2', 5-Дихлор-[N-(3,4-диметоксифенилсульфонил) -N((2R)(2-метоксикарбонил)пирролидинокарбонилметил)] -2- аминобензофенон.
Это соединение получают из кислоты, приготовленной в примерах 10 и 11, этап А (3 г), к которой добавляют 1,2 г метил (D))-пролината и 2,8 г БОФ в 10 мл ДХМ в присутствии 1,15 г триэтиламина. Смесь оставляют на 1 ч при комнатной температуре и затем разбавляют ДХМ, органическую фазу промывают карбонатом натрия и гидросульфатом калия, сушат и концентрируют. Неочищенный продукт хроматографируют на двуокиси кремния при элюировании смесью ДХМ/этилацетат (95/5 по объему). Полученный продукт затем перекристаллизовывают из смеси ДХМ/изопропиловый эфир.
Т. пл. 140-141oC
α
В) 5-Хлор-3- (2-хлорфенил) -1- (3,4-диметоксифенилсульфонил) -3-гидрокси-2- [(2R)(2-метоксикарбонил)пирролидинокарбонил]индолин, цисизомер.
1,5 г полученного на предыдущей стадии соединения кипятят в течение ночи с обратным холодильником в 5 мл ДХМ в присутствии 360 мг ДБУ. Смесь хроматографируют на окиси алюминия. Смесь ДХМ/этилацетат (95/5, по объему) элюирует наименее полярную фракцию (m = 300 мг), которую дважды перекристаллизовывают из смеси ДХМ/изопропиловый эфир.
Т. пл. 186-188oC
α
Это соединение представляет собой энантиомер, полученный из (D)-пролина, описанного в примере 27.
Примеры 32 и 32а.
N-Метил-N-метоксикарбонилметил-5-хлор-3-(2-хлорфенил) -1-(4-этоксифенилсульфонил)-3-гидрокси-2-индолин-карбоксамид, трансизомер и цисизомер.
А) 2', 5-Дихлор-[N-(4-этоксифенилсульфонил) -N-(N'-метил-N'-метоксикарбонилметил)карбамоилметил] -2-аминобензофенон.
5,7 г 2',5-дихлор-4-этокси-2-фенилсульфонамидобензофенона растворяют в атмосфере аргона в 40 мл ДМФ и добавляют 400 мг 80%-го гидрида натрия при 0oC, через 15 мин добавляют 4,3 г метил-N-(бромацетил)саркосината. Спустя 48 ч экстрагируют обычным способом и затем очищают хроматографированием на двуокиси кремния при элюировании смесью ДХМ/этилацетат (90/10, по объему) и перекристаллизацией из смеси ДХМ/изопропиловый эфир.
Т. пл. 158-160oC
В) N-Метил-N-метоксикарбонилметил-5-хлор-3-(2-хлорфенил)- 1- (4-этоксифенилсульфонил) -3-гидрокси-2-индолинкарбоксамид, трансизомер
1 г соединения, полученного на предыдущей стадии, растворяют в 4 мл ДХМ и обрабатывают в течение 90 мин при комнатной температуре с помощью 312 мг ТБД. Добавляют раствор гидросульфата калия, ДХМ испаряют в вакууме, смесь экстрагируют этилацетатом, а экстракт промывают и сушат над сульфатом магния. Целевой продукт получают хроматографией на силикагеле при элюировании ДХМ/этилацетат (90/10 по объему).
m 590 мг
Т. пл. 168-171oC после перекристаллизации из смеси ДХМ/ гексан
C) N-метил-N-метоксикарбонилметил-5-хлор-3-(2-хлорфенил) -1- (4-этоксифенилсульфонил) -3-гидрокси-2-индолинкарбоксамид, цисизомер.
2,96 г соединения, полученного на этапе А, суспендируют в 20 мл метанола и 10 мл ТГФ, добавляется 100 мг метилата натрия, после чего смесь оставляют на 7 ч в холодильнике. Добавляют воду, смесь нейтрализуют раствором гидросульфата калия, и часть метанола выпаривают в вакууме. После экстракции этилацетатом остаток хроматографируют на окиси алюминия и затем элюируют смесью ДХМ/этилацетат (80/20, по объему). Получают 850 мг целевого продукта, который перекристаллизовывают из смеси ДХМ/изопропиловый эфир.
ЯМР-спектр дается на фиг.6.
При использовании методов, аналогичных описанным выше, были получены промежуточные соединения (VI) для синтеза соединений (I), соответствующих изобретению.
Полученные соединения (VI) описываются в табл.1.
Полученные соединения (I) описываются в табл. 2.
/3/ Соединение (см. табл.1) характеризуется величиной вращения плоскости поляризации:
α
Для каждого соединения формулы /1/, имеющего значения заместителей R'1, R'5, R'2 и NR6R7, ( см.табл.2), указан изомер цис-и изомертранс.
Пример 34.
Анализ
Рассчитано,%: C 55,54; H 4,24; N 3,93
Установлено,%: C 55,72; H 4,57; N 3,83 1
ЯМР-спектр при 200 МГц (ДМСО : 2,5 м.д.)
Пример 34 поясняет фиг.5.
Пример 38.
0,7-1,1 м.д. : м: 6H: 2CH3(Et)
2-4 м.д. : м: 17Н:2CH2 (Et), 2CH2-N, N-CH3, 2OCH3
5,2-5,7 м.д. : 3с: 1H:H (индолин)
6,2-8,2 м.д. : м: 11Н:ОН+ ароматика
Пример 39.
0,3-1,2 м.д. : м: 3Н: CH3(Et)
1,5-4,3 м.д. : м: 15Н: CH2-CO, CH2 (Et), CH2-N, 2OCH3, CO2CH3
5,2-5,6 м.д.: 3с: 1Н:H (индолин)
6,2-8,2 м.д. :м: 11Н: ОН + ароматика
Пример 40.
0,8-1,1 м.д. : м: 3Н: CH3 (Et)
2,2-3,9 м.д. : м: 15Н: CH2CO, CH2(Et), CH2N, CO2CH3, 2OCH3,
5,3-5,7 м.д. : 2c: 1H:H (индолин)
6,6-8,2 м.д. : м: 11Н: ОН: + ароматика
Пример 63.
0,4-1 м.д. : расщепл, т: 3Н:CH2-CH2-CH3
5 м.д. : м: 2Н: CH2-CH2-CH3
2,5-4,4 м.д. : м: 13Н: CH2-CH2-CH3, NCH2, COOCH3, 2OCH3
5,2-5,8 м.д. : уш.с: 1Н:H (индолин)
6,5-8,3 м.д. :м: 11Н: ОН + ароматика
Пример 66.
от 0 до 1,5 м.д. :м: 3Н:CH2-CH3,
2,3-5,8 м.д. : м: 14Н: CH2-CH3, NCH2COOCH3, 2OCH3, H(индолин)
6,1-8,3 м.д. :м: 11Н: CH + ароматика.
Пример 75.
1,95 м.д.: уш.с: 2Н: NH2
от 2,7 до 5,3 м.д.: м: 12Н: 2OСН3, 2NCH2, H(индолин), CH 11Н2
от 6 до 8,3 м.д. : м: 11Н: CH + ароматика.
Пример 76a.
альфа25 D = +102o (с = 0,35 : хлороформ)
Пример 76b.
альфа25 D =-158o (с = 0,2, хлороформ)
Некоторые соединения, соответствующие изобретению, описанные в табл.2, используются для получения других соединений, относящихся к изобретению. Например, соединение 41 было получено из соединения 39 обработкой в основной среде в метанольной смеси MeOH/H2О. Соединение 49 было получено из соединения 41 обработкой водным аммиаком в присутствии ДИРЕА и БОФ.
Пример 77.
N-Этил-N-(2-аминоэтил) -5-хлор-3- (2-хлорфенил) -1-(3,4-диметиксифенилсульфонил) -3-гидрокси-2-индолинкарбоксамид, (цисизомер)
500 мг соединения 49 растворяют в 10 мл ацетонитрила и 10 мл воды и добавляют 252 мг пиридина и 380 мг бис(Трифторацетокси) йодбензола.
После перемешивания в течение 2 ч смесь поглощают раствором хлористоводородной кислоты, экстрагируют эфиром, подщелачивают разбавленным раствором гидроокиси натрия, экстрагируют с помощью ДХМ, и экстракт сушат и концентрируют. Образуется масло, и целевой продукт затем кристаллизуют из эфира.
m 150 мг
Т.пл. 164oC
Пример 78.
N-Этил-N-[(lS) (1-этоксикарбонил) этил] -5-хлор-3-(2-хлорфенил) -1- (3,4-диметоксифенилсульфонил) -3-гидрокси-2-индолинкарбоксамид (цисизомер)
А) N-[2-(2-Хлорфенилкарбонил)-5-хлорфенил] -N(3,4-диметоксифенилсульфонил) хлорангидрид аминоуксусной кислоты.
Смесь, содержащую 11 г кислоты, полученной в примерах 10 и 11, этап А, и 5 г тионилхлорида в 10 мл хлороформа, нагревают в течение 1 ч при 60oC. Смесь оставляют до принятия комнатной температуры, концентрируют в вакууме, и остаток растворяют в ДХМ (дважды). Получается желтое масло, которое используется как таковое на следующем этапе.
ИК: 1800 см-1 (C=O)
В) 2' ,5-Дихлор-N-(3,4-диметоксифенилсульфонил)-N-[N-этил-N'-((1S)(1-этоксикарбонил)этил)этоксикарбамоил-метил]2-аминобензофенон.
Получение этого соединения проводилось в соответствии с работой J.Org. Chem. 1985, 50, 945-950.
5,15 г (L)-Boc (N-Et)AlaOEt: растирают с 10 мл трифторуксусной кислоты при 0oC для удаления группы Вос. Смесь концентрируют в вакууме, растворяют в 20 мл ДХМ, охлаждают до -78oC и добавляют 2 эквивалента ТЭА и хлорангидрид кислоты, полученный на предыдущем этапе, растворяют в ДХМ. Через 18 ч выдержки при комнатной температуре смесь экстрагируют с помощью ДХМ, экстракт промывают водой и затем хроматографируют на двуокиси кремния с использованием для элюирования смеси ДХМ/этилацетат (90/10, по объему). Целевой продукт кристаллизуют из изопропилового эфира.
Т.пл. 112oC
m 8 г
С) N-Этил-N-[(1S))(1-этоксикарбонил)этил] -5-хлор-3- (2-хлорфенил) -1-(3,4-диметоксифенилсульфонил) -3-гидрокси-2-индолинкарбоксамид (цисизомер)
Соединение, полученное на предшествующей стадии, перемешивают в течение 18 ч при комнатной температуре в 10 мл ТГФ и 20 мл этанола в присутствии 1,46 г ДБУ. Смесь концентрируют в вакууме, остаток поглощают в ДХМ, промывают водой, концентрируют, и продукт хроматографируют на окиси алюминия с элюированием смесью этилацетат /ДХМ/10/90 по объему)
ЯМР
О- 0,9 м.д. : расщепл, д.: 3Н:CH-CH3
0,9-1,7 м.д. :м: 6Н: 2CH3(этил)
от 2,6 до 5,8 м.д. :м: 12Н: 2OCH3, NCH2, OCH2, NCH, COCH
6,1 до 8,3 м.д. :м: ЛН: ОН -1OН ароматика.
Примеры 79 и 80.
N, N-ди(2-метоксикарбонил-этил)-5-хлор-3- ( 2-хлорфенил) -1-(3,4-диметоксифенилсульфонил)-3-гидрокси-2-индолинкарбоксамид, цисизомер и трансизомер.
A) N,N-ди(2-метоксикарбонилэтил)бензиламин
Получение осуществляется в соответствии с работой J.Am.Chem.Soc. 1950, 72, 3298.
107 г бензиламина в 200 мл этанола охлаждают на ледяной бане и к ним медленно добавляют 172,2 метилакрилата в 250 мл этанола. После выдерживания при комнатной температуре в течение 13 дней растворитель испаряют в вакууме, и затем часть маслянистого остатка перегоняют.
Т. кип. = 135-140oC при 0,6 мм рт.ст.
m 30 г
ИК: 1730 см-1
В) N,N-(2-метоксикарбонилэтил)амин
27,9 г амина, полученного на предшествующем этапе, вводят в 500 мл метанола, перемешивают с 3 г 5%-ного палладия на угле и обрабатывают в течение 1 ч под давлением в атмосфере водорода. Смесь фильтруют через Целит® , промывают метанолом, и растворитель выпаривают в вакууме, остаточное масло используют непосредственно на следующем этапе.
C) N,N-ди(2-метоксикарбонилэтил)бромацетамид
Смесь, содержащую 14,3 г амина, полученного на предшествующем этапе, 100 мл ДХМ и 10,6 мл ТЭА, охлаждают на ледяной бане, по каплям добавляют 15,3 г бромацетилбромида, и смесь оставляют на 48 ч при комнатной температуре при перемешивании. Смесь экстрагируют ДХМ, экстракт промывают водой, после чего осуществляют хроматографию на двуокиси кремния с элюированием смесью ДХМ/метанол (97/3, по объему)
m 15,9 г
ИК: 1650 см-1 и 1730 см-1
Д) 2',5-Дихлор-[N-(3,4-диметоксифенилсульфонил)-N-[(N,N-ди (2-метоксикарбонилэтил))карбамоилметил]]2-амино-бензофенон
14,3 г 2',5-дихлор-2-(3,4-диметоксифенилсульфонамидо) -бензофенона помещают в 180 мл ДМФ и добавляют порциями 1,1 г гидрида натрия. После перемешивания в течение 1 ч при комнатной температуре смесь охлаждают на ледяной бане и к ней добавляют 14,3 г продукта, полученного на предшествующей стадии, после чего смесь оставляют перемешиваться в течение 72 ч при комнатной температуре. Смесь экстрагируют с помощью ДХМ, экстракт промывают водой, затем хроматографируют на двуокиси кремния с элюированием смесью ДХМ/этилацетат (93/7, по объему)
m 28,4 г
Т.пл. 130oC
Е) N,N-Ди[(2-метоксикарбонил)этил]-5-хлор-3-(2-хлорфенил) -1-(3,4-диметоксифенилсульфонил) -3-гидрокси-индолинкарбоксамид, цисизомер.
12 г соединения, полученного на предшествующем этапе, смешивают с 0,930 г метилата натрия в 150 мл метанола при 0oC, и смесь затем оставляют на ночь при комнатной температуре при перемешивании. Реакционную смесь нейтрализуют добавлением 5%-ного KHSO4, после чего растворитель выпаривают в вакууме. Остаток хроматографируют на окиси алюминия с элюированием смесью ДХМ/этилацетат (8/2 по объему). Извлекают 2,4 г целевого продукта, который кристаллизуют из метанола.
Т. пл. 175oC
F) N,N-Ди(2-метоксикарбонил)этил] -5-хлор-3-(2-хлор-фенил) -1-(3,4-диметоксифенилсульфонил) -3-гидрокси-2-индолинкарбоксамид, трансизомер.
Продолжают хроматографию предшествующего этапа и элюирование осуществляют смесью ДХМ/метанол (9,5/0,5 по объему). Получают 1,82 г трансизомера, который кристаллизуют из изопропилового эфира.
Т. пл. 85oC
Примеры 81, 82 и 83.
2-((2R)2-Карбамоилтиазолидинокарбонил) -5-хлор-3- (2-хлор-фенил) -1- (3,4-диметоксифенилсульфонил) -3-гидроксииндолин, (цисизомер: два соединения и трансизомер).
A) (L)4-Тиазолидинкарбоксамид
Это соединение получается в соответствии с работой J.Med. Chem. 1981, 24, 692.
В) 2', 5-Дихлор[N-(3,4-диметоксифенилсульфонил)-N-(((2R) 2-карбамоил)тиазолидинокарбонилметил)] 2-аминобензофенон.
Это соединение получают обычными методами из кислоты, полученной в примерах 10-11, этап А).
Т. пл. 125oC после кристаллизации из эфира.
C) 2-((2R)2-Карбамоилтиазолидинкарбонил)-5-хлор -3-(2-хлорфенил) -1-(3,4-диметоксифенилсульфонил) -3-гидроксииндолин.
4,3 г продукта, полученного на этапе В), в 90 мл метанола циклизуют при комнатной температуре в присутствии 1 г ДБУ. Смесь концентрируют, остаток поглощают водой и ДХМ, декантируют, промывают KHSO4 и затем сушат и концентрируют. Остаток хроматографируется на окиси алюминия при элюировании смесью ДХМ/метанол (97/3 по объему). Соединение получают в цисформе (смесь 2 диастереоизомеров): 1,5 г и затем в трансформе (смесь 2 диастереоизомеров):
m 1 г
а) Цисфракцию кристаллизуют из смеси метанол/ДХМ с целью получения цис соединения 1.
Т.пл. 176oC после кристаллизации из изопропилового эфира,
αD = +57o (с = 0,1, хлороформ)
в) Кристаллизационные растворы от продукта предыдущей стадии хроматографируют на двуокиси кремния при элюировании смесью этилацетат/ДХМ (30/70 по объему). Полученное циссоединение 2 перекристаллизовывают из эфира.
Т.пл. 205oC
αD = -185o (с = 0,3, хлороформ)
с) Трансфракцию (смесь двух диастереоизомеров) получают перекристаллизацией из изопропилового эфира.
Т.пл. 170o
Примеры 84, 85, 86 и 86а
5-Хлор-3-(2-хлорфенил) -1- (3,4-диметоксифенилсульфонил) -3- гидрокси-2-[(2S) 2-N,N-диметилтиокарбамоил -пирролидинокарбонил]индолин, цисизомер (2 соединения), (трансизомер, 2 соединения)
A) (L) N,N-диметил(N'-Воc)-пролинтиоамид
Это соединение получается согласно работе J. Mod. Chem. 1989, 2178.
2,36 г N,N-диметил (N-Воc) пролинамида нагревают в безводном толуоле в атмосфере аргона при 80oC в течение 4 ч в присутствии 2,3 г реактива Лавессона. Через 24 ч растворитель испаряют и добавляют изопропанол. Образующийся осадок декантируют, изопропанол выпаривают, а остаток хроматографируют на двуокиси кремния с элюированием смесью гексан/этилацетат/ (30/70 по объему). Полученный продукт перекристаллизовывают в холодном виде из смеси ДХМ/ изопропиловый эфир (30/70 по объему)
Т.пл. 62oC
В) 2', 5-Дихлор-[N-(3,4-диметокснфенилсульфонил)-N- ((2S) (2- N',N'-диметилтиокарбамоил)пирролидинокарбонилметил)] 2-аминобенэофенон
3 г продукта, полученного на предшествующем этапе, растворяют в 10 мл ДХМ и обрабатывают при 0oC в течение 2 ч с помощью 10 мл трифторуксусной кислоты. Смесь выпаривают до сухого состояния, затем добавляют 20 мл ДХМ и 6,1 г кислоты, порученной в примерах 10 и 11, этап A) - при 0oC и смесь нейтрализуют действием 3 г ДИПЭА. 5,15 г БОФ растворяют в 30 мл ДХМ, и этот раствор добавляют в течение 30 мин при 0oC к полученному ранее раствору, pH поддерживают нейтральным добавлением ДИПЭА, и смесь оставляют при перемешивании при 0oC на 3 ч. После выдерживания в течение ночи при комнатной температуре смесь экстрагируют обычным способом, затем хроматографируют на двуокиси кремния с элюированием смесью ДХМ/этилацетат. G85/15, по объему). Полученный продукт перекристаллизовывают из изопропилового эфира.
Т.пл. 182-185oC
αD = -72o (с = 0,32, хлороформ)
C) 5-Хлор-3- (2-хлорфенил) -1-(3,4-диметоксифенилсульфонил) -3-гидрокси-2-((2S)2-N, N диметилтиокарбамоил -пирролидинкарбонил)индолин (цисизомерия: 2 соединения и трансизомер: 2 соединения)
3,8 г соединения, полученного на предыдущем этапе, растворяют в 15 мл ДХМ, и смесь нагревают с обратным холодильником в течение 36 ч в присутствии 850 мг ДБУ. Образующиеся различные изомеры разделяют последовательными хроматографическими прогонами на двуокиси кремния.
а) При использовании смеси ДХМ/этилацетат (85/15, по объему) целевое соединение элюируют сначала в виде смеси 2 цисдиастереоизомеров. Наименее растворимый диастереоизомер дважды кристаллизуют из смеси ДХМ/изопропиловый эфир/метанол и затем перекристаллизовывают из минимального количества ДМФ при 60 С с последующим добавлением 2 объемов этанола.
Т.пл. 270oC
αD = -278oC (с = 1, хлороформ)
в) Поглощают кристаллизационные жидкости от смеси предыдущего этапами второй цисдиастереоизомер кристаллизуют из смеси ДХМ/изопропиловый эфир.
Т.пл. 249-251oC
αD +42 (с = 0,22, хлороформ)
с) Хроматографические фракции, элюированные последними, а также и кристаллизационные маточные жидкости этапов а) и в) соединяют и хроматографируют снова на двуокиси кремния с элюированием смесью гексан/этилацетат (20/80, по объему). Выделяют первую фракция, которую трижды перекристаллизовывают из смеси ДХМ/изопропиловый эфир, а нерастворившееся вещество остается на бумаге после каждой перекристаллизации. Таким образом получают трансизомер 1.
Т.пл. 191-193oC
αD = +74,5o (с = 0,2, хлороформ)
d) Вторая фракция содержит трансизомер 2, который подвергается перекристаллизации из смеси ДХМ/изопропиловый эфир и кристаллизуется с 1/3 моля изопропилового эфира.
Т.пл. 170oC
αD = -266o (С = 0,14, хлороформ)
Примеры 87, 88 и 89.
2-((2S) -2-Карбамоилпирролидинокарбонил) -5-хлор-3-циклогексил-1- (3,4-диметоксифенилсульфонил) -3-гидрокси-индолин, (цисизомеры: 2 соединения, трансизомер)
А) 5-Хлор-2-[N-(3,4-диметоксифенилсульфонил) амино]-циклогексилфенон.
Раствор 35,6 г 2-амино-5-хлорциклогексилфенона и 39,5 г 3,4-диметоксифенилсульфонилхлорида в 340 мл пиридина перемешивают при комнатной температуре в течение 24 ч. Растворитель выпаривают в вакууме, а остаток затем промывают водой и кислотным раствором (0,5 NHCl). Целевой продукт кристаллизуют из этанола.
Т.пл. 135oC
m 56,1 г
В) 2-[N-Бензилоксикарбонилметил-N(3,4-диметоксифенилсульфонил) амино] -5-хлорциклогексилфенон
3,2 г гидрида натрия порциями добавляют к 52,6 г соединения, полученного на предыдущем этапе, в 520 мл ДМФ и смесь перемешивают при комнатной температуре в течение 1 ч. После охлаждения на ледяной бане по каплям добавляют 21 мл бензилоксикарбонилметилбромида, и смесь перемешивают при комнатной температуре в течение 24 ч. Растворитель выпаривают в вакууме, а остаток растворяют в воде. Его экстрагируют с помощью ДХМ, и экстракт промывают водой, полученный продукт используется непосредственно на следующем этапе.
С) N-(4-Хлор-2-циклогексилкарбонил)фенил -N-(3,4-ди-метоксифенилсульфонил) аминоуксусная кислота
Соединение, полученное на предшествующей стадии, вводят вместе с 3,9 г 5%-го палладия на угле в 700 мл уксусной кислоты в токе водорода (1 атмосфера). В конце реакции палладий фильтруют на Целите и промывают горячей уксусной кислотой, растворитель выпаривают в вакууме, а остаток растворяют в воде. Его экстрагируют ДХМ и экстракт промывают водой и затем концентрированным раствором NaHCO3. Полученный остаток хроматографируют на двуокиси кремния при элюировании смесью ДХМ/метанол (97/3 по объему). Целевой продукт кристаллизуют из этанола.
Т.пл. 160oC
m 22,4 г
D) 5-хлор-[N-(3,4-диметоксифенилсульфонил)-N-((2S) -2-карбамоилпирролидинкарбонилметил)] 2-аминоциклогексилфенон
Смесь, содержащую 9,92 г кислоты, полученной на предыдущем этапе, 3 г (L)-пролинамидгидрохлорида и 3,5 мл ДИПЭА в 75 мл ДХМ охлаждают до 0oC. добавляют 8,84 г БОФ в растворе ДХМ и pH поддерживают при 7 добавлением ДИПЭА. Смесь оставляют перемешиваться на 24 ч при комнатной температуре. Смесь экстрагируют ДХМ, и экстракт промывают насыщенным раствором NaHCO3, соленым раствором, 5%-ным раствором KHSO4 и снова соленым раствором. Продукт хроматографируют на двуокиси кремния при элюировании смесью ДХМ/ метанол (96/4 по объему). Целевой продукт затвердевает в изопропиловом эфире.
Т.пл. 110oC
m 7,3 г
αD = -53,9o (с = 1, хлороформ)
Е) 2-((2S) -2-Карбамоилпирролидинкарбонил) -5-хлор-3-циклогексил-1- (3,4-диметоксифенилсульфонил) -3-гидроксииндолин, цисизомер (2 соединения), трансизомер.
5,9 г соединения, полученного на предшествующей стадии, и 1,67 г ДБУ помещают в 60 мл метанола и перемешивают 48 ч при 0oC. Растворитель выпаривают в вакууме, добавляют воду, смесь экстрагируют ДХМ, а экстракт затем промывают 5%-ным раствором KHSO4. Продукт хроматографируют на окиси алюминия с элюированием смесью ДХМ/метанол (98/2 по объему).
а) Наименее полярная фракция содержит 2 цисизомера. Эта фракция подвергается перекристаллизации из метанола. Первое полученное таким образом соединение (цис 1) очищают с использованием ВЭЖХ.
Т.пл. 185oC
Перекристаллизацией маточных растворов из метанола получают второе соединение (цис 2). Чистота ВЭЖХ: 75% (он содержит 25% цис 1)
Т.пл. 132oC
в) Наиболее полярная фракция содержит трансизомер, в качестве, по-видимому, единственного соединения, которое выделяют перекристаллизацией из метанола.
Т.пл. 240oC
αD = -55,1o (с = 1, хлороформ)
Пример 89а.
2-((2S) -2-Карбамоилпирролидинокарбонил) -5-хлор-3-циклогексил -1-(3,4-диметоксифенилсульфонил) -3-гидроксииндолин, цисизомер (2 соединения), трансизомер.
При использовании методики, аналогичной описанной для примеров 87, 88 и 89, получается соединение с использованием соединений (D) - пролинового ряда.
Соединение, полученное после кристаллизации из смеси ДХМ/метанол, имеет трансконфигурации.
Т.пл. 238oC
αD = +164o (с = 0,245, хлороформ/метанол, 8,2 по объему).
ЯМР-спектр этого соединения и соединения, описанного на этапе E в) предшествующего примера, являются идентичными.
Пример 90.
5-Хлор-3- (2-хлорфенил) -1-(3,4-диметоксифенилсульфонил) -2-[(2S) -2-(N-метиламинокарбонил) пирролидинокарбонил] -3-гидроксииндолин, цисизомер.
920 мг соединения, полученного в примере 28, вводят при перемешивании в 20 мл ДХМ, содержащих 371 мг БОФ, в течение 15 мин, затем пропускают ток монометиламина в течение 10 мин, после чего перемешивание продолжают еще 30 мин. Смесь растворяют в воде, декантируют, промывают гидросульфатом калия и карбонатом натрия, сушат и концентрируют. Остаток хроматографируют на двуокиси кремния с элюированием смесью ДХМ/метанол (97,5/2,5 по объему). Полученный целевой продукт кристаллизуют из смеси изопропиловый эфир/ДХМ.
m 750 мг
Т.пл. = 158oC
αD = -216o (с = 0,3, хлороформ)
Действуя в соответствии с методиками, описанными в предшествующих примерах (примеры 27-31 и 90) при использовании производных (L) - пролина (за исключением противоположных указаний), были получены другие промежуточные соединения (VI) для синтеза соединений (1), соответствующих изобретению.
Полученные соединения (VI) описываются в табл. 3.
Полученные соединения (1) описываются в табл. 4.
Соединение примера 107 является энантиомером соединения примера 106.
Соединение примера 108а было получено из соединения примера 28 реакцией с гидрохлоридом гидроксиламина в ДМФ и действием реактива БОФ в присутствии ДИПЭА.
Некоторые соединения, соответствующие изобретению, описанные в табл. 4, используются для получения других соединений. Так, соединение примера 99 дает возможность получить соединение примера 101, затем примера 103 и, наконец, соединения примера 104.
Пример 109.
2-((2S, 4S) 4-Азидо-2-(метоксикарбонил)пирролидинокарбонил) -5-хлор-3-(2-хлорфенил) -1-(3,4-диметоксифенилсульфонил) -3-гидроксииндолин, цисизомер.
А) 2', 5-Дихлор-2)-N-(3,4-диметоксифенилсульфонил)-N- ((2S, 4R) 4-гидрокси-2-(метоксикарбонил)пирролидинокарбонилметил)] 2-аминобензофенон
15 г кислоты, полученной в примерах 10 и 11, этап А), и 6,25 г метил-(2S,4R) 4-гидроксипролинат гидрохлорида охлаждают до 0oC в 150 мл ДХМ в присутствии 7,4 ДИПЭА. По каплям в течение 30 мин добавляют 12,7 г БОФ в 30 мл ДХМ и добавляют количество ДИПЭА, необходимое для нейтрализации раствора. После выдерживания в течение ночи при комнатной температуре смесь экстрагируют обычным образом и хроматографируют на двуокиси кремния с элюированием смесью ДХМ/ этилацетат (60/40 по объему). Целевой продукт кристаллизуют из смеси ДХМ/эфир/изопропиловый эфир.
Т.пл. 128-131oC
αD = +8,5o (с = 0,38, хлороформ)
В) 2', 5-Дихлор-[N-(3,4-диметоксифенилсульфонил)-N-((2S, 4R) 4-мезилокси-2- (метоксикарбонил) пирролидинокарбонилметил] 2-аминобензофенон
2 г соединения, полученного на предшествующей стадии, растворяют при 0oC в 10 мл ДХМ. Добавляют 550 мг триэтиламина и затем 550 мг метансульфонилхлорида, и смесь оставляют на 20 ч при 0o. Добавляют воду и промывают 0,5 N хлористоводородной водой и затем раствором бикарбоната натрия сушат над сульфатом магния и выпаривают. Полученное масло используется непосредственно на следующем этапе.
C) [N-((2S, 4S)4-азидо-2-(метоксикарбонил) пирролидинокарбонилметил) -N-(3,4/диметоксифенилсульфонил] 2-амино-2',5-дихлорбензофенон
11 г продукта, полученного на предыдущем этапе, нагревают в 60 мл ДМСО при 80-90oC в присутствии 2,7 г азида натрия в течение 18 ч. Смесь выливают в воду, экстрагируют этилацетатом, промывают водой, сушат и подвергают хроматографированию на двуокиси кремния с элюированием смесью пентан/этилацетат (5o/50 по объему). Получается масло (10 г)
αD = -25,5o (с 0,39, хлороформ, 26oC)
D) 2-((2S,4S)4-Азидо-2(метоксикарбонил)пирролидинокарбонил) -5-хлор-3-(2-хлорфенил) -1-(3,4-диметоксифенилсульфонил) -3-гидроксииндолин, цисизомер.
3,38 г продукта, полученного на предыдущем этапе, циклизуют в обычных условиях в присутствии ДБУ. Получают целевой продукт, который подвергают перекристаллизации из смеси ДХМ/изопропиловый эфир.
m 755 мг
Т.пл. 200-202oC
αD = -176o (с 0,21, хлороформ, 26oC).
Пример 110.
2-[(2S,4S)(N-Бензилоксикарбонил-N-метил) 4-амино-2-(метоксикарбонил)пирролидинокарбонил]-5-хлор-3- (2-хлорфенил) -1-(3,4-диметоксифенилсульфонил)-3-гидроксииндилин, цисизомер.
А) Метиловый эфир (N-Вос)-4-гидроксипролина.
Исходным веществом является хлоргидрат метилового эфира (2S,4R) 4-гидроксипролина.
19 г этого соединения суспендируют в 100 мл ТГФ, добавляют 22,9 г (Вос)2O, после чего смесь охлаждают до 0oC. По каплям добавляют 21,2 г триэтиламина в 25 мл ТГФ, и затем смесь перемешивают в течение 12 ч при 0oC и 4 ч при 60oC. Добавляют воду, смесь экстрагируют этилацетатом, промывают водой, раствором гидросульфата калия (4 раза), водой и затем соленой водой. Растворитель выпаривают и выделяют масло (21,6 г), которое содержит небольшое количество (Воc)2О.
В) Метиловый эфир (2S,4R)(N-Вос)-4-мезилокси-пролина
Раствор, содержащий 22,9 г продукта, полученного на предыдущем этапе в 250 мл ДХМ, охлаждают до 0oC. По каплям добавляют 22,9 г мезилхлорида в 10 мл ДХМ, затем по каплям добавляют 9,4 г триэтиламина в 100 мл ДХМ, и смесь оставляют на ночь для доведения ее до комнатной температуры. Смесь выпаривают до сухого состояния, добавляют воду, смесь экстрагируют этилацетатом, промывают водой и соленой водой и сушат над сульфатом магния. После второго выпаривания образуется масло, которое непосредственно используется на следующем этапе.
C) Метиловый эфир (2S,4S)(N-Вос)-4-азидопролина
Это соединение получается из соединения, полученного на этапе В.
15,2 г метилового эфира (N-Вос)-4-мезилоксипролина растворяют в 70 мл ДМСО, и раствор нагревают при 90oC в течение 5 ч в присутствии 3,05 г азида натрия. Смесь охлаждают, добавляют воду, смесь экстрагируют этилацетатом, промывают водой, затем соленой водой и сушат над MgSO4. Полученное масло очищается хроматографированием на двуокиси кремния при элюировании смесью этилацетат/гексан (40/60 по объему)
αD = -37,8o (с 3, хлороформ)
αD = -36,6o (с 2,8, хлороформ) согласно D.J.Abraham и др. J.Med.Chem. 1983, 549, 26.
D) Метиловый эфир (2S,4S)(N-Вос)-4-аминопролина
8,45 г соединения, полученного на этапе С, растворяют в 100 мл метанола, добавляют 500 мг 10%-ного Pd/C,и смесь подвергают гидрогенизации при 40oC в течение 18 ч. Катализатор отфильтровывают, половину метанола выпаривают, добавляют 100 мл 0,5 N НС1, затем выпаривают оставшуюся часть метанола, а непрореагировавшее исходное вещество экстрагируют этилацетатом. Водную фазу обрабатывают карбонатом натрия, а фракцию, содержащую целевой продукт (m 4,35 г), экстрагируют этилацетатом.
Е) Метиловый эфир (2S,4S)(N-Вос)-4-(N'-бензилоксикарбониламино)пролина.
Неочищенный продукт, полученный на предыдущем этапе, растворяют в 15 мл эфира и 15 мл ДХМ при 0o. Добавляют 2,3 г ДИПЭА и затем 3,03 г бензилхлорформиата в 5 мл ДХМ в течение 70 мин при 0oC. Спустя 3 ч растворители выпаривают в вакууме при комнатной температуре, добавляют воду и этилацетат, и органическую фазу промывают последовательно раствором гидросульфата калия (3 раза), водой (3 раза), раствором карбоната натрия (3 раза), водой (3 раза) и соленой водой. Продукт подвергают хроматографированию на двуокиси кремния элюированием смесью гексан/этилацетат (40/60 по объему) с целью получения целевого продукта.
αD = -16,4o (с 0,3, хлороформ)
F) Метиловый эфир (2S,4S)(N-Вос)-(N'-бензилоксикарбонил-N'-метил) 4-аминопролина
2 г соединения, полученного на предыдущей стадии, растворяют в 20 мл ДМФ при 0oC в атмосфере аргона в присутствии 2,25 г йодистого метила. По порциям добавляется 170 мг 80%-ного гидрида натрия, после чего смесь перемешивают в течение 90 мин при 0oC. Смесь экстрагируют водой и этилацетатом, органическую фазу промывают водой, затем соленой водой. Продукт подвергают хроматографированию на двуокиси кремния элюированием смесью гексан/этилацетат (50/50 по объему). Получают 1,55 г целевого продукта.
αD = -38,8o (с = 0,38, хлороформ)
G) 2',5-Дихлор-[(2S,4S)N-(3,4-диметоксифенилсульфонил) -N(N'-бензилоксикарбонил-N'-метил) 4-амино-2-метоксикарбонил) пирролидинокарбонилметил]2-аминобензофенон.
Этот продукт получают с использованием обычных методов
αD = -22,4 o (с = 0,37, хлороформ)
Н) 2-[((2S,4S)N-Бензилоксикарбонил-N-метил)4-амино-2- (метоксикарбонил) пирролидинокарбонил] -5-хлор-3- (2-хлорфенил) -1-(3,4-диметоксифенилсульфонил) -3-гидроксииндолин, цисизомер
Этот продукт получается циклизацией в присутствии ДБУ в соответствии с обычными методами. Образованные кристаллические вещества кристаллизуют из смеси ДХМ/изопропиловый эфир.
Т.пл. 129oC
αD = -129o (с = 0,321, хлороформ)
Изомерная чистота по методу ВЭЖХ составляет 99%
Соединения, полученные в примерах 109 и 110, используются для получения соединений, соответствующих изобретению, описанных в табл.5.
Соединение примера 112 дает возможность последовательно получить соединения примеров 115 и 116, описанные в табл. 6 ниже, а соединение примера 114 дает возможность получить соединение примера 116а.
Соединение примера 116а может быть получено либо конверсией примера 114, либо из (2S,4S)-(N-Вос)-4-(диметиламино)пролинамида, получение которого осуществляется следующим образом.
1) Метил (2S,4S)-(N-Вос)-4-аминопролинат готовится из метил (2S,4S)-4-азидопролината в соответствии с работой J.R.Well в J.Org.Chem. 1991, 56, 3009.
2) Метил (2S,4S)-(N-Вос)-4-(диметиламино)-пролинат.
4 г соединения, полученного на стадии 1), растворяют в 50 мл ацетонитрила, добавляют 12,8 мл 30%-ного формалина и затем, спустя 5 мин, 3 г цианоборогидрата натрия. После взаимодействия реагентов в течение 2 ч добавляется уксусная кислота для доведения pH раствора до 6. Спустя 3 ч ацетонитрил испаряют, добавляют воду, карбонат калия и твердый хлористый натрий, и смесь экстрагируют 4-мя объемами этилацетата. Органическую фазу выпаривают, остаток растворяют в 1 N хлористоводородной кислоте и осуществляют экстракцию этилацетатом. К водной фазе добавляют твердый карбонат натрия, затем твердый хлорид натрия и проводят экстракцию этилацетатом. После выпаривания остаток подвергают хроматографированию на силикагеле элюированием смесью ДХМ/метанол (95/5, по объему), и твердеющее масло выделяют.
m 2,1 г
ИК (ДХМ): 1755-1см , 1695-1см
3) 534 мг эфира, полученного на стадии 2), растворяют в 4 мл метанола и обрабатывают гидроокисью натрия (116 мг) в 1 мл воды в течение 48 ч при комнатной температуре. Смесь подкисляют до pH 3,5 с 0,5 H соляной кислотой и выпаривают до сухого состояния. Проводят азеотропную сушку остатка в присутствии бензола (5 раз) и затем остаток сушат в вакууме в течение 8 ч. После этого добавляют 2 мл ДМФ и 3 мл ДХМ, и смесь охлаждают до 0oC. Для нейтрализации реакционной смеси добавляются 865 мг БОФ и ДИПЭА. Через 15 мин через нее пропускают ток газообразного аммиака - дважды в течение 30 мин. После выдерживания в течение 2 ч при комнатной температуре ДХМ испаряют, добавляют карбонатную воду и хлорид натрия, и смесь экстрагируют 4-мя объемами этилацетата. После выпаривания остаток подвергают хроматографированию на двуокиси кремния. Смесью (ДХМ/метанол/NH4 ОН; 84,5/15/0,5 по объему) элюируют твердое вещество (m 185 мг), которое перекристаллизовывают из смеси ДХМ/изопропиловый эфир.
Т.пл. 183-186oC
αD = -63,1 o (с 0,24, хлороформ)
Пример 117.
Декарбоксилирование N-(2-карбоксиэтил)-N-этил-3-(2- хлорфенил) -5-хлор-1- (3,4-диметоксифенилсульфонил) -3-гидрокси-2-индолинкарбоксамида, цисизомер.
630 мг соединения, полученного в примере 41, растворяют в 20 мл ТГФ в атмосфере аргона и затем добавляют 101 мг N-метилморфолина при -15oC и 118 мг изобутилхлорформиата. После перемешивания в течение 5 мин добавляют 127 мг N-гидрокси-2-пиридинтиона и 101 мг ТФУ, смесь выдерживают при -15oC при перемешивании 15 мин, а затем добавляют 900 мг третбутилмеркаптана, и смесь оставляют для доведения ее до комнатной температуры. Затем реакционную смесь облучают в течение 1 ч 30 вольфрамовой лампой накала (150 Вт). Смесь концентрируют, поглощают водой, экстрагируют ДХМ, и экстракт сушат и концентрируют. Остаток подвергают хроматографии на двуокиси кремния с элюированием ДХМ/этилацетат (95/5 по объему). Получают целевой продукт.
m 300 мг
Т.пл. 215oC
Это соединение аналогично соединению примера 125, описанного в Европейской Патентной Заявке ЕР 469984. Оно имеет цисконфигурацию по отношению к связи 2,3 индолина и используется как исходный продукт.
Пример 118.
Декарбоксилирование 2-((2R) 2-карбоксиметил пирролидинокарбонил) -5-хлор-3-(2-хлорфенил) -1-(3,4-диметоксифенилсульфонил) -3-гидроксииндолина, цисизомер.
Работают по методике предыдущего примера, но используя соединение примера 102.
Полученный продукт перекристаллизовывают из смеси ДХМ/изопропиловый эфир.
Т.пл. 215-220oC
αD = -214,5o (с = 0,2, хлороформ)
Это соединение представляет собой 2((2$)2-метилпирролидинокарбонил) -5-хлор-3-(2-хлорфенил) -1-(3,4-диметоксифенилсульфонил)-3-гидроксииндолин, цисизомер.
Пример 119.
Декарбоксилирование 2-(2-карбоксипирролидинокарбонил) -5-хлор-3-(2-хлорфенил) -1-(3,4-диметоксифенилсульфонил) -3-гидроксииндолина, цисизомер
Работают по методике предыдущего примера с использованием соединения, полученного в примере 28, в качестве исходного вещества. Полученный продукт перекристаллизовывают из смеси изопропиловый эфир-ДХМ.
Т.пл. 263oC
αD = -201,5o (с 0,2, хлороформ)
Это соединение представляет собой 2-пирролидинокарбонил-5-хлор-3-(2-хлорфенил) -1-(3,4-диметоксифенилсульфонил) -3-гидроксииндолин, цисизомер.
Примерами фармацевтических композиций могут быть следующие галеновые формы:
Желатиновая капсула емкостью 480 мг
1) Активное начало - 5,0 мг
Кукурузный крахмал - 137,45 мг
Кристаллическая лактоза - 320,75 кг
Тальк - 9,6 мг
Коллоидная безводная двуокись кремния - 2,4 мг
Стеарат магния - 4,8 мг
2) Активное начало - 100,0 мг
Кукурузный крахмал - 108,96 кг
Кристаллическая лактоза - 257,24 мг
Тальк - 9,6 мг
Безводная коллоидная двуокись кремния - 2,4 мг
Стеарат магния - 4,8 мг
Ампула объемом - 10 мл
1. Активное начало - 1 мг
Полиэтиленгликоль 400 - 1,5 г
2. Активное начало - 10 мг/мл
Полиэтиленгликоль 400 - 4 г
Результаты биологических испытаний полученных соединений приведены в табл.7.
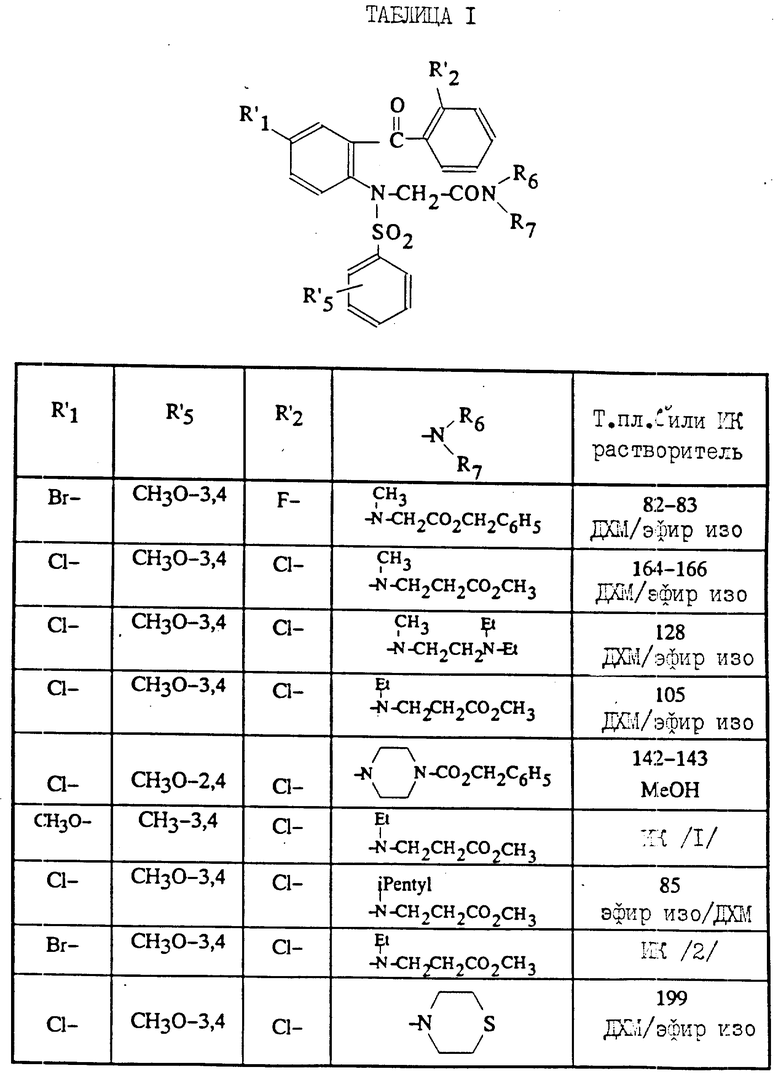
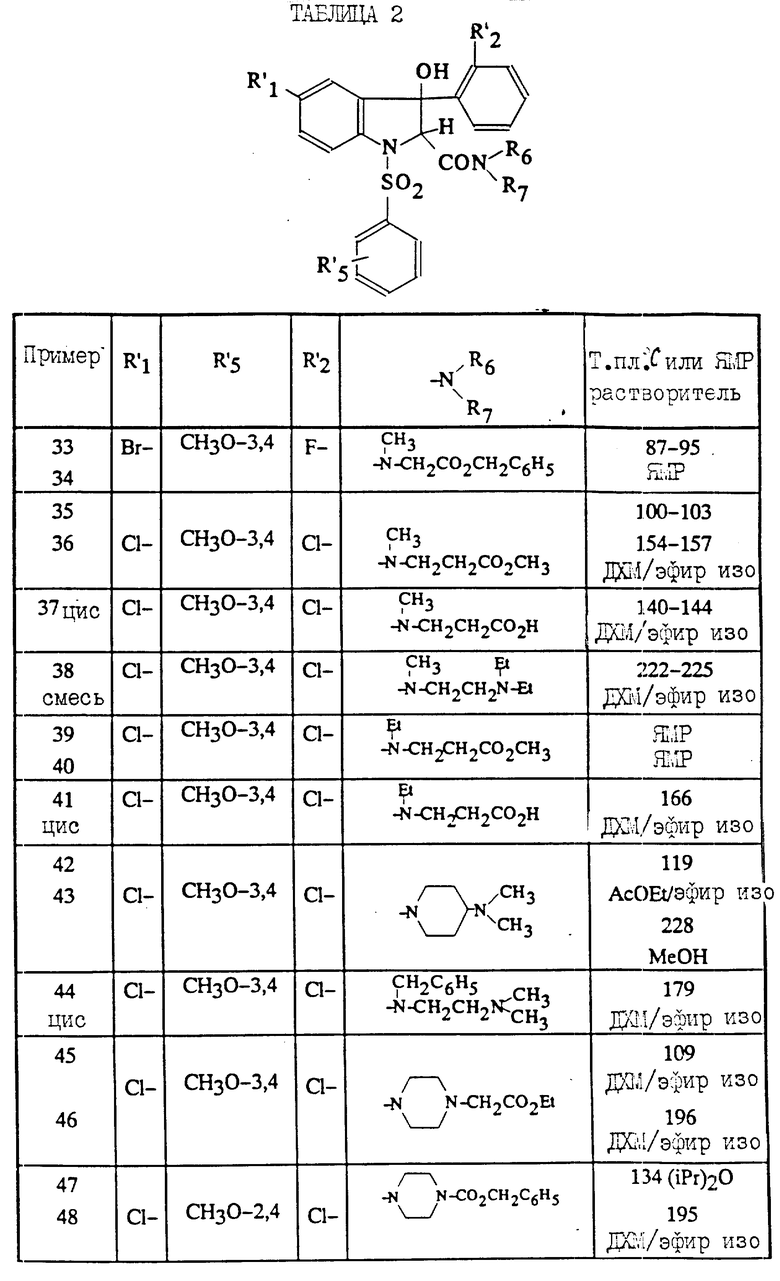
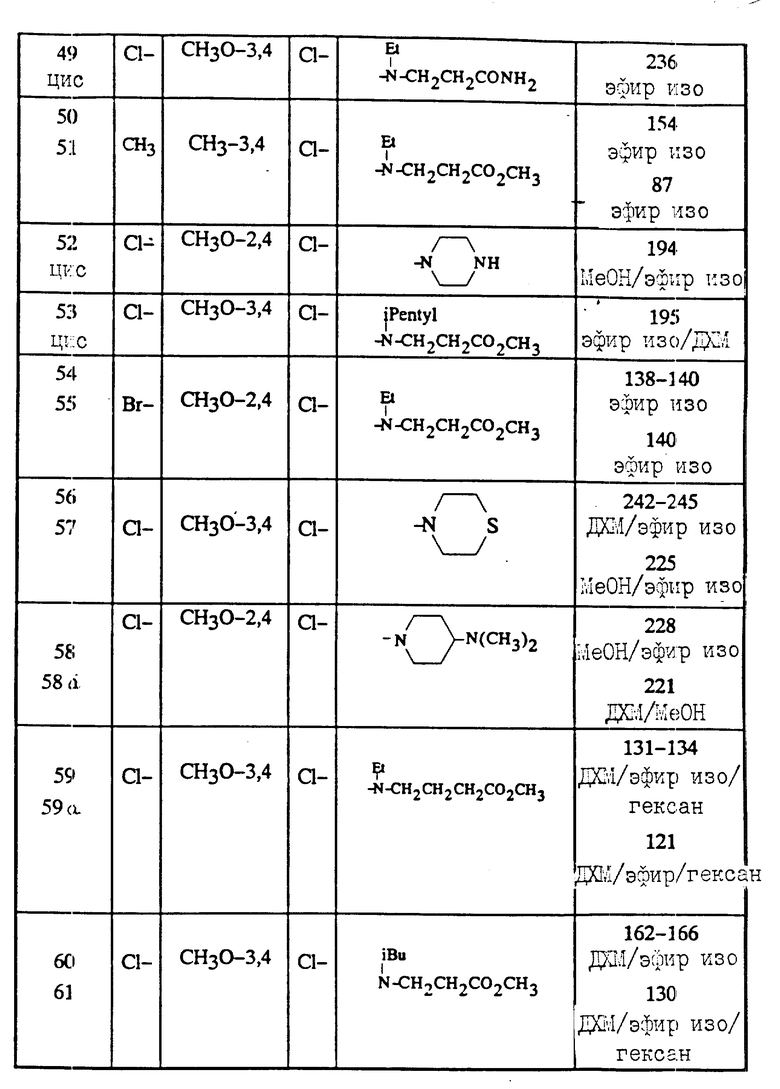
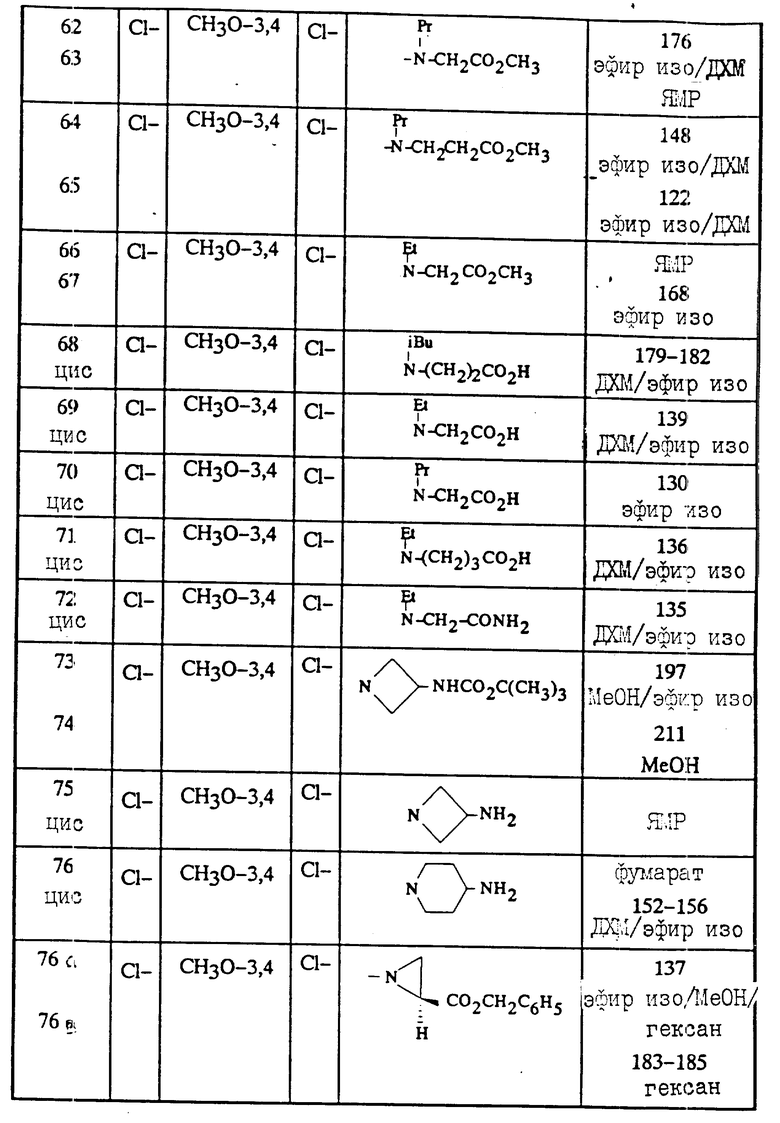
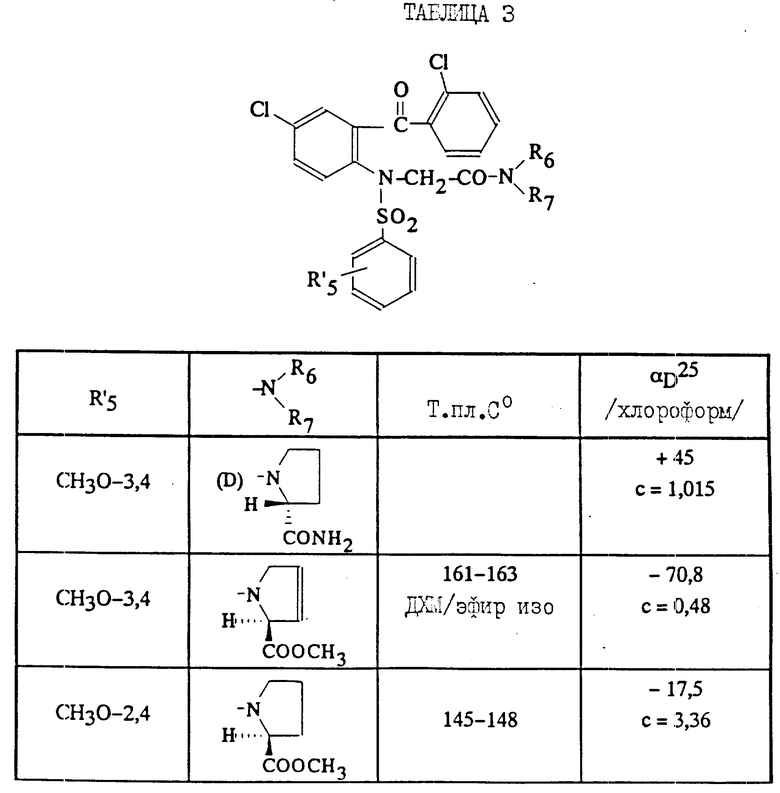
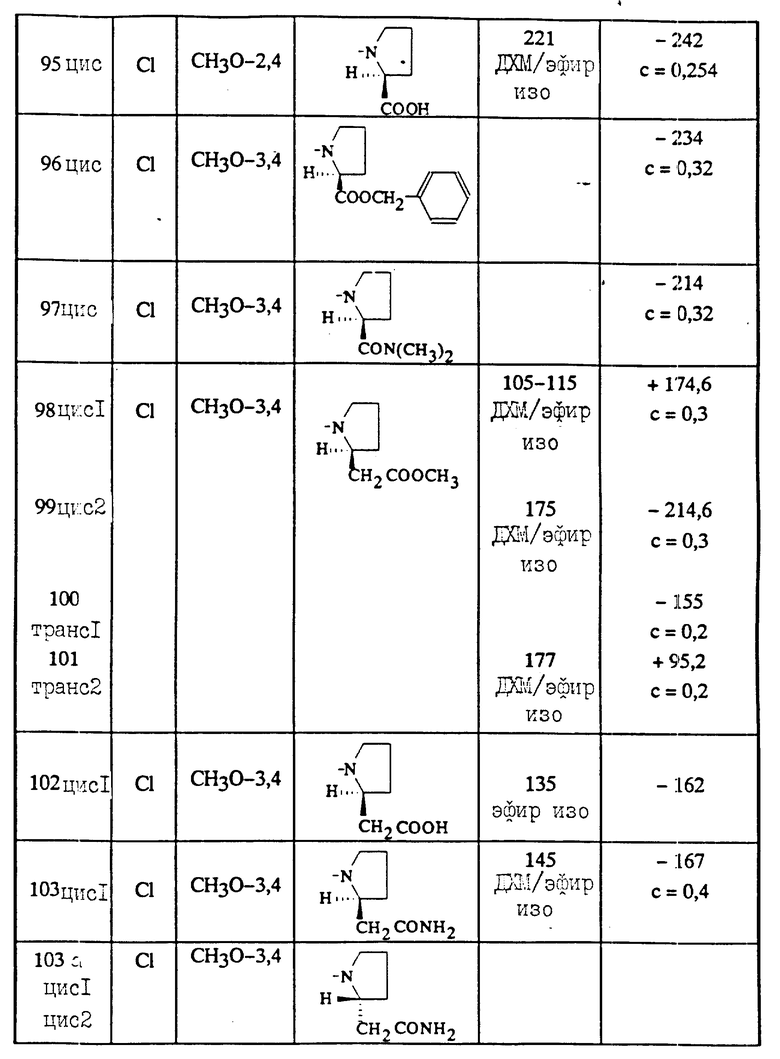
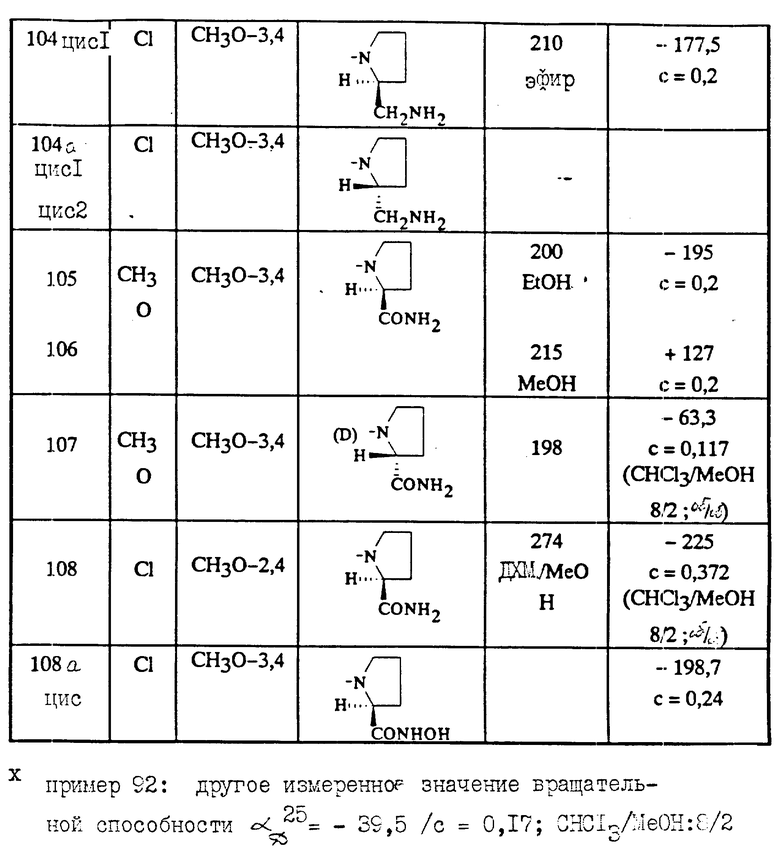
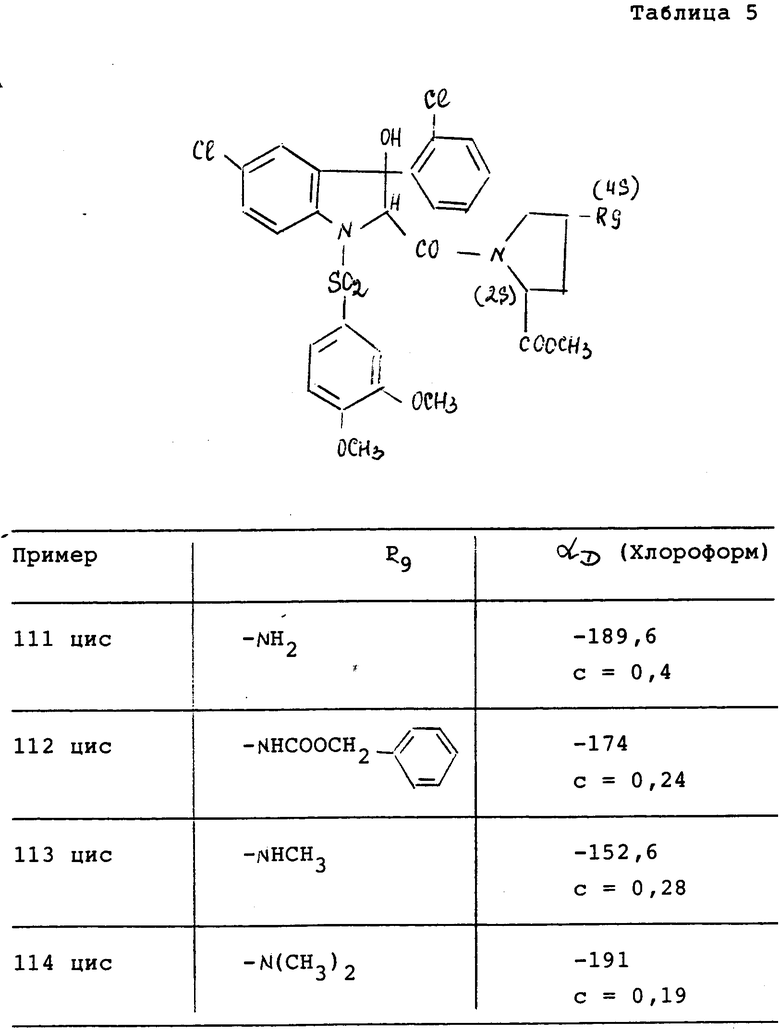
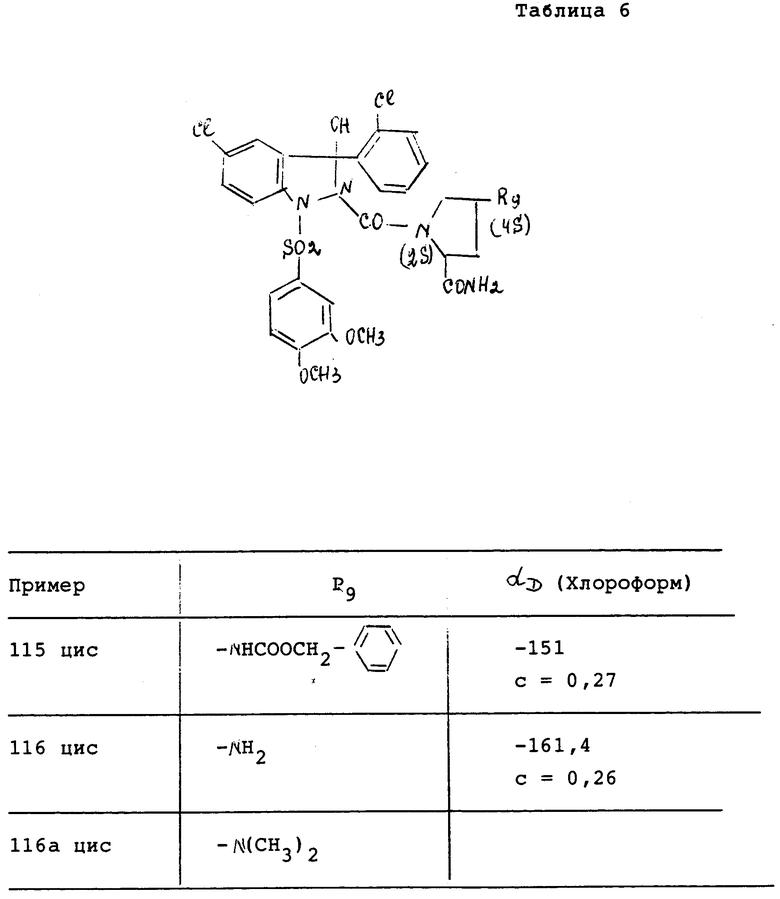
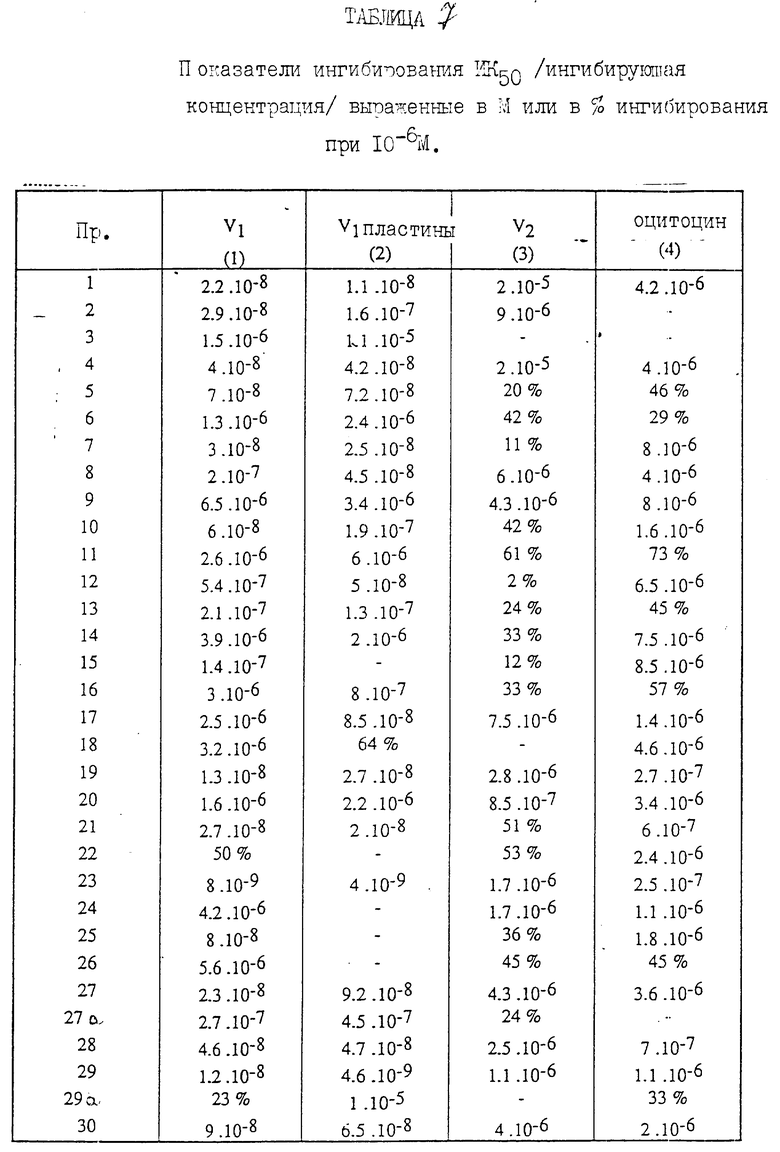
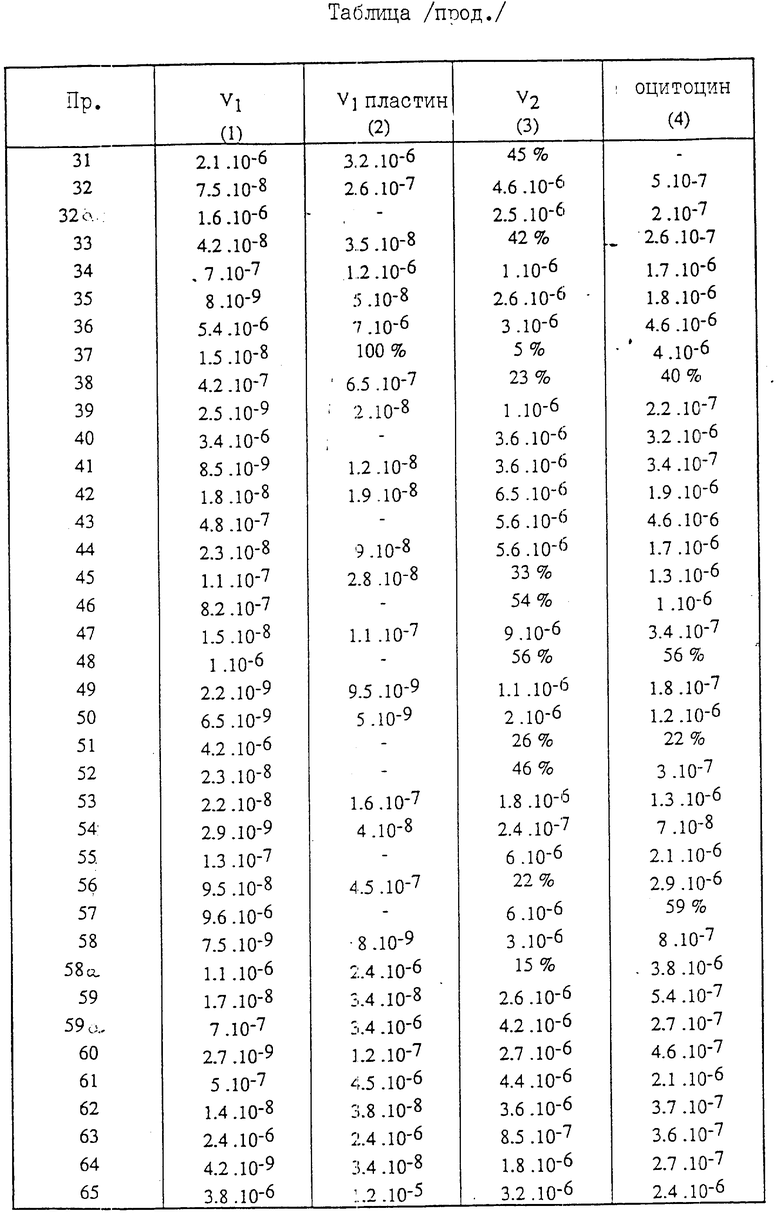
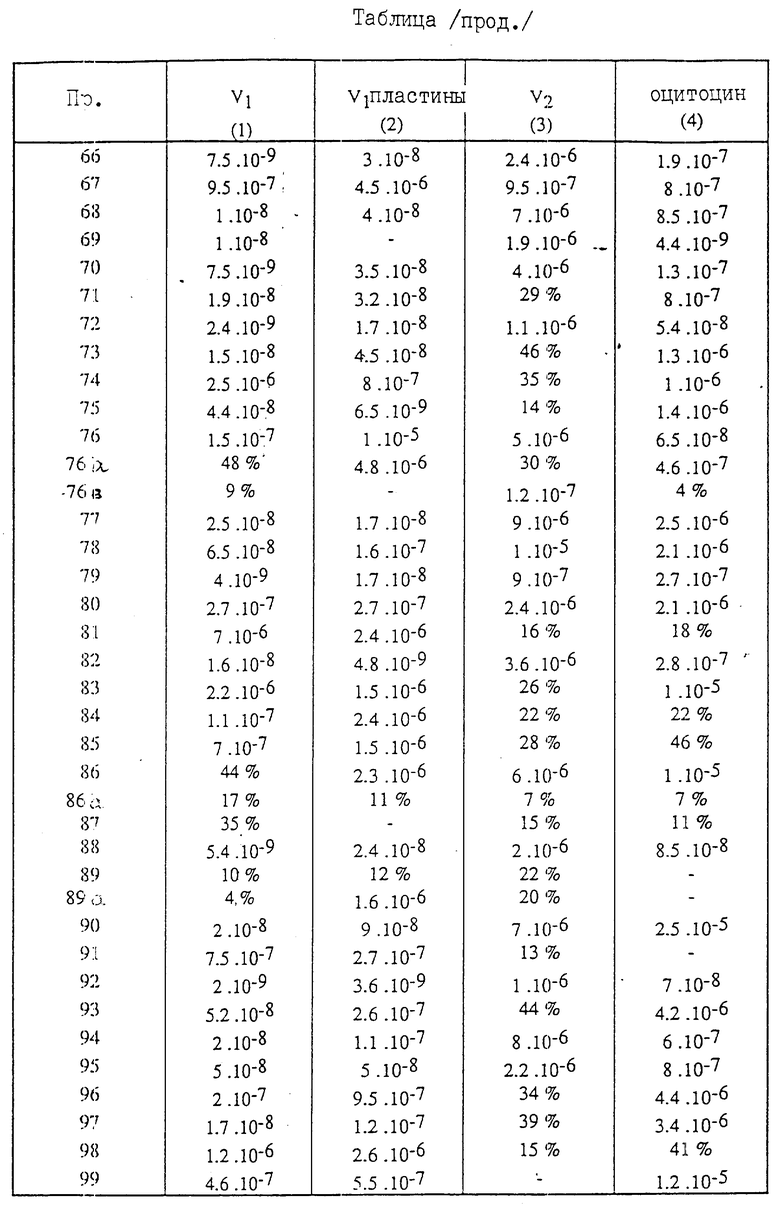
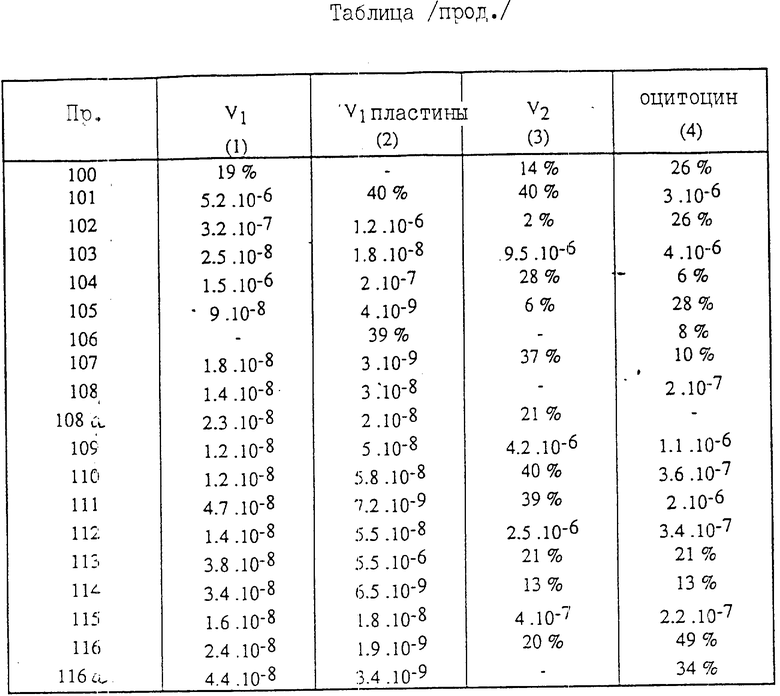
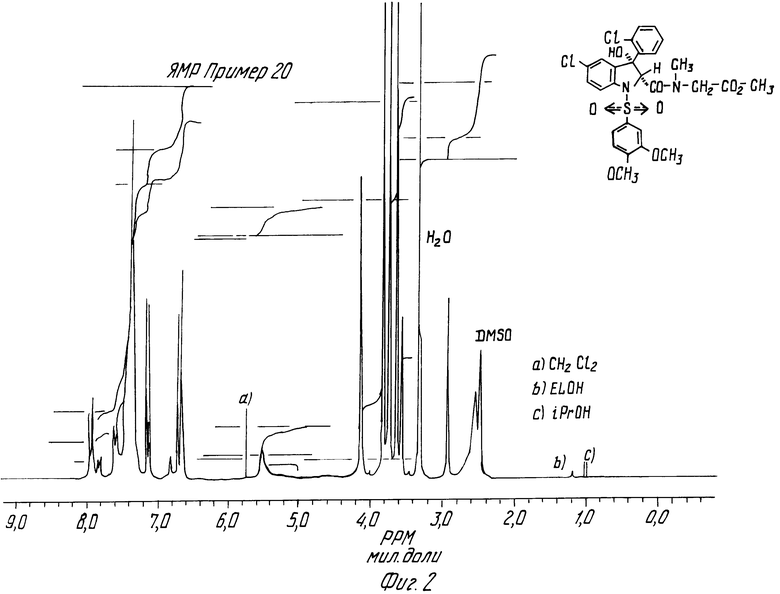
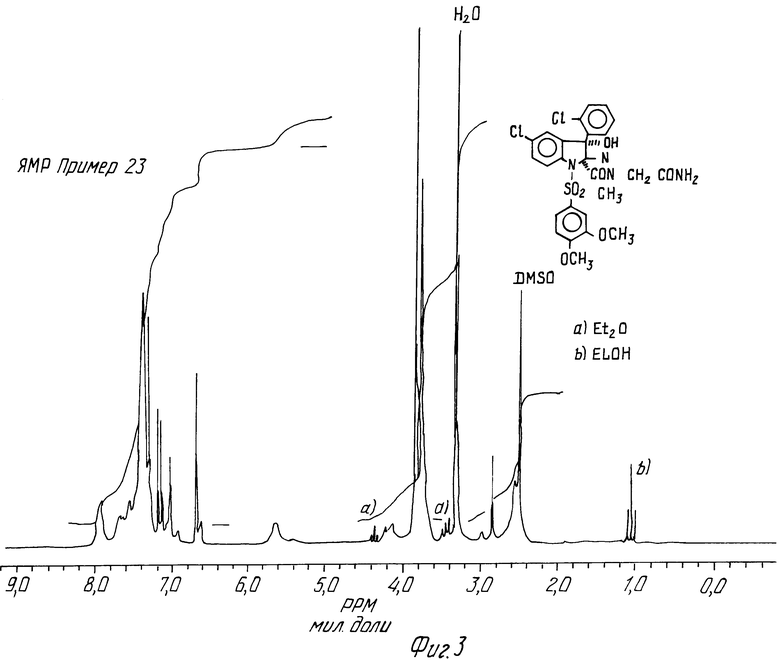
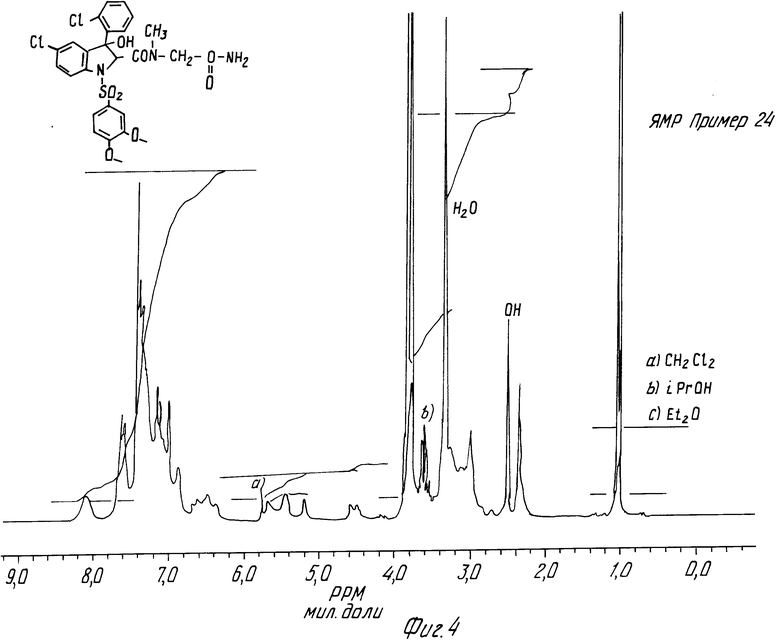
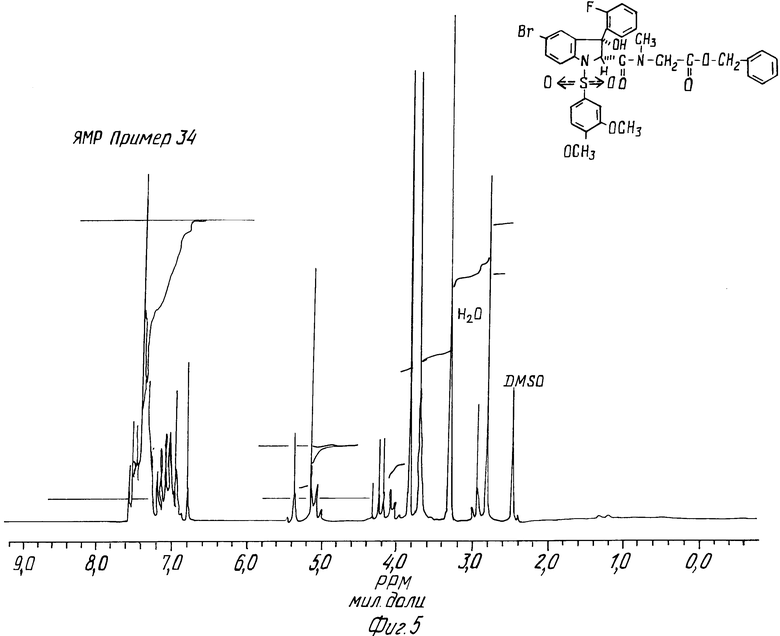
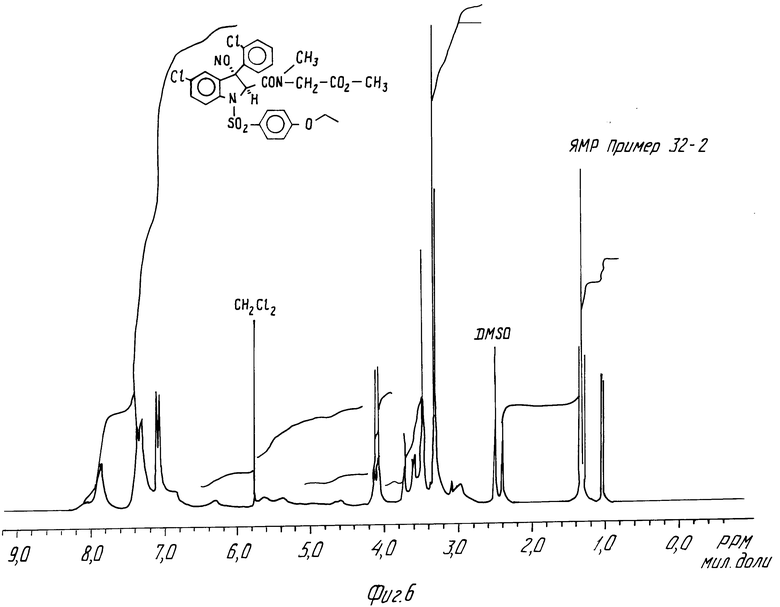
Изобретение относится к новым индолиновым производным формулы, к способу их получения и к фармацевтическим композициям на их основе. Эти соединения могут быть использованы при лечении расстройств центральной нервной системы, сердечно-сосудистой системы и желудочной системы у человека и животных. 4 с. и 10 з.п. ф-лы, 7 табл., 6 ил.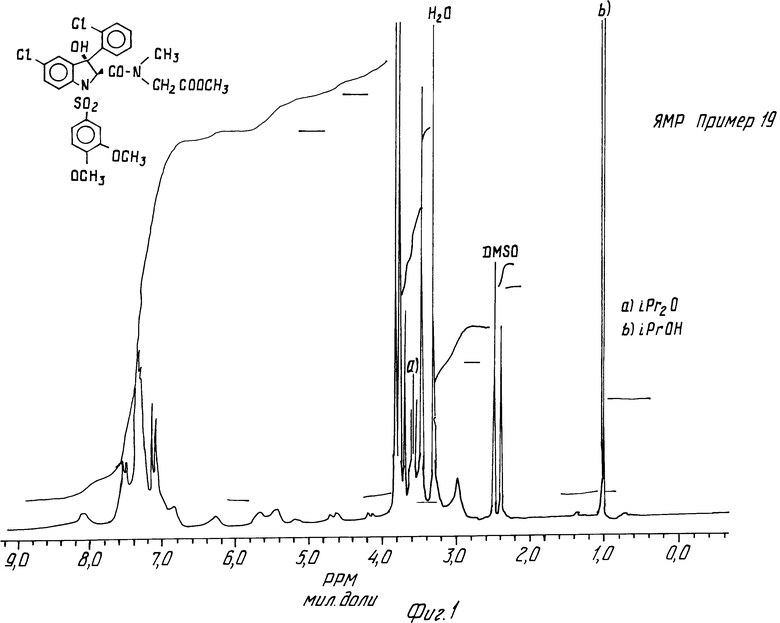
в которой R1 галоген, С1 С4-алкил, С1 - С4-алкокси;
R2 С1 С6-алкил, С3 С7 - циклоалкил, фенил, незамещенный или замещенный одной или несколькими группами С1 С4-алкил или галогеном;
R3 водород;
R4 карбамоильная группа формулы CONR6R7;
R5 С1 С4-алкил, фенил, незамещенный или замещенный одним или несколькими заместителями, выбираемыми из галогена, С1 С4-алкила, С1 С4-алкокси;
R6 С1 С6-алкил или R6 такой же, как R7;
R7 группа пиперидин-4-ил, незамещенная или замещенная на азоте С1 С4-алкилом, С1 С4-алкоксикарбонилом, группа (СН2)r, которая сама замещена группой 2-, 3- или 4-пиридил или группой амино, свободной или замещенной одним или двумя С1 С4-алкилами, карбоксильной группой, группой С1 - С4-алкоксикарбонил, карбамоильной группой, свободной или замещенной одним или двумя С1 С4-алкилами, или R6 и R7 образуют вместе с атомом азота, с которым они связаны, гетероцикл, выбираемый из морфолина, тиоморфолина, тиазолидина или 2,2-диметилтиазолидина, пиперазина, незамещенного или замещенного в положении 4 группой R8'', ненасыщенного 5-членного цикла, содержащего единственный атом азота, замещенного радикалом R8, или насыщенного 3-, 4-, 5- или 6-членного цикла, содержащего единственный атом азота и замещенного радикалами R8 и R9;
R8 R'8 или группа (CH2)r, которая сама замещена гидроксилом или аминогруппой, свободной или замещенной одним или двумя С1 С4-алкилами;
R'8 группа (СН2)q, которая сама замещена карбоксильной группой, С1 С4-алкоксикарбонильной группой, бензилоксикарбонильной группой, карбамоильной группой, свободной или замещенной гидроксилом или одним или двумя С1 С4-алкилами, или аминокарботиоильной группой, свободной или замещенной одним или двумя С1 С4-алкилами;
R''8 R'8 или группа (СН2)2NH2, свободная или замещенная одним или двумя С1 С4-алкилами;
R9 водород, группа (СН2)rNR1 0R1 1, группа азидо или группа NH-бензилоксикарбонил;
R1 0 и R1 1 независимо друг от друга водород или С1 С4-алкил;
n 0, 1 или 2;
m 0, 1 или 2;
q 0, 1, 2 или 3;
r 0, 1, 2 или 3 при условии, что r ≠ 0, когда R8 и R9 находятся в альфа-положении к амидному межцикличному азоту,
или их возможные соли.
С4-диалкиламино.
в которой R1, R2 и n имеют значения, определенные в п.1,
с сульфонильным производным формулы III
Hal-SO2-(CH2)m-R5,
в которой Hal галоген, предпочтительно хлор или бром;
m и R5 имеют значения, определенные в п.1,
полученное соединение формулы IV
обрабатывают галогенированным производным формулы V
Hal' СН2СОA,
в которой Hal' галоген, предпочтительно бром;
A или группа NR6R7, или группа OR, в которой R - третичный бутил или бензил,
с получением соединения формулы VI'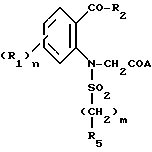
которое подвергают циклизации в основной среде, если A - NR6R7, с получением целевого продукта, либо в случае, когда A OR, подвергают операции снятия защитной группы с получением соответствующей кислоты формулы VI''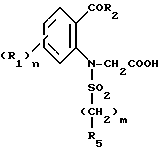
или ее хлорангидрида формулы VI'''
которые подвергают обработке амином HNR6P7 для получения амидного производного формулы VI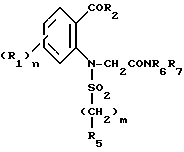
которое подвергают указанной циклизации с получением целевого продукта формулы I, разделяют при необходимости полученный продукт на цис- и транс-изомеры и выделяют энантиомеры.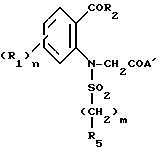
где A' группа, выбираемая из NR6R7;
R1 галоген, С1 С4-алкил, С1 - С4-алкокси;
R5 С1 С4-алкил, фенил, незамещенный или замещенный одним или несколькими заместителями, выбираемыми из галогена, С1 С4-алкила;
R6 С1 С4-алкил или R6 такой же, как R7;
R7 группа пиперидин-4-ил, незамещенная или замещенная на азоте С1 С4-алкилом, С1 С4-алкоксикарбонилом, группа (СН2)r, которая сама замещена группой 2-, 3- или 4-пиридил или группой амино, свободной или замещенной одним или двумя С1 С4-алкилами, карбоксильной группой, группой С1 - С4-алкоксикарбонил, карбамоильной группой, свободной или замещенной одним или двумя С1 С4-алкилами, или R6 и R7 образуют вместе с атомом азота, с которым они связаны, гетероцикл, выбираемый из морфолина, тиоморфолина, тиазолидина или 2,2-диметилтиазолидина, пиперазина, незамещенного или замещенного в положении 4 группой R''8, ненасыщенного 5-членного цикла, содержащего единственный атом азота, замещенного радикалом R8, или насыщенного 3-, 4-, 5- или 6-членного цикла, содержащего единственный атом азота и замещенного радикалами R8 и R9;
R8 R'8 или группа (CH2)r, которая сама замещена гидроксилом или аминогруппой, свободной или замещенной одним или двумя С1 С4-алкилами;
R'8 группа (СН2)q, которая сама замещена карбоксильной группой, С1 С4-алкоксикарбонильной группой, бензилоксикарбонильной группой, карбамоильной группой, свободной или замещенной гидроксилом или одним или двумя С1 С4-алкилами;
R''8 R'8 или группа (СН2)2NH2, свободная или замещенная одним или двумя С1 С4-алкилами;
R9 водород, группа (СН2)rNR1 0R1 1, группа азидо или группа NH-бензилоксикарбонил;
R1 0 и R1 1 независимо друг от друга водород или С1 С4-алкил;
n 0, 1 или 2;
m 0, 1 или 2;
q 0, 1, 2 или 3;
r 0, 1, 2 или 3 при условии, что r ≠ 0, когда R8 или R9 находятся в альфа-положении к амидному межцикличному азоту,
или их возможные соли.
| US, патент, 3838167, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

