Изобретение относится к фармацевтическим составам. Более конкретно, это изобретение относится к орально или ректально вводимым составам биологически активного вещества, в частности белковых веществ (веществ, напоминающих белок).
В медицинской практике на протяжении многих лет прописывают или советуют принимать множество биологически активных веществ для лечения или профилактики широкого спектра заболеваний или состояний. Одним из наиболее известных, но не единственным, назначаемым биологически активным веществом белковой природы является инсулин, который используется при контроле различных форм диабета.
Возможно наиболее легким способом приема любого лекарственного средства является пероральный прием. Такой способ приема лекарственного средства, которое может быть в виде сиропа, эликсира, таблеток, капсул, гранул, порошков или любых других удобных составов, обычно прост, и с точки зрения пациента такой способ введения является наиболее удобным и приятным. К сожалению, с точки зрения медикаментозного лечения и профилактики предпочтительный путь приема белковоподобных препаратов и других биологически активных веществ включает прохождение вещества через желудок, среда которого вредна для многих веществ, включая белки. Поскольку кислотная, гидролитическая и протеолитическая среда желудка эффективно воздействует на переваривание белковоподобных веществ и превращение их в аминокислоты и олигопептиды с последующей их ассимиляцией, то не удивительно, что только очень незначительная часть любого из широкого множества биолонически активных веществ при пероральном приеме остается жизнеспособной после прохождения через желудок для того, чтобы быть воспринятой организмом в тонком кишечнике.
В результате, как это могут подтвердить многие диабетики, многие белковые медикаменты должны вводиться парентерально, часто подкожно, внутримышечно или внутривенно путем инъекций, что влечет за собой массу неудобств, дискомфорта и трудностей для больного.
Этот случай не является частным, поскольку число заболеваний, нуждающихся в контроле, осуществляемом путем введения белковоподобных веществ, может быть весьма расширено. Например, от сахарного диабета страдает множество людей во всем мире. В частности, из-за большого числа людей, страдающих сахарным диабетом в той или иной форме, существует необходимость создания пероральных форм инсулина, которые каким-то образом защищены от вредного воздействия среды желудка. Хотя предпринимались ранее различные попытки создания таких составов, заявители не уверены в том, что к настоящему моменту какой-либо из составов прототипа имеет какое-либо коммерческое значение. К имевшимся ранее предложениям, о которых осведомлены заявители, относятся следующие.
WO-A-8701035 относится к парентеральным составам жирорастворимых лекарственных препаратов и витаминов; составы содержат "псевдомицеллы".
WO-A-8705505 раскрывает композиции, которые можно принимать перорально, а именно композиции инсулина, нанесенные на твердые частицы из водного препарата; частицы с нанесенным на них инсулином затем покрыты жиром.
Патент США N 4849405 раскрывает композиции инсулина для перорального приема; по описанию композиции представляют собой двухфазные препараты, и очевидно, что обе фазы являются водными, с фазами, которые для эффективного хранения разделены коацерватной системой.
Патент EP-A-0140085 описывает препараты, содержащие лекарственное средство в пузырьке жира.
Shichiri et al. (Acta diabet. lat., 15, 175-183 (1978)) раскрывает мицеллы инсулина вода-масло-вода.
Патенты США NN 4784845 и 4816247 раскрывают эмульсионные композиции для парентерального введения гидрофобных лекарственных препаратов.
Патент Японии JP-A-55017328 раскрывает эмульсии вода-масло-вода, содержащие инсулин, для перорального приема.
Патент EP-A-0366277, опубликованный 2 мая 1990 года, относится к усовершенствованным фармацевтическим составам, которые можно вводить перорально или ректально. Более конкретно, EP-A-0366277 раскрывает фармацевтический состав, содержащий микроэмульсию, состоящую из гидрофильной фазы и гидрофобной фазы, в которой
(A) гидрофильная фаза диспергирована в гидрофобной фазе,
(B) гидрофильная фаза содержит биологически активное вещество и
(C) гидрофобная фаза содержит хиломикрон или материал, из которого хиломикрон образуется in vivo.
Гидрофильная фаза может содержать физиологически приемлемый растворитель для биологически активного вещества, такой как вода. Полагают, что при введении биологически активного вещества, ассоциированного с хиломикронами или с веществами, образующими хиломикроны, оно захватывается ворсинками и микроворсинками стенки кишечника, откуда секретируется в хилезные сосуды тонкого кишечника и кишечную лимфу и затем дренируется в лимфатические протоки и, в конечном итоге, в циркулирующий поток крови.
Как известно, хиломикроны содержат липид-холестериновое ядро или матрицу, окруженную мембраной, содержащую фосфолипидный монослой, усеянный протеинами (Redgrave in Gastrointestinal Physiology IV, International Review of Physiology, Volume 28, 103-130. Young, J.A., Ed., University Park Press. Baltimore, 1983). Таким образом, очевидно, что Европейская патентная заявка обеспечивает биологически активное вещество в гидрофобном ядре.
Это изобретение относится к отличному подходу к разрешению проблемы перорального (или ректального) введения биологически активного соединения(ий), в частности белков (протеинов). Установлено, что белковоподобные соединения, включающие, но не ограничивающиеся инсулином (бычьим, свиным, человека или другим), гормон роста (человека или какой-либо другой), кальцитонин (лосося или других), эритропоэтин (человека или другой), могут быть введены перорально, будучи ассоциированными с одним или более фосфолипидами или другим соединениями, включенными в образование лецитина (предшественниками лецитина). Ассоциация может быть отнесена к типу "фосфолипо-протеиновой" (такой как "фосфолипо-инсулиновая") в тех случаях, когда биологически активное соединение представляет собой белок (является белковоподобным).
Согласно первому аспекту настоящего изобретения обеспечивается фармацевтическая композиция, содержащая белковоподобное или другое биологически активное соединение и лецитин или предшественник лецитина.
Белковоподобное соединение может быть замещено (или дополнено) другим биологически активным соединением: механизм действия в этих случаях, как полагают, аналогичен механизму, изложенному выше. Биологически активное соединение и лецитиновый предшественник могут, как правило, находиться в форме какого-либо ассоциата друг с другом.
Лецитин может интегрироваться в хиломикроны, в частности в их мембраны. Использование других соединений или их предшественников, которые могут аналогичным путем интегрироваться, также находится в рамках настоящего изобретения. Дискуссия в этой спецификации, относящаяся к лецитину или его предшественникам, может быть применима к этому более общему классу соединений mutatis mutandis.
Предшественник лецитина может образовывать лецитин в кишечном эпителии человека или животных, и, следовательно, белковоподобное соединение может быть ассоциировано с лецитином с образованием лецитин-протеинового комплекса (такого, как лецитин-инсулиновый комплекс). Лецитин, образующийся таким путем в кишечном эпителии, может образовывать поверхностную мембрану хиломикронов, а также покрывать до 80% поверхности аполипопротеинов, таких как апопротеин-A, -B, -C и -E. Таким образом, возможно используя подходящие усилители адсорбции для фосфолипо-протеиновых комплексов, можно добиться абсорбции комплекса в кишечном эпителии, лецитин при этом синтезируется (и комплекс превращается в лецитин-протеиновый комплекс), а затем лецитин может покрывать хиломикроновые ядра так же, как и ядра тех апопротеинов, которые прикреплены к хиломикронам. Затем лецитин может выделяться в лимфатических сосудах, дренироваться в лимфатические протоки (а те лецитин-протеиновые комплексы, которые еще остаются прикрепленными к хиломикронам, могут формировать часть рудимента хиломикрона), направляется по каналам в печень и из печени выделяется в циркулирующую кровь. Как полагают, лецитин посредством такой системы (такого механизма) эффективно переносит протеин в общую систему циркуляции.
In vivo лецитин может образовываться различными путями. Некоторые из них состоят в следующем. Во-первых, может быть использован α-глицериновый путь метаболизма, в этом случае в качестве предшественника используют sn-глицерин-3-фосфат так же, как и фосфатидаты и диглицериды. Во-вторых, лецитин может быть синтезирован под действием холинфосфотрансферазы, в этом случае в качестве предшественников используют холин, фосфохолин, цитидин, дифосфохолин и диглицериды. В-третьих, лецитин может быть синтезирован из других фосфатидов, таких как фосфатидилэтаноламин. В-четвертых, лецитин может быть синтезирован из триглицерида и, как полагают, продуктов распада лецитина или в процессах трансэтерификации.
Термин "биологически активное вещество" включает, в частности, фармацевтически активные белковоподобные вещества. Белковоподобным веществом может быть чистый белок (протеин), или оно может содержать белок, например, как гликопротеин, содержащий и протеиновый, и сахарный остатки. Вещество может быть полезно при лечении человека и в ветеринарии как при лечении, так и для профилактики заболеваний или симптомов или может быть полезно в косметических целях или при диагностике. Примеры белковоподобных биологических веществ, которые могут обеспечивать составы для перорального или ректального введения согласно настоящему изобретению, включают белковые гормоны, такие как инсулин, кальцигонин и гормон роста, выделенные из человека или животных или полученные полусинтетическим путем или полностью синтезированные, эритропоэтин или гематопоэтин, активаторы профибринолизина и их предшественники, такие как t-РА, урокиназа, проурокиназа и стрептокиназа, интерфероны, включая человеческий интерферон-альфа, интерферон-бета, интерлейкины, включая ИЛ-1, ИЛ-2, ИЛ-3, ИЛ-4 и ИЛ-5, колониестимулирующие факторы, включая G-КСФ и GM-КСФ, и факторы крови, включая фактор VIII.
Следует, однако, подчеркнуть, что изобретение не ограничивается составами на основе белковоподобных соединений, многие небелковые фармацевтические средства могут быть с успехом составлены при использовании настоящего изобретения. Например, противовоспалительные препараты нестероидной природы (NSAIDs), такие как индометацин и другие средства, включая гентамицин, могут быть приготовлены согласно настоящему изобретению.
Однако, что касается соединения в составе биологически активного вещества с, например, фосфолипидами в данном изобретении, то желательно, чтобы активное вещество не относилось к веществам, которые образуют необратимые ковалентные связи с фосфолипидами или, как полагают, и с любыми другими компонентами состава, поскольку это может в определенных обстоятельствах уменьшить биологическую активность и/или пригодность. Говоря об этом, не имеют в виду, что существует какая-то проблема при приготовлении состава согласно настоящему изобретению из каких-либо активных молекул, о которых шла речь выше. Ассоциация активного соединения с лецитином или предшественником лецитина может происходить в природе за счет образования нековалентного комплекса. Такой комплекс может включать водородные связи, образовываться за счет ван-дер-Ваальсовского взаимодействия, ионных взаимодействий и/или липид-липидных взаимодействий.
Хотя и не делается предположение о том, что существует какой-либо конкретный ограниченный размер молекулы биологически активных веществ, которые могут быть включены в состав согласно настоящему изобретению, очевидно из примерного, но не ограничивающего выбора биологически активного вещества, приведенного выше, что изобретение, в частности, подходит для включения в состав макромолекул. Молекулярная масса таких макромолекул может составлять примерно 1 кДа или примерно 5 кДа, примерно 10 кДа или выше, или даже примерно 15 кДа или выше. Кроме того, хотя и не предполагается, что гидрофильность или гидрофобность (липофильность) биологически активного вещества представляет особо критичный параметр, согласно изобретению легко получают составы гидрофильных молекул, таких как инсулин, кальцитонин (в особенности кальцитонин лосося), гормоны роста или соматотропин (в особенности соматотропин свиньи), каждый из которых (в особенности кальцитонин лосося) настолько гидрофилен, что становится гигроскопичным.
Количество биологически активного вещества, присутствующего в составе, может зависеть от природы вещества и должно соответствовать такому количеству, которое используется для приготовления лекарственного средства согласно рецептуре применяемых на практике, удобных для введения количеств. Принимая во внимание эти рассуждения, составы согласно настоящему изобретению могут содержать от 1 μг, 10 μг, 0,1 мг или 1 мг на литр до 1, 10 или 100 г на литр.
Настоящее изобретение включает производные или составные части, или группы фосфолипидов или других соединений, способных действовать как предшественники для синтеза in vivo лецитина в кишечном эпителии человека или животного, лецитин, в свою очередь, образует, по крайней мере, часть мембраны на ядре хиломикрона. Полагают, что в зависимости от условий введения интегрированные в мембрану соединения вызывают интеграцию ассоциированного биологически активного вещества с лецитиновой мембраной, покрывающей, например, ядро хиломикрона, первичной основой мембраны, как обсуждалось выше, служит фосфолипид. Поскольку хиломикронные мембраны богаты фосфолипидом, то фосфолипиды представляют собой весьма подходящий материал для приготовления составов с биологически активными веществами согласно настоящему изобретению.
Предшественники лецитина не должны служить причиной загрязнения биологически активного вещества; например, сообщалось о том, что некоторые жирные кислоты, такие как олеиновая и стеариновая, могут оказывать вредное воздействие на свиной соматотропин, так что некая, но определенная степень внимания должна быть проявлена при выборе фосфолипидов, производных фосфолипидов или других мембранно-интегрирующих веществ или предшественников, которые будут использоваться в составах. Этот выбор, однако, может быть легко осуществлен специалистом в данной области.
Фосфолипиды - это предшественники лецитина. Фосфолипиды - это триглицериды, в которых одну из эфирных групп возможно образует замещенная фосфорная кислота. Для использования в настоящем изобретении предпочтительны фосфолипиды, имеющие следующую общую формулу: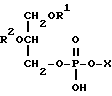
в которой каждый R1 и R2 независимо друг от друга представляет ацильную группу, содержащую, например, 10, 12 или от 14 до 26 атомов углерода, которая возможно моно- или полиненасыщена, а X представляет атом водорода или фосфолипидную головную группу.
Головной фосфолипидной группой может быть любая группа, способная образовывать физиологически приемлемый фосфолипид. Примеры фосфолипидов включают
диацил фосфатидил глицериды, такие как димиристоил фосфатидил глицерид (DPMG), дипальмитоил фосфатидил глицерид (DPPG) и дистеароил фосфатидил глицерид (DSPG),
диацил фосфатидил холины, такие как димиристоил фосфатидилхолин (DPMC), дипальмитоил фосфатидилхолин (DPPC) и дистеароил фосфатидилхолин (DSPC),
диацил фосфатидиновая кислота, такая как димиристоил фосфатидиновая кислота (DPMA), дипальмитоил фосфатидиновая кислота (DPPA) и дистеароил фосфатидиновая кислота (DSPA), и
диацил фосфатидилэтаноламины, такие как димиристоил фосфатидил этаноламин (DPME), дипальмитоил фосфатидил этаноламин (DPPE) и дистеароил фосфатидил этаноламин (DSPE).
Другие примеры включают, но ими не ограничиваются, производные этаноламина (такие как упомянутый выше фосфатидил этаноламин или цефалин), серин (такое как фосфатидил серин) и 3'-О-лизилглицерид (такое как 3'-О-лизил-фосфатидил глицерид).
К одной фосфолипидной головной группе может быть прикреплено более одной фосфатидильной группы: например, две фосфатидильные группировки могут быть прикреплены к одному остатку глицерина, как в дифосфатидилглицериде или кардиолипине. В тех случаях, когда X представляет атом водорода, фосфолипид - это фосфатидиновая кислота, такая как бимиристоил L-α-фосфатидиновая кислота.
Фосфолипиды, пригодные в настоящем изобретении, включают синтетические и природные фосфолипиды как в виде отдельных компонентов, так и в виде смеси двух или более компонентов. Получение видимо чистых природных фосфолипидов трудно достижимо, если даже они фактически представляют один вид фосфолипидов, но этот фактор, как полагают, не является критическим параметром для целей настоящего изобретения.
К особенно предпочтительным фосфолипидам относятся 1,2-димиристоил-sn-глице рин-3-фосфохолин, который может быть в виде моногидрата, и L-α-фосфатидиновой кислоты бимиристоил, который может быть в форме натриевой соли.
Вместо или в добавление могут быть использованы другие предшественники лецитина.
В композициях настоящего изобретения биологически активное вещество может быть ассоциировано с предшественником лецитина. Хотя точная природа этого ассоциата не представляется существенной для целей настоящего изобретения, полагают, что такая ассоциация обусловлена нековалентными взаимодействиями, в частности они возникают за счет образования водородных связей и гидрофобного взаимодействия, большинство за счет липопротеинов, традифионно присутствующих в хиломикроне или других фосфолипидных мембранах.
В присутствии одного или более поверхностно-активных веществ с высоким гидрофил-липофильным балансом (HLB), таких как те, которые имеют значение HLB выше 10 или даже выше 14, биологически активное вещество, ассоциированное с предшественником лецитина, может образовывать гидрофильный комплекс, который легко переходит в энтероциты (клетки эпителиальной стенки кишок). В энтероцитах предшественник лецитина используется для синтеза лецитина. На этом этапе предшественник и ассоциированное с ним биологически активное вещество по-видимому обходит лизосому и превращается в комплекс биологически активного вещества и мембранно-интегрированного соединения (такого как лецитин). Такого типа комплекс может замещать или дополнять лецитин, который образует внешний слой мембраны, покрывающей примерно 80% или более поверхности хиломикронного ядра.
В циркулирующей крови белки и фосфолипиды могут обмениваться на другие липопротеины. Так, по крайней мере, часть белка, введенного согласно настоящему изобретению, может проциркулировать в кровь в качестве фосфолипо-протеина, отделившегося от хиломикрона, с которым он был ранее или первоначально ассоциирован. Некоторые фосфолипо-протеины могут выделяться в циркулирующую кровь в свободном виде, а часть из них может проходить через печень, будучи прикрепленными к рудиментам хиломикрона. Скорость и степень обмена фосфолипид - протеин может подвергаться воздействию различных факторов, включая такой, как изменение длины фосфолипидной цепи.
Составы, соответствующие настоящему изобретению, могут, как правило, также содержать гидрофильную жидкость, которая должна быть водосодержащей и может быть водой, вполне можно использовать физиологический раствор или фосфатно-солевой буфер. Может присутствовать смешивающийся с водой растворитель, например, для облегчения формирования состава. Для этих целей можно использовать этанол или другие подходящие растворители. Природа растворителя, который используется, зависит от активного вещества. Гидрофильная жидкость может быть представлена смесью вода:растворитель, например, в объемных соотношениях от 0,5:1 до 2:1, хотя присутствие неводного растворителя не обязательно, но предпочтительно.
Широкий и предпочтительный процентный состав (который, как правило, весо/весовой процентный состав, но может быть также весо/объемным процентным составом или объем/объемным процентным составом) компонентов дан в табл. 1 при условии, что общее содержание никогда не превышает 100%.
Составы, соответствующие настоящему изобретению, могут содержать гидрофильное поверхностно-активное вещество (например, со значением НLВ, большим 10). Они могут оказывать промотирующее воздействие на образование комплекса между биологически активным веществом и лецитиновым предшественником (в особенности при синтезе ими лецитиновых предшественников в эпителии кишок человека и некоторых животных, таких как свинья) и/или на придание гидрофильного характера такому комплексу. Гидрофильные поверхностно-активные вещества могут присутствовать в количестве до 10% (весо/объемных или объем/объемных), предпочтительно от 1 до 5%, как правило, от 1,5 до 4%, например, они могут присутствовать в количестве примерно 2,4-2,5%.
Еще один компонент, присутствие которого очень часто бывает желательным, -это ингибитор протеазы, который может находиться в виде одного или больше индивидуальных ингибиторов протеазы. Ингибиторы протеазы, пригодные для использования в настоящем изобретении, можно разделить на две категории. Первая категория - это ингибиторы протеазы, которые пригодны при лимитировании или предотвращении деструкции биологически активного вещества в тех случаях, когда оно является белковоподобным соединением. Такими ингибиторами протеазы должны быть вещества, оказывающие ингибирующее действие на протеолитические ферменты, обнаруживаемые в желудочно-кишечном тракте, такие как трипсин, химотрипсин и карбоксипептидаза. В случае инсулина ингибиторами протеазы должны быть, как правило, ингибиторы класса ферментов, известные под названием инсулиназы, которые включают фермент транс-сульфатазу. Подходящими источниками ингибиторов трипсина могут служить вытяжки из соевых бобов или яичного белка (овомукоид-яйцевая слизь). Во-вторых, если в составах, соответствующих настоящему изобретению, присутствует апопротеин, то желательно добавлять ингибиторы протеазы для уменьшения степени деструкции апопротеина до его достижения слизистой кишечника. В общем говоря, подобные ингибиторы протеазы могут быть использованы как для защиты белковоподобных биологически активных веществ, так и как ингибитор протеазы, выполняя обе функции. Ингибиторы протеазы могут быть добавлены к ассоциату или комплексу, образованному биологически активным веществом и мембранным интегратором или предшественником (например, фосфолипидами); для удобства их можно добавлять в гидрофильную фазу, в которой присутствуют две фазы. Выбор количества ингибитора протеазы, которое добавляют, легко может быть осуществлен специалистом в данной области, но, как правило, он должен находиться в количестве примерно 0,1% весо/объемный или даже 0,5% весо/объемных. Апротинин может быть добавлен в количестве, составляющем до 10 миллионов IU (IU - международная единица), предпочтительно от 0,5 до 5 миллионов IU, например 3 миллиона IU, но точное количество, которое используют, может зависеть от фактической активности биологически активного вещества.
Во многих случаях может оказаться предпочтительнее вводить комплекс биологически активного вещества с мембранным интегратором или предшественником (в предпочтительном варианте - это фосфолипо-протеиновый комплекс) вместе с необязательным, но предпочтительным гидрофильным поверхностно-активным веществом и ингибитором протеазы, суспендированными или приготовленными в виде эмульсии или микроэмульсии, содержащей липофильное вещество, включающее поверхностно-активное вещество с низким значением HLB (например, имеющее значение HLB менее 4). Липофильное вещество может (но не обязательно) включать те вещества, которые, как известно, образуют хиломикроны in vivo, к таким веществам относятся, но ими не ограничиваются, холестерин, эфиры холестерина, лецитин и/или другие фосфолипиды, или насыщенные, или моно- или полиненасыщенные жирные кислоты (например, содержащие от 16 до 24 атомов углерода), возможно этерифицированные в глицериновый эфир с образованием моно-, ди- или триглицеридов. В альтернативном варианте в основном гидрофильный фосфолипо-протеиновый (или другой) комплекс может быть просто смешан с подходящими маслами, в частности с растительными маслами, такими как жирное масло со средней цепочкой (МСТ) или любое другое подходящее масло, плюс одно или более подходящее поверхностно-активное вещество, имеющее низкое значение HLB (например, менее 4). Подходящие поверхностно-активные вещества включают лизолецитин, производные лизолецитина и другие, в основном, липофильные вещества.
Гидрофильный фосфолипо-протеин (или другие комплексы) может быть покрыт соответствующей энтеросолюбильной оболочкой и может приниматься внутрь перорально. Однако на основании экспериментов предполагают, что предпочтительнее смешать комплекс с подходящим маслом или предшественниками так, чтобы доставить активное вещество к ворсинкам тонкого кишечника, откуда оно адсорбируется и дренируется в лимфатическую систему.
Широкий и предпочтительный процентный состав (который, как правило, весо/весовой процентный состав, но может быть весо/объемным или даже объем/объемным) липофильного вещества для общих целей дан в табл. 2 при условии, что общее количество никогда не превышает 100%.
Может присутствовать в композициях смешивающийся в некоторой степени с липофильной фазой органический растворитель для облегчения образования состава. Природа такого растворителя будет зависеть от других присутствующих веществ. Очень часто подходит этанол. Количество растворителя может составлять от 5 до 50% объем/объемных в расчете на объем липофильной фазы.
Когда фосфолипо-протеиновый комплекс или другой комплекс получают в виде эмульсии или микроэмульсии с липофильной фазой, как это обсуждалось выше (обычно, как эмульсия вода в масле), не существенно присутствуют или нет любые другие ингредиенты, хотя на практике обычно бывает очень удобно, когда добавлены другие ингредиенты. Возможным ингредиентом является стабилизатор для биологически активного вещества. Точная природа этого стабилизатора, если он присутствует в составе, будет, конечно, зависеть от природы самого биологически активного вещества. Например, имеется ряд хорошо известных стабилизаторов инсулина, которые можно успешно включать в составы, содержащие инсулин, соответствующие настоящему изобретению. К ним относятся гидроксипропилцеллюлоза (HPC), соли кальция и лимонная кислота и ее соли. Соли кальция, как известно, не только стабилизируют инсулин, но также оказывают дополнительное положительное действие, связанное с увеличением пористости клеточных мембран, ускоряя тем самым посадку активного вещества на клетки стенки кишечника. Поскольку биологически активное вещество добавляют в гидрофильную фазу, то предпочтительно стабилизатор также добавлять в эту фазу. Количество стабилизатора также должно зависеть от его природы и природы биологически активного вещества, выбор концентрации стабилизатора может быть легко осуществлен специалистом в данной области, но чаще всего она составляет 1 или 2% весо/объемных.
В некоторых случаях бывает желательным включение эмульгирующих добавок, представляющих традиционные эмульгирующие добавки, которые используют при приготовлении эмульсий. Некоторые эмульгирующие добавки являются поверхностно-активными веществами, а поверхностно-активные вещества, которые пригодны для использования в этих целях, не ограничиваются какими-либо конкретными значениями HLB. К полезным эмульгирующим добавкам относятся холестерин, стеариновая кислота, стеарат натрия, пальмитиновая кислота, пальмитат натрия, олеат натрия, моноолеат глицерина, полиоксиэтилена 50 стеарат, полиоксиэтилена 40 стеарат, полисорбат 20, полисорбат 40, полисорбат 60, полисорбат 80, пропилен гликоля диацетат, пропиленгликоля моностеарат.
Эмульгирующие добавки могут присутствовать как в липофильной, так и в гидрофильной фазе. Количество присутствующей эмульгирующей добавки, если в ней есть необходимость, должно быть просто достаточно для поддержания адекватного получения стабильного состава. Точное количество может быть установлено специалистом в данной области. Вообще говоря, они могут быть использованы в количестве от 0 до 15% весо/объемных, например от 0,1 до 5% в/о в расчете на весь состав. Она может быть использована для обеспечения ее содержания в гидрофильной фазе от 1 до 5%. Обнаружено, что, в частности, уместно добавлять полисорбат 80 к липофильной фазе, а полиоксиэтилена 40 стеарат к гидрофильной фазе.
Составы, соответствующие настоящему изобретению, могут содержать различные консерванты. Антиоксиданты и противомикробные средства представляют две особенно пригодные категории консервантов. Антиоксиданты, в частности, пригодны потому, что некоторые соединения, полезные для создания составов настоящего изобретения, склонны к автоокислительной деструкции. Хотя эта проблема может быть разрешена путем приготовления составов, соответствующих настоящему изобретению, в атмосфере инертного газа, такого как азот, но это неудобно и дорого, и поэтому зачастую предпочитают добавлять химические антиоксиданты. К подходящим фармацевтически приемлемым антиоксидантам относятся пропилгаллат, бутилированный гидроксианизол, бутилированный гидрокситолуол, аскорбиновая кислота или аскорбат натрия, DL- или D-α-токоферол и DL- или D-α-токоферола ацетат. Антиоксидант, если он присутствует, можно добавлять в составы, соответствующие настоящему изобретению, в количестве до, например, 0,1% весо/объемного, предпочтительно от 0,0001 до 0,3%. Фаза, соответствующая антиоксиданту, зависит от природы антиоксиданта. Как правило, такие антиоксиданты, как α-токоферол, могут быть соответственно включены в гидрофобную фазу, в то время как гидрофильные антиоксиданты типа аскорбиновой кислоты можно включать в гидрофильную фазу. Кунжутное масло предпочтительно в виде рафинированного химически масла может быть добавлено к составам настоящего изобретения, поскольку оно обладает антиоксидантной активностью. Кунжутное масло имеет еще одно преимущество, связанное с тем, что оно улучшает запах составов, повышая тем самым восприимчивость больного. Кунжутное масло может присутствовать в количестве от 0,1 до 3% весо/объемных, предпочтительно от 5 до 20% весо/объемных, в конечном жидком составе. Его следует добавлять в липофильную фазу.
К антимикробным консервантам, которые могут быть использованы, как правило, в количестве до примерно 3 % весо/объемных, предпочтительно от примерно 0,5 до 2,5%, от общего количества, относятся метилпарабен, этилпарабен, пропилпарабен, бутилпарабен, фенол, дегидроуксусная кислота, фенилэтиловый спирт, бензоат натрия, сорбиновая кислота, тимол, натрия дегидроацетат, бензиловый спирт, крезол, пара-хлоро-мета-крезол, хлоробутанол, ртутьфенилацетат, ртутьфенилборат, ртутьфенилнитрат, бензалкония хлорид и антимикробные средства могут быть добавлены в любую из фаз, когда это требуется или удобно.
Хотя это и не существенно, но может быть практичным или удобным для улучшения транс-лимфатической абсорбции фосфолипо-протеина или других комплексов у человека или некоторых других видов, чтобы составы настоящего изобретения были в двухфазной форме. Двухфазные системы, соответствующие настоящему изобретению, включают системы вода-в масле (т.е. гидрофильная фаза в липофильной), вода-в масле-в воде и масло-в воде-в масле.
Двухфазные системы можно получать, как правило, при тщательном смешении гидрофильной и липофильной фаз. Двухфазные системы, соответствующие настоящему изобретение, могут представлять эмульсии или микроэмульсии. Объем: объемное соотношение гидрофильной фазы и липофильной фазы может, как правило, находиться в пределах от 0,2:1 до 5:1, обычно от 0,5:1 до 2:1.
Для получения эмульсий или микроэмульсий временами необходимо использовать два различных поверхностно-активных вещества (сурфактанта): одно - гидрофильное с высоким значением гидрофильно-липофильного баланса HLB и второе - более липофильное (как описано выше) с низким значением HLB. Гидрофильные сурфактанты, полезные для целей настоящего изобретения, в тех случаях, когда они присутствуют, имеют высокие значения HLB, равные по крайней мере 10, или очень высокие значения HLB, равные по крайней мере 17 и возможно достигающие 20. Липофильные поверхностно-активные вещества, используемые в настоящем изобретении, имеют низкие значения HLB, например меньше 10. Предпочтительно, когда липофильный сурфактант имеет значение HLB меньше 7 или даже меньше 4. Общее правило, которым следует руководствоваться при выборе каждого из сурфактантов, используемых при получении составов настоящего изобретения, заключается в следующем: поверхностно-активные вещества следует выбирать среди сурфактантов, классифицируемых как анионные или неионные. Эти сурфактанты, в частности, пригодны для использования в фармацевтических системах благодаря их совместимости, стабильности и нетоксичности. К поверхностно-активным веществам, используемым в настоящем изобретении для различных целей, относятся следующие: жирные кислоты с длинной C16-C24-цепочкой, например пальмитиновая кислота, стеариновая кислота и олеиновая кислота; эфиры жирных кислот с длинной C16-C24-цепочкой, например натрия пальмитат, натрия стеарат и натрия олеат; натрия лаурилсульфат; эфиры жирных кислот полиэтиленгликоля, например моно- или ди-стеарат полиэтиленгликоля; пропиленгликоль и эфиры жирных кислот и пропиленгликоля, например пропиленгликоля моностеарат; глицерин и моно- или полиглицериды жирных кислот, например глицерина моностеарат; эфиры жирных кислот и полиоксиэтилена, простые эфиры и амины, например моно- и ди-стеарат полиоксиэтилена, и лауриловый эфир полиоксиэтилена; эфиры полиоксиэтилена и сорбитана, например монолаурат полиоксиэтиленсорбитана, монопальмитат, моностеарат или моноолеат; полиоксиэтиленалкилфенолы и алкилфениловые эфиры; полиоксиэтиленкасторовое масло; сорбитановые эфиры жирных кислот; полисорбаты; стеарилмин; триэтаноламинолеат; растительные масла, например кунжутное масло или кукурузное масло; холестерин и трагакант.
Поверхностно-активными веществами, составляющими предмет выбора, будут, конечно, те, которые пригодны в фармацевтических составах и которые имеют соответственно низкие значения ЛД50. Далее следует перечень некоторых примерных сурфактантов со значениями HLB и ЛД50 в тех случаях, когда они известны.
Примеры подходящих сурфактантов с высокими значениями HLB приведены в табл. 3.
Примеры подходящих сурфактантов с низкими значениями HLB приведены в табл. 4.
Следует отметить, что в настоящем изобретении зачастую можно использовать смеси поверхностно-активных веществ вместо одного поверхностного вещества. Например, вместо одного гидрофильного поверхностно-активного вещества может быть использована смесь двух или более относительно гидрофильных поверхностно-активных веществ: однако эффективное значение HLВ смеси должно быть больше 10. Под "эффективным значением" HLB подразумевается, что гидрофильно-липофильный баланс смеси поверхностно-активных веществ должен быть эквивалентен одному поверхностно-активному веществу, имеющему значение HLB, превышающее 10. Аналогично, смеси липофильных поверхностно-активных веществ могут быть использованы вместо одного липофильного поверхностно-активного вещества. В этом случае эффективное значение HLB липофильных поверхностно-активных веществ должно быть меньше 10.
Выбор концентрации поверхностно-активного вещества, которое используют в составах настоящего изобретения, принадлежит специалисту в данной области. Естественно, что точные количества, которые должны быть оптимальными для каждого случая, в очень сильной степени зависят от природы поверхностно-активных веществ и от того, какие иные ингредиенты присутствуют в составах. Тем не менее следующее положение может служить общим руководством при выборе концентрации поверхностно-активного вещества: количества гидрофильного поверхностно-активного вещества, как правило, должно находиться в интервале (рассчитано на общий объем состава) от 0,1 г до 50 г на литр, обычно предпочтителен интервал от 0,5 до 25 г на литр, а оптимальное количество часто варьируется в пределах от 1 до 10 г на литр. Липофильное поверхностно-активное вещество обсуждалось выше в связи с масляной фазой микроэмульсии. Оно, как правило, должно присутствовать в количестве от 0,1 г до 100 г на литр, интервал от 0,5 до 50 г на литр предпочтителен, а оптимальное количество часто варьируется в пределах от 2 до 25 г на литр, эти величины также рассчитывались, исходя из общего объема состава.
Композиции, соответствующие настоящему изобретению, могут быть получены наиболее широко путем смешения ингредиентов. В соответствии с вторым аспектом настоящего изобретения обеспечивается способ приготовления состава, описанного выше, способ включает смешение ингредиентов состава.
Как правило, сначала предпочитают осуществить смешение активного (обычно белковоподобного) соединения и предшественника лецитина. Это дает возможность сформировать "фосфолипо-протеиновый" комплекс в предпочтительных вариантах осуществления изобретения.
Как обсуждалось выше, некоторые составы, соответствующие настоящему изобретению, включают две фазы. Предпочтительный способ приготовления таких композиций включает
(i) создание гидрофильной фазы, содержащей биологически активное вещество и предшественник лецитина;
(ii) формирование двухфазной системы, включающей гидрофильную фазу, сопряженную с одним или более веществами, усиливающими абсорбцию.
Благодаря свойственной некоторым эмульсиям и микроэмульсиям термодинамической стабильности жидкие составы, соответствующие настоящему изобретению, могут быть приготовлены простым смешением гидрофильной и липофильной фаз, которые в свою очередь могут быть получены при смешении соответствующих им ингредиентов вместе. Кинетические же рассмотрения, однако, приводят к предположению о том, что с практической точки зрения необходимо выполнить определенные стадии для быстрого и эффективного образования эмульсионных и микроэмульсионных составов, соответствующих настоящему изобретению. В частности, в течение или после соединения гидрофильной и липофильной фаз микроэмульсия может быть быстро создана при использовании микрофлюидизатора, а эмульсия может быть приготовлена с использованием подходящей аппаратуры, которая обеспечивает тщательное смешивание.
Следует отметить, что некоторые составы, соответствующие настоящему изобретению, в особенности двухфазные составы, существуют предпочтительно в жидком состоянии. Однако они могут быть превращены в твердый продукт, порошкообразные формы стандартными методами. Обычно жидкие формы могут быть нанесены на твердые, порошкообразные носители в псевдоожиженном слое или при использовании аналогичного оборудования (такого как SPIR-A-FLOW оборудования, это обозначение относится к торговой марке). Порошок или гранулы, получаемые в результате подобной операции, могут быть упакованы в капсулы из твердого желатина, которые затем могут быть при необходимости покрыты энтеросолюбильной оболочкой. В альтернативном варианте полученные порошок или гранулы могут быть превращены в гранулы размером примерно 1-2 мм, которые можно покрыть энтеросолюбильной оболочкой и поместить в капсулы из твердого желатина. Жидкие составы могут быть также помещены в капсулы из мягкого желатина, которые, если это необходимо и возможно, покрывают энтеросолюбильной оболочкой. Известны подходящие материалы для энтеросолюбильных оболочек, например, эту информацию можно получить из Remington's Pharmaceutical Sciences, 15th Edition, с. 1614, 1615 (1975); 2nd Edition, с. 116, 117, 371-374 (1976); и Hagers Handbuch der Plasmazeutischen Praxie, 4th Edition, Volume 7a (Springer Verlag 1971), с. 739-742 и 776-778.
Таким образом, составы, соответствующие настоящему изобретению, могут вводиться перорально и с использованием широкого выбора выпускных форм, например в виде жидкости, в мягких желатиновых капсулах, в твердых желатиновых капсулах, в прессованных таблетках, которые могут быть покрыты энтеросолюбильными оболочками, и других пероральных форм. Далее, высокий уровень содержания в плазме, как и высокий уровень на рецепторах предполагаемой мишени свидетельствуют о том, что биологически активные вещества, вводимые средствами настоящего изобретения, обладают высокой биологической доступностью, а активное вещество биоактивно.
При ректальном введении жидкие или твердые составы могут вводиться в виде клизмы или суппозитория. Основой для суппозитория может служить масло какао или другой подходящий материал.
Согласно еще одному аспекту изобретения обеспечивается способ лечения людей или животных, состоящий в пероральном или ректальном введении состава, соответствующего первому пункту изобретения. В частности, изобретение распространяется на лечение диабета, состоящее в пероральном или ректальном введении состава, соответствующего настоящему изобретению, в котором биологически активное вещество - это инсулин.
Изобретение также распространяется на использование ингредиентов составов, соответствующих первому пункту изобретения, для получения перорально или ректально вводимых составов для лечения или профилактики заболеваний, которые лечатся или регулируются биологически активным веществом.
В частности, инсулин может быть использован в приготовлении состава, предназначаемого для лечения или контроля диабета. Кальцитонин лосося может быть использован для лечения случаев, связанных с костным обменом (например, случая костного заболевания Педжета - деформирующей остеодистрофии), острой гиперкальциеэмии, связанной со злокачественными заболеваниями и остеопорозом. Эритропоэтин может быть использован при лечении анемии, вызванной хроническим применением либо экстракорпорально приборов почечного диализа, либо противораковой химиотерапии, или других случаев. Свиной соматотропин можно вводить свиньям в целях сокращения времени, необходимого для выращивания свиней и возможно для снижения толщины слоя сала. Гормон роста человека может быть использован при лечении детей с заторможенным ростом.
Далее изобретение будет проиллюстрировано рядом неограничивающих его примеров.
Пример 1. В данном примере используют в качестве биологически активного вещества инсулин и получают состав, содержащий фосфолипо-инсулиновый комплекс.
При комнатной температуре бычий инсулин (от 2 до 20 мг кристаллического или порошкообразного бычьего инсулина с активностью примерно 22-26 IU на мг) добавляют к 1-3 г лецитина из соевых бобов, лецитина из яичного желтка, L-α-фосфатидиновой кислоты бимиристоила (натриевая соль) и/или моногидрату 1,2-димиристоил-sn-глице рин-3-фосфохолина и растворяют или в 0,9%-ном растворе бензилового спирта или в 0,9%-ном растворе хлорида натрия (150 мл) в присутствии 400 мг апротинина (3000000 ед. инактиватора Калликреина), доводят до pH 2,3 раствором лимонной кислоты при комнатной температуре. К полученному фосфолипид-инсулиновому комплексу добавляют неионное поверхностно-активное вещество, предпочтительно полиокси-40-стеарат (4 г).
При осторожном, но постоянном перемешивании при комнатной температуре полученный выше фосфолипо-инсулиновый раствор медленно добавляют в раствор с масляной фазой, содержащий прехиломикроновые комплексы (представляют комплексы фосфатидилхолина, моно-, ди- и/или триглицерида, холестерина и других), неионное поверхностно-активное вещество со значением HLB, меньшим 4, и один или более антиоксидантов.
Приведенная выше эмульсия вода-в масле пропускается через микрофлюидизатор при температуре 5-10oC при 100000 PSI (7031 кг/см2) или более в два последовательных приема. Таким образом получают микроэмульсионную (вода-в масле) форму фосфолипо-инсулинового комплекса. Каждые 400 мл этой микроэмульсии содержат следующие ингредиенты:
Химический состав - На 400 мл микроэмульсии
Бычий инсулин - 87,00 IU
Димиристоил-глицерин-фосфохолин - 1,0 г
Апротинин - 2000000 кIU
Полиокси-40-стерат - 2,9 г
Холестерин - 11,6 г
Моноолеат глицерина - 10,6 г
Олеат - 92,5 г
Полисорбат 80 - 6,8 г
d-альфа-токоферол - 1,2 г
Лимонная кислота - 0,9 г
Физиологический солевой раствор - до 400 мл
Пример 2. В качестве биологически активного соединения в этом примере используют эритропоэтин (ЭПО), и фосфолипо-эритропоэтиновый комплекс получают в соответствии с описанием примера 1. Также половину ЭПО непосредственно добавляют в водную фазу двухфазной (микроэмульсия) системы. Так, в этом примере, белковоподобное соединение не только связывают с фосфолипидом, но и добавляют непосредственно в водную фазу микроэмульсионной системы.
Эритропоэтин получен от фирмы Chugai Pharmaceutical Company, Ltd. of Tokyo, Japan ( Лот N R 9H05). В одной аликвоте содержалась концентрация белка, равная 0,936, измерение проводилось методом аминокислотного анализа, а определенная методом PR-ВДЖХ она равнялась 1,018 мг, и специфическая активность in vivo составила 180,000 IU в фосфатном буфере с pH 7,2. При комнатной температуре ЭПО-аликвоту разделили на две половины. К первой половине ЭПО-аликвоты добавили от 0,004 до 0,07 мг 1,2-димири стоил-sn-глицерин-фосфохолин моногидрата на 1000 IU ЭПО, и полученную смесь растворили в 50 мл 0,9%-ного физиологического солевого раствора, pH раствора довели до 7,3, добавляя 0,1 М фосфатный буфер (pH 7,8) в присутствии 3000-4000 IU апротинина (на 1000-15000 IU ЭПО). Оставшуюся половину ЭПО растворили в 100 мл физиологического солевого раствора, довели pH раствора до 7,3, как указывалось выше, и затем апротинин (как указано выше) и неионное поверхностно-активное вещество со значением HLB 7, такое как полиокси-40-стеарат, добавили при концентрациях от 0,0044 до 0,00044 мг на 1000 IU ЭПО. В вышеприведенном растворе могут быть растворены при комнатной температуре стабилизаторы эмульсии и вещества, повышающие вязкость, такие как гидроксипропилцеллюлоза-SL.
При осторожном, но постоянном перемешивании при комнатной температуре фосфолипо-ЭПО-комплекс и ЭПО-содержащий "водно-фазный" раствор медленно добавляют в маслянно-фазный раствор, содержащий холестерин, лецитин, моноолеат глицерина (неионное поверхностно-активное вещество со значением HLB, меньшим 4) и антиоксиданты.
Приведенную выше вода-в-масле эмульсию пропустили один раз через эмульгатор-микрофлюидизатор, охлаждаемый льдом. На каждую 1000 IU ЭПО полученная микроэмульсия вода-в масле ЭПО может содержать следующие ингредиенты:
Химический состав - мг/1000 IU ЭПО
ЭПО (Лот N R 9H05) - 1000 IU
1,2-димиристоил-sn-глице рин-фосфохолин моногидрат - 0,0056
Гидроксипропилцеллюлоза-SL - 0,880
Полиокси-40-стеарат - 0,440
Апротинин - 3,000 кIU
Холестерин - 1,880
Лецитин из яичного желтка - 3,800
Моноолеат глицерина - 1,680
d-α-токоферол - 1,180
Олеиновая кислота - 15,000
Твин-80 - 1,06
Этанол - 7,00*
* (в основном должен быть испарен).
Пример З. В этом примере получены фосфолипо-соматотропиновый комплекс (pST) и состав, соответствующий настоящему изобретению. Кристаллический порошок (pST Лот N 7368C-250) свиного соматотропина (pST) получен от American Cyanamid Company, Agricultural Research Division, of Princeton, New Iersey, USA. При комнатной температуре pST добавили к 0,9%-ному солевому раствору, содержащему фосфолипиды, такие как моногидрат 1,2-димиристоил-sn-глицерин-фосфохолина, L-альфа-фосфатидиновой кислоты бимиристоил (натриевая соль), лецитин яичного желтка и/или лецитин соевых бобов. Также добавили неионное поверхностно-активное вещество со значением HLB выше 7 в присутствии апротинина, ингибитора трипсина в физиологическом растворе.
pH водно-фазного раствора доводят, если в этом есть необходимость, до 7,2, добавляя NaCl/10 мМ натрийфосфатный буфер. При осторожном, но постоянном перемешивании при комнатной температуре вышеприведенный фосфолипо - pST раствор медленно добавляют в масляно-фазный раствор, содержащий смесь лецитин-холестерин-моноолеатглицерина; полученную смесь дважды на льду пропускают через эмульгатор-микрофлюидизатор. Каждый мл pST микроэмульсии содержит следующие ингредиенты:
Химический состав - мг/мл pST эмульсии
Лецитин - 12,5
Холестерин - 30,48
Соевый лецитин - 187,43
Моногидрат 1,2-димири стоил-sn-глицерин-3-фосфохолина - 1,91
Натриевая соль L-альфа-фосфатидиновой кислоты бимиристоила - 0,095
Олеиновая кислота - 242,29
d-альфа-токоферол - 7,62
Глицерин-1-моноолеат - 27,81
Гидроксипропилцеллюлоза-L - 14,48
Полиокси-40-стеарат - 7,62
Апротинин - 2850 кIU
Твин-80 - 17,91
pH доводят до 7,2 натрийфосфатным буфером (10 мМ).
Пример 4. Рекомбинантный гормон роста человека (r-hGH; серия N 9-08 P-508-2, 16 августа 1989) получили от Smith Klein Beecham of Philadelphia, r-hGH включили в фармацевтический состав, как и в других примерах. В частности, r-hGH связали и растворили в 0,9%-ном растворе NaCl, производное фосфолипида в воде добавили для получения водорастворимого фосфолипо-r-hGH-комплекса. Последний затем медленно добавляли в масляную фазу, как и ранее.
Масляная фаза состояла из лецитина яичного желтка, холестерина, глицерин-1-моноолеата, d-альфа-токоферола, раствора антиоксидантов и Твина-80. Эту эмульсию "вода-в масле" пропустили один раз через гомогенизатор-микрофлюидизатор на льду. Каждый мл r-hGH микроэмульсии содержит следующие ингредиенты:
Химический состав - мг/мд r-hGH эмульсии
Лецитин яичного желтка - 37,50
L-альфа-фосфатидиновой кислоты бимиристоил - 0,095
Моногидрат 1,2-димири стоил-sn-глицерин-3-фосфолина - 1,91
Холестерин - 30,48
Моноолеат глицерина - 27,81
Олеиновая кислота - 242,30
Твин-80 - 17,91
Вода - 119,05
pH довели до 7,2, добавляя 10 мМ натрийфосфатный буфер. (Отметим, что в этом примере ингибитор трипсина апротинин не был добавлен).
Пример 5. В этом примере соединили с лецитином яичного желтка кальцитонин лосося (получен Rorer Central Research of Hirsham, Pennsylvania, USA; NPD# 8906046, NPP#211) в присутствии апротинина в 0,9%-ном солевом растворе, pH довели до 2, добавляя лимонную кислоту и аскорбиновую кислоту. Таким путем был сформирован "водорастворимый" фосфолипид-sCT-апротининовый комплекс и его перевели в микроэмульсию вода-в масле, как описано в примере 1. Каждая порция состава, в которой было 100 IU sCT, содержала следующие ингредиенты:
Химический состав - мг/100 IU sCT
Лецитин яичного желтка - 13,016
Апротинин - 1500 IU
sCT - 100 IU
Полиокси-40-стеарат - 2,646
Гидроксипропилцеллюлоза-L - 5,026
Бензоат натрия - 1,587
Лимонная кислота - 1,720
Аскорбиновая кислота - 1,244
Холестерин - 10,582
d-альфа-токоферол - 0,265
Моноолеат глицерина - 9,418
Олеиновая кислота - 84,127
Твин-80 - 6,217
Пропилпарабан - 0,529
Метилпарабан - 1,852
Масло семян кунжута (химически чистое) - 2,646
Антиоксиданты - 1,111
Этанол* - 41,336
Деионизованная вода - 66,137
(* большая часть этанола испарена).
Пример 6. Порошковую или гранулированную форму препарата примера 1 можно получить путем напыления вышеупоминаемой микроэмульсии на фармакологически инертный носитель, такой как карбоксиметилцеллюлоза-Ca, желатин, гидроксипропилцеллюлоза-L, альгиновая кислота или смесь этих веществ. Порошковый (или гранулированный) инсулинсодержащий препарат пакуют в капсулы из твердого желатина размером N 1. Получают следующий состав:
Химические составляющие - мг на капсулу (приблизительно)
Бычий инсулин - 4,5-6,0 IU
Димиристоил-глицерин-фосфохолин* - 2,2
Апротинин - 0,14 кIU
Полиокси-40-стеарат - 1,2
Холестерин - 5,2
Олеиновая кислота - 42,0
Моноолеат глицерина - 4,8
Твин-80 - 3,1
Витамин Е - 0,55
Карбоксиметилцеллюлоза-Са - 90,9+
Альгиновая кислота - 45,5+
Желатин - 22,7+
Гидроксипропилцеллюлоза-L - 25,2+
Каждая капсула размером N 1 из твердого желатина содержит до 250 мг.
* 1,2-димиристоил-sn-глице рин-3-фосфохолина моногидрат.
+ Эти вещества практически не поглощаются в человеческом организме, и поэтому представляют фармакологически инертные, неактивные вещества.
Биологический пример A.
Клиническое исследование провели на группе наблюдаемых диабетиков, используя пероральную систему доставки лекарственного препарата, полученного в примере 1 (лецитин - бычий инсулин -апротинин). Состав примера 1, как было установлено, оказался эффективен как препарат, снижающий системный сахар крови (см. табл. 5).
Следует отметить, что, как правило, гипогликемическое действие состава лецитин - бычий инсулин после его перорального приема наступало достаточно быстро - через 30-90 мин, содержание сахара в крови слегка поднималось через 120 мин или около после перорального приема инсулина в таком составе и затем снова снижалось через 3 ч или более, таким образом, этот инсулиновый состав для перорального приема обладает двухфазным действием. При связывании липофильных участков бычьего инсулина фосфолипидами, такими как лецитин, образуется фосфолипо-инсулиновый комплекс (который затем становится гидрофильным соединением подобно другим липопротеинам в организме человека). Часть этого фосфолипо-инсулинового комплекса, по-видимому, сразу же вызывает гипогликемическую реакцию при непосредственном поглощении из гастрокишечной системы человека, оставшаяся часть комплекса (т.е. та часть, которая не провзаимодействовала с периферическими инсулиновыми рецепторами), по-видимому, направляется в печень, хранится временно в печени и затем выделяется в циркулирующую кровь, вызывая вторичную реакцию инсулиновых рецепторов.
Биологический пример B.
В этом исследовании приняли участие восемь здоровых, нормальных мужчин-добровольцев. После ночного голодания в первый день обследования субъекты получили следующие назначения лекарственного препарата:
Субъект - Исследуемые дозы ЭПО-препарата (IU)
A - ЭПО внутривенное вливание 10 IU/кг
B - ODD - ЭПО, per os 20 IU/кг
С - Плацебо, per os 0 IU/кг
D - ЭПО внутривенное вливание 10 IU/кг
E - ODD - ЭПО, per os 15 IU/кг
F - Плацебо, per os 0 IU/кг
G - ODDS - ЭПО, per os 20 IU/кг
H - ODDS - ЭПО, per os 15 IU/кг
Образцы крови отбирались через 0, 0,5, 1, 2, 3, 4, 6, 10 и 14 ч после введения препарата. Определяли число ретикулоцитов (%) и содержание ЭПО в плазме методом радиоиммунного анализа. На второй день и на третий день каждый субъект получил прописанный закодированный препарат в 8 утра, 14 и 22, и после ночного голодания на четвертый день каждый субъект был обследован снова после принятого курса исследуемых препаратов так же, как и выше.
Число ретикулоцитов, в особенности на четвертый день, возросло после приема ЭПО перорально 20 IU/кг и после приема 15 IU/кг так же, как и в случае внутривенного вливания ЭПО в количестве 10 IU/кг. Число ретикулоцитов постепенно падало в группе, получавшей плацебо.
В первый день измеренное методом радиоиммунного анализа содержание ЭПО в плазме крови заметно возросло в обоих случаях 20 IU/кг и 15 IU/кг ЭПО перорально так же, как и после внутривенного вливания 10 IU/кг ЭПО. В группе, принимавшей плацебо, снова наблюдали постепенное снижение содержания ЭПО в плазме крови в исследуемый период. Доставка ЭПО составами примера 2 при пероральном приеме была эффективна и биодоступна человеческому организму.
Биологический пример C.
Состав, полученный в примере 3 (комплекс димиристоилглицерино фосфохолин-фосфатидиновый бимиристоил - pSCT), интрадуоденально влили группе свиней, находящихся в сознании. Провели серийные анализы крови на содержание сахара и радиоиммунный анализ плазмы на содержание pST до и после введения состава примера 3. Нижеприведенные результаты указывают на соотносительный рост сахара в крови и увеличение измеренного методом радиоиммунного анализа содержания pST.
Содержание глюкозы в крови и pST в плазме (RIA) после интрадуоденального вливания перорального pST свиньям приведено в табл. 6.
Содержание сахара в крови и pST в плазме после интрадуоденального вливания перорального pST приведено в табл. 7.
Биологический пример D.
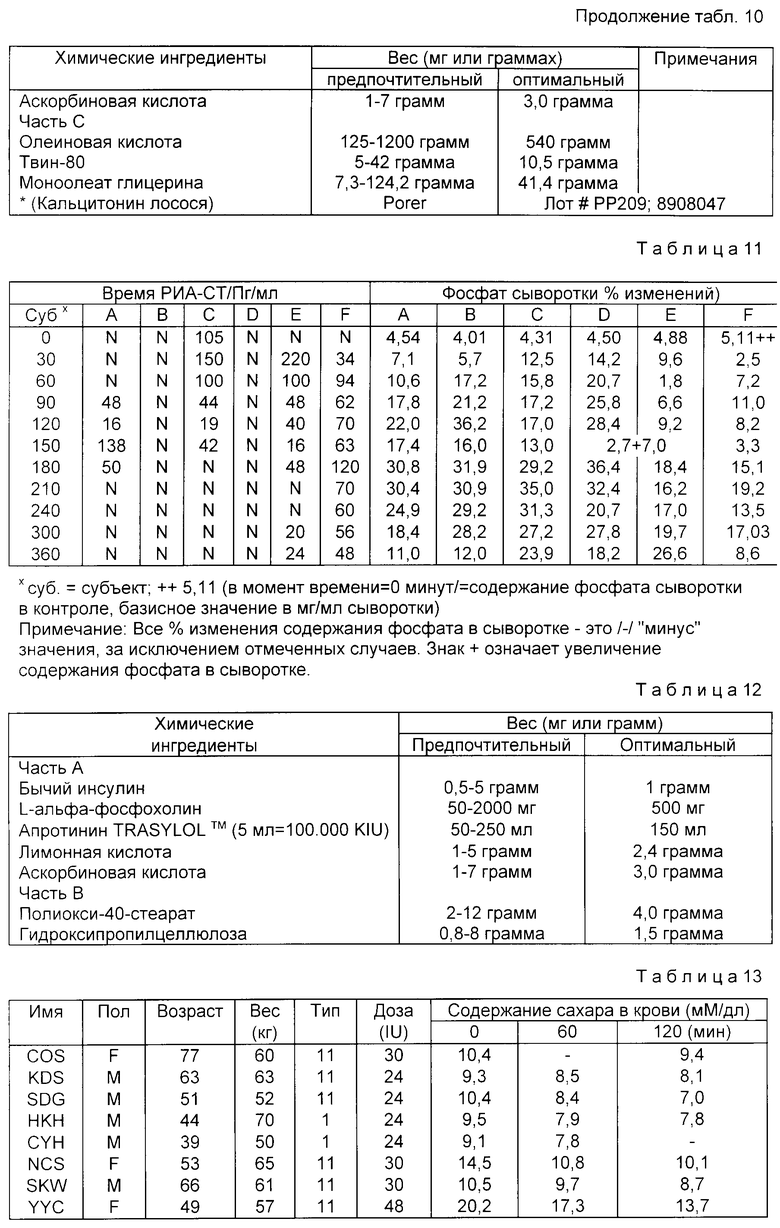
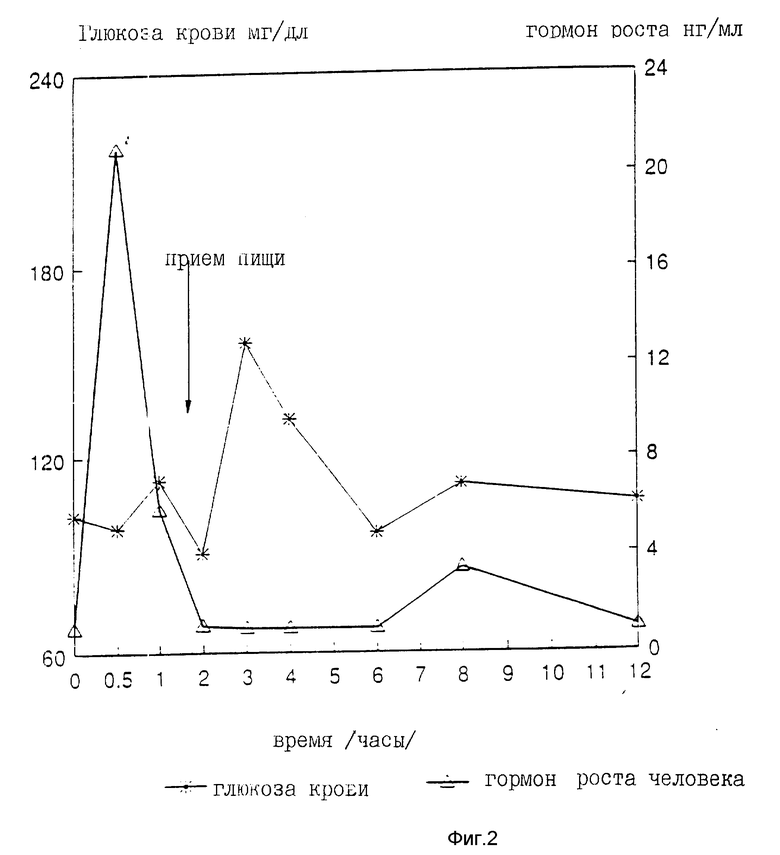
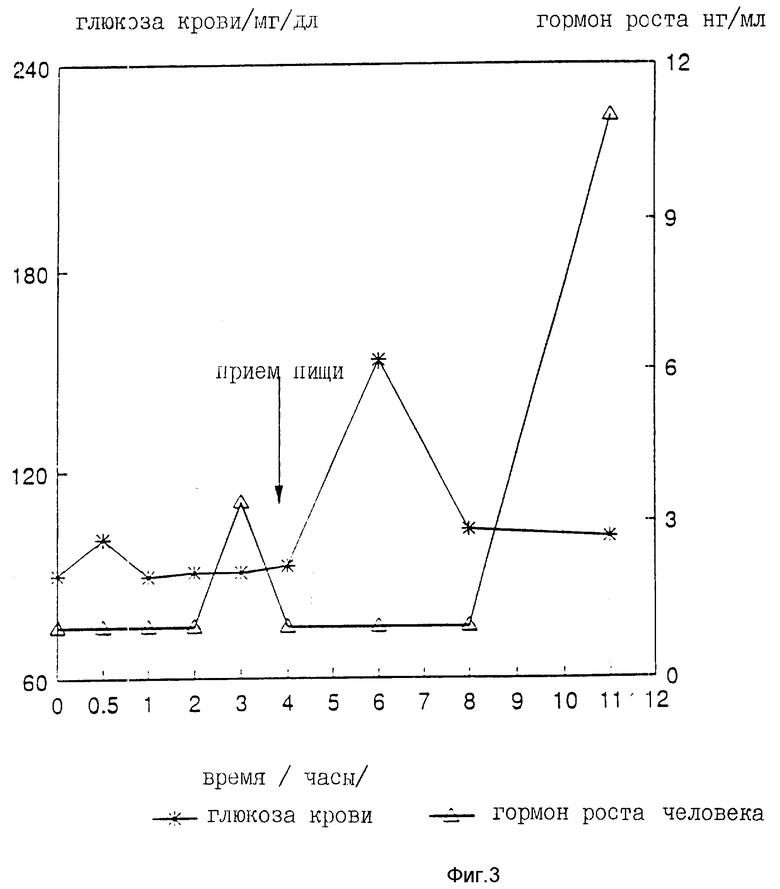
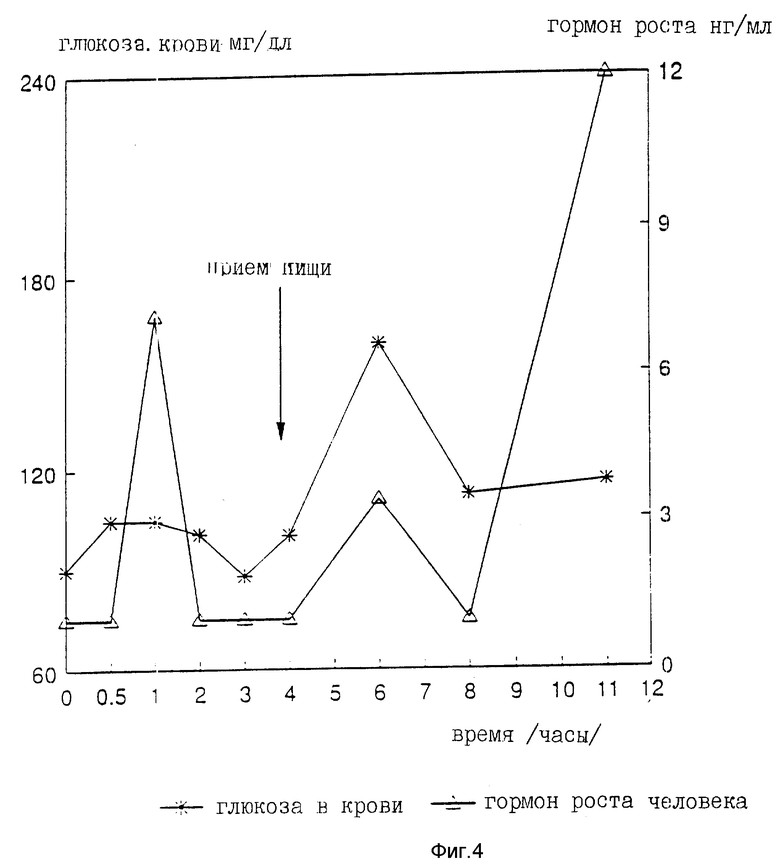
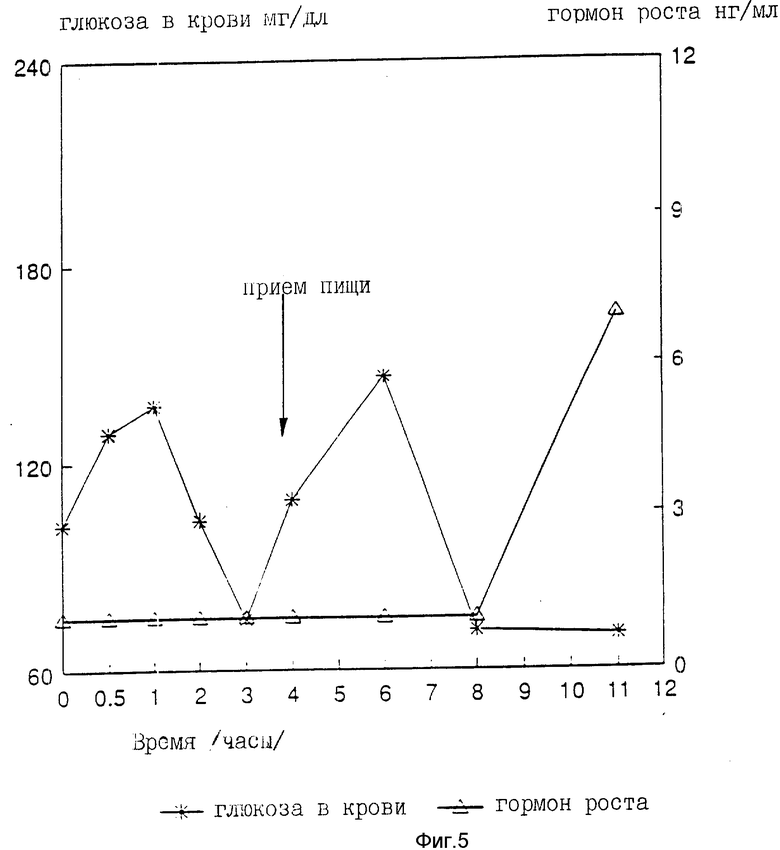
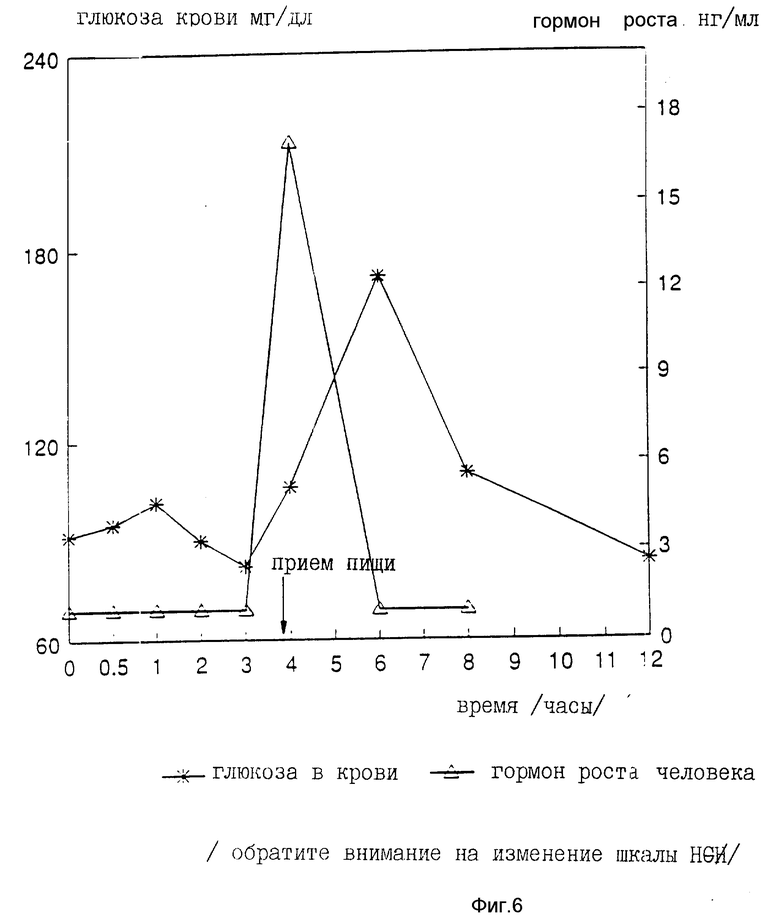
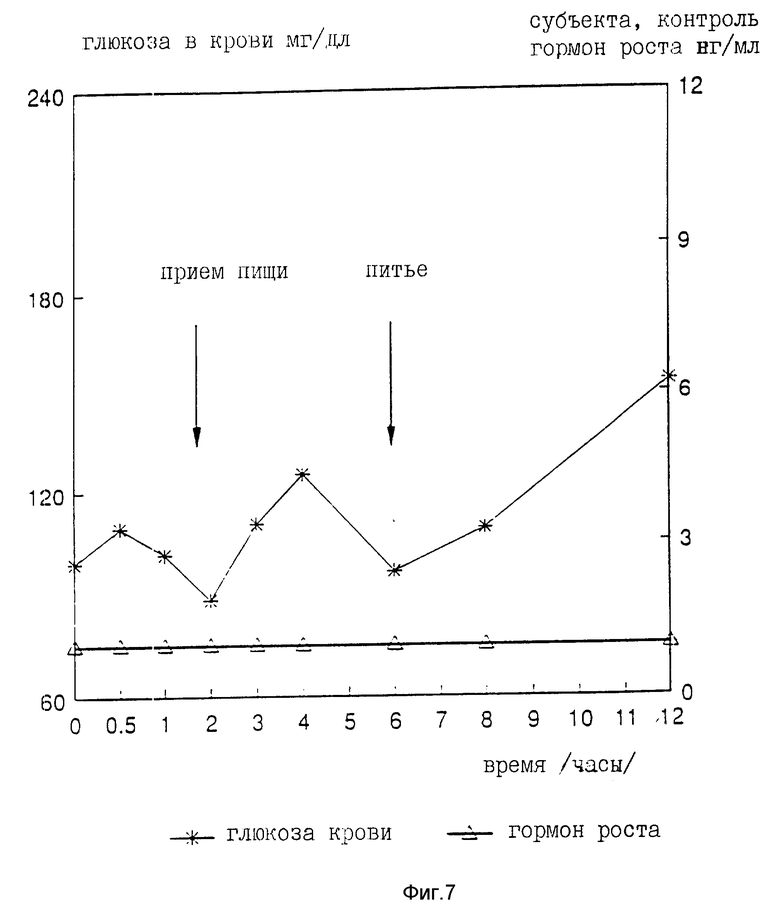
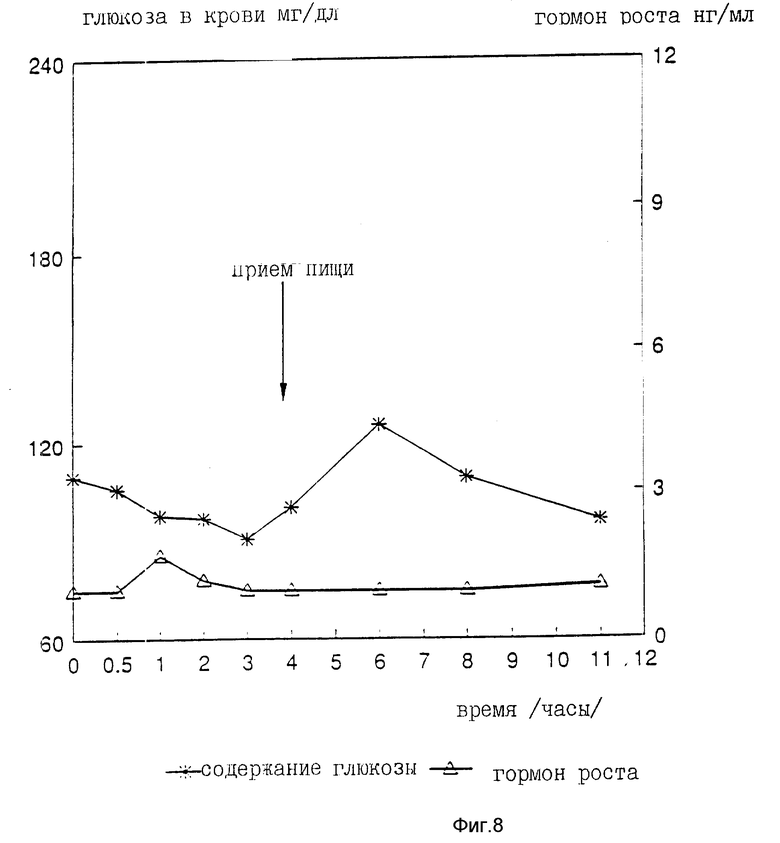
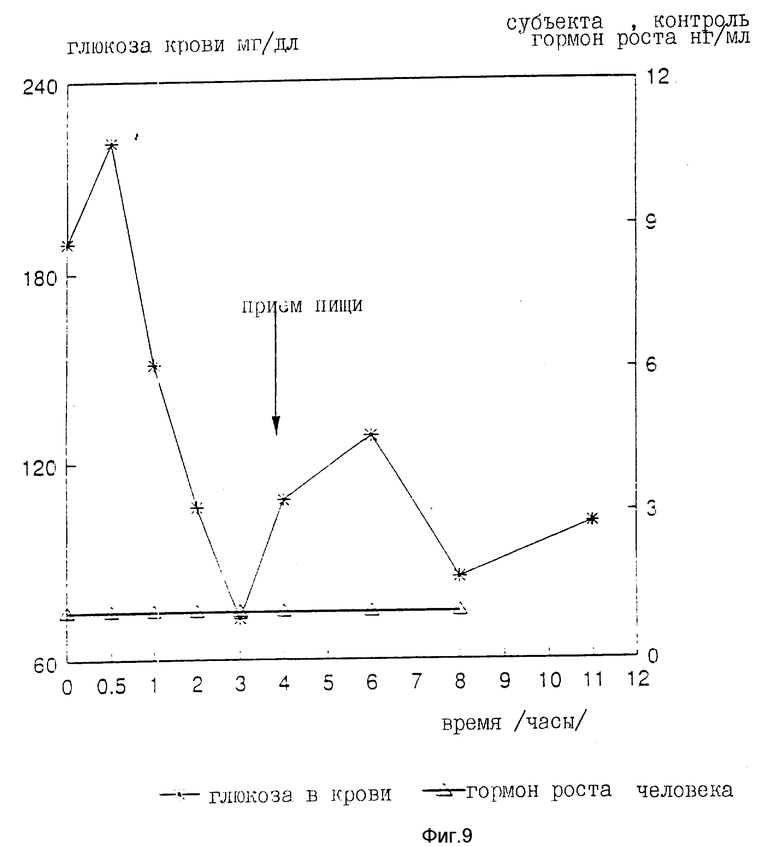
На девяти здоровых молодых мужчинах-добровольцах исследовали клиническую биоактивность (измеряемую по изменению содержания в крови сахара, индуцированному пероральным приемом r-hGH) состава, полученного в примере 4 (включающего комплекс фосфолипид r-гормон роста человека).
Топография исследуемых субъектов (все субъекты - мужчины) приведена в табл. 8.
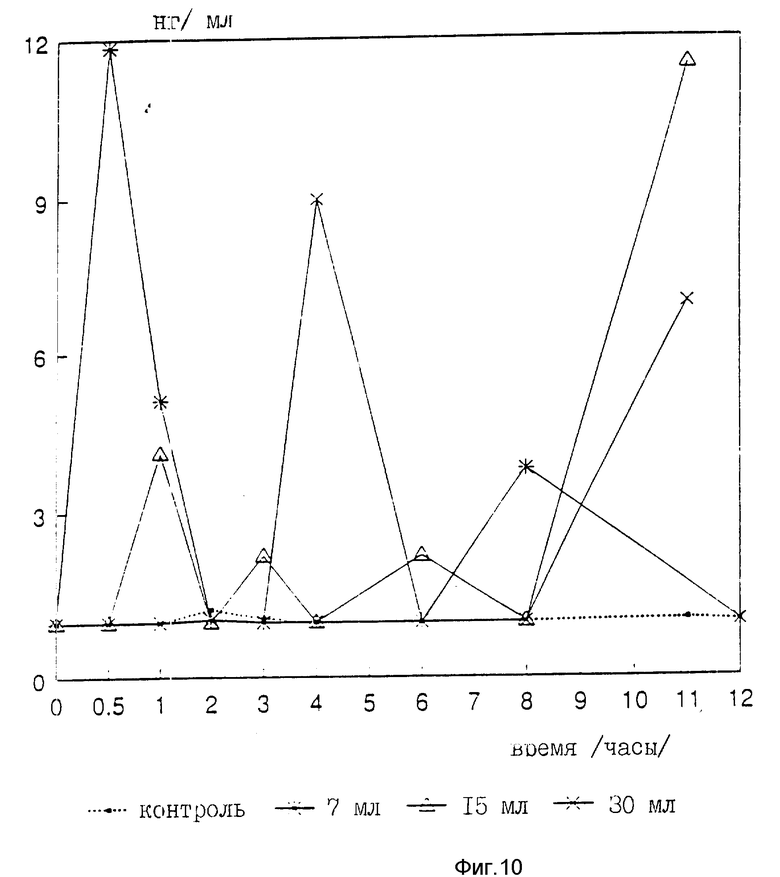
Были измерены изменения содержания в крови сахара и hGH, индуцированные пероральным приемом r-hGH этими девятью субъектами при дозах 0, 7, 15 и 30 мл (что соответствует 0, 7, 15 и 30 мг перорального r-hGH состава). Измерения проводились с использованием коммерческого набора обработанной ЭДТА плазмы. Результаты, полученные с каждым субъектом, проиллюстрированы в индивидуальном порядке на фиг. 1-9. Среднее значение дозаотвечаемость на пероральный прием r-hGH содержания hGH в плазме, измеренного методом радиоиммунного анализа, показана на фиг. 10. Наблюдали "диабетогенное" действие и повышение измеряемого методом радиоиммунного анализа содержания hGH в плазме у всех добровольцев, получавших активный препарат, как это уже наблюдалось при введении других препаратов, соответствующих настоящему изобретению, таких как пероральная форма pST у свиней и пероральная форма инсулина у диабетиков.
Итак, снова, по-видимому, r-hGH, вводимый перорально (в виде фосфолипо-r-hGH-комплекса), абсорбируется и затем индуцирует биологическое воздействие hGH у людей. Он становится биодоступным в циркулирующей крови обычно через 0,5-4 ч после перорального приема, полагают, что hGH проходит через каналы в печень, откуда он выделяется и становится доступным в циркулирующей крови снова после 8-12 (или более) ч после перорального приема препарата. Это двухфазное действие hGН или точнее двухфазный эффект в биодоступности hGН (дополнительно в биоактивности) несомненно зависит от вводимой дозы: при относительно низкой дозировке препарата пероральный hGН был биологически доступен в течение 0,5-2 ч после перорального приема и снова биодоступен через 8 ч или около, в то время как при более высоких дозах (30 мг hGH) начальный пик биодоступности имел место примерно через 4 ч, а второй пик наступал через 11 ч после перорального приема.
Рекомендованная доза составляет 15 мг на человека. В нашем исследовании 7 мг также оказались весьма эффективной дозой. 1-10 мг на человека, в особенности 3-5 мг на человека, может быть подходящей терапевтической дозой.
Биологический пример E.
Состав примера 5 (перорально доставляемая форма кальцитонина лосося: ODDS - sCT) был изучен на группе из пяти молодых мужчин-добровольцев. Демографическая картина этих добровольцев приведена в табл. 9.
Вкратце - каждый субъект голодал в течение ночи и либо капсулы ODDS - sCT вводили per os либо CALSY NARTM (препарат кальцитонина лосося для инъекций) вводили подкожно в 6 утра в день исследования. Образцы венозной крови отбирали через постоянный катетер, установленный в предплечной вене, через 0, 60, 90, 120, 150, 180, 210, 240, 300 и 360 мин после введения исследуемого медикамента. Сразу же после взятия крови на анализ определяли содержание фосфата в сыворотке, содержание кальцитонина лосося в плазме определяли методом радиоиммунного анализа на обработанной ЭДТА плазме.
У всех субъектов наблюдали заметное снижение содержания фосфата в сыворотке, и такие изменения в содержании фосфата в сыворотке после перорального приема состава примера 5 (либо 300 IU или 600 IU ODDS - sCT капсулы) были аналогичны явлениям (снижение содержания фосфата в сыворотке), наблюдаемым у субъектов после подкожной инъекции кальцитонина. Таким образом, установлено, что действие перорально введенного ODDS - sCT в количестве 300 IU и 600 IU на содержание фосфата в сыворотке в основном аналогично действию 100 IU кальцитонина лосося, вводимого в виде инъекции sCT. Следовательно, составы, соответствующие настоящему изобретению, также высокоэффективны.
Пример 7. Комплекс фосфолипо-кальцитонин лосося получили путем смешения 1,2-димиристоил-sn-глице рин-3-фосфатидиновой кислоты моногидрата и L-α-фосфатидилхолин бимиристоила (натриевая соль) в присутствии апротинина в 0,9%-ном изотоническом солевом растворе, реакцию проводили при комнатной температуре в течение 30 мин. Фосфолипиды, которые, как известно, участвуют в L-α-фосфатидилглицериновом пути синтеза фосфатидилхолина в эпителиальном слое тонкого кишечника, по-видимому, нековалентно связываются с липофильными участками кальцитонина лосося in vivo.
Упомянутый выше комплекс фосфолипо-кальцитонин лосося суспендировали и смешали с раствором, содержащим поверхностно-активное вещество со значением HLB, равным 14 или выше, (полиокси-40-стеарат) в присутствии вещества, повышающего вязкость, (загустителя) и стабилизатора для суспендированного фосфолипо-кальцитонина (например, менее 5%, предпочтительно менее 2%, гидроксипропилцеллюлозы). pH довели до примерно 2, добавляя концентрированные растворы лимонной и аскорбиновой кислот (хотя можно использовать для регулирования pH и другие кислотные добавки).
Полученный выше раствор суспендировали в три или четыре объема олеиновой кислоты (или любой другой жирной кислоты, содержащей в углеродной цепочке 16 или более атомов углерода) в присутствии поверхностно-активного вещества со значением HLB менее 4 или равным 4. Жирные кислоты с числом атомов углерода, равным 16 или более, могут действовать как "экспандер", а также как усилители трансмембранной абсорбции фосфолипо-кальцитонина и/или энтеросолюбильные оболочки для фосфолипо-кальцитонина. Однако в абсолютном плане эта комбинация олеиновая кислота/поверхностно-активное вещество не существенна.
В табл. 10 приведен фактический химический состав препарата фосфолипо-кальцитонин лосося, соответствующего данному примеру.
Процедура приготовления препарата заключалась в следующем.
Компоненты части A тщательно перемешали и оставили стоять при комнатной температуре на 30 или более мин. Смешали и подготовили часть B, а затем часть A суспендировали в части B при осторожном перемешивании при комнатной температуре. pH состава довели до 2, добавляя лимонную и аскорбиновую кислоты.
Часть C, масляный раствор приготовили путем смешения. При осторожном перемешивании подготовленные часть A и часть B вылили в часть C.
Смешанный таким образом раствор может быть напылен на примерно такую же по массе порошкообразную компоненту, представляющую либо карбоксиметилцеллюлозу-Ca и альгиновую кислоту, либо альгиновую кислоту и желатин. Покрытый высушенный порошок можно упаковать в капсулы из твердого желатина, которые можно назначать больным и здоровым субъектам для перорального приема. В альтернативном варианте жидкая форма может приниматься перорально сама по себе или может быть предварительно упакована в капсулы из мягкого желатина.
Биологический пример E.
В этом тестировании принимали участие шесть молодых мужчин-добровольцев. После ночного голодания в 5.30 утра четыре субъекта приняли перорально препарат лосося, приготовленный в примере 7: два субъекта получили капсулу, содержащую 300 IU кальцитонина лосося из примера 7, а два других субъекта получили 600 IU перорального кальцитонина лосося из примера 7. Два субъекта получили инъекцию CALCYNAR - торговая марка кальцитонина лосося для инъекций, 100 IU подкожно. Фосфат сыворотки и содержание кальцитонина в плазме, определяемое методом радиоиммунного анализа, определяли за 30 мин до ввода препарата, в момент принятия препарата и через 30, 60, 90, 120, 150, 180, 210, 240 и 300 мин после введения препарата.
Изменения (% к контролю) содержания фосфата в сыворотке и кальцитонина лосося sCT, измеряемого методом РИА (г/мл) в плазме у людей, после перорального приема кальцитонина лосося (пример 7) и после подкожного введения CALCYNAR приведены в табл. 11.
Код для дозировок изучаемого препарата:
Субъект A = 600 IU перорально sCT из примера 1, per os,
Субъект B = 300 IU, per os,
Субъект C = 100 IU CALCYNAR, подкожная инъекция,
Субъект D = 600 IU, per os,
Субъект E = 100 IU CALCYNAR, подкожная инъекция,
Субъект F = 300 IU, per os.
Препарат кальцитонина лосося, соответствующий примеру 7,принимаемый перорально группой здоровых мужчин, был биологически активен в отношении снижения сывороточного фосфата в такой же степени или большей, как и вводимый подкожно кальцитонин лосося. Однако содержание sCT, измеряемое методом РИА, не всегда определялось согласно настоящей методологии после перорального приема как 600 IU, так и 300 IU кальцитонина лосося, полученного в примере 7. При связывании sCT с фосфолипидами могут происходить некоторые антигенные изменения, в связи с этим sCT не всегда может быть определен с помощью существующих антител, специфических для sCT, используемых в радиоиммунном анализе.
Пример 8. Препарат инсулина (бычьего инсулина) для перорального приема приготовили смешением L-α-фосфохолина (предшественник лецитина) с инсулином в присутствии подходящего поверхностно-активного вещества и апротинина в изобутиловом спирте (0,9%). Полученный таким образом нековалентно связанный комплекс суспендировали в воде и МСТ (триглицериды средней длины) масле для последующего приема группой диабетиков.
В табл. 12 приведен химический состав препарата.
Состав был получен следующим образом.
Часть A при комнатной температуре тщательно смешали при осторожном перемешивании. Часть B получили растворением полиокси-40-стеарата и гидроксипропилцеллюлозы полностью в 100 мл деионизованной воды. Затем часть A добавили к части B при осторожном перемешивании, смешивая при комнатной температуре.
Этот состав инсулина можно давать диабетикам для перорального приема как таковой или его можно суспендировать в 10-30 мл МСТ (триглицериды средней длины) масле, которое может действовать как энтеросолюбильное покрытие и экспандер объема, промотируя желудочное опорожнение состава через привратник (пилорус) в двенадцатиперстную-тощую-подвздошную кишки.
Биологический пример G.
В этом тестировании приняли участие десять диабетиков. Натощак рано утром в день исследования каждый диабетик принял перорально препарат инсулина из примера 8; содержание сахара в крови определяли в течение 2 ч (см. табл. 13).
Препарат инсулина примера 8 при пероральном приеме был эффективен при контроле гипергликемии у обоих типов диабетиков.
Использование: в медицине, для увеличения биологической доступности активных веществ. Сущность изобретения: фармацевтический состав содержит гидрофильную и липофильную фазы. Гидрофильная фаза содержит воду и биологически активное вещество, связанное в нековалентный комплекс с летицином или соединением, способным служить предшественником лецитина in vivo. Липофильная фаза содержит соединение, образующее матрицу хиломикрона, лецитин или фосфолипид, и липофильное поверхностно-активное вещество. Объемное соотношение гидрофильной и липофильной фаз находится в пределах от 0,2:1 до 5:1. Фармацевтический состав является нелипосомным. Биологически активное вещество является белковым веществом, выбранным из группы, включающей инсулин, эритропоэтин, соматотропин свиньи, гормон роста человека и кальцитонин лосося. Способ получения фармацевтического состава включает приготовление гидрофильной и липофильной фаз, которые тщательно смешивают с получением нелипосомного состава. 3 с. и 22 з. п. ф-лы, 13 табл., 10 ил.
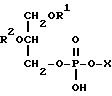
где R1 и R2 независимо друг от друга ацильная группа, содержащая 14 26 атомов углерода, каждая из которых может являться моно- или полиненасыщенной, а Х атом водорода или фосфолипидная головная группа.
| WO, заявка, 88/06881, кл | |||
| Устройство для сортировки каменного угля | 1921 |
|
SU61A1 |
